
Assessment of the Infant with Acute Metabolic Problems · hyperammonemia, the organic acide ......
16
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 21, No. 1 Copyright © 1991, Institute for Clinical Science, Inc. Assessment of the Infant with Acute Metabolic Problems HOBART E. WILTSE, M.D., P h .D. Department o f Pediatrics, University o f Nebraska College o f Medicine Omaha, NB 68198-2165 ABSTRACT Those inborn errors of metabolism which characteristically produce acute illness during the first year of life often resemble infectious illnesses in their nonspecific modes of presentation. Grouping of signs and symp- toms into prototypes, followed by an active search for clinical and labora- tory clues with higher specificity, often proves helpful in making an appro- priate choice of confirmatory diagnostic tests. Introduction The diagnosis of inborn errors of metabolism causing acute disease during the neonatal period or later infancy can be a daunting task. Experience has pro- vided clinicians with the symptom recog- nition skills generally needed for initiat- ing a metabolic investigation, and recent laboratory innovations have brought us improved tools for confirmatory diag- nosis. Differential diagnosis, the inter- mediate step between diagnostic suspi- cion and confirmation, usually constitutes the difficult step because of complexity and rarity of the diseases and poor specificity of their early manifesta- tions. This review has as its purpose helping the clinician, or clinician-in- training, to achieve maximum success in the identification of those metabolic dis- eases of infancy which are life-threaten - ing and potentially treatable, and espe- cially those which are relatively common in occurrence. Most clinicians rely less on extensive knowledge of diseases within this group than on general famil- iarity with important prototypes, such as galactosemia, maple syrup urine disease, hyperammonemia, the organic acide- mias, and the beta oxidation defects. Diagnostic strategies can appropriately be organized around these prototypes. The reader seeking more detailed information about the clinical presenta- tion of these diseases than found here is referred to the landmark reviews by Burton 8 and by Greene and coworkers .19 The Initial Clues to Metabolic Illness The symptoms and signs which first call attention to the possibility of an inherited metabolic disease in an acutely ill infant are likely to be nonspecific and perhaps more suggestive of infection than disordered metabolism. A search should be undertaken for more specific chemical clues at the same time as the bacteriologic studies, since valuable time can be lost if consideration of metabolic disease is delayed until the infant is 40 0091-7370/91/0100-0040 $02.00 © Institute for Clinical Science, Inc.
-
Upload
hoangthuan -
Category
Documents
-
view
213 -
download
0
Transcript of Assessment of the Infant with Acute Metabolic Problems · hyperammonemia, the organic acide ......

ANNALS O F CLINICAL AND LABORATORY SCIEN CE, Vol. 21, No. 1Copyright © 1991, Institute for Clinical Science, Inc.
Assessment of the Infant with Acute Metabolic Problems
HOBART E. W ILTSE, M .D ., Ph .D.
Department o f Pediatrics, University o f Nebraska College o f Medicine
Omaha, NB 68198-2165
ABSTRACT
Those inborn errors of m etabolism which characteristically produce acute illness during the first year of life often resem ble infectious illnesses in their nonspecific modes of presentation. Grouping of signs and symptoms into prototypes, followed by an active search for clinical and laboratory clues with higher specificity, often proves helpful in making an appropriate choice of confirmatory diagnostic tests.
Introduction
T he d iagnosis of in b o rn e rro rs of metabolism causing acute disease during the neonatal period or later infancy can be a daunting task. Experience has provided clinicians with the symptom recognition skills generally needed for initiating a metabolic investigation, and recent laboratory innovations have brought us im proved tools for confirm atory diagnosis. D ifferential diagnosis, the in te rm ediate step betw een diagnostic suspic io n a n d c o n f i r m a t i o n , u s u a l ly constitutes the difficult step because of complexity and rarity of the diseases and poor specificity of their early manifestations. This review has as its purpose help ing the clinician, or clinician-in- training, to achieve maximum success in the identification of those metabolic diseases of infancy which are life-threatening and potentially treatable, and especially those which are relatively common in occurrence. Most clinicians rely less on ex ten s iv e know ledge of d iseases
within this group than on general familiarity with im portant prototypes, such as galactosemia, maple syrup urine disease, hyperam m onem ia, the organic acidemias, and the beta oxidation defects. Diagnostic strategies can appropriately be organized around these prototypes.
The rea d e r seeking m ore d e ta iled information about the clinical presentation of these diseases than found here is re fe rre d to the landm ark review s by Burton8 and by G reene and coworkers.19
The Initial Clues to M etabolic Illness
The symptoms and signs which first call a tte n tio n to the possib ility of an inherited metabolic disease in an acutely ill infant are likely to be nonspecific and perhaps m ore suggestive of infection than disordered metabolism. A search should be undertaken for more specific chemical clues at the same time as the bacteriologic studies, since valuable time can be lost if consideration of metabolic d isease is de layed u n til the infant is
400091-7370/91/0100-0040 $02.00 © Institute for Clinical Science, Inc.

ASSESSMENT O F THE INFANT WITH ACUTE METABOLIC PROBLEMS 41
found not to have septicemia. Vomiting, in terruption of feedings, and the commonly used infusions of 10 percent glucose can norm alize an elevated blood ammonia in an infant with a urea cycle defect, or cause galactose to disappear from the urine in a neonate with galactosemia, or glutaric acid to disappear from the urine of an infant with glutaric acidemia type I. A blood transfusion given to a jaundiced infant m ight interfere with the diagnosis of galactosem ia by enzym e assay on the ery th rocy tes, unless the neonatal metabolic screening panel has been carried out prior to transfusion.
In table I are listed examples of the im portant bu t non-specific clues which can alert one to the presence of acute metabolic disease. Such clues offer little help in indicating directions for the metabo lic s tu d ie s , and th e p h y sic ian is therefore advised to explore actively for additional clues having greater specificity. These m ight be found in the clinical presentation itse lf or in the results of preliminary screening tests readily available in the clinical laboratory (table II).
Based on abnorm alities which m ight be found in the preliminary screening, table III offers suggestions for differential diagnosis. The age group organization of this table is based upon a consideration that acute metabolic symptoms in the neonate tend to be provoked by intolerance to protein or carbohydrate in the feedings, whereas the older infant m ore typically develops sym ptom s in
TABLE IValuable But Nonspecific Clues
to Metabolic Disease
Poor feeding VomitingJaundice HepatomegalyHypoglycemia HypotoniaSeizures ApneaFailure to thriveFamily history of neonatal or Infantile death
TABLE IIInitial Screening Tests from the
Clinical Laboratory
Blood:Glucose pH, electrolytes Ammonia Liver enzymes *Complete blood count with differential
Urea nitrogen Uric acid Lactic acid
Urine, Qualitative Tests for:Acetone GlucoseReducing sugar Methylmalonic acidKetoacids (2,4-dinitrophenylhydrazine) Sulfur-containing amino acids
(cyanide - nltroprusside)Tyrosine derivatives (nltrosonaphthol)
* Alanine aminotransferase (ALT)Asparate transamlnase(AST))Lactate dehydrogenase (LD)
response to the aggravated fasting and a c u te c a tab o lism w h ich accom pany an in te rcu rre n t illness w ith vom iting and fever.
Beyond Screening—Selection of More Specialized Laboratory Tests
Prototype diseases are listed in table IV and the confirmatory tests listed in table V. A physician dealing with one of the diagnostic problems in table I might use table II for preliminary screening. If this leads to a close fit with one or more of the diagnostic clues in table III, table IV can be used for developing diagnostic hypo theses and tab le V for se lec ting appropriate laboratory tests. Several of the procedures listed in table V (e.g., quantitative amino acid analysis, organic acid screen, and acyl carnitine profile) are notably efficient screening tools in th e ir own right, offering considerable p o ten tia l for y ield ing inform ation of diagnostic value, even if the original suspicion which p rom pted the tes t is not supported.

