
AXONAL MICROTUBULES OF CRAYFISH AND SPINY LOBSTER … · Mince and homogenize cords in PEG/PIPES,...
16
J. Cell Sd. 71, 1-15 (1984) Printed in Great Britain © The Company of Biologists Limited 1984 AXONAL MICROTUBULES OF CRAYFISH AND SPINY LOBSTER NERVE CORDS ARE DECORATED WITH A HEAT-STABLE PROTEIN OF HIGH MOLECULAR WEIGHT ROBERT H. WARREN Department of Anatomy and Cell Biology, School of Medicine, University of Miami, Miami 33101, Florida, U.SA. SUMMARY Axons of crayfish and spiny lobster ventral nerve cords contain large numbers of microtubules that are decorated with fine filaments. These microtubules can be stabilized in permeabilized axons using buffers that contain either polyethylene glycol or glycerol/dimethyl sulphoxide. In the former, the stabilized microtubules retain their filaments and their normal spacing; in the latter, the filaments are stripped off and the bare microtubules collapse onto one another. This observation has been used as the basis for a method of identifying some of the proteins that make up the filaments. Axons are first permeabilized and stabilized in either buffer and then treated with a microtubule- depolymerizing buffer. The axons treated first with polyethylene glycol release tubulin and sig- nificant quantities of microtubule-associated proteins (MAPs), while the axons pre-treated with glycerol release tubulin and only traces of associated proteins. One of the proteins released in largest quantity along with tubulin from polyethylene glycol-treated axons is a high molecular weight, heat- stable MAP that co-electrophoreses with MAP-2 from mammalian brain. This same protein co- purifies with tubulin that is obtained from crayfish nerve cords by two cycles of polymerization and depolymerization. It is concluded that this protein is a component of the filaments that decorate the axonal microtubules of the crayfish and spiny lobster. INTRODUCTION Neuronal microtubules are characterized by the presence of associated proteins, or MAPs, that were originally defined as proteins co-purifying with tubulin from mammalian brain during one or more cycles of polymerization/depolymerization in vitro. The two principal categories of MAPs in mammalian brain are high molecular weight proteins (300 to 350X10 3 ), termed MAPs 1 and 2 (Borisy et al. 1975), and a complex of proteins in the range of 55 to 62X 10 3 termed tau (Weingarten, Lockwood, H wo & Kirschner, 1975). These proteins are associated with cytoplasmic microtubules (Connolly & Kalnins, 1980), and MAP-2 in particular is a component of the filaments that project from microtubules (Dentler, Granett & Rosenbaum, 1975). The presence of MAPs in solutions of tubulin in vitro shifts the equilibrium towards polymerization (Murphy, Vallee & Borisy, 1977) and MAPs may help to regulate the extent of tubulin polymerization inside the cell. There is evidence that MAP-2 can bind to non-tubulin constituents of the cytoskeleton (Griffith & Pollard, 1978; Pytela & Wiche, 1980; Sattilaro, Dentler & LeCluyse, 1981; Bloom & Vallee, 1983), but the significance of such interactions in the functioning of the cytoskeleton remains to be defined.
Transcript of AXONAL MICROTUBULES OF CRAYFISH AND SPINY LOBSTER … · Mince and homogenize cords in PEG/PIPES,...

J. Cell Sd. 71, 1-15 (1984)Printed in Great Britain © The Company of Biologists Limited 1984
AXONAL MICROTUBULES OF CRAYFISH AND SPINY
LOBSTER NERVE CORDS ARE DECORATED WITH A
HEAT-STABLE PROTEIN OF HIGH MOLECULAR
WEIGHT
ROBERT H. WARRENDepartment of Anatomy and Cell Biology, School of Medicine, University of Miami,Miami 33101, Florida, U.SA.
SUMMARY
Axons of crayfish and spiny lobster ventral nerve cords contain large numbers of microtubules thatare decorated with fine filaments. These microtubules can be stabilized in permeabilized axons usingbuffers that contain either polyethylene glycol or glycerol/dimethyl sulphoxide. In the former, thestabilized microtubules retain their filaments and their normal spacing; in the latter, the filamentsare stripped off and the bare microtubules collapse onto one another. This observation has been usedas the basis for a method of identifying some of the proteins that make up the filaments. Axons arefirst permeabilized and stabilized in either buffer and then treated with a microtubule-depolymerizing buffer. The axons treated first with polyethylene glycol release tubulin and sig-nificant quantities of microtubule-associated proteins (MAPs), while the axons pre-treated withglycerol release tubulin and only traces of associated proteins. One of the proteins released in largestquantity along with tubulin from polyethylene glycol-treated axons is a high molecular weight, heat-stable MAP that co-electrophoreses with MAP-2 from mammalian brain. This same protein co-purifies with tubulin that is obtained from crayfish nerve cords by two cycles of polymerization anddepolymerization. It is concluded that this protein is a component of the filaments that decorate theaxonal microtubules of the crayfish and spiny lobster.
