
ATM kinase inhibition in glial cells activates the innate … kinase inhibition in glial cells...
9
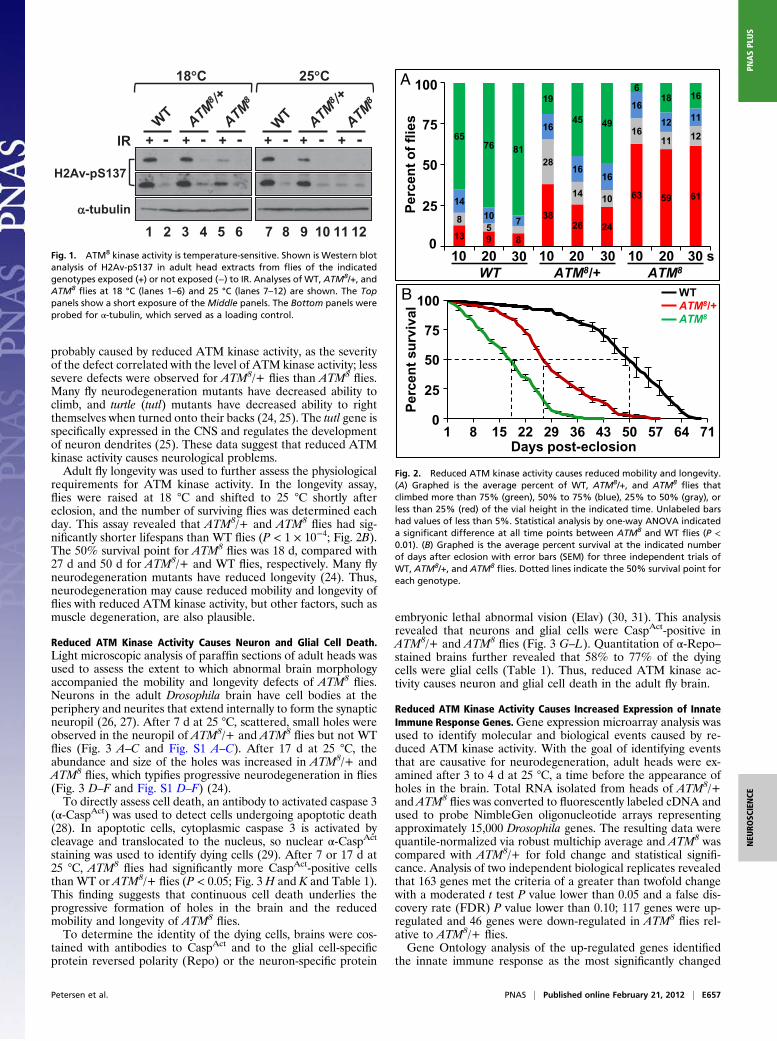
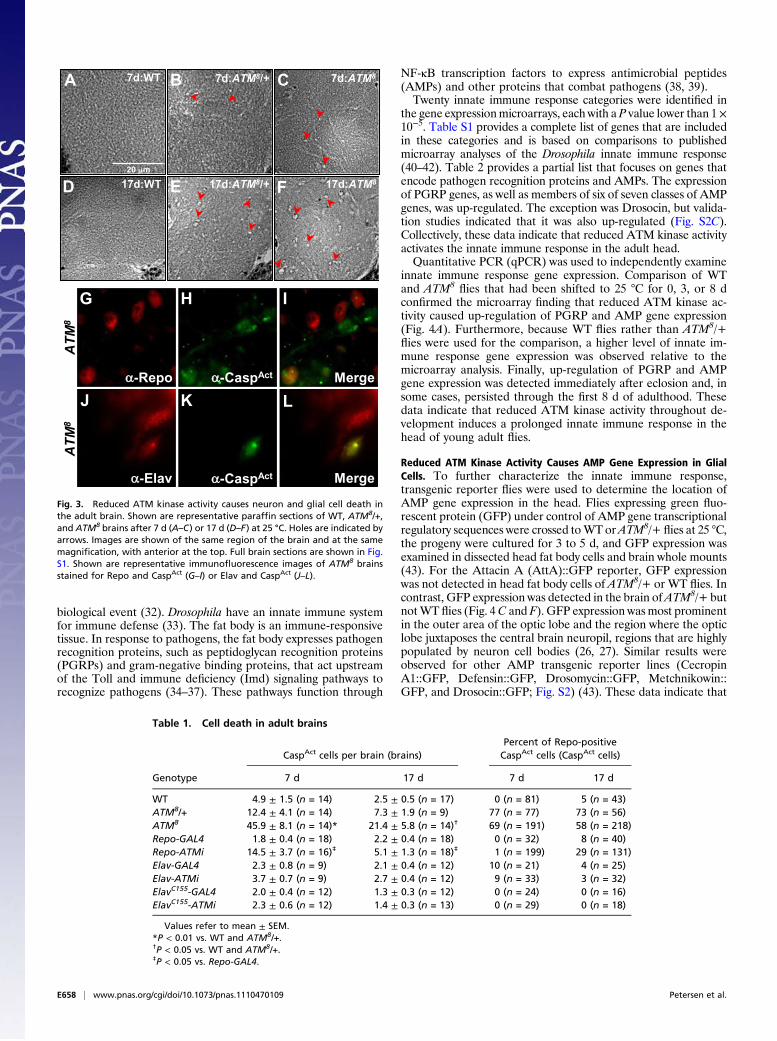
ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila Andrew J. Petersen, Stacey A. Rimkus, and David A. Wassarman 1 Department of Cell and Regenerative Biology, University of Wisconsin School of Medicine and Public Health, Madison, WI 53706 Edited by Ulrike A. Heberlein, University of California, San Francisco, CA, and approved January 30, 2012 (received for review June 29, 2011) To investigate the mechanistic basis for central nervous system (CNS) neurodegeneration in the disease ataxia–telangiectasia (A- T), we analyzed flies mutant for the causative gene A-T mutated (ATM). ATM encodes a protein kinase that functions to monitor the genomic integrity of cells and control cell cycle, DNA repair, and apoptosis programs. Mutation of the C-terminal amino acid in Drosophila ATM inhibited the kinase activity and caused neuron and glial cell death in the adult brain and a reduction in mobility and longevity. These data indicate that reduced ATM kinase activ- ity is sufficient to cause neurodegeneration in A-T. ATM kinase mutant flies also had elevated expression of innate immune re- sponse genes in glial cells. ATM knockdown in glial cells, but not neurons, was sufficient to cause neuron and glial cell death, a re- duction in mobility and longevity, and elevated expression of in- nate immune response genes in glial cells, indicating that a non– cell-autonomous mechanism contributes to neurodegeneration in A-T. Taken together, these data suggest that early-onset CNS neu- rodegeneration in A-T is similar to late-onset CNS neurodegenera- tion in diseases such as Alzheimer’s in which uncontrolled inflam- matory response mediated by glial cells drives neurodegeneration. A taxia–telangiectasia (A-T) is a multisystem disease charac- terized by neurodegeneration in the central nervous system (CNS) (1–3). Progressive neurodegeneration occurs during childhood and mainly affects Purkinje and granule cells in the cerebellum. A-T is caused by mutation of the A-T mutated (ATM) gene, which encodes a serine/threonine protein kinase of the PI3K-related kinase (PIKK) family (4). ATM functions to ensure genome integrity by controlling the cell cycle, DNA re- pair, and apoptosis programs in response to DNA damage (5, 6). ATM is recruited to sites of DNA damage, where autophos- phorylation activates the kinase activity to allow phosphorylation of many downstream targets, including histone H2AX (7–9). Insights into why ATM loss causes neurodegeneration have come from analyses of ATM-deficient animals. Analyses of postmortem human A-T brains, as well as mouse and fly models of A-T, indicate that ATM protects postmitotic neurons from degeneration by suppressing cell cycle reentry (10, 11). ATM- deficient neurons reenter the cell cycle and may die rather than arrest or divide because of excessive DNA damage. Many aspects of neurodegeneration are not understood, including whether it is solely caused by ATM loss in neurons (12). In neurodegenerative diseases such as Alzheimer’s and Parkinson disease, prolonged activation of microglia—the resident innate immune cells in the CNS—is thought to cause neurotoxicity (13–15). In addition, in neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), dysfunction of astrocytes, which serve to maintain neuron metabolism and neurotransmitter release, drives neurodegen- eration after onset (16). In the case of A-T, abnormal microglial cell morphology is observed in ATM knockout (KO) mice, and cultured astrocytes from ATM KO mice show markers of oxida- tive and endoplasmic reticulum stress and exhibit a senescence- like growth defect (17, 18). Furthermore, several lines of evidence suggest that systemic inflammation contributes to pathogenesis in patients with A-T (19, 20). Thus, ATM loss in glial cells may contribute to neurodegeneration in A-T. To investigate this possi- bility, we analyzed Drosophila melanogaster brains in which ATM kinase activity was reduced in all cells or ATM expression was specifically reduced in glial cells or neurons. Results ATM Kinase Activity Is Temperature-Sensitive in ATM 8 Flies. Because ATM is essential for fly viability to the adult stage, a temperature- sensitive allele of ATM (ATM 8 ) was used to investigate the role of ATM in adult flies (21). ATM 8 /ATM 8 flies (hereafter referred to as ATM 8 ) developed to the adult stage when raised at 18 °C but not at 25 °C. Thus, to characterize adult phenotypes, ATM 8 flies were raised at 18 °C and shifted to 25 °C shortly after eclosion. ATM 8 contains a leucine to phenylalanine mutation of the C-terminal amino acid, which is predicted to affect kinase activity but not protein stability (21, 22). Western blot analysis for histone H2Av phosphorylation (H2Av-pS137) revealed that ATM kinase activity in ATM 8 adult fly heads was temperature-sensitive (Fig. 1). H2Av is the Drosophila homologue of mammalian H2AX, an ATM substrate in response to ionizing radiation (IR)-induced DNA damage (23). At 18 °C, IR-induced H2Av phosphorylation was not affected in ATM 8 /+ flies but was highly reduced in ATM 8 flies relative to wildtype (WT) flies. At 25 °C, IR-induced H2Av phosphorylation was slightly reduced in ATM 8 /+ flies and was below the limit of detection in ATM 8 flies. These data indicate that ATM 8 kinase activity is temperature-sensitive and that ATM kinase activity in adult heads is correlated with ATM 8 allele dose. Reduced ATM Kinase Activity Reduces Mobility and Longevity. As previously reported by Pedersen et al., ATM 8 flies had mobility defects (21). They were less able to walk, fly, and right themselves when turned onto their backs. A climbing assay was used to quantify mobility. When tapped to the bottom of a vial, flies naturally respond by climbing to the top, referred to as negative geotaxis. Ten seconds after being tapped to the bottom of a vial, 65% of WT flies had climbed to the top quarter of the vial; however, only 19% of ATM 8 /+ and 6% of ATM 8 flies had climbed to the top quarter of the vial (Fig. 2A). Similar effects were ob- served if climbing was assessed at different heights in the vial or if climbing was assessed after longer recovery times (20 or 30 s), suggesting that the climbing defect was not a result of in- capacitation from the tapping action. The climbing defect was Author contributions: A.J.P., S.A.R., and D.A.W. designed research; A.J.P. and S.A.R. per- formed research; A.J.P. contributed new reagents/analytic tools; A.J.P., S.A.R., and D.A.W. analyzed data; and A.J.P., S.A.R., and D.A.W. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. Data deposition: The data reported in this paper have been deposited in the Gene Ex- pression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE34315). 1 To whom correspondence should be addressed. E-mail: [email protected]. See Author Summary on page 4039 (volume 109, number 11). This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10. 1073/pnas.1110470109/-/DCSupplemental. E656–E664 | PNAS | Published online February 21, 2012 www.pnas.org/cgi/doi/10.1073/pnas.1110470109
Transcript of ATM kinase inhibition in glial cells activates the innate … kinase inhibition in glial cells...

