
CIDM-PH ANNUAL REPORT - WSLHD
76
2014 Centre for Infectious Diseases and Microbiology – Public Health Westmead Hospital, Sydney CIDM-PH ANNUAL REPORT
Transcript of CIDM-PH ANNUAL REPORT - WSLHD

2014
Centre for Infectious Diseases and Microbiology – Public Health Westmead Hospital, Sydney
CIDM-PH ANNUAL REPORT

1 CIDM-PH ANNUAL REPORT 2014
Centre for Infectious Diseases & Microbiology – Public Health (CIDM-PH)
Level 3, Institute of Clinical Pathology & Medical Research
Westmead Hospital. PO Box 533, Wentworthville
New South Wales, Australia 2145
Ph: (02) 9845 9870
Fax: (02) 9893 8659
Email: [email protected]
Website: www.cidmpublichealth.org

2 CIDM-PH ANNUAL REPORT 2014
CONTENTS
C IDM- PH Overv iew
3-4
2014 Abstracts, Projects & Works in Progress
Antibiotic resistance surveillance & rapid diagnostics 5-6
Bacteriology epidemiology & disease surveillance 7-26
Fungal epidemiology & invasive infections 27-42
Medical Entomology 43-50
Viral epidemiology & disease surveillance 51-59
CIDM- PH Management St ructu re
Organizational Chart 60
Grants
Current Grants in 2014 61-64
Publ ica t ions
2014 CIDM-PH Senior Investigator Publications 65-70
Educat ion & Tra i n ing
2014 Workshops & Training 71
Newsl e t te rs & Prom ot ion
2014 CIDM-PH Newsletters & Brochure 72-74
Contacts
75

3 CIDM-PH ANNUAL REPORT 2014
CIDM-PH OVERVIEW
Review of CIDM-PH research in 2014-2015
A/Professor Vitali Sintchenko
The Centre for Infectious Diseases and Microbiology-Public Health (CIDM-PH) is an interdisciplinary research
group within the Microbiology and Infectious Diseases Departments of the Western Sydney LHD and
Pathology West-ICPMR. Our goals are to improve prevention and control of communicable diseases of public
health importance by:
developing, evaluating and implementing innovative pathogen detection and typing methods;
integrating enhanced laboratory surveillance data and bioinformatics analyses into early warning systems and laboratory networks to advance public health research;
identifying evidence-based, cost-effective strategies to translate research findings into public health and clinical practice.
Senior investigators in CIDM-PH have been successful in building the network of collaborative research and
in improving surveillance and management of diseases with epidemic potential with invaluable support from
the NSW Health Population Health and Health Services Research Support Program. The CIDM-PH Colloquium
2014 aims at (1) showcasing achievements in translational research conducted by CIDM-PH investigators,
post-doctoral researchers and students and (b) planning future research directions in accordance with
current NSW Population Health Surveillance Strategy (NSW 2011 to 2020). The CIDM-PH, in collaboration
with Marie Bashir Institute of the University of Sydney, has secured funding from the NSW Ministry of Health
for the NSW Centre for Public Health Pathogen Genomics. Figure below describes enablers of this new
initiative.

4 CIDM-PH ANNUAL REPORT 2014
CIDM-PH OVERVIEW
Collaborations between CIDM-Public Health (CIDM-PH) and the Marie Bashir
Institute for Infectious Diseases and Biosecurity (MBI)
Professor Tania Sorrell
CIDM-PH is a key collaborator of MBI, with several members, competitive grants, post-doctoral scientists and
post-graduate students in common. CIDM-PH constitutes the major centre for translation of public health
microbiology research into public health actions in MBI and in NSW. The translation of the new genomic
information into public health action has been a major initiative in 2014, with the award of a $1million grant
to the MBI-CIDM-PH collaboration by the Office of Health & Medical Research, NSW Ministry of Health.
Program in Public Health pathogen Genomics.
Pathogen genomics is being heralded as the future of public health and clinical microbiology yet much
remains to be discovered, translated, integrated, interpreted and validated before the technology is
introduced broadly into diagnostics and personalised medicine (e.g., drug choices). Not least is the major and
pressing issue of bioinformatics analysis, interpretation and sharing of the large amounts of data generated
by this technology. Public Health Microbiology is currently the area most suited to application of whole
genome sequencing (WGS). This program aims to fill the current gap in assisting translation and
implementation of research findings into public health and infection control capabilities in NSW and beyond.
The main outcomes will be (i) establishment of the Centre for Public Health Pathogen Genomics as a NSW
(and National) Health Centre of Excellence, (ii) implementation of novel genomic methods of pathogen
tracking and analysis into communicable disease control, and (iii) the sustained diffusion of innovation into
other NSW centres of translational research and public health pathology providers.

5 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS ANTIBIOTIC RESISTANCE SURVEILLANCE & RAPID DIAGNOSTICS
Rapid quantification of bacteraemia and reliable detection of abR genes with high
predictive value - new opportunities for diagnosis - a practical how-to explanation
Dr Andrew Ginn
Early appropriate antibiotic treatment reduces mortality in severe sepsis but current methods to identify
bacterial infections and associated antibiotic resistance still generally rely on bacterial culture. However,
conventional cultures, even when positive, provide no information about infection intensity and no data at
all in the critical early hours of therapy. Automated nucleic acid extraction technology and real-time PCR can
enable detection and quantifying bacterial load in blood stream infection in approximately 4 hours. Modern
diagnostics also promise rapid antibiotic resistance gene detection, but the apparent diversity of relevant
resistance genes in the Enterobacteriaceae is a problem. Local surveys and analysis of publicly available
datasets suggest that the resistance gene pool is dominated by a relatively small subset of genes, with a very
high positive predictive value for phenotype, over 96% in the case of third-generation cephalosporin
resistant Escherichia coli and Klebsiella pneumoniae. In a country like Australia, with a background
prevalence of resistance to third-generation cephalosporins of 5-10%, this equates to a negative predictive
value of >99.5% for non-susceptibility and is therefore suitable for diagnostic application.

6 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS ANTIBIOTIC RESISTANCE SURVEILLANCE & RAPID DIAGNOSTICS
Inferring antibiotic resistance data from genomic data
Dr Sally Partridge
Antibiotic resistance, especially Gram-negative multi-resistance to most (or even all) available antibiotics, is an increasing global public health problem. Whole genome sequencing is being applied to look at resistance but several factors complicate the extraction of useful information from these data. These include the sheer number of known resistance genes that can confer resistance to each antibiotic, the importance of minor variations that can change the resistance phenotype conferred, nomenclature problems and the complexity of resistance genetics. Multi-resistance in Gram-negative bacteria is often due to "mobile" resistance genes that have been transferred from the chromosome of one bacterial species by a variety of different mobile genetic elements and transferred to plasmids. These plasmids can then transfer sets of resistance genes and associated mobile elements, which tend to accrete in large complex multi-resistance regions, between bacterial cells, including those of different species. While resistance genes can be identified in contigs from short-read data, accurate annotation is needed to identify exactly which variant is present. Making the most of sequence data for epidemiological tracking and understanding co-selection and co-transfer generally also requires assembly of resistance regions and whole plasmids, but multiple repeats (usually mobile elements) confound assembly from short reads, so that long-read methods are more suited. Identifying mutations in chromosomal genes that can contribute to resistance to certain antibiotics (e.g. porin mutations in Klebsiella pneumoniae) may also be important.

7 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Effectiveness of hospital-wide MRSA infection control policies differs
by ward specialty
Sadsad R, Sintchenko V, McDonnell G, Gilbert GL.
Methicillin-resistant Staphylococcus aureus (MRSA) is a major cause of preventable nosocomial infections and is endemic in hospitals worldwide. The effectiveness of infection control policies varies significantly across hospital settings. The impact of the hospital context towards the rate of nosocomial MRSA infections and the success of infection control is understudied. We conducted a modelling study to evaluate several infection control policies in surgical, intensive care, and medical ward specialties, each with distinct ward conditions and policies, of a tertiary public hospital in Sydney, Australia. We reconfirm hand hygiene as the most successful policy and find it to be necessary for the success of other policies. Active screening for MRSA, patient isolation in single-bed rooms, and additional staffing were found to be less effective. Across these ward specialties, MRSA transmission risk varied by 13% and reductions in the prevalence and nosocomial incidence rate of MRSA due to infection control policies varied by up to 45%. Different levels of infection control were required to reduce and control nosocomial MRSA infections for each ward specialty. Infection control policies and policy targets should be specific for the ward and context of the hospital. The model we developed is generic and can be calibrated to represent different ward settings and pathogens transmitted between patients indirectly through health care workers. This can aid the timely and cost effective design of synergistic and context specific infection control policies. Published in PLoS ONE 2013;8(12):e83099.

8 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Added value of environmental sampling in nosocomial MRSA outbreak
investigations with a novel rapid high-resolution typing system
Matthew V. N. O’Sullivan, Marija Parisa, Nicole Tolhurst, Kathy Dempsey, Jo Tallon, Rachel Paton,
Fei Zhou, Vitali Sintchenko3 and Lyn Gilbert
Objective: The role of environmental sampling in investigations of nosocomial outbreaks of MRSA remains
controversial. This study investigated the utility of environmental sampling when combined with highly
discriminatory typing using a novel 19-target binary typing system in a setting of high MRSA endemicity in a
large tertiary University Hospital in Sydney, Australia. Methods: A point prevalence survey of MRSA carriage
was conducted on 3 adjacent surgical wards in response to a high rate of clinical infections. Subsequently,
selected environmental locations and staff were also screened. All MRSA isolates were typed using a 19-
target binary typing system on a multiplex PCR/reverse line blot assay platform, which is rapid, high-
throughput, inexpensive and has similar discriminatory power to pulse field gel electrophoresis. Results:
There were 76 patients in the wards at the time of the survey, of whom 10 were already known to be
infected or colonized with MRSA. Fourteen patients were found to be colonized with MRSA for the first time
during the survey. Thirty-seven returned negative screens and 15 were not screened because they refused or
were unavailable at the time. Of the 52 who were negative or not screened, 3 became colonized or infected
with MRSA during the next 3 months. Twelve of 144 (8%) environmental samples and 1 of 21 (6%) staff nasal
swabs were positive for MRSA. Seventeen binary types were found amongst the 37 isolates (27 from
patients; 10 from staff or environment) available for typing; 5 of these 17 types were represented by
multiple isolates (range 2-9) while 12 were singletons. Geographic clustering was evident within types,
indicating links between patient and environmental isolates. Conclusion: This rapid, highly discriminatory
and inexpensive binary typing system for MRSA redefines the role of environmental sampling in establishing
links between environmental contamination with MRSA and patient colonization.
Presented at the 10th International Meeting on Microbial Epidemiological Markers ( IMMEM-10), Inst itut Pasteur, Paris, France, October 2013.

9 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Fluoroquinolone resistance in non multidrug-resistant tuberculosis - a surveillance
study in New South Wales Australia and a review of global resistance
Ho J, Jelfs P, Sintchenko V.
BACKGROUND: Fluoroquinolones (FQs) are used for drug-susceptible tuberculosis (TB) in patients unable to
tolerate first-line agents. Current trials are also investigating these drugs in empiric first-line TB therapy, to
improve outcomes and allow for shortened treatment regimens. Widespread FQ use in the community has
resulted in FQ resistance in many microorganisms, including Mycobacterium tuberculosis. Despite this, FQ
drug susceptibility testing (DST) is rarely performed in non-multidrug-resistant TB (non-MDR-TB). METHODS:
We conducted a 1-year surveillance study of FQ resistance on all MTB isolates from New South Wales (NSW),
Australia. In addition, we performed a literature review of previous studies assessing FQ resistance in non-
MDR-TB to summarize the global extent of this resistance pattern. RESULTS: Two (0.6%) out of 357 MTB
isolates from NSW were found to be FQ-resistant. One isolate was an MDR strain (11% of all MDR-TB). The
other was isoniazid-monoresistant (0.3% of all non-MDR-TB). Eleven studies from 10 countries had
performed FQ resistance surveillance on non-MDR-TB. In the majority of these studies, FQ resistance was
found to be low (mean 1%; 95% confidence interval 0.2-2%). CONCLUSIONS: FQ resistance in non-MDR-TB is
uncommon in NSW, Australia. The existing global evidence suggests that FQ resistance remains largely
confined to MDR-TB strains. In the majority of TB endemic regions, however, FQ resistance in non-MDR-TB
has not been assessed. Knowledge of the prevalence of FQ resistance in MTB is essential to guide the
rational use of these drugs, including their feasibility as first-line agents.
Published in the International Journal of Infectious Diseases 2014;26:149-53.

10 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Phenotypically occult multidrug-resistant Mycobacterium tuberculosis – dilemmas
in diagnosis and treatment
Ho J, Jelfs P, Sintchenko V.
Objectives: The clinical significance of the emergence of Mycobacterium tuberculosis (MTB) isolates that contain rpoB mutations (genotypic resistance), but are phenotypically susceptible to rifampicin (RIF GR PS), remains uncertain. The aim of this study was to determine the prevalence of MTB cases that demonstrate this discordant rifampicin resistance pattern and to establish whether these patients have poorer treatment outcomes with rifampicin-based regimens. Methods: rpoB sequencing was performed on all MTB isolates demonstrating phenotypic resistance to one or more first-line antituberculosis agents (excluding rifampicin). Rifampicin MICs were determined for rpoB mutation-positive isolates and clinical case notes were reviewed to identify treatment outcomes in these patients. Results: Of the 214 phenotypically drug (excluding rifampicin)-resistant isolates tested, 5 contained rpoB mutations (4 isoniazid resistant and 1 pyrazinamide resistant). These isolates demonstrated elevated rifampicin MICs (low-level resistance), despite testing susceptible using phenotypic broth-based methods. One patient experienced a relapse of tuberculosis (TB) 2 years after completion of a rifampicin-containing regimen. These findings are consistent with a recent study that reported treatment failure with rifampicin-based regimens in patients with isoniazid-resistant MTB and genotypic rifampicin resistance. Conclusions: While MTB RIF GR PS strains remain relatively uncommon,they can be associated with low-level rifampicin resistance and poorer treatment outcomes with rifampicin-based regimens. This recently recognized form of multidrug-resistant TB should be adequately detected and managed. Published in Journal of Antimicrobial Chemotherapy 2013; 68:2915-2920.

11 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Microbiological and epidemiological features of MDR-TB outbreak in Mongolia
Ulziijargal Gurjav, Buyankhishig Burneebaatar, Baasansuren Ekhembayar, Grant Hill-Cawthorne, Ben Marais, Vitali Sintchenko Background: Mongolia has the fifth highest incidence of tuberculosis (TB) in the Western Pacific Region with high rates of multi-drug resistant tuberculosis (MDR-TB). Numbers escalated rapidly since 2006, mainly due to improved case detection. However, the harsh climate and crowded living conditions may facilitate spread. The aim of this study was to examine molecular epidemiology of Mycobacterium tuberculosis (MTB) in Mongolia. Materials and methods: Data from the National TB program and 66 MDR-TB isolates from the national TB laboratory in Ulaanbaatar, collected between January and December 2012. All MTB isolates were screened for 24-loci mycobacterial interspersed repetitive unit (MIRU) typing and sequenced the gyrA gene to identify fluoroquinolone resistance. Results: Among all MDR-TB cases, 68% were from Ulaanbaatar; most were young (41% 15-29yrs of age) males (70% male). The MTB population of MDR-TB cases consisted of Beijing (86%), Latin-American Mediterranean (8%), Haarlem (5%) and NEW-1 (2%) lineages, respectively. Six MIRU clusters were detected, 5 belonging to Beijing. Two Beijing lineage clusters made up 42% (28/66) of all isolates suggesting recent transmission within the community. Alarmingly one of the Beijing clusters was pre-Extensively Drug Resistant TB with a non-synonymous mutation on codon 94 of gyrA. We also identified a potential outbreak in homeless shelter; 7/10 isolates belonged to a single Beijing cluster and 5/7 isolates shared an identical phenotypic drug resistance profile. Conclusion: Beijing lineage is overrepresented among MDR-TB cases in Mongolia, particularly among young people, with evidence of recent transmission. A likely homeless shelter outbreak indicates the need for routine TB surveillance in these populations. Presented at the Australian Society for Microbiology Annual Meeting, Melbourne, July 2014

12 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
New insights into molecular epidemiology of Mycobacterium tuberculosis in the
most populous state of Australia
Ulziijargal Gurjav, Peter Jelfs, Andrea Bustamante, Basel Suliman, Vitali Sintchenko
Background: Mycobacterial interspersed repetitive unit-variable tandem repeat analysis (MIRU-VNTR) has been applied to examine population dynamics and clustering rates of Mycobacterium tuberculosis (MTB). The aim of this study was to investigate the dynamics of MTB epidemiology since 2006-2008 and the relative impact of common lineages of MTB. Methods: Total 930 culture confirmed tuberculosis cases identified in 2010, 2011 and 2012 in New South Wales (NSW), Australia were analyzed. MTB isolates were prospectively genotyped by 24 loci MIRU-VNTR method and lineages were assigned using miruvntrplus.org.. The associations between MTB lineages, patient demographics, sites of infection and drug resistance were explored using descriptive statistics. Results: While the proportion of Beijing lineage isolates (28.2%) has not significantly changed since previous report (Gallego et al. 2009), the frequency of East African Indian (EAI) and Central Asian lineages (Delhi/CAS) has increased up to 28.7% and 12.7%, respectively. Cases due to Beijing lineage were more likely to be associated with respiratory disease and drug resistance (p<0.05) whereas EAI strains and Delhi/CAS were associated with non-respiratory TB (p<0.001 and p<0.05, respectively). Age of patients was significantly associated with lineage (p<0.0001). In particular, the age of those who affected with Delhi/CAS (mean age 37) was significantly less than of those affected by MTB Beijing (mean age 42, p<0.05) or MTB EAI (mean age 44, p<0.01). Further the age of those affected by Harleem lineage (mean age 59, p<0.05) was significantly higher than people affected by any other lineages detected in NSW. Recent evidence from China suggested that MTB Beijing with 223325173533 MIRU-VNTR allele can be highly transmissible and related to multi-drug resistance (Hu et al. 2011). We found 85 cases of MTB with these MIRU-VNTR alleles in our patient population. 10 MTB isolates were monoresistant to any first line tuberculosis drugs and 1 isolate was multi-drug resistant. Conclusion: East African Indian strains of Mycobacterium tuberculosis recently overtook Beijing family as a prevalent cause of tuberculosis in New South Wales, Australia. Despite this change in molecular epidemiology of MTB the rates of recent transmission in NSW remain low. Presented at the 44th World Conference on Lung Health, International Union Against Tuberculosis and Lung
Disease. October 2013. Paris, France

