
Cdsco Sahni
35
Recombinant Therapeutic Proteins Presentation By: S.P. Shani Deputy Drugs Controller (I)
-
Upload
pratima-doshi-mehta -
Category
Documents
-
view
56 -
download
3
description
Biotech Industry Basic
Transcript of Cdsco Sahni

Recombinant
Therapeutic Proteins
Presentation By:
S.P. Shani
Deputy Drugs Controller
(I)

Regulation for r-DNA Technology based
Therapeutic proteins - Indian Scenario
The Guidelines on Similar Biologics was prepared by CDSCO and DBT laid down the regulatory pathway for similar biologic claiming to be similar to an already authorized reference biologic.
CDSCO is the national regulatory authority in India that evaluates safety, efficacy and quality of drugs in the country.
DBT through Review Committee on Genetic Manipulation/ Institutional Biosafety Committee is responsible for overseeing the development and preclinical evaluation of recombinant biologics.

Definition of New Drug
• As per under Rule 122-E New Drug is defined “A drug, including bulk drug substance which has not been used in the country to any significant extent under the conditions prescribed, recommended or suggested in the labeling thereof and has not been recognized as effective and safe by the licensing authority mentioned under Rule 21 for the proposed claims.
• A drug already approved by the Licensing Authority mentioned in Rule 21 for certain claims , which is now proposed to be marketed with modified or new claims , namely , indications, dosage, dosage form ( including sustained release dosage form) and route of administration.

