
Chapter 292 Antimicrobial Agents
33
Antimicrobial agents are essential in the therapy of bacterial infections. The approach to antimicrobial therapy is outlined in Chapter 289 (Principles of Anti-Infective Therapy), providing the clinician with an overview of the selection of agents based on the characteristics of infected children with respect to their pathogens and antibiotic susceptibilities, sites of infection, drug absorption, distribution and elimination, comorbidities, and a consideration of the benefits versus the risks of antimicrobial therapy. In this chapter, the agents themselves are discussed, providing a background on mechanism of action, spectrum of antibacterial activity, antibiotic resistance, and current clinical use. A more detailed discussion for specific infections is found in each chapter describing that infection. Table 289-1 provides more detailed information on the pharmaco- dynamics of the antibiotics of various classes, whereas Table 292-1 provides a summary of the pharmacokinetics, tissue distribution, metabolisms, and excretion of commonly used antimicrobial agents within each of the antibiotic classes. T able 292-2 provides spect ra of activities of antibiotics. Appendices 292-1 and 292-2 provide dosages of antibiotics. CELL WALL-ACTIVE AGENTS The synthesis of the bacterial cell wall is remarkably complicated and still not fully understood. 1–3 Several steps are involved in cell wall creation, from the synthesis of precursors within the bacterial cytoplasm to the intricate construction of a lattice-like structure around the organism that maintains cell shape and osmotic integrity. Many of these steps have been exploited as targets of currently available antimicrobial agents and others provide potential targets for ongoing anti-infective research (Figure 292-1). Gram-negative cell walls consist of inner (plasma) and outer membranes, and are more complicated than those of gram-positive organisms that contain a single membrane. The saccharide precursors of cell walls, N -acetylmuramic acid (MurNAc), and N -acetylglucosamine (GlcNAc) are modified enzymatically by a series of steps, with MurNAc acquiring a side chain consisting of five peptides, incorporating D-alanine, D-alanine as the terminal two amino acids in this chain. This MurNAc-pentapeptide is subsequently attached to a GlcNAc saccharide unit, completing the disaccharide pentapeptide building block required for cell wall peptidoglycan formation (Figure 292-1). Agents that inhibit these initial steps have been identified in a research setting but most are not currently under investigation as clinically important targets. 4 The disaccharide pentapeptide building block is subsequently transferred SECTION B Anti-Infective Therapy 1420 PART IV Laboratory Diagnosis and Therapy of Infectious Diseases APPENDIX 291-1. Usual Dosing, Therapeutic T argets, and Suggestions for Maintenan ce Dosing of Selected Antimicrobial Agents in Individuals with Impaired Renal Function a —Continued Adjustment for Renal Failure: Creatinine Clearance (mL/min) Drug Usual Dose Target > 50 10–50 <10 Adjustment for Dialysis Ritonavir 400 mg/m 2 NC NC NC H/P/C: none q12 hours PO Saquinavir 50 mg/kg q8h PO NC (no data) NC (no data) NC (no data) H/P/C: none (no data) C, continuous arteriovenous or venovenous hemofiltration, usually with dialysis: CLCr, creatinine clearance (calculated or measured); ESRD, end-stage renal disease; H, hemodialysis (unless otherwise stated, the referred dose after dialysis is a full dose for systemic administration; HIV, human immunodeficiency virus; IV, intravenous; MD, multiple dose, traditional aminoglycoside administration; NC, no change; OD, once-daily aminoglycoside administration; P, continuous peritoneal dialysis (doses are given systemically (mg/kg) or instilled into dialysate (mg/L)); doses are not for treatment of peritonitis; PO, orally. Data from Arnoff GR, Berns JS, Brier ME, et al. Drug Prescribing in Renal Failure, 4th ed. Philadelphia, American College of Physicians, 1999; Jayasekara D, Aweeka FT, Rodriguez R, et al. Antiviral therapy for HIV patients with renal insufficiency. J Acquir Immune Defic Syndr Hum Retroviral 1999;21:384–395; and multiple other sources. a Data are based primarily on studies in adults. Doses are for children beyond the neonatal period and for maintenance of systemic drug levels after an initial loading dose (which is standard in patients with impaired renal function). Doses are for intravenous use unless indicated; they may differ from those recommended by manufacturers (see package inserts) and in many cases are not approved for children. Doses are based on ideal body weight (with maximal doses for drugs at 50 kg) and the calculated (0.55 μ Ht (cm) ÷ serum Cr) or measured creatinine clearance. These dose projections are only guidelines; measurement of serum drug concentrations must be used when feasible, especially if toxicities are related to accrual of drug (e.g., aminoglycosides, vancomycin, flucytosine). For prolonged therapy with nephrotoxic agents in all children, creatinine clearance should be recalculated and drug levels monitored weekly. When supporting evidence is not available, the recommendation is an extrapolation from the drug’s pharmacokinetic properties, and “no data” appears next to the recommendation. When dialysis does not affect the disposition of the drug, “none” is used to indicate that the drug should be dosed according to the recommendations for the appropriate creatinine clearance. b Serum drug concentrations should be monitored. Concurrent administration with penicillins may result in subtherapeutic gentamicin or tobramycin. Peritoneal absorption increases with inflammation. c Peritoneal absorption is generally good. d Active metabolite also accumulates in ESRD. The dose should be further reduced for hepatic and renal failure. e To treat urinary tract infection in ESRD, the dose should be as for normal renal function. f Drugs with renal and hepatic excretion require little change unless both mechanisms are impaired. g Neurotoxicity can occur, especially in ESRD. h Ototoxicity can occur with prolonged high doses in ESRD. i Doxycycline is the preferred member of the tetracycline class for use in individuals with impaired renal function. j Dosage adjustment is necessary because metabolite accumulates in ESRD. k Serum trough concentrations should be monitored. l Serum drug concentrations are the best guide. m Serum drug concentrations should be monitored; bone marrow suppression is more common in azotemic individuals. n b -cyclodextrin, the vehicle for the oral and intravenous preparation, is cleared by the kidneys and accumulates in significant renal failure after intravenous administration. Intravenous dosing should be avoided because. o Acyclovir impairs urate secretion and can cause gout; uric acid levels should be monitored. p The maintenance dose is half the induction dose; bone marrow suppression is more common in azotemic individuals. q Neurotoxicity is especially common in ESRD. CHAPTER 2 9 2 Antimicrobial Ag ents John S. Bradley and Jason Sauberan
-
Upload
carmen-vargas -
Category
Documents
-
view
277 -
download
0
Transcript of Chapter 292 Antimicrobial Agents

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 1/33
Antimicrobial agents are essential in the therapy of bacterialinfections. The approach to antimicrobial therapy is outlined inChapter 289 (Principles of Anti-Infective Therapy), providing the
clinician with an overview of the selection of agents based on thecharacteristics of infected children with respect to their pathogensand antibiotic susceptibilities, sites of infection, drug absorption,distribution and elimination, comorbidities, and a consideration of the benefits versus the risks of antimicrobial therapy. In this chapter,the agents themselves are discussed, providing a background onmechanism of action, spectrum of antibacterial activity, antibioticresistance, and current clinical use. A more detailed discussion forspecific infections is found in each chapter describing that infection.Table 289-1 provides more detailed information on the pharmaco-dynamics of the antibiotics of various classes, whereas Table 292-1provides a summary of the pharmacokinetics, tissue distribution,metabolisms, and excretion of commonly used antimicrobial agentswithin each of the antibiotic classes. Table 292-2 provides spectra of
activities of antibiotics. Appendices 292-1 and 292-2 provide dosagesof antibiotics.
CELL WALL-ACTIVE AGENTS
The synthesis of the bacterial cell wall is remarkably complicated andstill not fully understood.1–3 Several steps are involved in cell wallcreation, from the synthesis of precursors within the bacterialcytoplasm to the intricate construction of a lattice-like structurearound the organism that maintains cell shape and osmotic integrity.Many of these steps have been exploited as targets of currentlyavailable antimicrobial agents and others provide potential targets forongoing anti-infective research (Figure 292-1). Gram-negative cell
walls consist of inner (plasma) and outer membranes, and are morecomplicated than those of gram-positive organisms that contain asingle membrane.
The saccharide precursors of cell walls, N -acetylmuramic acid(MurNAc), and N -acetylglucosamine (GlcNAc) are modifiedenzymatically by a series of steps, with MurNAc acquiring a sidechain consisting of five peptides, incorporating D-alanine, D-alanine asthe terminal two amino acids in this chain. This MurNAc-pentapeptideis subsequently attached to a GlcNAc saccharide unit, completingthe disaccharide pentapeptide building block required for cell wallpeptidoglycan formation (Figure 292-1). Agents that inhibit theseinitial steps have been identified in a research setting but most are notcurrently under investigation as clinically important targets.4 Thedisaccharide pentapeptide building block is subsequently transferred
S E C T IO N B Anti-Infective Therapy1420
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
APPENDIX 291-1. Usual Dosing, Therapeutic Targets, and Suggestions for Maintenance Dosing of Selected Antimicrobial Agents in Individuals with
Impaired Renal Functiona—Continued
Adjustment for Renal Failure:
Creatinine Clearance (mL/min)
Drug Usual Dose Target > 50 10–50 < 10 Adjustment for Dialysis
Ritonavir 400 mg/m2 NC NC NC H/P/C: noneq12 hours PO
Saquinavir 50 mg/kg q8h PO NC (no data) NC (no data) NC (no data) H/P/C: none (no data)
C, continuous arteriovenous or venovenous hemofiltration, usually with dialysis: CLCr, creatinine clearance (calculated or measured); ESRD, end-stage renal disease; H,
hemodialysis (unless otherwise stated, the referred dose after dialysis is a full dose for systemic administration; HIV, human immunodeficiency virus; IV, intravenous; MD, multiple
dose, traditional aminoglycoside administration; NC, no change; OD, once-daily aminoglycoside administration; P, continuous peritoneal dialysis (doses are given systemically
(mg/kg) or instilled into dialysate (mg/L)); doses are not for treatment of peritonitis; PO, orally.
Data from Arnoff GR, Berns JS, Brier ME, et al. Drug Prescribing in Renal Failure, 4th ed. Philadelphia, American College of Physicians, 1999; Jayasekara D, Aweeka FT,
Rodriguez R, et al. Antiviral therapy for HIV patients with renal insufficiency. J Acquir Immune Defic Syndr Hum Retroviral 1999;21:384–395; and multiple other sources.
aData are based primarily on studies in adults. Doses are for children beyond the neonatal period and for maintenance of systemic drug levels after an initial loading dose (which is
standard in patients with impaired renal function). Doses are for intravenous use unless indicated; they may differ from those recommended by manufacturers (see package inserts)
and in many cases are not approved for children. Doses are based on ideal body weight (with maximal doses for drugs at 50 kg) and the calculated (0.55 μ Ht (cm) ÷ serum Cr)
or measured creatinine clearance. These dose projections are only guidelines; measurement of serum drug concentrations must be used when feasible, especially if toxicities are
related to accrual of drug (e.g., aminoglycosides, vancomycin, flucytosine). For prolonged therapy with nephrotoxic agents in all children, creatinine clearance should be
recalculated and drug levels monitored weekly. When supporting evidence is not available, the recommendation is an extrapolation from the drug’s pharmacokinetic properties, and
“no data” appears next to the recommendation. When dialysis does not affect the disposition of the drug, “none” is used to indicate that the drug should be dosed according to the
recommendations for the appropriate creatinine clearance.bSerum drug concentrations should be monitored. Concurrent administration with penicillins may result in subtherapeutic gentamicin or tobramycin. Peritoneal absorption increases
with inflammation.cPeritoneal absorption is generally good.dActive metabolite also accumulates in ESRD. The dose should be further reduced for hepatic and renal failure.eTo treat urinary tract infection in ESRD, the dose should be as for normal renal function.f Drugs with renal and hepatic excretion require little change unless both mechanisms are impaired.gNeurotoxicity can occur, especially in ESRD.hOtotoxicity can occur with prolonged high doses in ESRD.iDoxycycline is the preferred member of the tetracycline class for use in individuals with impaired renal function.
jDosage adjustment is necessary because metabolite accumulates in ESRD.k Serum trough concentrations should be monitored.lSerum drug concentrations are the best guide.mSerum drug concentrations should be monitored; bone marrow suppression is more common in azotemic individuals.nb -cyclodextrin, the vehicle for the oral and intravenous preparation, is cleared by the kidneys and accumulates in significant renal failure after intravenous administration.
Intravenous dosing should be avoided because.oAcyclovir impairs urate secretion and can cause gout; uric acid levels should be monitored.pThe maintenance dose is half the induction dose; bone marrow suppression is more common in azotemic individuals.qNeurotoxicity is especially common in ESRD.
C H A P T E R 292
Antimicrobial Agents
John S. Bradley and Jason Sauberan
8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 2/33
Antimicrobial Agents C H A P T E R 292 142
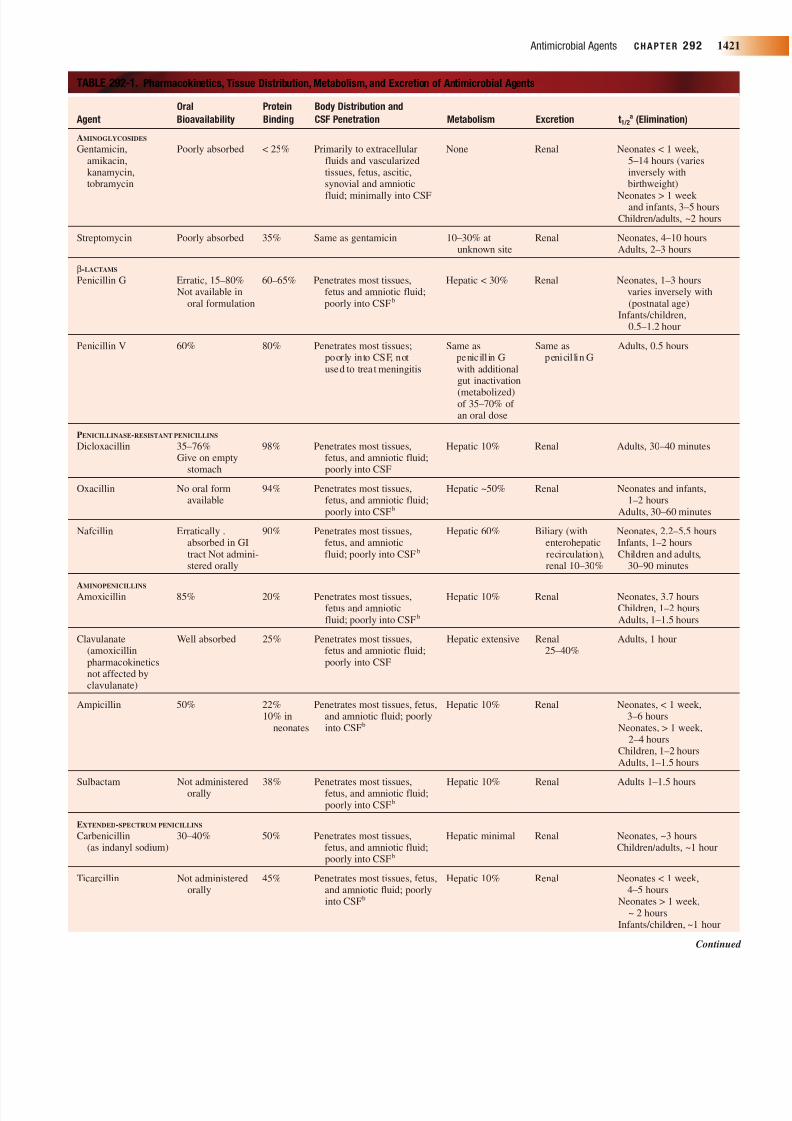
TABLE 292-1. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Antimicrobial Agents
Oral Protein Body Distribution and
Agent Bioavailability Binding CSF Penetration Metabolism Excretion t1/2a (Elimination)
AMINOGLYCOSIDES
Gentamicin, Poorly absorbed < 25% Primarily to extracellular None Renal Neonates < 1 week,amikacin, fluids and vascularized 5–14 hours (varieskanamycin, tissues, fetus, ascitic, inversely withtobramycin synovial and amniotic birthweight)
fluid; minimally into CSF Neonates > 1 week and infants, 3–5 hours
Children/adults, ~2 hours
Streptomycin Poorly absorbed 35% Same as gentamicin 10–30% at Renal Neonates, 4–10 hoursunknown site Adults, 2–3 hours
b-LACTAMS
Penicillin G Erratic, 15–80% 60–65% Penetrates most tissues, Hepatic < 30% Renal Neonates, 1–3 hoursNot available in fetus and amniotic fluid; varies inversely with
oral formulation poorly into CSFb (postnatal age)Infants/children,
0.5–1.2 hour
Penicillin V 60% 80% Penetrates most tissues; Same as Same as Adults, 0.5 hourspoorly into CSF, not penicill in G penicil lin Gused to treat meningitis with additional
gut inactivation
(metabolized)of 35–70% of an oral dose
PENICILLINASE-RESISTANT PENICILLINS
Dicloxacillin 35–76% 98% Penetrates most tissues, Hepatic 10% Renal Adults, 30–40 minutesGive on empty fetus, and amniotic fluid;
stomach poorly into CSF
Oxacillin No oral form 94% Penetrates most tissues, Hepatic ~50% Renal Neonates and infants,available fetus, and amniotic fluid; 1–2 hours
poorly into CSFb Adults, 30–60 minutes
Nafcillin Erratically . 90% Penetrates most tissues, Hepatic 60% Biliary (with Neonates, 2.2–5.5 hoursabsorbed in GI fetus, and amniotic enterohepatic Infants, 1–2 hourstract Not admini- fluid; poorly into CSFb recirculation), Children and adults,stered orally renal 10–30% 30–90 minutes
AMINOPENICILLINSAmoxicillin 85% 20% Penetrates most tissues, Hepatic 10% Renal Neonates, 3.7 hours
fetus and amniotic Children, 1–2 hoursfluid; poorly into CSFb Adults, 1–1.5 hours
Clavulanate Well absorbed 25% Penetrates most tissues, Hepatic extensive Renal Adults, 1 hour(amoxicillin fetus and amniotic fluid; 25–40%pharmacokinetics poorly into CSFnot affected byclavulanate)
Ampicillin 50% 22% Penetrates most tissues, fetus, Hepatic 10% Renal Neonates, < 1 week,10% in and amniotic fluid; poorly 3–6 hours
neonates into CSFb Neonates, > 1 week,2–4 hours
Children, 1–2 hoursAdults, 1–1.5 hours
Sulbactam Not administered 38% Penetrates most tissues, Hepatic 10% Renal Adults 1–1.5 hoursorally fetus, and amniotic fluid;poorly into CSFb
EXTENDED-SPECTRUM PENICILLINS
Carbenicillin 30–40% 50% Penetrates most tissues, Hepatic minimal Renal Neonates, ~3 hours(as indanyl sodium) fetus, and amniotic fluid; Children/adults, ~1 hour
poorly into CSFb
Ticarcillin Not administered 45% Penetrates most tissues, fetus, Hepatic 10% Renal Neonates < 1 week,orally and amniotic fluid; poorly 4–5 hours
into CSFb Neonates > 1 week,~ 2 hours
Infants/children, ~1 hour
Continue

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 3/33
S E C T IO N B Anti-Infective Therapy1422
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
TABLE 292-1. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Antimicrobial Agents—Continued
Oral Protein Body Distribution and
Agent Bioavailability Binding CSF Penetration Metabolism Excretion t1/2a (Elimination)
Piperacillin Not administered 15–20% Penetrates most tissues, fetus, Hepatic minimal Renal, Neonates, 2–3 hoursorally and amniotic fluid; poorly biliary Infants/children, 0.5–1 hour
into CSFb < 20% Adults, 0.5 hour (increasesto 1–1.5 hours for high
dose due to saturation of hepatobiliary excretion(dose-dependent t1/2))
Tazobactam Not administered 20–23% Penetrates most tissues, fetus, Hepatic minimal Renal Infants, 1.6 hours(piperacillin kinetics orally and amniotic fluid; poorly Children/adults,are unaffected into CSFb 45 minutes–1 hourby tazobactam)
CEPHALOSPORINS
FIRST-GENERATION
Cefadroxil Well absorbed 20% Penetrates most tissues, None Renal (slower Adult, 1–2 hoursfetus and amniotic fluid; excretion rateminimally into CSF than cephalexin)
Cefazolin Not administered 80% Penetrates most tissues, None Renal Neonates, 3–5 hoursorally fetus, and amniotic fluid; Adult, 1.5–2.5 hours
minimally into CSF
Cephalexin Well absorbed; 6% Penetrates most tissues, fetus, None Renal, Neonates, 5 hoursΠwith food and amniotic fluid; some biliary Infants, 2.5 hours
minimallyinto CSF Children/adults, 1 hour
Cephradine Well absorbed 10% Penetrates most tissues; None Renal, Children/adults, ~1 hourΠwith food fetus, and amniotic fluid; some biliary
minimally into CSF
SECOND-GENERATION
Cefaclor Well absorbed 25% Penetrates most tissues; Unknown Renal Adults, 0.5–1 hourunknown fetal; amniotic, (nonrenal:and CSF distribution elimination at
unknown sitein renal failure)
Cefprozil 95% 36% Penetrates middle-ear fluids Unknown Renal, Infants/children,and tonsillar, adenoidal, nonrenal 30% 1.5–2 hoursskin, and soft tissues well; Adults, 1–1.5 hoursunknown fetal, amnioticand CSF distribution
Cefuroxime 37% (as axetil); 50% Penetrates most tissues, None Renal Neonates, 3–6 hoursØ to 52% when fetus, and amniotic fluid; Infants/children, 1.5–2 hoursgiven with food minimally into CSF Adults, 1.2 hours
Cefoxitin Not administered 75% Penetrates most tissues, Hepatic minimal Renal Neonates, 1.4 hoursorally fetus, and amniotic fluid; Infants/children/adults,
minimally into CSFa ~45 minutes
Loracarbef 90% but can 25% Penetrates most tissues, None Renal Children/adults, ~ 1 hourwith food unknown fetal, amniotic
and CSF distribution
THIRD-GENERATION
Cefdinir 16–21% cap; 60–70% Penetrates most tissues; None Renal Adults, 1.7 hours25% suspension unknown fetal, amniotic
and CSF distribution
Cefixime 40–50% 65–70% Not well studied Unknown Renal, biliary Adults, 3–4 hours
Cefoperazone Not administered 90% Penetrates most tissues, Hepatic < 20% Biliary, Neonates, 6–10 hoursorally fetus, and amniotic fluid; renal (varies inversely with
minimally into CSFa 20–30% postnatal age)Infants/children,
2.2–2.3 hoursAdults, ~2 hours

