
Chemical Modification 1998
28
CHEMICAL MODIFICATIONS OF PROTEINS: A REVIEW GARY E. MEANS' Dept. of Biochemistry The Ohio State IJniversity 484 W. 121h Ave. Columbus. OH 43210 AND ROBERT E. FEENEY Dept. of Food Science and Technology University of California, Davis One Shields Ave. Davis. CA 9561 6 Received for Publication September 15, 1997 Accepted for Publication January 31, 1998 ABSTRACT The chemical modfications of proteins are reviewed. Subjects include: (1) modiJcations done to s~udy protein function; (2) two naturally occurring modijfications: carbonylamine reactions and reactions with nitric oxide; (3) applications for bioconjugation and mass spectral analysis; and (4) modfications offoodproteins. INTRODUCTION The chemical modification of proteins is almost an ancient art. A wide variety of procedures for protein modification were developed and used for many years prior to any interest in, or significant understanding of the chemical basis of those processes. Chemical modifications of animal skins (i.e., hides) to preserve and stabilize them for use as clothing and other purposes has, for example, been around from the very beginning of recorded time. Interest in quantative determinations of proteins and their various constituent amino acids was a major impetus for many early studies of protein modification. As a result of new methodologies developed during and shortly after World War I1 (Olcott and Fraenkel-Conrat 1947; Balls and Jansen 1952), many additional ITo whom correspondence should be addressed. Journal of Food Biochemistry 22 (1998) 399-425. All Righrs Reserved. "Copyright I998 by Food & Nutrition Press, Inc.. Trumbuli, Connecticut. 399
-
Upload
bhanuprasadb -
Category
Documents
-
view
33 -
download
0
description
article
Transcript of Chemical Modification 1998

CHEMICAL MODIFICATIONS OF PROTEINS: A REVIEW
GARY E. MEANS'
Dept. of Biochemistry The Ohio State IJniversity
484 W. 121h Ave. Columbus. OH 43210
AND
ROBERT E. FEENEY
Dept. of Food Science and Technology University of California, Davis
One Shields Ave. Davis. CA 9561 6
Received for Publication September 15, 1997 Accepted for Publication January 31, 1998
ABSTRACT
The chemical modfications of proteins are reviewed. Subjects include: (1) modiJcations done to s~udy protein function; (2) two naturally occurring modijfications: carbonylamine reactions and reactions with nitric oxide; (3) applications for bioconjugation and mass spectral analysis; and (4) modfications of foodproteins.
INTRODUCTION
The chemical modification of proteins is almost an ancient art. A wide variety of procedures for protein modification were developed and used for many years prior to any interest in, or significant understanding of the chemical basis of those processes. Chemical modifications of animal skins (i.e., hides) to preserve and stabilize them for use as clothing and other purposes has, for example, been around from the very beginning of recorded time.
Interest in quantative determinations of proteins and their various constituent amino acids was a major impetus for many early studies of protein modification. As a result of new methodologies developed during and shortly after World War I1 (Olcott and Fraenkel-Conrat 1947; Balls and Jansen 1952), many additional
ITo whom correspondence should be addressed.
Journal of Food Biochemistry 22 (1998) 399-425. All Righrs Reserved. "Copyright I998 by Food & Nutrition Press, Inc.. Trumbuli, Connecticut. 399

40() G.E. MEANS and R.E. FEENEY
studies were initiated in the late 1940’s and early 1950’s (Fraenkel-Conrat et af. 1949; Fraenkel-Conrat and Feeney 1950). Further impetus was generated in succeeding years by the developement of many new methodologies, such as automated amino acid analysis (Moore and Stein 1963), automated sequencing (Edman and Begg 1967) gel-electrophoresis (Shapiro et af. 1967) and by the increased availability of radioisotopically labelled reagents.
Between 1960 and 1980, thousands of reports were published using chemical modification to identify amino acid residues required for the catalytic activities of enzymes and the various functional properties of other proteins. Several books (Means and Feeney 1971; Glazer et al. 1975; Lundblad and Noyes 1984) and reviews (Widder and Green 1985; Pfleiderer 1985; Means and Feeney 1990) were also published describing procedures for the chemical modification of proteins and their uses.
Today the many different uses of chemical modification include determina- tion of relative reactivities of side chain groups, the identification and quantitation of individual amino acid residues required for biological activity, the development of affinity and mechanism-based reagents for pharmaceutical uses, cross-linking reagents, special techniques for bioprosthesis, blocking reagents for peptide synthesis, reagents for specific cleavages of peptide bonds, bioconjugation, and special adjunctive modifications for analytical purposes, such as mass spectrome- try.
GENERAL CHEMISTRY
Chemical modification of proteins is based on differences in the reactivities of the individual amino acid side chains. These include the imidazole moiety of histidine, the indole of tryptophan, the p-hydroxyphenyl of tyrosine, the thioether of methionine, the thiol group of cysteine, the disulfide bond of cystine, the carboxyl groups of glutamic and aspartic acids, of carboxyl-terminal amino acids, and amino groups of both lysine and amino-terminal amino acids. These side chain groups react at different rates, not only with different reagents, but also under different conditions (e.g., pH), and also due to differences in their locations and specific environments within a particular protein. A few examples of current interest will be described.
Affinity Labels and Mechanism-based Inhibitors (Suicide Reagents)
Affinity labeling has become one of the most widely used techniques for the cr-iemical modification of enzymes and other proteins with specific ligand binding sites. An overall definition might be that affinity labeling is a technique designed

CHEMICAL MODIFICATIONS OF PROTEINS 40 1
to selectively modify side chain groups in, or close to, the active sites or other specific binding sites of enzymes and other proteins. In the case of an enzyme, reaction with a group in or near the active site is usually achieved by the use of a reagent with a structure that resembles a substrate or, sometimes, a reversible inhibitor of that enzyme. Due to their resemblance to a substrate, affinity labels usually interact strongly with the active site. Upon so doing, a chemically reactive group in the affinity label is then able to react with and chemically label a nearby side chain group. a-Haloketones, which can, potentially, react with several different kinds of nucleophilic side chains, and photo-activatable aryl aides, which can react with an even wider variety of side chains upon activation, are probably the most widely used reactive groups for affinity labeling. Several important reviews of affinity labeling have been published (Jakoby and Wilchek 1977; Coleman 1989).
With mechanism-based inhibitors, chemically reactive agents generated by the catalytic actions of target enzymes react with and chemically alter side chain groups in or near the active sites of those enzymes. The requirement for a catalytic transformation, in addition to a specific binding interaction, can make such reagents very specific. Due to their high specificity, mechanism-based inhibitors are particularly interesting as potential pharmacological agents. Many mechanism- based enzyme inhibitors have been described and the subject has been frequently reviewed (Walsh 1984; Pratt 1992).
Cross-linking and Immobilization
These two modifications have much in common. Cross-linking usually refers to processes wherein one protein is covalently attached to another, wherein two or more side chain groups in a single protein are covalently linked, or wherein proteins are linked covalently to another soluble substance. Immobilization usually refers to many similar processes wherein proteins are attached to some type of insoluble material. Chemically cross-linked agaroses and dextrans, a wide variety of synthetic organic polymers, and silica gel are some of the most common insoluble supports used for protein immobilization.
Crosslinking is sometimes used to determine distances, and other kinds of spatial relationships, between the components of simple protein oligomers, large multiprotein complexes, in biological membranes and other types of macromo- lecular assemblies. It is also commonly used to effect the coupling of proteins with different biological, chemical, or physical activities so as to create new multifunctional molecular entities. The so-called immunotoxins, for example, combine the toxic effects of certain protein toxins with the highly specific recognition and binding properties of antibodies (Lambert et af. 1985; Marsh 1988).
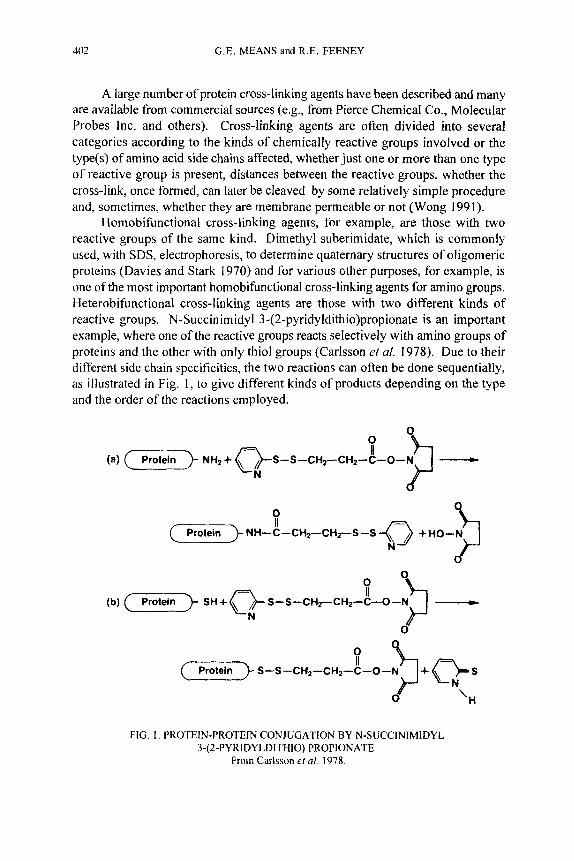
402 G.E. MEANS and R.E. FEENEY
A large number of protein cross-linking agents have been described and many are available from commercial sources (e.g., from Pierce Chemical Co., Molecular Probes Inc. and others). Cross-linking agents are often divided into several categories according to the kinds of chemically reactive groups involved or the type(s) of amino acid side chains affected, whether just one or more than one type of reactive group is present, distances between the reactive groups, whether the cross-link, once formed, can later be cleaved by some relatively simple procedure and, sometimes, whether they are membrane permeable or not (Wong 1991).
Homobifunctional cross-linking agents, for example, are those with two reactive groups of the same kind. Dimethyl suberimidate, which is commonly used, with SDS, electrophoresis, to determine quaternary structures of oligomeric proteins (Davies and Stark 1970) and for various other purposes, for example, is one of the most important homobifunctional cross-linking agents for amino groups. Heterobifunctional cross-linking agents are those with two different kinds of reactive groups. N-Succinimidyl 3-(2-pyridyldithio)propionate is an important example, where one of the reactive groups reacts selectively with amino groups of proteins and the other with only thiol groups (Carlsson et af. 1978). Due to their different side chain specificities, the two reactions can often be done sequentially, as illustrated in Fig. 1, to give different kinds of products depending on the type and the order of the reactions employed.
II S-S -CH2-CH2-C-O-N
‘H
FIG. I . PROTEIN-PROTEIN CONJUGATION BY N-SUCCINIMIDYL 3-(2-PYRIDYLDITHIO) PROPlONATE
From Carlsson et al. 1978.

