
BY The Rockefeller Institute New York, New York) · THE VERATRINE ALKALOIDS XxX11. THE STRUCTURE OF...
17
THE VERATRINE ALKALOIDS XxX11. THE STRUCTURE OF VERATRAMINE BY WALTER A. JACOBS AND YOSHIO SAT0 (From the Laboratories of The Rockefeller Institute for Medical Research, New York, New York) (Received for publication, February 20, 1951) Veratramine, CUHBOZN, has been shown (1, 2) to be an unsaturated secondary base with two acylatable hydroxyl groups and with at least one double bond demonstrable by its hydrogenation to dihydroveratra- mine. Three other points of unsaturation have been inferred from its composition and the benzenoid character of its ultraviolet absorption spectrum. A similar curve obtained with the dihydro derivative has shown that the reactive double bond is not conjugated with the benzene ring. The general steroid structure of veratramine is suggested by its empirical formula and by its association with other steroid bases. How- ever, some modification of such a structure is required to permit a ben- zenoid ring, such as a simple rearrangement of an angular methyl group or, among other possibilities, its participation in a ring D enlargement. Of the two hydroxyl groups the position of one on CZ3has been made fairly certain by the isolation of 3-methyl-5-hydroxypyridine (3) from the dehydrogenation products of veratramine. As presented below, the iden- tity of this product has since been confirmed by synthesis. The remaining hydroxyl group has been assumedto be the usual 3-hydroxyl by analogy between the veratramine glucoside veratrosine (4) and the jervine glu- coside, pseudojervine. Other evidence for the 3 position has now been obtained. Although our earlier attempt to apply t.he Oppenauer method to vera- tramine miscarried, we have since succeeded in using this method for its conversion to what may be interpreted as the CY,P-unsaturated ketone, L\4-veratram&%one. This interpretation is supported by the ultraviolet absorption spectra curves, Fig. 1. Although the ketone, which was sepa- rated through alumina, was not obtained as the crystalline base, it was readily characterized as the crystalline hydrochloride. The presence of a CO group was confirmed by the formation of an oxime, which was not obtained in crystalline form. With aluminum isopropoxide the ketone was reduced to a crystalline derivative, presumably A4-veratramine, which also yielded a crystalline hydrochloride and gave an absorption spectrum curve (Fig. 1) similar to that of veratramine. In contrast to the latter, 71 by guest on June 18, 2019 http://www.jbc.org/ Downloaded from
Transcript of BY The Rockefeller Institute New York, New York) · THE VERATRINE ALKALOIDS XxX11. THE STRUCTURE OF...

THE VERATRINE ALKALOIDS
XxX11. THE STRUCTURE OF VERATRAMINE
BY WALTER A. JACOBS AND YOSHIO SAT0
(From the Laboratories of The Rockefeller Institute for Medical Research, New York, New York)
(Received for publication, February 20, 1951)
Veratramine, CUHBOZN, has been shown (1, 2) to be an unsaturated secondary base with two acylatable hydroxyl groups and with at least one double bond demonstrable by its hydrogenation to dihydroveratra- mine. Three other points of unsaturation have been inferred from its composition and the benzenoid character of its ultraviolet absorption spectrum. A similar curve obtained with the dihydro derivative has shown that the reactive double bond is not conjugated with the benzene ring. The general steroid structure of veratramine is suggested by its empirical formula and by its association with other steroid bases. How- ever, some modification of such a structure is required to permit a ben- zenoid ring, such as a simple rearrangement of an angular methyl group or, among other possibilities, its participation in a ring D enlargement.
Of the two hydroxyl groups the position of one on CZ3 has been made fairly certain by the isolation of 3-methyl-5-hydroxypyridine (3) from the dehydrogenation products of veratramine. As presented below, the iden- tity of this product has since been confirmed by synthesis. The remaining hydroxyl group has been assumed to be the usual 3-hydroxyl by analogy between the veratramine glucoside veratrosine (4) and the jervine glu- coside, pseudojervine. Other evidence for the 3 position has now been obtained.
Although our earlier attempt to apply t.he Oppenauer method to vera- tramine miscarried, we have since succeeded in using this method for its conversion to what may be interpreted as the CY,P-unsaturated ketone, L\4-veratram&%one. This interpretation is supported by the ultraviolet absorption spectra curves, Fig. 1. Although the ketone, which was sepa- rated through alumina, was not obtained as the crystalline base, it was readily characterized as the crystalline hydrochloride. The presence of a CO group was confirmed by the formation of an oxime, which was not obtained in crystalline form. With aluminum isopropoxide the ketone was reduced to a crystalline derivative, presumably A4-veratramine, which also yielded a crystalline hydrochloride and gave an absorption spectrum curve (Fig. 1) similar to that of veratramine. In contrast to the latter,
71
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