ATM kinase inhibition in glial cells activates the innateimmune response and causes neurodegenerationin DrosophilaAndrew J. Petersen, Stacey A. Rimkus, and David A. Wassarman1
Department of Cell and Regenerative Biology, University of Wisconsin School of Medicine and Public Health, Madison, WI 53706
Edited by Ulrike A. Heberlein, University of California, San Francisco, CA, and approved January 30, 2012 (received for review June 29, 2011)
To investigate the mechanistic basis for central nervous system(CNS) neurodegeneration in the disease ataxia–telangiectasia (A-T), we analyzed flies mutant for the causative gene A-T mutated(ATM). ATM encodes a protein kinase that functions to monitorthe genomic integrity of cells and control cell cycle, DNA repair,and apoptosis programs. Mutation of the C-terminal amino acid inDrosophila ATM inhibited the kinase activity and caused neuronand glial cell death in the adult brain and a reduction in mobilityand longevity. These data indicate that reduced ATM kinase activ-ity is sufficient to cause neurodegeneration in A-T. ATM kinasemutant flies also had elevated expression of innate immune re-sponse genes in glial cells. ATM knockdown in glial cells, but notneurons, was sufficient to cause neuron and glial cell death, a re-duction in mobility and longevity, and elevated expression of in-nate immune response genes in glial cells, indicating that a non–cell-autonomous mechanism contributes to neurodegeneration inA-T. Taken together, these data suggest that early-onset CNS neu-rodegeneration in A-T is similar to late-onset CNS neurodegenera-tion in diseases such as Alzheimer’s in which uncontrolled inflam-matory response mediated by glial cells drives neurodegeneration.
Ataxia–telangiectasia (A-T) is a multisystem disease charac-terized by neurodegeneration in the central nervous system
(CNS) (1–3). Progressive neurodegeneration occurs duringchildhood and mainly affects Purkinje and granule cells in thecerebellum. A-T is caused by mutation of the A-T mutated(ATM) gene, which encodes a serine/threonine protein kinase ofthe PI3K-related kinase (PIKK) family (4). ATM functions toensure genome integrity by controlling the cell cycle, DNA re-pair, and apoptosis programs in response to DNA damage (5, 6).ATM is recruited to sites of DNA damage, where autophos-phorylation activates the kinase activity to allow phosphorylationof many downstream targets, including histone H2AX (7–9).Insights into why ATM loss causes neurodegeneration have
come from analyses of ATM-deficient animals. Analyses ofpostmortem human A-T brains, as well as mouse and fly modelsof A-T, indicate that ATM protects postmitotic neurons fromdegeneration by suppressing cell cycle reentry (10, 11). ATM-deficient neurons reenter the cell cycle and may die rather thanarrest or divide because of excessive DNA damage. Many aspectsof neurodegeneration are not understood, including whether it issolely caused by ATM loss in neurons (12). In neurodegenerativediseases such as Alzheimer’s and Parkinson disease, prolongedactivation of microglia—the resident innate immune cells in theCNS—is thought to cause neurotoxicity (13–15). In addition, inneurodegenerative diseases such as amyotrophic lateral sclerosis(ALS), dysfunction of astrocytes, which serve to maintain neuronmetabolism and neurotransmitter release, drives neurodegen-eration after onset (16). In the case of A-T, abnormal microglialcell morphology is observed in ATM knockout (KO) mice, andcultured astrocytes from ATM KO mice show markers of oxida-tive and endoplasmic reticulum stress and exhibit a senescence-like growth defect (17, 18). Furthermore, several lines of evidencesuggest that systemic inflammation contributes to pathogenesisin patients with A-T (19, 20). Thus, ATM loss in glial cells may
contribute to neurodegeneration in A-T. To investigate this possi-bility, we analyzed Drosophila melanogaster brains in which ATMkinase activity was reduced in all cells or ATM expression wasspecifically reduced in glial cells or neurons.
ResultsATM Kinase Activity Is Temperature-Sensitive in ATM8 Flies. BecauseATM is essential for fly viability to the adult stage, a temperature-sensitive allele of ATM (ATM8) was used to investigate the role ofATM in adult flies (21). ATM8/ATM8
flies (hereafter referred toas ATM8) developed to the adult stage when raised at 18 °C butnot at 25 °C. Thus, to characterize adult phenotypes, ATM8
flieswere raised at 18 °C and shifted to 25 °C shortly after eclosion.ATM8 contains a leucine to phenylalanine mutation of the
C-terminal amino acid, which is predicted to affect kinase activitybut not protein stability (21, 22). Western blot analysis for histoneH2Av phosphorylation (H2Av-pS137) revealed that ATM kinaseactivity in ATM8 adult fly heads was temperature-sensitive (Fig.1). H2Av is the Drosophila homologue of mammalian H2AX, anATM substrate in response to ionizing radiation (IR)-inducedDNA damage (23). At 18 °C, IR-induced H2Av phosphorylationwas not affected in ATM8/+ flies but was highly reduced in ATM8
flies relative to wildtype (WT) flies. At 25 °C, IR-induced H2Avphosphorylation was slightly reduced in ATM8/+ flies and wasbelow the limit of detection in ATM8
flies. These data indicatethat ATM8 kinase activity is temperature-sensitive and that ATMkinase activity in adult heads is correlated with ATM8 allele dose.
Reduced ATM Kinase Activity Reduces Mobility and Longevity. Aspreviously reported by Pedersen et al., ATM8
flies had mobilitydefects (21). They were less able to walk, fly, and right themselveswhen turned onto their backs. A climbing assay was used toquantify mobility. When tapped to the bottom of a vial, fliesnaturally respond by climbing to the top, referred to as negativegeotaxis. Ten seconds after being tapped to the bottom of a vial,65% of WT flies had climbed to the top quarter of the vial;however, only 19% ofATM8/+ and 6% ofATM8
flies had climbedto the top quarter of the vial (Fig. 2A). Similar effects were ob-served if climbing was assessed at different heights in the vial or ifclimbing was assessed after longer recovery times (20 or 30 s),suggesting that the climbing defect was not a result of in-capacitation from the tapping action. The climbing defect was
Author contributions: A.J.P., S.A.R., and D.A.W. designed research; A.J.P. and S.A.R. per-formed research; A.J.P. contributed new reagents/analytic tools; A.J.P., S.A.R., and D.A.W.analyzed data; and A.J.P., S.A.R., and D.A.W. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Ex-pression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE34315).1To whom correspondence should be addressed. E-mail: [email protected].
See Author Summary on page 4039 (volume 109, number 11).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110470109/-/DCSupplemental.
E656–E664 | PNAS | Published online February 21, 2012 www.pnas.org/cgi/doi/10.1073/pnas.1110470109
probably caused by reduced ATM kinase activity, as the severityof the defect correlated with the level of ATM kinase activity; lesssevere defects were observed for ATM8/+ flies than ATM8
flies.Many fly neurodegeneration mutants have decreased ability toclimb, and turtle (tutl) mutants have decreased ability to rightthemselves when turned onto their backs (24, 25). The tutl gene isspecifically expressed in the CNS and regulates the developmentof neuron dendrites (25). These data suggest that reduced ATMkinase activity causes neurological problems.Adult fly longevity was used to further assess the physiological
requirements for ATM kinase activity. In the longevity assay,flies were raised at 18 °C and shifted to 25 °C shortly aftereclosion, and the number of surviving flies was determined eachday. This assay revealed that ATM8/+ and ATM8
flies had sig-nificantly shorter lifespans than WT flies (P < 1 × 10−4; Fig. 2B).The 50% survival point for ATM8
flies was 18 d, compared with27 d and 50 d for ATM8/+ and WT flies, respectively. Many flyneurodegeneration mutants have reduced longevity (24). Thus,neurodegeneration may cause reduced mobility and longevity offlies with reduced ATM kinase activity, but other factors, such asmuscle degeneration, are also plausible.
