
Chapter-1 Introduction to Biological importance of...
55
Chapter-1 Introduction to Biological importance of Benzimidazoles and Benzisoxazoles
Transcript of Chapter-1 Introduction to Biological importance of...

Chapter-1
Introduction to Biological importance of
Benzimidazoles and Benzisoxazoles

Chapter-1
1
1.1 Introduction to Heterocyclic Chemistry
The organic compounds have enormous diversity of structures; many of these structures
contain ring systems. If the ring system is made up of atoms of carbon and at least one
element other than carbon, the compound is classified as heterocyclic. The elements that
present most commonly, together with carbon, in ring systems are nitrogen, oxygen and
sulfur. About half of the known organic compounds have structures that incorporate at
least one heterocyclic component. A cyclic organic compound containing all carbon
atoms in ring formation is referred to as a carbocyclic compound. And, if at least one
atom other than carbon forms a part of the ring system then it is designated as a
heterocyclic compound.1-4
Heterocyclic chemistry is a vast and expanding area of chemistry because of the obvious
applications of compounds derived from heterocyclic rings in pharmacy, medicine,
agriculture and other fields. The chemistry of heterocyclic compounds is as relevant as
that of alicyclic or aromatic compounds. Their study is of great interest both from the
theoretical as well as practical standpoint.
The literature on heterocyclic compounds is very vast. They may be classified into
alicyclic and aromatic heterocyclic compounds. The alicyclic heterocycles are the cyclic
analogues of amines, ethers, thioethers, amides, etc. The aromatic heterocyclic
compounds in contrast are those which have a heteroatom in the ring and behave in a
manner similar to benzene in some of their properties. A heterocyclic ring may comprise

Chapter-1
2
of three or more atoms which may be saturated or unsaturated. Also the ring may contain
more than one heteroatom; this may be similar or dissimilar.
Heterocyclic compounds have a wide range of applications: they are predominant among
the types of compounds used as pharmaceuticals,
as agrochemicals, as veterinary
products, used as optical brightening agents, as antioxidants, as corrosion inhibitors and
as additives with a variety of other functions.5
Also, many dyestuffs and pigments have
heterocyclic structures.6
One of the reasons for the widespread use of heterocyclic
compounds is that their structures can be subtly manipulated to achieve the required
modification in function. Many heterocycles can be fitted into one of a few broad groups
of structures that have overall similarities in their properties but significant variations
within the group. Such variations include differences in acidity or basicity, difference in
susceptibility to attack by electrophiles or nucleophiles, and different polarity. The
possible structural variations include the change of one heteroatom for another in a ring
and the different positioning of the same heteroatoms within the ring.
The number of possible heterocyclic systems are reported in the field of synthetic organic
chemistry. An enormous number of heterocyclic compounds are known and the number
is increasing very rapidly. The literature on the subject is correspondingly vast and there
are three major divisions in organic chemistry namely, aliphatic, aromatic and
heterocyclic, the last one is the biggest. Over six million compounds are recorded in
chemical abstracts and approximately half of them are heterocyclic.

Chapter-1
3
An example of the way in which heterocycles are used is provided by an account of the
development of a new systemic fungicide (1), 7
the pyrimidine ring was incorporated into
this structure because a related compound (2) proved to be too lipophilic. The water
solubility and the transport of the fungicide through the plant were improved by replacing
a benzene ring by the more polar heterocycle.
Many heterocyclic compounds are biosynthesized by plants and animals and are
biologically active. Over millions of years these organisms have been under intense
evolutionary pressure, and their metabolites may be used to advantage: for example, as
toxins to ward off predators, or as coloring agents to attract mates or pollinating insects.
Some heterocycles are fundamental to life, such as haeme derivatives in blood and the
chlorophyll present in the plants is essential for photosynthesis. Similarly, the paired
bases found in RNA and DNA are heterocycles, as the sugars in combination with
phosphates provide the backbones and determine the topology of these nucleic acids.
Dye stuffs of plant origin include indigo blue, used to dye jeans. A poison of detective
novel fame is strychnine, obtained from the plant resin curare. The biological properties
of heterocycles in general make them one of the prime interests of the pharmaceutical and
biotechnology industries.

Chapter-1
4
There are many thousands of other heterocyclic compounds, both natural and synthetic
which have major importance, not only in medicine but also in other activities known to
man. Small wonder than that, their chemistry forms a major part of both undergraduate
and post graduate curricula.
Some heterocyclic compounds are shown below.
A brief introduction to applications of benzimidazole and benzisoxazole derivates in the
area of medicinal chemistry is discussed in the following sections.

Chapter-1
5
1.2 Introduction to Benzimidazoles
Benzimidazoles are involved in a great variety of biological processes. Some of their poly
functional derivatives have been proved to possess antibacterial, fungicidal and
antihelmintic activities. Therefore, substituted benzimidazoles have attracted the interest
of various research groups, especially, it has been reported that the introduction of the
substitution at 1, 2 and 5 positions of the benzimidazole ring is very important for their
pharmacological effects.
One of the main goals of medicinal chemistry research and drug discovery is to provide a
rational basis for the design of new medicinal agents. Organic compounds and their
reactions have been utilized by people since antiquity. When leaves or tree bark or plant
roots were mixed with water to make a medicinal potion. A complex mixture of organic
products is actually extracted for its biologically active components. In 1960s, a broad
spectrum group of drugs, known as benzimidazoles, were discovered with a big- gang
having specific activity. Due to the increasing demand for bioactive molecules, organic
chemists are increasingly required to synthesize new compounds of biological interest.
There has been an unlimited expansion of molecular diversity in synthetic organic
compounds by the application of combinatorial methodology. The benzimidazole nucleus
is an important pharmacophore in drug discovery8 and it is a fused aromatic imadozole
ring where a benzene ring is fused to 4 and 5 positions of an imidazole ring.
Benzimidazoles are very useful intermediates for the development of molecules of
pharmaceutical or biological interest. Substituted benzimidazole derivatives have found

Chapter-1
6
applications as in diverse therapeutic agents including antiulcer, antihelmintic,
antihypertensive, anticoagulant, antiallergic, analgesic, anti-inflammatory, antipyretic,
antibacterial, antifungal, antiviral, antiparasitic, antioxidant, anticancer and
antianxiolytic. Because of their significant medicinal importance, the synthesis of
substituted benzimidazoles is listed with various effects on human body and are used to
treat multiple system disorders.
Presence of benzimidazole nucleus in numerous categories of therapeutic agents such as
antimicrobials, antivirals, antiparasites, anticancer, anti-inflammatory, antioxidants,
proton pump inhibitors, antihypertensives, anticoagulants, immunomodulators, hormone
modulators, CNS stimulants as well as depressants, lipid level modulators, antidiabetics,
etc. has made it an indispensable anchor for development of new therapeutic agents.
Varied substitutents around the benzimidazole nucleus have provided a wide spectrum of
biological activities. Importance of this nucleus in some activities like, Angiotensin I
(AT1) receptor antagonism and proton-pump inhibition is reviewed separately in
literature. Even some very short reviews on biological importance of this nucleus are also
known in literature. However, owing to fast development of new drugs possessing
benzimidazole nucleus many research reports are generated in short span of time. So,
there is a need to couple the latest information with the earlier information to understand
the current status of benzimidazole nucleus in medicinal chemistry research. In the
present chapter, various derivatives of benzimidazole with different pharmacological
activities are described on the basis of substitution pattern around the nucleus with an aim
to help medicinal chemists for developing SAR on benzimidazole derived compounds for

Chapter-1
7
each activity. This discussion will further help in the development of novel
benzimidazole compounds.
Benzimidazole nucleus can be termed ‘Master Key’ as it is an important core in many
compounds acting at different targets to elicit varied pharmacological properties. Though
all seven positions in the benzimidazole nucleus can be substituted with a variety of
chemical entities, most of the biologically active benzimidazole compounds are based on
the functional groups bearing at 1, 2 and/or 5 (or 6) positions. Accordingly, the
compounds may be mono-, di- or tri-substituted derivatives of the nucleus. In the present
study, various benzimidazole based compounds are designed, synthesized and evaluated
and categorized on the basis of their biological activities. The major activities include
antihypertensive, anti-inflammatory, antibacterial, antifungal, anthelmintic, antiviral,
antioxidant, antiulcer, antitumor, pyschoactivity, etc.
1.2.1 Antimicrobial activity
Antimicrobial agents constitute a diverse group of chemical entities acting against varied
kinds of microbes including bacteria, protozoa, helminths (worms), fungi and viruses.
Various research groups have evaluated antibacterial, antiprotozoal, anthelmintic and/or
antifungal activities concomitantly while evaluation of antiviral compounds remains
solitary. Hence, in the present section compounds having antibacterial, antiprotozoal,
antihelmintic and antifungal activities are discussed collectively under the heading of
antimicrobials while antiviral compounds are discussed independently. Most of the
research activities on development of antimicrobials from benzimidazole nucleus have

Chapter-1
8
been taken up after the year 2000. Iwahi and Satoh have reported 2-(substituted pyridyl
methylsulfinyl) benzimidazole (3) as antibacterial agent against Campylobacter pylori.9
Modifications in (3) led to development of similar compound (4) having antibacterial
activity against C. pylori equivalent to omeprazole.10
Coupling of 2-
alkylthiobenzimidazole with β-lactam ring has produced compound (5) wherein
antibacterial and antifungal activities were dependent upon nature of R group.11
Holloway et al. have discovered 2-iminobenzimidazoles as antibacterial agents acting
through inhibition of Trypanothione reductase (a bacterial enzyme).12
1-Substituted
benzimidazole compounds have been found to exhibit poor antimicrobial properties.13
However, substitution at both 1- and 2-positions of benzimidazole has produced potent
antimicrobials. Semicarbazide, thiosemicarbazide and carbamate substituent at 1-position
along with a methyl group at 2-position has yielded compounds which have potent
bactericidal activities.14-16
Attachment of other heterocylces like chromane, β -lactam, thiadiazole and oxadiazole to
benzimidazole nucleus resulted in hybrid compounds (6-8) having potent antibacterial
and/or antifungal properties.17-21

