
Aspects of Organoselenium Chemistry - University of Adelaide · 2014. 1. 15. · ABSTRACT iii...
246
Aspects of Organoselenium Chemistry A thesis presented for the degree of Doctor of Philosophy by Virginia R. Ward B.Sc. (Hons) School of Chemistry and Physics November 2012
Transcript of Aspects of Organoselenium Chemistry - University of Adelaide · 2014. 1. 15. · ABSTRACT iii...

Aspects of
Organoselenium Chemistry
A thesis presented for the degree of
Doctor of Philosophy
by
Virginia R. Ward
B.Sc. (Hons)
School of Chemistry and Physics
November 2012

TABLE OF CONTENTS
ABSTRACT iii
STATEMENT OF ORIGINALITY v
ACKNOWLEDGEMENTS vi
ABBREVIATIONS vii
1 INTRODUCTION 1
1.1 SELENIUM 1
1.2 ORGANOSELENIUM CHEMISTRY 5
1.2.1 Selenium dioxide 5 1.2.2 Electrophilic Selenium Reagents 7 1.2.3 Nucleophilic Selenium Reagents 15 1.2.4 Radical Chemistry of Organoselenium Compounds 21 1.2.5 The Selenoxide syn-Elimination 26 1.2.5 Biotransformation of Organoselenium Compounds 29
2 THE AMIDOSELENATION OF ALKENES 32
2.1 INTRODUCTION 32
2.2 INVESTIGATION OF THE FORMATION OF THE trans-OXAZOLINE (2.9) 34
2.3 ONE-POT PREPARATION OF -AMIDO SELENIDES 40
2.4 ALTERNATIVE SOLVENTS FOR THE AMIDOSELENATION REACTION 46
2.5 TWO-STEP PREPARATION OF -AMIDO SELENIDES 49
2.6 PREPARATION OF THE trans-OXAZOLINE (2.9) 56
3 CYCLISATION OF -AMIDOALKYL PHENYL SELENIDES 59
3.1 INITIAL ATTEMPTS TO OPTIMISE THE FORMATION OF N-ACYLAZIRIDINES 59
3.2 CYCLISATION OF -AMIDO SELENIDES AT LOW TEMPERATURE 67
3.3 SUMMARY OF RESULTS FROM THE CYCLISATION OF -AMIDO SELENIDES 76
3.4 FACTORS DETERMINING THE FORMATION OF 3- VERSUS 5-MEMBERED RINGS 77
3.5 OCCURRANCE AND UTILITY OF N-ACYLAZIRIDINES 79
4 AMIDOSELENATION via ADDITION OF ‘PHENYLSELENENYL PERCHLORATE’ 83
4.1 INTRODUCTION 83
4.2 PREPARATION OF -(PHENYLSELANYL)CYCLOHEXYL AMIDES 86
4.3 ALTERNATIVES TO THE -AMIDO SUBSTITUENT 88

5 CLOSER EXAMINATION OF A SELENOXIDE AND A SELENONE 92
5.1 PREPARATION OF N-[2-(PHENYLSELENINYL)CYCLOHEXYL]BENZAMIDE
AND N-[2-(PHENYLSELENONYL)CYCLOHEXYL]BENZAMIDE 92
5.2 HYDROGEN-BONDING IN THE SELENIDE (2.5), SELENOXIDE (5.1) AND SELENONE (5.9) 98
5.3 NMR-SCALE OXIDATION OF N-[2-(PHENYLSELANYL)CYCLOHEXYL]BENZAMIDE (2.5) 101
6 PREPARATION AND CYCLISATION OF -HYDROXY SELENIDES 107
6.1 INTRODUCTION 107
6.2 ATTEMPTED ONE-POT PREPARATION OF 2-PHENYLOXETANE 110
6.3 PREPARATION AND ATTEMPTED CYCLISATION OF 3-PHENYL-3-PHENYLSELENOPROPANOL 112
6.3 PREPARATION AND CYCLISATION OF -HYDROXY SELENIDES BEARING A PRIMARY SELENIUM MOIETY 115
6.4 OXETANES IN NATURAL PRODUCTS AND DRUG DESIGN 124
7 EXPERIMENTAL 128
7.1 GENERAL EXPERIMENTAL 128
7.2 WORK DESCRIBED IN CHAPTER 2 131
7.3 WORK DESCRIBED IN CHAPTER 3 160
7.4 WORK DESCRIBED IN CHAPTER 4 182
7.5 WORK DESCRIBED IN CHAPTER 5 187
7.6 WORK DESCRIBED IN CHAPTER 6 196
REFERENCES 212
PUBLICATIONS 230

