
Chimeras of the Flp and Cre Recombinases: Tests of the Mode of ...
22
Chimeras of the Flp and Cre Recombinases: Tests of the Mode of Cleavage by Flp and Cre A. C. Shaikh and Paul D. Sadowski* Department of Molecular and Medical Genetics, University of Toronto, Toronto, M5S 1A8 Canada The Flp and Cre recombinases are members of the integrase family of tyrosine recombinases. Each protein consists of a 13 kDa NH 2 -terminal domain and a larger COOH-terminal domain that contains the active site of the enzyme. The COOH-terminal domain also contains the major determinants for the binding specificity of the recombinase to its cognate DNA binding site. All family members cleave the DNA by the attach- ment of a conserved nucleophilic tyrosine residue to the 3 0 -phosphate group at the sites of cleavage. In order to gain further insights into the determinants of the binding specificity and modes of cleavage of Flp and Cre, we have made chimeric proteins in which we have fused the NH 2 -terminal domain of Flp to the COOH-terminal domain of Cre (‘‘Fre’’) and the NH 2 -terminal domain of Cre to the COOH-terminal domain of Flp (‘‘Clp’’). These chimeras have novel binding specificities in that they bind strongly to hybrid sites con- taining elements from both the Flp and Cre DNA targets but poorly to the native target sites. In this study we have taken advantage of the unique binding specifici- ties of Fre and Clp to examine the mode of cleavage by Cre, Flp, Fre and Clp. We find that the COOH-terminal domain of the recombinases deter- mines their mode of cleavage. Thus Flp and Clp cleave in trans whereas Cre and Fre cleave in cis. These results agree with the studies of Flp and with the cocrystal structure of Cre bound to its DNA target site. They disagree with our previous findings that Cre could carry out trans clea- vage. We discuss the variations in the experimental approaches in order to reconcile the different results. # 2000 Academic Press Keywords: Flp; Cre; site-specific recombination; DNA binding; chimeric proteins *Corresponding author Introduction The integrase family of conservative site-specific recombinases is comprised of over 100 members (Abremski & Hoess, 1992; Argos et al., 1986; Esposito & Scocca, 1997; Nunes-Duby et al., 1998). These proteins all use a conserved catalytic tryo- sine residue to break and covalently attach to their target sequences at specific phosphodiester bonds (Craig, 1988; Evans et al., 1990; Gronostajski & Sadowski, 1985; Landy, 1989; Sadowski, 1993, 1995). The target sequences contain two inverted binding sites each of which binds a molecule of the recombinase. Although cleavage can take place in a dimeric structure consisting of two recombinase molecules bound to one target sequence (Andrews et al., 1987; Lee et al., 1994; Qian et al., 1990), recom- bination takes place in a synaptic structure consist- ing of two target sites each bound by two molecules of recombinase (Amin et al., 1991; Guo et al., 1997, 1999; Hamilton & Abremski, 1984; Hoess et al., 1990b). The crystal structure of the Cre synapse reveals an extensive series of cyclic NH 2 - terminal and COOH-terminal interactions that establish both ‘‘cross-core’’ interactions between the two protomers bound to the same lox site and ‘‘synaptic’’interactions between the Cre molecules bound to the two lox sites in the synapse (Guo et al., 1997). Two of the most extensively characterized mem- bers of the integrase family are the Flp protein encoded by the 2 m plasmid of yeast (Sadowski, 1995) and the Cre protein of the bacteriophage P1 (Hoess & Abremski, 1990). The first step in the recombination reaction is the site-specific binding E-mail address of the corresponding author: [email protected] doi:10.1006/jmbi.2000.3967 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 302, 27–48 0022-2836/00/010027–22 $35.00/0 # 2000 Academic Press
Transcript of Chimeras of the Flp and Cre Recombinases: Tests of the Mode of ...

doi:10.1006/jmbi.2000.3967 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 302, 27±48
Chimeras of the Flp and Cre Recombinases: Tests ofthe Mode of Cleavage by Flp and Cre
A. C. Shaikh and Paul D. Sadowski*
Department of Molecular andMedical Genetics, University ofToronto, Toronto, M5S 1A8Canada
E-mail address of the [email protected]
0022-2836/00/010027±22 $35.00/0
The Flp and Cre recombinases are members of the integrase family oftyrosine recombinases. Each protein consists of a 13 kDa NH2-terminaldomain and a larger COOH-terminal domain that contains the active siteof the enzyme. The COOH-terminal domain also contains the majordeterminants for the binding speci®city of the recombinase to its cognateDNA binding site. All family members cleave the DNA by the attach-ment of a conserved nucleophilic tyrosine residue to the 30-phosphategroup at the sites of cleavage.
In order to gain further insights into the determinants of the bindingspeci®city and modes of cleavage of Flp and Cre, we have made chimericproteins in which we have fused the NH2-terminal domain of Flp to theCOOH-terminal domain of Cre (``Fre'') and the NH2-terminal domain ofCre to the COOH-terminal domain of Flp (``Clp''). These chimeras havenovel binding speci®cities in that they bind strongly to hybrid sites con-taining elements from both the Flp and Cre DNA targets but poorly tothe native target sites.
In this study we have taken advantage of the unique binding speci®ci-ties of Fre and Clp to examine the mode of cleavage by Cre, Flp, Fre andClp. We ®nd that the COOH-terminal domain of the recombinases deter-mines their mode of cleavage. Thus Flp and Clp cleave in trans whereasCre and Fre cleave in cis. These results agree with the studies of Flp andwith the cocrystal structure of Cre bound to its DNA target site. Theydisagree with our previous ®ndings that Cre could carry out trans clea-vage. We discuss the variations in the experimental approaches in orderto reconcile the different results.
# 2000 Academic Press
Keywords: Flp; Cre; site-speci®c recombination; DNA binding; chimericproteins
*Corresponding authorIntroduction
The integrase family of conservative site-speci®crecombinases is comprised of over 100 members(Abremski & Hoess, 1992; Argos et al., 1986;Esposito & Scocca, 1997; Nunes-Duby et al., 1998).These proteins all use a conserved catalytic tryo-sine residue to break and covalently attach to theirtarget sequences at speci®c phosphodiester bonds(Craig, 1988; Evans et al., 1990; Gronostajski &Sadowski, 1985; Landy, 1989; Sadowski, 1993,1995). The target sequences contain two invertedbinding sites each of which binds a molecule of therecombinase. Although cleavage can take place ina dimeric structure consisting of two recombinasemolecules bound to one target sequence (Andrews
ing author:
et al., 1987; Lee et al., 1994; Qian et al., 1990), recom-bination takes place in a synaptic structure consist-ing of two target sites each bound by twomolecules of recombinase (Amin et al., 1991; Guoet al., 1997, 1999; Hamilton & Abremski, 1984;Hoess et al., 1990b). The crystal structure of the Cresynapse reveals an extensive series of cyclic NH2-terminal and COOH-terminal interactions thatestablish both ``cross-core'' interactions betweenthe two protomers bound to the same lox site and``synaptic''interactions between the Cre moleculesbound to the two lox sites in the synapse (Guoet al., 1997).
Two of the most extensively characterized mem-bers of the integrase family are the Flp proteinencoded by the 2 m plasmid of yeast (Sadowski,1995) and the Cre protein of the bacteriophage P1(Hoess & Abremski, 1990). The ®rst step in therecombination reaction is the site-speci®c binding
# 2000 Academic Press

28 Chimeras of the Flp and Cre Recombinases
of the recombinase to its cognate recognition site,the FRT site for the Flp protein and the lox site forthe Cre protein (Figure 1(a) and (b)). Although thesequences of the sites differ, they share an identicalorganization consisting of two inverted 13 bpsequences (``symmetry elements'') surrounding an8 bp core region (Hoess et al., 1984, 1986; Lee &Saito, 1998; Vetter et al., 1983).
Early gel mobility shift and footprinting exper-iments established the 13 bp symmetry elements asthe recognition elements for Flp and Cre (Andrewset al., 1987; Mack et al., 1992). Hoess et al. (1990a)showed that the NH2-terminal domain of Cre pro-tected the core region of the lox site whereas theCOOH-terminal domain bound to the symmetryelement. Missing contact probing of a single sym-metry element showed that the core-proximal 4 bpof the symmetry were important for the binding ofintact Cre but not Cre25, the COOH-terminaldomain (Hoess et al., 1990a). The core-distal 9 bpwere needed for binding of both intact Cre andCre25 (Shaikh, 1997). Crosslinking studies usingthe NH2 and COOH-terminal domains of Flpshowed a similar bipartite structure of the FRTsymmetry element. The core-proximal 4 bp wereimportant for the binding of the NH2-terminaldomain, P13 and the COOH-terminal domain, P32bound to the core-distal 9 bp (Panigrahi &Sadowski, 1994).
In order to gain further insight into the mechan-ism of binding and cleavage by the Flp and Crerecombinases, we have constructed chimerasbetween the two recombinases. The Fre proteincontains the NH2-terminal 13 kDa of Flp fused tothe COOH-terminal 25 kDa of Cre. The Clp proteincontains the NH2-terminal 13 kDa of Cre fused tothe 32 kDa COOH-terminal domain of Flp. Each ofthe chimeras binds site-speci®cally to hybrid sitescomposed of sequence elements from the targetsite of Flp and Cre, the FRT and lox sites. Thesenovel binding speci®cities enabled us to examinethe mode of cleavage by Flp, Cre, Fre and Clp.
The members of the integrase family of recombi-nases all employ a conserved nucleophilic tyrosinethat breaks the scissile phosphodiester bond andcovalently attaches the protein to the 30-phosphorylend at the site of the break (Argos et al., 1986;Esposito & Scocca, 1997; Nunes-Duby et al., 1998).However the family members display a diversityof mechanisms of cleavage (Jayaram, 1997;Jayaram & Lee, 1995). Cis cleavage means that themolecule of the recombinase that donates the cata-lytic tyrosine is bound immediately adjacent to thescissile bond. Trans cleavage implies that therecombinase molecule that donates the tyrosine isnot bound next to the scissile bond but resides else-where in the synaptic structure.
The Flp recombinase was the ®rst to be studiedin this respect and all tests have showed that itcleaves in trans (Chen et al., 1992; Lee et al., 1994,1999; Pan et al., 1993). This implies the active siteof Flp is formed by contributions from two Flpprotomers: the Flp molecule bound next to the
bond to be cleaved activates that bond to receivethe catalytic tyrosine from another Flp protomerbound to the target FRT sequence. While initialexperiments on the l integrase were compatiblewith a trans mode of cleavage (Han et al., 1993),subsequent experiments using Holliday junctionsgave unequivocal evidence of cis cleavage (Nunes-Duby et al., 1994). The XerC/XerD recombinasealso gave clear evidence for cis cleavage(Arciszewska & Sherratt, 1995). Our initial comple-mentation experiments also indicated that the Crerecombinase could cleave in trans (Shaikh &Sadowski, 1997). However, the cocrystal structureof Cre that was covalently attached to its DNA tar-get showed that the cleavage had occurred in cis(Guo et al., 1997).
The studies showing trans cleavage by Flp andCre made use of complementation experiments inwhich a given molecule of recombinase was posi-tioned upon a full or half-target site (Chen et al.,1992; Shaikh & Sadowski, 1997). Trans cleavagewas demonstrated when one molecule of recombi-nase donated its catalytic tyrosine to anotherrecombinase protomer that was unable to cleavedue to a mutation and was bound to a differentsite.
Such complementation experiments are subjectto the possible artifact that trans cleavage occursbecause the design of the experiment precludes ciscleavage. The experiments with l integrase andHolliday junctions gave unequivocal resultsbecause they made use of two integrases (l andHK022) of different binding speci®cities and theauthors were thereby able to position the proteinsat known sites in the Holliday substrate (Nunes-Duby et al., 1994). Likewise the XerC and XerDproteins have different binding speci®cities andtherefore one can be certain of their positionduring the cleavage experiment (Arciszewska &Sherratt, 1995).
In the present study we have made use of thechimeric recombinases Clp and Fre to re-examinethe mode of cleavage by Cre and Flp. We exploitedthe altered binding speci®cities of the Clp and Freproteins to position them on hybrid bindingelements called lrt and fox. We isolated heterodi-meric complexes containing one molecule of eachrecombinase and assayed for covalent attachmentof the recombinases to the target site. We ®nd thatthe Cre and Fre proteins cleave in cis whereas theFlp and Clp proteins cleave in trans. We concludethat the COOH termini of the Cre and Flp proteinsdetermine their mode of cleavage.
Results
Production of chimeric proteins Fre and Clp
Proteolysis of the Cre and Flp proteins hadsuggested a bidomainal structure for each protein(Hoess et al., 1990a; Pan & Sadowski, 1993; Panet al., 1991). Furthermore, previous studies (Hoesset al., 1990a; Panigrahi & Sadowski, 1994; Shaikh,