Chapter-1
9
Fusion of 5- or 6-membered heterocycle at N1–C bond of benzimidazole nucleus has
produced tricyclic derivatives of benzimidazole like triazino[1,2-a]benzimidazole (9)
bearing fluorophenyl group as moderately antibacterial,22
1,2,4-triazolo[2,3-
a]benzimidazole bearing short alkyl and alkenyl groups (10) as potent
antimycobacterial23
and pyridobenzimidazole derivatives (11) as antifungal.24,25
Recently,
Kuarm et al. have reported benzimidazo[1,2-c]quinazolin-5-yl chromene derivative (12)
as antibacterial agent.26
Replacement of chloro groups with bromo groups converted the
molecule to antifungal. Varied 2, 5- and 2, 6-disubstituted benzimidazole derivatives
have also been explored for antimicrobial activities.
A critical analysis of these variedly substituted derivatives has revealed that either of the
1 and 2 positions of benzimidazole nucleus should bear a bulky electronic and lipophillic

Chapter-1
10
group while the other should have a small alkyl substituent for the optimum antimicrobial
activity. Further, a small lipophillic group containing a heteroatom at 5/6-position incurs
additional activity.
1.2.2 Antioxidant activity
The drugs possessing antioxidant and free radical scavenging activity have been
implicated in treatment of various diseases like cancer which are directly related to lack
of antioxidant capacity of organism. Cole et al. (1974) reported 5-hydroxybenzimidazole
and 5-hydroxy-2-methylbenzimidazole as effective antioxidants.27
Incorporation of
thiadiazoles, triazoles and their open chain counterparts, that is, thiosemicarbazides at 1-
position of benzimidazole incurs antioxidant activity. Further substitution of varied aryl
and alkyl substituents on these heteronuclei at 1-position has also yielded potent
antioxidants (13-15). Amongst these, semicarbazide derivatives produced stronger
inhibitory effects on lipid peroxidation levels as well as DPPH model.28, 29
Fused thiazolo[3,2-a]benzimidazoles substituted at 3-position by amino methyl group
(16) inhibited the oxidation of adrenaline to adrenochrome by preventing the formation of

Chapter-1
11
superoxide radical.30
Anisimova et al. reported a series of 2-(heteroaryl)imidazo[1,2-
a]benzimidazoles possessing 1-methylbenzimidazol-2-yl and 5-bromo-2-thienyl at R2
with varied dialkylaminoalkyl substituents at R1 as antioxidants in in vitro model of
ascorbate dependent lipid peroxidation model.31
Subsequently, the same research group
disclosed N-acylmethyl derivatives of 9H-2,3-dihydroimidazo and 10H-2,3,4,10-
tetrahydropyrimido[1,2-a]benzimidazole with varied substitutents at 1-position to possess
weak antioxidant activity.32
In continuation on their work on imidazobenzimidazole,
hydroxyl group in aroyl moiety are reported to possess high antioxidant activity.33
However, 2,2,2-trichloro-1-hydroxyethyl group at 3-position weakens the antioxidant
potential which complies with their earlier reports.34
A halogenophenyl group at 2-
position incurs moderate antioxidant activity with fluorine producing the maximally
active compound from the series.35
Recently, cyclization of dialkylaminoethyl at 1-position to 4-substituted piperazines and
piperidines (17) have been investigated for antioxidant activity.36
Schiff’s bases of
benzimidazole (18) has been found to exhibit high lipid peroxidation inhibitory activity
which increased with lipophilicity. A 4-carboxamidobenzimidazole analog (19) was
identified as potent hydroxyl radical scavenging property through poly (ADP-Ribose)
polymerase (PARP) inhibition.37

Chapter-1
12
1.2.3 Anti-inflammatory activity
Control of inflammation has become prime importance due to its association with
numerous diseased states like Alzheimer’s disease, asthma, atherosclerosis, Crohn’s
disease, gout, multiple sclerosis, osteoarthritis, psoriasis, rheumatoid arthritis, diabetes
mellitus, carcinoma, bacterial or viral infections, etc. which result in chronic
inflammation.38,39
The most common and widely explored points for control of
inflammation include inflammatory mediators like plasma proteases, prostaglandins,
leukotrienes, histamine, serotonin, nitric oxide, interleukins 1–16 (IL-1 to IL-16), tumour
necrosis factor-α (TNF-α), chemokines (CXC, CC and C subsets) and colony stimulating
factors (CSF).40–42
These mediators are produced through various processes involving
cyclooxygenases, caspases and kinases like cyclin dependent kinases (CDK1 and CDK5),
mitogen activated protein kinase 38 (MAP38), c-Jun N-terminal kinase (JNK), serine
threonine kinases (IKK1 and IKK2), interleukin receptor associated kinase 4 (IRAK-4),
Janus kinases (JAK1- JAK3 and Tyk2), kinase insert domain receptor (KDR),
lymphocyte specific kinase (Lck), spleen tyrosin kinase (Syk) and TNF-α kinase
(TNFK).43,44

Chapter-1
13
A large number of chemical entities derived from diverse group of heterocyclic nuclei are
reported to inhibit or block the inflammatory process at one or other stages. The search
for anti-inflammatory compounds derived from benzimidazole nucleus is as old as the
age of modern medical chemistry. Though a good number of research groups have
reported various benzimidazole derivatives having well to excellent anti-inflammatory
activity but no such molecule has made its way to the clinics so far. A number of
compounds targeting the kinases are currently undergoing clinical trials related to
inflammation and autoimmunity.45
Benzimidazole nucleus substituted at 1-position with varied heterocycles has produced
potent anti-inflammatory compounds. Sabat et al. have synthesized a series of 1-
(substituted pyrimidin-2-yl) benzimidazoles of which compound (20) has elicited anti-
inflammatory effect by blocking activity of Lck.46
Another similar compound (21) in
which pyrimidine is replaced by thiophene has been identified as moderately potent
inhibitor of IKK-3 kinase with pIC50 of 5.4.47
Based on extensive SAR studies, it has
been found that replacement of the amide moiety in (21) by the nitrile group increases
inhibitory effect on IKK-3. Further, substitution at 6-position in the benzimidazole has
resulted in compound (22) as potent inhibitor of JAK3 which is expressed in high levels
in natural killer cells, platelets, thymocytes, mast cells and inducible T and B cells.48
Buckley et al. have brought another similar 1,6-disubstituted compound as highly potent
IRAK4 inhibitor having good TNF-α inhibition.49

Chapter-1
14
Based on the moderate anti-inflammatory and analgesic activities of thiabendazole (a
well known anthelmintic), the Pharmaceutical Research Centre at Kanebo Ltd. (Japan)
have developed a series of 2-(2-pyridinyl) benzimidazoles by isosteric replacement of
thiazole ring in the lead.50–52
From a series of fifty five compounds, 2-(5-ethyl-2-
pyridinyl) benzimidazole (23) (KB-1043) is found to have anti-inflammatory, analgesic
and antipyretic activities better than phenylbutazone and tiaramide. Moreover, it has
slightly less gastrointestinal irritation and therapeutic index 2–3 times better than the
reference compounds. Recently, Achar et al. have synthesized novel 2-(substituted
phenyl) aminomethyl benzimidazoles and evaluated using carrageenan-induced paw
edema model. The compound (24) has emerged as potent compound (81% protection)
and the activity is further improved (89% inhibition) by placement of bromo group at 6-
position (25).53
A high throughput screening (HTS) of small molecules followed by SAR studies at
Amgen Inc. has identified (26) as potent inhibitor of IRAK4.54
A recent patent also
discloses similar N-acyl 2-aminobenzimidazole derivative with varied aroyl and

Chapter-1
15
heteroaroyl substituents at 2-position exemplified by compound (27) as potent IRAK4
inhibitor.55
Another HTS at Abbott Corporate has identified a new series of 1,2-
disubstituted benzimidazole derivatives through binding studies of CXCL10 to CHO cell
membranes.56
The compound (28) proved maximally active. Further, substitution with
methoxy group at 4-position of benzimidazole nucleus retained the activity, which
however decreased with substitution at 5- and 6-positions.
A structure based design of 2-methyl-N-substituted benzimidazole bearing varied sugar
moieties (29) has been reported to have significant anti-inflammatory activity dependent
on the kind and the linked-position of the sugar conjugated to the nucleus.57
Simultaneous
substitutions at 2- and 5-positions of benzimidazole nucleus have fancied many research
groups to develop novel anti-inflammatory drugs. Taking benoxaprofen as lead, Dunwel
et al.58
have synthesized (30) by bioisosteric replacement of benzoxazole nucleus with
benzimidazole. However, it did not reduce the inflammation in rat paw edema model
probably due to lower solubility or altered drug-receptor interactions. Subsequently,
Evans et al. from the same laboratory synthesized an exhaustive series of 72

Chapter-1
16
benzimidazole derivatives and tested on rat adjuvant arthritis screen.59
Only two
compounds (31 and 32) have been found to exhibit activity comparable to indomethacin.
The activity exhibited by compounds derived from varied substituents all around the
benzimidazole nucleus has prompted many research groups to synthesize fused
benzimidazole compounds. Toja et al. have synthesized for the first time fused imidazole
derivatives (benzimidazoles or naphthimidazoles) substituted at 1 and 2 positions as non-
acidic anti-inflammatory agents.60
SAR studies on about 50 compounds revealed that
electron rich groups such as –OCH3, –OC2H5, –NHCH3, –N(CH3)2 on para position in
phenyl ring at 2-position of the nucleus increases the activity. Compounds with
substituent at 1-position exhibit stronger anti-inflammatory potency. In addition to good
anti-inflammatory, analgesic and antipyretic activities, all active compounds also lack
ulcerogenic properties. The compounds (33) and (34) have emerged as the most potent
compounds. Sondhi et al. have synthesized and screened some tricyclic benzimidazole
derivatives and revealed that pyrimido[1,6-a]benzimidazole derivative (35) controls the
inflammation and pain not better than ibuprofen but pyrazolo[1,2-a]benzimidazole (36)

