
Patient of Myotonic Dystrophy presented with Celebellar Infarction

Journal of Clinical Neuroscience 20 (2013) 1002–1006
Contents lists available at SciVerse ScienceDirect
Journal of Clinical Neuroscience
journal homepage: www.elsevier .com/ locate/ jocn
Clinical Study
Cardiologic predictors of sudden death in patients with myotonic dystrophy type 1
Vidosava Rakocevic Stojanovic, Stojan Peric ⇑, Teodora Paunic, Sanja Pavlovic, Edita Cvitan, Ivana Basta,Marina Peric, Milena Milicev, Dragana LavrnicNeurology Clinic, Clinical Centre of Serbia, School of Medicine, University of Belgrade, Dr Subotica 6, 11000 Belgrade, Serbia
a r t i c l e i n f o
Article history:Received 11 February 2012Accepted 29 September 2012
Keywords:ElectrocardiographyMyotonic dystrophy type 1PredictorsSudden deathSurvival
0967-5868/$ - see front matter � 2012 Elsevier Ltd. Ahttp://dx.doi.org/10.1016/j.jocn.2012.09.014
⇑ Corresponding author. Tel.: +381 11 3064230; faxE-mail address: [email protected] (S. Peric).
a b s t r a c t
The aim of this study was to analyze survival, causes of death and cardiologic predictors of sudden deathin a large cohort of patients with myotonic dystrophy type 1 (DM1). The study was comprised of 171adult DM1 patients hospitalized at the Neurology Clinic in a 20-year period. Severe electrocardiographic(ECG) abnormality included at least one of the following: rhythm other than sinus, PR interval ofP240 ms, QRS complex duration of 120 ms or more, and second-degree or third-degree atrioventricular(AV) block. Survival data were analyzed by the Kaplan–Meier test, log–rank test and Cox regression anal-ysis. During the mean follow-up period of 9.4 ± 5.4 years, a pacemaker was implanted in 5.8% of DM1patients and 14% of patients died. The mean age at death was 55.6 ± 12.5 years. The most common causesof death in our cohort were sudden death (41.7%) and respiratory failure (29.2%). The presence of palpi-tations (hazard ratio [HR] = 4.7, p < 0.05) and increased systolic blood pressure (HR = 9.8, p < 0.05) weresignificant predictors of sudden death. Among ECG parameters, severe ECG abnormality (HR = 4.7,p < 0.05), right bundle branch block (RBBB; HR = 3.9, p < 0.05) and bifascicular block (HR = 5.8, p < 0.05)were significant predictors of sudden death.
� 2012 Elsevier Ltd. All rights reserved.
1. Introduction
Myotonic dystrophy type 1 (DM1) is the most common form ofmuscular dystrophy in adults, with a worldwide prevalence of 1 to35 in 100,000.1–3 DM1 is an autosomal dominant multisystem dis-order.4 Heart involvement is well recognized in DM15 and the mostfrequent cardiac abnormalities are conduction defects and arrhyth-mias.6–11
Life expectancy is reduced in patients with DM1, with mortalityincreased five to seven times compared with the general popula-tion.1,3,7 Common causes of premature mortality are respiratoryfailure and sudden death.1,3,7,12,13 Life-threatening respiratoryinvolvement often develops in a late stage of disease.11 Suddendeath in DM1 is traditionally associated with severe cardiac con-duction disturbances and heart block.1,12,14 Ventricular tachyar-rhythmias may also significantly contribute to sudden death inDM1.1,7,12,13,15 Recently, atrial tachyarrhythmia has emerged asan independent risk factor for sudden death.7 Non-cardiac causesof sudden death, such as embolic cerebral accident and massivepulmonary embolism, have also been suggested in DM1,13 butthese have not been supported by autopsy results.14,16 Althoughcauses of death are thoroughly described in DM1 patients, it is stillcontroversial which parameters are good predictors of lethal
ll rights reserved.