72 VERATRINE ALKALOIDS. xxx11
it gave with trichloroacetic acid a dull purple color resembling that ob- tained with A4-stenols. This product was possibly a mixture of the 3(a)- and 3(p)-hydroxy epimers and a separation was not attempted.
In the above transformations it appeared that the hydroxyl on CB re- mained unaffected. Its secondary character, however, was shown in studies with dihydroveratramine, in which it again appeared to be less
1
4.
3.
bJ F l-l
2
1
FIG. 1. +, A”-veratramin-‘&one hydrochloride; l , the same with 1 equivalent of NaOH; 0, A’-veratramine; X, veratramine; in ethanol.
readily oxidized than the 3-hydroxyl group. With aluminum tert-butox- ide, which can be assumed to act as it does with veratramine, only di- hydroveratramin-S-one ([a], = +58.9”) was obtained. The same mono- keto base was also obtained as the first product of the action of chromic oxide on the dihydro base ([aID = +53.6”). The ultraviolet absorption curve is shown in Fig. 2. The substance yielded an OX&W which did not crystallize as the free base but formed a crystalline hydrochloride. On acylation a diacetyldihydroveratramin-3-one was obtained, as is consist- ent with the retention of the 23-hydroxyl group. This derivative did
0.* '5
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

W. A. JACOBS AND Y. SAT0 73
not crystallize, but on saponification gave a crystalline N-acetyldihydro- veratramin-J-one.
The difference in the observed molecular rotation between dihydrovera- tramin-&one ([& = +53.6”) and dihydroveratramine ([cY], = +27.4’) is about +108”, while the recorded increment, on going from a 3(p)- stanol to a 3-stanone, is about +73” (5). This is a difference which will require ultimate clarification. The marked dextro change in rotation on
\,
FIG. 2. l , dihydroveratramin-3-one; 0, dihydroveratramin-3,23-dione; f, di- hydroveratramine; in ethanol.
passing from veratramine of [a], = -69’ to its A4-ketone of [c&, = +69” is suggestive of what has been experienced with A4-3-stenones obtained from As-3(p)-stenols.
More vigorous oxidation of dihydroveratramine with chromic oxide yielded the diketo derivative dihydroveratramin-3, %-okne which was separated through alumina and isolated first as the hydrochloride. The absorption spectrum curve obtained with it is shown in Fig. 2. Attempts to obtain a dioxime from thii substance proved unsatisfactory. The product isolated was amorphous and the figure of 7.75 for N (the average of two preparations) lies between the figures calculated for a monooxime
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

74 VERATRINE ALKALOIDS. XXX11
and a dioxime, 6.63 and 9.61 respectively. This result indicates a hin- dered character for the 23-carbonyl group as it occurs in the veratramine derivative. In many alkylated 3-ketopyrrolidine and ketopiperidine de- rivatives, however, oximes have been recorded and seem to be formed without difficulty. It appears that the hindered reactivity on carbon 23 observed in veratramine may be caused by the other structural features present in the latter and it is desirable to make a subsequent check on such lack of reactivity on model substances.
Since veratramine on dehydrogenation yields 3-methyl-5-hydroxypyri- dine and not the 3-methyl-6-ethyl-5-hydroxypyridine obtained from jer- vine, and since there was no evidence of the formation of an amount of methylethylpyridine such as is observed with other veratrine bases, it is possible that its benzenoid structure may be responsible. To explain these observations, the structure as shown in Formula I has been under
T
consideration, in which ring D has become enlarged and benzenoid by participation of the angular methyl group. The usual side chain attached to this ring could on dehydrogenation adhere to the latter and the rupture would occur between Cm and CB. The resulting basic side chain frag- ment would be a /3-picoline derivative as obtained. The participation of the angular group (6) in the production of a new ring has been suggested to explain the formation of certain hydrocarbons under the conditions of high temperature dehydrogenation. It is not certain, however, whether or how such a ring enlargement is possible under biological conditions, and further work is necessary to verify the correctness of this structure.
The hindered character observed with the second carbonyl group in the diketone has now reopened the question of the interpretation of the inert carbonyl group of jervine. In the latter this has been placed ten- tatively on C11,l but in the highly substituted piperidine side chain its inertness could possibly also be explained now by its involvement of C&. In such a case, the double bond conjugated with it could be a Azot2*) or A24 bond and it is possible that the assumed oxidic group of jervine is to be found elsewhere than on the side chain. Reinvestigation of these
1 In a personal communication Professor L. F. Fieser of Harvard University has questioned the correctness of placing the CO at C&l, on the basis of studies in his laboratory.
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