All vaccines and r-DNA products are New Drugs
vide G.S.R. 45 (E) dated 24/01/2011.
122-A clearance required for imported r-DNA
products
122-B clearance required for Indigenously
manufactured r-DNA products.
Regulatory Requirement for Market Authorization of r-DNA
products
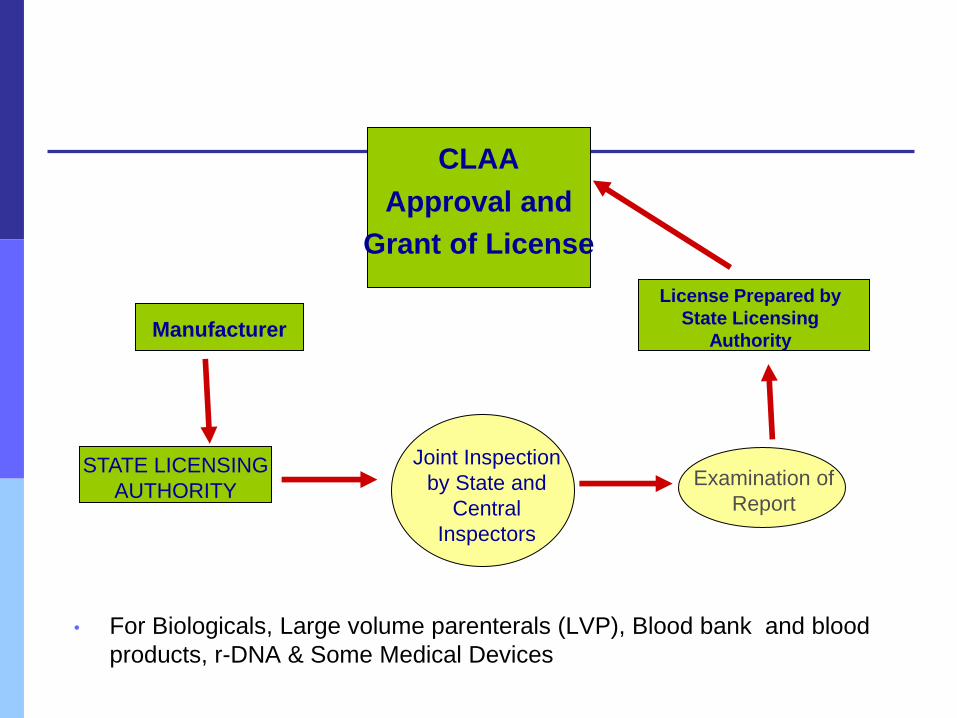
Central Licensing*
• For Biologicals, Large volume parenterals (LVP), Blood bank and blood
products, r-DNA & Some Medical Devices
STATE LICENSING
AUTHORITY
CLAA
Approval and
Grant of License
Joint Inspection
by State and
Central
Inspectors
Manufacturer
License Prepared by
State Licensing
Authority
Examination of
Report
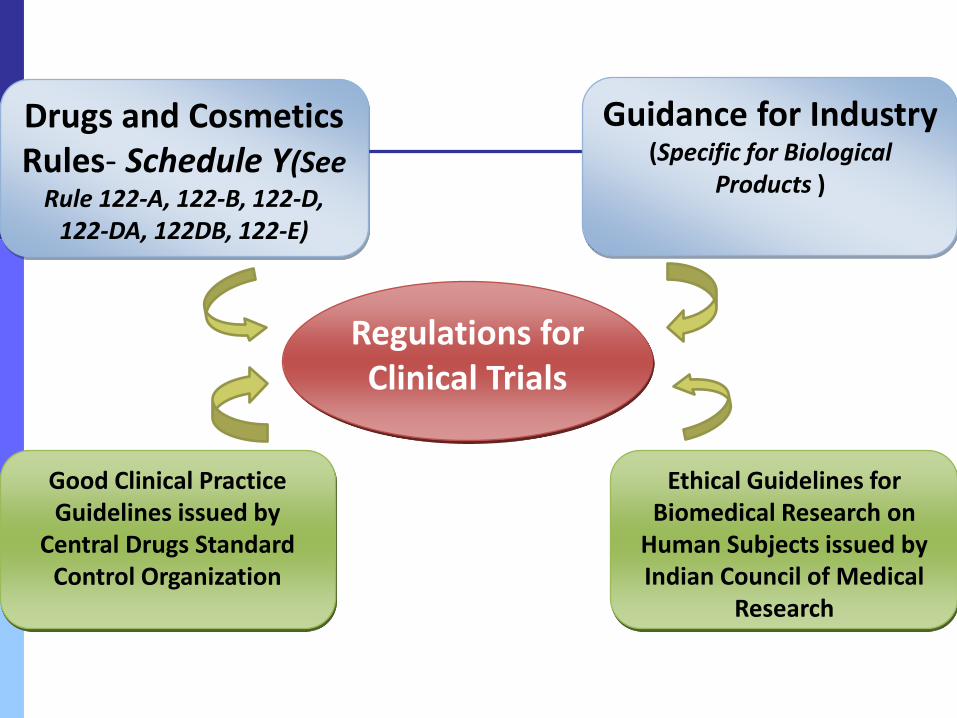
Regulations for Clinical Trials
Drugs and Cosmetics Rules- Schedule Y(See
Rule 122-A, 122-B, 122-D, 122-DA, 122DB, 122-E)
Guidance for Industry (Specific for Biological
Products )
Good Clinical Practice Guidelines issued by
Central Drugs Standard Control Organization
Ethical Guidelines for Biomedical Research on
Human Subjects issued by Indian Council of Medical
Research
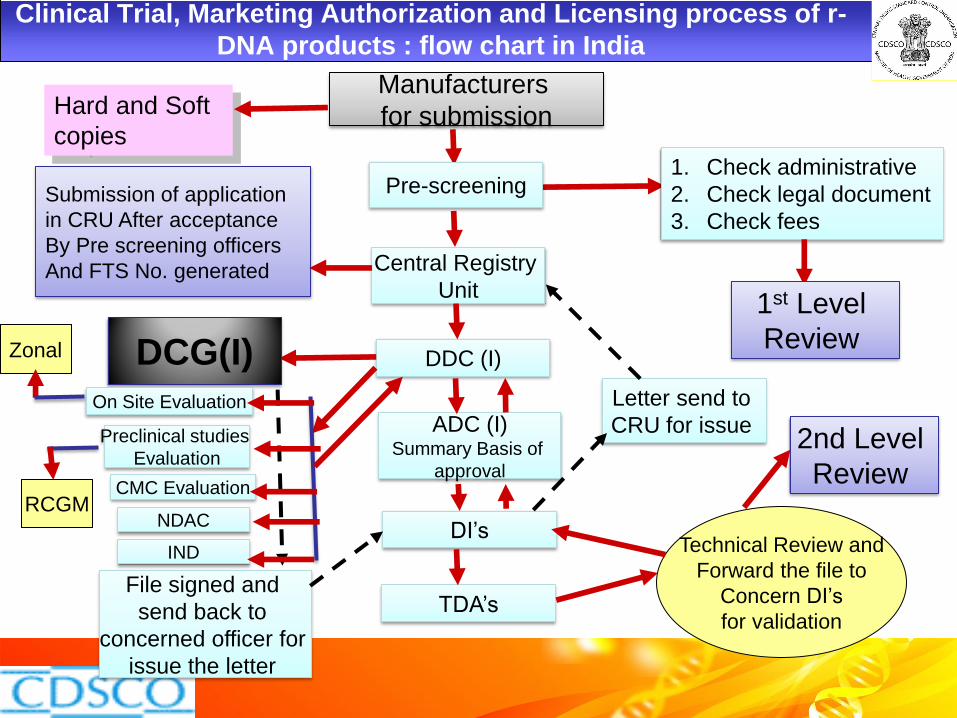
Technical Review and
Forward the file to
Concern DI’s
for validation
Manufacturers
for submission
Submission of application
in CRU After acceptance
By Pre screening officers
And FTS No. generated
Clinical Trial, Marketing Authorization and Licensing process of r-
DNA products : flow chart in India
Pre-screening 1. Check administrative
2. Check legal document
3. Check fees
1st Level
Review
Central Registry
Unit
DDC (I)
ADC (I) Summary Basis of
approval
DI’s
TDA’s
DCG(I)
File signed and
send back to
concerned officer for
issue the letter
Letter send to
CRU for issue 2nd Level
Review
On Site Evaluation
CMC Evaluation
NDAC
Zonal
Hard and Soft
copies
IND
Preclinical studies
Evaluation
RCGM