INTRODUCTION
Neuronal microtubules are characterized by the presence of associated proteins, orMAPs, that were originally defined as proteins co-purifying with tubulin frommammalian brain during one or more cycles of polymerization/depolymerization invitro. The two principal categories of MAPs in mammalian brain are high molecularweight proteins (300 to 350X103), termed MAPs 1 and 2 (Borisy et al. 1975), and acomplex of proteins in the range of 55 to 62X 103 termed tau (Weingarten, Lockwood,H wo & Kirschner, 1975). These proteins are associated with cytoplasmic microtubules(Connolly & Kalnins, 1980), and MAP-2 in particular is a component of the filamentsthat project from microtubules (Dentler, Granett & Rosenbaum, 1975). The presenceof MAPs in solutions of tubulin in vitro shifts the equilibrium towards polymerization(Murphy, Vallee & Borisy, 1977) and MAPs may help to regulate the extent of tubulinpolymerization inside the cell. There is evidence that MAP-2 can bind to non-tubulinconstituents of the cytoskeleton (Griffith & Pollard, 1978; Pytela & Wiche, 1980;Sattilaro, Dentler & LeCluyse, 1981; Bloom & Vallee, 1983), but the significance ofsuch interactions in the functioning of the cytoskeleton remains to be defined.

2 R. H. Warren
Axons of the ventral nerve cord of the crayfish and related crustacean species areespecially rich in microtubules, and there have been several studies of microtubulesubstructure and organization in these species. Particular attention has been focusedupon the fine, irregular filaments that project from the surfaces of the microtubules(Burton & Fernandez, 1973; Burton & Hinkley, 1974; Warren & Rubin, 1978; Ochs& Burton, 1980). These filaments appear to interconnect the axonal microtubules ina cytoskeletal lattice inside the axon. It has been suggested that the filaments on themicrotubules could play a role in axonal transport (Fernandez, Burton & Samson,1971), but there is no evidence providing direct support for such a function. Therehave been preliminary reports (Ochs, Ochs & Burton, 1980) concerning the possibleprotein constituents of the microtubular filaments in crayfish axons, but they haveremained poorly characterized. The present project was undertaken in order to deter-mine if the MAPs of crayfish neurotubules could be more clearly defined and, inparticular, to see if any of the proteins making up the filaments projecting from theaxonal microtubules could be identified. The project was facilitated by the discoverythat the axonal microtubules in permeabilized axons can be stabilized in two differentforms: with and without the attached filaments. By comparison of the proteins thatare released along with tubulin when microtubules stabilized in the two differentforms are intentionally depolymerized, evidence has been obtained that indicates thata protein similar to MAP-2 of mammalian brain is a constituent of the filamentsdecorating the crayfish microtubules.
MATERIALS AND METHODS
AnimalsCrayfish (Pmcambarus clarkii) were obtained from Carolina Biological Supply, Burlington,
North Carolina. Spiny lobsters (Panulirus argus) were obtained from local fishermen in Miami.Both species were kept in aquaria in the laboratory and fed until used for experiments.
Preparation of nerve cordsThe removal and desheathing of nerve cords from spiny lobsters were carried out as previously
described (Warren & Rubin, 1978), using a balanced salt solution to bathe the isolated cords.Crayfish nerve cords were dissected in a similar fashion using van Herreveld's (1936) salt solution.
Media for axonal permeabilization and microtubule stabilizationPEG/PIPES medium. 0 1 M - P I P E S (1,4-piperazinediethanesulphonic acid), 3 % polyethylene
glycol (M, 20000), 0-1% Triton X-100, 2mM-EGTA (ethyleneglycol-bis(/3-aminoethyl ether)-AVV,Ar',JV'-tetraacetic acid), 0-5mM-MgCl2, 0-5 mM-ATP, 0-2min-dithiothreitol (DTT),O'l mM-phenylmethylsulphonyl fluoride (PMSF), pH 6-9. Nerve cords were permeabilized in thismedium after soaking for half an hour at room temperature.
GSD medium. Glycerol (50%, v/v), 0-25 M-sucrose, 10% (w/v) dimethyl sulphoxide(DMSO), 5 mM-MgCl2 , and S mM sodium phosphate buffer (pH 7-0). Nerve cords were soaked inthis medium for a minimum of 24 h at room temperature before use.
Purification of tubulinBovine brain tubulin was purified using the method of Shelanski, Gaskin & Cantor (1973). Two
cycles of polymerization were carried out, after which the protein was stored at — 70 °C before use.

Crayfish microtubule-associated protein 3
Tubulin was purified from Procambarus nerve cords using a method modified from that of Burton& Hinkley (1974). Nerve cords were dissected from IS large crayfish and rinsed with calcium-freevan Herreveld's salt solution. The cords were then minced in the following buffer: 0 - 1 M - M E S(2-[Af-morpholino]ethanesulphonate), 1 mM-EGTA, O5mM-MgCl2l 0-047M-NaCl, 1 mM-GTP(pH6-5) and immediately homogenized with three strokes in a small glass Dounce homogenizer.The homogenates were then treated according to the following protocol: (1) centrifugation at100 000 £ for 30min at 4°C; (2) addition to supernatant of GTP to 2-5 min and Taxol to 1 /M,followed by incubation for 45 min at 28 °C; (3) centrifugation at 45 000 giot 45 min at 28 °C to obtain1-cycle tubulin. To obtain 2-cycle tubulin, the pellet from step (3) was re-dissolved in the startingbuffer without additional GTP, and then steps (l)-(3) were repeated.