: +381 11 2684577.
outcome. The identification of significant mortality predictorsmay have a direct influence on the assessment, follow up and treat-ment of cardiac involvement in DM1.
The aim of this study was to analyze survival and causes ofdeath, as well as to identify cardiologic and electrocardiographicpredictors of sudden death, in a large cohort of patients withDM1 during a 20-year period.
2. Patients and method
This retrospective study was comprised of 211 consecutiveadult patients primarily diagnosed with DM1 who were hospital-ized at the Neurology Clinic in Belgrade from January 1, 1990, untilDecember 31, 2009. There were no patients with the congenitalform of DM1. Fourteen patients were excluded from the study afternegative gene analysis.17 In addition to typical clinical and electro-myographic data, genetic diagnosis of CTG repeat expansion wasobtained from patients or from their first-degree relatives.17 De-tailed clinical and electrocardiographic data were available for171 patients (Table 1) and they did not differ from the DM1 groupas a whole in any of the investigated parameters. The study wasapproved by the Ethical Board of the Neurology Clinic.
Medical records from the first admission to our clinic were usedto obtain baseline data. The severity of disease was assessedaccording to the Muscular Impairment Rating Scale (MIRS).18 Theform of disease was defined as childhood-juvenile (onset between
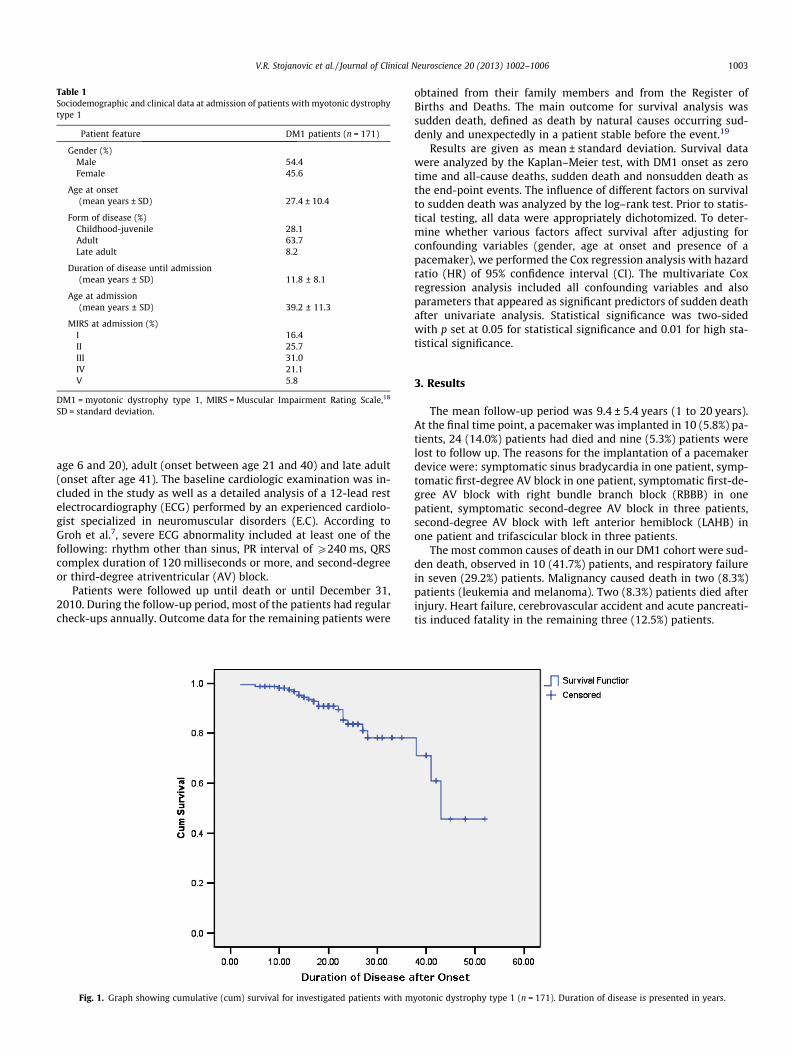
Table 1Sociodemographic and clinical data at admission of patients with myotonic dystrophytype 1
Patient feature DM1 patients (n = 171)
Gender (%)Male 54.4Female 45.6
Age at onset(mean years ± SD) 27.4 ± 10.4
Form of disease (%)Childhood-juvenile 28.1Adult 63.7Late adult 8.2
Duration of disease until admission(mean years ± SD) 11.8 ± 8.1
Age at admission(mean years ± SD) 39.2 ± 11.3
MIRS at admission (%)I 16.4II 25.7III 31.0IV 21.1V 5.8
DM1 = myotonic dystrophy type 1, MIRS = Muscular Impairment Rating Scale,18
SD = standard deviation.