Chapter-1
17
analogs exhibit the activity equivalent to ibuprofen.61,62
A similar pyrimido[1,2-
a]benzimidazole derivative has also been found to have very weak anti-inflammatory and
analgesic activities.63
Further exploration of pyrimidobenzimidazoles has led to a series of
pyrimido[5`,4`:5,6]pyrimido[1,2-a]benzimidazol-5-ones as potent orally active specific
inhibitors of LcK. SAR studies have revealed the compound (37) as the most potent
compound.64
Very recently, Shen et al. have discovered a novel compound (38) by fusion
of an imidazole nucleus with phenyl ring of an active metabolite isolated from
fermentation broth of fungus Curvularia verruculosa having strong inhibitory activity
against TNF-α transcription.65
A series of 1-acyl-2-alkylthio-1,2,4-triazolo[3,2-
a]benzimidazole derivatives is reported from which the compound (39) exhibits the most
potent anti-inflammatory and analgesic activities reiterating the importance of amino
group at 2-position of benzimidazole nucleus.66
It has also exhibited a superior
gastrointestinal (GI) safety profile compared to indomethacin.

Chapter-1
18
The salient structural features of a typical benzimidazole based anti-inflammatory
compound as suggested by critical analysis of chemical structures of various such
compounds is that either of the 1 and 2 positions of the nucleus should bear a bulky,
lipophilic aryl/heteroaryl moiety appropriately substituted with alkyl, electronic or
heterocyclic groups while the tolerable substituents at 5 or 6-position of the nucleus
should have small electronic groups like halogens, nitro, amino, methyl or lower alkoxy
to mildly substituted or unsubstituted aryl or aralkyl groups.
1.2.4 Antihypertensive activity
Benzimidazole nucleus has been explored well for development of antihypertensive
drugs. Many benzimidazole based compounds act as antihypertensives by intercepting
with Renin–Angiotensin System (RAS). Angiotensin II (Ang II) is an octapeptide which
is active pressor produced by RAS cascade. Angiotensinogen, a polypeptide, is cleaved
by rennin to produce a decapeptide, Ang I, which is further acted upon by Angiotensin
converting enzyme (ACE) to generate Ang II. The latter acts on angiotensin receptor 1
(AT1) resulting in vasoconstriction, Na+ retention and aldosterone release to cause
hypertensive action. The various strategies to control these actions of Ang II include

Chapter-1
19
blocking production of Ang II through use of Renin and ACE inhibitors or blocking
binding of Ang II to AT1 receptors. Inhibition at the receptor level has proved maximally
safe, specific and effective. Hence, most of the research and development activities on
producing antihypertensives have been targeted towards development of AT1 receptor
blockers.67
One of the first reports discloses 2-butyl-benzimidazole-7-carboxylic acid
derivative (40) as a potent AT1 receptor antagonist.68
Optimization of the functional groups around the nucleus has produced CV-11974 which
reduces blood pressure in dose-dependent manner by blocking AT1 receptors in a non-
competitive manner due to slow dissociation from AT1 receptors.69-71
It is significantly
more active than losartan and EXP3174.72
Esterification of 7-carboxyl group has
culminated in discovery of orally active and long acting AT1 receptor blocker,
candesartan cilexitil73-77
which is now commercially available. It has triggered a spurt in
research activities to explore all seven positions of the benzimidazole nucleus by various
research groups to develop more potent compounds. In general, it has been found that the
position 4 must remain unsubstituted for favourable interaction of N-3 of the nucleus
with H-bond donor site in AT1 receptor while position 1 is reserved for biphenyl moiety.

Chapter-1
20
Replacement of biphenyl moiety with other moieties in the compounds (41-43) has
produced the compounds with varied potencies.78-82
Replacement of tetrazole moiety with varied acidic heterocylces like oxathiadiazole,
oxatriazole, oxadiazolone, oxadiazolidindione, thiazolone, oxathiadiazolone, etc. has
produced compounds with varied activity.83-85
However, none of the compounds were
found to be potent. Incorporation or substitution of tetrazole with a carboxyl group in the
molecule has produced insurmountable and orally active antagonists.86
A –COOH group
at 7-position provides potent compounds. Further esterification of this acidic function
improves the oral bioavailability as indicated by clinical use of candesartan cilexitil.
Recently, Kuroita et al. have disclosed 5-methyl-2-oxo-1,3-dioxol-4-yl methyl esters
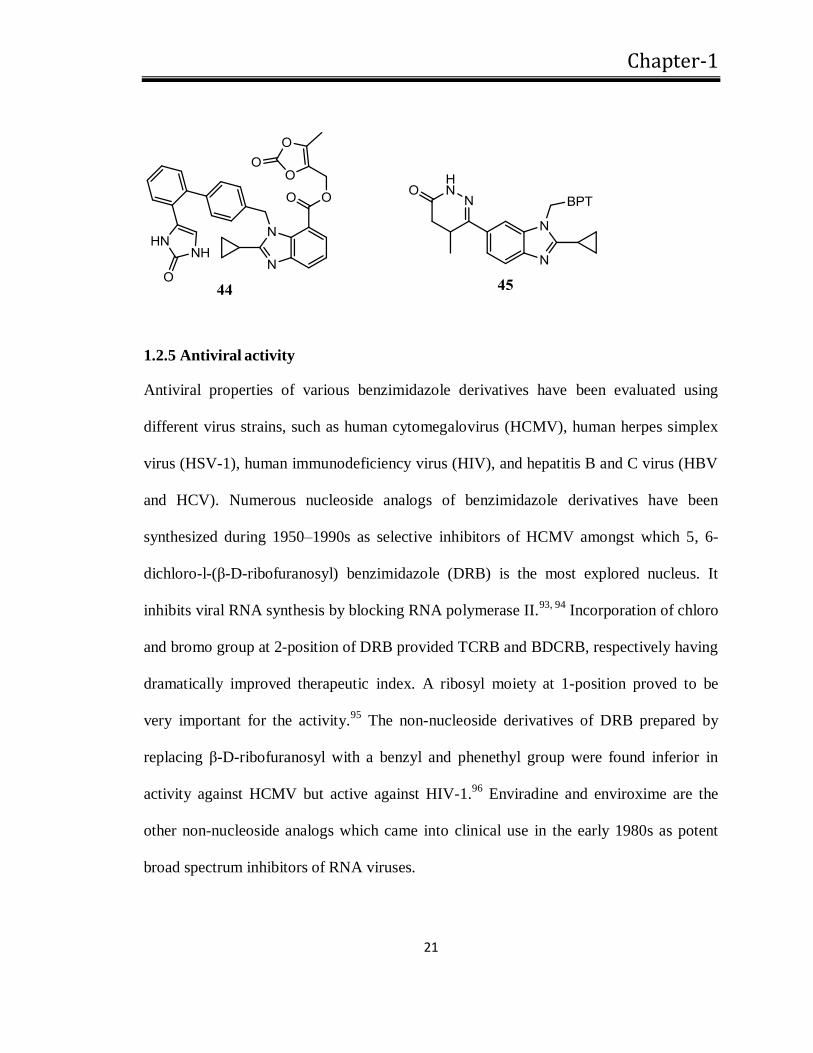
analogs (44) as potent orally active antagonists of Ang II.87-89
Telmisartan is an orally
active, potent and insurmountable AT1 selective antagonist that is formed by a bulky
lipophillic group at 6-position.90,91
Substitution with pyridazinone moiety also produces a
potent benzimidazole derived compound (45).92
Chapter-1
21
1.2.5 Antiviral activity
Antiviral properties of various benzimidazole derivatives have been evaluated using
different virus strains, such as human cytomegalovirus (HCMV), human herpes simplex
virus (HSV-1), human immunodeficiency virus (HIV), and hepatitis B and C virus (HBV
and HCV). Numerous nucleoside analogs of benzimidazole derivatives have been
synthesized during 1950–1990s as selective inhibitors of HCMV amongst which 5, 6-
dichloro-l-(β-D-ribofuranosyl) benzimidazole (DRB) is the most explored nucleus. It
inhibits viral RNA synthesis by blocking RNA polymerase II.93, 94
Incorporation of chloro
and bromo group at 2-position of DRB provided TCRB and BDCRB, respectively having
dramatically improved therapeutic index. A ribosyl moiety at 1-position proved to be
very important for the activity.95
The non-nucleoside derivatives of DRB prepared by
replacing β-D-ribofuranosyl with a benzyl and phenethyl group were found inferior in
activity against HCMV but active against HIV-1.96
Enviradine and enviroxime are the
other non-nucleoside analogs which came into clinical use in the early 1980s as potent
broad spectrum inhibitors of RNA viruses.