13 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Added value of whole genome sequencing for management of highly drug
resistant tuberculosis
Outhred A, Jelfs P, Suliman B, Hill-Cawthorne GA, Crawford ABH, Marais BJ, Sintchenko V.
Objectives Phenotypic drug susceptibility testing (DST) for Mycobacterium tuberculosis (MTB) typically takes
at least one week from initiation to completion for each round of testing, which is often performed
sequentially for first- and second-line drugs. In addition, second-line DST is confined to a reference-
laboratory setting, often poorly validated and not necessarily reproducible between laboratories. These
issues can compromise the timeliness and appropriateness of clinical decision-making for highly resistant
strains of MTB. Following a fatal case of extensively drug resistant (XDR) tuberculosis, we investigated the
potential benefit of using whole-genome sequencing to generate an in silico drug susceptibility profile.
Methods The clinical course of the patient was reviewed, focusing on the times at which phenotypic DST
data became available to the attending physician, and the changes made to the therapeutic regimen at those
times. Whole genome sequencing was performed on the earliest available isolate from the patient, and
following read mapping to a reference genome, variants associated with drug resistance were identified.
Results The final DST report, including second-line drugs, was issued 10 weeks after patient presentation,
and 8 weeks after initial growth of MTB. In the interim, the patient received a compromised regimen that
had the potential to select for further resistance. The in silico susceptibility profile, although extrapolated
from evolving evidence in the literature, nonetheless provided comparable or superior data to the DST
results for second-line drugs. Conclusions We propose routine whole genome sequencing of all multi-drug
resistant Mycobacterium tuberculosis isolates when possible. This will improve individual patient care,
monitor transmission events at the population level and advance our understanding of resistance-associated
mutations.
Published on the Journal of Antimicrobial Chemotherapy 2014 (in press)

14 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Global population structure and evolution of Bordetella pertussis and their
relationship with vaccination
Marieke J Bart, Simon R Harris, Abdolreza Advani, Yoshichika Arakawa, Daniela Bottero, Valérie Bouchez, Pamela K Cassiday, Chuen-Sheue Chiang, Tine Dalby, Norman K. Fry, María Emilia Gaillard, Marjolein van Gent,, Nicole Guiso, Hans O Hallander, Eric T. Harvill, Qiushui He, Han GJ van der Heide, Kees Heuvelman, Daniela F Hozbor, Kazunari Kamachi, Gennady I Karataev, Ruiting Lan, Anna Lutyoska, Ram P Maharjan,
Jussi Mertsola, Tatsuo Miyamura, Sophia Octavia, Michael A. Quail, Vitali Sintchenko, Paola Stefanelli, M LuciaTondella, Raymond SW Tsang, Yinghua Xu, Shu-Man Yao,Shumin Zhang, Julian Parkhill, Frits R. Mooi
Bordetella pertussis causes pertussis, a respiratory disease that is most severe for infants. Vaccination was introduced in the 1950s, and in recent years, a resurgence of disease was observed worldwide, with significant mortality in infants. Possible causes for this include the switch from whole-cell vaccines (WCVs) to less effective acellular vaccines (ACVs), waning immunity, and pathogen adaptation. Pathogen adaptation is suggested by antigenic divergence between vaccine strains and circulating strains and by the emergence of strains with increased pertussis toxin production. We applied comparative genomics to a worldwide collection of 343 B. pertussis strains isolated between 1920 and 2010. The global phylogeny showed two deep branches; the largest of these contained 98% of all strains, and its expansion correlated temporally with the first descriptions of pertussis outbreaks in Europe in the 16th century. We found little evidence of recent geographical clustering of the strains within this lineage, suggesting rapid strain flow between countries. We observed that changes in genes encoding proteins implicated in protective immunity that are included in ACVs occurred after the introduction of WCVs but before the switch to ACVs. Furthermore, our analyses consistently suggested that virulence-associated genes and genes coding for surface-exposed proteins were involved in adaptation. However, many of the putative adaptive loci identified have a physiological role, and further studies of these loci may reveal less obvious ways in which B. pertussis and the host interact. This work provides insight into ways in which pathogens may adapt to vaccination and suggests ways to improve pertussis vaccines. Published in mBio 2014;5(2):e01074-14.

15 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Multiple independent emergence and rapid expansion of pertactin-deficient
Bordetella pertussis in Australia
Lam C, Octavia S, Ricafort L, Sintchenko V, Gilbert GL, Wood N, McIntyre P, Marshall H, Guiso N, Keil AD, Lawrence A, Robson J, Hogg G, Lan R.
Acellular vaccines against Bordetella pertussis were introduced in Australia in 1997. By 2000, these vaccines had replaced whole-cell vaccines. During 2008-2012, a large outbreak of pertussis occurred. During this period, 30% (96/320) of B. pertussis isolates did not express the vaccine antigen pertactin (Prn). Multiple mechanisms of Prn inactivation were documented, including IS481 and IS1002 disruptions, a variation within a homopolymeric tract, and deletion of the prn gene. The mechanism of lack of expression of Prn in 16 (17%) isolates could not be determined at the sequence level. These findings suggest that B. pertussis not expressing Prn arose independently multiple times since 2008, rather than by expansion of a single Prn-negative clone. All but 1 isolate had ptxA1, prn2, and ptxP3, the alleles representative of currently circulating strains in Australia. This pattern is consistent with continuing evolution of B. pertussis in response to vaccine selection pressure. Published in Emerging Infectious Diseases 2014; 20(4):626-633.

16 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Investigating genome reduction of Bordetella pertussis using a multiplex PCR-
basedreverse line blot assay (mPCR/RLB)
Lam C, Octavia S, Sintchenko V, Gilbert GL, Lan R.
BACKGROUND: The genetic composition of the bacterium causing whooping cough, Bordetella pertussis, has
been investigated using microarray studies in order to examine potential genetic contributors to the disease
re-emergence in the past decade. Regions of difference (RDs) have been previously identified as clusters of
genes flanked by insertion sequences which are variably present in different sets of isolates, and have also
been shown to be potential markers of B. pertussis evolution. This study used microarray data to identify
and select a panel of RDs; primers and probes for these RDs were then designed to test for the presence or
absence of these regions in a novel and less expensive multiplex PCR-based reverse line blot (mPCR/RLB)
assay. By comparing the presence or absence of RDs, we aimed to determine the genomic variability of a
diverse collection of B. pertussis strains and how they have changed over time. RESULTS: A B. pertussis
specific mPCR/RLB using 43 genes representing 30 RDs, was developed and used to characterise a set of 42
B. pertussis isolates. When mapped against the previously identified evolutionary relationships of the strains,
the losses of two RDs - BP0910A - BP00930 and BP1948-BP1962 - were found to be associated with
significant events in B. pertussis history: the loss of BP0910A - BP00930 coincided with introduction of whole
cell vaccines in the 1950s while that of BP1948-BP1962 occurred after the introduction of acellular vaccines.
The loss of BP1948-BP1962 also coincided with expansion of the most recent B. pertussis strains.
CONCLUSIONS: The mPCR/RLB assay offers an inexpensive and fast method of determining the gene content
of B. pertussis strains and also confirms that gene losses are an ongoing feature of B. pertussis evolution.
Published in BMC Research Notes 2014;7:27.

17 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Temporal dynamics of Mycobacterium tuberculosis genotypes in New South
Wales, Australia
Gurjav U, Jelfs P, McCallum N, Marais BJ, Sintchenko V.
BACKGROUND: Molecular epidemiology of Mycobacterium tuberculosis, its transmission dynamics and
population structure have become important determinants of targeted tuberculosis control programs. Here
we describe recent changes in the distribution of M. tuberculosis genotypes in New South Wales (NSW),
Australia and compared strain types with drug resistance, site of disease and demographic data. METHODS:
We evaluated all culture-confirmed newly identified tuberculosis cases in NSW, Australia, from 2010-2012.
M. tuberculosis population structure and clustering rates were assessed using 24-loci Mycobacterial
interspersed repetitive unit (MIRU) analysis and compared to MIRU data from 2006-2008. RESULTS: Of 1177
tuberculosis cases, 1128 (95.8%) were successfully typed. Beijing and East African Indian (EAI) lineage strains
were most common (27.6% and 28.5%, respectively) with EAI strains increasing in relative abundance from
11.8% in 2006-2008 to 28.5% in 2010-2012. Few cases of multi-drug resistant tuberculosis were identified
(18; 1.7%). Compared to 12-loci, 24-loci MIRU provided improved cluster resolution with 695 (61.6%) and
227 (20.1%) clustered cases identified, respectively. Detailed analysis of the largest cluster identified (an 11
member Beijing cluster) revealed wide geographic diversity in the absence of documented social contact.
CONCLUSIONS: EAI strains of M. tuberculosis recently overtook Beijing family as a prevalent cause of
tuberculosis in NSW, Australia. This lineage appeared to be less commonly related to multi-drug resistant
tuberculosis as compared to Beijing strain lineage. The resolution provided by 24-loci MIRU typing was
insufficient for reliable assessment of transmissions, especially of Beijing family strains.
Published in BMC Infectious Diseases 2014;14:455.

18 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Quantitative estimation of MRSA strain-typing system stability using Kaplan-Meier
survival analysis
O’Sullivan MV, Zhou F, Sintchenko V, Gilbert GL.
Knowledge concerning stability is important in the development and assessment of microbial molecular typing systems and is critical for the interpretation of their results. Typing system stability is usually measured as the fraction of isolates that change type after several in vivo passages, but this does not necessarily reflect in vivo stability. The aim of this study was to utilize survival analysis to provide an informative quantitative measure of in vivo stability and to compare the stabilities of various techniques employed in typing methicillin-resistant Staphylococcus aureus (MRSA). We identified 100 MRSA pairs (isolated from the same patient>1 month apart) and typed them using multilocus sequence typing (MLST), phage-derived open reading frame (PDORF) typing, toxin gene profiling (TGP), staphylococcal cassette chromosome mec (SCCmec) subtyping, pulsed-field gel electrophoresis (PFGE), and spa sequence typing. Discordant isolate pairs, belonging to different MLST clonal complexes, were excluded, leaving 81 pairs for analysis. The stabilities of these methods were examined using Kaplan-Meier survival analysis, and discriminatory power was measured by Simpson’s index of diversity. The probability percentages that the type remained unchanged at 6 months for spa sequence typing, TGP, multilocus variable number of tandem repeats analysis (MLVA), SCCmec subtyping, PDORF typing, and PFGE were 95, 95, 88, 82, 71, and 58, respectively, while the Simpson’s indices of diversity were 0.48, 0.47, 0.70, 0.72, 0.89, and 0.88, respectively. Survival analysis using sequential clinical isolates adds an important quantitative dimension to the measurement of stability of a microbial typing system. Of the methods compared here, PDORF typing provides high discriminatory power, comparable with that of PFGE, and a level of stability suitable for MRSA surveillance and outbreak investigations.
Published in Journal of Clinical Microbiology 2013; 51(1):112-116.

19 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Use of the 5’ Untranslated Region and VP1 Region to Examine the Molecular
Diversity in Enterovirus B Species
Zhou F, Wang Q, Sintchenko V, Gilbert GL, O’Sullivan MVN, Iredell JR, Dwyer DE.
Human enteroviruses evolve quickly. The 5' untranslated region (UTR) is fundamentally important for
efficient viral replication and for virulence; the VP1 region correlates well with antigenic typing by
neutralization, and can be used for virus identification and evolutionary studies. In order to investigate the
molecular diversity in EV-B species, the 5' UTR and VP1 regions were analysed for 208 clinical isolates from a
single public-health laboratory (serving New South Wales, Australia), representing 28 EV-B types. Sequences
were compared with the 5' UTR and VP1 regions of 98 strains available in GenBank, representing the same
28 types. The genetic relationships were analysed using two types of software (mega and BioNumerics). The
sequence analyses of the 5' UTR and VP1 regions of 306 EV-B strains demonstrated that: (i) comparing the
two regions gives strong evidence of epidemiological linkage of strains in some serotypes; (ii) the
intraserotypic genetic variation within each gene reveals that they evolve distinctly largely due to their
different functions; and (iii) mutation and possible recombination in the two regions play significant roles in
the molecular diversity of EV-B. Understanding the tempo and pattern of molecular diversity and evolution is
of great importance in the pathogenesis of EV-B enteroviruses, information which will assist in disease
prevention and control.
Published in Journal of Medical Microbiology 2014;63:1339-1355.

20 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Improved identification of rapidly growing mycobacteria by a 16S-23S internal
transcribed spacer region PCR and capillary gel electrophoresis
Gray T, Kong F, Jelfs P, Sintchenko V, Chen SCA
The identification of rapidly growing mycobacteria (RGM) remains problematic because of evolving taxonomy, limitations of current phenotypic methods and absence of a universal gene target for reliable speciation. This study evaluated a novel method of identification of RGM by amplification of the mycobacterial 16S-23S rRNA internal transcribed spacer (ITS) followed by resolution of amplified fragments by capillary gel electrophoresis (CGE). Nineteen American Type Culture Collection (ATCC) Mycobacterium strains and 178 clinical isolates of RGM (12 species) were studied. All RGM ATCC strains generated unique electropherograms with no overlap with slowly growing mycobacteria species, including M. tuberculosis. A total of 47 electropherograms for the 178 clinical isolates were observed allowing the speciation of 175/178 (98.3%) isolates, including the differentiation of the closely related species, M. massiliense (M. abscessus subspecies bolletii) and M. abscessus (M. abscessus sensu stricto). ITS fragment size ranged from 332 to 534 bp and 33.7% of clinical isolates generated electropherograms with two distinct peaks, while the remainder where characterized with a single peak. Unique peaks (fragment lengths) were identified for 11/12 (92%) RGM species with only M. moriokaense having an indistinguishable electropherogram from a rarely encountered CGE subtype of M. fortuitum. We conclude that amplification of the 16S-23S ITS gene region followed by resolution of fragments by CGE is a simple, rapid, accurate and reproducible method for species identification and characterization of the RGM.
Published in PLoS One 2014;9(7):e102290.

21 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
No innovation without evaluation: Added value of prospective multiple-locus
variable-number tandem-repeat analysis (MLVA) for surveillance of Salmonella
enterica serovar Typhimurium
Vitali Sintchenko, Qinning Wang, Peter Howard, Nadine Holmes, Sophie Octavia, Ruiting Lan, Grant Hill-Cawthorne
Objective: Subtyping of Salmonella enterica serovar Typhimurium (STM) by multiple-locus variable-number tandem-repeat analysis (MLVA) can assist in identifying hidden clusters of community infections, as well as sources of endemic and outbreak cases. This public health surveillance study examined the temporal and spatial population dynamics of STM infection using prospective STM MLVA typing. Methods: All STM referred to the New South Wales Enteric Reference Laboratory during the 5-year period between January 2008 and December 2012, were fingerprinted using MLVA-5 and phage typed. Clusters of ≥5 isolates with the same MLVA pattern collected over 4 weeks were followed up epidemiologically and their temporal and spatial densities measured. Results: A total of 8,936 human isolates were analysed. The STM spectrum was dominated by PT170 (44.6% of all isolates) and PT135 (13.9%). The gradual increase in the number of STM cases during the study was not related to changes in the number of clusters or their size. 1562 MLVA patterns were observed and 14 ‘endemic’ patterns were responsible for 45-50% of cases. However, the ratio between novel and endemic patterns remained constant. The Simpson’s index of diversity was 0.109 for PT135 and 0.172 for PT170. 88 STM clusters were observed ranging in size from 5 to 262 cases and in duration from 4 to 25 weeks; 43 clusters had novel MLVA types and 23 represented recurrences of previously recorded MLVA types. Re-occurring clusters were larger than initial clusters and less spatially dense (P<0.001). 32 clusters were investigated. The relapse rate for clusters that did or did not trigger any public health actions was similar (34%). However, the former reoccurred on average 9 weeks later than clusters that were not investigated epidemiologically (P<0.001). Gene loss and acquisition in the emerging STM 2-7-6-11-0212 clone of PT170 (Figure) will be discussed. Conclusions: Prospective STM MLVA typing improves the resolution and timeliness of public health surveillance. Sustained levels of STM diversity were accompanied by an increasing incidence of human infections and the endemicity of PT170 clones. Monitoring ratios of novel versus re-occurring MLVA types offers a new benchmark for assessing impacts of public health interventions.
Presented at the 10th International Meeting on Microbial Epidemiological Markers
(IMMEM-10), Institut Pasteur, Paris, France.

22 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Inaccurate ascertainment of morbidity and mortality due to influenza in
administrative databases, a population-based record linkage study
Muscatello DJ, Amin J, MacIntyre CR, Newall AT, Rawlinson WD, Gilmour R, Sintchenko V, Thackway S.
BACKGROUND: Historically, counting influenza recorded in administrative health outcome databases has been considered insufficient to estimate influenza attributable morbidity and mortality in populations. We used database record linkage to evaluate whether modern databases have similar limitations. METHODS: Person-level records were linked across databases of laboratory notified influenza, emergency department (ED) presentations, hospital admissions and death registrations, from the population (∼6.9 million) of New South Wales (NSW), Australia, 2005 to 2008. RESULTS: There were 2568 virologically diagnosed influenza infections notified. Among those, 25% of 40 who died, 49% of 1451 with a hospital admission and 7% of 1742 with an ED presentation had influenza recorded on the respective database record. Compared with persons aged ≥65 years and residents of regional and remote areas, respectively, children and residents of major cities were more likely to have influenza coded on their admission record. Compared with older persons and admitted patients, respectively, working age persons and non-admitted persons were more likely to have influenza coded on their ED record. On both ED and admission records, persons with influenza type A infection were more likely than those with type B infection to have influenza coded. Among death registrations, hospital admissions and ED presentations with influenza recorded as a cause of illness, 15%, 28% and 1.4%, respectively, also had laboratory notified influenza. Time trends in counts of influenza recorded on the ED, admission and death databases reflected the trend in counts of virologically diagnosed influenza. CONCLUSIONS: A minority of the death, hospital admission and ED records for persons with a virologically diagnosed influenza infection identified influenza as a cause of illness. Few database records with influenza recorded as a cause had laboratory confirmation. The databases have limited value for estimating incidence of influenza outcomes, but can be used for monitoring variation in incidence over time.
Published in PLoS ONE 2014;9(5):e98446.