W. A. JACOBS AND Y. SdTO 75
points with jervine is now in progress and it is planned to discuss in a later paper other data which have been obtained.
Before the above observations had been made with veratramine, the attempt was made to determine directly a position for the reactive double bond which would be compatible with a regular steroid cyclopentanoper- hydrophenanthrene ring system. By oxidation of triacetylveratramine with perbenzoic acid an oxido derivative, C33H4606N, was obtained which can be interpreted as triacetyld ,6-oxidoveratramine. The findings sug- gested that epimeric oxides might have resulted from the addition to the double bond, but no real attempt was made to isolate sterically pure indi- vidual substances. On saponification the product yielded a presumed N-acetyl-5,6-oxidoveratramine. With dilute acid the oxide group of the tri- acetyl derivative was opened with satisfactory yields to the glycol, tri- acetyl-5,6-dihydroxyveratramine. As was to be expected from the tertiary character of a 5-hydroxyl group, only a 6-hydroxyl should be readily ac- ylated and would explain the formation of a tetraacetyl-5,6-dihydroxy- veratramine.
Attempts to rupture the glycol linkage with periodic acid were unsuc- cessful. This recalls the experience of Wintersteiner and Moore (7), who also found that secondary tertiary glycols of some stenols do not react with this reagent under conditions similar to ours.2 The assumption of the usual five-membered ring D would allow for a Are bond which on oxi- dation would yield a 16,17-oxido compound and from this a secondary tertiary glycol. To test such a possibility it was planned to reduce tri- acetyloxidoveratramine with lithium aluminum hydride (8) to the as- sumed 17-hydroxy compound, to dehydrate this to a Al7 derivative, and then t,o accomplish the final oxidative removal of the side chain. This reagent yielded a substance (m.p. 178-182”) which was separated through alumina; its analysis indicated it to be the desired tertiary alcohol with the N-acetyl group reduced to N-ethyl, C33H.,905N. In view of the later results this could now be interpreted as an N-ethyl-5-hydroxydihydrovera- tramine-3,d3-diacetate. A second substance was obtained of higher melt- ing point, but its analysis indicated the composition C&H4906N, which
2 The inertness to periodic acid appears to rule out the possibility that we have been dealing with a primary tertiary glycol. Such a glycol would be the hydrolytic product of a 20,21-oxide which could have been formed by oxidation of an original Az0(21) bond if present in veratramine. This structure was under consideration to explain not only the formation of the dehydrogenation product hydroxypicoline, but also the series of reactions from veratramine leading to the isomer which gives the Rosenheim reaction. In such a series of reactions oxidation of the OH on carbon 23 to CO would involve rearrangement of a A e”(zl) bond to AzO(zz) to conjugate with the carbonyl. Greater objection applies to the assumption of an original A26@7) bond which could similarly yield a primary tertiary glycol.
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from
76 VERATRINE ALKALOIDS. XXX11
suggested that mere hydrolytic cleavage of the oxide group had taken place, together with reduction of the N-acetyl to N-ethyl, to yield N- ethyl-5,6-dihydroxydihydroveratramine-3, W-diacetute (?) .
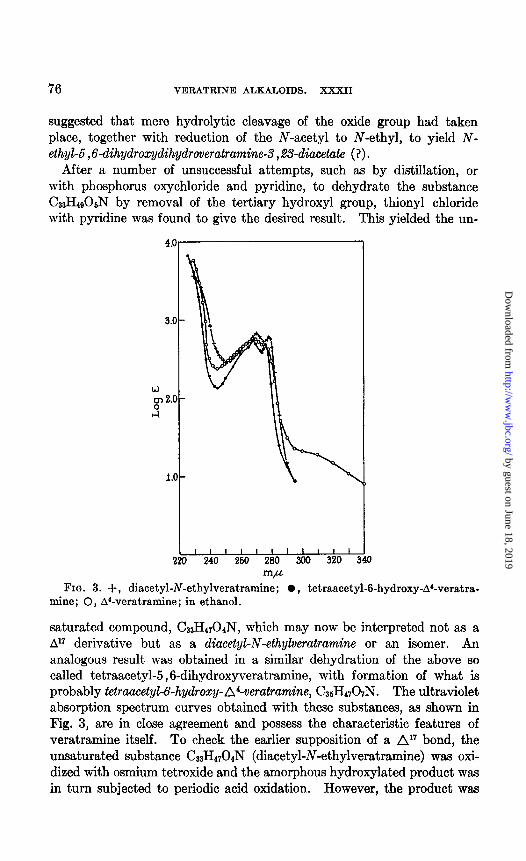
After a number of unsuccessful attempts, such as by distillation, or with phosphorus oxychloride and pyridine, to dehydrate the substance G3H490SN by removal of the tertiary hydroxyl group, thionyl chloride with pyridine was found to give the desired result. This yielded the un-
220 240 260 280 300 320 !
n-w
FIG. 3. +, diacetyl-N-ethylveratramine; 0, tetraacetyl-6-hydroxyd’-veratra- mine; 0, A’-veratramine; in ethanol.
saturated compound, CS3H4r04N, which may now be interpreted not as a A” derivative but as a diacetyl-N-ethylveratramine or an isomer. An analogous result was obtained in a similar dehydration of the above so called tetraacetyl-5,6-dihydroxyveratramine, with formation of what is probably tetraucetyL6-hydroxy-A4-veratramine, C36H4707N. The ultraviolet absorption spectrum curves obtained with these substances, as shown in Fig. 3, are in close agreement and possess the characteristic features of veratramine itself. To check the earlier supposition of a Al7 bond, the unsaturated substance G3H4704N (diacetyl-N-ethylveratramine) was oxi- dized with osmium tetroxide and the amorphous hydroxylated product was in turn subjected to periodic acid oxidation. However, the product was
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