Chimeras of the Flp and Cre Recombinases 29
1997) as well as the recent cocrystal structure ofCre (Guo et al., 1997), suggested that each domainhad a unique role in DNA binding. Therefore wedecided to attempt to construct novel recombinasesby fusing the NH2-terminal domains of Flp andCre to the COOH-terminal domain of Cre and Flp,respectively. We made a plasmid construct thatfused the DNA encoding the NH2-terminal domainof Flp (P13) to the COOH-terminal domain of Cre(Cre25) to give the Fre chimera. Another plasmidencoded Clp, a chimera in which the NH2-terminaldomain of Cre (Cre13) was fused to the COOH-terminal domain of Flp (P32, see Figure 2(a) and(b)).
As can be seen in Figure 2(c) and (d), both chi-meras were produced in large amounts whenexpressed as His-tagged proteins transcribed fromthe phage T7 promoter. Each protein was con-veniently puri®ed using nickel-af®nity chromatog-raphy (see Materials and Methods).
Substrates used to characterize binding ofnative and chimeric recombinases
Before attempting complementations betweenCre or Flp and Fre or Clp, we used gel mobilityshift assays to characterize the binding activities ofthe chimeras on a variety of substrates. Theseexperiments provided an estimate of the relativebinding af®nity of each hybrid protein for the loxand FRT sites and allowed us to design compositebinding substrates that were used later in the testsof the mode of cleavage.
The target sequence of Cre is called lox(Figure 1(a)) and consists of two inverted 13 bpsymmetry elements (inverted arrows) surroundingan 8 bp core region (open box). The sequences ofthe lox symmetry elements are shown in green,with the core-proximal 4 bp underlined. The loxcore region is shown in magenta. The two cleavagesites for Cre are within the core and are 6 bp apart(vertical arrows, Figure 1(a)) The minimal recombi-nation sequence for Flp is called FRT (Figure 1(b))and has a similar organization to lox. The FRTsymmetry elements are shown in red with thecore-proximal 4 bp underlined. The 8 bp coreregion is in blue and the cleavage sites for Flp are8 bp apart (vertical arrows, Figure 1(b)). Previousfootprinting and cross-linking studies hadsuggested that the NH2-terminal domains of Flpand Cre contacted the core-proximal nucleotidesand the COOH-terminal domains contacted thecore-distal regions of the symmetry elements(Hoess et al., 1990a; Panigrahi & Sadowski, 1994;Shaikh, 1997). We therefore reasoned that hybridsymmetry elements of the FRT and lox sites mightprovide unique binding targets for the Fre and Clpchimeras (see Figure 3).
The test substrates ArkZ, ArkP, ArkF, ArkL andfox2b (Figure 1(c)-(g)), contained portions of bothlox and FRT as indicated. A hybrid symmetryelement consisting of the core-proximal 4 bp of theright-hand FRT symmetry element and the core-
distal 9 bp of the lox symmetry element is calledfox. The hybrid symmetry element consisting of thecore-proximal 4 bp of the lox symmetry elementand the core-distal 9 bp of the FRT symmetryelement is called lrt.
Binding of proteins to the ArkZ substrate
The substrate ArkZ (Figure 1(c)) contains asingle Cre binding element and a Flp bindingelement ¯anking a lox core. We incubated ArkZwith Fre, Clp, Cre, Flp, Cre25 and P32, and ana-lyzed the complexes using gel mobility shiftassays. As expected, Cre and Flp each formed asingle complex (cI Cre and cI Flp) caused by bind-ing of a single protein molecule to its cognate sym-metry element (Figure 4, lanes 2 and 3, 6 and 7).Interestingly, incubation of ArkZ with both pro-teins produced the expected cI complexes of Creand Flp, but very little of the mixed complex II(cII, lanes 12 and 13). The amount of complex for-mation can be compared with the binding of thesame amounts of Cre and Flp to the full-lox andfull-FRT sites (Figure 5(a) and (b)), sites shown inFigure 1(a) and (b), where both proteins readilygenerate dimeric complexes (cII) and where Crealso generates higher order (HO) complexes thatare most likely the result of protein-dependentsynaptic assemblies (Shaikh, 1997; Wierzbicki et al.,1987).
The His-Fre and His-Clp proteins bound weaklyto the ArkZ substrate but, like their parentalrecombinases, generated only complex Is (Figure 4,lanes 4 and 5, 8 and 9). Note that the presence ofthe His-tag on both chimeras allowed us to resolvethe migration of the four different protein-depen-dent complex Is in the polyacrylamide gel. Sinceonly the COOH-terminal domains of both Cre andFlp are able to form protein:DNA complexes invitro (Hoess et al., 1990a; Pan & Sadowski, 1993),the Fre and Clp proteins were most likely interact-ing with the core-distal lox and FRT elements inthe substrate. To be sure that the ArkZ substratewas capable of interacting with the COOH-term-inal domains of Fre and Clp, we incubated the sub-strate with isolated Cre25 or His-P32 peptide. Notonly did Cre25 and His-P32 generate monomercomplexes with the ArkZ substrate, but each pro-tein demonstrated apparently greater af®nity forthe binding sites in this substrate than the compar-able chimera (Figure 4, lanes 20-23). As the nativeCre and Flp proteins have a ®ve to ten times great-er af®nity for their cognate sites than their COOH-terminal domains (Hoess et al., 1990a; Pan &Sadowski, 1993), the reduced binding af®nity ofthe Fre and Clp proteins for the lox and FRTelements may represent an inhibitory effect of theheterologous NH2-terminal domain on the COOH-terminal binding domains of Cre and Flp in thechimeric proteins (see Discussion).
As noted above, incubation with Cre and Flprevealed the complex Is of each protein as well as

Figure 1 (legend opposite)
30 Chimeras of the Flp and Cre Recombinases
a weak slower migrating complex, cII (Figure 4,lanes 12 and 13). Binding of a molecule of Cre orFlp seems to exclude binding of a second molecule.Alternately, the absence of cII may re¯ect theabsence of cooperativity between the two proteins(see Discussion). A small amount of mixed com-plex II was observed when His-Fre and Flp wereincubated together with ArkZ (Figure 4, lanes 16and 17). The slightly slower migration of this com-plex II is due to the presence of the additional His-
tag in the Fre-Flp mix. In contrast, Cre and His-Fregenerated no mixed complex IIs (Figure 4, lanes 10and 11) and a barely detectable amount of cI withHis-Fre. Similar results were observed with a mix-ture of Flp and His-Clp (Figure 4, lanes 18 and 19).We had expected the mixture of Cre and His-Clpto generate a heterodimeric complex II, but onlycomplex I of each protein was observed (Figure 4,lanes 14 and 15), perhaps due to the very weakbinding of Clp to the substrate.

Chimeras of the Flp and Cre Recombinases 31
Binding of proteins to the lox substrate
We next used a full-lox site (Figure 1(a)) to testthe abilities of Fre and Clp to bind to the lox site.As expected, Cre readily forms monomer (cI),dimer (cII) and higher order (HO) complexes on itscognate site (Figure 5(a), lanes 2 and 3). The Flpprotein forms the analogous complex I and II onits cognate FRT site, though little higher ordercomplex is seen (Figure 5(b)). As with the singlelox element-containing site ArkZ, the His-Fre pro-tein bound weakly to the lox site compared to Creand the Cre25 peptide (Figure 5(a), lanes 4 and 5,lane 7). Despite the decreased binding af®nity ofHis-Fre for the lox site, the binding of the chimerawas cooperative, as formation of complex II (cIIFre) was favored over complex I (cI Fre). In con-trast to the binding of Cre25, protein-protein inter-actions between Fre monomers also generatedsome higher order complexes.
The Clp protein was unable to bind the lox site,consistent with both the absence of a Cre COOH-terminal binding domain in the fusion protein andthe inability of the NH2-terminal domain to conferaf®nity to the heterologous COOH-terminaldomain (Figure 5(a), lane 6).
Binding of the proteins to the ArkP substrate
We then examined the ability of the native andchimeric recombinases to bind to the ArkP sub-strate (Figure 6). The substrate contains a lox core¯anked by the left lox symmetry element and a foxelement (Figure 1(d)). The fox element contains the4 bp FRT core-proximal sequence and the 9 bpcore-distal sequence of the lox site. This fox sym-metry element should provide the binding regionsfor both the P13 and Cre25 portions of the Fre chi-mera.
Neither Flp nor Clp was able to bind to theArkP substrate because of the absence of the core-distal sequence of the FRT symmetry element inthe substrate (Figure 6, lanes 8-10).
The modi®ed lox symmetry element contained inthe ArkP substrate dramatically affected Cre bind-
Figure 1. The lox and FRT-based substrates used in this sEach was 35 nucleotides in length. Where required, the 50-eSymmetry elements (horizontal arrows) are derived from tregions are underlined. lox and FRT core regions (open boxthe putative cleavage sites by Cre or Flp on the native lox anfor the Cre protein contains two identical, inverted 13 bp loThe cleavage sites are 6 bp apart. (b) Full-FRT DNA substinverted 13 bp FRT symmetry elements (a and b) that ¯anapart. The two symmetry elements differ by a single base-pThe hybrid substrate consists of a lox core region ¯anked bymetry element from the FRT site. (d) ArkP DNA substrate. Tby the left symmetry element of the lox site and a composiproximal 4 bp of the FRT a symmetry element and the coresubstrate. The hybrid substrate consists of a lox core region ¯composite binding element, lrt, which is derived from the ccore-distal 9 bp of the FRT symmetry element. (f) ArkL DNAregion ¯anked by the left symmetry element of the FRT sitesubstrate. The substrate contains two inverted 13 bp fox sym
ing (Figure 6, lanes 2 and 3). The degree of coop-erativity previously observed with Cre binding tothe full-lox substrate (Figure 5(a), lanes 2 and 3)was diminished as the Cre-bound complexesfavored the monomeric cI over the dimeric cII. Fur-thermore, no Cre-dependent higher order com-plexes were observed. In contrast, the His-Freprotein exhibited cooperativity (Figure 6, lanes 4and 5) and bound with two to four times higheraf®nity to the ArkP substrate than to the full-loxsubstrate (Figure 5(a), lanes 4 and 5). Incubationwith both Cre and His-Fre generated a complex IIband (cII Mix, Figure 6, lanes 6 and 7) that was dis-tinct from both the Cre and Fre homodimer com-plexes. The intermediate migration of the mixedcomplex between the homodimer complexes of Creand His-Fre was consistent with the binding of onemonomer each of Cre and His-Fre to the lox andfox elements of the ArkP substrate. Indeed, the het-erodimeric cII of Cre and His-Fre was favored overeach of the homodimer species (Figure 6, cf. lanes2, 4 and 6).
Binding of proteins to the ArkF substrate
We designed a second hybrid substrate to testthe binding of the native and chimeric proteins.The ArkF substrate was similar to the ArkP sub-strate described above, except that the fox site wasreplaced with another composite symmetryelement, lrt (Figure 1(e)). This lrt element containedthe core-proximal region of a lox symmetryelement and the core-distal region of an FRTelement and therefore contained the recognitionelements for both the NH2 and COOH-terminaldomains of the Clp protein. We performed gelmobility shift assays with this substrate using theCre, Flp, Cre25, His-P32, His-Fre and His-Clp pro-teins and the results are shown in Figure 7.
Cre generated copious amounts of the mono-meric cI and, interestingly, formed some dimericcII as well (Figure 7, lanes 2 and 3) when higherlevels of Cre protein were used. As with the singlelox symmetry element-containing ArkZ substrate,
tudy. The sequences of the oligonucleotides are shown.nd was labeled as described in Materials and Methods.he lox site (green) or the FRT site (red). Core-proximales) are magenta and light blue. Vertical arrows indicated FRT sites. (a) Full-lox DNA substrate. The cognate site
x symmetry elements that ¯ank an 8 bp lox core region.rate. The cognate site for the Flp protein contains twok an 8 bp FRT core region. The cleavage sites are 8 bpair in the core-proximal region. (c) ArkZ DNA substrate.the left symmetry element of the lox site and the a sym-he hybrid substrate consists of a lox core region ¯anked
te binding element, fox, which is derived from the core--distal 9 bp of the lox symmetry element. (e) ArkF DNAanked by the left symmetry element of the lox site and aore-proximal 4 bp of the lox symmetry element and the
substrate. The hybrid substrate consists of an FRT coreand the composite binding element, fox. (g) fox2b DNA
metry elements ¯anking an FRT core region.