23 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Utility of multilocus sequence typing (MLST) for inferring prevalent Salmonella
Enterica serotypes
Cristina Sotomayor, Peter Howard, Qinning Wang and Vitali Sintchenko
Background: Salmonella has been one of the most common agents responsible for outbreaks of foodborne diseases worldwide and in Australia. Historically, these bacteria have been classified into serotypes according their somatic and flagella antigens. However, recently there have been calls to replace serotyping with multilocus sequence typing (MLST) based classification which recognizes evolutionary groupings. This study aimed to examine the capacity of MLST to differentiate S.enterica serotypes using isolates identified and characterised in the NSW Enteric Reference Laboratory (ERL). Methods: 127 randomly selected Salmonella enterica isolates representing 9 most common serovars observed in NSW between 2010 and 2012 were used in the study. The MLST method was employed targeting housekeeping aroC, dnaN, hemD, hisD, purE, sucA, and thrA gene fragments. Sequence types (ST) were assigned using the MLST server (University College, Cork). Results: Isolates of Salmonella enterica serovar Typhimurium (STM), the most common serovar among observed in NSW (91/127), were all classified as ST-19. However, STM monophasic isolates were assigned to ST-85 and 34. S. enteritidis isolates were mainly grouped into ST-11; ST-183 and 180 also were found. S. Saint Paul was classified as ST-50; S. Bovimorbificans belonged to ST-1499 and 377. S. Virchow was defined as ST-16, S. Infantis as ST-32, S. Birkenhead as ST-424, and S. Stanley as ST-29, respectively. S. Montevideo was associated with ST-138 and 316. No isolates of different serotypes classified in the same ST were identified. Conclusions: Clearly identifiable ST can accurately differentiate the most common serotypes of S. enterica observed in NSW. MLST can be an appropriate alternative to serotyping in the classification of S. enterica for diagnostic and surveillance purposes and could be considered as a method to identify evolutionary clusters as well as to display diversity among serovars. However, the discriminatory power of MLST-7 for detecting community outbreaks remains limited. Presented at the Australian Society for Microbiology Annual Meeting, Melbourne, July 2014

24 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Proof-of-concept study for successful inter-laboratory comparison of MLVA results
Jonas T Larsson1, Mia Torpdahl1, MLVA working group2, Eva Møller Nielsen and MLVA Working group: Anna Aspán, Sophie Bertrand, Chien-Shun Chiou, Chelsey Goodman, Max Heck, Lester Hiley, Katie Hopkins, Geoff Hogg, Eija Hyytia-Trees, Hidemasa Izumiya, Cecilia Jernberg, Simon Le Hello, Bjørn-Arne Lindstedt,
Burkhard Malorny, Deirdre Prendergast, Catherine Ragimbeau, Vitali Sintchenko, Anthony Smith, Gitte Sørensen, Erhard Tietze.
Multiple-locus variable-number of tandem repeats analysis (MLVA) is widely used for typing of pathogens. Methods such as MLVA based on determining DNA fragment size by the use of capillary electrophoresis have an inherent problem as a considerable offset between measured and real (sequenced) lengths is commonly observed. This discrepancy arises from variation within the laboratory set-up used for fragment analysis. To obtain comparable results between laboratories using different set-ups, some form of calibration is a necessity. A simple approach is to use a set of calibration strains with known allele sizes and determine what compensation factors need to be applied under the chosen set-up conditions in order to obtain the correct allele sizes. We present here a proof-of-concept study showing that using such a set of calibration strains makes inter-laboratory comparison possible. In this study, 20 international laboratories analysed 15 test strains using a five-locus Salmonella enterica serovar Typhimurium MLVA scheme. When using compensation factors derived from a calibration set of 33 isolates, 99.4% (1,461/1,470) of the MLVA alleles of the test strains were assigned correctly, compared with 64.8% (952/1,470) without any compensation. After final analysis, 97.3% (286/294) of the test strains were assigned correct MLVA profiles. We therefore recommend this concept for obtaining comparable MLVA results.
Published in Eurosurveillance 2013; 18(35):pil=20566.

25 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Pathogen Genome Bioinformatics
Sintchenko V, Roper MPV. Recent advances in DNA sequencing technology have made the whole-genome sequencing of pathogens in a clinically relevant turn-around-time both technically and economically feasible. The DNA sequencing of pathogens with epidemic potential offers new and exciting opportunities for high-resolution public health surveillance. This chapter outlines major methods and bioinformatics tools for pathogen genome characterization, the identification of infectious disease clusters, as well as for genomics-guided bio-surveillance. Existing challenges are also considered. Published in: Trent RJA (Ed). Clinical Bioinformatics (2nd Edition). Humana Press, Springer Science 2014. PP. 173-93. ISBN 978-1-4939-0846-2

26 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS BACTERIOLOGY EPIDEMIOLOGY & DISEASE SURVEILLANCE
Medical microbiology informatics
Rhoads DD, Sintchenko V, Rauch CA, Pantanowitz L. The medical microbiology laboratory has responsibilities ranging from characterizing the causative agent in a patient’s infection to helping detect global disease outbreaks. All of these processes are becoming increasingly more intimately partnered with informatics. Effectively using informatics tools can produce more accurate, rapid and automated results while decreasing laboratory workload, which can lead to a better and more scalable product at a decreased cost. Informatics is poised to be increasingly relevant in medical microbiology with the advent of total laboratory automation, complex instrument interfaces, electronic health records, clinical decision support tools, and the clinical implementation of microbial whole genome sequencing. This review discusses the diverse informatics aspects that are relevant to the medical microbiology laboratory including the following: the microbiology laboratory information system, decision support tools, expert systems, instrument interfaces, total laboratory automation, telemicrobiology, automated image analysis, nucleic acid sequence databases, electronic reporting of infectious agents to public health agencies, and disease outbreak surveillance. The breadth and utility of informatics tools used in medical microbiology have made them indispensable to contemporary clinical and laboratory practice. Continued advances in technology and development of these informatics tools will further improve patient and public healthcare in the future. Published in Clinical Microbiology Reviews 2014;27(4):1025-1047.

27 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
The Scedosporium aurantiacum genome sequencing project
Ramsperger M1, Pérez-Bercoff Å2, Firacative C1, Papanicolau A3, Patel HR2, Huttley GA2, Nevalainen H4 & Meyer W1
1 Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Sydney Medical School-Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, University of Sydney, Westmead Millennium Institute, Westmead NSW Australia; 2John Curtin School of Medical Research, Australian National University, Canberra ACT, Australia; 3CSIRO Ecosystem Sciences, Black Mountain Labs, Canberra, ACT, Australia; 4 Macquarie University, Sydney, NSW, Australia
Scedosporium aurantiacum is a fungus commonly present in urban environments and acts as an important opportunistic pathogen, causing infections in both immunocompromised and immunocompetent humans and animals. Virulence studies using the Galleria mellonella (greater wax moth) larval model have identified strain-specific differences in virulence. To establish a molecular basis for the origins of virulence and the genomic differences between strains, we have sequenced and assembled the first genome of S. aurantiacum (strain WM 09.24). For genome annotation and the development of gene models we performed whole-genome alignment against the closest annotated genome publicly available, Trichoderma virens. However, this species was too genetically diverged, with only 30% homology present between the two genomes. Neither direct-gene projection nor BLAST searches against the nr/nt database were effective in annotating the S. aurantiacum genome. To overcome this genome annotation problem we performed RNA-seq of strain WM 09.24 under different growth conditions to maximize the diversity of transcribed genes. The RNA-seq data identified genes and intron-exon boundaries within the genome. To allow for the assembly of chromosomes PFGE is currently underway. In addition the annotated genome of strain WM 09.24 is currently used to annotate three additional S. aurantiacum strains with varying levels of virulence to enable a whole-genome comparison to identify genetic signatures that are associated with virulence.
International Mycology Congress IMC10 Bangkok, Thailand 3-8.8.2014

28 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Cryptococcus: an opportunistic or primary pathogen?
Popchai Ngamskulrungroj1, Sirada Kaocharoen2,3, Young Hwa Choi4,5, Ariya Chindamporn2, Kyung J. Kwon-Chung5, Wieland Meyer3
1. Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand 2. Mycology Laboratory, King Chulalongkorn Memorial Hospital, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand 3. Molecular Mycology Research Laboratory, CIDM, Sydney Medical School - Westmead Hospital, The University of Sydney, Westmead Millennium Institute, Westmead, New South Wales, Australia 4. Department of Pulmonary and Critical Care Medicine, Ajou University School of Medicine, Suwon, Korea. 5. Molecular Microbiology Section, Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, USA
Cryptococcosis is a basidiomycetous yeast infection caused by Cryptococcus neoformans-gattii species complex which comprises of two sibling species, Cryptococcus neoformans and Cryptococcus gattii. Since the beginning of the acquired immune deficiency syndrome (AIDS) pandemic in 1980s, prevalence of cryptococcosis has increased dramatically. More than 95% of cryptococcosis was AIDS-associated thus cryptococcosis was considered as an opportunistic infection. However, over the years, this paradigm has been challenged by several epidemiological studies reporting non-AIDS-associated cryptococcosis. Firstly, in 1999, more than 200 cases of cryptococcosis were reported on the Vancouver Island, Canada where most patients (62%) were non-HIV. Secondly, in 2008, Chang et al. reported that most (91.5%) of 129 cryptococcosis cases from China occurred in immunocompetent patients. Finally, in 2010, an epidemiological survey of cryptococcosis in Korea revealed 77.4% of the 62 cases were non-HIV patients. Genetic susceptibility to cryptococcosis of the Far East Asian bloodline was suspected. However, this figure is converted in a study from Thai patients which are close siblings of the Far East Asian bloodline where only 6% of the 113 cryptococcosis cases were non-HIV patients. Thus, the genetic susceptibility hypothesis is inconclusive and further studies are needed.
International Mycology Congress IMC10 Bangkok, Thailand 3-8.8.2014

29 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Tracing the global spread of human pathogens
Wieland Meyer1
1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Westmead
Millennium Institute, Sydney Medical School-Westmead Hospital, Marie Bashir Institute for Infectious
Diseases and Biosecurity, University of Sydney, Westmead, NSW, Australia
Human pathogenic fungi are becoming an increasing global problem, due to the growing human mobility, global trade and interconnectivity between different parts of human society. A number of outbreaks including the famous ongoing outbreak of Cryptococcosis on Vancouver Island have recently been described. To gain better insights into the outbreak dynamic and the spread of human/animal fungal pathogens it is important to understand the population genetic structure and molecular epidemiology of human/animal pathogenic fungi. This talk will review the molecular methods currently available to trace the spread of human and animal pathogens, including PCR-fingerprinting, Amplified fragment length polymorphism (AFLP), Multilocus Microsatellite Typing (MLMT), multilocus sequence type analysis (MLST and whole genome sequencing (WGS). Examples on how the different molecular techniques have been applied in different scenarios, including Cryptococcus gattii, Scedosporium aurantiacum and Pneumocystis jirovecii will be discussed. The global Multilocus Sequence typing database for human/animal pathogenic fungi established as part of the International Society of Human and Animal Mycology (ISHAM) working groups “Genotyping of Cryptococcus neoformans and Cryptococcus gattii” and “Pseudallescheria/Scedosporium infections”, accessible via mlst.mycologylab.org or www.isham.org, will be presented. This database features specific MLST databases for: Cryptococcus neoformans (formerly housed at mslt.net); Cryptococcus gattii; Scedosporium aurantiacum, Scedosporium apiospermum, Scedosporium boydii and Pneumocystis jirovecii. The MLST databases provide standardized genetic loci, primer and PCR amplification information, and allows for the identification of allele (AT) and sequence types (ST) facilitating a globally standardised strain typing system to enable an early public health response to emerging outbreaks. International Mycology Congress IMC10 Bangkok, Thailand 3-8.8.2014

30 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Virulence of Cryptococcus gattii VGIII isolates in a Galleria mellonella insect model
William King1, 2, Aziza Khan1, Carolina Firacative1 and Wieland Meyer1.
1Molecular Mycology Research Laboratory, Centre for Infectious Disease and Microbiology, Sydney Medical School-Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, The University of Sydney, Westmead Millennium Institute, Sydney, Australia 2University of Technology, Sydney The emergence of Cryptococcus gattii as a primary pathogen has sparked concerns for the social impacts it can have on human health and health services. Although four major molecular types of this species have been identified, VGI to VGIV, the majority of virulence studies have been focused on VGII because of an ongoing outbreak caused by this molecular type on Vancouver Island, Canada and the Pacific Northwest, USA. However, progressive studies focusing on the other molecular types are needed. This study aims to determine the virulence of a number of VGIII strains, whose molecular type was confirmed by URA5-RFLP analysis, by inoculating each strain in the larvae of the wax moth Galleria mellonella. Forty-two strains were chosen on the basis of previously determined multi locus sequence typing (MLST) data, with each strain representing a different MLST genotype. Three strains WM1635, WM11.950 and WM11.124 were identified as having the same virulence as the highly virulent VGIIa strain, CDCR265, which previously caused the majority of the cases reported in the Vancouver Island outbreak. Westmead Hospital week, Westmead, Australia 27-28.8.2014

31 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Galleria mellonella model identifies highly virulent strains among all major
Cryptococcus gattii molecular types
Carolina Firacative1,2 and Wieland Meyer1*
1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Sydney Medical School – Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, The University of Sydney, Westmead Millennium Institute, Sydney, Australia; 2Grupo de Microbiología, Instituto Nacional de Salud, Bogotá, Colombia. *Corresponding author: [email protected] Background. Worldwide cryptococcosis is mainly caused by Cryptococcus neoformans. However, the number of cases due to C. gattii is increasing, affecting mainly immunocompetent hosts. By different molecular methods C. gattii has been divided into four major molecular types VGI to VGIV, which differ in their host range, epidemiology, antifungal susceptibility and geographic distribution. Besides studies on the Vancouver Island outbreak strains, which showed that the sub-genotype VGIIa is highly virulent compared to the sub-genotype VGIIb, little is known about the virulence of the other major molecular types. Aim. To investigate the virulence potential of C. gattii strains of all major molecular types and different MLST genotypes, by comparing their pathogenesis and assessing the cryptococcal virulence factors in an invertebrate model of infection. Methods. Galleria mellonella larvae were inoculated with ten globally selected strains per molecular type, which were previously studied by MLST. Larvae were checked daily, deaths were recorded and survival plots were constructed. Cells of all C. gattii strains were isolated from the larvae after inoculation and cell and capsule sizes were measured. Cell and capsule sizes of the strains before inoculation were also measured. Melanin production and growth at 37º C was determined in all strains. Results. Among the studied strains, one VGII, one VGIII and one VGIV strains were more virulent (p-value <0.05) than the highly virulent Vancouver Island outbreak strain VGIIa (CDCR265); eleven strains (four VGI, two VGII, four VGIII and one VGIV) had similar virulence (p-value >0.05) and twenty-five (six VGI, six VGII, five VGIII and eight VGIV) were less virulent (p-value <0.05). Overall, strains of the molecular type VGIV showed to be the leats virulent. Cell and capsule size of all strains increased drastically during larvae infection (p-value <0.001). Melanin production was directly associated with the level of virulence of the strains: highly virulent strains produced more melanin that low virulent strains (p-value <0.05). All strains had the ability to growth at 37º C and there was no difference among the strains regarding their growth rate (p-value >0.05). Discussion/Conclusion. The results indicate that all major C. gattii molecular types exhibit a range of virulence, with the potential of some strains amongst them being highly virulent. As reported in C. neoformans, the present study supports the fact that fungal virulence factors like capsule, melanin and growth at physiological temperature, which are involved in mammalian pathogenesis, are also needed for G. mellonella infection and killing. This study highlights the relevance to determine the major molecular type/genotype of the isolates as part of the epidemiology of Cryptococcus and cryptococcosis and warrants further studies to identify specific genotypic traits of highly virulent C. gattii strains that are associated with their virulence and which are relevant for the development and progress of cryptococcosis. 9th International Congress of Cryptococcus and Cryptococcosis, Amsterdam, The Netherland, 15-19.52014

32 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Molecular epidemiology of clinical Cryptococcus gattii isolates from Colombia
Carolina Firacative1,2, Jairo Lizarazo3, Patricia Escandón2, Clara Inés Agudelo2, Wieland Meyer1, Elizabeth Castañeda2, *
1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Sydney Medical School – Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, The University of Sydney, Westmead Millennium Institute, Sydney, Australia; 2Microbiology Group, Instituto Nacional de Salud, Bogotá, Colombia; 3Neurology Department, Hospital Universitario Erasmo Meoz, Cúcuta, Norte de Santander, Colombia. *Corresponding author: Elizabeth Castañeda, Grupo de Microbiología, Instituto Nacional de Salud, Avenida calle 26 # 51-20, Bogotá Colombia. Phone: +57 1 217 2408, e-mail: [email protected]
Background. Cryptococcosis caused by Cryptococcus gattii is endemic in several countries and affects mostly immunocompetent patients, both their pulmonary and central nervous systems. Worldwide the number of cases of this mycosis is increasing mainly because C. gattii is expanding its environmental niche, which leads to a wider geographic distribution. In Colombia, a national surveillance on cryptococcosis, including demographic, clinical and microbiologic data, is being conducted since 1997, although molecular characterization of the isolates using MLST has not been done. Aim. To characterize by molecular methods the clinical isolates of C. gattii recovered in Colombia from 1997 until 2010, to provide insights into the molecular epidemiology of this important pathogen in the country and to contribute to the general knowledge of Cryptococcus and cryptococcosis in the world. Methods. From 1.207 surveys analyzed, 43 C. gattii cases from 15 departments were reported, with the majority of the cases (n=15) from Norte de Santander. Among all isolates, four were recovered from HIV patients, one from a patient with arthritis and one with diabetes. The remaining 37 patients did not have or report any risk factor. Molecular type of the isolates was determined using PCR fingerprinting with the primer (GTG)5. Mating type a or alpha was determined using specific primers. Multilocus Sequence Typing (MLST) was carried out using the ISHAM consensus MLST typing scheme for C. neoformans/C. gattii species complex, which includes seven genetic loci, CAP59, GPD1, LAC1, PLB1, SOD1, URA5 and the IGS1 region. Results. The molecular type VGII was the most frequent among the isolates (55.8%), followed by VGIII (27.9%), and VGI (16.3%). Among the patients with risk factors associated with the development of cryptococcosis, the molecular type VGII and VGI were identified in two and one HIV+ patients, respectively. One VGII isolate was recovered from a patient with arthritis and one VGIII isolate from a patient with diabetes. Mating type was determined as alpha in 23 (53.5%) isolates and as a in 20 (46.5%). Among 42 isolates, 16 MLST sequence types (ST) were identified: two STs amongst VGI isolates, nine amongst VGII isolates, with ST25 being the most common one (68%), and five amongst VGIII isolates. Eight STs of the VGII isolates (ST8, ST12, ST31, ST46, ST321, ST322, ST323 and ST324) and three of the VGIII isolates (ST59, ST64 and ST85) were identified for the first time in this study. The obtained MLST data were incorporated into the C. neoformans/C. gattii databases accessible at http://mlst.mycologylab.org.