Polyacrylamide gel electrophoresisOne-dimensional gels were run according to the method of Laemmli (1970), using an acrylamide
concentration of 10% (w/v), or according to the microslab method of Matsudairi & Burgess (1978),with a linear gradient of 5 % to 20 % acrylamide. Two-dimensional gels were run using the methodof O'Farrell (1975) as modified by Wilson, Hall, Stone & Rubin (1977).
Methods for electron microscopyNerve cords were fixed, embedded and sectioned for transmission electron microscopy as
described previously (Warren & Rubin, 1978). Negative staining was carried out on axoplasm fromPanulirus axons prepared in two ways. In order to examine axonal microtubules with filamentsattached, fresh axoplasm was extruded from cut nerve-cord segments into a small drop of PEG/PIPES medium on a surface of Parafilm. This was collected and dispersed gently in amicrohomogenizer. A portion of the homogenate was placed on the surface of a Formvar-coatedelectron microscope (EM) grid for 3-5 min, rinsed with the medium, fixed with 3 % glutaraldehydein 0-1 M-cacodylate buffer (pH 7-0), rinsed with distilled water, and stained with 1 % (w/v) aqueousuranyl acetate. To examine axonal microtubules stripped of filaments, nerve cords were extractedwith GSD medium for a minimum of 24 h and the axoplasm was then extruded onto a coated EMgrid and processed as for the first procedure.
Desheathed bundles of crayfish nerve pre-treated with either PEG/PIPES or GSD medium wereprepared for quick-freezing by fixation in 3 % glutaraldehyde followed by rinsing in distilled waterand then 10% (v/v) methanol in distilled water. The nerves were frozen against a copper blockcooled with liquid helium using a device patterned after the 'slammer' first developed by Heuser etal. (1979). The frozen nerve segments were stored in liquid nitrogen until they were cleaved at— 150°C and etched for 5 min at — 95 °C. The etched surfaces were replicated with carbon andplatinum using a rotary stage in a Balzers freeze-etch device, and the replicas were cleaned withClorox before examination in the electron microscope.
RESULTS
Nerve organization
The organization of inter-ganglionic nerve axons in spiny lobster and crayfishventral nerve cords has been described in detail (Fernandez et al. 1971; Burton &Hinkley, 1974; Warren & Rubin, 1978). The most significant feature is that the axonsare filled with microtubules, but entirely lack neurofilaments (Figs 1-3). Fine,irregular filaments extend from the surfaces of the microtubules and interconnectadjacent microtubules. The only other organelles present are mitochondria and clearvesicles that are generally located around the peripheries of the axons.
Stabilization of microtubules in extracted nerve axons
Two different protocols were used to stabilize the microtubular cytoskeleton innerve axons after disruption of the membranes of the axons and ensheathing glial cells.

R. H. Warren
Figs 1-3
Crayfish microtubule-associated protein
<- " »
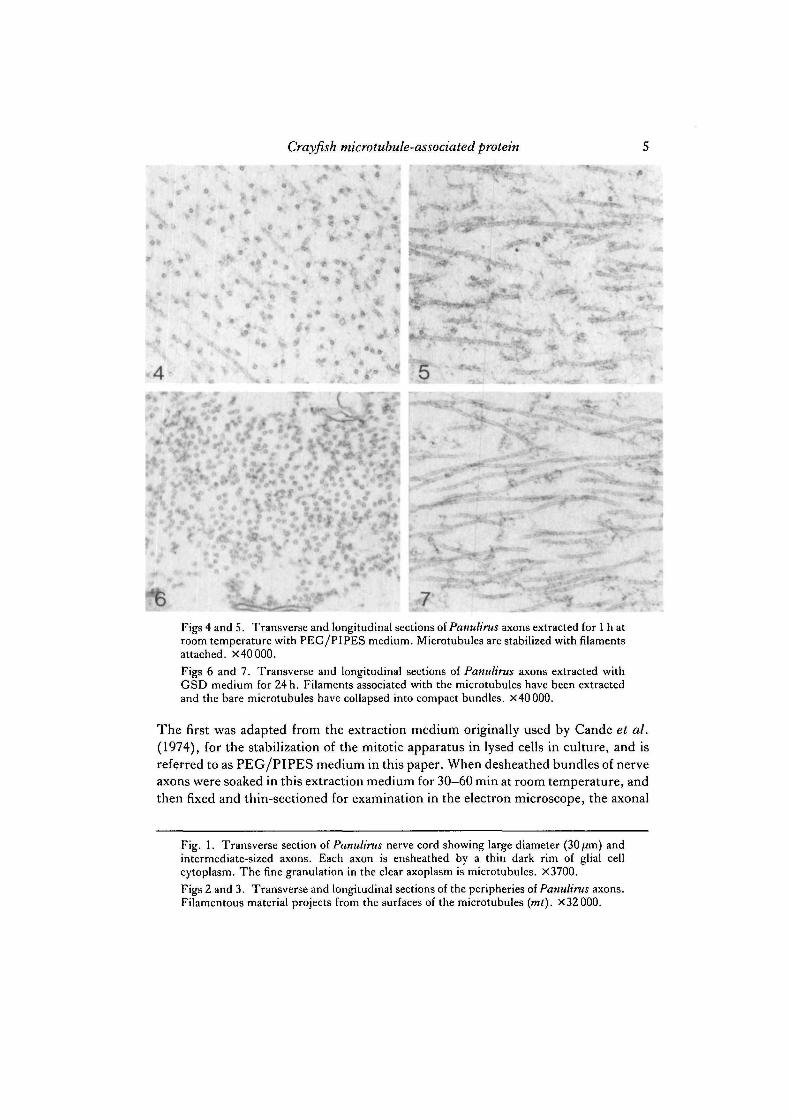
Figs 4 and 5. Transverse and longitudinal sections of Panulirus axons extracted for 1 h atroom temperature with PEG/PIPES medium. Microtubules are stabilized with filamentsattached. X40000.