Figure 2. Peptide maps of proteins used in this study and the puri®cation of Fre and Clp. (a) Peptide maps of Flpand Cre. Flp and Cre are divided into NH2-terminal domains called P13 and Cre13 and COOH-terminal regionscalled P32 and Cre25. The amino acid residues included in each region are shown. The four conserved catalytic resi-dues are shown for Flp and Cre. (b) Peptide maps of Fre and Clp. The chimeras were constructed by swapping theheterologous NH2-terminal domains between Cre and Flp. The Fre protein results from the fusion of the NH2-term-inal domain of Flp and the COOH-terminal domain of Cre while the Clp protein results from the fusion of the NH2-terminal domain of Cre to the COOH-terminal domain of Flp. The amino acid residues from each parent protein areshown. The four conserved catalytic residues in the two chimeric proteins are shown with Cre or Flp numbering.(c) Puri®cation of His-tagged Clp and Fre. Puri®cation of His-Clp and His-Fre from an induced culture by Ni-NTA-af®nity chromatography. Marker lanes contain standards of the indicated molecular mass (kDa). Load lanes representthe soluble fraction of the total cell protein that was loaded on the Ni-NTA-agarose column. Pass lanes represent the¯ow-through. His-Clp was eluted in the 150 to 200 mM imidazole-containing fractions. His-Fre protein was eluted by175 and 200 mM imidazole. Both proteins were greater than 90 % pure at this point. Clp and Fre-containing fractionswere concentrated by spin dialysis as described in Materials and Methods.
32 Chimeras of the Flp and Cre Recombinases
Cre25 generated a single complex with the ArkFsubstrate and His-Fre bound weakly to form amonomeric cI (Figure 7, lanes 10-13).
No complex was formed when the ArkF sub-strate containing the lrt symmetry element wasincubated with Flp (Figure 7, lanes 8 and 9). Incontrast, the His-Clp protein formed copiousamounts of monomer complexes with the ArkFsubstrate (Figure 7, lanes 4 and 5): by phosphori-mager quanti®cation, the amount of complex I gen-erated by Clp on the lrt site was about 20 timesthat observed in the Clp reaction with the FRTsymmetry element in the ArkZ substrate (cf.Figure 4, lanes 8 and 9). Clp formed no dimericcomplex II, showing that the protein does not bindto the lox symmetry element of the ArkF substrate.
The incubation of Cre and His-Clp with theArkF substrate generated abundant amounts of aheterodimeric complex (cII Mix) in addition to theindividual complex I bands attributable to thebinding of Cre and His-Clp (Figure 7, lanes 6 and7). The mixed complex II consists of one Cre mono-mer bound to the lox symmetry element and oneHis-Clp protein bound to the lrt symmetry elementin the ArkF substrate.
Binding of proteins to the ArkL substrate
While both the ArkP and ArkF substrates wereessentially modi®ed lox substrates, it was also ofinterest to examine the binding of the chimeric pro-teins to an altered FRT substrate. The ArkL sub-

Figure 3. Scheme of binding of native and chimeric recombinases to symmetry elements. The two domains areshown in black (Cre) or gray (Flp). The sequences of the symmetry elements (horizontal arrows) are shown for thenative lox site (left top) or FRT site (left bottom) with the core-proximal 4 bp underlined. Binding of the chimericrecombinases to the hybrid symmetry elements is shown on the right. Vertical arrows indicate cleavage sites.
Chimeras of the Flp and Cre Recombinases 33
strate contains the FRT b symmetry element, theFRT core and a fox symmetry element (Figure 1(f)).We conducted gel mobility shift assays to measurethe binding of Cre, Flp, Cre25, His-P32 and thetwo chimeric proteins to the ArkL substrate.
As expected, both Flp and His-Fre formed onlycomplex Is on the ArkL substrate (Figure 8, lanes2-5). The Cre25 and His-P32 proteins also gener-ated a single complex band by binding to the foxand the FRT symmetry elements contained in theArkL substrate (Figure 8, lanes 8-10). The His-P32peptide exhibited a decreased binding af®nity tothe ArkL substrate compared to the Flp protein, asobserved previously with the FRT symmetryelement contained in the ArkZ substrate. In con-trast, the His-Fre chimera bound the ArkL DNAwith a higher af®nity than the Cre25 peptide.
Both Cre and His-Clp exhibited weak inter-actions with the substrate (Figure 8, lanes 11-14).Modi®cations of the core-proximal nucleotides ofthe lox symmetry element severely impaired thebinding of Cre, while the absence of the core-proxi-mal nucleotides of the lox symmetry in the FRTsymmetry element severely impaired the bindingof the Clp.
A mixed complex II band (cII Mix) was observedwhen the Flp and His-Fre proteins were incubatedwith the ArkL substrate (Figure 8, lanes 6 and 7).This heterodimeric complex II re¯ects the bindingof one monomer each of Flp and His-Fre to theFRT and fox symmetry elements of ArkL.
The combined results of the binding assays illus-trate the altered binding af®nity of the Fre and Clpchimeras compared to the Cre and Flp proteins.Cre and Flp bound with high af®nity to the loxand FRT sites. In contrast, the Fre and Clp chi-meras bound weakly to these symmetry elements.The composite symmetry elements fox and lrt,were high-af®nity binding sites for the Fre and Clpproteins. Native Cre and Flp recombinases boundweakly to these novel composite target symmetryelements. The Cre25 and P32 proteins bound theirrespective native and composite symmetryelements with equal af®nity; hence the sequence ofthe core-proximal nucleotides in a target symmetryelement did not affect the binding of the isolatedCOOH-terminal peptides of Cre or Flp. Finally,incubation of the native and chimeric recombinaseswith the hybrid substrate containing both the cor-responding native and composite target symmetryelements generated distinct mixed dimer com-plexes. The combinations of Cre and His-Fre withthe ArkP substrate, Cre and His-Clp with the ArkFsubstrate and the incubation of Flp and His-Frewith the ArkL substrate, are all examples of suchmixed reactions combining a native protein with achimeric protein that generate heterodimeric com-plexes.
Mode of cleavage: experimental design
Previous experiments designed to illustrate transcleavage by Cre and Flp made use of a comple-
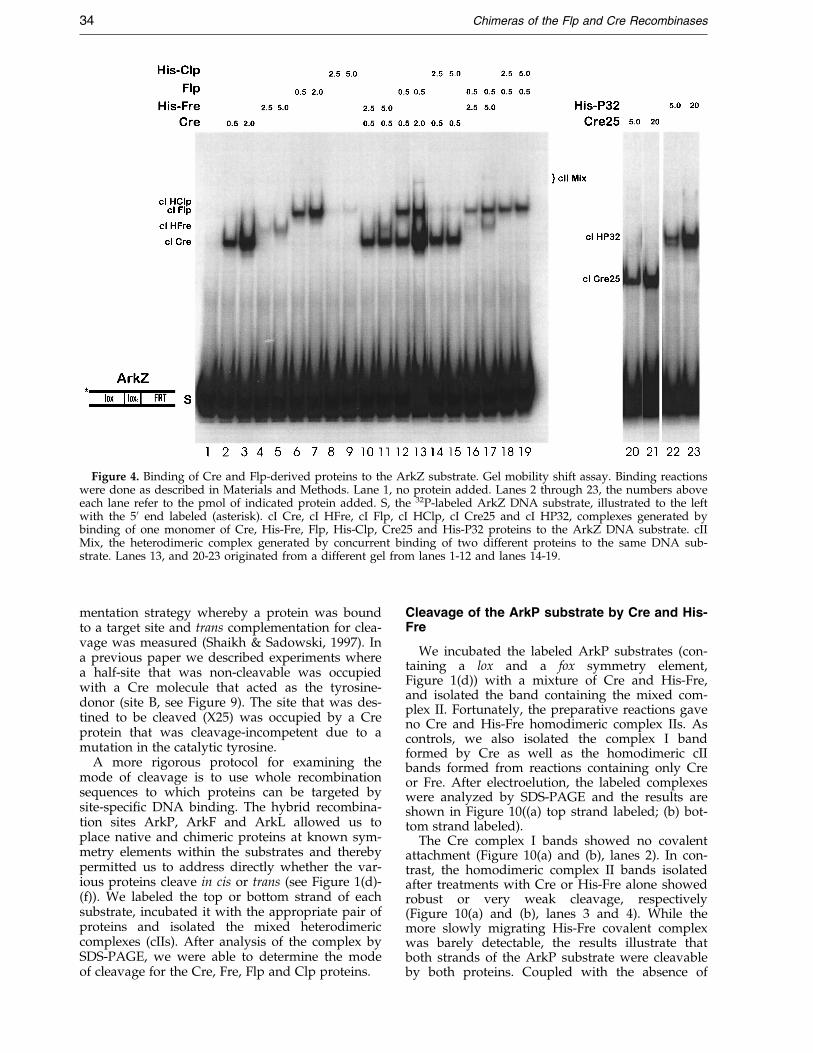
Figure 4. Binding of Cre and Flp-derived proteins to the ArkZ substrate. Gel mobility shift assay. Binding reactionswere done as described in Materials and Methods. Lane 1, no protein added. Lanes 2 through 23, the numbers aboveeach lane refer to the pmol of indicated protein added. S, the 32P-labeled ArkZ DNA substrate, illustrated to the leftwith the 50 end labeled (asterisk). cI Cre, cI HFre, cI Flp, cI HClp, cI Cre25 and cI HP32, complexes generated bybinding of one monomer of Cre, His-Fre, Flp, His-Clp, Cre25 and His-P32 proteins to the ArkZ DNA substrate. cIIMix, the heterodimeric complex generated by concurrent binding of two different proteins to the same DNA sub-strate. Lanes 13, and 20-23 originated from a different gel from lanes 1-12 and lanes 14-19.
34 Chimeras of the Flp and Cre Recombinases
mentation strategy whereby a protein was boundto a target site and trans complementation for clea-vage was measured (Shaikh & Sadowski, 1997). Ina previous paper we described experiments wherea half-site that was non-cleavable was occupiedwith a Cre molecule that acted as the tyrosine-donor (site B, see Figure 9). The site that was des-tined to be cleaved (X25) was occupied by a Creprotein that was cleavage-incompetent due to amutation in the catalytic tyrosine.
A more rigorous protocol for examining themode of cleavage is to use whole recombinationsequences to which proteins can be targeted bysite-speci®c DNA binding. The hybrid recombina-tion sites ArkP, ArkF and ArkL allowed us toplace native and chimeric proteins at known sym-metry elements within the substrates and therebypermitted us to address directly whether the var-ious proteins cleave in cis or trans (see Figure 1(d)-(f)). We labeled the top or bottom strand of eachsubstrate, incubated it with the appropriate pair ofproteins and isolated the mixed heterodimericcomplexes (cIIs). After analysis of the complex bySDS-PAGE, we were able to determine the modeof cleavage for the Cre, Fre, Flp and Clp proteins.
Cleavage of the ArkP substrate by Cre and His-Fre
We incubated the labeled ArkP substrates (con-taining a lox and a fox symmetry element,Figure 1(d)) with a mixture of Cre and His-Fre,and isolated the band containing the mixed com-plex II. Fortunately, the preparative reactions gaveno Cre and His-Fre homodimeric complex IIs. Ascontrols, we also isolated the complex I bandformed by Cre as well as the homodimeric cIIbands formed from reactions containing only Creor Fre. After electroelution, the labeled complexeswere analyzed by SDS-PAGE and the results areshown in Figure 10((a) top strand labeled; (b) bot-tom strand labeled).
The Cre complex I bands showed no covalentattachment (Figure 10(a) and (b), lanes 2). In con-trast, the homodimeric complex II bands isolatedafter treatments with Cre or His-Fre alone showedrobust or very weak cleavage, respectively(Figure 10(a) and (b), lanes 3 and 4). While themore slowly migrating His-Fre covalent complexwas barely detectable, the results illustrate thatboth strands of the ArkP substrate were cleavableby both proteins. Coupled with the absence of
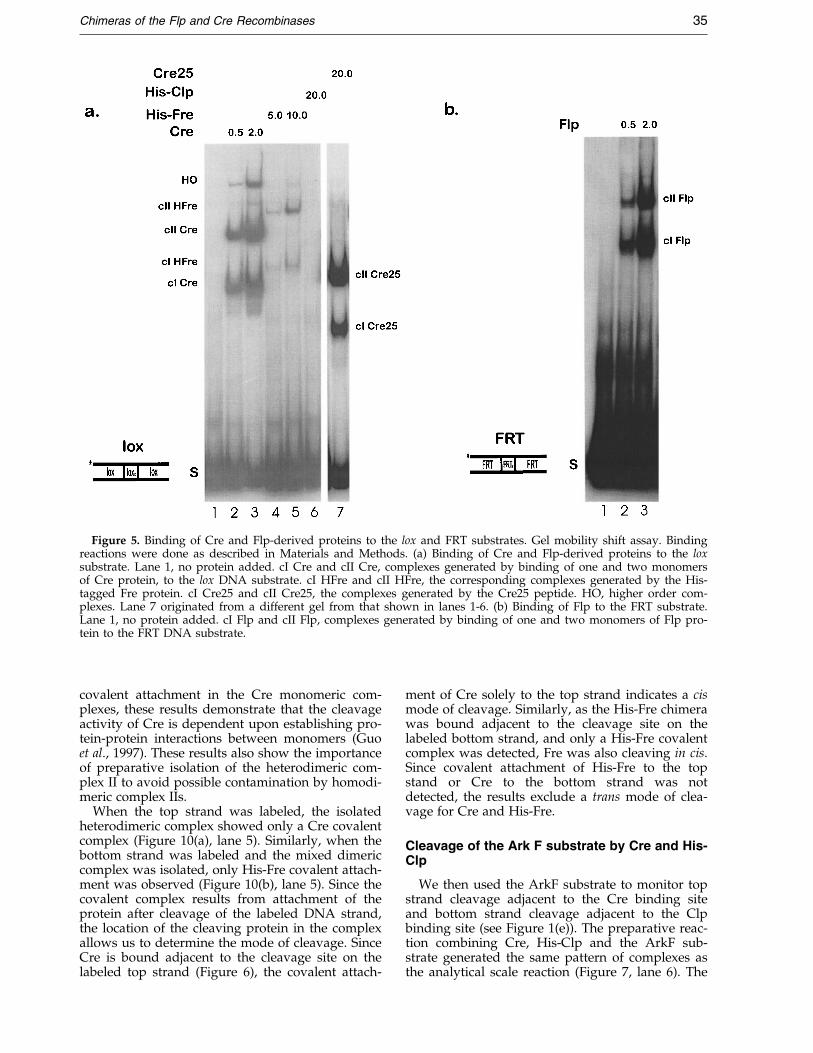
Figure 5. Binding of Cre and Flp-derived proteins to the lox and FRT substrates. Gel mobility shift assay. Bindingreactions were done as described in Materials and Methods. (a) Binding of Cre and Flp-derived proteins to the loxsubstrate. Lane 1, no protein added. cI Cre and cII Cre, complexes generated by binding of one and two monomersof Cre protein, to the lox DNA substrate. cI HFre and cII HFre, the corresponding complexes generated by the His-tagged Fre protein. cI Cre25 and cII Cre25, the complexes generated by the Cre25 peptide. HO, higher order com-plexes. Lane 7 originated from a different gel from that shown in lanes 1-6. (b) Binding of Flp to the FRT substrate.Lane 1, no protein added. cI Flp and cII Flp, complexes generated by binding of one and two monomers of Flp pro-tein to the FRT DNA substrate.
Chimeras of the Flp and Cre Recombinases 35
covalent attachment in the Cre monomeric com-plexes, these results demonstrate that the cleavageactivity of Cre is dependent upon establishing pro-tein-protein interactions between monomers (Guoet al., 1997). These results also show the importanceof preparative isolation of the heterodimeric com-plex II to avoid possible contamination by homodi-meric complex IIs.
When the top strand was labeled, the isolatedheterodimeric complex showed only a Cre covalentcomplex (Figure 10(a), lane 5). Similarly, when thebottom strand was labeled and the mixed dimericcomplex was isolated, only His-Fre covalent attach-ment was observed (Figure 10(b), lane 5). Since thecovalent complex results from attachment of theprotein after cleavage of the labeled DNA strand,the location of the cleaving protein in the complexallows us to determine the mode of cleavage. SinceCre is bound adjacent to the cleavage site on thelabeled top strand (Figure 6), the covalent attach-
ment of Cre solely to the top strand indicates a cismode of cleavage. Similarly, as the His-Fre chimerawas bound adjacent to the cleavage site on thelabeled bottom strand, and only a His-Fre covalentcomplex was detected, Fre was also cleaving in cis.Since covalent attachment of His-Fre to the topstand or Cre to the bottom strand was notdetected, the results exclude a trans mode of clea-vage for Cre and His-Fre.
Cleavage of the Ark F substrate by Cre and His-Clp
We then used the ArkF substrate to monitor topstrand cleavage adjacent to the Cre binding siteand bottom strand cleavage adjacent to the Clpbinding site (see Figure 1(e)). The preparative reac-tion combining Cre, His-Clp and the ArkF sub-strate generated the same pattern of complexes asthe analytical scale reaction (Figure 7, lane 6). The