V.R. Stojanovic et al. / Journal of Clinical Neuroscience 20 (2013) 1002–1006 1003
age 6 and 20), adult (onset between age 21 and 40) and late adult(onset after age 41). The baseline cardiologic examination was in-cluded in the study as well as a detailed analysis of a 12-lead restelectrocardiography (ECG) performed by an experienced cardiolo-gist specialized in neuromuscular disorders (E.C). According toGroh et al.7, severe ECG abnormality included at least one of thefollowing: rhythm other than sinus, PR interval of P240 ms, QRScomplex duration of 120 milliseconds or more, and second-degreeor third-degree atriventricular (AV) block.
Patients were followed up until death or until December 31,2010. During the follow-up period, most of the patients had regularcheck-ups annually. Outcome data for the remaining patients were
Fig. 1. Graph showing cumulative (cum) survival for investigated patients with m
obtained from their family members and from the Register ofBirths and Deaths. The main outcome for survival analysis wassudden death, defined as death by natural causes occurring sud-denly and unexpectedly in a patient stable before the event.19
Results are given as mean ± standard deviation. Survival datawere analyzed by the Kaplan–Meier test, with DM1 onset as zerotime and all-cause deaths, sudden death and nonsudden death asthe end-point events. The influence of different factors on survivalto sudden death was analyzed by the log–rank test. Prior to statis-tical testing, all data were appropriately dichotomized. To deter-mine whether various factors affect survival after adjusting forconfounding variables (gender, age at onset and presence of apacemaker), we performed the Cox regression analysis with hazardratio (HR) of 95% confidence interval (CI). The multivariate Coxregression analysis included all confounding variables and alsoparameters that appeared as significant predictors of sudden deathafter univariate analysis. Statistical significance was two-sidedwith p set at 0.05 for statistical significance and 0.01 for high sta-tistical significance.
3. Results
The mean follow-up period was 9.4 ± 5.4 years (1 to 20 years).At the final time point, a pacemaker was implanted in 10 (5.8%) pa-tients, 24 (14.0%) patients had died and nine (5.3%) patients werelost to follow up. The reasons for the implantation of a pacemakerdevice were: symptomatic sinus bradycardia in one patient, symp-tomatic first-degree AV block in one patient, symptomatic first-de-gree AV block with right bundle branch block (RBBB) in onepatient, symptomatic second-degree AV block in three patients,second-degree AV block with left anterior hemiblock (LAHB) inone patient and trifascicular block in three patients.
The most common causes of death in our DM1 cohort were sud-den death, observed in 10 (41.7%) patients, and respiratory failurein seven (29.2%) patients. Malignancy caused death in two (8.3%)patients (leukemia and melanoma). Two (8.3%) patients died afterinjury. Heart failure, cerebrovascular accident and acute pancreati-tis induced fatality in the remaining three (12.5%) patients.
yotonic dystrophy type 1 (n = 171). Duration of disease is presented in years.