CHEMICAL MODIFICATIONS OF PROTEINS 403
Immobilization of proteins on chemically cross-linked dextrans, agarose, and a variety of other materials is commonly used to prepare supports for various kinds of affinity chromatography. Relatively simple “activation” procedures are available for coupling proteins to some of these materials and “preactivated” derivatives of others, usually also with some kind of so-called “spacer arm”, are available from a number of commercial sources. Antibodies, for example, can be readily immobilized on cyanogen bromide-activated Sepharose CL4B or a variety of similar materials and used for the affinity chromatography of their specific antigens. The same or similar procedures can also be used to immobilize antigens, which can then be used for the affinity chromatography of antibodies. Avidin, protein A, various lectins, protein inhibitors of certain enzymes, regulatory subunits and other kinds of enzyme effectors (e.g. calmodulin, thioredoxin, a-lactalbumin, etc.), and many other proteins have been similarly immobilized and used for the affinity chromatography of various biomolecules with which they specifically interact. Detailed descriptions of the most important supports, “spacer-arms”, activation and coupling procedures, elution conditions and other salient features of affinity chromatography have been thoroughly described elsewhere (Porath and Kristiansen 1975; Hermanson et al. 1992).
Proteins (e.g., enzymes) are also frequently immobilized to facilitate their separation from soluble components of reaction solutions and, in many cases, to increase their stability. Immobilized enzyme bioreactors of various types are frequently used in both the research laboratory and in industry for the conversion of readily available (e.g., cheap) materials into relatively less available (i.e., more expensive) products and, to a lesser extent, in medicine (i.e., kidney dialysis machines and other extra- and intracorporeal devices). In the first two cases, the immobilized enzyme or enzymes are packed into a column or otherwise confined to some kind of reactor, through which a solution of the enzyme’s substrate (i.e., the cheap material) is continuously passed. The product, or products, of the enzyme’s reaction, uncontaminated by the enzyme itself, are then collected and isolated from the emerging solution. The production of high fructose corn syrup, which is currently the largest commercial application of an immobilized enzyme, is accomplished by the use of various immobilized forms of glucose (xylose) isomerase. Many excellent books and other types of reviews of enzyme immobili- zation have been published (Laskin 1985; Mosbach 1987).
Reductive Alkylation of Proteins
Reductive alkylation is an important procedure for the modification of amino groups in proteins. It proceeds readily under mild conditions, affects only amino groups, and, although it increases their size, has relatively little effect on their pK, values or electrostatic charges (Means and Feeney 1968; Jentoft and Dearborn

404 G.E. MEANS and R.E. FEENEY
1979; etc.). It involves the condensation of an amino group with a carbonyl compound to give an imine (i.e., a Schiff base) which is then reduced by sodium borohydride or another mild reducing agent (Eq. I) .
0 H R' II PI' 9 I t
I I @NH* + RT-C -R"- N + - c -0- -8-
R ' R' H R' I BH4- 4- / ?H,O I 1
I \ 1 1 @ N - - c - H + @ - N = c 6 @N+-c -OH
H R" H R t l H R"
With formaldehyde, the resulting methylamino groups are then usually converted rapidly into dimethylamino groups by reaction with additional formaldehyde and reducing agent. Although sodium borohydride was used initially, and is still useful as the reducing agent in some cases, sodium cyanoborohydride and certain amine boranes are usually employed today due to their greater stability at neutral and lower pH (Means and Feeney 1968; Jentoft and Dearborn 1979; Cabacungan et af. 1982; Wong et al. 1984).
Because the dimethylamino groups produced by reductive methylation are small and retain the same positive charge as the amino groups they replace, they are, arguably, the most subtle modification of such groups, Reductive methylation thus appears to have few structural effects on proteins and any observed effects are more easily interpreted than would be the case with other modification procedures. Because its effects are so minor, reductive methylation is sometimes used to facilitate physical studies of protein structure. Reductive alkylations using carbonyl compounds larger and more complicated than formaldehyde, on the other hand, are more often employed to purposely alter the properties of proteins. The following are intended to illustrate some important applications of reductive a I ky lation.
Radioisotopically labeled dimethylamino derivatives of proteins, for example, are often used as models of the native proteins. 3H- and I4C-labeled derivatives of several proteins, usually prepared by reductive methylation with 'H-labeled sodium borohydride or ''C-labeled formaldehyde, respectively, are available from several commercial sources. Because it is a relatively simple procedure and is likely to have few effects on the properties of most proteins, radiolabeling by reductive