Figure 6. Binding of Cre and Flp-derived proteins to the ArkP sub-strate. Gel mobility shift. Com-plexes named as in Figures 4 and5. Note that the heterodimeric cII
is resolved from each of the of thehomodimeric cIIs.
36 Chimeras of the Flp and Cre Recombinases
SDS-PAGE analysis of the isolated mixed com-plexes and the complex I bands formed by Cre andClp is shown in Figure 11.
No covalent complexes were observed in the Creand His-Clp complex I bands isolated after treat-ment with single proteins. This again shows thedependence of the catalytic activity on the for-mation of multimeric protein-DNA complexes.
Most surprisingly, the isolated mixed dimercomplex in which the top strand was labeledshowed, in addition to Cre-dependent covalentattachment, a large excess of the His-Clp covalentcomplex (Figure 11(a), lane 6). These results,coupled with the binding assignments of Cre andClp on the ArkF substrate, demonstrate that Crecleaves the top strand in cis, while the Clp chimerais cleaving the same site in trans. A number ofadditional bands appear that are also seen inassays of covalent attachment by Flp (Figure 12).These most likely result from proteolytic fragmentsof Clp covalently attached to the cleaved labeledstrand. By phosphorimager analysis, we deter-mined that Clp-dependent covalent attachmentexceeded that of Cre by a factor of 10-15 times(data not shown). The Cre complex I band isolatedfrom a mixed reaction showed barely detectablelevels of covalent attachment (Figure 11(a), lane 4).However, the isolated His-Clp complex I band
showed a slightly greater amount of cleavage pro-duct than that observed from the isolated dimer(Figure 11(a), lane 5). As the complex I bands fromthe unmixed protein reactions showed no covalentattachment, the presence of cleavage in these com-plex Is may be attributable to the disassembly ofthe heterodimeric complex after cleavage.
The results for the isolated mixed dimeric com-plex II in which the bottom strand was labeled areshown in Figure 11(b). A Cre covalent complexwas detectable (Figure 11(b), lane 6). But by phos-phorimage quanti®cation, the level of Cre-depen-dent cleavage was of the order of 50 times lowerthan the amount of Cre covalent complex observedon the top strand. While the amount of Clp-depen-dent covalent complex far exceeded the amount ofCre cleavage product as before, the level of Clpcleavage was also reduced by 100-fold comparedto the top strand cleavage results. This reduction incleavage product from the Cre-His-Clp dimer wasalso evident in the complex I bands isolated fromthe mixed reaction (Figure 11(b), lanes 4 and 5).Here, the amount of Clp-dependent covalent com-plex was decreased by 100 times, while no clea-vage product was detectable from the isolated Crecomplex I band. However, despite the reducedlevels of cleavage by both Cre and Clp of the bot-tom strand, these results are consistent with trans
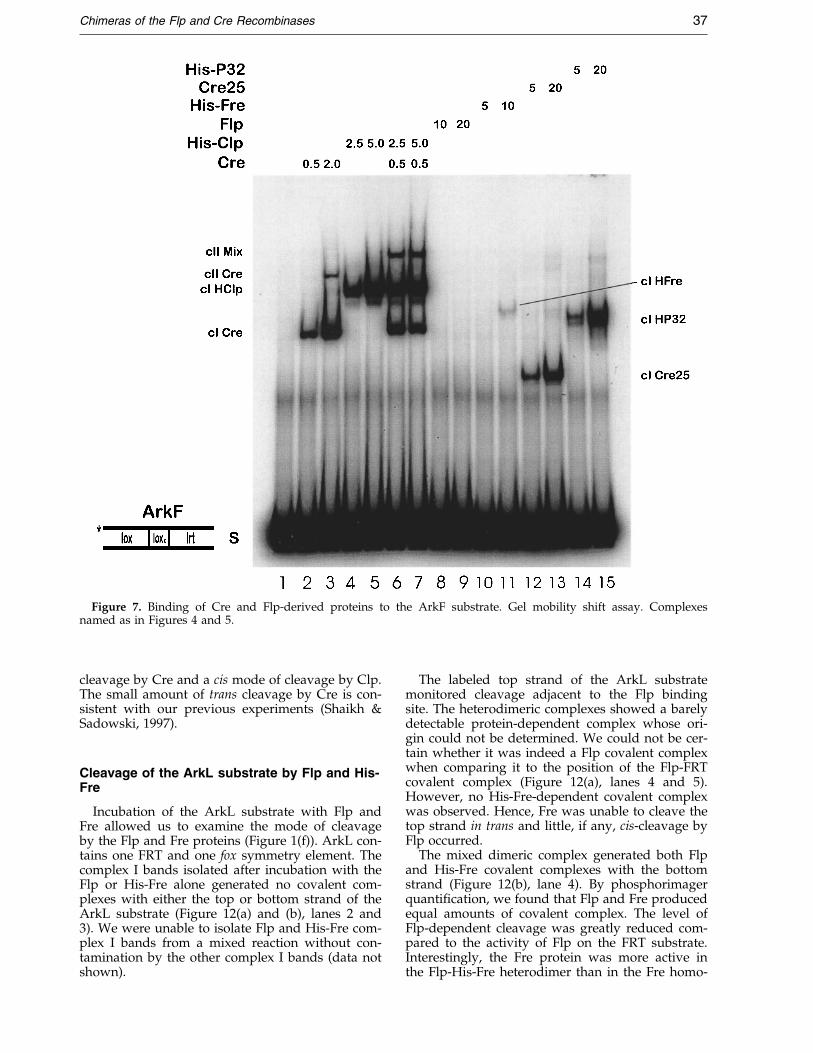
Figure 7. Binding of Cre and Flp-derived proteins to the ArkF substrate. Gel mobility shift assay. Complexesnamed as in Figures 4 and 5.
Chimeras of the Flp and Cre Recombinases 37
cleavage by Cre and a cis mode of cleavage by Clp.The small amount of trans cleavage by Cre is con-sistent with our previous experiments (Shaikh &Sadowski, 1997).
Cleavage of the ArkL substrate by Flp and His-Fre
Incubation of the ArkL substrate with Flp andFre allowed us to examine the mode of cleavageby the Flp and Fre proteins (Figure 1(f)). ArkL con-tains one FRT and one fox symmetry element. Thecomplex I bands isolated after incubation with theFlp or His-Fre alone generated no covalent com-plexes with either the top or bottom strand of theArkL substrate (Figure 12(a) and (b), lanes 2 and3). We were unable to isolate Flp and His-Fre com-plex I bands from a mixed reaction without con-tamination by the other complex I bands (data notshown).
The labeled top strand of the ArkL substratemonitored cleavage adjacent to the Flp bindingsite. The heterodimeric complexes showed a barelydetectable protein-dependent complex whose ori-gin could not be determined. We could not be cer-tain whether it was indeed a Flp covalent complexwhen comparing it to the position of the Flp-FRTcovalent complex (Figure 12(a), lanes 4 and 5).However, no His-Fre-dependent covalent complexwas observed. Hence, Fre was unable to cleave thetop strand in trans and little, if any, cis-cleavage byFlp occurred.
The mixed dimeric complex generated both Flpand His-Fre covalent complexes with the bottomstrand (Figure 12(b), lane 4). By phosphorimagerquanti®cation, we found that Flp and Fre producedequal amounts of covalent complex. The level ofFlp-dependent cleavage was greatly reduced com-pared to the activity of Flp on the FRT substrate.Interestingly, the Fre protein was more active inthe Flp-His-Fre heterodimer than in the Fre homo-
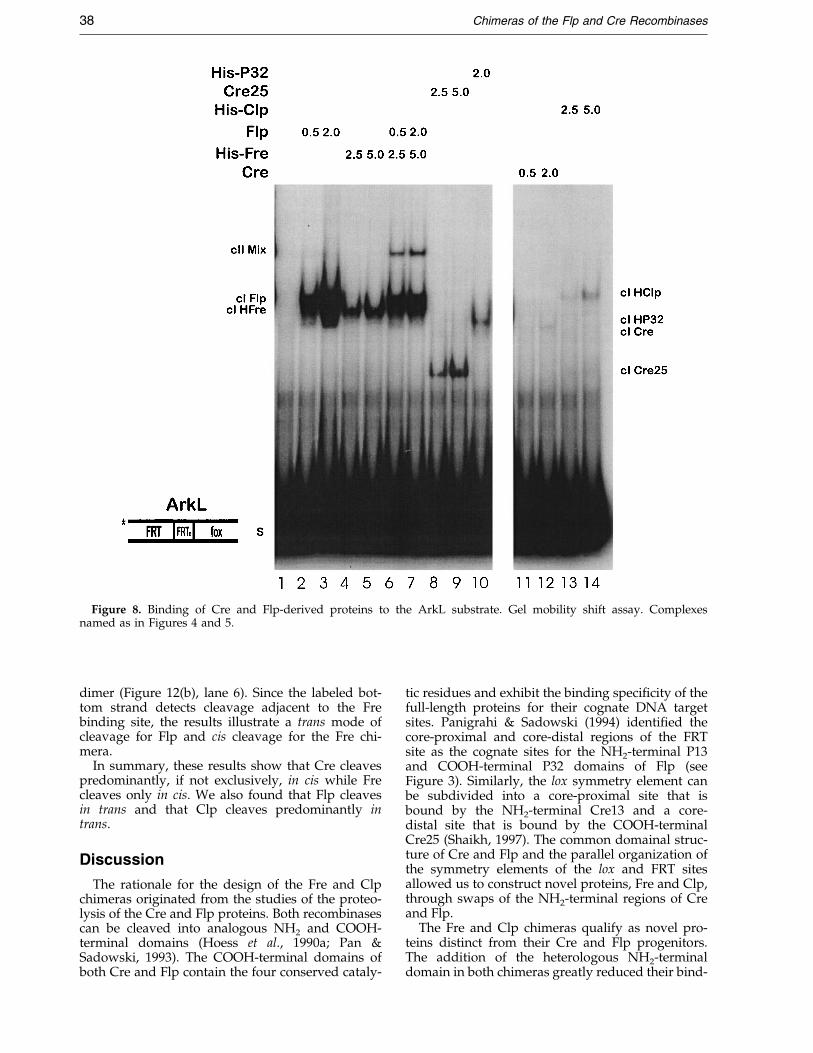
Figure 8. Binding of Cre and Flp-derived proteins to the ArkL substrate. Gel mobility shift assay. Complexesnamed as in Figures 4 and 5.
38 Chimeras of the Flp and Cre Recombinases
dimer (Figure 12(b), lane 6). Since the labeled bot-tom strand detects cleavage adjacent to the Frebinding site, the results illustrate a trans mode ofcleavage for Flp and cis cleavage for the Fre chi-mera.
In summary, these results show that Cre cleavespredominantly, if not exclusively, in cis while Frecleaves only in cis. We also found that Flp cleavesin trans and that Clp cleaves predominantly intrans.
Discussion
The rationale for the design of the Fre and Clpchimeras originated from the studies of the proteo-lysis of the Cre and Flp proteins. Both recombinasescan be cleaved into analogous NH2 and COOH-terminal domains (Hoess et al., 1990a; Pan &Sadowski, 1993). The COOH-terminal domains ofboth Cre and Flp contain the four conserved cataly-
tic residues and exhibit the binding speci®city of thefull-length proteins for their cognate DNA targetsites. Panigrahi & Sadowski (1994) identi®ed thecore-proximal and core-distal regions of the FRTsite as the cognate sites for the NH2-terminal P13and COOH-terminal P32 domains of Flp (seeFigure 3). Similarly, the lox symmetry element canbe subdivided into a core-proximal site that isbound by the NH2-terminal Cre13 and a core-distal site that is bound by the COOH-terminalCre25 (Shaikh, 1997). The common domainal struc-ture of Cre and Flp and the parallel organization ofthe symmetry elements of the lox and FRT sitesallowed us to construct novel proteins, Fre and Clp,through swaps of the NH2-terminal regions of Creand Flp.
The Fre and Clp chimeras qualify as novel pro-teins distinct from their Cre and Flp progenitors.The addition of the heterologous NH2-terminaldomain in both chimeras greatly reduced their bind-