Chapter-1
22
Amongst a series of benzimidazole derivatives bearing amidino group at 5-position and
varied heteronuclie such as pyridine, N-methyl-pyrrole or imidazole, the compound with
pyridine ring at 2-position (46) showed distinct and selective antiviral activity towards
RNA replicating enteroviruses. In contrast, pyrrole substituted compound (47) showed
prominent activity against other types of viruses especially adenovirus.97
SAR study on
2-naphthyl benzimidazoles with varied substituents at 5,6-positions of benzimidazole ring
and 4-position of naphthyl ring (48) suggested that electron releasing groups on
benzimidazole enhances the activity. An amino group on naphthalene ring yields a potent
antiviral compound. Replacement of amino with nitro and acetyl groups decreases the
activity significantly.98
Taking this 2-aryl benzimidazole as a lead, 2-biphenyl derivatives
of benzimidazoles were developed but most of the compounds except (49) and (50)
showed poor activities against all viruses tested.99
1H-Benzimidazole-4-carboxamide
derivatives bearing furyl at 2-postion and aryl moiety at carboxamide nitrogen possess
good inhibitory activity.100,101
Barreca et al. reported 1-benzyl-1,3-dihydro-2H-
benzimidazol- 2-ones as potential non-nucleoside reverse transcriptase inhibitors

Chapter-1
23
(NNRTIs) active against HIV-1.102
A 6-chloro-1-(2,6-difluorobenzyl)-substituted
derivative (51) was found to possess significant activity against HIV-1. Subsequently
molecular modeling studies, led to the rational discovery of N1-arylsulfonyl-1,3-dihydro-
2H-benzimidazol-2-one (52) as a novel template for design of new NNRTIs active
against wild-type and mutant strains of HIV-1.103
1.2.6 Antitumor activity
Cancer is one of the leading health hazards which is affecting a wide majority of people
in world population. Various anticancer agents (also referred as antitumor,
antiproliferative and antineoplastics) reported for treatment of varied kinds of cancers act
through different mechanisms. However, the major side effect associated with these
agents is cytotoxicity towards normal cells due to lack of selectivity for the abnormal
cells. Therefore search on anticancer agent has been in continuum since many years.
Benzimidazole being an isostere of purine based nucleic acid and an important scaffold in

Chapter-1
24
various biologically active molecules is widely explored for development of anticancer
agents.
Pyrrolo[1,2-a]benzimidazoles (53–55) is one of the early classes of anticancer agents
acting through cleavages of G and A bases and reductive alkylation of DNA.104–108
The
variedly substituted benzimidazole derivatives (56–58) are reported as cytotoxic against
lung and breast cancers.109,110
Ni et al. have developed some 2-(substituted quinolinon-3-
yl)benzimidazoles as serine/threonine checkpoint kinase (CHK-1) inhibitors for treatment
of cancer.111
The compound (59) has emerged as potent compound with subnanomolar
IC50 value. Neff et al. have reported another series (60) of CHK-1 inhibitors but all
compounds are found to have inhibitory activity significantly less than that of (59).112
Recent developments on 2-substituted benzimidazoles have revealed varied heterocycles
at 2-position to yield potent anticancer agents to various carcinoma cell lines. These

Chapter-1
25
include pyrimidine derivatives (61), 113
pyrazoline derivatives (62),114
and thiazole
derivatives (63).115
Further, 2-substituted benzimidazoles with chloro or carboxy group at
5-position having 4-amino-thioxothiazole (64), 4-oxothiazolidine (65), 4-
fluorobenzylidene (66) and cycloalkylidene are reported as potent antitumor agents.116
Planar fused benzimidazole analogs have the potential to get inserted into the space
between the base pairs of DNA resulting in DNA cleavage. Based on this mechanism, a
benzimidazo[1,2-a]quinoline derivative (67) has exerted potent activity on all cell lines
tested with IC50 values of 0.8–30 μM.117
Recently, more fused planar benzimidazole
derivatives have been reported to exhibit potent cytotoxicity. The examples include (68)
(pyrimido [1, 2-a] benzimidazole-3(4H)-one)118
and (69) (1,3-diarylpyrazino[1,2-
a]benzimidazole).119

Chapter-1
26
Huang et al. have synthesized their benzimidazole isosteres amongst which (70) was
found as the most potent anticancer synthetic precursor of bis(benzimidazoles) against
human A-549, BFTC-905, RD, MES-SA, and HeLa carcinoma cell lines.120
Benzimidazolyl- 1,2,4-triazino[4,5-a]benzimidazol-1-one (71) is another
bis(benzimidazole) analog having significant activity against multidrug- resistant P-
glycoprotein expressing cell lines.121
Two benzimidazole nuclei linked through a
thiophene ring have displayed moderate to strong antiproliferative effect toward a panel
of eight carcinoma cell lines. The most active compound (72) of the series is reported to
enter into live HeLa cells within 30 min, but did not accumulate in nuclei even after 2.5
h.122

Chapter-1
27
Taking Hoechst-33258 (a head-to-tail bis-benzimidazole wherein benzo ring a
benzimidazole nucleus is connected to the imidazole ring of the other nucleus through a
bond) and a head to head bis-benzimidazole (73), wherein two benzimidazole nuclei are
connected at either their benzo or imidazole rings through a bond as leads, Yang et al.
have designed and synthesized another series of symmetrical head-to-head bis-
benzimidazoles and found (74) to possess good antitumour activity.123
Very recently,
Singh and Tandon have modified Hoechst-33258 to synthesize another series of head-to-
tail bis-benzimidazole bearing aryl group at 2- position.124
The derivatives bearing
electron withdrawing groups like F (75) and Cl on the aryl ring exhibited potent
anticancer activity over the compounds bearing electron releasing groups.

Chapter-1
28
1.2.7 Anticoagulant activity
Thrombin causes proteolytic cleavage of fibrinogen, induces platelets activation and
triggers a wide range of effects secondary to thrombosis, for example, vascular smooth
muscle cell and fibroblast proliferation, monocyte chemotaxis, and neutrophil adhesion.
Inhibition of thrombin is an important mechanism for inhibition of coagulation.
Benzimidazole nucleus acts as an appropriate template to place the varied substitutents
required for interaction with thrombin. Hauel et al. have designed a series of
benzimidazole derivatives and arrived at BIBR 953 having excellent inhibitory potency
and tolerability.125
It was double prodrug BIBR 1048 that has exhibited good
pharmacokinetic properties and is in clinical evaluation. 1,2-Disubstituted benzimidazole
derivatives (76) possessing basic amine moieties have been reported as active site
directed thrombin inhibitors.126
Berlex Biosciences have reported tetra substituted
benzimidazole with naphthylamidine group at 1-position (77) as anticoagulant due to
factor Xa (fXa) inhibition. The activity is independent of the substituent at C-2 whereas
substitution of a nitro group at 4-position on the benzimidazole template affords potent
fXa inhibitor with excellent thrombin selectivity.127
Replacing the naphthylamidine
with differently substituted biphenylamidines caused a disappointing change in in vitro
profile. However, simplification of the naphthylamidine group to yield a
propenylbenzene group dramatically improved the potency and selectivity over the
unsubstituted naphthalene analogs.128

Chapter-1
29
Ueno et al. have conducted SAR studies leading to benzimidazole derivative (78) as
potent and selective factor Xa inhibitor possessing excellent anticoagulant activity with
no fatal acute toxicity.129
Inhibitor of factor VIIa/Tissue Factor (fVIIa/TF) complex is
another class of compounds for treatment of thromboembolic disease. The research group
at Celera Genomics has designed and optimized a series of benzimidazole derivatives
wherein compound (79) has emerged as safe anticoagulant but having less residence time
due to excessive glucuronidation.130
Further research into the compounds has led to
development of selective dicarboxylic acid analog (80) with pharmacokinetic profile
amenable to once daily subcutaneous dosing in humans.

Chapter-1
30
1.2.8 Psychoactive activity
The H3 receptors in CNS are associated with central disorders such as impaired cognitive
functions. A series of H3-antagonists composed of an imidazole ring connected through
an alkyl spacer to a 2-aminobenzimidazole moiety was designed and synthesized. Its
QSAR and quantitative structure-property relationship (QSPR) analysis suggested a three
carbon atoms chain length (81) optimum for the antagonistic activity.131
Replacement of
imidazole ring with piperidine and chlorophenoxy substituents retained the affinity for
H3 receptor inferring the importance of 2-aminobenzimidazole in receptor interactions.
The piperidine analog (82) showed good affinity for H3-receptor.132
1, 2-Disubstituted-5-
fluorobenzimidazole derivatives with aza-heterocycles (83) were evaluated to have potent
H3 antagonist activity.133
2-Aminobenzimidazole scaffold was also selected for

Chapter-1
31
development of H1-antihistaminic agents therapeutically used for insomnia. The varied
compounds evolved starting from a series of 2-aminobenzimidazoles include (84-
86).134,135
1.3 Introduction to Benzisoxazoles
Benzisoxazole is an aromatic organic compound with molecular formula C7H5NO
containing a benzene-fused isoxazole ring structure. Benzisoxazole has no household use.
It is used primarily in industry and research.
Being a heterocyclic compound, benzisoxazole finds use in research as a starting material
for the synthesis of larger, usually bioactive structures. It is found within the chemical
structures of pharmaceutical drugs such as the antipsychotic risperidone and
anticonvulsant zonisamide.