Diag
nosti
c Clue
s with
Grea
ter S
pecif
icity
for N
eona
te an
d Olde
r Infa
ntTA
BLE I
II
In the
Neo
nate
Possi
ble S
ignific
ance
Hype
rventi
lation
Acido
sis, h
ypera
mmon
emia
Hype
rammo
nemi
aUr
ea cy
cle de
fect, t
ransie
nt hy
peram
mone
mia o
f the n
ewbo
rn, or
ganic
acide
mia w
ith 2°
ele
vatio
n in N
H3, p
yruva
te ca
rboxy
lase d
eficie
ncy,
holoc
arbox
ylase
synth
etase
defic
iency
Metab
olic a
cidos
is wi
th inc
rease
d anio
n gap
Prop
ionic
acide
mia,
methy
lmalo
nic ac
idemi
a, iso
valer
ic ac
idemi
a, 1°
or 2°
lacti
c acid
osis
Lacti
c acid
osis
Pyruv
ate de
hydro
gena
se de
ficien
cy, p
yruva
te ca
rboxy
lase d
eficie
ncy,
biotin
idase
de
ficien
cy, h
oloca
rboxy
lase s
ynthe
tase d
eficie
ncy,
prop
ionic
acide
mia,
methy
lmalo
nic
acide
mia,
glyco
gen s
torag
e dise
ase,
fructo
se-1,
6-bisp
hosp
hatas
e defi
cienc
y, 3-h
ydrox
y- 3-f
nethy
lgluta
ryl C
oA ly
ase d
eficie
ncy,
glutar
ic ac
idemi
a II, r
espir
atory
chain
defec
ts (im
porta
nt no
n-ge
netic
alter
nativ
es: h
ypox
ia, liv
er ne
crosis
)Ke
tosis
Prop
ionic
acide
mia,
methy
lmalo
nic ac
idemi
a, iso
valer
ic ac
idemi
a, br
anch
ed-ch
ain
ketoa
clduri
a, fru
ctose
-1,6-
bisph
osph
atase
defic
iency
Neutr
open
ia a
Prop
ionic,
meth
ylmalo
nic, o
r isov
aleric
acide
mia;
lysinu
ric pr
otein
intole
rance
Hypo
therm
iaIso
valer
ic ac
idemi
a, Me
nkes
synd
rome
Distin
ctive
odor
Bran
ched
-chain
ketoa
ciduri
a, iso
valer
ic ac
idemi
a, glu
taric
acide
mia
IIJa
undic
e, he
patom
egaly
, diar
rhea,
eleva
tion
of live
r enz
ymes
, prol
onga
tion o
f prot
hromb
in tim
e and
partia
l throm
bopla
stin t
ime t
>,c,d.e
Galac
tosem
ia, ty
rosine
mia 1
, alph
a-1-a
ntitry
psin
defic
iency
, 3-hy
droxy
dicarb
oxyll
c acid
uria
Apne
a, se
izures
, hicc
uppin
g, my
oclon
ic jerk
ingNo
nketo
tic hy
pergl
ycine
mia
Dysm
orphic
featu
res in
asso
ciatio
n with
Pyruv
ate de
hydro
gena
se de
ficien
cy, g
lutari
c acid
emia
li, Zell
wege
r syn
drome
, neo
natal
hypo
tonia
and s
eizure
sad
renole
ukod
ystro
phy
conti
nued
WILTSE

TABL
E III c
ontin
ued
Diag
nosti
c Clue
s with
Grea
ter Sp
ecific
ity fo
r Neo
nate
and O
lder I
nfant
In the
Olde
r Infan
tPo
ssible
Sign
ifican
ce
Letha
rgy, c
oma,
seizu
res, a
nd ac
idosis
.He
redita
ry fru
ctose
intol
eranc
e, ure
a cyc
le de
fects,
lysin
uric p
rotein
intol
eranc
e.pr
ecipi
tated
by fo
od in
take,
often
3-me
thylcr
otony
l CoA
carbo
xylas
e defi
cienc
yas
socia
ted w
ith fo
od av
ersion
Letha
rgy, c
oma,
seizu
res w
ith pr
omine
nt2-
Methy
laceto
acety
l CoA
thiol
ase d
eficie
ncy,
interm
ittent
bran
ched
-chaln
ketoa
ciduri
a.ke
tosis,
asso
ciated
with
aggr
avate
d fas
ting
glutar
ic ac
idemi
a 1 (im
porta
nt no
n-ge
netic
alter
nativ
e: ke
totlc
hypo
glyce
mia)
Letha
rgy, c
oma,
seizu
res w
ith hy
pogly
cemi
aBe
ta ox
idatio
n defe
cts (p
articu
larly
medlu
m-ch
ain ac
yl Co
A de
hydro
gena
se de
ficien
cy).
and m
inima
l or a
bsen
t keto
sis (h
ypok
etotlc
3-hyd
roxy-3
-meth
ylglut
aryl C
oA ly
ase d
eficie
nce,
glutar
ic ac
idemi
a II (I
mport
ant n
on-
hypo
glyce
mia)
asso
ciated
with
aggr
avate
dge
netic
alter
nativ
e: hy
perin
sulin
ism)
fastin
gLe
igh di
seas
e (su
bacu
te ne
crotiz
ingPy
ruvate
dehy
droge
nase
defic
iency
f, co
mplex
1 & co
mplex
IV re
spira
tory c
hain
defec
t 0en
ceph
alomy
elopa
thy) U
biotin
idase
defic
iency
hDy
stonia
Lesc
h-Nyh
an di
seas
e, glu
taric
acide
mia 1
, hom
ocys
tlnuri
a k, m
ethylm
alonic
acide
mia1
Osteo
poros
isPr
opion
ic ac
idemi
a, lys
inuric
prote
in int
oleran
ce, h
omoc
ystln
uria
Rena
l tubu
lar re
abso
rptive
defec
tsTy
rosine
mia 1
, gala
ctose
mia,
hered
itary
fructo
se in
tolera
nce,
glyco
gen s
torag
e dise
ase
a J, P
ediat
r. 70
0:62-6
5,198
5bj
. Ped
iatr.
7 7 7:1
039-1
045,1
987.
cj. P
ediat
r. 77
7:313
-319,
1987.
d New
Engl.
J. Me
d. 32
7:101
4-102
1, 10
92-10
99, 1
989.
e J. In
her. M
etab.
DIs. 7
2:339
-342,1
989.
f Ped
iatric
s 79:3
70-37
3,198
7.0 J
. Inhe
r. Meta
b. Dis
. 72Ó
247-2
56, 1
989.
h Ped
iatr. R
es. 26
:260-2
66,19
89.
i Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e. 19
89, p
p. 86
9-888
.JJ.
Ped
latr.
174:3
40, 1
989.
kj. P
ediat
r. 7 7
3:863
-864,
1988.
1J. P
ediat
r. 77
3:102
2-102
7,198
8.
ASSESSMENT OF THE INFANT WITH ACUTE METABOLIC PROBLEMS