Figure 9. Rationale of the previous complementation test used (Shaikh & Sadowski, 1997, Reproduced withpermission from Journal of Biological Chemistry (1997) 272, 5695-5702). The cleavable X25 site is loaded with thecleavage-incompetent CreHis Y324C protein and the non-cleavable B site with a cleavage-competent Cre protein(top). After the two reactions are mixed, the Cre protein bound to the B site donates its tyrosine 324 which cleavesthe X25 site and covalently attaches the protein to the 32P-labeled top strand (middle). The radioactive label covalentcomplex is detected by SDS-PAGE (bottom). Squares, CreHis Y324C; triangles, CreHis; asterisk, 32P radioactive label.
Chimeras of the Flp and Cre Recombinases 39
ing af®nity for the cognate DNA target sites of Creand Flp. In addition, the composite recognition tar-get sites developed for Fre and Clp were speci®c foreach chimera and did not permit binding of the par-ental Cre and Flp proteins. Secondly, the Fre andClp proteins were not simply new versions of the
COOH-terminal peptides with relatively large NH2-terminal tags. Fre and Clp both exhibited greaterbinding af®nity for their cognate sites than the Cre25and P32 peptides. Furthermore, Clp showed a levelof cleavage activity similar to intact Flp and muchgreater than that generated by P32 (Shaikh, 1997).
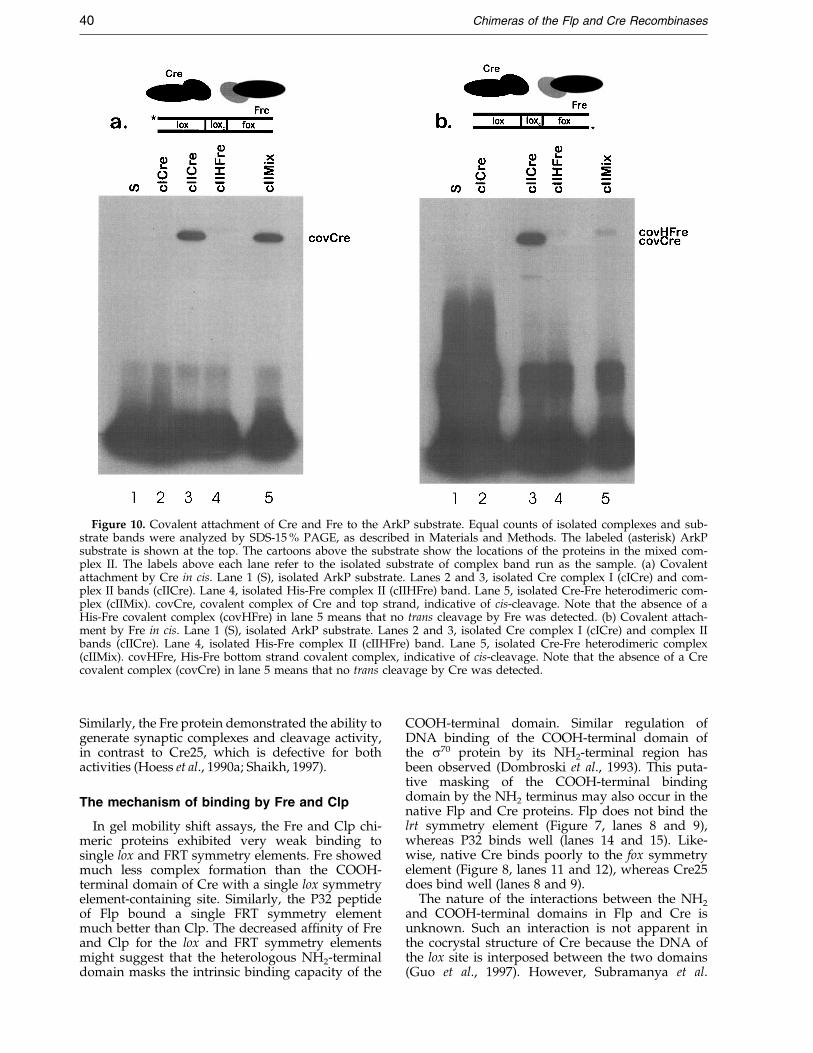
Figure 10. Covalent attachment of Cre and Fre to the ArkP substrate. Equal counts of isolated complexes and sub-strate bands were analyzed by SDS-15 % PAGE, as described in Materials and Methods. The labeled (asterisk) ArkPsubstrate is shown at the top. The cartoons above the substrate show the locations of the proteins in the mixed com-plex II. The labels above each lane refer to the isolated substrate of complex band run as the sample. (a) Covalentattachment by Cre in cis. Lane 1 (S), isolated ArkP substrate. Lanes 2 and 3, isolated Cre complex I (cICre) and com-plex II bands (cIICre). Lane 4, isolated His-Fre complex II (cIIHFre) band. Lane 5, isolated Cre-Fre heterodimeric com-plex (cIIMix). covCre, covalent complex of Cre and top strand, indicative of cis-cleavage. Note that the absence of aHis-Fre covalent complex (covHFre) in lane 5 means that no trans cleavage by Fre was detected. (b) Covalent attach-ment by Fre in cis. Lane 1 (S), isolated ArkP substrate. Lanes 2 and 3, isolated Cre complex I (cICre) and complex IIbands (cIICre). Lane 4, isolated His-Fre complex II (cIIHFre) band. Lane 5, isolated Cre-Fre heterodimeric complex(cIIMix). covHFre, His-Fre bottom strand covalent complex, indicative of cis-cleavage. Note that the absence of a Crecovalent complex (covCre) in lane 5 means that no trans cleavage by Cre was detected.
40 Chimeras of the Flp and Cre Recombinases
Similarly, the Fre protein demonstrated the ability togenerate synaptic complexes and cleavage activity,in contrast to Cre25, which is defective for bothactivities (Hoess et al., 1990a; Shaikh, 1997).
The mechanism of binding by Fre and Clp
In gel mobility shift assays, the Fre and Clp chi-meric proteins exhibited very weak binding tosingle lox and FRT symmetry elements. Fre showedmuch less complex formation than the COOH-terminal domain of Cre with a single lox symmetryelement-containing site. Similarly, the P32 peptideof Flp bound a single FRT symmetry elementmuch better than Clp. The decreased af®nity of Freand Clp for the lox and FRT symmetry elementsmight suggest that the heterologous NH2-terminaldomain masks the intrinsic binding capacity of the
COOH-terminal domain. Similar regulation ofDNA binding of the COOH-terminal domain ofthe s70 protein by its NH2-terminal region hasbeen observed (Dombroski et al., 1993). This puta-tive masking of the COOH-terminal bindingdomain by the NH2 terminus may also occur in thenative Flp and Cre proteins. Flp does not bind thelrt symmetry element (Figure 7, lanes 8 and 9),whereas P32 binds well (lanes 14 and 15). Like-wise, native Cre binds poorly to the fox symmetryelement (Figure 8, lanes 11 and 12), whereas Cre25does bind well (lanes 8 and 9).
The nature of the interactions between the NH2
and COOH-terminal domains in Flp and Cre isunknown. Such an interaction is not apparent inthe cocrystal structure of Cre because the DNA ofthe lox site is interposed between the two domains(Guo et al., 1997). However, Subramanya et al.
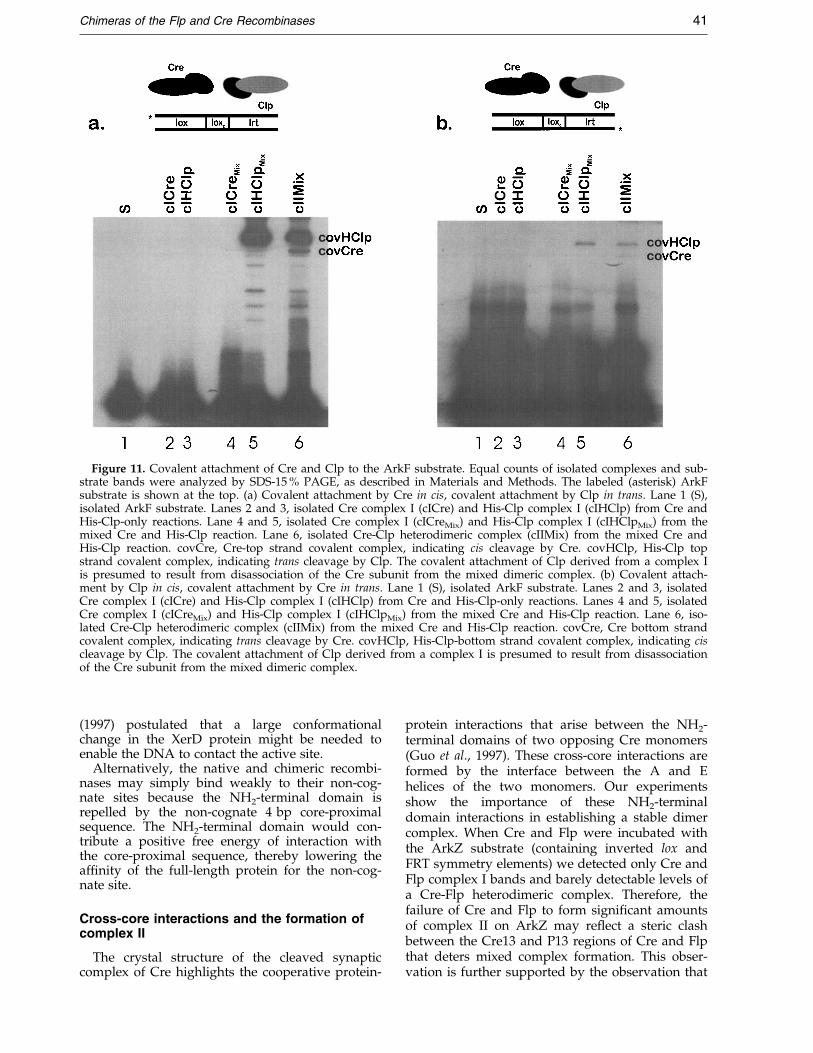
Figure 11. Covalent attachment of Cre and Clp to the ArkF substrate. Equal counts of isolated complexes and sub-strate bands were analyzed by SDS-15 % PAGE, as described in Materials and Methods. The labeled (asterisk) ArkFsubstrate is shown at the top. (a) Covalent attachment by Cre in cis, covalent attachment by Clp in trans. Lane 1 (S),isolated ArkF substrate. Lanes 2 and 3, isolated Cre complex I (cICre) and His-Clp complex I (cIHClp) from Cre andHis-Clp-only reactions. Lane 4 and 5, isolated Cre complex I (cICreMix) and His-Clp complex I (cIHClpMix) from themixed Cre and His-Clp reaction. Lane 6, isolated Cre-Clp heterodimeric complex (cIIMix) from the mixed Cre andHis-Clp reaction. covCre, Cre-top strand covalent complex, indicating cis cleavage by Cre. covHClp, His-Clp topstrand covalent complex, indicating trans cleavage by Clp. The covalent attachment of Clp derived from a complex Iis presumed to result from disassociation of the Cre subunit from the mixed dimeric complex. (b) Covalent attach-ment by Clp in cis, covalent attachment by Cre in trans. Lane 1 (S), isolated ArkF substrate. Lanes 2 and 3, isolatedCre complex I (cICre) and His-Clp complex I (cIHClp) from Cre and His-Clp-only reactions. Lanes 4 and 5, isolatedCre complex I (cICreMix) and His-Clp complex I (cIHClpMix) from the mixed Cre and His-Clp reaction. Lane 6, iso-lated Cre-Clp heterodimeric complex (cIIMix) from the mixed Cre and His-Clp reaction. covCre, Cre bottom strandcovalent complex, indicating trans cleavage by Cre. covHClp, His-Clp-bottom strand covalent complex, indicating ciscleavage by Clp. The covalent attachment of Clp derived from a complex I is presumed to result from disassociationof the Cre subunit from the mixed dimeric complex.
Chimeras of the Flp and Cre Recombinases 41
(1997) postulated that a large conformationalchange in the XerD protein might be needed toenable the DNA to contact the active site.
Alternatively, the native and chimeric recombi-nases may simply bind weakly to their non-cog-nate sites because the NH2-terminal domain isrepelled by the non-cognate 4 bp core-proximalsequence. The NH2-terminal domain would con-tribute a positive free energy of interaction withthe core-proximal sequence, thereby lowering theaf®nity of the full-length protein for the non-cog-nate site.
Cross-core interactions and the formation ofcomplex II
The crystal structure of the cleaved synapticcomplex of Cre highlights the cooperative protein-
protein interactions that arise between the NH2-terminal domains of two opposing Cre monomers(Guo et al., 1997). These cross-core interactions areformed by the interface between the A and Ehelices of the two monomers. Our experimentsshow the importance of these NH2-terminaldomain interactions in establishing a stable dimercomplex. When Cre and Flp were incubated withthe ArkZ substrate (containing inverted lox andFRT symmetry elements) we detected only Cre andFlp complex I bands and barely detectable levels ofa Cre-Flp heterodimeric complex. Therefore, thefailure of Cre and Flp to form signi®cant amountsof complex II on ArkZ may re¯ect a steric clashbetween the Cre13 and P13 regions of Cre and Flpthat deters mixed complex formation. This obser-vation is further supported by the observation that

