
BME 130 – Genomes Lecture 5 Genome assembly I The good old days.
28
BME 130 – Genomes Lecture 5 Genome assembly I The good old days
-
Upload
byron-hext -
Category
Documents
-
view
215 -
download
0
Transcript of BME 130 – Genomes Lecture 5 Genome assembly I The good old days.

BME 130 – Genomes
Lecture 5
Genome assembly IThe good old days

Administrivia
Homework 1 – on the website today, due Friday; homework policy
Student-led paper discussion; choose groups and pick paper
Guest lecture Friday – Bob Kuhn will demo the UCSC genome browser

Genomics in the newsGenomic Fossils Calibrate the Long-Term
Evolution of Hepadnaviruses
Citation: Gilbert C, Feschotte C (2010) Genomic Fossils Calibrate the Long-Term Evolution of Hepadnaviruses. PLoS Biol 8(9): e1000495. doi:10.1371/journal.pbio.1000495
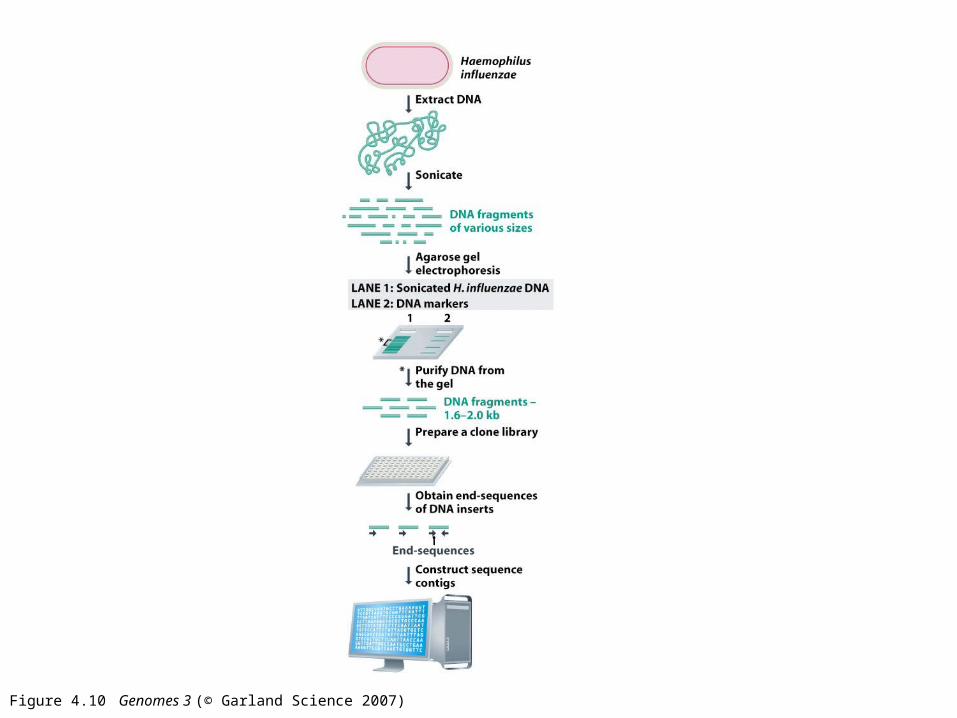
Figure 4.10 Genomes 3 (© Garland Science 2007)