Chapter-1
32
Its aromatic nature makes it relatively stable, although as a heterocycle, it has reactive
sites which allow for functionalization.
1.3.1 Benzisoxazoles as antimicrobial agents
6-Fluoro-4-piperidinyl-1,2-benzisoxazole derivatives 87 were evaluated for their efficacy
as antimicrobials136
in vitro by disc diffusion and microdilution method against
pathogenic strains such as Bacillus substilis, Escherichia coli, Pseudomonas fluorescens,
Xanthomonas campestris pvs, X. oryzae, Aspergillus niger, A. flavus, Fusarium
oxysporum, Trichoderma species, F. monaliforme, and Penicillium species.
In their attempt for synthesizing new antimicrobial compounds, they have synthesized,6-
fluoro-4-piperidiny 1,2-benzisoxazole amides 4(I–VI) and evaluated their efficacy as
antimicrobials in vitro by disc diffusion and microdilution methods against various
pathogenic strains. Nystatin was used as standard drug against fungi, streptomycin and
tetracycline and were tested against bacteria. In all determinations, tests were performed
in duplicate and the results were reported as mean of three determinations. The results
showed that, from the 6-fluoro-4-piperidinyl-1,2-benzisoxazole amide series, compounds
with chloro and ethoxy groups in the second position of the benzene ring showed

Chapter-1
33
significant inhibition. Interestingly the compound bearing pyridine ring in second
position showed significant activity whereas at third position, least inhibition was
observed.
1.3.2 Benzisoxazoles as anticonvulsant agents
Several 3-(sulfamoylmethyl)-1,2-benzisoxazole derivatives 88 were synthesized from 3-
(bromomethyl)-l,2-benzisoxazole by reaction with sodium bisulfite followed by
chlorination and amination. Some of them displayed marked anticonvulsant activity137
in
mice. The introduction of a halogen atom at 5th position of the benzisoxazole ring caused
increased activity and neurotoxicity; the substitution of a sulfonyl group caused
decreased activity. The activity of monoalkylated compounds might be the result of
biotransformation.
The study of N-substituted compounds revealed that the introduction of simple
monoalkyl substituents did not abolish activity, but when the substituent was an amino,
dimethylamino, benzyl or larger, the activity was generally lost. With the exception of
diethylamino and N-(methylpiperanyl) derivatives, the disubstituted compounds were
inactive in the anticonvulsant test.

Chapter-1
34
1.3.3 Benzisoxazoles as Inhibitors of Chaperone Heat Shock Protein 90
Heat shock protein 90 (Hsp90) is a molecular chaperone that is responsible for activating
many signaling proteins and is a promising target in tumor biology. Gopalsamy, et al
have identified small-molecule benzisoxazole derivatives as Hsp90 inhibitors.138
Crystallographic studies show that these compounds bind the ATP binding pocket
interacting with the Asp93.
All the compounds bind to the ATP binding pocket of Hsp90, they were found to be very
selective for Hsp90 inhibition and was not active when tested against a panel of kinases
like B-Raf, PKC, PKC-θ, PI3K-R, PDK-1, MK2, IKK-2, ActRIIB, and m-TOR (IC50
>20 µM). Overall, they have identified benzisoxazole derivatives 89 as potent and
selective inhibitors of molecular chaperone Hsp90. The hit to lead optimization was
guided by structure-based design facilitated by the cocrystallization efforts. Inhibitors
with improved physical properties resulting in enhanced potency in the cellular systems
were disclosed.

Chapter-1
35
1.3.4 Benzisoxazoles as antipsychotic agents
A series of 3-(l-substituted-4-piperidinyl)-1,2-benzisoxazoles 90 are synthesized and
tested for neuroleptic activity139
by climbing mice assay and inhibition of spiroperidol
binding. Structure activity relationships were studied by variation of the substituent on
the benzisoxazole ring with concomitant variation of four different 1-piperidinyl
substituents. Maximum neuroleptic activity was realized when there was a 6-fluoro
substituent on the benzisoxazole ring. The 1-piperidinyl substituent appeared less
significant, although in most cases, the (1,3-dihydro-2-oxo-2H-benzimidazol-l-yl) propyl
group imparted maximum potency. The most potent compound in both assays was 6-
fluoro-3-[1-[3-(1,3-dihydro-2-oxo-2H-benzimidazol-l-yl)propyl]-4- piperidinyl]-1,2-
benzisoxazole.
From the structure-activity relationships it becomes apparent that, for any given 1-
piperidinyl substituent, a fluorine in the 6-position of the benzisoxazole nucleus results in
maximum activity. It also appears that the presence of methoxy or hydroxy groups
greatly diminishes activity, with the 5,6-dimethoxy compounds being quite weak.
Intermediate in potency were the unsubstituted analogues and the 6-chloro derivatives,

Chapter-1
36
with the nuclear unsubstituted compounds being somewhat more potent. The two
compounds bearing 5-fluoro substituent showed activity similar to that of their 6-chloro
counterparts.
1.3.5 Benzisoxazoles as antithrombotic agents
A series of 3-(2-thienyl)- and 3-(1-imidazolyl)-1,2-benzisoxazoles 91 as well as some
isomeric benzoxazoles were synthesized and tested in vitro for their inhibitory effect on
arachidonic acid-induced human platelet aggregation.140
The most active compound (7-
methoxy-3-(2-thienyl)-1,2-benzisoxazole SC) was nearly 20-30-fold more potent than
acetylsalicylic acid in inhibiting platelet aggregation. Structure-activity relationships
within the series are briefly discussed.
In an attempt to understand the contribution of the cyclic structure at position 3 of the
benzisoxazole nucleus on biological activity, authors have synthesized some non-
thiophenic derivatives differing in the degree of aromaticity. Permutation of the
thiophene nucleus by a nitrogen ring such as imidazole resulted in a marked decrease in
anti-aggregating activity. On the other hand, the introduction of a π electron-rich group

Chapter-1
37
such as the para methoxy phenyl moiety retained to a large extent, the pharmacological
activity. It has long been accepted that one of the most widely employed criteria for the
quantitative assessment of aromaticity is the resonance energy. Available values for
benzene, thiophene and imidazole are respectively 152, 121 and 59 kJ mol-1, indicating
that imidazole has a lower degree of aromaticity than the other 2. However, the marked
increase in activity exhibited by the thiophene derivative SC, compared to the para-
methoxy phenyl analogue seems to indicate that aromaticity is not the only factor
involved in the biological response. The thiophenic sulfur atom might be important for
activity.
1.3.6 Benzisoxazoles as antiglycating agents
Shantharam, et al have synthesized a series of urea/thiourea derivatives of Gly/Pro
conjugated benzisoxazoles 92 and has been screened for their in vitro antiglycation
activity141
. Several compounds showed promising activity with IC50 < 5 μM compared to
standard rutin (IC50 = 41.9 μM). Further, it was found that compounds containing
methoxy and bromine substituents have exerted highly potent activity.
They have successfully synthesized a series of urea/thiourea derivatives of Gly/Pro
conjugated benzisoxazole with different functionalities. Some of the representatives of

Chapter-1
38
the series were identified as highly potent antiglycating agents. The antiglycating activity
of the synthesized compounds showed that urea and thiourea moieties play a major role
in enhancing the activity.
Further, it is interesting to note that OCH3 and Br act as active moieties in inhibiting the
glycation. Thus, nature of the substituent was found to be crucial to improve the activity.
This study extends the knowledge of different substituents at phenyl ring and also various
amino acids which might be of interest for the identification of more antiglycation agents.
1.4 Scope of the present work
Heterocyclic compounds have a wide range of applications: they are predominant among
the types of compounds used as pharmaceuticals,
as agrochemicals, as veterinary
products, used as optical brightening agents, as antioxidants, as corrosion inhibitors and
as additives with a variety of other functions. In particular, benzimidazoles are involved
in a great variety of biological processes. Some of their poly functional derivatives have
proved to possess antibacterial, fungicidal and antihelmintic activities. Therefore,
substituted benzimidazoles have attracted the interest of various research groups.
Similarly, benzisoxazole finds use in research as a starting material for the synthesis of
larger, usually bioactive structures. It is found within the chemical structures of
pharmaceutical drugs.
A series of pyridine conjugated benzimidazole derivatives were synthesized according to
Scheme 1 and evaluated for their antibacterial, antioxidant and anti-inflammatory
activities. The results showed that most of the tested compounds exhibited good to
moderate antimicrobial activity against some strains of Gram negative bacteria

Chapter-1
39
(Escherichia coli, Klebsiella pneumoniae, Salmonella typhi, Shigella flexineri) and Gram
positive bacteria- Bacillus subtilis. Further, the molecules were evaluated for antioxidant
assays such as DPPH scavenging, super oxide radical scavenging and hydroxyl radical
scavenging assays. Most of the compounds showed potent antioxidant activities. Also,
the synthesized compounds were screened for anti-inflammatory activities such as
lipoxygenase inhibition and indirect haemolytic assays, where compounds revealed good
activity.
Reagents and reaction conditions: i) NaOCH2CF3 ii) NaNO2, H2SO4 iii) H2SO4,
CH3OH. iv) NaBH4, Methanol. v) SOCl2, DCM. vi) Mercaptobenzimidazole, Methanol.
vii) 8 (R-X/R-SO2Cl), Toluene, 50% KOH, TBAB.
Scheme 1

Chapter-1
40
A series of benzisoxazole derivatives were synthesized according to Scheme 2 and
evaluated for their antibacterial, antioxidant and anti-inflammatory activities. The results
indicated that most of the compounds exhibited moderate antimicrobial activity against
Gram negative (Escherichia coli, Klebsiella pneumoniae, Salmonella typhi, Shigella
flexineri) and Gram positive (Bacillus subtilis) bacterial cultures. The molecules were
evaluated for antioxidant activities using 2,2-diphenyl-1-picrylhydrazyl, super oxide
radical and hydroxyl radical scavenging assays and most of them showed good
antioxidant activities. Also, the synthesized compounds were screened for anti-
inflammatory activities such as lipoxygenase inhibition and indirect haemolytic assays.
Reagents and reaction conditions: (a) MeONa/THF, 0 °C-RT, 8 h. (b)
LiOH/MeOH/H2O, 0°C-RT, 3h. (c) 5 (6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole
hydrochloride), EDC.HCl/HOBt/DIPEA/CH2Cl2, 0 °C-RT, 8h. (d) HCl/ether, 0 °C-RT,
1h. (e) 8 (RCOCl/RSO2Cl), TEA/EDC, 0 °C-RT, 3-4 h.
Scheme 2

Chapter-1
41
References:
1. Katritzky, R.; Boultan, J. A. (Eds). Academic press, New York, “Advances in
Heterocyclic chemistry”, 1963-1980, 1.
2. Weissberger,.; Wiley Interscience. New York, “The Chemistry of heterocyclic
compounds”, 1950-1975, 1.
3. Katritzky. R.; Academic press, New York, “Physical methods in Heterocyclic
Chemistry”, 1963 -1973, 1
4. Newkome,G.R.; Pandler,W.W. “Contemporary Heterocyclic Chemistry”, John
Wiley, New York, 1982.
5. Czarnik, A. W. top 20 ethical pharmaceuticals prescribed in the USA in 1994,
17 are heterocyclic compounds: “Acc. Chem. Res”, 1996, 29, 112.
6. Meth-Cohn, O. Reviews of several applications of Heterocyclic Compounds can
be found in “Comprehensive Heterocyclic Chemistry,” Vol 1, ed., Pergamon
Press, Oxford, 1984.
7. Clough, J. M.; Godfrey, C. R. A. the discovery of this fungicide is described by
“Chem. Br”, 1995, 466.
8. Spasov, A. R.; iezhitsa, I. N.; Bugaeva, L. I.; Anisimova, V. A. Khim-Farm.
Zhurn, 1999, 33, 6.
9. Iwahi, T.; Satoh, H. U.S. Patent 5013,743, 1991.
10. Masaki, M.; Yamakawa, T.; Nomura, Y.; Matsukura, H. U.S. Patent 5576,341,
1996.
11. Desai, K. G.; Desai, K. R. Bioorg. Med. Chem. 2006, 14, 8271.