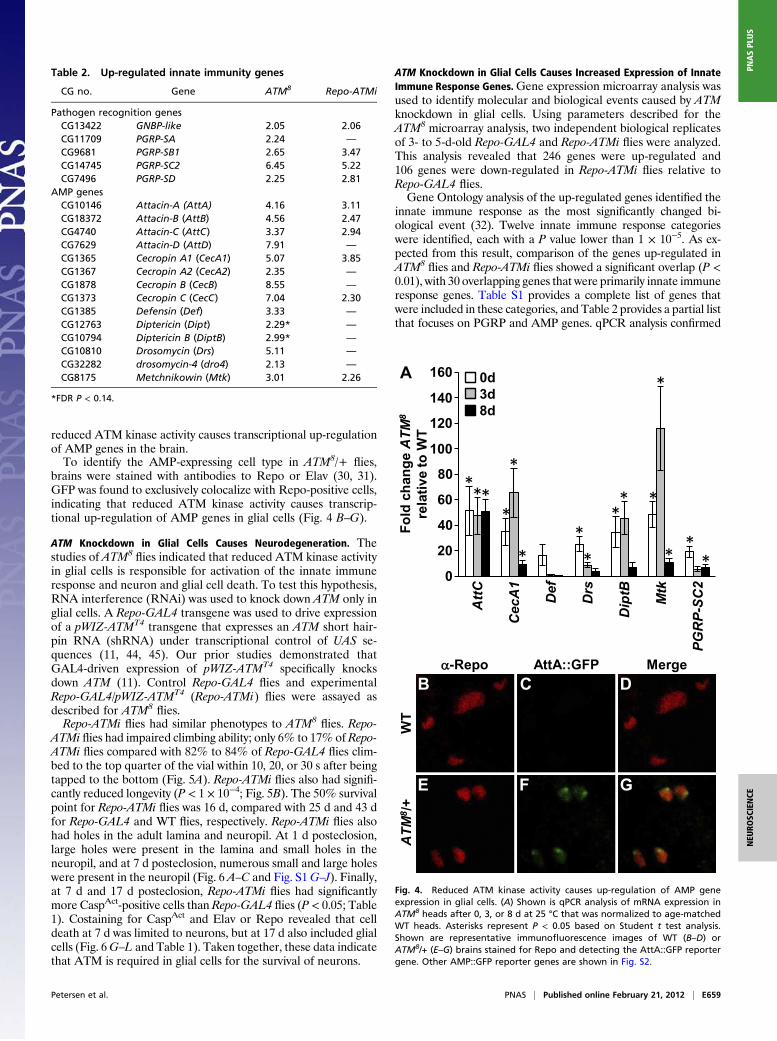
Reduced ATM Kinase Activity Causes Neuron and Glial Cell Death.Light microscopic analysis of paraffin sections of adult heads wasused to assess the extent to which abnormal brain morphologyaccompanied the mobility and longevity defects of ATM8
flies.Neurons in the adult Drosophila brain have cell bodies at theperiphery and neurites that extend internally to form the synapticneuropil (26, 27). After 7 d at 25 °C, scattered, small holes wereobserved in the neuropil of ATM8/+ and ATM8
flies but not WTflies (Fig. 3 A–C and Fig. S1 A–C). After 17 d at 25 °C, theabundance and size of the holes was increased in ATM8/+ andATM8
flies, which typifies progressive neurodegeneration in flies(Fig. 3 D–F and Fig. S1 D–F) (24).To directly assess cell death, an antibody to activated caspase 3
(α-CaspAct) was used to detect cells undergoing apoptotic death(28). In apoptotic cells, cytoplasmic caspase 3 is activated bycleavage and translocated to the nucleus, so nuclear α-CaspAct
staining was used to identify dying cells (29). After 7 or 17 d at25 °C, ATM8
flies had significantly more CaspAct-positive cellsthan WT or ATM8/+ flies (P < 0.05; Fig. 3 H and K and Table 1).This finding suggests that continuous cell death underlies theprogressive formation of holes in the brain and the reducedmobility and longevity of ATM8
flies.To determine the identity of the dying cells, brains were cos-
tained with antibodies to CaspAct and to the glial cell-specificprotein reversed polarity (Repo) or the neuron-specific protein
embryonic lethal abnormal vision (Elav) (30, 31). This analysisrevealed that neurons and glial cells were CaspAct-positive inATM8/+ and ATM8
flies (Fig. 3 G–L). Quantitation of α-Repo–stained brains further revealed that 58% to 77% of the dyingcells were glial cells (Table 1). Thus, reduced ATM kinase ac-tivity causes neuron and glial cell death in the adult fly brain.
Reduced ATM Kinase Activity Causes Increased Expression of InnateImmune Response Genes.Gene expression microarray analysis wasused to identify molecular and biological events caused by re-duced ATM kinase activity. With the goal of identifying eventsthat are causative for neurodegeneration, adult heads were ex-amined after 3 to 4 d at 25 °C, a time before the appearance ofholes in the brain. Total RNA isolated from heads of ATM8/+and ATM8
flies was converted to fluorescently labeled cDNA andused to probe NimbleGen oligonucleotide arrays representingapproximately 15,000 Drosophila genes. The resulting data werequantile-normalized via robust multichip average and ATM8 wascompared with ATM8/+ for fold change and statistical signifi-cance. Analysis of two independent biological replicates revealedthat 163 genes met the criteria of a greater than twofold changewith a moderated t test P value lower than 0.05 and a false dis-covery rate (FDR) P value lower than 0.10; 117 genes were up-regulated and 46 genes were down-regulated in ATM8
flies rel-ative to ATM8/+ flies.Gene Ontology analysis of the up-regulated genes identified
the innate immune response as the most significantly changed
-tubulin
H2Av-pS137
+ -+IR - -+ + -+- -+
1 2 3 4 5 6 7 8 9 10 11 12
18 C 25 C
Fig. 1. ATM8 kinase activity is temperature-sensitive. Shown is Western blotanalysis of H2Av-pS137 in adult head extracts from flies of the indicatedgenotypes exposed (+) or not exposed (−) to IR. Analyses of WT, ATM8/+, andATM8
flies at 18 °C (lanes 1–6) and 25 °C (lanes 7–12) are shown. The Toppanels show a short exposure of the Middle panels. The Bottom panels wereprobed for α-tubulin, which served as a loading control.
13 9 8
3826 24
63 59 61
85
28
14 10
1611 12
14
10 7
16
1616
16
12 11
6576 81
19
45 49
618 16
10 20 30 10 20 30 10 20 30 sWT ATM8/+ ATM8
100
75
50
25Per
cen
t o
f fl
ies
1 8 15 22 29 36 43 50 57 64 71
0
B
A
Per
cen
t su
rviv
al
Days post-eclosion
WTATM8/+ATM8
100
75
50
25
0
Fig. 2. Reduced ATM kinase activity causes reduced mobility and longevity.(A) Graphed is the average percent of WT, ATM8/+, and ATM8
flies thatclimbed more than 75% (green), 50% to 75% (blue), 25% to 50% (gray), orless than 25% (red) of the vial height in the indicated time. Unlabeled barshad values of less than 5%. Statistical analysis by one-way ANOVA indicateda significant difference at all time points between ATM8 and WT flies (P <0.01). (B) Graphed is the average percent survival at the indicated numberof days after eclosion with error bars (SEM) for three independent trials ofWT, ATM8/+, and ATM8
flies. Dotted lines indicate the 50% survival point foreach genotype.