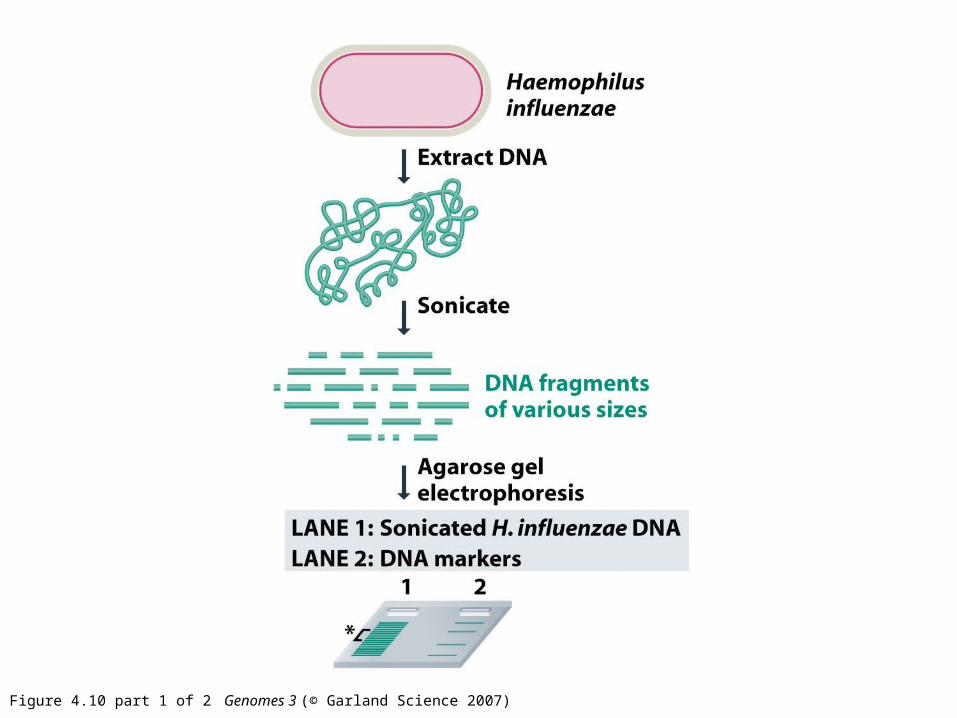
Figure 4.10 part 1 of 2 Genomes 3 (© Garland Science 2007)
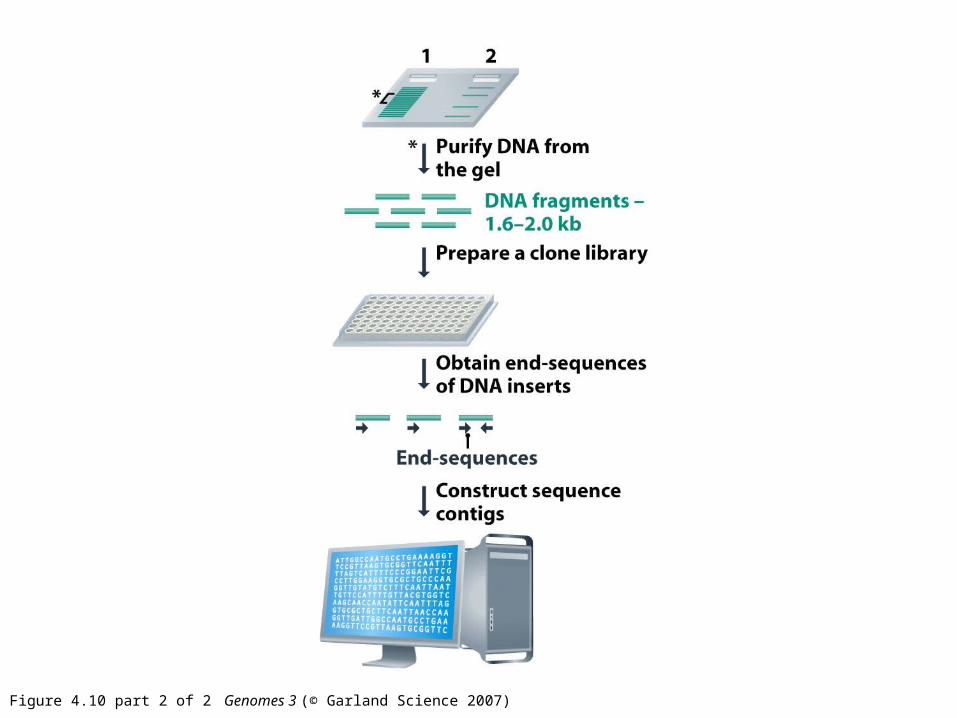
Figure 4.10 part 2 of 2 Genomes 3 (© Garland Science 2007)
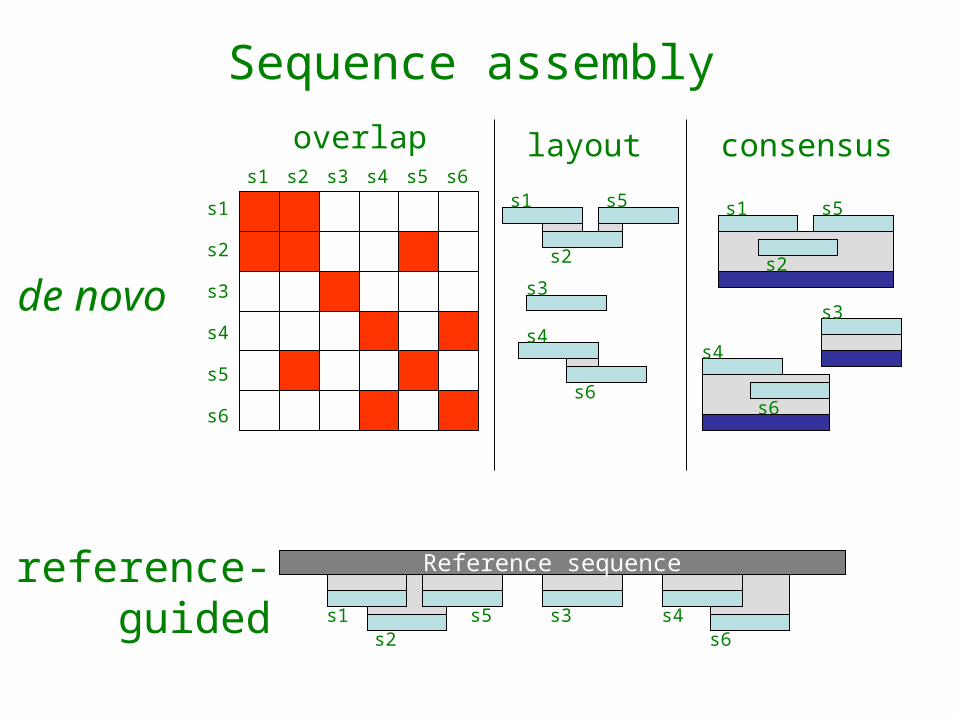
Sequence assembly
de novo
reference-guided
overlap layout consensus
s1
s2
s3
s4
s5
s6
s1 s2 s3 s4 s5 s6s1
s2
s5
s3
s4
s6
s1
s2
s5
s3
s4
s6
s1s2
s5 s3 s4s6
Reference sequence
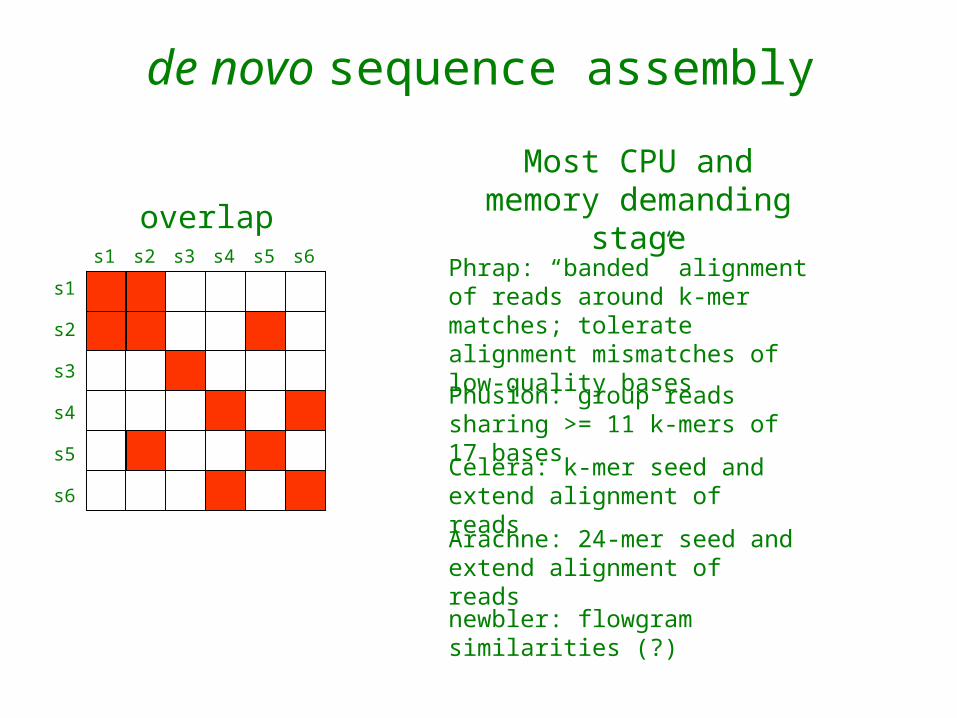
de novo sequence assembly
overlap
s1
s2
s3
s4
s5
s6
s1 s2 s3 s4 s5 s6
Most CPU and memory demanding
stage
Phusion: group reads sharing >= 11 k-mers of 17 bases
Phrap: “banded” alignment of reads around k-mer matches; tolerate alignment mismatches of low-quality bases
Celera: k-mer seed and extend alignment of reads
Arachne: 24-mer seed and extend alignment of reads
newbler: flowgram similarities (?)