iii
ABSTRACT
A range of-amidoalkyl phenylselenides were prepared in order to explore their
cyclisation via oxidation of the selenium moiety to the selenone followed by
intramolecular displacement. At first, the -amidoalkyl phenylselenides were
prepared in one-step from the alkenes. However, the one-step preparation was
complicated by side-reactions and a two-step method was found to give clean
reactions and higher yields of a wide range of the desired amido selenides.
Along with the expected oxazolines, isomeric N-acylaziridines were obtained from the
cyclisation reaction. Formation of N-acylaziridines by cyclisation of amides is
unusual, and variation of the conditions was explored in order to optimise this novel
aziridine-forming reaction. It was found that conducting the oxidation reaction at low
temperature favoured the aziridine products. In this way, the aziridines derived from
all prepared -amido selenides were obtained in good to excellent yield. From some
substrates, the aziridine was obtained as the exclusive product.
The low temperature generation of a selenone from the corresponding selenide had
not been reported previously. Experiments were carried out which provided
evidence for the supposition that the intermediate in the cyclisation reaction was the
selenone.
The preparation of -amido selenides was also investigated using silver ion to
sequester the halide of the selenium reagent, rendering the selenium species more
electrophilic and its addition to the alkene to give a seleniranium ion, irreversible.
The seleniranium ion was generated in the presence of nitrile to allow attack by the

iv
weak nitrile nucleophile upon the seleniranium ion, giving a nitrilium ion. With
addition of water to the nitrilium ion, -amido selenides were formed in moderate
yield. Thus, it was shown that the -amido selenides could be prepared without the
use of strong acid. Addition of azide to the nitrilium ion gave a tetrazole, which
demonstrated that this methodology could provide access to selenides substituted at
the -position with groups other than the amido group.
-Benzamidocyclohexyl phenyl selenoxide and -benzamidocyclohexyl phenyl
selenone were prepared, and hydrogen bonding in the two compounds was
examined spectroscopically. An X-ray crystal structure of the selenoxide showed
intermolecular hydrogen bonding between the amide hydrogen and the seleninyl
oxygen, in contrast to proposals in the literature that analogous selenoxides were
stabilised by intramolecular hydrogen bonding in the solid state.
Three -hydroxy selenides were prepared and their low-temperature oxidation and
cyclisation was explored with a view to obtaining the corresponding oxetanes. The
low-temperature procedure did not translate successfully to the cyclisation of -
hydroxy selenides to oxetanes, instead giving complex mixtures. However, with
reference to literature conditions for the preparation of methoxy-substituted oxetanes,
the -hydroxy selenides were cyclised to the corresponding oxetanes by oxidation in
methanol at room temperature, demonstrating that the scope of this method could be
widened to a more generalised preparation of oxetanes.

v
STATEMENT OF ORIGINALITY
I certify that this work contains no material which has been accepted for the award of
any other degree or diploma in any university or other tertiary institution and, to the
best of my knowledge and belief, contains no material previously published or written
by another person, except where due reference has been made in the text. In
addition, I certify that no part of this work will, in the future, be used in a submission
for any other degree or diploma in any university or other tertiary institution without
the prior approval of the University of Adelaide and where applicable, any partner
institution responsible for the joint-award of this degree.
I give consent to this copy of my thesis, when deposited in the University Library,
being made available for loan and photocopying, subject to the provisions of the
Copyright Act 1968.
I also give permission for the digital version of my thesis to be made available on the
web, via the University’s digital research repository, the Library catalogue and also
through web search engines, unless permission has been granted by the University
to restrict access for a period of time.
Virginia Ward November, 2012

vi
ACKNOWLEDGEMENTS
I thank David Ward for giving me the opportunity to work with this intriguing element,
for sagely guiding my experimental endeavours, for generously editing my thesis
chapters, and for being a steadfast presence throughout this long journey.
I have many happy memories of working in Lab 6, thanks to the good company of our
postdocs, Matt Lucas and Pasquale Razzino. Thanks also to Herbert Foo for much
helpful advice and assistance during my brief stay in Lab 3. I wish him a happy and
successful career.
Thanks to the staff of the Chemistry Department, particularly Phil Clements for his
expert assistance in obtaining NMR and mass spectra.
Many thanks to John Bowie and Simon Pyke for overseeing the final stages and
making it possible for me to complete this work.
And thanks to Tricia, Hugh, Vanessa, Edward and Graham, and to my mother for
their encouragement and the constant distractions.