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 4/33
Antimicrobial Agents C H A P T E R 292 142
TABLE 292-1. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Antimicrobial Agents—Continued
Oral Protein Body Distribution and
Agent Bioavailability Binding CSF Penetration Metabolism Excretion t1/2a (Elimination)
Cefotaxime Not administered 35–40% Penetrates most tissues, Hepatic Renal Neonates, 2–6 hoursorally fetus, and amniotic fluid; (varies inversely with
adequately into CSFb gestational andpostnatal age)
Infants/children,1–1.5 hoursOlder children/adults,
45 minutes–1 hour
Cefpodoxime 50% 20–30% Penetrates most tissues; None Renal Adults, 2–3 hoursunknown fetal, amniotic,and CSF distribution
Ceftazidime Not administered < 10% Penetrates most tissues, None Renal Neonates, 4–7 hoursorally fetus, and amniotic fluid; (varies inversely with
adequately into CSFb gestational age)Adults, 1.4–2 hours
Ceftibuten > 90% 65–77% Penetrates most tissues; Hepatic minimal Renal Children/adults,unknown fetal, amniotic, 1.5–2.5 hoursand CSF distribution
Ceftizoxime Not administered 31% Penetrates most tissues, None Renal Neonates, 2–4 hours
orally fetus, and amniotic fluid; Adults, 1–2 hoursminimally into CSFb
Ceftriaxone Not administered 95% Penetrates most tissues, None Renal, Neonates, 9–19 hoursorally fetus, and amniotic fluid; biliary Children, 4–7 hours
adequatelyinto CSFb Adults, 6–9 hours
FOURTH-GENERATION
Cefepime Not administered 20% Penetrates most tissues, Hepatic minimal Renal Neonates, 3–7 hoursorally fetus, and amniotic fluid; Children/adults, ~2 hours
adequatelyinto CSFb
OTHER b -LACTAMS, MONOBACTAMS
Aztreonam Not administered 50–70% Penetrates most tissues, Minimal hydrolysis Renal, Neonates < 1 week,orally fetus, and amniotic fluid; at unknown site biliary minor 6–10 hours
minimally into CSFb (varies inversely withbirthweight)
Neonate > 1 week,~ 3 hours
Children/adults,1.5–2 hours
CARBAPENEMS
Meropenem Not administered Minimal Penetrates most tissues, Renal, serum, Renal, Neonates, 2–3 hoursorally fetus and amniotic fluid; hepatic 20–25% biliary minor Infants, 1.5 hour
adequately into CSFb Adults, 1 hour
Imipenem (I) + Not administered 20% (I) Penetrates most tissues, fetus Renal, serum, Renal, biliary Neonates, 1.5–2.5 hourcilastatin (C) orally 40% (C) and amniotic fluid, hepatic 20–25% minor (cilastatin 3–8 hours)
adequately into CSF b but Infants/children,relatively contraindicated 1–1.4 hoursfor meningitis Adults, ~1 hour
Ertapenem Not administered 95% Penetrates interstitial fluids; Renal 20%, Renal, biliary Infants/children,orally unknown fetal, amniotic, hepatic minor minor 2.5 hours
and CSF distribution Adolescents/adults,
4 hours
CHLORAMPHENICOL SUCCINATE (INJECTION)
PO forms (base ~50% Widely distributed including Hepatic Renal (as Highly variable; see textand palmitate fetal, amniotic, and CSF succinate saltsalt) not and glucuronideavailable metabolite)
biliary minimal
FLUOROQUINOLONES AND QUINOLONES
Ciprofloxacin 60–80%; > 90% 20–40% Penetrates most tissues, fetus, Hepatic < 20% Renal, feces Neonates/infants/ in adolescents amniotic fluid; minimally children/adults,with CF into CSFb ~ 3–5 hours
Continue
8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 5/33
S E C T IO N B Anti-Infective Therapy1424
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
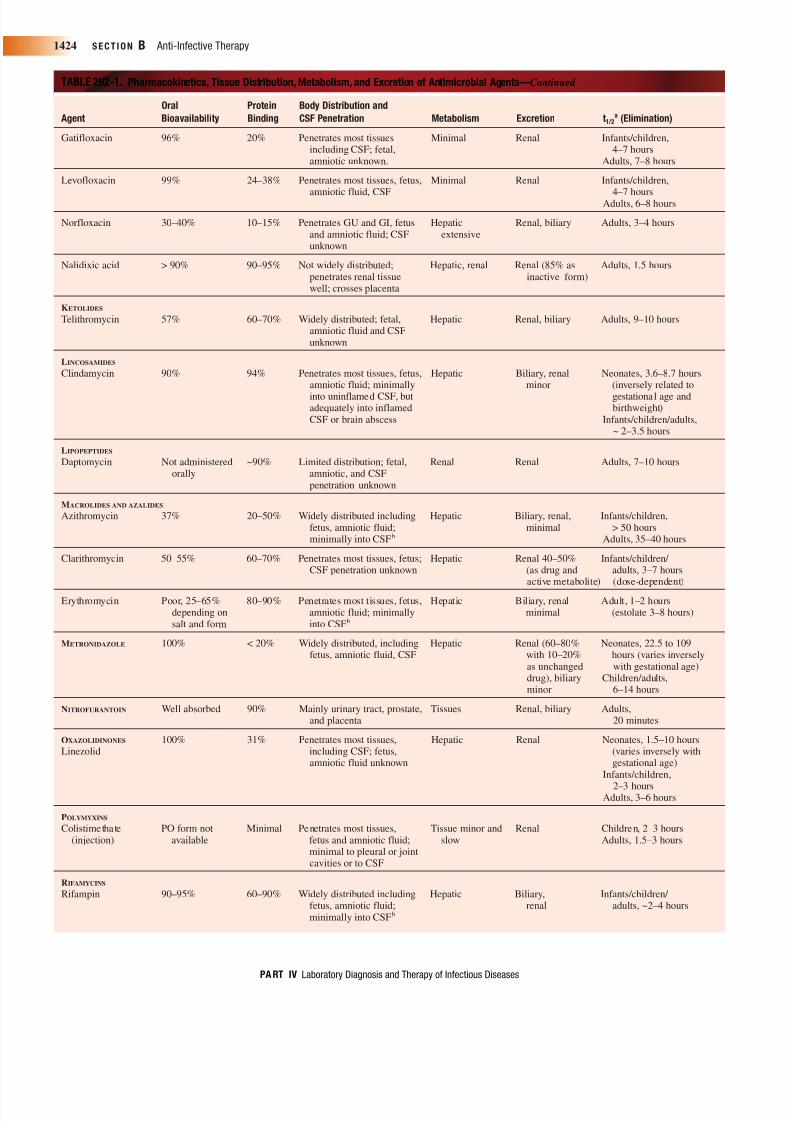
TABLE 292-1. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Antimicrobial Agents—Continued
Oral Protein Body Distribution and
Agent Bioavailability Binding CSF Penetration Metabolism Excretion t1/2a (Elimination)
Gatifloxacin 96% 20% Penetrates most tissues Minimal Renal Infants/children,including CSF; fetal, 4–7 hoursamniotic unknown. Adults, 7–8 hours
Levofloxacin 99% 24–38% Penetrates most tissues, fetus, Minimal Renal Infants/children,
amniotic fluid, CSF 4–7 hoursAdults, 6–8 hours
Norfloxacin 30–40% 10–15% Penetrates GU and GI, fetus Hepatic Renal, biliary Adults, 3–4 hoursand amniotic fluid; CSF extensiveunknown
Nalidixic acid > 90% 90–95% Not widely distributed; Hepatic, renal Renal (85% as Adults, 1.5 hourspenetrates renal tissue inactive form)well; crosses placenta
KETOLIDES
Telithromycin 57% 60–70% Widely distributed; fetal, Hepatic Renal, biliary Adults, 9–10 hoursamniotic fluid and CSFunknown
LINCOSAMIDES
Clindamycin 90% 94% Penetrates most tissues, fetus, Hepatic Biliary, renal Neonates, 3.6–8.7 hoursamniotic fluid; minimally minor (inversely related to
into uninflamed CSF, but gestational age andadequately into inflamed birthweight)CSF or brain abscess Infants/children/adults,
~ 2–3.5 hours
LIPOPEPTIDES
Daptomycin Not administered ~90% Limited distribution; fetal, Renal Renal Adults, 7–10 hoursorally amniotic, and CSF
penetration unknown
MACROLIDES AND AZALIDES
Azithromycin 37% 20–50% Widely distributed including Hepatic Biliary, renal, Infants/children,fetus, amniotic fluid; minimal > 50 hoursminimally into CSFb Adults, 35–40 hours
Clarithromycin 50–55% 60–70% Penetrates most tissues, fetus; Hepatic Renal 40–50% Infants/children/ CSF penetration unknown (as drug and adults, 3–7 hours
active metabolite) (dose-dependent)
Erythromycin Poor, 25–65% 80–90% Penetrates most tissues, fetus, Hepatic Biliary, renal Adult, 1–2 hoursdepending on amniotic fluid; minimally minimal (estolate 3–8 hours)salt and form into CSFb
METRONIDAZOLE 100% < 20% Widely distributed, including Hepatic Renal (60–80% Neonates, 22.5 to 109fetus, amniotic fluid, CSF with 10–20% hours (varies inversely
as unchanged with gestational age)drug), biliary Children/adults,minor 6–14 hours
NITROFURANTOIN Well absorbed 90% Mainly urinary tract, prostate, Tissues Renal, biliary Adults,and placenta 20 minutes
OXAZOLIDINONES 100% 31% Penetrates most tissues, Hepatic Renal Neonates, 1.5–10 hoursLinezolid including CSF; fetus, (varies inversely with
amniotic fluid unknown gestational age)Infants/children,
2–3 hoursAdults, 3–6 hours
POLYMYXINS
Colistimethate PO form not Minimal Penetrates most tissues, Tissue minor and Renal Children, 2–3 hours(injection) available fetus and amniotic fluid; slow Adults, 1.5–3 hours
minimal to pleural or jointcavities or to CSF
RIFAMYCINS
Rifampin 90–95% 60–90% Widely distributed including Hepatic Biliary, Infants/children/ fetus, amniotic fluid; renal adults, ~2–4 hoursminimally into CSFb

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 6/33
through the cell membrane to undergo further modification ultimatelyto create the peptidoglycan structure either outside the cell membrane(in gram-positive organisms), or between the inner plasma membraneand outer membrane within the cell wall (in gram-negativeorganisms). Linking of the disaccharide pentapeptide building blocksoccurs by transglycosylation, and creates repeating disaccharidesubunits (GlcNAc-MurNAc-pentapeptide) to produce long glycanchains.3 Vancomycin and related glycopeptide antibiotics inhibit thisstep in cell wall synthesis by binding to the terminal D-ala, D-ala of the pentapeptide attached to MurNAc, and interfering stericallywith the enzymatic function of the transglycosylase.5
The mature glycan chains containing the repeating disaccharidunits are subsequently linked by connecting the pentapeptides locateon the MurNAc units from adjacent glycan chains. In thtranspeptidation step, a stable bridge is created between glycan chainto form the two-dimensional peptidoglycan structure. The beta-lactamclass of antibiotics inhibits the transpeptidase function by bindincovalently to the active serine site of the enzyme responsible folinking the two pentapeptide arms from MurNAc units on adjacenglycan strands.6 The structure of enzymes that are responsible fotransglycosylation and transpeptidation varies somewhat betweebacteria. Fortunately, the active sites of these enzymes tend to be quit
Antimicrobial Agents C H A P T E R 292 142
TABLE 292-1. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Antimicrobial Agents—Continued
Oral Protein Body Distribution and
Agent Bioavailability Binding CSF Penetration Metabolism Excretion t1/2a (Elimination)
Rifaximin Poorly absorbed N/A Minimal systemic distribution Hepatic minimal Feces Minimal systemicdue to poor oral bioavail- absorptionability, but high intra-luminal GI concentrations
STREPTOGRAMINSQuinuprist in- Poorly absorbed 55–78% Penetrates most t issues; Hepatic, conversion Biliary, Adults, 0.85 hours (Q)
dalfopristin Not administered (Q) minimally into CSF; fetus, to several active renal ~15% 0.75 hours (D)orally 11–26% amniotic fluid unknown metabolites 2.5–3.5 hours (Q + m)
(D) ~1 hour (D + m):m = metabolites
SULFONAMIDES AND TRIMETHOPRIM
Sulfadiazine 100% 20% Widely distributed, including Hepatic wide Renal (free and Adults, 7–17 hoursfetus, amniotic fluid, CSF individual conjugated
variation forms)
Sulfamethoxazole 100% 65% Widely distributed, including Hepatic wide Renal (free and Adults, 9–12 hoursfetus, amniotic fluid, individual conjugatedCSF variation forms)
Sulfisoxazole 100% 85% Widely distributed, including Hepatic wide Renal (free and Adults, 5–8 hoursfetus, amniotic fluid, CSF individual conjugated
variation forms)Trimethoprim 100% ~45% Widely distributed, including Hepatic < 20% Renal Infants/children,
fetus, amniotic fluid, CSF 3–5.5 hoursAdults, 8–10 hours
TETRACYCLINES AND GLYCYLCYCLINES
Doxycycline 90–100% 82% Widely distributed including Hepatic Renal, biliary Adults, ~20 hoursfetus, amniotic fluid;minimally into CSFb
Minocycline 90–100% 76% Widely distributed, including Hepatic Biliary, Adults, 11–22 hoursfetus, amniotic fluid; minimal renalminimally into CSFb
Tetracycline (T), 75–80%; 65% (T) Widely distributed, including Hepatic Renal, biliary Adults, 7–10 hours (T)Demeclocycine (D) decreases 41–91% fetus, amniotic fluid; minimal Adults, 10–17 hours (D)
significantly (D) minimally into CSFb
with food
Tigecycline Limited, not 70–90% Widely distributed; fetal, Hepatic 5–20% Biliary, renal Adults, 40 hoursadministered amniotic fluid andorally CSF unknown
GLYCOPEPTIDES
Vancomycin Negligible 30% Penetrates most tissues, fetus, None Renal, biliary Neonates, 4–11 hoursamniotic fluid; adequately minimal (varies inversely withbut erratically into CSFb gestational age)
Infants, 2–4 hoursChildren, 2–2.5 hoursAdults, 4–6 hours
CF, cystic fibrosis; CSF, cerebrospinal fluid; GI, gastrointest inal; GU, genitourinary; IV, intravenous; PO, orally.aAgents with both minimal metabolism and urinary excretion will have a prolonged t 1/2 in a patient with renal impairment.bConcentration of drug in CSF significantly increased with inflamed meninges.
See references.170–177

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 7/33
conserved. An organism often contains several transpeptidases, eachresponsible for a different cell wall function, including repair,elongation, septation, and cell wall thickening, among others. Someof these enzymes appear to contain both transglycosylation andtranspeptidation functions. Historically, these enzymes were identifiedby penicillin attachment to them, and are also known as penicillin-binding proteins, or PBPs.
Beta-Lactam Antibiotics
The beta-lactam antibiotics all share the capacity to inhibit thetranspeptidase cross-linking of peptidoglycan in the final stepsof formation of the cell wall. Whereas the beta-lactam structureitself is consistent across all antibiotics in this class, the ring towhich the lactam moiety is fused is variable, with relatively smalldifferences in the composition of the ring allowing for variableactivity against the PBPs of both gram-positive and gram-negativebacteria (Figure 292-2). The addition of chemical “chains” to thering structures enhances activity against certain organisms, butsimultaneously may decrease activity against others. Differences inthe charges of the antibiotic molecule affect the ability of thecompound to reach and to bind to its target, particularly for gram-negative pathogens.
In general, the beta-lactam antibiotics are bactericidal with theconcentrations required for killing being very close to those requiredfor inhibition of growth. The maximal bactericidal effect occurs onrapidly growing bacteria; in stationary phase, this class of antibioticshas substantially less impact on the viability of organism.7
Resistance to Beta-Lactam Antibiotics
Probably just as ancient as the natural antibiotics are naturalmechanisms of resistance to them (Figure 292-3). Resistance to thebeta-lactam antibiotics occurs primarily in four ways: (1) enzymatichydrolysis of the beta-lactam ring by bacterial beta-lactamases,rendering the antibiotic harmless; (2) alterations in the structure of thetranspeptidase, so that binding of the antibiotic to the active serine siteof the transpeptidase does not occur; (3) efflux pumps that, in gram-negative organisms, quickly and efficiently remove the antibioticsfrom the periplasmic space before they can bind to the transpeptidases;
and (4) alterations in the gram-negative outer membrane proteins thatprevent the antibiotic from entering the periplasmic space. Each of these resistance mechanisms is variably effective and can lead eitherto profound resistance or merely to slightly increased resistance thathas no clinical impact. Unfortunately, some pathogens combineseveral resistance mechanisms, each creating incremental increases inbeta-lactam resistance, ultimately leading to the development of anorganism that is no longer susceptible to these antibiotics.
The chemical modifications of the beta-lactam ring structure thatprovide altered charges on the molecule can allow the new agent (e.g.,ampicillin) to enter the gram-negative bacterial periplasmic space, incontrast to an older agent (e.g., penicillin G) that could not. Sidechains can also create enhanced stability of the antibiotic against oneor more of the hundreds of beta-lactamases that have been identified.Unfortunately, new, more active and broader-spectrum beta-
lactamases are reported with disturbing regularity.8
Although manydifferent efflux pump systems exist, changes in the structure andcharge of the antibiotic can decrease the affinity of the antibiotic forthe pump, while hopefully not decreasing its affinity for the targettranspeptidase (see Chapter 290, Mechanisms of AntibioticResistance).
Penicillins
The penicillins are the most commonly used antibiotics in pediatrics,and can be broadly divided into four different groups: (1) naturalpenicillins; (2) penicillinase-stable penicillins; (3) aminopenicillins;and (4) extended-spectrum penicillins.
S E C T IO N B Anti-Infective Therapy1426
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
TABLE 292-2.
I. Cell Wall-Active Agents
A. Antibiotic Class: Transpeptidase Inhibitors
Beta-Lactam Antibiotics Spectrum of Activitya
PENICILLINS
Natural penicillins Penicillin G Gram-positivePenicillin V Streptococci
Benzathine penicillin G Groups A, B, C, G, FProcaine penicillin G Viridans groupBenzathine/procaine streptococci
penicillin G Streptococcus pneumoniaecombinations Enterococcus faecalisb
Enterococcus faeciumb
Actinomyces Bacillus anthracis
Listeria monocytogenesGram-negative
Eikenella corrodens
Neisseria meningitidis Neisseria gonorrhoeaePasteurella multocida
Borrelia burgdorferiSpirillum minusStreptobacillus
moniliformisTreponema pallidum
Leptospira speciesAnaerobes
Bacteroides andPrevotella species(non-beta-lactamase-producing strains)
Fusobacterium speciesVeillonella speciesClostridium species
Eubacterium speciesPeptococcus speciesPeptostreptococcus speciesPropionibacterium
species
Penicillinase-stable Methicillin Gram-positivepenicillins Oxacillin Streptococci (as above
Nafcillin for penicillins)Cloxacillin Staphylococcus aureusDicloxacillin (except MRSA)
Aminopenicillins Ampicillin Gram-positiveAmoxicillin Streptococci (as above
for penicillins) Enterococcus faecalisb
Enterococcus faeciumb
Listeria monocytogenes
Gram-negative Escherichia coli Haemophilus influenzae
Neisseria meningitidisAnaerobes
For ampicillin: as abovefor penicillins
Amoxicillin/clavulanate Adds activity toamoxicillin:
Staphylococcus aureus(except MRSA)
Haemophilus influenzae,beta-lactamase-producing strains
AnaerobesAs above for penicillins,
but now includes: Bacteroides and
Prevotella species,

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 8/33
Antimicrobial Agents C H A P T E R 292 142
TABLE 292-2.—Continued
I. Cell Wall-Active Agents
A. Antibiotic Class: Transpeptidase Inhibitors
Beta-Lactam Antibiotics Spectrum of Activitya
beta-lactamase-producing strains
Ampicillin/sulbactam Adds activity toampicillin:
Staphylococcus aureus(except MRSA)
Escherichia coli, beta-lactamase-producingstrains
Klebsiella speciesProteus mirabilisProteus vulgarisProvidencia rettgeri
Providencia stuartii Morganella morganii
AnaerobesAs above for penicillins,
but now includes: Bacteroides and
Prevotella species(beta-lactamase-producing strains)
Extended-spectrum Carbenicillin Gram-positivepencillins Ticarcillin Streptococci, as above
Piperacillin for penicillinsGram-negative
Escherichia coli
Haemophilus influenzaeProteus mirabilisProteus vulgaris
Morganella morganiiPseudomonas
aeruginosaProvidencia rettgeri
Enterobacter species
Anaerobes Bacteroides and
Prevotella species(non-beta-lactamase-producing strains)
Fusobacterium speciesVeillonella speciesClostridium species
Eubacterium speciesPeptococcus speciesPeptostreptococcus
species
Ticarcillin/clavulanate Adds beta-lactamase-Piperacillin/tazobactam producing strains of:
Staphylococcus aureus(except MRSA)
Escherichia coli Haemophilus influenzaeKlebsiella speciesSerratia marcescensCitrobacter species
Enterobacter speciesAnaerobes
Bacteroides andPrevotella species(including beta-lactamase-producingstrains)
Fusobacterium speciesVeillonella species
TABLE 292-2.—Continued
I. Cell Wall-Active Agents
A. Antibiotic Class: Transpeptidase Inhibitors
Beta-Lactam Antibiotics Spectrum of Activitya
Clostridium species Eubacterium speciesPeptococcus speciesPeptostreptococcus
species
CEPHALOSPORINS
First-generation Cephalothin Gram-positiveCephapirin StreptococciCefazolin Groups A, B, C, G, FCephalexin Viridans groupCephradine streptococciCefadroxil Streptococcus
pneumoniaeStaphylococcus aureus
(except MRSA)Gram-negative
Escherichia coli
Proteus mirabilis
Second-generation Cefamandole Gram-positiveCefuroxime StreptococciCefonicid Groups A, B, C, G, FCeforanide Viridans groupCefaclor streptococciCefoxitin StreptococcusCefotetan pneumoniae
Staphylococcus aureus(except MRSA)
Gram-negative Escherichia coli Haemophilus influenzae
(including beta-lactamase producingstrains)
Klebsiella species Moraxella catarrhalis
Neisseria gonorrhoeae Neisseria meningitidisProteus mirabilis
Providencia rettgeriSalmonella speciesShigella speciesAnaerobes
Bacteroides andPrevotella species(nonbeta lactamase-producing strains,except for cefoxitinand, to a lesser extent,cefotetan)
Fusobacterium speciesVeillonella species
Eubacterium species
Peptococcus speciesPeptostreptococcus specie
Third-generation Cefotaxime Gram-positiveCeftriaxone StreptococciCeftazidime Groups A, B, C, G, FCefoperazone Viridans groupCeftizoxime streptococciCefixime StreptococcusCefpodoxime pneumoniaeCeftibuten Staphylococcus aureusCefdinir (except MRSA)
Gram-negativeCitrobacter species
Enterobacter species
Continue