Generate alignments s1
s2
s5
s3
s4
s6
de novo sequence assembly
Wide range of strategies for the layout stage, many using mate-pair
information
s1
s2
s3
s4
s5
s6
s1 s2 s3 s4 s5 s6
Find connected
components
s1 s2
s3
s4
s5
s6
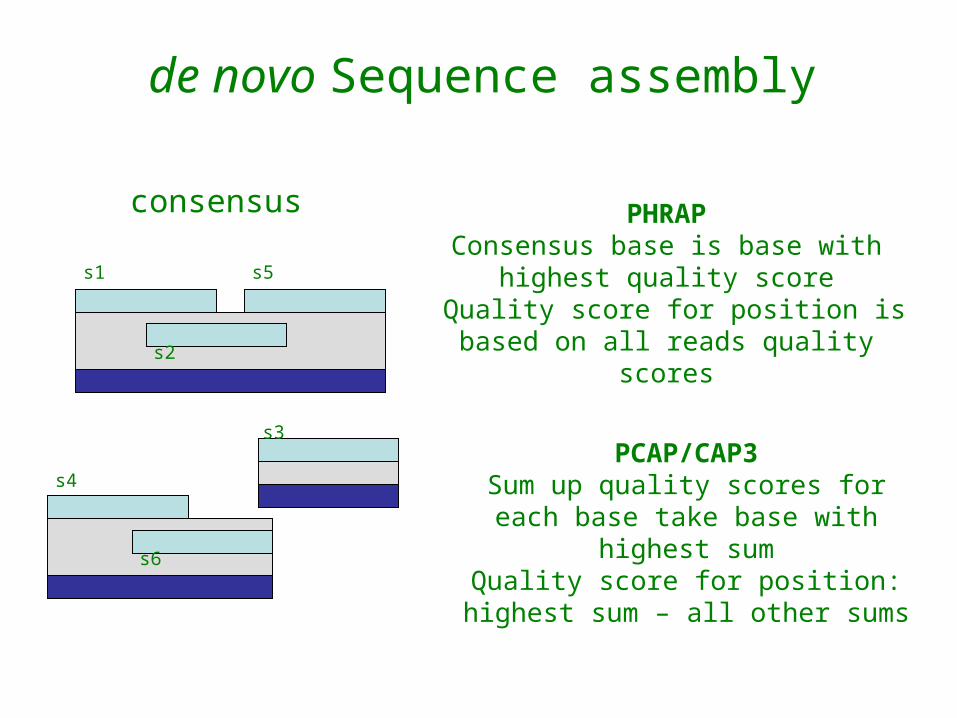
consensus
s1
s2
s5
s3
s6
de novo Sequence assembly
s4
PHRAPConsensus base is base with
highest quality score Quality score for position is based
on all reads quality scores
PCAP/CAP3Sum up quality scores for each
base take base with highest sumQuality score for position:
highest sum – all other sums
s1
s2
s5 s3 s4
s6
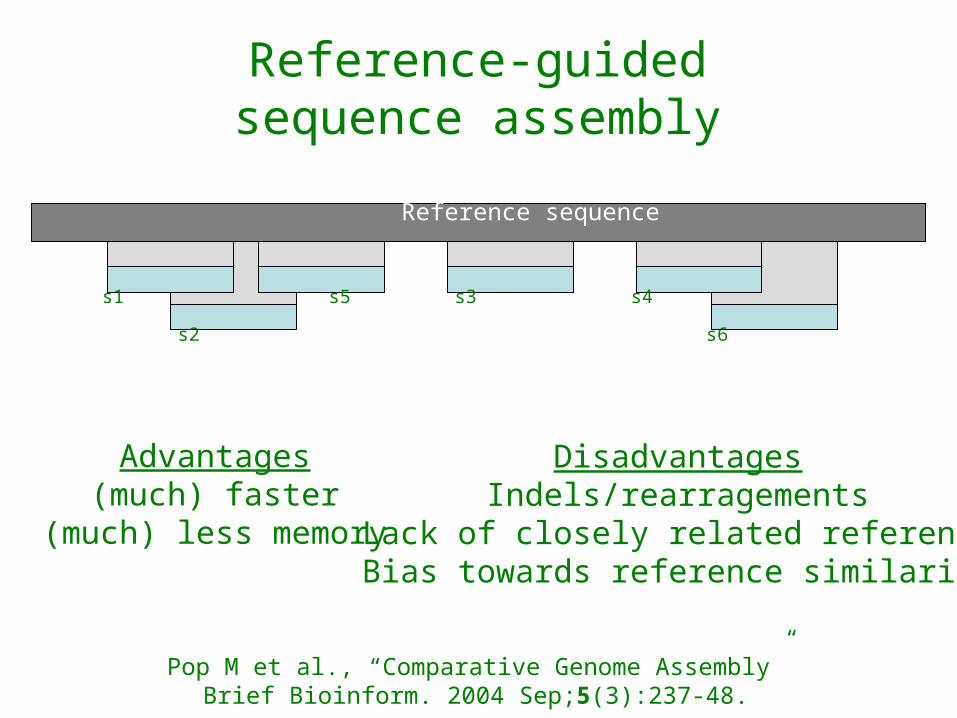
Reference sequence
Reference-guidedsequence assembly
Advantages(much) faster
(much) less memory
DisadvantagesIndels/rearragements
Lack of closely related referenceBias towards reference similarity
Pop M et al., “Comparative Genome Assembly”Brief Bioinform. 2004 Sep;5(3):237-48.