vii
ABBREVIATIONS
General Ac acetate
AIBN azobisisobutyronitrile
Bn benzyl, C6H5CH2
Bu3SnH tri-butyltin hydride
CH2Cl2 dichloromethane
CHCl3 chloroform
de diastereomeric excess
DMF dimethyl formamide
DMSO dimethyl sulfoxide
ee enantiomeric excess
Et2O diethyl ether
EtOAc ethyl acetate
EtOH ethanol
HMPA hexamethylphosphoramide
i-PrOH isopropanol
KOH potassium hydroxide
LDA lithium diisopropylamide
m-CPBA meta-chloroperbenzoic acid
Me methyl, CH3
MeOH methanol
MgSO4 magnesium sulfate
N2 nitrogen
NaBH4 sodium borohydride
NaCl sodium chloride

viii
NaH sodium hydride
NaHCO3 sodium hydrogen carbonate
Nu nucleophile
OTf trifluoromethanesulfonate, triflate
Ph phenyl, C6H5
Pr propyl, C3H7
r.t. room temperature
t-BuOK potassium tertiary-butoxide
TfOH trifluoromethanesulfonic acid, triflic acid
THF tetrahydrofuran
TLC thin layer chromatography
NMR d doublet
Hz hertz
m multiplet
MHz megahertz
ppm parts per million
q quartet
qn quintet
s singlet
sept septet
t triplet
chemical shift

ix
IR br broad
cm-1 wavenumbers (reciprocal centimeters)
KBr potassium bromide pressed disc
s strong
w weak
MS EI electron impact
ESI electrospray
FAB fast atom bombardment
HRMS high resolution mass spectrum
M molecular ion
m/z mass per unit charge

Chapter 1
1
1 INTRODUCTION
1.1 SELENIUM
Selenium was discovered in 1818 by Jöns Jakob Berzelius who observed a powdery
red deposit which precipitated from the burning of sulfur at his sulfuric acid plant in
the Swedish mining town of Fahlun.[1] Upon heating the red powder Berzelius noted
that it gave off a strong odour of decayed radishes.[1] The German chemist Martin
Heinrich Klaproth had observed a similar odour upon heating a sample of tellurium.
Subsequently, Berzelius showed that the tellurium sample must have been
contaminated with a new substance which he named ‘selenium’ after the moon to
recall its association with tellurium which Klaproth had named after the earth.[1]
The sixty-sixth most abundant crustal element, selenium is found principally in
association with the sulfides of chalcophyllic metals, clausthalite (PbSe) being the
most abundant selenium mineral.[2] Elemental selenium has three crystalline
allotropes: two red allotropes of puckered Se8 rings which are transformed with
heating to the more thermodynamically stable grey or black trigonal selenium, which
is made up of helical Sen chains.[3] Industrial uses of selenium include the
vulcanisation of rubber, the decolourisation of glass and, as cadmium selenide, in the
manufacture of ruby-coloured glass.[2] The photoconductive properties of trigonal
selenium formed the basis for its use in the first photocells[2] while the
photoconductive properties of amorphous selenium found application in the
development of xerography.[4-5]

Chapter 1
2
Plants take up inorganic selenium from the soil as selenite or selenate and
incorporate it into organic compounds such as amino acids - particularly
selenomethionine (1.1), selenocysteine (1.2), Se-methylselenocysteine (1.3), -
glutamyl-Se-methylselenocysteine (1.4) - and isoselenocyanates such as (1.5).[6]
Selenium–accumulating plants can take up selenium in higher proportion to the
selenium concentration in the soil. In the selenium-accumulators wheat,[7] brazil
nuts,[8] yeast[9] and mushrooms[6] the major proportion of the absorbed selenium is
incorporated as selenomethionine (1.1). Recognised as an essential amino acid in
1983 and regarded as the ‘nutritional form’ of selenium,[10] selenomethionine (1.1) is
incorporated non-specifically into proteins by the body in place of methionine.
Selenomethionine (1.1) is also converted to selenocysteine (1.2) which has its own
triplet code and is incorporated non-randomly into selenoproteins, and is therefore
considered to be the twenty-first genetically coded amino acid.[11] In garlic, onions,
broccoli and wild leeks, selenium is mainly incorporated into the amino acid Se-
methylselenocysteine[12] (1.3), or its -glutamyl- derivative (1.4), both of which are
metabolised to methylselenol.[8-9, 13]