Petersen et al. PNAS | Published online February 21, 2012 | E657
NEU
ROSC
IENCE
PNASPL
US
biological event (32). Drosophila have an innate immune systemfor immune defense (33). The fat body is an immune-responsivetissue. In response to pathogens, the fat body expresses pathogenrecognition proteins, such as peptidoglycan recognition proteins(PGRPs) and gram-negative binding proteins, that act upstreamof the Toll and immune deficiency (Imd) signaling pathways torecognize pathogens (34–37). These pathways function through
NF-κB transcription factors to express antimicrobial peptides(AMPs) and other proteins that combat pathogens (38, 39).Twenty innate immune response categories were identified in
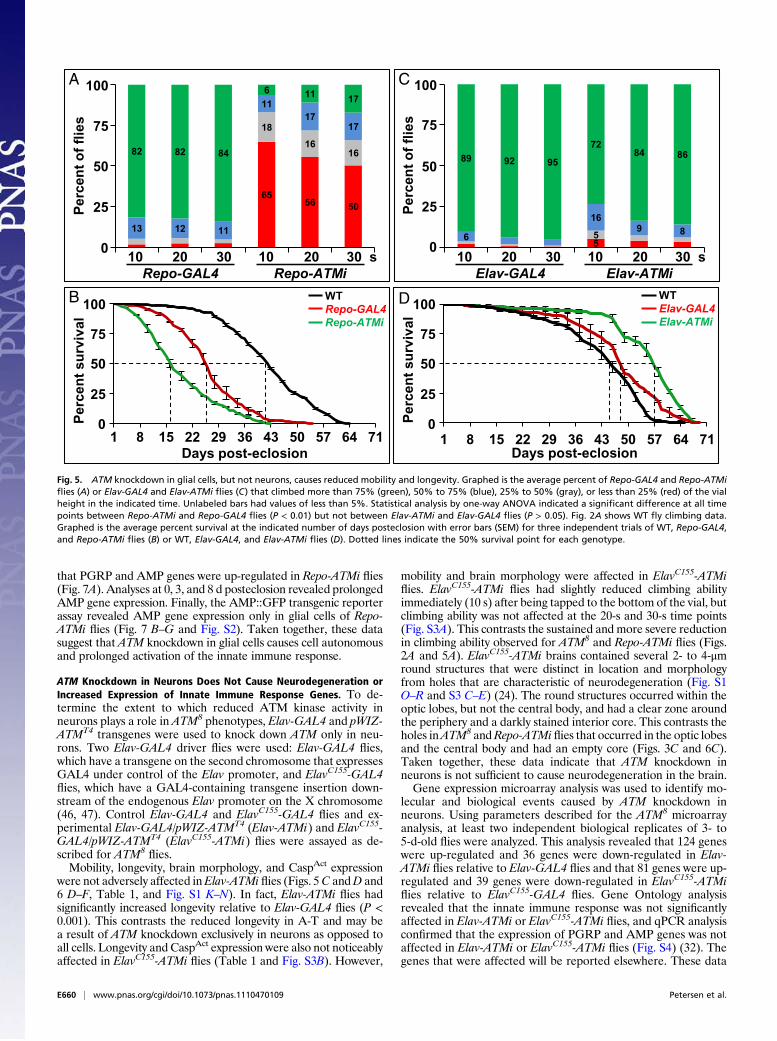
the gene expressionmicroarrays, each with a P value lower than 1×10−5. Table S1 provides a complete list of genes that are includedin these categories and is based on comparisons to publishedmicroarray analyses of the Drosophila innate immune response(40–42). Table 2 provides a partial list that focuses on genes thatencode pathogen recognition proteins and AMPs. The expressionof PGRP genes, as well as members of six of seven classes of AMPgenes, was up-regulated. The exception was Drosocin, but valida-tion studies indicated that it was also up-regulated (Fig. S2C).Collectively, these data indicate that reduced ATM kinase activityactivates the innate immune response in the adult head.Quantitative PCR (qPCR) was used to independently examine
innate immune response gene expression. Comparison of WTand ATM8
flies that had been shifted to 25 °C for 0, 3, or 8 dconfirmed the microarray finding that reduced ATM kinase ac-tivity caused up-regulation of PGRP and AMP gene expression(Fig. 4A). Furthermore, because WT flies rather than ATM8/+flies were used for the comparison, a higher level of innate im-mune response gene expression was observed relative to themicroarray analysis. Finally, up-regulation of PGRP and AMPgene expression was detected immediately after eclosion and, insome cases, persisted through the first 8 d of adulthood. Thesedata indicate that reduced ATM kinase activity throughout de-velopment induces a prolonged innate immune response in thehead of young adult flies.
Reduced ATM Kinase Activity Causes AMP Gene Expression in GlialCells. To further characterize the innate immune response,transgenic reporter flies were used to determine the location ofAMP gene expression in the head. Flies expressing green fluo-rescent protein (GFP) under control of AMP gene transcriptionalregulatory sequences were crossed toWT orATM8/+ flies at 25 °C,the progeny were cultured for 3 to 5 d, and GFP expression wasexamined in dissected head fat body cells and brain whole mounts(43). For the Attacin A (AttA)::GFP reporter, GFP expressionwas not detected in head fat body cells of ATM8/+ or WT flies. Incontrast, GFP expression was detected in the brain ofATM8/+ butnotWT flies (Fig. 4C and F). GFP expression wasmost prominentin the outer area of the optic lobe and the region where the opticlobe juxtaposes the central brain neuropil, regions that are highlypopulated by neuron cell bodies (26, 27). Similar results wereobserved for other AMP transgenic reporter lines (CecropinA1::GFP, Defensin::GFP, Drosomycin::GFP, Metchnikowin::GFP, and Drosocin::GFP; Fig. S2) (43). These data indicate that
Table 1. Cell death in adult brains
Genotype
CaspAct cells per brain (brains)Percent of Repo-positiveCaspAct cells (CaspAct cells)
7 d 17 d 7 d 17 d
WT 4.9 ± 1.5 (n = 14) 2.5 ± 0.5 (n = 17) 0 (n = 81) 5 (n = 43)ATM8/+ 12.4 ± 4.1 (n = 14) 7.3 ± 1.9 (n = 9) 77 (n = 77) 73 (n = 56)ATM8 45.9 ± 8.1 (n = 14)* 21.4 ± 5.8 (n = 14)† 69 (n = 191) 58 (n = 218)Repo-GAL4 1.8 ± 0.4 (n = 18) 2.2 ± 0.4 (n = 18) 0 (n = 32) 8 (n = 40)Repo-ATMi 14.5 ± 3.7 (n = 16)‡ 5.1 ± 1.3 (n = 18)‡ 1 (n = 199) 29 (n = 131)Elav-GAL4 2.3 ± 0.8 (n = 9) 2.1 ± 0.4 (n = 12) 10 (n = 21) 4 (n = 25)Elav-ATMi 3.7 ± 0.7 (n = 9) 2.7 ± 0.4 (n = 12) 9 (n = 33) 3 (n = 32)ElavC155-GAL4 2.0 ± 0.4 (n = 12) 1.3 ± 0.3 (n = 12) 0 (n = 24) 0 (n = 16)ElavC155-ATMi 2.3 ± 0.6 (n = 12) 1.4 ± 0.3 (n = 13) 0 (n = 29) 0 (n = 18)
Values refer to mean ± SEM.*P < 0.01 vs. WT and ATM8/+.†P < 0.05 vs. WT and ATM8/+.‡P < 0.05 vs. Repo-GAL4.
7d:WT
20 m
7d:ATM8
17d:ATM817d:WT
7d:ATM8/+
17d:ATM8/+
-Repo Merge-CaspAct
Merge-Elav -CaspAct
A C
F
B
ED
IHG
LKJ
AT
M8
AT
M8
Fig. 3. Reduced ATM kinase activity causes neuron and glial cell death inthe adult brain. Shown are representative paraffin sections of WT, ATM8/+,and ATM8 brains after 7 d (A–C) or 17 d (D–F) at 25 °C. Holes are indicated byarrows. Images are shown of the same region of the brain and at the samemagnification, with anterior at the top. Full brain sections are shown in Fig.S1. Shown are representative immunofluorescence images of ATM8 brainsstained for Repo and CaspAct (G–I) or Elav and CaspAct (J–L).
E658 | www.pnas.org/cgi/doi/10.1073/pnas.1110470109 Petersen et al.
reduced ATM kinase activity causes transcriptional up-regulationof AMP genes in the brain.To identify the AMP-expressing cell type in ATM8/+ flies,
brains were stained with antibodies to Repo or Elav (30, 31).GFP was found to exclusively colocalize with Repo-positive cells,indicating that reduced ATM kinase activity causes transcrip-tional up-regulation of AMP genes in glial cells (Fig. 4 B–G).
ATM Knockdown in Glial Cells Causes Neurodegeneration. Thestudies of ATM8
flies indicated that reduced ATM kinase activityin glial cells is responsible for activation of the innate immuneresponse and neuron and glial cell death. To test this hypothesis,RNA interference (RNAi) was used to knock down ATM only inglial cells. A Repo-GAL4 transgene was used to drive expressionof a pWIZ-ATMT4 transgene that expresses an ATM short hair-pin RNA (shRNA) under transcriptional control of UAS se-quences (11, 44, 45). Our prior studies demonstrated thatGAL4-driven expression of pWIZ-ATMT4 specifically knocksdown ATM (11). Control Repo-GAL4 flies and experimentalRepo-GAL4/pWIZ-ATMT4 (Repo-ATMi) flies were assayed asdescribed for ATM8
flies.Repo-ATMi flies had similar phenotypes to ATM8
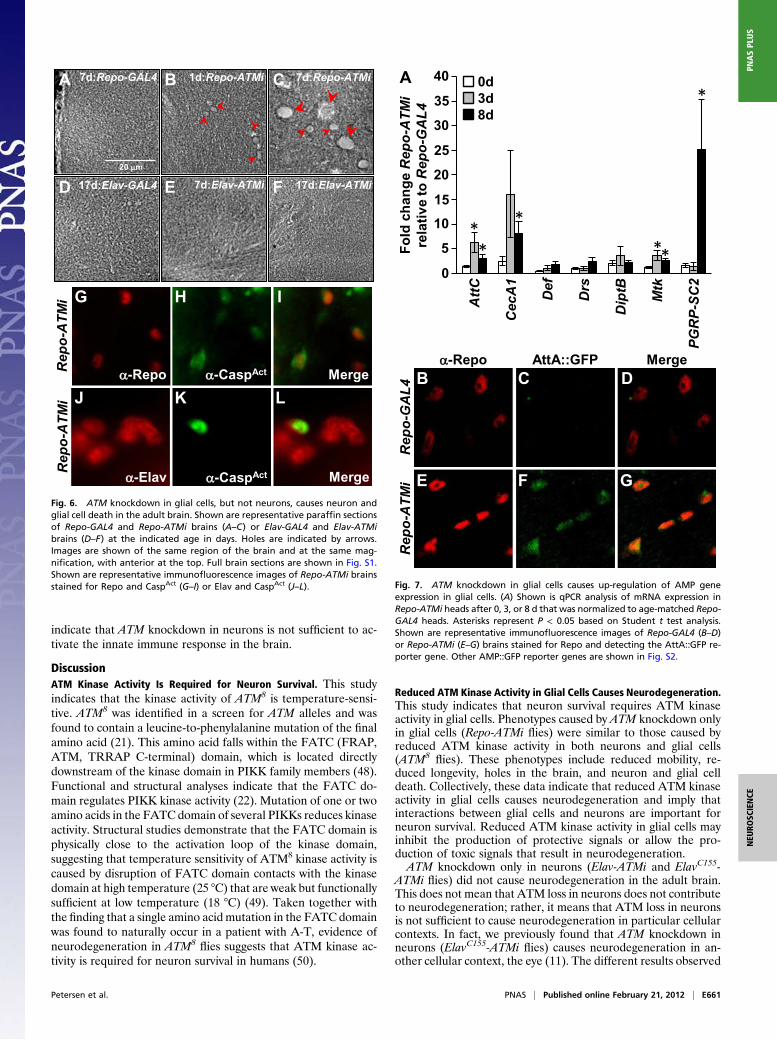
flies. Repo-ATMi flies had impaired climbing ability; only 6% to 17% of Repo-ATMi flies compared with 82% to 84% of Repo-GAL4 flies clim-bed to the top quarter of the vial within 10, 20, or 30 s after beingtapped to the bottom (Fig. 5A). Repo-ATMi flies also had signifi-cantly reduced longevity (P < 1 × 10−4; Fig. 5B). The 50% survivalpoint for Repo-ATMi flies was 16 d, compared with 25 d and 43 dfor Repo-GAL4 and WT flies, respectively. Repo-ATMi flies alsohad holes in the adult lamina and neuropil. At 1 d posteclosion,large holes were present in the lamina and small holes in theneuropil, and at 7 d posteclosion, numerous small and large holeswere present in the neuropil (Fig. 6 A–C and Fig. S1G–J). Finally,at 7 d and 17 d posteclosion, Repo-ATMi flies had significantlymore CaspAct-positive cells than Repo-GAL4 flies (P < 0.05; Table1). Costaining for CaspAct and Elav or Repo revealed that celldeath at 7 d was limited to neurons, but at 17 d also included glialcells (Fig. 6G–L and Table 1). Taken together, these data indicatethat ATM is required in glial cells for the survival of neurons.