W. A. JACOBS AND Y. SAT0 77
inhomogeneous and contained nitrogen (3.1 per cent). The retention of the side chain indicates that the new double bond is not at A17(20), but is compatible with a A4 or A 6 bond. Ozonolysis of the substance C&H4r04N yielded only an amorphous product which failed to give the Zimmermann reaction characteristic of a 17-ketosteroid. Other oxidative procedures were tried, but all yielded products in which the side chain N was retained.
Tribenzoylveratramine, described in the experimental part, was pre- pared in the course of these studies.
For the synthesis of S-methyl-5-hydroxypyridine, 3-methylpyridine-5- sulfonic acid, prepared from p-picoline by the procedure of McElvain and Goese (9), was fused with NaOH; the product was indistinguishable from the hydroxypicoline obtained on dehydrogenation of veratramine.
EXPERIMENTAL
A4-Veratramin-S-one-A solution of 0.2 gm. of veratramine in benzene was boiled down to about 10 cc. and treated with 1 gm. of aluminum tert-butoxide, followed by 7 cc. of dry acetone. The mixture, protected from moisture, was then refluxed. The copious gelatinous precipitate, which first formed slowly, diminished; after about 23 hours, the mixture was decomposed with excess dilute NaOH and repeatedly shaken with benzene. The extract was washed, dried with Na2S04, and concentrated. The colored residue, which contained acetone condensation products, was dissolved in benzene and chromat.ographed through 10 gm. of alumina in benzene. After continued elution with 60 cc. of benzene, which first re- moved the condensation products, the eluent was changed to 0.5 per cent methanol in benzene. After about 35 cc. had been used, solute suddenly emerged. The eluate, then collected in 5 cc. fractions, yielded succes- sively 23, 36, 39, 28, 21, and 10 mg. of product.
Attempts to obtain the crystalline base from these fractions were un- successful. The following analytical data were obtained with the resin dried at 110’ and 0.2 mm.
[a]: = +69” (c = 0.81’ in 95% ethanol) C2,H8,0aN. Calculated, C 79.55, H 9.16; found, C 78.73, H 8.89
The fourth fraction, dissolved in a small volume of methanol, when treated with a slight excess of HCl followed by ether, yielded the hydro- chloride as aggregates of minute delicate micro needles. It gradually softens to a melt at 216-232”.
For analysis it was dried at 110” and 1 mm.
CZ~H~O~N.HCI. Calculated, C 73.01, H 8.63; found, C 72.97, H 8.70
The oxime was prepared from the third fraction by heating for 2 hours
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

78 VERATRINE ALKALOIDS. xxx11
in methanol solution with an excess of hydroxylamine hydrochloride and sodium acetate. The mixt.ure, rendered ammoniacal and diluted, yielded the amorphous base, which was collected with water. This substance could not be crystallized.
C&H3802N2. Calculated. C 76.72, H 9.07, N 6.63 Found. “ 76.60, “ 9.09, “ 6.77
A second preparation of the ketone from 0.6 gm. of veratramine yielded through 30 gm. of alumina successive fractions of 44, 53, 62, 63, 61, 54, 31, and 10 mg. Of these, Fractions 2 to 6, or 293 mg., were used for the following preparation.
A4-Vera&amine-Aluminum isopropoxide was prepared from 1 gm. of amalgamated aluminum and 15 cc. of isopropanol and added to the above 293 mg. of the A4-veratraminone. The protected mixture was refluxed for 20 hours and after addition of another 20 cc. of isopropanol was gradu- ally distilled to small volume. The diluted concentrate was treated with excess NaOH and extracted with chloroform. The washed and dried ex- tract yielded on concentration a colored resin which was dissolved in benzene and chromatographed through 10 gm. of alumina. Since no elu- tion occurred with 60 cc. of benzene, the product was finally obtained after the use of about 45 cc. of 0.5 per cent methanol in benzene, which was collected in successive 3 to 4 cc. fractions of 18, 20, 20, 25, 25, 22, 21, 20, 15, 13, and 8 mg. All of these fractions slowly yielded a charac- teristic dirty purple color in the Rosenheim test with trichloroacetic acid.
Fractions 6, 7, and 8, on solution in a small volume of methanol and careful dilution, separated as microcrystalline globular clusters. The product (51 mg.) was collected and washed with 50 per cent methanol; it melted at 129-131.5’.
The filtrate, on dilution with the washings, yielded an additional 4 mg. of delicate micro needles which sintered at 122” and melted at 124-127”. The gradually developing Rosenheim tests with both of these fractions were indistinguishable. The color was a dirty deep purple. Veratra- mine itself gave no color.
For analysis and rotation the first fraction was dried at 110” and 0.2 mm.
[a]: = +40” (c = 0.85 in 95% ethanol) Cz?Hs902N. Calculated, C 79.16, H 9.60; found, C 78.80, H 9.55
The ultraviolet absorption curve shown in Fig. 1 was obtained with this substance.
Fractions 4 and 5, amounting to 50 mg., were converted to the hydro- chloride in methanol. Addition of ether yielded successive 30 and 14
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