Figure 12. Covalent attachment of Flp and Fre to the ArkL substrate. Equal counts of isolated complexes and sub-strate bands were analyzed by SDS-17 % PAGE. The labeled (asterisk) ArkL substrate is shown at the top. (a) Nocovalent attachment by Flp or Fre. Lane 1 (S), isolated ArkL substrate. Lanes 2 and 3, isolated Flp complex I (cIFlp)and His-Fre complex I (cIHFre) from Flp- and His-Fre-only reactions. Lane 4, isolated Flp-Fre heterodimeric complex(cIIMix) from the mixed Flp and His-Fre reaction. Lane 5, isolated complex II band of Flp (cIIFlpFRT) generated witha top strand labeled full-FRT substrate (see Figure 1(b)). Lane 6, isolated complex II band of His-Fre (cIIHFreFox2) gen-erated with a top strand labeled fox2b substrate. This substrate contains two inverted fox symmetry elements sur-rounding an FRT site core. (Figure 1(g)). covFlp, covalent complex generated by cleavage of the labeled DNA strandby Flp. covHFre, covalent complex generated by cleavage of the labeled DNA strand by His-Fre. Note that theabsence of covFlp and covHFre in lane 4 means no cis cleavage by Flp or trans cleavage by Fre occurred. (b) Covalentattachment by Fre in cis, covalent attachment by Flp in trans. Lane 1 (S), isolated ArkL substrate. Lanes 2 and 3, iso-lated Flp complex I (cIFlp) and His-Fre complex I (cIHFre) from Flp- and His-Fre-only reactions. Lane 4, isolated Flp-Fre heterodimeric complex (cIIMix) from the mixed Flp and His-Fre reaction. Lane 5, isolated complex II band of Flp(cIIFlpFRT) generated with a top strand labeled full-FRT substrate (Figure 1(b)). Lane 6, isolated complex II band ofHis-Fre (cIIHFreFox2) generated with a top strand labeled fox2b substrate. covFlp, covalent complex generated by clea-vage of the labeled DNA strand by Flp. covHFre, covalent complex generated by cleavage of the labeled DNA strandby His-Fre. Note that the presence of both covFlp and covHFre in lane 4 indicates cleavage by Flp in trans and clea-vage by Fre in cis.
42 Chimeras of the Flp and Cre Recombinases
incubation of the ArkZ substrate with Cre and P32from Flp or with Flp and Cre25 generated muchgreater levels of a mixed dimer complex (data notshown). On the other hand the failure to formmixed complexes on this substrate may simplyre¯ect the absence of cooperative interactionsbetween the heterologous NH2-terminal andCOOH-terminal domains of Flp and Cre.
In contrast, Cre and Clp readily formed mixeddimeric complexes when incubated with the ArkFsubstrate (containing inverted lox and lrt symmetryelements). Likewise, heterodimeric complexes were
easily seen when Flp and Fre were incubated withthe ArkL substrate (containing inverted FRT andfox symmetry elements). Although all three sub-strates would position the heterologous COOH-terminal domains of Cre and Flp opposite eachother, only the Cre�Clp and the Flp�Fre pairedmixtures would position the same NH2-terminaldomains together across the core. In the case ofArkF, a Cre13-Cre13 interaction is created betweenthe two protein monomers bound in an invertedcon®guration on the substrate. In the case of theArkL substrate, the two P13 domains are opposed.

Chimeras of the Flp and Cre Recombinases 43
Hence, the formation of the mixed dimer complexwas dependent upon presenting two identicalhomologous NH2-terminal domains cross-core atthe dimer interface.
The Cre cocrystal structure showed that the Credimer was also stabilized by the insertion of theCOOH-terminal N helix of the cleaving subunitinto a COOH-terminal hydrophobic pocket of theopposed, non-cleaving monomer. Although pairingof the heterologous NH2-terminal domains of Creand Fre seems unlikely, the binding of Cre and Freto the ArkP substrate (containing inverted lox andfox symmetry elements) resulted in the formationof a heterodimeric complex. It is possible that thiscomplex was stabilized through protein-proteininteractions involving the capture of the N helix ofCre by the Fre monomer bound across the core.Similarly, both Cre and Fre generated homodi-meric complex IIs on the ArkP substrate likely dueto protein-protein interactions between the COOH-terminal regions of the two proteins in the dimer.
Cre formed a small amount of homodimericcomplex with the ArkF substrate (containinginverted lox and lrt symmetry elements) despitegenerating only a monomeric complex with ArkZ(containing inverted lox and FRT symmetryelements). It is likely that this homodimeric com-plex is mediated by cross-core interactions betweentwo opposing Cre13 domains.
As observed from the crystal data and con®rmedby these results, Cre relies on major protein-proteininteractions between the NH2 and COOH-terminaldomains of Cre monomers to stabilize multi-pro-tein-DNA complexes. The Flp protein, however,does not show the same degree of binding coop-erativity that is shown by Cre (Ringrose et al.,1998). Formation of a stable heterodimeric complexwhen Flp and Fre were bound to the ArkL sub-strate (containing inverted FRT and fox symmetryelements) supports the idea that the P13-P13 inter-actions stabilize the mixed dimeric complex despitethe heterology between the COOH-terminaldomains bound cross-core on the same substrate.
The diminished level of cooperative cross-coreinteractions between the Flp COOH-terminaldomains may relate to the trans mode of cleavageby Flp. The Cre cocrystal shows that the N helix ofthe cleaving subunit is buried in a hydrophobicpocket of the non-cleaving Cre monomer situatedacross the core (Guo et al., 1997). The catalytic tyro-sine present in the M helix of the COOH-terminaldomain of the cleaving molecule of Cre hasattached to the scissile phosphate in cis. The VanDuyne group has modeled a trans cleaving mech-anism in which the N helix of the trans-cleavingmolecule (e.g. Flp) would have to be buried in cisto allow for donation of the active tyrosine in trans(Gopaul & Van Duyne, 1999; Guo et al., 1999). Thisarrangement might effectively remove the majorprotein-protein interaction that is evident betweenthe COOH-terminal domains of Cre and mayaccount for the apparent lack of cooperative inter-
actions between the COOH-terminal domains ofFlp and Clp.
Footprinting studies suggest that protein-proteininteractions between the NH2-terminal domains ofopposing Cre monomers trigger a conformationalchange in the COOH-terminal domain of Cre thatrepositions the entire protein on the site (Guo et al.,1997; Hoess et al., 1990a; Shaikh, 1997). Hence, thecore-proximal region of the lox site may function asa nucleation site for the NH2-terminal domain ofCre that allows communication between proteinmonomers leading to the formation of catalyticallyactive complexes. Similarly, the P13 domain of Flpis believed to induce a conformational change inthe COOH-terminal domain of Flp upon bindingthe FRT site (Panigrahi & Sadowski, 1994). Thecore-proximal region in the FRT site may also func-tion as an initiation site for complex formation bythe Flp protein.
The mode of cleavage
Here, we have used the chimeric Fre and Clpproteins to test the mode of cleavage by the nativeCre and Flp recombinases. We have also used thedistinct site-speci®c binding activities of all fourproteins to target them to speci®c symmetryelements in hybrid recombination sites. Further-more, by directly isolating the mixed dimeric com-plex, we were certain of the location of the twoproteins on the hybrid substrate and that only pro-teins in the mixed complex II could contribute tothe covalent complexes observed on subsequentSDS-PAGE gels. This strategy allowed us to deter-mine the mode of cleavage of both proteins in theheterodimeric complex. However, since the modeof cleavage was determined on dimeric complexescontaining single target DNA sites, we cannotassess how the formation of a synaptic complexconsisting of two sites might modify the mode ofcleavage.
Our results showed that the native Cre proteinin this protocol cleaved predominantly in ciswhereas the Flp protein cleaved almost entirely intrans. The chimeric Fre protein also cleaved in ciswhile the Clp protein cleaved mostly in trans. Ourresults illustrate that the COOH-terminal domaindetermined the mode of cleavage of the proteinand that the addition of the heterologous NH2-terminal domain in the chimeric proteins did notalter the mode of cleavage de®ned by the catalyticCOOH-terminal region of the protein.
In earlier experiments we showed that Crecleaved in trans (Shaikh & Sadowski, 1997). Whataccounts for the discrepancy between the two setsof experiments? Previously we made use of a half-site complementation strategy whereby a Cre pro-tein was pre-bound to a non-cleavable half-site andcleavage in trans was measured on another cleava-ble half-site (Figure 9). The protocol also made useof cleavage-incompetent Cre variants. The use oftyrosine-de®cient and His-tagged versions of Cre

44 Chimeras of the Flp and Cre Recombinases
and the non-cleavable half-lox sites may have con-tributed to the trans cleavage by Cre.
One of the half-lox sites, X25, was cleavable onlyby the non-His-tagged version of Cre in the half-site reactions. Therefore, the His-tagged version ofCre may have been unable to act in cis in reactionswith the X25 site alone. Furthermore, the otherhalf-lox site, B, was not cleavable by either versionof Cre. Hence, this site prevented cleavage in cis bythe protein bound to it and acted solely as a carrierof catalytically active Cre proteins to an assemblywith the Cre-X25 complex.
Most complementation experiments used to testthe cleavage mode of Cre or Flp also relied on theuse of protein variants that replace the catalytictyrosine with an amino acid that may not form ahydrogen bond in the active site (Chen et al., 1992;Gopaul et al., 1998; Guo et al., 1997, 1999). Whilethe Cre Y324F protein assumes the conformationsin crystal structures similar to that of wild-typeCre (Guo et al., 1999), the effect of the Y324Cmutation that we used may have allowed for transcomplementation to occur in vitro. The crystalstructure of a Cre synaptic complex shows that theactive site of the cleaving Cre monomer is com-posed of the conserved catalytic amino acid resi-dues Arg173, Arg292, His289 and Trp315surrounding the scissile bond, with the catalytictyrosine covalently attached at the cleavage site(Guo et al., 1997). The non-cleaving monomer ofCre shows a similar arrangement except that boththe histidine and tyrosine residues are shiftedaway from the scissile bond. If cis attack were pre-vented by the cleavage-incompetent nature of thescissile bond, then the catalytic tyrosine of the Cremonomer bound to one half-site may have beendirected towards a trans attack of an opposing clea-vable half-site. Likewise the Cre monomer boundto the X25 site may have also adopted the confor-mation required for the acceptance of an incomingcatalytic tyrosine residue.
It is possible that the original ®nding of transcleavage by the phage l integrase was attributableto the same explanation (Han et al., 1993). It shouldbe noted that the crystal structure of the COOH-terminal domain of the l integrase showed con-siderable mobility of the catalytic tyrosine thatmight lead to trans cleavage (Kwon et al., 1997).Construction of a Fre Y324C variant would allow atest of the prediction that such a mutation wouldchange the mode of cleavage by Cre on the ArkPsubstrate to trans.
It is important to note that the experiments inour previous paper did not rule out cis cleavage byCre (Shaikh & Sadowski, 1997). When both Creand His-Cre were bound to their respective half-sites, His-Cre cleaved in trans, but cleavage by Crewas also observed. The design of the experimentdid not allow us to distinguish cis from trans clea-vage by Cre. The present results from the mixedCre and Clp heterodimeric complex demonstratedthat Cre could cleave in cis or support trans clea-vage by Clp. Here, we show that although Cre
cleaves predominantly in cis it can also cleave intrans even when the experimental protocol allowscis cleavage.
Action of Cre and Fre on ArkP
When Cre and Fre were both bound to the ArkPsubstrate, both proteins cleaved in cis although Crecleaved more ef®ciently than Fre. This differencemay be related to the ef®ciency with which eachprotein activates the scissile phosphate bond. Alter-natively the substrate may be bent differently bythe respective protein. In the Cre and Fre heterodi-meric complex the homologous catalytically activeCOOH-terminal domains were positioned together.In the Cre cocrystal structure, the catalyticallyactive mixed dimeric complex is comprised ofcleaving and non-cleaving protein monomers (Guoet al., 1997). The cis-cleaving monomer inserts itsN-helix into a hydrophobic pocket in the non-cleaving monomer. Cre cleaves the top strand ofthe ArkP substrate in cis and covalently attaches itscatalytic tyrosine to the scissile phosphate adjacentto the Cre binding site. The N helix of the cleavingCre monomer could have been donated in trans tothe non-cleaving Fre protein. When Fre cleaves thebottom strand of ArkP in cis, its N helix might bedonated in trans to the COOH-terminal dockingsite in the non-cleaving Cre protein.
Assembly of Cre and Clp heterodimers on ArkF
The Cre and Clp heterodimeric complexes werecomprised of the cis-cleaving COOH-terminaldomain derived from Cre and a trans-cleavingCOOH-terminal domain of Flp. These complexesveri®ed a cis cleavage mode for Cre (Figure 11(a)).This may also mean that the N helix of Cre isdonated in trans to the non-cleaving Clp proteinbound cross-core on the substrate. This wouldrequire that the COOH-terminal domain of Clp(derived from Flp) must accept the heterologousCOOH-terminal N-helix from Cre.
The heterodimeric complex of Cre and Clp onthe ArkF substrate showed that Clp cleaved thetop strand in trans (Figure 11(b)). Since Clp isderived from the COOH-terminal domain of Flp,this result is consistent with the known cleavagemode of Flp. Surprisingly, Cre was able to activatethe scissile phosphate and to accept the incomingcatalytic tyrosine from a Flp-derived COOH termi-nus. The donation of the catalytic tyrosine of Clpto the acceptor Cre monomer might require a ciscollapse of the N helix in both Cre and its equival-ent in Clp (Gopaul & Van Duyne, 1999).
As both Cre cleavage in cis and Clp cleavage intrans occur at the top strand of the ArkF substrate,the scissile bond activated by the adjacently boundCre should be the normal Cre cleavage site. In con-trast, the putative cleavage site of the bottomstrand of the substrate is in fact a Cre cleavage site,which may be inef®ciently activated by the adja-cently bound Clp protein. This may have contribu-

