Author’s Accepted Manuscriptgenetics.wustl.edu/sjlab/files/2009/04/Schill_ibuprofen_ENS.pdf ·...
53
Author’s Accepted Manuscript Ibuprofen slows migration and inhibits bowel colonization by enteric nervous system precursors in zebrafish, chick and mouse Ellen Merrick Schill, Jonathan I. Lake, Olga A. Tusheva, Nandor Nagy, Saya K. Bery, Lynne Foster, Marina Avetisyan, Stephen L. Johnson, William F. Stenson, Allan M. Goldstein, Robert O. Heuckeroth PII: S0012-1606(15)30087-7 DOI: http://dx.doi.org/10.1016/j.ydbio.2015.09.023 Reference: YDBIO6922 To appear in: Developmental Biology Received date: 28 July 2015 Revised date: 31 August 2015 Accepted date: 7 September 2015 Cite this article as: Ellen Merrick Schill, Jonathan I. Lake, Olga A. Tusheva Nandor Nagy, Saya K. Bery, Lynne Foster, Marina Avetisyan, Stephen L. Johnson, William F. Stenson, Allan M. Goldstein and Robert O. Heuckeroth, Ibuprofen slows migration and inhibits bowel colonization by enteric nervou system precursors in zebrafish, chick and mouse, Developmental Biology http://dx.doi.org/10.1016/j.ydbio.2015.09.023 This is a PDF file of an unedited manuscript that has been accepted fo publication. As a service to our customers we are providing this early version o the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain www.elsevier.com/locate/developmentalbiology
-
Upload
nguyenthuy -
Category
Documents
-
view
227 -
download
0
Transcript of Author’s Accepted Manuscriptgenetics.wustl.edu/sjlab/files/2009/04/Schill_ibuprofen_ENS.pdf ·...

Author’s Accepted Manuscript
Ibuprofen slows migration and inhibits bowelcolonization by enteric nervous system precursorsin zebrafish, chick and mouse
Ellen Merrick Schill, Jonathan I. Lake, Olga A.Tusheva, Nandor Nagy, Saya K. Bery, LynneFoster, Marina Avetisyan, Stephen L. Johnson,William F. Stenson, Allan M. Goldstein, Robert O.Heuckeroth
PII: S0012-1606(15)30087-7DOI: http://dx.doi.org/10.1016/j.ydbio.2015.09.023Reference: YDBIO6922
To appear in: Developmental Biology
Received date: 28 July 2015Revised date: 31 August 2015Accepted date: 7 September 2015
Cite this article as: Ellen Merrick Schill, Jonathan I. Lake, Olga A. Tusheva,Nandor Nagy, Saya K. Bery, Lynne Foster, Marina Avetisyan, Stephen L.Johnson, William F. Stenson, Allan M. Goldstein and Robert O. Heuckeroth,Ibuprofen slows migration and inhibits bowel colonization by enteric nervoussystem precursors in zebrafish, chick and mouse, Developmental Biology,http://dx.doi.org/10.1016/j.ydbio.2015.09.023
This is a PDF file of an unedited manuscript that has been accepted forpublication. As a service to our customers we are providing this early version ofthe manuscript. The manuscript will undergo copyediting, typesetting, andreview of the resulting galley proof before it is published in its final citable form.Please note that during the production process errors may be discovered whichcould affect the content, and all legal disclaimers that apply to the journal pertain.
www.elsevier.com/locate/developmentalbiology

1
Title: Ibuprofen slows migration and inhibits bowel colonization by enteric nervous system
precursors in zebrafish, chick and mouse
Authors: 1Ellen Merrick Schill,
1Jonathan I. Lake,
1Olga A. Tusheva,
2,3Nandor Nagy,
4Saya K.
Bery, 5Lynne Foster,
1Marina Avetisyan,
6Stephen L. Johnson,
5William F. Stenson,
2Allan M.
Goldstein and 4Robert O. Heuckeroth
Affiliations: 1Department of Pediatrics,
5Department of Internal Medicine, and
6Department of
Genetics, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO
63110, U.S.A., 2Department of Pediatric Surgery, Massachusetts General Hospital, Harvard
Medical School, 55 Fruit St., Boston, MA 02114, U.S.A., 3Department of Human Morphology
and Developmental Biology, Faculty of Medicine, Semmelweis University, Budapest, Hungary,
4Department of Pediatrics, The Children’s Hospital of Philadelphia Research Institute and the
Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center,
3615 Civic Center Blvd, Philadelphia, PA 19104
Corresponding Author
Robert O. Heuckeroth M.D. Ph.D.
The Children’s Hospital of Philadelphia Research Institute
Perelman School of Medicine at the University of Pennsylvania
Abramson Research Center — Suite 1116I
3615 Civic Center Blvd.
Philadelphia, Pennsylvania 19104-4318, USA.
Phone: 215-590-1209; Fax: 215-590-3324; E-mail: [email protected].

2
Conflict of Interest: The authors have declared that no conflict of interest exists.
Key words: Enteric nervous system development, gene-environment interactions, migration
Abstract
Hirschsprung Disease (HSCR) is a potentially deadly birth defect characterized by the absence of
the enteric nervous system (ENS) in distal bowel. Although HSCR has clear genetic causes, no
HSCR-associated mutation is 100% penetrant, suggesting gene-gene and gene-environment
interactions determine HSCR occurrence. To test the hypothesis that certain medicines might
alter HSCR risk we treated zebrafish with medications commonly used during early human
pregnancy and discovered that ibuprofen caused HSCR-like absence of enteric neurons in distal
bowel. Using fetal CF-1 mouse gut slice cultures, we found that ibuprofen treated enteric neural
crest-derived cells (ENCDC) had reduced migration, fewer lamellipodia and lower levels of
active RAC1/CDC42. Additionally, inhibiting ROCK, a RHOA effector and known RAC1
antagonist, reversed ibuprofen effects on migrating mouse ENCDC in culture. Ibuprofen also
inhibited colonization of Ret+/- mouse bowel by ENCDC in vivo and dramatically reduced
bowel colonization by chick ENCDC in culture. Interestingly, ibuprofen did not affect ENCDC
migration until after at least three hours of exposure. Furthermore, mice deficient in Ptgs1 (COX
1) and Ptgs2 (COX 2) had normal bowel colonization by ENCDC and normal ENCDC migration
in vitro suggesting COX-independent effects. Consistent with selective and strain specific effects
on ENCDC, ibuprofen did not affect migration of gut mesenchymal cells, NIH3T3, or WT
C57BL/6 ENCDC, and did not affect dorsal root ganglion cell precursor migration in zebrafish.

3
Thus, ibuprofen inhibits ENCDC migration in vitro and bowel colonization by ENCDC in vivo
in zebrafish, mouse and chick, but there are cell type and strain specific responses. These data
raise concern that ibuprofen may increase Hirschsprung disease risk in some genetically
susceptible children.
Introduction
The enteric nervous system (ENS) is an intrinsic neuronal and glial network in the bowel
that controls intestinal function (Furness, 2012; Sasselli et al., 2012). Most ENCDC that give rise
to the ENS begin to delaminate from vagal neural tube by E8.5 in mice and prior to week four in
humans (Fu et al., 2004; Kapur et al., 1992). Sacral ENCDC also contribute to distal bowel ENS
(Kapur, 2000; Wang et al., 2011). Vagal ENCDC migrate to the foregut and then begin
coordinated proliferation, rostro-caudal migration, and differentiation, a process that normally
results in interconnected neurons and glia throughout the bowel by E13.5 in mice and by week 7
in humans. This migratory route is one of the longest traversed by any cell during development.
In one in 5000 human infants, ENCDC do not colonize the bowel completely, causing
Hirschsprung disease (HSCR), a condition defined by the absence of distal bowel enteric neurons
(Heuckeroth, 2013; Hirschsprung, 1888; McKeown et al., 2013; Skinner, 1996).
HSCR is potentially fatal because aganglionic bowel (i.e., bowel without neurons)
tonically contracts, causing constipation, bilious vomiting, abdominal distension, enterocolitis,
and sepsis (Dasgupta and Langer, 2004; Heuckeroth, 2013; Skinner, 1996). Most children with
HSCR (80%) have only a short region of aganglionic bowel (Amiel et al., 2008; Suita et al.,
2005) and even modestly increased ENCDC bowel colonization (e.g. 5-10% increase) might
have prevented HSCR. For children with extensive aganglionosis, having only slightly greater

4
bowel colonization by ENCDC could prevent the need for intravenous nutrition and associated
life-threatening infections. Therefore, it is valuable to identify non-genetic potentially avoidable
factors that influence ENCDC migration.
Because ENS development depends on many signaling pathways (Anderson et al., 2006;
Goldstein et al., 2013; Lake and Heuckeroth, 2013; Laranjeira and Pachnis, 2009), many
medicines may increase HSCR risk. One critical pathway includes RET receptor tyrosine kinase
(Pachnis et al., 1993; Schuchardt et al., 1994), the ligand glial cell line-derived neurotrophic
factor (GDNF), the co-receptor GFRα1 (Airaksinen and Saarma, 2002) and the small
RhoGTPases RAC1 and CDC42 that induce actin polymerization and reorganization at the
leading edge of migrating cells (de Curtis, 2008; Fukata et al., 2003; Goto et al., 2013; Ridley,
2006; Stewart et al., 2007; Vohra et al., 2007a). Among other roles, RAC1 induces lamellipodia,
promotes ENCDC migration in vitro and ex vivo and inhibits RHOA, a small GTPase (Stewart et
al., 2007; Wu et al., 2009). RHOA also inhibits RAC1 via its effector ROCK (Guilluy et al.,
2011; Nakayama et al., 2008). The complex signaling pathways needed for normal development
increase the vulnerability of ENS precursors to medicines that impact these and many other
signaling pathways.
By testing medicines commonly used during early human pregnancy, we discovered that
ibuprofen dramatically reduced bowel colonization by ENCDC in zebrafish and chick.
Furthermore, ibuprofen delayed Ret+/- mouse bowel colonization by ENCDC in vivo, but effects
were not observed in WT mice. Consistent with these observations, ibuprofen reduced migration
speed on 2-dimensional surfaces, reduced lamellipodia and reduced filamentous actin in murine
ENCDC. These changes correlated with reduced active RAC1/CDC42. Furthermore,
ibuprofen’s effects on migration could be prevented using a ROCK inhibitor. In contrast,

5
ibuprofen did not slow migration or reduce lamellipodia in mouse gut mesenchymal or NIH3T3
cells. Surprisingly, mice with mutations in Ptgs (i.e., cyclooxygenase, COX), ibuprofen’s main
therapeutic target, had normal ENCDC bowel colonization and normal migration in vitro.
Additionally, live imaging experiments demonstrated that ENCDC required at least three hours
of exposure to ibuprofen before ENCDC migration speed was impacted, a much slower time
course than would be expected for a solely COX dependent effect. Collectively these studies
suggest ibuprofen use during early pregnancy could increase HSCR risk in some genetically
susceptible children. This may be important since 23.5 % of women in the United State take
ibuprofen during early pregnancy (Thorpe et al., 2013).
Materials and Methods
Zebrafish
Wild type AB zebrafish, fertilized in vitro, were exposed to drugs from 34 to 96 hours
post fertilization (hpf) in E3 media with 1% DMSO (Murphey and Zon, 2006). At 96 hpf,
zebrafish were stained with HuC/D antibody to visualize neurons. Uncolonized gut was
measured from most distal HuC/D+ cell to end of bowel. Drugs (from Sigma, St. Louis) tested,
number of fish evaluated and drug concentrations are in Table 1.
Avian intestine organ culture
Fertilized White Leghorn chicken eggs from commercial breeders were maintained in a
37°C humidified incubator. E6 midgut plus hindgut from umbilicus to cloaca was pinned to
Sylgard, cultured in DMEM, 100 U/mL penicillin-G, 0.1 mg/mL streptomycin (Sigma), DMSO