33 CIDM-PH ANNUAL REPORT 2014
Discussion/Conclusion. The current study showed that the clinical C. gattii isolates from Colombia are genetically diverse. Despite the low number of isolates studied, several genotypes were identified within each molecular type, opposite to the less diverse and rather clonal C. gattii populations reported in other countries such as Canada, USA, Australia and Thailand, where few genotypes have been identified in a much larger number of isolates. The identification of the same ST in different departments, like the prevalent ST25 for VGII isolates found in 7 different departments, the ST51 and ST58 in VGI isolates, and the ST64, ST79 and ST146 in VGIII isolates, suggests the circulation of some genotypes in the country. The isolation of C. gattii predominantly from otherwise healthy hosts rather than from patients with impaired immune system supports the idea of C. gattii as primary pathogen and as being clinically more pathogenic than its sibling species C. neoformans. Our data is giving a more detailed picture of the molecular epidemiology of cryptococcosis in Colombia and includes the country as integral part of the global population genetics studies of the C. neoformans/C. gattii species complex.
9th International Congress of Cryptococcus and Cryptococcosis, Amsterdam, The Netherland, 15-19.52014

34 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Characterisation of Cryptococcus strains isolated from mammals living in an
environmental site with high cryptococcal presence in South-Western Australia
Patrizia Danesi,1 Richard Malik,2 Mark B. Krockenberger2 and Wieland Meyer1
Molecular Mycology Research Laboratory, Center for Infectious Diseases and Microbiology, Sydney Medical School-Westmead Hospital, Faculty of Veterinary Science, The University of Sydney, NSW, Australia Background: Cryptococcosis is one of the most important systemic fungal infections of mammals in Australia. Investigations of veterinary isolates1 and surveillance studies of captive koalas2 identified a wildlife park in Perth, WA with an exceedingly high environmental occurrence of Cryptococcus gattii. Furthermore, there is a high rate of nasal colonisation of animals in this location, particularly in koalas, with both subclinical and symptomatic disease. Aim: To further monitor the extent of colonization and the associated genetic variability within the Cryptococcus neoformans/gattii species complex in animals living in this park. Specifically, we were interested in whether colonisation of individual animals or focal environmental sites is clonal, or involved more than one cryptococcal strain/molecular type. Methods: In October 2012, 58 samples, comprising swabs from captive koalas (n=27), dingoes (n=2), a wombat (n=1) and the environment (n=18) were collected at Caversham Wildlife Park. After transport to the laboratory (at 5oC; for 5 days), swabs were inoculated onto birdseed agar plates and incubated at room temperature (approx. 21oC). The extent of cryptococcal colonisation or environmental presence was codified as being of low, intermediate and high grade according to the number of colonies showing a brown-colouring per plate (low < 50; intermediate 50-100; high grade >100 colonies).3 Following DNA extraction, restriction fragment length polymorphism (RFLP) analysis of the URA5 gene and a mating type specific PCR were performed to determine the molecular type and the mating type, respectively, for one colony from each plate, representing isolates from an individual animal or a single environmental site.4 Genetic variation among Cryptococcus isolates was studied using the ISHAM consensus multi-locus sequence type (MLST) scheme for the C. neoformans/C. gattii species complex.5 Additionally, to determine whether different Cryptococcus clones were present amongst brown colonies grown from a single plate from an individual animal or environmental site, 74 colonies isolated from four plates (n=18/plate from one koala; n=19/plate from another koala; n=20/plate from an environment sample; n=17/plate from a different environmental sample) were analysed by M13 PCR fingerprinting.

35 CIDM-PH ANNUAL REPORT 2014
Results: Positive cultures were obtained from 14 of the 27 koalas (52%) and from 10 of the 18 environment samples (56%). The extent of colonisation or environmental presence was low (8 koalas and 3 environmental samples), intermediate (5 environmental samples) and high grade (6 koalas and 2 environmental samples). Among the positive cultures, C. gattii VGII (n=16; 67%), C gattii VGI (n=5; 21%) and C. neoformans VNI (n=3; 12%) were identified. VNI was identified only from koalas. All isolates were of the α-mating type. MLST analysis revealed six distinct sequence types (ST) among the 24 isolates: ST7 (14), ST23 (2), ST48 (2), ST51 (1), ST58 (1) and ST 154 (4). PCR fingerprinting profiles showed identical patterns among colonies from the same plate for the four plates so analysed. Discussion & Conclusions: C. neoformans and C. gattii were both isolated from nasal swabs of koalas, whereas only C. gattii was found in the environment. The wide variety of MLST types of C. gattii isolates supported the possibility of genetic recombination at this location, consistent with earlier work2 and the previous finding of rare a mating types in this environment.2 On the other hand, PCR fingerprinting profiles suggest that within an individual animal or environmental site, clonal replication prevails, based on the number of plates studied. The persistent high environmental presence of VGII in Caversham Park, with corresponding high prevalence of asymptomatic nasal colonisation, subclinical infection and clinical disease in animals, especially koalas and wombats, emphasizes the need for on-going surveillance at this location. Clonal outbreaks might develop in this location after genetic recombination, possibly giving rise to a Vancouver Island-like outbreak in the vicinity. References 1. S McGill, R Malik, N Saul, S Beetson, C Secombe, I Robertson, P Irwin. Cryptococcosis. (2007) Medical Mycology 47: 625-639 2. F Carriconde, F Gilgado, I Arthur, D Ellis, R Malik, N van de Wiele, V. Clonality and α-a Recombination in the Australian Cryptococcus gattii VGII Population-An Emerging Outbreak in Australia. (2011) PloS One 6 (2), e16936 3. Connolly JH, Krockenberger MB, Malik R, Canfield PJ, Wigney DI and Muir DB. Asymptomatic carriage of Cryptococcus neoformans in the nasal cavity of the koala (Phascolarctos cinereus). (1999) Medical Mycology 37: 331–338 4. Meyer W, Aanensen DM, Boekhout T, Cogliati M, Diaz MR, Esposto MC, Fisher M, Gilgado F, Hagen F, Kaocharoen S, Litvintseva AP, Mitchell TG, Simwami SP, Trilles L, Viviani MA, Kwon-Chung J. Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. (2009) Medical Mycology 47: 561–570. 5. Meyer W, Castañeda A, Jackson S, Huynh M, Castañeda E, IberoAmerican Cryptococcal Study Group. Molecular typing of IberoAmerican Cryptococcus neoformans isolates. (2003) Emerging Infectious Diseases 9:189–195. 9th International Congress of Cryptococcus and Cryptococcosis, Amsterdam, The Netherland, 15-19.52014

36 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
State of barcoding of human pathogenic fungi
Wieland Meyer1
1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Westmead Millennium Institute, Sydney Medical School-Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, University of Sydney, Westmead, NSW, Australia, email: [email protected] Human and animal pathogenic fungi are a continuously growing threat worldwide. The diversity of yeasts and filamentous fungi causing novel life-threatening invasive infections in immunocompromised and immunocompetent hosts is constantly increasing. As such there is a growing need for a rapid and accurate identification of pathogens to enable early diagnosis to ensure targeted antifungal therapy. Traditional morphological and biochemical identification methods are time-consuming and require trained experts in mycology. Alternatively DNA barcoding is a practical approach for species identification and less demanding in terms of taxonomical expertise. However, wide spread application of DNA barcoding is challenged by a lack of quality controlled reference databases and the ever increasing division of fungal species/complexes. To assess these limitations an international consortium of medical mycology reference laboratories was formed with the aim to establish a quality controlled ITS database under the umbrella of the ISHAM working group on “DNA barcoding of human and animal pathogenic fungi”. This new database, containing 2800 ITS sequences representing 421 fungal species, provides the medical community with a freely accessible tool to rapidly and reliably identify most agents of mycoses, accessible at www.isham.org or http://its.mycologylab.org. Through this process the overall utility of the ITS as the current official barcode for identification of pathogenic fungi, at intra-and interspecies level, was evaluated. However, for a number of species complexes intra- and interspecies genetic distances are overlapping, revealing major limitations of the ITS region as universal fungal barcode. Data analysis highlighted important pathogenic fungal species, e.g. the dermatophytes and emerging yeast pathogens, for which additional molecular methods/genetic markers are required to reliably identify these strains from clinical and veterinary specimens. To overcome these limitations new genetic loci identified from whole genome data are currently under investigation to either be used as secondary or alternative fungal barcode.

37 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
The global MLST network
Wieland Meyer1, Carolina Firacative1,2, Luciana Trilles1,3, Vincent Robert4 and the ISHAM Working group for genotyping of C. neoformans and C. gattii 1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Sydney Medical School – Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, The University of Sydney, Westmead Millennium Institute, Sydney, Australia; 2Grupo de Microbiología, Instituto Nacional de Salud, Bogotá, Colombia; 3Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil; 4Centraalbureau voor Schimmelcultures, Utrecht, The Netherlands; *Corresponding author: [email protected] Background. As a result of the increasing number of cryptococcosis cases world-wide and the emergence of disease outbreaks a better understanding of the molecular epidemiology of the members of the C. neoformans/C. gattii species complex needed. A number of different typing techniques have divided the complex into 8 major molecular types: VNI, VNII, VNIII and VNIV for C. neoformans and VGI, VGII, VGIII and VGIV for C. gattii. Multilocus Sequence Typing (MLST) has gained increasing popularity in population genetic studies of this species complex. Aim. Establishment of a globally standardised typing scheme and associated database to enable online allele (AT) and sequence type (ST) assignment as basis for global population genetic analyses of these fungal pathogens. Methods. The in 2009 by the ISHAM working group for genotyping of C. neoformans and C. gattii established ISHAM MLST consensus scheme for the C. neoformans/C. gattii species complex, based on the analysis of seven unlinked genetic loci: CAP59, GPD1, IGS1, LAC1, PLB1, SOD1 & URA5, was used to investigate the global distribution of the seven major haploid molecular types of this species complex. Results. Since the standardisation of the MLST typing scheme the ISHAM consensus MLST typing schemes has been used in 12 published C. neoformans and 9 published C. gattii studies. However, a few studies still using an alternative MLST scheme, rendering those data from being included in a global population genetics analysis. After initial problems with the ST assignment, due to the deletion of gaps in the original data set housed at the mlst.net webpage of the Imperial College London, the data have been corrected and the new numbering has been published (Khayhan et al. PloSONE 2013;8(9):e72222). A new unified online database has been established at the University of Sydney, accessible at: mlst.mycologylab.org, allowing for the online identification of ATs and STs via single or multiple loci queries and the addition of new ATs and STs after double-checking by one of the three current database curators. An online strain database has been established containing all published MLST data. 1153 C. neoformans strains fall into 169 STs in 22 countries, with VNI having 86 STs in 22 countries, VNB 19 STs in 2 countries, VNII 14 STs in 10 countries and VNIV 50 STs in 5 countries’. 662 C. gattii strains fall into 152 STs in 39 countries, with VGI having 61 STs in 27 countries, VGII 45 STs in 15 countries, VGIII 30 STs in 7 countries and VGIV 14 STs in 6 countries. The highest genetic diversity amongst C. neoformans isolates is found in Italy 51 STs, France 47 STs, Thailand/Botswana 25 STs, and Uganda 17 STs. The highest genetic diversity amongst C. gattii isolates is found in Brazil 16 STs, Australia 10 STs, France 8 STs and Canada 7 STs.

38 CIDM-PH ANNUAL REPORT 2014
Discussion/Conclusions. The C. neoformans/C. gattii species complex shows and extensive genetic diversity, which can be accessed via on online centralized MLST database. Increasingly MLST loci are obtained from whole genome sequences (WGS), where MLST data show highly similar genetic relationships as the WGS data. While a large number of STs are restricted to certain countries, a number of STs types have a global distribution, including the minor Vancouver Island outbreak genotype VGIIb (ST7). These findings indicate that it is crucial to have an active global surveillance in place to allow for an active monitoring of potential outbreak or highly virulent strains. 9th International Congress of Cryptococcus and Cryptococcosis, Amsterdam, The Netherland, 15-19.52014

39 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Molecular epidemiology of nosocomial Pneumocystis jirevecii infections – lessons
learned from case clusters in renal patients
Wieland Meyer1*, Carolina Firacative1, Sharon Chen1 & Brian J Nankivell2
1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Westmead Millennium Institute, Sydney Medical School-Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, University of Sydney, Westmead, NSW, Australia, 2Departments of Renal Medicine,
Westmead Hospital, Westmead Australia, *contact email: [email protected] Pneumocystis jirovecii pneumonia (PJP) is an important infection-related complication, whose mode of transmission remains uncertain. We encountered a cluster of fulminant PJP in 14 transplant recipients
occurring 6.3 5.3 years (mean SD) after transplantation, well beyond our 6-month trimethoprim-sulfamethoxazole (TMP-SMZ) prophylaxis period. Additional clusters then emerged in seven other Sydney hospitals (n=30 patients), three distant regional hospitals (n=8), four large interstate transplant units (n=24) and seven other interstate hospitals (n=13) over 2 years, eventually controlled by reintroduction of blanket TMP-SMZ prophylaxis to potentially-exposed patients. Eight further cases presented following discontinuation of prophylaxis in our long-term patients (two hospitals), after being disease-free for 14 months. Prophylaxis was again restarted and continued indefinitely. A total of 96 PJP infections occurred in kidney (n=87), liver (n=4), liver-kidney (n=1), and kidney-pancreas (n=3) transplant recipients, and one lupus patient in 23 hospitals; resulting in 14 deaths and 10 kidney allograft failures. Four-loci MLST typing of known variable regions within the P. jirovecii genome was undertaken to define the outbreak’s molecular epidemiology and an online MLST database was established at mlst.mycologylab.org. MLST analysis of the concatenated DNA sequences revealed an initial outbreak genotype (sequence type 1 [ST1]), and two closely related genotypes differing only by a single nucleotide polymorphism in either the mtLSU (ST2) or ITS1/2
region (ST9), while the -tub and DHPS sequences were identical. The last genotype (ST10), which emerged 52 months after the index case, differed by 6 nucleotides. Meticulous contact tracing found co-localization of asymptomatic prodromal PJP patients within our clinic waiting areas, and inter-hospital transmission facilitated by travel of infected patients with local cross-infection in distant locations, suggesting person-to-
person transmission. Minimal and maximal PJP incubation periods were 124 83 to 172 71 days, respectively. Oropharyngeal washes from outpatient staff and ambient air samples were negative for P. jirovecii DNA. Cohort analysis (14 exposed clinic cases versus 324 unaffected control patients) identified independent risk factors including prior CMV infection (OR=65.9, 95%CI=7.9-550, P<0.001), underlying pulmonary disease (OR=10.1, 95%CI=2.3-45.0, P=0.002) and transplant dysfunction (OR=1.61 per10mls/min/1.73m2, 95%CI=1.15-2.25, P=0.006). The outbreaks were controlled by extension of trimethoprim/sulphamethoxazole prophylaxis to 12 months in recent recipients and reintroduction to all clinic patients. Prompt recognition and pre-emptive blanket PJP prophylaxis to all exposed recipients after the second confirmed case are recommended to limit outbreak escalation. Most contemporary PJP cases actually represent a public heath problem, rather than a reactivation of an intrinsic latent infection by reduced immunity. We suggest centralised genotyping of all isolates to identify related clusters, and mandatory reporting to allow appropriate preventative counter-measures.

40 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
The emerging human pathogen Scedosporium aurantiacum in the light of its
global epidemiology and the area of genomics.
Wieland Meyer1
1Molecular Mycology Research Laboratory, Centre for Infectious Diseases and Microbiology, Westmead Millennium Institute, Sydney Medical School-Westmead Hospital, Marie Bashir Institute for Infectious Diseases and Biosecurity, University of Sydney, Westmead, NSW, Australia, email: [email protected] Scedosporium spp. are amongst the most important fungal infections for patients with cystic fibrosis (CF). Invasive Scedosporium infections are also problematic after near-drowning accidents and in transplant patients. Scedosporium aurantiacum is an emerging pathogen of humans and animals causing a wide range of invasive infections. It is commonly present in urban environments. To identify possible environmental sources of infections and to determine the spread of this emerging pathogen a better understanding of its epidemiology is required. A total of 188 clinical, environmental and veterinary S. aurantiacum isolates from Australia, Europe, Asia and America were characterised by multilocus sequence typing (MLST). The scheme
is based on the combined sequence analysis of six genes: actin (ACT), calmodulin (CAL), elogation factor-1
(EF1 ), RNA polymerase subunit II (RPB2), manganese superoxide dismutase (SOD2), and -tubulin (TUB) at:
mlst.mycologylab.org. Among this geographically diverse collection of 188 strains 22 ACT, 8 CAL, 23 EF1 , 15 RPB2, 18 SOD2 and 12 TUB alleles resulting in 159 unique sequence types were identified. Allele and sequence types can be determined online at http://mlst.mycologylab.org. Phylogenetic analysis revealed separate clustering of the Australian and two subgroups among European samples. The high diversity among the Australian strains suggests that S. aurantiacum may have originated within Australia and was subsequently dispersed to other regions. A fact revealed by the close phylogenetic relationships between some of the Australian sequence types and those found in other parts of the world. Animal studies, using the Galleria mellonella model, have revealed virulence differences among strains. To establish a molecular basis for future studies to understand the origin of those differences, we have sequenced and assembled for the first time the genome of S. auarantiacum strain WM 09.24 using different genome assemblers. To project the genome annotation and develop gene models we performed whole-genome alignment with the next closest species for which an annotated genome was available, Trichoderma virens. However, this species is still so distant that only approximately 30 % of the two genomes could be aligned through whole-genome alignment. Therefore, neither direct-gene projection nor BLAST searches could be applied in order to annotate the S. aurantiacum genome. To overcome this problem we performed RNA-sequencing of strain WM 09.24 under different growth conditions (to maximise the number of transcribed genes), and used the JAMg pipeline to annotate the genome. The primary annotation of the genome of strain WM 09.24 resulted in 2809 scaffolds, 10.515 genes, 11,639 gene transcripts (mRNAs) and a total genome size of 40,888kb. The annotated genome of strain WM 09.24 will be used to annotate three additional S. aurantiacum strains with varying virulence to enable a whole-genome comparison to identify genes or genetic signatures that are associated with virulence.