Table 2Frequency of disturbances at initial electrocardiography in patients with myotonicdystrophy type 1 (n = 171)
ECG finding No. of patients Percentage (%)
Severe ECG abnormality 61 35.7
Sinus bradycardia <60 43 25.11st degree AV block 43 25.12nd degree AV block 2 1.2LBBB 10 5.8RBBB 28 16.4LAHB 12 7.0LPHB 1 0.6Bifascicular block 6 3.5Incomplete TFB 2 1.2
Early repolarization 6 3.5SVES 1 0.6Atrial fibrillation 5 2.9VES 3 1.8
Right axis deviation 4 2.3Left axis deviation 7 4.1Low P wave 8 4.7LV hypertrophy 2 1.2ST segment elevation 6 3.5Low T wave 10 5.8Abnormal Q wave 8 4.7U wave 1 0.6
ECG = electrocardiography, LAHB = left anterior hemiblock, LBBB = left bundlebranch block, LPHB = left posterior hemiblock, LV = left ventricle, RBBB = rightbundle branch block, SVES = supraventricular extrasystoles, TFB = trifascicularblock, VES = ventricular extrasystoles.
Table 3Predictors of sudden death in patients with myotonic dystrophy type 1 (n = 171)
Features at firstadmission
HR (95% CI)Univariateregression analysis
HR (95% CI)Multivariateregression analysis
Palpitations 4.7 (1.1–20.7)* n.s.Systolic hypertension 9.8 (1.0–96.2)* 43.5 (1.5–1000)*
Severe ECG abnormality 4.7 (1.2–18.5)* 15.2 (1.5–167.7)*
RBBB 3.9 (1.0–15.0)* 7.1 (1.0–52.6)*
Bifascicular block 5.8 (1.2–28.7)* n.s.
CI = confidence interval, ECG = electrocardiography, HR = hazard ratio, n.s. = non-significant, RBBB = right bundle branch block.* p < 0.05
1004 V.R. Stojanovic et al. / Journal of Clinical Neuroscience 20 (2013) 1002–1006
For 24 patients who died during the follow-up period, the meanage at death was 55.6 ± 12.5 (range, 22 to 74): 54.9 ± 8.7 for males(range, 34 to 65) and 57.3 ± 19.4 for females (range, 22 to 74). Themean survival period in the whole group of DM1 patients was41.0 ± 27.5 years with median survival of 43 years after disease on-set (Fig. 1).
The features of the investigated patients at the time of study en-try are presented in Table 1. Male patients had a survival time of42.2 ± 14.5 years from disease onset to sudden death in compari-son to female patients with 49.9 ± 18.5 years (p = 0.075). Patientswith the childhood-juvenile form of DM1 had a longer life expec-tancy to sudden death in comparison to those with the adult andlate-adult form (p = 0.055). The duration of disease, age at admis-sion and MIRS did not reduce survival time (p > 0.05). There wasno difference in survival between patients with and without apacemaker (p > 0.05). Two patients with pacemakers died, one ofthem suddenly.
Fainting was present in 31 (18.1%) patients at admission andwas not associated with shorter survival to sudden death(p > 0.05). The presence of palpitations (20.5% of patients) was re-lated to a shorter survival time to sudden death (43.1 ± 21.3 vs46.3 ± 11.7 years, p < 0.05) and it appeared as a predictor of suddendeath after univariate analysis (HR = 4.7, CI = 1.1–20.7, p < 0.05).Systolic blood pressure above 140 mmHg was found in eight(4.7%) patients and diastolic blood pressure above 90 mmHg in16 (9.4%) patients. Systolic hypertension was associated withworse survival (19.6 ± 5.1 vs 46.5 ± 28.1 years, p < 0.01) and was asignificant predictor of sudden death after univariate (HR = 9.8,CI = 1.0–96.2, p < 0.05) and multivariate (HR = 43.5, CI = 1.5–1000,p < 0.05) regression analysis. A systolic murmur (27.5% of patients)was not related to worse survival (p > 0.05).