ATM Knockdown in Glial Cells Causes Increased Expression of InnateImmune Response Genes.Gene expression microarray analysis wasused to identify molecular and biological events caused by ATMknockdown in glial cells. Using parameters described for theATM8 microarray analysis, two independent biological replicatesof 3- to 5-d-old Repo-GAL4 and Repo-ATMi flies were analyzed.This analysis revealed that 246 genes were up-regulated and106 genes were down-regulated in Repo-ATMi flies relative toRepo-GAL4 flies.Gene Ontology analysis of the up-regulated genes identified the
innate immune response as the most significantly changed bi-ological event (32). Twelve innate immune response categorieswere identified, each with a P value lower than 1 × 10−5. As ex-pected from this result, comparison of the genes up-regulated inATM8
flies and Repo-ATMi flies showed a significant overlap (P <0.01), with 30 overlapping genes that were primarily innate immuneresponse genes. Table S1 provides a complete list of genes thatwere included in these categories, and Table 2 provides a partial listthat focuses on PGRP and AMP genes. qPCR analysis confirmed
Att
C
Cec
A1
Def
Drs
Dip
tB
Mtk
PG
RP
-SC
2
0d3d8d
**
*
*
***
*
*
*
***
**
-Repo AttA::GFP Merge
A
B C D
E F G
WT
AT
M8 /
+F
old
ch
ang
e A
TM
8
rela
tive
to W
T
0
20
40
60
80
100
120
140
160
Fig. 4. Reduced ATM kinase activity causes up-regulation of AMP geneexpression in glial cells. (A) Shown is qPCR analysis of mRNA expression inATM8 heads after 0, 3, or 8 d at 25 °C that was normalized to age-matchedWT heads. Asterisks represent P < 0.05 based on Student t test analysis.Shown are representative immunofluorescence images of WT (B–D) orATM8/+ (E–G) brains stained for Repo and detecting the AttA::GFP reportergene. Other AMP::GFP reporter genes are shown in Fig. S2.
Table 2. Up-regulated innate immunity genes
CG no. Gene ATM8 Repo-ATMi
Pathogen recognition genesCG13422 GNBP-like 2.05 2.06CG11709 PGRP-SA 2.24 —
CG9681 PGRP-SB1 2.65 3.47CG14745 PGRP-SC2 6.45 5.22CG7496 PGRP-SD 2.25 2.81
AMP genesCG10146 Attacin-A (AttA) 4.16 3.11CG18372 Attacin-B (AttB) 4.56 2.47CG4740 Attacin-C (AttC) 3.37 2.94CG7629 Attacin-D (AttD) 7.91 —
CG1365 Cecropin A1 (CecA1) 5.07 3.85CG1367 Cecropin A2 (CecA2) 2.35 —
CG1878 Cecropin B (CecB) 8.55 —
CG1373 Cecropin C (CecC) 7.04 2.30CG1385 Defensin (Def) 3.33 —
CG12763 Diptericin (Dipt) 2.29* —
CG10794 Diptericin B (DiptB) 2.99* —
CG10810 Drosomycin (Drs) 5.11 —
CG32282 drosomycin-4 (dro4) 2.13 —
CG8175 Metchnikowin (Mtk) 3.01 2.26
*FDR P < 0.14.
Petersen et al. PNAS | Published online February 21, 2012 | E659
NEU
ROSC
IENCE
PNASPL
US
that PGRP and AMP genes were up-regulated in Repo-ATMi flies(Fig. 7A). Analyses at 0, 3, and 8 d posteclosion revealed prolongedAMP gene expression. Finally, the AMP::GFP transgenic reporterassay revealed AMP gene expression only in glial cells of Repo-ATMi flies (Fig. 7 B–G and Fig. S2). Taken together, these datasuggest that ATM knockdown in glial cells causes cell autonomousand prolonged activation of the innate immune response.
ATM Knockdown in Neurons Does Not Cause Neurodegeneration orIncreased Expression of Innate Immune Response Genes. To de-termine the extent to which reduced ATM kinase activity inneurons plays a role in ATM8 phenotypes, Elav-GAL4 and pWIZ-ATMT4 transgenes were used to knock down ATM only in neu-rons. Two Elav-GAL4 driver flies were used: Elav-GAL4 flies,which have a transgene on the second chromosome that expressesGAL4 under control of the Elav promoter, and ElavC155-GAL4flies, which have a GAL4-containing transgene insertion down-stream of the endogenous Elav promoter on the X chromosome(46, 47). Control Elav-GAL4 and ElavC155-GAL4 flies and ex-perimental Elav-GAL4/pWIZ-ATMT4 (Elav-ATMi) and ElavC155-GAL4/pWIZ-ATMT4 (ElavC155-ATMi) flies were assayed as de-scribed for ATM8
flies.Mobility, longevity, brain morphology, and CaspAct expression
were not adversely affected inElav-ATMi flies (Figs. 5C andD and6 D–F, Table 1, and Fig. S1 K–N). In fact, Elav-ATMi flies hadsignificantly increased longevity relative to Elav-GAL4 flies (P <0.001). This contrasts the reduced longevity in A-T and may bea result of ATM knockdown exclusively in neurons as opposed toall cells. Longevity and CaspAct expression were also not noticeablyaffected in ElavC155-ATMi flies (Table 1 and Fig. S3B). However,
mobility and brain morphology were affected in ElavC155-ATMiflies. ElavC155-ATMi flies had slightly reduced climbing abilityimmediately (10 s) after being tapped to the bottom of the vial, butclimbing ability was not affected at the 20-s and 30-s time points(Fig. S3A). This contrasts the sustained and more severe reductionin climbing ability observed for ATM8 and Repo-ATMi flies (Figs.2A and 5A). ElavC155-ATMi brains contained several 2- to 4-μmround structures that were distinct in location and morphologyfrom holes that are characteristic of neurodegeneration (Fig. S1O–R and S3 C–E) (24). The round structures occurred within theoptic lobes, but not the central body, and had a clear zone aroundthe periphery and a darkly stained interior core. This contrasts theholes inATM8 andRepo-ATMi flies that occurred in the optic lobesand the central body and had an empty core (Figs. 3C and 6C).Taken together, these data indicate that ATM knockdown inneurons is not sufficient to cause neurodegeneration in the brain.Gene expression microarray analysis was used to identify mo-
lecular and biological events caused by ATM knockdown inneurons. Using parameters described for the ATM8 microarrayanalysis, at least two independent biological replicates of 3- to5-d-old flies were analyzed. This analysis revealed that 124 geneswere up-regulated and 36 genes were down-regulated in Elav-ATMi flies relative to Elav-GAL4 flies and that 81 genes were up-regulated and 39 genes were down-regulated in ElavC155-ATMiflies relative to ElavC155-GAL4 flies. Gene Ontology analysisrevealed that the innate immune response was not significantlyaffected in Elav-ATMi or ElavC155-ATMi flies, and qPCR analysisconfirmed that the expression of PGRP and AMP genes was notaffected in Elav-ATMi or ElavC155-ATMi flies (Fig. S4) (32). Thegenes that were affected will be reported elsewhere. These data
1 8 15 22 29 36 43 50 57 64 711 8 15 22 29 36 43 50 57 64 71
WTRepo-GAL4Repo-ATMi
6556 50
18
1616
13 12 11
1117
17
82 82 84
6 11 17
10 20 30 10 20 30 sRepo-GAL4
Per
cen
t o
f fl
ies
100
75
50
25
0
Repo-ATMi
Per
cen
t su
rviv
al
WTElav-GAL4Elav-ATMi
556
169 8
89 92 95
7284 86
10 20 30 10 20 30 sElav-GAL4
Per
cen
t o
f fl
ies
Elav-ATMi
100
75
50
25
0
A
D
C
B
Per
cen
t su
rviv
al
Days post-eclosion
100
75
50
25
0
100
75
50
25
0
Days post-eclosion
Fig. 5. ATM knockdown in glial cells, but not neurons, causes reduced mobility and longevity. Graphed is the average percent of Repo-GAL4 and Repo-ATMiflies (A) or Elav-GAL4 and Elav-ATMi flies (C) that climbed more than 75% (green), 50% to 75% (blue), 25% to 50% (gray), or less than 25% (red) of the vialheight in the indicated time. Unlabeled bars had values of less than 5%. Statistical analysis by one-way ANOVA indicated a significant difference at all timepoints between Repo-ATMi and Repo-GAL4 flies (P < 0.01) but not between Elav-ATMi and Elav-GAL4 flies (P > 0.05). Fig. 2A shows WT fly climbing data.Graphed is the average percent survival at the indicated number of days posteclosion with error bars (SEM) for three independent trials of WT, Repo-GAL4,and Repo-ATMi flies (B) or WT, Elav-GAL4, and Elav-ATMi flies (D). Dotted lines indicate the 50% survival point for each genotype.