Chapter-1
42
12. Holloway, G. A.; Baell, J. B.; Fairlamb, A. H.; Novello, P. M.; Parisot, J. P.;
Richardson, J.; Watsona, K. G.; Street, I. P. Bioorg. Med. Chem. Lett. 2007, 17,
1422.
13. Guven, O. O.; Erdogan, T.; Goker, H.; Yıldız, S. Bioorg. Med. Chem. Lett.
2007, 17, 2233.
14. Fahmy, H. H.; El-masry, A.; Abdelwahed, S. H. A. Arch. Pharm. Res. 2001, 24,
27. 100.
15. Nofal, Z. M.; Fahmy, H. H.; Mohamed, H. S. Arch. Pharm. Res. 2002, 25, 250.
16. Nofal, Z. M.; Fahmy, H. H.; Mohamed, H. S. Arch. Pharm. Res. 2002, 25, 28.
17. Siva Kumar, B. V.; Vaidya, S. D.; Kumar, R. V.; Bhiryd, S. B.; Manr, R. B. Eur.
J. Med. Chem. 2006, 41, 599.
18. Ansari, K. F.; Lal, C. J. Chem. Sci. 2009, 121, 1017.
19. Ansari, K. F.; Lal, C. Eur. J. Med. Chem. 2009, 44, 4028.
20. Ansari, K. F.; Lal, C. Eur. J. Med. Chem. 2009, 44, 2294.
21. Gill, C.; Jadhav, G.; Shaikh, M.; Kale, R.; Shawalkar, A.; Nagargoje, D.;
Shiradkar, M. Bioorg. Med. Chem. Lett. 2008, 18, 6244.
22. Dolzhenko, A. V.; Chui, W.; Dolzhenko, A. V.; Chan, L. J. Fluor. Chem. 2005,
126, 759.
23. Mohamed, B. G.; Hussein, M. A.; AbdeI-Alim, A. M.; Hashem, M. Arch.
Pharm. Res. 2006, 29, 26.
24. Kitamura, A.; Someya, K.; Hata, M.; Nakajima, R.; Takemura, M. Antimicrob.
Agents Chemother. 2009, 53, 670.

Chapter-1
43
25. Takeshita, H.; Watanabe, J.; Kimura, Y.; Kawakami, K.; Takahashi, H.;
Takemura, M.; Kitamura, A.; Someya, K.; Nakajima, R. Bioorg. Med. Chem.
Lett. 2010, 20, 3893.
26. Kuarm, B. S.; Reddy, Y. T.; Madhav, J. V.; Crooks, P. A.; Rajitha, B. Bioorg.
Med. Chem. Lett. 2011, 21, 524.
27. Cole, E. R.; Crank, G.; Salam-Sheikh, A. J. Agr. Food Chem. 1974, 22, 918.
28. Kus, C.; Ayhan-Kilcigil, G.; Eke, B. C.; Iscan, M. Arch. Pharm. Res. 2004, 27,
156.
29. Kus, C.; Ayhan-Kılcıgil, G.; Ozbey, S.; Betu, F.; Kaynak, I.; Kaya, M.; Coban,
T.; Can-Eke, B. Bioorg. Med. Chem. 2008, 16, 4294.
30. Dianov, V. M. Pharm. Chem. J. 2007, 41, 308.
31. Anisimova, V. A.; Tolpygin, I. E.; Spasov, A. A.; Kosolapov, V. A.; Stepanov,
A. V.; Kucheryavenko, A. F. Pharm. Chem. J. 2006, 40, 261.
32. Anisimova, V. A.; Spasov, A. A.; Kucheryavenko, A. F.; Panchenko, T. I.;
Ostrovskii, O. V.; Kosolapov, V. A.; Larionov, N. P. Pharm. Chem. J. 2002, 36,
528.
33. Anisimova, V. A.; Tolpygin, I. E.; Spasov, A. A.; Kosolapov, V. A.; Stepanov,
A. V.; Orlova, A. A.; Naumenko, L. V. Pharm. Chem. J. 2007, 41, 126.
34. Anisimova, V. A.; Spasov, A. A.; Kosolapov, V. A.; Tolpygin, I. E.; Porotikov,
V. I.; Kucheryavenko, A. F.; Sysoeva, V. A.; Tibirkova, E. V.; Eltsova, L. V.
Pharm. Chem. J. 2009, 43, 491.

Chapter-1
44
35. Anisimova, V. A.; Spasov, A. A.; Tolpygin, I. E.; Minkin, V. I.; Chernikov, M.
V.; Yakovlev, D. S.; Stukovina, A. Y.; Goryagin, I. I.; Grechko, O. Y.;
Kirillova, N. V.; Kosolapov, V. A.; Tibirkova, E. V.; Salaznikova, O. A.;
Naumenko, L. V.; Gurova, N. A. Pharm. Chem. J. 2010, 44, 345.
36. Anisimova, V. A.; Spasov, A. A.; Tolpygin, I. E.; Chernikov, M. V.; Yakovlev,
D. S.; Goryagin, I. I.; Gurova, N. A.; Salaznikova, O. A.; Naumenko, L. V.;
Kosolapov, V. A.; Eltsova, L. V.; Kolobrodova, N. A. Pharm. Chem. J. 2010,
44, 241.
37. Kalai, T.; Balog, M.; Szabo, A.; Gulyas, G.; Jeko, J.; Sumegi, B.; Hideg, K. J.
Med. Chem. 2009, 52, 1619.
38. Medzhitov, R. Cell 2010, 140, 771.
39. Grivennikov, S.; Greten, F. R.; Karin, M. Cell 2010, 140, 883.
40. Nathan, C. Nature 2002, 420, 846.
41. Cronstein, B. N.; Weissmann, G. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 449.
42. Feghali, C. A.; Wright, T. M. Frontiers Biosci. 1997, 2, 12.
43. Bhagwat, S. S. Purinergic Signal. 2009, 5, 107.
44. Dinarello, C. A. Cell 2010, 140, 935.
45. Sondhi, S. M.; Singhal, N.; Johar, M.; Reddy, B. S. N.; Lown, J. W. Current.
Med. Chem. 2002, 9, 1054.
46. Sabat, M.; VanRens, J. C.; Laufersweiler, M. J.; Brugel, T. A.; Maier, J.;
Golebiowski, A.; De, B.; Easwaran, V.; Hsieh, L. C.; Walter, R. L.; Mekel, M.
J.; Evdokimov, A.; Janusz, M. J. Bioorg. Med. Chem. Lett. 2006, 16, 5973.

Chapter-1
45
47. Bamborough, P.; Christopher, J. A.; Cutler, G. J.; Dickson, M. C.; Mellor, G.
W.; Morey, J. V.; Patel, C. B.; Shewchuk, L. M. Bioorg. Med. Chem. Lett. 2006,
16, 6236.
48. Chen, J. J.; Thakur, K. D.; Clark, M. P.; Laughlin, S.; George, K. M.; Bookland,
R. G.; Davis, J. R.; Cabrera, E. J.; Easwaran, V.; De, B.; Zhang, Y. G. Bioorg.
Med. Chem. Lett. 2006, 16, 5633.
49. Buckley, G. M.; Ceska, T. A.; Fraser, J. L.; Gowers, L.; Groom, C. R.;
Higueruelo, A. P.; Jenkins, K.; Mack, S. R.; Morgan, T.; Parry, D. M.; Pitt, W.
R.; Rausch, O.; Richarda, M. D.; Sabin, V. Bioorg. Med. Chem. Lett. 2008, 18,
3291.
50. Tsukamoto, G.; Yoshino, K.; Kohno, T.; Ohtaka, H.; Kagaya, H.; Ito, K. J. Med.
Chem. 1980, 23, 734.
51. Ito, K.; Kagaya, H.; Fukuda, T.; Yoshino, K.; Nose, T. Arzneimittel-forschung
1982, 32, 49.
52. Ito, K.; Kagaya, H.; Satoh, I.; Tsukamoto, G.; Nose, T. Arzneimittel-forschung
1982, 32, 117.
53. Achar, K. C. S.; Hosamani, K. M.; Seetharamareddy, H. R. Eur. J. Med. Chem.
2010, 45, 2048.
54. Powers, J. P.; Li, S.; Jaen, J. C.; Liu, J.; Walker, N. P. C.; Wang, Z.; Wesche, H.
Bioorg. Med. Chem. Lett. 2006, 16, 2842.
55. Frenkel, A. D.; Lively, S. E.; Powers, J. P.; Smith, A.; Sun, D.; Tomooka, C.;
Wang, Z. U.S. Patent. US 7635774, 2009.