41 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
Genome analysis of Scedosporium aurantiacum
Meyer W1, Pérez-Bercoff Å1,2, Ramsperger M2, Firacative C1, Papanicolau A2, Patel HR2, Elbourne L3, Huttley GA2, and Nevalainen H3 1 Molecular Mycology Research Laboratory, CIDM, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, University of Sydney, Westmead Millennium Institute, Westmead, NSW, Australia; 2 John Curtin School of Medical Research, Australian National University, Canberra ACT, Australia; 3 Department of Chemistry and Biomolecular Sciences, Macquarie University Sydney, NSW, Australia E-mail: [email protected] Abstract. Scedosporium aurantiacum is an emerging pathogen of humans and animals. It is commonly present in urban environments. Animal studies, using the Galleria mellonella model, have revealed virulence differences among strains. To establish a molecular basis for future studies to understand the origin of those differences, we have sequenced and assembled for the first time the genome of S. auarantiacum strain WM 09.24 using different genome assemblers. To project the genome annotation and develop gene models we performed whole-genome alignment with the next closest species for which an annotated genome was available, Trichoderma virens. However, this species is still so distant that only approximately 30 % of the two genomes could be aligned through whole-genome alignment. Therefore, neither direct-gene projection nor BLAST searches could be applied in order to annotate the S. aurantiacum genome. To overcome this problem we performed RNA-sequencing of strain WM 09.24 under different growth conditions (to maximise the number of transcribed genes), and used the JAMg pipeline to annotate the genome. The annotated genome of strain WM 09.24 was used to identify functional genes in will is also being used to annotate three additional S. aurantiacum strains with varying virulence to enable a whole-genome comparison to identify genes or genetic signatures that are associated with virulence. Third meeting of the ECMM/ISHAM Working group on Fungal respiratory infections in cystic fibrosis, Angers, France, 5-6, June 2014

42 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS FUNGAL EPIDEMIOLOGY & INVASIVE INFECTIONS
An update on the Scedosporium aurantiacum MLST analysis
Meyer W1, Schwabenbauer K1, Harun A1,2 & Firacative C1
1 Molecular Mycology Research Laboratory, CIDM, Marie Bashir Institute for Infectious Diseases and Biosecurity, Sydney Medical School-Westmead Hospital, University of Sydney, Westmead Millennium Institute, Westmead, NSW, Australia; 2School of Medical Sciences, Universiti Sains Malaysia, Kota Bharu, Kelantan, Malaysia. E-mail: [email protected] Abstract. Scedosporium spp. are amongst the most important fungal infections for patients with cystic fibrosis (CF). Invasive Scedosporium infections are also problematic after near-drowning accidents and in transplant patients. Scedosporium aurantiacum is an emerging human pathogen causing a wide range of invasive infections. To identify possible environmental sources of infections and to determine the spread of this emerging pathogen a better understanding of its epidemiology is required. A total of 188 clinical, environmental and veterinary S. aurantiacum isolates from Australia, Europe, Asia and the America were characterised by multilocus sequence typing (MLST). The scheme is based on the combined sequence
analysis of six genes: actin (ACT), calmodulin (CAL), elogation factor-1 (EF1 ), RNA polymerase subunit II
(RPB2), manganese superoxide dismutase (SOD2), and -tubulin (TUB) at: mlst.mycologylab.org. Among this
geographically diverse collection 21 ACT, 8 CAL, 23 EF1 , 13 RPB2, 18 SOD2 and 12 TUB alleles resulting in 159 unique sequence types were identified. Allele and sequence types can be determined online at http://mlst.mycologylab.org. Phylogenetic analysis revealed separate clustering of the Australian and European samples. The high diversity among the Australian strains suggests that S. aurantiacum may have originated within Australia and was subsequently dispersed to other regions. A fact revealed by the close phylogenetic relationships between some of the Australian sequence types and those found in other parts of the world. DNA Barcoding workshop CBS, Utrecht the Netherlands, 22-23.4.2014

43 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
Detection of knockdown resistance (kdr) in Cimex lectularius and Cimex
hemipterus (Hemiptera: Cimicidae)
Kai Dang1, 2, Cheryl S Toi1, David G Lilly1, Chow-Yang Lee3, Richard Naylor4, Apiwat Tawatsin5,
Usavadee Thavara5, Wenjun Bu2 and Stephen L. Doggett1
1Department of Medical Entomology, Pathology West, ICPMR, Westmead Hospital, Westmead, NSW, Australia, 2145 2Institute of Entomology, Nankai University, Tianjin, China, 300071 3Urban Entomology Laboratory, Vector Control Research Unit, School of Biological Sciences, Universiti Sains Malaysia, Penang, Malaysia, 11800 4Prior's Loft, Coleford Road, Tidenham, Chepstow, Monmouthshire, UK, NP16 7JD 5National Institute of Health, Department of Medical Sciences, Nonthaburi, Thailand, 11000 Worldwide, there are many reports of pyrethroid resistance in bed bugs (Cimex spp.). Previous studies on
the Common bed bug, Cimex lectularius L., have identified two mutations (V419L and L925I) in the voltage-
gated sodium channel (VGSC) gene responsible for knockdown resistance (kdr). However, nothing is known
on possible kdr mutations in Australian strains of C. lectularius or on the Tropical bed bug, C. hemipterus F.
This study aimed to identify the status of kdr mutations in Australian C. lectularius strains, and C. hemipterus
in Australia and international strains. Samples of C. lectularius were obtained from 24 sites across Australia,
while the C. hemipterus were sourced from Australia, as well as Africa, India, Malaysia and Thailand. DNA
was extracted, purified and examined for the kdr-related genes by PCR and Sanger sequencing. In C.
lectularius field populations, the haplotypes A (neither V419L nor L925I), B (L925I only), and C (V419L and
L925I) were found, with most (88%) of the field populations being haplotype B. A novel mutation, I936F, was
identified in an ‘Adelaide’ strain (initially identified as haplotype A), which may be linked with kdr-type
resistance. In C. hemipterus, the V419L and L925I mutations were not detected, however, three novel
mutations, M918I (Methionine to Isoleucine), D953G (Aspartic acid to Glycine) and L1014F (Leucine to
Phenylalanine), were identified. Samples from Thailand have the three mutations, while samples from
Australia and India have both M918I and L1014F mutations. Only the L1014F mutation was evident in the
samples from Malaysia and Africa. The M918I and L1014F were assumed to be kdr mutations and contribute
to the high pyrethroid resistance in C. hemipterus. Further studies are in process to determine the non-kdr
type resistance mechanisms.
8th International Conference on Urban Pests, Zurich, Switzerland, 20 - 23 July 2014

44 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
Medical Entomology in Australia, 1964-2014
Stephen L Doggett
Department of Medical Entomology, CIDMLS, Pathology West – ICPMR Westmead, Westmead Hospital,
Westmead, NSW 2145.
Australia has had a rich history in the field of Medical Entomology, in both practical and applied research,
particularly over the last 50 years, which has placed the nation on the world stage. Innovations in taxonomy
has seen the change from the classical taxonomists, including Marks, Dobrotworsky and Lee (mosquitoes),
Kettle, Dyce and Debenham (Ceratopogonidae) and ticks (Roberts), to the development of proteomics
(including Bryan, Sweeney and Foley on mosquitoes), to modern molecular based taxonomy (Beebe again on
mosquitoes). The emphasis of research over the last 50 years in Medical Entomology has focused on
mosquitoes due to their nuisance biting and propensity to transmit infectious agents. This includes the
mapping of malaria vectors (Sweeney and colleagues), mosquito biology (notably Russell & Kay),
investigations into mosquito borne disease, surveillance and vector competence (most notably Marshal,
Mackenzie & colleagues, Cloonan, Russell & colleagues, and van den Hurk), saltmarsh mosquito
management (Dale and Easton in runnelling, Whelan and Muller in practical control), constructed wetlands
design to minimise vectors (Russell and Webb), repellents (Frances), and to modelling (Williams). In more
recent years, many of the research initiatives relating to Dengue and the control of its main vector, Aedes
aegypti, have been led by Ritchie and colleagues, with the development of such innovations as lethal and
biodegradable ovitraps, and new surveillance technology. More recently, Gates Foundation money has been
provided to Scott and colleagues to investigate the use of the intracellular bacteria, Wolbachia, to minimise
Dengue transmission. Outside of mosquitoes, researchers such as Tovey have investigated dust mites, while
various workers have examined ticks in their impact upon human morbidity (Graves on rickettsia, Russell et
al. on Lyme disease, van Nunen on mammalian meat allergies). Geary has led the way in providing a world
class pathology service and was the first to employ disinfected maggots for wound debridement. Finally, I
have proudly worked with a team who were the first in the world to develop an industry standard to combat
the global bed bug resurgence. This presentation will focus on the Medical Entomology researchers, their
research and achievements, during the 50 year history of the Australian Entomological Society.
Annual Conference of the Australian Entomological Society, Canberra, ACT, 28 September – 1 October 2014

45 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
High phylogenetic diversity of the cat flea (Ctenocephalidesfelis) at two
mitochondrial DNA markers Andrea Lee Lawrence1, Graeme Brown1, Derek Spielman1, Bryce Peters2, Victoria Morin-Adeline1, Jan Slapeta1 1The University of Sydney, Faculty of Veterinary Science Australia 2University of Technology, Faculty of Science, Sydney, Australia The cat flea, Ctenocephalides felis (Siphonaptera: Pulicidae) (Bouché), is the most common flea species found
on cats and dogs worldwide. We investigated the genetic identity of the cosmopolitan subspecies C. felis felis
and evaluated diversity of cat fleas from Australia, Fiji, Thailand and Seychelles using mtDNA sequences from
cytochrome c oxidase subunit I (cox1) and II (cox2) genes. Bothcox1 and cox2 confirmed the high
phylogenetic diversity and paraphyletic origin of C. felis felis. The African subspecies C. felis strongylus
(Jordan) is nested within the paraphyletic C. felis felis. The south East Asian subspecies C. felis orientis
(Jordan) is monophyletic and is supported by morphology. We confirm that Australian cat fleas belong to C.
felis felis and show that in Australia they form two distinct phylogenetic clades, one common with fleas from
Fiji. Using a barcoding approach, we recognize two putative species within C. felis (C. felis and C. orientis).
Nucleotide diversity was higher in cox1 but COX2 outperformed COX1 in amino acid diversity. COX2 amino
acid sequences resolve all phylogenetic clades and provide an additional phylogenetic signal. Both cox1 and
cox2 resolved identical phylogeny and are suitable for population structure studies of Ctenocephalides
species.
The Australian Society for Parasitology Annual Conference, Canberra, ACT, 30 June – 3 July 2014

46 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
The importance of methodology and strain selection when determining efficacy of
insecticides against bed bugs
David Lilly Stephen Doggett and Cameron Webb
Department of Medical Entomology, University of Sydney and PathologyWest – ICPMR Westmead,
Westmead Hospital, Westmead, NSW, Australia, 2145
Selection of an appropriate bioassay technique and insect strain(s) are known to be important factors when
attempting to accurately detect and monitor for insecticide resistance or define the efficacy of an insecticide.
Recent studies with both susceptible and resistant strains of the common bed bug, Cimex lectularius, have
indicated these principles similarly apply to bed bugs and must be considered prior to undertaking diagnostic
bioassays. Age, access to a blood meal, and the period since repletion may all influence the outcomes of
bioassays with bed bugs. Dry residual deposits of insecticides, in particular those of neonicotinoids, also have
the potential to overestimate resistance ratios or provide a false negative indication of efficacy when viewed
in comparison to more applicable topical or wet residual exposure methods. Resistance monitoring of
Australian field strains has also revealed that a wide spectrum in the magnitude of resistance can exist
between strains that express identical resistance mechanisms, and that laboratory strains held in culture for
long periods of time may lose resistance or change resistant genotypic frequencies. When factored in to the
proliferation of field strains with various combinations of multiple and/or cross resistance mechanisms, this
clearly presents a challenge to product manufacturers, registration bodies, and pest managers as to how
they can ensure the experimental methodology and strain selected is most appropriate for the desired
purpose or outcome. The results of laboratory investigations to provide informed guidance on
recommended ‘best practise’ bioassays with bed bugs will be presented.
Annual Meeting of the Entomological Society of America, Portland, Oregon, USA, 15-19 November 2014

47 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
Implications of insecticide resistance in Cimex lectularius (Hemiptera: Cimicidae),
in Australia
David G. Lilly1, Myron P. Zalucki2, Christopher J. Orton3, Richard C. Russell1, Cameron E. Webb1
and Stephen L. Doggett1
1Department of Medical Entomology, University of Sydney and PathologyWest – ICPMR Westmead, Westmead Hospital, Westmead, NSW, Australia, 2145,
2School of Biological Sciences, The University of Queensland, St Lucia, QLD, Australia, 4072 3School of Biological Earth and Environmental Sciences, University of NSW, NSW, Australia, 2052 Insecticide resistance in bed bugs (Cimex lectularius L. and C. hemipterus F.) has been nominated as a major
factor in the pest’s resurgence. Various studies across Europe, Asia and North America have demonstrated
resistance to the pyrethroids, carbamates and, to a lesser extent, the organophosphates. Resistance has
been suspected in Australia, with anecdotal reports of poor product performance. Laboratory studies
applying insecticides via topical application have demonstrated insecticide resistance in a previously
suspected-resistant field strain of bed bugs, with substantial differences in LD50 values when compared to a
susceptible strain. The resistance factors for each compound were: permethrin = 1.4 million, deltamethrin =
430,000, bendiocarb = 240, pirimiphos-methyl = 2.8, imidacloprid = 2.7. Thus resistance was confirmed in the
field-collected strain with the pyrethroids and carbamates, but not the organophosphates or neonicotinoids
(the small number being unrelated to resistance). With demonstrated resistance in bed bugs, professional
pest management operations require the development of new strategies to combat the pest. Regulatory
authorities must similarly consider the implications of resistance to multiple insecticide groups when
registering new products utilizing either existing or new modes of action. One of the gaps in our current
knowledge is an investigation of the drivers of this resistance and further research is required to elucidate
the breadth, magnitude and mechanisms of the resistance observed.
8th International Conference on Urban Pests, Zurich, Switzerland, 20 - 23 July 2014

48 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
An assessment of new approaches to arbovirus surveillance in NSW
Cheryl S Toi1, John Haniotis1, Cameron E Webb1,2, John Clancy1and Stephen L Doggett1
1Department of Medical Entomology, CIDMLS, Pathology West – ICPMR Westmead, New South Wales 2Marie Bashir Institute for Infectious Diseases and MicrobiologyUniversity of Sydney, New South Wales Until recently, the arbovirus surveillance in NSW has relied on cell culture of homogenates from mosquitoes
with definitive identification by FC-ELISA. While a reliable method, the turnaround time is relatively slow. A
new approach to arbovirus surveillance has been proposed to reduce laboratory turnaround times. Rather
than testing the mosquitoes, nucleic acid preservation cards (FTA), onto which trapped mosquitoes may
have expectorated virus, are tested using molecular assays. To complement this laboratory method has been
newly developed “passive traps” that allow for longer deployment in the field than the traditional “once per
week” approach using EVS traps. During the 2012-2014 surveillance period, we compared the two
approaches to both mosquito collection and arbovirus detection. Preliminary investigations indicated that
the use of FTA cards in EVS traps deployed for a single night per week provided as efficient approach to
arbovirus detection. Ross River and Barmah Forest viruses were all detected from FTA cards. Further work is
required to ensure appropriate quality assurance of molecular methods and to determine the implications of
new approaches as an early warning system for the issue of public health messages.
11th Mosquito Control Association of Australia Conference, Mandurah, Western Australia, 7-10 September
2014

49 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
Can social media extend the reach of public health messages?
Cameron E Webb1,2
1Department of Medical Entomology, CIDMLS, Pathology West – ICPMR Westmead, New South Wales 2Marie Bashir Institute for Infectious Diseases and MicrobiologyUniversity of Sydney, New South Wales Increasing the exposure of public health messages is critical. This is particularly the case for mosquito-borne
disease where advice on personal protection measures often informs the first line of defence against biting
mosquitoes. Traditional media has been the mainstay of communication efforts by local authorities but
could the use of social media provide a new vehicle for disseminating information and engaging with the
wider community? The aims of this study were to determine if promotion and engagement via social media
influenced how online information is accessed. A range of social media platforms, particularly Twitter, were
employed to disseminate public health messages and engage the community and traditional media outlets.
The total weekly exposure of “tweets” was measured for six months with approximately 40,000 people per
week received tweets with maximum exposure of almost 190,000 people in a single week. Engagement with
the accounts of traditional media (e.g. radio, print, television, online) was found to be the main route to
increased exposure and, subsequently, to increased access of public health information online. With the
increasing accessibility of the community to online resources via smartphones, researchers and public health
advocates must develop strategies to effectively use social media. Many people now turn to social media as
a source of news and information and those in the field of public health, as well as entomological research
more generally, must take advantage of these new opportunities.
Annual Meeting of the Entomological Society of America, Portland, Oregon, USA, 15-19 November 2014

50 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS MEDICAL ENTOMOLOGY
Managing mosquitoes in urban wetlands: Strategic control and social
communications
Cameron E Webb1,2 and Swapan Paul3
1Department of Medical Entomology, CIDMLS, Pathology West – ICPMR Westmead, New South Wales 2Marie Bashir Institute for Infectious Diseases and MicrobiologyUniversity of Sydney, New South Wales 3Sydney Olympic Park Authority, Sydney Olympic Park, New South Wales Mosquito management programs in urban wetlands need to find a balance between reducing nuisance-
biting and mosquito-borne disease risk with wetland conservation and rehabilitation. Community
engagement is critical. The mosquito management program at Sydney Olympic Park has been in place since
1998. The region contains extensive estuarine wetlands that provide ideal habitats for the saltmarsh
mosquito, Aedes vigilax. The region also has a growing residential and working population as well as
attracting millions of visitors each year. A coordinated treatment program associated with a carefully
designed surveillance program, including timely notifications to the public and stakeholders, ensures that
mosquito treatment is undertaken with minimal or no interruption to the community. Operational
efficiencies have ensured that both peaks in mosquito abundance and frequency of wetland treatments have
been minimised. Community engagement is a priority, not only with regard to mosquito awareness but
broader wetland conservation within the region. With increasing accessibility of online resources via
smartphones, local authorities must develop strategies to effectively use social media for communications.
In particular, the ability to easily engage with traditional media outlets through a network of social media
platforms further increases exposure beyond online communities. Examples from this region will be
discussed with implications for state and national community engagement strategies.
11th Mosquito Control Association of Australia Conference, Mandurah, Western Australia, 7-10 September
2014

51 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Pandemic clinical case definitions are non-specific: multiple respiratory viruses
circulating in the early phases of the 2009 influenza pandemic in New South
Wales, Australia.
Ratnamohan VM, Taylor J, Zeng F, McPhie K, Blyth CC, Adamson S, Kok J, Dwyer DE1.
1Centre for Infectious Diseases and Microbiology Laboratory Services, Institute of Clinical Pathology and Medical Research, Westmead Hospital, Westmead, New South Wales, Australia. [email protected]. BACKGROUND: During the early phases of the 2009 pandemic, subjects with influenza-like illness only had laboratory testing specific for the new A(H1N1)pdm09 virus. FINDINGS: Between 25th May and 7th June 2009, during the pandemic CONTAIN phase, A(H1N1)pdm09 virus was detected using nucleic acid tests in only 56 of 1466 (3.8%) samples meeting the clinical case definition required for A(H1N1)pdm09 testing. Two hundred and fifty-five randomly selected A(H1N1)pdm09 virus-negative samples were tested for other respiratory viruses using a real-time multiplex PCR assay. Of the 255 samples tested, 113 (44.3%) had other respiratory viruses detected: rhinoviruses 63.7%, seasonal influenza A 17.6%, respiratory syncytial virus 7.9%, human metapneumovirus 5.3%, parainfluenzaviruses 4.4%, influenza B virus 4.4%, and enteroviruses 0.8%. Viral co-infections were present in 4.3% of samples. CONCLUSIONS: In the very early stages of a new pandemic, limiting testing to only the novel virus will miss other clinically important co-circulating respiratory pathogens.

