
Chemically Modified Oligonucleotides: Synthesis, Physicochemical · PDF fileChemically...
68
Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 973 Chemically Modified Oligonucleotides: Synthesis, Physicochemical and Biochemical Properties of their Duplexes with DNA and RNA BY PUSHPANGADAN INDIRA PRADEEPKUMAR ACTA UNIVERSITATIS UPSALIENSIS UPPSALA 2004
Transcript of Chemically Modified Oligonucleotides: Synthesis, Physicochemical · PDF fileChemically...

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Science and Technology 973
Chemically ModifiedOligonucleotides: Synthesis,
Physicochemical and BiochemicalProperties of their Duplexes with
DNA and RNA
BY
PUSHPANGADAN INDIRA PRADEEPKUMAR
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2004


To My Parents

The Original Publications
This thesis is based on the following original publications referred by Roman numerals. I Zamaratski, E.; Ossipov, D.; Pradeepkumar, P. I.; Amirkhanov,
N. V.; Chattopadhyaya, J. The 3'-modified antisense oligos pro-mote faster hydrolysis of the target RNA by RNase H than the natural counterpart. Tetrahedron 2001, 57, 593-606.
II Pradeepkumar, P. I.; Zamaratski, E.; Foldesi, A.; Chat-topadhyaya, J. Transmission of the conformational information in the antisense/RNA hybrid duplex influences the pattern of the RNase H cleavage reaction. Tetrahedon Lett. 2000, 41, 8601-8607.
III Pradeepkumar, P. I.; Zamaratski, E.; Foldesi, A.; Chat-topadhyaya, J. Conformation-specific cleavage of antisense oli-gonucleotide-RNA duplexes by RNase H J. Chem. Soc., Perkin Trans. 2, 2001, 402-408.
IV Pradeepkumar, P. I.; Chattopadhyaya J. Oxetane modified an-tisense oligonucleotides promote RNase H cleavage of the com-plementary RNA strand in the hybrid duplex as efficiently as the native, and offer improved endonuclease resistance. J. Chem. Soc., Perkin Trans. 2, 2001, 2074-2083.
V Pradeepkumar, P. I.; Amirkhanov, N. V.; Chattopadhyaya, J. Antisense oligonucleotides with oxetane-constrained cytidine enhance heteroduplex stability, elicit satisfactory RNase H re-sponse as well as show improved resistance to both exo and en-donucleases. Org. Biomol. Chem. 2003, 1, 81-92.
VI Pradeepkumar, P. I.; Cheruku, P.; Plashkevych, O.; Acharya, P.; Gohil, S.; Chattopadhyaya, J. Synthesis, physicochemical and biochemical studies of 1',2'-oxetane constrained adenosine and guanosine modified oligonucleotides, and their comparison with those of the corresponding cytidine and thymidine analogs. 2004 J. Am. Chem. Soc. (under review)

VII Opalinska, J. B.; Kalota, A.; Rodriquez, L.; Henningson, C.; Gif-ford, L. K.; Lu, P.; Jen, K-Y.; Paradeepkumar, P. I.; Barman, J.; Kim, T. K.; Swider, C.; Chattopadhyaya, J.; Gewirtz, A. M. Ra-tionally targeted, conformationally-constrained, oxetane modi-fied oligonucleotides are highly efficient gene silencing mole-cules. 2004 Proc. Natl. Acad. Sci. USA (under review)
VIII Ossipov, D.; Pradeepkumar, P. I.; Holmer, M.; Chattopadhyaya, J. Synthesis of [Ru(phen)2DPPZ]2+-tethered oligo-DNA and stud-ies on the metallointercalation mode into the DNA duplex. J. Am. Chem. Soc. 2001, 123, 3551-3562.
IX Boon, E. M.; Barton, J. K.; Pradeepkumar, P. I.; Isaksson, J.; Petit, C.; Chattopadhyaya, J. An electrochemical probe of DNA stacking in an antisense oligonucleotide containing C3'-endo- locked sugar. Angew. Chem. Int. Ed. 2002, 41, 3402-3405.

Contents
1. Introduction.........................................................................................................1 1.1 Impact of enabling oligonucleotide chemistry.............................................1 1.2 Overview of this thesis ................................................................................2
2. Synthesis, physicochemical and biochemical studies of chemically modified oligonucleotides and their duplexes with DNA and RNA...............................3 2.1 Antisense oligonucleotides (AONs) as therapeutic agents ..........................3
2.1.1 AONs: mechanism of action and pharmaceutical challenges..........3 2.1.2 Limitations of phosphorothioate AONs and other backbone
modified AONs ..............................................................................4 2.1.3 Chemically modified AONs retaining natural PO-backbones.........5
2.1.3.1 The 2'-modified AONs .....................................................6 2.1.3.2 AONs modified with conformationally constrained
bicyclic and tricyclic nucleosides.....................................7 2.1.3.3 AONs conjugated with chromophores and other
functionalities...................................................................9 2.1.4 Present work..................................................................................11
2.1.4.1 Physicochemical properties of dipyridophenazine (DPPZ) and phenazine (PZN) conjugated AON/RNA hybrids (Paper I) .........................................................................11
2.1.4.2 RNase H eliciting power of chromophore tethered AON/RNA hybrids and exonuclease stability of conjugated AONs (Paper I)............................................13
2.1.4.3 Synthesis of 1',2'-oxetane-modified phosphoramidite building blocks (Papers III, V and VI)...........................15
2.1.4.4 Conformation of oxetane-modified nucleosides and CD spectra of their AON/RNA hybrid duplexes (Papers II-IV and VI) ...........................................................................17
2.1.4.5 Thermostability and thermodynamics of oxetane-modified AON/RNA hybrids (Papers II-VI)..................18
2.1.4.6 Endonuclease, exonuclease, and serum stability of oxetane-modified AONs (Papers IV-VI) .......................21
2.1.4.7 RNase H cleavage pattern and extent of RNA hydrolysis in oxetane-modified AON/RNA hybrids (Papers II-VI) 22
2.1.4.8 Michaelis-Menten kinetics of RNase H cleavage in oxetane-modified AON/RNA hybrids (Papers V and VI).......................................................................................24
2.1.4.9 Gene down-regulation using oxetane-modified AONs in cellular system (Paper VII) ............................................26
2.2 Elucidation of metallointercalation mode in DNA/DNA duplexes by DNase 1 footprinting................................................................................28 2.2.1 DNase 1 footprinting.....................................................................28

2.2.2 Present work..................................................................................29 2.2.2.1 Elucidation of metallointercalation mode in
[Ru(phen)2(DPPZ)]2+ conjugated ODN/DNA duplexes (Paper VIII)....................................................................29
2.3 Nucleic acid mediated charge transport as a tool to probe base stacking perturbations.............................................................................................31 2.3.1 Charge transport in nucleic acids and electrochemical detection ..31 2.3.2 Present work..................................................................................32
2.3.2.1 Electrochemical detection of structural influence of a single oxetane-T unit in DNA/DNA and DNA/RNA duplexes (Paper IX) ......................................................32
3. Summary ...........................................................................................................34
4. Future perspectives...........................................................................................36
5. Acknowledgements ...........................................................................................39
6. Summary in Swedish ........................................................................................41
7. References..........................................................................................................42

Abbreviations
A Adenin-9-yl or adenosine Ac Acetyl ANA Arabinonucleic acid AON Antisense oligonucleotide B Any nucleobase Bz Benzoyl C Cytosin-1-yl or cytidine CD Circular dichroism CPG Controlled pore glass CT Charge transport DNase 1 Deoxyribonuclease 1 DMF N,N-dimethylformamide DPC Diphenylcarbamoyl dmf N,N-dimethylforamamidine DMTr 4,4'-dimethoxytrityl DNA Deoxyribonucleic acid DPPZ Dipyridophenazine ds Double stranded EDTA Ethylenediaminetetraacetic acid G Guanin-9-yl or guanosine HIV Human immunodeficiency virus i-Pr Isopropyl i-Bu Isobutyryl K Kelvin Km Michaelis constant kcat Turnover number LB Leucomethylene blue LNA Locked nucleic acid mRNA Messenger RNA MB Methylene blue MD Molecular dynamics NaHMDS Sodium bis(trimethylsilyl)amide NMR Nuclear magnetic resonance N-E type North-East type NOE Nuclear overhauser effect ODN Oligodeoxynucleotide Oxetane (1',3'-O-anhydro-β-D-psicofuranosyl) nucleosides P Pseudorotational phase angle Pac Phenoxyacetyl PAGE Polyacrylamide gel electrophoresis PNA Peptide nucleic acid

PO Phosphodiester PS Phosphorothioate phen 1,10-phenanthroline PZN Phenazine PZNM Phenazinium QRT-PCR Quantitative realtime-polymerase chain reaction RISC Ribonucleic acid induced silencing complex RNA Ribonucleic acid RNAi Ribonucleic acid interference RNase H Ribonuclease H siRNA Small interfering ribonucleic acids SVPDE Snake venom phosphodiesterase SQRM Self-quenching reporter molecule T Thymin-1-yl or thymidine TFA Trifluroacetic acid TFO Triplex forming oligonucleotide Tm Melting temperature TBAF n-Tetrabutylammonium fluoride THF Tetrahydrofuran TIPDSCl2 1,3-dichloro-1,1',3,3'-tetraisopropyldisiloxane Tol 4-Toluoyl ss Single stranded U Uracil-1-yl or uridine UV Ultraviolet Vmax Maximum velocity ∆G Free energy of a process ∆H Enthalpy of a process ∆S Entropy of a process φm Puckering amplitude


1
1. Introduction
1.1 Impact of enabling oligonucleotide chemistry The first chemically synthesized oligonucleotide was a thymidine dimer by Michelson and Todd in 1955.1 Pioneering works of Khorana,2-8 Letsinger,9-14 Reese15-20 and Caruthers21-24 have pushed the frontier of knowledge of the oligonucleotide synthesis to the present state-of-the-art. However, search for new and improved solid phase methodologies is still of considerable inter-est.25-30 Two discoveries made the indispensability of oligonucleotides in research and therapy are the polymerase chain reaction (PCR) by Kary B Mullis31-33 and the halting of viral replication by oligonucleotides demon-strated by Zamecnik and Stephenson in 1978.34-35 The latter discovery util-izes oligonucleotides complementary to the mRNA transcribing from a gene, where the externally supplied oligodeoxynucleotide binds to the mRNA by Watson-Crick base pairing36 and prevents the translation (mRNA to protein) by varying mechanisms of action.37-45 This approach is called antisense tech-nology and the oligodeoxynucleotide used for this purpose is termed an-tisense oligonucleotide (AON). The rationality of antisense approach at least in theory gives an opportunity to target any mRNA transcribed from disease causing genes and this has fuelled considerable therapeutic interests in oli-gonucleotides.39-46 AONs also provided new gene knock-out tools in func-tional genomics as well as target validation tools in drug discovery.47-48 Moreover, oligonucleotides find applications in diagnostics (microarrays)49-
53 and nanotechnology.54-56 Besides AONs, several other utilities of oligonucleotides have emerged
with possible pharmaceutical applications.42,43 Oligonucleotides called tri-plex forming oligonucleotides (TFOs)57-62 can be targeted to the double stranded DNA where they bind to the major groove of the DNA double helix by Hoogsteen base pairing.63 This approach of gene down-regulation is called the antigene technology.57-62 Although this method offers gene knock off using minimum amount of TFOs, the difficulties in target accessibility owing to the proteins associated with genome, and the need of homo-purine tracks in the gene for triplex formation limit the applications of antigene technology.43, 64 The discovery of catalytic RNAs called Ribozymes65-71 in early 1980s and the discovery of catalytic DNAs called DNAzymes72-80 in mid 1990s have also opened new window for oligonucleotide based thera-peutics. Protein binding oligodeoxynucleotide duplexes called "decoys" have shown potential in targeting transcription factors.81,82 Oligonucleotides con-taining d(CpG) dinucleotides (CpG DNA) exhibit several immunological responses and are being developed as therapeutic agents and adjuvants for various diseases.83-87 Recently, the short double stranded RNA duplexes called small interfering RNAs (siRNA) have been successfully used to si-lence gene function utilizing naturally occurring mechanism called RNA

2
interference (RNAi).88-97 This has given tremendous boost to the RNA based oligonucleotide therapeutics.42,43,98-100
Among the all aforementioned ways, making use of oligonucleotides as gene silencing agents, in pharmacological perspective, antisense technology is the most matured one.41-45,64,101 This is evident from the fact that one oli-gonucleotide drug called Vitravane developed by ISIS Pharmaceuticals, USA to treat the inflammatory viral infection of the eye caused by cy-tomegalovirus (CMV) has already been introduced to the market.43,45 An-other drug called Genasense (Genta Inc, USA) targeting Bcl-2, a protein expressed in cancer cells to protect them against chemotherapy is waiting for FDA approval.64,102 However, there is still doubt remaining about the "real" mechanism in which these drugs work.46,100 Other 20 AONs are in different phases of clinical trials.45
Although the natural phosphodiester (PO) oligonucleotides are easy to synthesize, their use is limited as they are degraded by intracellular endo- and exonucleases.103-109 Moreover, the degradation products of the PO-oligonucleotides are highly toxic.110-112 This has warranted the chemical modification of oligonucleotides in order to utilize them for therapeutic ap-plications.113-116
1.2 Overview of this thesis In this thesis we present the utilities of chemically modified oligonucleotides as potential antisense agents. Two different types of modifications of the AONs have been attempted: (1) Conjugation of chromophores such as dipyridophenazine (DPPZ), phenazine (PZN), phenazinium (PZNM), [Ru(phen)2(DPPZ)]2+ etc. at the 3'- or 5'-ends or middle of oligonucleotides. (2) AONs incorporated with North-East conformationally constrained 1',2'-oxetane nucleosides [(1',3'-O-anhydro-β-D-psicofuranosyl) nucleosides]. These modified AONs have shown tight binding to the target RNA, en-hanced nuclease resistance, RNase H recruitment capability and non-toxicity. A combination of 3'-DPPZ and oxetane-cytidine modified AONs have been found to be efficient gene silencing agents in cellular system. We have also explored the structural perturbations brought by these chemical modifications in DNA/DNA and DNA/RNA duplexes utilizing spectro-scopic (UV and CD), enzymatic (DNase 1) and charge transport (CT) stud-ies.

3
2. Synthesis, physicochemical and biochemical studies of chemically modified oligonucleotides and their duplexes with DNA and RNA
2.1 Antisense oligonucleotides (AONs) as therapeutic agents
2.1.1 AONs: mechanism of action and pharmaceutical challenges AONs can down-regulate genes of interest using different mechanism of actions.37-45 They can for example alter or stop splicing by binding to pre-messenger RNA in the nucleus or sterically block the ribosomal read-through by strong binding to the mRNA (steric-blocker AONs).117-120 An-other and the most important mechanism is the recruitment of an enzyme called RNase H to cleave the mRNA in AON/mRNA hybrid duplex (RNase H dependent AONs).121-127 Antisense inhibition based on RNase H activation has obvious advantages in terms of efficiency and dosage. This is because once the RNA target is cleaved by RNase H (permanent inactivation of the message) the AON is released and can find other copies of the target (Figure 1). Thus using minimum amount of AON, maximum efficiency can be achieved. 45
RNase H is an enzyme ubiquitous in all cells.128,129 The enzyme is present in the nucleus and the cytoplasm.130 It produces oligonucleotides with 5'-phosphate and 3'-hydroxy groups as final cleavage products.130 Bivalent cations such as Mg2+ and Mn2+ are found to be necessary cofactors for en-zymatic activity.130-132 The enzyme is widely present in various organisms130, including retroviruses, as a domain of the reverse transcriptase.133,134 The RNase H1 from Escherichia coli is the most characterized enzyme in this family.135-138 Even though the physiological functions of E. coli RNase H1 have not been understood clearly, it has been suggested to be involved in the DNA replication and repair.139-145 Recent isolation of the human RNase H1 and RNase H2 highlights the importance of the development of the antisense drugs utilizing this mechanism of action.146-150 Although RNase H binds to DNA/DNA and RNA/RNA hybrids (in a lesser extent) it cleaves only the RNA strand in the DNA/RNA hybrid.151,152 A four deoxynucleotide stretch in an AON/RNA hybrid is necessary for the cleavage by the enzyme.152 This might account for the "low stringency" of this enzyme.153 Various structural and functional studies have underscored the importance of an optimal minor groove width (close to A type helix), 2'-OH group in the RNA strand, charged backbone, β-linkage of the nucleobases and flexibility of the DNA/RNA helix at the cleavage centre are the essential requirements for the efficient RNase H cleavage. 125,154-161
The problems with employing AONs for therapeutic purposes are mani-fold: (i) the AON should be stable in the cellular media and serum, (ii) it should be easily deliverable, (iii) it should find the target (specificity) and

4
exert the desirable biological effect using minimal dosage, (iv) it should elicit RNase H cleavage, (v) it should not exert any toxicity, (vi) the AON
Figure 1. The RNase H promoted cleavage of the target mRNA through the forma-tion of AON/RNA Hybrid Duplex. The kinetic scheme of the RNase H hydrolysis is shown in the bottom part of the cartoon, where D is AON (antisense Oligo); R is the target RNA; Kd1 is the equilibrium constant of dissociation of the heteroduplex DR; Kd2 is equilibrium constant of dissociation of the substrate-enzyme complex DRE
should have favorable pharmacokinetic properties (tissue and cellular distri-bution), and (vii) the synthesis should be cheap and straightforward. The last 25 years of research has shown that achieving all these properties by chemi-cal modification at the backbone, sugar and nucleobase is indeed a challeng-ing task. 38,41-46 Furthermore, finding the right target sequence in a mRNA for AON hybridization42,162-164 and finding suitable delivery agents165-168 pose serious challenges in the future application of the antisense technology.
2.1.2 Limitations of phosphorothioate AONs and other backbone modified AONs
The first generation AONs, the phosphorothioates (PS),169,170 where one of the non-bridging oxygen atom in the phosphodiester linkage is substituted by sulfur atom, have so far been widely used for clinical trials against many targets,45 but with little success.46,100 The phosphorous centre in PS oligos is chiral, generating many diastereomers, and imparts nuclease resistance to PS-AONs.169 It should be noted that among the Rp and Sp diastereomers only the Sp isomer is nuclease resistant.171 They also have excellent RNase H acti-vation (at lower AON concentration) and some favorable pharmacokinetic properties.45,169 However, at high AON concentration they inhibit RNase H activity.151 Furthermore, PS modification makes the AON/RNA duplex lose 0.5-1.5 oC in melting temperature (Tm) per PS linkage,172,173 which hampers

5
their sequence specificity and sequence accessibility. They have also shown severe non-specific interactions with proteins41,174-176 which makes the cor-rect interpretation of the antisense effect caused by these AONs doubtful and problematic.175 PS-AONs, compared to the PO-counterparts, have one to three orders of magnitude higher binding affinity for various cellular pro-teins especially the heparin binding proteins and to many cellular receptors.41 The proteins that have strong affinity to PS-AONs include basic fibroblast growth factor (bFGF)177 and acidic fibroblast growth factor (aFGF), vascular endothelial growth factor (VEGF)177,178 and its receptors and the epidermal growth factor receptor (EGFR).179 They also have high affinity for cell sur-face heparin binding proteins such as fibronectin, laminin180 and Mac-1(CD11-CD18).181 PS-AON can also bind to CD4,182 HIV-glycoprotein 120,183 HIV-reverse transcriptase and protein kinases.184 Many other classes of such proteins are still to be identified.41 The binding of PS oligonucleo-tides to such proteins is mostly sequence independent and length depend-ant.41 In vivo, immune stimulation and prolongation of activated partial thromboplastin time (aPPT) have been observed after the systematic admini-stration of all PS-AONs.185 This kind of toxicity associated with PS-AONs prompted chemists to search for other chemical modifications.
The backbone modifications such as methylphosphonate186-188 and peptide nucleic acids (PNA)189-194 although enhanced the nuclease resistance, they failed to recruit RNase H for the target RNA cleavage. The presence of neu-tral backbone in these modifications also reduces their aqueous solubility and cellular internalization.188,193,195 The boranophosphate modification196-198 has shown enhanced RNase H eliciting power but the low binding affinity to the target hampers its antisense potential. Among the numerous backbone modifications introduced,199 two modifications extensively studied in vitro and in vivo are the N3'-P5' phosphoramidate based AONs200,201 and the mor-pholino oligonucleotides.202,203 Both of these modifications do not support RNase H cleavage.115 However, they have exhibited excellent target affinity and nuclease resistance.200-203 Consequently, they have been successfully employed as steric-blockers in cellular systems.204-207 Efficient delivery of morpholino AONs is still found to be problematic.38 Recently phos-phonoacetate and thiophosphonoacetate AONs have been reported as nucle-ase resistant and RNase H compatible backbone modifications.208,209 How-ever, their target RNA affinity was found to be much lower than that found for the isosequential PS and PO counterparts.209 This makes them less attrac-tive in antisense research.
2.1.3 Chemically modified AONs retaining natural PO-backbones Studies employing various backbone modified AONs have revealed that to retain RNase H activity and to limit the toxicity, the natural phosphate back-bone should be retained in an AON. This has prompted chemists to modify AONs at the base or sugar moieties. Another approach is the conjugation of

6
intercalators and chemical functionalities at the 3'- or 5'-end or middle of AONs.
Among the various nucleobase modified AONs reported,210 the in vitro and in vivo gene down-regulation has been shown only for very limited base modified AONs. This includes AONs containing 2'-deoxyuridine and 2'-deoxycytidine nucleosides bearing propyne group at C-5211,212 and the re-cently reported 9-(aminomethoxy)phenoxazine analogue of cytosine (G-clamp).213,214 They have shown enhanced binding affinity towards RNA, exonuclease resistance, RNase H activation and efficient gene silencing in cellular systems.210-214 However, the in vivo studies showed that the C-5 pro-pynyl pyrimidine modified AONs are highly hepatotoxic.215 The in vivo stud-ies with G-clamp modification are not yet available.
In recent years the modification of the sugar moiety in AONs has been in-tensively pursued to achieve the properties of an ideal antisense drug.216,217 Among the numerous modifications reported only few of them have been studied thoroughly64 as antisense agents and have shown promising in vitro and in vivo antisense activity.
2.1.3.1 The 2'-modified AONs The 2'-modifications in the sugar such as 2'-F,218 2'-O-Me,219 2'-O-MOE (methoxyethyl),219 2'-O-AP (aminopropyl),220 2'-O-DMAOE (dimethylami-noethyl),221 2'-O-DMAP (dimethylaminopropyl),217 2'-O-DMAEOE (di-methylaminoethyloxyethyl),222 2'-O-NMA (N-methylacetamido)217 have been shown to increase the target affinity with RNA by 1 to 3 ºC per modifi-cation in their AON/RNA hybrids.217 However, complete modifications of AONs with these 2'-substituted nucleotides failed to elicit RNase H activa-tion.217 This is due to the fact that once incorporated into AONs, they lock the sugar conformation into N-form and make the resulting AON/RNA hy-brid into a rigid RNA/RNA type duplex.217,223 It should, however, be noted that the monomer 2'-O-alkyl modified units in solution have shown only little preference for N-conformer.224 The exonuclease stability of 2'-O-Me modification was lower than that of the PS-AON where as the 2'-O-MOE modified AONs have shown similar stability to that of PS-AONs.217 The highest nuclease stability has been displayed by 2'-O-AP modified AON, which was 6-8 times higher than the PS-AON.220 Among the above men-tioned 2'-modified AONs the most widely studied as antisense agents are the 2'-O-Me and 2'-O-MOE modified AONs.45,115,225 The AONs fully modified with these units have been successfully used as steric blockers as well as agents for correcting aberrations in splicing.225-227 To achieve RNase H acti-vation using these 2'-modifications the gapmer (chimeric) oligos have been constructed.217,151,152,225 They constitute the second-generation AONs. A chimera consists of 3 to 4 modified nucleotides at both ends of the AON with a stretch of unmodified nucleotides (4-8) in the middle. Although these constructs could elicit RNase H, the efficiency was 3 times lower than the