6
(5 µM), +/- ibuprofen (250 µM) for 48 hours and then processed for immunocytochemistry. 5-
ethynyl-2’-deoxyuridine (EdU) (10 µM, Invitrogen) was added 3 hours before fixation.
Mice
Mice used include WT CF-1 (Charles River Laboratories), WT C57BL/6J (The Jackson
Laboratory), Ptgs1tm1Unc
(called Ptgs1 or Cox1, RRID:MGI_4366280), Ptgs2tm1Unc
(called Ptgs2
or Cox2, RRID:MGI_4366244) (C57BL/6J background) (Langenbach et al., 1995; Morham et
al., 1995), Sox10tm1Weg
(called Sox10+/- or Sox10 LacZ, RRID:MGI_4437131) (C3H
background) (Britsch et al., 2001) (from M. Wegner (Friedrich Alexander University Erlangen
Nuremberg, Erlangen, Germany) and M. Southard-Smith (Vanderbilt University, Nashville,
Tennessee, USA)), and Rettm1Jmi
(Ret (tau-EGFP-myc), Ret-TGM, RRID:MGI_3623107), called
Ret+/-, C57/B6J background)(Enomoto et al., 2001). The day of the vaginal plug was called
E0.5. Mice were genotyped by PCR as described in the references above.
Midgut Slice Explant Culture
E12.5 small bowel (midgut) was cut into 300-500 micron slices. These slices were
cultured on fibronectin-coated (250 µg/mL; Life Technologies) plastic or glass Lab-Tek
Permanox chamber slides (ThermoFisher) in OptiMem (Life Technologies), 2 mM L-glutamine
(Life Technologies), 100 IU/mL penicillin, 100 µg/mL streptomycin (Life Technologies).
Ibuprofen stock (1 g/L) was prepared fresh in media daily. Unless noted, ibuprofen was added
when slices were plated. Four hours after plating, GDNF (100 ng/mL final concentration) was
added to cultures, which were then maintained for an additional 16 hours (37oC, 5% CO2) after
GDNF addition. When needed, 10 µM bromodeoxyuridine (BrdU) was added four hours before

7
fixation. Y-27632 (5 µM, Sigma, St. Louis MO) in DMSO was added four hours after plating
slices. 16, 16-dimethyl prostaglandin E2 and 16, 16-dimethyl prostaglandin F2 (Cayman
Chemicals, Ann Arbor, MI) dissolved in ethanol were added (final concentration 1 µM) at the
same time as GDNF (for 16, 16-dimethyl prostaglandin E2) or at plating (for 16, 16-dimethyl
prostaglandin F2). Control cultures received equivalent diluent volumes.
Immunoselected ENCDC Culture
E12.5 CF-1 small and large bowel were treated with collagenase (0.2 mg/mL) and
dispase (0.2 mg/mL), triturated, resuspended in media (Neurobasal® (Life Technologies), B27®
(50x, Life Technologies), 2 mM L-glutamine, 100 IU/mL penicillin, 100 µg/mL streptomycin),
and then exposed to rabbit anti-p75NTR (anti-nerve growth factor receptor, P75, EMD
Millipore, 1:1000, one hour, 4oC), followed by goat anti-rabbit coupled paramagnetic beads
(anti-rabbit IgG MicroBeads1:50, Miltenyi Biotec, GmbH) (30 min, 4oC). ENCDC (p75NTR
positive) were isolated with MACS columns (Miltenyi Biotec, GmbH), before culturing on poly-
d-lysine (100 µg/mL, Sigma, St. Louis) and laminin (20 µg/mL, BD Biosciences) coated glass
chamber slides (Sato and Heuckeroth, 2008). Cells were treated with GDNF (50 ng/mL) with or
without ibuprofen (250 µM) at plating and then grown 48 hours before fixation.
In Vivo Mouse Ibuprofen Treatment
Pregnant mice were fed Lab Diet 5053 (Test Diet, Richmond, IN) with or without 375
parts per million ibuprofen from E8.5 to until analysis.

8
Embryo Sections
10 µm sagittal E12.5 CF-1 mouse sections were prepared from immersion fixed (4%
paraformaldehyde overnight, rotating, 4oC), sucrose treated (30% overnight), frozen (Optimal
Cutting Temperature Compound, Tissue-Tek) mice.
Immunohistochemistry
Fixed zebrafish (2% paraformaldehyde, two hours, 25oC) were washed in PBS, stained
with mouse anti-HuC/HuD (16A11) antibody (overnight, 4oC) and examined after Alexa Fluor®
anti-mouse 594 secondary (1:250, Life Technologies) (two hours, 25oC) treatment.
Mouse cells were fixed (4% paraformaldehyde, 25oC) for 20 minutes (slice and
dissociated culture) or 30 minutes (whole bowel)), washed (PBS or Tris-buffered saline plus
0.1% Triton-X (TBST)), and blocked (4% normal donkey serum/TBST, 1 hour, 25oC or
overnight 4oC) before primary antibodies (Table 2) were applied in TBST (4
oC, overnight). TuJ1
and Phalloidin were applied only two hours at 25oC. Samples were washed in PBS or TBST
before incubating with secondary antibodies (Alexa Fluor® Donkey anti-Rabbit, Donkey anti-
goat, Donkey anti-mouse 488, 594, or 647nm, Invitrogen, 1:400) and DAPI (100 ng/mL, 4’,6–
diamidine–2–phenylidole–dihydrochloride, Vector Labs) in TBST (1 hour, 25oC) and then
washing again in PBS or TBST before mounting (50% glycerol).
Avian bowel was fixed (4% paraformaldehyde in PBS, 1 hour), rinsed (PBS), incubated
in 15% sucrose/PBS (overnight, 4°C), then in 7.5% gelatin,15% sucrose/PBS (37°C, 1 hour),
before freezing (−50°C, 2-Methylbutane, Sigma). 10 µm frozen sections on Probe On Plus slides

9
(Fisher Scientific) were stained with ENCDC specific anti-chicken N-cadherin antibody (Nagy et
al., 2012) Nuclei were DAPI stained. EdU was detected using Clik-iT EdU Imaging Kit
(Invitrogen).
Active RAC1/CDC42 and RHOA Measurements
NIH3T3 (ATCC) for scratch test were grown on glass 8-well chamber slides to 100%
confluence in DMEM (high glucose), 10% fetal calf serum (FCS), 100 IU/mL penicillin, and 100
µg/mL streptomycin. 250 µM ibuprofen was added two hours before scratching to remove some
cells using a 10 µL pipette tip. Media was changed after the scratch. For RAC1/CDC42 and
RHOA studies, NIH3T3 were starved (18 hours in 1 % FCS, then 24 hours in 0 % FCS), then
stimulated with 100 ng/mL RAC1/CDC42 Activator II (3 min, Cytoskeleton, Denver) or 100
µg/mL Rho Activator I (30 min, Cytoskeleton, Denver). During stimulation, RAC1/CDC42
control cells were kept in 0% FCS media while RHOA control cells were in 1 % FCS media.
After culture ENCDC and NIH3T3 were fixed (4% paraformaldehyde, 20 min, 25oC),
permeabilized in TBST (55oC, one hour) and incubated with 20 µg GST-tagged PAK-PBD
protein (PBD-GST) (Cytoskeleton, Denver) or 100 µg GST-tagged Rhotekin-RBD protein
(RBD-GST) (Cytoskeleton, Denver) (1 hour, 4oC) in 100 µl TBST. Cells were PBS washed,
post-fixed (4% paraformaldehyde, 15 min, 25oC), and washed again (PBS) before anti-GST
immunohistochemistry.
Microscopy and Analysis
Micrographs were obtained with 1) Olympus BX60 or IX71, Axiocam CCD, Axiovision
software, 2) Olympus FV1000 confocal with Fluoview software, 3) Zeiss Axio Imager.A2,

10
AxioCam MRm Rev.3, ZEN software, 4) Zeiss LSM 710 confocal, Zen software, 5) Nikon
Eclipse 80i, Spot camera, software version 3.3.1 (Diagnostic Instruments). FIJI (NIH ImageJ)
and Photoshop CS6 (Adobe) were used to process images (Schindelin et al., 2012) including
only cropping, stitching (Preibisch et al., 2009), rotating, centering, and uniform adjustments of
brightness, contrast and saturation. Confocal images show flattened Z–stacks. Time-lapse
imaging used an AxioObserver.Z1 microscope (Zeiss) with motorized stage and incubator with
temperature and CO2 controls. Live-imaging was at 37oC and 5% CO2.
ENCDC colonization of mouse bowel was measured from cecal tip to most distal TuJ1+
cell or neurite. For midgut slices, migration was measured in octants from slice edges to the
most distal ENCDC or mesenchymal cell (Supplemental Figure 1). Octants adjacent to other
slices were not evaluated. ENCDC were RET+ by immunohistochemistry. Mesenchymal cells
were identified by absent RET immunostaining and an actin-rich cytoskeleton after Alexa488-
phalloidin staining. Each slice was considered a single replicate.
Lamellipodia were defined as regions of obvious F-actin ruffling wider than the cell body
and were analyzed in phalloidin-stained ENCDC furthest from gut slices (Supplemental Figure
1). The longest neurite in RET+ cells were measured using FIJI (NIH ImageJ). Cell speed was
measured in time-lapse images using MTrackJ manually (Meijering et al., 2012).
Immunofluorescent pixel intensity was measured using FIJI on images taken at fixed exposure
times. Only RET+ cells that migrated furthest from slices were analyzed for fluorescence
intensity. All samples for which fluorescence intensity was compared were stained on the same
chamber slide.

11
Statistical Analysis
SigmaPlot 11 (Systat Software), student’s t-test or one-way ANOVA was used for
comparisons. All studies include at least three biological replicates. Non-parametric data were
analyzed by ranks. Post-hoc Holm-Sidak or Dunn’s tests were used for multiple comparisons.
Data are plotted as mean +/- SEM unless noted. P<0.05 was considered significant.
Approvals
All studies were approved by Animal Studies Committee at Washington University
School of Medicine, Institutional Animal Care and Use Committee at The Children’s Hospital of
Philadelphia, and by Massachusetts General Hospital’s Institutional Subcommittee on Research
Animal Care.
Results
Ibuprofen inhibited enteric nervous system development in zebrafish
Zebrafish embryos were exposed to selected medicines that are used by > 0.5 % of
women during early pregnancy (Thorpe et al., 2013) (Table 1). Fish were treated during the
period that ENCDC migrate through bowel (34 to 96 hours post fertilization (hpf)) and then
stained with Elavl3/4 (HuC/D) antibody to show enteric neurons (Kuhlman and Eisen, 2007;
Lake et al., 2013). Ibuprofen and acetylsalicylic acid specifically reduced bowel colonization by
ENCDC at concentrations found in human blood with therapeutic dosing (Table 1). We focused
on ibuprofen because it inhibited bowel colonization by ENCDC in a dose dependent manner
(Figure 1 A-E) and because acetylsalicylic acid caused death at 660 µM (i.e., twice the dose that
caused ENS defects) suggesting more generalized toxicity. In contrast, at an ibuprofen dose that