TABL
E IV
Major
Dise
ases
and
Reco
mmen
ded T
ests
Disea
sePr
omine
nt Fe
atures
Tests
Ornit
hine t
ransc
arbam
ylase
defic
iency
ab,c
,d.e
Hype
rammo
nemi
a, se
vere
in ma
les,
varia
ble in
fema
lesQu
antita
tive a
mino
acids
(bloo
d)
orotic
acid
(urine
) all
opurl
nol c
halle
nge (
urine
)Ar
ginino
succ
lnic a
ciduri
a bHy
peram
mone
mia,
abno
rmal
hair
Quan
titativ
e ami
no ac
ids (b
lood)
qu
antita
tive a
mino
acids
(urin
e) oro
tic ac
id (ur
ine)
Trans
ient h
ypera
mmon
emia
of ne
wborn
wHy
peram
mone
mia
Quan
titativ
e ami
no ac
ids (b
lood)
oro
tic a
dd (u
rine)
Lysinu
ric pro
tein i
ntoler
ance
9Hy
peram
mone
mia,
failur
e to t
hrive
, os
teopo
rosis
hQu
antita
tive a
mino
acids
(bloo
d)
quan
titativ
e ami
no ac
ids (u
rine)
orotic
acid
(urine
)Pr
opion
ic ac
idemi
a iAc
idosis
, keto
sis, h
ypera
mmon
emia,
ne
utrop
enia
Quan
titativ
e ami
no ac
ids (b
lood)
ca
rnitin
e, tot
al an
d free
(bloo
d)
organ
ic ac
id sc
reen (
urine
)Me
thylm
alonic
acide
mia i
Acido
sis, k
etosis
, hyp
eramm
onem
ia,
neutr
open
iaQu
antita
tive a
mino
acids
(bloo
d)
carni
tine,
total
and f
ree (b
lood)
org
anic
acid
scree
n (uri
ne)
Isova
leric
acide
mia i
Acido
sis, k
etosis
, hyp
eramm
onem
ia,
neutr
open
ia, pu
ngen
t odo
rQu
antita
tive a
mino
acids
(bloo
d)
carni
tine,
total
and f
ree (b
lood)
org
anic
acid
scree
n (uri
ne)
Pyruv
ate de
hydro
gena
se de
ficien
cy k
Lacti
c acid
osis,
Leigh
ence
phalo
pathy
Lacta
te/py
ruva
te rat
io (b
lood)
qu
antita
tive a
mino
acids
(bloo
d)
organ
ic ac
id sc
reen (
urine
) py
ruvate
dehy
droge
nase
(enz
yme)
conti
nued
WILTS E

TABL
E IV (
conti
nued
)Ma
jor D
iseas
es an
d Rec
omme
nded
Tests
Disea
sePr
omine
nt Fe
atures
Tests
Pyruv
ate ca
rboxy
lase d
eficie
ncy *
Lacti
c acid
osis,
hype
rammo
nemi
a, he
patom
egaly
Lacta
te/py
ruva
te rat
io (b
lood)
qu
antita
tive a
mino
acids
(bloo
d) or
ganic
add
scree
n (uri
ne)
pyruv
ate ca
rboxy
lase (
enzy
me)
Holoc
arbox
ylase
synth
etase
defic
iency
iEa
rly on
set, l
actic
acido
sis, k
etosis
, hy
peram
mone
mia,
coma
, seiz
ures,
alope
cia, fr
eque
nt Inf
ectio
nsLa
ctate/
pyru
vate
ratio
(bloo
d) or
ganic
acid
scree
n (uri
ne)
biotin
idase
(enz
yme)
holoc
arbox
ylase
synth
etase
(en
zyme
)Bio
tinida
se de
ficien
cy1
Late
onse
t, lac
tic ac
idosis
, keto
sis,
hype
rammo
nemi
a, co
ma, s
eizure
s, alo
pecia
, freq
uent
infec
tions
Lacta
te/py
ruva
te rat
io (b
lood)
orga
nic ac
id sc
reen (
urine
) bio
tinida
se (e
nzym
e)Fru
ctose
-1,6-b
ispho
spha
tase d
eficie
ncy m
Hypo
glyce
mia,
lactic
acido
sis,
ketos
is, hy
perur
icemi
aLa
ctate/
pyru
vate
ratio
(bloo
d)
quan
titativ
e ami
no ac
ids (b
lood)
orga
nic ac
id sc
reen (
urine
) fru
ctose
-1,6-b
lspho
spha
tase
(enzy
me)
Hered
itary
fructo
se in
tolera
nce m
,nHy
pogly
cemi
a, lac
tic ac
idosis
, hyp
eruric
emia,
av
ersion
to sw
eets
Lacta
te/py
ruva
te rat
io (b
lood)
qu
antita
tive a
mino
acids
(bloo
d) ora
l fruc
tose t
oleran
ce te
st (b
lood)
orga
nic ac
id sc
reen (
urine
) fru
ctose
-l-ph
osph
ate al
dolas
e (en
zyme
)3-M
ethylc
roton
yl Co
A ca
rboxy
lase
defic
iency
°Hy
pogly
cemi
a, ac
idosis
, com
aOr
ganic
acid
scree
n (uri
ne)
3-Meth
ylcrot
onyl-
CoA
carbo
xylas
e (en
zyme
)
conti
nued

Major
Dise
ases
and
Reco
mmen
ded T
ests
TABL
E IV (
conti
nued
)
Disea
se
Prom
inent
Featu
res
Tests
Glyc
ogen
stora
ge ty
pe I p
Glyc
ogen
stora
ge ty
pe III
p
Glyc
ogen
stora
ge ty
pe IV
pGl
ycog
en st
orage
type
VI (li
ver
phos
phory
lase o
r pho
spho
rylas
e kin
ase d
eficie
ncy)
p
3-Hyd
roxy-3
-meth
ylglut
aryl C
oA
lyase
defic
iency
aw
Gluta
ric ac
idemi
a, typ
e II t
Resp
irator
y cha
in de
fects
(comp
lex I)
*
Resp
irator
y cha
in de
fects
(comp
lex IV
) u
Bran
ched
-chaln
ketoa
cidurl
a w*
Hypo
glyce
mia,
lactic
acido
sis,
hype
rurice
mia,
marke
d hep
atome
galy
Mild
hypo
glyce
mia,
lactic
acido
sis,
hype
rurice
mia,
hepa
tomeg
alyPro
gressi
ve ci
rrhos
is with
early
fatal
outco
meRe
lative
ly mi
ld hy
pogly
cemi
a and
he
patom
egaly
Hypo
glyce
mia,
hype
rammo
nemi
a, ac
idosis
with
out k
etosis
Hepa
tomeg
aly, h
ypoto
nia, h
ypog
lycem
ia,
acido
sis w
ithou
t keto
sisLa
ctic a
cidos
is, hy
poton
ia, ea
rly de
ath,
Leigh
ence
phalo
pathy
uLa
ctic a
cidos
is, m
yopa
thy, r
enal
dysfu
nctio
n, Le
igh en
ceph
alopa
thy v
Ketos
is, co
ma, s
eizure
s, op
hthalm
opleg
ia,
distin
ctive
odor
Gluc
ose-6
-phos
phata
se (e
nzym
e)
Debra
nche
r enz
yme (
enzy
me)
Bran
ching
enzy
me (e
nzym
e)Ph
osph
orylas
e (en
zyme
) Ph
osph
orylas
e b ki
nase
(enz
yme)
Orga
nic a
dd sc
reen (
urine
) 3-h
ydrox
y-3-m
ethylg
lutary
l-CoA
lya
se (e
nzym
e)Or
ganic
acid
scree
n (uri
ne)
Lacta
te/py
ruva
te rat
io (b
lood
organ
ic ac
id sc
reen (
urine
)La
ctate/
pyru
vate
ratio
(bloo
d)
quan
titativ
e ami
no ac
ids (u
rine)
organ
ic ac
id sc
reen (
urine
)Qu
antita
tive a
mino
acids
(bloo
d)
organ
ic ac
id sc
reen (
urine
) br
anch
ed-ch
ain al
pha-
keto
acid
dehy
droge
nase
(enz
yme) co
ntinu
ed
WILTSE