E660 | www.pnas.org/cgi/doi/10.1073/pnas.1110470109 Petersen et al.
indicate that ATM knockdown in neurons is not sufficient to ac-tivate the innate immune response in the brain.
DiscussionATM Kinase Activity Is Required for Neuron Survival. This studyindicates that the kinase activity of ATM8 is temperature-sensi-tive. ATM8 was identified in a screen for ATM alleles and wasfound to contain a leucine-to-phenylalanine mutation of the finalamino acid (21). This amino acid falls within the FATC (FRAP,ATM, TRRAP C-terminal) domain, which is located directlydownstream of the kinase domain in PIKK family members (48).Functional and structural analyses indicate that the FATC do-main regulates PIKK kinase activity (22). Mutation of one or twoamino acids in the FATC domain of several PIKKs reduces kinaseactivity. Structural studies demonstrate that the FATC domain isphysically close to the activation loop of the kinase domain,suggesting that temperature sensitivity of ATM8 kinase activity iscaused by disruption of FATC domain contacts with the kinasedomain at high temperature (25 °C) that are weak but functionallysufficient at low temperature (18 °C) (49). Taken together withthe finding that a single amino acid mutation in the FATC domainwas found to naturally occur in a patient with A-T, evidence ofneurodegeneration in ATM8
flies suggests that ATM kinase ac-tivity is required for neuron survival in humans (50).
Reduced ATM Kinase Activity in Glial Cells Causes Neurodegeneration.This study indicates that neuron survival requires ATM kinaseactivity in glial cells. Phenotypes caused by ATM knockdown onlyin glial cells (Repo-ATMi flies) were similar to those caused byreduced ATM kinase activity in both neurons and glial cells(ATM8
flies). These phenotypes include reduced mobility, re-duced longevity, holes in the brain, and neuron and glial celldeath. Collectively, these data indicate that reduced ATM kinaseactivity in glial cells causes neurodegeneration and imply thatinteractions between glial cells and neurons are important forneuron survival. Reduced ATM kinase activity in glial cells mayinhibit the production of protective signals or allow the pro-duction of toxic signals that result in neurodegeneration.ATM knockdown only in neurons (Elav-ATMi and ElavC155-
ATMi flies) did not cause neurodegeneration in the adult brain.This does not mean that ATM loss in neurons does not contributeto neurodegeneration; rather, it means that ATM loss in neuronsis not sufficient to cause neurodegeneration in particular cellularcontexts. In fact, we previously found that ATM knockdown inneurons (ElavC155-ATMi flies) causes neurodegeneration in an-other cellular context, the eye (11). The different results observed
Rep
o-A
TM
i
G H I
J LK
FD 17d:Elav-GAL4 17d:Elav-ATMiE 7d:Elav-ATMi
A B C
20 m
7d:Repo-GAL4 7d:Repo-ATMi1d:Repo-ATMiR
epo
-AT
Mi
-Repo Merge-CaspAct
Merge-Elav -CaspAct
Fig. 6. ATM knockdown in glial cells, but not neurons, causes neuron andglial cell death in the adult brain. Shown are representative paraffin sectionsof Repo-GAL4 and Repo-ATMi brains (A–C) or Elav-GAL4 and Elav-ATMibrains (D–F) at the indicated age in days. Holes are indicated by arrows.Images are shown of the same region of the brain and at the same mag-nification, with anterior at the top. Full brain sections are shown in Fig. S1.Shown are representative immunofluorescence images of Repo-ATMi brainsstained for Repo and CaspAct (G–I) or Elav and CaspAct (J–L).
Rep
o-G
AL
4R
epo
-AT
Mi E
DCB
GF
0
5
10
15
20
25
30
35
40
Att
C
Cec
A1
Def
Drs
Dip
tB
Mtk
PG
RP
- SC
2
Fo
ld c
han
ge
Rep
o-A
TM
ire
lati
ve to
Rep
o-G
AL
4
0d3d8d
*
*
*
***
A
-Repo AttA::GFP Merge
0
5
10
20
30
40
15
25
35
Fig. 7. ATM knockdown in glial cells causes up-regulation of AMP geneexpression in glial cells. (A) Shown is qPCR analysis of mRNA expression inRepo-ATMi heads after 0, 3, or 8 d that was normalized to age-matched Repo-GAL4 heads. Asterisks represent P < 0.05 based on Student t test analysis.Shown are representative immunofluorescence images of Repo-GAL4 (B–D)or Repo-ATMi (E–G) brains stained for Repo and detecting the AttA::GFP re-porter gene. Other AMP::GFP reporter genes are shown in Fig. S2.
Petersen et al. PNAS | Published online February 21, 2012 | E661
NEU
ROSC
IENCE
PNASPL
US

in the brain and the eye may result from different levels of ATMknockdown. Alternatively, the different results may result fromunique properties of neurons, glial cells, or other cells in the brainor eye. This alternative explanation is attractive because, in A-Tand other neurodegenerative diseases, neuron types differ in theirsusceptibility to degeneration, with Purkinje and granule neuronsbeing particularly susceptible in A-T (1, 2).
Reduced ATM Kinase Activity Triggers an Innate Immune Response inGlial Cells. This study indicates that glial cells in the DrosophilaCNS produce an innate immune response. This adds glial cells inthe CNS to the existing list of innate immune response-compe-tent cells that includes surface epithelia and the fat body in adultlabellar glands, midgut, malpighian tubules, trachea, and maleand female reproductive tracts (43, 51). However, among theidentified types of glial cells, the relevant type remains to bedetermined (52, 53). Additionally, the involvement of the Tolland Imd pathways in the glial cell innate immune responseremains to be determined. None of the components of thesepathways, including the NF-κB genes that are transcriptionallyup-regulated in response to microbial infection, was found tohave altered expression in the ATM8 or Repo-ATMi microarrays(40, 41). Activation of AMP gene expression independently ofthe Toll and Imd pathways is not unprecedented. FOXO andGATA transcription factors have been shown to mediate theinnate immune response independently of the pathogen-re-sponsive innate immunity pathways (54, 55). Future studies willassess the details of the glial cell innate immune response.Increased expression of innate immune response genes may
directly result from reduced ATM kinase activity or indirectlyresult from increased susceptibility to infection or altered physi-ology. Data presented here are consistent with a model in whichATM directly represses the innate immune response throughprotein phosphorylation. However, there are no obvious candi-dates for substrates. ATM-dependent phosphorylation is essen-tial for activation of NF-κB in response to DNA damage (56).Although NF-κB proteins are also activated in the innate immuneresponse, results presented here suggest that ATM-dependentphosphorylation inhibits NF-κB proteins, as reduced ATM kinaseactivity activated the innate immune response. In regard to anindirect mechanism, AMP gene expression in glial cells has notbeen observed in response to infection or to physiological per-turbations such as starvation, altered circadian rhythm, and al-tered sleep pattern that up-regulate AMP gene expression in thefat body (42, 57, 58). However, AMP gene expression in glial cellsmay not have been adequately examined under these conditions.Finally, prolonged expression of AMP genes in ATM8
flies arguesagainst a burst of pathogen growth that is fought back, but it doesnot rule out persistent infection. Future studies will establish therole of ATM in regulating the glial cell innate immune response.
Activation of Innate Immune Response May Be a Common Feature ofNeurodegeneration. This study revealed a correlation betweenneurodegeneration and the innate immune response in a fly modelof the human neurodegenerative disease A-T. A link betweenneurodegeneration and the innate immune response may be acommon phenomenon in flies. Innate immune response genes areup-regulated in a fly model of Parkinson disease, and the Tollpathway mediates neurodegeneration in a fly model of Alz-heimer’s disease (59, 60). Moreover, studies in mammalian sys-tems have uncovered considerable evidence linking the innateimmune response and neurodegeneration in a variety of humanneurological disorders, including Alzheimer’s disease, Parkinsondisease, Huntington disease, multiple sclerosis, and ALS (14, 15,61). In mammals, microglial cells are responsible for the innateimmune response. They recognize pathogens through their Toll-like receptors, leading to release of proinflammatory cytokinesincluding Tumor necrosis factor-alpha (TNF-α) (62). Current
thinking is that prolonged activation of microglial cells is a causa-tive factor for neurodegeneration. For example, in Alzheimer’sdisease, amyloid-β activatesmicroglia to release neurotoxic factorssuch as TNF-α and reactive oxygen species, and in Parkinsondisease, overactivated microglia produce reactive oxygen speciesin response to damaged ascending dopaminergic neurons (63, 64).The discovery of a glial cell immune response inDrosophilamakesthis model organism well suited for studying the effects of neu-roinflammation on neuron survival.