Chimeras of the Flp and Cre Recombinases 45
ted to the inef®cient trans cleavage by Cre and ciscleavage by Clp at this site. Although we considerit unlikely, it is possible that cleavages by Cre andClp at this site occur after redistribution of the pro-tein(s) that occurs after isolation of the mixed com-plex II.
Action of Fre and Flp on ArkL
In contrast to the robust cleavage activitydemonstrated by both Cre and Clp in the heterodi-meric complex with the ArkF substrate, the Flpand Fre heterodimeric complex on the ArkL sub-strate showed weak catalytic activity of both pro-tein partners. The amount of covalent attachmentof Flp to the bottom strand of the ArkL substratewas much lower than that generated by Flp on thefull-FRT substrate. This may have been due to adecreased ability of Fre to activate the scissile bondfor cleavage or to a defect in accepting the catalytictyrosine in the active pocket from either a cis ortrans-cleavage mode. However, it should be notedthat Cre had no dif®culty in accepting the catalytictyrosine from Clp in trans. Therefore, we favor theformer explanation.
In none of these experiments have we deter-mined the actual location of the scissile bond.Although it is reasonable to assume that the pro-tein bound next to the scissile bond would deter-mine the position of cleavage, it is possible that thesource of the catalytic tyrosine could also play arole. For example, in the Cre and Clp heterodimer,it is likely that the scissile bond is in the ``normal''position on the top strand for cis cleavage by Cre.However, trans cleavage by Clp might shift the siteof cleavage by the incoming tyrosine residue. Simi-larly, Flp cleavage at the bottom strand of theArkL substrate might be inef®cient if Fre had acti-vated the phosphodiester bond one nucleotidecore-proximal to the expected site of trans cleavageby Flp. The cleavage sites for Flp in the FRT siteare 8 bp apart whereas the scissile bonds for Cre inthe lox site are 6 bp apart. These differences in spa-cing may have in¯uenced cross-core interactionsbetween the native recombinases and the chimerasand hence the ef®ciency and sites of cleavage.Unfortunately, the SDS-PAGE analysis we used todistinguish the covalent complexes of the differentproteins was not sensitive enough to determine thesite of cleavage to one nucleotide resolution.
Materials and Methods
Enzymes
All enzymes were obtained from New England Bio-Labs and used according to the manufacturer's instruc-tions.
Plasmids
The pShe2, pShe3, pShe5, pShe6, pShe11 and pLD3plasmids have been described previously (Shaikh, 1997;
Shaikh & Sadowski, 1997). All plasmids were preparedusing the Qiagen plasmid isolation kit.
Oligonucleotides
Oligonucleotides were synthesized at the Hospital forSick Children/Pharmacia Biotechnology Service Centerat the Banting Institute, University of Toronto or at Dal-ton Chemical Laboratories, Toronto. They were puri®edusing the OPC cartridge method. Where needed the oli-gonucleotide was 50-labeled with [g-32P]ATP with T4polynucleotide kinase. After phenol/chloroform-extrac-tion, the oligonucleotide was separated from unincorpo-rated nucleotides using a BioRad P6 spin column(BioRad). It was then annealed to the appropriate comp-lementary oligonucleotide by heating and slow coolingin 0.1 M NaCl, 5 mM MgCl2.
Proteins
Native Cre protein was puri®ed from an induced cul-ture containing pShe11 and Cre25 from an induced cul-ture containing pShe5. His-tagged P32 and native Flpwere puri®ed from induced cultures containing pShe2and pLD3, respectively. Puri®cation conditions for allproteins have been described previously (Shaikh, 1997;Shaikh & Sadowski, 1997).
Construction of His-Fre expression vector
The Fre protein is a chimera of the NH2-terminaldomain of Flp, P13, residues 1-123, and the COOH-term-inal domain of Cre, Cre25, residues 119-343 (Figure 2(a)and (b)). The P13-coding region of Flp was isolated byPCR using as template the pShe2 plasmid that containsthe Flp NH2-terminal coding region. The 50 NH2-terminalprimer, FLPN, was 44 nucleotides long and hybridized tothe start of the P13 coding region. This primer containedan NdeI restriction site (underlined) for later cloning stepsand had the sequence: 50 TAGGGCAGCCATATGCCA-CAATTTGATATATTATGTAAAACACC 30.
The COOH-terminal primer was P13B and hybridizedto the sequence encoding the amino acid residues 117-123 of the P13 coding region. This primer contained aBamHI restriction site for later cloning steps (underlined)and had the sequence: 50 TCTAGGGGATCCGCAAAC-TACTTACAATATCAGT 30.
PCR reactions contained 10 mM KCl, 20 mM Tris-HCl(pH 8.8), 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1 % (v/v)Triton X-100, (ThermoPol buffer, New England BioLabs),1 mM each primer FLPN and P13B, 0.8 pM pShe3 tem-plate, and 400 mM each dNTP (Pharmacia) in a totalreaction volume of 100 ml. Reaction mixtures were pre-heated to 95 �C for four minutes, two units of Vent DNApolymerase were added and the 95 �C incubation wascontinued for 1.5 minutes. Cycling occurred in threestages. The ®rst stage consisted of two cycles of 50 �C,one minute, 72 �C, 42 seconds, 95 �C, 30 seconds. Thesecond stage consisted of 30 cycles of 65 �C, one minute,72 �C, 42 seconds, 95 �C, 30 seconds. The third stage con-sisted of a 65 �C, one minute annealing step and a 72 �C,®ve minutes extension. Enzyme, dNTPs and oligonucleo-tide primers were removed by the QIAquick column.The puri®ed 365 bp PCR product was double-digestedwith NdeI and BamHI enzymes, and was puri®ed by theQIAquick column as before. The pShe6 vector, contain-ing the full Cre coding sequence fused to a 10� HisNH2-terminal tag, was digested with BamHI. The 710 bp

46 Chimeras of the Flp and Cre Recombinases
DNA fragment that contains the sequences from aminoacid 119 of Cre to beyond the stop codon for Cre inpShe6 (Cre25-coding fragment) was isolated from a 1.0 %(w/v) agarose gel and puri®ed using the QIAquick col-umn. Similarly, the pShe6 plasmid was digested withNdeI and BamHI enzymes, and the 5.7 kbp vector frag-ment was isolated and puri®ed as before. The 360 bpNdeI-BamHI digested PCR product, the 710 bp BamHI-BamHI Cre25-coding fragment and the NdeI-BamHIdigested She6 vector fragment were ligated togetherusing T4 DNA ligase. An aliquot of the ligation mixturewas used to transform competent XL1-Blue cells (Strata-gene: F0 LacIq/recA1 hsdR17 (rK
ÿ mK�)) as described by
Sambrook et al. (1989). Resulting colonies from overnightgrowth on LB-ampicillin agar plates were picked andgrown in liquid medium. DNA from individual isolateswas puri®ed as described by Sambrook et al. (1989).A unique Bsu36 restriction site in the P13-coding regionand a unique XhoI restriction site in the Cre25-codingregion were used in restriction analysis of the DNA andcon®rmed that all clones contained both the P13 PCRproduct and the Cre25-coding fragment as inserts in thecorrect orientation. One such isolate was sequenced toverify the accuracy of the PCR and the cloning. Thedesired plasmid, encoding the P13-Cre25 chimera, Fre, inframe with an NH2-terminal 10� His-tag, was denotedpShe21 and maintained in both the XL1-Blue strain andthe expression strain BL21 (DE3, pLysS), (Studier et al.,1990). The predicted molecular mass of His-Fre isapproximately 41 kDa (38 kDa Fre � 3 kDa His leadersequence).
Construction of His-Clp expression vector
The Clp protein is a chimera of the NH2-terminaldomain of Cre, Cre13, residues 1-122, and the COOH-terminal domain of Flp, P32, residues 124-423 (Figure 2).The Cre13-coding region of Cre was isolated by PCRusing the pShe6 plasmid as template. The 50 NH2-term-inal primer, NCC, was 32 nucleotides long and hybri-dized to the 10� His-leader sequence upstream of thestart codon of the Cre gene in pShe6. The primercontained an NcoI restriction site (underlined) forlater cloning steps and had the sequence:50 TAGGGCTACCATGGGCCATCATCATCATCATC 30.
The 30 COOH-terminal primer was Cre13B and hybri-dized to the sequence encoding amino acid residues 116-122 of the Cre protein. Cre13B was 43 nucleotides longand contained sequences that encode the amino acidresidues 124-128 of the Flp protein as well as a BstBIrestriction site (underlined) immediately 50 to the start ofthe Cre portion (italics) of the primer. The sequence ofthis primer was: 50 TCTAGGTTCGAACTG-TAATTGTTTTCGGATCCGCCGCATAACC 30.
A two-stage PCR was done as described before usingVent DNA polymerase, these two primers and pShe6 asthe template. The ®rst stage consisted of 30 cycles of68 �C, one minute, 72 �C, 42 seconds, 95 �C, 30 seconds.The second stage consisted of a 68 �C, one minutehybridization and a 72 �C, ®ve minutes extension. ThePCR product was puri®ed and the 455 bp Cre13-PCRproduct was digested with NcoI and BstBI. The pShe2plasmid encodes the COOH-terminal P32 fragment ofFlp in frame with an NH2-terminal His-tag. This plasmidwas digested with NcoI and then BstBI and the 5.8 kbpDNA vector fragment isolated from an agarose gel. The450 bp NcoI-BstBI digested PCR product and the 5.8 kbpNcoI-BstBI digested pShe2 vector were ligated together.
An aliquot of this mixture was transformed into the XL1-Blue strain and the desired plasmid, encoding the Cre13-P32 chimera, Clp, in frame with an NH2-terminal 10�His-tag, was denoted pShe20 and maintained in both theXL1-Blue strain and the BL21 expression strain. The pre-dicted molecular mass of the chimera is approximately48 kDa (45 kDa Clp � 3 kDa His leader sequence).
Expression and purification of His-Fre and His-Clp
The BL21 strains transformed with the Fre and Clpexpression vectors were grown at 37 �C and proteinexpression induced with 1 mM isopropyl-b-D-thiogalac-toside for 30 minutes at 37 �C; the culture was shifted toroom temperature for four hours. When produced in5 ml cultures the proteins were at least 85 % soluble asassayed by SDS-PAGE and Coomassie Blue stainingafter sonication and low-speed centrifugation. The cellpellet from a 500 ml culture was resuspended in threevolumes of sonication buffer (50 mM sodium phosphate(pH 8.0), 300 mM NaCl). Sonication was done with six20 second bursts (40 % gain, Vibra Cell sonicator) on icewith two minute intervals between bursts. The sonicatewas centrifuged at 100,000 g for one hour at 4 �C. Allsubsequent manipulations were done at 4 �C. The super-natant was applied to a 2 ml Ni-NTA agarose column(Qiagen) previously equilibrated in wash buffer (50 mMsodium phosphate (pH 8.0), 300 mM NaCl, 10 % glycer-ol). The column was washed with ®ve column volumesof wash buffer and then successively with three columnvolumes of wash buffer containing 50 mM, 75 mM,100 mM and 125 mM imidazole to remove proteinsbinding non-speci®cally to the column. The His-Fre pro-tein was eluted from the column with 175 and 200 mMimidazole in wash buffer. Similarly, the His-Clp proteinwas eluted between 150 and 200 mM imidazole-contain-ing buffer (Figure 2(c)). Both proteins were greater than95 % pure as assayed by SDS-PAGE and migrated aspredicted by their respective molecular mass. The yieldsof His-Fre and His-Clp were 10 mg/l of induced culture.Fractions were pooled and the imidazole-containing buf-fer was exchanged for a buffer containing 20 mM Hepes(pH 7.8), 0.1 mM EDTA and 0.1 mM (NH4)2SO4 using10DG desalting columns (BioRad). Initially, both chi-meric proteins caused a non-speci®c stimulation of Crebinding as assayed in gel mobility shift assays (data notshown). This activity was removed by passing the His-Fre and His-Clp samples through a Centricon 50 fol-lowed by a Centricon 10 spin-concentrator (Millipore).Even though the SDS-PAGE pro®le did not differ fromthe original eluted fractions, apparently the molecularmass sieves used in the concentrators removed factorsthat stimulated Cre binding. These His-Fre and His-Clpsamples were resuspended in buffer containing 20 mMHepes (pH 7.8), 0.1 mM EDTA and 0.1 mM (NH4)2SO4
and their concentrations were determined by the Brad-ford assay (Bradford, 1976) with reagents and IgG stan-dard from BioRad. Both proteins were stored at ÿ70 �C.
Gel mobility shift analysis of proteins
Gel mobility shift assays were used to test the abilityof His-Fre, His-Clp, Cre and Flp, and the COOH-terminal peptides of Flp and Cre to bind to a number ofsubstrates shown in Figure 1 (Andrews et al., 1987;Wierzbicki et al., 1987). The design of the substrates isdescribed in detail in Results. Substrates were singly50-end labeled and the binding reactions were performed