Chapter 1
3
Selenium was recognised as an essential element in 1957.[14] In areas where the
soil is low in selenium, deficiency of the element manifests as a cardiomyopathic
condition known as ‘Keshan disease’ in humans and nutritional muscular dystrophy
or ‘white muscle disease’ in calves and lambs.[15] Twenty-five mammalian
selenoproteins have been identified.[16] Three whose activity has been elucidated
are glutathione peroxidase, thioredoxin reductase and iodothyronine deiodinase.
Glutathione peroxidase is important for oxidative defense, having a selenium atom at
its active site and acting as a scavenger of hydroperoxides.[17] Thioredoxin
reductase reduces disulfide bonds and the oxidised state of vitamin C and catalyses
the reduction of thioredoxin while iodothyronine deiodinase regulates thyroid
hormone metabolism by converting thyroxine to triiodothyronine.[18]
The anticarcinogenic potential of selenium was first noted almost 100 years ago.[19]
However, research was inhibited by a limited understanding of the safe dosage and
the nature of the most appropriate form to administer. Recent epidemiological
studies indicate an inverse relationship between selenium status and the risk of a
range of cancer types[18] while human and animal trials using selenium
supplementation provide strong indications that selenium plays an important role in
protecting against and reversing the early stages of cancer.[17-19] There is evidence
that it is small selenium-containing metabolites such as methylselenol, rather than
selenium-containing enzymes, which are active in cancer prevention.[12-13, 20-21]

Chapter 1
4
The propensity of selenium (II) organic compounds to undergo oxidation to selenium
(IV) by a variety of oxidants and their subsequent ease of reduction back to the
divalent state affords organoselenium compounds potential as modulators of the
redox environment of cells. Thus, a number of selenium-containing compounds
have been developed and explored for their antioxidant, antitumour and antiinfective
properties and other types of biological activity.[22-23] Selenium-containing
compounds which show high antioxidant activity include the clinically useful

Chapter 1
5
glutathione peroxidase mimetic ebselen (1.6),[24] the related cationic compound
(1.7),[25] which exhibits glutathione peroxidase-like activity in vitro, and the selenium
analogue (1.8) of the body’s most important cell membrane antioxidant, -
tocopherol.[26] The high activity of compound (1.11) as an intracellular redox cycler
was attributed to having more than two redox centres in the molecule and
particularly, the quinone-selenide moiety.[27] This compound (1.11) exhibits
considerable cytotoxicity against tumor lines in cell culture.[27] Activity in cell culture
indicates potential for selenosartans (1.9) as anti-hypertensive agents[28] while
compound (1.10) demonstrates high superoxide anion and hydrogen peroxide
scavenging ability in vitro as well as bactericidal properties and wound healing in
vivo.[29]
1.2 ORGANOSELENIUM CHEMISTRY
Organoselenium chemistry has many parallels with organosulfur chemistry.
However, because of the greater polarisability of its electrons, weaker C-Se bonds
and the greater capacity of selenium for hypervalency, transformations of selenium
compounds and reagents often occur with greater ease and under milder conditions
than those of its chalcogen relative.
1.2.1 SELENIUM DIOXIDE The unique qualities of organoselenium reagents and compounds were poorly
appreciated until the 1970s, prior to which the main selenium reagent with wide
application in organic chemistry was selenium dioxide, utilised for the oxidation of
methyl or methylene groups - to a double bond or aromatic ring.[30-31]

Chapter 1
6
The application of selenium dioxide for the oxidation of aldehydes and ketones to
glyoxals and diketones,[32] and for the transformation of alkenes to allylic alcohols,[33]
was first reported in the 1930s. However, the reaction mechanisms were not
elucidated until forty years later when Sharpless et al. determined that both
oxidations involved a seleninic acid intermediate, (1.12) and (1.13), the first reaction
proceeding via a Pummerer rearrangement[34] and the second by an ene reaction
followed by a [2,3] sigmatropic shift (Scheme 1.1).[35]
Subsequent to these mechanistic studies was the recognition by Sharpless et al. of
the potential of the selenoxide syn-elimination (vide infra) as a powerful method for
the introduction of a double bond.[36] This facile elimination reaction was first noted
in 1970 by Jones, Mundy and Whitehouse.[37] Sharpless et al.[38] proved the syn-
nature of the reaction and showed it to be effective for the conversion of epoxides to
allylic alcohols.[36] The generality of this method for introducing a double bond
combined with the mild conditions under which it proceeds inspired a surge of
interest in organoselenium reagents

Chapter 1
7
1.2.2 ELECTROPHILIC SELENIUM REAGENTS The addition of an electrophilic selenium reagent to an alkene is one of a wide range
of methods for the introduction of a selenium moiety into a molecule. The facile
addition of such reagents to alkenes, first reported in 1958,[39] did not receive much
attention until the 1970s renaissance of organoselenium chemistry. Over the
following two decades, methods were developed for the preparation of -hydroxy,[40-
41] -azido,[40, 42] -alkoxy[42] and -acetoxy selenides[43] from alkenes via addition of
phenylselenenyl halide.
The addition of the pseudohalides phenylselenenyl chloride or bromide to an alkene
gives a -halo selenide in equilibrium with a seleniranium ion. The reaction of the
seleniranium ion with an external nucleophile affords a -substituted selenide. In the
presence of a suitably positioned internal nucleophile, a cyclic product is formed
(Scheme 1.2).