Major
Dise
ases
and
Reco
mmen
ded T
ests
TABL
E IV (
conti
nued
)
Disea
sePr
omine
nt Fe
atures
Tests
Gluta
ric ac
idemi
a, typ
e 1 y^
aaEp
isodic
ketoa
cidos
is, hy
pogly
cemi
a, hy
peram
mone
mia,
dysto
niaQu
antita
tive a
mino
acids
(bloo
d) or
ganic
acid
scree
n (uri
ne)
Gluta
ryl C
oA de
hydro
gena
se
(enzy
me)
2-Me
thylac
etoac
etyl C
oA th
lolas
e de
ficien
cy *
Ketoa
cidos
is, hy
pergl
ycine
mia,
hema
temes
ls, co
maQu
antita
tive a
mino
acids
(bloo
d) or
ganic
acid
scree
n (uri
ne)
2-me
thylac
etoac
etyl C
oA th
iolas
e (en
zyme
)Me
dium-
chaln
acyl
CoA
dehy
droge
nase
de
ficien
cy bb
Hypo
ketot
lc hy
pogly
cemi
a, dlc
arbox
yllc a
ciduri
a, 2°
carni
tine
deple
tion,
hepa
tomeg
alyCa
rnitin
e, tot
al an
d free
(bloo
d) org
anic
acid
scree
n (uri
ne)
acyl
carni
tine p
rofile
(urin
e) me
dlum-
chain
acyl
CoA
dehy
droge
nase
(enz
yme)
DNA s
tudy w
ith po
lymera
se ch
ain
reacti
on am
plific
ation
Long
-chaln
acyl
CoA
dehy
droge
nase
de
ficien
cy t>b
Hypo
ketot
lc hy
pogly
cemi
a, he
patom
egaly
, ca
rdiom
yopa
thy (fe
w ca
ses)
Carni
tine,
total
and f
ree (b
lood)
acyl
carni
tine p
rofile
(bloo
d) or
ganic
acid
scree
n (uri
ne)
Short
-chaln
acyl
CoA
dehy
droge
nase
de
ficien
cy bb
Musc
le we
akne
ss, e
pisod
ic hy
pogly
cemi
a an
d acid
osis
(few
case
s, va
riable
ma
nifes
tation
s)Ca
rnitin
e, tot
al an
d free
(bloo
d) ac
yl ca
rnitin
e prof
ile (b
lood)
orga
nic ac
id sc
reen (
urine
) ac
yl ca
rnitin
e prof
ile (u
rine)
Prima
ry ca
rnitin
e defi
cienc
y cc
Hypo
ketot
lc hy
pogly
cemi
a, he
patom
egaly
, co
ma (f
ew ca
ses)
Carni
tine,
total
and f
ree (b
lood)
acyl
carni
tine p
rofile
(urin
e) conti
nued
-aASSESSMENT OF THE INFANT WITH ACUTE METABOLIC PROBLEMS
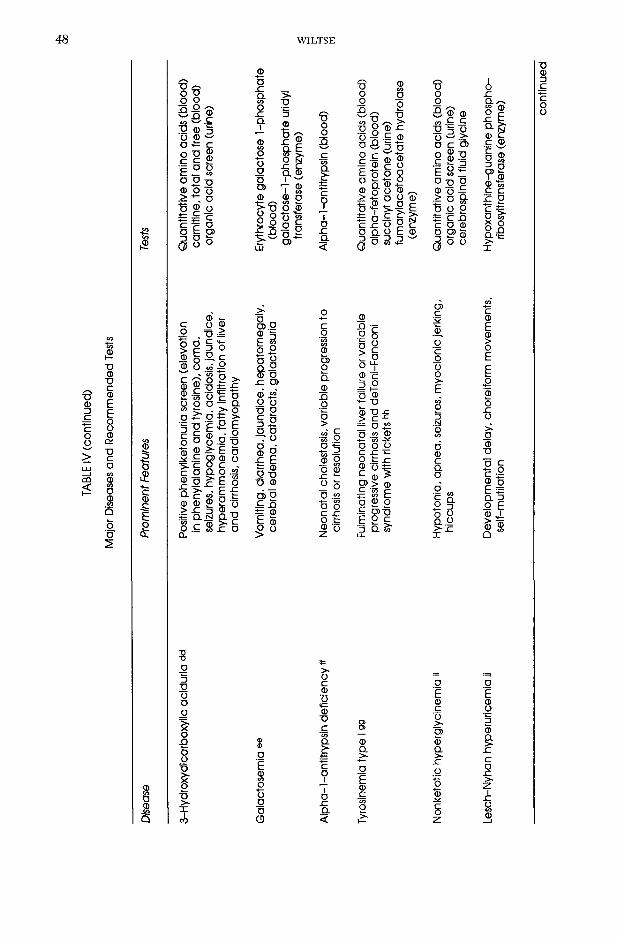
Major
Dise
ases
and
Reco
mmen
ded T
ests
TABL
E IV (
conti
nued
)
Disea
sePr
omine
nt Fe
atures
Tests
3-Hyd
roxyd
icarbo
xylic
acidu
ria dd
Positi
ve ph
enylk
etonu
ria sc
reen (
eleva
tion
in ph
enyla
lanine
and t
yrosin
e), co
ma,
seizu
res, h
ypog
lycem
ia, ac
idosis
, jaun
dice,
hype
rammo
nemi
a, fat
ty inf
iltrati
on o
f live
r an
d cirrh
osis,
cardi
omyo
pathy
Quan
titativ
e ami
no ac
ids (b
lood)
ca
rnitin
e, tot
al an
d free
(bloo
d)
organ
ic ac
id sc
reen (
urine
)
Galac
tosem
ia ®e
Vomi
ting,
diarrh
ea, ja
undic
e, he
patom
egaly
, ce
rebral
edem
a, ca
tarac
ts, ga
lactos
uria
Eryth
rocyte
galac
tose
1-pho
spha
te (bl
ood)
galac
tose-1
-phos
phate
uridy
l tra
nsfer
ase (
enzy
me)
Alph
a-l-a
ntitry
psin
defic
iency
Neon
atal c
holes
tasis,
varia
ble pr
ogres
sion t
o cir
rhosis
or re
solut
ionAlp
ha-1-
antitr
ypsln
(bloo
d)
Tyros
inemi
a typ
e 199
Fulm
inatin
g neo
natal
liver
failur
e or v
ariab
le pro
gress
ive ci
rrhos
is and
deTo
ni-Fa
ncon
l sy
ndrom
e with
ricke
ts hh
Quan
titativ
e ami
no ac
ids (b
lood)
alp
ha-fe
toprot
ein (b
lood)
succ
inyl a
ceton
e (uri
ne)
fumar
ylace
toace
tate h
ydrol
ase
(enzy
me)
Nonk
etotic
hype
rglyc
inemi
a «Hy
poton
ia, a
pnea
, seiz
ures,
myoc
lonic
jerkin
g, hic
cups
Quan
titativ
e ami
no ac
ids (b
lood)
org
anic
add
scree
n (uri
ne)
cereb
rospin
al flu
id gly
cine
Lesc
h-Nyh
an hy
perur
icemi
a ii
Deve
lopme
ntal d
elay,
chore
iform
mov
emen
ts,
self-m
utllat
lonHy
poxa
nthine
-guan
ine ph
osph
o-
ribos
yltran
sferas
e (en
zyme
) conti
nued
WILTSE