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 9/33
S E C T IO N B Anti-Infective Therapy1428
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
TABLE 292-2.—Continued
I. Cell Wall-Active Agents
A. Antibiotic Class: Transpeptidase Inhibitors
Beta-Lactam Antibiotics Spectrum of Activitya
Escherichia coli
Haemophilus influenzae(including beta-lactamase-producingstrains)
Klebsiella species Morganella morganii Neisseria gonorrhoeae
(including beta-lactamase-producingstrains)
Neisseria meningitidisProteus mirabilisProteus vulgaris
Providencia rettgeriProvidencia stuartiiSerratia marcescens
For ceftazidime andcefoperazone:
Pseudomonas aeruginosa
Anaerobes Bacteroides and
Prevotella species(non-beta-lactamase-producing strains)
Fusobacterium species Eubacterium speciesPeptococcus species
Fourth-generation Cefepime Gram-positiveStreptococci
Groups A, B, C, G, FViridans group
streptococciStreptococcus
pneumoniaeStaphylococcus aureus
(except MRSA)Gram-negativeAs above for third-
generation cephalo-sporins, but includingPseudomonas aeruginosa)
Anaerobes Bacteroides andPrevotella species
(nonbeta lactamase-producing strains)
Fusobacterium speciesVeillonella species
Eubacterium speciesPeptococcus species
Fifth-generation Ceftobiprole As above for fourth-generationcephalosporins, butalso includes MRSAstrains of Staphylococcus aureus
CARBAPENEMS Imipenem (with Gram-positivecilastatin) StreptococciMeropenem Groups A, B, C, D, G, FErtapenem Viridans group
streptococciStreptococcus
pneumoniae Enterococcus faecalis
Staphylococcus aureus(except MRSA)
TABLE 292–2.—Continued
I. Cell Wall-Active Agents
A. Antibiotic Class: Transpeptidase Inhibitors
Beta-Lactam Antibiotics Spectrum of Activitya
Gram-negative Acinetobacter speciesCitrobacter species
Enterobacter species Escherichia coli
(including ESBL-producing strains)
Gardnerella vaginalis Haemophilus influenzaeKlebsiella species
(including ESBL-producing strains)
Morganella morganii
Proteus vulgarisProvidencia rettgeriPseudomonas aeruginosa
(except ertapenem)Serratia speciesAnaerobes
Bifidobacterium species
Clostridium species Eubacterium speciesPeptococcus speciesPeptostreptococcus
speciesPropionibacterium
species Bacteroides and
Prevotella species,(including beta-lactamase-producingstrains)
Fusobacterium species
MONOBACTANS Aztreonam Gram-negativeCitrobacter species
Enterobacter species
Escherichia coli Haemophilus influenzae
(including beta-lactamase-producing strains)
Klebsiella speciesProteus mirabilis
Pseudomonasaeruginosa
Serratia species
B. Antibiotic Class: Transglycosylase Inhibitors
Spectrum of Activitya
GLYCOPEPTIDES Vancomycin Gram-positiveTeicoplanin (not Streptococciavailable in the United Groups A, B, C, G, F
States) Viridans groupstreptococciStreptococcus
pneumoniae Enterococcus faecalisb
Enterococcus faeciumb
Staphylococcus aureus(including MRSA, butnot vancomycin-intermediate orvancomycin-resistantstrains)
Staphylococcusepidermidis
Actinomyces species

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 10/33
Antimicrobial Agents C H A P T E R 292 142
TABLE 292–2.—Continued
I. Cell Wall-Active Agents
B. Antibiotic Class: Transglycosylase Inhibitors
Lactobacillus species Listeria monocytogenesAnaerobesClostridium dif cile
II. Cell Membrane Active Agents
A. Antibiotic Class: Lipopeptides
Spectrum of Activitya
Daptomycin Staphylococcus aureus(including methicillin-resistant andvancomycin-resistantstrains)
Enterococcus faecalis(vancomycin-susceptibleand resistant strains)
Enterococcus faecium(vancomycin-susceptibleand resistant strains)
Streptococci
Groups A, BViridans group
streptococci
B. Antibiotic Class: Polymyxins
Colistin Enterobacter aerogenes Escherichia coli
Klebsiella pneumoniaePseudomonas aeruginosa
Actinobacter speciesCitrobacter species
Haemophilus speciesSalmonella speciesShigella species
III. Ribosome-Active Agents
A. Antibiotic Class: Macrolides
MACROLIDES Erythromycin Gram-positiveCorynebacterium
diphtheriaeCorynebacterium
minutissimum
Listeria monocytogenesStaphylococcus aureusStreptococcus
pneumoniaeStreptococcus pyogenes
Gram-negative Bordetella pertussis Legionella pneumophila Neisseria gonorrhoeae
Other pathogens
Chlamydia trachomatis Entamoeba histolytica Mycoplasma pneumoniae
Treponema pallidumUreaplasma urealyticum
Clarithromycin Gram-positiveStaphylococcus aureusStreptococcus
pneumoniaeStreptococcus pyogenes
Gram-negative Haemophilus influenzae Moraxella catarrhalis Helicobacter pylori
TABLE 292-2.—Continued
III. Ribosome-Active Agents
A. Antibiotic Class: Macrolides
Other pathogens Mycoplasma pneumoniaeChlamydophila pneumonia
Mycobacterium aviumcomplex
AZALIDES Azithromycin Gram-positiveStaphylococcus aureus
StreptococciGroups A, B C, F, GViridans group
streptococciStreptococcus
pneumoniae
Bordetella pertussisGram-negative
Haemophilus influenzae Haemophilus ducreyi
Moraxella catarrhalis Neisseria gonorrhoeae
Other pathogensChlamydophila pneumonia
Chlamydia trachomatis Legionella pneumophila
Mycoplasma hominis Mycoplasma pneumoniaeUreaplasma urealyticum
KETOLIDES Telithromycin Gram-positiveStaphylococcus aureusStreptococci Groups A, C
and G Viridans groupstreptococci
Gram-negative Haemophilus influenzae Moraxella catarrhalis
Other pathogens Bordetella pertussis Mycoplasma pneumoniae
Legionella pneumophila
Chlamydophila pneumoniae
B. Antibiotic Class: Tetracyclines
TETRACYCLINES Tetracycline Gram-positiveMinocycline Actinomyces speciesDoxycycline Gram-negative
Vibrio cholerae Brucella speciesCampylobacter speciesFrancisella tularensis
Listeria monocytogenesYersinia pestis
Neisseria meningitidis Neisseria gonorrhoeaeOther pathogens
Borrelia recurrentisChlamydophila psittaciChlamydia trachomatis
Mycoplasma pneumoniaeUreaplasmaTreponema pallidum
Entamoeba species
GLYCYLCYCLINES Tigecycline Gram-positiveStreptococci
Groups A, BViridans group
streptococciStreptococcus
pneumoniae Enterococcus faecalis
Enterococcus faecium
Continue

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 11/33
S E C T IO N B Anti-Infective Therapy1430
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
TABLE 292-2.—Continued
III. Ribosome-Active Agents
B. Antibiotic Class: Tetracyclines
Staphylococcus aureus Listeria monocytogenes
Clostridium perfringensPeptostreptococcus speciesGram-negative
Acinetobacter baumannii Aeromonas hydrophilaCitrobacter freundii
Citrobacter koseri Enterobacter cloacae Enterobacter aerogenes
Escherichia coliKlebsiella oxytocaKlebsiella pneumoniae
Pasteurella multocidaSerratia marcescensStenotrophomonas
maltophilia Bacteroides speciesOther pathogensChlamydia trachomatis
Mycoplasma pneumoniae
Ureaplasma Mycobacterium abscessus
Mycobacterium chelonae Mycobacterium fortuitum
C. Antibiotic Class: Lincosamides
LINCOSAMIDES Clindamycin Gram-positiveStreptococci
Groups A, BStreptococcus
pneumoniae
Staphylococcus aureusAnaerobes
Bacteroides fragilis
Prevotellamelaninogenica
Fusobacterium species
Peptococcus speciesPeptostreptococcus
species Actinomyces speciesClostridium perfringensPropionibacterium
species
D. Antibiotic Class: Aminoglycosides
AMINOGLYCOSIDES Streptomycin Gram-negative Brucella speciesFrancisella species
Mycobacteriumtuberculosis
Gentamicin Gram-positiveNetilmicin Staphylococcus aureus
Tobramycin Gram-negativeAmikacin Escherichia coli
Klebsiella species Enterobacter speciesSerratia speciesCitrobacter species
Morganella morganii Acinetobacter speciesProvidencia speciesProteus mirabilisProteus vulgarisPseudomonas aeruginosa
Paromomycin Entamoeba histolytica Dientamoeba fragilis
Cryptosporidium species
TABLE 292-2.—Continued
III. Ribosome-Active Agents
E. Antibiotic Class: Oxazolidinones
OXAZOLIDINONES Linezolid Gram-positiveStreptococci
Groups A, BViridans group
streptococciStreptococcus
pneumoniaeStaphylococcus aureus
Enterococcus faecium
Enterococcus faecalis
F. Antibiotic Class: Streptogramins
STREPTOGRAMINS Quinupristin/ Gram-positivedalfopristin Streptococci
Groups A, BStaphylococcus aureus
Enterococcus faecium
IV. Nucleic Acid-Active Antibiotics
A. Antibiotic Class: Rifamycins
RIFAMYCINS Rifampin Gram-positive
Staphylococcus aureusGram-negative
Neisseria meningitidis Haemophilus influenzaeOther
Mycobacteriumtuberculosis
Mycobacterium aviumcomplex
Rifabutin MycobacteriumRifapentine tuberculosis
Mycobacterium aviumcomplex
Rifaximin Susceptible atconcentrations achievedwithin the
gastrointestinal lumen:Campylobacter
Escherichia coliSalmonella speciesShigella speciesVibrio speciesYersinia species
B. Antibiotic Class: Quinolones
QUINOLONES Nalidixic acid Gram-negative Escherichia coli Enterobacter species Morganella morganiiProteus mirabilisProteus vulgarisProvidencia rettgeri
FLUOROQUINOLONES
Ciprofloxacin Gram-positiveStreptococcus pyogenesStreptococcus pneumoniae
Staphylococcus aureus Enterococcus faecalis Bacillus anthracis
Gram-negative Aeromonas species Acinetobacter species
Escherichia coliKlebsiella pneumoniae
Enterobacter cloacae
Citrobacter diversusCitrobacter freundiiCampylobacter jejuni

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 12/33
Natural Penicillins
Natural penicillins are the natural products of Penicillium chrysogenumIt is likely that penicillins evolved millions of years ago as result ocompetition for survival between single-cell organisms.9 Flemingobservations in the 1920s led to the identification of penicillin, anthe discovery of the mechanism by which Penicillium killed othebacteria, paving the way for the modern era of antibacterial therapyThe basic structure of penicillin, 6-aminopenicillanic acid, characteristic of the lactam ring fused to a larger ring structure tcreate a penam nucleus that is the basic structure of all penicillins (seFigure 292-2). Of the natural penicillins, only penicillin G (crystallinpenicillin G, benzyl penicillin G) and penicillin V (phenoxymethypenicillin) are currently available commercially. Penicillin G available in both oral and parenteral formulations. For intramuscula
injection, penicillin G is also available in repository forms of the druProcaine penicillin G and benzathine penicillin G both have muclonger serum elimination half-lives as a result of prolonged absorptiofrom the muscle injection site, compared with crystalline penicillin GHowever, the peak serum concentrations of the repository forms openicillin G are considerably lower than those achieved witintravenous administration of crystalline penicillin G. Therefore, thonly situations in which the repository forms of penicillin are effectivare those in which the organisms are exquisitely susceptible tpenicillin, in tissues with good perfusion. For those receiving comparable mg/kg dosages, procaine penicillin has a half-life oapproximately 12 hours and achieves peak serum concentrations oabout 2 μg/mL, compared with a half-life of 30 to 50 minutes focrystalline penicillin G, and achieved peak serum concentrations oapproximately 20 μg/mL. Benzathine penicillin G yields even lowe
serum concentrations (only about 1.5 mg/mL), but may remain abov0.2 μg/mL for 3 weeks or longer. Combinations of procaine anbenzathine penicillin, either in equal amounts, or as a 3:(benzathine:procaine) mixture are also available.
In clinical practice, although active against a wide range of bacteri(see Table 292-2), the natural penicillins are most widely used fotreatment and prevention of infections caused by streptococcPharyngitis, lower respiratory tract infection, skin and skin structurinfections, and bloodstream infection (BSI) caused by group Astreptococcus (Streptococcus pyogenes) are effectively treated witpenicillin. The in vitro susceptibility of these organisms has remaineunchanged over the past several decades,10 although the efficacy in thtreatment of streptococcal pharyngitis in more recent studies is lesthan expected, for reasons that are not known.11 Intramuscul
Antimicrobial Agents C H A P T E R 292 143
TABLE 292-2.—Continued
IV. Nucleic Acid-Active Antibiotics
B. Antibiotic Class: Quinolones
Proteus mirabilisProteus vulgarisProvidencia rettgeri
Providencia stuartiiSerratia marcescensPseudomonas aeruginosa
Morganella morganiiSalmonella speciesShigella species
Haemophilus influenzae Haemophilus
parainfluenzae
Moraxella catarrhalis Neisseria gonorrhoeaeb
Pasteurella multocida
Vibrio speciesYersinia enterocolitica
Other pathogens Legionella pneumophila
Levofloxacin Gram-positiveGatifloxacin Streptococci
Group AViridans group
streptococciStreptococcus
pneumoniae Enterococcus faecalis
Staphylococcus aureus Actinomyces species Bacillus anthracis
Listeria monocytogenesGram-negative
Acinetobacter species Escherichia coli Enterobacter speciesKlebsiella speciesProteus speciesProvidencia species
Serratia marcescensCitrobacter species
Morganella morganiiPseudomonas aeruginosa
Haemophilus influenzae Moraxella catarrhalisAnaerobesClostridium perfringensOther pathogens
Legionella pneumophila Mycoplasma pneumoniae
Chlamydophila pneumoniae
C. Antibiotic Class: Nitroimadazoles
NITROIMADAZOLES Metronidazole AnaerobesClostridium species
Eubacterium speciesPeptococcus speciesPeptostreptococcus
species Bacteroides fragilis
Fusobacterium species
D. Antibiotic Class: Sulfonamides
SULFONAMIDES SulfisoxazoleSulfamethoxazole
SULFA IN COMBINATION Sulfamethoxazole plus Gram-positiveWITH ANOTHER trimethoprim StreptococcusANTIMICROBIAL AGENT pneumoniaeb
Gram-negative Escherichia coli
TABLE 292–2.—Continued
IV. Nucleic Acid-Active Antibiotics
G. Antibiotic Class: Sulfonamides
Klebsiella species Enterobacter species Morganella morganii
Proteus mirabilisProteus vulgarisShigella species
Haemophilus influenzeOther pathogensPneumocystis jirovecii
Sulfadiazine plus Toxoplasma gondiipyramethamine Plasmodium species
ESBL, extended-spectrum beta-lactamases; MRSA, methicillin-resistantStaphylococcus aureus.aA majority of strains of the listed bacteria are susceptible; however, some organisms
within the group may be less susceptible or resistant to one or more agents listed.
Susceptibility pattern for each pathogen and antibiotic may be available to
physicians through local health care institutions.bImportant exceptions exist

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 13/33
S E C T IO N B Anti-Infective Therapy1432
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
injections of benzathine penicillin every 3 to 4 weeks are effective inthe prevention of rheumatic fever due to the prolonged tonsillar tissueconcentrations of penicillin G.
Empiric penicillin therapy of infections suspected to be caused byStreptococcus pneumoniae is no longer recommended as a result of widespread decreased susceptibility to penicillin of pneumococci.Alterations in the structure of several pneumococcal PBPs yieldspenicillin-nonsusceptible organisms, which has forced the medicalcommunity to use other, more active beta-lactam agents or agentsfrom other antibiotic classes. However, if culture results documentsusceptibility, penicillin still represents highly effective therapy.
Most anaerobes, with the exception of beta-lactamase-producingstrains of Bacteroides sp. and Prevotella sp. are highly susceptible topenicillin G. However, due to the common presence of Bacteroides
fragilis among the anaerobes present in intra-abdominal infections and
Prevotella melaninogenica among the organisms causing sinus-relatedbrain abscesses, other anaerobic agents are preferred to treat infectionsat these sites.
Penicillin G continues to play a role in the treatment of infectionscaused by other alpha- and beta-hemolytic streptococci, most of whichremain very susceptible. For life-threatening infections such asbacterial endocarditis, susceptibility testing should be performed toensure that the organisms do not exhibit penicillin tolerance, whichmay decrease the chances of treatment success using standarddosages.
Penicillin G has also been effective therapy of less commoninfections, including diphtheria, naturally occurring anthrax, actino-mycosis, leptospirosis, and syphilis.
Penicillinase-Resistant Penicillins
This class of semisynthetic penicillins was created to meet thechallenge of the development of penicillin-resistant Staphylococcusaureus. The bulky side chains prevent the staphylococcal beta-lactamases from binding to and hydrolyzing the lactam ring of themolecule. However, these antibiotics are only resistant to thestaphylococcal penicillinases, and not to the beta-lactamases of gram-negative organisms, to which they remain quite vulnerable. Theseantibiotics are also not active against methicillin-resistant strainsof S. aureus (MRSA) due to the presence of a transpeptidase (PBP 2a)that is not bound and inactivated by any currently available beta-lactam antibiotic.
In clinical practice, these antibiotics are used to treat infectionscaused by susceptible strains of S. aureus. They are available in both
Transpeptidase Penicillins Cephalosporins Carbapenems Monobactams
PBP(s)
mraY Tunicamycin
Mureidomycin Liposidomycin
Pacidomycin 5B-D liposidomycin analogs
murG Ramoplanin
murA
murB
Vancomycin Lipophilic vancomycins Moenomycin 5E moenomycin analog Transglycosylase
UDP-GlcNAc
UDP-MurNAc
murC
UDP-MurNAc
D-ala-D-ala
P
murD
ddlA/B
mur I
murF
UDP-MurNAc
D-glu L-glu
alr
L-ala Lipid II
Lipid I
D-ala
murE
UDP-MurNAc UDP-MurNAc
MurNAc P-P
MurNAc
GLcNAc
P-P
Stage Icytoplasm
UDP, uridine diphosphate; MurNAc, N-Acetylmuramic acid; GLcNAc,N-Acetylglucosamine; PBP(s), penicillin binding proteins (transpeptidase);L-ala, L-alanine; D-ala, D-alanine.
Stage IImembrane
Stage IIImembrane
MurNAc
GLcNAc
P-P
WallP
Figure 292-1. The peptidoglycan synthesis pathway in cell wall formation. (Redrawn with modification from Wong VK, Pompliano DL. Peptidoglycan
biosynthesis: unexploited targets within a familiar pathway. In: Rosen BP, Mobashery S (eds) Resolving the Antibiotic Paradox. New York, Kluwer
Academic/Plenum Publishers, 1998, pp 197–217.)
N
Carbacephem(loracarbef)
ON
Carbapenem(meropenem)
O
N
Cephem(ceffriaxone, cefotaxime)
O
R
R
R
S
N
Monobactam
O
R
R
SO3HN
Penam(penicillin, ampicillin)
OR
S
N
Clavam(clavulanate)
O
O
Figure 292-2. Beta-lactam antibiotic structures.

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 14/33
Antimicrobial Agents C H A P T E R 292 143
parenteral and oral formulations. Recently, with the emergence of community-associated (CA) MRSA, their long-standing role in theempiric therapy of presumed staphylococcal infections is nowcompromised. For susceptible strains of S. aureus, however, theyremain among the safest and most effective therapeutic agentsavailable.
Aminopenicillins
This class of semisynthetic penicillins contains an amino substitutionin the phenyl acetamido side chain of the penam nucleus, providing apolar charge on the molecule that allows antibiotics of this class
to demonstrate activity against gram-negative pathogens, includin Escherichia coli and Haemophilus influenzae (see Table 292-2However, this class of antibiotics is not stable to the staphylococcapenicillinases, or to the hundreds of different beta-lactamases thgram-negative pathogens may produce. The activity against othegram-positive organisms, such as group A and group B streptococcis still very good, and activity against most enterococci is equivalento or better than penicillin G.
As a means of enhancing the activity of the aminopenicillinagainst beta-lactamase-producing pathogens, the concurrent use of second beta-lactam agent that binds irreversibly to a pathogen’s betalactamase has led to success in the therapy of infections cause
Teichoric acid
GRAM-POSITIVE BACTERIAL CELL WALL
Peptidoglycan
Cytoplasmic membrane
A
Figure 292-3. Structure of bacterial cell walls of
gram-positive and gram-negative bacteria.
GRAM-NEGATIVE BACTERIAL CELL WALL
Peptidoglycan
Lipopolysaccharide
Cytoplasmicinner membrane
Outermembrane
Porinprotein
Effluxpump
Functional proteinwithin membrane
Functional proteinwithin membrane
N-acetylglucosamine
N-acetylmuramic acidB