52 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Evaluation of the Sofia Influenza A + B fluorescent immunoassay for the rapid
diagnosis of influenza A and B.
Hazelton B1, Nedeljkovic G, Ratnamohan VM, Dwyer DE, Kok J. 1Centre for Infectious Diseases and Microbiology Laboratory Services, Institute of Clinical Pathology and Medical Research, Westmead Hospital, Westmead, New South Wales, Australia. Rapid influenza diagnostic tests (RIDTs) can facilitate the appropriate prescription of antivirals for influenza, obviate the need for unnecessary testing and antibacterial agents and allow the implementation of infection control measures. However, the reported sensitivities and specificities of different RIDTs vary widely in clinical settings, as does assay ability to distinguish between influenza types and subtypes. To evaluate the performance of the Sofia Influenza A + B fluorescent immunoassay (FIA) for the detection of influenza A and B during the 2013 Southern Hemisphere influenza season, a total of 209 consecutive respiratory tract swabs from adult patients with an influenza-like illness were tested by both Sofia Influenza A + B and an in-house real-time, reverse transcription-polymerase chain reaction (RT-PCR) assay. Compared to RT-PCR, the sensitivity and specificity of the Sofia Influenza A + B FIA for detection of influenza A was 72.4% and 98.3%, respectively. Too few influenza B positive samples were available during the study to accurately assess the Sofia's performance for influenza B detection. The sensitivity of Sofia Influenza A + B FIA for both influenza A and B detection correlated with the amount of influenza RNA present in the sample as indicated indirectly by the RT-PCR cycle threshold (Ct ). In conclusion, the Sofia Influenza A + B FIA continues to perform well as a RIDT with the circulating influenza strains of the 2013 Southern Hemisphere influenza season.

53 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Viral respiratory infections among Hajj pilgrims in 2013.
Barasheed O1, Rashid H, Alfelali M, Tashani M, Azeem M, Bokhary H, Kalantan N, Samkari J,
Heron L, Kok J, Taylor J, El Bashir H, Memish ZA, Haworth E, Holmes EC, Dwyer DE, Asghar A, Booy
R; Hajj Research Team.
National Centre for Immunisation Research and Surveillance (NCIRS), The Children's Hospital at Westmead, Westmead, NSW, Australia, [email protected]. Middle East respiratory syndrome coronavirus (MERS-CoV) has emerged in the Arabian Gulf region, with its epicentre in Saudi Arabia, the host of the 'Hajj' which is the world's the largest mass gathering. Transmission of MERS-CoV at such an event could lead to its rapid worldwide dissemination. Therefore, we studied the frequency of viruses causing influenza-like illnesses (ILI) among participants in a randomised controlled trial at the Hajj 2013. We recruited 1038 pilgrims from Saudi Arabia, Australia and Qatar during the first day of Hajj and followed them closely for four days. A nasal swab was collected from each pilgrim who developed ILI. Respiratory viruses were detected using multiplex RT-PCR. ILI occurred in 112/1038 (11%) pilgrims. Their mean age was 35 years, 49 (44%) were male and 35 (31%) had received the influenza vaccine pre-Hajj. Forty two (38%) pilgrims had laboratory-confirmed viral infections; 28 (25%) rhinovirus, 5 (4%) influenza A, 2 (2%) adenovirus, 2 (2%) human coronavirus OC43/229E, 2 (2%) parainfluenza virus 3, 1 (1%) parainfluenza virus 1, and 2 (2%) dual infections. No MERS-CoV was detected in any sample. Rhinovirus was the commonest cause of ILI among Hajj pilgrims in 2013. Infection control and appropriate vaccination are necessary to prevent transmission of respiratory viruses at Hajj and other mass gatherings

54 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Outcomes of influenza A(H1N1)pdm09 virus infection: results from two
international cohort studies.
Lynfield R1, Davey R2, Dwyer DE3, Losso MH4, Wentworth D5, Cozzi-Lepri A6, Herman-Lamin K5,
Cholewinska G7, David D8, Kuetter S9, Ternesgen Z10, Uyeki TM11, Lane HC2, Lundgren J12,
Neaton JD5; INSIGHT Influenza Study Group.Collaborators (259)
BACKGROUND: Data from prospectively planned cohort studies on risk of major clinical outcomes and prognostic factors for patients with influenza A(H1N1)pdm09 virus are limited. In 2009, in order to assess outcomes and evaluate risk factors for progression of illness, two cohort studies were initiated: FLU 002 in outpatients and FLU 003 in hospitalized patients. METHODS AND FINDINGS: Between October 2009 and December 2012, adults with influenza-like illness (ILI) were enrolled; outpatients were followed for 14 days and inpatients for 60 days. Disease progression was defined as hospitalization and/or death for outpatients, and hospitalization for >28 days, transfer to intensive care unit (ICU) if enrolled from general ward, and/or death for inpatients. Infection was confirmed by RT-PCR. 590 FLU 002 and 392 FLU 003 patients with influenza A (H1N1)pdm09 were enrolled from 81 sites in 17 countries at 2 days (IQR 1-3) and 6 days (IQR 4-10) following ILI onset, respectively. Disease progression was experienced by 29 (1 death) outpatients (5.1%; 95% CI: 3.4-7.2%) and 80 inpatients [death (32), hospitalization >28 days (43) or ICU transfer (20)] (21.6%; 95% CI: 17.5-26.2%). Disease progression (death) for hospitalized patients was 53.1% (26.6%) and 12.8% (3.8%), respectively, for those enrolled in the ICU and general ward. In pooled analyses for both studies, predictors of disease progression were age, longer duration of symptoms at enrollment and immunosuppression. Patients hospitalized during the pandemic period had a poorer prognosis than in subsequent seasons. CONCLUSIONS: Patients with influenza A(H1N1)pdm09, particularly when requiring hospital admission, are at high risk for disease progression, especially if they are older, immunodeficient, or admitted late in infection. These data reinforce the need for international trials of novel treatment strategies for influenza infection and serve as a reminder of the need to monitor the severity of seasonal and pandemic influenza epidemics globally

55 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
First outbreak of locally acquired hepatitis E virus infection in New South Wales,
Australia
Yapa C, Furlong C, Rosewell A, Ward K, Adamson S, Kok J, Bowden S, Shadbolt C, Smedley L, Ferson M, Staff M, Sheppeard V, McAnulty J BACKGROUND: Foodborne transmission of hepatitis E virus (HEV) has been previously reported in developed countries but not in Australia, where laboratory testing for HEV is generally only performed in persons with a history of overseas travel. In March 2014, two cases of HEV infection in patients who had not travelled overseas, but who were part of a larger group that dined at a single restaurant, were reported. We initiated an investigation to determine the source and extent of the outbreak. METHODS: In Australia, laboratory diagnoses of HEV infection (by IgG seroconversion, detection of HEV-specific IgM or nucleic acid, or isolation in culture), are notifiable to the public health authority. We interviewed notified HEV infected cases about potential risk factors including overseas travel, foods consumed and occupational history in the 2 to 9 weeks before symptom onset. Further cases were identified by serological testing and interviewing co-diners of cases, and by testing patients in whom viral hepatitis screening was requested at a major private and public reference laboratory, where hepatitis A, B, C, Epstein-Barr virus and cytomegalovirus infection had been excluded. Reported foods eaten by cases were compared with those reported by uninfected co-diners. Restaurant associated cases were genotyped using virus isolated from serum. Implicated foods were traced back to the source farm. RESULTS: Thirty-five cases were notified from October 2013 to June 2014. Eighteen cases reported eating at the implicated restaurant during their incubation periods. Pork liver pâté was the only food consumed by all 16 cases with complete food histories, compared with seven uninfected co-diners (RR 3.0, CI 0.3 – 33.5). Cases reported no other common food exposures. HEV was detected by nucleic acid testing in 11 of 18 cases - all were genotype 3. Sequencing showed 95 - 99% sequence similarity. Pork livers were traced back to a single farm. CONCLUSIONS: This is the first HEV outbreak in Australia associated with consumption of contaminated pork products. Clinicians in areas of low endemicity should consider the diagnosis of HEV in patients presenting with a compatible illness in the absence of overseas travel history. Further work is warranted on the prevalence of HEV in pork liver products, which in all cases should be thoroughly cooked.

56 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Viral exanthems
Caitlin L. Keighley1,2, Rebecca B. Saunderson3, Jen Kok1,4,5, Dominic E. Dwyer1,4,5 Purpose of review Determining the viral aetiology of a rash presents significant diagnostic challenges. We review contemporary literature on viral exanthems, and suggest a structured approach to aid diagnosis. Recent findings Strains responsible for, and the clinical presentation of enteroviral infections have diverged from classic descriptions. The causative relationship between antibiotic administration and rash in Epstein-Barr virus infection has been recently questioned. Major measles virus outbreaks have recently occurred in Europe and the United States of America. The largest Ebola virus outbreak in West Africa has resulted in importation of the virus to other countries and secondary local transmission. Autochthonous transmission of Chikungunya virus has occurred in non-endemic areas, including Europe, the Caribbean and Americas. Zika virus has re-emerged in the Pacific with local transmission from imported cases. Climate change, global warming and spillover of zoonotic viruses are contributing to the emergence and spread of viral diseases. Summary Important clues to the diagnosis of viral exanthems include their distribution and morphology, geographic location, and potential exposure to vector-borne or blood-borne viruses. Diagnosis is commonly made via serology, nucleic acid tests, or rarely, viral culture. Skin biopsy is not usually required. In general, viral exanthems are self-limiting and treatment is supportive.

57 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Detection of Influenza A and B with the Alere™ i Influenza A & B: A Novel
Isothermal Nucleic Acid Amplification Assay
Briony HAZELTONa*, Timothy GRAYa, Jennifer HOa, V. Mala RATNAMOHANa, Dominic E. DWYER
a,b,c, Jen KOK
We evaluated the Alere™ i Influenza A&B (Alere™ iNAT), a new rapid isothermal nucleic acid amplification assay for the detection of influenza A and B viruses. Using reverse transcriptase polymerase chain reaction (RT-PCR) as the reference standard, Alere™ iNAT detected 77.8% influenza A positives samples versus 71.4% and 44.4% for the Quidel Sofia® Influenza A+B fluorescent immunoassay and BinaxNOW® Influenza A & B immunochromatographic assay, respectively. For influenza B, Alere™ iNAT detected 75% of those positive with RT-PCR, versus 33.3% and 25.0% for Sofia® and BinaxNOW®, respectively. The specificity of Alere™ iNAT was 100% for influenza A and 99% for influenza B. Alere™ iNAT is a promising new rapid influenza diagnostic assay with potential point-of-care applications.

58 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Phylogenetic analysis of human rhinoviruses collected over four successive years
in Sydney, Australia
V Mala Ratnamohana, Frank Zenga, Linda Donovana, C Raina MacIntyreb, Jen Kokacd* and Dominic
E Dwyeracd
Background: Human rhinoviruses (HRV) cause a wide spectrum of disease, ranging from a mild influenza-like illness (ILI) to severe respiratory infection. Molecular epidemiological data is limited for HRV circulating in the Southern Hemisphere. Methods: Combined nose and throat swabs or nasopharyngeal aspirates collected from individuals with ILI were tested for HRV using real-time reverse-transcriptase polymerase chain reaction (RT-PCR). Sequencing data of 5’UTR and VP4/VP2 coding regions on RT-PCR positive specimens were analyzed. Results: HRV was detected by real-time PCR in 20.9% (116/555) of samples tested. Phylogenetic analysis of 2 genes, 5’UTR and VP4/VP2 on HRV-positive samples collected over 4 years (2006-2009) was concordant in the grouping of HRV A and B species. Discordance in the grouping of the newly described HRV C species was seen between the VP4/VP2 and 5’UTR genes. Eighty percent (16/20) of sequences that grouped as HRV C in the VP4/VP2 tree clustered as HRV A, alongside some previously described C strains as sub species C/A. Discordant branching was seen within HRV A group: two sequences clustering as A in the VP4/VP2 tree branched within the C/A sub-species in the 5’UTR tree, one sequence showed identity to different HRV A strains in the two genes. The prevalence of HRV C and C/A species was greater in pediatric compared to adult patients (47.9% vs. 25.5%, p=0.032). Conclusion: HRV are a common cause of respiratory infections, and HRV C is present in the Southern Hemisphere. Sequencing of multiple HRV regions may be necessary to determine exact phylogenetic relationships.

59 CIDM-PH ANNUAL REPORT 2014
ABSTRACTS, PROJECTS & WORKS IN PROGRESS VIRAL EPIDEMIOLOGY & DISEASE SURVEILLANCE
Enhanced detection of measles virus infection following successful development
of a real-time reverse transcriptase PCR in respiratory and urine samples using the
Cepheid SmartCycler®
Kyra Y. L. Chua1#,, Kiran Thapa1, Chaturangi M. Yapa2, Lucy K. Somerville1, Sharon C-A Chen1,3, Dominic E. Dwyer1,3,4, Vicky Sheppeard2, Jen Kok1,3,4 Despite the World Health Organization (WHO)-reported elimination of measles in Australia, importation of cases especially in travellers from Asia continues in Sydney, Australia’s largest city. Laboratory confirmation supports clinico-epidemiological evidence of measles virus infection, and is needed to establish elimination. Nucleic acid testing (NAT) with a random access real-time reverse transcriptase polymerase chain reaction (RT-PCR) assay using the SmartCycler® platform for the detection of measles virus from combined nose and throat swabs (NTS), nasopharyngeal aspirates (NPA) and urine samples was developed. One hundred samples collected from patients with suspected measles were tested in parallel using immunofluorescence antigen (IFA) detection and NAT with SmartCycler® and LightCycler® RT-PCR platforms. Where available, the performance of measles virus-specific IgM and IgG serology was also assessed. Using LightCycler® RT-PCR as the reference standard, measles virus was detected in 35 clinical samples. There was 100% concordance between the results of the SmartCycler® and the LightCycler®-based RT-PCR. The sensitivity and specificity of IFA compared to NAT was 34.3% and 96.5%, respectively. Testing urine in addition to NTS did not improve diagnostic yield. Measles-specific IgM was detected in 16/24 (66.7%) patients with specimens that were measles NAT positive. The performance of the SmartCycler® is comparable to the LightCycler® in the detection of measles virus. However, IFA had poor sensitivity and should not be used to confirm measles virus infection where NAT is available.

60 CIDM-PH ANNUAL REPORT 2014
CIDM-PH MANAGEMENT STRUCTURE

61 CIDM-PH ANNUAL REPORT 2014
CIDM-PH 2014 CURRENT GRANTS
TITLE INVESTIGATORS GRANT TYPE
INSTITUTION PERIOD
National Health and Medical Research Council (NHMRC) Project Grants
Black Death genomics and the evolution of pathogen virulence
Holmes E Poinar H Iredell J
Project
USYD
2014-2017
Signalling pathways and fungal virulence – the inositol polyphosphate kinase pathway in Cryptococcus neoformans
Djordjevic Saiardi Sorrell Lev
Project
USYD
2014-2016
The true burden of nosocomial staphylococcal disease: Genomic markers of transmission of methicillin-sensitive and –resistant Staphylococcus aureus
O’Sullivan MVN Gilbert GL Sinchenko V Holmes N van Hal S
Project
USYD
2014-2016
Rapid prediction of antibiotic resistancein the Enterobacteriaceae: making use of restricted diversity in mobile resistance gene pools
Iredell J Partridge S
Project USYD 2013-2015
Ecological effects of antibiotics
Iredell J Paulson I Partridge S Seppelt I Holmes A
Project USYD 2013-2015
Molecular epidemiology and high resolution surveillance of Salmonella enterica serovar Typhimurium in Australia
Lan R Sintchenko V Tanaka M Octavia S
Project USYD 2013-2015
Reducing the prevalence of TB in a highly endemic setting by community-wide active case finding: "turning off the tap"
Marks G Nguyen L Dinh S Nhung NV Sintchenko V Fox G Wood J
Project USYD
2013-2015
Mechanisms and markers of tuberculosis transmission within Australia
Sintchenko V Marais B Gilbert GL
Project USYD
2013-2015