There are three Competent Authorities involved in
approval process namely :
a) Review Committee on Genetic Manipulation
(RCGM)/IBSC under Department of Biotechnology
(DBT), Ministry of Science and Technology.
b) Genetic Engineering Appraisal Committee (GEAC)
under the Ministry of Environment and Forests
(MoEF) and
c) Central Drugs Standard Control Organization
(CDSCO) under Ministry of Health & Family
Welfare
Competent Authorities

a) Review Committee on Genetic Manipulation (RCGM) under
Department of Biotechnology (DBT), Ministry of Science
and Technology – is responsible for authorizing
import/export for research and development and review of
data up to preclinical evaluation.
b) Genetic Engineering Appraisal Committee (GEAC) under
the Ministry of Environment and Forests (MoEF) - is
responsible for review and approval of activities involving
large scale use of genetically engineered organisms
(LMOs) and products thereof in R&D, industrial production,
environmental release and field applications.
Functions of Competent Authorities

c) Central Drugs Standard Control Organization (CDSCO)
under Ministry of Health & Family Welfare headed by DCG
(I) - It is the apex regulatory body and is responsible for
the approval of new drugs, for grant of import/export
license, clinical trial approval and permission for marketing
and manufacturing.
State Licensing Authority works with CDSCO in each state
and is responsible for issuance of license to manufacture
similar biologics in India.
Functions of Competent Authorities…contd