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 15/33
by these organisms. These concurrently used agents, called beta-lactamase inhibitors, or “suicide” beta-lactams, have little antibioticactivity on their own as they have been selected for avid bindingcharacteristics to specific beta-lactamases, rather than to PBPs.However, just as diversity exists in the affinity of binding of penicillinto the target PBPs of various pathogens, diversity also exists in thebinding affinity of each beta-lactamase inhibitor to the beta-lactamases of different organisms. Currently, clavulanate is pairedwith amoxicillin in an oral formulation in the United States (and a
parenteral formulation in other parts of the world), and ampicillin ispaired with sulbactam in a parenteral preparation (see Table 292-2).The clinical uses of ampicillin and amoxicillin are extensive. In
addition to treatment of streptococcal infections, the enhanced activityagainst E. coli compared with penicillin G permits ampicillin andamoxicillin to be used widely for the treatment of urinary tractinfections (UTIs) and gastrointestinal infections. Ampicillin is themost bactericidal agent for susceptible Enterococcus. Unfortunately,the development of resistance in Escherichia coli, Shigella, andSalmonella has limited the usefulness of aminopenicillins againstthese pathogens. The addition of sulbactam to ampicillin allowsactivity against a much broader array of beta-lactamase-producingorganisms, including staphylococci, many enteric gram-negativebacilli, and Bacteroides fragilis. This allows for the treatment of skinand structure infections as well as intra-abdominal infections.
The addition of clavulanate to amoxicillin allows activity againstbeta-lactamase-producing strains of Haemophilus influenzae and
Moraxella catarrhalis as well as Staphylococcus aureus. Thiscombination increases the clinical usefulness of amoxicillin in thetreatment of community-acquired upper and lower respiratory tractinfections (e.g., acute otitis media, sinusitis, and pneumonia), in add-ition to skin and skin structure infections.
Extended-Spectrum Penicillins
These semisynthetic penicillins are designed to increase activityagainst the gram-negative pathogens, including Klebsiella, Entero-bacter , and, for some agents, Pseudomonas (see Table 292-2). Thetwo major classes are the carboxypenicillins, represented by ticarcillinand carbenicillin and the acylureidopenicillins, represented bypiperacillin. Although the spectrum of activity of these antibiotics has
clearly been enhanced beyond the aminopenicillins, they remainsusceptible to hydrolysis by many beta-lactamases, including those of staphylococcus. Similar to the aminopenicillins, activity of these drugshas been enhanced by pairing them with beta-lactamase inhibitors,resulting in ticarcillin-clavulanate and piperacillin-tazobactam.
The clinical uses of these antibiotics reflect their broad activityagainst gram-negative enteric bacilli and Pseudomonas aeruginosa.Whereas carbenicillin is an oral agent and is only used to treat UTIs,ticarcillin and piperacillin are given parenterally, and are used fortherapy of a broad variety of serious gram-negative infections. Cur-rently, ticarcillin is used most commonly when paired withclavulanate, and piperacillin when paired with tazobactam, whichincreases their activity to include many beta-lactamase-producingorganisms, including many gram-negative enteric bacilli, Bacteroides
fragilis, Prevotella melaninogenica, and S. aureus (see Table 292-2).
This allows for successful therapy for many skin and skin structureinfections, intra-abdominal infections, and, most importantly, gram-positive and gram-negative hospital-associated infections, includingwound infections, UTIs, and pneumonia. The extended spectrum-pectrum penicillins retain reasonably good activity against ampicillin-susceptible strains of Enterococcus.
Cephalosporins
Cephalosporins, like the penicillins, are beta-lactam antibiotics foundin nature. Cephalosporin C, the precursor molecule for antibioticsused in humans, was originally isolated from Cephalosporium acre-monium. Successive modifications of the cephem ring structure have
resulted in “generations” of cephalosporin antibiotics. There is noofficial scientific designation of generations; rather, the description of enhanced activity of the second generation over the first was createdas a marketing tool.12 However, the ability to distinguish the relativeactivity of the large number of cephalosporin antibiotics by generationis useful for the practitioner (see Table 292-2).13
In general, the first-generation cephalosporins (represented bycefazolin intramuscularly (IM)/intravenously (IV) and cephalexinorally (PO)) are active against gram-positive pathogens, group A
streptococcus, and penicillinase-producing Staphylococcus aureus(methicillin-susceptible strains) (MSSA), which has led to their usefor skin and skin structure infections and surgical prophylaxis, as wellas in invasive infections caused by these organisms. Although theyare better tolerated than the penicillinase-stable penicillins (e.g.,methicillin), they are somewhat less active in vitro against S. aureus,and may not be as effective in the treatment of serious infections. Of importance is the uniform lack of activity of all cephalosporins againstenterococci.
In addition to gram-positive activity, the cephalosporins are alsoactive against many strains of Escherichia coli, allowing treatment of urinary tract and intestinal infections. However, increasing resistanceto first-generation cephalosporins in gram-negative organisms duringthe past few decades has limited the usefulness of these agents in thetreatment of hospital-associated infections.
The second-generation cephalosporins have enhanced activityagainst gram-negative pathogens as well as demonstrating enhancedstability against beta-lactamases compared with first-generationagents (see Table 292-2). This increases the spectrum of activity of these agents to include many enteric gram-negative bacilli, and beta-lactamase-negative and positive strains of Haemophilus influenzae.While the activity of second-generation agents is enhanced for gram-negative organisms, the activity against staphylococci is decreased,although not sufficiently to lead to clinical failures in treatment of mildto moderate staphylococcal infections. This broad spectrum of activityallows for single-drug therapy of staphylococcal, streptococcal, and
Haemophilus influenzae infections in children. Due to poorpenetration of the first- and second-generation cephalosporins intocerebrospinal fluid (CSF), limitations exist in the treatment of invasive BSIs caused by Streptococcus pneumoniae and H. influenzaewith these agents. Within the second generation of agents, all of which
share the cephem ring structure (see Figure 292-2), are both truecephalosporins and the cephamycins. The cephamycins wereoriginally isolated from Streptomyces sp., and contain an additionalside chain that enhances stability to beta-lactamases, providing theseagents (cefoxitin and cefotetan) with improved activity against
Bacteroides fragilis. As such, these agents are effective in thetreatment of intra-abdominal infections given reasonable activityagainst gram-positive organisms (except enterococci), gram-negativeenteric bacilli, and anaerobes. Oral second-generation agents arewidely used for the treatment of upper and lower respiratory tractinfections in children, given their activity against streptococci and H.influenzae. However, with increasing beta-lactam resistance in S.
pneumoniae caused by changes in the PBP structures of thesepathogens, treatment failures of pneumococcal infections with the oralsecond-generation cephalosporins occur more commonly than at the
time these agents were first introduced.The third-generation cephalosporins have further enhanced gram-
negative activity, which extends to Pseudomonas aeruginosa forceftazidime and cefoperazone, but at the expense of a further decreasein activity against staphylococci. As with other cephalosporins,activity against enterococci is still lacking. Enhanced activity againstenteric gram-negative bacilli has led to successful therapy of UTIs andmany nosocomial infections. Therapy of infections caused by Entero-bacter, Serratia, and Citrobacter , which have the ability to producechromosomally mediated (inducible) beta-lactamases, may fail due tothe selection of organisms at the site of infection that constitutivelyhyperproduce these enzymes, and are resistant to third-generationagents.14 The third-generation cephalosporins are, in general, alsohydrolyzed by the extended-spectrum beta-lactamases (ESBLs)
S E C T IO N B Anti-Infective Therapy1434
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 16/33
produced most commonly by Escherichia coli and Klebsiella sp.15 Theactivity of the third-generation agents is superb against virtually allstrains of H. influenzae. These agents, in general, achieve CSFconcentrations that are effective in treatment of bacterial meningitiscaused by all three major pediatric pathogens: H. influenzae, Strep-tococcus pneumoniae, and Neisseria meningitidis. Of note, certainpenicillin-resistant strains of S. pneumoniae have decreasedsusceptibility to these cephalosporins and have been associated withclinical and microbiologic failure at tissue sites with decreased
antibiotic penetration, such as the central nervous system (CNS).
16, 17
However, the most active of the third-generation cephalosporinsagainst S. pneumoniae, ceftriaxone and cefotaxime, have not beenassociated with treatment failure of respiratory tract infections causedby penicillin-resistant strains when appropriate dosing regimens areused. None of the third-generation agents should be consideredoptimal for the treatment of infections caused by Staphylococcusaureus as other cephalosporins and penicillinase-stable penicillins aremore active against this pathogen.
Of the third-generation agents, ceftriaxone has a prolonged serumhalf-life compared with the other agents, allowing for its once-dailyuse. The infrequent dosing and the ability to use either intramuscularor intravenous routes of administration have allowed for outpatienttherapy of serious, invasive infections at a point when the child’scondition is stable.18
The fourth-generation cephalosporin, cefepime, maintains activityagainst Pseudomonas aeruginosa, displays enhanced stability to theampC chromosomal beta-lactamases of Enterobacter, Serratia, andCitrobacter species, while retaining signicant (but not optimal)activity against Staphylococcus aureus (see Table 292-2). This broadactivity allows for empiric therapy of neutropenic children with fever,and allows for treatment of a wide variety of nosocomial gram-negativeinfections.19–22 However, lack of activity against beta-lactamase-positive strains of Bacteroides fragilis and against Enterococcuslimits the ability to treat intra-abdominal infections using this singleagent.
The fth generation of cephalosporins, still in clinical trials,combines the activity of the third- and fourth-generation cephalo-sporins with the rst documented in vitro activity of any beta-lactamagent against CA-MRSA. These agents have been designed to bind toand inactivate PBP2a, which confers resistance in MRSA to all other
currently available beta-lactam agents.23
Carbapenems
These agents, also naturally occurring, were initially isolated from aspecies of Streptomyces, with the beta-lactam moiety contained withina carbapenem nucleus (see Figure 292-2). They demonstrate thebroadest spectrum of activity of all of the beta-lactam antibiotics andcurrently include imipenem, meropenem, and ertapenem. They areactive against both gram-positive pathogens, including staphylococciand streptococci (with moderate activity against ampicillin-susceptible enterococci), and gram-negative pathogens, including P.aeruginosa for imipenem and meropenem, with enhanced stabilityagainst both the chromosomal ampC beta-lactamases of Enterobacter,
Serratia, and Citrobacter species and the ESBLs of E. coli andKlebsiella (see Table 292-2). They are highly active against anaerobicorganisms, including beta-lactamase-producing strains of Bacteroidesand Prevotella. Of these agents, the antibacterial spectrum of activityof imipenem and meropenem is similar, whereas ertapenem matchesthe activity against enteric bacilli, but is not as potent againstPseudomonas aeruginosa. Imipenem is paired with cilastatin, a renaldehydropeptidase inhibitor that inhibits the destruction of imipenemby renal tubular enzymes providing both an increase in the serum half-life of imipenem and a decrease in the renal toxicity of the compound.Imipenem use was associated with unexpected seizures in an open,noncomparative clinical trial in children with meningitis,24 probablyattributable to competitive inhibition of the inhibitory CNS neuralpathways. Therefore, meropenem, which does not produce clinically
detectable CNS side effects, is the preferred carbapenem agent fotreatment of CNS infections, including meningitis, brain abscesepidural abscess, and subdural empyema. Ertapenem has the moprolonged serum half-life of the carbapenems, and requires only oncedaily dosing in older children (13 years of age) and once- or twice-aday dosing in younger children. These agents are all used primarily fonosocomial infections or infections in immunocompromised hoswhen the exceptionally broad spectrum of activity is essential. Datsupport clinical and microbiologic ef cacy in pneumonia, UTI
wound infections, bone and joint infections, and skin and skistructure infections. Imipenem and meropenem are reasonablsingle-drug empiric therapy of fever and neutropenia in immunocompromised children.19 As with the later-generation cephalosporinthey provide good, but not optimal, activity against Staphylococcuaureus. They provide the best activity of all beta-lactam agents againpathogens harboring either chromosomally mediated ampC betalactamases or ESBLs. In addition, single-agent therapy of appendicithas been documented to be effective, and allows for the possibilitof convalescent outpatient therapy.25 Use of such broad-spectrumagents must be weighed against the risk of promoting resistance anprofoundly altering normal flora.
Monobactams
This unique beta-lactam structure is a naturally occurring antibiotiisolated from Chromobacterium sp.; it is not fused to an adjacent ringAztreonam, the only available agent in this class, has been highlmodied chemically with side chains,26 and demonstrates gramnegative activity comparable with the third-generation cephalosporinbut without signicant gram-positive or anaerobic activity. Clinicuse in pediatrics is limited, and occurs primarily in communityacquired infections in which enteric gram-negative organisms arsuspected or proven pathogens.
Glycopeptide Antibiotics
This class of antibiotics interferes with cell wall formation in the stepthat create the glycan chains prior to cross-linking the chains in th
formation of peptidoglycan (see Figure 292-1). These antibiotics hava large, complex structure that consists of multiple peptides linketogether into three rings, with various side-chain substitutionincluding large saccharide moieties attached to the central polycyclstructure. Strong hydrogen bonds occur between the glycopeptidantibiotic and the terminal D-alanine, D-alanine dipeptide of thpentapeptide side chains of the MurNAc subunits of the glycan chainOnce bound, the glycopeptides sterically prevent the tranglycosylation steps required for lengthening the chain and thsubsequent cross-linking.5 Glycopeptide antibiotics are primarilactive against gram-positive organisms, in which the cell waconstruction occurs outside the cell membrane (see Figure 292-3Little activity is demonstrated against gram-negative organismas the large structure does not easily cross the gram-negative outemembrane, preventing contact with enzymes responsible fo
transglycosylation in the periplasmic space. Recent documenteresistance in gram-positive pathogens to vancomylycin has led tintense investigation of derivatives of vancomycin and teicoplanin, glycopeptide antibiotic available outside the United States.
Vancomycin
Vancomycin is a natural product, originally isolated from Streptomycesp. in 1956. Vancomycin is the only glycopeptide currently availabin the United States. Originally developed to treat staphylococcinfections, vancomycin was rarely used following the availability othe penicillinase-stable penicillins, which were better toleratedHowever, with the rst appearance of healthcare-associate(HA)-MRSA four decades ago, vancomycin played a continuing ro
Antimicrobial Agents C H A P T E R 292 143

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 17/33
in the treatment of nosocomial Staphylococcus aureus infections.Recently, with the increasing prevalence of CA-MRSA, vancomycin isnow routinely used in the empiric therapy of serious and life-threatening staphylococcal infections.
Some concern exists for the degree of bactericidal activity andclinical efficacy demonstrated by vancomycin compared with thepenicillinase-stable penicillins for treatment of MSSA. Based on theseconsiderations and greater toxicity of vancomycin compared withbeta-lactam agents, vancomycin is not generally the preferred therapy
in infections caused by MSSA. Newer glycopeptides and lipopeptideshave shown increased in vitro activity against S. aureus compared withvancomycin, although prospective, controlled clinical data todocument improved outcomes are not available, particularly forchildren.5,27,28
Resistance to vancomycin has developed in several ways. In Enterococcus sp., vanA resistance, that leads to virtual completeresistance to vancomycin, occurs with a transmissible set of 7 genes whichencode a series of biologic functions. These genes allow Enterococcus tosense the presence of vancomycin, to cleave the D-alanine, D-alaninedipeptide from the pentapeptide chain, and to substitute enzymatically D-alanine, D-lactate at the terminus of the pentapeptide, resulting in a 1000-fold decrease in binding of vancomycin.28–30 The new pentapeptideappears to be as viable a precursor for peptidoglycan formation as theoriginal pentapeptide. Enterococcusalso has less common mechanisms of resistance.29 The vanA resistance mechanism has now been detected in4 adult patients with staphylococcal infections, creating vancomycin-resistant S. aureus (VRSA). A more common resistance mechanism of S. aureus to vancomycin, producing intermediately susceptible strains orVISA, is proliferation of the D-alanine, D-alanine glycan structures in adisorganized, thickened cell wall, leading to increases in the bindingand trapping of vancomycin to nonfunctional dipeptides.5,31,32 Undervancomycin pressure, these strains, which are present within every largepopulation of staphylococci, are selected out. This mechanism of resistance to vancomycin, called heteroresistance, is either stable orunstable.33
Three new glycopeptide antibiotics, dalbavancin, telavancin, andoritavancin, have demonstrated clinical efficacy in small clinical trials,but none is yet approved for use. Modifications of the glycopeptide toenhance binding to targets, to increase stability of antibiotic bindingby creating glycopeptide dimers, and to anchor the glycopeptide to the
cell membrane have all been successful strategies at enhancing theactivity of this class of agents.
Clinical uses of vancomycin include therapy of gram-positiveinfections in children who are penicillin-allergic, therapy of infectionscaused by Streptococcus pneumoniae that are resistant to penicillin,34
and therapy of infections caused by MRSA. Treatment of Clostridiumdifficile infections with orally administered vancomycin is highlyeffective, but it has not been recommended for the past decade due tothe emergence of vancomycin-resistant enterococci (VRE) followingtherapy. However, in selected cases of metronidazole failure,vancomycin represents effective alternative therapy.
A common reaction may occur with the rapid infusion of vancomycin, the red-man or red-neck syndrome, characterized byflushing and hypotension. This reaction is histamine-mediated and notimmunoglobulin E-mediated and is distinct from anaphylaxis. The
risk of this reaction varies directly with the rapidity of the vancomycininfusion; therefore, each dose of vancomycin is usually infused over1 hour. For children who develop this reaction, prolonging the infusionor pretreating with antihistamines may allow continuation of therapywith vancomycin.
Cell Membrane Active Antibiotics
Daptomycin
Daptomycin, a natural product derived from Streptomyces sp., is anovel lipopeptide antibiotic that is rapidly bactericidal based on effectson the gram-positive cell membrane. It has a unique structure that
consists of 13 amino acids, with 9 peptides linked together in a ringstructure, attached to a lipophilic tail that inserts into the cellmembrane. The mechanism of action of daptomycin is not wellunderstood, but it appears that depolarization of the membrane occursas the antibiotic polymerizes within the bacterial cell membrane,producing channels in the membrane that result in leakage of cellcontents, inhibition of protein, DNA and RNA synthesis, and celldeath. Daptomycin is one of the most rapidly bactericidal antibioticsagainst Staphylococcus aureus based on in vitro assays.35 It is active
on a wide variety of gram-positive organisms, including MSSA,MRSA, VRSA, streptococci, and enterococci (including VRE) (seeTable 292-2).
Clinical use of daptomycin has focused on MRSA infections thatare unresponsive to vancomycin. Efficacy has been demonstrated inadults for skin and skin structure infections, and for BSI. Surprisingly,daptomycin is not effective for the treatment of pneumonia, based onclinical trials in which response rates were not equivalent tocomparator agents, and in which relatively low concentrations of daptomycin were found in bronchial-alveolar epithelial lining fluidand lung parenchyma.36 No pharmacokinetic or clinical study has beenreported in children. The prolonged half-life in adults of 8 to 9 hoursallows for once-daily dosing.
In the first human clinical trials several years ago, daptomycin wasassociated with a high incidence of myalgias and muscle weaknessaccompanied by elevations in serum creatine kinase. This adverseeffect appears to depend on the frequency of dosing, with less adverseeffect noted with decreased frequency of dosing. While toxicity wasprohibitive when daptomycin was administered every 8 hours, it hadno greater toxicity than comparator agents when administered toadults in a dose of 4 mg/kg once daily.37 With these concerns in mind,it is currently recommended to monitor creatine kinase levels weeklyduring therapy.
Colistin
Colistin is a polymyxin antibiotic that is a natural product isolatedfrom Bacillus polymyxa, and consists of 10 linked peptides in a ringstructure attached to a fatty-acid side chain. Colistin is also knownas polymyxin E, structurally similar to polymyxin B, which is used
extensively as a topical agent. The polymyxin antibiotics were firstdiscovered in 1947, with the first use of colistin in the United Statesin 1959.38,3 The active antibiotic, colistin, is available as a sodiummethanesulfonate salt, known as colistimethate. The dosage iscalculated as the colistin base, at 2.5 to 5.0 mg/kg per day given in 2to 4 divided doses.39 Colistimethate is hydrolyzed to colistin, but therate and extent of hydrolysis, and the contributions of biologic activityof the parent compound and products of metabolism are not wellknown. The mechanism of bactericidal activity against thecytoplasmic membrane is based on cationic detergent activity, bindingto lipopolysaccharides in the outer cell membrane, and displacingcalcium and magnesium. Permeability changes subsequently occurin the membrane, disrupting the osmotic gradient of the cell andimpacting cellular metabolism and nucleic acid synthesis.40
The antibiotic has significant toxicity and should only be used
when no other agents are active or available. Toxicities are primarilyrenal and neurologic. Renal side effects include decreased urine outputwith elevated blood urea nitrogen and serum creatinine, proteinuria,hematuria, and acute tubular necrosis. Because the drug is eliminatedby renal excretion, it is imperative to assess renal function closelyduring therapy, decreasing the dosage if any degree of renalinsufficiency is noted. Drug accumulation and additional renal toxicityoccur if dosing is not altered when renal insufficiency first occurs. Therenal toxicity is usually reversible if detected early. Neuromuscularside effects are evident in up to 50% of patients treated, most oftenmanifest as oral and perioral paresthesias, weakness, lethargy,confusion, ataxia, and respiratory muscle paralysis.39,40 Colistin alsocrosses the placenta and has been shown to be teratogenic in certainlaboratory animals following drug exposures in pregnant animals.
S E C T IO N B Anti-Infective Therapy1436
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 18/33
Due to the toxicity of this antibiotic, clinical use of parenteralcolistimethate is limited to therapy of infections caused by multidrug-resistant gram-negative pathogens for which no other options areavailable. Other clinical uses include parenteral therapy in ventilatedpatients with nosocomial multidrug-resistant gram-negative pulmo-nary infections41 and aerosolized colistimethate in children with cysticfibrosis as adjunctive antimicrobial therapy.42 There is anecdotalexperience with aerosol use in adults with ventilator-associatedpneumonia.
RIBOSOME-ACTIVE ANTIBIOTICS
The bacterial ribosome, highly conserved over millions of years, haslong been a target of antibiotics. As our ability to understand thefunction of the ribosome has increased, and with recent advances inour ability to visualize ribosomal structures with crystallography, ourknowledge of the mechanism of activity of both older and neweragents and our ability to design more effective antibiotics has im-proved substantially. The ribosome contains a 30S and a 50S subunit,each comprised of rRNA and ribosomal proteins. Several sites havebeen documented to be antibiotic targets on each of the subunits andat the junction of the subunits. Targets include: the entry site of mRNA, the initial recognition and binding site of tRNAs, the site of attachment of the tRNA-peptide chain where peptide bonds areformed (the peptidyl transferase center), and the exit channel of thegrowing polypeptide.43–45 The critical chemical and structuralrelationships between the ribosomal rRNA and the peptidyltransferase center, which promote the chemical reactions to create anew peptide, as well as movement of mRNA and the newly formedpeptide through the ribosome provide opportunities for interference inbacterial protein synthesis.
Macrolides
The currently available macrolides consist of the erythromycins,azithromycin, clarithromycin, and telithromycin. All share structuralsimilarities which include a 14-membered lactone ring (erythromycin,clarithromycin, telithromycin) or 15-membered ring (azithromycin),
all of which bind to at least one site in common within the peptide exittunnel of the ribosome: domain V of the 23S RNA within the 50Ssubunit. Binding to a specific adenine residue of the rRNA, A2058,within this channel prevents the orderly movement of protein out of the ribosome.46–49
Clarithromycin is structurally very similar to erythromycin, withonly the addition of a single methyl group to the erythromycin ring,primarily conferring improved stability to gastric acid. Whereaserythromycin, clarithromycin, and azithromycin contain a cladinosecarbohydrate attached to the lactone ring, telithromycin substitutes aketone in this position (hence the name, ketolide) while adding highlycharged side chains to the C11 and C12 positions. These changesimprove the binding characteristics to both the peptide tunnel bindingsite within domain V, and create an additional unique binding site atadjacent domain II within the ribosome, improving activity against
many macrolide-resistant gram-positive organisms. Azithromycin isstructurally similar to erythromycin, but contains a 15-membered(rather than 14-membered) ring with the addition of a nitrogen atomwithin the ring itself, structurally changing the drug from a macrolideto an azalide, but containing the same side chain-attachedcarbohydrate moieties as erythromycin. This change improves gram-negative activity as well as increasing gastric acid stability. Thedegradation products of azithromycin provide far less stimulationof gastric motility, improving the tolerability of azithromycin overerythromycin.
In general, the macrolides are inhibitory to bacteria, notbactericidal, and therefore are not commonly employed in thetreatment of serious and life-threatening infections when otherbactericidal agents can be used. The various macrolide agents have
different binding affinities for their ribosomal targets in differenorganisms. The binding is generally reversible, with a more prolonged ratof dissociation off the ribosome potentially adding to a more prolongepostantibiotic effect seen with some macrolides (Table 289-1).46
The macrolides are most active against gram-positive cocci anbacilli, and, to a lesser extent, gram-negative bacilli (see Table 292-2Some of these agents are also active against spirochetes and certaimycobacteria. Pathogens that lack a formal cell wall and are nosusceptible to beta-lactam antibiotics often remain susceptible t
macrolides, including Mycoplasma and Ureaplasma species.All of the macrolides achieve high intracellular antibioticoncentrations within phagocytic cells. These concentrations are oftemuch higher than those measured in serum, providing access of thesantibiotics to infected tissue spaces by means of neutrophils anestablishing higher tissue concentration than antibiotics that enteprimarily by diffusion alone. However, much of the macrolide present in an intracellular location, allowing less free drug available texpose extracellular pathogens. Due to high intracellular concentrations, the macrolides are particularly effective therapy againsusceptible intracellular pathogens.
In general, this class of drugs is well tolerated. However, thgastrointestinal side effects of erythromycin may be problematic isome children. Clarithromycin, azithromycin, and telithromycin arall better tolerated than erythromycin. With the exception oazithromycin, this class of antibiotics is metabolized by hepaticytochrome P-450 system, and drug–antibiotic interactions should bconsidered as they may increase or decrease the macrolide anconcurrent drug concentrations.50 Azithromycin has demonstrateminimal drug–drug interactions, and may represent the preferremacrolide in certain situations, particularly for immunocompromisechildren receiving multiple concurrent medications.
Not surprisingly, resistance to the macrolides is documented whemolecular changes occur at the critical ribosomal attachment sitemost commonly a mono- or dimethylation of the A2058 adeninbinding site. The methyltransferase enzymes encoded by grampositive organisms are most often inducible, but may be constitutivelproduced, leading to high-level resistance to erythromycin, clarithromycin, and azithromycin, and decreased susceptibility to telithromycin. Less frequent alterations at this site also impact binding, witeither substitution of guanine for adenine, or structural changes in th
L4 ribosomal protein.51 Efflux pumps represent another commomechanism of resistance in gram-positive pathogens, includinpneumococcus, group A streptococcus, and Staphylococcus aureuThe most common pumps are active against erythromycin, clarithromycin, and azithromycin, whereas most of the strains harboring thespumps remain susceptible to telithromycin.52,53
Erythromycin
Erythromycin, a natural product isolated from Saccharopolysporerythraea (formerly Streptomyces) in 1949, was first approved foclinical use in 1952. Erythromycin is degraded by gastric acid, and halong been associated with stimulation of motilin receptors in thstomach and possibly in the colon, leading to adverse gastrointestinside effects, including cramping and diarrhea.54, 55 Many preparation
have attempted to bypass exposure of erythromycin to gastric acidthereby avoiding products of macrolide hydrolysis. These preparationinclude enteric coating of orally administered tablets, delayed-releasformulations, polymer coating of beads, and various formulations osalts and esters.56 The lactobionate salt used for intravenouadministration of erythromycin can produce phlebitis at the site oinjection.
Erythromycin is used for the treatment of group A streptococcinfections in children who are penicillin-allergic. Erythromycin is aalternative treatment for both streptococcal pharyngitis anstreptococcal or staphylococcal impetigo. The clinical use oerythromycin for respiratory tract infections caused by Streptococcu
pneumoniae has been greatly diminished by the development owidespread resistance to the macrolides.34 Empiric therapy of uppe
Antimicrobial Agents C H A P T E R 292 143