Figs 6 and 7. Transverse and longitudinal sections of Panulirus axons extracted withGSD medium for 24 h. Filaments associated with the microtubules have been extractedand the bare microtubules have collapsed into compact bundles. X4000Q.
The first was adapted from the extraction medium originally used by Cande et al.(1974), for the stabilization of the mitotic apparatus in lysed cells in culture, and isreferred to as PEG/PIPES medium in this paper. When desheathed bundles of nerveaxons were soaked in this extraction medium for 30—60 min at room temperature, andthen fixed and thin-sectioned for examination in the electron microscope, the axonal
Fig. 1. Transverse section of Panulirus nerve cord showing large diameter (30 jUm) andintermediate-sized axons. Each axon is ensheathed by a thin dark rim of glial cellcytoplasm. The fine granulation in the clear axoplasm is microtubules. X3700.
Figs 2 and 3. Transverse and longitudinal sections of the peripheries of Panulirus axons.Filamentous material projects from the surfaces of the microtubules (mt). X 32 000.

H R. H. Warren
microtubular apparatus was stabilized in a form very similar to that of unextractedaxons (Figs 4, 5). Microtubules were present in their normal number anddistribution. Most significantly, the filaments still decorated the surfaces of themicrotubules. The microtubules were stable in PEG/PIPES medium for 3h orlonger.
9A
£•«- --w^pp%^
Figs 8-11

Crayfish microtubule-associated protein 7
Table 1. Identification of crayfish microtubule-associated proteins by depolymeriza-tion of stabilized microtubules in pellets of nerve homogenates
Excise nerve cord in Ca2+-free saline
A. Stabilization of axonal MTs withfilaments intact:
Mince and homogenize cordsin PEG/PIPES, room temp.
\
B. Stabilisation of axonal MTs withoutfilaments:
Soak intact cords in GSD for 1-5days, then mince and homogenizeroom temp.
\ sCentrifuge at 12 000 #lOmin, room temp.
1Rinse pellet with fresh PEG/PIPES or GSD and centrifuge as above. Repeat to remove loosely
associated proteins from stabilized fragments of axonal MTs, room temp.
1Disperse washed pellet in MT-depolymerizing solution
30min, on ice(0-1 M - P I P E S or MES buffer,
1 mM-CaCh, 1 mM-MgCl20-2 mM-DTT, 0-1 mM-PMSF
pH6-9)
1Centrifuge at 100 000 g
30min, 4°C
1Dialyse supernatant against ammonium acetate buffer (pH7), lyophylize, and run on
1- or 2-dimensional gel
Fig. 8. A-c. Negatively stained preparation of microtubules extruded from Panulirusaxons extracted for 1 h with PEG/PIPES medium. The surfaces of the microtubules areobscured by adherent amorphous or filamentous material. X100 000.
Fig. 9. A-C. Negatively stained preparation of microtubules extruded from Panulirusaxons extracted for 24 h with GSD medium. The bare surfaces of the microtubules arevisible after removal of the filaments. X100 000.
Fig. 10. Quick-frozen and deep-etched axoplasm from Procambarus following extractionfor 1 h in PEG/PIPES medium and fixation in glutaraldehyde. The surfaces of themicrotubules (mt) are obscured by adherent material, and they are spaced well apart byfilamentous linkages ( / ) . X80 000.
Fig. 11. Quick-frozen and deep-etched axon from Procambarus following extraction for24 h in GSD medium and fixation in glutaraldehyde. The surfaces of the microtubules arerelatively bare, and the microtubules have collapsed into closely packed bundles. X 80 000.

8 R. H. Warren
Different results were obtained if desheathed nerve bundles were soaked in anextraction medium (GSD) based on the glycerol/DMSO microtubule-stabilizationmedium of Filner & Behnke (1973). If extraction times in this medium were extendedto 24 h or more, the axonal cytoplasm was thoroughly extracted and the filamentsdecorating the microtubular surfaces were removed (Figs 6, 7). This resulted in thecollapse of the microtubular cytoskeleton into a compact bundle within the core of theaxon, allowing microtubules to come into close contact with one another. This com-paction is never seen in fresh-fixed or PEG/PIPES-stabilized axoplasm.