7
isosequential native counterpart.45,152,225 Owing to the unfavorable pharma-cokinetics, in many of the in vitro and in vivo studies PS backbone is re-tained in these gapmer AONs.225 These PS-MOE/DNA/MOE gapmers have shown excellent gene silencing effect in cellular systems.45,225
The first completely sugar modified AONs, which have shown the RNase H activation are the arabinonucleic acids (ANA) and 2'-F-ANAs.228-230 The RNase H activation can be corroborated to the similarity in the global helical conformation and the flexibility of ANA/RNA and F-ANA/RNA hybrid duplexes to that of the native AON/RNA hybrid.231,232 Furthermore, the 2'-OH and 2'-F atom project in the major groove of the ANA/RNA or F-ANA/RNA duplex preventing the steric clash with RNase H in the minor groove.232 The ANA imparts slight destabilisation (0.5 ºC in Tm/modification) of the hybrid duplex while F-ANA causes slight gain in thermostability (1.2 oC /modification) compared to the native hybrid du-plex.228-230 The mixmer F-ANA/RNA hybrids were better substrate for RNase H than the fully modified F-ANA/RNA hybrids.233 Both ANA and F-ANA failed to show exonuclease stability similar to that of PS-AONs.229 This is probably the reason why in two recent reports F-ANA chimeras con-sisting of full PS backbone have been used for gene down-regulation.233,234 No in vivo antisense studies are hitherto available for these modified AONs.
In addition to ANA and F-ANA, among the sugar modified AONs, the cyclohexene nucleic acids have shown RNase H recruitment only in the presence of large excess of the enzyme.235 Therefore the kcat for RNase H cleavage was 600 fold lower compared to the isosequential unmodified counterpart.236 Also it is noteworthy that there are no reports available in the literature showing antisense inhibition by cyclohexene nucleic acids in cellu-lar systems.
2.1.3.2 AONs modified with conformationally constrained bicyclic and tricyclic nucleosides
In conformationally constrained bicyclic and tricyclic nucleosides the sugar moieties are being locked into the different poles of the pseudorotational cycle (Figure 2).237-276 It is well known that the incorporation of N-conformationally constrained nucleosides (–1º < P < 34º)277,278 into an AON can impart enhanced stability to the corresponding AON/RNA hybrid due to their ability to drive the AON/RNA hybrid to more stable RNA/RNA type duplex.223 This has been clearly demonstrated for one of the early members of this family the methanocarbathymidines (2 and 3 in Figure 2).243-246 The incorporation of (N)-methanocabathymidine (2) (i.e. the sugar is in the N-form) into AON causes increase in the thermostability (0.8 to 2.2 ºC/modification) of the resulting AON/RNA hybrid duplexes,243,245 whereas the introduction of (S)-methanocarbathymidine (3) (i.e. the sugar is in the S-
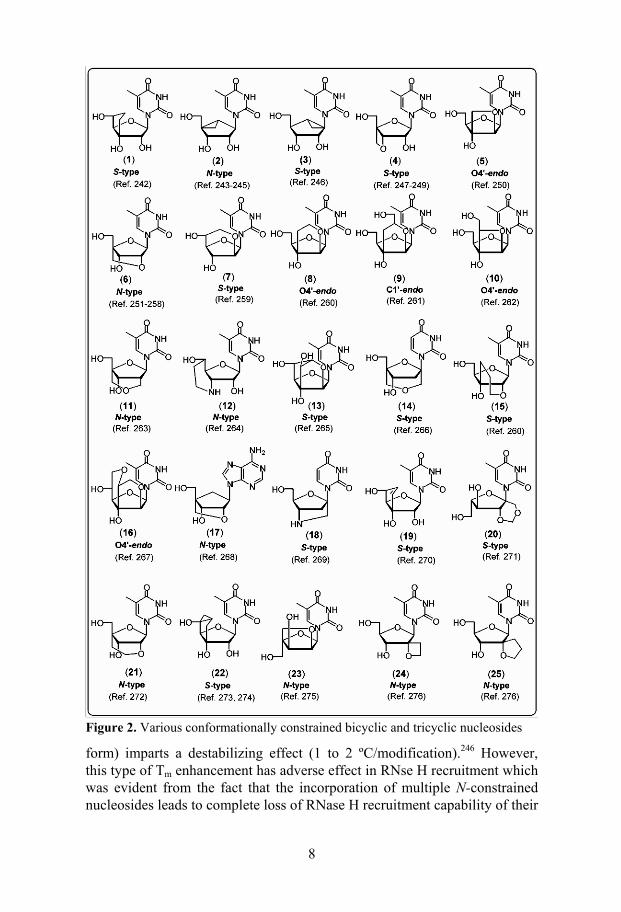
8
Figure 2. Various conformationally constrained bicyclic and tricyclic nucleosides
form) imparts a destabilizing effect (1 to 2 ºC/modification).246 However, this type of Tm enhancement has adverse effect in RNse H recruitment which was evident from the fact that the incorporation of multiple N-constrained nucleosides leads to complete loss of RNase H recruitment capability of their

9
AON/RNA hybrids.245,125,237,238 Neither systematic studies of RNase H acti-vation nor gene down-regulation properties of many AONs [with the excep-tion of locked nucleic acid (LNA) (6)257] modified with conformationally constrained nucleosides are available for comparing their antisense poten-tials with other chemically modified AONs.
The most widely studied and promising AONs in this class are oligos in-corporated with N-conformationally constrained LNA monomers introduced by Wengel et al.251 and Imanishi et al.252 Various stereoisomers and ana-logues of LNA have also been reported.257 The incorporation of β-D-LNA imparts an unprecedented stability enhancement (2 to 10 ºC/modification) to their AON/RNA hybrids.251,257,258 Structural studies have shown that incor-poration of just three β-D-LNA units was sufficient to drive the conforma-tion of a 9mer AON/RNA hybrid into a RNA/RNA type duplex.279,280 Such duplexes however failed to elicit RNase H cleavage. A recent systematic study from Erdmann's group has revealed that RNase H activation can be achieved using LNA/DNA mixmer or LNA/DNA/LNA gapmer AON con-structs.281 However, a deoxynucleotide gap size of 6 units only barely acti-vates RNase H.281 Efficient RNase H activation, however, was achieved only by increasing the gap size to 8 to 10 nucleotides.281 A similar strategy has been employed in a recent report using ethylene-bridged nucleic acid (ENA, 21) for the efficient gene down-regulation in cellular systems.282 Considering the endonuclease vulnerability of such gapmers,283-287 the use of such lengthy gaps could be harmful for in vivo applications. A recent report demonstrated the utility of α-L-LNA (23) to circumvent this problem.288 End blocking of AONs with three β-D-LNA units helped to achieve serum stability better than the 2'-O-Me modification and PS oligos.281 Various LNA/DNA mix-mers and LNA/DNA/LNA gapmers were reported to be very efficient an-tisense molecules in cellular systems.257,281,289 Two in vivo antisense studies have so far been reported for β-D-LNA incorporated AONs.290,291 Fully modified LNA-AONs have been used as steric blockers and decoys for effi-cient gene silencing.257
Recently AONs modified with tricyclo DNA monomers (22 in Figure 2)273,274 have been reported for correcting aberrant splicing of pre-mRNA in cellular systems.292,293 Although tricyclo-DNA increases the RNA target affinity (1 ºC/modification) and serum stability they failed to recruit RNase H.273,274
2.1.3.3 AONs conjugated with chromophores and other functionalities Conjugation of various chromophores, hydrophobic moieties and other chemical functionalities294-296 to AONs has emerged as a promising area in the antisense research in view of their potential ability to form stable AON/RNA duplexes with preserved helical structural parameters297,298 of the unconjugated counterpart, their utility for cellular delivery,166 stability to-wards various exonucleases299,300 and their ability to modulate pharmacoki-

10
netic properties.295 However, only few 295of these conjugated AONs have been investigated on their ability to activate RNase H and in vitro and in vivo gene down-regulation capabilities.
Conjugation of polyaromatic compounds such as phenanthridinium,301 phenazinium,302,303 phenazine,304 acridine,305-307 ethidium,308 pyrene,309,310 flourescein,309 anthraquinone,311,312 stilbene,313 anthracene314,315 to the 5'- or 3'-end or middle of AONs has been shown to increase the target RNA bind-ing affinity by 5 to 30 ºC. The thermostability of these conjugated AON/RNA duplexes depends on the aromatic nature of the conjugated chromophore, nature of linker and the site of conjugation.316 Acridine conju-gated AONs were found to be better substrate for RNase H than the uncon-jugated counterpart.317 Studies from our lab have shown the effect of various 5'-chromophores in enhancing thermostability and RNase H potential in short AON/RNA hybrids.298
In order to enhance the AON delivery into cells and to improve pharma-cokinetic properties, various hydrophobic moieties such as cholesterol,318-320 cholic acid,321 adamantane,322 lipids,323 dimethoxytrityl group324 and ibupro-fen325 have been conjugated to AONs. Among these, the cholesterol conju-gated AONs have been found to be most promising.295,326 Cholesterol conju-gated AONs not only enhanced cell permeability and tissue distribution but also enhanced RNase H promoted cleavage.327 Several cellular and in vivo studies have demonstrated that 5'- or 3'- or bis-cholesterol conjugated AONs are more efficient antisense agents than their unconjugated counter-parts.295,328
To facilitate AON internalization in cells via receptor mediated endocyto-sis vitamins such as folic acid329 or α-tocopherol330 and multivalent carbohy-drate clusters331 as well as glycopeptides332 have been conjugated to AONs. Another successful approach to enhance the cellular permeation properties is the conjugation of various peptides to AONs.166,295 Two widely studied ex-amples are 13-amino acid sequence (Tat) from HIV Tat protein 333and a 16-amino acid sequence (Ant) from Drosophila antennapedia homeotic pro-tein.334 Conjugation of various cationic modifications and polyamines has also been reported.295,335 In this line, the C-branched spermine tethered oli-gonucleotides have been reported from our laboratory.336
Another application of conjugation chemistry in AON is the metal as-sisted hydrolytic cleavage337,338 of the target RNA and the photocrosslinking of the AON/ODN to the target.339 Conjugation of various mRNA cleaving agents based on Cu2+,340 Zn2+,341-343 Eu3+,344 Fe2+,345 Th3+,346 Lu3+ 346,347 has been reported. These AONs are generally called as artificial nucleases.337,338 The photocrosslinking agents such as psoralen348and various Pt2+ 349 as well as Ru2+ 350 complexes have also been conjugated to AONs in order to modu-late gene expression via translational arrest. However, the cellular and in vivo studies employing the AONs appended with these artificial nucleases and crosslinking agents are still scarce in the literature.295

11
2.1.4 Present work This is comprised mainly of two parts. The first part consists of 3'- and 5'-conjugation of AONs and exploration of the physicochemical properties as well as RNase H recruitment capabilities of their AON/RNA hybrids. The second part deals with the synthesis and antisense properties of 1',2'-oxetane-modified AONs.
2.1.4.1 Physicochemical properties of dipyridophenazine (DPPZ) and phenazine (PZN) conjugated AON/RNA hybrids (Paper I)
(This work has been done in collaboration with Dr. E. Zamaratski and Dr. D. Ossi-pov)
In this study we have explored the utility of short 9mer AONs (27-30 and 38-41) conjugated with various 3'- and 5'-chromophores such as phenazine (PZN)351 dipyridophenazine (DPPZ)352 as antisense reagents (Table 1). Three different RNA targets (42-44) of varying self-aggregation properties (charac-terized by UV and CD spectroscopy) have been used in order to evaluate the kinetic accessibility of conjugated AONs to the folded targets. All those target RNAs have the same complementary sequence for AON hybridiza-tion.
The 11mer RNA (42) is found to be non-aggregated in our temperature dependent UV353 and CD studies. The 17mer low-aggregated RNA (43) did not show any significant change in hyperchromicity on UV melting. This directed us to use CD spectroscopy, which is reported to be more sensitive in finding some nucleic acid structural transitions,354 to characterize the in-tramolecular association of this target. Temperature and concentration de-pendent CD studies have clearly shown the self-aggregation (bimolecular process) of this target and allowed us to calculate the ∆Gº298 of duplex for-mation which was found to be –34.7 kJ/mol (Figures 5, 3B, 4A and 4B in Paper 1). The 17mer highly-aggregated RNA (44) has shown clear sigmoi-dal transition in UV-melting (∆Gº298 = –38.3 kJ/mol) as well as in the tem-perature dependent CD studies (Table 1, Figures 2, 3C, 4C and 4D in Paper 1).
When evaluated for the target affinity with the non-aggregated 11mer tar-get (42) all the PO (27-30) and PS (38-41) AONs have shown enhanced binding affinity, which was evident from Tm and ∆Gº298 (Table 3 and 4 in Paper 1). Although the pairing entropy of the chromophore conjugated AON/RNA hybrids was found to have increased compared to the unconju-gated AON (26 and 37)/RNA (42) hybrids, the large gain in enthalpy helped to overcome the unfavourable entropy effects. Among all the modified hy-brid duplexes in the PO and PS series, the most stabilized ones were those formed by 3'-DPPZ conjugated AONs (30 and 41). This is evident from 8.5 ºC gain in ∆Tm and –8.1 kJ/mol gain in ∆Gº298 for 3'-DPPZ conjugated AON
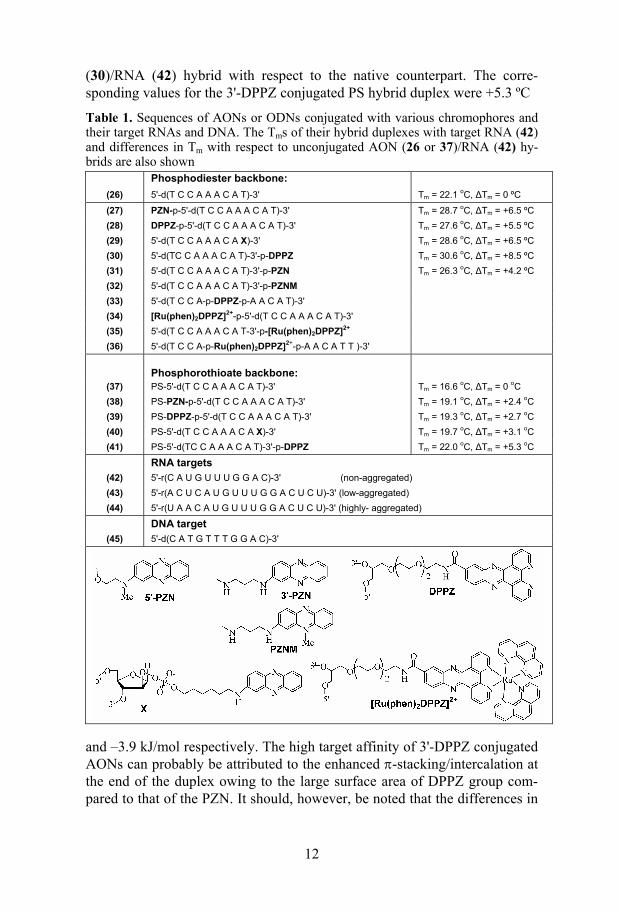
12
(30)/RNA (42) hybrid with respect to the native counterpart. The corre-sponding values for the 3'-DPPZ conjugated PS hybrid duplex were +5.3 ºC
Table 1. Sequences of AONs or ODNs conjugated with various chromophores and their target RNAs and DNA. The Tms of their hybrid duplexes with target RNA (42) and differences in Tm with respect to unconjugated AON (26 or 37)/RNA (42) hy-brids are also shown
Phosphodiester backbone:
(26) 5'-d(T C C A A A C A T)-3' Tm = 22.1 oC, ∆Tm = 0 ºC
(27) PZN-p-5'-d(T C C A A A C A T)-3' Tm = 28.7 oC, ∆Tm = +6.5 ºC (28) DPPZ-p-5'-d(T C C A A A C A T)-3' Tm = 27.6 oC, ∆Tm = +5.5 ºC (29) 5'-d(T C C A A A C A X)-3' Tm = 28.6 oC, ∆Tm = +6.5 ºC (30) 5'-d(TC C A A A C A T)-3'-p-DPPZ Tm = 30.6 oC, ∆Tm = +8.5 ºC (31) 5'-d(T C C A A A C A T)-3'-p-PZN Tm = 26.3 oC, ∆Tm = +4.2 ºC (32) 5'-d(T C C A A A C A T)-3'-p-PZNM (33) 5'-d(T C C A-p-DPPZ-p-A A C A T)-3' (34) [Ru(phen)2DPPZ]2+-p-5'-d(T C C A A A C A T)-3' (35) 5'-d(T C C A A A C A T-3'-p-[Ru(phen)2DPPZ]2+ (36) 5'-d(T C C A-p-Ru(phen)2DPPZ]2+-p-A A C A T T )-3'
Phosphorothioate backbone:
(37) PS-5'-d(T C C A A A C A T)-3' Tm = 16.6 oC, ∆Tm = 0 oC (38) PS-PZN-p-5'-d(T C C A A A C A T)-3' Tm = 19.1 oC, ∆Tm = +2.4 oC (39) PS-DPPZ-p-5'-d(T C C A A A C A T)-3' Tm = 19.3 oC, ∆Tm = +2.7 oC (40) PS-5'-d(T C C A A A C A X)-3' Tm = 19.7 oC, ∆Tm = +3.1 oC (41) PS-5'-d(TC C A A A C A T)-3'-p-DPPZ Tm = 22.0 oC, ∆Tm = +5.3 oC
RNA targets
(42) 5'-r(C A U G U U U G G A C)-3' (non-aggregated) (43) 5'-r(A C U C A U G U U U G G A C U C U)-3' (low-aggregated) (44) 5'-r(U A A C A U G U U U G G A C U C U)-3' (highly- aggregated)
DNA target
(45) 5'-d(C A T G T T T G G A C)-3'
and –3.9 kJ/mol respectively. The high target affinity of 3'-DPPZ conjugated AONs can probably be attributed to the enhanced π-stacking/intercalation at the end of the duplex owing to the large surface area of DPPZ group com-pared to that of the PZN. It should, however, be noted that the differences in

13
the linker or chemical moieties used for the conjugation of DPPZ and PZN may also contribute to the variation in duplex stability.
The Tm values of the hybrid duplexes formed by AONs (27-30) with low-aggregated RNA (43) were found to be very close to the Tm of their duplexes with the non-aggregated RNA target (42). However, the Tms of PS-AONs (38-41) with RNA (43) were somewhat lower compared to their binding affinity with the 11mer RNA (42). No duplex was detected for the unconju-gated PS-AON (37) with the low-aggregated RNA target. This can be corre-lated to the ability of the chromophore conjugated AONs to break the secon-dary structure of the target and to form the hybrid duplex, which is supported by a free energy comparison. The ∆Gº298 of PS-AON (37)/RNA (42) hybrid is –29.5 kJ/mol which is lower than the ∆Gº298 (–34.7 kJ/mol) for the self-aggregation process of the target RNA (42). Moreover these data support the fact that the conjugation of DPPZ or PZN to AONs increases their target accessibility. When the conjugated AONs were hybridized to the highly-aggregated RNA target (44), all the Tms observed by UV melting studies were close to the Tm of the target itself (Table 5 in Paper 1), which made it difficult to estimate the extent of AON/RNA duplex formation.
The CD spectra of all the 5'- and 3'-conjugated DPPZ/PZN-AON (27-30 and 38-41)/RNA (42) hybrids were found to be very similar to the unconju-gated AON (26 and 37)/RNA (42) hybrid duplexes (Figure 6 in Paper 1). This clearly demonstrates that the conjugation of chromophores does not alter the global helical conformation of AON/RNA hybrids thereby making them possible substrates for RNase H.
2.1.4.2 RNase H eliciting power of chromophore tethered AON/RNA hybrids and exonuclease stability of conjugated AONs (Paper I)
All the 3'- and 5'-conjugated AON/RNA hybrids were found to be better substrates for RNase H than their native counterparts irrespective of the self-aggregation nature of the target RNAs (Figures 7-9 in Paper 1). The extent of hydrolysis was found to be maximum for 3'-DPPZ conjugated AON (91% of RNA was cleaved after 2h incubation with RNase H) which has also shown site-specific cleavage of the target (Figure 3). The extent of cleavage of the target RNA was slightly lower for the hybrid duplexes formed by PS-AONs (38-41) compared to their PO counterparts. This can probably be at-tributed to the low AON/RNA population owing to the lower thermody-namic stability of the PS-AON/RNA hybrids.
These results indicate that the kinetic accessibilities of the conjugated AONs to the various aggregated RNAs (43 and 44) are the same as that to-wards the non-aggregated RNA (42). The reason can probably be the role that RNase H plays in the formation of heteroduplexes as reported recently on some hairpin RNA targets.355 Since in all our studies the target was not saturated by AON because of their equimolar ratio and lower Tms of AON/RNA hybrids (< 30 oC), the population of AON/RNA hybrids should
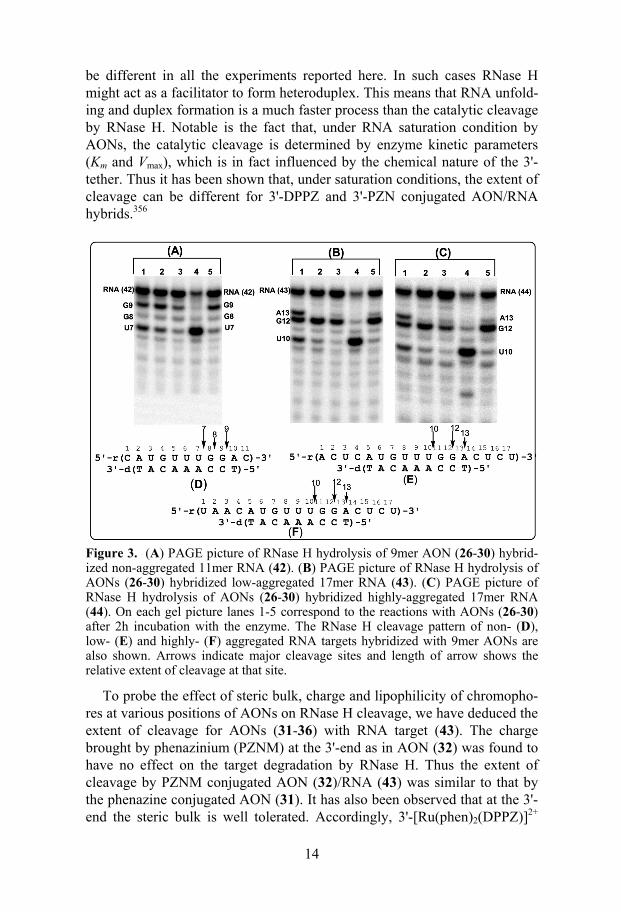
14
be different in all the experiments reported here. In such cases RNase H might act as a facilitator to form heteroduplex. This means that RNA unfold-ing and duplex formation is a much faster process than the catalytic cleavage by RNase H. Notable is the fact that, under RNA saturation condition by AONs, the catalytic cleavage is determined by enzyme kinetic parameters (Km and Vmax), which is in fact influenced by the chemical nature of the 3'-tether. Thus it has been shown that, under saturation conditions, the extent of cleavage can be different for 3'-DPPZ and 3'-PZN conjugated AON/RNA hybrids.356
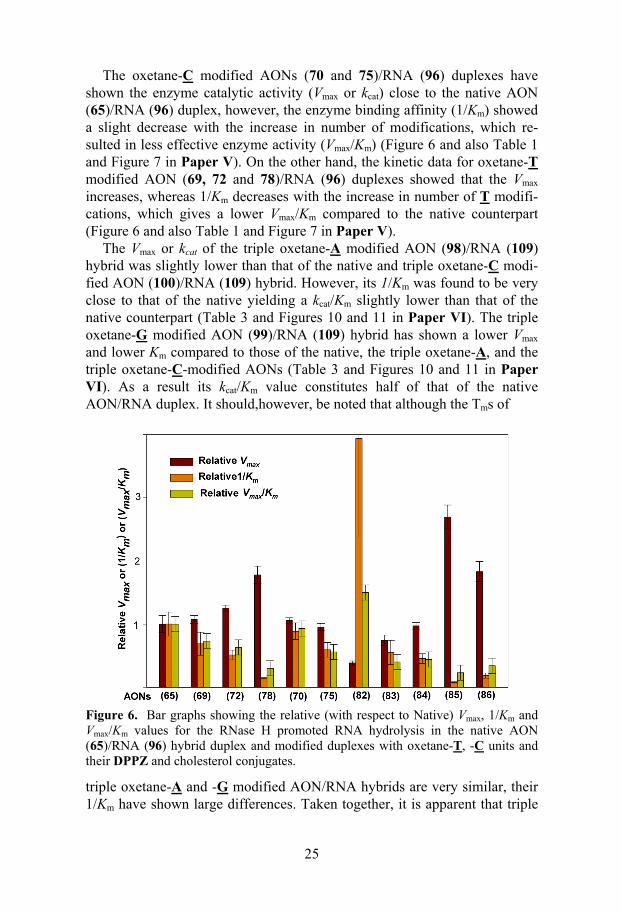
Figure 3. (A) PAGE picture of RNase H hydrolysis of 9mer AON (26-30) hybrid-ized non-aggregated 11mer RNA (42). (B) PAGE picture of RNase H hydrolysis of AONs (26-30) hybridized low-aggregated 17mer RNA (43). (C) PAGE picture of RNase H hydrolysis of AONs (26-30) hybridized highly-aggregated 17mer RNA (44). On each gel picture lanes 1-5 correspond to the reactions with AONs (26-30) after 2h incubation with the enzyme. The RNase H cleavage pattern of non- (D), low- (E) and highly- (F) aggregated RNA targets hybridized with 9mer AONs are also shown. Arrows indicate major cleavage sites and length of arrow shows the relative extent of cleavage at that site.
To probe the effect of steric bulk, charge and lipophilicity of chromopho-res at various positions of AONs on RNase H cleavage, we have deduced the extent of cleavage for AONs (31-36) with RNA target (43). The charge brought by phenazinium (PZNM) at the 3'-end as in AON (32) was found to have no effect on the target degradation by RNase H. Thus the extent of cleavage by PZNM conjugated AON (32)/RNA (43) was similar to that by the phenazine conjugated AON (31). It has also been observed that at the 3'-end the steric bulk is well tolerated. Accordingly, 3'-[Ru(phen)2(DPPZ)]2+