12
reduced bowel colonization by ENCDC (25 µM), fish looked grossly normal (Figure 1A - D).
Body length was slightly reduced (Figure 1A, C, F), but dorsal root ganglia (DRG), another
neural crest-derivative, were in a normal position after ibuprofen treatment (Figure 1A, C, G)..
Furthermore fish treated with 25 µM ibuprofen had a normal number of DRG neurons per
ganglion (DMSO 2.2 +/- 0.09, ibuprofen 2.1 +/- 0.1, p = 0.3, 2 tailed t-test). These data suggest
that ibuprofen inhibits zebrafish bowel colonization by ENCDC at concentrations that do not
globally disrupt development or block migration of other neural crest derivatives.
Ibuprofen inhibits ENCDC colonization of chick bowel in organ culture
To determine if ibuprofen affects ENS development in other species we cultured E6
chick hindgut for 48 hours with or without ibuprofen (250µM) and then stained with N-cadherin
antibody (Figure 2A-B). This ibuprofen concentration was selected based on work in mouse
culture (Figure 3) where higher ibuprofen doses are needed to affect ENCDC migration. This
ibuprofen level is comparable to human serum levels after consuming three over-the-counter
ibuprofen tablets (i.e., 600 mg, a commonly used dose) (Ritschel and Kerns, 2009). Remarkably,
ibuprofen-treated hindgut was almost devoid of ENCDC, whereas control bowel was almost
fully colonized by ENCDC (n=6 per treatment group). Proliferation of ENCDC was also
reduced by ibuprofen based on EdU incorporation into N-Cadherin expressing cells at the
leading edge of the migration wavefront (N-Cadherin+ EdU+/N-Cadherin x 100: Control 58 +/-
0.06%, ibuprofen 38 +/- 0.05%, P = 0.03, n = 6/group, t-test) as was proliferation in gut
mesenchymal cells (DAPI+ N-Cadherin- EdU+/DAPI+ N-Cadherin- x 100: Controls 20 +/-
1.5%, ibuprofen 13 +/- 0.9%, P = 0.03, n = 6/group, t-test). Thus in chick and in fish, ibuprofen

13
dramatically reduced colon colonization by ENCDC. In chick, this was accompanied by reduced
proliferation of ENCDC and surrounding mesenchymal cells.
Ibuprofen slows ENCDC colonization of mouse bowel in vivo
To determine if ibuprofen affects mammalian ENCDC development, we fed ibuprofen to
two mouse lines with genetic defects that predispose to HSCR-like disease. Pregnant mice with
Ret or Sox10 mutations received ibuprofen containing or control chow from E8.5 to E12.5. We
selected these dates because E8.5 is one day before ENCDC enter fetal bowel and by E12.5
ENCDC have normally migrated to mid-colon. We then stained the bowel with an antibody to
neuron specific β3 tubulin (TuJ1) (Figure 2C-F). Ibuprofen-exposed Ret+/- mice had a small,
but significant reduction in bowel colonization by ENCDC (% colon colonization: untreated
Ret +/− 73.6 ± 3.9%, n=4; ibuprofen treated Ret+/- 65.2 ± 5.2% n=9, P<0.042, t-test). In
contrast, colon colonization by ENCDC was normal in ibuprofen treated E12.5 Sox10+/- mice
and in E12.5 WT littermates of Sox10+/- and Ret+/- animals (% colon colonization: untreated
Sox10+/- 59.4 ± 2.4% n=19; ibuprofen Sox10+/- 60.3 ± 2.3% n=13, p=0.79, t-test and data not
shown) (Figure 2F). These data show that in Ret+/- mice, ibuprofen can delay ENCDC
colonization of fetal bowel in vivo, although the effect is more subtle than in chick and fish.
Ibuprofen specifically inhibited murine ENCDC migration in vitro
To gain insight into why ibuprofen slowed bowel colonization by ENCDC, we cultured
E12.5 CF-1 mouse gut slices on fibronectin coated dishes in the presence or absence of ibuprofen
(Fu et al., 2013; Fu et al., 2010; Lake et al., 2013; Wang et al., 2010). Addition of GDNF to the
culture media enhanced ENCDC migration onto the dishes permitting drug effects on ENCDC

14
migration, proliferation, cell death and differentiation to be evaluated. After 16 hours in culture,
ENCDC were identified by RET immunoreactivity. RET is expressed by all ENS precursors and
differentiated enteric neurons in vitro (Heuckeroth et al., 1998; Pachnis et al., 1993). ENCDC
migration was assessed by measuring the distance between gut slice edges and the most distant
ENCDC (Supplemental Figure 1). 250 µM ibuprofen reduced the distance ENCDC migrated
from gut slices, but migration distance was normal at lower ibuprofen concentrations (50 µM,
100 µM) (Figure 3A-B, I).
Since ENCDC migration might be affected by the number of migrating ENCDC and by
differentiation (Lake et al., 2013), we determined if ibuprofen altered ENCDC proliferation,
apoptosis, or differentiation. Double label immunohistochemistry for RET (expressed in
ENCDC and enteric neurons) plus BrdU, RET plus activated caspase-3, and RET plus TuJ1
(neuron specific β3 tubulin antibody that identifies early neurons) demonstrated ibuprofen had
no effect on murine ENCDC proliferation, apoptosis, or differentiation (Figure 3C-H and 3J-L).
These data suggest ibuprofen specifically affects murine ENCDC migration without altering
survival, proliferation or differentiation.
NIH 3T3 and gut mesenchymal cell migration was not affected by ibuprofen
To determine if ibuprofen affects migration of other cells, we performed “scratch test
migration assays” on confluent NIH3T3 fibroblasts. After two hours of ibuprofen treatment, we
created a wound to induce migration (Figure 4 A-D) and imaged for 24 hours. Ibuprofen did not
slow NIH3T3 migration (P = 0.75) (Figure 4 A-E). To determine if ibuprofen effects require
specialized media or factors produced by bowel, we examined CF-1 mesenchymal cells that
migrated from the gut along with ENCDC (Supplemental Figure 1). Like NIH3T3, gut

15
mesenchymal cell migration was not affected by ibuprofen (Figure 4G). These data suggest that
ENCDC migration is especially sensitive to ibuprofen, compared to other cell types.
Ibuprofen-exposed ENCDC have reduced lamellipodia and filamentous actin
To assess if ibuprofen affected ENCDC morphology, we cultured CF-1 mouse fetal
bowel slices with or without ibuprofen for 16 hours and stained with phalloidin. Fewer
ibuprofen treated migrating ENCDC had lamellipodia than control ENCDC at all doses tested
(Figure 5A-F, I). In contrast, ibuprofen did not reduce well-formed lamellipodia in NIH 3T3
(Figure 4F) nor in gut mesenchymal cells (Figure 4H). This finding may underlie ibuprofen
effects on ENCDC migration because lamellipodia are thin membranous actin projections that
facilitate cell migration (Hall, 2005).
Lamellipodia formation is driven by actin polymerization and cross-linking. To assess
filamentous actin (F-actin), we quantified fluorescence intensity of Alexa488-phalloidin stained
ENCDC. Ibuprofen treated ENCDC had reduced Alexa488 staining suggesting that ibuprofen
affects ENCDC actin dynamics (Figure 5A-F, J, K). As actin polymerization is also important
for neurite growth and we assessed neurite length and found that ibuprofen reduced neurite
length by 36 % when dissociated ENCDC differentiate in vitro (Figure 5G-H, L). This contrasts
with studies showing ibuprofen enhanced neurite growth in corticospinal neurons, dorsal root
ganglion cells (Fu et al., 2007) and raphespinal neurons (Wang et al., 2009) via PPARγ and
reduced RHOA/ROCK signaling (Dill et al., 2010). Developing enteric neurons, however, have
much less PPARγ than dorsal root ganglion neurons (Figure 6) providing a possible explanation
for different ibuprofen effects. Collectively these data suggest ibuprofen slows ENCDC

16
migration and reduces neurite length in ENCDC-derived neurons by altering actin cytoskeletal
dynamics.
Ibuprofen slows ENCDC migration but effects are delayed
To gain insight into the mechanism by which ibuprofen slows ENCDC migration, we
performed time-lapse microscopy on gut slice cultures with ibuprofen added, at the initiation of
imaging (Figure 7A-D, Supplemental Movie 1-2). Analysis of sequential images showed that
ENCDC persistence (net distance migrated / total distance migrated) was unaffected by
ibuprofen (data not shown), but ENCDC migration speed was reduced. Interestingly, the effect
on migration speed was only apparent after 3 hours of ibuprofen (i.e., during “imaging interval
4”, Figure 7A, E, Supplemental Movies 1-2). This is much longer than would be expected if
ibuprofen effects were mediated by COX inhibition, the primary therapeutic effect of ibuprofen
(Peppelenbosch et al., 1993; Vane, 1971). One possible explanation for delayed ibuprofen
responsiveness of ENCDC is that prolonged contact with tissue culture surfaces changes
ENCDC biology so that these cells become ibuprofen sensitive. To test this hypothesis, we
added ibuprofen just as gut slices were placed in culture (i.e. four hours before initiation of
imaging (Supplemental Movie 3)) and observed that ibuprofen-mediated effects on ENCDC
migration were already apparent when imaging began. This suggests that the delayed response of
ENCDC to ibuprofen was not dependent on a change in ENCDC phenotype after migration onto
the culture dish, but instead is consistent with the hypothesis that ibuprofen effects on ENCDC
migration were COX-independent.

17
Ptgs1 -/- Ptgs2 -/- mice have normal appearing ENCDC migration in vitro and in vivo
To directly test if reduced COX activity affected ENCDC, we cultured mid-small bowel
slices from E12.5 Ptgs1-/- Ptgs2-/- (i.e., Cox1-/- Cox2-/-) mice on fibronectin and found no
difference in ENCDC migration or lamellipodia compared to WT (Figure 8A-B, E-F). We also
stained WT and Ptgs1 -/- Ptgs2 -/- E12.5 bowel with TuJ1antibody and found equivalent colon
colonization by ENCDC (Figure 8C-D, G). Finally, we tried to rescue ibuprofen effects on CF-1
ENCDC using stable analogs of two prostaglandins (PGE2 and PGF2) reported to facilitate cell
migration (16,16-dimethyl PGE2 or 16, 16-dimethyl PGF2 respectively), but did not observe any
effect on migration or lamellipodia (Figure 8H-K). These data also suggest that ibuprofen
effects on ENCDC migration may be COX-independent.
One caveat is that Ptgs mutant animals were on a C57BL/6 genetic background. We
therefore cultured E12.5 small bowel slices from C57BL/6 mice on fibronectin in media with
GDNF +/- ibuprofen. Ibuprofen did not reduce ENCDC migration from WT C57BL/6 gut
(Distance of most distant ENCDC from gut slice: WT 335 +/- 7 µm; 250 µM ibuprofen 343 +/- 7
µm, 4 biological replicates, 128 control slices, 161 ibuprofen slices, P = 0.39, t-test), however,
when this experiment was performed using C57BL/6 Ret+/- gut explants, ENCDC migration
was reduced by ibuprofen (untreated: 349 +/- 7 µm, 250 µM ibuprofen 324 +/- 7 µm, 4
biological replicates 125 control slices, 147 ibuprofen slices, P = 0.015, t-test), consistent with
our in vivo results. These data suggest that the C57BL/6 genetic background makes ENCDC
more resistant to ibuprofen effects on migration compared to CF-1.

18
Ibuprofen reduces RAC1 activity in migrating murine ENCDC
Reduced F-actin, lamellipodia, and neurite growth in ibuprofen treated ENCDC
suggested reduced RAC1 or increased RHOA activity (Bryan et al., 2005; Hall, 2005; Koh,
2006; Sato and Heuckeroth, 2008; Wang et al., 2003). We attempted to measure active RAC1
and RHOA in migrating ENCDC using sensitive G-LISA kits (Ferri et al., 2014; Nini and
Dagnino, 2010), but could not obtain reproducible results. Furthermore, we wanted to measure
RAC1 and RHOA activity only in cells that migrated furthest from cultured gut slices (i.e., distal
ENCDC, Supplemental Figure 1) because these cells were examined for all other studies.
Therefore we used a validated “in situ” method to analyze active RAC1/CDC42 or active
RHOA based on binding to p21 PAK binding domain (PBD) or rhotekin binding domain (RBD)
respectively, fused to glutathione-S-transferase (GST) (Supplemental Figure 2) (Li et al., 2002;
Lindsley et al., 2011). Gut slices were cultured with or without ibuprofen, fixed and incubated
with PBD-GST or RBD-GST followed by GST immunohistochemistry. Analysis of mean
fluorescence intensity in ENCDC that migrated furthest from the gut edge demonstrated less
PDB-GST immunoreactive protein in ibuprofen-treated ENCDC, suggesting less active
RAC1/CDC42 than in control ENCDC (Figure 9A-H, Supplemental Figure 2A, C). In contrast,
active RHOA levels were similar in ibuprofen-treated and control ENCDC based on RBD-GST
binding (Figure 9I-P, Supplemental Figure 2 B, D). Collectively these data suggest ibuprofen
hinders ENCDC migration by reducing active RAC1 without affecting RHOA activity.