The differences between axonal microtubules stabilized in PEG/PIPES or GSDmedium were also examined using negative staining and quick-freezing techniques.Negative-stain preparations yielded images such as those shown in Figs 8 and 9. Thesurfaces of microtubules stabilized with PEG/PIPES medium were obscured by acoating of amorphous or filamentous material. After GSD treatment, the surfaces ofthe microtubules were much cleaner, and it was often possible to detect the granulartexture of the wall of the microtubule. Similar results were obtained when extractedaxons were quick-frozen against a copper block cooled with liquid helium andsubsequently fractured and deep-etched. Axoplasm stabilized with PEG/PIPES(Fig. 10) presented a cytoskeleton in which microtubules were widely spaced, coatedwith surface material, and interconnected by long filaments bridging the spaces be-tween the microtubules. In axoplasm stabilized with GSD, the microtubular latticehad collapsed and the surfaces of the microtubules appeared to be much cleaner.
Release of microtubule-assodated proteins from stabilized nerve microtubules bydepolymerization
The observation that microtubules stabilized in the glycerol-based medium lackedthe filamentous projections seen on the PEG/PIPES-stabilized microtubules led to
Fig. 12. Proteins released by microtubule depolymerization from pellets of Procambarusnerve homogenates previously treated with either PEG/PIPES or GSD microtubule-stabilization media. Lane A, bovine brain tubulin standard, twice-purified, showingdoublet tubulin bands of 55x\(fiMt and a complex of HMW-MAPs at about 300X103.Lane B, tubulin and associated proteins released from pellets stabilized in PEG/PIPESmedium, including a complex that comigrates with the HMW brain MAPs. Lane C,tubulin and associated proteins released from pellets stabilized in GSD medium.Arrowheads indicate protein bands that are greatly reduced in proportion to the amountspresent in the PEG/PIPES sample of lane B. Note especially the reduction in the HMWcomplex. Lane D, Proteins of chicken intestinal brush borders (courtesy of David Burgess)used as molecular weight standards. The molecular weights of myosin (200X103), villin(95X103) and actin (43X103) are indicated. Microgel, 5% to 20% acrylamide gradient,with urea.
Fig. 13. Two-dimensional gel of a sample identical to lane B. (Fig. 12; PEG/PIPESmicrotubule stabilization). The major spots are the central tubulin doublet and a highmolecular weight band (arrowhead).
Fig. 14. Two-dimensional gel of a sample identical to lane c (Fig. 12; GSD microtubulestabilization). Even though the sample is loaded more heavily than the sample of Fig. 13,the amount of protein in the high molecular weight spot (arrowhead) is barely detectablein comparison to the heavily loaded tubulin spots.

Crayfish microtubule-associated protein 9
the development of a procedure for characterization of the proteins in the filaments.This protocol, which is outlined in Table 1, involves extracting and stabilizing theentire nerve cord in the appropriate medium, and then homogenizing it to release thestabilized microtubules from the axons. The microtubules (or fragments thereof) andcellular debris are pelleted and washed in the stabilization buffer. The pellet is thentreated with a microtubule-depolymerizing buffer containing calcium and cooled inan ice bath. The rationale for this step is that selective depolymerization of themicrotubules in the pellet will release tubulin and any associated proteins that are
A W C I T
200
95
13
43
12 Ir
14
Figs 12-14

m R. H. Warren
Mr
200
HMW
95
16 B
B
15Fig. 15. Comparison of Procambarus nerve cord tubulin and bovine brain tubulin, aftertwo cycles of purification for both. Lane A, intestinal brush border molecular weightstandard. Lane B, twice-cycled crayfish nerve tubulin (t), with an HMW band as the majorcopurifying protein, and a minor band that is probably actin (a). Lane c, twice-cycledbovine brain tubulin, with MAP-2 band that co-migrates with crayfish HMW band (8 %polyacrylamide gel).
Fig. 16. Heat stability of crayfish HMW-MAP. Lane A, crayfish neurotubulin after onecycle of purification in the presence of Taxol. Lane B, one-cycle crayfish neurotubulin afterthe sample was heated in boiling water for lOmin and then centnfuged at 100000^ forISmin to remove denatured protein. Only the HMW-MAP band remains (8%polyacrylamide gel).

Crayfish microtubule-associated protein 11
bound to the microtubules. After a final spin at 100 000# to remove particulatematerial, the supernatant can be run on one- or two-dimensional polyacrylamide gels.A comparison of the proteins released from PEG/PIPES-stabilized microtubules(bearing attached filaments) with those released from GSD-stabilized microtubules(lacking attached filaments) should reveal proteins that are potentially components ofthe filaments attached to the microtubules in fresh-fixed or PEG/PIPES-stabilizednerves. After using this procedure, four such proteins were found that appear to besignificantly reduced or absent from GSD-treated pellets (Fig. 12). Of greatestinterest is a complex of high molecular weight proteins that comigrate with the MAP-1 and MAP-2 bands of bovine brain tubulin. This 300X 103/35Ox 103Mr complex isa prominent constituent of the proteins released from PEG/PIPES-stabilizedmicrotubules but is almost entirely absent from the supernatants of GSD-stabilizedmicrotubules. Similar results were obtained when the microtubule-depolymerizationsupernatants were compared in two-dimensional polyacrylamide gels (Figs 13, 14).