15
conjugated AON (35) has shown cleavage characteristics similar to the 3'-DPPZ conjugated AON(30)/RNA(43) duplex. However, the conjugation of bulky chromophores such as [Ru(phen)2(DPPZ)]2+ at the 5'-end or middle of the AONs has deleterious effects on RNase H binding and cleavage. Since in all our AON /RNA hybrids, the RNase H cleavage is localized at the 5'-end, the bulky substituents might have interfered with the catalytic centre of the enzyme. On the other hand the presence of bulky groups at the middle of the AON/RNA hybrids might interfere with the RNase H binding which in turn slows down the cleavage rate.
When tested for 3'-exonuclease tolerance (using snake venom phosphodi-esterase, SVPDE), all the 3'-conjugated PZN and DPPZ AONs have shown enhanced resistance towards the cleavage by the enzyme compared to their native PO and PS counterparts (Figure 10 in Paper 1). This is evident from the fact that under experimental conditions native PO-AON had a half life of 4 min, however, 3'-PZN-AON (29) and 3'-DPPZ-AON (30) conjugated AONs did not show any sign of degradation after 2h of incubation with the enzyme. We believe that the presence of the bulky chromophore and flexible linker in the 3'-conjugated AONs prevents the effective binding of SVPDE for cleavage reaction. The nuclease resistance offered by the 3'-PZN conju-gation as in AON (29) was 6 times higher than that shown by the 3'-DPPZ.
2.1.4.3 Synthesis of 1',2'-oxetane-modified phosphoramidite building blocks (Papers III, V and VI)
(A part of this work has been carried out in collaboration with Mr. P. Cheruku)
The synthetic strategy employed for the synthesis of North-East conformationally constrained 1-(1',3'-O-anhydro-β-D-psicofuranosyl)thymine /cytosine [oxetane-T (46) and oxetane-C (47)] nucleosides (Figure 4) and their corresponding phosphoramidite building blocks 54a-b is depicted in Scheme 1. The protected sugar, 6-O-(4-toluoyl)-1,2:3,4-di-O-isopropylidene-β-D-psicofuranose (50)357 was coupled with persilylated thymine/N4-benzoylcytosine in presence of trimethylsilyl triflu-romethanesulfonate (TMSOTf) as Lewis acid catalyst to afford an anomeric mixture of nucleosides358,359 which on 1'-O-mesylation yielded pure β isomers 51a-b. The deprotection of isopropylidene group using 90% triflu-roacetic acid (TFA) in water followed by cyclisation using NaH in DMF afforded 52a-b. The complete deprotection of 52a-b using NH3/MeOH gave the oxetane nucleosides 46 and 47. The 6'-OH of 46 was protected with DMTr group to give 53a. The exocyclic NH2 group of 47 was protected first by isobutyryl group (iBu) and then subjected to dimethoxytritylation. The resulted 53a-b were phosphitylated23 to furnish 54a-b, the building blocks required for their incorporation into the AONs (66-86). The 4'-succinate 55 required for the CPG derivatization23 has also been prepared from 53b.

16
Figure 4. The structure of oxetane-T (46), -C (47), -A (48) and -G (49) nucleo-sides. The 3JH-4',H-5' coupling constants obtained from 1H-NMR spectra recorded at 600 MHz (DMSO-d6 + methanol-d4) as well as the pseudorotational phase angles (P) and puckering amplitudes (Φm) deduced from the ab initio 6-31G* Hartree-Fock optimized molecular geometries are also shown.
Scheme 1. Synthesis of oxetane-T and oxetane-C phosphoramidite building blocks.
Since the attempted coupling of compound 50 with persilylated purine nucleobases was unsuccessful an alternative strategy has been devised for the synthesis of 9-(1',3'-O-anhydro-β-D-psicofuranosyl)adenine/guanine [oxetane-A (48) and oxetane-G (49)] nucleosides (Figure 4) and their corresponding phosphoramidite building blocks 64a-b (Scheme 2). The syn-thesis started with the conversion of 50 to 57, a possible key intermediate in the nucleobase coupling, using a reported procedure.360 Since the efforts to couple the silylated purine nucleobase derivatives with 57 were unsuccess-ful, we have decided to transform 57 into the bromosugar 58 followed by immediate coupling of the crude 58 with the silylated N6-benzoyladenine and N2-acetyl-O6-diphenylcarbamoylguanine in presence of SnCl4 as Lewis acid catalyst. This afforded the β-nucleosides 59a and 59b (after a short treatment with TFA to remove the DPC from the G-derivative). The β configurations of the products were determined by NOE measurements. The N9-connectivity of the base in 59b was evidenced by the chemical shift of C5 of the base at 122.0 ppm in the 13C-NMR spectra, which is characteristic for the N9-isomer.361 Methanolysis of 59a-b followed by simultaneous protection of 4'- and 6'-hydroxyl groups using 1,3-dichloro-1,1',3,3'-tetraisopropyldisiloxane (TIPDSCl2) furnished the precursors, 60a-b re-

17
quired for the oxetane ring formation. The cyclisation was achieved by the treatment of 60a-b with NaHMDS in THF at 4 ºC, giving 61a-b. The exo-cyclic amino group of 61a was protected by phenoxyacetyl group and that of 61b by dimethylformamidine group to furnish 62a-b. The removal of the TIPDS group from 62a-b followed by one-pot dimethoxytritylation afforded 63a-b. Compounds 63a-b were transformed to their corresponding phos-phoramidites 64a-b, the building blocks required for the solid phase synthe-sis of the modified AONs (98-99, 101-103 and 105-107).
Scheme 2. Synthesis oxetane-A and oxetane-G building blocks
2.1.4.4 Conformation of oxetane-modified nucleosides and CD spectra of their AON/RNA hybrid duplexes (Papers II-IV and VI)
(A part of this work has been carried out in collaboration with Dr. O. Plashkevych)
The molecular structures of the oxetane-T (46), -C (47), -A (48), and -G (49) monomer units have been studied by means of high-field 1H-NMR, theoretical ab initio and MD simulations. The combined experimental and theoretical studies have demonstrated that the oxetane-fused furanose ring is indeed locked in the typical North-East-type conformation. The 3JH-4',H-5'
values of the oxetane-modified compounds 46-49 are ~8.6 Hz (Figure 4 and also Table 5 in Paper VI) and found to be temperature independent, which is a clear indication of locked North-type conformation278,362 of the sugar moieties of the oxetane-C, -T, -A and -G nucleosides. The dependence 3JH-
3',H-4' vs. 3JH-4',H-5' (Figures 12 and 13 in Paper VI) shows the combination of these coupling constants for the oxetane-modified nucleosides to be typical for P = 20-60o range which unambiguously points to the North-East type conformation (Table 6 and Figure 12 in Paper VI). The pseudorotational phase angle (P) and puckering amplitude (φm) for the ab initio optimized geometries (6-31G* HF) are varying from 39.8º < P < 42.8º, 35.1º < φm <

18
36.6º for all four oxetane-modified nucleosides (Figure 4 and also Tables 6 and 7 in Paper VI). The last 100 picoseconds (ps) of 0.5 nanosecond (ns) MD simulation starting from respective ab initio geometries have shown accessible conformational range of P and φm to be 16º < P < 56º, 23º < φm < 41º for the oxetane-modified sugars (Table 7 and Figure 14 in Paper VI).
The CD spectra of oxetane-T, -C, -A and -G modified AON/RNA hy-brids were found to be very similar to those of the native AON/RNA hybrids (Figure 1 in Paper 1, Figures 8, 9 and 10 in Paper IV and Figure 3 in Paper VI). It should be noted that incorporation of up to 6 oxetane units as in 20mer AON (103)/RNA (109) hybrid duplex failed to produce alteration in the CD spectrum. From these results, it is evident that CD has failed to de-tect the local conformational changes brought by the N-E-type oxetane-modification(s), which has in fact been sensed by RNase H as it can be seen from the cleavage patterns (Section 2.1.4.7).
2.1.4.5 Thermostability and thermodynamics of oxetane-modified AON/RNA hybrids (Papers II-VI)
A systematic study of various oxetane-T modified 15mer AON/RNA hybrid duplexes has revealed that oxetane-T introduction into AONs causes ~ 6 oC drop in Tm per modification of their AON/RNA hybrids with respect to the native hybrid duplex (Table 2). The corresponding drop in net free energy (∆∆Go) is ~ 9 kJ/mol (Table 1 in Paper IV). However, the incorporation of oxetane-C units causes only ~ 3 ºC drop in the thermostability of their AON/RNA duplexes (Table 2). It should, however, be noted that the pres-ence of mismatches in the same sequence leads to ~ 10 oC drop of Tm per each mismatch (Table 2). On the other hand oxetane-A and -G modified 20mer AON (98-99 and 101-103)/RNA (109) hybrids showed a melting temperature (Tm) similar or very close to that of the native AON (97)/RNA (109) hybrid (Table 2). This clearly shows that the oxetane-pyrimidine units have considerable destabilizing effect on the thermostability of the AON/RNA hybrids compared to their purine counterparts.
A comparison of ∆Hº, which is a measure of hydrogen bonding and stack-ing interactions,363 of the oxetane-T modified and mismatched AON/RNA hybrids underscores the difference between the effect of the oxetane-T on duplex stability compared to the effect of mismatches. AON (76)/RNA (96) hybrid duplex with three oxetane-T modifications has a ∆Hº value of –355 ± 26 kJ/mol, while the corresponding AON (90)/RNA (96) hybrid duplex with three mismatches has a ∆Hº of only –233 ± 14 kJ/mol. The ∆Hº of native duplex is –494 ± 32 kJ/mol (Table 1 in Paper IV). This shows that the en-thalpy of the oxetane-T modified duplex lies in between those of the mis-matched and native duplexes and this large difference in enthalpy clearly
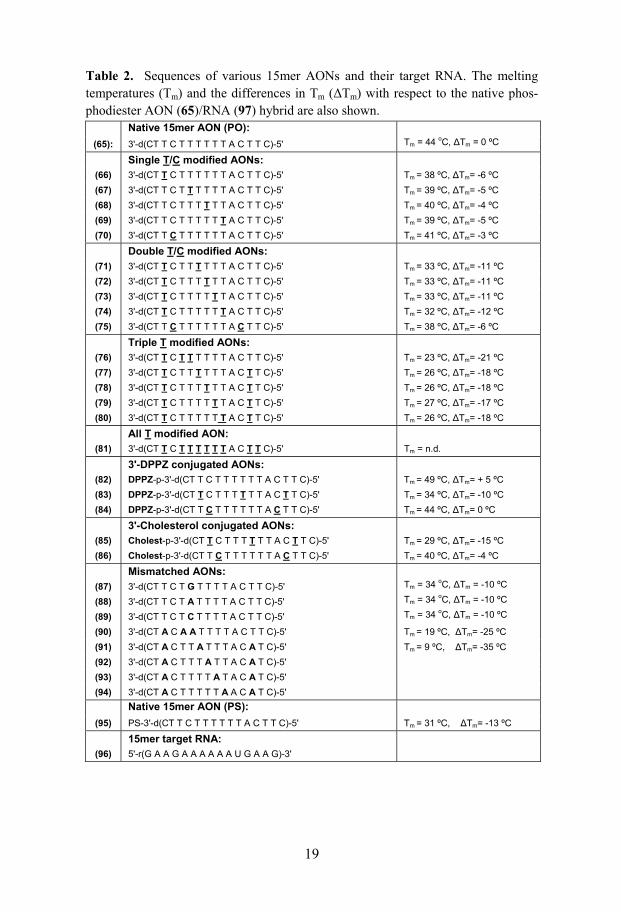
19
Table 2. Sequences of various 15mer AONs and their target RNA. The melting temperatures (Tm) and the differences in Tm (∆Tm) with respect to the native phos-phodiester AON (65)/RNA (97) hybrid are also shown.
Native 15mer AON (PO):
(65): 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 44 oC, ∆Tm = 0 ºC
Single T/C modified AONs: (66) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 38 ºC, ∆Tm= -6 ºC (67) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 39 ºC, ∆Tm= -5 ºC (68) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 40 ºC, ∆Tm= -4 ºC (69) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 39 ºC, ∆Tm= -5 ºC (70) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 41 ºC, ∆Tm= -3 ºC
Double T/C modified AONs: (71) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 33 ºC, ∆Tm= -11 ºC (72) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 33 ºC, ∆Tm= -11 ºC (73) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 33 ºC, ∆Tm= -11 ºC (74) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 32 ºC, ∆Tm= -12 ºC (75) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 38 ºC, ∆Tm= -6 ºC
Triple T modified AONs: (76) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 23 ºC, ∆Tm= -21 ºC (77) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 26 ºC, ∆Tm= -18 ºC (78) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 26 ºC, ∆Tm= -18 ºC (79) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 27 ºC, ∆Tm= -17 ºC (80) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = 26 ºC, ∆Tm= -18 ºC
All T modified AON: (81) 3'-d(CT T C T T T T T T A C T T C)-5' Tm = n.d.
3'-DPPZ conjugated AONs: (82) DPPZ-p-3'-d(CT T C T T T T T T A C T T C)-5' Tm = 49 ºC, ∆Tm= + 5 ºC (83) DPPZ-p-3'-d(CT T C T T T T T T A C T T C)-5' Tm = 34 ºC, ∆Tm= -10 ºC (84) DPPZ-p-3'-d(CT T C T T T T T T A C T T C)-5' Tm = 44 ºC, ∆Tm= 0 ºC
3'-Cholesterol conjugated AONs: (85) Cholest-p-3'-d(CT T C T T T T T T A C T T C)-5' Tm = 29 ºC, ∆Tm= -15 ºC (86) Cholest-p-3'-d(CT T C T T T T T T A C T T C)-5' Tm = 40 ºC, ∆Tm= -4 ºC
Mismatched AONs: (87) 3'-d(CT T C T G T T T T A C T T C)-5' Tm = 34 oC, ∆Tm = -10 ºC
(88) 3'-d(CT T C T A T T T T A C T T C)-5' Tm = 34 oC, ∆Tm = -10 ºC
(89) 3'-d(CT T C T C T T T T A C T T C)-5' Tm = 34 oC, ∆Tm = -10 ºC
(90) 3'-d(CT A C A A T T T T A C T T C)-5' Tm = 19 ºC, ∆Tm= -25 ºC (91) 3'-d(CT A C T T A T T T A C A T C)-5' Tm = 9 ºC, ∆Tm= -35 ºC (92) 3'-d(CT A C T T T A T T A C A T C)-5' (93) 3'-d(CT A C T T T T A T A C A T C)-5' (94) 3'-d(CT A C T T T T T A A C A T C)-5'
Native 15mer AON (PS): (95) PS-3'-d(CT T C T T T T T T A C T T C)-5' Tm = 31 ºC, ∆Tm= -13 ºC
15mer target RNA:
(96) 5'-r(G A A G A A A A A A U G A A G)-3'
20
Table 3. Sequences of various 20mer AONs and their target RNA. The melting temperatures (Tm) and the differences in Tm (∆Tm) with respect to the native phos-phodiester AON (97)/RNA (109) hybrid are also shown.
Native 20mer AON (PO):
(97) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm= 74 ºC, ∆Tm = 0 ºC
20mer A/ T / C/ G modified AONs: (98) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 73 ºC, ∆Tm = -1 ºC (99) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 74 ºC, ∆Tm = 0 ºC (100) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 61 ºC, ∆Tm= -13 ºC (101) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 73 ºC, ∆Tm = -1 ºC (102) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 73 ºC, ∆Tm = -1 ºC (103) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 74 ºC, ∆Tm = 0 ºC (104) 3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 59 ºC, ∆Tm= -15 ºC
3'-DPPZ conjugated AONs: (105) DPPZ-p-3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 75 ºC, ∆Tm = +1 ºC (106) DPPZ-p-3'-d(T T A C G T A C A G T G T C C G C C C T)-5' Tm = 76 ºC, ∆Tm = +2 ºC
3'-C A G modified AON: (107) 3'-d(C A G C G T A C A G T G T C C G C C C C)-5'
Native 20mer AON (PS): (108) PS-3'-d(T T A C G T A C A G T G T C C G C C C T)-5'
20mer target RNA: (109) 5'-r(A A U G C A U G U C A C A G G C G G G A)-3'
underlines the fact that oxetane-T effect is different from that caused by mismatches. Additional supports are provided by RNase H assays (Section 2.1.4.7) and charge transport studies (Section 2.3.2.1).
The loss of Tm of the AON/RNA hybrid duplexes owing to the introduc-tion of oxetane-T or -C units into 15mer-AONs has been partly or fully re-gained (+6 to +8 ºC) by the conjugation of non-toxic DPPZ group at the 3'-end of the AON as exemplified in AON (83 and 84)/RNA (96) hybrid du-plexes which also gave enhanced protection of AONs against 3'-exonucleases and nucleases in human serum (Section 2.1.4.6). However, the Tm enhancement effect of the 3'-DPPZ group has been considerably reduced in case of oxetane-A and -G modified 20mer AON (105 and 106)/RNA (109) hybrids.
To shed more light on the differential binding affinities of the oxetane- purine and oxetane-pyrimidine modified AONs to the target RNA (109), a comparison of their thermodynamic parameters has been carried out (Table 1 in Paper VI). It has emerged that the triple oxetane-G modified AON (99)/RNA (109) duplex has the ∆Ηº, ∆Sº and ∆Gº very close to that of the native AON (97)/RNA (109) duplex. However, ∆Ηο and ∆Sº of the triple oxetane-A modified AON (98)/RNA (109) duplex are slightly lower than those of the native duplex and the triple oxetane-G modified hybrid du-plexes. This amounts to 5.5 kJ/mol drop in ∆Gº for the triple oxetane-A modified AON (98)/RNA (109) hybrid duplex compared to that of the na-tive. The triple oxetane-C modified AON (100)/RNA (109) as well as the

21
triple oxetane-T modified AON (104)/RNA (109) duplexes have shown a large enthalpic destabilization and a slight entropic stabilization, which re-sulted in the net loss of –32.5 kJ/mol of ∆Gº for triple oxetane-C and –37.2 kJ/mol for triple oxetane-T modified hybrids compared to that of the native duplex. Apparently, the large drop in enthalpy of the oxetane-modified pyrimidine AON/RNA duplexes contributes to the destabilization of the duplex which reflects in the weakening of the stacking and hydrogen bond-ing interactions. But it is not clear which force is dominantly disturbed dur-ing the duplex formation. Clearly, more structural studies are needed to make a definite conclusion in this issue.
2.1.4.6 Endonuclease, exonuclease, and serum stability of oxetane-modified AONs (Papers IV-VI)
The oxetane-C modified AONs (70 and 75), as well as the oxetane-T modi-fied AONs (68 and 78), have shown enhanced tolerance towards endonucle-ase (DNase 1) degradation: the single modification gives ~ 2-3 fold protec-tion from cleavage by DNase 1 and the double modification gives ~ 4 fold protection in comparison to that of the native AON (65) (Figure 11 in Paper IV and Figure 3 in Paper V).
Surprisingly, when tested for endonuclease resistance, it was found that the triple oxetane-A (98) and triple oxetane-G (99) modified AONs offer no resistance to the nucleolytic cleavage (Figure 4 in Paper VI). The half-life of the AON (103) with the three A and three G units was found to be about 2h which is about half of that observed for the triple oxetane-C modified AON (100) studied under similar conditions. These results reveal the high susceptibility of the oxetane-purine modified AONs towards the DNase 1 promoted cleavage.
Literature search has revealed that in addition to the minor groove width and DNA flexibility364,365, the local sequence preference is crucial for the proper alignment of the phosphodiester bond for the cleavage reaction by DNase 1. Three nucleotides towards the 5'- and 3'-end of the cleavage site have significant influence on the cleavage properties. Based on the cleavage characteristics of various DNA duplexes, Herrera and Chaires366 found that among those 6 nucleotides around the cleavage site, the nucleotide se-quences at position 3 (denoted as –3) and 2 (denoted as –2) towards the 5'-end (Figure 4 in Paper VI) and nucleotide at position 2 (denoted as +2) to-wards the 3'-end of the cleavage site are very crucial in determining the ma-jor cleavage sites and cleavage rates of DNA duplexes by DNase 1.366 This is because the modifications at position –3 cause steric clash with arginine-41 of the enzyme and hamper the cleavage rate.365,366 Tyrosine-76 has shown favorable interactions with T/C moieties at the position –2. At the position +2 towards 3'-end presence of T is highly disfavored.366
For all our sequences, it was found that the cleavage occurs one or two nucleotides away from the site of the oxetane-modification. This indicates