W. A. JACOBS AND Y. SAT0 79
mg. fractions of small micro needles, which were dried for analysis at 110” and 0.2 mm.
ClrHasOzN.HC1. Calculated, C 72.68, H 9.04; found, C 72.85, H 9.17
Dihydroveratramin-S-one-A solution of 0.4 gm. of dihydroveratramine in a mixture of 36 cc. of acetic acid and 4 cc. of Hz0 was treated with 0.4 gm. of chromic oxide in 2 cc. of HzO. The clear solution gradually deep- ened in color at room temperature, and after 80 minutes it was diluted and extracted with chloroform. The latter was washed twice with small portions of Hz0 and then with excess dilute NaOH and HzO. The dried solution on concentration yielded 0.38 gm. of a slightly yellow resin which was dissolved in benzene and chromatographed through 20 gm. of AlzOa. After elution with 85 PC. of benzene, which removed only traces of sub- stance, 0.5 per cent of methanol in benzene was used and collected in 10 cc. fractions. An eluate suddenly emerged in Fraction 9 and was recov- ered as a colorless resin. Fraction 9 weighed 10 mg., Fraction 10, 44 mg., Fraction 11, 80 mg., Fraction 12, 74 mg., Fraction 13, 40 mg., Frac- tion 14, 13 mg., and Fraction 15, 3.5 mg. Fraction 10 yielded 28 mg. of needles from dilute acetone. Similarly, Fractions 11 and 12 together gave 97 mg. and Fractions 13 and 14, 33 mg.
The substance formed needles which progressively softened above 113” and melted at 123-125”. Its ultraviolet absorption spectrum is shown in Fig. 2.
[a]: = +54” (c = 0.907 in 95% ethanol) [ol]i = +53.6” (c = 1.11 in chloroform)
For analysis the substance was dried at 110” and 0.2 mm.
CWHMO~N. Calculated. C 79.16, H 9.60 Found. “ 78.57, “ 9.48
‘( 79.24, (( 9.48
0.1 gm. of dihydroveratramine was refluxed for 19 hours with 1 gm. of aluminium tert-butoxide in a mixture of 10 cc. of benzene and 6 cc. of acetone. After decomposition with Hz0 and addition of excess NaOH, the reaction product was extracted with benzene. The washed and dried extract yielded an oily resin containing acetone condensation products. This was chromatographed in benzene through 5 gm. of alumina. After elution with about 70 cc. of benzene, which removed the condensation products, 0.5 per cent methanol in benzene was used.
After 10 cc. more emerged, the oxidation product suddenly appeared. Successive 5 cc. fractions then progressively yielded 7, 26, 17, 7, and 2 mg. of resin. Fractions 2 and 3 were combined and crystallized readily from dilute acetone. The product, 31 mg. of needles, melted at 119-
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

80 VERATRINE ALKALOIDS. xxx11
124” after preliminary softening and was indistinguishable from that ob- tained with chromic oxide.
[a]: = +53.9” (c = 0.81 in chloroform) Found, C 79.09, H 9.46
The Oxime-A methanolic solution of 40 mg. of the chromic acid prod- uct, was refluxed for several hours with hydroxylamine hydrochloride and sodium acetate. On concentration and careful dilution with water, it crystallized as small leaflets or flat needles and proved to be the hydro- chloride of the oxime. All attempts to crystallize the base were unsuc- cessful. The salt did not exhibit a sharp melting point. Most of it softened to a melt at about 194-215”, but a portion appeared to retain birefrigence to 260-270”.
For analysis the salt was dried at 110” and 0.2 mm.
CnlHloOzN~.HC1. Calculated. C 70.31, H 8.97, N 6.08 Found. “ 70.74, I‘ 9.07, “ 6.42
N-Acetyldihydroveratramin-S-one and the 2%Acetate-A solution of 48 mg. of the chromic acid product in 2 cc. of acetic anhydride was refluxed for 20 minutes and concentrated to dryness in vacua A solution of the resinous residue in methanol was treated with excess dilute NaOH and, after occasional warming, the mixture was left at room temperature over- night. Although some crystals appeared above a colored resin, the mix- ture was extracted with chloroform. The extract was washed succes- sively with water, dilute HzS04, and water and dried. The product was obtained in successive slowly crystallizing fractions from et.hanol-ether as compact microcrystalline aggregates which melted at 191-194”.
C20H,103N. Calculated, C 77.11, H 9.16; found, C 77.32, H 9.03
In an experiment in which the reaction mixture was concentrated and the excess reagent removed by distillation with alcohol, the resinous resi- dues failed to crystallize. The product was obtained from acetone. Hz0 as a resinous powder which analysis showed to be the diacetyl derivative.
For analysis it was dried at 110” and 0.2 mm.
CaIH,30,N. Calculated, C 75.40, H 8.78; found, C 74.96, H 8.87
Dihydroveratramin-3,2%dione-A solution of 1.3 gm. of dihydrovera- tramine in a mixture of 100 cc. of acetic acid and 10 cc. of Hz0 was treated at room temperature with 13 cc. of Kiliani’s solution. The mixture gradually deepened in color and after about 5 minutes was warmed and kept at 50-55” for 13 hours, during which the reagent was gradually used up. The mixture, after dilution and addition of Na acetate in excess of the H$SOd requirement, was extracted with chloroform as in the previous
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