62 CIDM-PH ANNUAL REPORT 2014
National Health and Medical Research Council (NHMRC) Project Grants
Microevolution &transmission of MRSA in a hospital setting
Gilbert Sintchenko O’Sullivan Iredell
Project USYD 2011-14
DNA Barcoding of pathogenic fungi as basis for the development of novel standardised diagnostic tools
Meyer Robert Ellis Chen
Project
USYD
2012-14
Uncovering the genetic basis for fungal virulence
Meyer Huttley Sorrell Nevalainen Kwon-Chung
Project
USYD 2012-14
Infectious Disease Informatics
Sintchenko Career Development Award
USYD 2011–14
Critical Infections Iredell Practitioner Fellowship
USYD 2011-14
Cell therapy for the prevention and treatment of invasive filamentous fungal infections in immunocompromised patients
Gottlieb D Sorrell TC
Project
USYD
2012-15
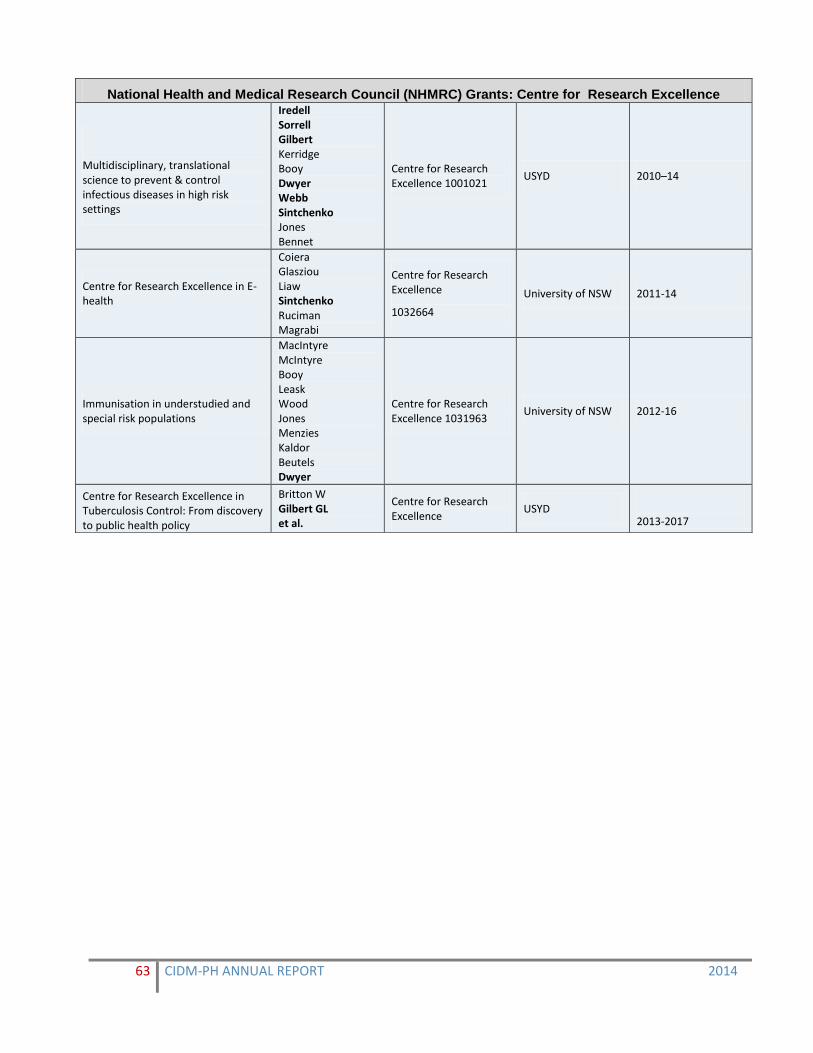
63 CIDM-PH ANNUAL REPORT 2014
National Health and Medical Research Council (NHMRC) Grants: Centre for Research Excellence
Multidisciplinary, translational science to prevent & control infectious diseases in high risk settings
Iredell Sorrell Gilbert Kerridge Booy Dwyer Webb Sintchenko Jones Bennet
Centre for Research Excellence 1001021
USYD 2010–14
Centre for Research Excellence in E-health
Coiera Glasziou Liaw Sintchenko Ruciman Magrabi
Centre for Research Excellence
1032664
University of NSW 2011-14
Immunisation in understudied and special risk populations
MacIntyre McIntyre Booy Leask Wood Jones Menzies Kaldor Beutels Dwyer
Centre for Research Excellence 1031963
University of NSW 2012-16
Centre for Research Excellence in Tuberculosis Control: From discovery to public health policy
Britton W Gilbert GL et al.
Centre for Research Excellence
USYD
2013-2017

64 CIDM-PH ANNUAL REPORT 2014
Non – NHMRC/ARC Grants ACGR
Population Health and Health Services Research Support Program
Gilbert Iredell Sorrell Dwyer Sintchenko Meyer Webb Douglas O’Sullivan
Research Support Grant
NSW Ministry of Health
Western Sydney Local Health District
2013-16
Staying Strong and Staying On! – A Psychosocial Screening Tool to Improve Uptake of HCV Treatment
M Douglas
Research Grants Program
Gilead Australia Fellowship
USYD
2014
A Pilot Study of Fenofibrate to Improve HCV Treatment Response
M Do ug las
Research Grant
Australian Centre for HIV and Hepatitis Virology Research (ACH
2)
USYD
2014
International Grants
Prediction of possible endemic areas for Cryptococcus neoformans/C. gattii in Colombia: ecological modelling of risk areas
Escandon Castaneda Meyer
Project Grant Colciencias, Colombia
Instituto Nacional de Salud, Bogotá, Colombia
2012-14
Functional genomic analyses of emerging Cryptococcus subtypes in North America
Keim Engelthaler Lockhart Meyer Thompson
Project Grant NIH USA 2012-14
Grants (not part of Aust Competitive grants register, but peer-reviewed)
Sydney Emerging Infections and Biosecurity Project
Sorrell Establishment S Med School Foundation
SEIB (University of Sydney)
2011-15

65 CIDM-PH ANNUAL REPORT 2014
CIDM-PH 2014 SENIOR INVESTIGATOR PUBLICATIONS
1. Read SA, Tay E, Shahidi M, George J, Douglas MW. Hepatitis C virus infection mediates cholesteryl ester synthesis to facilitate infectious particle production. J Gen Virol. 2014 Sep;95(Pt 9):1900-10. doi: 10.1099/vir.0.065300-0. Epub 2014 May 24
2. Sublette VA, Hopwood M, George J, Smith SK, Nicholson Perry K, McCaffery K, Douglas MW. Instrumental support to facilitate hepatitis C treatment adherence: Working around shortfalls in shared-care. Psychol Health Med. 2014 Jul 7:1-12.
3. Young S, Ojaimi S, Dunckley H, Douglas MW, Kok J, Fulcher DA, Lin MW, Swaminathan S. Vancomycin-associated drug reaction with eosinophilia and systemic symptoms syndrome. Intern Med J. 2014 Jul;44(7):694-6. doi: 10.1111/imj.12462
4. Shahidi M, Tay ES, Read SA, Ramezani-Moghadam M, Chayama K, George J, Douglas MW. Endocannabinoid CB1 antagonists inhibit hepatitis C virus production, providing a novel class of antiviral host-targeting agents. J Gen Virol. 2014 Nov;95(Pt 11):2468-79. doi: 10.1099/vir.0.067231-0. Epub 2014 Jul 22
5. Eltahla AA, Tay E, Douglas MW, White PA. Cross-genotypic examination of hepatitis C virus polymerase inhibitors reveals a novel mechanism of action for thumb binders. Antimicrob Agents Chemother. 2014 Dec;58(12):7215-24. doi: 10.1128/AAC.03699-14. Epub 2014 Sep 22
6. Beard MR, French R, Gowans EJ, Helbig KJ, Eyre NM, Douglas MW et al. (2014) A Summary of the 20th
International Symposium on Hepatitis C Virus and Related Viruses. Gastroenterology 147(1):e1-4
7. Eslam M, Leung R, Romero-Gomez M, Mangia A, Irving WL, Sheridan D, Douglas MW et al. (2014) IFNL3 polymorphisms predict response to therapy in chronic hepatitis C genotype 2/3 infection. J Hepatol. 2014 Aug;61(2):235–41.
8. O'Connor KS, Parnell G, Patrick E, Ahlenstiel G, Suppiah V, van der Poorten D, Read SA, Leung R, Douglas MW, Yang JY, Stewart GJ, Liddle C, George J, Booth DR (2014) Hepatic metallothionein expression in chronic hepatitis C virus infection is IFNL3 genotype-dependent. Genes and Immunity 15(2): 88-94
9. Douglas MW, Esmaili S, George J. (2014) A new role for IKK- in hepatitis C virus induced lipogenesis. Hepatology 59(5):2046-9
10. Sivagnanam S, van der Poorten D and Douglas MW. (2014) Hepatic lesions and eosinophilia in an urban dweller. Liver International 34(4):643.
11. O'Connor KS, Ahlenstiel G, Suppiah V, Schibeci S, Ong A, Leung R, van der Poorten D, Douglas MW, Weltman MD, Stewart GJ, Liddle C, George J, Booth DR. (2014) IFNL3 mediates interaction between innate immune cells: Implications for hepatitis C virus pathogenesis. Innate Immunity 20(6):598-605
12. Barasheed O, Rashid H, Alfelali M, Tashani M, Azeem M, Bokhary H, Kalantan N, Samkari J, Heron L, Kok J, Taylor J, El Bashir H, Memish ZA, Haworth E, Holmes EC, Dwyer DE, Asghar A, Booy R; Hajj Research Team. Viral respiratory infections among Hajj pilgrims in 2013. Virol Sin. 2014 Nov 14. [Epub ahead of print
13. Borges AH, O'Connor JL, Phillips AN, Baker JV, Vjecha MJ, Losso MH, Klinker H, Lopardo G, Williams I, Lundgren JD, Dwyer DE for the INSIGHT SMART and ESPRIT Study Groups and the SILCAAT Scientific Committee. Factors associated with D-dimer levels in HIV-infected individuals. PLoS One. 2014; 9(3):e90978. doi: 10.1371/journal.pone.0090978.
14. Branley JM, Weston KM, Englund J, Roy B, Dwyer DE, Sorrell TC. Clinical features of endemic community-
acquired psittacosis. New Microbes and New Infections 2014; doi:10.1002/2052-2975.29.
15. MacIntyre CR, Wang Q, Rahman BR, Seale H, Ridda I, Gao Z, Yang P, Shi W, Pang X, Zhang Y, Moa A Dwyer DE. Efficacy of face masks and respirators in preventing upper respiratory tract bacterial colonisation and co-infection in hospital health care workers. Preventive Medicine 2014; 62: 1-7.

66 CIDM-PH ANNUAL REPORT 2014
16. MacIntyre CR, Wang Q, Rahman BR, Seale H, Ridda I, Gao Z, Yang P, Shi W, Pang X, Zhang Y, Moa A Dwyer DE. Efficacy of face masks and respirators in preventing upper respiratory tract bacterial colonisation and co-infection in hospital health care workers – Authors reply. Preventive Medicine 2014; DOI: 10.1016/j.ypmed.2014.05.024.
17. Shi W, Cui S, Peng X, Dwyer DE, Huang F, Wang Q, Pang X, Deng Y. Evaluation of the FluA-Ag rapid assay for
detection of influenza A viruses of human, avian and swine origin. Clinical Laboratory 2014; 60:297-300.
18. Haskelberg H, Pocock N, Amin J, Ebeling PR, Emery S, Carr A, Dwyer DE; STEAL study investigators. Hip structural parameters over 96 weeks in HIV-infected adults switching treatment to tenofovir-emtricitabine or abacavir-lamivudine. PLoS One. 2014; 9(4):e94858. doi: 10.1371/journal.pone.0094858.
19. Barasheed
O, Rashid H, Heron L, Ridda I, Haworth E, Nguyen-Van-Tam J, Dwyer DE, Booy R, on behalf of the
Hajj Research Team. Influenza vaccination among Australian Hajj pilgrims: uptake, attitudes and barriers. Journal of Travel Medicine 2014; doi: 10.1111/jtm.12146.
20. Cheng AC, Dwyer DE, Holmes M, Irving L, Brown S, Waterer G, Korman T, Hunter C, Hewagama S, Friedman
ND, Wark P, Simpson G, Upham J, Bowler S, Senenayake S, Kotsimbos T, Kelly P. Influenza epidemiology, vaccine coverage and vaccine effectiveness in sentinel Australian hospitals in 2012: the Influenza Complications Alert Network (FluCAN). Communicable Diseases Intelligence. Commun Dis Intell Q Rep. 2014 Jun 30;38(2):E143-9
21. Lynfield R, Davey R, Dwyer DE, Losso MH, Wentworth D, Cozzi-Lepri A, Herman-Lamin K, Cholewinska G, David
D, Kuetter S, Ternesgen Z, Uyeki TM, Lane HC, Lundgren J, Neaton JD for the INSIGHT Influenza Study Group. Outcomes of influenza A(H1N1)pdm09 virus infection: Results from two international cohort studies. PLoS One 2014;9(7):e101785. doi:10.1371/journal.pone.0101785. eCollection 2014.
22. Dixit R, Khandaker G, Hay P, McPhie K, Taylor J, Rashid H, Heron L, Dwyer DE, Booy R. A randomized study of
standard versus double dose oseltamivir for treating influenza in the community. Antiviral Therapy 2014; doi: 10.3851/IMP2807.
23. Haskelberg H, Cordery DV, Amin J, Kelleher AD, Cooper DA, Emery S, Dwyer DE on behalf of the STEAL Study Group. HLA alleles association with changes in bone mineral density in HIV-1-infected adults changing treatment to tenofovir-emtricitabine or abacavir-lamuvudine. PLoS One. 2014; 9(3):e93333. doi: 10.1371/journal.pone.0093333
24. Hazelton B, Nedeljkovic G, Ratnamohan VM, Dwyer DE, Kok J. Evaluation of the Sofia A+B fluorescent
immunoassay for the rapid diagnosis of influenza A and B. Journal of Medical Virology 2014; doi:10.1002/jvm.23976.
25. Simpson S, Taylor B, Burrows J, Burrows S, Dwyer DE, Taylor J, Ponsonby AL, Blizzard L, Dwyer T, Pittas F, van
der Mei I. EBV and HHV6 reactivation is infrequent and not associated with MS clinical course. Acta Neurologica Scandinavica 2014. doi:10.1111/ane.12268.
26. Ratnamohan VM, Taylor J, Zeng F, McPhie K, Blyth CC, Adamson S, Kok J, Dwyer DE. Pandemic clinical case
definitions are non-specific: Multiple respiratory viruses circulating in the early phases of the 2009 influenza pandemic in New South Wales, Australia. Virology Journal 2014; 11:113. doi:10.1186/1743-422X-11-113.
27. Zhou F, Wang Q, Sintchenko V, Gilbert GL, O’Sullivan MVN, Iredell JR, Dwyer DE. Use of the 5’ untranslated
region and VP1 region to examine molecular diversity in enterovirus B species. Journal of Medical Microbiology 2014 Jul 18. pii: jmm.0.074682-0. doi: 10.1099/jmm.0.074682-0.

67 CIDM-PH ANNUAL REPORT 2014
28. Nordell AD, McKenna M, Borges ÁH, Duprez D, Neuhaus J, Neaton JD, Dwyer DE and the INSIGHT SMART, ESPRIT Study Groups and SILCAAT Scientific Committee. Severity of cardiovascular disease outcomes among patients with HIV is related to markers of inflammation and coagulation. Journal of the American Heart Association 2014;3(3):e000844. doi: 10.1161/JAHA.114.000844.
29. Azeem M, Tashani M, Barasheed O, Heron L, Hill-Cawthorne GA, Haworth E, Rashid H,
Dwyer DE, Booy R.
Knowledge, Attitude and Practice (KAP) survey concerning antimicrobial use among Australian Hajj pilgrims. Infectious Disorders – Drug Targets 2014;Jul 13.
30. Ridda
I, Heywood
AE, Hueston L, Dwyer
DE, MacIntyre
CR. The burden of pertussis in patients with and without
recurrent ischaemic vascular events. Infectious Disorders – Drug Targets 2014.
31. Barasheed O, Wang M, Rashid H, Booy R, El Bashir H, Haworth E, Ridda I, Holmes E, Dwyer DE, Nguyen-Van-Tam J, Memish ZA, Heron L. A cluster-randomised controlled trial to test the effectiveness of facemasks in preventing respiratory virus infection among Hajj pilgrims. Journal of Epidemiology and Global Health 2014.
32. Lam C, Octavia S, Sintchenko V, Gilbert GL, Lan R. Investigating genome reduction of Bordetella pertussis using a multiplex PCR-based reverse line blot assay (mPCR/RLB). BMC Res Notes. 2014 Oct 15;7:727. doi: 10.1186/1756-0500-7-727
33. Hooker LC, Mayes C, Degeling C, Gilbert GL, Kerridge IH Don't be scared, be angry: the politics and ethics of Ebola. Med J Aust. 2014 Sep 15;201(6):352-4
34. Gilbert GL. One moment doctor! Have you forgotten hand hygiene? Med J Aust. 2014 Sep 1;201(5):265-6. PMID: 25163373
35. Zhou F, Wang Q, Sintchenko V, Gilbert GL, O'Sullivan MV, Iredell JR, Dwyer DE. Use of the 5' untranslated region and VP1 region to examine the molecular diversity in enterovirus B species. J Med Microbiol. 2014 Oct;63(Pt 10):1339-55. doi: 10.1099/jmm.0.074682-0. Epub 2014 Jul 18. PMID: 25038138
36. Chan WSW, Chua SC, Gidding HF, Ramjan D, Wong MYW, Olma T, Thomas L, Gilbert GL. Rapid identification of group B streptococcus carriage by PCR to assist in the management of women with prelabour rupture of membranes in term pregnancy. ANZ J Obstet Gynaecol 2014;52;138-45
37. Lochindarat S, Teeratakulpisarn J, Warachit B, Chanta C, Thapa K, Gilbert GL, Wangroongsarb Y, et al. Bacterial etiology of empyema thoracis and parapneumonic pleural effusion in Thai children aged less than 16 years. Southeast Asian J Trop Med Pub Health 2014;45:442-6.
38. Xue G, Wang Q, Yan C, Jeoffreys N, Wang L, Gilbert GL, Sun H. Molecular characterizations of Mycoplasma pneumoniae positive specimen collected from Australia and China. J Clin Microbiol 2014; 52:1478-82
39. Chen X-Y, Wang B, Yang W, Kong F-R, Li C-Y, Sun Z-G, Jelfs P, Gilbert GL. Application of rolling circle amplification for direct detection of rpoB gene mutations in Mycobacterium tuberculosis directly from clinical specimens J Clin Microbiol. 2014;52:1540-48.
40. Lam C, Octavia S, Ricafort L, Sintchenko V, Gilbert GL, Wood N, et al. Rapid increase in pertactin deficient Bordetella pertussis isolates, Australia. Emerg Infect Dis 2014;20:626-633
41. One moment doctor! Have you forgotten hand hygiene? Gilbert GL. Med J Aust 2014;200:508-9.
42. Read P, Jeoffreys N, Tagg K, Guy RJ, Gilbert GL, Donovan B. Azithromycin resistant syphilis-causing strains in Sydney: prevalence and risk factors. J Clin Microbiol 2014;52:2776-81.
43. Hazelton B, Thomas LC, Olma T, Kok J, O'Sullivan M, Chen S, Iredell JR. Rapid and Accurate Direct Antibiotic Susceptibility Testing of Blood Culture Broths using MALDI Sepsityper™ combined with the BD Phoenix™ Automated System. J Med Microbiol. 2014 Sep 11. pii: jmm.0.075580-0. doi: 10.1099/jmm.0.075580-0. [Epub ahead of print]
44. Zhou F, Wang Q, Sintchenko V, Gilbert GL, O'Sullivan MV, Iredell JR, Dwyer DE Use of the 5' untranslated region and VP1 region to examine the molecular diversity in enterovirus B species. J Med Microbiol. 2014 Oct;63(Pt 10):1339-55. doi: 10.1099/jmm.0.074682-0. Epub 2014 Jul 18.