Major
Dise
ases
and
Reco
mmen
ded T
ests
TABL
E IV (
conti
nued
)
Disea
sePr
omine
nt Fe
atures
Tests
Zellw
eger
synd
rome k
kHig
h fore
head
, epic
antha
l folds
, cata
racts
, Bru
shfie
ld sp
ots, h
ypoto
nia, n
eona
tal se
izures
Very-
long-
chain
fatty
acid
analy
sis
(bloo
d)pla
smalo
gen (
blood
) dih
ydrox
yace
tone p
hosp
hate
acylt
ransfe
rase (
enzy
me)
subc
ellula
r loca
lizati
on of
catal
ase
(enzy
me)
Neon
atal a
dreno
leuko
dystr
ophy
kkHy
poton
ia an
d seiz
ures
Very-
long-
chain
fatty
acid
analy
sis
(bloo
d)pla
smalo
gen (
blood
) dih
ydrox
yace
tone p
hosp
hate
acylt
ransfe
rase (
enzy
me)
subc
ellula
r loca
lizati
on of
catal
ase
(enzy
me)
Menk
es sy
ndrom
e «Ne
onata
l hyp
otherm
ia an
d hyp
erbilir
ubine
mia,
abno
rmal
hair,
cereb
ral de
gene
ration
Copp
er (b
lood)
cerul
oplas
min (
blood
)
a New
Engl.
J. Me
d. 32
2:165
2-165
5, 19
90.
b Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 198
9, pp
.629-6
63.
cj. P
ediat
r. ? 7
5:415
-417.
1989.
d j. P
ediat
r. 7 7
5:611
-614,
1989.
e Ped
iatr.
Res.
26:27
-82, 1
989.
f J. P
ediat
r. 70
7:712
-719,
1985.
9 Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e., 19
89, p
p. 24
97-25
13.
h New
Engl.
J. Me
d. 37
2:290
-294,
1985
. ' S
crive
r, C.R.
: The
Meta
bolic
Basis
of In
herite
d Dise
ase,
1989
, pp.
821-8
44.
) Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis o
f Inhe
rited D
iseas
e, 19
89, pp
. 791
-819.
k Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis o
f Inhe
rited D
iseas
e, 19
89, p
p. 86
9-888
.' S
crive
r, C.R.
: The
Meta
bolic
Basis
of In
herite
d Dise
ase,
1989
, pp.
2083
-2103
. m S
crive
r, C.R.
: The
Meta
bolic
Basis
of In
herite
d Dise
ase,
1989,
pp. 3
99-42
4.n P
ediat
rics 8
5:600
-603,1
990.
° J, In
her. M
etab.
Dis. 7
2:339
-341,1
989.
p Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis o
f Inhe
rited D
iseas
e, 19
89, p
p. 42
5-452
.co
ntinu
ed
ASSESSMENT OF THE INFANT WITH ACUTE METABOLIC PROBLEMS

Major
Dise
ases
and R
ecom
mend
ed Te
stsTA
BLE I
V (co
ntinu
ed)
q J. In
her. M
etab.
Dis.73
:156-1
64,19
90.
'J. In
her.
Metab
. DIs.
7 7:76
-87,19
88.
s J. In
her.
Metab
. Dis.
9:225
-233,1
986.
t Ped
iatr.
Res.
27:31
1-315
,1990
.u J
. Ped
iatr.
770:8
4-87,1
990.
v Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 19
89, p
p. 84
5-853
.w S
crive
r, C.R.
: The
Meta
bolic
Basis
of In
herite
d Dise
ase,
1989
, pp.
671-6
92.
x Scrì
ver, C
.R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 198
9, pp
. 791
-819.
y J. P
ediat
r. 77
4:983
-989,
1989.
z P
ediat
rics 8
3:228
-234,1
989.
aa Sc
river,
C.R.:
The M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 19
89, pp
. 845
-853.
bb Sc
river,
C.R.:
The M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 19
89, p
p. 88
9-914
.cc
New.
Eng
. J. M
ed. 3
79:13
31-13
36,19
88.
dd j.
Pedia
tr. 7 7
0:387
-392,
1990.
e« sc
river,
C.R.:
The M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 19
89, p
p. 45
3-480
.ff S
crive
r, C.R.
: The
Meta
bolic
Basis
of In
herite
d Dise
ase,
1989,
pp. 2
409-2
437.
99 Sc
river,
C.R.:
The M
etabo
lic Ba
sis o
f Inhe
rited D
iseas
e, 19
89, p
p. 54
7-562
.hh
J. Pe
diatr.
7 72:7
34-73
9,198
8. « S
crive
r, C.R.
: The
Meta
bolic
Basis
of In
herite
d Dise
ase,
1989
, pp.
743-7
53.
ii Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis o
f Inhe
rited D
iseas
e, 19
89, pp
. 100
7-102
8, kk
Scriv
er, C.
R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 19
89, p
p. 14
79-15
09.
il Scri
ver, C
.R.: T
he M
etabo
lic Ba
sis of
Inhe
rited D
iseas
e, 19
89, pp
. 141
1-143
1.

ASSESSMENT O F THE INFANT WITH ACUTE METABOLIC PROBLEMS 51
TABLE VSpecialized Diagnostic Procedures from Clinical, Reference, or Research Laboratory
Blood1. Lactate/pyruvate ratio2. Quantitative amino acids3. Carnitine, free and total4. Acyl carnitine profile5. Alpha-fetoproteln6. Alpha-l-antitrypsin7. Erythrocyte galactose-l-phosphate8. Very-long-chain fatty acid analysis9. Plasmalogen10. Copper11. Ceruloplasmin12. Oral fructose tolerance test
Urine13. Quantitative amino acids14. Orotic acid15. Organic acid screen16. Acyl carnitine profile17. Succlnyl acetone18. Allopurinol challenge0
Enzyme Assays19. Biotinidase20. Holocarboxylase synthetase21.3-Methylcrotonyl-CoA carboxylase22. Galactose-l-phosphate uridyltransferase23. Pyruvate dehydrogenase24. Pyruvate carboxylase25. Fructose-1,6-blsphosphatase26. Fructose-1-phosphate aldolase27. Glucose-6-phosphatase28. Debrancher enzyme29. Branching enzyme30. Phosphorylase31. Phosphorylase b kinase32. Branched-chaln alpha-keto acid dehydrogenase33. Glutaryl CoA dehydrogenase34. 2-Methylacetoacetyl CoAthlolase35. 3-Hydroxy-3-methylglutaryl-CoA lyase36. Medium-chain acyl CoA dehydrogenase37. Fumarylacetoacetate hydrolase38. Hypoxanthine-guanine phosphoribosyltransferase39. Dihydroxyacetone phosphate acyltransferase40. Subcellular localization of catalase
SerumFibroblastsLeukocytes, fibroblastsErythrocytesFibroblastsbFibroblastsLiverLiverLiverLiverLeukocytes, fibroblastsLiverLiverLeukocytes, fibroblasts Leukocytes, fibroblasts FibroblastsLeukocytes, fibroblasts Leukocytes, fibroblasts Liver, fibroblasts Erythrocytes Fibroblasts Fibroblasts
Other41. Cerebrospinal fluid glycine42. DNA study with polymerase chain reaction amplification«:
aNew Engl. J. Med. 322:1641-1645.1990. <=J. Clin. Invest. 86:1000-1013, 1990.bAn unusual sibship has been reported (Pediatr. Res. 24.-95-100, 1988) in which pyruvate dehydro
genase activity was normal In cultured skin fibroblasts but virtually undetectable in liver, heart, and skeletal muscle. In most instances, fibroblast assay avoids the problems of enzyme lability encountered with tissue assay of pyruvate dehydrogenase.
Discussion metabolic diseases will often reveal nonspecific abnorm alities o f in te rest, bu t
Analysis of the plasma amino acids in distinctive or pathognom onic patternsinfants with these acutely symptomatic are found rather infrequently. Examples