19
ROCK inhibition rescues ibuprofen effects on ENCDC migration in vitro
RAC1 and RHOA are regulated in complex ways as they control actin cytoskeleton to
alter cell morphology (Guilluy et al., 2011; Hall, 2005; Parri and Chiarugi, 2010). We
hypothesized that if ibuprofen slowed ENCDC migration and reduced lamellipodia via reduced
RAC1 activation, it might be possible to rescue ibuprofen’s effects by inhibiting ROCK, a
RHOA effector kinase that inhibits RAC1. To test this hypothesis, CF-1 midgut slices were
cultured with the ROCK inhibitor Y-27632 (Figure 10). This drug increased migration of
ibuprofen-treated ENCDC to the level seen in control cells, but did not affect ENCDC migration
in the absence of ibuprofen (Figure 10M). These data suggest that excess RHOA/ROCK activity
relative to RAC1/CDC42 may underlie ibuprofen’s effect on ENCDC migration (Figure 11).

20
Discussion
Despite surgical treatment available for Hirschsprung disease since 1948 (Swenson and
Bill, 1948), approximately 5% of children with HSCR die at an early age (Rescorla et al., 1992;
Suita et al., 2005) and > 40% have problems after surgery (El-Sawaf et al., 2013; Menezes et al.,
2008). It would therefore be ideal if HSCR could be prevented from occurring in the first place.
We now believe this may be possible in some cases by optimizing maternal nutrition, health, and
medication use before conception and during early pregnancy. If non-genetic risk factors were
identified, targeted advice could be provided to families at “high genetic risk” for HSCR. From
this standpoint it is particularly valuable to identify common and avoidable exposures.
Using zebrafish to test medicines commonly used by pregnant women we discovered that
ibuprofen causes HSCR-like absence of neurons in developing fish bowel in vivo. Studies in
mice also show ibuprofen can slow colonization of fetal bowel in vivo. Most dramatically,
ibuprofen treatment of fetal chick bowel led to almost complete absence of colon colonization by
ENCDC. Detailed analysis of actively migrating mouse ENCDC confirmed ibuprofen slowed
migration, reduced lamellipodia, reduced filamentous actin, and reduced RAC1 activation.
These in vitro and in vivo data suggest ibuprofen use during the period that ENCDC colonize
fetal bowel (e.g. week 3-8 of human gestation) might increase HSCR risk.
Human studies are now needed to extend this work. Although ibuprofen reduced
ENCDC colonization of fetal bowel in three species, the dose needed and severity of inhibitory
effects differed between species. The reason for interspecies and interstrain differences is not
known, but may reflect differences in drug metabolism, the presence of alternative migration

21
modes, target sensitivity, or intracellular signaling. One valuable approach will be human HSCR
epidemiologic studies examining maternal medication use in early pregnancy.
To put the mouse in vivo studies in context, Ret+/- mice fed 375 ppm ibuprofen in chow
had a 12% reduction in ENCDC colonization of colon at E12.5. This dose was selected because
it suppresses COX-dependent inflammation (Lim et al., 2000; Ritschel and Kerns, 2009) and
gives serum levels (60-70 µM) (Morihara et al., 2005) within the human therapeutic dosing
range (Ritschel and Kerns, 2009). Because ibuprofen use in early pregnancy is common (almost
1 in 4) (Thorpe et al., 2013) and HSCR relatively rare (about 1:5000) (Amiel et al., 2008),
ibuprofen alone is unlikely to cause HSCR in humans even at higher doses. Ibuprofen might,
however, increase HSCR occurrence in the context of underlying genetic risk. This is important
because known genetic causes for HSCR are partially penetrant suggesting gene-gene or gene-
environment interactions cause HSCR. This was demonstrated dramatically in mice by the
observation that Ret+/- mice never have distal bowel aganglionosis (McCallion et al., 2003), but
develop HSCR-like disease more readily than WT when other mutations or non-genetic risk
factors are present (Arnold et al., 2009; Carrasquillo et al., 2002; Fu et al., 2010; Gunadi et al.,
2014; Lake et al., 2013; McCallion et al., 2003; Phusantisampan et al., 2012; Wallace and
Anderson, 2011). In contrast to mice, humans with a single inactive RET allele (i.e., ~30% of
HSCR cases) have about a 50% chance of having HSCR (Amiel et al., 2008). The vast majority
of these children have only a small region of distal aganglionic bowel, suggesting that even small
changes in bowel colonization efficiency by ENCDC could have an important effect on HSCR
occurrence. For example, in mice, male sex only slightly reduces bowel colonization by
ENCDC similar to the effect of ibuprofen (Vohra et al., 2007b), but in humans the male sex
increases HSCR occurrence four-fold.

22
From the standpoint of human teratogenicity, it may be important that ibuprofen effects
on ENCDC appear likely to be COX-independent. COX (PTGS) enzymes that make
prostaglandins (Vane, 1971) are the primary therapeutic target of ibuprofen and other NSAIDs,
but Ptgs1-/- Ptgs2-/- mice had normal bowel colonization by ENCDC and normal ENCDC
migration in vitro. Furthermore, prolonged ibuprofen exposure and high doses were needed to
slow migration. NSAIDs should inhibit COX enzymes much faster (with effects apparent within
10 minutes) and at low micromolar doses (IC50 = 2.1 µM for PTGS1 and 1.6 µM for PTGS2)
(Peppelenbosch et al., 1993; Tegeder et al., 2001). One caveat is that our Ptgs mutant mice were
C57BL/6 genetic background, which appears more resistant to ibuprofen effects than CF-1.
The precise mechanisms through which ibuprofen inhibits ENCDC migration remain
uncertain, but our observations suggest ibuprofen treated ENCDC have abnormal regulation of
actin dynamics. Reduced neurite growth in ibuprofen treated ENCDC was not anticipated since
ibuprofen increased neurite growth in several other neuron types (Fu et al., 2007; Wang et al.,
2009). We hypothesize this cell-type specific difference occurs because unlike other neurons
tested, ENCDC have low levels of PPARγ that is required for ibuprofen to reduce RHOA
activity and enhance neurite growth (Dill et al., 2010). The lack of ibuprofen effect on NIH 3T3
or CF-1 gut mesenchymal cell migration and lamellipodia further highlights how cell-type
specific gene expression impacts biology and emphasizes that extrapolation from one cell type to
another may be misleading.
Our studies suggest ibuprofen reduces active RAC1/CDC42 in migrating ENCDC. This
fits with emerging literature on the role of small RhoGTPases in neural crest-derived cell
migration. Elegant in vivo FRET studies showed RAC1 promotes ENCDC chain migration
through fetal bowel (Goto et al., 2013). Reduced RAC1 activity in ibuprofen treated ENCDC

23
provides a reasonable explanation for reduced migration and fewer lamellipodia. The ability of
ROCK inhibition to prevent ibuprofen induced changes in migration and lamellipodia, without
any apparent alteration of RHOA activity, may occur because RAC1 and RHOA usually inhibit
each other and ROCK is a RHOA effector kinase (Figure 9) (Nakayama et al., 2008; Sordella
and Van Aelst, 2008).
Conclusions: These data demonstrate that ibuprofen, a medication commonly used in the first
trimester of pregnancy, inhibits ENCDC migration and might increase HSCR risk in genetically
susceptible individuals. This study extends our previous observations demonstrating
environmental risk factors like vitamin A deficiency (Fu et al., 2010) and mycophenolate (Lake
et al., 2013) can cause HSCR-like disease in mice, especially when combined with predisposing
genetic changes. Human epidemiological studies are necessary to determine the extent to which
ibuprofen contributes to HSCR risk.

24
Acknowledgements
We thank Dr. Tatyana Svitkina for insightful guidance about the actin cytoskeleton, Dr.
Allen Mitchell for sharing unpublished data about medicine use in early pregnancy, Ryo Hotta,
Ming Fu, Elizabeth Wright-Jin, Rajarshi Sengupta, and Alisha Jamil, and the Mouse Genetics
Core at Washington University School of Medicine for assistance and advice. This work was
supported by Irma and Norman Braman Endowment (ROH), Suzi and Scott Lustgarten Center
Endowment (ROH), The Children’s Hospital of Philadelphia Research Institute (ROH), The
Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital
(grant nos. CH-II-1008-123, CH-II-2010-390, MD-II-2013-269) (ROH), NIH grants RO1
DK087715 (ROH), R01 GM059688 (SLJ), R37 DK33165 (WFS), RO1 DK080914 (AMG),
Burroughs Wellcome Fund Clinical Scientist Award in Translational Research (grant no.
1008525) (ROH), NIH F30 DK100101 (EMS) and by the NIH Medical Scientist Training
Program Training Grant T32 GM07200.
Author Contributions
EMS, JIL, NN, WFS, SLJ, AMG, and ROH designed experiments. EMS, OAT, NN,
SKB, MA, and LF collected data. EMS, OAT, AMG, NN, SKB, and ROH analyzed data. EMS
and ROH wrote the manuscript. All authors reviewed and edited.

25
References:
Airaksinen, M.S., Saarma, M., 2002. The GDNF family: signalling, biological functions and
therapeutic value. Nat Rev Neurosci 3, 383-394.
Aluri, J.B., Stavchansky, S., 1993. Determination of guaifenesin in human plasma by liquid
chromatography in the presence of pseudoephedrine. J Pharm Biomed Anal 11, 803-808.
Amiel, J., Sproat-Emison, E., Garcia-Barcelo, M., Lantieri, F., Burzynski, G., Borrego, S., Pelet,
A., Arnold, S., Miao, X., Griseri, P., Brooks, A.S., Antinolo, G., de Pontual, L., Clement-Ziza, M.,
Munnich, A., Kashuk, C., West, K., Wong, K.K., Lyonnet, S., Chakravarti, A., Tam, P.K.,
Ceccherini, I., Hofstra, R.M., Fernandez, R., 2008. Hirschsprung disease, associated
syndromes and genetics: a review. J Med Genet 45, 1-14.
Anderson, R.B., Newgreen, D.F., Young, H.M., 2006. Neural crest and the development of the
enteric nervous system. Adv Exp Med Biol 589, 181-196.
Arnold, S., Pelet, A., Amiel, J., Borrego, S., Hofstra, R., Tam, P., Ceccherini, I., Lyonnet, S.,
Sherman, S., Chakravarti, A., 2009. Interaction between a chromosome 10 RET enhancer
and chromosome 21 in the Down syndrome-Hirschsprung disease association. Hum Mutat
30, 771-775.
Baselt, R.C., 1982. Disposition of Toxic Drugs and Chemicals in Man. Biomedical
Publications, Seal Beach, CA, U.S.A.
Britsch, S., Goerich, D.E., Riethmacher, D., Peirano, R.I., Rossner, M., Nave, K.A., Birchmeier,
C., Wegner, M., 2001. The transcription factor Sox10 is a key regulator of peripheral glial
development. Genes Dev 15, 66-78.
Bryan, B., Cai, Y., Wrighton, K., Wu, G., Feng, X.H., Liu, M., 2005. Ubiquitination of RhoA by
Smurf1 promotes neurite outgrowth. FEBS Lett 579, 1015-1019.
Carrasquillo, M.M., McCallion, A.S., Puffenberger, E.G., Kashuk, C.S., Nouri, N., Chakravarti, A.,
2002. Genome-wide association study and mouse model identify interaction between RET
and EDNRB pathways in Hirschsprung disease. Nat Genet 32, 237-244.
Dasgupta, R., Langer, J.C., 2004. Hirschsprung disease. Curr Probl Surg 41, 942-988.
de Curtis, I., 2008. Functions of Rac GTPases during neuronal development. Dev Neurosci
30, 47-58.
Dill, J., Patel, A.R., Yang, X.L., Bachoo, R., Powell, C.M., Li, S., 2010. A molecular mechanism
for ibuprofen-mediated RhoA inhibition in neurons. J Neurosci 30, 963-972.
El-Sawaf, M., Siddiqui, S., Mahmoud, M., Drongowski, R., Teitelbaum, D.H., 2013. Probiotic
prophylaxis after pullthrough for Hirschsprung disease to reduce incidence of enterocolitis:
A prospective, randomized, double-blind, placebo-controlled, multicenter trial. J Pediatr
Surg 48, 111-117.
Enomoto, H., Crawford, P.A., Gorodinsky, A., Heuckeroth, R.O., Johnson, E.M., Jr., Milbrandt,
J., 2001. RET signaling is essential for migration, axonal growth and axon guidance of
developing sympathetic neurons. Development 128, 3963-3974.
Ferri, N., Panariti, F., Ricci, C., Maiocchi, G., Corsini, A., 2014. Aliskiren inhibits prorenin-
induced human aortic smooth muscle cell migration. Journal of the renin-angiotensin-
aldosterone system : JRAAS.
Fu, M., Landraville, S., Agapova, O.A., Wiley, L.A., Shoykhet, M., Harbour, J.W., Heuckeroth,
R.O., 2013. Retinoblastoma protein loss in the enteric nervous system causes selective
defects and early death. The Journal of Clinical Investigation In press.