68 CIDM-PH ANNUAL REPORT 2014
45. Shoma S, Kamruzzaman M, Ginn AN, Iredell JR, Partridge SR. Characterization of multidrug-resistant Klebsiella pneumoniae from Australia carrying blaNDM-1. Diagn Microbiol Infect Dis. 2014 Jan;78(1):93-7
46. Watts MR, James G, Sultana Y, Ginn AN, Outhred AC, Kong F, Verweij JJ, Iredell JR, Chen SC, Lee R. A loop-mediated isothermal amplification (LAMP) assay for Strongyloides stercoralis in stool that uses a visual detection method with SYTO-82 fluorescent dye. Am J Trop Med Hyg. 2014 Feb;90(2):306-11
47. Gilroy N, Iredell J. The clinical and public health challenge of Gram-negative resistance in Australasia. Future Microbiol. 2014 Jan;9(1):17-20
48. Ginn AN, Wiklendt AM, Zong Z, Lin RT, Teo JW, Tambyah PA, Peterson LR, Kaul K, Partridge SR, Iredell JR. Prediction of major antibiotic resistance in Escherichia coli and Klebsiella pneumoniae in Singapore, USA and China using a limited set of gene targets. Int J Antimicrob Agents. 2014 Jun;43(6):563-5
49. Tagg KA, Iredell JR, Partridge SR. Complete Sequencing of IncI1 Sequence Type 2 Plasmid pJIE512b Indicates Mobilization of blaCMY-2 from an IncA/C Plasmid. Antimicrob Agents Chemother. 2014 Aug;58(8):4949-4952
50. Guo X, Dillon BB, Ginn AN, Wiklendt AM, Partridge SR, Iredell JR. Simple multiplex real-time PCR for rapid detection of common 16S rRNA methyltransferase genes. Diagn Microbiol Infect Dis. 2014 Jun 5. pii: S0732-8893(14)00242-9
51. Lizarazo J, Escandón P, Agudelo CI, Firacative C, Meyer W, Castañeda E. Retrospective Study of the Epidemiology and Clinical Manifestations of Cryptococcus gattii Infections in Colombia from 1997-2011. PLoS Negl Trop Dis. 2014 Nov 20;8(11):e3272. doi: 10.1371/journal.pntd.0003272. eCollection 2014 Nov
52. Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014 Oct;27(4):980-1024. doi: 10.1128/CMR.00126-13
53. Vandeputte P, Ghamrawi S, Rechenmann M, Iltis A, Giraud S, Fleury M, Thornton C, Delhaès L, Meyer W, Papon N, Bouchara JP. Draft Genome Sequence of the Pathogenic Fungus Scedosporium apiospermum. Genome Announc. 2014 Oct 2;2(5). pii: e00988-14. doi: 10.1128/genomeA.00988-14
54. Engelthaler DM, Hicks ND, Gillece JD, Roe CC, Schupp JM, Driebe EM, Gilgado F, Carriconde F, Trilles L, Firacative C, Ngamskulrungroj P, Castañeda E, dos Santos Lazera M, Melhem MSC, Pérez-Bercoff Å, Huttlley G, Sorrell TC, Voelz K, May RC, Fisher MC, Thompson GR, III, Lockhart SR, Keim P, Meyer W (2014). Cryptococcus gattii in North American Pacific Northwest: whole-population genome analysis provides insights into species evolution and dispersal. mBio 5(4):e01464-14. doi:10.1128/mBio.01464-14
55. Thompson III GR, Albert N, Hodge G, Wilson MD, Sykes JE, Bays DJ, Firacative C, Meyer W, and Kontoyiannis DP (2014): Phenotypic Differences of Cryptococcus Molecular Types: implications for virulence in a Drosophila model of infection. Infect. Immun. published ahead of print 5 May 2014, Doi:10.1128/IAI.01805-14
56. Trilles L, Wang B, Firacativa C, dos Santos Lazera M, Wanke B, Meyer W (2014): Identification of the major molecular types of Cryptococcus neoformans and Cryptococcus gattii by hyberbranched rolling circle amplification. PLoSONE 9(4):e94648 (DOI: 10.1371/journal.pone.0094648)
57. Chen S, Nankivel B, Firacative C, Kable K, Marriott D, McDonald P, Meyer W, Chapman J (2014): Hospital-acquired Pneumocystis pneumonia: a renewed concern? Microbiology Australia 35(1): 57-59.
58. Maduro AP, Mansinho K, Teles F, Silva I, Meyer W, Martins ML, Inacio J (2014): Insights on the genotype distribution among Cryptococcus neoformans and C. gattii Portuguese clinical isolates. Current Microbiology 68:199-203.
59. Robert V, Vu
D, Amor ABH, van de Wiele N, Brouwer C, Jabas B, Szoke S, Dridi A, Triki M, Daoud
S, Chouchen O,
Vaas L, de Cock A, Stalpers J, Stalpers D, Verkleij G, Groenwald M, Borges dos Santos F, Stegehuis G, Li W, Zhang R, Ma J, Zhou M, Pérez Gorjón S, Eurwilaichit L, Ingsriswang S, Hansen K, Schoch C, Robbertse B, Irinyi
L,
Meyer W, Cardinali G, Hawskworth DL, Taylor J, Crous
P (2013): MycoBank gearing up for new horizons. IMA
Fungus 4(2): 371-379.

69 CIDM-PH ANNUAL REPORT 2014
60. Zhou F, Wang Q, Sintchenko V, Gilbert GL, O’Sullivan MV, Iredell JR, and Dwyer DE. Use of the 5’ Untranslated Region and VP1 Region to Examine the Molecular Diversity in Enterovirus B Species. J Medical Microbiol, 2014 Jul 18; doi: 10.1099/jmm.0.074682-0.
61. Holmes N, Turnidge J, Munckhof W, Robinson JO, Korman T, O'Sullivan MV, Anderson T, Roberts R, Warren S, Coombs G, Tan H, Gao W, Johnson P, and Howden B. Genetic and molecular predictors of high vancomycin minimum inhibitory concentration in Staphylococcus aureus bacteremia isolates. J Clin Microbiol 2014; 52(9):3384-93.
62. Groves M, O'Sullivan MVN, Brouwers H, Chapman T, Abraham S,; Trott D, Coombs G, Skov R, Jordan D. Staphylococcus aureus ST398 detected in pigs in Australia. J Antimicr Chemother 2014. 69(5)1426-8.
63. Lam C, Octavia S, Sintchenko V, Gilbert GL, Lan R Investigating genome reduction of Bordetella pertussis using a multiplex PCR-based reverse line blot assay (mPCR/RLB). 2014 Oct 15;7:727. doi: 10.1186/1756-0500-7-727
64. Rhoads DD, Sintchenko V, Rauch CA, Pantanowitz L.Clinical microbiology informatics. Clin Microbiol Rev. 2014
Oct;27(4):1025-47. doi: 10.1128/CMR.00049-14
65. Gurjav U, Jelfs P, McCallum N, Marais BJ, Sintchenko V. Temporal dynamics of Mycobacterium tuberculosis genotypes in New South Wales, Australia. BMC Infectious Diseases 2014;14:455.
66. Gray T, Kong F, Jelfs P, Sintchenko V, Chen SCA. Improved identification of rapidly growing mycobacteria by a
16S-23S internal transcribed spacer region PCR and capillary gel electrophoresis. PLoS One 2014;9(7):e102290.
67. Muscatello DJ, Amin J, MacIntyre CR, Newall AT, Rawlinson WD, Gilmour R, Sintchenko V, Thackway S. Inaccurate ascertainment of morbidity and mortality due to influenza in administrative databases, a population-based record linkage study. PLoS ONE 2014
68. Ho J, Jelfs P, Sintchenko V. Fluoroquinolone resistance in non multidrug-resistant tuberculosis - a surveillance
study in New South Wales Australia and a review of global resistance. International Journal of Infectious Diseases 2014
69. Gray TJ, Kong F, Jelfs P, Sintchenko V, Chen SCA. Improved identification of rapidly growing mycobacteria by a
16S-23S internal transcribed spacer region PCR and capillary gel electrophoresis. PLoS One 2014
70. Marieke J Bart, Simon R Harris, Abdolreza Advani, Yoshichika Arakawa, Daniela Bottero, Valérie Bouchez,
Pamela K Cassiday, Chuen-Sheue Chiang, Tine Dalby, Norman K. Fry, María Emilia Gaillard, Marjolein van Gent,,
Nicole Guiso, Hans O Hallander, Eric T. Harvill, Qiushui He, Han GJ van der Heide, Kees Heuvelman, Daniela F Hozbor, Kazunari Kamachi, Gennady I Karataev, Ruiting Lan, Anna Lutyoska, Ram P Maharjan, Jussi Mertsola, Tatsuo Miyamura, Sophia Octavia, Michael A. Quail, Vitali Sintchenko, Paola Stefanelli, M Lucia Tondella, Raymond SW Tsang, Yinghua Xu, Shu-Man Yao,Shumin Zhang, Julian Parkhill, Frits R. Mooi.
. Global population
structure and evolution of Bordetella pertussis and their relationship with vaccination. MBio 2014;5(2):e01074-14.
71. Lev S, Crossett B, Cha SY, Desmarini D, Li C, Chayakulkeeree M, Wilson CF, Williamson PR, Sorrell TC, Djordjevic JT. Identification of Aph1, a phosphate-regulated, secreted, and vacuolar acid phosphatase in Cryptococcus neoformans. MBio. 2014 Sep 16;5(5):e01649-14. doi: 10.1128/mBio.01649-14.
72. Halliday CL, Sorrell TC, Chen SC. Detection of multiple fungal species in blood samples by real-time PCR: an interpretative challenge. J Clin Microbiol. 2014 Sep;52(9):3515-6. doi: 10.1128/JCM.01685-14.
73. Banerjee A, Mor SM, Kok J, Sorrell TC, Hill-Cawthorne GA. Vulnerability, hysteria and fear - conquering Ebola virus. Med J Aust. 2014 Sep 15;201(6):320-1.
74. Branley JM, Weston KM, England J, Dwyer DE, Sorrell TC. Clinical features of endemic community-acquired psittacosis. New Microbes New Infect. 2014 Jan;2(1):7-12. doi: 10.1002/2052-2975.29. Epub 2014 Jan 13.

70 CIDM-PH ANNUAL REPORT 2014
75. Saijo, T., Chen, J., Chen, S., Rosen, L., Yi, J., Sorrell, T., Bennett, J., Holland, S., Browne, S., Kwong-Chung, K. (2014). Anti-GM-CSF autoantibodies are a risk factor for central nervous system infection by cryptococcus gattii in otherwise immunocompetent patients. mBio, Accepted for publication 11/2/14.
76. Biswas, C., Zuo, X., Chen, S., Schibeci, S., Forwood, J. Jolliffe, K., Sorrell, T., Djordjevic, J. (2014). Functional
disruption of yeast metacaspase, Mca1, leads to miltefosine resistance and inability to mediate miltefosine-induced apoptotic effects. Fungal Genetics and Biology, 67, 71-81.
77. Gray T.J. and Webb C.E. (2014) A review of the epidemiological and clinical aspects of West Nile virus.
International Journal of General Medicine, 7:193-203.
78. Lilly D.G., Zalucki M., Orton C.J. Russell R.C., Webb C.E. and Doggett S.L. (2014). Confirmation of Insecticide Resistance in Cimex lectularius L. (Hemiptera: Cimicidae) in Australia. Austral Entomology, early view. DOI: 10.1111/aen.12098
79. Watts MR, Chan RCF, Cheong EYL, Brammah S, Clezy KR, Tong C, Marriott D, Webb CE, Chacko B, Tobias V, Outhred AC, Field AS, Prowse MV, Bertouch JV, Stark D, and Reddel SW. (2014). Anncaliia algerae Microsporidial Myositis. Emerging Infectious Diseases 20: 185-191.
BOOK CHAPTERS
1. Sintchenko V, Roper MPV. Pathogen Genome Bioinformatics. In: Trent RJA (Ed). Clinical Bioinformatics, Methods in
Molecular Biology, vol.1168 (2nd
Edition). Humana Press, Springer Science 2014. PP. 173-93. ISBN 978-1-4939-0846-2
2. Webb C.E. (2014). Insect repellents derived from Australian plants and implications for public health messages. In. Insect Repellents Handbook, Second Edition (Ed M. Debboun et al) CRC Press [Publication Date: 5 September 2014].

71 CIDM-PH ANNUAL REPORT 2014
CIDM-PH 2014 EDUCATION & TRAINING
CIDM-Public Health holds a number of seminars, workshops, and symposia each
year. Programs and selected PowerPoint presentations are available at:
www.cidmpublichealth.org
7th April 2014 THE PHYLOGENY AND METAGENOMICS OF MICROBIAL DARK MATTER SEMINAR Speaker: A/Professor Aaron E. Darling, ithree institute, University of Technology Sydney
20th June 2014 SYMPOSIUM: LABORATORY DIAGNOSIS AND SURVEILLANCE OF DRUG-RESISTANT INFECTIONS SYMPOSIUM Speakers: Prof Tom Gottlieb, Infectious Diseases Concord Hospital, Clinical School, University of Sydney, Prof Jon Iredell, CRE in Critical Infection, CIDM Westmead Hospital, MBI USYD, Dr Andrew Ginn, CRE in Critical Infection, CIDM Westmead Hospital, Prof Ben Howden, Infectious Diseases and Microbiology, Austin Health, MDU, University of Melbourne, Prof Iain Gosbell, Microbiology and Infectious Diseases, Liverpool Hospital, School of Medicine, University of Western Sydney, Dr Nadine Holmes, CIDM Westmead Hospital, MBI USYD, Prof David Lewis, Western Sydney Sexual Health, Westmead Hospital, Sydney Medical School, University of Sydney, A/Prof Monica Lahra, SEALS Pathology, Faculty of Medicine, University of NSW, Ms Kaitlin Tagg, CRE in Critical Infection, CIDM Westmead Hospital, University of Sydney, A/Prof Sharon Chen, Director CIDMLS, Westmead Hospital, Dr Elena Martinez, TB-CRE Centenary Institute, CIDM Westmead Hospital, Dr Kyra Chua, CIDM Westmead Hospital
12th September 2014 CHASING THE TIGER AND THE PURSUIT OF INNOVATIVE SOLUTIONS TO MOSQUITO-BORNE DISEASE THREATS SYMPOSIUM
Speakers: Assoc. Prof Dina Fonseca, Rugters University, NJ, USA, Assoc. Prof Andrew van den Hurk, QLD Health and University of Queensland, Dr Cameron Webb, Pathology West – ICPMR Westmead and University of Sydney, Stephen Doggett, Pathology West – ICPMR Westmead, Dr Cheryl Toi, Pathology West – ICPMR Westmead
10th October 2014 WHOLE GENOME SEQUENCING IN CLINICAL AND PUBLIC HEALTH MICROBIOLOGY SYMPOSIUM Speakers: Prof Vitali Sintchenko, Centre for Infectious Diseases and Microbiology – Public Health, Dr Amy Jennison, Forensic and Scientific Services, Queensland Department of Health, Professor Eddie Holmes, Marie Bashir Institute for Infectious Diseases and Biosecurity and The University of Sydney, Dr Grant Hill-Cawthorne, Marie Bashir Institute for Infectious Diseases and Biosecurity and School of Public Health, University of Sydney, Dr Qinning Wang, Centre for Infectious Diseases and Microbiology – Public Health, Dr Connie Lam, Centre for Infectious Diseases and Microbiology – Public Health, Dr Torsten Seemann, Microbiological Diagnostic Unit, University of Melbourne, Dr Nadine Holmes, Centre for Infectious Diseases and Microbiology – Public Health and The University of Sydney, Dr Rosemarie Sadsad, Centre for Infectious Diseases and Microbiology – Public Health, Dr Nico Petty, The ithree Institute, University of Technology Sydney, Dr Sally Partridge, CRE in Critical Infections, Westmead Millenium Institute, University of Sydney, Dr Sophie Octavia, University of NSW

72 CIDM-PH ANNUAL REPORT 2014
CIDM-PH 2014 NEWSLETTERS
Broad Street Pump Newsletters
The following Broad Street Pump newsletters were distributed during 2014, and are available on the
CIDM-Public Health website www.cidmpublichealth.org
June 2014 Issue 35
Hospitals in the community - a matter of public health
L Gilbert
Preventing infections Bacteraemia Battle
L Gilbert (International Innovation)
Implementation of MRSA Control Bundle
K Dempsey & J Tallon
Innovations for MRSA control in a Neonatal Intensive Care Unit
K Dempsey & J Tallon
MRSA strain typing project related abstracts
MV O’Sullivan, AN Pinto & R Sadsad
August 2014 Issue 36
What can Australia learn from the introduction of chikungunya virus to North America?
Cameron E Webb
An assessment of new approaches to arbovirus surveillance in NSW
Cheryl S Toi
Medical Entomology in Australia, 1964-2014
Stephen L Doggett
Implications of insecticide resistance in Cimex lectularius (Hemiptera: Cimicicdae), in Australia
David G Lilly
Detection of knockdown resistance (kdr) in Cimex lectularius and Cimex hemipterus (Hemiptera: Cimicidae)
Kai Dang
Managing mosquitoes in urban wetlands: Strategic control and social communications
Cameron E Webb
High phylogenetic diversity of the cat flea (Ctenocephalidesfelis) at two mitochondrial DNA markers
Andrea Lee Lawrence
Solving the common mystery of the cat flea and their role in public health risks
Andrea Lee Lawrence

73 CIDM-PH ANNUAL REPORT 2014
CIDM-PH 2014 NEWSLETTERS
Broad Street Pump Newsletters
The following Broad Street Pump newsletters were distributed during 2014, and are available on the
CIDM-Public Health website www.cidmpublichealth.org
November 2014 Issue 37
Zoonotic infection meets health inequality: the emergence of Ebola in West Africa
Dr Grant Hill-Cawthorne
Unprecendented Ebolavirus disease outbreak demands concerted international and local responses
Prof Lyn Gilbert
Don’t panic about Ebola’s spread, here’s what we can do instead
Dr Grant Hill-Cawthorne
Prof Lyn Gilbert
Quarantine works against Ebola but over-use risks disaster
Dr Grant Hill-Cawthorne

74 CIDM-PH ANNUAL REPORT 2014
CIDM-PH 2014 PROMOTION
2014 CIDM-PH Brochure:
A copy of the 2014 CIDM-PH Brochure can be found at the CIDM-PH Website:
www.cidmpublichealth.org

75 CIDM-PH ANNUAL REPORT 2014
CONTACT US
CIDM-PH Contact Information
A/PROF VITALI SINTCHENKO
CIDM-PH DIRECTOR
MRS LOU ORSZULAK
CIDM-PH PROJECT OFFICER
Email: [email protected]
Tel: (612) 9845 6255
Email: [email protected]
Tel: (612) 9845 9870
Centre for Infectious Diseases & Microbiology – Public Health (CIDM-PH)
Street Address: CIDM-PH, Level 3
Institute of Clinical Pathology & Medical Research
Westmead Hospital
Hawkesbury Road
Westmead
Postal Address: PO Box 533, Wentworthville
New South Wales, Australia 2145
Ph: (02) 9845 9870
Fax: (02) 9893 8659
Website: www.cidmpublichealth.org
Email: [email protected]


