CHEMICAL MODIFICATIONS OF PROTEINS 405
methylation should also be considered as an alternative to more commonly used procedures for labeling proteins by radioiodination (Rice and Means I97 I ; Tack er al. 1980). Although the specific activities obtained by reductive methylation are usually lower, the radioactive half-lives are much, much, longer, which is almost always a significant advantage (Means and Feeney 1995). As cost is also usually an important consideration, it should be noted that both 'H-labeled sodium borohydride and i4C-labeled formaldehyde are relatively inexpensive as compared to other radiolabeling agents.
'H- and 'T-labelled dimethylamino groups have also been introduced into a number of proteins by reductive methylation via the use of appropriately enriched precursors (i.e., 'H-sodium borohydride and i3C-formaldehyde) and characterized by 'H- or I3C-NMR. Due to the relatively wide distribution of their chemical shifts, it has been possible to characterize individual ' Wabe led dimethylamino groups in several such proteins with respect to their chemical shifts, their pK, values and general environments (Jentoft and Dearborn 1979; Jentoft and Rayford 1989; Zhang and Vogel 1993). Due to their similarities, such information is thought to apply, more-or-less, to the unmodified amino groups of those proteins. Changes in chemical shift accompanying the interactions of ligands, like those that accompany changes in pH, can also be used in some cases to locate and characterize protein-ligand binding sites.
Rayment and coworkers (Rypniewski et al. 1993), have shown that reductive methylation can be used to promote the crystallization of proteins that may otherwise be difficult to crystallize and, in the relatively small number of cases examined so far, without a detectable effect on their three-dimensional structures. The high yields with which reductive methylations usually proceed and the absence of any other reaction products, are particularly important in such cases for maintaining the homogeneity needed for high resolution x-ray structure determinations. Reasons for the altered crystallization properties are still subject to speculation but may have something to do with the reduced rotational mobility of dimethylamino as compared to unsubstituted amino groups, their poorer solvation, andor their stronger electrostatic interactions with oppositely charged groups (Baxter and Byvoet 1975).
Many other types of alkylamino derivatives of proteins can be prepared similarly by reductive alkylation with one or more of the many aldehydes and ketones that are readily available. Relatively large, hydrophobic, alkyl substituents can be obtained, for example, using octanal, dodecylaldehyde, and various other large, hydrophobic, aldehydes. Such derivatives are known to adsorb relatively strongly to certain kinds of hydrophobic surfaces, to interact significantly with oil in water emulsions, and to have increased solubilities in organic solvents (Fretheim etal. 1979; Wu and Means 1981; Wirth et al. 1991). Other carbonyl compounds have been used to introduce fluorescent labels into proteins, to facilitate their radioiodination (Panuska and Parker 1987) and for a wide variety of other purposes
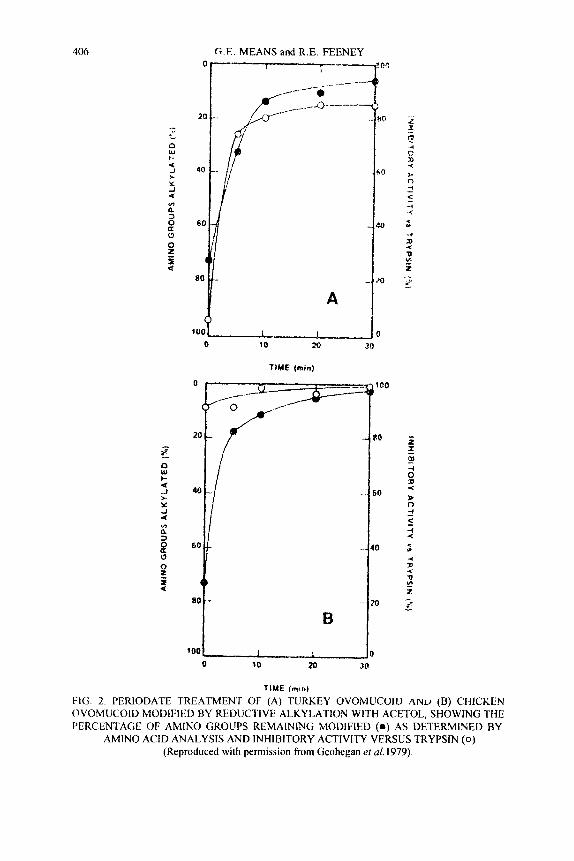
406 G E. MEANS and R.E. FEENEY
TIME fmin)
TIME (min)
FIG 2. PERIODATE TREATMENT OF (A) TURKEY OVOMUCOIU AND (B) CHICKEN OVOMUCOID MODIFIED BY REDUCTIVE ALKYLATION WITH ACETOL, SHOWING THE PERCENTAGE OF AMINO GROUPS REMAINING MODIFIED (0 ) AS DETERMINED BY
AMINO ACID ANALYSIS AND INHIBITORY ACTIVITY VERSUS TRYPSIN ( 0 )
(Reproduced with permission from Geohegan eta/. 1979).

CHEMICAL MODIFICATIONS OF PROTEINS 407
(King et al. 1977; Brown et ul. 1988; Lee et al. 1989). Reductive alkylation with a-hydroxyaldehydes, glyoxal, acetol, and others,
affords 2-hydroxyalkylamino moieties that may subsequently be dealkylated by mild treatment with sodium periodate (Geoghegan et al. 1979). Because sodium periodate reacts substantially faster with vicinal amino alcohols than with vicinal diols, the alkylation and its subsequent reversal are applicable even to glycoproteins. The trypsin inhibitory activity of turkey ovomucoid, which is a glycoprotein with an essential amino group, was lost upon reductive alkylation with acetol (i.e. 2-hydroxyacetone), for example, but was almost completely restored by its subsequent treatment with periodate (Fig. 2 ) and with only a 2% decrease in its carbohydrate content.
Glucose and many related sugars, which are either a-hydroxyaldehydes or ketones, can be coupled similarly to proteins by reductive alkylation and later removed by treatment with sodium periodate (Marsh et al. 1977; Wong et al. 1985). Coupling rates are usually quite slow, due to the prevalence of hemiacetal forms, and are related to concentrations of the free aldehyde forms. Pentoses, where there are usually higher percentages of the free aldehydes, usually react more rapidly than hexoses and monosaccharides, and di- tri- and higher oligosaccharides (Schwartz and Gray 1977; Wong et al. 1985).
PROTEIN MODIFICATIONS OCCURRING IN VIVO AND IN FOODSTUFF
Protein synthesis and protein degradation are highly controlled and coordinated in most cells. Some proteins are degraded soon after their synthesis whereas others persist for the life of a cell. Some of the structural characteristics that determine whether a protein will persist or undergo rapid degradation are known and the principal mechanisms by which they are degraded are also known (Hershko 1988). Genetic, nutritional, and environmental factors all appear to be involved with protein turnover. A wide variety of physical and chemical modifications, denaturation, partial proteolysis, reactions with cellular oxidants, and many other kinds of covalent modifications, are among the things that predispose cellular proteins to degradation (Stadtman 1990). Long-lived extracellular proteins appear to undergo most of the same reactions but, in many cases, without any comparable turnover. Carbonyl compounds, including glucose, and nitric oxide are two of the substances known to effect in vivo protein modifications, albeit of a very different sort.

408 G.E. MEANS and R.E. FEENEY
Carbonyl-amine Reactions
Glucose and other physiologically important sugars react readily with the amino groups of proteins according to Eq. 2 (Feeney and Whitaker 1982; Led1 and Schleicher 1990).
Aldose Aldimine Amadori 1 -Amino-1 deoxy Intermediate Ketose
The first step is reversible and dependent on the free aldehyde forms of the participating sugars (i.e. -0.001% in the case of glucose). The second step, known as an Amadori rearrangement, appears to depend on steady-state levels of Schiff base forms and is, practically, irreversible. With relatively long-lived proteins, like hemoglobin and serum albumin, their Amadori products (sometimes called fructoseamine derivatives) accumulate and reflect the protein's cumulative exposure to various sugars (Bum 1981). Because the levels of those products, below -5% of the total hemoglobin in a normal individual but approximately double that in an uncontrolled diabetic, are easy to determine and provide a relatively long-term measure of glucose exposure, they are commonly used to diagnose cases of diabetes and also as a means to monitor the blood-sugar concentrations of diabetics on various experimental and/or therapeutic regimens.
In still longer-lived proteins, like collagen and lens crystallins, the Amadori products appear to undergo still further reaction, sometimes involving oxidation, a second amino acid side chain and, perhaps, other cellular constituents, to give an unknown number of products collectively refered to as advanced glycosylation end-products (AGES). E-N-Carboxymethyllysine and pentosidine, a fluorescent, cross-linked, derivative of both lysine and arginine residues, are the most thoroughly characterized (Sell and Monnier 1989; Dyer et af. 1991). They have been found in collagen, especially that from older animals or patients, and in several other long-lived proteins. Both are also produced during the in v i m exposure of various proteins to glucose and other reducing sugars. Rates of formation vary according to the particular sugars and their concentrations.
Although glucose is the most abundant monosaccharide under physiological conditions, and is thought to be the principal contributor to nonenzymatic glycosylation, it exists to a greater extent than others as a mixture of unreactive hemiacetal forms. Studies have shown, for example, that ribose and other pentoses are far more effective precursors of pentosidine, although their involvement remains uncertain due to the low concentrations of pentoses under physiological conditions. The formation of pentosidine and other AGES from glucose, other

CHEMICAL MODIFICATIONS OF PROTEINS 409
hexoses, pentoses, or whatever, is thought to account for some of the observed stiffening, the decreased digestibility, and other age-related changes in collagen and other extracellular proteins. An accelerated formation of those products in the tissues of diabetics is also thought to be responsible for some of the chronic complications of that condition.
Reactions with Nitric Oxide
Until about ten years ago, the biological importance of nitric oxide was thought to be related entirely to its presence in ‘‘polluted’’ urban air. Since then, it has been shown to be an important physiological regulator, a new kind of “second messenger”; possibly a neurotransmitter, “the retrograde messenger”; an important component of the immune system; and a key intermediate in the pharmacological mechanisms of many vasoactive drugs (Ignarro 1989; Snyder 1992). Although a number of proteins are affected by nitric oxide, the manner by which they are affected and the mechanisms by which it exerts its physiological effects are not well understood.
Its short physiological life-time, and the brief duration of its effects, appear to be due to rapid reactions with superoxide ion, oxygen, and, perhaps, other physiological oxidants (Beckman 1996). The simultaneous production of nitric oxide and superoxide ion by activated macrophages is thought to result in a very rapid formation of peroxinitrite ion (Eq. 3),
N O + Oi- + ONOO- (3 1
which is a very strong oxidant, a potent cytotoxic agent, an inhibitor of the mitochondria1 ATPase, cytochrome P450, several other key enzymes, and one of the immune systems responses to foreign substances. Nitric oxide, superoxide ion and peroxinitrite are all cytotoxic and together may be considered the immune system’s chemical defense system. They may also be responsible, however, for some of the long-term changes associated with a number of pathological conditions including, for example, stroke, heart disease, and pulmonary edema. In dilute buffer solutions, peroxynitrite can either dismutate to oxygen and nitrite (Eq. 4), to nitrogen dioxide and a highly reactive hydroxyl radical (Eq. 5), or rearrange to nitrate ion (Eq. 6).
ONOO -+ O2 + NO; (4)
ONOO- + NO, + *OH ( 5 )