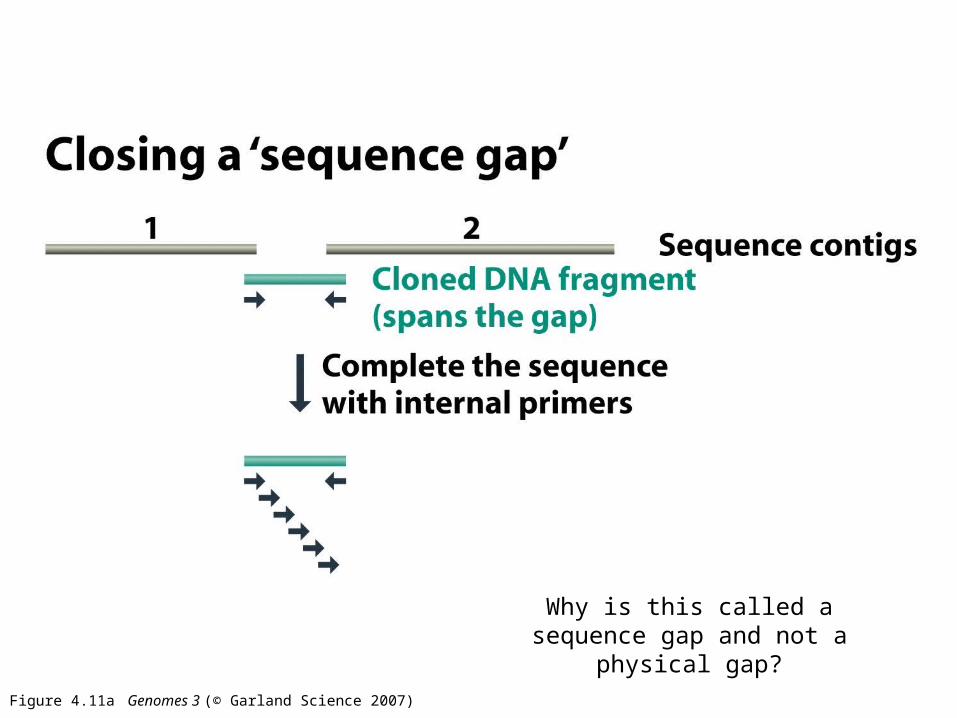
Figure 4.11a Genomes 3 (© Garland Science 2007)
Why is this called a sequence gap and not a physical gap?

Closing a physical gap means finding a physical clone to
sequence that will span the gap
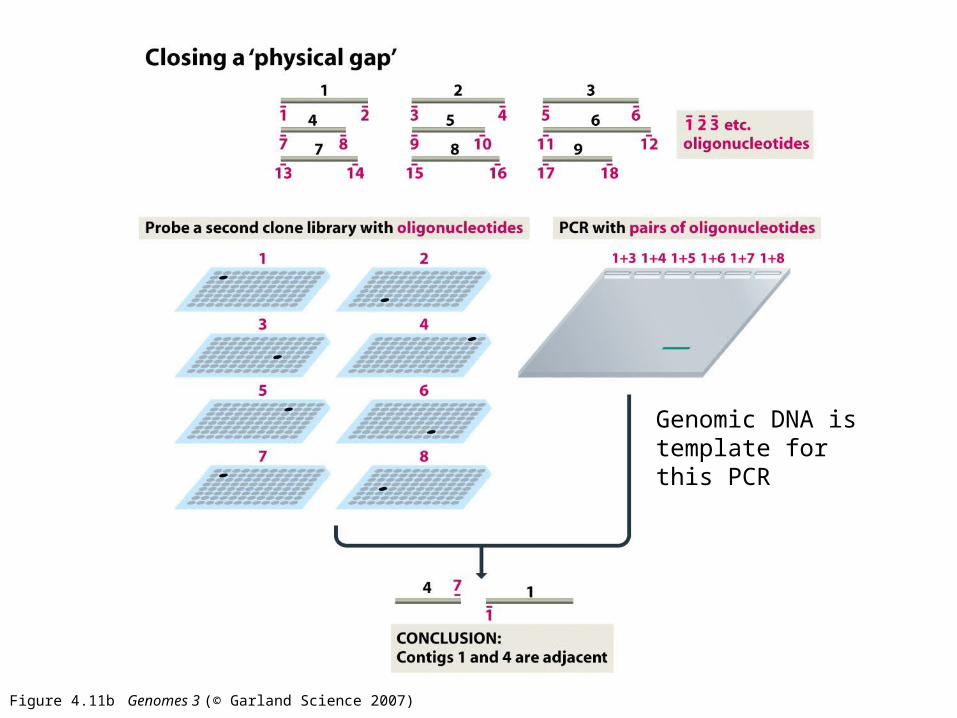
Figure 4.11b Genomes 3 (© Garland Science 2007)