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 19/33
respiratory tract infections (otitis media and sinusitis) or lowerrespiratory tract infections (pneumonia) potentially caused by S.
pneumoniae has a relatively high likelihood of failure, particularly inyounger children who are at risk of infections caused by antibiotic-resistant strains. For upper respiratory tract infections, erythromycinhas inadequate activity against H. influenzae, and must be paired withanother agent such as a sulfonamide for empiric therapy. Macrolidetherapy for atypical pneumonia caused by Mycoplasma pneumoniae,Chlamydophila pneumoniae, or Legionella pneumophila remains
effective for the vast majority of these pathogens.Erythromycin and azithromycin are the preferred antibiotics fortreatment of Campylobacter gastroenteritis. Erythromycin alsoremains the most appropriate therapy for diphtheria (Corynebacteriumdiphtheriae). Erythromycin, clarithromycin, or azithromycin isrecommended for treatment or prophylaxis of pertussis ( Bordetella
pertussis). Azithromycin is preferred for treatment or prophylaxisfor pertussis in neonates, based on concerns for the developmentof pyloric stenosis.57 Efficacy of erythromycin has also beendemonstrated in infections caused by Chlamydia trachomatis,including neonatal conjunctivitis and pneumonia, as well as urogenitalinfections during pregnancy. Erythromycin is also active in vitroagainst Ureaplasma urealyticum, but the role of erythromycin in thetreatment of neonatal respiratory tract infections caused by thisorganism is not well defined.58
Clarithromycin
With the improved activity demonstrated against H. influenzae andimproved tolerability compared with erythromycin, treatment of respiratory tract infections is the most common clinical use forclarithromycin. Food and Drug Administration (FDA)-approvedindications include pharyngitis/tonsillitis, acute otitis media, acutemaxillary sinusitis, and community-acquired pneumonia caused bysusceptible strains of S. pneumoniae, H. influenzae, Moraxella catar-rhalis, Mycoplasma pneumoniae, Chlamydia pneumoniae, and
Legionella pneumophila. For S. pneumoniae, strains that are resistantto erythromycin from either methyltranferase or efflux mechanismsare also resistant to clarithromycin. The activity of clarithromycinagainst H. influenzae is only moderate, but in noninferiority trials of clarithromycin in the treatment of respiratory tract infections, the
microbiologic and clinical efficacy was not significantly less than thatof other approved agents.
Clarithromycin is one of the most effective macrolides fortreatment and prevention of disseminated mycobacterial infectionsdue to Mycobacterium avium-intracellulare complex (MAC) inhuman immunodeficiency virus (HIV)-positive hosts. Although notwell studied in normal immunocompetent children, clarithromycinmay play a role in the treatment of cervical adenitis and pneumoniacaused by MAC (or other nontuberculosis mycobacteria proved tobe susceptible in vitro), in conjunction with other antibiotics and/orsurgery.
Clarithromycin also plays a role in the treatment of Helicobacter pylori infections (the primary cause of duodenal ulcers) incombination with amoxicillin and lansoprazole, or omeprazole, or incombination with ranitidine.59 Clarithromycin has demonstrated
efficacy similar to erythromycin in pertussis infections in smallclinical trials, and is considered as one of three first-line drugs.Although approved for treatment of skin and skin structure infections,clarithromycin is not often used for this indication as other more cost-effective or more active agents are available in the treatment of infections caused by Staphylococcus aureus. Similarly, other beta-lactam and macrolide antibiotics are preferred for the treatment of streptococcal pharyngitis.
Azithromycin
Azithromycin has among the highest intracellular concentrations of the macrolides and provides the most prolonged tissue concentrationsat the site of infection, allowing the antibiotic to be provided for very
short courses for respiratory tract infections. The activity againstStreptococcus pneumoniae is similar to erythromycin and clar-ithromycin. Erythromycin-resistant strains of S. pneumoniae are alsoresistant to azithromycin. Activity against H. influenzae is moderate,with increased activity in vitro compared with erythromycin, but withdecreased in vitro activity compared with clarithromycin andtelithromycin. However, the impact of relatively small differences insusceptibility in vitro is likely to be offset by higher concentrations of antibiotic at an intracellular site of infection. Azithromycin is also active
against the pathogens causing atypical pneumonia (see Table 292-2).Azithromycin is far better tolerated than erythromycin, and can begiven once daily, and comes in both oral and intravenous formulations.Based on noninferiority clinical trials, azithromycin has beenapproved for treatment of streptococcal pharyngitis, acute otitis media,sinusitis, and community-acquired pneumonia in children. Because of prolonged tissue concentrations, particularly using larger azithromycindosages, 5-day, 3-day, and 1-day treatment courses have been shownto be comparable for clinical and microbiologic outcomes to 10-daytreatment courses of comparator beta-lactam antibiotics in acute,uncomplicated otitis media. However, as the dosage increases, thegastrointestinal tolerability of the antibiotic decreases, with vomitingand diarrhea occurring in about 10% of children receiving 30 mg/kgas a single dose.60 Although clinical data on single-dose treatmentcourses for otitis media are available, data exist only for treatmentcourses of 3 and 5 days for sinusitis, and for 5 days for treatment of community-acquired pneumonia and streptococcal pharyngitis. Thedosage for treatment of streptococcal pharyngitis is 12 mg/kg per dayonce daily for 5 days, which is larger than that for otitis and providesa total dosage of 60 mg/kg for this infection.
Azithromycin has the widest use in children in the treatment of upper and lower respiratory tract infections.61 However, other useshave been documented in clinical trials, although FDA approval formany of these infections has not been requested. Treatment of pertussis has been shown to be effective in small trials.62 Based onconcerns for pyloric stenosis caused by erythromycin in young infants,azithromycin is the recommended macrolide therapy or prophylaxisfor infants under 1 month of age and is considered on an equal footingwith erythromycin and clarithromycin in older individuals.57
Azithromycin is also used in the treatment of sexually transmittedinfections, including Chlamydia trachomatis-caused infections
(urethritis, cervicitis, and lymphogranuloma venereum), chancroid,granuloma inguinale, and gonorrhea.63
Similar to clarithromycin, azithromycin has been shown to play arole in the prophylaxis and therapy of MAC infections in HIV-positivepersons.64 Azithromycin may also have a role in therapy of cutaneousand lymph node infection caused by these pathogens in normal children.
Azithromycin has enhanced activity compared with the othermacrolides against many gastrointestinal pathogens, including
Escherichia coli, Salmonella, Shigella, and Campylobacter .65 In vitroactivity demonstrated against Salmonella is particularly advantageousgiven the intracellular location of the pathogen in this infection.66 Withwidespread resistance among gastrointestinal pathogens to beta-lactam antibiotics, fluoroquinolones, and trimethoprim-sulfamethoxa-zole in certain parts of the world, the utility of empiric therapy of traveler’s diarrhea with azithromycin has increased.67
Azithromycin is the only antibiotic that has been prospectivelyevaluated for the treatment of cat-scratch lymphadenitis caused by
Bartonella henselae and is one of the preferred therapies for thisinfection.68 However, the clinical response to treatment of lymph nodedisease is not dramatic, and azithromycin has not been evaluatedprospectively for the treatment in other tissue sites of infection, suchas liver, bone, or CNS.
Telithromycin
Telithromycin, a semisynthetic macrolide, has the same ring structureas erythromycin, but with the substitution of the cladinose side chainwith a ketone group (enhances gastric acid stability), and with theaddition of a cyclic carbamate group providing a second link between
S E C T IO N B Anti-Infective Therapy1438
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 20/33
the C11 and C12 positions of the ring. In addition, a long alkyl-arylextension side chain has been added to the lactone ring, whichprovides for two additional binding sites for telithromycin to theribosome. The ketone substitution also appears to prevent theinduction of methyltransferase activity that has been associated withmono- or di-methylation of the primary macrolide binding site in thepeptide tunnel and loss of antibiotic activity of other macrolides.53, 69
As a result, telithromycin has improved activity against manyerythromycin-resistant strains of S. pneumoniae and group A strepto-
coccus compared with the other macrolides. However, telithromycinhas only moderate activity against H. influenzae strains, equivalent toclarithromycin. Like clarithromycin, in clinical trials of respiratorytract infections designed to demonstrate noninferiority, telithromycinwas statistically similar to comparators (amoxicillin, amoxi-cillin/clavulanate, clarithromycin, or cefuroxime).
An unusual visual side effect of telithromycin, a decreased abilityto accommodate visually and to release accommodation, may affectup to 2% of certain population groups. For these few people, it ismomentarily difficult to focus on distant images if one has beenfocusing on a close image, and vice versa.
At present, limited pharmacokinetic, safety, and clinical efficacydata exist in children and the antibiotic is only FDA-approved foradults 18 years of age and older.
TetracyclinesThe tetracyclines were also originally derived as natural products fromStreptomyces spp., with discovery in the 1940s and subsequent availa-bility of two agents by 1948, chlortetracycline and oxytetracycline.70
The tetracycline antibiotics bind reversibly to the ribosome at theacceptor site (A site) where the amino acid-charged tRNA binds to theribosome immediately adjacent to the site on the ribosome holding themRNA strand.
The aminoacyl tRNA binds together at the A site with anelongation factor (EF-Tu) and guanosine triphosphate (GTP), whichsupplies the energy required to drive protein synthesis. The proteinsynthesis step includes the chemical reaction to attach the amino acidto the growing peptide chain, together with changes in theconformation of the ribosome that are associated with movement of the protein chain and the mRNA through the ribosome, followed by
the subsequent release of the “empty” tRNA.44 It appears that the flat,four-ring structure characteristic of the tetracyclines binds to at leasttwo locations within the ribosome. Binding at the classically recog-nized A site appears to prevent movement of the tRNA/mRNA/EF-Tucomplex into the “P site” (peptidyl site) by steric hindrance, whichprevents elongation of the growing peptide. Binding to a second site inthe 30S ribosome may stabilize the ribosome in an inappropriateconformation at the crucial site of recognition of the aminoacyl tRNAsanticodon with the corresponding codon within the mRNA, therebypreventing placement of the correct amino acid in the elongatingchain.71 Inhibition of peptide formation by tetracyclines occurs afterthe binding of the tRNA to the complex and after expenditure of GTP-mediated energy, presenting the bacteria with an energy cost inaddition to blocking the synthesis of a new protein.
The tetracyclines are effective against many gram-positive and
gram-negative bacteria as well as against cell wall-deficient pathogens( Mycoplasma, Rickettsia) and certain single-cell parasites (see Table292-2). Eukaryotic cells have elongation factors different thanbacteria, and are therefore not susceptible to the protein synthesisinhibition activity of this class of antibiotics. The tetracyclines are, ingeneral, bacteriostatic due to the reversible nature of binding to theribosome. The tetracyclines enter the gram-negative cell wall throughouter membrane porin proteins and are sufficiently lipophilic to allowpassage through the cytoplasmic membrane of both gram-negative andgram-positive bacteria.
Resistance to tetracyclines occurred quickly following theiravailability, primarily based on efflux pumps and, to a lesser extent, onthe presence of ribosomal protection proteins.70 These resistancemechanisms are present on plasmids, conjugative transposons and
integrons, allowing free exchange of resistance determinants betweea wide range of bacteria. Over 200 different efflux pumps have beecharacterized, most of which are active against tetracycline, somof which are also active against minocycline, and fewer still also activagainst tigecycline. The ribosomal protection proteins have sequenchomologies with bacterial elongation factors present in thtRNA/mRNA/EF-Tu complex. It is believed that, as these protectioproteins themselves bind to the ribosome, changes in the conformatioat the tetracycline binding site occur, preventing the binding o
tetracycline but not interfering with protein synthesis. Resistance athe second 30S ribosomal binding site has also been described, due tbase substitutions in the rRNA of the 30S unit.
Advances in the design of the structure of the early tetracyclineled to doxycycline (in 1967) and minocycline (in 1972), both of whicprovided a greater spectrum of activity and improved solubilitycreating improvements in both oral and parenteral preparations (seTable 292-2). Doxycycline and minocycline may be taken with foo(with the exceptions noted below), as the absorption of these drugs inot significantly decreased with meals. In most cases, doxycycline anminocycline demonstrate increased activity against gram-positivorganisms, and decreased activity against gram-negative organismcompared with tetracycline. Activity against Enterococcus faeciumbut not E. faecalis, was achieved with newer agents. Minocyclinhowever, demonstrates improved activity against gram-negativorganisms, including Haemophilus influenzae, Moraxella catarrhali
Escherichia coli, and Klebsiella compared with tetracycline, but onlfair activity against Salmonella and Shigella spp. and Pseudomonaaeruginosa. Tigecycline increases the spectrum of activity againmany enteric gram-negative bacilli and anaerobes, includin
Bacteroides fragilis, but still lacks a high degree of activity againP. aeruginosa (see Table 292-2).
Historically clinical use in pediatrics has been limited by thbinding of tetracyclines to teeth and bones in growing childrenPermanent staining of the teeth (not affecting the structural integrity othe tooth) and enamel hypoplasia occurs with any tetracyclinantibiotic, with the degree of staining directly proportional to thnumber of tetracycline courses prescribed. A single course of therapis not associated with clinically detectable changes.72, 73 Stabcalcium complexes can also develop in bone, and reversible decreasein long-bone growth rates in juvenile animals have been observed. Th
clinical impact of these observations for children is not well definebut has been a cause for concern, limiting the use of tetracyclines tchildren 8 years of age and older.74 In addition, the tetracyclines crosthe placenta to expose the fetus; skeletal embryopathy in experimentanimals has been noted. The oral tetracylines cannot be taken witdairy products due to the insoluble chelation complexes that form witcalcium; similar complexes form with magnesium and iron ionWhen ingested with foods containing these ions, absorption from thgastrointestinal tract is blocked.
In adults and older children, the tetracyclines have been used fothe treatment of mild to moderate respiratory tract infections, skiand skin structure infections (most commonly acne), and sexualltransmitted infections. Some agents in this class demonstrate activitagainst strains of CA-MRSA,75 penicillin-resistant pneumococci, anVRE, and have been used in the treatment of these infection
However, few prospective, comparative data are available to assess thefficacy of this class of antibiotics against these pathogens, with thexception of recent studies on tigecycline.
Important infectious diseases for which the tetracyclines remaifirst-line therapy include infections caused by the Rickettsia (monotably Rocky Mountain spotted fever), tularemia (oral therapy foless severe infections), brucellosis (with rifampin), cholera, chlamydigenital infections, and Lyme disease ( Borrelia burgdorferi) in oldechildren.
Tigecycline
Tigecycline is a chemically modified minocycline, with the addition oa t -butylglycylamido side chain to the C9 carbon of the “D” tetra
Antimicrobial Agents C H A P T E R 292 143