410 G.E. MEANS and R.E. FEENEY
Under physiological conditions it also reacts rapidly with thiols to give disulfides (Eq. 7) and, to a lesser extent, with the tyrosine residues of proteins to give 3- nitrotyrosine residues (van der Vliet et al. 1996) (Eq. 8).
ONOO- + 2 RSH -+ RSSR + NO; + H,O (7)
ONOO- + Tyr + 3-Nitro-Tyr + OH- (8)
3-Nitrotyrosine has been detected in human urine, a variety of other physiological fluids, and elevated levels have been associated with several human diseases (e.g., atherosclerosis, respiratory distress syndrome, sepsis, etc.) thought to involve increased NO synthesis and/or oxidative stress.
Nitric oxide, at moderate concentrations, reacts rapidly with oxygen to give nitrogen dioxide via nitrosyldioxyl radical and dinitrogen tetroxide intermediates (Eq. 9). At much lower concentrations, like those thought to exist under physiological conditions (i.e., low micromolar range), however, this reaction is very slow. In the presence of thiols, at concentrations similar to those found in most cells, the nitrosyldioxyl radical, dinitrogen tetroxide, andor other closely related species (e.g., N,03) appear to be efficiently intercepted to produce S- nitrosothiols (Goldstein and Czapski 1996) (Eq. 10).
N O + 0, -+ OONO + NO-+ ONOONO -+ 2NO; (9)
RSH + [ NO, 3 -+ RSNO + H,O (10)
Alternative mechanisms for the formation of S-nitrosothiols involving the formation of radical intermediates and their subsequent reaction with oxygen (Eq. 11) and other one electron acceptors (Eq. 12) may also be important under physiological conditions (Beckman 1996).
RSH + N O -+ [RSN -OH] + 0 2 -+ RSNO + 0;- -I- H’ (1 1)
Nitric oxide synthase, which has an essential iron-heme moiety and requires thiols for activity, may in fact account for the formation of S-nitrosothiols by the latter mechanism along with its better known formation of nitric oxide.
An S-nitroso derivative of serum albumin has been detected in the blood stream at approximately 7 pM and appears to account for a majority of the vascular “NO” (Keaney et al. 1993). Under those conditions, it is thought to be relatively

CHEMICAL MODIFICATIONS OF PROTEINS 41 1
stable, more so than most simple S-nitrosothiols, and to serve as either a vascular reservoir or carrier of NO. Glutathione and other low molecular weight thiols have been shown to promote vasodilation by S-nitrososerum albumin due, probably, to an accelerated rate of transnitrosation (Eq. 13).
Serum Albumin-SNO + GSH -i.r Serum Albumin-SH + GSNO (13)
S-Nitrososerum albumin may also be formed by transnitrosation at high levels of low molecular weight S-nitrosothiols (Zhang and Means 1996). Cysteine p93 of hemoglobin appears to undergo similar nitrosation-den itrosation with glutathione and other low molecular weight thiols in red blood cells. Conformation changes accompanying its oxygenation and deoxygenation appear to be linked to its uptake and release of “NO’, and vice versa, in a manner so as to effect vascular dilation and increased oxygen delivery to hypoxic tissues (Stamler et al. 1997).
S-Nitrosoglutathione and other low molecular weight S-nitrosothiols have been detected in the blood stream and in neural tissue, and have physiological effects very similar to those of nitric oxide. Those physiological effects: smooth muscle relaxation, inhibition of platelet aggregation, and others, are usually attributed to the nitric oxide released upon their homolytic decomposition (Eq. 14).
RSNO (cuz+icu+) R S + N O + RSNO -+ RSSR + N O (14) 4
Homolyses can be accelerated by some transition metal cations, which, however, are not readily available under physiological conditions, but are otherwise usually quite slow (Butler and Williams 1993). The effects of S-nitrosothiols on blood pressure, smooth muscle relaxation, guanylate cyclase activity, and so on, however, are immediate and rather short in duration. Transnitrosations between low molecular weight thiols, S-nitrosothiols, protein thiol and S-nitrosothiol groups (Eq. 15), on the other hand, are very fast under physiological conditions and might easily account for the rapidity of those physiological effects (Park et al. 1993).
Protein-SH + GSNO + Protein-SNO + GSH (15)
Like protein phosphorylation and other regulatory posttranslational modifications, S-nitrosation is completely reversible and might be directed primarily to particular thiol groups due to their specific environments and bring about allosteric changes that affect the activities of enzymes and other biologically important proteins.
The relaxation of smooth muscles by nitric oxide is thought to be the result of its activation of guanylate cyclase and the resulting increases in cytoplasmic cyclic-GMP. A soluble, cytoplasmic form of that enzyme with two closely related but different subunits, a large number of essential sulfhydryf groups, and an

412 G.E. MEANS and R.E. FEENEY
associated Fez'-heme moiety is thought to be the principal receptor for nitric oxide in those cells. Increases of approximately 400-fold in activity are thought to result from conformational changes accompanying nitric oxides binding to the Fe2'-heme moiety (Stone and Marletta 1995). Subsequent dissociation of nitric oxide and its ensuing reactions with oxygen, superoxide ion, or other cellular oxidants (Eq. 3, 9, and 16) are usually thought to account for a subsequent relatively rapid deactiva- tion of that enzyme.
N O -+ Fe3+ + H 2 0 -+ NO; + Fe2+ + 2 H' (16)
The activities of creatine kinase, aconitase, protein kinase C, and other enzymes that are also affected by nitric oxide and so-called NO-donor compounds (e.g. sodium nitroprusside, S-nitroso-N-acetylpenicillamine, etc.) but have no iron- heme moieties cannot be accounted for in the same way, nor can the rapid activation of guanylate cyclase by various S-nitrosothiols be attributed to their slow homolytic release of nitric oxide (Eq. 14). All of the affected enzymes do, however, have multiple thiol groups and, for most or possibly all of them, the thiol groups are required for their biological activity. The nitrosation of those thiol groups might therefore account for the effects of nitric oxide and NO-donors on those biological activities.
In proteins with multiple thiol groups, S-nitrosation can also effect the formation of disulfide bonds between neighboring thiol groups (Eq. 17). For low molecular weight dithiol compounds, like dithiothreitol and 6,8-thioctic acid,
disulfide formation proceeds immediately after S-nitrosation with rates that depend primarily on the proximity of the thiol and S-nitrosthiol groups (Le er al. 1997). A similar transient S-nitrosation and subsequent formation of disulfide bonds between neighboring thiol groups might account for some transient or temporary changes in proteins upon their exposure to nitric oxide and NO-donors like, for example, the activation and subsequent rapid deactivation of guanylate cyclase in the presence of such compounds.
CHEMICAL MODIFICATIONS OF FOOD PROTEINS
Chemical modifications of food proteins are very limited due to concerns

CHEMICAL MODIFICATIONS OF PROTEINS 413
about safety and consumer acceptance. Overt modification procedures are not really possible at this time due to the publics fear of “chemicals” or anything that might be considered “unnatural”. A wide variety of inadvertent protein modifica- tions do, however, occur during the normal preparation, processing and storage of foods and, almost always, have important effects on the final products. Cooking, or heating for other purposes, whether by the consumer or as part of commercial processing, for example, brings about a wide variety of modifications that are, for the most part, acceptable to the general public.
Hydrolysis of peptide bonds, especially those of aspartic acid residues, deamidation of asparagine, p-elimination of cystine (Eq. 18), of glycosylated and
0 0 = C H ~ H ~ - S - S - C H ~ < : * C H 2 = C Z
3 CH-CH~-S-S-C H ~ X H z
,”, (18) z C x H 2 <r 0 Z C H X H 2 - P < $ ‘,CH4H2-S-S
H20
S H 2 0 S
phosphorylated serine and threonine residues, oxidation of histidine, methionine, cysteine, tryptophan and tyrosine residues are all prominent at high temperatures (Daniel et al. 1996). The heating time and temperature, the pH and other conditions affect the extent of each reaction. Disulfide interchange reactions, wherein thiol groups react with disulfide bonds to give new thiols and new disulfides (Eq. 19) or wherein hydrosulfide ions (HS), produced by the p- elimination of cystine residues, react with disulfide bonds to produce new thiols and new disulfide bonds (Eq. 19), are also important at high temperatures.
,S-S’ + RS- -+ ,S-SR + -S’ R = H or Cys (19)
In the presence of glucose and other reducing sugars, the Maillard reaction, or so-called nonenzymatic browning, proceeds via Schiff base formation with the lysine residues of proteins and low molecular weight amines that may be present and a variety of condensation-elimination intermediates similar to those involved in in vivo nonenzymatic glycosylation, pentosidine formation, etc. The final products, pyrroles, hrans, etc., however, appear to be quite different, probably much more varied, and are thought to be responsible for some of the characteristic colors, flavors, and aromas of cooked foods (Led1 and Schleicher 1990).