All recombinant biopharmaceuticals are considered as a
new drugs and their approval is regulated by provisions
of Drugs and Cosmetics Act and allied rules (Rule
122DA, 122DAA, 122E, 122A, 122B and Schedule Y
specially) as well as the “Guidance for Industry”
developed by the CDSCO.
Ministry of Health & Family Welfare's constituted New
Drug Advisory Committee (NDAC) on 31/03/2012 to
advise Drugs Controller General (India) in matters
related to approval of New Drugs including
biopharmaceutical products and Clinical Trials.
Recombinant Therapeutic Proteins

Similar Biologic can only be developed against an
authorized reference biologic that has been approved using
a complete data package in India.
In case the reference biologic is not authorized in India, it
should have been licensed and marketed for atleast 4 years
with significant safety and efficacy data.
In case of no medicine or only pallitive therapy is available
or in national healthcare emergency, this period of 4 years
may be reduced or waived off.
Scope

Any product can be considered as Similar Biologic only if it
is proven to be similar using extensive quality
characterization against reference biologic. Further product
development should only be considered once the similarity
of the product/molecule is demonstrated in quality.
Scope

Filing of Information as per CTD
Module I: Administration/Legal Information Module II: Overall Summaries Module III: Quality Information (Chemical, Pharmaceutical and Biological) Module IV: Non-Clinical Information Module V: Clinical Information
For Biological section Comprehensive Information is asked as per
CTD Module:-

ADOPTION OF COMMON TECHNICAL DOCUMENTS
In order to harmonize the documentation submission for the purpose of
• Clinical Trial
• Marketing Authorisation and Licensing
• Post Approval Changes in biological products
Comprehensive guidelines for submission of applications as per Common
Technical Document (CTD) has been implemented and same has been posted on
official website (www.cdsco.nic.in) on 4th Dec, 2008
These guidelines facilitate
1. Submission of Clinical Trial application for Evaluating Safety and Efficacy.
2. Requirement for permission of new drugs approval.
3. Post approval changes in biological products: Quality safety and Efficacy
Documents
4. Preparation of the quality information for drug submission for new drug
approvals: Biotechnological / Biological Product.

The CTD comprises of five modules which are as follows :-
• Module I : Administrative/Legal Information
• Module II : Summaries
• Module III : Quality Information (Chemical, Pharmaceutical and Biological)
• Module IV : Non-Clinical Information
• Module V : Clinical Information
Various SOPs’ and checklists for all the critical function of dossier review
have been introduced in the system. A prescreening mechanism is in place which
facilitates the stake holder for the submission of the application in the appropriate
manner.
The staff have been trained in the GCP, GMP and dossier evaluation in
order to update the knowledge.
Contd…

Common application prescribed for imported vaccine/r-DNA product
and indigenously manufactured vaccine/r-DNA product.
Application made as per Form-44 for import or manufacture or
undertake clinical trial.
Form-44 also accompanied with information as per Schedule-Y
(Requirement and Guidelines on Clinical Trial for import and
manufacture of Vaccine/r-DNA product).
Need for generation of Safety / Efficacy data as per GCP
Guidelines under Schedule ‘Y’
Regulatory requirements for clearance under Rule 122A/B
(New Drugs)
(Imported/Indigenously manufactured)

Drugs and Cosmetic Act Prescribes various applications Forms and
approvals & license certificates
• FORM 44 (See rules 122A, 122B, 122D and 122 DA)
Application for grant of permission to import or
manufacture a New Drug or to undertake clinical trial.
• FORM 40 (Rule 24-A)
Application for issue of Registration Certificate for
import of drugs into India under the Drugs and
Cosmetics Rules 1945
• FORM 27D (Rule 75)
Application for grant or renewal of license to
manufacture for sale or for distribution of vaccine

• SCHEDULE D(I)
(Rule 21 (d) and Rule 24 A)
Information and undertaking required to be submitted by
the manufacturer or his authorized agent with the
Application Form for a Registration Certificate. The format
shall be properly filled in for each application in Form 40..
• SCHEDULE D (II)
(Rule 21 (d) and Rule 24 A)
Information required to be submitted by the manufacturer
or his authorized agent with the Application Form for the
registration of a bulk drug/formulation/special product for its
import into India.
Contd…