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 21/33
cycline ring.76 Tigecycline is not affected by the majority of effluxpumps and ribosomal protection proteins which decrease the activityof other tetracyclines. It has a higher binding affinity to the ribosomalbinding site than the previous tetracyclines70 and has a broaderspectrum of activity than any other tetracycline agent (see Table 292-2). In rat models, bone discoloration was documented, suggesting thattigecycline forms calcium complexes in bone similar to othertetracycline antibiotics.
In adults, tigecycline is approved for treatment of complicated
skin and skin structure infections and complicated intra-abdominalinfections given its activity against enteric gram-negative bacilli andanaerobes, including Bacteroides fragilis. Tigecycline retains activityagainst the agents of atypical pneumonia that is equivalent to, or betterthan, earlier tetracyclines.
The ultimate clinical role of tigecycline has yet to be defined in thetreatment of nosocomial infections caused by multidrug-resistantgram-negative and gram-positive organisms that remain susceptible totigecycline. For children, the risks of bone toxicity and tooth stainingneed to be balanced with the benefits of therapy, particulary for drug-resistant pathogens. For situations in which no alternatives exist, thetetracyclines represent effective therapy.
Lincosamides
The lincosamides are naturally occurring compounds derived fromStreptomyces spp. Clindamycin, approved in 1966, is the only lincos-amide available in the United States and is a semisynthetic derivativeof lincomycin. The lincosamide antibiotics bind to the 50S subunit ata site which overlaps both the A and P sites on the ribosome,preventing the docking of charged tRNAs and their movement throughthe peptidyl transferase center, thus inhibiting the formation of protein. The P-site attachment of clindamycin occurs at the sameribosomal structural bases as the macrolide binding sites (A2058,A2059), explaining the competitive inhibition between binding of thetwo classes of antibiotics for the ribosome, as well the resistance thatoccurs to both antibiotics by altering a single base.49 The lincosamidesare generally considered bacteriostatic, although bactericidal activitycan be demonstrated against certain organisms at antibiotic concen-trations 2 to 4 times the minimum inhibitory concentration (MIC). 77
Resistance to the lincosamides occurs primarily for bacteriaconstitutively producing the methyltransferase that mono- ordimethylates the A2058 adenine present at the outlet of the ribosomalpeptidyl tranferase center. This inducible enzyme is most often onlyactivated in the presence of the appropriate substrate, usually amacrolide. In contrast, the lincosamides do not appear to induce themethylase enzyme. Therefore, organisms which have inducibleresistance should remain susceptible to clindamycin, even followingexposure to the antibiotic. However, genetically altered strains thatconstitutively produce methylase occur at a rate of approximately onein 107 Staphylococcus aureus organisms, raising the concern that forserious infections involving greater than 107 organisms, selection of constitutive mutants may occur during therapy, with subsequenttreatment failure.52 The commonly encountered efflux pumps that areactive against the macrolides are not active against clindamycin.
High intracellular concentrations of clindamycin in phagocyticcells are believed to be beneficial in certain clinical infections.78
However, no prospectively collected data have confirmed the benefit inclinical or microbiologic cure of infection either as a result of improved intracellular killing of organisms such as staphylococci, orimproved delivery of clindamycin to the site of infection throughphagocytic cell migration.
Clinical use of clindamycin has changed substantially over the pastdecade. It has commonly been used for its activity against anaerobesin the treatment of intra-abdominal infections such as appendicitis,although the percentage of strains of Bacteroides fragilis resistant toclindamycin is increasing.79 Deep head and neck space infections,and aspiration pneumonia (with or without empyema), also causedprimarily by anaerobes and gram-positive cocci, continue to be
excellent uses of clindamycin. Less common uses in the past havebeen for treatment of gram-positive cocci, such as failures of penicillinin group A streptococcal pharyngitis, and for treatment of S. aureusinfections in children who could not tolerate beta-lactam antibiotics.
In the early 1990s, the emergence of penicillin and macrolide-resistant Streptococcus pneumoniae causing upper and lowerrespiratory tract infections promoted the use of clindamycin in thetreatment of mild to moderate respiratory tract infections in children.Although no formal, randomized prospective comparative studies
were performed in acute otitis media, sinusitis, and pneumonia,clindamycin has been recommended for treatment of penicillin-resistant pneumococci in these situations.80
Since the mid-1990s, with the dramatic emergence of CA-MRSA,the use of clindamycin for skin and skin structure infections and boneand joint infections has also increased substantially. Although mostpublished data on the efficacy of clindamycin in the treatment of MRSA are retrospective, clindamycin appears to be effective for thesepathogens.81,82 While some regions of the United States in whichMRSA is prevalent have documented decreasing resistance toclindamycin due to spread in the community of certain clindamycin-susceptible clones,83 other areas may experience increasingclindamycin resistance.
The ability of clindamycin to target ribosomal protein productionhas led investigators to consider its use in the treatment of toxin-mediated infections caused by Staphylococcus aureus (toxic shock syndrome) and Streptococcus pyogenes (toxic shock-like illness),either alone or in combination with cell wall active antibiotic agents.In vitro data and retrospectively analyzed human data suggest somebenefit of combined therapy.84
The principal adverse event associated with clindamycin is a directfunction of its activity against normal anaerobic gastrointestinal flora– diarrhea.85 Clostridium difficile-mediated pseudomembranous colitisis a potential complication of virtually any broad-spectrum antibiotic,including clindamycin. Accurate, prospectively collected data on theincidence of C. difficile-positive enterocolitis are not available for clin-damycin-treated children, but increasing reports of enterocolitis havenot occurred with the increased use of clindamycin for pneumococcalor staphylococcal infections.
Aminoglycosides
The aminoglycoside class of antibiotics is comprised of manyantibiotics originally derived from organisms, including Streptomycesspp. and Micromonospora spp. In general, these antibiotics contain a2-deoxystreptamine ring attached to two or three additional moieties,most often amino sugars, all connected together by glycosidiclinkages. Substitutions at up to 10 different positions on the three ringsor their associated amino groups have led to the creation of severalaminoglycoside antibiotics. However, given the nephrotoxicity andototoxicity inherent in the aminoglycosides, little recent activity hasoccurred in the development of newer agents by the pharmaceuticalindustry. All aminoglycosides currently available in the United Statesare generic, with gentamicin, tobramycin, and amikacin representingthose most often used in children.
All of the aminoglycoside antibiotics share a common binding
region within the 30S ribosome. This region is located at the peptidyltransferase center where charged tRNAs are first recognized andattach to the A site. With aminoglycoside binding close to the A site,conformational changes occur at the ribosomal tRNA docking sitewhich creates enhanced affinity for tRNA binding, including incorrectbinding of noncognate tRNAs which do not match the correspondingcodon on the mRNA. With attachment of incorrect tRNAs, misreadingoccurs and amino acid sequences in the resulting peptide are incorrect,leading to the creation of nonfunctional proteins. With binding of theaminoglycoside, proofreading for the accuracy of the attached tRNAis also compromised, as subsequent conformational changes thatshould occur in the 30S unit to allow for exact recognition of theattached tRNA cannot take place.43,44,86 The structures of two of thelarger aminoglycosides, streptomycin and spectinomycin, allow for
S E C T IO N B Anti-Infective Therapy1440
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 22/33
additional binding sites within the 30S ribosome. Streptomycinattaches at four different domains within the 30S rRNA, as well ashaving a unique attachment to one of the ribosomal proteins.Spectinomycin, a larger structure with fused rings, appears to bind atthe A site in a unique manner which also blocks the movement of theaminoacyl tRNA/peptidyl tRNA/EF-Tu complex from the A site to theP site during the peptidyl transferase reaction. With the production of abnormal proteins that may be incorporated into cellular structuressuch as the cell membrane, increased permeability of the membrane
occurs. With increased permeability, the aminoglycosides demonstrateenhanced entry into the cells, allowing further saturation of amino-glycoside binding sites on the ribosome, thus preventing the formationof new, functional ribosomes, which ultimately results in cell death.The aminoglycosides are bactericidal, and show concentration-dependent killing of bacteria (Table 289-1).
Resistance to aminoglycosides occurs primarily with the acquisi-tion of a variety of aminoglycoside-modifying enzymes, many of whichare carried on plasmids and transposons for efficient spread betweenbacteria. The most common are acetyltransferases, adenyltransferases,and phosphotransferases.86 Many different efflux pump systems alsoplay a major role in aminoglycoside resistance, particularly in gram-negative bacilli.86
Of interest, the Actinomyces species from which the aminoglyco-sides are derived contain resistance mechanisms to the antibiotics theyproduce. Ribosomal RNA methylase genes of this genus that conferresistance to aminoglycosides were not believed to have clinicalrelevance, but have recently been identified in clinical isolates of enteric gram-negative bacilli and Pseudomonas aeruginosa.87
Although this mechanism of resistance is currently uncommon, thesegenes now have the potential to spread quickly within clinical settings.
The clinical use of aminoglycosides occurs primarily for thetreatment of gram-negative facultative bacillary infections – from thepremature neonate through the adolescent.88,89 The antibiotics arestrongly polar, with high solubility in water, but with poor solubility inlipids resulting in poor penetration into the CNS, vitreous, bronchialsecretions, and saliva. However, these antibiotics are concentrated inthe proximal renal tubules and excreted in urine and achieve urinarytract concentrations up to 100 times the serum concentration. Due tothe toxicity of the aminoglycosides at serum concentrations whichare only 5- to 10-fold above the bacterial MICs, they are not usually
used as the sole agents for treatment of serious infections. Theaminoglycosides are frequently paired with a beta-lactam antibiotic tocreate synergistic antibacterial activity and potentially to retard theemergence of antibiotic resistance, although the clinical impact of combination therapy has not been well demonstrated outside theimmunocompromised host.90 Empiric and definitive therapy of early-and late-onset neonatal septicemia with gentamicin-containingcombinations for enteric gram-negative infections is still consideredappropriate three decades following the first recommendations in thisage group, although well-controlled, prospective, comparative studieshave not generally been performed.88,89,91 Therapy of nosocomialinfections with aminoglycoside-containing regimens is still acceptablein institutions in which nosocomial pathogens remain susceptible tothe aminoglycosides.92
In gram-positive infections, the aminoglycosides add enhanced
bacterial killing to cell wall active agents, particularly in the treatmentof serious infections.93 Enterococcal infections are treated withampicillin or vancomycin in combination with gentamicin in order toachieve bactericidal activity. The combination penicillin plus gentamicin,or nafcillin plus gentamicin, are considered the most effective therapyfor infective endocarditis caused by susceptible strains of viridansstreptococci and Staphylococcus aureus, respectively.94
Empiric therapy of nosocomial infections and infections inimmunocompromised hosts has included combination regimenscontaining aminoglycosides in addition to agents active against gram-positive and frequently anaerobic bacteria.18,92 Aminoglycosides arealso used in combination with other agents in the treatment of intra-abdominal infections. However, the cationic charges on the moleculechange as the pH changes in infected tissues, with the acidic environ-
ment of an abscess decreasing the ability of these antibiotics to entebacterial cells (as documented by MICs that can be higher than thossafely achievable in serum).95 In addition, the active transport oaminoglycosides through the inner cell membrane into the cytoplasmof the bacteria requires an oxygen-dependent transport system nopresent in anaerobes, explaining the lack of activity of this class oagent in the treatment of anaerobic infections.
Given the high level of activity against certain gram-negativpathogens, the aminoglycosides still represent preferred therapy fo
tularemia and plague.Streptomycin remains highly active against most strains o Mycobacterium tuberculosis requiring parenteral inpatient therapywhich allows for use in treatment of multidrug-resistant tuberculosiStreptomycin may be particularly useful in the treatment of serioulife-threatening tuberculosis, including tuberculous meningitiAlthough originally approved for use in 1952 and previouslimportant for the treatment of infections caused by Brucella spp. anFrancisella spp., streptomycin has been replaced by gentamicibecause of concerns for streptomycin toxicity.
Paromomycin is an oral, nonabsorbable aminoglycoside. Thagent is effective in the treatment of intestinal protozoal infectionincluding amebiasis and cryptosporidiosis.
Inhaled aminoglycoside agents are used clinically for persons witcystic fibrosis. Achieving high enough antibiotic concentrations irespiratory tract secretions and at the bronchial mucosa to be effectivagainst Pseudomonas aeruginosa is not usually possible witparenterally administered aminoglycosides. However, with inhaletobramycin, high concentrations at the site of infection are achievabland can be provided without concern for nephro- or ototoxicity.96 Thuse of aerosolized aminoglycosides in patients with pneumonia causeby other multidrug-resistant gram-negative pathogens has not beeinvestigated systematically.
The toxicity of the aminoglycosides is directed primarily at thkidney and eighth cranial nerve. Nephrotoxicity is dose-dependent anis primarily tubular. Monitoring renal function or aminoglycosidserum concentrations during therapy allows early detection odecreased renal function attributable to the aminoglycosideOtotoxicity can be either cochlear (with hearing loss) or vestibula(with vertigo). Aminoglycoside toxicity correlates with increasinserum trough concentrations as a function of overall drug exposur
rather than the peak serum concentration achieved during therapy.97
Providing children with a single daily dose of aminoglycosides tdecrease nephrotoxicity while maintaining or increasing efficacy hanot been widely accepted due to the lack of convincing, welcontrolled studies in children. However, limited data which do exist ichildren, taken together with convincing data in adults, suggest thonce-daily dosing should become routine for children.97–99
While streptomycin was the first aminoglycoside available for usethe toxicity and rapid development of resistance in bacteria limited itlong-term viability. Kanamycin was approved for use in 1957 but wareplaced by the less nephrotoxic gentamicin in 1963 as a more broadlactive antibiotic against resistant gram-negative pathogenTobramycin was approved in 1968 and provided a small bupredictable increase in in vitro activity against P. aeruginosa with concomitant small decrease in activity against enteric gram-negativ
bacilli. For most situations, these small differences in in vitrsusceptibility do not translate into differences in clinical efficacyAmikacin, a semisynthetic derivative of kanamycin, was approved i1972, and offered enhanced activity against many gentamicin- antobramycin-resistant pathogens, in addition to providing serumantibiotic concentrations approximately three- to fourfold greater thathose achievable with gentamicin or tobramycin. Amikacin is alsuseful in the treatment of infections caused by some strains onontuberculous mycobacteria.
Streptogramins A and B
The streptogramins are two of a series of naturally occurrinantibiotics isolated from Streptomyces pristinaespiralis. Tw
Antimicrobial Agents C H A P T E R 292 144

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 23/33
antibiotics have been modified from the naturally occurring pristino-mycins I and IIA to create the semisynthetic antibiotics, quinupristin(streptogramin B) and dalfopristin (streptogramin A), respectively.These two antibiotics are present in the FDA-approved combination,Synercid, in a 70:30 ratio.100 Quinupristin and dalfopristin havecompletely different chemical structures, each with a distinct butoverlapping binding region within the P site of the peptidyl transferasecenter in the ribosome. Binding of dalfopristin to this region producesa conformational change that significantly increases the affinity of
binding of quinupristin, in part explaining the synergy observed whenthe combination is used. Dalfopristin inhibits protein synthesis byinterfering with substrate attachment to both A and P sites of the 50Ssubunit, while quinupristin blocks peptide bond synthesis duringelongation by causing incorrect positioning of the peptidyl tRNA atthe P site.101 Once elongation has been initiated, quinupristin triggersthe premature release of the elongating peptide chain. Quinupristinbinds to the peptidyl transferase site at a similar location as themacrolides and lincosamides; methylation of the A2058 adenine atthis important binding site prevents binding of quinupristin as well asthe macrolides and clindamycin, and creates the macrolide-lincosamide-streptogramin (MLS) resistance phenotype.
Unfortunately, many mechanisms of bacterial resistance limit theclinical utility of quinupristin/dalfopristin, with resistance developingin some patients soon after starting therapy. Alterations of target bindingsites, efflux removal of antibiotics from cytoplasm, and enzymaticalteration of the antibiotic structure have all been reported.102
Although each of the streptogramins independently producesinhibitory effects on protein synthesis, the combination is bactericidalfor a number of gram-positive pathogens, including S. aureus, but notenterococci.
The agents have been available in Europe for several years astopical therapy, but with the development and spread of enterococcithat are resistant to vancomycin, beta-lactams, and aminoglycosides,an urgent need for quinupristin/dalfopristin was well documented.Clinical use is largely limited to serious infections caused by vanco-mycin-resistant strains of Enterococcus faecium. The combination isnot active against the more common enterococcal pathogen,
Enterococcus faecalis. Although quinupristin/dalfopristin is alsoFDA-approved for treatment of skin and skin structure infectionscaused by Staphylococcus aureus, there are better evaluated and
tolerated agents for therapy of staphylococcal infections in children.Quinupristin/dalfopristin significantly inhibits cytochrome P-450
CYP 34A function and may impact serum concentrations of concomitant drugs which are eliminated by this pathway. Phlebitiswas a major side effect in quinupristin/dalfopristin-treated patients,occurring in almost half of the adults receiving therapy. Thiscombination agent has not been systematically studied for childrenunder 16 years of age.
Oxazolidinones
Linezolid is the first antibiotic in the oxazolidinone class to be usedin children. This class of antibiotics is unique in that members arenot natural products, but were discovered as one of a number of
compounds created as potential monoamine oxidase-inhibitingagents.103 However, these compounds also demonstrated antibacterialactivity, although early compounds were associated with significanttoxicity. Subsequent chemical modifications led to the developmentof linezolid, which demonstrated a reasonable balance betweenantimicrobial activity, clinical efficacy, and acceptable clinicaltoxicity. The oxazolidinones have a unique mechanism of action onthe ribosome, distinct from all other classes of antimicrobial agents.The antibiotic binds to an area close to the ribosomal peptidyltransferase center and inhibits the initiation of protein synthesis bypreventing the formation of the initiation complex of fMet-tRNA/elongation factors/mRNA/GTP at the peptidyl transferasecenter. The movement of the complex from the ribosome’s “A site” of tRNA attachment to the peptidyl “P site” is blocked and coupling
of amino acids and lengthening of the peptide chain cannotoccur.104–106
Linezolid demonstrates bactericidal activity against Streptococccus pneumoniae , but bacteriostatic activity against S. aureus and Enterococcus species. However, limited studies comparingvancomycin, a bactericidal agent (with limitations in activity ininfected lung), with linezolid in the treatment of nosocomialpneumonia caused by MRSA suggest equivalent if not improvedoutcomes.107 Activity of linezolid is primarily limited to gram-positive
bacteria, due to the presence of an efflux pump (AcrAB) presentin many gram-negative organisms. Linezolid is not active against Mycoplasma or Ureaplasma.108
Resistance to linezolid has been described and consists of structural changes at the linezolid binding site that preventattachment.109 These relatively infrequent changes, which tend tooccur more frequently with more prolonged therapy, are primarilysingle-base changes in the rRNA occurring around the peptidyltransferase site. No transferable resistance has been described.
Linezolid has been studied clinically for nosocomial andcommunity-acquired pneumonia, and for complicated anduncomplicated skin and skin structure infections.110, 111 The clinicaland microbiologic response rates for each of these tissue-specificinfections in children were equivalent to comparator agents, usuallyvancomycin. The in vitro activity and clinical efficacy of linezolid inthe treatment of infections caused by penicillin- and macrolide-resistant pneumococci, VRE, MRSA, and VRSA support a role forlinezolid when other, better-studied agents are not available or are nottolerated.103, 104 The most common current clinical use of linezolid inchildren is for the treatment of MRSA causing skin or respiratory tractinfections. Little data other than case reports are available for the useof linezolid in treatment of CNS infections.112
Data on pharmacokinetics for both parenterally and orallyadministered linezolid are available in children, including prematureneonates.113 Linezolid is virtually 100% absorbed by the oral route,allowing equivalent mg/kg dosing for both intravenous and oralformulations. Linezolid is not metabolized by the cytochrome P-450system, and does not induce this enzyme or compete with other drugsin P-450-mediated metabolism.
Some concerns have been raised regarding hematologic toxicity,including neutropenia and thrombocytopenia, which was found to be
dependent on dose and duration of therapy.103 However, prospective,comparative data in the pediatric FDA registration studies failed todemonstrate a significant difference in toxicity compared with otheragents. Also of concern in the early pediatric clinical trials of linezolidwas the possibility of hypertension due to effects of the antibiotic’smild monoamine oxidase (MAO) inhibitor activity. However, no MAOinhibitor side effects were noted in any phase I to III pediatric trial,permitting children to remain on usual diets while receivingtreatment.114
NUCLEIC ACID-ACTIVE ANTIBIOTICS
RNA Polymerase
RifamycinsThe rifamycins, rifampin (also called rifampicin), rifabutin,rifapentine, and the oral nonabsorbed rifaximin, are all semisyntheticderivatives of natural products of Streptomyces spp. The intracellulartarget for the rifamycins is DNA-dependent RNA polymerase and hasbeen well defined on a molecular level.115 During the creation of anRNA strand from the bacterial DNA strand, the polymerase requiresfunctional channels within the enzyme for both the DNA templatestrand and complementary strand, as well as for the elongating RNAstrand. As the RNA bases dock sequentially at the active site withinthe polymerase, the newly created RNA strand moves forward, onebase at a time. The rifamycins bind in the channel occupied by thenewly created RNA strand, approximately 2 to 3 bases downstream
S E C T IO N B Anti-Infective Therapy1442
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 24/33
from the active site, blocking the RNA strand from moving out of the polymerase and terminating the further elongation of that RNAsegment as it also uncouples the RNA strand from the active site. 115, 116
Although the actual active site of the polymerase is highly conservedbetween bacterial species, there is some diversity between bacteriaaround the active site with respect to the RNA polymerase structure.This diversity alters the binding affinity for rifampin across bacterialand mycobacterial species and provides one basis for the variablesusceptibility to this class of agents. Rifampin demonstrates
bactericidal activity against susceptible organisms.Given the large size of the rifamycin molecule and the restrictedbinding pocket on the polymerase, it is not surprising that manydifferent single amino acid substitutions from mutational events willlead to stable resistance with little impact on the viability of theorganism. In S. aureus, at least 18 genotypes resistant to rifampin havebeen characterized, some of which contain multiple basesubstitutions.117 Each of these mutations is likely to affect either thebinding affinity or the access of rifampin to the binding pocket to adifferent degree. Most of mutations causing rifampin resistance alsoproduce resistance to rifabutin and rifapentine.118
The rifamycins have a remarkably broad spectrum of activity thatincludes Staphylococcus, Streptococcus pyogenes, Neisseria spp. ,
Haemophilus influenzae, Campylobacter jejuni, Helicobacter pylori,Chlamydia trachomatis, both tuberculous and nontuberculous
Mycobacterium spp., Aspergillus spp., Naegleria fowleri , andToxoplasma gondii (see Table 292-2). Rifabutin has increased in vitroactivity against M. tuberculosis compared with rifampin, but theclinical relevance of this finding has not been documented to result inimproved microbiologic outcomes in treated patients.119 In addition,rifabutin has increased activity against MAC compared withrifampin.120, 121 Rifapentine has both increased in vitro activity against M. tuberculosis in addition to an extended serum elimination half-life(16 hours) compared with rifampin (2 to 3 hours).
Rifamycins achieve high intracellular concentrations, between5- and 20-fold greater than extracellular concentrations.120 This likelyexplains the effectiveness of this class in the therapy for mycobacterialinfections, which may be sequestered intracellularly.
Of the rifamycins, rifampin is the most common agent in clinicaluse in pediatrics. Rifabutin and rifapentine were developed fortreatment of mycobacterial infections, rather than bacterial infections,
and neither is currently FDA-approved for use in children. Rifampin isused in children as part of combination therapy for tuberculosis (seeChapter 134, Mycobacterium tuberculosis). Rifampin can also be usedin combination therapy for treatment of nontuberculous mycobacterialinfections causing adenitis, cellulitis, or pneumonia. For treatment of bacterial infections, rapid emergence of resistance in virtually everypathogen dictates a need for combination antibiotic therapy.Combination therapy may eradicate rifampin-resistant strains beforethey become clinically apparent. Rifampin has been used in thetherapy of Bartonella henselae infections (cat-scratch disease),multidrug-resistant Staphylococcus aureus infections (particularlyMRSA), and multidrug-resistant pneumococcal infections, includingmeningitis.122 Although not well studied in prospective comparativeclinical trials, the excellent tissue penetration characteristics of rifampin may provide improved clinical outcomes in deep bone and
joint infections and in foreign-body or device infections associatedwith intravascular catheters or implanted devices compared with beta-lactam antibiotic therapy alone. As single-drug therapy, rifampin isonly used in the eradication of colonization of Neisseria meningitidisand in the prophylaxis of close contacts of patients or family memberswith invasive meningococcal infections.
Rifabutin has documented efficacy in both prophylaxis and therapyof MAC (in combination with other agents) in HIV-infectedpatients.119–121,123 Only limited data are available in children, either HIV-infected or immunocompetent children with MAC infections.124,125 Theextended half-life of rifapentine provides a rationale for once-weeklytherapy, although the failure rate in those with more serious tuberculosiswas slightly higher in those treated with rifapentine compared withrifampin.119 Rifapentine has not been evaluated in children.
Rifaximin is an oral agent that is not absorbed from thgastrointestinal tract and produces high intraluminal antibioticoncentrations with little systemic toxicity. Microbiologic activity habeen demonstrated in vitro at achievable intraluminal antibioticoncentrations against Escherichia coli, Salmonella, Shigella, VibriYersinia, and Campylobacter species. Although the susceptibility othese enteric pathogens is about 32 to 64 μg/mL, a concentration noachievable in tissues, the concentrations achieved in the gastrointestintract are as high as 8000 μg/mL, providing an antibiotic exposure tha
is well above that required for an antibacterial effect.
126
Although thantibiotic is currently only approved for traveler’s diarrhea in adults, 1
an oral nonabsorbable, broad-spectrum agent for other gastrointestinapathogens could have considerable value for children.
The most common clinical side effect of treatment with rifampin nausea, which appears to decrease as the treatment course progresseThe most common adverse effect of treatment is hepatotoxicity, aassessed by elevation of hepatic transaminase levels. Most childrereceiving rifampin for tuberculosis therapy also receive othehepatotoxic agents, preventing an accurate assessment of the role orifampin as a single agent. Children receiving multiple hepatotoxagents should be assessed at regular intervals for evidence of ongoinhepatic injury.
The drug–drug interaction profile of the rifamycins frequentlcomplicates clinical management. The P-450 CYP3A system activated by the rifamycins. If the child is receiving other drug(metabolized by this system, decreased, potentially ineffective serumconcentrations of that drug may be present. In addition, if thconcurrent drug represents a competitive substrate for P-45metabolism, increased and potentially toxic serum concentrations othat drug may result from decreased metabolism.120 The potency oCYP3A induction is greatest with rifampin and least with rifabutinRifabutin is also a CYP3A substrate itself, resulting in higher rifabutiserum concentrations when given concomitantly with CYP3Ainhibitors.
DNA-Dependent DNA Polymerase
Quinolones
The quinolones are a diverse group of antibiotics that target DNAsynthesis and are active against a wide range of bacteria, Mycoplasmand Chlamydia spp., and, for some agents, mycobacteria.128–131 Thfirst of the quinolone antibiotics was discovered during thcommercial preparation of the antimalarial agent chloroquine, and wasubsequently modified for antibacterial use in humans.128 The firapproved quinolone agent, nalidixic acid, has a limited gram-negativspectrum of activity and a poor pharmacokinetic profile for treatmenof invasive infections. With chemical modifications to the basiquinolone (and the closely related naphthyridone) ring structure, thspectrum of activity and pharmacokinetics have been greatlenhanced, allowing for once-daily dosing for infections in mandifferent tissue sites. The 6-fluoroquinolones (beginning witnorfloxacin and ciprofloxacin) demonstrated a significanimprovement in antibacterial activity, and virtually all subsequentl
approved compounds are fluoroquinolones.The quinolones all interfere with DNA synthesis by interferin
with two closely related type II topoisomerase enzymes involved iDNA synthesis: DNA gyrase and topoisomerase IV.132–134 Each othese enzymes is comprised of 4 subunits: DNA gyrase contains subunits of GyrA and 2 of GyrB, whereas topoisomerase IV consistof 2 subunits of ParC and 2 of ParE (in Staphylococcus aureus, thestopoisomerase IV subunits are termed GrlA and GrlB). The DNAgyrase is responsible for uncoiling DNA ahead of the replication forto allow for DNA strand replication by DNA polymerase, or for thcreation of RNA strands by RNA polymerase. In the process ouncoiling and coiling, strands of DNA are cut and then religated bthese enzymes in order to maintain a stable double-helix structure. Thtopoisomerase IV appears to be primarily responsible for stabilizin
Antimicrobial Agents C H A P T E R 292 144