W. A. JACOBS AND Y. SAT0 81
case. The resulting 0.9 gm. of yellow resin was chromatographed in benzene through 40 gm. of alumina. After elution with four successive 25 cc. portions of benzene, substance began to emerge and was collected in 25 cc. fractions. Fractions through 11 yielded respectively 20, 25, 25, 25, and 15 mg. of resin. These fractions were combined and brought to a small volume in 95 per cent ethanol and treated with a slight. excess of HCl (1.19). The solution, which became yellow on addition of ether, yielded the hydrochloride as micro needles. The collected material amounted to 60 mg. and was followed by an additional 10 mg. fraction.
The analytical data were obtained with unrecrystallized product from a preliminary experiment, dried at 100” and 0.2 mm.
C2,Has0~NCl. Calculated. C 73.01, H 8.63, Cl 7.99 Found. “ 72.48, “ 8.46, “ 7.75
The 60 mg. fraction dissolved in a small volume of 95 per cent ethanol was treated with excess ammonia, followed by careful dilution. The base readily crystallized as micro needles or leaflets and was washed with 50 per cent ethanol. Yield, 38 mg. It melted at 116-122.5’ after prelim- inary softening; its ultraviolet absorption spectrum is shown in Fig. 2.
[a]: = +56” (c = 0.77 in 95% ethanol) ClrHarOrN. Calculated. C 79.55, H 9.16
Found. (a) “ 79.49, I‘ 8.80 (b) “ 79.52, “ 9.05 (4 “ 79.66, “ 8.96
All attempts to form a dioxime gave unsatisfactory results. In one case the diketone was heated in methanol with hydroxylamine hydro- chloride and Na acetate for about 5 hours. A crystalline product could not be obtained. The concentrated mixture on dilution yielded material which coagulated as a resin. This was collected with water. An attempt to reprecipitate it from ethanol with Hz0 resulted in a colloidal suspension which coagulated only on addition of NaCl solution, and filtered very slowly. Similar material was obtained when the reaction occurred at 100” for 47 hours.
The following analysis was obtained in each case with substances dried at 100” and 0.2 mm.
C~THH~SOZNN~. Calculated. N 6.63 CaHaoOzNs. “ “ 9.61
Found. (a) N 7.92 ‘I @I “ 7.58
Triacetyl-6,6-oxidoveratramine-A chilled solution of 1 gm. of triacetyl- veratramine in 15 cc. of chloroform was treated with 0.3 gm. of perbenzoic acid in 4 cc. of moist chloroform. After 18 hours at 5” the mixture was
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

82 VERATRINE ALKALOIDS. xxx11
washed successively with dilute Na&03 and water and dried with Na2SOI. After removal of the solvent, 1.12 gm. of partly crystalline residue resulted. This was dissolved in benzene-petroleum ether (5: 1) and chromatographed through 34 gm. of alumina. Since about 225 cc. of this solvent mixture removed only traces, elution was continued with benzene-ether (1: 1) and then pure ether, which yielded two main successive fractions.
The substances which emerged first melted at 203-205”, followed by material which melted at 196-199”. Although the division was not clear cut, the same result was obtained in separate experiments.
After recrystallization from ether-petroleum ether the melting points were, respectively, 208-211” and 198-199” after preliminary softening. Since the analytical data obtained with both were in close agreement, it is possible that they consist of isomeric a- and P-epoxides.
The following rotations were obtained with samples which were not recrystallized to optical constancy.
a. [a]: = +43” (c = 1.46 in chloroform) 0. [c&f = +33” (c = 1.18 “ “ 1 CsrH,bOsN. Calculated. C 71.82, H 8.23
a. Found. (a) “ 71.77, “ 8.15 “
@I “ 72.03, “ 8.25 8. (‘ (a) “ 72.01, “ 8.19
‘I (b) “ 71.56, “ 8.05
N-Acetyl-6,6-oxidoveratramine-A solution of 45 mg. of triacetyloxido- veratramine ((r form) in 5 cc. of 5 per cent methanolic KOH was refluxed gently for 30 minutes. After dilution with Hz0 the precipitated sub- stance was extracted with ether and crystallized from ether-methanol, from which it formed stout columns or rods melting at 167-172”.
C&HllO,N. Calculated, C 74.48, H 8.84; found, C 74.21, H 8.74
Triacetylveratramine Glycol-O.2 gm. of the epoxide (CY form) was dis- solved in 10 cc. of acetone and diluted by cautious addition of 6 cc. of HzO. This was followed by 0.3 cc. of 10 per cent H$Od and 2 cc. of acetone to clear up the turbidity. After 48 hours at room temperature, a portion of the acetone was removed in vacua. Addition of Hz0 caused a copious precipitate; this was extracted with ether and the extract was washed in turn with 5 per cent NaHCOs solution and HzO. In some ex- periments involving larger amounts of substance chloroform was used for the extraction. Removal of the solvent gave 0.15 gm. of partly crystal- line material, which melted at 270-281” after preliminary softening. This was purified through alumina and was obtained by elution with benzene- chloroform (1: 1) or with chloroform alone. It crystallized from dilute methanol as platelets or small rods which melted at 281-284”.
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