26
Fu, M., Sato, Y., Lyons-Warren, A., Zhang, B., Kane, M.A., Napoli, J.L., Heuckeroth, R.O., 2010.
Vitamin A facilitates enteric nervous system precursor migration by reducing Pten
accumulation. Development 137, 631-640.
Fu, M., Tam, P.K., Sham, M.H., Lui, V.C., 2004. Embryonic development of the ganglion
plexuses and the concentric layer structure of human gut: a topographical study. Anat
Embryol (Berl) 208, 33-41.
Fu, Q., Hue, J., Li, S., 2007. Nonsteroidal anti-inflammatory drugs promote axon
regeneration via RhoA inhibition. J Neurosci 27, 4154-4164.
Fukata, M., Nakagawa, M., Kaibuchi, K., 2003. Roles of Rho-family GTPases in cell
polarisation and directional migration. Curr Opin Cell Biol 15, 590-597.
Furness, J.B., 2012. The enteric nervous system and neurogastroenterology. Nat Rev
Gastroenterol Hepatol 9, 286-294.
Ghobadi, C., Mirhosseini, N., Shiran, M.R., Moghadamnia, A., Lennard, M.S., Ledger, W.L.,
Rostami-Hodjegan, A., 2009. Single-dose pharmacokinetic study of clomiphene citrate
isomers in anovular patients with polycystic ovary disease. J Clin Pharmacol 49, 147-154.
Goldstein, A.M., Hofstra, R.M., Burns, A.J., 2013. Building a brain in the gut: development of
the enteric nervous system. Clin Genet 83, 307-316.
Goto, A., Sumiyama, K., Kamioka, Y., Nakasyo, E., Ito, K., Iwasaki, M., Enomoto, H., Matsuda,
M., 2013. GDNF and endothelin 3 regulate migration of enteric neural crest-derived cells
via protein kinase A and Rac1. J Neurosci 33, 4901-4912.
Govindan, V.M., Faulstich, H., Wieland, T., Agostini, B., Hasselbach, W., 1972. In-vitro effect
of phalloidin on plasma membrane preparation from rat liver. Die Naturwissenschaften 59,
521-522.
Guilluy, C., Garcia-Mata, R., Burridge, K., 2011. Rho protein crosstalk: another social
network? Trends Cell Biol 21, 718-726.
Gunadi, Kapoor, A., Ling, A.Y., Rochadi, Makhmudi, A., Herini, E.S., Sosa, M.X., Chatterjee, S.,
Chakravarti, A., 2014. Effects of RET and NRG1 polymorphisms in Indonesian patients with
Hirschsprung disease. J Pediatr Surg 49, 1614-1618.
Hall, A., 2005. Rho GTPases and the control of cell behaviour. Biochem Soc Trans 33, 891-
895.
Heuckeroth, R.O., 2013. Hirschsprung disease, in: Faure, C., DiLorenzo, C., Thapar, N. (Eds.),
Pediatric neurogastroenterology : gastrointestinal motility and functional disorders in
children. Springer, New York, pp. 271-283.
Heuckeroth, R.O., Lampe, P.A., Johnson, E.M.J., Milbrandt, J., 1998. Neurturin and GDNF
promote proliferation and survival of enteric neuron and glial progenitors in vitro.
Developmental Biology 200, 116-129.
Hilbert, J., Moritzen, V., Parks, A., Radwanski, E., Perentesis, G., Symchowicz, S.,
Zampaglione, N., 1988. The pharmacokinetics of loratadine in normal geriatric volunteers.
The Journal of international medical research 16, 50-60.
Hirschsprung, H., 1888. Stuhlträgheit Neugeborener in Folge von Dilatation und
Hypertrophie des Colons. Jahrbuch für Kinderheilkunde und physische Erziehung (Berlin)
27, 1-7.
Kapur, R.P., 2000. Colonization of the murine hindgut by sacral crest-derived neural
precursors: experimental support for an evolutionarily conserved model. Dev Biol 227,
146-155.

27
Kapur, R.P., Yost, C., Palmiter, R.D., 1992. A transgenic model for studying development of
the enteric nervous system in normal and aganglionic mice. Development 116, 167-175.
Koh, C.G., 2006. Rho GTPases and their regulators in neuronal functions and development.
Neurosignals 15, 228-237.
Kuhlman, J., Eisen, J.S., 2007. Genetic screen for mutations affecting development and
function of the enteric nervous system. Dev Dyn 236, 118-127.
Lake, J.I., Heuckeroth, R.O., 2013. Enteric nervous system development: migration,
differentiation, and disease. Am J Physiol Gastrointest Liver Physiol 305, G1-24.
Lake, J.I., Tusheva, O.A., Graham, B.L., Heuckeroth, R.O., 2013. Hirschsprung-like disease is
exacerbated by reduced de novo GMP synthesis. J Clin Invest 123, 4875-4887.
Langenbach, R., Morham, S.G., Tiano, H.F., Loftin, C.D., Ghanayem, B.I., Chulada, P.C., Mahler,
J.F., Lee, C.A., Goulding, E.H., Kluckman, K.D., Kim, H.S., Smithies, O., 1995. Prostaglandin
synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and
indomethacin-induced gastric ulceration. Cell 83, 483-492.
Laranjeira, C., Pachnis, V., 2009. Enteric nervous system development: Recent progress and
future challenges. Auton Neurosci 151, 61-69.
Li, Z., Aizenman, C.D., Cline, H.T., 2002. Regulation of rho GTPases by crosstalk and neuronal
activity in vivo. Neuron 33, 741-750.
Lim, G.P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., Tran, T., Ubeda, O., Ashe, K.H.,
Frautschy, S.A., Cole, G.M., 2000. Ibuprofen suppresses plaque pathology and inflammation
in a mouse model for Alzheimer's disease. J Neurosci 20, 5709-5714.
Lindsley, T.A., Shah, S.N., Ruggiero, E.A., 2011. Ethanol alters BDNF-induced Rho GTPase
activation in axonal growth cones. Alcohol Clin Exp Res 35, 1321-1330.
McCallion, A.S., Stames, E., Conlon, R.A., Chakravarti, A., 2003. Phenotype variation in two-
locus mouse models of Hirschsprung disease: tissue-specific interaction between Ret and
Ednrb. Proc Natl Acad Sci U S A 100, 1826-1831.
McKeown, S.J., Stamp, L., Hao, M.M., Young, H.M., 2013. Hirschsprung disease: a
developmental disorder of the enteric nervous system. Wiley interdisciplinary reviews.
Developmental biology 2, 113-129.
Meijering, E., Dzyubachyk, O., Smal, I., 2012. Methods for cell and particle tracking. Methods
Enzymol 504, 183-200.
Menezes, M., Pini Prato, A., Jasonni, V., Puri, P., 2008. Long-term clinical outcome in patients
with total colonic aganglionosis: a 31-year review. J Pediatr Surg 43, 1696-1699.
Morham, S.G., Langenbach, R., Loftin, C.D., Tiano, H.F., Vouloumanos, N., Jennette, J.C.,
Mahler, J.F., Kluckman, K.D., Ledford, A., Lee, C.A., Smithies, O., 1995. Prostaglandin
synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83, 473-482.
Morihara, T., Teter, B., Yang, F., Lim, G.P., Boudinot, S., Boudinot, F.D., Frautschy, S.A., Cole,
G.M., 2005. Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic
alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer's
models. Neuropsychopharmacology 30, 1111-1120.
Murphey, R.D., Zon, L.I., 2006. Small molecule screening in the zebrafish. Methods 39, 255-
261.
Nagy, N., Burns, A.J., Goldstein, A.M., 2012. Immunophenotypic characterization of enteric
neural crest cells in the developing avian colorectum. Dev Dyn 241, 842-851.

28
Nakayama, M., Goto, T.M., Sugimoto, M., Nishimura, T., Shinagawa, T., Ohno, S., Amano, M.,
Kaibuchi, K., 2008. Rho-kinase phosphorylates PAR-3 and disrupts PAR complex formation.
Dev Cell 14, 205-215.
Nini, L., Dagnino, L., 2010. Accurate and reproducible measurements of RhoA activation in
small samples of primary cells. Anal Biochem 398, 135-137.
Pachnis, V., Mankoo, B., Costantini, F., 1993. Expression of the c-ret proto-oncogene during
mouse embryogenesis. Development 119, 1005-1017.
Parri, M., Chiarugi, P., 2010. Rac and Rho GTPases in cancer cell motility control. Cell
Commun Signal 8, 23.
Peppelenbosch, M.P., Tertoolen, L.G., Hage, W.J., de Laat, S.W., 1993. Epidermal growth
factor-induced actin remodeling is regulated by 5-lipoxygenase and cyclooxygenase
products. Cell 74, 565-575.
Phusantisampan, T., Sangkhathat, S., Phongdara, A., Chiengkriwate, P., Patrapinyokul, S.,
Mahasirimongkol, S., 2012. Association of genetic polymorphisms in the RET-
protooncogene and NRG1 with Hirschsprung disease in Thai patients. J Hum Genet 57, 286-
293.
Preibisch, S., Saalfeld, S., Tomancak, P., 2009. Globally optimal stitching of tiled 3D
microscopic image acquisitions. Bioinformatics 25, 1463-1465.
Rescorla, F.J., Morrison, A.M., Engles, D., West, K.W., Grosfeld, J.L., 1992. Hirschsprung's
disease. Evaluation of mortality and long-term function in 260 cases. Arch Surg 127, 934-
941; discussion 941-932.
Ridley, A.J., 2006. Rho GTPases and actin dynamics in membrane protrusions and vesicle
trafficking. Trends Cell Biol 16, 522-529.
Ritschel, W.A., Kerns, G.L., 2009. Handbook of basic pharmacokinetics... including clinical
applications. , 7th ed. American Pharmacists Association, Washington, D.C.
Sasselli, V., Pachnis, V., Burns, A.J., 2012. The enteric nervous system. Dev Biol 366, 64-73.
Sato, Y., Heuckeroth, R.O., 2008. Retinoic acid regulates murine enteric nervous system
precursor proliferation, enhances neuronal precursor differentiation, and reduces neurite
growth in vitro. Dev Biol 320, 185-198.
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S.,
Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J.Y., White, D.J., Hartenstein, V., Eliceiri, K.,
Tomancak, P., Cardona, A., 2012. Fiji: an open-source platform for biological-image analysis.
Nat Methods 9, 676-682.
Schuchardt, A., D'Agati, V., Larsson-Blomberg, L., Constantini, F., Pachnis, V., 1994. Defects
in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret.
Nature 367, 380-383.
Skinner, M., 1996. Hirschsprung's Disease. Curr. Probl. Surg. 33, 391-461.
Sordella, R., Van Aelst, L., 2008. Dialogue between RhoA/ROCK and members of the Par
complex in cell polarity. Dev Cell 14, 150-152.
Stewart, A.L., Young, H.M., Popoff, M., Anderson, R.B., 2007. Effects of pharmacological
inhibition of small GTPases on axon extension and migration of enteric neural crest-
derived cells. Dev Biol 307, 92-104.
Suita, S., Taguchi, T., Ieiri, S., Nakatsuji, T., 2005. Hirschsprung's disease in Japan: analysis of
3852 patients based on a nationwide survey in 30 years. J Pediatr Surg 40, 197-201;
discussion 201-192.