Genomic DNA is template for this PCR
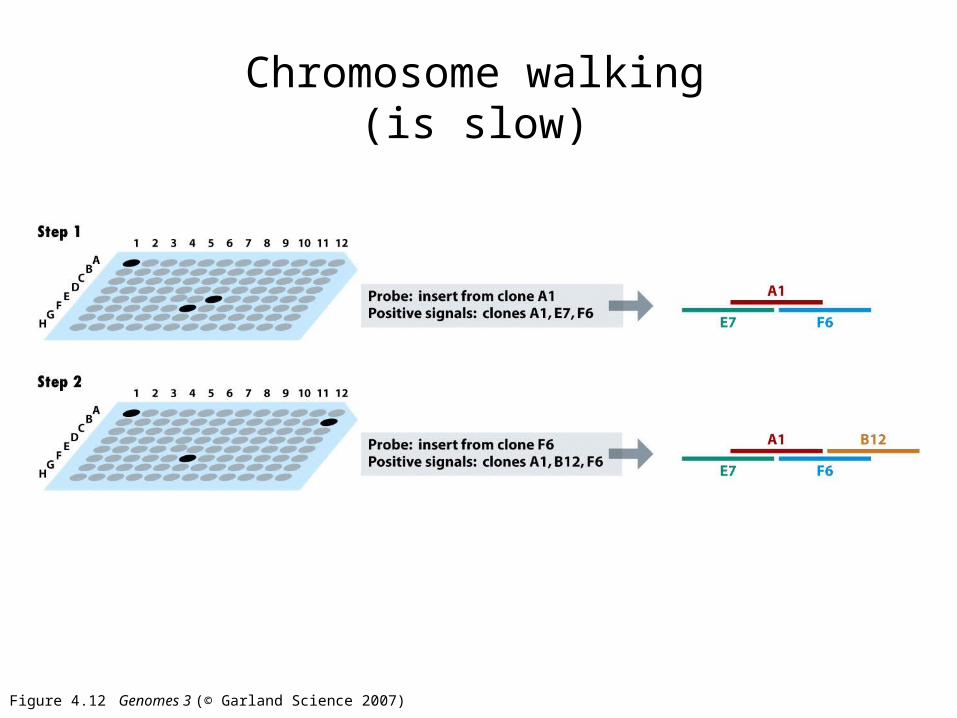
Figure 4.12 Genomes 3 (© Garland Science 2007)
Chromosome walking(is slow)
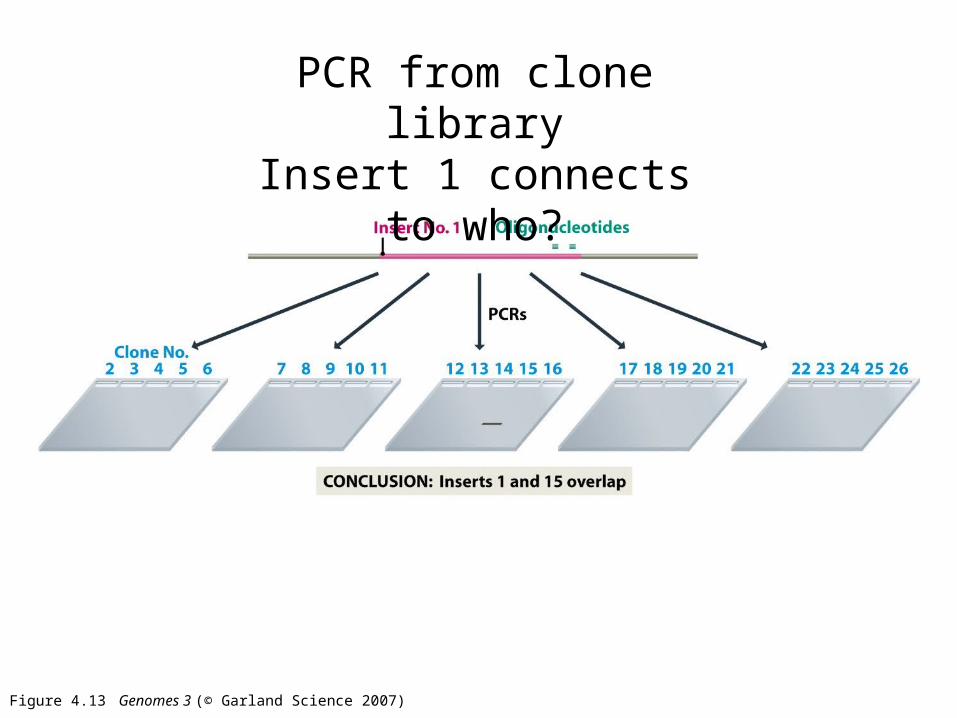
Figure 4.13 Genomes 3 (© Garland Science 2007)
PCR from clone libraryInsert 1 connects to who?