Chapter 1
8
For terminal alkenes, the addition of phenylselenenyl halide at low temperature
generally gives the anti-Markovnikov product which isomerises to the Markovnikov
product, via the seleniranium ion, upon warming.[44-45] Electronic factors
predominate in the reactions of tri- and tetra-substituted alkenes,[45-46] and styrene
and its derivatives,[45, 47-48] from which the Markovnikov adduct is the favoured
product, even at low temperature.
The preference for Markovnikov addition can be overridden where there is an oxygen
atom that can coordinate to the selenium atom of the seleniranium ion
intermediate.[43, 49-52] In the addition of phenylselenenyl chloride to an allylic alcohol
or allylic acetate in aqueous acetonitrile, ‘PhSeOH’ can add with anti-Markovnikov
orientation.[51-52] It has been proposed that coordination of the seleniranium ion to
the hydroxyl or carbonyl oxygen weakens the C-Se bond, promoting nucleophilic
attack at the -carbon.[51-53] In the addition of phenylselenenyl chloride to ,-
unsaturated carbonyl compounds, interaction between the carbonyl oxygen and the
selenium of the seleniranium ion leads to the predominance of the -phenylseleno
regioisomer[54] (Scheme 1.3).

Chapter 1
9
Replacement of the halide with a non-nucleophilic counterion such as
trifluoroacetate,[55] hexafluorophosphate,[56] hexafluoroantimonate[56] or
tetrafluoroborate[57] generates a more electrophilic selenium reagent. These
reagents can be prepared in situ from the phenylselenenyl halide and a silver salt or
by addition of the silver salt to the -halo selenide adduct. The diminished
nucleophilicity of these counterions allows the reaction of the seleniranium ion with
less reactive nucleophiles such as carbamates and cyanamide.[57]
Toshimitsu et al.[58] found that the amidoselenenylation of electron-rich alkenes such
as tri- and tetra-substituted alkenes with phenylselenenyl halide and a nitrile was low-
yielding and attributed this to the stabilisation of the intermediate seleniranium ion by
the electron-donating substituents, reducing its reactivity toward nucleophilic attack.
Amidoselenation of electron-rich alkenes using the 2-pyridylseleno group gave the
desired products in high yield as a result of an increase in the reactivity of the
seleniranium ion.[58] The binary reagent PhSeCl-ZnCl2 facilitates the
chloroselenenylation of electrophilic olefins such as the fumarate diester (1.14),
giving the adduct (1.15) in excellent yield[59] (Scheme 1.4).

Chapter 1
10
Although the seleniranium ion intermediates are not usually observed in addition
reactions of electrophilic selenium, Schmid and Garratt[56] showed that stable
seleniranium salts (1.16) could be generated by the addition of silver
hexafluorophosphate or hexafluoroantimonate to the 4-tolylselenenyl chloride
adducts of ethylene and 2-butene, or by the addition of tolylselenenyl
hexafluorophosphate or hexafluoroantimonate to the alkene. The seleniranium ions
(1.16) were found to be stable at low temperature. Treatment of the seleniranium
ions (1.16) with chloride ion generated the -chloro selenide adducts (1.17) (Scheme
1.5).
Denmark and Edwards[48] observed by NMR the formation of the seleniranium ion
formed from the addition of the methyl ester (1.18) to a solution of phenylselenenyl
hexafluoroantimonate at -70°C.[48] Treatment of the seleniranium ion (1.19) with
tetra-n-butyl ammonium chloride generated the -chloro selenide adduct (1.20) along
with starting material (Scheme 1.6).

Chapter 1
11
Cross-over experiments[48, 60] have shown that the formation of the seleniranium ion
is reversible. Addition of 4-(2-methylphenyl)-3-butenoic acid to a solution of the
phenylselenenyl chloride adduct (1.21) of 4-phenyl-3-butenoic acid led to a mixture of
the addition products (1.21) and (1.22).[48] Reversing the order of addition of the
acids gave the same mixture of products[48] (Scheme 1.7).
NMR analysis[48] of the addition of phenylselenenyl bromide to 4-phenyl-3-butenoic
acid showed that the equilibrium between the adduct and the alkene was affected by

Chapter 1
12
temperature, with the equilibrium shifting toward starting material as the temperature
was increased.[48]
The evolution of chiral electrophilic selenium reagents began with the binaphthyl
diselenide (1.23) described by Tomoda and Iwaoka in 1988.[61]
Methoxyselenenylation of alkenes with this reagent resulted in diastereomeric
excesses of up to 49%.[61] Following this were the reports of C2-symmetric reagents
such as (1.24) developed by Deziel,[62] the diferrocenyl reagent (1.25) prepared by
Uemura et al.,[63] the D-mannose-derived reagent (1.26) designed by Tomoda et al.[64]
and a range of diselenides of type (1.27) synthesised by Wirth et al.[23, 65] Reagents
(1.24), (1.26) and (1.27) share the common feature of a heteroatom that is able to
coordinate to the selenium atom, inducing a conformational rigidity in the molecule.