Since the results of the microtubule-depolymerization method described abovecannot be taken as a conclusive demonstration that any of the released proteins aremicrotubule-associated proteins in vivo, an additional approach has been used. Thisinvolved the purification of tubulin directly from fresh, homogenized, crayfish nervecords using a two-cycle, Taxol-driven, polymerization/depolymerization protocol inorder to determine if any of the four MAP candidates defined above also co-purifywith tubulin. The results (Fig. IS) show that crayfish tubulin, which is highly purifiedafter two cycles of polymerization, contains significant quantities of a high molecularweight MAP (HMW-MAP) that comigrates with the HMW protein released fromaxons treated with PEG/PIPES medium and with the MAP-2 complex accompany-ing bovine brain tubulin. As a further test of the similarity of the crayfish HMW-MAPto MAP-2 of bovine brain, the purified crayfish neurotubulin containing the HMW-MAP was heated in a boiling water bath. As shown in Fig. 16, the crayfish HMW-MAP is resistant to denaturation by this treatment.
DISCUSSION
The most direct evidence that crayfish nerve axons contain a high molecular weightmicrotubule-associated protein is the appearance of the HMW band in the one-dimensional gels of the twice-cycled crayfish axonal tubulin. It should be noted thatpurification of the tubulin samples run on these gels was carried out using amicrotubule polymerization buffer that contained Taxol but not glycerol. The factthat Taxol has been shown to stabilize the interaction of MAPs and microtubules(Albertini, Herman & Sherline, 1984) provides additional support for the conclusionthat the HMW protein associated with the crayfish tubulin is a MAP rather than aprotein that associates non-specifically with the tubulin during polymerization. Thesimilarity of the crayfish MAP to MAP-2 of mammalian brain is suggested by theapparently similar molecular weights of the two proteins when they are co-electrophoresed and by the observation that the crayfish HMW-MAP is heat-stable,a property also characteristic of brain MAP-2 (Vallee, 1980).

12 R.H. Warren
The experiments carried out on the axons extracted with the PEG/PIPES andGSD microtubule-stabilization buffers have provided another means of identifyingaxonal microtubule-associated proteins and have also allowed a correlation to be madebetween the MAPS and the filaments that decorate the axonal microtubules. The keyto the latter is the observation that axons extracted in GSD medium display baremicrotubules in contrast to the decorated microtubules in axons extracted withPEG/PIPES medium. There may be several factors that contribute to the differencesin microtubule and filament organization seen with these two media. Since the extrac-tion times in the GSD medium are prolonged, the loss of filaments could arise fromproteolysis. The short extraction times and the presence of protease inhibitor mayprevent this with the PEG/PIPES protocol. Aside from possible effects of proteases,the results with the glycerol-based medium are consistent with previous observationsthat tubulin purified from brain homogenates in buffer with glycerol contains 5—10 %microtubule-associated protein (Cleveland, Hwo & Kirschner, 1977), as opposed to25 % MAPs in tubulin re-assembled in buffer without glycerol (Borisy et al. 1975).This phenomenon is not well understood, but may be related to the ability of glycerolto stabilize microtubules even in the total absence of MAPs.
The validity of the microtubular depolymerization-release method for the identi-fication of MAPs rests upon the assumption that the microtubules in the per-meabilized axons are sufficiently stabilized to survive tissue homogenization, pelletingand washing before they are intentionally depolymerized to release tubulin andMAPs. For axons extracted with the GSD medium, this criterion is easily met sincemicrotubules are stable indefinitely at room temperature in GSD medium(unpublished observations). Regarding the axons extracted with the PEG/PIPESmedium, previous study (Warren & Rubin, 1978) has shown that microtubules werestable in this medium for 3 h or more at room temperature. This allows sufficient timefor the nerves to be homogenized, pelleted, resuspended in the same medium, andpelleted one or two more times. Although these pellets have not been fixed andexamined in the electron microscope for the present study, they presumably containintact microtubules, since subsequent treatment with cold and calcium releases a largeamount of tubulin from the pellet in comparison to the previous washing steps.Similarly, the GSD-treated homogenates also release significant quantities of tubulinduring the depolymerization step, indicating that the microtubules are not irrevers-ibly stabilized by the GSD medium.
Of the proteins that are released from PEG/PIPES- and GSD-stabilized pelletsalong with tubulin during the microtubule-depolymerization step, it is necessary todetermine which are bonafide MAPs attached to the microtubules in the pellet andwhich are non-specifically solubilized from the pellet by the depolymerization buffer.The chief criterion is that the protein must be released in significantly greater quantityfrom the PEG/PIPES preparation than from the sample treated with GSD. Compari-sons of the gel patterns of the proteins released with tubulin from both PEG/PIPESand GSD pellets indicates that there are at least four such proteins, and any one or allof them could be associated with the microtubular filaments. There could, of course,be additional proteins that have not been detected. For example, permeabilization and

Crayfish microtubule-associated protein 13
extraction with PEG/PIPES medium could wash out some proteins that are function-ally associated with the axonal microtubules in vivo but only loosely bound. Alter-natively, either of the extraction media could render a MAP insoluble so that it wouldnot be released during the microtubule depolymerization step. None of thesepossibilities can be ruled out at present. Of the four proteins that do appear to bereleased uniquely from axonal pellets stabilized in PEG/PIPES medium, the HMWprotein is the most likely to be a constituent of the microtubular filaments since it alsoco-purifies with axonal tubulin prepared by the polymerization/depolymerizationmethod. Within the limits of the sensitivity of the staining methods used, none of theother three potential MAPs was detected in gels of the twice-cycled axonal tubulin.