Materials and MethodsDrosophila Genetics. Flies were maintained on standard molasses medium at25 °C unless otherwise stated. For all experiments, WT flies were w1118.pWIZ-ATMT4 is described by Rimkus et al. (2008) (11). ATM8, Repo-GAL4, andElav-GAL4 flies were obtained from the Bloomington Stock Center; ElavC155-GAL4 flies were obtained from Barry Ganetzky (University of Wisconsin,Madison, WI); and AMP::GFP flies were obtained from Ylva Engstrom(Stockholm University, Stockholm, Sweden) (12, 43, 44, 46, 47). Repo-ATMiflies were generated by recombining the Repo-GAL4 and the pWIZ-ATMT4
transgenes onto the same chromosome. To generate ATM8flies, ATM8/TM3
flies were raised at 18 °C and their progeny were screened for the absence ofTM3. For ATM8 experiments, flies were cultured at 18 °C until 0 to 3 dposteclosion and then transferred to 25 °C for the indicated time.
Western Blot Analysis. For analysis of ATM kinase activity, vials containing 10flies (4–5 d old) were untreated or irradiated with 50 Gy by using a Mark 1irradiator. Flies were allowed to recover for 30 min and heads were isolatedby manual dissection and homogenized in Laemmli sample buffer (Bio-Rad).Head lysates were fractionated on polyacrylamide gels and probed withH2Av-pS137 (1:500; Rockland) and α-tubulin (1:10,000, Sigma) antibodies.The secondary antibody was α-rabbit IgG horseradish peroxidase (1:5,000; GEHealthcare/Amersham).
Mobility Assay. A climbing assay was used to quantify mobility. Groups of 20adultflies of the indicated genotypewere aged for 3 d andwere placed in 2.5-cm by 9.5-cm (diameter by height) plastic vials with graduationmarks at 25%,50%, and 75% of the vial height. Vials were sealed with Parafilm. Flies weretapped to the bottom of the vial and videotaped for 30 s. The tape waspaused at 10, 20, and 30 s, and the number of flies in each quadrant wascounted. For each genotype, four independent replicates were averaged andplotted as a percentage of flies in each quadrant at the indicated time points.
Longevity Assay. Longevityassayswereperformed intriplicatewithcontrol andexperimental genotypes at the same time. For each of the triplicate experi-ments, 100 flies were examined (five vials of 20 flies each).ATM8,ATM8/+, andw1118
flieswere raised at 18 °C and transferred to 25 °C at 0 to 1 d posteclosion.All other flies were collected 0 to 3 d posteclosion at 25 °C. The day of col-lection was designated day 1, and the number of surviving flies was counteddaily until all flies had died. Flies were transferred to new vials approximatelyevery 3 d. The number of surviving flies for each genotypewas averaged, witherrors representing the SEM between replicates. Statistical significance wasdetermined by using log-rank analysis and the χ2 statistic.
Paraffin Sectioning. Fly heads were hand dissected and incubated in ethanol:chloroform:acetic acid (6:3:1) at room temperature overnight. Heads werethen incubated in 70% ethanol, processed into paraffin, sectioned at 5 μm,and stained with hematoxylin (Harris modified with acetic acid; Fisher) andeosin (Eosin Y powder; Polysciences) by using standard procedures (65).
Microarray Analysis. For each genotype, approximately 150 flies (approxi-mately equalnumbers ofmales and females)were collectedandaged for 3d at25 °C. To collect head, flies were transferred to 1.5 mL tubes and frozen inliquid N2, tubeswere shaken vigorously to shear heads frombodies, and headswere isolated from other body parts by sequential use of US Standard no. 25(710 μm) and no. 40 (425 μm) sieves. Total RNA was isolated from heads byusing TRI reagent (Sigma). The resulting RNA was resuspended in 40 μLdiethylpyrocarbonate-treated water and subjected to DNase treatment usingthe TURBODNA-free kit (Ambion). The resulting RNAwas ammonium acetateprecipitated and resuspended in 20 μL of diethylpyrocarbonate-treated wa-ter. Typically, 40 μg of RNA was recovered per sample. RNA (1 mg/mL) wasshipped at −80 °C to NimbleGen (Reykjavik, Iceland) for probe generation andanalysis on D.melanogaster 4 × 72-plex arrays, containing a minimum of fourprobes per gene for 15,473 genes. The data were analyzed by using the
E662 | www.pnas.org/cgi/doi/10.1073/pnas.1110470109 Petersen et al.

ArrayStar program (DNAStar). Pair files were used as input, and normalizationwas conducted by using robust multichip average processing and quantilenormalization. Two replicates were performed for all genotypes except Elav-GAL4 and Elav-ATMi flies, which had four replicates. All replicates were usedto generate fold changes and statistical confidence limits. Before statisticalanalysis, genes with a low coefficient of variation (<0.05) were filtered. Initialstatistical analysis identified genes with a P value lower than 0.05 by using amoderated t test analysis with no additional corrections. Further statisticalanalysis using the FDR test was used to generate the final list of genes, withthe final criteria being a greater than twofold change in expression, a mod-erated t test P < 0.05, and an FDR P < 0.10. Raw and normalized microarraydata have been deposited in the Gene Expression Omnibus database (acces-sion no. GSE34315).
qPCR. Flies were collected as indicated for the microarrays and aged for thestated times. The 0-d time point indicates that RNA was processed on the dayof collection. Heads were isolated from approximately 25 flies per genotype,and RNA was isolated using the RNeasy Plus Mini Kit (Qiagen). cDNA wasgenerated with an iScript cDNA Synthesis Kit (Bio-Rad). Real-time qPCR wascarried out as described by Katzenberger et al. (2006) (66). Primer sequencesare provided in Table S2.
Immunofluorescence Microscopy. For the AMP::GFP experiments, brains weredissected from flies aged 4 to 6 d at 25 °C. For the CaspAct experiments, brainswere dissected, fixed for 15 min in fresh 4% formaldehyde, washed in 1× PBSsolution with 0.3% Triton-X, and incubated in primary antibody overnight at4 °C. Primary antibodies used were α-Repo (1:200; Developmental StudiesHybridoma Bank), α-Elav (1:200; Developmental Studies Hybridoma Bank),and α-CaspAct (1:50; Millipore). Fluorescently labeled secondary antibodiesused were α-mouse rhodamine (1:300; Invitrogen), α-rat rhodamine (1:200;Invitrogen), and α-rabbit Alexa Fluor 488 (1:300; Invitrogen). Brains weremounted in Vectashield (Vector Laboratories) and imaged on a Zeiss Axiovert200M invertedmicroscope or a Bio-RadMRC-1024 confocal microscope (W.M.Keck Laboratory for Biological Imaging, University ofWisconsin,Madison,WI).
ACKNOWLEDGMENTS. We thank Grace Boekhoff-Falk, Barry Ganetzky, andRandy Tibbetts for providing advice throughout the course of this research;Ylva Engstrom for providing AMP reporter flies; Satoshi Kinoshita for paraffinsectioning; Matt Wagoner and Jean-Yves Sgro for assistance analyzing themicroarray data; Becky Katzenberger for technical support; and the twoanonymous reviewers for thoughtful comments on the manuscript. This workwas supported by National Institutes of Health (NIH) Grant R01 NS059001 (toD.A.W.) and a predoctoral fellowship from NIH Training Grant T32 GM08688(to A.J.P.).
1. Sedgwick RP, Boder E (1960) Progressive ataxia in childhood with particular referenceto ataxia-telangiectasia. Neurology 10:705–715.
2. Bundey S (1994) Clinical and genetic features of ataxia-telangiectasia. Int J Radiat Biol66(6 suppl):S23–S29.
3. McKinnon PJ (2012) ATM and the molecular pathogenesis of ataxia telangiectasia.Ann Rev Pathol Mech Dis 7:303–321.
4. Savitsky K, et al. (1995) A single ataxia telangiectasia gene with a product similar toPI-3 kinase. Science 268:1749–1753.
5. Derheimer FA, Kastan MB (2010) Multiple roles of ATM in monitoring andmaintaining DNA integrity. FEBS Lett 584:3675–3681.
6. Bhatti S, et al. (2011) ATM protein kinase: The linchpin of cellular defenses to stress.Cell Mol Life Sci 68:2977–3006.
7. Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecularautophosphorylation and dimer dissociation. Nature 421:499–506.
8. Matsuoka S, et al. (2007) ATM and ATR substrate analysis reveals extensive proteinnetworks responsive to DNA damage. Science 316:1160–1166.
9. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylateshistone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467.
10. Yang Y, Herrup K (2005) Loss of neuronal cell cycle control in ataxia-telangiectasia:a unified disease mechanism. J Neurosci 25:2522–2529.
11. Rimkus SA, et al. (2008) Mutations in String/CDC25 inhibit cell cycle re-entry andneurodegeneration in a Drosophila model of Ataxia telangiectasia. Genes Dev 22:1205–1220.
12. Biton S, Barzilai A, Shiloh Y (2008) The neurological phenotype of ataxia-telangiectasia: Solving a persistent puzzle. DNA Repair (Amst) 7:1028–1038.
13. Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology inParkinson’s disease. Mov Disord 26:6–17.