414 G.E. MEANS and R.E. FEENEY
The alkaline conditions required to solubilize many plant proteins also bring about some inadvertant p-elimination of cystine and of 0-glycosylated serine and threonine residues (Eq. 20).
>CH-CH,-S-S-CH,-CH<OH--+ >C=CH, + -S-S-CH,-CH< (20) dehydroalanine
Under those conditions, the resulting “dehydroalanine” residues, which are highly reactive a$-unsaturated carbonyl compounds, undergo nucleophilic addition, primarily with the lysine and cysteine residues of other peptide chains (Eq. 21,22), to give cross-linked products containing the newly formed, “unnatural”, or, at least, rare amino acids, lysinoalanine and cystathionine, respectively.
>C=CH, + NH,-(CH,),CH< -t >CH-CH,-NH-(CHJ,CH< ( 2 I ) lysinoalanine
Although some questions have been raised concerning their safety, formation of such cross-links imparts a certain amount of rigidity to those proteins and is very important for the proper texture of some food protein products (Whitaker and Feeney 1983).
Recent advances in our understanding of nitric oxide’s role as a physiological regulator have also lead to some understanding of nitrite’s effects on “cured” meats, and other food products, including some tentative ideas concerning the molecular basis of its bacteriostatic effects. A denatured nitroso adduct of myoglobin, and lesser amounts of other iron’’-heme proteins, are thought to account for the typical pink color of most cured meats. They are thought to be formed from the heat denatured proteins and nitrite ion under the influence of various endogenous reductants, NADH, reduced flavins, cysteine, mitochondria1 respiratory enzymes, etc.; added reductants, ascorbate, cysteine, hydroquinone, etc.; or due to limited bacterial growth (Cornforth 1996).
The bacteriostatic effects of nitrite are thought to be due to its slow conversion into nitric oxide andor other closely related species. Nitric oxide is known to be an inhibitor of several important enzymes but there is still no detailed understanding of how it may inhibit microbial growth. Peroxynitrite, which is thought to be the principle cytotoxic agent of the immune system (Eq. 3), does not appear to be involved. None of the reductants usually required to produce the characteristic color of cured meats can, by itself, reduce nitrite ion to nitric oxide,

CHEMICAL MODIFICATIONS OF PROTEINS 415
but, nitrite ion can, by itself, react directly with some iron"-heme proteins to give corresponding nitric oxide adducts (Eq. 23) .
2 Heme-Fe2' + NO; + 2 H' - Heme-Fe2'-NO + Heme-Fe3+ + H,O (23)
The iron"'-heme proteins that are also formed can then be reduced directly or indirectly by one of those reductants. Its bacteriostatic effects have also been attributed to the low concentrations of S-nitrosothiols, thought to be formed from endogenous thiols and low concentrations of nitrous acid in equilibrium with the added nitrite ion. Low concentrations of S-nitrosocysteine and other S-nitroso- thiols can be detected in nitrite-treated meat products and are known to be strongly cytostatic (Byler et ul. 1983). The mechanism of their inhibitory effects are, again, not really known but they are thought to affect primarily membrane or cellular thiol groups or, perhaps, some key iron-containing proteins (Morris et al. 1984).
Acylations of soy protein and many other common plant proteins by succinic anhydride and some other dicarboxylic acid anhydrides are known to improve many of their functional properties (Feeney and Whitaker 1985; Kinsella 1978). Succinylation of soy protein, for example, enhanced its foam capacity, its emulsifying activity, and the emulsion stability. Its isoelectric point was reduced and its tolerance to Ca2+ ion was increased but its solubility was decreased below its new isoelectric point (Kinsella 1978).
Covalent attachment of amino acids to the side chains of proteins has been studied as a means to increase the nutritional value of some proteins and is of particular interest for those that are naturally deficient in one or more essential amino acids. Increasing the methionine content of casein by this means, for example, gave a product that produced faster growth rates in rats (Puigserver et uf. 1979).
Some Current Developing Areas
Bioconjugation. In the first issue of the new journal, Bioconjugate Chemistry, Claude Meares described bioconjugation as "the joining of two molecular functions by chemical or biological means" and listed the following topics: Conjugation of.. . antibodies (and their fragments) nucleic acids and their analogs (a-anomers, phosphonates,. . .), liposomal components, other biologically active molecules (receptor-binding proteins, polysaccharides, hormones, peptides, ...) with each other or with any molecular groups that add useful properties ... drugs, radionuclides, toxins, fluorophores, photoprobes, inhibitors, enzymes, haptens, ligands, etc. (Meares 1990).

416 G.E. MEANS and R.E. FEENEY
The chemical conjugation of proteins to other proteins and to a wide range of other substances is currently an active and expanding area of research with many different applications. One of the more interesting applications has to do with the attachment of chelating agents to antibodies. By the use of such conjugates, radioactive metal ions that bind to the introduced chelating groups can be delivered to a wide variety of predetermined physiological targets as determined by the specificity of the particular antibodies. After delivery to a group of malignant cells, the radioactivity can then be used, for example, either to detect the presence, the exact anatomical location and distribution, or to specifically kill those cells (Li et al. 1994).
Conjugation of polyethylene glycol to proteins is now commonly used to provide them with biocompatible protective coatings. Those polyethylene glycol conjugates usually elicit a diminished immune response, a reduced rate of kidney clearance, and, therefore, have increased serum life-times. Several different types of polyethylene glycol reagents have been developed that are suitable for selective coupling to either the amino, sulfhydryl, or carboxyl groups of proteins, which also vary in their chain lengths and average molecular weights, whether they are branched or straight chained, and have minimal effects on the overall structure and other properties of most proteins. Full functional activity has been reported with moderate conjugation with some proteins. For example, conjugation of horse cytochrome C with six residues gave a product with full functional activity (Mabrouk et a). 1994).
Conjugation of other biocompatible materials to proteins can also be used sometimes to increase their physiological life-times and to alter their tissue distributions. Conjugating succinylated gelatin to superoxide dismutase, an enzyme deficiency associated with some cases of amyotrophic lateral sclerosis (ALS), for example, increases its circulating half-life from -4.5 min to -30 min in mice (Kojima et al. 1993). Conjugation of a maleic acid-styrene copolymer to the anticancer peptide neocarzinostatin also increases its circulatory stability, causes its selective accumulation in certain types of tumor tissues, and, thereby, increases its cancer cell cytotoxicity (Maeda and Konno 1997).
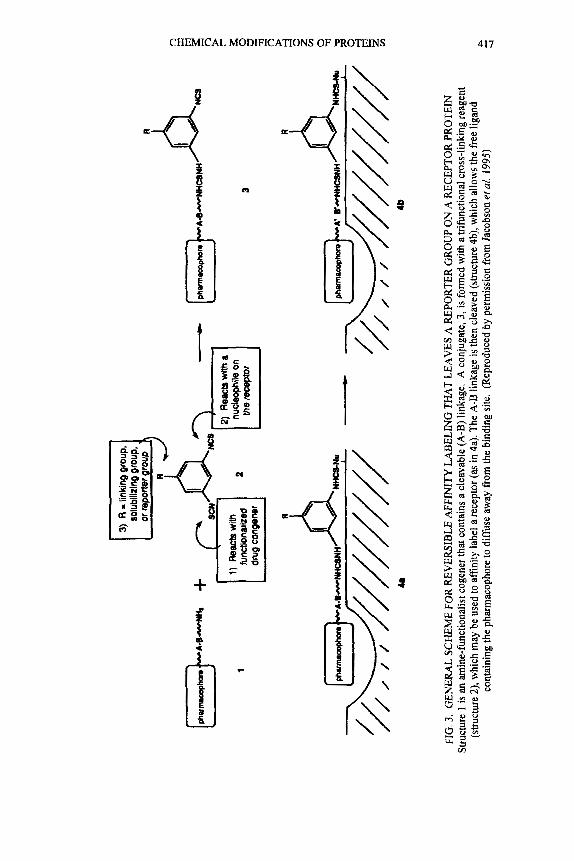
Another active area is the reversible affinity labelling of receptors (Jacobsen el al. 1995). In a study with reversible affinity labelling of A,- Adenosine receptor in bovine bovine membranes, cleavable disulfides or ester linkages of a trifunctional phenyl ring and a pharmacophore were used (Fig. 3). Here the receptor is labelled with the phenyl ring, while the affinity part is removed.
Conjugation of polystyrene-co-maleic acid half-n-butylate (SNA) with an anticancer agent, the peptide Neocarzinostatin, (NCS), formed the conjugate called SMANCS. The NCS portion of the conjugate SMANCS inhibits cancer cell cytotoxicity at pM range, thereby belonging to the most potent class of cytotoxic agents (Maeda and Konno 1997).
1
soiu
biliz
lng
grou
p,
A
A
R
3
A
4b
4a
FIG
. 3.
GEN
ERA
L SC
HEM
E FO
R RE
VER
SIBL
E A
FFIN
ITY
LA
BELI
NG
TH
AT
LEA
VES
A R
EPO
RTER
GRO
UP O
N A
REC
EPTO
R PR
OTE
IN
Stru
ctur
e 1 is
an
min
e-fu
nctio
nalis
t cog
ener
that
con
tain
s a cl
eava
ble
(A-B
) lin
kage
. A c
onju
gate
, 3, i
s for
med
with
a tr
ifunc
tiona
l cro
ss-li
nkin
g rea
gent
(s
truct
ure 2
), w
hich
may
be
used
to a
ffini
ty la
bel a
rece
ptor
(as i
n 4a
). Th
e A-B
link
age
is th
en c
leav
ed (s
truct
ure 4
b), w
hich
allo
ws t
he fr
ee li
gand
co
ntai
ning
the p
harm
acop
hore
to d
iffus
e aw
ay fr
om th
e bi
ndin
g sit
e. (
Repr
oduc
ed by
per
miss
ion
from
Jaco
bson
er a
l. 19
95)
c
P 4