Chapter 1
13
The stereoselective step in the addition of electrophilic selenium reagents to
unsymmetrical alkenes and trans-alkenes is the formation of a diastereomeric
seleniranium cation.[64, 66] A more electrophilic selenium reagent, achieved by using
a less nucleophilic counterion, is more reactive toward the alkene and allows the
addition to occur at a lower temperature, which contributes to a higher
stereoselectivity as equilibration between the two seleniranium ion diastereomers is
inhibited.[64] Strong interaction between the heteroatom and the selenium atom of
reagents (1.24), (1.26) and (1.27) enhances asymmetric induction by bringing the
chiral source close to the reaction centre[64] and by stabilising the seleniranium ion,
inhibiting equilibration of the two diastereomers.[67] Greater bulkiness and rigidity of
the selenium reagent also contribute to a higher facial selectivity.[64, 67] The nature
of the counterion has also been found to affect yield as well as stereoselectivity.[67]
The electrophilic selenium reagent (1.28) was effective in inducing chirality in the
carboselenenylation of a range of styrene derivatives with heterocyclic aromatic
compounds and electron-rich benzene derivatives (Scheme 1.8).[68]

Chapter 1
14
The carbocycle (1.29) and related structures were prepared in up to 98% ee through
the reaction of the corresponding alkene (e.g. 1.30) with the selenenyl triflate (1.31).
In some cases cyclisation was facilitated by a Lewis acid to shift the equilibrium from
the methoxy selenide toward the reactive seleniranium intermediate (Scheme 1.9).[62]

Chapter 1
15
Stereoselective addition of the electrophilic selenium compound (1.33) was utilised
by Wirth et al.[69] in a synthesis of the lignan (+)-samin (1.32). The reaction of
selenenyl triflate (1.33) with the alkene (1.34) followed by addition of 2,3-butadien-1-
ol gave the lignan (1.32) in 85% ee after radical cyclisation and cleavage of the
TBDMS-group (Scheme 1.10).
Using a chiral electrophilic selenium reagent (1.35) with enhanced rigidity, the
isoquinoline alkaloid (-)-(S)-salsolidine (1.36) was synthesised in 90% ee via
selenocyclisation of carbamate (1.37) followed by removal of the protecting group[67]
(Scheme 1.11).
1.2.3 NUCLEOPHILIC SELENIUM REAGENTS A selenium moiety can be introduced into an organic molecule via nucleophilic attack
by a selenolate anion. Reduction of diselenides or elemental selenium produces

Chapter 1
16
selenolate anions, excellent nucleophiles whose reactivity depends on the conditions
under which they are generated.
The reduction of diphenyl diselenide with sodium borohydride in ethanol gives the
complexed selenolate anion, Na+[PhSeB(OEt)3]-,[70] which will readily displace a
halide or sulfonate[71] or open an epoxide.[36] Its nucleophilicity is improved in a less
protic environment such as when it is generated in dimethyl formamide.[72]
The uncomplexed selenolate anion, RSe-Na+ or RSe-K+ can be generated from
diaryl and dialkyl diselenides by reduction with sodium in THF/HMPA,[73] or with
sodium hydride or potassium hydride respectively in THF or DMF.[74-75] This anion is
a more potent nucleophile than the borane complex Na+[PhSeB(OEt)3]-,[73] and will
cleave an ester or lactone at the carbinol carbon in high yield under mild
conditions.[73, 75-76]
The reduction of diphenyl diselenide with lithium aluminium hydride generates a
selenolate ion having Lewis acid character due to the oxygenophilic nature of the
aluminium.[77] The anion is effective in the ring-opening of oxetanes and oxolanes,
providing access to - and -phenylselenenyl alcohols.[77] Diisobutylaluminium
phenylselenolate (i-Bu2AlSePh) reacts regioselectively with ,-unsaturated acetals,
giving exclusively 1-alkoxy-3-phenylseleno-1-alkenes, and 3-phenylselenoalkanals
after hydrolysis[78] and also exclusively affords 1,1-disubstituted ethenes upon
reaction with terminal acetylenes[79] (Scheme 1.12).