• FORM 8 (Rule 24)
Application for license to import drugs (Vaccine)
(excluding those specified in Schedule X) to the Drugs and
Cosmetics Rules, 1945
• FORM 9 ( Rule 24)
Form of undertaking to accompany an application for an import license
Contd…

Drugs and Cosmetic Act Prescribes various applications Forms and
approvals & license certificates
• FORM 45 (See rules 122 A, 122 D and 122 DA)
Permission to import Finished Formulation of the
New Drug.
• FORM 46 (See rules 122 B, 122 D and 122 DA)
Permission / Approval for manufacture of new drug
formulation

Drugs and Cosmetic Act Prescribes various applications Forms and
approvals & license certificates
• FORM 41 (Rule 27 A)
Registration Certificate to be issued for import of drugs
into India under Drugs and Cosmetics Rules, 1945
• FORM 10 (Rules 23 and 27)
Licence to import drugs (excluding those specified in
Schedule X) to the Drugs and Cosmetic Rules, 1945

• FORM 28D (Rule 76)
Licence to manufacture for sale or for distribution of Large Volume
Parenterals / Sera and Vaccines specified in Schedules C and C(I)
excluding those specified in Schedule X
a. Condition of License (Rule 78)
[(p) The license shall comply with the requirements of
[Good Laboratory Practise as laid down in Schedule L-I
and} Good Manufacturing Practices as laid down in
Schedule M.]
b. Rule 79
Inspection before grant or renewal of license
Contd…

CMC Evaluation as per Bio Similar Guidelines:
Development of Similar Biologics
1. Selection of Reference Biologic
RMP should be licensed in India and should be innovator product.
In case the reference biologic is not marketed in India, the reference
biologic should have been licensed and widely marketed for 4 years post
approval in innovator jurisdiction in a country with well established
regulatory framework.
The same reference biologic should be used throughout the studies
supporting the safety, efficacy and quality of the product
Dosage form, strength and route of administration of the similar biologic
should be the same as that of the reference biologic.
The active substance (active ingredient) of the reference biologic and that
of the similar biologic must be shown to be similar

2. Manufacturing Process
Molecular Biology Considerations
Details of Host cell cultures (including viral clearance), vectors, gene sequences,
promoters etc.
Details of Post-translational modifications ((glycosylation, oxidation, deamidation,
phosphorylation etc.)
Fermentation Process Development
Three batches of reproducible fermentation data at pilot scale
Carried out in controlled and monitored environment.
Fermentation kinetics data
Data to verify that the specific protein yield (amount of protein per unit cell mass) remains
constant for all fermentation batches.
Overall productivity is reproducible and scalable.
Downstream Process Development
Steps involved in purification of protein.
Batch size for protein purification.
Consistency of recovery in 3 consecutive batches of purification from 3 independent
batches of fermentation.

3. Product Characterization
Structural and Physicochemical Properties:
Determination of primary and higher order structure of the product
The target amino acid sequence of the similar biologic should be confirmed
and is expected to be the same as for the reference biologic.
In cases, where post translational modifications are taking place, these
modifications need to be identified and quantified.
In case any significant differences are found, these should be
scientifically justified and critically examined in preclinical studies and
clinical trials.
Biological Characterization:
Biological assays will be required to characterize the activity and establish
the product’s mechanism of action and clinical effects (in units of activity).
Assays should be calibrated against an international or national reference
standard, where available and appropriate. If no such standards are
available, an internal reference standard must be established as per the
ICH guidelines. If the methods of bioassay(s) are documented in the
specification, test(s) can be conducted accordingly.