418 G.E. MEANS and R.E. FEENEY
Chemical Derivation for Analysis by Mass Spectrometry
The union of chemical derivation of proteins with mass spectrometry is providing important novel ways for characterization. Several approaches that have been developed use derivatives directly on the mass spectrometry probe. In some of these studies, characterization is done with compounds that seem to tether (capture) individual molecules from mixtures and release them as desired (eg., laser induced) in the mass spectrophotometric apparatus. With the proper derivatives, spacial calculations of the proteins in sites can be determined. A variety of techniques is possible.
One procedure (Ching et al. 1996) uses polymers as surface based tethers with photolytic triggers. These enable laser-induced release/desorption of covalently bound molecules upon photolytic cleavage. A defined portion of the tether (“tail”) remains attached to the biomolecule as a probe. In one such experiment (Ching et al. 1996), an 1 8-residue peptide (N-terminus of human p- casein) was attached to such a tether probe, irradiated with coherent UV light, and released with two reporter tails. These tails had a mass predicted by tether formation at two primary amino groups and subsequent photolytic cleavage at the intended site (Fig. 4).
A related procedure used an antibody on the mass spectrometry probe to remove selectively targeted substances. In one study, affinity mass spectrometry was done directly on frozen liver sections and purified bile duct epithelial cells from patients with primary biliary cirrhosis (Yip el al. 1996). Signals associated with a specific antigen were extensively increased by an enzyme (phosphatase) conjugated to a second antibody directed at the initial specific absorbing antibody (Fig. 5).
FUTURE APPLICATIONS
Some Possible Extensions
As with all science, there should be many developments of chemical modification in the future. Progress in the modifications in organic solvents should occur. Special derivatives for industrial uses (e.g., enzymes) should receive attention. More specific methods for determinations are desirable. Chemical modifications of genetically engineered mutants can be useful (e.g., for introduced suflhydryls). Special procedures might be needed for chemically synthesized peptides or proteins with unnatural side chains or nonpeptide-bond linkages.

CHEMICAL MODIFICATIONS OF PROTEINS 419
FIG. 4. MODULAR DESIGN OF BIOMOLECULAR TETHER-PROBE DEVICES Each device is defined by three components attached to a specified surface architecture. A photolytic component (1) is placed at different relative positions (indicated by dotted arrows) within a variable length spacer arm (2) terminating wtih a bioconjugation component (3) for covalent attachment through specific sites on the target biomolecule surface. This device is designed so that laser irradation induces a photolytic event that triggers uncoupling of the tethered biomolecule in a manner that leaves the dissociated biopolymer with one or more reported tails. (Reproduced by permission from
Ching efal . 1996).
Food Usages
As compared to modifications for pharmaceutical uses, modified food proteins are likely to be consumed in much larger quantities and without direct medical advice. Because of the possible long-term deleterious effects of consumption of chemically modified food proteins, their use is not generally accepted at this time. Hopefully, future uses will occur with proper research. With at least one modification, the reductive dimethylation of amino groups of lysines, derivatized lysines were extensively utilized in animal trials (Lee et al. 1978).
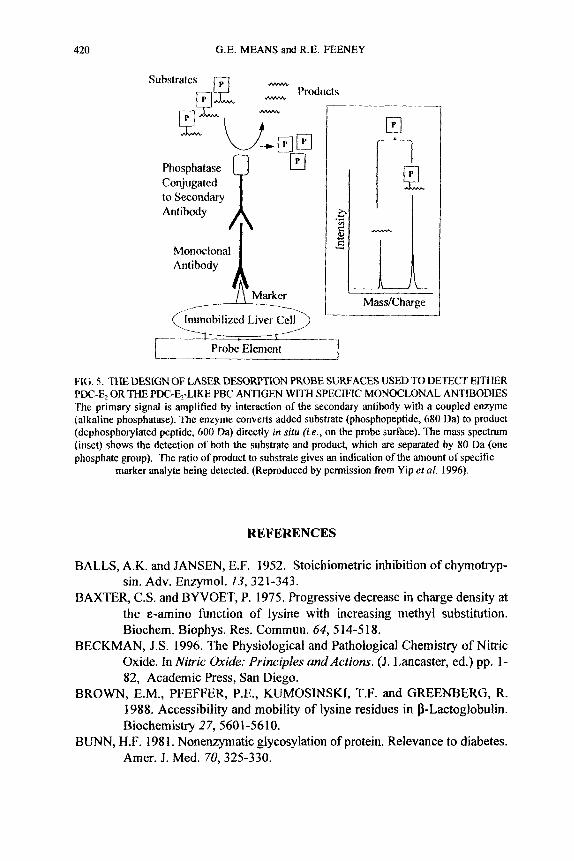
420 G.E. MEANS and R.E. FEENEY
Substrates Products
n
El P Phosphatase Conjugated tosecondary I htibody A
Monoclonal I Antibody
,
FIG. 5. THE DESIGN OF LASER DESORF'TXON PROBE SURFACES USED TO DETECT EITHER
The primary signal is amplified by interaction of the secondary antibody with a coupled enzyme (alkaline phosphatase). The enzyme converts added substrate (phosphopeptide, 680 Da) to product (dephosphorylated peptide, 600 Da) directly in situ f ie . , on the probe surface). The mass spectrum (inset) shows the detection of both the substrate and product, which are separated by 80 Da (one phosphate group). The ratio of product to substrate gives an indication of the amount of specific
marker analyte being detected. (Reproduced by permission from Yip et al. 1996).
PDC-E, OR THE PDC-El-LIKE PBC ANTIGEN WITH SPECIFIC MONOCLONAL ANTIBODIES
REFERENCES
BALLS, A.K. and JANSEN, E.F. 1952. Stoichiometric inhibition of chymotryp- sin. Adv. Enzymol. 13, 321-343.
RAXTER, C.S. and BYVOET, P. 1975. Progressive decrease in charge density at the &-amino function of lysine with increasing methyl substitution. Biochem. Biophys. Res. Commun. 64,5 14-5 18.
BECKMAN, J.S. 1996. The Physiological and Pathological Chemistry of Nitric Oxide. In Nitric Oxide: Principles and Actions. (J. Lancaster, ed.) pp. 1 - 82, Academic Press, San Diego.
BROWN, E.M., PFEFFER, P.E., KUMOSMSKI, T.F. and GREENBERG, R. 1988. Accessibility and mobility of lysine residues in P-Lactoglobulin. Biochemistry 27,5601-5610.
BUNN, H.F. 198 1. Nonenzymatic glycosylation of protein. Relevance to diabetes. Amer. J. Med. 70, 325-330.