ECG findings at admission are presented in the Table 2. Patientswith severe ECG abnormalities had shorter survival to suddendeath compared with those without (38.5 ± 16.4 vs47.4 ± 31.5 years, p < 0.05). Severe ECG abnormality was also a sig-nificant predictor of sudden death after univariate (HR = 4.7,
CI = 1.2–18.5, p < 0.05) and multivariate (HR = 15.2, CI = 1.5–167.7, p < 0.05) analysis. Among conduction disturbances, we iden-tified RBBB (36.4 ± 11.1 vs 48.3 ± 21.5 years, p < 0.05) and bifascic-ular block (34.5 ± 11.0 vs 47.6 ± 21.8 years, p < 0.05) to beassociated with shorter survival to sudden death. Both appearedas significant predictors of sudden death (HR = 3.9, CI = 1.0–15.0,p < 0.05 and HR = 5.8, CI = 1.2–28.7, p < 0.05, respectively) afterunivariate analysis, while RBBB was also an independent predictorafter multivariate analysis (HR = 7.1, CI = 1.0–52.6, p < 0.05). First-degree AV block and left bundle branch block (LBBB) did not affectsurvival to sudden death in our cohort of DM1 patients (p > 0.05).The log–rank test was not performed for arrhythmias and other ob-served ECG changes because of the small number of observedcases.
Significant predictors of sudden death in our patients with DM1are summarized in Table 3
4. Discussion
During a mean follow-up period of 9 years, pacemakers wereimplanted in almost 6% of our DM1 patients. The proportion of pa-tients with pacemakers varies greatly in different DM1 cohortsdepending on the different criteria for its application. In a studyby Breton and Mathieu, criteria similar to ours were used and pace-makers were implanted in 3% of patients during a 12-year period.15
In a study by Laurent et al., pacemakers were implanted in 49% of100 patients during a 6-year period20 as the authors used the strictelectrophysiologic criterion of a Hiss-ventricle (HV) interval pro-longation above 70 milliseconds.13 Official recommendations ofthe American College of Cardiology/American Heart Associationare to place pacemakers in DM1 patients with an HV interval above100 milliseconds (class IIa recommendation) and in those with anyAV block regardless of symptoms, as the progression of cardiacconduction disturbances in this disease is unpredictable (class IIbrecommendation).21 However, pacemakers did not improve sur-vival in the study performed by Groh et al7 which is in accordancewith our data. Sudden death has previously been described in DM1patients with pacemakers1,7,12,13 raising the question of ventriculartachyarrhythmias as a cause of sudden cardiac death and of cardio-verter-defibrillator (ICD) implantation. However, there are no clearcriteria for the implantation of ICD in DM1 patients and the benefitof its application is still not clear.20,22 Controversies about ICDimplantation in DM1 patients have increased since one DM1 pa-tient died suddenly because of inappropriate stimulation by ICD.7
Furthermore, ventricular tachyarrhythmia was not detected inthe pacemaker memory of several DM1 patients who died sud-denly.20 Thus, some authors have stated that embolic cerebral acci-dent, massive pulmonary embolism and acute decompensation ofchronic heart failure may be potential causes of death at least insome patients with DM1.13
The two most common causes of death in our DM1 cohort weresudden death (42%) and respiratory failure (29%). In previous

V.R. Stojanovic et al. / Journal of Clinical Neuroscience 20 (2013) 1002–1006 1005
studies, sudden death is usually reported to occur in one-third ofall deaths in DM1 patients, while neuromuscular respiratory weak-ness is attributed to 40–50% of deaths.1,7,12,22,23 A strong connec-tion between respiratory failure and arrhythmia has beendescribed.11 The risk of conduction disorders is almost three timeshigher in patients with restrictive lung disease. Thus, a pneumo-nologist who establishes a diagnosis of restrictive lung diseaseshould refer the patient to a cardiologist.11 Mörner proposed thatin patients with severe conduction disturbances, additional hypox-ia or limited oxygen supply can provide a substrate for cardiacdeath without evidence for infarction.9