22
that the presence of the oxetane ring at the cleavage site hampers the cleav-age activity of DNase 1. However, the extent of cleavage appeared to be dependent on the particular sequence surrounding the site of the cleavage as the purines and pyrimidines seems to be interacting differently with the en-zyme. This is probably because the oxetane ring in case oxetane-purines is further away from the nucleobases than in case of the pyrimidines and the amino acids of the DNase 1 do not encounter any steric clash with these rings. These results lead us to conclude that to get effective endonuclease resistance with the oxetane-purine units, extensive (probably every alternate nucleotide) modifications of the AON strand are needed. This however may slow down the RNase H recruiting capability. Another viable approach can be the use of a mixture of the oxetane-purines and a minimum amount of the oxetane-pyrimidines.
To investigate the tolerance of the oxetane-modifications towards an ex-onuclease (SVPDE), we have synthesized an AON (107) with 3 consecutive oxetane-C, -A and -G units at the 3'-end. Incubation with the SVPDE showed that the presence of the oxetane-modified units at the 3'-end offers resistance towards cleavage: 36% of AON (107) was left after 24h incuba-tion with the enzyme, while the native PO-AON (97) was completely de-graded in less than 2h of incubation (Figure 5 in Paper VI). The same AON showed half-life of around 9h in human blood serum where major degrading enzymes are exonucleases. Complete exonuclease protection was achieved in case of the 3'-DPPZ conjugated AONs (83, 84, 105 and 106, Figure 4 in Paper V and Figure 5 in Paper VI), which were even found to have slightly better stability than the native PS-AON in human serum (Figure 6 in Paper VI).
2.1.4.7 RNase H cleavage pattern and extent of RNA hydrolysis in oxetane-modified AON/RNA hybrids (Papers II-VI)
(A part of this work has been done in collaboration with Dr. E. Zamaratski)
When tested for RNase H1 cleavage, all the oxetane-T and -C modified 15mer AON (66-80)/RNA (96) hybrids and their 3'-DPPZ conjugated AON (83 and 84)/RNA (96) hybrids were found to be substrate for the enzyme in a manner comparable to that of the native hybrid AON (65)/RNA (96) du-plex (Table 2, Figures 2-7 in Paper IV and Figure 6 in Paper V). The 20mer AON (98-102)/RNA (109) hybrids modified with oxetane-A/-G/-C units and their 3'-DPPZ conjugated AON (105 and 106)/RNA (109) hybrids were also found to be substrate for RNase H to an extent comparable to that of the native AON (97)/RNA (109) hybrid duplex (Table 2 and Figures 7-9 in Pa-per VI)
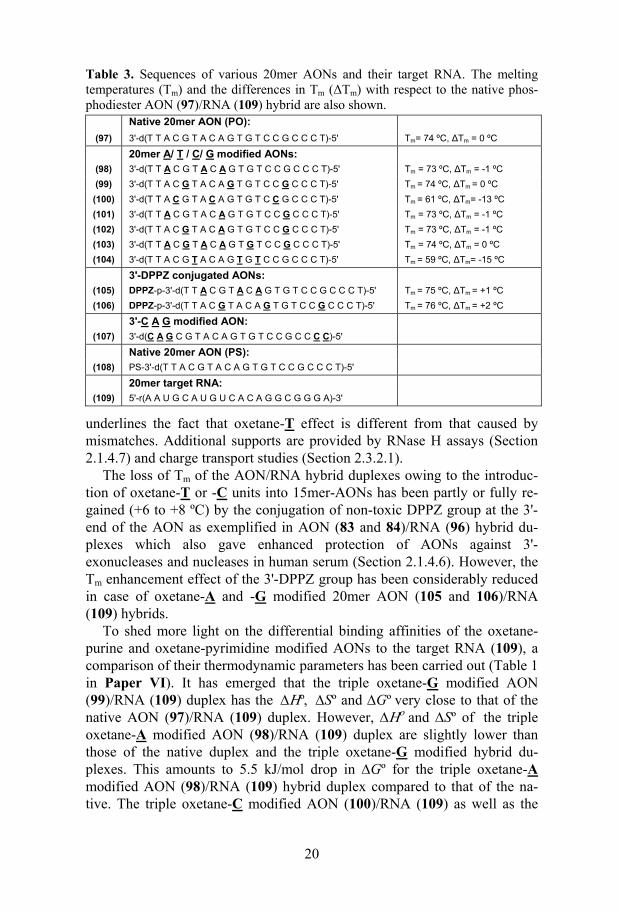
Interestingly, in the oxetane-modified AON/RNA hybrids a region of 5 nucleotides in the RNA strand towards the 3'-end from the site opposite to the oxetane-modification, was found to be resistant towards RNase H pro-
23
moted cleavage (Figure 5 and also Figure 2 in Paper II, Figures 2-5 in Pa-per IV, Figure 6 in Paper V and Figures 7 and 8 in Paper VI). This is pre-sumably owing to the local steric and structural alterations brought about by the N-E-conformationally constrained oxetane-modification, thereby pre-venting the flexibility and accessibility required for the RNase H cleavage. Since the sites just after the 5 nucleotides were accessible for enzymatic cleavage (Figure 5), and the fact that the binding and cleavage sites are dif-ferent for RNase H,367 it is evident that the structurally altered duplex region was suitable for enzyme binding but not for cleavage. By suitably placing just three oxetane-T modifications we have shown that a single cleavage site can be engineered in the 15mer AON/RNA hybrid duplex (Figure 5). This work clearly shows that the conformational constrain introduced at the oli-gonucleotide level, leads to microenvironmental conformational alterations in the neighboring 4 nucleotides toward the 5'-end of the AON strand. RNase H can effectively map this local conformational transmission, whereas CD
fail to show any conformational heterogeneity in the helical structure.
Figure 5 (A): Autoradiograms of 20% denaturing PAGE, showing the cleavage kinetics of 5'-32P-labelled target RNA (96) by RNase H in the native AON (65)/RNA (96) and oxetane T modified AON (67, 72 and 79)/RNA (96) hybrid duplexes. (B): RNase H cleavage pattern of hybrid duplexes. The vertical arrows show the RNA cleavage sites and relative length of an arrow shows the relative extent of cleavage at that site. The boxes represent regions, which are resistant to RNase H cleavage in the duplexes owing to oxetane-modifications.

24
We have found only two exceptions out of 27 examples for the above mentioned 5 nucleotide footprinting by RNase H. One was in the DPPZ con-jugated AON (83)/RNA (96) duplex (Figure 7 in Paper IV) and the other was in the case of 20mer AON (103)/RNA (109) (Figure 8 in Paper VI), where we have observed a cleavage in the anticipated footprint region. The reason for this unusual behavior of RNase H is not clear.
Our studies have clearly shown that only 4 deoxynucleotide gaps are needed to promote efficient RNase H cleavage of the target RNA in oxetane-modified AON/RNA hybrids. After this systematic evaluation of RNase H tolerance towards local conformational alterations, a report281 has shown that in the case of N-type conformationally constrained β-D-LNA incorporated AON/RNA hybrids, at least 6 deoxynucleotide gaps are needed in the AON strand to initiate the RNase H cleavage. Efficient cleavage was only achieved when the gap size was increased to 8-10 nucleotides. This supports the fact that LNA incorporation imposes more structural constraints to AON/RNA hybrids than the oxetane moiety which in fact was evident from CD and NMR studies.279,280 The incorporation of just three β-D-LNA units into a 9 mer AON/RNA has resulted in clear A-type duplex which was con-firmed by CD and NMR.279,280 This is contrary to the behaviour of our N-E constrained oxetane-incorporated AON/RNA hybrids (Section 2.1.4.4).
To distinguish the RNase H recognition and cleavage of the oxetane-T modification from those of mismatches, we have systematically incorporated and studied single and triple mismatches at exactly the same places where the oxetane-T was introduced (Table 2). Single mismatched AON (87-89)/RNA (96) hybrids have shown additional cleavages compared to that found in the corresponding oxetane-T modified AON (67)/RNA (96) hy-brids. The additional cleavage site is situated in the footprinting region of the latter (Figure 4, Paper IV). Interestingly, none of the triple mismatched AON (90-94)/RNA (96) hybrids, except the AON (90)/RNA (96) hybrid where mismatches are concentrated at the end, was able to evoke RNase H promoted cleavage (Figure 6, Paper V). This further supports the evidence obtained from Tm analysis, thermodynamics (Section 2.1.4.5) and charge transport studies (Section 2.3.2.1) that the structural effect of oxetane-T is quite different from that caused by the mismatches.
2.1.4.8 Michaelis-Menten kinetics of RNase H cleavage in oxetane-modified AON/RNA hybrids (Papers V and VI)
(A part of this work has been done in collaboration with Dr. N.V. Amirkhanov)
A comparison of kinetic parameters of RNase H cleavage of the various AON/RNA hybrids incorporated with oxetane-T/-C /-A /-G units has shed light on the RNase H recruiting capability of AONs in depth in terms of sub-strate affinity and cleavage activity.
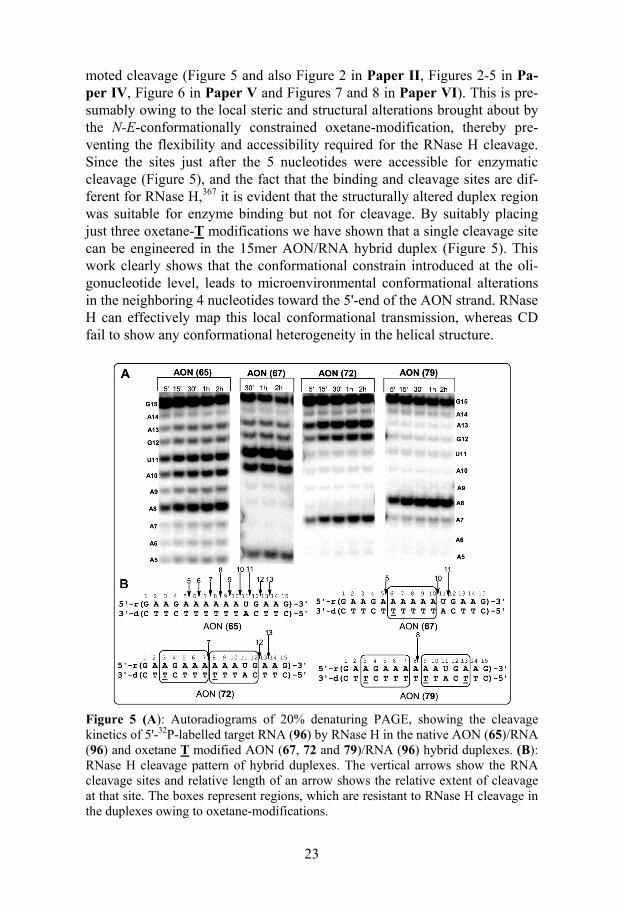
25
The oxetane-C modified AONs (70 and 75)/RNA (96) duplexes have shown the enzyme catalytic activity (Vmax or kcat) close to the native AON (65)/RNA (96) duplex, however, the enzyme binding affinity (1/Km) showed a slight decrease with the increase in number of modifications, which re-sulted in less effective enzyme activity (Vmax/Km) (Figure 6 and also Table 1 and Figure 7 in Paper V). On the other hand, the kinetic data for oxetane-T modified AON (69, 72 and 78)/RNA (96) duplexes showed that the Vmax increases, whereas 1/Km decreases with the increase in number of T modifi-cations, which gives a lower Vmax/Km compared to the native counterpart (Figure 6 and also Table 1 and Figure 7 in Paper V).
The Vmax or kcat of the triple oxetane-A modified AON (98)/RNA (109) hybrid was slightly lower than that of the native and triple oxetane-C modi-fied AON (100)/RNA (109) hybrid. However, its 1/Km was found to be very close to that of the native yielding a kcat/Km slightly lower than that of the native counterpart (Table 3 and Figures 10 and 11 in Paper VI). The triple oxetane-G modified AON (99)/RNA (109) hybrid has shown a lower Vmax and lower Km compared to those of the native, the triple oxetane-A, and the triple oxetane-C-modified AONs (Table 3 and Figures 10 and 11 in Paper VI). As a result its kcat/Km value constitutes half of that of the native AON/RNA duplex. It should,however, be noted that although the Tms of
Figure 6. Bar graphs showing the relative (with respect to Native) Vmax, 1/Km and Vmax/Km values for the RNase H promoted RNA hydrolysis in the native AON (65)/RNA (96) hybrid duplex and modified duplexes with oxetane-T, -C units and their DPPZ and cholesterol conjugates.
triple oxetane-A and -G modified AON/RNA hybrids are very similar, their 1/Km have shown large differences. Taken together, it is apparent that triple

26
oxetane-A and-C modified AON/RNA hybrids are better substrates for RNase H than the triple oxetane-G modified AON/RNA hybrid.
The reason for different behavior of the oxetane-T and -C modified AON/RNA hybrids toward RNase H cleavage at the high substrate concen-tration (well reflected in the Vmax) can be related to the Tm of their AON/RNA duplexes. All the oxetane-T modified AON/RNA hybrids are destabilized in a more pronounced manner (∆Tm ~ 6 ºC per modification) than the oxetane-C modified duplexes (∆Tm ~ 3 ºC per modification) and the native counterpart. This gives a less stable enzyme/substrate complex, and obviously a less stable enzyme/product complex for the oxetane-T modified AONs, which leads to lower enzyme binding affinity and high catalytic turnover of the enzyme. The similar reason can be attributed to the high RNase H recruitment of methylphosphonate chimeras369 and boranophos-phate AON/RNA hybrids.197
A direct correlation between kinetic parameters Vmax or 1/Km or Vmax/Km of RNase H with the Tm values of the corresponding oxetane-C and oxetane-T modified AON/RNA duplexes supports the above proposed explanation (Figure 9 in Paper V). The [log(Vmax)] for the substrates (i.e. duplex), con-taining oxetane-C or -T modifications is found to be linearly dependent on the Tm of AON/RNA duplex, and it decreases with increase of the melting temperature (Figure 9A in Paper V). The linear correlation was also found for the [log(1/Km)] and [log(Vmax/Km)] vs. Tm where the values of the kinetic parameters are increasing with the increase of Tm (Figure 9B and C in Paper V).
The introduction of the π electron rich DPPZ or highly hydrophobic cho-lesterol residues into the oxetane-C or -T modified AON/RNA hybrids has shown different influence on the 1/Km and Vmax or kcat of the RNase H (Fig-ure 6). DPPZ conjugation as in AONs (83 and 84) gave lower or similar Vmax and lower 1/Km of the RNase H in comparison with the native counterpart. In contradistinction, conjugation of hydrophobic cholesterol residue to AONs (85 and 86) gave less binding affinity and more catalytic activity of RNase H.
2.1.4.9 Gene down-regulation using oxetane-modified AONs in cellular system (Paper VII)
(This work has been carried out in collaboration with Dr. J. Opalinska and Prof. A. Gewirtz at the University of Pennsylvania School of Medicine)
Our in vitro studies have revealed that many of the desirable properties of an ideal antisense reagent such as optimal target binding, excellent RNase H activation, exo- and endonuclease resistance can be achieved using AONs modified with oxetane-cytidine units and 3'-DPPZ group with intact natural PO-backbone. This has prompted us to investigate the gene silencing effects of such constructs in cellular systems (K562 human leukemia cells) and to

27
compare their efficiency with unmodified isosequential PS-AONs. The tar-get chosen was c-myb mRNA. This encodes a protein, which is a regulator of cell-cycle transition and cellular maturation, primarily in haematopoietic cells, as well as in other cell types.370
The AONs were targeted to two different sites of c-myb mRNA. One site was chosen arbitrarily; the AON (110) and its isosequential PS-AON (113) are complementary to this site (Table 4). The other site was identified using a "rational" strategy based on predicted hybridization accessibility after a physical mapping of the mRNA with self quenching reporter molecule (SQRM).371 AON (111), AON (112) and their unmodified isosequential PS-AON (114) correspond to the rationally designed sequences. For control experiments, scrambled AONs (115 and 116) have been employed.
Table 4. AONs used for gene silencing in K562 cell line. (110) 5'-d(T A T G C T G T G C C G G G G T C T T C G G G C)-3'-DPPZ (111) 5'-d(A C A G A C C A A C G T T T C G G A C C G T A T T T C T G T)-3'-DPPZ (112) 5'-d(A C A G A C C A A C G T T T C G G A C C G T A T T T C T G T)-3'-DPPZ (113) PS-5'-d(T A T G C T G T G C C G G G G T C T T C G G G C)-3' (114) PS-5'-d(A C A G A C C A A C G T T T C G G A C C G T A T T T C T G T)-3' (115) 5'-d(T C T T C A G A G A C T G C G A C A T A G C G C)-3'-DPPZ (116) 5'-d(A C C G T C C A T C G T G T A G C A A C C T T A G C A A G T)-3'-DPPZ
The effectiveness of targeted versus random, phosphorothioate and oxetane-modified AONs on silencing c-myb gene expression has been evaluated at mRNA and protein levels. In the case of an arbitrarily chosen 24mer sequence, the PS-AON (113) appeared to perform somewhat better than oxetane-modified AON (110) (~ 40% vs ~ 20% reduction of c-myb mRNA respectively) (Figure 3 in Paper VII). The AONs (111, 112 and 114) targeted according to the mapping procedure were much more efficient in diminishing the mRNA than the randomly chosen AONs (110 and 113). Here also the rationally designed PS-AON (113) performed somewhat better than the corresponding oxetane molecules (111 and 112) yielding mRNA reduction of ~ 85% and ~ 70% respectively. Note, that although AON (111) and AON (112) were of identical sequence, AON (111) contained 4 oxetane-C units, while AON (112) contained 5 such modifications. Nevertheless, their biological activity, as determined by ability to diminish c-myb mRNA levels did not appear to differ significantly when measured by the QRT-PCR.372
We have also examined the effect of the AONs tested on c-myb protein expression by Western Blotting (Figure 4 in Paper VII). Diminution of the c-myb protein after treatment with oxetane-C modified AON (110) is found to be ~ 20% compared with untreated cells, which correlates well with its effect on the mRNA, and is very similar to that of a scrambled control AON (115). The corresponding thioate AON (113), which suppressed c-myb mRNA levels to ~ 40%, effected a substantial reduction of the protein as well. Once again in agreement with their effects on the mRNA levels, the

28
rationally designed antisense 30mers were highly efficient in decreasing protein levels in treated cells. AONs (111 and 112) suppressed the produc-tion of c-myb protein to ~ 70% compared to untreated controls. No protein was detected in cells treated with PS-AON (114).
In QRT-PCR and Western Blotting experiments described above, the PS-AONs (113 and 114) appeared to outperform the oxetane-modified mole-cules (110-112) with respect to suppression of mRNA and protein expres-sion. We thought that this could probably be due to the varying extent of AON penetration and accumulation into the cell after nucleoporation. To check whether this assumption is true, we have quantified the intracellular concentration of PS and the oxetane-modified AONs. The slot blot has shown that the oxetane-modified AONs were delivered ~ 6 times less effi-ciently in the cytoplasm and inside the nucleus than in the case of their thioate counterparts (Figure 5 in Paper VII). These results suggest that if one considers the amount of material actually delivered into the cells, the oxetane-modified AONs were ~ 5-6 times more potent than the correspond-ing PS molecules.
In conclusion, our studies clearly demonstrated that oxetane-C modified molecules are able to hybridize to their intended targets in living cells, and that they are highly efficient antisense agents. The AON design based on the RNA mapping procedure outperformed the one with the random design. Based on the amount of AON uptake after delivery, it is evident that the oxetane-modified AONs are more efficient antisense agents than the isose-quential PS analogues. The presence of PO backbone in all of our AONs studies gives additional advantages as far as the nonspecific biological ef-fects associated with PS-AONs are concerned. We believe that appropriately directed, oxetane-modified AONs could well prove a significant addition to the armamentarium of anticancer drugs, and in other diseases where gene silencing is expected to lead to useful therapeutic consequences.
2.2 Elucidation of metallointercalation mode in DNA/DNA duplexes by DNase 1 footprinting
2.2.1 DNase 1 footprinting Footprinting is a method to assess the sequence specific interactions of a ligand with DNA duplex.373 It is basically a protection assay in which cleav-age of DNA is inhibited at certain positions owing to the binding of small ligands or proteins to the helix.364,373 This was first demonstrated in 1978 in the case of protein DNA interactions.374 Later it has been developed as a valuable tool to find the interactions of many ligands into DNA duplex.373 Various enzymic and chemical cleavage agents such as DNase 1,364 DNase II,375 copper phenanthroline,376 methediumpropyl-EDTA.Fe(II) (MPE),377 uranyl photocleavage378 and hydroxyl radicals379 have been used in footprint-ing experiments. Among those, DNase 1 has been emerged as the most commonly used reagent for footprinting because of its low cost and ease of