CHEMICAL MODIFICATIONS OF PROTEINS 42 1
BUTLER, A.R. and WILLIAMS, D.L.H. 1993. The physiological role of nitric oxide. Chem. SOC. Reviews 22,233-241.
BYLER, D.M., GOSSER, D.K. and SUSI, H. 1983. Spectroscopic estimation of the extent of S-nitrosothiol formation by nitrite action on sulfhydryl groups. J. Agric. Food Chem. 31,523-527.
CABACUNGAN, J .C., AHMED, A.I. and FEENEY, R.E. 1982. Amhe boranes as alternative reducing agents for reductive ablation of proteins. Analyt. Biochem. 124,272-278.
CARLSSON, J., DREVIN, H. and AXEN, R. 1978. Protein thiolation and reversible protein-protein conjugation. N-Succinimidyl3-(2-pyridyldi- thio) propionate, a new heterobihctional reagent. Biochem. J. J 73, 723.
CHING, J., VOIVODOV, K.I. and HUTCHENS, T.W. 1996. Polymers as surface-based tethers with photolytic triggers enabling laser-induced release-desorption of covalently bound molecules. Bioconjugate Chemistry 7(5), 525-528.
CHONG, P.C.S. and HODGES, R.S. 1981. J. Biol. Chem. 256,5064-5070. COLEMAN, R.F. 1989. Protein Function - A Practical Approach. (T.E. Creigh-
ton, ed.) pp. 77-99, IRL Press, Oxford. CORNFORTH, D. 1996. Role of nitric oxide in treatment of foods. In Nitric
Oxide: Principres and Actions. ( J . Lancaster, ed.) pp. 259-287, Academic Press.
DANIEL, R.M., DINES, M. and PETACH, H.H. 1996. The denaturation and degradation of stable enzymes at high temperatures. Biochem. J. 317, I - ll.
DAVIES, G.E. and STARK, G.R. 1970. Use of dimethyl suberimidate, a cross- linking reagent, in studying the subunit structures of oligomeric proteins. Roc. Natl. Acad. Sci. USA. 66,651-656.1.
DYER, D.G., BLACKLEDGE, J.A., THORPE, S.R. and BAYNES, J.W. 1991. Formation of pentosidine during nonenzymatic browning of proteins by glucose. J. Biol. Chem. 266, 11654-1 1660.
EDMAN, P. and BEGG, G. 1967. A protein sequenator. Eur. J. Biochem. 1,80- 91.
FEENEY, R.E. and WHITAKER, J.R. 1982. The Maillard Reaction and its prevention. In Food Protein Deterioration: Mechanisms and Functional- ity, (J.P. Cherry, ed.) Am. Chem. SOC. Symposium Series 206,203-229.
FEENEY, R.E. and WHITAKER, J.R. 1985. Seed storage proteins. In New Protein Foods Vol. 5 (A.M. Altschul and H.L. Wilcke, eds.) pp. 181 -2 19, Academic Press, New York.
FRAENKEL-CONRAT, H., BEAN, R.S. and LINEWEAVER, H. 1949. Essential groups for the interaction of ovomucoid (egg white trypsin inhibitor) and trypsin, and for tryptic activity. J. Biol. Chem. 177,385-403.

422 G.E. MEANS and R.E. FEENEY
FRAENKEL-CONRAT, H. and FEENEY, R.E. 1950. The metal-binding activity of conalbumin. Arch. Biochem. 29, 101-1 13.
FRAENKEL-CONRAT, H. and OLCOTT, H.S. 1948. The reaction of formalde- hyde with proteins. V. Cross-linking between amino and primary amide or guanidyl groups. J . Am. Chem. SOC. 70,2673-2684.
FRETHEIM, K., IWAI, S. and FEENEY, R.E. 1979. Extensive modification of protein amino groups by reductive addition of different sized substituents. Int. J. Peptide Protein Res. 14,45 1-456.
GEOGHEGAN, K.F., YBARRA, D.M. and FEENEY, R.E. 1979. Reversible reductive alkylation of amino groups in proteins. Biochemistry 18, 5392- 5399.
GLAZER, A.N., DELANGE, R.J. and SIGMAN, D.S. 1975. Chemical modifica- tion of proteins. Laboratory techniques in biochemistry and molecular biology (T.S. Work and E. Work, eds.) American Elsevier Publishing Co., New York.
GOLDSTEIN, S. and CZAPSKI, G. 1996. Mechanism of nitrosation of thiols and amines by oxygenated NO solutions - The nature of the nitrosating intermediates. J. Amer. Chem. SOC. 118, 3419-3425.
HERMANSON, G.T., MALLIA, A.K. and SMITH, P.K. 1992. Immobilized Aflnity Ligand Techniques. Academic Press, San Diego.
HERSHKO, A. 1988. Ubiquitin-mediated protein degradation. J. Biol. Chem. 263,
IGNARRO, L.J. 1989. Endothelium-derived nitric oxide: actions and properties.
JACOBSON, K.A., FISCHER, B. and XIAO-DUO, J. 1995. “Cleavable trifunctional” approach to receptor affinity labeling: chemical regenera- tion of binding to A- I-adenosine receptors. Bioconjugate Chem. 6,255- 263.
JAKOBY, W.B. and WILCHEK, M. (eds.) 1977. Methods Enzymol. Vol. 46, Academic Press, New York.
JENTOFT, N. and DEARBORN, D.G. 1979. Labeling of proteins by reductive methylation using sodium cyanoborohydride. J. Biol. Chem. 254, 4359- 4365.
JENTOFT, J.E. and RAYFORD, R. 1989. Comparison of the fragment from a human IgGl and its CH2, pFc’, and tFc’ subfragments. A study using reductive methylation and I3C NMR. Biochemistry 28, 3250-3257.
KEANEY, J.F., SIMON, D.I., STAMLER, J.S., JARAKI, O., SCHARFSTEIN, J., VITA, J.A. and LOSCALZO, J. 1993. NO forms an adduct with serum albumin that has endothelium-derived relaxing factor-like properties. J. Clin. Invest. 1582- 1589.
15237- 15240.
FASEB J. 3, 3 1-36.

CHEMICAL MODIFICATIONS OF PROTEINS 423
KING, T.P., KOCHOUMIAN, L. and LICHTENSTEIN, L.M. 1977. Preparation and immunochemical properties of methoxypolyethylene gl ycol-coupled and N- carboxymethylated derivatives of ragweed pollen allergen, antigen E. Arch. Biochem. Biophys. 178,442-450.
KINSELLA, J.E. 1978. Texturized proteins: fabrication, flavoring, and nutrition. CRC Crit. Rev. Food Sci. Nutr. 10, 147-207.
KOJIMA, Y., HARUTA, A., IMAI, T., OTAGIRI, M. and MAEDA, H. 1993. Conjugation of Cu,Zn-superoxide dismutase with succinylated gelatin: pharmacological activity and cell-lubricating function. Bioconjugate Chem. 4,490-498.
LAMBERT, J.M., SENTER, P.D., YOUNG, A.Y.Y., BLATTLER, W.A. and GOLDMACHER, V.S. 1985. Purified immunotoxins that are reactive with human lymphoid cells. J. Biol. Chem. 260, 12035-12041.
LASKIN, A.I. 1985. Enzymes and Immobilized Cells in Biotechnology. Benjamin/Cummings Inc., Menlo Park.
LE, M., ZHANG, H. and MEANS, G.E. 1997. The decomposition of S-nitrosated dithols. Bioorg. Med. Chem. Lett. 7, 1393-1398.
LEDL, F. and SCHLEICHER, E. 1990. New aspects of the Maillard Reaction in foods and in the human body. Angew. Chem. Int. Ed. Engl. 29, 565-594.
LEE, H.S., SEN, L.C., CLIFFORD, A.J., WHITAKER, J.R. and FEENEY, R.E. 1978. Effect of reductive alkylation of epsilon-amino groups of lysyl residues of casein on its nutritive value in rats.
LEE, R.T., WONG, T.-C., LEE, R., YUE, L. and LEE, Y.C. 1989. Efficient coupling of glycopeptides to proteins with a heterobifunctional reagent. Biochemistry 28, 1856-1861.
LI, M., MEARES, C.F., ZHONG, G., MIERS, L., XIONG, C. and DENARDO, S. 1994. Labeling monoclonal antibodies with 'Oyttrium- and ' I ' indium- DOTA chelates: A simple and efficient method. Bioconjugate Chem. 5, 101.
LUNDBLAD, R.L. and NOYES, C.M. 1984. Chemical Reagents for Protein Mod$cation. Vols. 1 and 2, CRC Press, Boca Raton, FL.
MABROUK, P.A. 1994. Effect of pegylation on the structure and function of horse cytochrome C. Bioconjugate Chem. 5,236-241.
MAEDA, H. and KONNO, T. 1997. Metamorphosis of neocarzinostatin to SMANCS: chemistry, biology, pharmacology, and clinical effect of the first prototype anticancer polymer therapeutic. In Neocarzinostatin: The Past, Present, and Future of an Anticancer Drug (H. Maeda, K. Ed0 and N. Ishida, eds.) pp, 227-267, Springer-Verlag, Tokyo.
MARSH, J.W. 1988. Antibody-mediated routing of diphtheria toxin in murine cells results in a highly efficacious immunotoxin. J. Biol. Chem. 263, 15993- 15999.