52 WILTSE
of distinctive findings include elevated branched-chain amino acids and especially L -allo iso leucine44 in b ranched- chain ketoaciduria, argininosuccinic acid and perhaps c itru lline in argininosuccinic aciduria, m ethionine and homocystine in homocystinuria, and 2-aminoaci- dipic acid in glutaric aciduria type I.
Nonspecific elevations in plasma glyc in e a re c h a ra c te r is t ic a lly fo und in p ro p io n ic ac id em ia , m e th y lm a lo n ic acidemia, isovaleric acidemia, 2-methyl- acetoacetyl CoA lyase deficiency, and lysinuric protein intolerance. E levated glycine levels have also been observed in children treated with valproic acid. For these reasons, a diagnosis of nonketotic hyperglycinemia requires demonstration of elevated cerebrospinal fluid glycine levels and absence of distinctive organic acids in the urine, in addition to elevation in p lasm a g lycine .38 The plasm a alanine tends to elevate in parallel with lactate and pyruvate in several conditions, such as hereditary fructose intolerance, fructose-1, 6-bisphosphatase deficiency, glycogen storage disease type I, and py ruvate carboxylase deficiency. Elevations in alanine may characteristically occur in patients with urea cycle defects and lysinuric protein intolerance, without simultaneous increases in lactate and pyruvate. Low alanine levels may be found in branched-chain ketoaciduria and in the syndrome of ketotic hypoglycemia of childhood.
Tyrosine and m ethionine tend to elevate nonspecifically in infants with hepatocellular disease. Alone, these abnormalities would not support a diagnosis of tyrosine type I, and this diagnosis should be based on finding succinylacetone in the u rin e and an abnorm al assay for fumarylacetoacetate hydrolase. Brusilow and Horwich7 have called attention to the significance of glutamine as a protective b u t sa tu ra tab le “bu ffe r” against excessive ammonia accumulation in the
urea cycle disorders. Accordingly, a high plasm a glutam ine may have the same significance as a high ammonia in a child with a defect in ureagenesis or lysinuric protein intolerance, or it may serve as an early w arning of im pending hyperam - monemic coma.
A urinary amino acid study may be preferable to a blood study when argininosuccinic aciduria is suspected; the low renal threshold for argininosuccinic acid causes it to appear in urine in large amounts even at low blood levels. This substance has a tendency to form cyclic anhydrides under conditions of analysis, causing it to be misidentified unless the chrom atographer is experienced. A urinary study may be particularly informative in lysinuric p ro te in in to lerance, dem onstrating large amounts of lysine, arginine, and o rn ith ine in contrast to their low levels in plasma.
Pyruvate is more likely than lactate to be the m etabolite of d irect in terest in the diseases being considered here, but m e a su re m e n ts o f la c ta te a lone w ill usually suffice. W ith the exception of severe pyruvate carboxylase deficiency, where the lactate/pyruvate ratio may be e levated , d epartu res from the norm al ratio are unusual in m etabolic disease. By contrast, the ratio is often elevated in p a t i e n ts w ith c i r c u la to ry fa i lu re , asphyxia, o r shock, all of w hich are u su a lly d is t in g u is h a b le on c lin ic a l grounds from metabolic conditions.
Analysis of urinary organic acids by means of combined capillary gas chromatography/mass spectrometry has emerged as a remarkably powerful screening and diagnostic tool. The m ethod possesses excellent sensitivity and specificity for pathologic organic acids, their glycine adducts, and the dicarboxylic acids characteristic of medium-chain acyl CoA dehydrogenase deficiency and other defects in beta oxidation. One should recognize that dicarboxylic aciduria can occur innocendy

ASSESSM ENT O F THE INFANT WITH ACUTE METABOLIC PROBLEMS 53
in normal neonates13 and in infants given feeding formulas containing medium-chain triglycerides. Unusual but non-pathologic organic acids have also been found in the urine of infants with gastroenteritis.30
Another highly significant innovation is the acyl carnitine profile obtained by means of fast atom bom bardm ent/m ass spectrometry. This has proven particularly useful in the diagnosis of organic acidem ias and b e ta oxidation defects th rough th e ir characteristic carnitine esters. Octanoyl carn itine , pathognomonic of medium-chain acyl CoA dehydrogenase deficiency, is readily dem onstrable in urine by this technique. In the few years since this procedure became available, m edium-chain acyl CoA dehydrogenase deficiency has become a relatively common diagnosis, with an incidence now es tim a ted at 0.4 to 1 p e r 10,000. Long-chain acyl CoA dehydrogenase deficiency requires a plasma sample for diagnosis, rather than urine, and is considerably more rare.
On a case-by-case basis, the diagnostician must weigh the need for diagnostic certainty (as this relates to prognosis, treatm ent options, and genetic advice) a g a in s t th e r e la t iv e a v a ila b il ity or unavailability of confirm atory enzym e assays. The need for confirmatory assay will vary greatly. The diagnosis of pyruvate dehydrogenase deficiency on clinical grounds alone would be very uncerta in p r io r to assay. By c o n tra s t , a confident clinical diagnosis of isovaleric acidemia might be made on the basis of the clinical presentation, urinary organic acid study, and urinary acyl carnitine profile. A confirmatory enzyme assay for o rn ith ine transcarbam ylase deficiency requires liver3 or duodenal23 biopsy, but a newly described allopurinol challenge te s t24 m akes i t p o ss ib le to id en tify women who are heterozygous for this x- linked defect without an invasive biopsy. A significant innovation is the use of
desoxynbonucleic acid (DNA) analysis for confirming the diagnosis of medium- chain acyl CoA dehydrogenase deficiency.56 The unusual high frequency of a single m u ta tion accounting for the defect makes this procedure feasible.
References
1. A mir, N ., E l p e l e g , O . N ., S h a l e v , R. S ., and C h r i s t e n s e n , E .: G lu ta r ic a c id u ria ty p e I: Enzym atic and neuroradiologic investigations of two kindreds. J. Pediatr. 114:983 - 989, 1989.
2. A r b o u r , L ., R o s e n b l a t t , B., C l o w , C ., and W il s o n , G. N .: P o sto p era tiv e dy sto n ia in a female pa tien t w ith hom ocystinuria. J. Pediatr. 113:863-864 , 1988.
3. A r n , P. H . , H a u s e r , E. R ., T h o m a s , G. H ., H e r m a n , G., H e s s , D ., and B r u s il o w , S. W.: H yperam m onem ia in w om en with a m utation at th e o rn ith in e ca rb am o y ltran sferase locus: a cause of postpartum coma. New Engl. J. Med. 322:1652-1655, 1990.
4. B arash , V , M a n d e l , H ., Se l l a , S ., and G e ig er , R .: 3-Hydroxy-3-m ethylglutaryl-coenzym e A lyase deficiency—biochem ical studies and family investigation o f four generations. J. Inher. M etab. Dis. i3 :1 5 6 -1 6 4 , 1990.
5 . B a u m g a r t n e r , E . , Su o r m a l a , T , W ic k , H ., P r o bst , A., B l a u e n s t e in , , U., B a c h m a n n , C., and V e s t , M .: Biotinidase defic iency-a cause of su b a c u te n e c ro tiz in g e n c e p h a lo m y e lo p a th y (Leigh sy n d ro m e)-rep o rt of a case with lethal outcom e. Pediatr. Res. 26:260-266 , 1989.
6. B e r g m a n , I ., F in e g o l d , D . , G a r t n e r , J. C ., Z it e l l i, B . J., C l a a s e n , D . , Sc a r a n o , J., Ro e , C . R ., St a n l e y , C ., and G o o d m a n , S. I.: Acute profound dystonia in infants with glutaric acidemia. Pediatrics 83:228— 234, 1989.
7. B r u s il o w , S. and H o r w ic h , A.: U rea cycle enzym es. In: Scriver, C. R., ed. The Metabolic Basis o f Inherited Disease, 6th ed. New York, M cGraw-Hill, 1989, pp. 629-663.
8. B u r t o n , B . K.: In b o rn erro rs o f m etabolism : The clinical diagnosis in early infancy. Pediatrics 79:359-369, 1987.
9 . C a r p e n t e r , T. O . , L e v y , H . L ., H o l t r o p , M . D ., S h ih , V. E ., an d A n a st , C . S .: L ysin u ric p r o te in in t o le r a n c e p r e s e n t in g as c h i ld h o o d o steo p o r o sis . N e w E n g . J. M ed . 312:2 9 0 —294 ,1985.
10. Cox, D .: A lp h a-l-an titry p sin deficiency. In: S c river, C. R ., ed . T h e M e ta b o lic B a sis o f Inherited Disease , 6 th ed. New York, McGraw- Hill, 1989, pp. 2409-2437.
11. D a n k s , D .: D isorders of copper transport. In: S c riv er, C. R ., ed . The M e ta b o lic B a sis o f Inherited Disease, 6th ed. New York, McGraw- Hill, 1989, pp. 1411-1431.
12. D a nn e r , D . and E lsas, L .: D isorders o f branched ch a in a m in o a c id an d k e to acid m eta b o lism . In:

54 WILTSE
S criv e r, C. R ., e d . The M e ta b o lic B asis o f Inherited Disease, 6 th ed. New York, McGraw- Hill, 1989, pp. 671-692 .
13. D o w n in g , M ., R o s e , P., B e n n e t t , M ., M a n n i n g , N., and POLLITT, R.: G eneralized dicar- boxylic a c id u ria -a comm on finding in neonates. J. Inher. M etab. Dis. 12:321-324, 1989.
14. EDSTROM, C. S.: H e red ita ry fructose in to le rance in th e vom iting infant. Pediatrics 85:6 0 0 - 603, 1990.
15. E s q u iv e l , C. O., Iw a tsu k i, S ., G o r d o n , R. D., M a r s h , W. W ., Ko n e r u , B ., M a k o w k a , L ., T z a k is , A . G ., T o d o , S . , and St a r z l , T. E .: Indications for pediatric liver transplantation. J. Pediatr. H J : 1039-1045, 1987.
16. F u j ii , T., It o , M ., O k u n o , T ., M u t o h , K ., N is h ik o m o r i, R., and M ik aw a , H.: Complex I (reduced n ico tinam ide-adenine d inucleo tide- c o e n z y m e Q re d u c ta s e ) d e f ic ie n c y in tw o p a tie n ts w ith p ro b ab le L eigh syndrom e. J. P ed iatr 116:8 4 - 87, 1990.
17. G ib s o n , K ., B r e u e r , J., Ka is e r , K ., N y h a n , W ., M c C o y , E . , F e r r e ir a , P ., G r e e n e , C . , B l i t z e r , M . , S h a p i r a , E . , R e v e r t e , F . , C o n d e , C ., B a g n e l l , P ., and C o l e , D .: 3- H ydroxy-3-m ethylglu taryl coenzym e A lyase defiency—report of five new patients. J. Inher. M etab. Dis. 11:7 6 -8 7 , 1988.
18. G it z e l m a n n , R., St e in m a n , B ., an d van d e n B e r g h e , G .: D iso rd er s o f fru ctose m eta b o lism . In: S criver, C. R., e d . The Metabolic Basis o f Inherited Disease, 6 th ed . N e w York, M cG raw - H ill, 1989, pp. 399-424.
19. G r e e n e , C. L ., B litzer , M. G ., and S h a p ir a , E.: Inborn errors o f metabolism and Reye synd ro m e : d i f f e r e n tia l d iag n o s is . J. P e d ia tr . 773:156-159, 1988.
20. G o l d s m it h , L . and La b e r g e , C.: Tyrosinemia and re la ted d isorders. In : Scriver, C. R ., ed. The M etabolic Basis o f Inherited Disease, 6th e d . N e w Y ork, M c G ra w -H il l , 1989 , p p . 547-562 .
21. G o o d m a n , S. and F r e r m a n , F.: Organic acidemias due to defects in lysine oxidation-2-ketoa- d ip ic a c id e m ia a n d g lu ta r ic a c id e m ia . In : S c riv er, C . R ., ed . The M e ta b o lic B asis o f Inherited Disease, 6 th ed. New York, McGraw- H ill, 1989, pp. 845-853.
22. H a g e n f e l d t . L . , v o n D ö b e l n , U ., H o l m e , E ., A l m , J ., B r a n d b e r g , G ., E n o c k s s o n , E ., and L in d e b e r g , L .: 3-Hydroxydicarboxylic ac- id u r ia -a fatty acid oxidation defect w ith severe prognosis. J. Pediatr. 116:387-392 , 1990.
23. H a m a n o , Y., K o d a m a , H . , F u jik a w a , Y., T an a k a , Y., N is h im u r a , K ., and Ya n a g isa w a , M.: Use of im m unocytochem ical analysis of a duodenal biopsy specim en to identify a carrier of o rn ith in e tran sca rb am y lase deficiency. N ew Engl. J. M ed. 378:1521-1523, 1988.
24. H a u s e r , E. R ., F in k e l s t e in , J. E ., V a l l e , D ., and B r u s il o w , S. W.: Allopurinol-induced oro- tidinuria: a tes t for m utations at the ornithine c a rb am o y ltran sfe ra se locus in w om en. N ew Engl. J. M ed. 322:1641-1645, 1990.
25. H ayasaka , K ., B r o w n , G ., D a n k s , D ., D r o s t e , M ., and Ka d e n b a c h , B .: Cytochrom e c oxidase deficiency in subacute necrotizing encephalopathy (Leigh’s syndrome). J. Inher. M etab. Dis. 12:247-256 , 1989.
26. H e l d e n r e ic h , R., N a to w ic z , M ., H a in l in e ,B. E ., B e r m a n , P., K e l l e y , R. I., H il l m a n , R. E ., and B erry, G. T .: Acute extrapyram idal syndrom e in m ethylm alonic acidem ia: "m eta bolic s troke” involving the globus pallidus. J. Pediatr. 773:1022-1027, 1988.
27. H e r s , H .-G ., V an H o o f , F ., and d e B arsy, T.: Glycogen storage diseases. In: Scriver, C. R., ed. The M etabolic Basis o f Inherited Disease, 6 th ed . N ew York, M cG raw -H ill, 1989, pp . 425-452 .
28. H u d a k , M. L., Jo n e s , M. D ., and B r u s il o w ,S. W .: D ifferentiation o f transien t hyperam m onem ia o f th e new born and urea cycle enzym e defec ts by c lin ical p re sen ta tio n . J. P ed iatr. 107:7 12-719 , 1985.
29. H u t c h in s o n , R. J., B u n n e l l , K ., andTH O ENE, J. G.: S u p p ress io n o f g ra n u lo p o ie tic p ro g en ito r c e l l p r o l i f e r a t i o n b y m e t a b o l i t e s o f t h e b ran ch ed -ch a in am in o acids. J. Pediatr . 106:6 2 -65, 1985.
30. Kay, M . A ., O ’B r ie n , W., K e s s l e r , B ., M c V i e , R ., U r s in , S . , D ie t r ic h , K ., and M c C a b e , E. R. B .: Transient organic aciduria and m eth em oglobinem ia w ith acute gastroenteritis. Pediatrics 85 :589- 592, 1990.
31. Ker r , D ., B erry, S ., L u s k , M., H o , L ., and Pa t e l , M .: A defic iency o f b o th su b u n its o f pyruvate dehydrogenase which is not expressed in fibroblasts. Pediatr. Res. 24 :95-100, 1988.
32. K r e t z sc h m a r , H. A ., D e A r m o n d , S. J., Ko c h , T . K . , Pa t e l , M . S . , N e w t h , C . J. L . , Sc h m i d t , K . A ., a n d Pa c k m a n , S .: P y ru v a te d e h y d ro g e n a se c o m p lex d e fic ie n c y as a ca u se o f su b a cu te n e c ro tiz in g en cep h a lo p a th y (L e ig h d is ease ). P ed ia tr ics 79:370— 373, 1987.
33. La r g il l ie r e , C ., H o u s s in , D ., G o t t r a n d , F , M a th e y , C ., C h e c o u r y , A ., A l a g il l e , D ., and F a r r ia u x , J. P.: L iver transplantation for orn ith in e transcarbam ylase deficiency in a girl. J. Pediatr. 115:415-417 , 1989.
34. La y w a r d , E ., T a n n e r , M ., Po l l it t , R., and B a r t l e t t , K .: Isolated biotin-resistant 3-m eth- ylcrotonyl-CoA carboxylase deficiency p resen ting as a Reye syndrom e-like illness. J. Inher. M etab. Dis. 72:339-341, 1989.
35. La z a r o w , P. and M o se r , H.: D isorders of p e roxisome biogenesis. In: Scriver, C . R., ed. The M etabolic Basis o f In h erited D isease, 6 th ed. New York, M cGraw-Hill, 1989, pp. 1479-1509.
36. L o e h r , J ., G o o d m a n , S . , an d F r e r m a n , F . : Glutaric acidemia type II-heterogeneity of clinical and biochem ical phenotypes. Pediatr. Res. 27:311-315, 1990.
37. N a g a o , M ., T s u c h iy a m a , A ., A o y a m a , T . , M o r i , T . , and O y a n a g i, K .: Secondary carnitine deficiency in th e new born period in twins of a m other w ith partial ornithine transcarbam ylase deficiency. J. Pediatr. 775:611—614, 1989.