29
use.364 DNase 1 footprinting has been successfully employed for identifying the DNA binding sites for several ligands including actinomycin,380,381 mithramycin,382 quinoxalin antibiotics,383 daunomycin,384 noglamycin,385 various minor groove binding agents,386 hairpin polyamides387 and triplex forming oligonucleotides.388
DNase 1 is a double strand specific endonuclease, which introduces single strand nicks into the phosphodiester backbone. Single stranded DNA is cleaved at least four order of magnitudes more slowly.389 The cleavage pro-duces 3'-OH and 5'-phosphate in the DNA strand. The enzyme requires diva-lent cations for its activity and it shows optimal cleavage in the presence of Mg2+ or Ca2+.390 The DNA-binding surface of DNase 1 covers a complete turn of DNA helix.391 The crystal structure of the DNase 1 with various short oligonucleotide duplexes365,391 has shown that DNase 1 binds to the DNA duplex by inserting an exposed loop into the DNA minor groove, interacting with the phosphate group as well as the walls of the groove. The drugs that modify DNA conformation upon binding can enhance the enzyme activity in region surrounding their binding sites.392,393 The importance of local se-quence preference in dictating DNase 1 cleavage is discussed in section 2.1.4.6.
2.2.2 Present work
2.2.2.1 Elucidation of metallointercalation mode in [Ru(phen)2(DPPZ)]2+ conju-gated ODN/DNA duplexes (Paper VIII)
(Synthesis and physicochemical studies have been carried out by Dr. D. Ossipov and Mr. M. Holmer)
We have employed the unique property of DNase 1 cleavage to find the met-allointercalation mode of various [Ru(phen)2(DPPZ)]2+ ODN (34-36)/DNA (45) duplexes. In all the [Ru(phen)2(DPPZ)]2+-conjugated ODNs (34-36) the complex is tethered to the oligo using a linker attached to the DPPZ group (Table 1). The UV melting studies have shown that the 5'- (∆Tm = +22 ºC), 3'- (∆Tm = +12.8 ºC) and middle (∆Tm = +19.4 ºC) Ru2+-conjugated ODNs have shown dramatic increase in duplex stability with the complementary DNA (45) (Table 4 in Paper VIII). The CD spectra of the 5'- modified ODN (34)/DNA (45) and middle-modified ODN (36)/DNA (45) duplexes were quite different from that of the unconjugated duplex whereas the 3'-conjugated ODN (35)/DNA (45) has shown similar CD behavior as that of the native (Figure 5 in Paper VIII). These data show that various Ru2+-conjugated ODN/DNA duplexes have different conformations and modes of intercalation of the metal complex. This has prompted us to use the DNase 1 footprinting assay to find the intercalation mode of the [Ru(phen)2(DPPZ)]2+ complex into ODN/DNA duplexes.
The DNase 1 digestion showed that the extent of hydrolysis (after 15 min.) of the Ru2+-conjugated duplexes decreases in the following order: na-
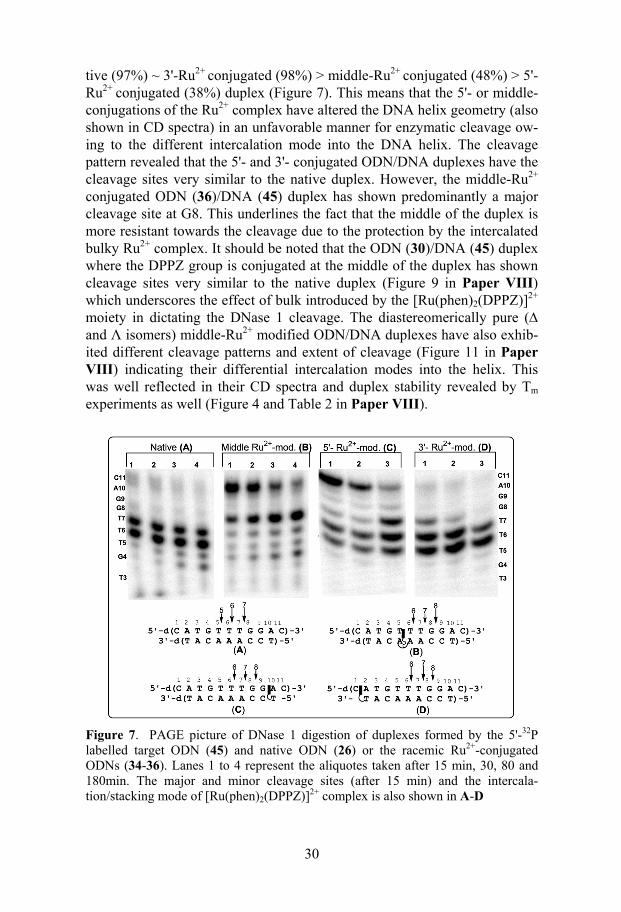
30
tive (97%) ~ 3'-Ru2+ conjugated (98%) > middle-Ru2+
conjugated (48%) > 5'- Ru2+
conjugated (38%) duplex (Figure 7). This means that the 5'- or middle-conjugations of the Ru2+ complex have altered the DNA helix geometry (also shown in CD spectra) in an unfavorable manner for enzymatic cleavage ow-ing to the different intercalation mode into the DNA helix. The cleavage pattern revealed that the 5'- and 3'- conjugated ODN/DNA duplexes have the cleavage sites very similar to the native duplex. However, the middle-Ru2+
conjugated ODN (36)/DNA (45) duplex has shown predominantly a major cleavage site at G8. This underlines the fact that the middle of the duplex is more resistant towards the cleavage due to the protection by the intercalated bulky Ru2+ complex. It should be noted that the ODN (30)/DNA (45) duplex where the DPPZ group is conjugated at the middle of the duplex has shown cleavage sites very similar to the native duplex (Figure 9 in Paper VIII) which underscores the effect of bulk introduced by the [Ru(phen)2(DPPZ)]2+ moiety in dictating the DNase 1 cleavage. The diastereomerically pure (∆ and Λ isomers) middle-Ru2+ modified ODN/DNA duplexes have also exhib-ited different cleavage patterns and extent of cleavage (Figure 11 in Paper VIII) indicating their differential intercalation modes into the helix. This was well reflected in their CD spectra and duplex stability revealed by Tm experiments as well (Figure 4 and Table 2 in Paper VIII).
Figure 7. PAGE picture of DNase 1 digestion of duplexes formed by the 5'-32P labelled target ODN (45) and native ODN (26) or the racemic Ru2+-conjugated ODNs (34-36). Lanes 1 to 4 represent the aliquotes taken after 15 min, 30, 80 and 180min. The major and minor cleavage sites (after 15 min) and the intercala-tion/stacking mode of [Ru(phen)2(DPPZ)]2+ complex is also shown in A-D

31
Taken together, our DNase 1 protection assays and physicochemical stud-ies of the various [Ru(phen)2(DPPZ)]2+-conjugated AONs suggest the fol-lowing: (i) in the 5'- and the middle-Ru2+ modified ODN/DNA duplexes the metal complex is intercalated into the internal base pairs of the duplex thus making very stable ODN/DNA duplexes (ii) in the 3'-Ru2+-conjugated ODN/DNA duplex the complex [Ru(phen)2(DPPZ)]2+ is stacked out of the duplex offering a less stable π−π interaction (Figure 8 in Paper VIII).
2.3 Nucleic acid mediated charge transport as a tool to probe base stacking perturbations
2.3.1 Charge transport in nucleic acids and electrochemical detection Various physicochemical and biochemical studies have unambiguously con-firmed that DNA in solution is a medium for charge transport (CT).394-398 The most widely studied example of DNA mediated CT is the transport of positive charge (holes) in the DNA duplexes in solution.394 These studies have exploited the oxidation of Gs in the DNA strand in vicinity to a hole injector, which is usually an appended photooxidant.394,395 The holes are being transported based on a hopping mechanism where the Gs in the se-quence act as stepping stones.394 In model systems, it has been demonstrated that hole transport can occur up to 200Å (20 nm).396,399 This type of hole transport in DNA has great biological importance as it is involved in the oxidative damage of DNA in human genome.400 The second way in which CT occurs in DNA is the movement of excess electrons through the double helix.398 This type of CT is also of extreme biological importance since it is being used by the enzyme DNA photolyase401 to repair UV induced lesions (thymine dimers).402 Unfortunately little is known about the mechanism in which excess electron transport occurs in DNA duplexes.
Various studies have revealed that DNA mediated CT through electroni-cally coupled π-stack of the base pairs is exquisitely sensitive to the DNA sequence dependent conformation, dynamics and local flexibility.394,395 Those studies have well demonstrated the sensitivity of the long range hole transport towards mismatches,403DNA bulges,404 and protein-induced struc-tural distortions.405,406 All external or internal alterations distorting the π-stacking or dynamics407 eventually cause diminution in CT. Systematic stud-ies408,409 from Barton's group have shown the influence of the base pair life-time of the various mismatches in dictating the long range guanine oxida-tion. Since many of the mismatches are well stacked in the helix,409,410 this study underscores the importance of the dynamics influencing the CT rather than the thermodynamics of base pairing.
DNA mediated CT can be detected using an electrochemical assay in or-der to probe the variations in DNA structure and stacking.411-417 In these ex-periments oligonucleotide duplexes containing thiolinker are attached to the gold surface and a redox active DNA intercalator methylene blue (MB) is

32
bound noncovalently to the top layer of the DNA film (Figure 8). The reduc-tion of the intercalator to leucomethylene blue (LB) serves as probe for base pair stacking within the film. The signal from MB as result of CT can be amplified in an electrocatalytic cycle in which the LB is used as a catalyst for the reduction of the species [Fe(CN)6]3- diffusing in solution outside the DNA film (Figure 8). Once reoxidised by ferricyanide, MB is available for
Figure 8. Electrochemical detection of DNA-mediated CT using a DNA-modified gold electrode. Electrons flow through the well-stacked DNA to reduce methylene blue (MB+) to leucomethylene blue (LB). LB can easily reduce Fe(CN)63- and regenerate MB+ that can continue in the catalytic cycle, thus repeated interrogation of the DNA monolayer is achieved.
subsequent electrochemical reduction and the catalytic cycle continues. Even small perturbations in base pair stacking diminish the efficiency of MB re-duction.414,415 Using this electrocatalytic assay, all single mismatches, includ-ing GA and GT, are clearly distinguished from Watson-Crick base paired DNA bases.413-415 Furthermore, this sensitive assay has been employed to detect the base perturbational effects due to the interactions of proteins such as M HhaI with DNA helix.415
2.3.2 Present work
2.3.2.1 Electrochemical detection of structural influence of a single oxetane-T unit in DNA/DNA and DNA/RNA duplexes (Paper IX)
(This work has been carried out in collaboration with Dr. E. M. Boon and Prof. J. K. Barton of California Institute of Technology)
In order to probe the local structural perturbations (stacking/dynamics) ow-ing to the N-E conformationally constrained oxetane-T units in DNA/DNA and DNA/RNA duplexes, we have exploited the sensitive electrochemical

33
assay to detect DNA mediated CT. The homo- and heteroduplexes fabricated on the gold surface used in this study are depicted in Table 5. Table 5. Various DNA/DNA and DNA/RNA duplexes used for the CT studies using chronocoulometry
(117): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCTTTTTTACTTC)
(123): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCT ATTTTACTTC)
(118): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCTTTTTTACTTC)
(124):SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCT GTTTTACTTC)
(119): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCTTTTTTACTTC)
(125): SHi-5'-d(AGTACAGTCATCGCG) 3'-d(TCATGTCAGTAGCGC)
(120): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCTTTTTTACTTC)
(126):SHi-5'-d(AGTACAGTCATCGCG) 3'-d(TCATGTCAGTAGCGC)
(121): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCTTTTTTACTTC)
(127): SHi-5'-d(AGTACAGTCATCGCG) 3'-r(TCATGTCAGTAGCGC)
(122): SHi-5'-d(GAAGAAAAAATGAAG) 3'-d(CTTCTCTTTTACTTC)
(128):SHi-5'-d(AGTACAGTCATCGCG) 3'-r(TCATGTCAGTAGCGC)
iSH-5' stands for SH(CH2)2CONH(CH2)6NHCO attached to the 5'-OH of the DNA strand.
The chronocoulometric measurements showed that the CT through single oxetane-T modified DNA/DNA duplexes (118-121) is very similar to that of the single mismatched duplexes (122-124) which is in fact very low com-pared to the native duplex (117) (Figure 2A and 2B in Paper IX). However, when CT was measured in the oxetane-T modified DNA/RNA duplex (128), the electrochemical signal was not diminished and it was very similar to that of the native duplex (127) (Figure 2C in Paper IX). This clearly shows the different local structural influences (stacking/dynamics) of oxetane-T in homo- and heteroduplexes. It should be noted that the oxetane-T incorpora-tion in ODNs resulted in the drop of Tm by ~ 6 ºC of their duplexes with DNA or RNA (Section 2.1.4.5). This suggests that thermodynamics of du-plex formation is not the deciding factor in DNA mediated electron transfer efficiency. The most likely explanation is the correlation between the duplex dynamics change upon modification and the time scale for the electrochemi-cal measurements (10-2 s).408 Our NMR and MD studies of the DNA/DNA duplex have shown that stacking is intact in spite of the oxetane- T modifica-tion, however, the dynamics of base pair opening at the site of modification has significantly increased.368 This supports the aforementioned hypothesis. In the DNA/DNA duplex (B-type helix) introduction of single N-E-conformationally constrained oxetane-T can induce more dynamics com-pared to a similar T incorporation in a DNA/RNA duplex (close to A type helix). In other words, the oxetane-T is better accommodated in the DNA/RNA duplex than in DNA/DNA duplex. Thus the CT studies underscore the sensitivity of the electrochemical detection in deciphering the local struc-tural perturbations in DNA/DNA or DNA/RNA helices as a result of the incorporation of a conformationally constrained nucleotide.

34
3. Summary
In this thesis, we have presented the synthesis, physicochemical and bio-chemical properties of two types of chemically modified oligonucleotides.
The first set is comprised of 9mer AONs conjugated with DPPZ, PZN, PZNM, [Ru(phen)2(DPPZ)]2+ at the 3'- or 5'-end or middle of the oligo chain. Conjugation of the chromophores at the 3'- or 5'-end enhanced the target RNA binding affinity (2 to 8.5 ºC) and RNase H recruitment capabili-ties compared to those of the native counterpart. CD spectra showed that the chromophore conjugated AON/RNA hybrid duplexes have the global helical conformation similar to that of the native hybrid duplex. The 3'-conjugation was found to be more effective in enhancing RNA binding affinity, RNase H activation and 3'-exonuclease resistance. Steric bulk, lipophilicity and charge of the chromophore at the 3'-end of the AON were well tolerated by RNase H. 3'-DPPZ has emerged as the most promising non-toxic chromophore in this series of modifications. It was also found that the cleavage rate of the target RNA in the AON/RNA hybrid duplex is independent of the complex-ity of the folding of the target, thereby suggesting that the kinetic accessibil-ity of the target RNA by AONs is the same under our model condition, irre-spective of the level of folding of the target RNA. This also suggests that the RNase H promoted cleavage is a slower process than the unfolding of RNA and the formation of the AON/RNA hybrid duplex.
We have employed DNase 1 footprinting to shed light on the metal-lointercalation mode of [Ru(phen)2(DPPZ)]2+ into short ODN/DNA du-plexes. These studies have revealed that in the case of the 5'-end or middle-Ru2+ modified ODN/DNA duplexes, the metal complex is intercalated into the internal base pairs of the duplex, thus making very stable ODN/DNA duplexes. However, in the case of the 3'-Ru2+-conjugated ODN/DNA duplex, the [Ru(phen)2(DPPZ)]2+ is being stacked out of the duplex by π−π interac-tion.
The second set encompasses a new class of AONs containing N-E con-formationally constrained nucleoside units (Oxetanes). The phosphoramidite building blocks of oxetane-T, -C, -A and G nucleosides have been synthe-sized and incorporated in 15mer or 20mer AONs. The NMR, ab initio and MD studies unambiguously confirmed the N-E conformation of the oxetane nucleosides. The introduction of oxetane-T and -C units reduced the Tm by ~ 6º and ~ 3 ºC/modification, respectively, of the AON/RNA hybrids, whereas the incorporation of the corresponding oxetane-A and-G units into AONs did not alter the thermostability in comparison with that of the native hybrid duplex. All oxetane-modified mixmer AON/RNA hybrid duplexes were, however, found to be excellent substrates for RNase H cleavage. A system-atic RNase H assay has shown that only four deoxynucleotide long gaps are needed in an oxetane-modified AON/RNA hybrid to elicit RNase H activity.

35
The local conformational alterations owing to the oxetane incorporation into the AON/RNA hybrids were insensitive to CD spectroscopy, however, sensed by RNase H, which was evident from the cleavage pattern. Micha-elis-Menten kinetics of the RNase H cleavage showed that Vmax and Km in-crease with increase in the number (one to three) of oxetane-T modifications in the AONs, indicating higher catalytic activity and lower enzyme binding affinity of the respective oxetane-modified AON/RNA hybrids. The triple oxetane-A modified AON/RNA duplex has Vmax slightly lower than those of the native and the triple oxetane-C modified AON /RNA hybrids. However, the Km of the RNase H for the oxetane-A modified AON/RNA duplex is very close to that for the native AON/RNA hybrid yielding a kcat/Km close to that of the native hybrid duplex. The triple oxetane-G modified AON/RNA duplex showed low Km and Vmax compared to the native counterpart, result-ing in a kcat/Km value being half of that of the native AON/RNA hybrid. The loss in Tms of the oxetane-T and -C modified 15mer AON/RNA hybrids has been partly or fully compensated by the introduction of non-toxic 3'-DPPZ group, which has also enhanced the AON stability against 3'-exonuclease and nucleases present in human serum to an extent similar or better than that of the PS-AONs. The 3'-end blocking of AONs with oxetane units has also enhanced their stability in human serum. The presence of 1 to 3 oxetane-T and -C units in AONs imparts 3 to 4 fold enhancement in endonuclease sta-bility compared to those of the native, oxetane-A and -G incorporated coun-terparts.
The efficacy of our AON design has been successfully verified by target-ing the protooncogene c-myb mRNA in K562 human leukemia cells. The designed oligo constructs with the oxetane-C and 3'-DPPZ were found to be non-toxic and down-regulated c-myb in a very effective manner. The QRT-PCR and Western Blotting (the "gold standard" in antisense efficacy175) have shown that rationally designed oxetane-C modified AONs were highly effi-cient in diminishing the c-myb mRNA (85% has been reduced) and the c-myb protein (70% of its expression was found to be halted) of the targeted gene. Based on the amount of AON uptake after delivery, determined by slot blot, it was apparent that the oxetane-modified AONs are 5-6 times more efficient antisense agents than the corresponding isosequential PS analogues. Thus we have achieved a balance between target binding affinity, RNase H activation as well as exo- and endonuclease resistance with the oxetane-modified AONs retaining the natural PO backbone.
The electrochemical assay based on exquisitely sensitive nucleic acid me-diated charge transport (CT) has revealed that oxetane-T units cause more stacking perturbations in a DNA/DNA duplex than in a DNA/RNA duplex thereby underlying the better accommodative nature of N-E constrained oxetane-T unit in a DNA/RNA duplex than in DNA/DNA duplex.

36
4. Future perspectives
Sequence specific knock-down of mRNA by oligonucleotides has the poten-tial to be one of the dominating utilities among the numerous applications of the chemically synthesized oligonucleotides. Irrespective of the approach, whether it is the antisense or ribozyme or DNAzyme or siRNA, the problems these technologies have to overcome for their clinical use include delivery and stability of the oligonucleotide inside the cell (pharmacokinetics), se-quence-specific targeting (pharmacodynamics), and efficacy (pharmacology) are essentially the same. Although the last two decades of research in the antisense field has addressed many of these issues, the final solution for the highly efficient bona fide antisense drug has never been achieved. Also, it is noteworthy that each of these technologies has its own merits and demerits, and it is not wise to anticipate that one will supplant the other technology (for critical comparisons see refs 42, 43, 64, 418).
Using the oxetane-modification, we have achieved many desirable an-tisense properties of oligonucleotides such as optimal target binding, RNase H recruitment, exo and endonuclease stability, and serum stability. More-over, restricting the RNase H cleavage sites by suitably placing the oxetane-modifications without loosing the overall cleavage efficiency has reduced the off-target effects of the AONs. The in vivo studies are due to start in leu-kemia infected mice using an injectable formulation for systematic delivery, we expect that the toxicity profiles of the oxetane-modified AONs would be reduced due to the absence of PS backbone. Whether our oligo design with oxetane modifications would enhance the pharmacokinetic properties of the AONs remains to be seen.
To further widen the applicability of oxetane-modified AONs, the synthe-sis and evaluation of N-E conformationally-constrained azetidine, thiatane and carbocyclic analogues of oxetane nucleosides are being intensively pur-sued in our laboratory. The synthesis of the azetidine analogues has already been achieved (in collaboration with D. Honcharenko). This is clearly the most attractive member of the family because we should be able to achieve two or three goals in one shot: (1) we have already observed that the fu-ranose-fused 1',2'-azetidine ring indeed holds the furanose conformation locked into the N-E-type pucker as found for the corresponding oxetane ana-logues, (2) as the endocyclic NH of the 1',2'-azetidine ring can be protonated under physiological conditions, it can partially neutralize the negative charge of the internucleotidic phosphate, and should eventually lead to a more stabi-lized AON/mRNA hybrid duplex, which may assist cellular penetration more effectively than what we have seen for the corresponding oxetane ana-logues, (3) The NH group of the azetidine ring should provide a centre to append chromophores, lipophilic moietes or amino-linkers through the nitro-gen atom to modulate the hydration/hydrophobicity or the steric require-

37
ments of the minor groove of an AON/RNA duplex. A similar approach has been recently exploited by Wengel et al. to functionalize 2'-amino-LNA.419
In addition to developing therapeutically potential AONs, one of the ma-jor applications of our findings will be the utilization of oxetane-modifications and its analogues and the conjugation chemistry in the design and synthesis of single-stranded (ss) and double-stranded (ds)-siRNAs and DNAzymes. Since ds-siRNAs demands an A-type helix for efficient gene silencing,95 introduction of oxetane or azetidine blocks, owing to their N-type conformation, would be ideal to preserve the A-type helix geometry. In addition, it can impart enhanced nuclease stability. In an oxetane- or azetidine-modified siRNA/mRNA duplex, the oxetane or azetidine ring will be projected into the minor groove of the duplex where it can modulate the hydration, hydrophobicity and electronegativity of modified siRNA/mRNA duplex for effective RNAi. Another advantage of the oxetane- or azetidine-modified ds-siRNA duplexes will be due to its optimal duplex stability, the easy unwinding of the ds-siRNA will be possible, which in fact is expected to play an important role for finding the correct mRNA target. We can also modulate the target affinity by incorporating oxetane-purine or -pyrimidine modifications. This will further facilitate the release of RISC* from the mRNA target for the catalytic turnover97 of the antisense strand, thereby the dose response vis-à-vis toxicity of the siRNA molecules can be kept under control. The conjugation of non-toxic chromophores such as DPPZ at the 3'-end of the antisense strand of ds-siRNA would not only impart exonuclease and serum stability but also enhance the target specificity and accessibility. The modification of binding arms and catalytic core of DNAzymes using oxetane or azetidine units could evoke more catalytic activitiy, endonuclease resistance and thereby efficient gene silencing.
The emergence of toxicity and bioavailability profiles of AONs in animal models and clinical trials has augmented our understanding of the design of antisense reagents. Now it is widely conceived that to reduce the off-target effects and non-antisense effects (mainly protein binding and immunostimu-lation by interferon activation), in addition to choosing the right sequence and chemical modification, the concentration of the antisense agent should be kept at minimum.42,175 The state-of-the-art antisense agent should work in nanomolar or subnanomolar concentration. This can only be achieved through the controlled and sustained delivery of antisense agent in vivo, which is indeed a critical bottleneck in oligonucleotide-based therapeu-tics.165,420 Although naked oligonucleotides can reach their biological targets in vivo, they are relatively rapidly eliminated from the blood stream due to extensive liver and kidney uptake. They also can localize in endosome and lysosome, which prevents the release of oligonucleotides and that is consid-ered to be the dead end of the oligonucleotide based drug.. Although numer-ous viral and synthetic delivery systems (complexation and conjugation ap-proaches) have been developed to increase the cell permeability and

38
bioavailability of the antisense agents for in vitro and in vivo use, none of these approaches has been found successful in the clinic.42 Therefore finding a viable solution for the delivery will continue to pose serious challenges to oligonucleotide based therapeutics.
We believe that the conjugation of the chemical functionalities with bio-degradable linkers will be dominating in addressing the delivery issues. These conjugates include: (i) carbohydrate conjugates with multivalent car-bohydrate recognition motif for cellular receptors, (ii) cationic amphiphiles containing cholesterol or cholic acid scaffolds linked via alkylamino side chains, (iv) oligopeptides, (v) dendrimers, (vi) biodegradable polymers. Al-though many of these moieties have been conjugated to AONs, further chemical modifications are warranted in those conjugates to achieve efficient delivery.
Modulation of pharmacokinetic properties of oligonucleotides by chemi-cal modification should be addressed more carefully to enhance the effi-ciency of the antisense reagent.
Identification of the correct mRNA target for oligonucleotide based therapeutics is important because this would enable gene down-regulation with highly target specific antisense agent using lowest possible concentra-tion. Several approaches163,421 to this problem have been tried simply by trial and error using oligonucleotide “walks” 5' to 3' down the target mRNA se-quence,422 computer modelling,423 hybridization of RNA to random oligonu-cleotides arrayed on glass slides,424 and using random oligonucleotide librar-ies to identify RNase H cleavable sites.425 However, none of these methods has proven to be ideal for the target selection. We believe that the simple and elegant approaches like the use of Self Quenching Reporter Molecule (SQRM),371 which we have utilized in identifying the c-myb mRNA targets may find wide applicability in future. It should also be noted that what is optimal target for an AON may not be an ideal target for siRNA. This has been reflected in some recent studies.418
Before claiming the antisense potential of a chemically modified oligonu-cleotide, the in vitro antisense screening of the oligonucleotides has to be carried out using right control sequences as suggested by Stein et al.175 The most important thing here is to show the down-regulation of mRNA (if RNA cleavage is expected) by RT-PCR and the down-regulation of protein by Western-Blot as we have shown for the oxetane-modified AONs. The bio-logical end points can never be considered as the proof for antisense effi-cacy.
Taken together, we believe the scientific cross talk between chemistry, biology and medicine will pave the way for the bona fide antisense drugs in near future.