Although MAP-2 was originally described in preparations from mammalian brain,very similar proteins have been found in other tissues (Sherline & Schiavone, 1978;Valdivia et al. 1982; Weatherbee et al. 1982), and it has been suggested (Cleveland,Spiegelman & Kirschner, 1979) that MAPs, like tubulin, are conserved proteins. Thepresent observations support that concept and suggest that a protein with molecularweight and heat stability similar to MAP-2 is a component of the filaments attachedto axonal microtubules in crayfish. The localization of brain MAP-2 in the filamentsattached to brain microtubules has been established definitively by experiments inwhich the addition of purified MAP-2 to bare microtubules restores the filaments tothe surfaces of the microtubules (Kim, Binder & Rosenbaum, 1979; Zingsheim,Herzog & Weber, 1979). Similar experiments have not been done with the crayfishsystem, however, since the HMW-MAP has not been purified. Regarding the functionof the crayfish axonal MAPs, it has been proposed (Fernandez et al. 1971) that themicrotubular filaments in the crayfish axons serve to maintain the microtubules in alattice with a preferred spacing between adjacent microtubules. This is supported byan observation in another study that axoplasm can be extruded from crayfish axonsinto PEG-PIPES medium without losing its integrity (Warren & Rubin, 1978) as longas the microtubular filaments remain intact, and by the observation in this study thatthe microtubular lattice collapses in GSD medium after removal of the filaments.
While the structural function of the filaments in maintaining the microtubularlattice seems well-defined, evidence for involvement of the filaments and HMW-MAPs in dynamic activities such as axonal transport remains elusive. Several featuresof arthropod nerve axons make them attractive for use in studies of axonal transport.These features include their accessibility and often large diameter, their lack of amyelin sheath, and the relative simplicity of the internal cytoskeleton that containsmicrotubules but no neurofilaments. Recently, these types of nerves have been ex-ploited as permeabilized models in which to reactivate vesicular translocations similarto those occurring during transport in intact axons (Forman, Brown & Livengood,1983; Adams & Bray, 1983). The success of this new approach to the analysis ofstructure and function in axons suggests that it will be possible before long to obtaininformation about the specific functions of the microtubule-associated proteins andto integrate this into a broader understanding of transport and microtubule-relatedintracellular movement in general.
This work was supported by a research grant from the National Parkinson Foundation.

14 R.H. Warren
REFERENCES
ADAMS, R. J. & BRAY, D. (1983). Particle movement in Arthropod axons. J . Cell Biol. 97, 4a.ALBERTINI, D. F., HERMAN, B. & SHERLINE, P. (1984). In vivo and in vitro studies on the role of
HMW-MAPs in Taxol-induced microtubular bundling. Eur.J. Cell Biol. 33, 134-143.BLOOM, G. S. & VALLEE, R. B. (1983). Association of microtubule-associated protein 2 (MAP
2) with microtubules and intermediate filaments in cultured brain cells. J. Cell Biol. 96,1523-1531.
BORJSV, G. G., MARCUM, J. M., OLMSTED, J. B., MURPHY, D. B. & JOHNSON, K. A. (1975).Preparation of tubulin and associated high molecular weight proteins from porcine brain andcharacterization of microtubule assembly in vitro. Ann. Nevi York Acad. Sci. 253, 107-132.
BURTON, P. R. & FERNANDEZ, H. L. (1973). Delineation by lanthanum of filamentous elementsassociated with the surfaces of axonal microtubules. J . Cell Sci. 12, 567-583.
BURTON, P. R. & HINKLEY, R. E. (1974). Further electron microscopic characterization of axo-plasmic microtubules of the ventral nerve cord of the crayfish. J. submicrosc. Cytol. 6, 311-326.
CANDE, W. Z., SNYDER, J., SMITH, D., SUMMERS, K. & MCINTOSH, J. R. (1974). A functional
mitotic spindle prepared from mammalian cells in culture. Proc. natn. Acad. Sci. U.SA. 71,1559-1563.
CLEVELAND, D. W., HWO, S.-Y. & KIRSCHNER, M. W. (1977). Microtubule associated proteinthat induces assembly of microtubules from purified tubulin. J. molec. Biol. 116, 207-226.
CLEVELAND, D. W., SPIEGELMAN, B. M. & KIRSCHNER, M. W. (1979). Conservation of
microtubule associated proteins. J. biol. Chem. 254, 12670-12678.CONNOLLY, J. A. & KALNINS, V. I. (1980). The distribution of tau and HMW microtubule
associated proteins in different cell types. Expl Cell Res. 127, 341-350.DENTLER, W. L., GRANETT, S. & ROSENBAUM, J. L. (1975). Ultrastructural localization of the
high molecular weight proteins associated with in wfn)-assembled brain microtubules. J. CellBiol. 65, 237-241.
FERNANDEZ, H. L., BURTON, P. R. & SAMSON, F. E. (1971). Axoplasmic transport in the crayfishnerve cord. J. Cell Biol. 51, 176-192.