In our group of DM1 patients, the mean age at death was56 years, which is in line with previous studies.1,3,7,12,20,23 In ourcohort, male patients died of sudden death 8 years earlier on aver-age than female patients. Male sex has already been reported tocontribute to conduction abnormalities and to be an unfavorableprognostic factor for survival in DM1 patients.3,8,11,16 Our patientswith the childhood-juvenile form of DM1 had increased life expec-tancy, which has been previously published.3,12
Our results showed that symptoms and routine cardiologicexaminations may be significant predictors of sudden death inDM1. Patients with palpitations had increased risk for suddendeath. Thus, every DM1 patient with palpitations should undergoa thorough cardiologic investigation. Conversely, the presence offainting was not suggestive of lethal outcome. The reason for thismay be that fainting is not specific for cardiac conduction distur-bances and arrhythmias, but also may be related to low bloodpressure and/or orthostatic hypotension, which have been de-scribed to be frequent in DM1 patients.24 Our patients with sys-tolic pressure above 140 mmHg had an increased risk of dyingsuddenly. Systemic hypertension is known to be one of the mostimportant causes of pathologic left ventricular hypertrophy in thegeneral population and these patients are at increased riskof sudden cardiac death, probably because of ventriculararrhythmias.25
Cardiac conduction abnormalities are reported in 30–75% ofDM1 patients7,10,11,20 which is in accordance with our data of35.7% patients with severe ECG abnormalities. These disturbancesare related to subendocardial fibrosis and fatty infiltration as partof the multisystemic fibrotic pathology observed in DM1.9,14,26 Se-vere ECG abnormality was a significant predictor of sudden deathin our DM1 patients, which is in accordance with study by Grohet al.7 Thus, baseline ECG may be significant in the identificationof DM1 patients who are at risk of sudden death.
A first-degree AV block was the most common conduction dis-turbance in our cohort of patients, as well as in others.11,20,26 Pre-vious studies showed that PR interval prolongation is a goodpredictor of sudden death in DM1.7,9,15 Although the slow progres-sion of an AV block is considered to be the predominant mode ofmortality in DM1,7,27 our study did not identify first-degree AVblock as a predictor of sudden death. This could be due to substan-tial individual variations, as some DM1 patients may present witha quick and unpredictable progression of AV conduction abnormal-ity.7,12 Also, some of our patients with first-degree AV blocks hadan implanted pacemaker, which probably affected their outcomes.
A few studies have shown that QRS interval prolongation is alsoa good predictor of sudden death in DM1.7,9,15 Surprisingly, wefound RBBB, but not LBBB, to be a predictor of sudden death inDM1 patients, although some previous studies have stated thatcardiologic patients with LBBB, but not with RBBB, suffer a greaterrisk for mortality.28 However, the study by Morin et al. showedthat sudden death risk was associated with increasing QRS dura-tion independent of the presence or absence of LBBB.29 Further-more, the study by Adesanya et al. revealed that increasing QRSduration was an independent predictor of cardiac mortality inpatients with RBBB.30 It is also noteworthy that patients with RBBB
and ST segment elevation in V1–V3 ECG chest leads (Brugada syn-drome) have a high risk of sudden cardiac death secondary to ven-tricular tachycardia and fibrillation.31 One French study showed an80-fold higher incidence of Brugada syndrome in DM1 comparedwith the normal population, and this was not explained by achance association of a mutation in the sodium channel, voltage-gated, type 5, alpha subunit (SCN5A) gene.32 Preliminary resultsprovide evidence that a missplicing of a voltage-gated sodiumchannel of the cardiac type (SCN5A) may be responsible for thepathogenesis of the conduction block in DM1.33 Since ST elevationwas observed in 3.5% of our DM1 patients, we believe that evenRBBB, especially in association with right side ST elevation, canbe a significant predictor of sudden death in DM1. The absenceof a correlation between sudden death on one side and second-de-gree AV block and LBBB on the other may be explained by the rel-atively low frequency of these disturbances in our DM1 cohort.Also, a significant number of these patients had implantedpacemakers.