IV. A. JACOBS AND Y. S.4TO 83
CaaH,,OrN. Calculated. C 69.57, H 8.32 Found. (a) “ 69.88, “ 8.34
@I “ 69.69, “ 8.36
In a later experiment, in which 3:5 gm. of triacetylveratramine were oxidized, 3.35 gm. of epoxide were obtained which were used directly without attempting separation into possible cr and /3 forms. This mate- rial, on hydrolysis as above to the glycol, yielded 2.53 gm. of crude mate- rial which was chromatographed through 55 gm. of alumina. Elution first with benzene-chloroform (3:l) yielded a fraction which crystallized from methanol and consisted of unchanged oxide, m.p. 206-209”.
Found, C 72.45, H 8.21
Subsequent elution with benzene-chloroform and then with chloroform alone gave successive fractions of 0.425 and 0.8 gm. of glycol which, after recrystallization from dilute methanol, were indistinguishable. The first fraction melted at 282-284’ and its rotation was [cr]:’ = +51.3” (c = 1.5 in chloroform).
The second fraction melted at 286-290” and showed [(Y]:” = $52’ (c = 1.44 in chloroform).
Tetrczacetyl-6,6-dihydroxyveratramine-The glycol (31 mg.) was dissolved in 1 cc. of acetic anhydride and refluxed for 45 minutes. The reagent was removed in vacw, and the residue crystallized gradually from an ether- petroleum ether mixture. It formed rosettes of needles which melted at 167-172”.
For analysis it was dried at 80” and 0.2 mm.
CasHdeOsN. Calculated, C 68.71, H 8.07; found, C 69.19, H 8.08
Lithium Aluminum Hydride Reduction of Epoxide-To a stirred solu- tion of 1.825 gm. of the triacetyl oxido compound in a mixture of 100 cc. of ether and 30 cc. of benzene were added dropwise during 30 minutes 130 cc. of an ethereal solution of 1.53 gm. of the hydride. The mixture was gently refluxed for 35 minutes. Ice water was then cautiously added, followed by 10 per cent H&J04 until the milky aqueous layer became clear and acid to Congo red. The upper phase yielded no significant residue. When the aqueous phase was made alkaline with ammonia, a copious pre- cipitate resulted which was extracted with chloroform. The extract, washed and dried in the usual manner, yielded 1.21 gm. of a resin. The latter was dissolved in 10 cc. of pyridine and treated with 20 cc. of acetic anhydride for acetylation. After 48 hours at room temperature, a por- tion of the solvent was removed in vacua under 35” and then poured into 200 cc. of H20. Some precipitation occurred, but the amount was greatly increased by careful addition of ammonia. The flocculent substance was
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

84 VERATRINE ALKALOIDS. XXX11
washed with water and dried. It weighed 1.35 gm. Its solution in 10 cc. of benzene was chromatographed through 50 gm. of alumina and yielded three components. The first emerged by elution with benzene and benzene-ether (4:l) and melted at 178-182” after recrystallization from methanol.
[cy]: = +40” (c = 0.75 in chloroform)
The analytical data obtained are in agreement with the requirements for N-ethyl-5-hydroxydihydroveratramine-3,23-diacetate.
CaaH,pOaN. Calculated. C 73.43, H 9.15, (N)CzHs 5.38 Found. ‘I 73.70, “ 8.93, I‘ 4.54
A second fraction, eluted with ether, melted at 177-178” after recrystal- lization from ether-petroleum ether. Unfortunately, its optical rotation was not obtained and the question of its relationship to the first sub- stance was not decided.
Found, C 73.49, H 9.16, (N)GHs 4.97
The (N)CzHs group is produced by reduction of the N-acetyl radical. The third component was obtained by elution with ether containing
1 per cent of methanol. It melt.ed at 221.5-226.5”. From its later emer- gence and from the analytical data, this substance appears to be the diol formed by hydrolysis of the oxidic group.
[a]: = +24” (c = 0.38 in chloroform) Cs3H,00eN. Calculated, C 71.30, H 8.89; found, C 71.04, H 8.64
Dehydration of Reduction Product-To a solution of 115 mg. of the pre- vious 5-hydroxy derivative in 1 cc. of dry pyridine 0.1 cc. of thionyl chlo- ride was carefully added with chilling. The mixture was allowed to come to room temperature and, after 10 minutes, was slowly treated with wa- ter and cooled. A mixture of the hydrochloride and the base gradually crystallized. This melted partly at 147-160” and partly at a considerably higher temperature. It was dissolved in ether and a little methanol and concentrated to crystallization. Ether was then added; the insoluble crystals melted 198-205” with preliminary softening and contained halo- gen .
Ca;H,lO,N-HCI. Calculated, C 71.00, H 8.67; found, C 71.17, H 8.62
The mother liquor was concentrated to dryness and again treated with ether, when more of the sparingly soluble salt separated. The filtrate, after concentration and addition of petroleum ether, crystallized as flat hexagonal platelets which melted at 158-162”.
The higher melting substance, when dissolved in chloroform and washed
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