39
5. Acknowledgements This work has been carried out at the Department of Bioorganic Chemistry, Uppsala University (Sept 1998 - May 2004). I am greatly indebted to many people without whose support and encouragement this thesis would not have been possible. To begin with, I would like to express my sincere gratitude to Prof. Jyoti Chat-topadhyaya, my supervisor, for accepting me as a Ph.D. student at his state-of-the-art laboratory and introducing me to the chemical, biochemical and biophysical aspects of nucleic acids, and also for his thoughtful guidance and support throughout the years. I also would like to thank him for encouraging me to follow the emerging literature on RNA interference, DNAzymes and drug delivery, which helped me to see the research in much larger perspective. I would like to express my sincere thanks to my former and present colleagues at the Department of Bioorganic Chemistry: Dr. Edouard Zamaratski, the best collaborator I have ever had, for patiently teaching me many practical aspects in the research and also for sharing with me those "rule of thumbs" in various practical problems of oligonucleotide chemistry. Dr. Andras Foldesi, for being really nice and an accessible senior colleague. His help with the NMR analysis and also with the critical reading of this thesis are highly appreciated. My friend Dr. Parag Acharya for his timely help during my stay in Uppsala, espe-cially when I fell sick during my first Swedish winter. Dr. Oles Plashkevych for being pleasant and approachable, for successful collabora-tions and also for helping me to fix the computers at the lab and at home. Dr. Nariman Amirkhanov for showing me "what professionalism really is" and also for teaching me the various practical aspects of the enzyme-kinetics. Dr. Dimitri Ossipov for the fruitful collaboration. Pradeep Cheruku, for being good and "cool" collaborator. Jharna Barman for cleaning up the mess that I have created at the work place many times and also for her help in the research. Johan Isaksson for fruitful collaborations and the help with the Swedish summary. Dr. Nitin Puri and Dr. Mrinal Kanti Kundu for being friendly and also for making me to settle down quickly in the lab. Melcer Holmer, Oommen Varghese, Brian Lawrence, Anna Trifonova, Gopal Datta, Catherine Petit, Subhrangsu Chatterjee, Xiaoguang Luo, Dmitry Honcharenko, Irina Velikian, Kartik Babu, Puneet Sreevastava, Wimal Pathmasiri, Peter Agback, Tatiana Maltseva, Jan Milecki, Christophe Thibaudeau and Malgorzata Bogucka for their contribution to keeping a lively atmosphere at the Department. I am grateful to Prof. Goverdhan Mehta (Indian Institute of Science) for introducing me to the research in Organic Chemistry and also for his support. I am indebted to Mr. Pushkar Dandekar for inspiring me to come to Sweden. I am still unable to cope with his untimely death in a car accident in Lund. My special thanks also due to: Mr. Suresh Gohil, Swedish University of Agricultural Sciences, for his help with Mass Spectrometry, especially when we were in a desperate need. Prof. Leif Kirsebom, Dr. Mathias Brännvall, Dr. Santanu Dasgupta, and Mr. Klas I. Udekwu at the Department of Cell and Molecular Biology for finding time to answer my “simple” questions in molecular biology.

40
Dr. Devapriya Choudhury for giving me "right advices at the right time". Jaana Öst, Pernilla Brohage and Rabab Elkarib for their kind assistance and com-petent administrative works. I would like to thank all our external collaborators, in particular Prof. Alan Gewirtz and Dr. Joana Opalinska at the University of Pennsylvania School of Medicine, Prof. Jacqueline Barton and Dr. Elizebeth Boon at the California Institute of Tech-nology, Dr. Sudhir Agrawal and Dr. Ekambar Kandimalla at the Hybridon Inc. for expanding the horizons of "our Chemistry." My sincere thanks are also due to: Susan Bergman for being cordial and also for arranging nice summer and Christmas parties. Dr. Girish Ganesan and his wife Sampreetha for the Friday dinners, and for their friendly and warm company. Dr. Ajey Jacob for his friendship and regular telephone conversations, since our first meeting at the airport during our first journey to Sweden. I also thank Ajey for fuel-ling my reading habits on socio-political topics. Bharath Bikkaji for his friendship, late night discussions about everything under and above the Sun and also for keeping my ego in check. Sandipta for being friendly and for providing many nice lunches and dinners. All my friends in Flogsta especially Gunnel, Yara, Stefan, Joan, Magie, Kemenes, David, George, Olivier, Tingting, Ingrid, Amela, Anika, Diana, Gosta, Thobias, Sara and Yaqing for making my stay in the corridor pleasant and memorable. Many thanks to friends at the “Art of Living Foundation” for keeping me in touch with the Pranayama. "Sernanders Krog" Staffan for serving the ''spiciest cuisine in Uppsala'' late at nights. I would like to thank all my Indian friends in Sweden, in particular Nelson, Kiran, Prabhakar, Prasad, Aji, Dan, Rajesh, Gopan, Vinod, Sabil, Rituparna, Deepa, Vasu and Abhiman for helping to keep my Indianness intact. My special thanks to Drs. Rajeev Thottapillil, Asok Swain, Dileepkumar, Ramaswamy, Sitaram, Thomas, Ma-halingam, Murugaiah, Ragoth, Biplab, Suparna, Ambujom, Mangala, Chandra, Meena, Vijayalakshmi, Snoopy, Sasikumar, Mrs. Saramma Varkey, Mrs. Sobhana Nair and Mr. Sreekumar and their families for being my friends and well wishers. I am grateful to my teachers in India especially Mrs. V. Sujatha (Govt. High School, Ayirooppara, Trivandrum), Mr. Jayakumar (Institute of Physics, Trivandrum), Prof. R. Narayanaswamy and Prof. M.S. Suseelan (University College, Trivandrum), Prof. V.N. Rajasekharan Pillai, Dr. Ibnu Saud, Dr. Pius Kuruvilla, Dr. Sabu Thomas, Dr. C.V. Asokan and Dr. C.T. Aravindakumar (Mahatma Gandhi University, Kottayam) for their support, advice and care which enabled me to pursue the Ph.D. studies. I would like to thank all my Indian friends around the world for being good friends. I do not have enough words to express my gratitude to my Parents, my Brother, Sister and their respective families for their love and for their support in my endeav-our. I am especially grateful to my Brother who was the first to teach me Chemistry in the "proper way". He also introduced me to many topics ranging from the Ve-danta to the Tao of Physics and showed me the value of "Silence". Finally, I would like to thank Uppsala University, Swedish Research Council (Vetenskapsrådet), Swedish Foundation for Strategic Research (SSF) and Philip Morris USA Inc. for financially supporting our research.

41
6. Summary in Swedish
Denna avhandling bygger på nio artiklar vilka behandlar syntes, fysikoke-miska- och biologiska egenskaper hos två typer av kemiskt modifierade oli-gonukleotider som har potential att nedreglera genuttryck: (i) Den första typen består av antisense oligonukleotider (AONs) konjugerade med olika kromoforer med olika storlek, laddning och π-elektron densitet. Konjugering av kromoforen till 3'- och 5'-ändarna ökade bindningsaffiniteten för mål-RNA:t och aktiveringen av RNase H jämfört med den ursprungliga sekven-sen, utan att för den skull förändra den globala konformation hos AON/RNA hybriden. 3'-dipyridophenazine (DPPZ) har visat sig vara mest lovande icke-toxiska kromoforen i denna serie. (ii) Den andra typen utgörs av en ny klass AONs som innehåller North-East konformationellt låsta 1',2'-oxetane-nukleosider. Introduktionen av oxetane-T och -C enheter sänker smältpunk-ten för en AON/RNA hybrid med 6° respektive 3 °C/modifikation, medan motsvarande oxetane-A och -G inte förändrar stabiliteten. De modifierade AONs har visat sig vara mer stabila i serum jämfört med motsvarande omo-difierade sekvens där oxetane-T och -C var mer resistenta mot endonuklea-ser än oxetane-A och -G. Samtliga blandade sekvenser av AON/RNA hybri-der visade sig vara utmärkta substrat för RNase H, vilket har undersökts med Michaelis-Menten kinetik. De oxetane modifierade AONs nedreglerar effek-tivt den proto-onkogena c-myb mRNA i K562 stammen av mänskliga leu-kemiceller, vilket analyserats med QRT-PCR och Western blot. Antalet upp-tagna AONs efter administration har visat att oxetane-modifierade AONs är 5-6 gånger mer effektiva än motsvarande isosekvensiella phosphothioate analoger. Elektrokemisk assay baserad på känslig nukleinsyramedierad laddningstransport (CT) har visat att introduktionen av oxetane-T skapar större störningar i stackningen i en DNA/DNA helix än i en DNA/RNA he-lix.

42
7. References
1. Michelson, A. M.; Todd. A. R. J. Chem. Soc. 1955, 2632. 2. Khorana, H. G.; Razzell, W. E.; Gilham, P. T.; Tener G. M.; Pol. E. H. J. Am.
Chem. Soc. 1957, 79, 1002. 3. Khorana. H. G. Pure Appl. Chem. 1968, 17, 349. 4. Khorana. H. G. Biochem. J. 1968, 109, 709. 5. Agarwal, K. L.; Yamazaki, A.; Cashion, P. J.; Khorana, H. G. Angew. Chem.
Int. Ed. 1972, 11, 451. 6. Agarwal, K. L.; Buchi, H.; Caruthers, M. H.; Gupta, N.; Khorana, H. G.;
Kleppe, K.; Kumar, A.; Ohtsuka, E.; RajBhandary, U. L.; van de Sande, J. H.; Sgaramella, V.; Weber, H.; Yamode, T. Nature 1970, 227, 27.
7. Smith, M.; Rammler, D. H.; Goldberg, I. H.; Khorana, H. G. J. Am. Chem. Soc. 1961, 84, 430.
8. Brown, E. L.; Belagaje, R.; Ryan, M. J.; Khorana, H. G. Methods Enzymol. 1979, 68, 109.
9. Letsinger, R. L.; Kornet, M. J. J. Am. Chem. Soc. 1963, 385, 3045. 10. Letsinger, R. L.; Mahadevan, V. J. Am. Chem. Soc. 1965, 87, 3526. 11. Letsinger, R. L.; Ogilvie, K. K. J. Am. Chem. Soc. 1967, 89, 801. 12. Letsinger, R. L.; Ogilvie, K. K.; Miller, P. S. J. Am. Chem. Soc. 1969, 91,
3360. 13. Letsinger, R. L.; Lunsford, W. B. J. Am. Chem. Soc. 1976, 98, 3655. 14. Letsinger, R. L.; Kornet, M. J.; Mahadevan, V.; Jerina, D. M. J. Am. Chem.
Soc. 1964, 86, 5163. 15. Reese, C. B. Tetrahedron 1978, 34, 3143. 16. Reese, C. B.; Saffhill, R. Chem. Commun. 1968, 767. 17. Cusack, N. J.; Reese, C. B.; van Boom, J. H. Tetrahedron Lett. 1973, 2209. 18. Chattopadhyaya, J. B.; Reese, C. B. Tetrahedron Lett. 1979, 5059. 19. Chattopadhyaya, J. B.; Reese, C. B. Nucleic Acids Res. 1980, 8, 2039. 20. Reese, C. B. Tetrahedron 2002, 58, 8893. 21. Beaucage, S. L.; Caruthers, M. H. Tetrahedron Lett. 1981, 22, 1859. 22. McBride, L. J.; Caruthers, M. H. Tetrahedron Lett. 1983, 24, 245. 23. Caruthers, M. H. Acc. Chem. Res. 1991, 24, 278. 24. Beaucage, S. L.; Iyer, R. P. Tetrahedron 1992, 48, 2223. 25. Garegg, P. J.; Regberg, T.; Stawinski, J.; Strömberg, R. Chem. Scripta 1985,
25, 280. 26. Garegg, P. J.; Lindl, I.; Regberg, T.; Stawinski, J.; Strömberg, R. Tetrahedron
Lett. 1986, 27, 4051. 27. Stawinski, J.; Karaszewski. A. Acc. Chem. Res. 2002, 35, 952. 28. Scaringe, S. A.; Wincott, F. E.; Caruthers, M. H. J. Am. Chem. Soc. 1998,
120, 11820. 29. Micura, R. Angew. Chem. Int. Ed. 2002, 41, 2265. 30. Scaringe, S. A. Methods 2001, 23, 206.

43
31. Saiki, R. K.; Gelfand, D. H.; Stoffel, S.; Scharf, S. J.; Higuchi, R.; Horn, G. T.; Mullis, K. B.; Erlich, H. A. Science 1998, 239, 487.
32. Mullis, K. B.; Faloona, F. A. Methods Enzymol. 1987, 155, 335. 33. Metzker, M. L.; Caskey, T, C. in Nature Encyclopedia of Life Sciences, Na-
ture Publishing Group, London, 2001. (doi:10.1038/npg.els.0000998) 34. Zamecnik, P. C.; Stephenson, M. L. Proc. Natl. Acad. Sci. USA 1978, 75, 280. 35. Stephenson, M. L.; Zamecnik, P. C. Proc. Natl. Acad. Sci. USA 1978, 75, 285. 36. Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737. 37. Baker, B. F.; Monia, B. P. Biochim. Biophys. Acta 1999, 1489, 3. 38. Dias, N.; Stein, C. A. Mol. Cancer. Ther. 2002, 1, 347. 39. Crooke, S.T. Methods Enzymol. 2000, 313, 3. 40. Crooke, S.T. Biochim. Biophys. Acta 1999, 1489, 31. 41. Lebedeva, I.; Stein C. A. Annu. Rev. Pharmacol. 2001, 41, 403. 42. Scherer, L. J.; Rossi, J. J. Nat. Biotechnol. 2003, 21, 1457. 43. Opalinska, J. B.; Gewirtz, A. M. Nat. Rev. Drug. Discov. 2002, 1, 5303. 44. Agrawal, S.; Kandimalla, E. R. Mol. Med. Today. 2000, 6, 72. 45. Crooke, S. T. Annu. Rev. Med. 2004, 55, 61. 46. Dove, A. Nat. Biotechnol. 2002, 20, 121. 47. Bennet, C. F.; Cowsert, L. M. Biochim. Biophys. Acta. 1999, 1489, 19. 48. Lee, L. K.; Roth, C. M. Curr. Opin. Biotechnol. 2003, 14, 505. 49. Niemeyer, C. M.; Blohm, D. Angew. Chem. Int. Ed. 1999, 38, 2865. 50. Lander, E. S. Nat. Genet. 1999, 21, 3. 51. Cheung, V. G.; Morley, M.; Aguilar, F.; Massimi, A.; Kucherlapati, R.;
Childs, G. Nat. Genet. 1999, 21, 15. 52. Lipshutz, R. J.; Fodor, S. P.; Gingeras, T. R.; Lockhart, D. J. Nat. Genet.
1999, 21, 20. 53. Mantripragada, K. K.; Buckley, P. G.; de Ståhl, T. D.; Dumanski, J. P. Trends
Genet. 2004, 20, 87. 54. Seeman, N. C. Nature 2003, 421, 427. 55. Seeman, N. C. Angew. Chem. Int. Ed. 1998, 37, 3220. 56. Wengel, J. Org. Biomol. Chem. 2004, 2, 277. 57. Hélène, C. Eur. J. Cancer 1991, 27, 1466 58. Thoung, N. T.; Hélène, C. Angew. Chem. Int. Ed. 1993, 32, 666. 59. Buchini, S.; Leumann, C. J. Curr. Opin. Chem. Biol. 2003, 7, 717. 60. Praseuth, D.; Guieysee, A. L.; Hélène, C. Biochim. Biophys. Acta. 1999, 1489,
181. 61. Sun, J.-S.; Garestier, T., Hélène, C. Curr. Opin. Struct. Biol. 1996, 6, 327. 62. Vasquez, K. M.; Wilson, J. Trends Biochem. Sci. 1998, 23, 4. 63. Hoogsteen, K. Acta Crystallogr. 1959, 12, 822. 64. Opaliska, J. B.; Gewirtz, M. Sci. STKE 2003, 47. (DOI:
10.1126/stke.2003.206.pe47) 65. Chech, T. R.; Zaug, A. J.; Grabowski, P. J. Cell 1981, 27, 487. 66. Kruger, K.; Grabowski, P. J.; Zaug, A. J.; Sands, J.; Gottschling, D. E.;
Chech, T. R. Cell 1982, 31, 147.

44
67. Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. Cell 1983, 35, 849.
68. Doudna, J. A.; Chech, T. R. Nature 2002, 418, 222. 69. James, H. A.; Gibson, I. Blood 1998, 91, 371. 70. Sullenger, B. A.; Gilboa, E. Nature 2002, 418, 252. 71. Michienzi, A.; Rossi, J. J. Methods Enzymol. 2001, 341, 581. 72. Breaker, R. R.; Joyce, G. F.; Santoro, S. W. Chem. Biol. 1994, 1, 223. 73. Santoro,; Joyce, G. F. Proc. Natl. Acad. Sci. USA 1997, 94, 4262. 74. Santoro, S. W.; Joyce, G. F. Biochemistry 1998, 37, 13330. 75. Coppins, R. L.; Silverman, S. K. Nat. Struct. Mol. Biol. 2004, 11, 270. 76. Warashina, M.; Kuwabara, T.; Nakamatsu, Y.; Taira, K. Chem. Biol. 1999, 6,
237. 77. Dass, C. R.; Saravolac, E. G.; Li, Y.; Sun, L. Q. Antisense Nucleic Acid Drug
Dev. 2002, 12, 289. 78. Cairns, M J.; Saravolac, E. G.; Sun, L. Q. Curr. Drug Targets 2002, 3, 269. 79. Vester, B.; Lundberg, L. B.; Sørensen, M. D.; Babu, B. R.; Douthwaite, S.;
Wengel, J. J. Am. Chem. Soc. 2002, 124, 13682. 80. Schubert, S.; Gül, D. C.; Grunert, H. P.; Grunert, H.; Erdmann, V. A.; Kur-
reck, J. Nucleic Acids Res. 2003, 31, 5982. 81. Sharma, H. W.; Perez, J. R.; Higgins-Sochaski, K.; Hsiao, R.; Narayanan, R.
Anticancer Res. 1996, 6, 61. 82. Cho-Chung, Y. S.; Park, Y. G.; Lee, Y. N. Curr. Opin. Mol. Ther. 1999, 1,
386. 83. Krieg, A. M. Biochim. Biophys. Acta 1999, 1489, 107. 84. Krieg, A. M. Curr. Oncol. Rep. 2004, 6, 88. 85. Agrawal, S.; Kandimalla, E. R. Trends Mol. Med. 2002, 8, 114. 86. Agrawal, S.; Kandimalla, E. R. Ann. N.Y. Acad. Sci. 2003, 1002, 30. 87. Wang, D.; Li, Y.; Yu, D.; Song, S. S.; Kandimalla, E. R.; and Agrawal, S. Int.
J. Oncol. 2004, 24, 901. 88. Fire, A.; Xu, S.; Motegomery, M. K.; Kostas, S. A.; Driver, S. E.; Mello, C.
C. Nature 1998, 391, 806. 89. Elbashir, S. M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl,
T. Nature. 2001, 411, 494. 90. McManus, M. T.; Sharp, P. A. Nat. Rev. Genet. 2002, 3, 737. 91. Bernstein, E.; Caudy, A. A.; Hammond, S. M.; Hannon, G. J. Nature 2001,
409, 363. 92. Hutvagner, G.; Zamore, P. D. Curr. Opin. Genet Dev. 2002, 12, 225. 93. Hannon, G. Nature 2002, 418, 244. 94. Chiu, Y-L.; Rana, T. M. Mol. Cell. 2002, 10, 549. 95. Chiu, Y-L.; Rana, T. M. RNA 2003, 9, 1034. 96. Dykxhoorn, D. M.; Novina, C. D.; Sharp, P. A. Nat. Rev. Mol. Cell Biol.
2003, 4, 457. 97. Hutvagner, G.; Zamore, P. D. Science 2002, 297, 2056. 98. Dorsett, Y.; Tuschl, T. Nat. Rev. Drug. Discov. 2004, 3, 318.

45
99. Check, E. Nature 2003, 425, 10. 100. Robison, R. PLoS Biology 2004, 2, 18. (DOI: 10.1371/journal.pbio.0020028) 101. Koehn, R. K.; Rossi, J. J.; Stein C. A. Science 2001, 293, 1047. 102. Dias, N.; Stein C. A. Eur. J. Pharm. Biopharm. 2002, 54, 263. 103. Wickstrom, E. J. Biochem. Biophys. Methods 1986, 13, 97. 104. Akthar, S.; Kole, R.; Juliano, R. L. Life Sci. 1991, 49, 1793. 105. Eder, P. S.; DeVine, R. J; Dagle, J. M; Walder, J. A. Antisense Res. Dev.
1991, 1, 141. 106. Vlassov, V. V.; Yakubov, L. A. in Oligonucleotides: Antisense Inhibitors of
Gene Expression, Cohen, J. S., Ed., Macmillan, London, 1989, 243. 107. Temsamani, J.; Tang, J.-Y.; Padmapriya, A.; Kubert, M.; Agrawal, S. Antisen-
se Res. Dev. 1993, 3, 277. 108. Shaw, J.-P.; Kent, K.; Fishback, J.; Froehler, B. Nucleic Acids Res. 1991, 19,
747. 109. Nicklin, P. L.; Ambler, J.; Mitchelson, A.; Bayley, D.; Phillips, J. A.; Craig,
S. J.; Monia, B. P. Proc. Natl. Acad. Sci. USA 1997, 16, 1145. 110. Vaerman, J. L.; Mouerau, P.; Deldime, F.; Lewalle, P.; Lammineur, C.
Morschhauser, F.; Martiat, P. Blood 1997, 90, 331. 111. Koziolkiewicz, M.; Gendaszewska, E.; Maszewska, M.; Stein, C. A.; Stec, W.
J. Blood 2001, 98, 995. 112. Kara, J.; Duschinsky, R. Biochim. Biophys. Acta 1969, 186, 223. 113. Uhlmann, E.; Peyman, A. Chem. Rev. 1990, 90, 543. 114. De Mesmaeker, A.; Häner, R.; Martin, P.; Moser, H. E. Acc. Chem. Res. 1995,
28, 366. 115. Kurreck, J. Eur. J. Biochem. 2003, 270, 1628. 116. Braasch, D. A.; Corey, D. R. Biochemistry 2002, 41, 4503. 117. Bertrand, J. R.; Imbach, J. L.; Paoletti, C. Biochem. Biophys. Res. Commun.
1989, 164, 311. 118. Blacke, K. R.; Murakami, A.; Spitz, S. A. Biochemistry 1985, 24, 6139. 119. Boiziau, C.; Kurfurst, R.; Cazenave, C. Nucleic Acids Res. 1991, 19, 1113. 120. Johansson, H. E.; Belsham, G. J.; Sproat, B. S. Nucleic Acids Res. 1994, 22,
4591. 121. Haeuptle, M. T.; Frank, R.; Dobberstein, B. Nucleic Acids Res. 1986, 14,
1427. 122. Minshull, J.; Hunt, T. Nucleic Acids Res. 1986, 14, 6433. 123. Cazenave, C.; Loreau, N.; Thuong, N. T. Nucleic Acids Res. 1987, 15, 4717 124. Walder, R. Y.; Walder, J. A. Proc. Natl. Acad. Sci. USA 1988, 85, 5011. 125. Zamaratski, E.; Pradeepkumar, P. I.; Chattopadhyaya, J. Biochem. Biophys.
Methods 2001, 48, 189. 126. Chiang, M. Y.; Chan, H.; Zounes, M. A. J. Biol. Chem. 1991, 266, 18162. 127. Giles, R. V.; Ruddell, C. J.; Spiller, D. G. Nucleic Acids Res. 1995, 23, 954. 128. Stein, H.; Hausen, P. Science 1969, 166, 393. 129. Hausen, P.; Stein, H. Eur. J. Biochem. 1970, 14, 278.