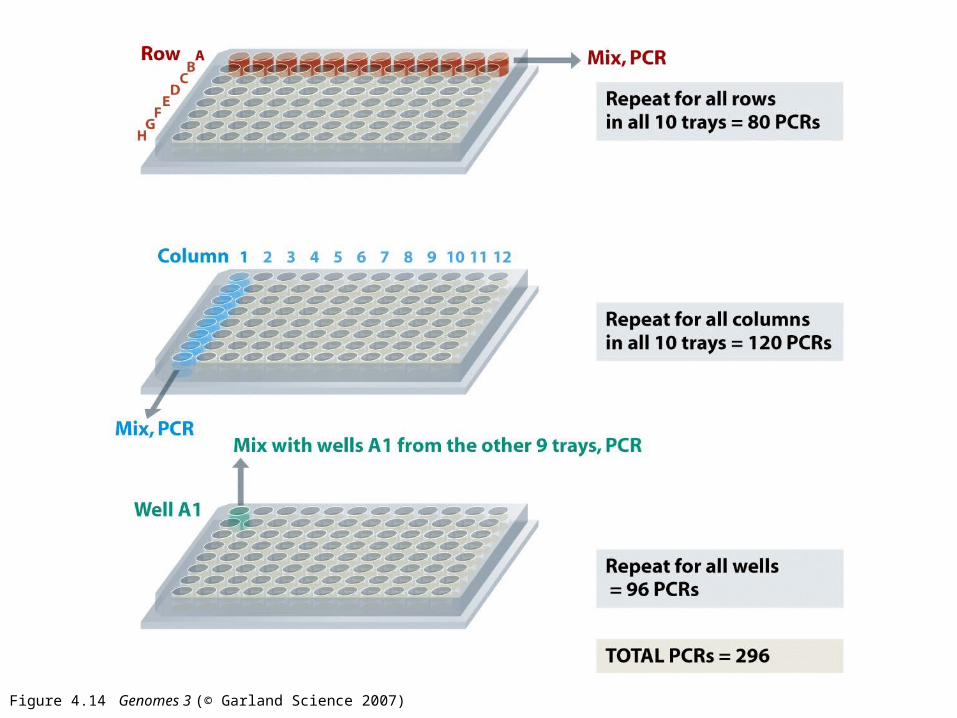
Figure 4.14 Genomes 3 (© Garland Science 2007)
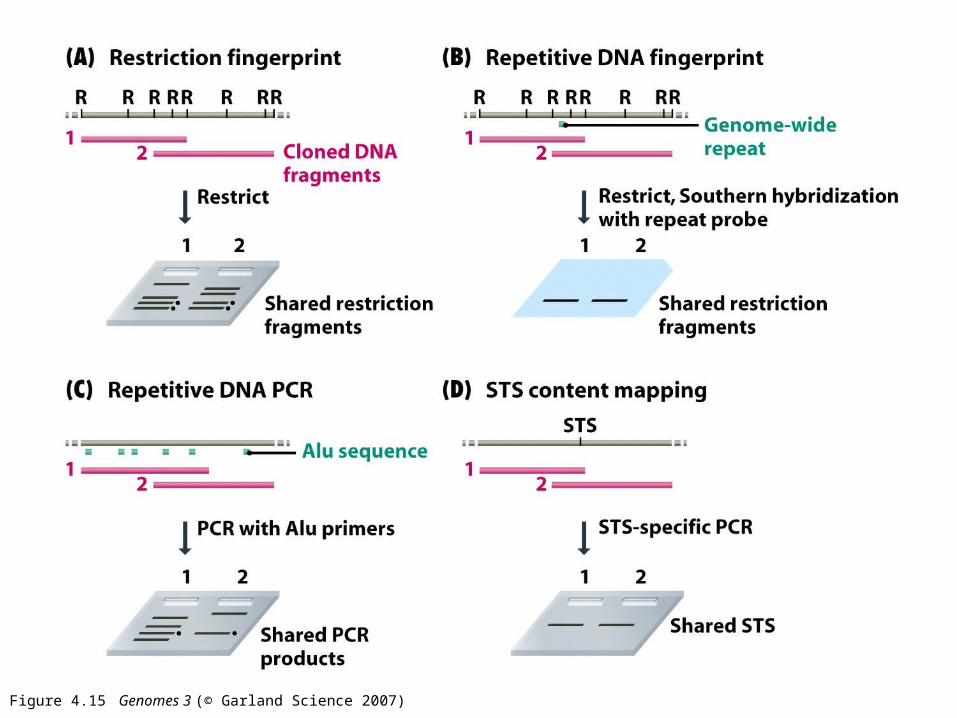
Figure 4.15 Genomes 3 (© Garland Science 2007)
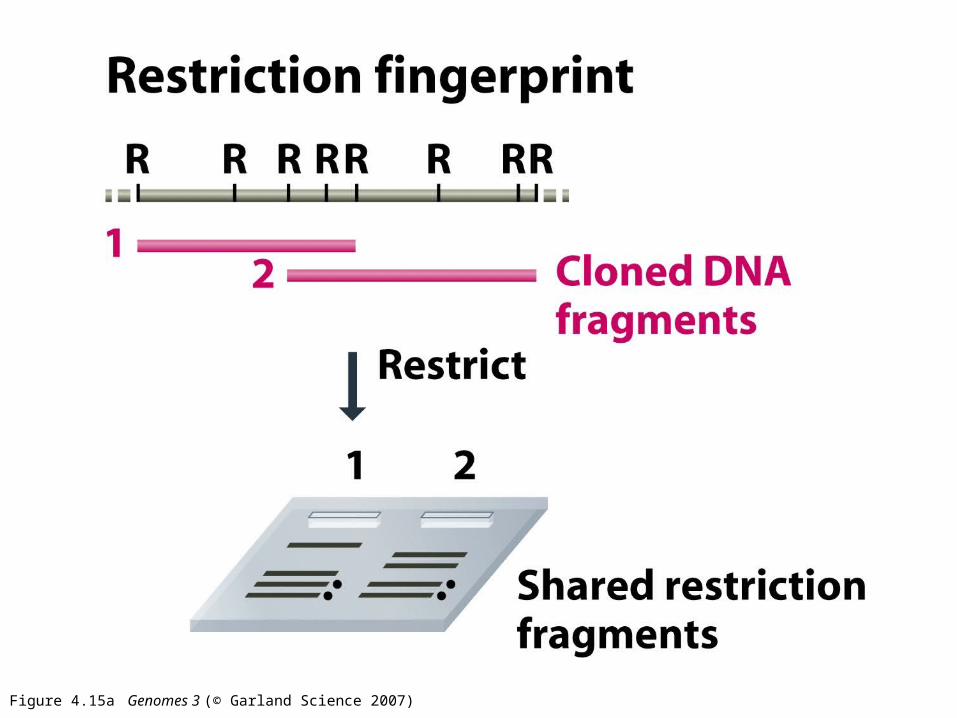
Figure 4.15a Genomes 3 (© Garland Science 2007)
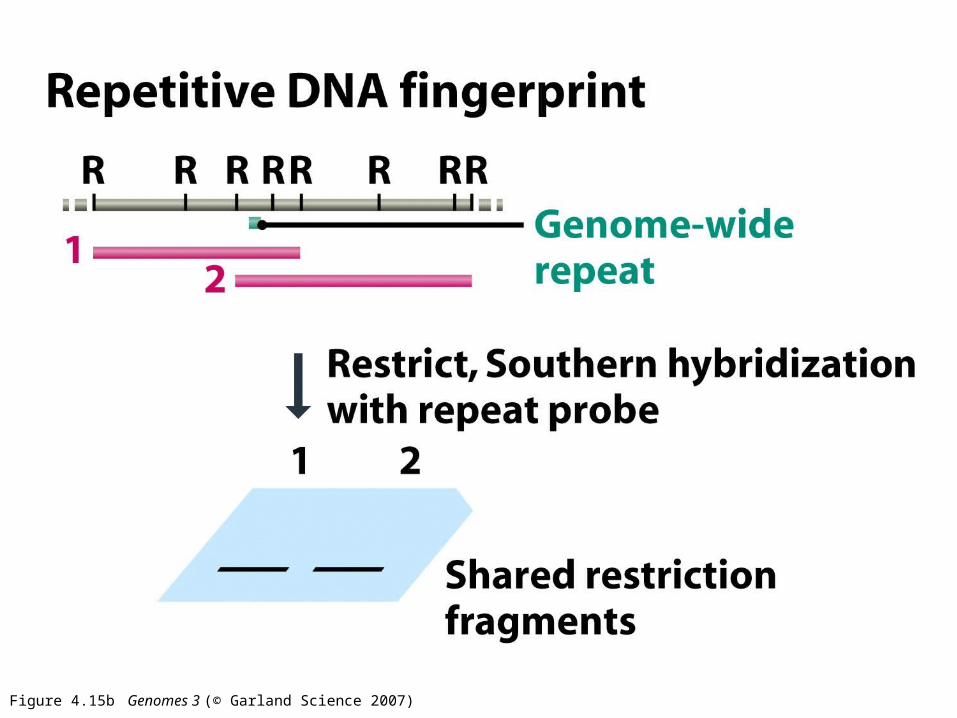