29
Swenson, O., Bill, A.H., Jr., 1948. Resection of rectum and rectosigmoid with preservation of
the sphincter for benign spastic lesions producing megacolon; an experimental study.
Surgery 24, 212-220.
Tegeder, I., Pfeilschifter, J., Geisslinger, G., 2001. Cyclooxygenase-independent actions of
cyclooxygenase inhibitors. FASEB J 15, 2057-2072.
Thorpe, P.G., Gilboa, S.M., Hernandez-Diaz, S., Lind, J., Cragan, J.D., Briggs, G., Kweder, S.,
Friedman, J.M., Mitchell, A.A., Honein, M.A., National Birth Defects Prevention, S., 2013.
Medications in the first trimester of pregnancy: most common exposures and critical gaps
in understanding fetal risk. Pharmacoepidemiol Drug Saf 22, 1013-1018.
Vane, J.R., 1971. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-
like drugs. Nat New Biol 231, 232-235.
Vohra, B.P., Fu, M., Heuckeroth, R.O., 2007a. Protein kinase Czeta and glycogen synthase
kinase-3beta control neuronal polarity in developing rodent enteric neurons, whereas
SMAD specific E3 ubiquitin protein ligase 1 promotes neurite growth but does not
influence polarity. J Neurosci 27, 9458-9468.
Vohra, B.P., Planer, W., Armon, J., Fu, M., Jain, S., Heuckeroth, R.O., 2007b. Reduced
endothelin converting enzyme-1 and endothelin-3 mRNA in the developing bowel of male
mice may increase expressivity and penetrance of Hirschsprung disease-like distal
intestinal aganglionosis. Dev Dyn 236, 106-117.
Wallace, A.S., Anderson, R.B., 2011. Genetic interactions and modifier genes in
Hirschsprung's disease. World J Gastroenterol 17, 4937-4944.
Wang, H., Hughes, I., Planer, W., Parsadanian, A., Grider, J.R., Vohra, B.P., Keller-Peck, C.,
Heuckeroth, R.O., 2010. The timing and location of glial cell line-derived neurotrophic
factor expression determine enteric nervous system structure and function. J Neurosci 30,
1523-1538.
Wang, H.R., Zhang, Y., Ozdamar, B., Ogunjimi, A.A., Alexandrova, E., Thomsen, G.H., Wrana,
J.L., 2003. Regulation of cell polarity and protrusion formation by targeting RhoA for
degradation. Science 302, 1775-1779.
Wang, X., Budel, S., Baughman, K., Gould, G., Song, K.H., Strittmatter, S.M., 2009. Ibuprofen
enhances recovery from spinal cord injury by limiting tissue loss and stimulating axonal
growth. J Neurotrauma 26, 81-95.
Wang, X., Chan, A.K., Sham, M.H., Burns, A.J., Chan, W.Y., 2011. Analysis of the sacral neural
crest cell contribution to the hindgut enteric nervous system in the mouse embryo.
Gastroenterology 141, 992-1002 e1001-1006.
Wu, Y.I., Frey, D., Lungu, O.I., Jaehrig, A., Schlichting, I., Kuhlman, B., Hahn, K.M., 2009. A
genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461,
104-108.

30
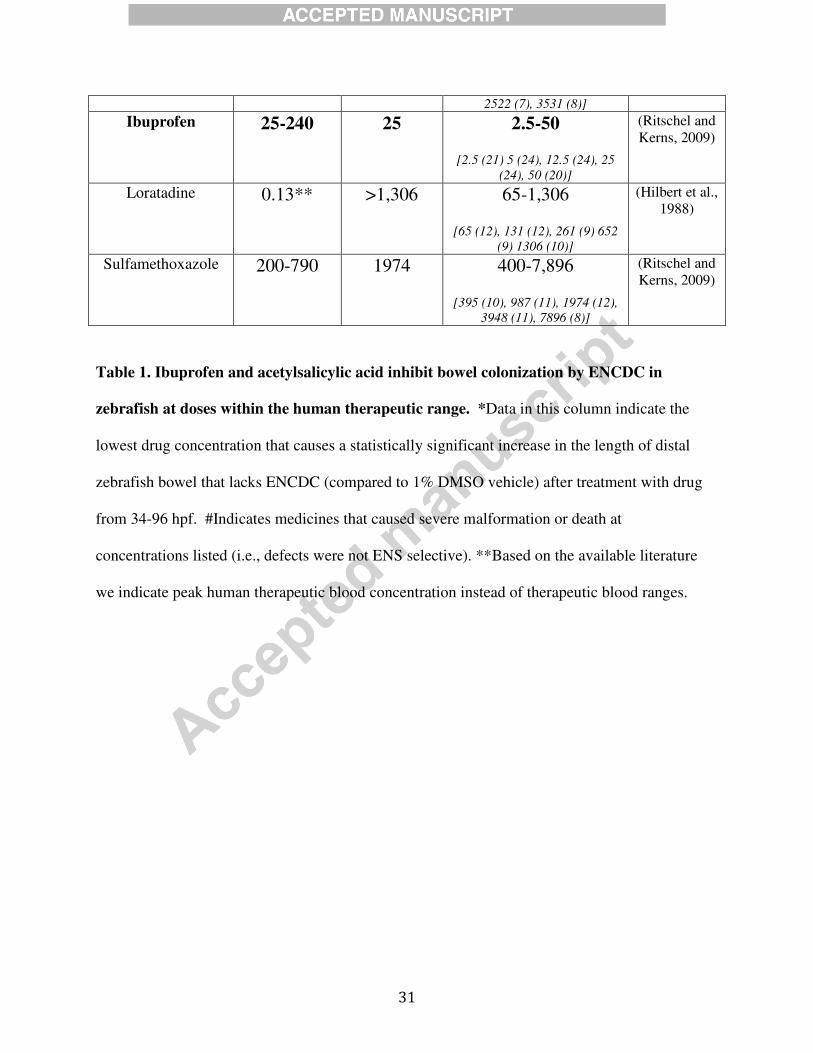
Table 1. Medicines tested for an effect on zebrafish ENS development
Medicine
Human
therapeutic
blood
concentration
(µM)
Lowest
concentration
that affects
ENCDC*
(µM)
Concentration range tested
(µM)
[Actual concentrations
tested (number evaluated
at each concentration)] Reference
Acetaminophen 65-130 2,315 331-3,308
[331 (9), 662 (8), 1650 (8),
2315 (7) 3308 (7)]
(Ritschel and
Kerns, 2009)
Acetylsalicylic
Acid 110-1,700 333 56-666
[56 (21), 111(22), 222 (22),
333 (24), 666 (7)]
(Ritschel and
Kerns, 2009)
Caffeine 10-50 257 5-1,030
[5 (21), 51 (15), 257 (17), 514
(18), 1030 (16)]
(Baselt,
1982)
Chlorpheniramine 0.013-0.025 256# 13-1,279
[13 (6), 26 (9), 128 (10), 256
(5), 1279 (6)]
(Ritschel and
Kerns, 2009)
Clomiphene 0.050** 83# 17-836
[(17 (9) 83 (≥5), 167 (≥5) 836
(≥5)]
(Ghobadi et
al., 2009)
Dextromethorphan 0.74-1.3 368 18-1,840
[18 (9), 37 (9) 184 (7) 368 (7),
1840 (≥5)]
(Ritschel and
Kerns, 2009)
Diphenhydramine 0.034-0.34 103# 17-343
[17 (9) 34 (9) 103 (≥5), 172
(≥5), 343 (≥5)]
(Ritschel and
Kerns, 2009)
Doxylamine 0.3** 1,287# 129-1,287
[129 (8), 257 (8) 1287 (6)]
(Ritschel and
Kerns, 2009)
Erythromycin 0.68-3.4 >1,363 14-1363
[14 (7), 68 (9), 136 (9), 681 (8),
1363 (9)]
(Ritschel and
Kerns, 2009)
Guaifenesin 7.6** >3,31 252-3,531
[252 (5), 504 (9), 1009 (9),
(Aluri and
Stavchansky,
1993)
31
2522 (7), 3531 (8)]
Ibuprofen 25-240 25 2.5-50
[2.5 (21) 5 (24), 12.5 (24), 25
(24), 50 (20)]
(Ritschel and
Kerns, 2009)
Loratadine 0.13** >1,306 65-1,306
[65 (12), 131 (12), 261 (9) 652
(9) 1306 (10)]
(Hilbert et al.,
1988)
Sulfamethoxazole 200-790 1974 400-7,896
[395 (10), 987 (11), 1974 (12),
3948 (11), 7896 (8)]
(Ritschel and
Kerns, 2009)
Table 1. Ibuprofen and acetylsalicylic acid inhibit bowel colonization by ENCDC in
zebrafish at doses within the human therapeutic range. *Data in this column indicate the
lowest drug concentration that causes a statistically significant increase in the length of distal
zebrafish bowel that lacks ENCDC (compared to 1% DMSO vehicle) after treatment with drug
from 34-96 hpf. #Indicates medicines that caused severe malformation or death at
concentrations listed (i.e., defects were not ENS selective). **Based on the available literature
we indicate peak human therapeutic blood concentration instead of therapeutic blood ranges.

32
Table 2. Primary and secondary antibodies for immunohistochemistry or immunoselection
Antibody Concentration Catalog # Source
Rabbit anti-Tuj1 1:10,000 PRB-435P Covance*; RRID:AB_10063850
Goat anti-RET 1:100 GT15002 Neuromics; RRID:AB_2179886
Mouse anti-BRDU
(Conjugated to Alexa-594)
1:100 A21304 Invitrogen; RRID:AB_221472
Rabbit anti-cleaved
caspase-3
1:100 9661 Cell Signaling Technology;
RRID:AB_2314091
Mouse anti-PPARγ 1:100 sc-7273 Santa Cruz; RRID:AB_628115
Phalloidin (Conjugated to
Alexa-488)
1:50 A12379 Invitrogen; RRID:AB_2315147
Rabbit anti-GST 1:100 sc-35614 Santa Cruz; RRID:AB_647587
Mouse anti-vinculin 1:100 V9131 Sigma; RRID:AB_477629
Mouse anti-HuC/HuD
(Biotin Conjugate (16A11))
1:800 A21272 Invitrogen; RRID:AB_10375876
Mouse anti-chicken N-
cadherin antibody (clone
6B3)
1:5 6B3 Developmental Studies
Hybridoma Bank, Iowa;
RRID:AB_528118
Rabbit anti-nerve growth
factor (NGF-receptor), P75
1:1000 AB1554 EMD Millipore
RRID:AB_90760
Anti-rabbit IgG
MicroBeads
1:50 130-048-602 Miltenyi Biotec
RRID:AB_244362
Alexa Fluor® 594 Donkey
anti-mouse
1:400
A21203 Molecular Probes (Invitrogen);
RRID: AB_141633
Alexa Fluor® 594 Donkey
anti-rabbit
1:400 A21207 Life Technologies:
RRID:AB_10049744
Alexa Fluor® 488 Donkey
anti-rabbit
1:400 A21206 Life Technologies;
RRID:AB_10049650
Alexa Fluor® 594 Donkey
anti-goat
1:400 A11058 Life Technologies;
RRID:AB_10563390
Alexa Fluor® 647 Donkey
anti-rabbit
1:400 A31573 Life Technologies;
RRID:AB_10561706
* Covance was acquired by BioLegend in 2014 (new catalog # 802001; RRID not yet assigned)
RRID = Research Resource Identifiers (https://www.force11.org/node/4856)