Chapter 1
17
In a biphasic solution of diethyl ether and 10% hydrochloric acid, diphenyl diselenide
can be reduced by zinc dust.[80-81] In the acidic aqueous phase, aziridines can be
activated and undergo ring opening by the selenolate anion. A range of chiral -
seleno amines were obtained from unprotected chiral aziridines in this way[81]
(Scheme 1.13).
A mixture of diorganyl diselenide, tertiary alkyl halide and zinc in dichloromethane
gives the tertiary-substituted organyl selenides in good to excellent yield. This
reaction is selective for tertiary halides; the phenylseleno- group substitutes for the
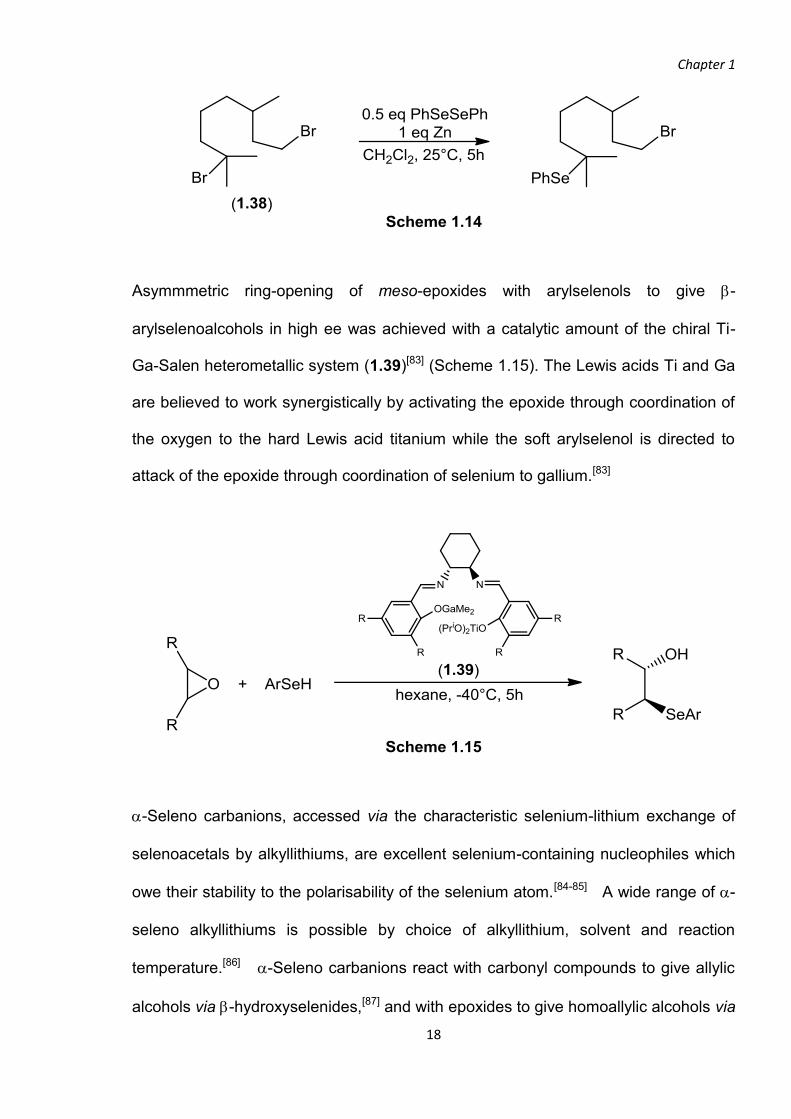
tertiary bromide of (1.38), leaving the primary bromide intact[82] (Scheme 1.14).
Chapter 1
18
Asymmmetric ring-opening of meso-epoxides with arylselenols to give -
arylselenoalcohols in high ee was achieved with a catalytic amount of the chiral Ti-
Ga-Salen heterometallic system (1.39)[83] (Scheme 1.15). The Lewis acids Ti and Ga
are believed to work synergistically by activating the epoxide through coordination of
the oxygen to the hard Lewis acid titanium while the soft arylselenol is directed to
attack of the epoxide through coordination of selenium to gallium.[83]
-Seleno carbanions, accessed via the characteristic selenium-lithium exchange of
selenoacetals by alkyllithiums, are excellent selenium-containing nucleophiles which
owe their stability to the polarisability of the selenium atom.[84-85] A wide range of -
seleno alkyllithiums is possible by choice of alkyllithium, solvent and reaction
temperature.[86] -Seleno carbanions react with carbonyl compounds to give allylic
alcohols via -hydroxyselenides,[87] and with epoxides to give homoallylic alcohols via