46
130. Kanaya, S.; Ikehara, M. in Subcell. Biochemistry, Proteins: Structure, Func-tion, and Engineering, Biswas B. B. and Roy B., Ed., Plenum Press, New York, 1995, 377.
131. Berkower, I.; Leis, J.; Hurwitz, J. J. Biol. Chem. 1973, 248, 5914. 132. Stavrianopoulos, J. G.; Gambino-Giuffrida, A.; Chargaff, E. Proc. Natl. Acad.
Sci. USA 1976, 73, 1087. 133. Wintersberger, U. Pharmac. Ther. 1990, 48, 259. 134. Hughes, S. H.; Arnold, E.; Hostomsky, Z. in Ribonuclease H, Crouch R. J.
and Toulme J. J., Ed., INSERM, Paris, 1998, Ch. 10, 195. 135. Kogoma, T.; Foster, P. L. in Ribonuclease H, Crouch R. J. and Toulme J. J.,
Ed., INSERM, Paris, 1998, Ch. 2, 39. 136. Katayanagi, K.; Miyagawa, M.; Matsushima, M.; Ishikawa, M.; Kanaya, S.;
Ikehara, M.; Matsuzaki, T.; Morikawa, K. Nature 1990, 347, 306. 137. Yang, W.; Hendrickson, W. A.; Crouch, R. J.; Satow, Y. Science 1990, 249,
1398. 138. Katayanagi, K.; Miyagawa, M.; Matsushima, M.; Ishikawa, M.; Kanaya, S.;
Nakamura, H.; Ikehara, M.; Matsuzaki, T.; Morikawa, K. J. Mol. Biol. 1992, 223, 1029.
139. Casregola, S.; Khidhir, M.; Holland, I. B. Mol. Gen. Genet. 1987, 209, 494. 140. Itoh, T.; Tomizawa, J. Proc. Natl. Acad. Sci. USA 1980, 77, 2450. 141. Dasgupta, S.; Masukata, H.; Tomizawa, J. Cell 1987, 51, 1113. 142. Ogawa, T.; Pickett, G. G.; Kogoma, T.; Kornberg, A. Proc. Natl. Acad. Sci.
USA 1984, 81, 1040. 143. Kogoma, T.; Subia, N. L.; Meyenberg, K. Mol. Gen. Genet. 1985, 200, 103. 144. Horiuchi, T.; Maki, H.; Sekiguchi, M. Mol. Gen. Genet. 1985, 195, 17. 145. Hong, X.; Kogoma, T. J. Bacteriol. 1993, 175, 6731. 146. Frank, P.; Braunshofer-Reiter, C.; Wintersberger, U.; Grimm, R.; Busen. W.
Proc. Natl. Acad. Sci. USA 1988, 95, 12872. 147. Cerritelli, S. M.; Crouch, R. J. Genomics 1988, 53, 300. 148. Wu, H. J.; Lima, W. F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S. T. J. Biol.
Chem. 2004., Article in the press, DOI: 10.1074/jbc.M311683200. 149. Wu, H. J.; Lima, W. F.; Crooke, S. T. J. Biol. Chem. 1999, 272, 6146. 150. Lima, W. F.; Wu, H.; Crooke, S. T. Methods Enzymol. 2001, 341, 430. 151. Crooke, S. T.; Lemonidis, K. M.; Neilson, L.; Griffey, R.; Lesnik, E. A.; Mo-
nia, B. P. Biochem. J. 1995, 312, 599. 152. Lima, W. F.; Crooke, S. T. Biochemistry 1997, 36, 390. 153. Stein, C. A. Pharmacol. Ther. 2000, 85, 231. 154. Morikawa, K.; Katayanagi, K. in Ribonuclease H, Crouch R. J. and Toulme J.
J., Ed., INSERM, Paris, 1998, Ch. 9, 181. 155. Sarafianos, S. G.; Das, K.; Tantillo, C.; Clark, A. D.; Ding, J.; Whitcomb, J.
M.; Boyer, P. L.; Hughes, S. H.; Arnold, E. EMBO J. 2001, 20, 1449. 156. Tonelli, M.; Ulyanov, N. B.; Billeci, T. M.; Karwowski, B.; Guga, P.; Stec,
W. J.; James, T. L. Biophys. J. 2003, 85, 2525.

47
157. Mangos, M. M.; Min, K.-L.; Viazovkina, E.; Galarneau, A.; Elzagheid, M. I.; Parniak, M. A.; Damha, M. J. J. Am. Chem. Soc. 2003, 125, 654.
158. Minasov, G.; Teplova, M.; Nielsen, P.; Wengel, J.; Egli, M. Biochemistry 2000, 39, 3525.
159. Salazar, M.; Fedoroff, O. Y.; Miller, J. M.; Ribeiro, N. S.; Reid, B. R. Bio-chemistry 1993, 32, 4207.
160. Fedoroff, O. Y.; Salazar, M.; Reid, B. R. J. Mol. Biol. 1993, 233, 509. 161. Nishizaki, T.; Iwai, S.; Ohtsuka, E.; Nakumara, H. Biochemistry 1997, 36,
2577. 162. Sohail, M. Drug. Discov. Today 2001, 6, 1260. 163. Sohail, M. Sothern, E. M. Adv. Drug. Deliv. Rev. 2000, 44, 23. 164. Frier, S. M. in Antisense Drug Technology: Principles, Strategies and Appli-
cations, Crooke, S. T., Ed., Marcel Dekker, New York, 2001, Ch. 5, 107. 165. Wolff, J. A. Nat. Biotechnol. 2002, 20, 768. 166. Lebedeva, I.; Benimetskaya, L.; Stein C. A.; Vilenchik, M. Europ. J. Pharm.
Biopharm. 2000, 50, 101. 167. Hughes, J.; Astriab, A.; Yoo, H.; Alahari, S.; Liang, E.; Serguev, D.; Shaw, B.
R.; Juliano, R. L. Methods Enzymol. 2000, 313, 342. 168. Hughes, D. M.; Hussain, M.; Nawaz, Q.; Sayyed, P.; Akthar, S. Drug. Discov.
Today 2001, 6, 303. 169. Eckstein, F. Antisense Nucleic Acids Drug Dev. 2000, 10, 117. 170. De Clerq, E.; Eckstein, F.; Merigan, T. C. Science 1969, 165, 1137. 171. Koziolkiewicz, M.; Wojcik, M.; Kobylanska, A.; Karwowski, B.; Rebowska,
B.; Guga, P.; Stec, W. J. Antisense Nucleic Acids Drug Dev. 1997, 7, 43. 172. Eckstein, F. Ann. Rev. Biochem. 1985, 54, 367. 173. Stein, C. A.; Subasinghe, C.; Shinozuka, K.; Cohhen, J. S. Nucleic Acids Res.
1988, 16, 3209. 174. Levin, A. A. Biochim. Biophys. Acta. 1999, 1489, 69. 175. Stein, C. A. J. Clin. Investig. 2001, 108, 641. 176. Levin, A. A.; Henry, S. P.; Monteith, D.; Templin, V. in Antisense Drug
Technology: Principles, Strategies and Applications, Crooke, S. T., Ed., Mar-cel Dekker, New York, 2001, Ch. 9, 201.
177. Guvakova, M. A.; Yakubov, L. A.; Vlodavsky, I.; Tonkinson, J. L.; Stein, C. A. J. Biol. Chem. 1995, 270, 2620.
178. Fennewald, S. M.; Rando, R. F. J. Biol. Chem. 1995, 270, 21718. 179. Rockwell, P.; O'Connor, W. J.; King, K.; Goldstein, N. I.; Zhang, L. M.; Stein
C. A. Proc. Natl. Acad. Sci. USA 1997, 94, 6523. 180. Khaled, Z.; Benimetskaya, L.; Zeltser, R.; Khan, T.; Sharma, H. W.; Naraya-
nan, R.; Stein, C. A. Nucleic Acids Res. 1996, 24, 737. 181. Benimetskaya, L.; Loike, J. D.; Khaled, Z.; Loike, G.; Silverstein, S. C.; Cao
L.; el Khoury, J.; Cai, T. Q.; Stein, C. A. Nat. Med. 1997, 4, 414. 182. Yakubov, L.; Khaled, Z.; Zhang, L. M.; Truneh, A.; Vlassov, V.; Stein, C. A.
J. Biol. Chem. 1993, 268, 18818.

48
183. Stein, C. A.; Cleary, A. M.; Yakubov, L.; Lederman, S. Antisense. Res. Dev. 1993, 3, 19.
184. Majundar, C.; Stein, C. A.; Cohen, J. S.; Broder, S.; Wilson, S. H. Biochemis-try 1989, 28, 1340.
185. Henry, S. P.; Giclas, P. C.; Leeds, J.; Pangburn, M.; Auletta, C.; Levin, A. A.; Kornbrust, D. J. J. Pharmacol. Exp. Ther. 1997, 281, 810.
186. Miller, P. S. in Oligodeoxynucleotides: Antisense Inhibitors of Gene Expres-sion, Cohen, J. S., Ed., CRC Press, Boca Raton, FL, 1989, 79.
187. Furdon, P. J.; Dominski, Z.; Kole, R. Nucleic Acids Res. 1989, 22, 9193. 188. Giles, R. V.; Tidd, D. M. Nucleic Acids Res. 1992, 20, 763. 189. Nielsen, P. E.; Egholm, M.; Berg, R. H.; Buchardt, O. Science 1991, 254,
1497. 190. Larsen, H. J.; Bentin, T.; Nielsen, P. E. Biochim. Biophys. Acta 1999, 1489,
159. 191. Nielsen, P. E. Methods Enzymol. 2001, 313, 156. 192. Koppelhus, U.; Nielsen, P. E. in Antisense Drug Technology: Principles,
Strategies and Applications, Crooke, S. T., Ed., Marcel Dekker, New York, 2001, Ch. 14, 359.
193. Uhlman, E.; Peyman, A.; Breipohl, G.; Will, D. W. Angew. Chem. Int. Ed. 1998, 37, 2796.
194. Ganesh, K. N.; Nielsen, P. E. Curr. Org. Chem. 2000, 4, 916. 195. Corey, D. R. Trends Biotechnol. 1997, 15, 224. 196. Shaw, B. R.; Dobrikov, M.; Wang, X.; Wan, J.; He, K.; Lin, J. L.; Li, P.; Rait
V.; Sergueeva, Z. A.; Sergueev, D. Ann. N.Y. Acad. Sci. 2003, 1002, 12. 197. Rait, V. K.; Shaw, B. R. Antisense Nucleic Acid Drug Dev. 1999, 9, 53. 198. Shaw, B. R.; Sergueev, D.; He, K.; Porter, K.; Summers, J.; Sergueeva, Z.;
Rait V. Methods Enzymol. 2000, 313, 226. 199. Micklefield, J. Curr. Med. Chem. 2001, 8, 1157. 200. Gryaznov, S. M.; Lloyd, D. H.; Chen, J. K.; Schultz, R. G.; DeDionisio, L. A.;
Ratmeyer, L.; Wilson, W. D. Proc. Natl. Acad. Sci. USA 1995, 92, 5798. 201. Gryaznov, S. M. Biochim. Biophys. Acta 1999, 1489, 131. 202. Summerton, J.; Weller, D. Antisense Nucleic Acid Drug Dev. 1997, 7, 187. 203. Summerton, J. Biochim. Biophys. Acta. 1999, 1489, 141. 204. Skorski, T.; Perrotti, D.; Nieborowsa-Skorska, M.; Gryaznov, S.; Calabretta,
B. Proc. Natl. Acad. Sci. USA 1997, 94, 3966. 205. Faira, M.; Spiller, D. G.; Dubertret, C.; Nelson, J. S.; White, M. R. H.;
Scherman, D.; Hélène, C.; Giovannangeli, C. Nat. Biotechnol. 2001, 19, 40. 206. Heasman, J. Dev. Biol. 2002, 243, 209. 207. Nasevicius, A.; Ekker, S. C. Nat. Genet. 2000, 26, 216. 208. Dellinger, D. J.; Sheehan, D. M.; Christensen, N. K.; Lindberg, J. G.;
Caruthers, M. H. J. Am. Chem. Soc. 2003, 125, 940. 209. Sheehan, D.; Lunstad, B.; Yamada, C. M.; Stell B. G.; Caruthers, M. H.;
Dellinger, D. J. Nucleic Acids Res. 2003, 31, 4109. 210. Herdewijn, P. Antisense Nucleic Acid Drug Dev. 2000, 10, 297.

49
211. Gutierrez, A. J.; Matteucci, M. D.; Grant, D.; Matsumura, S.; Wagner, R. W.; Froehler, B. C. Biochemistry 1997, 36, 743.
212. Wagner, R. W.; Matteucci, M. D.; Grant, D.; Huang, T.; Froehler, B. C. Nat. Biotechnol. 1996, 14, 840.
213. Flanagan, W. M.; Wolf, J. J.; Olson, P.; Grant, D.; Lin, K.-Y.; Wagner, R. W.; Matteucci, M. D. Proc. Natl. Acad. Sci. USA 1999, 96, 3513.
214. Maier, M. A.; Leeds, J. M.; Balow, G.; Springer, R. H.; Bharadwaj, R.; Manoharan, M. Biochemistry 2002, 41, 1323.
215. Shen, L.; Siwkowski, A.; Wancewicz, E. V.; Lesnik, E.; Butler, M.; Witchell, D.; Vasquez, G.; Ross, B.; Acevedo, O.; Inamati, G.; Sasmor, H.; Manoharan, M.; Monia, B. P. Antisense Nucleic Acid Drug Dev. 2003, 13, 129.
216. Freier, S. M.; Altmann, K. H. Nucleic Acids Res. 1997, 25, 4429. 217. Manoharan, M. Biochim. Biophys. Acta 1999, 1489, 117. 218. Kawasaki, A. M.; Casper, M. D.; Freier, S. M.; Lesnik, E. A.; Zounes, M. C.;
Cummins, L. L.; Gonzalez, C.; Cook, P. D. J. Med. Chem. 1993, 36, 831. 219. Martin, P. Helv. Chim. Acta 1995, 78, 486. 220. Griffey, R. H.; Monia, B. P.; Cummins, L. L.; Frier, S. M.; Greig, M. J.; Gui-
nosso, C. J.; Lesnik, E.; Manalili, S. M.; Mohan, V.; Owens, S.; Ross, B. R.; Sasmor, H.; Wancewicz, E.; Wieler, K.; Wheeler, P. D.; Cook, P. D. J. Med. Chem. 1996, 39, 5100.
221. Prakash, T. P.; Kawasaki, A. M.; Vasquez, G.; Fraser, A. S.; Casper, M. D.; Manoharan, M. Nucleosides Nucleotides 1999, 18, 1381.
222. Prakash, T. P.; Kawasaki, A. M.; Fraser, A. S.; Vasquez, G.; Manoharan, M. J. Org. Chem. 2002, 67, 357.
223. Acharya, P.; Cheruku, P.; Chatterjee, S.; Acharya, S.; Chattopadhyaya J. J. Am. Chem. Soc. 2004, 126, 2862.
224. Kawai, G.; Yamamoto, Y.; Amimura, T.; Masegi, T.; Sekine, M.; Hata, T.; Iimori, T.; Watanabe, T.; Miyazawa, T.; Yokoyama, S. Biochemistry 1992, 31, 1040.
225. Dean, N. M.; Butler, M.; Monia, B. P.; Manoharan, M. in Antisense Drug Technology: Principles, Strategies and Applications, Crooke, S. T., Ed., Mar-cel Dekker, New York, 2001, Ch. 12, 319.
226. Baker, B. F.; Lot, S. S.; Condon, T. P.; Cheng-Flournoy, S.; Lesnik, E. A.; Sasmor, H. M.; Bennett, C. F. J. Biol. Chem. 1997, 272, 11994.
227. Sierakowska, H.; Sambade, M.; Agrawal, S.; Kole, R. Proc. Natl. Acad. Sci. USA 1996, 93, 12840.
228. Damha, M. J.; Wilds, C. J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M. A. J. Am. Chem. Soc. 1998, 120, 12976.
229. Noronha, A. M.; Wilds, C. J.; Lok, C-N.; Viazovkina, K.; Arion, D.; Parniak, M. A.; Damha, M. J. Biochemistry 2000, 39, 7050.
230. Wilds, C. J.; Damha, M. J. Nucleic Acids Res. 2000, 28, 3625. 231. Trempe, J. F.; Wilds, C. J.; Denisov, A. Y.; Pon, R. T.; Damha, M. J.; Gehring
K. J. Am. Chem. Soc. 2001, 123, 4896. 232. Mangos, M. M.; Damha, M. J. Curr. Top. Med. Chem. 2002, 2, 1147.

50
233. Lok, C. N.; Viazovkina, E.; Min, K. L.; Nagy, E.; Wilds, C. J.; Damha, M. J.; Parniak, M. A. Biochemistry 2002, 41, 3457.
234. Lacombe, J.; Viazovkina, E.; Bernatchez, P. N.; Galarneau, A.; Damha, M. J.; Sirois, M. G. Can. J. Physiol. Pharmacol. 2002, 80, 951.
235. Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, F.; Rosemeyer, H.; Seela, F.; Van Aerschot, A.; Herdewijn, P. J. Am. Chem. Soc. 2000, 122, 8595.
236. Verbeure, B.; Lescrinier, E.; Wang, J.; Herdewijn, P. Nucleic Acids Res. 2001, 29, 4941.
237. Herdewijn, P. Biochim. Biophys. Acta 1999, 1489, 167. 238. Kværnø; L.; Wengel, J. Chem. Commun. 2001, 16, 1419. 239. Leumann, C. Bioorg. Med. Chem. 2002, 10, 841. 240. Meldgaard, M.; Wengel, J. J. Chem. Soc., Perkin Trans. 1 2000, 3539. 241. Herdewijn, P. Liebigs Ann. Chem. 1996, 1337. 242. Tarkoy, M.; Bolli, M.; Schweizer, B.; Leumann, C. Helv. Chim. Acta. 1993,
76, 481. 243. Altman, K.-H.; Kesselring, R.; Francotte, E.; Rihs, G. Tetrahedron Lett. 1994,
35, 2331. 244. Siddiqui, M. A.; Ford, H.; George, C.; Marquez, V. E. Nucleosides Nucleo-
tides 1996, 15, 235. 245. Marquez, V. E.; Siddiqui, M. A.; Ezzitouni, A.; Russ, P.; Wang, J.; Wanger,
W. R.; Matteucci, M. D. J. Med. Chem. 1996, 39, 3739. 246. Altman, K.-H.; Imwinkelried, R.; Kesselring, R.; Rihs, G. Tetrahedron Lett.
1994, 35, 7625. 247. Obika, S.; Moira, K.; Nambu, D.; Imanishi, T. Chem. Commun. 1997, 1643. 248. Obika, S.; Moira, K.; Hari, Y.; Imanishi, T. Bioorg. Med. Chem. Lett. 1999, 9,
515. 249. Obika, S.; Moira, K.; Nambu, D.; Hari, Y.; Itoh, H.; Imanishi, T. Tetrahedron
2002, 58, 3039. 250. Christensen, N. K.; Petersen, M.; Nielsen, P.; Jacobsen, J. P.; Olsen, C. E.;
Wengel, J. J. Am. Chem. Soc. 1998, 120, 5458. 251. Koshkin, A.; Singh, S. K.; Nielson, P., Rajwanshi, V. K.; Kumar R.; Meld-
gaard, M.; Wengel, J. Tetrahedron 1998, 54, 3607. 252. Obika, S.; Nanbu, D.; Hari, Y.; Morio, K.; In, Y.; Ishida, T.; Imanishi, T.
Tetrahedron Lett. 1997, 38, 8735. 253. Wengel, J. Acc. Chem. Res. 1999, 32, 301. 254. Koshkin, A. A.; Rajwanshi, V. K.; Wengel, J. Tetrahedron Lett.1998, 39,
4381. 255. Singh, S. K.; Nielsen, P.; Koshkin, A. A.; Wengel, J. Chem. Commun. 1998,
455. 256. Singh, S. K.; Kumar, R.; Wengel, J. J. Org. Chem.1998, 63, 10035. 257. Petersen, M.; Wengel, J. Trends Biotechnol. 2003, 21, 74. 258. Wengel, J. in Antisense Drug Technology: Principles, Strategies and Applica-
tions, Crooke, S. T., Ed., Marcel Dekker, New York, 2001, Ch. 13, 339.