Chapter-1
46
56. Hayes, M. E.; Wallace, G. A.; Grongsaard, P.; Bischoff, A.; George, D. M.;
Miao, W.; McPherson, M. J.; Stoffel, R. H.; Green, D. W.; Roth, G. P. Bioorg.
Med. Chem. Lett. 2008, 18, 1573.
57. El-Nezhawy, A. O. H.; Gaballah, S. T.; Radwan, M. A. A.; Baiuomy, A. R.;
Abdel- Salam, A. M. E. Med. Chem. 2009, 5, 558.
58. Dunwel, D. W.; Evans, D.; Smith, C. E.; Williamson, W. R. N. J. Med. Chem.
1975, 18, 692.
59. Evans, D.; Hicks, T. A.; Williamson, W. R. N.; Dawson, W.; Meacockz, S. C.
R.; Kitchen, E. A. Eur. J. Med. Chem. 1996, 3, 635.
60. Toja, E.; Selva, D.; Schiatti, P. J. Med. Chem. 1984, 27, 610.
61. Sondhi, S. M.; Rajvanshi, S.; Johar, M.; Bharti, N.; Azam, A.; Singh, A. K. Eur.
J. Med. Chem. 2002, 37, 835.
62. Sondhi, S. M.; Rani, R.; Singh, J.; Roy, P.; Agrawal, S. K.; Saxena, A. K.
Bioorg. Med. Chem. Lett. 2010, 20, 2306.
63. Shaaban, M. R.; Saleh, T. S.; Mayhoub, A. S.; Mansour, A.; Farag, B. M.
Bioorg. Med. Chem. 2008, 16, 6344.
64. Martin, M. W.; Newcomb, J.; Nunes, J. J.; Boucher, C.; Chai, L.; Epstein, L. F.;
Faust, T.; Flores, S.; Gallant, P.; Gore, A.; Gu, Y.; Hsieh, F.; Huang, X.; Kim, J.
L.; Middleton, S.; Morgenstern, K.; Oliveira-dos-Santos, A.; Patel, V. F.;
Powers, D.; Rose, P.; Tudor, Y.; Turci, S. M.; Welcher, A. A.; Zack, D.; Zhao,
H.; Zhu, L.; Zhu, X.; Ghiron, C.; Ermann, M.; Johnston, D.; Saluste, C. P. J.
Med. Chem. 2008, 51, 1637.

Chapter-1
47
65. Shen, Y.; Boivin, R.; Yoneda, N.; Du, H.; Schiller, S.; Matsushima, T.; Goto,
M.; Shirota, H.; Gusovsky, F.; Lemelin, C.; Jiang, Y.; Zhang, Z.; Pelletier, R.;
Ikemori- Kawada, M.; Kawakami, Y.; Inoue, A.; Schnaderbeck, M.; Wang, Y.
Bioorg. Med. Chem. Lett. 2010, 20, 3155.
66. Mohamed, B. G.; Abdel-Alim, A. M.; Hussein, M. A. Acta Pharm. 2006, 56, 31.
67. Naik, P.; Murumkar, P.; Giridhar, R.; Yadav, M. R. Bioorg. Med. Chem. 2010,
18, 8418.
68. Kubo, K.; Inada, Y.; Kohara, Y.; Sugiura, Y.; Ojima, M.; Itoh, K.; Furukawa,
Y.; Nishikawa, K.; Nakat, T. J. Med. Chem. 1993, 36, 1772.
69. Kubo, K.; Kohara, Y.; Imamiya, Y.; Sugiura, Y.; Inada, Y.; Furukawa, Y.;
Nishikawa, K.; Naka, T. J. Med. Chem. 1993, 36, 2182.
70. Vanderheyden, P. M. L.; Fierens, F. L. P.; Vauquelin, G.; Verheijen, I. J. Clin.
Basic Cardiol. 2002, 5, 75.
71. Ojima, M.; Inada, Y.; Shibouta, Y.; Wada, T.; Sanada, T.; Kubo, K.; Nishikawa,
K. Eur. J. Pharmacol. 1997, 319, 137.
72. Shibouta, Y.; Inada, Y.; Ojima, M.; Wada, T.; Noda, M.; Sanada, T.; Kubo, K.;
Kohara, Y.; Naka, T.; Nishikawa, K. J. Pharmacol. Exp. Ther. 1993, 266, 114.
73. Ogihara, T.; Nagano, M.; Higaki, J.; Kohara, K.; Mikami, H. J. Cardiovasc.
Pharmacol. 1995, 26, 490.
74. Kubo, K.; Kohara, Y.; Yoshimura, Y.; Inada, Y.; Shibouta, Y.; Furukawa, Y.;
Kato, T.; Nishikawa, K.; Naka, T. J. Med. Chem. 1993, 36, 2343.
75. Morimoto, S.; Ogihara, T. Cardiovasc. Drug Rev. 1994, 12, 153.

Chapter-1
48
76. Inada, Y.; Wada, T.; Shibouta, Y.; Ojima, M.; Sanada, T.; Ohtsuki, K.; Itoh, K.;
Kubo, K.; Kohara, Y.; Naka, T.; Nishikawa, K. J. Pharmacol. Exp. Ther. 1994,
268, 1540.
77. Kohara, Y.; Imamiya, E.; Kubo, K.; Wada, T.; Inada, Y.; Naka, T. Bioorg. Med.
Chem. Lett. 1903, 1995, 5.
78. Thomas, A. P.; Allott, C. P.; Gibson, K. H.; Major, J. S.; Masek, B. B.; Oldham,
A. A.; Ratcliffe, A. H.; Roberts, D. A.; Russell, S. T.; Thomason, D. A. J. Med.
Chem. 1992, 35, 877.
79. Palkowitz, A. D.; Steinberg, M. I.; Zimmerman, K. M.; Thrasher, K. J.; Hauser,
K. L.; Boyd, D. B. Bioorg. Med. Chem. Lett. 1995, 5, 1015.
80. Xu, J. Y.; Ran, Q.; Hua, W. Y.; Wu, X. M.; Wang, Q. J.; Zhang, J. Chinese
Chem. Lett. 2007, 18, 251.
81. Wienen, W.; Hauel, N.; Van Meel, J. C. A.; Narr, B.; Ries, U. J.; Entzeroth, M.
Br. J. Pharmacol. 1993, 110, 245.
82. Yagupolskii, L. M.; Fedyuk, D. V. Tetrahedron Lett. 2000, 41, 2265.
83. Kohara, Y.; Kubo, K.; Imamiya, E.; Wada, T.; Inada, Y.; Naka, T. J. Med.
Chem. 1996, 39, 5228.
84. Soll, R. M.; Kinney, W. A.; Primeau, J.; Garrick, L.; McCaully, R. J.; Coltsky,
T.; Oshiro, G.; Park, C. H.; Hartupee, D.; White, V.; McCallum, J.; Russo, A.;
Dinish, J.; Wodian, A. Bioorg. Med. Chem. Lett. 1993, 3, 757.

Chapter-1
49
85. Ferrari, B.; Tailadas, J.; Perreaunt, P.; Berhart, C.; Gougat, J.; Guiraudou, P.;
Cazaubon, C.; Roccon, A.; Nisato, D.; Le Fur, G.; Breliere, J. C. Bioorg. Med.
Chem. Lett. 1994, 4, 45.
86. Cho, N.; Kubo, K.; Furuya, S.; Sugiura, Y.; Yasuma, T.; Kohara, Y.; Ojima, M.;
Inada, Y.; Nishikawa, Y.; Naka, T. Bioorg. Med. Chem. Lett. 1994, 4, 35.
87. Kuroita, T.; Sakamoto, H.; Ojima, M. U.S. Patent. US 2009/0270464, 2009.
88. Kuroita, T.; Ojima, M.; Ban, J. U.S. Patent. US 2009/0054502, 2009.
89. Kuroita, T.; Sakamoto, H.; Igawa, H.; Sasaki, M.; Asano, K.; Maekawa, T.; Fuji,
K. U.S. Patent. US7803940, 2010.
90. Ries, U. J.; Mihm, G.; Narr, B.; Hasselbach, K. M.; Wittneben, H.; Entzeroth,
M.; VanMeel, J. C. A.; Wienen, W.; Hauel, N. H. J. Med. Chem. 1993, 36,
4040.
91. Wienen, W.; Entzeroth, M.; van Meel, J. C. A.; Stangier, J.; Busch, U.; Ebner,
T.; Schmid, J.; Lehmann, H.; Matzek, K.; Kempthorne-Rawson, J.; Gladigau,
V.; Hauel, N. H. Cardiovas. Drug Rev. 2000, 18, 127.
92. Dorsch, D.; Mederski, W. W. K. R.; Beier, N.; Lues, I.; Minck, K. O.; Schelling,
P. Bioorg. Med. Chem. Lett. 1994, 4, 1297.
93. Tamm, I.; Nemes, M. M.; Osterhout, S. J. Exp. Med. 1960, 111, 339.
94. Chodosh, L. A.; Fire, A.; Samuels, M.; Sharp, P. A. J. Biol. Chem. 1989, 264,
2250.
95. Townsend, B. L.; Devivar, R. V.; Turk, S. R.; Nassiri, M. R.; Drach, J. C. J.
Med. Chem. 1996, 38, 4098.