14. Amor S, Puentes F, Baker D, van der Valk P (2010) Inflammation in neurodegenerativediseases. Immunology 129:154–169.
15. González-Scarano F, Baltuch G (1999) Microglia as mediators of inflammatory anddegenerative diseases. Annu Rev Neurosci 22:219–240.
16. Ilieva H, Polymenidou M, Cleveland DW (2009) Non-cell autonomous toxicity inneurodegenerative disorders: ALS and beyond. J Cell Biol 187:761–772.
17. Kuljis RO, Xu Y, Aguila MC, Baltimore D (1997) Degeneration of neurons, synapses,and neuropil and glial activation in a murine Atm knockout model of ataxia-telangiectasia. Proc Natl Acad Sci USA 94:12688–12693.
18. Liu N, et al. (2005) ATM deficiency induces oxidative stress and endoplasmic reticulumstress in astrocytes. Lab Invest 85:1471–1480.
19. McGrath-Morrow SA, et al. (2010) Elevated serum IL-8 levels in ataxia telangiectasia. JPediatr 156:682–684, e1.
20. Westbrook AM, Schiestl RH (2010) Atm-deficient mice exhibit increased sensitivity todextran sulfate sodium-induced colitis characterized by elevated DNA damage andpersistent immune activation. Cancer Res 70:1875–1884.
21. Pedersen M, Tiong S, Campbell SD (2010) Molecular genetic characterization ofDrosophila ATM conserved functional domains. Genome 53:778–786.
22. Lempiäinen H, Halazonetis TD (2009) Emerging common themes in regulation ofPIKKs and PI3Ks. EMBO J 28:3067–3073.
23. Joyce EF, et al. (2011) Drosophila ATM and ATR have distinct activities in theregulation of meiotic DNA damage and repair. J Cell Biol 195:359–367.
24. Lessing D, Bonini NM (2009) Maintaining the brain: Insight into humanneurodegeneration from Drosophila melanogaster mutants. Nat Rev Genet 10:359–370.
25. Bodily KD, Morrison CM, Renden RB, Broadie K (2001) A novel member of the Igsuperfamily, turtle, is a CNS-specific protein required for coordinated motor control. JNeurosci 21:3113–3125.
26. Cardona A, et al. (2010) An integrated micro- and macroarchitectural analysis of theDrosophila brain by computer-assisted serial section electron microscopy. PLoS Biol 8:e1000502.
27. Pereanu W, Kumar A, Jennett A, Reichert H, Hartenstein V (2010) Development-basedcompartmentalization of the Drosophila central brain. J Comp Neurol 518:2996–3023.
28. Fan Y, Bergmann A (2010) The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ 17:534–539.
29. Kamada S, Kikkawa U, Tsujimoto Y, Hunter T (2005) Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-likeprotein(s). J Biol Chem 280:857–860.
30. Xiong WC, Okano H, Patel NH, Blendy JA, Montell C (1994) repo encodes a glial-specific homeo domain protein required in the Drosophila nervous system. Genes Dev8:981–994.
31. Soller M, White K (2004) ELAV. Curr Biol 14:R53.32. Ashburner M, et al.; The Gene Ontology Consortium (2000) Gene ontology: tool for
the unification of biology. Nat Genet 25:25–29.33. Lemaitre B, Hoffmann J (2007) The host defense of Drosophila melanogaster. Annu
Rev Immunol 25:697–743.34. Lemaitre B, et al. (1995) A recessive mutation, immune deficiency (imd), defines two
distinct control pathways in the Drosophila host defense. Proc Natl Acad Sci USA 92:9465–9469.
35. De Gregorio E, Spellman PT, Tzou P, Rubin GM, Lemaitre B (2002) The Toll and Imdpathways are the major regulators of the immune response in Drosophila. EMBO J 21:2568–2579.
36. Levashina EA, Ohresser S, Lemaitre B, Imler JL (1998) Two distinct pathways cancontrol expression of the gene encoding the Drosophila antimicrobial peptidemetchnikowin. J Mol Biol 278:515–527.
37. Werner T, et al. (2000) A family of peptidoglycan recognition proteins in the fruit flyDrosophila melanogaster. Proc Natl Acad Sci USA 97:13772–13777.
38. Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventralregulatory gene cassette spätzle/Toll/cactus controls the potent antifungal responsein Drosophila adults. Cell 86:973–983.
39. Dushay MS, Asling B, Hultmark D (1996) Origins of immunity: Relish, a compound Rel-like gene in the antibacterial defense of Drosophila. Proc Natl Acad Sci USA 93:10343–10347.
40. De Gregorio E, Spellman PT, Rubin GM, Lemaitre B (2001) Genome-wide analysis ofthe Drosophila immune response by using oligonucleotide microarrays. Proc NatlAcad Sci USA 98:12590–12595.
41. Irving P, et al. (2001) A genome-wide analysis of immune responses in Drosophila.Proc Natl Acad Sci USA 98:15119–15124.
42. McDonald MJ, Rosbash M (2001) Microarray analysis and organization of circadiangene expression in Drosophila. Cell 107:567–578.
43. Tzou P, et al. (2000) Tissue-specific inducible expression of antimicrobial peptidegenes in Drosophila surface epithelia. Immunity 13:737–748.
44. Sepp KJ, Schulte J, Auld VJ (2001) Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev Biol 238:47–63.
45. Lee YS, Carthew RW (2003) Making a better RNAi vector for Drosophila: Use of intronspacers. Methods 30:322–329.
46. Yao KM, White K (1994) Neural specificity of elav expression: Defining a Drosophilapromoter for directing expression to the nervous system. J Neurochem 63:41–51.
47. Luo L, Liao YJ, Jan LY, Jan YN (1994) Distinct morphogenetic functions of similar smallGTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion.Genes Dev 8:1787–1802.
48. Jiang X, Sun Y, Chen S, Roy K, Price BD (2006) The FATC domains of PIKK proteins arefunctionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J Biol Chem 281:15741–15746.
49. Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O (2005) Three-dimensionalstructure and regulation of the DNA-dependent protein kinase catalytic subunit(DNA-PKcs). Structure 13:243–255.
50. Cavalieri S, et al. (2006) ATM mutations in Italian families with ataxia telangiectasiainclude two distinct large genomic deletions. Hum Mutat 27:1061.
Petersen et al. PNAS | Published online February 21, 2012 | E663
NEU
ROSC
IENCE
PNASPL
US

51. Ferrandon D, et al. (1998) A drosomycin-GFP reporter transgene reveals a local
immune response in Drosophila that is not dependent on the Toll pathway. EMBO J
17:1217–1227.52. Doherty J, Logan MA, Taşdemir OE, Freeman MR (2009) Ensheathing glia function as
phagocytes in the adult Drosophila brain. J Neurosci 29:4768–4781.53. Hartenstein V (2011) Morphological diversity and development of glia in Drosophila.
Glia 59:1237–1252.54. Becker T, et al. (2010) FOXO-dependent regulation of innate immune homeostasis.
Nature 463:369–373.55. Senger K, Harris K, Levine M (2006) GATA factors participate in tissue-specific
immune responses in Drosophila larvae. Proc Natl Acad Sci USA 103:15957–
15962.56. Miyamoto S (2011) Nuclear initiated NF-κB signaling: NEMO and ATM take center
stage. Cell Res 21:116–130.57. Bauer M, et al. (2006) Purine and folate metabolism as a potential target of sex-
specific nutrient allocation in Drosophila and its implication for lifespan-reproduction
tradeoff. Physiol Genomics 25:393–404.58. Zimmerman JE, et al. (2006) Multiple mechanisms limit the duration of wakefulness in
Drosophila brain. Physiol Genomics 27:337–350.
59. Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ (2005) Genetic andgenomic studies of Drosophila parkin mutants implicate oxidative stress and innateimmune responses in pathogenesis. Hum Mol Genet 14:799–811.
60. Tan L, Schedl P, Song HJ, Garza D, Konsolaki M (2008) The Toll—>NFkappaB signalingpathway mediates the neuropathological effects of the human Alzheimer’s Abeta42polypeptide in Drosophila. PLoS ONE 3:e3966.
61. Arroyo DS, Soria JA, Gaviglio EA, Rodriguez-Galan MC, Iribarren P (2011) Toll-likereceptors are key players in neurodegeneration. Int Immunopharmacol 11:1415–1421.
62. Parkhurst CN, Gan WB (2010) Microglia dynamics and function in the CNS. Curr OpinNeurobiol 20:595–600.
63. Morales I, Farías G, Maccioni RB (2010) Neuroimmunomodulation in the pathogenesisof Alzheimer’s disease. Neuroimmunomodulation 17:202–204.
64. Miller RL, James-Kracke M, Sun GY, Sun AY (2009) Oxidative and inflammatorypathways in Parkinson’s disease. Neurochem Res 34:55–65.
65. Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S (1997) The swiss cheesemutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci17:7425–7432.
66. Katzenberger RJ, Marengo MS, Wassarman DA (2006) ATM and ATR pathways signalalternative splicing of Drosophila TAF1 pre-mRNA in response to DNA damage. MolCell Biol 26:9256–9267.
E664 | www.pnas.org/cgi/doi/10.1073/pnas.1110470109 Petersen et al.











![· Web view[TYPESETTER: websum: The cytoplasmic DNA sensor cGAS detects DNA in ruptured micronuclei and activates an innate immune response.] [TYPESETTER: GEO accession code GSE100771]](https://static.fdocuments.in/doc/165x107/5aded9017f8b9af05b8ba5c6/viewtypesetter-websum-the-cytoplasmic-dna-sensor-cgas-detects-dna-in-ruptured.jpg)