51
259. Rajwanshi, V. K.; Kumar, R.; Kofod-Hansen, M. J. Chem. Soc., Perkin Trans. 1 1999, 1407.
260. Nielsen, P.; Pfundheller, H. M.; Olsen, C. E.; Wengel, J. J. Chem.Soc., Perkin Trans. 1 1997, 3423.
261. Raunkjær, M.; Olsen, C. E.; Wengel, J. J. Chem. Soc., Perkin Trans. 1 1999, 2543.
262. Christensen, N. K.; Andersen, A. K. L.; Schultz, T. R. Nielsen, P. Org. Bio-mol. Chem. 2003, 1, 3738.
263. Wang, G.; Giradet, J–L.; Gunic, E. Tetrahedron 1999, 55, 7707. 264. Wang, G. Tetrahedron Lett. 1999, 40, 6343. 265. Raven, J.; Thorup, N.; Nielsen, P. J. Chem. Soc., Perkin Trans. 1 2001, 1855. 266. Kværnø, L.; Wengel, J. J. Org. Chem. 2001, 66, 5498. 267. Nielsen, P.; Petersen, M.; Jacobsen, J. P. J. Chem. Soc., Perkin Trans. 1 2000,
3706. 268. Kim, H. S.; Jacobson, K. A. Org. Lett. 2003, 5, 1665. 269. Kværnø; L.; Wightman, R. H.; Wengel, J. J. Org. Chem. 2001, 66, 5106. 270. Ravn, J.; Nielsen, P. J. Chem. Soc., Perkin Trans. 1 2001, 985. 271. Zou, R.; Matteucci, M. D. Bioorg. Med. Chem. Lett. 1998, 8, 3049. 272. Morita, K.; Takagi, M.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.;
Ishikawa, T.; Imanishi, T.; Koizumi, M. Bioorg. Med. Chem. Lett. 2003, 11, 2211.
273. Steffens, R.; Leumann, C. J. Helv. Chim. Acta. 1997, 80, 2426. 274. Renneberg, D.; Leumann, C. J. J. Am. Chem. Soc. 2002, 124, 5993. 275. Sorensen, M. D.; Kværnø, L.; Bryld, T.; Hakansson, A. E.; Verbeure, B.;
Gaubert, G. J. Am. Chem. Soc. 2002, 124, 2164. 276. Babu, B. R.; Keinicke, L.; Petersen, M.; Nielsen, C.; Wengel, J. Org. Biomol.
Chem. 2003, 1, 3514. 277. de Leeuw, H. P. M., Haasnoot, C. A. G.; Altona, C. Isr. J. Chem. 1980, 20,
108. 278. Thibaudeau, C.; Chattopadhyaya, J. Stereoelectronic Effects in Nucleosides
and Nucleotides and their Structural Implications. Department of Bioorganic Chemistry ([email protected]), Uppsala University Press; Sweden 1999 (ISBN 91-506-1351-0)
279. Bondensgaard, K.; Petersen, M.; Singh, S. K.; Rajwanshi, V. K.; Wengel, J.; Jacobsen, J. P. Chem. Eur. J. 2000, 6, 2687.
280. Petersen, M.; Bondensgaard, K.; Jacobsen, J. P. J. Am. Chem. Soc. 2002, 124, 5974.
281. Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V. A. Nucleic Acids Res. 2002, 30, 1911.
282. Takagi, M.; Morita, K.; Nakai, D.; Nakagomi, R.; Tokui, T.; Koizumi, M.; Biochemistry 2004, Article in press, DOI: 10.1021/bi035847x
283. Sands, H.; Gorey-Feret, L. J.; Ho, S. P.; Bao, Y.; Cocuzza, A. J.; Chidester, D.; Hobbs, F. W. Mol. Pharmacol. 1995; 47, 636.

52
284. Miyao, T.; Takakura, Y.; Akiyama, T.; Yoneda, F.; Sezaki, H.; Hashida, M. Antisense Res. Dev. 1995; 5, 115.
285. Uhlmann, E.; Peyman, A.; Ryte, A.; Schmidt, A.; Buddecke, E. Methods Enzymol. 2000, 313, 268.
286. Peyman, A.; Helsberg, M.; Kretzschmar, G.; Mag, M.; Ryte, A.; Uhlmann, E. Antiviral Res. 1997, 33, 135.
287. Monia, B. P.; Johnston, J. F.; Ecker, D. J.; Zounes, M. A.; Lima, W. F.; Frier, S. M. J. Biol. Chem. 1992, 267, 19954.
288. Frieden, M.; Christensen, S. M.; Mikkelsen, N. D.; Rosenbohm, C.; Thrue, C. A.; Westergaard, M.; Hansen, H. F.; Oerum, H.; Koch, T. Nucleic Acids Res. 2003, 31, 6325.
289. Braasch, D. A.; Liu, Y.; Corey, D. R. Nucleic Acids Res. 2002, 30, 5160. 290. Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsen, T.; Hökfelt, T.;
Broberger, C.; Porreca, F.; Koshkin, A.; Jacobsen, M. H.; Wengel, J. Proc. Natl. Acad. Sci. USA 2000, 97, 5633.
291. Fluiter, K.; ten Asbroek A. L. M. A.; de Wissel, M. B.; Jakobs, M. E.; Wis-senbach, M.; Olsson, H.; Olsen, O.; Oerum, H.; Baas, F. Nucleic Acids Res. 2003, 31, 953.
292. Ittig, D.; Liu, S.; Schumperli, D.; Leumann, C. J. Nucleic Acids Res. 2004, 32, 346.
293. Renneberg, D.; Bouliong, E.; Reber, U.; Schumperli, D.; Leumann, C. J. Nu-cleic Acids Res. 2002, 30, 2751.
294. Beaucage, S. L.; in DNA and aspects of Molecular Biology. Kool, E. T., Ed., Pergamon, Oxford, UK, 1999, Ch 7.06, 153.
295. Manoharan, M. Antisense Nucleic Acid Drug Dev. 2002, 12, 103. 296. Manoharan, M in Antisense Drug Technology: Principles, Strategies and
Applications, Crooke, S. T., Ed., Marcel Dekker, New York, 2001, Ch. 16, 391.
297. Maltseva, T. V.; Agback, P.; Repkova, M. N.; Venyaminova, A. G.; Ivanova, E. M.; Sandström, A.; Zarytova, V. F., Cattopadhyaya J. Nucleic Acids Res. 1994, 22, 5590.
298. Puri, N.; Chattopadhyaya, J. Nucleosides Nucleotides 1999, 18, 2785. 299. Boutorin, A. S.; Guskova, L. V.; Ivanova, E. M.; Kobetz, N. D.; Zarytova, V.
F.; Ryte, A. S.; Yurchenko, L. V.; Vlassov, V. V. FEBS Lett. 1989; 254, 129. 300. Ryte, A. S.; Karamyshev, V. N.; Nechaeva, M. N.; Guskova, Z. V.; Ivanova,
E. M.; Zarytova, V. F.; Vlassov, V. V. FEBS Lett. 1992, 299, 124. 301. Letsinger, R.; Scott, M. E. J. Am. Chem. Soc. 1981, 103, 7394. 302. Lokhov, S.; Podyminogin, M. A.; Sregeev, D. S.; Silnikov, V. N.; Kutyavin, I.
V.; Sishkin, G. V.; Zarytova, V. P. Bioconjugate Chem. 1992, 3, 414. 303. Balbi, A.; Sottofattori, E.; Grandi, T.; Mazzei, M. Tetrahedron 1994, 50,
4009. 304. Ossipov, D.; Chattopadhyaya, J. Tetrahedron 1998, 54, 5667. 305. Asseline, U.; Delarue, M.; Lancelot, G.; Toulme, F.; Thuong, N. T.; Mon-
tenay-Garestier, T.; Hélène, C. Proc. Natl. Acad. Sci. USA 1984, 81, 3297.

53
306. Asseline, U.; Bonfils, E.; Dupret, D.; Thuong, N. T. Bioconjugate Chem. 1996, 7, 369.
307. Fukui, K.; Tanaka, K. Nucleic Acids Res. 1996, 24, 3962. 308. Asseline, U.; Thuong, N. T. Nucleosides Nucleotides 1991, 10, 359. 309. Telser, J.; Cruickshank, K. A.; Morrison, L. E.; Netzel, T. L. J. Am. Chem.
Soc. 1989, 111, 9428. 310. Christensen, U. B.; Pedersen, E. B. Helv. Chim. Acta 2003, 86, 2090. 311. Lin, K.-Y.; Matteucci, M. Nucleic Acids Res. 1991, 19, 3111. 312. Ono, A.; Dan, A.; Matsuda, A. Bioconjugate Chem. 1993, 4, 499. 313. Herrlein, M. K.; Letsinger, R. L. Angew. Chem. Int. Ed. 1997, 36, 599. 314. Casale, R.; McLaughlin, L. W. J. Am. Chem. Soc. 1990, 112, 5264. 315. Yamana, K.; Aota, R.; Nakano, H. Tetrahedron Lett. 1995, 36, 8427. 316. Asseline, U.; Thuong, N. T.; Hélène, C. New J. Chem. 1997, 21, 5. 317. Cazevane, C.; Loreau, N.; Thuong, N. T.; Toulme, J. J.; Hélène, C. Nucleic
Acids Res. 1987, 15, 4717. 318. Letsinger, R. L.; Zhang, G.; Sun, D. K.; Ikeushi, T.; Sarin, P. S. Proc. Natl.
Acad. Sci. USA 1989, 86, 6553. 319. de Smidt, P.; Doan, T.; de Falco, S.; van Berekel, T. Nucleic Acids Res. 1991,
19, 4695. 320. Krieg, A. M, Tonkinson, J.; Matson, S.; Zhao, Q.; Saxon, M.; Zhang, L. M.;
Bhanja, U.; Yakubov, L.; Stein, C. A. Proc. Natl. Acad. Sci. USA 1993, 90, 1048.
321. Starke, D.; Lischka, K.; Pagels, P.; Uhlmann, E.; Kramer, W.; Wess, G.; Pet-zinger, E. Bioorg. Med. Chem. Lett. 2001, 11, 945.
322. Habus, I.; Zhao, Q.; Agrawal, S. Bioconjugate Chem. 1995, 6, 327. 323. Manoharan, M.; Tivel, K.; Inamati, G.; Monia, B. P.; Dean, N.; Cook, P. D.
Nucleosides Nucleotides 1995, 14, 969. 324. Misiura, K.; Wojcik, M.; Krakowiak, A.; Koziolkiewicz, M.; Pawlowska, Z.;
Pluskota, E.; Cierniewiski, C. S.; Stec, W. J. Acta Biochim. Pol. 1998, 27. 325. Manoharan, M.; Inamati, G. B.; Lesnik, E. A.; Sioufi, N. B.; Freier, S. M.
ChemBioChem 2002, 3, 1257. 326. Crooke, S. T.; Graham, M. J.; Zuckerman, J. E.; Brooks, D.; Conklin, B. S.;
Cummins, L. L.; Greig, M. J.; Guinosso, C. J.; Kornbrust, D.; Manoharan, M.; Sasmor, H. M.; Schleich, T.; Tivel, K. L.; Griffey, R. H. J. Pharmacol. Exp. Ther. 1996, 277, 923.
327. Godard, G.; Boutorine, A. S.; Saison-Behmoaras, E.; Hélène, C. Eur. J. Bio-chem. 1995, 232, 404.
328. Desjardins, J.; Mata, J.; Brown, T.; Graham, D.; Zon, G.; Iversen, P. J. Drug Targeting 1995, 2, 477.
329. Li, S.; Deshmukh, H. M.; Huang, L. Pharm. Res. 1998, 15, 1540. 330. Moore, V. A.; Dunnion, D. J.; Brown, T.; Irwin, W. J.; Akthar, S. Biochem.
Pharmacol. 1997, 53, 1223. 331. Maier, M.; Yannopoulos, C. G.; Mohamed, N.; Roland, A.; Fritz, H.; Mohan,
V.; Just, G.; Manoharan, M. Bioconjugate Chem. 2003, 14, 18.

54
332. Duff, R. J.; Deamond, S. F.; Roby, C.; Zhou, Y.; Ts'o, P. O. Methods Enzy-mol. 2000, 313, 297.
333. Langel, U.; Bartfai, T.; Pooga, M.; Valkna, A.; Saar, K.; Hallbrink, M. PCT Int. Appl. 1999, WO, 61. (The Perkin-Elmer Corporation, USA)
334. Astriab-Fischer, A.; Sergueev, D. S.; Fischer, M.; Ramsayshaw, B.; Juliano, R. L. Biochem. Pharmacol. 2000, 60, 83
335. Prakash, T. P.; Barawkar, D. A.; Kumar, V.; Ganesh, K. N. Bioorg. Med. Chem. Lett. 1994, 4, 1733.
336. Sund, C.; Puri, N.; Chattopadhyaya, J. Tetrahedron 1996, 52, 12275. 337. Trawick, B. N.; Daniher, A. T.; Bashkin, J. K. Chem. Rev. 1998, 98, 939. 338. Komiyama, M.; Sumaoka, J.; Kuzuya, A.; Yamamoto, Y. Methods Enzymol.
2001, 341, 455. 339. Boutorine, A. S.; Brault, D.; Takasugi, M.; Delgado, O.; Hélène, C. J. Am.
Chem. Soc. 1996, 118, 9469. 340. Bashkin, J. K.; Frolova, E. I.; Sampath, U. J. Am. Chem. Soc. 1994, 116,
5981. 341. Astrom, H.; Williams, N. H.; Strömberg, R. Org. Biomol. Chem. 2003, 1,
1461. 342. Putnam, S.; Bashkin, J. Chem. Commun. 2000, 767. 343. Whitney, A.; Gavory, G.; Balasubramanian, S. Chem. Commun. 2003, 36. 344. Hall, J.; Husken, D.; Häner, R. Nucleic Acids. Res. 1996, 24, 3522. 345. Dreyer, G.B.; Dervan, P. B. Proc. Natl. Acad. Sci. USA 1985, 82, 968. 346. Matsumura, K.; Endo, M.; Komiyama, M. Chem. Commun. 1994, 2019. 347. Komiyama, M.; Takeda, N.; Shigekawa, H. Chem. Commun. 1999, 1443. 348. Puri, N.; Majumdar, A.; Cuenoud, B.; Natt, F.; Martin, P.; Boyd, A.; Miller,
P. S.; Seidman, M. M. J. Biol. Chem. 2001, 276, 28991. 349. Sharma, S.; McLaughlin, L. W. J. Am. Chem. Soc. 2002, 124, 9658. 350. Ossipov, D.; Gohil, S.; Chattopadhyaya, J. J. Am. Chem. Soc. 2002, 124,
13416. 351. Zamaratski, E.; Chattopadhyaya, J. Tetrahedron 1997, 53, 10409. 352. Ossipov, D.; Zamaratski, E.; Chattopadhyaya, J. Helv. Chim. Acta 1999, 82,
2186. 353. Marky, L. A.; Breslauer, K. J. Biopolymers 1987, 26,1601. 354. Davis, T. M.; McFail-Isom, L.; Keane, E.; Williams, L. D. Biochemistry 1998,
37, 6975. 355. Li, J.; Wartell, R. M. Biochemistry 1998, 37, 5154. 356. Amirkhanov, N. V.; Zamaratski, E.; Chattopadhyaya, J. Tetrahedron Lett.
2000, 42, 489. 357. Prinsbe, E. J.; Smejkal, J.; Verheyden J. P. H.; Moffatt, J. G. J. Org. Chem.
1976, 41, 1836. 358. Sarma, M. S. P.; Megati, S. R.; Klein S.; Otter, B. A. Nucleosides Nucleotides
1995, 14, 393. 359. Hrebabecky, H.; Farkas, J. Collect. Czech. Chem. Commun. 1974, 39, 1098.

55
360. Roivainen, J.; Vepsalainen, J.; Azhayev, A.; Mikhailopulo, I. A. Tetrahedron Lett. 2002, 43, 6553.
361. Garner, P.; Ramakanth, S. J. Org. Chem. 1988, 53, 1294. 362. Acharya, P.; Isaksson, J.; Pradeepkumar, P. I.; Chattopadhyaya, J. Collect.
Czech. Chem. Commun. 2002, 5, 99. 363. Kool, E. T. Annu. Rev. Biochem. 2002, 71, 191. 364. Fox, K. R. in Methods in Mol. Biol., Fox, K. R., Ed., Humana Press Inc., To-
towa, NJ, 1997, 90, 1. 365. Suck, D. J. Mol. Reco. 1994, 7, 65. 366. Herrera, J. E.; Chaires, J. B. J. Mol. Biol. 1994, 236; 405. 367. Kanaya, E.; Kanaya, S. Eur. J. Biochem. 1995, 231, 557. 368. Isaksson, J.; Plashkevych, O.; Pradeepkumar, P. I.; Petit, C.; Chattopadhyaya,
J. 2004. (Manuscript) 369. Giles, V.; Tidd, D. M. Anti-Cancer Drug Des. 1992, 7, 37. 370. Baer, M. R.; Augustinos, P.; Kinniburgh, A. Blood 1992, 79, 1319. 371. Sokol, D. L.; Zhang, X.; Lu, P.; Gewirtz, A. M. Proc. Natl. Acad. Sci. USA
1998, 95, 11538. 372. Wilhelm, J.; Pingoud, A. ChemBioChem 2003, 4,1120. 373. Fox, K. R.; Waring, M. J. Methods Enzymol. 2001, 340, 412. 374. Galas, D. J.; Schmitz, A. Nucleic Acids Res. 1978, 5, 3157. 375. Cons, B. M. G.; Fox, K. R. FEBS Lett. 1990, 264, 100. 376. Sigman, D. S. Biochemistry 1990, 29, 9097. 377. Hertzberg, R. P.; Dervan, P. B. Biochemistry 1984, 23, 3934. 378. Nielsen, P. E.; Jeppesen, C.; Buchardt, O. FEBS Lett. 1988, 235, 122. 379. Portugal, J.; Waring, M. J. FEBS Lett. 1987, 225, 195. 380. Lane, M. J.; Dabrowiak, J. C.; Vournakis, J. N. Proc. Natl. Acad. Sci. USA
1983, 80, 3260. 381. Fox, K. R.; Waring, M. J. Nucleic Acids Res. 1984, 12, 9271. 382. Fox, K. R.; Howarth, N. R. Nucleic Acids Res. 1985, 13, 8695. 383. Low, C. M.; Drew, H. R.; Waring, M. J. Nucleic Acids Res. 1984, 12, 4865. 384. Chaires, J. B.; Herrera, J. E.; Waring, M. J. Biochemistry 1990, 29, 6145. 385. Fox, K. R.; Waring, M. J. Biochemistry 1986, 25, 4349. 386. Abu-Daya, A.; Brown, P. M.; Fox, K. R. Nucleic Acids Res. 1995, 23, 3385. 387. Weyermann, P.; Dervan, P. B. J. Am. Chem. Soc. 2002, 124, 6872. 388. Keppler, M. D.; James, P. L.; Neidle, S.; Brown, T.; Fox, K. R. Eur. J. Bio-
chem. 2003, 270, 4982. 389. Kunitz, M. J. Gen. Physiol. 1950, 33, 349. 390. Price, P. A. J. Biol. Chem. 1975, 250, 1981. 391. Suck, D.; Oefner, C. Nature 1986, 321, 620. 392. Waterloh, K.; Fox, K. R. J. Biol. Chem. 1991, 266, 6381. 393. Waterloh, K.; Fox, K. R. Nucleic Acids Res. 1991, 19, 6719. 394. Delaney, S.; Barton, J. K. J. Org. Chem. 2003, 68, 6475. 395. Boon, E. M.; Barton, J. K. Curr. Opin. Struct. Biol. 2002, 12, 320. 396. Schuster, G. B. Acc. Chem. Res. 2000, 33, 253.

56
397. Giese, B. Annu. Rev. Biochem. 2002, 71, 51. 398. Carrel, T.; Behrens, C.; Gierlich. J. Org. Biomol. Chem. 2003, 1, 2221. 399. Holmlin, R. E.; Dandliker, P. J.; Barton, J. K. Angew. Chem. Int. Ed. 1997,
36, 2715. 400. Giese, B. Curr. Opin. Chem. Biol. 2002, 12, 320. 401. Sancar, A. Biochemistry 1994, 33, 2. 402. Carrel, T. Angew. Chem. Int. Ed. 1995, 34, 2491. 403. Kelley, S. O.; Holmlin, R. E.; Stemp, E. D. A.; Barton, J. K. J. Am. Chem.
Soc. 1997, 119, 9867. 404. Hall, D. B.; Barton, J. K. J. Am. Chem. Soc. 1997, 119, 5045. 405. Rajski, S. R.; Barton, J. K. Biochemistry 2001, 40, 5556. 406. Rajski, S. R.; Kumar, S.; Roberts, R. J.; Barton, J. K. J. Am. Chem. Soc. 1999,
121, 9695. 407. Wan, C.; Fiebig, T.; Kelley, S. O.; Treadway, C. R.; Barton, J. K.; Zewail, A.
H. Proc. Natl. Acad. Sci. USA. 1999, 96, 6014. 408. Bhattacharya, P. K.; Barton, J. K. J. Am. Chem. Soc. 2001, 123, 8649. 409. Bhattacharya, P. K.; Cha, J.; Barton, J. K. Nucleic Acids Res. 2002, 30, 4740. 410. Arnold, F. H.; Wolk, S.; Cruz, P.; Tinoco, I. J. Biochemistry 1987, 26, 4068. 411. Drummond, T. G.; Hill, M. G.; Barton, J. K. Nat. Biotechnol. 2003, 21, 1192. 412. Kelley, S.O.; Jackson, N. M.; Hill, M. G.; Barton, J. K. Angew. Chem. Int. Ed.
1999, 38, 941. 413. Boon, E. M.; Ceres, D. M.; Drummond, T. G.; Hill, M. G.; Barton, J. K. Nat.
Biotechnol. 2000, 18, 1096. 414. Kelley, S. O.; Boon, E. M.; Barton, J. K.; Jackson, N. M.; Hill, M. G. Nucleic
Acids Res. 1999, 27, 4830. 415. Boon, E. M.; Salas, J. E.; Barton, J. K. Nat. Biotechnol. 2002, 20, 282. 416. Boon, E. M.; Barton, J. K. Bioconjugate Chem. 2003, 14, 1140. 417. Boon, E. M.; Jackson, N. M.; Whightman, M. D.; Kelley, S. O.; Hill, M. G.;
Barton, J. K. J. Phys. Chem. B. 2003, 107, 11805. 418. Achenbach, T. V.; Brunner, B.; Heermeier, K. ChemBioChem 2003, 4, 928. 419. Sørensen, M. D.; Petersen, M.; Wengel, J. Chem. Commun. 2003, 2130. 420. Wang, L.; Prakash, R. K.; Stein, C. A.; Koehn, R. K.; Ruffner, D. E. Antisense
Nucleic Acid Drug Dev. 2003, 13, 169. 421. Sczakiel, G.; Far, R. K. Curr. Opin. Mol. Ther. 2002, 4, 149. 422. Monia, B. P.; Sasmor, H.; Johnston, J. F.; Freier, S. M.; Lesnik, E. A.; Muller,
M.; Geiger, T.; Altmann, K. H.; Moser, H.; Fabbro, D. Proc. Natl. Acad. Sci. USA 1996, 93, 15481.
423. Kretschmer-Kazemi Far, R.; Sczakiel, G. Nucleic Acids Res. 2003, 31, 4417 424. Milner, N.; Mir, K. U.; Southern, E. M. Nat. Biotechnol. 1997, 15, 537. 425. Ho, S. P.; Bao, Y.; Lesher, T.; Malhotra, R.; Ma, L. Y.; Fluharty, S. J.; Sakai,
R. R. Nat. Biotechnol. 1998, 16, 59.


Acta Universitatis UpsaliensisComprehensive Summaries of Uppsala Dissertations
from the Faculty of Science and TechnologyEditor: The Dean of the Faculty of Science and Technology
Distribution:Uppsala University Library
Box 510, SE-751 20 Uppsala, Swedenwww.uu.se, [email protected]
ISSN 1104-232XISBN 91-554-5957-9
A doctoral dissertation from the Faculty of Science and Technology, UppsalaUniversity, is usually a summary of a number of papers. A few copies of thecomplete dissertation are kept at major Swedish research libraries, while thesummary alone is distributed internationally through the series ComprehensiveSummaries of Uppsala Dissertations from the Faculty of Science and Technology.(Prior to October, 1993, the series was published under the title “ComprehensiveSummaries of Uppsala Dissertations from the Faculty of Science”.)


