Figure 4.15b Genomes 3 (© Garland Science 2007)
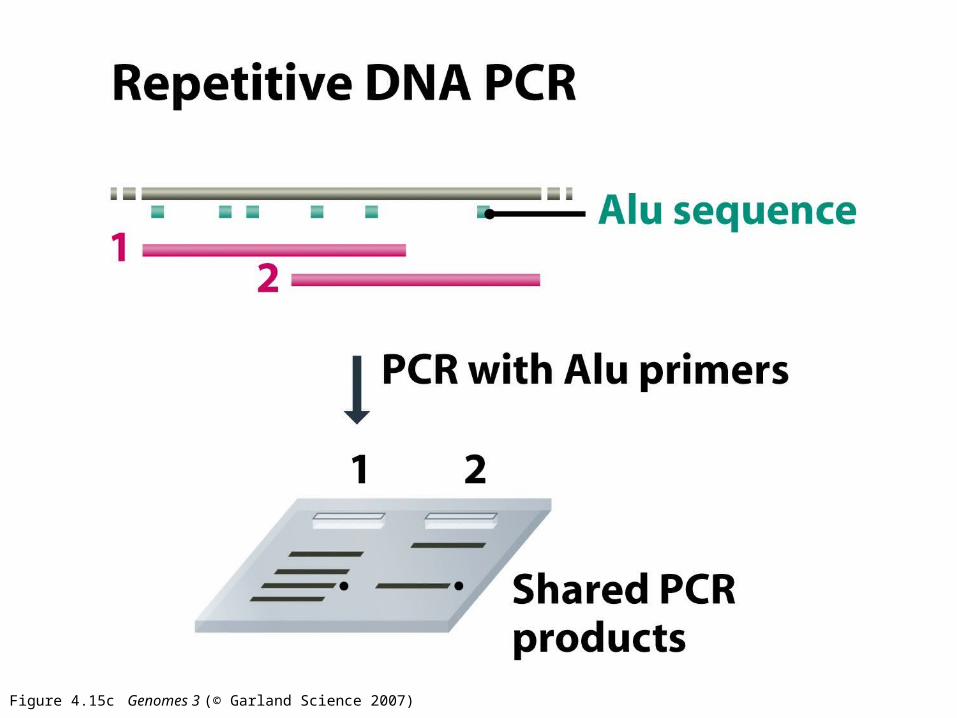
Figure 4.15c Genomes 3 (© Garland Science 2007)

Figure 4.15d Genomes 3 (© Garland Science 2007)
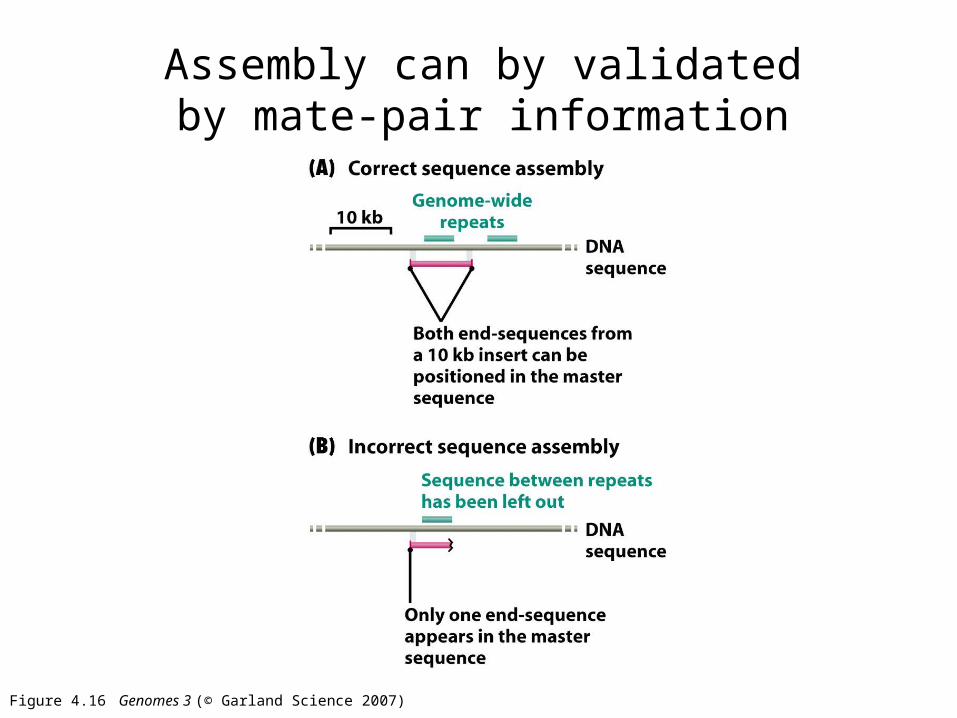
Figure 4.16 Genomes 3 (© Garland Science 2007)
Assembly can by validated by mate-pair information