ASSESSM ENT OF THE INFANT WITH ACUTE METABOLIC PROBLEMS 5538. N y h a n , W .: N onketotic hyperglycinem ia. In:
S c riv er, C . R ., ed . T h e M e ta b o lic B asis o f Inherited Disease, 6th ed. New York, McGraw- Hill, 1989, pp. 7 4 3 - 753.
39. O g ie r , H ., L o m b e s , A ., Sc h o l t e , H. R., P o l l - T h e , B. T., F a r d e a u , M , A l c a r d i, J., V ig n e s ,B ., N ia u d e t , P., and Sa u d u b r a y , J. M .: de Toni-Fanconi-D ebre syndrom e with Leigh syndrom e revealing severe m uscle cytochrom e C oxidase deficiency. J. P ed ia tr. 112:7 3 4 -7 3 9 , 1988.
40. R o b i n s o n , B.: L actic acidem ia. In : Scriver,C. R ., ed. The M etabolic Basis o f Inherited D isease, 6th ed. New York, M cGraw-Hill, 1989, pp. 8 6 9 - 888.
41. Ro e , C . and C o a t e s , P.: Acyl-CoA dehydrogenase deficiencies. In: Scriver, C . R . , ed . The M etabolic Basis o f In h erited D isease, 6 th ed. New York, M cGraw-Hill, 1989, pp. 889-914.
42. Ro s e n b e r g , L. and F e n t o n , W.: D isorders of propionate and m ethylm alonate m etabolism . In: S c riv er, C . R ., ed . T he M e ta b o lic B asis o f Inherited Disease, 6th ed. New York, McGraw- Hill, 1989, pp. 821-844.
43. R u s s e l l , G. J., F it z g e r a l d , J. F., and C lar k , J. H .: F u lm in an t h e p a tic fa ilu re . J. P ed iatr. I i i :3 1 3 —319, 1987.
44. S c h a d e w a l d t , P., H a m m e n , H . , D a l l e -F e s t e ,C ., an d WENDEL, U .: O n th e m ec h a n ism o f L- a llo is o le u c in e fo r m a tio n —s tu d ie s o n a h e a lth y s u b je c t a n d in f ib r o b la s ts fro m n o r m a ls a n d p a t ie n ts w ith m a p le sy ru p d is e a s e . J. In h er . M etab . D is . 13:137-150, 1990.
45. S e g a l , S.: D isorders of galactose metabolism. In: Scriver, C . R ., ed. The M etabolic Basis o f Inherited Disease, 6th ed. New York, McGraw- Hill, 1989, pp. 453-480.
46. SlMELL, O.: Lysinuric p rotein intolerance. In: S c riv er, C. R ., ed . T h e M e ta b o lic B asis o f Inherited Disease, 6th ed. New York, McGraw- Hill, 1989, pp. 2497-2513.
47. Sp e r l , W. and R e n ie r , W .: Definition of Leigh syndrom e (letter). J. Pediatr. 114:340, 1989.
48. St a r zl , T. E ., D e m e t r is , A. J., and V a n T h ie l ,
D .: L iver transp lan tation . New Eng. J. M ed. 322:1014-1021, 1092-1099, 1989.
49. St o u t , F. and C ask ey , C .: H ypoxanthine phos- p h o rib o sy ltran sfe rase d e fic ien cy —th e L esch- Nyhan syndrom e and gouty arthritis. In: Scriver,C. R., ed. The M etabolic Basis o f Inherited D isease, 6 th ed. New York, McGraw-Hill, 1989, pp. 1007-1028.
50. S w ee tm a n , L .: B ran ch ed chain o rgan ic acidurias. In: Scriver, C. R., ed. The M etabolic Basis o f Inherited Disease, 6th ed. N ew York, McGraw-Hill, 1989, pp. 791-819.
51. T u c h m a n , M., T sa i, M., H o l z k n e c h t , R., and B r u s il o w , S.: Carbam yl phosphate synthetase and o rn ith in e tran sca rb am y lase ac tiv ities in e n zy m e-d e fic ien t h u m an liv e r m easu red by radiochrom atography and correlated w ith ou tcome. Pediatr. Res. 26 :77-82 , 1989.
52. T r e e m , W . R ., S t a n l e y , C . A ., F i n e g o l d ,D. N., H a l e , D. E ., and C o a t e s , P. M.: Primary carnitine deficiency due to a failure of carn itine tran sp o rt in kidney, m uscle, and fibroblasts. New Eng. J. M ed. 37.9:1331-1336, 1988.
53. v a n Sp r o n s e n , F ., B e r g e r , R., S m it , G., d e K l e r k , J . , D u r a n , M ., B ij l e v e l d , C ., v a n F a a s s e n , H . , S l o o f , M ., and H e y m a n s , H .: Tyrosinaemia type I-o rth o to p ic liver transplantation as th e only definitive answer to a m etabolic as well as an oncological problem . J. Inher. M etab. Dis. 12:339-342 , 1989.
54. W o l f , B. and H e a r d , G. S.: D isorders of biotin metabolism. In: Scriver, C. R ., ed. The M etabolic Basis o f Inherited Disease, 6 th ed. New York, M cGraw-Hill, 1989, pp. 2083-2103.
55. W y so c k i, S. J. and H a h n e l , R.: 3-Hydroxy-3- m ethylglutaryl-coenzym e A lyase deficiency: a review. J. Inher. M etab. Dis. 9:225-233, 1986.
56. Yo k o t a , I., Ya s u h ir o , I ., C o a t e s , P., and Ta n a k a , K . : M olecular basis of m edium -chain acyl- coenzyme A dehydrogenase deficiency—an A to G transition at position 985 that causes a lysine- 304 to glutam ate substitution in the m ature p rotein is the single prevelan t m utation. J. Clin. Invest. 86:1000—1013, 1990.


