424 G.E. MEANS and R.E. FEENEY
MARSH, J.W., DENIS, J. and WRISTON, J.C. 1977. Glycosylation of Eschericia coli L- Asparaginase. J. Biol. Chem. 252,7678-7684.
MEANS, G.E. and FEENEY, R.E. 1968. Reductive alkylation of amino groups in proteins. Biochemistry 7,2 192-2200.
MEANS, G.E. and FEENEY, R.E. 1971. Chemical Mod9cation of Proteins. Holden-Day, San Francisco.
MEANS, G.E. and FEENEY, R.E. 1990. Chemical modifications of proteins: history and applications. Bioconjugate Chem. I , 2-12.
MEANS, G.E. and FEENEY, R.E. 1995. Reductive alkylation of proteins. Analyt. Biochem. 224,2-16.
MEARES, C.L. 1990. Editorial: introduction to bioconjugate chemistry. Bioconjugate Chem. I, 1.
MOORE, S. and STEIN, W.H. 1963. Chromatographic determination of amino acids by the use of automatic recording equipment. Methods Enzymol.
MORRIS, S.L., WALSH, R.C. and HANSEN, J.N. 1984. Identification and characterization of some bacterial membrane sulfhydryl groups which are targets of bacteriostatic and antibiotic action. J. Biol. Chem. 259, 13590- 13594.
MOSBACH, K. 1987. Immobilized enzymes and cells. Part C, Methods Enzymol. 136, Academic Press, New York.
OLCOTT, H.S. and FRAENKEL-CONRAT, H. 1947. Specific group reagents for proteins. Chem. Rev. 41, 151-197.
PANUSKA, J.R. and PARKER, C.W. 1987. Radioiodination of proteins by reductive alkylation. Analyt. Biochem. 160, 192-201.
PARK, J.W., BJLLMAN, G.E. and MEANS, G.E. 1993. Transnitrosation as a predominant mechanism in the hypotensive effect of S-nitrosoglutathione. Biochem. Mol. Biol. Interntl. 30, 885-891.
PFLEIDERER, G. 1985. Chemical modifications of proteins. In Modern Methods in Protein Chemistry (H. Tschesche, ed.) Walter de Gryter, Berlin.
PORATH, J. and KRISTIANSEN, T. 1975. Biospecijic Afinity Chromatography and Refuted Methods. The Proteins. 3rd Ed., (H. Neurath and R.L. Hill, eds.) pp. 95-178, Academic Press, New York.
PRATT, R.F. 1992. On the definition and classification of mechanism-based enzyme inhibitors. Bioorg. Med. Chem. Lett. 2, 1323-1326.
PUIGSERVER, A.J., SEN, L.C., CLIFFORD, A.J., FEENEY, R.E. and WHITA- KER, J.R. 1979. Covalent attachment of amino acids to casein. 2. Bioavailability of methionine and N-acetylmethionine covalently linked to casein. J. Agric. Food Chem. 27(6), 1286-1293.
RICE, R.H. and MEANS, G.E. 1971. Radioactive labeling of proteins in vitro. J. Biol. Chem. 246, 83 1-832.
6,819-831.

CHEMICAL MODIFICATIONS OF PROTEINS 425
RYPNIEWSKI, W.R., HOLDEN, H. and RAYMENT, I. 1993. Structural conse- quences of reductive methylation of lysine residues in hen egg white Iysozyme: an x-ray analysis at 1.8-A resolution. Biochemistry 32, 9851- 9858.
SCHWARTZ, B.A. and GRAY, G.R. 1977. Proteins containing reductively aminated disaccharides. Arch. Biochem. Biophys. 181,542-549.
SELL, D.R. and MONNIER, V.M. 1989. Structure elucidation of a senescence cross-link from human extracellular matrix. J. Biol. Chem. 264,21597- 21602.
SHAPIRO, A.L., VENUELA, E. and MAIZEL, J.V. 1967. Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacryl- amide gel. Biochem. Biophys. Res. Commun. 28,815-820.
SNYDER, S.H. 1992. Nitric oxide: First in a new class of neurotransmitters? Science 257,494-496.
STADTMAN, E.R. 1990. Covalent modification reactions are marking steps in protein turnover. Biochemistry 29, 6323-633 I .
STAMLER, J.S., JIA, L., EU, J.P., MCMAHAN, T.J., DEMCHENKO, I.T., BONAVENTURA, J., GERNERT, K. and PIANTADOSI, C.A. 1997. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276,2034-2037.
STONE, J.R. and MARLEITA, M.A. 1995. Heme stoichiometry of heterodimeric soluble guanylate cyclase. Biochemistry 34,4668-4674.
TACK, B.F., DEAN, J., EILAT, D., LORENZ, P.E. and SCHECHTER, A.N. 1980. Tritium labeling of proteins to high specific radioactivity by reductive methylation. J. Biol. Chem. 255, 8842-8847.
VAN DER VLIET, A., EISERICH, J.P., KAUR, H., CROSS, C.E. and HALLI- WELL, B. 1996. Nitrotyrosine as biomarker for reactive nitrogen species. Methods Enzymol. 269, 175-1 84.
WALSH, C.T. 1984. Suicide substrates, Mechanism-based enzyme inactivators: recent developments. Ann. Rev. Biochem. 53,493-535.
WHITAKER, J.R. and FEENEY, R.E. 1983. Chemical and physical modification of proteins by the hydroxide ion. CRC Crit Rev. Food Sci. Nutr. 19, 173.
WlDDER, K.J. and GREEN, R. 1985. Drug and enzyme targeting, part A, Methods Enzymol. 112.
WIRTH, P., SOUPPE, J., TRITSCH, D. and BIELLMANN, J.F. 1991. Chemical modification of horseradish peroxidase with ethanal-methoxypolyethylene glycol: Solubility in organic solvents, activity and properties. Bioorg. Chem. 19, 133-142.
WONG, S.S. 1991. Chemistry of Protein Conjugation and Cross-Linking. CRC Press. Boca Raton.

426 G.E. MEANS and R.E. FEENEY
WONG, W.S.D., KRISTJANSSON, M.M., OSUGA, D.T. and FEENEY, R.E. 1985. 1 - Deoxyglycitolation of protein amino groups and their regenera- tion by periodate oxidation. Int. J . Peptide Protein Res. 26, 55-62.
WONG, W.S.D., OSUGA, D.T. and FEENEY, R.E. 1984. Pyridine borane as a reducing agent for proteins. Analyt. Biochem. 139, 58-67.
WU, H.-L. and MEANS, G.E. 1981. Immobilization of proteins by reductive alkylation with hydrophobic aldehydes, Biotechnol. Bioengineer. 23, 855- 861.
YIP, T.T., VAN DE WATER, J., GERSHWIN, M.E., COPPEL, R.L. and HUTCH- ENS, T.W. 1996. Cryptic antigenic determinants on the extracellular pyruvate dehydrogenase compleximimeotope found in primary biliary cirrhosis. A probe by affinity mass-spectrometry. J. Biol Chem.
ZHANG, H. and MEANS, G.E. 1996. S-nitrosation of serum albumin: Spectropho- tometric determination of its nitrosation by simple S-nitrosothiols. Analyt. Biochem. 237, 141-144.
ZHANG, M. and VOGEL, H.J. 1993. Determination of the side chain pKa values of the lysine residues in calmodulin. J. Biol. Chem. 268, 22420-22428.
271(51), 32825-32833.


