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 25/33
the newly created strands of DNA as they separate from templatestrands following replication. The quinolones bind to these enzymesat specific nucleic acid strand attachment sites, producingconformational changes in the DNA gyrase/DNA or topoisomeraseIV/DNA complexes and stabilizing them, thus “freezing” the complex.This leads to an inability to translocate the entire DNA replicationcomplex along the DNA strand, halting the process of nucleic acidreplication. Binding of the fluoroquinolones leads to single- anddouble-strand DNA breaks without religation, and the subsequent
release of DNA fragments into the cell, which leads to cell death inways that are not well understood. The DNA gyrase/DNA replicationcomplex may also be involved in altering the formation and stabilityof DNA loops involved in the transcription process, representinganother mechanism by which fluoroquinolones impact cell function.
Although quinolones may bind to some extent to both of theseenzymes, structural characteristics of each quinolone create uniquebinding characteristics to each of these two enzymes for every drugin this class. The differences in binding to DNA gyrase andtopoisomerase IV lead to differences in the susceptibility of organismsto the various quinolones as well as differences in the development of resistance. In general, the quinolones preferentially bind to DNAgyrase in gram-negative bacteria, and to topoisomerase IV in gram-positive bacteria, although some of the newer quinolones bind to bothenzymes equally well. Binding to DNA gyrase, occurring before thereplication fork of DNA synthesis, produces a more rapid effect on theformation of DNA, whereas binding to the topoisomerase IV afterstrand duplication has occurred creates a less immediate effect on celldeath. However, as both enzymes function to nick and religate strandsof DNA during the coiling/uncoiling process, the quinolone effectsafter binding to either, or both enzymes, can lead to cell death. Theseagents display bactericidal activity against bacterial pathogens, withdose-dependent pharmacodynamic activity best described by the ratioof AUC:MIC (the ratio of the area under the serum concentrationversus time curve to the MIC of the antibiotic for that organism).135
As with many antibiotics, resistance can develop in many differentways, and resistance from multiple different mechanisms can beadditive. The most common resistance occurs from alterations in theamino acids present at certain critical and virtually identical sites onthe two enzymes that prevent avid binding of the quinolone to DNAgyrase, and topoisomerase IV, respectively. Single gene mutations
leading to these amino acid changes are primarily found in thequinolone resistance-determining regions of gyrA and parC .136
Additional but less common mutations have been detected in gyrBand parE . For Escherichia coli, accumulating additional amino acidchanges leads to increasing resistance. The first-step mutation in E.coli most often occurs in the gyrA subunit of DNA gyrase, leading toa mild-to-moderate increase in the MIC, depending on the quinolonebeing assessed. A second-step mutation, usually in parC , generallyleads to resistance that cannot be overcome at achievable tissueconcentrations. For Streptococcus pneumoniae and Staphylococcusaureus, the first-step mutation most often occurs in the parC (or GrlA)subunit of topoisomerase IV.136, 137 Depending on both drug andbacteria, mutations in one of the subunit sites can lead to substantialincreases in resistance for a particular drug, but for drugs that bind toboth gyrase and topoisomerase IV sites, a mutation at one site does not
effectively raise the MIC to a level that usually leads to treatmentfailure. When two or more mutations occur that affect binding to bothenzymes, clinical failures become more common.
Efflux pumps are also effective mechanisms to prevent quinolonebinding intracellularly to the gyrase and topoisomerase targets. Effluxpumps affect intracellular concentrations of different quinolones todiffering degrees.137 In addition, newly described mutations inaminoglycoside acetyltransferases confer the ability to acetylateciprofloxacin, decreasing its activity against E. coli two- to fourfold,with the modified enzyme still retaining activity against theaminoglycosides. This mechanism of resistance is transferable to otherbacteria, in contrast to the gyrA or parC mutations which only spreadhorizontally from patient to patient. It is not known how problematicthese strains will become.138
Safety issues for children have been a concern for this entire classof agents. Although nalidixic acid was approved for children by theFDA in 1963, more extensive animal toxicology studies were availablefor the agents that followed. Preclinical juvenile animal toxicity datafor ciprofloxacin in the late 1970s suggested the potential for damageto joints in young children. Therefore, no routine pediatric drugdevelopment was undertaken for this compound or any otherfluoroquinolone until the need for potential use of these agents forchildren was demonstrated to the FDA in 1997.139 No quinolone-
associated arthropathy has yet been documented clearly in children foragents available in the United States. Long-term toxicity studies arecurrently in progress, but are not of sufficient size to be able to detector define rates of rare adverse effects on joints or tendons. It isencouraging, nonetheless, that lack of reported toxicity from currentpublished data and from data presented to the FDA suggests that jointtoxicity is not a common problem in children, if it exists at all.140
Nalidixic Acid, Norfloxacin
Nalidixic acid was approved in 1963 for use in children down to3 months of age for the treatment of UTI. Norfloxacin, used fortreatment of UTIs in children in other areas of the world, is not FDA-approved for children in the United States. With the FDA approval of ciprofloxacin for children with UTIs in 2004, ciprofloxacin is thepreferred quinolone agent for these infections when a fluoroquinoloneis needed, based on the quality of prospectively collected dataregarding both safety and efficacy in children.
Ciprofloxacin
Ciprofloxacin was one of the first of the 6-fluoroquinolones to beFDA-approved for adults, but large clinical trial investigations inchildren did not begin until the late 1990s, with the exception of studies in children with cystic fibrosis. Ciprofloxacin was recentlystudied for complicated UTI, and was the first 6-fluoroquinolone to beapproved for use in pediatrics. Although all of the fluoroquinoloneshave a bitter taste, a tolerable suspension formulation of ciprofloxacinis available for children. Current usage of the fluoroquinolones islimited, based on concerns of cartilage toxicity, noted above. The
antimicrobial activity of ciprofloxacin is provided in Table 292-2, andincludes most of the enteric gram-negative bacilli as well asPseudomonas aeruginosa. Activity against the gram-positivepathogens and anaerobic pathogens is generally only fair.
Current clinical use of ciprofloxacin centers on gram-negativeinfections for which no other oral antibiotic agent is available.141–143 Inthis setting, ciprofloxacin and other fluoroquinolones representeffective therapy in which the risk/benefit assessment favors treatmentwith an oral quinolone agent rather than parenteral therapy with anonquinolone agent. These infections may be located in virtually anytissue site except the CNS. Examples of infections, many of which arehospital-associated, include complicated UTI, bone or joint infection(including those caused by P. aeruginosa), soft-tissue infection, andlower respiratory tract infection. Many of these children havecomorbid conditions that have created a need for previous courses of
antibiotics and therefore the selection of multidrug-resistantpathogens. Because these infections are uncommon, no prospective,randomized, controlled clinical trials of use of ciprofloxacin inchildren are available. In addition, some reluctance may exist on thepart of the pharmaceutical industry to perform these studies due topotential toxicity. FDA approval has also been given to ciprofloxacinfor the treatment of inhalational anthrax in children.
Quinolones achieve effective concentrations in the gastrointestinaltract and have an advantage over beta-lactam antibiotics in that theyalso achieve high intracellular antibiotic concentrations, particularlyeffective in the treatment of Salmonella infections. Ciprofloxacin hasbeen studied in children for shigellosis and salmonellosis, andprovides equivalent or superior rates of eradication compared withstandard agents.67,144,145
S E C T IO N B Anti-Infective Therapy1444
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 26/33
With the high bioavailability of orally administered fluoro-quinolones and activity against most gram-negative pathogens forimmunocompromised children, selective use of these agents (withor without additional oral therapy for gram-positive pathogens) inlow risk children with fever and neutropenia is a promising area of clinical investigation.146
Levofloxacin
Levofloxacin was the first available agent in a series of newer
fluoroquinolone agents with enhanced activity against gram-positivepathogens, including Streptococcus pneumoniae and Staphylococcusaureus. The antibiotic has been clinically effective in treatment of upper and lower respiratory tract infections in adults,147 withcompleted clinical trials in children in both acute otitis media andcommunity-acquired pneumonia. No safety issues of significantconcern have been raised in either adult or pediatric clinical programs.No joint-related toxicity was noted in children, with a sufficientlylarge number of subjects prospectively studied to detect a 1% adverseevent rate. The most acknowledged clinical need for levofloxacin inchildren is for the treatment of unresponsive or recurrent otitis mediadue to resistance of pneumococci to beta-lactam and macrolideagents.148 However, use for therapy of serious infections is appropriatein situations for which no other class of active parenteral agent exists,or use as oral therapy of infections in situation for which only
nonquinolone parenteral therapies exist.143
Trovafloxacin, Gatifloxacin, Moxifloxacin, Gemifloxacin
Advancing generations of fluoroquinolones demonstrate increasedactivity against Streptococcus pneumoniae compared with earlierfluoroquinolones.130, 149 None of these newer agents has been approvedfor use in children, although trovafloxacin and gatifloxacin have beenevaluated in pediatric clinical trials, with documented clinical andmicrobiologic success. Trovafloxacin remains FDA-approved but notwidely available due to concerns for hepatotoxicity in critically illadults.
Nitroimidazoles
Metronidazole
Metronidazole is a synthetic nitroimidazole antibiotic with a poorlydefined mechanism of action despite decades of extensive use.Originally introduced for the treatment of Trichomonas infections(hence the original trade name, Flagyl), use in pediatrics has beenmost extensive for treatment of anaerobic infections, despite the factthat the antibiotic has never been approved by the FDA for use inchildren for these indications. The agent is taken up by cells by passivediffusion and the nitro side chain on the imidazole ring is reducedintracellularly by the pyruvate-ferredoxin reductase complex into atoxic nitro radical that reacts with DNA, leading to DNA strandbreaks, helix destabilization, and ultimately cell death in both dividingand nondividing cells.150,151 Metronidazole demonstrates con-centration-dependent bactericidal activity. One of the major
metabolites, a hydroxy derivative, retains significant antibioticactivity. Metronidazole is only active against anaerobic bacteria andcertain protozoal parasites, including Trichomonas, Entamoeba, andGiardia (Table 292-2).
In addition to direct bactericidal activity, metronidazole alsoappears to have direct effects on decreasing neutrophil generation of hydrogen peroxide and hydroxyl radicals, which may lead todecreased inflammation at the site of infection. In addition,metronidazole may inhibit lymphocyte transformation and granulomaformation.152 The clinical relevance of these observations is notknown.
Although resistance to metronidazole is uncommon, a number of bacteria, including some strains of Bacteroides fragilis, contain nimgenes which code for an inactivating enzyme that may exist on both
plasmids and within chromosomes. This enzyme effectively reducethe nitro group on metronidazole into a stable and inactive amine.150
Metronidazole is available in intravenous, oral tablet, and capsulformulations, and topical formulations. Although metronidazole has very bitter taste and is not well tolerated when pulverized and placein suspension, the absorption from the gastrointestinal tract excellent, with > 90% bioavailability. Although rectal administratioyields approximately 70% to 80% bioavailability, this route oadministration has never gained widespread acceptance in children
Metronidazole provides excellent therapeutic concentrations in a widrange of tissue sites, including CSF, in which concentrations are verclose to those achieved in serum.151 Although the serum eliminatiohalf-life is approximately 8 hours, early studies performed in adulfor FDA approval used 6-hour dosing regimens, which unfortunatelremains the current FDA dosing recommendation in the package labeThe observed half-life with oral administration appears to be longethan that found for IV administration, for unknown reasons. For mosclinical situations, based on the pharmacodynamics of antibiotexposure, dosing every 8 hours should be adequate. One of the metabolites of metronidazole that displays significant antibacterial activithas a more extended half-life, averaging 11 to 13 hours, providinanother rationale for less frequent dosing.
Clinical uses in pediatrics focus on the treatment of anaerobiinfections.153 Given the excellent tissue penetration characteristics ometronidazole, activity against all susceptible anaerobes is achievabin most tissue sites. However, data from randomized, prospectivclinical trials may not be available for many sites of infections. For aninfection that may also involve facultative or aerobic organismadditional antibiotics are necessary.
Metronidazole is bactericidal for Bacteroides spp. and has beeused extensively in the treatment of intra-abdominal infectionincluding complicated appendicitis, penetrating injury to the boweand colitis. These infections often involve multiple susceptiblanaerobic species, including B. fragilis. Other mixed aerobic/anaerobinfections include deep head and neck space infections (e.g., parapharyngeal abscesses, Ludwig angina) and necrotizing fasciitis/cellulit(e.g., necrotizing synergistic fasciitis, Fournier gangrene, omphalitisClostridia spp. are also susceptible to metronidazole, and can beffectively treated when causing deep-tissue infections. Sompenicillin-susceptible anaerobic gram-positive cocci are no
susceptible to metronidazole.Metronidazole, in combination with other antibiotics and proto
pump inhibitors, is part of a treatment regimen for Helicobacter pylormediated ulcer disease. In addition, some benefit in the treatment oCrohn disease may occur with metronidazole,154 as the antibiotic ananti-inflammatory properties of the agent may both play a role.
Given the excellent penetration into CSF and bactericidal capacitymetronidazole treatment of anaerobic organisms causing meningitis oventriculitis (traumatic, postsurgical, or nosocomial), or treatment oanaerobic brain abscesses is effective. Prospective, comparative dato document these indications are not available.
Metronidazole can also be used for female genital tract infectionincluding bacterial vaginosis, and as part of antimicrobial therapy opelvic inflammatory disease.
One of the most common pediatric uses for metronidazole is in th
treatment of Clostridium difficile enterocolitis. Following the documented increases in vancomycin resistance of gastrointestinal traflora with the use of oral vancomycin in adults for C. difficile colitimetronidazole became the drug of choice in therapy for children awell as adults. Clinical response rates for C. difficile infection arequivalent, comparing oral metronidazole therapy with oral vancomycin therapy.155
Entamoeba histolytica trophozoites (but not cysts) are susceptibto metronidazole, allowing therapy for both intestinal and extraintestinal amebiasis, including amebic liver abscess. Metronidazole one of the drugs of choice for treatment of Giardia intestinainfections and is an alternative treatment for Dientamoeba infectionFor sexually active adolescent females and males, metronidazolstill remains effective therapy for Trichomonas infections.156
Antimicrobial Agents C H A P T E R 292 144

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 27/33
the synthesis of thymidine, providing both synergistic activity againstmost pathogens, and decreasing the risk of development of resistanceto either the sulfa agent or the trimethoprim. Trimethoprim displaysbactericidal activity against bacteria, as does the combination.Sulfamethoxazole as a single agent is not currently manufactured foruse in the United States.159
Current clinical uses of TMP-SMX are somewhat limited in thetreatment of UTI due to increasing resistance of E. coli to thiscombination, but TMP-SMX therapy is effective for regions of the
world in which susceptibility remains high and for children with adocumented UTI in whom the susceptibility test results demonstratesusceptibility. The rate of pneumococcal resistance to sulfa in mostareas of the United States is greater than 40%, precluding it as first-line therapy of acute otitis media, sinusitis, or community-acquiredpneumonia. Despite in vitro susceptibility of S. pyogenes to TMP-SMX, the use of this agent in the treatment of streptococcalpharyngitis is not recommended due to failures in microbiologiceradication of the organism.
Increasing use of TMP-SMX in the treatment of CA-MRSAinfections has occurred recently, although prospective, controlledstudies were never performed for staphylococcal skin and skinstructure infections prior to original antibiotic approval, or since theemergence of CA-MRSA. In vitro susceptibility of CA-MRSA toTMP-SMX is almost universal.160
TMP-SMX is also being used more frequently for hospital-associated infections caused by some multidrug-resistant entericgram-negative bacilli such as Enterobacter and Klebsiella species andStenotrophomonas maltophilia (which remains susceptible). TMP-SMX is generally not bactericidal for high-density inocula of gram-negative bacilli. Although prospective comparative studies are notavailable, TMP-SMX is less toxic than the polymyxins (colistin), andmay provide an adequate clinical response for infections in most tissuesites, including meningitis, in situations for which no other well-evaluated therapy options exist.
Gastrointestinal infections caused by Salmonella and Shigella spp.and enteropathogenic strains of Escherichia coli were often treatedwith TMP-SMX in the past; however, the value of TMP-SMX hasdecreased as resistance in these gastrointestinal pathogens hasincreased.67,127 The intracellular activity of TMP-SMX has beenparticularly advantageous in the treatment of susceptible strains of
Salmonella, given the intracellular location of the organisms duringinfection. Susceptibility of enteric pathogens should be assessedbefore TMP-SMX is selected for therapy.
Other specific, less common infections remain effectively treatedwith TMP-SMX, including brucellosis and nocardiosis. Prophylaxisand treatment of Pneumocystis carinii (P. jiroveci) pneumonia inimmunocompromised children (HIV-infected, or selected oncologypatients) remains highly effective.
Sulfadiazine plus Pyrimethamine
This combination is active in a number of protozoan parasitic diseases,including toxoplasmosis and malaria. Clinical use in children isprimarily limited to congenital toxoplasmosis, with treatment startingas soon as physiologic jaundice has resolved, and to older children
who have documented toxoplasmosis associated with immunedeficiencies (primarily HIV-related).
Miscellaneous
Nitrofurantoin
Nitrofurantoin is a unique antibiotic, characterized by a hydantoin ringwith a nitro-substituted furanyl side chain that is metabolized withinthe bacteria to produce reactive compounds that are bactericidal. Themechanism of antibacterial activity is not well understood, butpresumably occurs by altering ribosomal proteins and other importantintracellular structures. Both gram-positive bacteria (including
Sulfonamides (Single Agents or in Combination with
Trimethoprim or Pyrimethamine)
Sulfisoxazole, Sulfamethoxazole, Sulfadiazine
The sulfonamide class was one of the first available antibiotics forhuman use, but due to widespread resistance in common bacterialpathogens after decades of extensive use, applications for this class of agents are now focused in a few areas of remaining effectiveness.
However, for pathogens that remain susceptible to sulfonamides,either used alone or in combination with trimethoprim, these agentshave a long history of efficacy with reasonable safety. Currentlyavailable sulfonamide agents are synthetic derivatives of the firstdescribed compound, sulfanilamide. At the time of discovery, sulfaagents were active against staphylococci and a wide range of gram-negative pathogens. However, due to a high rate of resistance to sulfaagents when used alone, these agents are now almost entirely used incombination with trimethoprim for the treatment or prophylaxis of bacterial infections. Sulfadiazine is currently used in combination withpyrimethamine for the treatment of toxoplasmosis, but it is no longerused to treat bacterial infections.
Of historical interest, with the success of sulfisoxazole followingits approval in 1953, many sulfa-sulfa combinations and sulfa-erythromycin combinations were approved to take advantage of thedifferent pharmacokinetic properties of the various sulfa agents, andthe complementary antibacterial spectrum achieved with otherantibiotic classes. Trisulfapyrimidines, or “triple-sulfa” agents,contained sulfa drugs of varying serum elimination half-lives(sulfadiazine, sulfamerazine, and sulfamethazine), whereas sulfi-soxazole-erythromycin combinations were active at that time againstotitis media pathogens, Streptococcus pneumoniae and H. influenzae.
The sulfonamides are bacteriostatic by means of competitiveinhibition of para-aminobenzoic acid, utilized by dihydropteroatesynthase in the synthesis of dihydrofolic acid, a precursor of purinebases in the formation of nucleic acid. As bacteria synthesize folicacid, they are susceptible to this class of compounds.
Sulfa agents are well absorbed from the gastrointestinal tract andare, in general, metabolized by the liver and excreted by the kidney.Both sulfisoxazole and sulfamethoxazole are highly protein-bound(70% to 80%), which raised concerns about the use of these agents in
the neonate with hyperbilirubinemia. Due to sulfonamide binding toalbumin, bilirubin may be displaced from its albumin binding sites,causing kernicterus as a result of subsequent bilirubin binding to CNStissue. Although sulfonamides may be contraindicated in ill, acidoticinfants with hyperbilirubinemia, no cases of kernicterus have beendocumented in full-term, well-appearing infants. Despite FDAlabeling cautions against the use of this class of agents in infants under2 months of age, they are safe in nonacidotic term infants as early asthe second week of life, as physiologic neonatal jaundice is resolving.
An intense immune-mediated separation of skin at the dermal–epidermal junction that may also involve separation of the respiratoryand gastrointestinal tract mucosa is the most severe hypersensitivityreaction to sulfonamides. Variously called Stevens–Johnson syndrome,toxic epidermal necrolysis, or erythema multiforme major, the spectrumof reactions varies from a cutaneous blistering rash to severe, extensive,
life-threatening sloughing of skin, respiratory tract and gastrointestinaltract mucosa.157 At the first sign of a skin reaction in a child receivingsulfa therapy, the child should be evaluated, the sulfonamidediscontinued, and careful observation begun.
Trimethoprim plus Sulfamethoxazole (TMP-SMX)
Trimethoprim, available as a single agent, but used far morecommonly in children in combination with sulfamethoxazole, actsat the metabolic step following that inhibited by sulfonamides inthe synthesis of purine bases.158 It prevents the formation of tetrahydrofolic acid from dihydrofolic acid by binding to andreversibly inhibiting dihydrofolate reductase. The combination of sulfamethoxazole and trimethoprim blocks two consecutive steps in
S E C T IO N B Anti-Infective Therapy1446
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 28/33
Antimicrobial Agents C H A P T E R 292 144
APPENDIX 292-1. Dosage of Antibacterial Drugs for Infants, Older Children, and Adolescents
Drug Generic (Trade) Route Dosage (per kg/day)a Commentsa
Aminoglycosidesb
Amikacin (Amikin) IV, IM 15–22.5 mg divided into 2–3 doses
Gentamicin (Garamycin) IV, IM 7–8 mg divided into 1–3 doses Cystic fibrosis 7–10 mg
Intraventricular 1–2 mg per dose (adult 3 mg)
Kanamycin (Kantrex) IV, IM 15–22.5 mg divided into 2–3 doses(adult maximum 1.5 g/day)
Neomycin (numerous) PO 50–100 mg divided into 3–4 doses Minimally absorbed from GI tract(adult, 4–8 g/day)
Streptomycin IM, IV 20–30 mg divided into 2 doses (adult 1–2 g/day)
Tobramycin (Nebcin) IV, IM 7–8 mg divided into 1–3 doses Cystic fibrosis 7–10 mg
Beta-lactam Agents
Monolactams
Aztreonam (Azactam) IV, IM 90–120 mg divided into 4 doses(adult 3–6 g/day, maximum 8 g/day)
Carbapenems
Imipenem-cilastatinc
(Primaxin) IV, IM 60–100 mg divided into 4 doses (adult 1–4 g/day) Seizures at high doses; not usedfor meningitis
Meropenem (Merrem) IV, IM 60 mg divided into 3 doses (adult 3–6 g/day) 120 mg for meningitis
Ertapenem (Invanz) IV, IM 30 mg divided into 2 doses, maximum 1 g/day(adult 1 g/day)
Cephalosporinsc Methicillin-resistant staphylococciare resistant to all currentlyavailable cephalosporins
Cefadroxil (Duricef, Ultracef) PO 30 mg divided into 2 doses(adult 1–2 g/day, maximum 4 g/day)
Cefaclor (Ceclor) PO 40 mg divided into 2 or 3 doses(adult 1–1.5 g/day, maximum 4 g/day)
Cefazolin (Kefzol, Ancef) IV, IM 50–100 mg divided into 3 doses
(adult 1.5–6 g/day, maximum 12 g/day)
Cefixime (Suprax) PO 8 mg divided into 1 or 2 doses (adult 400 mg/day) Poor antistaphylococcal activity
Cefoperazone (Cefobid) IV, IM 100–150 mg divided into 2 or 3 doses(adult 2–4 g/day, maximum 12 g/day)
Cefotaxime (Claforan) IV, IM 50–180 mg divided into 3 or 4 doses 200–300 mg divided into 4 doses(adult 3–6 g/day, maximum 12 g/day) for meningitis
Cefoxitin (Mefoxin) IV, IM 80–160 mg divided into 4–6 doses(adult 4–6 g/day, maximum 12 g/day)
Cefpodoxime proxetil (Vantin) PO 10 mg divided into 2 doses(adult 200–400 mg/day, maximum 800 mg/day)
Cefprozil (Cefzil) PO 15–30 mg divided into 2 doses (adult 0.5–1 g/day,maximum 1 g/day)
Ceftazidime IV, IM 100–150 mg divided into 3 doses 200–300 mg for serious(Fortaz, Tazicef, Tazidime) (adult 3–6 g/day, maximum 6 g/day) Pseudomonas infection
Ceftibuten (Cedax) PO 9 mg divided into 1 or 2 doses (adult 400 mg/day)
Cefepime (Maxipime) IV, IM 100–150 mg divided into 2–3 doses(adult 2–6 g/day, maximum 6 g/day)
Cefdinir (Omnicef) PO 14 mg divided into 1 or 2 doses,maximum 600 mg/day
Ceftizoxime (Cefizox) IV, IM 150–200 mg divided into 3 doses(adult 3–6 g/day, maximum 12 g/day)
Ceftriaxone (Rocephin) IV, IM 50–75 mg divided into 1 or 2 doses 100 mg for meningitis(adult 1–2 g/day, maximum 4 g/day)
Continue