Chimeras of the Flp and Cre Recombinases 47
in 20 ml containing 50 mM Tris-HCl (pH 7.4), 30 mMNaCl, 2 mM MgCl2, 3 % glycerol and 0.33 mg/ml ofsonicated and denatured calf thymus DNA as competi-tor. Binding reactions were done with 0.077 pmol of sub-strate and incubated at 30 �C for 45 minutes with Cre,Cre25, His-Fre, Flp, His-P32 peptide and His-Clp. Actuallevels used for each protein are described in Results andthe Figures. Reactions were ended by the addition of2 ml of binding dye solution (1 mM Tris-HCl (pH 7.4),0.1 mM EDTA, 10 mg/ml BSA, 2 % glycerol, and 0.01 %(w/v) xylene cyanol and bromophenol blue dyes) andthen run on a non-denaturing 10 % (w/v) polyacryl-amide gel at 4 �C (Andrews et al., 1987).
Isolation and analysis of complexes
Analytical binding reactions were scaled up for pre-parative reactions from 20 to 50 ml containing 100 mMTris-HCl (pH 7.4) 60 mM NaCl, 4 mM MgCl2, 6 % gly-cerol and 0.66 mg/ml of sonicated and denatured calfthymus DNA as competitor. Binding reactions weredone with 0.385 pmol of substrate, and were incubatedat 30 �C for 45 minutes and run on a polyacrylamide gelas described. For all scaled up reactions, the lower levelsof protein used in the analytical reactions were increasedby a factor of 5 for reactions containing both one andtwo proteins (see Results). After electrophoresis, sub-strate or complexes were located by a brief exposure ofthe wet gel to X-ray ®lm. Complexes were electroelutedfrom the gel slices into Centricon 10 spin units (Milli-pore) in SDS reservoir buffer (25 mM Tris-HCl (pH 6.8),250 mM glycine, 0.1 % (w/v) SDS); the sample volumewas reduced to about 40 ml by centrifugation. SDSsample buffer was added to the samples. The sampleswere heated to 95 � for ®ve minutes and equal counts ofeach sample were analyzed by either 15 % or 17 % poly-acrylamide SDS-PAGE. The gel was soaked in 50 %methanol, 20 % glycerol, dried and exposed to X-ray®lm.
Acknowledgements
This work was supported by a grant from the MedicalResearch Council of Canada. A.C.S. was supported by aUniversity of Toronto Open Scholarship. We thankLinda Beatty, Linda Lee, Barbara Funnell, Andy Beckerand Tom Yager for helpful discussions.
References
Abremski, K. E. & Hoess, R. H. (1992). Evidence for asecond conserved arginine residue in the integrasefamily of recombination proteins. Protein Eng. 5, 87-91.
Amin, A., Roca, H., Luetke, K. & Sadowski, P. D. (1991).Synapsis, strand scission, and strand exchangeinduced by the FLP recombinase: analysis withhalf-FRT sites. Mol. Cell. Biol. 11, 4497-4508.
Andrews, B. J., Beatty, L. G. & Sadowski, P. D. (1987).Isolation of intermediates in the binding of the FLPrecombinase of the yeast plasmid 2-micron circle toits target sequence. J. Mol. Biol. 193, 345-358.
Arciszewska, L. K. & Sherratt, D. J. (1995). Xer site-speci®c recombination in vitro. EMBO J. 14, 2112-2120.
Argos, P., Landy, A., Abremski, K., Egan, J. B.,Haggard-Ljungquist, E., Hoess, R. H., Kahn, M. L.,Kalionis, B., Narayana, S. V. & Pierson, L. S. D.,et al. (1986). The integrase family of site-speci®crecombinases: regional similarities and global diver-sity. EMBO J. 5, 433-440.
Bradford, M. M. (1976). A rapid and sensitive methodfor the quantitation of microgram quantities of pro-tein using the principle of protein-dye binding.Anal. Biochem. 72, 248-254.
Chen, J. W., Lee, J. & Jayaram, M. (1992). DNA cleavagein trans by the active site tyrosine during Flprecombination: switching protein partners beforeexchanging strands. Cell, 69, 647-658.
Craig, N. L. (1988). The mechanism of conservative site-speci®c recombination. Annu. Rev. Genet. 22, 77-105.
Dombroski, A. J., Walter, W. A. & Gross, C. A. (1993).Amino-terminal amino acids modulate sigma-factorDNA-binding activity. Genes Dev. 7, 2446-2455.
Esposito, D. & Scocca, J. J. (1997). The integrase familyof tyrosine recombinases: evolution of a conservedactive site domain. Nucl. Acids Res. 25, 3605-3614.
Evans, B. R., Chen, J. W., Parsons, R. L., Bauer, T. K.,Teplow, D. B. & Jayaram, M. (1990). Identi®cationof the active site tyrosine of Flp recombinase. Poss-ible relevance of its location to the mechanism ofrecombination [published erratum appears in J. Biol.Chem. (1991) Apr 15; 266(11):7312]. J. Biol. Chem.265, 18504-18510.
Gopaul, D. N. & Van Duyne, G. D. (1999). Structureand mechanism in site-speci®c recombination. Curr.Opin. Struct. Biol. 9, 14-20.
Gopaul, D. N., Guo, F. & Van Duyne, G. D. (1998).Structure of the Holliday junction intermediate inCre-loxP site-speci®c recombination. EMBO J. 17,4175-4187.
Gronostajski, R. M. & Sadowski, P. D. (1985). The FLPrecombinase of the Saccharomyces cerevisiae 2 micronplasmid attaches covalently to DNA via a phospho-tyrosyl linkage. Mol. Cell. Biol. 5, 3274-3279.
Guo, F., Gopaul, D. N. & Van Duyne, G. D. (1997).Structure of Cre recombinase complexed with DNAin a site-speci®c recombination synapse. Nature,389, 40-46.
Guo, F., Gopaul, D. N. & Van Duyne, G. D. (1999).Asymmetric DNA bending in the Cre-loxP site-speci®c recombination synapse. Proc. Natl Acad. Sci.USA, 96, 7143-7148.
Hamilton, D. L. & Abremski, K. (1984). Site-speci®crecombination by the bacteriophage P1 lox-Cre sys-tem. Cre-mediated synapsis of two lox sites. J. Mol.Biol. 178, 481-486.
Han, Y. W., Gumport, R. I. & Gardner, J. F. (1993).Complementation of bacteriophage lambda inte-grase mutants: evidence for an intersubunit activesite. EMBO J. 12, 4577-4584.
Hoess, R. H. & Abremski, K. (1990). The Cre-lox recom-bination system. In Nucleic Acids and MolecularBiology (Eckstein, F. & Lilley, D. M. J., eds), vol. 4,pp. 99-109, Springer-Verlag, Berlin.
Hoess, R. H., Abremski, K. & Sternberg, N. (1984). Thenature of the interaction of the P1 recombinase Crewith the recombining site loxP. Cold Spring HarborSymp. Quant. Biol. 49, 761-768.
Hoess, R. H., Wierzbicki, A. & Abremski, K. (1986). Therole of the loxP spacer region in P1 site-speci®crecombination. Nucl. Acids Res. 14, 2287-2300.
Hoess, R. H., Abremski, K., Irwin, S., Kendall, M. &Mack, A. (1990a). DNA speci®city of the Cre recom-

48 Chimeras of the Flp and Cre Recombinases
binase resides in the 25 kDa carboxyl domain of theprotein. J. Mol. Biol. 216, 873-882.
Hoess, R. H., Wierzbicki, A. & Abremski, K. (1990b).Synapsis in the Cre-lox Site Speci®c RecombinationSystem. In Structure & Methods (Sarma, R. H. &Sarma, M. H., eds), vol. 1, pp. 203-213, HumanGenome Initiative & DNA Recombination, AdeninePress.
Jayaram, M. (1997). The cis-trans paradox of integrase[see comments]. Science, 276, 49-51.
Jayaram, M. & Lee, J. (1995). Return to sobriety after thecatalytic party. Trends Genet. 11, 432-433.
Kwon, H. J., Tirumalai, R., Landy, A. & Ellenberger, T.(1997). Flexibility in DNA recombination: structureof the lambda integrase catalytic core. Science, 276,126-131.
Landy, A. (1989). Dynamic, structural, and regulatoryaspects of lambda site-speci®c recombination. Annu.Rev. Biochem. 58, 913-949.
Lee, G. & Saito, I. (1998). Role of nucleotide sequencesof loxP spacer region in Cre-mediated recombina-tion. Gene, 216, 55-65.
Lee, J., Whang, I., Lee, J. & Jayaram, M. (1994). Directedprotein replacement in recombination full sitesreveals trans-horizontal DNA cleavage by Flprecombinase. EMBO J. 13, 5346-5354.
Lee, J., Jayaram, M. & Grainge, I. (1999). Wild-type Flprecombinase cleaves DNA in trans. EMBO J. 18,784-791.
Mack, A., Sauer, B., Abremski, K. & Hoess, R. (1992).Stoichiometry of the Cre recombinase bound to thelox recombining site. Nucl. Acids Res. 20, 4451-4455.
Nunes-Duby, S. E., Tirumalai, R. S., Dorgai, L., Yagil, E.,Weisberg, R. A. & Landy, A. (1994). Lambda inte-grase cleaves DNA in cis. EMBO J. 13, 4421-4430.
Nunes-Duby, S. E., Kwon, H. J., Tirumalai, R. S.,Ellenberger, T. & Landy, A. (1998). Similarities anddifferences among 105 members of the Int family ofsite-speci®c recombinases. Nucl. Acids Res. 26, 391-406.
Pan, G. & Sadowski, P. D. (1993). Identi®cation of thefunctional domains of the FLP recombinase. Separ-ation of the nonspeci®c and speci®c DNA-binding,cleavage, and ligation domains. J. Biol. Chem. 268,22546-22551.
Pan, G., Luetke, K. & Sadowski, P. D. (1993). Mechan-ism of cleavage and ligation by FLP recombinase:classi®cation of mutations in FLP protein by in vitro
complementation analysis. Mol. Cell. Biol. 13, 3167-3175.
Pan, H., Clary, D. & Sadowski, P. D. (1991). Identi®-cation of the DNA-binding domain of the FLPrecombinase. J. Biol. Chem. 266, 11347-11354.
Panigrahi, G. B. & Sadowski, P. D. (1994). Interaction ofthe NH2- and COOH-terminal domains of the FLPrecombinase with the FLP recognition targetsequence. J. Biol. Chem. 269, 10940-10945.
Qian, X. H., Inman, R. B. & Cox, M. M. (1990). Protein-based asymmetry and protein-protein interactionsin FLP recombinase-mediated site-speci®c recombi-nation. J. Biol. Chem. 265, 21779-21788.
Ringrose, L., Lounnas, V., Ehrlich, L., Buchholz, F.,Wade, R. & Stewart, A. F. (1998). Comparative kin-etic analysis of FLP and cre recombinases: math-ematical models for DNA binding andrecombination. J. Mol. Biol. 284, 363-384.
Sadowski, P. D. (1993). Site-speci®c genetic recombina-tion: hops, ¯ips and ¯ops. FASEB J. 7, 760-767.
Sadowski, P. D. (1995). The Flp recombinase of the 2-micron plasmid of Saccharomyces cerevisiae. Prog.Nucl. Acid Res. Mol. Biol. 51, 53-91.
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Molecu-lar Cloning: A Laboratory Manual, 2nd edit., ColdSpring Harbor Laboratory Press, Cold Spring Har-bor, NY.
Shaikh, A. C. (1997). Assays of the functional domainsof the Cre and Flp recombinases. MSc thesis,University of Toronto.
Shaikh, A. C. & Sadowski, P. D. (1997). The Cre recom-binase cleaves the lox site in trans. J. Biol. Chem.272, 5695-5702.
Studier, F. W., Rosenberg, A. H., Dunn, J. J. &Dubendorff, J. W. (1990). Use of T7 RNA polymer-ase to direct expression of cloned genes. MethodsEnzymol. 185, 60-89.
Subramanya, H. S., Arciszewska, L. K., Baker, R. A.,Bird, L. E., Sherratt, D. J. & Wigley, D. B. (1997).Crystal structure of the site-speci®c recombinase,XerD. EMBO J. 16, 5178-5187.
Vetter, D., Andrews, B. J., Roberts-Beatty, L. &Sadowski, P. D. (1983). Site-speci®c recombinationof yeast 2-micron DNA in vitro. Proc. Natl Acad. Sci.USA, 80, 7284-7288.
Wierzbicki, A., Kendall, M., Abremski, K. & Hoess, R.(1987). A mutational analysis of the bacteriophageP1 recombinase Cre. J. Mol. Biol. 195, 785-794.
Edited by M. Gottesman
(Received 13 April 2000; received in revised form 5 June 2000; accepted 13 June 2000)


