Ventricular arrhythmias were less frequent than supraventricu-lar arrhythmias in our DM1 patients at baseline, which is similar toprevious data published by Laurent et al.20 According to Groh et al.,atrial tachyarrhythmia is the only independent predictor of bothsudden death and death from progressive neuromuscular respira-tory failure.7 On the other hand, although ventricular tachyar-rhythmia was the most commonly observed pathologic rhythmin DM1 patients seen on ECG immediately before sudden death,it was not a significant predictor of sudden or respiratory deathin these patients.7 In our study, survival analysis was not per-formed for supraventricular and ventricular arrhythmias as theywere infrequent.
The main limitation of this study was the relatively small num-ber of outcome events. Thus, further multicentre studies areneeded to confirm our findings and to investigate other potentialpredictors of sudden death in DM1.
5. Conclusion
In our group of DM1 patients the mean age at death was56 years. Sudden death was observed in almost half of the DM1 pa-tients, and death from respiratory failure in almost one-third of allpatients. The most significant predictors of sudden death in DM1were increased systolic blood pressure, severe ECG abnormality,the presence of bifascicular block and patient complaints of palpi-tations. Even patients with RBBB were at an increased risk of sud-den death.
Acknowledgment
This study was supported by the Ministry of Science of theRepublic of Serbia (Grant number 175083).
References
1. Mathieu J, Allard P, Porvin P, et al. A 10-year study of mortality in a cohort ofpatients with myotonic dystrophy. Neurology 1999;52:1658–62.
2. Emery AE. Population frequencies of inherited neuromuscular diseases – aworld survey. Neuromusc Disord 1991;1:19–29.
3. Mladenovic J, Pekmezovic T, Todorovic S, et al. Survival and mortality ofmyotonic dystrophy type 1 (Steinert’s disease) in the population of Belgrade.Eur J Neurol 2006;13:451–4.
4. Osborne RJ, Lin X, Welle S, et al. Transcriptional and post-transcriptional impactof toxic RNA in myotonic dystrophy. Hum Mol Genet 2009;18:1471–81.
5. Harper P. Myotonic dystrophy. 3rd ed. London: WB Saunders; 2001.6. Sovari AA, Bodine CK, Farokhi F. Cardiovascular manifestations of myotonic
dystrophy-1. Cardiol Rev 2007;15:191–4.7. Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden
death in myotonic dystrophy type 1. N Engl J Med 2008;358:2688–97.8. Cudia P, Bernasconi P, Chiodelli R, et al. Risk of arrhythmia in type I myotonic
dystrophy: the role of clinical and genetic variables. J Neurol Neurosurg Psychiat2009;80:790–3.

1006 V.R. Stojanovic et al. / Journal of Clinical Neuroscience 20 (2013) 1002–1006
9. Mörner S, Lindqvist P, Mellberg C, et al. Profound cardiac conduction delaypredicts mortality in myotonic dystrophy type 1. J Intern Med 2010;268:59–65.
10. McNally EM, Sparano D. Mechanism and management of the heart in myotonicdystrophy. Heart 2011;97:1094–100.
11. Kaminsky P, Poussel M, Pruna L, et al. Organ dysfunction and musculardisability in myotonic dystrophy type 1. Medicine 2011;90:262–8.
12. De Die-Smulders CE, Howeler CJ, Thijs C, et al. Age and causes of death in adultonset myotonic dystrophy. Brain 1998;121:1557–63.
13. Lazarus A, Varin J, Babuty D, et al. Long-term follow-up of arrhythmias inpatients with myotonic dystrophy treated by pacing. J Am Coll Cardiol2002;40:1645–52.