33
Figure Legends
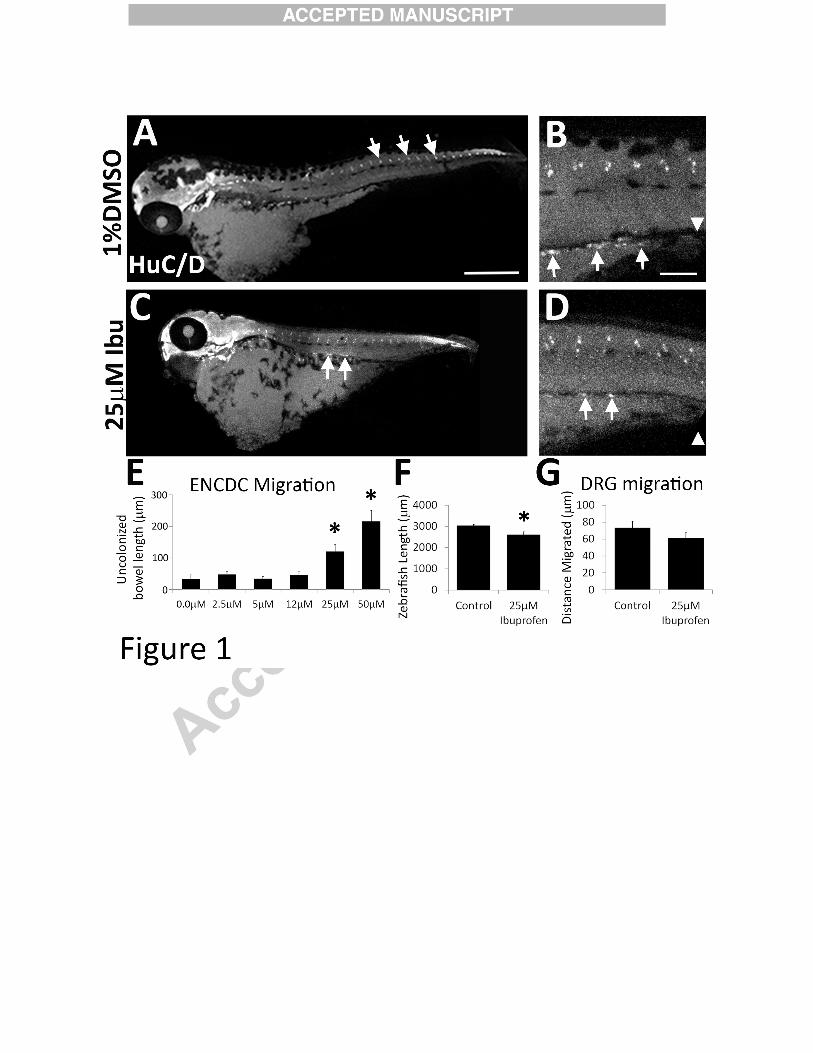
Figure 1:
Ibuprofen reduced ENCDC colonization of zebrafish bowel. (A-D) Zebrafish were treated
with vehicle (1%DMSO) or ibuprofen (Ibu) from 34-96 hpf and then stained with HuC/D
antibody. (A) White arrows indicate dorsal root ganglia. (C) White arrows highlight enteric
neurons. Scale bar=500µm. (B, D) Higher magnification of fish midsection. Scale bar=100µm.
(E) Length of bowel from most distal HuC/D+ cell (arrows in B, D) to bowel terminus
(arrowheads in B, D). *P<0.05 (ANOVA on Ranks) (F) Length of whole zebrafish (1% DMSO
n=4, 25 µM Ibuprofen n=6). *P=0.025 (t-test). (G) Distance between DRG and dorsal zebrafish
edge (1% DMSO n=3, 25 µM ibuprofen n=4). *P>0.05 (t-test).
Figure 2:
Ibuprofen inhibited hindgut colonization by chick ENCDC ex vivo and mouse ENCDC in
vivo. (A, B) Ibuprofen (250 µM) almost completely blocked E6 chick colon colonization by N-
cadherin+ (green) ENCDC during 48 hour culture. (Scale bar=200 µm, n=6 per group). Insets
show the migration wavefront with EdU (red) and N-cadherin (green) immunohistochemistry.
(Scale bar=35 µm). mg=midgut, hg=hindgut. (C-E) Ibuprofen feeding from E8.5 to E12.5
reduced Ret +/- mouse colon colonization by ENCDC visualized with TuJ1 antibody (red). Scale
bars=400 µm. (E, F) Quantitative data show the proportion of the colon colonized by ENCDC
(i.e., length of colon containing ENCDC divided by total colon length) (Control n=4, ibuprofen
n=9). *P =0.042 (t-test). (F) Sox10 +/- colon was normally colonized by ENCDC when dams
were fed ibuprofen from E8.5 to E12.5 (Control n = 19, ibuprofen n=13). P=0.79 (t-test).

34
Figure 3:
Ibuprofen reduced murine ENCDC migration, but did not affect proliferation, caspase-3
activation, or neuronal differentiation. E12.5 CF-1 midgut slices cultured with or without
ibuprofen for 16 hours after GDNF addition were stained for (A, B) RET (red), phalloidin
(green), DAPI (blue) (scale bar=200µm), (C, D) RET (green), BrdU (red), DAPI (blue) (Scale
bar=100µm), (E, F) RET (red), cleaved-caspase 3 (green), DAPI (blue) (Scale bar=100µm), or
(G, H) RET (green), TuJ1 (red), DAPI (blue) (scale bar=100µm). (I) Ibuprofen reduced the
distance ENCDC migrated from gut explants at 250 µM and 500 µM. Control n=275 slices, 29
biological replicates; 50 µM ibuprofen n = 48 slices, 3 biological replicates; 100 µM ibuprofen n
= 55 slices, 3 biological replicates; 250 µM ibuprofen n=111 slices, 5 biological replicates; 500
µM ibuprofen n = 102 slices, 8 biological replicates) but not at lower ibuprofen concentrations.
**P<0.001 (ANOVA). (J) Mean percentage BrdU+ ENCDC (RET+ cells) (Control n=21 slices,
7 biological replicates; ibuprofen n=9 slices, 3 biological replicates). P=0.24 (t-test). (K) Mean
percentage cleaved-caspase 3+ ENCDC (RET+ cells) (Control n=15 slices, ibuprofen n=15
slices; both 3 biological replicates). P=0.36 (t-test). (L) Mean percentage TuJ1+ ENCDC (RET+
cells) (Control n=19 slices, ibuprofen n=23 slices; both 3 biological replicates). P=0.35 (t-test).
Figure 4:
Ibuprofen did not affect migration or lamellipodia of NIH 3T3 or mouse gut mesenchymal
cells. Confluent NIH 3T3 were pretreated with 250 µM ibuprofen (2 hours) before making a
scratch to remove some cells. (A, B) NIH3T3 immediately after the scratch was made. (C, D)
Two hours after the scratch. Cells were imaged every 5 minutes for 24 hours 250 µM

35
ibuprofen did not affect (E) average speed of gap closure (Control n=5 assays, ibuprofen; n=7
assays), P=0.76 (t-test) or (F) percentage of NIH 3T3 with lamellipodia (Control n=8 assays,
ibuprofen; n=9 assays). P=0.75 (t-test). (G, H) 250 µM ibuprofen did not affect distance CF-1
mesenchymal cells migrated from gut explants (Control n=19 slices, ibuprofen n=24 slices; 3
biological replicates). P=0.57 (t-test) or the percentage of mesenchymal cells with lamellipodia
after 16 hours (Control=9 slices, ibuprofen=9 slices; 3 biological replicates). P=0.52 (t-test).
Figure 5:
Ibuprofen reduced lamellipodia in migrating murine ENCDC and reduced neurite length
in differentiating murine enteric neurons. (A-F) E12.5 CF-1 midgut slices were cultured with
250 µM ibuprofen or control media for 16 hours after GDNF addition and then stained for (A, D)
RET, (B, E) Alexa488-phalloidin, and (C, F) DAPI (merged image). Arrows highlight well-
formed lamellipodia. (G, H) Dissociated immunoselected E12.5 CF-1 ENCDC cultured 48 hours
were stained for RET (red), TuJ1 (green) and DAPI (blue). Scale bars=50 µm. (I, J). (I) The
percentage of migrating ENCDC with lamellipodia was reduced by 50 µM or higher ibuprofen
concentrations. RET+ ENCDC most distant from the gut slices were analyzed. (Control n=61
slices, 13 biological replicates; 50 µM Ibuprofen n = 9 slices, 3 biological replicates; 100 µM
ibuprofen n=9 slices, 3 biological replicates, 250 µM ibuprofen n=27 slices, 9 biological
replicates; 500 µM ibuprofen n = 12 slices, 4 biological replicates). *P <0.05, **P <0.001,
(ANOVA, Holm-Sidak method). (J) Ibuprofen reduced mean Alexa488-phalloidin fluorescence
intensity for RET+ ENCDC (Control n=232 cells, ibuprofen n=236 cells; both 4 biological
replicates). *P<0.001 (Mann-Whitney Rank Sum). (K) Ibuprofen reduced longest neurite in

36
cultured differentiating ENCDC (Control n=50, ibuprofen n=76, both 3 biological replicates).
*P=0.009 (Mann-Whitney Rank Sum).
Figure 6:
E12.5 CF-1 mouse ENCDC had less immunoreactive PPARγγγγ than E12.5 dorsal root
ganglion (DRG) neurons. Sagittal sections were stained with antibodies to PPARγ (red) and
neuron specific beta 3 tubulin (green, TuJ1). (A-D) PPARγ is readily detectable in DRG
neurons. The boxed region in C contains DRG neurons that are immunoreactive for both PPARγ
and TuJ1 antibodies (yellow cells) and is enlarged in D to show DRG neurons immunoreactive
for PPARγ staining (red cells, white arrows). (E-H) Developing enteric neurons (TuJ1+ cells
highlighted with arrows in F) have much lower levels of immunoreactive PPARγ (i.e., no yellow
cells in the region of the ENS in G). The boxed region in G is enlarged in H to show the lack of
PPARγ staining (red) in the ENCDC. Arrows highlight the region of the developing ENS.
Images are from the same fetal mouse stained on the same slide. Scale bar = 100µm.
Figure 7:
Ibuprofen had delayed effects on murine ENCDC migration. (A) Experimental paradigms.
E12.5 CF-1 midgut slices were cultured on fibronectin. Ibuprofen was added four hours after
plating (ibuprofen late) or at plating (ibuprofen). Time lapse images were then obtained every
two minutes for four hours. (B-D) DIC images show final time points from time-lapse movies.
Tracks show trajectories for single cells that migrated from explants. Immunohistochemistry
after imaging confirmed tracked cells expressed RET. Scale bar = 100µm. (E) Mean ENCDC
migration speed during each one hour interval. Ibuprofen slowed ENCDC speed, but not until 3-