W. A. JACOBS AND Y. SAT0 85
with NaG03 solution, yielded on concentration the same substance, which now melted at 164-168”.
CZaHI~OIN. Calculated. C 75.97, H 9.08 Found. (a) “ 75.37, “ 8.96
0) “ 76.14, “ 8.97
In later preparations the base was directly obtained by washing a chloroform extract of the reaction product with alkali.
The ultraviolet absorption spectrum is given in Fig. 3. Tetraacetyl-6-hydroxy-A4-veratramine-Tetraacetyldihydroxyveratramine
(60 mg.) was treated in 1 cc. of pyridine with 0.1 cc. of thionyl chloride and worked up as in the previous case. Crystallization from ether- petroleum ether gave needles or rods which melted at 247-252” with some preliminary sintering.
C&HUO~N. Calculated, C 70.80, H 7.98; found, C 70.30, H 7.80
The ultraviolet absorption curve is shown in Fig. 3. Tribenzoylveratramine-To a solution of 0.25 gm. of dried veratramine
in a mixture of 3 cc. of benzene and 3 cc. of dry pyridine 0.2 cc. of benzoyl chloride was carefully added. The mixture was heated at 95-100” for 1 hour. The benzoic anhydride which crystallized was filtered and the fil- trate concentrated in vacua The residue was broken up with petroleum ether and collected. It formed clusters of platelets, m.p. 151-154”, from 95 per cent ethanol.
CtsH,OaN. Calculated, C 79.86, H 7.12; found, C 79.40, H 7.15
S-MethykLhydroxypyridine-To 15 gm. of NaOH, heated in a silver crucible to about 150”, 3 cc. of Hz0 were carefully added. When the temperature had reached about 180”, 5 gm. of 3-methylpyridine-5-sul- fonic acid (9) was stirred into the melt. As the temperature of the mix- ture was raised, the thick paste gradually melted at about 210-220” and suddenly resolidified at 240” to a hard cake. About 25 minutes elapsed from the addition of the sulfonic acid till the completion of the heating. The cake was dissolved in 275 cc. of HZO; the solution was acidified with dilute HCl until slightly acid (pH 5 to 6) and extracted with ether. The washed and dried extract yielded on concentration a residue which was crystallized from benzene. It formed heavy compact crystals which melted at 140-141’. The mixed melting point with the hydroxypico- line obtained from veratramine was 140-142’. Its ultraviolet absorption curve was indistinguishable from that obtained with the veratramine pro- duct. The same color was produced with the Folin-Denis reagent and in other ways it agreed in properties.
CBH,ON. Calculated, C 66.04, H 6.46; found, C 65.84, H 6.35
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

86 VERATRINE ALKALOIDS. XXX11
All analytical data have been obtained by Mr. D. Rigakos of this lab oratory.
SUMMARY
Veratramine shows resemblances to other steroid bases, which suggests that it possesses in part a 3(P)-hydroxy-AK-steno1 structure. The product of the Oppenauer reaction appears to be in part a A4-3-stenone, which in turn can be reduced to A4-veratramine. The second hydroxyl is also secondary and located on carbon atom 23 and is less readily oxidized. This permits the stepwise oxidation of dihydroveratramine to dihydro- veratramin-3-one and to dihydroveratramin-3,23-dione in which the 23- CO group is much less reactive. It is possible that the benzenoid struc- ture of veratramine occurs in an enlarged ring D, which could explain the production of 5-hydroxy-3-methylpyridine on dehydrogenation. The iden- tity of this product has now been confirmed by synthesis.
Other transformation products of veratramine are described, the nature of which is suggested on the above structural basis.
The reduced reactivity of the CO group at position 23 raises the ques- tion whether the inert CO group of jervine is also to be found on carbon 23 instead of on carbon 11, as previously assumed.
BIBLIOGRAPHY
1. Saito, K., Bull. Chem. Sot. Japan, 16, 22 (1949). 2. Jacobs, W. A., and Craig, L. C., J. Biol. Chem., 169,666 (1946). 3. Jacobs, W. A., and Sato, Y., J. Biol. Chem., 181, 55 (1949). 4. Jacobs, W. A., and Craig, L. C., J. Biol. Chem., 166,566 (1944). 5. Barton, D. H. R., and Cox, J. D., J. Chem. Sot., 783 (1948). 6. Bergmann, E., J. Am. Chem. Sot., 60,2306 (1938). 7. Wintersteiner, O., and Moore, M., J. Am. Chem. Sot., 73,1923 (1959). 8. Plattner, A., Heusser, H., and Kulkarni, A. B., Helu. chim. acta, 31,1822 (1948). 9. McElvain, S. M., and Goese, M. A., J. Am. Chem. Sot., 66,2233 (1943).
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from

Walter A. Jacobs and Yoshio SatoTHE STRUCTURE OF VERATRAMINETHE VERATRINE ALKALOIDS: XXXII.
1951, 191:71-86.J. Biol. Chem.
http://www.jbc.org/content/191/1/71.citation
Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
alerts to choose from all of JBC's e-mailClick here
ml#ref-list-1
http://www.jbc.org/content/191/1/71.citation.full.htaccessed free atThis article cites 0 references, 0 of which can be
by guest on June 18, 2019http://w
ww
.jbc.org/D
ownloaded from


