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 29/33
S E C T IO N B Anti-Infective Therapy1448
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
APPENDIX 292-1. Dosage of Antibacterial Drugs for Infants, Older Children, and Adolescents —Continued
Drug Generic (Trade) Route Dosage (per kg/day)a Commentsa
Cefuroxime (Zinacef) IV, IM 100–240 mg divided into 3 doses (adult 2.25–4.5 g/day, Should not be used if meningitismaximum 9 g/day) is possible
Cefuroxime axetil (Ceftin) PO 30–40 mg divided into 2 or 3 doses,maximum 1 g/day
Cephalexin (Keflex) PO 25–50 mg divided into 4 doses (adult 1–4 g/day) 100 mg for osteoarticular
infections
Cephradine (Anspor, Velosef) PO 25–50 mg divided into 3–4 doses (adult 1–4 g/day) 100 mg for osteoarticularinfections
Loracarbef (Lorabid) PO 15–30 mg divided into 2 doses (adult 800 mg/day) 30 mg/kg for AOM
Penicillins
Penicillin G and V
Penicillin G, crystalline K or Na IV, IM 100,000–250,000 units divided into 4–6 doses Contains Na or K,(adult 8–24 million units/day, maximum 1.68 mEq/1,000,000 units80 million units/day)
Penicillin G, procaine IM 25,000–50,000 units divided into 1–2 doses(adult 600,000–1.2 million units/day,maximum 4.8 million units/day)
Penicillin G, benzathine IM 50,000 units/kg once (adult 1.2–2.4 million units μonce)(Bicillin LA)
Penicillin V K (numerous) PO 25–50 mg divided into 3 or 4 doses, 1600 units = 1 mg; optimalmaximum 3 g/day (adult 0.5–2 g/day, administration on emptymaximum 7.2 g/day) stomach
Penicillinase-resistant penicillins Methicillin-resistant staphylococciare resistant to all penicillins
Oxacillin (Bactocill) IV, IM 150–200 mg divided into 4–6 doses(adult 2–6 g/day, maximum 12 g/day)
Nafcillin (Unipen, Nafcil) IV, IM 100–200 mg divided into 4–6 doses(adult 2–6 g/day, maximum 12 g/day)
Dicloxacillin (Dynapen, Pathocil) PO 12.5–25 mg divided into 4 doses 100 mg for osteoarticular(adult 0.5–2 g/day, maximum 4 g/day) infections
Aminopenicillins
Ampicill in IV, IM 100–200 mg divided into 4 doses 200–400 mg divided into 4 doses(adult 4–8 g/day, maximum 14 g/day) for meningitis
Ampicillin-sulbactam (Unasyn) IV 100–200 mg ampicillin component dividedinto 4 doses (adult 4–8 g day)
Ampicillin/ampicillin trihydrate PO 50 mg divided into 4 doses(Principen) (adult 1–4 g/day, maximum 14 g/day)
Amoxicillin (Amoxil) PO 40–100 mg divided into 2–3 doses(adult 1–3 g/day)
Amoxicillin-clavulanate PO 14:1 formulation: 90 mg amoxicillin component Twice-daily dosing studied for(Augmentin) divided into 2 doses efficacy only in AOM
7:1 formulation: 25–45 mg amoxicillincomponent divided into 2 doses
4:1 formulation: 20–40 mg amoxicillin
component divided into 3 doses
Broad-spectrum penicillins
Carbenicillin PO 30–50 mg divided into 4 doses, maximum 3 g/day
Piperacillin IV 200–300 divided into 3–4 doses(adult 6–18 g/day, maximum 24 g/day)
Piperacillin-tazobactam (Zosyn) IV 240 mg piperacillin component divided into 4 doses(adult 12–18 g/day)
Ticarcillin-clavulanate IV 200–300 mg ticarcillin component divided Cystic fibrosis 300–600 mginto 4–6 doses (adult 12–18 g/day, maximum 24 g/day) divided into 4 doses

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 30/33
Antimicrobial Agents C H A P T E R 292 144
APPENDIX 292-1. Dosage of Antibacterial Drugs for Infants, Older Children, and Adolescents —Continued
Drug Generic (Trade) Route Dosage (per kg/day)a Commentsa
Chloramphenicol b IV 50–75 mg divided into 4 doses 75–100 mg for meningitis(Chloromycetin) sodium-succinate (adult maximum 4 g/day)
Fluoroquinolones/Quinolones Arthropathy is a potentialfluoroquinolone-class side effectinchildren
Nalidixic acid PO 55 mg divided into 4 doses (adult 2–4 g/day)
Norfloxacin (Noroxin) PO 9–14 mg divided into 2 doses(adult 400–800 mg/day, maximum 1.2 g/day)
Ciprofloxacin (Cipro) PO 20–40 mg divided into 2 doses, maximum 2 g/dayIV 20–30 mg divided into 2–3 doses,
maximum 1.2 g/day
Levofloxacin (Levaquin) PO, IV 20 mg divided into 2 doses(children < 5 years); 10 mg in once-daily dose(children > 5 years) (adult 500–750 mg/day)
Ketolides
Telithromycin (Ketek) PO Adult dose 800 mg once daily
Lincosamides
Clindamycin (Cleocin) IM, IV 20–40 mg divided into 3–4 doses(adult 900 mg–2.7 g/day, maximum 4.8 g/day)
PO 10–30 mg divided into 3–4 doses(adult 600 mg–1.8 g/day, maximum 2.7 mg/day)
Lipopeptides
Daptomycin (Cubicin) IV Adult dose 4–6 mg/kg once daily
Macrolides/Azalides
Erythromycin (numerous) PO 20–40 mg divided into 2–4 doses Available as base, stearate, ethyl(adult 1–2 g/day, maximum 4 g/day) succinate, and estolate
preparationsand aserythromycin-sulfisoxazole
IV 20–40 mg divided into 4 doses(adult 1–2 g/day, maximum 4 g/day)
Clarithromycin (Biaxin) PO 15 mg divided into 2 doses (adult 0.5–1 g/day)
Azithromycin (Zithromax) PO, IV Otitis: 10 mg/kg per day loading dose on day 1,then 5 mg/kg per day μ 4 days; or 10 mg/kgper day μ 3 days; or 30 mg/kg μ 1
Pharyngitis: 12 mg/kg per day μ 5 daysGastroenteritis: 10 mg/kg per day μ 3 daysPneumonia: 10 mg/kg per day μ 5 days
Methenamine mandelate PO 50–75 mg divided into 2–4 doses (adult 2–4 g/day) For UTI prophylaxis(Mandelamine)
Metronidazole (Flagyl) PO 15–35 mg divided into 3 dosesIV 30–40 mg divided into 3 doses
(adult 1–2 g/day, maximum 4 g/day)
Niftofurantoin (Furadantin) PO 5–7 mg divided into 4 doses (adult 200–400 mg/day, 1–2 mg/kg/daily for UTImaximum 7 mg/kg per day) prophylaxis
PolymyxinsColistimethate (Coly-Mycin M) IV, IM 5–7 mg divided into 3 doses Reserved for multidrug-resistant
gram-negative pathogensbecause of neuro- andnephrotoxicity
Quinupristin-dalfopristin IV 15–22.5 mg divided into 2–3 doses(Synercid) (adult dose same)
Rifamycins
Rifampin (Rifadin) PO, IV 10–20 mg divided into 1–2 doses (adult 600–1200 mg/day)
Rifaximin (Xifaxan) PO Adult dose 600 mg/day divided into 3 doses

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 31/33
S E C T IO N B Anti-Infective Therapy1450
PA RT IV Laboratory Diagnosis and Therapy of Infectious Diseases
APPENDIX 292–1. Dosage of Antibacterial Drugs for Infants, Older Children, and Adolescents —Continued
Drug Generic (Trade) Route Dosage (per kg/day)a Commentsa
Sulfonamides
Sulfadiazine PO 120–150 mg divided into 4 doses (adult 2–4 g/day)
Sulfisoxazole (Gantrisin) PO 120–150 mg divided into 4 doses
Trimethoprim-sulfamethoxazole IV, PO 8–12 mg TMP divided into 2 doses (adult 160–320 mg 20 mg TMP divided into 4 doses
(TMP-SMX) (Bactrim, Septra) TMP/day, maximum 15 mg TMP/kg per day) for Pneumocystis cariniipneumonia
Tetracyclines Tetracycline-class antibiotics stainunerupted teeth; use in children< 8 years of age if benefitsexceed risks
Tetracycline (numerous) PO 25–50 mg divided into 4 doses (adult 1–2 g/day)
Minocycline (Minocin) PO, IV 4 mg divided into 1–2 doses (adult 200 mg/day,maximum 400 mg/day)
Doxycycline (numerous) PO, IV 2–4 mg divided into 1–2 doses (adult 100–200 mg/day) IV use as a 2-hour infusion
Tigecycline (Tygacil) IV Adult dose 50 mg twice daily
Vancomycinb (Vancocin) IV 40 mg divided into 3–4 doses Consider dose of 60 mg/kg per dayfor meningitis
PO 40 mg divided into 4 doses Not absorbed from GI tract(adult, 500 mg divided into 4 doses)
Intraventricular 1–5 mg daily (adult, 3–5 mg daily)
AOM, acute otitis media; GI, gastrointestinal; IV, intravenous; IM, intramuscular; PO, orally; UTI, urinary tract infection.aDoses for children are listed as the number of dosing units (e.g. mg, mg, etc.) per kilogram per day along with an absolute maximum dose if known. The corresponding parenthetic
adult doses are the common adult dose range (not per kg per day, unless specified) followed by the adult maximum dose, if known.bDoses are adjusted according to serum concentration.cIn patients with a history of immediate hypersensitivity (anaphylaxis) to penicillin, other penicillins, cephalosporins, or carbapenems should not be used.
Reference for Appendix 292-1:178
APPENDIX 292-2. Part 1 Table of Antibiotic Dosages for Neonates
Dosages (mg/kg per day) and Intervals of Administration
Chronologic Age < 28 days
Bodyweight < 2000 g Bodyweight > 2000 g
Chronologic
Antibiotics Route 0–7 days old 8–28 days old 0–7 days old 8–28 days old Age > 28 days
Amoxicillin/clavulanate PO 30 div q 12 hours 30 div q 12 hours 30 div q 12 hours
Ampicillin IV, IM 100 div q 12 hours 150 div q 8 hours 150 div q 8 hours 200 div q 6 hours 200 div q 6 hours
Azithromcyina PO 5 q 24 hours 10 q 24 hours 5 q 24 hours 10 q 24 hours 10 q 24 hours
IV 5 q 24 hours 10 q 24 hours 5 q 24 hours 10 q 24 hours 10 q 24 hours
Cefazolin IV, IM 50 div q 12 hours 50 div q 12 hours 50 div q 12 hours 75 div q 8 hours 75 div q 8 hours
Cefepimeb IV, IM 60 div q 12 hours 60 div q 12 hoursb 60 div q 12 hours 60 div q 12 hoursb 150 div q 12 hours
Cefotaximec IV, IM 100 div q 12 hours 150 div q 8 hours 100 div q 12 hours 150 div q 8 hours 150 div q 8 hours
Ceftazidimec IV, IM 100 div q 12 hours 150 div q 8 hours 100 div q 12 hours 150 div q 8 hours 150 div q 8 hours
Ceftriaxonec IV, IM 25 q 24 hours 50 q 24 hours 25 q 24 hours 50 q 24 hours 50 q 24 hours
Cefuroxime IV, IM 100 div q 12 hours 150 div q 8 hours 150 div q 8 hours 150 div q 8 hours 150 div q 8 hours
Clindamycin IV, IM, PO 10 div q 12 hours 15 div q 8 hours 15 div q 8 hours 20 div q 6 hours 30 div q 6 hours
Erythromycin ethyl PO 20 div q 12 hours 30 div q 8 hours 20 div q 12 hours 40 div q 8 hours 40 div q 8 hourssuccinate

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 32/33
staphylococci and streptococci) and gram-negative bacteria ( E. coli,Klebsiella and Citrobacter spp.) are susceptible. Mechanisms of resistance have not been well defined, but no cross-resistance occursto other antibiotic classes. Recent data from the United States onsusceptibility of pediatric uropathogens document that, overall,resistance to nitrofurantoin across all pathogens and pediatric agegroups is approximately 5% to 10%.161
Nitrofurantoin was originally FDA-approved in 1953 for thetreatment of uncomplicated UTIs in adults and children 12 years of age and older. Nitrofurantoin is well absorbed orally and is rapidlycleared from the serum, producing subtherapeutic concentrations in
serum, but bactericidal concentrations in urine. Currently availabformulations include a rapidly absorbed monohydrate salt, monohydrate salt in slow-release matrix, and crystalline nitrofurantoithat is also more slowly absorbed. Dosages should be decreased ichildren with any degree of renal insufficiency.
In clinical practice, this agent has been used for both treatment anlong-term prophylaxis of UTI.162,163 Current clinical use is primarilfor prophylaxis, but few prospective comparative data are availablon which to base these recommendations.164,165 Serious pulmonartoxicities, both acute and chronic, were reported in the decadfollowing availability of nitrofurantoin.166,167 Acute lung injury, felt t
Antimicrobial Agents C H A P T E R 292 145
APPENDIX 292-2. Part 1 Table of Antibiotic Dosages for Neonates—Continued
Dosages (mg/kg per day) and Intervals of Administration
Chronologic Age < 28 days
Bodyweight < 2000 g Bodyweight > 2000 g
Chronologic
Antibiotics Route 0–7 days old 8–28 days old 0–7 days old 8–28 days old Age > 28 days
Linezolid IV, PO 20 div q 12 hours 30 div q 8 hours 30 div q 8 hours 30 div q 8 hours 30 div q 8 hours
Meropenemc IV 40 div q 12 hours 60 div q 8 hours 40 div q 12 hours 60 div q 8 hours 60 div q 8 hours
Metronidazole IV, PO 7.5 div q 24 hours 15 div q 12 hours 15 div q 12 hours 30 div q 12 hours 30 div q 6 hours
Nafcillin, oxacillin IV 50 div q 12 hours 75 div q 8 hours 75 div q 8 hours 150 div q 6 hours 150 div q 6 hours
Penicill in G, benzathine IM 50,000 units 50,000 units 50,000 units 50,000 units 50,000 unitsq 24 hours q 24 hours q 24 hours q 24 hours q 24 hours
Penicillin G, crystalline IV 100,000 units div 225,000 units div 150,000 units div 200,000 units 200,000 unitsq 12 hour q 8 hours q 8 hours div q 6 hours div q 6 hours
Penicillin G, procaine IM 50,000 units 50,000 units 50,000 units 50,000 units 50,000 unitsq 24 hours q 24 hours q 24 hours q 24 hours q 24 hours
Piperacillin-tazobactam IV 100 div q 12 hours 300 div q 8 hours 200 div q 12 hours 300 div q 8 hours 400 div q 6 hours
Rifampin IV, PO 10 q 24 hours 10 q 24 hours 10 q 24 hours 10 q 24 hours 10 q 24 hours
Ticarcillin IV, IM 150 div q 12 hours 225 div q 8 hours 225 div q 8 hours 300 div q 6 hours 300 div q 6 hours
Ticarcilllin-clavulanate IV 150 div q 12 hours 225 div q 8 hours 225 div q 8 hours 300 div q 6 hours 300 div q 6 hours
IV, intravenous; IM, intravenous; PO, orally.aAzithromycin dose for pertussis should be 10 mg/kg once daily for the entire 5-day treatment course, whereas for other infections, 10 mg/kg is given on the first day, followed by
5 mg/kg for 4 subsequent days.bCefepime should be given at 60 mg/kg per day div q12 hours for the first 2 weeks of life, after which the dosing increases to 100 mg/kg per day div q12 hours (or 150 mg/kg per
day div q8 hours for Pseudomonas infections, or for meningitis).cHigher dosage may be needed for meningitis.
Reproduced from Bradley JS, Nelson JD. Nelson’s Pocketbook 2006–2007. Buenos Aires, ACINDES, 2006, with permission.
APPENDIX 292-2. Part 2 Antibiotics for Neonates Dosed According Only to Age
Dosage (mg/kg/DOSE)by Gestational Age PLUS Weeks of Life
Antibiotics Route of Administration <26 Wks 27–34 Wks 35–42 Wks >43 Wks
Amikacina IV, IM 7.5 q 24 hours 7.5 q 18 hours 10 q 12 hourse 10 q 8 hourse
Gentamicinb IV, IM 2.5 q 24 hoursd 2.5 q 18 hoursd 2.5 q 12 hourse 2.5 q 8 hourse
Tobramycinb IV, IM 2.5 q 24 hoursd 2.5 q 18 hoursd 2.5 q 12 hourse 2.5 q 8 hourse
Vancomycinc IV 15 q 24 hours 15 q 18 hoursf 15 q 12 hoursf 15 q 8 hoursf
aDesired serum concentrations: 20-30 mg/mL (peak), <10 mg/mL (trough).bDesired serum concentrations: 5-12 mg/mL (peak), <2.0 mg/mL (trough).cDesired serum concentrations: 20-40 mg/mL (peak), <10-15 mg/mL (trough).dAlternative regimen: ≤29 wk: postnatal age 0-7 days, 5 mg/kg q48h; 8-28 days, 4 mg/kg q36 h; ≥29 d, 4 mg/kg q24h
30-34 wks: postnatal age 0-7 days, 4.5 mg/kg q36h; ≥8 days, 4 mg/kg q24heOnce daily dosing regimen (amikacin, 15 mg/kg; gentamicin, tobramycin, 4 mg/kg) should be effective and safe, but not prospectively evaluated.f At 28 days of age (4 weeks), vancomycin is dosed at 20 mg/kg/dose. The interval remains the same.

8/2/2019 Chapter 292 Antimicrobial Agents
http://slidepdf.com/reader/full/chapter-292-antimicrobial-agents 33/33
be immune-mediated, and chronic fibrosis have been reported inadults during long-term therapy. The rate and severity of thesemultiple pulmonary toxicities have not been evaluated prospectively inchildren. However, no reports of pulmonary toxicity in young childrenreceiving nitrofurantoin prophylaxis for UTI have been published inthe past two decades, despite continued widespread use. In addition topulmonary toxicity, hemolysis secondary to glucose-6-phosphatedehydrogenase deficiency has also been documented.
MethenamineMethenamine hippurate is used exclusively for the prevention of UTI.Initially available in 1967 and FDA-approved for patients down to12 years of age, methenamine is a salt that ultimately exerts anantibacterial effect in the urine as it becomes converted intoformaldehyde. This effect occurs when the urine pH is below 5.5.Formaldehyde has nonspecific bactericidal activity on both gram-positive and gram-negative bacteria. However, as the generation of formaldehyde is dependent on an acidic urinary pH, methenaminemay not be active in situations in which a more alkaline pH is createdby diet, or by urea-splitting bacteria in the urine, such as Proteusand Pseudomonas. A recent review of previously published data onclinical trials of methenamine prophylaxis, some of which werepublished as early as 1966, was not done in a prospective, randomized,blinded, fashion with well-defined endpoints.168 However, pooledanalyses suggested a treatment benefit for prophylaxis of bacteriuria,although a significant failure rate was still present in adults withacidified urine and documented adequate urinary formaldehydeconcentrations.168,169 The clinical use of methenamine in children islimited to situations in which neither antimicrobial agents norintermittent catheterization techniques in children with urinarybladder voiding dysfunction have been effective at preventingrecurrent symptomatic UTIs. Microbiologic failures during methen-amine prophylaxis are not uncommon.
Fungal pathogens are an increasingly recognized complication of organ transplantation and the ever more potent chemotherapeuticregimens for childhood malignancies. For over 40 years, there hasbeen limited progress in the treatment of invasive fungal infectionsand the field of pediatric antifungal therapy has been largely ignored.Although conventional amphotericin B deoxycholate was the drug of choice for many invasive fungal infections for years, newer, safer, and
more effective agents have relegated it to antiquity. Lipid formulationsof amphotericin B have reduced the toxicity of conventionalamphotericin B and these agents have a role in the management of several specific diseases, such as zygomycosis and others. The newertriazoles, voriconazole and posaconazole, have expanded our optionsfor therapy against mold infections such as invasive aspergillosis. Theechinocandins offer a new class of antifungal therapy where for thefirst time there is no cross-reactivity with a human substrate, leadingto minimal toxicities.
There are now numerous nuances to choosing the correct anti-
ment of a solid understanding of differences in the pharmacokineticsof these drugs in children and adults, which results in more optimaldosing.
ANTIFUNGAL AGENTS FOR TREATMENT OF SUPERFICIAL
INFECTIONS
Among the therapeutic options available for treatment of fungal
infections of the hair, skin, and nails, few have been tested in children,and fewer are approved for use in children younger than 13 years of age. Furthermore, extrapolating data from adults treated for dermato-mycoses to infants and children may not be justified. For example,Trichophyton tonsurans, although uncommon in adults, is the mostcommon cause of tinea capitis and tinea corporis in children.
The use of topical agents should be confined to infections of theepidermis, hair, and nails. The choice of treating superficial fungalinfections with a topical or a systemic agent depends on the fungalpathogen, the site of infection, and the extent of the lesion (see Chapter255, Dermatophytes and Other Superficial Fungi). For example, asystemic antifungal drug is almost always used for ringworm of thescalp, nails, palms, or soles; creams or solutions are preferred forfissured or intertriginous areas; and sprays are not recommended forthe face.
Over-the-Counter Preparations
Undecylenic acid is an unsaturated fatty acid antifungal agent avail-able as an ointment, powder, or liquid. Its therapeutic efficacy wasfirst recognized in the 1940s in treating superficial fungal diseasesaffecting military troops. However, undecylenic acid has little efficacywhen compared with newer agents. Tolnaftate has similar activityagainst dermatophytes, with cure rates of 60% to 90%, but tinea pedis,tinea cruris, and tinea corporis resolve spontaneously in 20% to 30%of cases.1 Tinea capitis and tinea unguinum are frequently resistant,probably because of poor penetration of tolnaftate into involvedareas. The advantages of these compounds are that they are safe,inexpensive, and rarely cause local irritation.
Topical Polyenes
Nystatin is a polyene antifungal agent named after New York state. Itbinds to ergosterol in the fungal cell membrane and causes changes incell permeability and, eventually, cell lysis. Nystatin is useful for thetreatment of oral, mucosal, and cutaneous infections caused byCandida. It is available as a suspension, powder, cream, or ointment(100 000 U/mL or U/g, respectively); a pastille (200 000 U) to bedissolved in the mouth; an oral tablet (500 000 U); and a vaginal tablet(100 000 U). Oral forms are administered four or more times daily andare well tolerated, although large oral doses of the suspension cancause nausea. Nystatin is not effective against the dermatophytes.
Azoles
Clotrimazole is an imidazole with broad-spectrum activity againstCandida and the dermatophytes T. tonsurans, T. rubrum, T. menta-grophytes, Epidermophyton floccosum, and Microsporum canis (Table293-1). This antifungal agent is also effective against Malassezia
furfur, the cause of tinea versicolor. Clotrimazole is one of the fewtopical antifungal agents studied in children. The 1% cream, lotion,and solution are available over the counter for the treatment of tineapedis, tinea cruris, and tinea corporis. High concentrations of clotri-mazole are achieved topically, but systemic absorption is negligible.
S E C T IO N B Anti-Infective Therapy1452
C H A P T E R 293
Antifungal Agents
William J. Steinbach and Christopher C. Dvorak


