Figure 4.16a Genomes 3 (© Garland Science 2007)
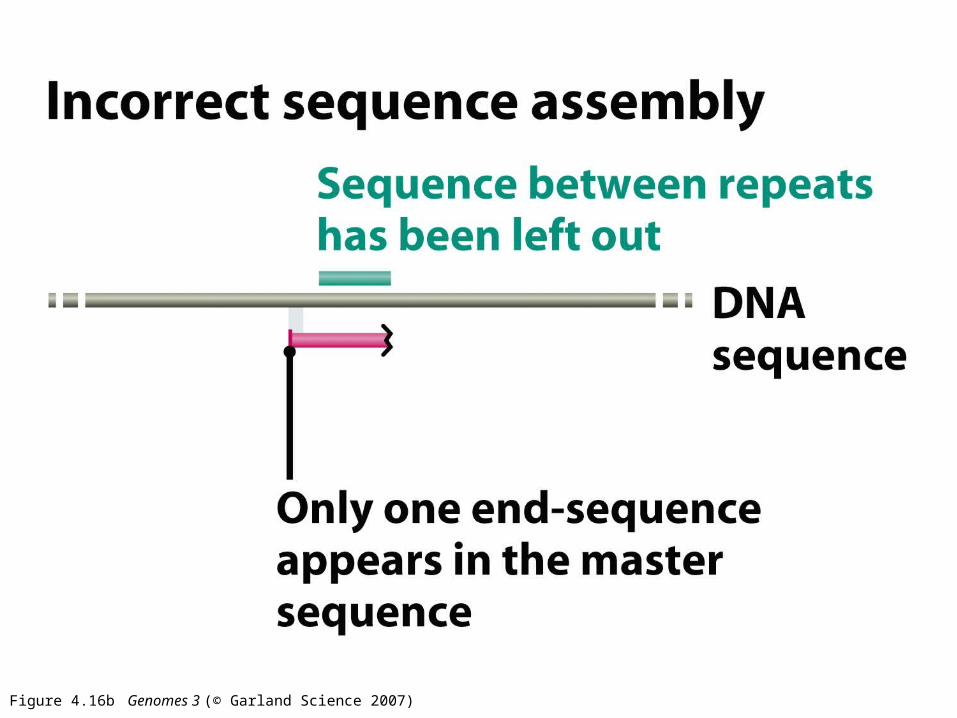
Figure 4.16b Genomes 3 (© Garland Science 2007)
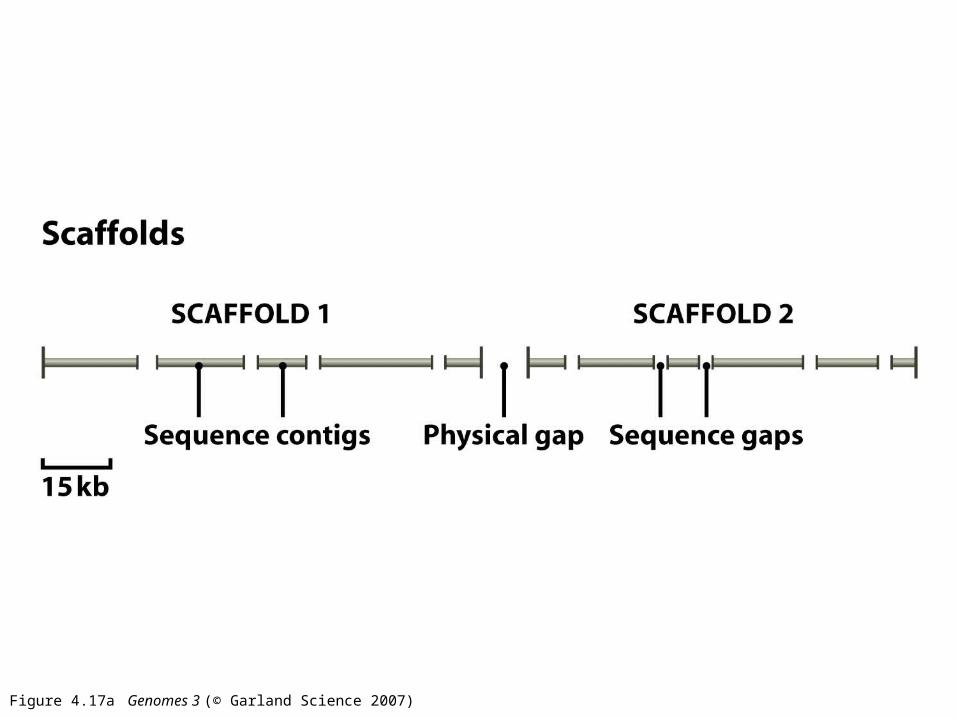
Figure 4.17a Genomes 3 (© Garland Science 2007)

Figure 4.17b Genomes 3 (© Garland Science 2007)

Figure 4.18 Genomes 3 (© Garland Science 2007)


