Chapter-1
50
96. Porcari, A. R.; Devivar, R. V.; Kucera, L. S.; Drach, J. C.; Townsend, L. B. J.
Med. Chem. 1998, 41, 1252.
97. Starcevic, K.; Kralj, M.; Ester, K.; Sabol, I.; Grce, M.; Pavelic, K.; Zamola, G.
K. Bioorg. Med. Chem. 2007, 15, 4419.
98. Vitalea, G.; Cartaa, A.; Lorigaa, M.; Pagliettia, G.; La Collab, P.; Busonerab, B.;
Collub, D.; Loddob, R. Med. Chem. 2008, 4, 605.
99. Vitale, G.; Corona, P.; Loriga, M.; Carta, A.; Pagliettia, G.; Collab, P. L.;
Busonera, B.; Marongiu, E.; Collu, D.; Loddo, R. Med. Chem. 2009, 5, 507.
100. Cheng, J.; Xie, J. T.; Luo, X. J. Bioorg. Med. Chem. Lett. 2005, 15, 267.
101. Zhang, Z. L.; Sun, Z. J.; Xue, F.; Luo, X. J.; Xiu, N. Y.; Teng, L.; Peng, Z. G.
Chinese Chem. Lett. 2009, 20, 921.
102. Barreca, M. L.; Rao, A.; Luca, L. D.; Zappala, M.; Monforte, A. M. J. Med.
Chem. 2005, 48, 3433.
103. Barreca, M. L.; Rao, A.; Luca, L. D.; Iraci, N.; Monforte, A. M.; Maga, G.;
Clercq, E. D.; Annecouque, C. P.; Balzarinic, J.; Chimirri, A. Bioorg. Med.
Chem. Lett. 2007, 17, 1956.
104. Schulz, W. G.; Islam, I.; Skibo, E. B. J. Med. Chem. 1995, 38, 109.
105. Islam, I.; Skibo, E. B.; Dorr, R. T.; Alberts, D. S. J. Med. Chem. 1991, 34, 2954.
106. Skibo, E. B.; Schulz, W. G. J. Med. Chem. 1993, 36, 3050.
107. Skibo, E. B.; Islam, I.; Heileman, M. J.; Schulz, W. G. J. Med. Chem. 1994, 37,
78.
108. Boruah, R. C.; Skibo, E. B. J. Med. Chem. 1994, 37, 1625.

Chapter-1
51
109. El-Naem, Sh. I.; El-Nazhawy, A. O.; El-Diwani, H. I.; Abdel Hamid, A. O.
Arch. der Pharm. 2003, 336, 7.
110. Ramla, M. M.; Omar, M. A.; Tokuda, H.; El-Diwani, H. I. Bioorg. Med. Chem.
2007, 15, 6489.
111. Ni, Z. J.; Barsanti, P.; Brammeier, N.; Diebes, A.; Poon, D. J.; Ng, S.; Pecchi,
S.; Pfister, K.; Renhowe, P. A.; Ramurthy, S.; Wagman, A. S.; Bussiere, D. E.;
Le, V.; Zhou, Y.; Jansen, J. M.; Ma, S.; Gesner, T. G. Bioorg. Med. Chem. Lett.
2006, 16, 3121.
112. Neff, D. K.; Dutra, A. L.; Blevitt, J. M.; Axe, F. U.; Hack, M. D.; Buma, J. C.;
Rynberg, R.; Brunmark, A.; Karlsson, L.; Breitenbucher, J. G. Bioorg. Med.
Chem. Lett. 2007, 17, 6467.
113. Abdel-Mohsen, H. T.; Ragab, F. A. F.; Ramla, M. M.; El Diwani, H. I. Eur. J.
Med. Chem. 2010, 45, 2336.
114. Shaharyar, M.; Abdullah, M. M.; Bakht, M. A.; Majeed, J. Eur. J. Med. Chem.
2010, 45, 114.
115. Luo, Y.; Xiao, F.; Qian, S.; Lu, W.; Yang, B. Eur. J. Med. Chem. 2011, 46, 417.
116. Refaat, H. M. Eur. J. Med. Chem. 2010, 45, 2949.
117. Hranjec, M.; Pavlovi, G.; Marjanovi, M.; Kralj, M.; Zamola, G. K. Eur. J. Med.
Chem. 2010, 45, 2405.
118. Neochoritis, C. G.; Tzitzikas, T. Z.; Tsoleridis, C. A.; Stephanatou, J. S.;
Kontogiorgis, C. A.; Hadjipavlou-Litina, D. J.; Papadopoulou, T. C. Eur. J.
Med. Chem. 2011, 46, 297.

Chapter-1
52
119. Demirayak, S.; Kayagil, I.; Yurttas, L. Eur. J. Med. Chem. 2011, 46, 411.
120. Huang, S. T.; Hseib, I. J.; Chena, C. Bioorg. Med. Chem. 2006, 14, 6106.
121. Styskala, J.; Styskalova, L.; Slouka, J.; Hajduch, M. Eur. J. Med. Chem. 2008,
43, 449.
122. Stolic, I.; Miškovic, K.; Magdaleno, A.; Silber, A. M.; Piantanida, I.; Bajic, M.;
Obrovac, L. G. Bioorg. Med. Chem. 2009, 17, 2544.
123. Yang, Y. H.; Cheng, M. S.; Wang, Q. H.; Nie, H.; Liao, N.; Wang, J.; Chen, H.
Eur. J. Med. Chem. 2009, 44, 1808.
124. Singh, M.; Tandon, V. Eur. J. Med. Chem. 2011, 46, 659.
125. Hauel, N. H.; Nar, H.; Priepke, H.; Ries, U.; Stassen, J. M.; Wienen, W. J. Med.
Chem. 2002, 45, 1757.
126. Takeuchi, K.; Bastian, J. A.; Gicord-Moore, D. S.; Harper, R. W.; Miller, S. C.;
Mullaney, J. T.; Sall, D. J.; Smith, G. F.; Zhangy, M.; Fisher, M. J. Bioorg. Med.
Chem. Lett. 2000, 10, 2347.
127. Zhao, Z.; Arnaiz, D.; Griedel, B.; Sakata, S.; Dallas, J.; Whitlow, M.; Trinh, L.;
Post, J.; Liang, A.; Morrissey, M.; Shaw, K. J. Bioorg. Med. Chem. Lett. 2000,
10, 963.
128. Shaw, K. J.; Guilford, W. J.; Griedel, B. D.; Sakata, S.; Trinh, L.; Wu, S.; Xu,
W.; Zhao, Z.; Morrissey, M. M. Bioorg. Med. Chem. Lett. 2002, 12, 1311.
129. Ueno, H.; Katoh, S.; Yokota, K.; Hoshi, J. I.; Hayashi, M.; Uchida, I.; Aisaka,
K.; Hase, Y.; Cho, H. Bioorg. Med. Chem. Lett. 2004, 14, 4281.

Chapter-1
53
130. Kolesnikov, A.; Rai, R.; Young, W. B.; Mordenti, J.; Liu, L.; Torkelson, S.;
Shrader, W. D.; Leahy, E. M.; Hu, H.; Gjerstad, E.; Janc, J.; Katz, B. A.;
Sprengeler, A. P. Bioorg. Med. Chem. Lett. 2006, 16, 2243.
131. Mor, M.; Bordi, F.; Silva, C.; Rivara, S.; Zuliani, V.; Vacondio, F.; Rivara, M.;
Barocelli, E.; Bertoni, S.; Ballabeni, V.; Magnanini, F.; Impicciatore, M.; Plazzi,
P. V. Bioorg. Med. Chem. 2004, 12, 663.
132. Rivara, M.; Zuliani, V.; Cocconcelli, G.; Morini, G.; Comini, M.; Rivara, S.;
Mor, M.; Bordi, F.; Barocelli, E.; Ballabeni, V.; Bertonib, S.; Plazzia, P. V.
Bioorg. Med. Chem. 2006, 14, 1413.
133. Aslanian, R.; Zhu, X.; Vaccaro, H. A.; Shih, N. Y.; Piwinski, J. J.; Williams, S.
M.; West, R. E. Bioorg. Med. Chem. Lett. 2008, 18, 5032.
134. Coon, T.; Moree, W. J.; Li, B.; Yu, J.; Zamani-Kord, S.; Malany, S.; Santos, M.
A.; Hernandez, L. M.; Petroski, R. E.; Sun, A.; Wen, J.; Sullivan, S.; Haelewyn,
J.; Hedrick, M.; Hoare, S. J.; Bradbury, M. J.; Crowe, P. D.; Beaton, G. Bioorg.
Med. Chem. Lett. 2009, 19, 4380.
135. Erb, K. L.; Ravula, S. B.; Yu, J.; Kord, S. Z.; Moree, W. J.; Petroski, R. E.;
Wen, J.; Malany, S.; Hoare, S. R. J.; Madan, A.; Crowe, P. D.; Beaton, G.
Bioorg. Med. Chem. Lett. 2010, 20, 2916.
136. Priya, B. S.; Basappa.; Swamy, S. N.; Rangappa, K. S. Bioorg. Med. Chem.
2005, 13, 2623.
137. Uno, H.; Kurokawa, M.; Masuda, Y.; Nishimura, H. J. Med. Chem. 1979, 22,
180.

Chapter-1
54
138. Gopalsamy, A.; Shi, M.; Golas, J.; Vogan, E.; Jacob, J.; Johnson, M.; Lee, F.;
Nilakantan, R.; Petersen, R.; Svenson, K.; Chopra, R.; Tam, M. S.; Wen, Y.;
Ellingboe, J.; Arndt, K.; Boschelli, F. J. Med. Chem. 2008, 51, 373.
139. Strupczewski, J. T.; Allen, R. C.; Gardner, B. A.; Schmid, B. L.; Stache, U.;
Glamkowski, E. J.; Jones, M. C.; Ellis, D. B.; Huger, F. P.; Dunn, R. W. J. Med.
Chem. 1985, 28, 761-767.
140. Nuhrich, A.; Varache-Lembege, M.; Renard, P.; Devaux, G. Eur. J. Med. Chem.
1994, 29, 75-82.
141. Shantharam, C. S.; Suyoga Vardhan, D. M.; Suhas, R.; Sridhara, M. B.; Channe
Gowda, D. Eur. J. Med. Chem. 2013, 60, 325-332.


