37
4 hours after ibuprofen exposure (Control n=130 cells, Ibuprofen n=180 cells, ibuprofen late
n=92 cells; all 3 biological replicates). *P<0.05 (ANOVA on ranks).
Figure 8:
Ibuprofen effects on murine ENCDC migration and lamellipodia do not appear to be
cyclooxygenase dependent. Ptgs1-/- Ptgs2-/- mice have normal ENCDC migration in vitro and
normal bowel colonization in vivo. (A, B) E12.5 WT C57BL/6 and Ptgs1-/- Ptgs2-/- midgut
slices were cultured 16 hours after GDNF addition and then stained with RET antibody (red),
phalloidin (green), and DAPI (blue). Scale bar=50 µm. (C, D) E12.5 bowel from WT and Ptgs1-
/- Ptgs2-/- mice was stained with Tuj1 antibody. Scale bar=500 µm. Ptgs1-/- Ptgs2-/- mutations
did not affect (E) the mean distance that ENCDC migrated from gut slices (WT n=57 slices, 5
embryos and Ptgs1-/- Ptgs2-/- n=48, slices, 7 embryos), P=0.55 (t-test), or (F) the mean
percentage of ENCDC with a well-formed lamellipodia (WT n=11 slices, 4 embryos and Ptgs1-/-
Ptgs2-/- n=10 slices, 5 embryos), P=0.38 (t-test), or (G) the extent of bowel colonization by
ENCDC in vivo (WT n=6 and Ptgs1-/- Ptgs2-/- n=5), P=0.23 (t-test). Only ENCDC most distant
from gut slices were evaluated for lamellipodia. (H-K) Stable Prostaglandin E2 (PGE2) and
Prostaglandin F2 (PGF2) analogs did not rescue ibuprofen-induced ENCDC migration defects.
E12.5 CF-1 midgut slices were cultured in media with GDNF with or without ibuprofen for 16
hours and then stained for RET, F-actin (phalloidin) and with DAPI. (H, I) Distance from the gut
slice edge to the most distant RET+ ENCDC was measured. Neither 16, 16-dimethyl PGE2 (H)
nor 16, 16-dimethyl PGF2 (I) restored ENCDC migration to control levels in the presence of
ibuprofen, but 16,16-dimethyl PGE2 (H) did reduce ENCDC migration in the absence of
ibuprofen. (For PGE2 studies: Control n = 165 slices, ibuprofen n = 112 slices, 16, 16-dimethyl

38
PGE2 n = 137 slices, ibuprofen plus 16, 16-dimethyl PGE2 n = 53 slices; *P<0.05 compared to
Control. #P<0.05 compared to Prostaglandin E2 (ANOVA). For PGF2 studies: Control n =103
slices, ibuprofen n = 96 slices, 16, 16-dimethyl PGF2 n = 127 slices, ibuprofen plus PGF2 n =
107 slices; *P<0.05 compared to Control. #P<0.05 compared to Prostaglandin F2 (ANOVA on
Ranks)) (J, K) The percentage of ENCDC with well-formed lamellipodia was determined by
examining phalloidin stained ENCDC that had migrated furthest from the gut slice edge. Neither
16, 16-dimethyl PGE2 nor 16, 16-dimethyl PGF2 rescued lamellipodia in the presence of
ibuprofen (For PGE2 studies: Control n = 27 slices, ibuprofen n = 18 slices, PGE2 n = 27 slices,
ibuprofen plus PGE2 n =18 slices) *P<0.05 compared to Control. #P<0.05 compared to
Prostaglandin E2 (ANOVA on Ranks). For PGF2 studies: Control n = 105 slices, ibuprofen n =
83 slices, 16, 16-dimethyl PGF2 n = 105 slices, ibuprofen plus 16, 16-dimethyl PGF2 n =93
slices *P<0.05 compared to Control. #P<0.05 compared to Prostaglandin F2 (ANOVA)).
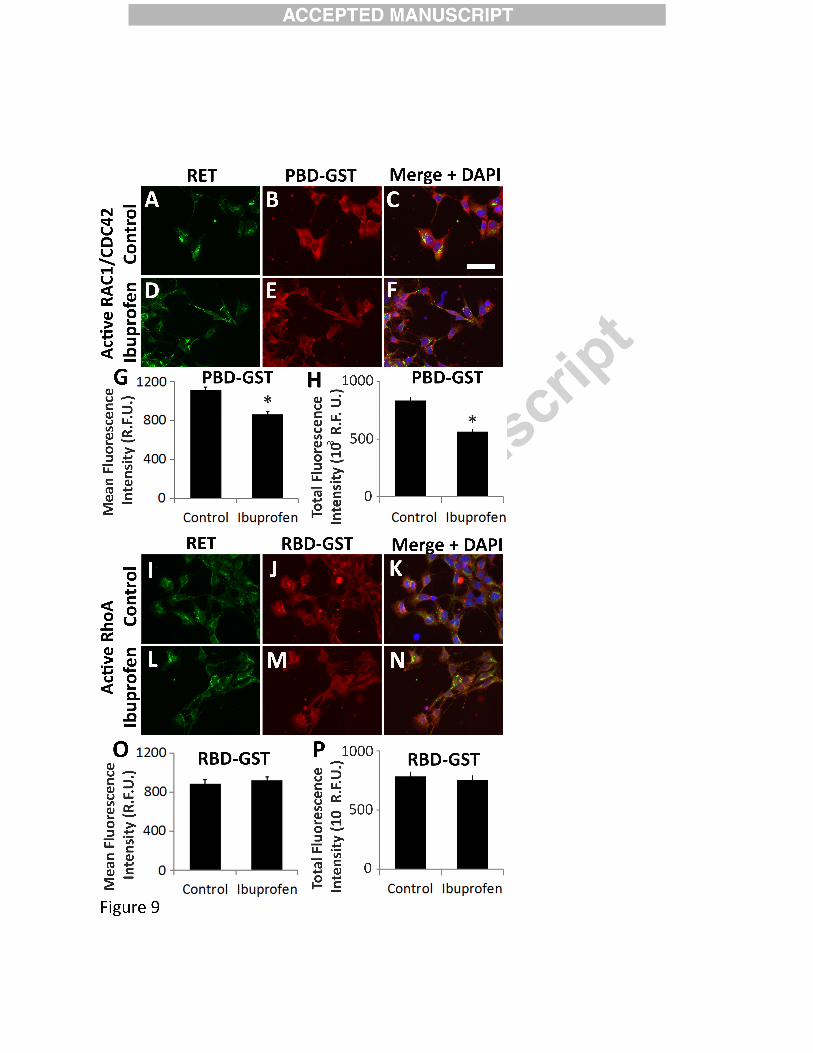
Figure 9:
Ibuprofen reduced active RAC1/CDC42, but did not affect active RHOA in migrating
murine ENCDC. E12.5 CF-1 midgut slices cultured 16 hours with GDNF were fixed,
permeabilized and incubated with (A-H) PBD-GST that binds active GTP-RAC1 and GTP-
CDC42, or (I-P) with RBD-GST that binds active GTP-RHOA. Cultures were then stained for
(A, D, I, L) RET (green), (B, E, J, M) GST (red), and (C, F, K, N) DAPI (blue; merged image).
Scale bar=50µm. (G, O) Mean fluorescence intensity for PBD-GST (G) and RBD-GST (O) for
ENCDC that migrated furthest from gut slices. (G) 250 µM Ibuprofen treated ENCDC had less
bound PBD-GST indicating reduced active RAC1/CDC42 (Control n=243 cells, ibuprofen
n=205 cells, both 4 biological replicates); *P<0.001 (Mann-Whitney Rank Sum), but did not

39
reduce (O) RBD-GST bound to active RHOA in actively migrating ENCDC (Control n=144
cells, ibuprofen n=136 cells, both 4 biological replicates). P=0.408 (Mann-Whitney Rank Sum).
(H, P) Because ENCDC shape is altered by ibuprofen and this may change mean fluorescence
intensity per pixel, total fluorescence intensity (mean fluorescence intensity/pixel x pixels/cell)
was determine for (H) active RAC1/CDC42 (via PGD-GST binding) (Control n=243 cells,
ibuprofen n=205 cells, both 4 biological replicates, and (P) active RHOA (via RBD-GST
binding) (Control n =144 cells, ibuprofen n=136 cells, both 4 biological replicates) per cell.
*P<0.001, Mann-Whitney Rank Sum. These data indicate that ibuprofen reduced total cellular
levels of F-actin and active RAC1/CDC42 without affecting active RHOA levels. Only ENCDC
furthest from the gut slice edge were analyzed.
Figure 10:
ROCK inhibition rescued ibuprofen effects on murine ENCDC migration. (A-L) E12.5 CF-
1 midgut slices were cultured with or without 250µM ibuprofen four hours before GDNF and 5
µM Y-27632 were added. Sixteen hours later cultures were fixed and stained for (A, D, G, J)
RET (red), (B, E, H, K) phalloidin (green), and (C, F, I, L) DAPI (blue; merged image). Scale
bar = 50 µm. (M) Distance from the gut slice edge to the most distant RET+ ENCDC was
measured. Y-27632 increased migration of ibuprofen treated ENCDC, but not control ENCDC
(Control n =43 slices, ibuprofen n=48 slices, Y-27632 n=54 slices, ibuprofen + Y-27632 n=59
slices, all from 7 biological replicates). *P<0.05 compared to control, #P<0.05 compared to
ibuprofen + Y-27632 (ANOVA on ranks).
Figure 11:

40
Model. RAC1 organizes the actin cytoskeleton to form lamellipodia and support migration.
Ibuprofen reduced ENCDC RAC1 activation. ROCK is a RHOA effector that inhibits RAC1. Y-
27632 inhibits ROCK to permit RAC1 activation, enhancing ENCDC migration.
Supplemental Figure Legends
Supplemental Figure 1: (Additional file 1: Schill Supplemental Figure 1.pdf)
Midgut slice explant culture and analysis. E12.5 CF-1 mouse midgut slices were cultured for
16 hours on fibronectin coated dishes in media with GDNF. ENCDC migrate from gut slices
onto the culture dish under these conditions. For analyses where we report the “Distance
migrated” by ENCDC from the slice, the slice area was divided into octants (dotted lines) and
the distance from the gut slice edge to the most distant ENCDC (RET immunoreactive cell, red)
was measured for each octant. Cell morphology was also assessed in “Distal ENCDC” (i.e., the
RET+ cells that had migrated furthest from the slice edge). Mesenchymal cells were identified
by their lack of RET immunoreactivity and their actin-rich cytoskeleton (stained green with
Alexa488-phalloidin). Phalloidin is a fungal bicyclic heptapeptide that binds filamentous, but
not G-actin (Govindan et al., 1972). The distance migrated by mesenchymal cells was
determined by measuring the distance from the gut slice edge to the most distant RET negative
cell with distinctive intense phalloidin staining.
Supplemental Figure 2: (Additional file 6: Schill Supplemental Figure 6.pdf)

41
PBD-GST and RBD-GST fusion proteins bind to cultured NIH 3T3 fibroblasts as predicted
using well-established methods to activate RAC1/CDC42 or RHOA. NIH 3T3 were serum
starved and then either (A, B) left unstimulated, (C) treated with RAC1/CDC42 Activator II to
activate RAC1 and CDC42, or (D) treated with RHO Activator I to activate RHOA. Cells were
fixed and treated with either PBD-GST (A, C) or RBD-GST (B, D) as in Figure 9. All cells were
then stained with antibodies to GST to visualize active RAC1/CDC42 or RHOA. Stimulated
cells had greater GST fluorescence compared to unstimulated cells for PBD-GST and RBD-GST
fusion proteins as expected after stimulation. Scale bar = 20 µm.
Supplemental Movies 1-3:
(Additional file 6: Movie 1 Control.avi)
(Additional file 7: Movie 2 Delayed ibuprofen.avi)
(Additional file 8: Movie 3 Ibuprofen.avi)
Time lapse images of migrating ENCDC. E12.5 CF-1 midgut slice explants were cultured for
4 hours before GDNF addition. Time lapse imaging began when GDNF was added and
continued for 4 hours. DIC images include a time stamp in “hours:minutes” in the upper left
hand corner.
Movie 1: No added ibuprofen.
Movie 2: Ibuprofen added just before imaging began.
Movie 3: Ibuprofen added 4 hours before imaging began.

42
Schill et. al. Highlights
1. Ibuprofen inhibits ENS precursor migration in zebrafish, chick and mouse.
2. Ibuprofen reduces lamellipodia and F-actin in cultured murine ENS precursors.
3. Ibuprofen reduces RAC1 activation in migrating ENS precursors.
4. Ibuprofen effects on ENS precursor migration appear to be COX independent.
5. Ibuprofen is commonly used in early pregnancy and might increase Hirschsprung risk.