FILNER, P. & BEHNKE, O. (1973). Stabilization and isolation of brain microtubules with glyceroland dimethylsulfoxide.J. Cell Biol. 59, 99a.
FORMAN, D. S., BROWN, K. J. & LIVENGOOD, D. R. (1983). Fast axonal transport in per-meabilized lobster giant axons is inhibited by vanadate. J. Neurosci. 3, 1279-1288.
GRIFFITH, L. M. & POLLARD, T. D. (1978). Evidence for actin filament-microtubule interactionmediated by microtubule-associated proteins. J . Cell Biol. 78, 958-965.
HEUSER, J. E., REESE, T. S., DENNIS, M. J., JAN, Y., JAN, L. & EVANS, L. (1979). Synapticvesicle exocytosis captured by quick freezing correlated with quantal transmitter release. J. CellBiol. 81, 275-300.
KIM, H., BINDER, L. I. & ROSENBAUM, J. L. (1979). The periodic association of MAP2 with brainmicrotubules in vitro. J. Cell Biol. 80, 266-276.
LAEMMLI, Y. K. (1970). Cleavage of structural proteins during the assembly of the head ofbacteriophage T4. Nature, Land. 227, 680-685.
MATSUDAIRI, P. T. & BURGESS, D. R. (1978). SDS microslab linear gradient polyacrylamide gelelectrophoresis. Analyt. Biochem. 87, 386-396.
MURPHY, D. B., VALLEE, R. B. & BORISY, G. G. (1977). Identity and polymerization-stimulationactivity of the nontubulin proteins associated with microtubules. Biochemistry 16, 2598-2605.
OCHS, R. L. & BURTON, P. R. (1980). Distribution and selective extraction of filamentous com-ponents associated with axonal microtubules of crayfish nerve cord. J. Ultrastruct. Res. 73,169-182.
OCHS, R. L., OCHS, D. L. & BURTON, P. R. (1980). Axons of crayfish nerve cord containintracellular hemocyanin.J. Cell Biol. 87, 73a.
O'FARRELL, P. H. (1975). High resolution two dimensional electrophoresis of proteins. J. biol.Chem. 250, 4007-4021.
PYTELA, R. & WICHE, G. (1980). High molecular weight polypeptides (270,000-340,000) fromcultured cells are related to hog brain microtubule-associated proteins but copurify with inter-mediate filaments. Proc. natn. Acad. Sci. U.SA. 77, 4808-4812.

Crayfish microtubule-associated protein 15
SATTILARO, R. F., DENTLER, W. L. & LECLUVSE, E. L. (1981). Microtubule-associated proteins(MAPs) and the organization of actin filamenents in vitro. J. Cell Biol. 90, 467-472.
SHELANSKI, M. L., GASKIN, F. & CANTOR, C. R. (1973). Microtubule assembly in the absenceof added nucleotides. Proc. natn. Acad. Set. U.SA. 70, 765-768.
SHERLINE, P. & SCHIAVONE, K. (1978). High molecular weight MAPs are part of the mitoticspindle. J . Cell Biol. 77, R9-R12.
VALDIVIA, M. M., AVILA, J., COLL, J., CALACO, C. & SANDOVAL, I. (1982). Quantitation andcharacterization of the microtubule-associated MAP2 in porcine tissues and its isolation fromporcine (PK 15) and human (HeLa) cell lines. Biochem. biophys. Res. Commun. 105, 1241-1249.
VALLEE, R. B. (1980). Structure and phosphorylation of microtubule-associated protein 2 (MAP2). Proc. natn. Acad. Sri. U.SA. 77, 3206-3210.
VAN HERREVELD, A. (1936). A physiological solution for freshwater Crustaceans. Proc. Soc. exp.Biol. Med. 36, 428-432.
WARREN, R. H. & RUBIN, R. W. (1978). Microtubules and actin in giant nerve fibers of the spinylobster, Panulirus argus. Tissue & Cell 10, 687-698.
WEATHERBEE, J. A., SHERLINE, P., MASCARDO, R. N., IZANT, J. G., LUFTIG, R. B. & WEIHUNG,R. R. (1982). Microtubule-associated proteins of HeLa cells: heat stability of the 200,000 mol wtHeLa MAPs and detection of the presence of MAP-2 in HeLa cell extracts and cycledmicrotubules. 7. Cell Biol. 92, 155-163.
WEINGARTEN, M. D., LOCKWOOD, A. H., Hwo, S.-Y. & KIRSCHNER, M. W. (1975). A proteinfactor essential for microtubule assembly. Proc. natn. Acad. Sci. U.SA. 72, 1858-1862.
WILSON, D. L., HALL, M. E., STONE, G. C. & RUBIN, R. W. (1977). Some improvements in two-dimensional gel electrophoresis of proteins: protein mapping of eukaryotic tissue extracts. Analyt.Biochem. 83, 33-44.
ZINGSHEIM, H. P., HERZOG, W. & WEBER, K. (1979). Differences in surface morphology ofmicrotubules reconstituted from pure brain tubulin using two different microtubule-associatedproteins: high molecular weight MAP-2 proteins and tau proteins. Eur.jf. Cell Biol. 19, 175-183.
(Received 6 March 1984-Accepted 18 April 1984)



