Immunological Characterization:
Evaluation by characterization(antibody or antibody-derived product);
comparison to reference biologic with respect to specificity, affinity, binding
strength and Fc function; and evaluation by animal studies
Purity and Impurities:
Product related variants (e.g., glycoforms, isomers etc.)
Product related impurities (e.g., aggregated, oxidized or deamidated
product)
Host cell related impurities (e.g., host cell protein, host cell DNA etc.)
Process related impurities (residual media components, resin leachates
etc.)
Differences observed in the purity and impurity profiles of the similar
biologic relative to the reference biologic should be evaluated to assess
their potential impact on safety and efficacy.

Specifications:
Acceptance limits should be set based on reference biologic data and data
from sufficient number of batches from preclinical or clinical batches.
Stability
Stability studies on drug substance and drug product should be carried out
using containers and conditions that are representative of the actual storage
containers and conditions, according to relevant guidelines (e.g. ICH
Q5C10, WHO TRS 82211).
Accelerated and stressed studies comparing the similar biologic to the
reference biologic

Preclinical studies evaluation as per Bio
Similar Guidelines The preclinical studies should be conducted prior to the initiation of any clinical after
getting approval from the RCGM.
The preclinical study design may vary depending upon the clinical parameters such
as therapeutic index, the type and number of indications applied.
The dosage form, strength and route of administration of the similar biologic should
be the same as that of the reference biologic and in case of any differences in these
parameters, it should be justified.
Following studies are required for preclinical evaluation:
Pharmacodynamic Studies
In vitro studies: (e.g. cell proliferation assays receptor binding assays).
In vivo studies:
In vivo evaluation of biological/ Pharmacodynamic activity may be dispensable if in
vitro assays are available, which are known to reliably reflect the clinically relevant
Pharmacodynamic activity of the reference biologic. In cases where the in-vitro
assays do not reflect the Pharmacodynamic, In vivo studies should be performed.

Toxicological Studies
In case of in vivo toxicity studies, at least one repeat dose toxicity study in a relevant
species is required to be conducted. The duration of the study would be generally not
less than 28 days with 14 days recovery period. However the duration may vary
depending on the dosage and other parameters on case by case basis.
Regarding the animal models to be used, the applicant should provide the scientific
justification for the choice of animal model(s) based on the data available in scientific
literature. However if the relevant animal species is not available and has been
appropriately justified, the toxicity studies need to be under taken in two species i.e.
one rodent and other non rodent species, as per the requirements of Schedule Y12
with due permission from the RCGM.
The dose should be calculated based on the therapeutic dose of the reference
biologic.
Other toxicity studies, including safety pharmacology, reproductive toxicity,
mutagenicity and carcinogenicity studies are not generally required for evaluation of a
similar biologic unless warranted by the results from the repeat dose toxicological
studies.

Immune Responses in Animals
Antibody response to the similar biologic should be compared to that
generated by the reference biologic in suitable animal model. The test
serum samples should be tested for reaction to host cell proteins.
Immunogenicity testing should be included as part of the sub-chronic
repeat dose study while developing the protocols.
The other parameters for evaluating immune toxicity include immune
complexes in targeted tissues may be considered while evaluating
histopathology observations, etc.

Guidance for
Industry • Submission of Clinical Trial
Application for Evaluating Safety
and Efficacy
• Requirements for permission of
New Drugs Approval
• Post approval changes in
biological products:
Quality safety and Efficacy
Documents • Preparation of the Quality
Information for Drug Submission
for New Drug Approval:
Biotechnological/Biological
Products

Post Approval Changes in Biological Products: Quality
Safety and Efficacy Documents (Document No. - PAC/
1108, Version – 1.1)
This section in the document Guidance of Industry deals with
requirements fulfilled by Indian firm before implementing the
following changes:
Notifiable Change e.g.: Generation of new Master Cell Bank (MCB)
from the same expression construct with same or closely related cell
line, change in approved therapeutic indication etc.
Supplement e.g.: change in the test specification of drug products/
drug substances
Annual Notification. e.g. Generation of a new Working Cell Bank
(WCB) etc.
Questions?

Thank You


