14. Nguyen HH, Wolfe JT, Holmes Jr DR, et al. Pathology of the cardiac conductionsystem in myotonic dystrophy: a study of 12 cases. J Am Coll Cardiol1988;11:662–71.
15. Breton R, Mathieu J. Usefulness of clinical and electrocardiographic data forpredicting adverse cardiac events in patients with myotonic dystrophy. Can JCardiol 2009;25:e23–7.
16. Sabovic M, Medica I, Logar N, et al. Relation of CTG expansion and clinicalvariables to electrocardiogram conduction abnornalities and sudden death inpatients with myotonic dystrophy. Neuromusc Disord 2003;13:822–6.
17. Prior TW. Technical standards and guidelines for myotonic dystrophy type 1testing. Genet Med 2009;11:552–5.
18. Mathieu J, Boivin H, Meunier D, et al. Assessment of a disease-specific muscularimpairment rating scale in myotonic dystrophy. Neurology 2001;56:336–40.
19. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminaryreport: effect of encainide and flecainide on mortality in a randomized trial ofarrhythmia suppression after myocardial infarction. N Engl J Med1989;321:406–12.
20. Laurent V, Pellieux S, Corcia P, et al. Mortality in myotonic dystrophy patientsin the area of prophylactic pacing devices. Int J Cardiol 2011;150:54–8.
21. Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines fordevice-based therapy of cardiac rhythm abnormalities: a report of theAmerican College of Cardiology/American Heart Association Task Force on
Practice Guidelines: developed in collaboration with the American Associationof Thoracic Surgery and Society of Thoracic Surgeons. Circulation2008;117:e350–408.
22. Bhakta D, Groh M, Shen C, et al. Increased mortality with left ventricularsystolic dysfunction and heart failure in adults with myotonic dystrophy type1. Am Heart J 2010;160:e1.
23. Groh WJ, Groh MR, Shen C, et al. Survival and CTG repeat expansion in adultswith myotonic dystrophy type 1. Muscle Nerve 2011;43:648–51.
24. Rakocevic-Stojanovic V, Milovanovic B, Ivic N, et al. Cardiac autonomic nervoussystem in patients with myotonic dystrophy type 1. Acta Myol 2007;26:112–4.
25. Saadeh AM, Jones JV. Predictors of sudden cardiac death in never previouslytreated patients with essential hypertension: long-term follow-up. J HumHypertens 2001;15:677–80.
26. Lindqvist P, Morner S, Olofsson BO, et al. Ventricular dysfunction in type 1myotonic dystrophy: electrical, mechanical or both. Int J Cardiol2010;143:378–84.
27. Nazarian S, Bluemke DA, Wagner KR, et al. QRS prolongation in myotonicmuscular dystrophy and diffuse fibrosis on cardiac magnetic resonance. MagnReson Med 2010;64:107–14.
28. Brenyo A, Zareba W. Prognostic significance of QRS duration and morphology.Cardiology J 2011;18:8–17.
29. Morin D, Oikarinen L, Viitasalo M, et al. QRS duration predicts sudden cardiacdeath in hypertensive patients undergoing intensive medical therapy: the LIFEstudy. Eur Heart J 2009;30:2908–14.
30. Adesanya CO, Yousuf KA, Co C, et al. Is wider worse? QRS duration predictscardiac mortality in patients with right bundle branch block. Ann NoninvasiveElectrocardiol 2008;13:165–70.
31. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of thesecond consensus conference. Circulation 2005;111:659–70.
32. Wahbi K, Fressart V, Becane HM, et al. High prevalence of Brugada syndrome inpatients with Steinert’s disease. Medgen 2009;21:423 abstract S9-04.
33. Kokunai Y, Kino Y, Moyi L et al. Altered splicing of cardiac sodium channel inheart muscles of myotonic dystrophy type 1. IDMC-8 2011;91 abstract P10.


