Chapter 1
19
-hydroxy selenides,[88] and provide a path to homologation of oxetanes to
tetrahydrofurans.[89] The unusual nucleophilicity of 2-lithio-2-selenopropanes toward
hindered carbonyl compounds enables the preparation of hydroxy selenides from
hindered ketones such as 2,2,6,6,-tetramethylcyclohexanone and their subsequent
transformation to hindered epoxides and olefins (Scheme 1.16).[90]
-Seleno carbanions are also derived from selenides possessing an -hydrogen via
deprotonation with non-nucleophilic bases, although potential substrates are limited
to those substituted with an anion-stabilising group and the reactions can be slow
and low-yielding.[91] However, deprotonation of polystyrene-supported selenides has
been successfully carried out with LDA or butyllithium.[92-93] After reaction of the
carbanion with an electrophile and transformation of the substrate via the

Chapter 1
20
stereospecific selenoxide syn-elimination the polymer-supported selenide is easily
regenerated for reuse[92-93] (Scheme 1.17).
Complexation of an -seleno alkyllithium with a chiral ligand and trapping of the
diastereomeric complex with an electrophile can lead to enantiomeric enrichment in
the product.[94-96] Thus, in the presence of a bisoxazoline, axially chiral
benzylidenecyclohexanes are produced in good yield and high ee via the
enantioselective reaction of an -seleno carbanion and a cyclohexanone followed by
stereospecific syn-elimination[97] (Scheme 1.18).

Chapter 1
21
1.2.4 RADICAL CHEMISTRY OF ORGANOSELENIUM COMPOUNDS
An organoselenium compound containing divalent selenium is stable to many
conditions and can withstand further manipulation until the selenium moiety is
removed reductively or oxidatively. Reductive cleavage of the selenium moiety can
be achieved with Raney nickel[98] or, more generally, with tributyltin or triphenyltin
hydride, by homolytic substitution at selenium with tributyltin or triphenyltin radical
and abstraction of hydrogen from the tin hydride by the carbon-centred radical
intermediate.[99-100] In the presence of a double or triple bond, a new carbon-carbon
bond can form faster than abstraction of hydrogen from the tin hydride, either inter- or
intramolecularly (Scheme 1.19).
Selenides as radical precursors offer the advantage over the alternative bromides in
being able to withstand attack by a nucleophile to which a bromide would be
vulnerable. For example, selenoesters as precursors of acyl radicals are less
electrophilic, and therefore more stable, than acyl bromides. The acyl radicals

Chapter 1
22
generated from tri-n-butyltin hydride treatment of phenyl selenoesters (1.39) undergo
free radical polycyclisations faster than both hydrogen atom abstraction from the tin
hydride and decarbonylation.[101] Under high dilution conditions, the acyl radicals
generated from selenoesters (1.40) and (1.42) undergo intramolecular addition to the
activated alkenyl group providing the macrocycles (1.41) and (1.43)[102] (Scheme
1.20).
Homolytic substitution at selenium with tributyltin radical is approximately three
orders of magnitude faster than the reaction with a sulfur analogue.[103-104] Thus,
N,Se- and O,Se-acetals are deselenated much more rapidly than desulfurisation of
the corresponding N,S- and O,S-acetals and are effective precursors to -N, and -O

Chapter 1
23
radicals. N,Se- and O,Se-acetals are also preferable to the corresponding -bromo
amides and ethers due to their greater stability.[105-106] Alkoxymethyl radicals such
as (1.44), generated via tributyltin hydride treatment of O,Se-acetals, cyclise to
tetrahydrofurans and tetrahydropyrans in good to excellent yield.[106] The N,Se-
acetal (1.45) is efficiently reduced with Bu3SnH/AIBN or allylated with methyl 2-
[(tributylstannyl)methyl]prop-2-enoate, whereas attempted allylation of the analogous
N,S-acetal gave no reaction[105, 107] (Scheme 1.21).
Photolysis of alkyl phenyl selenides generally favours cleavage of the alkyl C-Se
bond due to its lower bond dissociation energy. The efficiency of this reaction can
be improved through optimisation of the reaction conditions.[108] Photolysis of 1-
naphthyl alkyl selenides in an oxygen atmosphere under conditions optimised with
respect to irradiation wavelength, temperature, substrate concentration and solvent

Chapter 1
24
gives the corresponding carbonyl compounds in excellent yield[109] (Scheme 1.22).
Cleavage of the alkyl C-Se bond is further favoured if the alkyl radical fragment is
stabilised, such as with an active methylene moiety. Thus, photolysis of
phenylselenomalonates in the presence of alkenes or alkynes provides the radical
addition products in high yield[110-111] (Scheme 1.22).
Ogawa et al. have exploited the carbon-radical-capturing ability of diphenyl diselenide
in the four-component coupling of unsaturated compounds leading to
cyclopentanes.[112] In the reaction of diphenyl diselenide with ethyl propiolate, tert-
butyl acrylate and 2-methoxypropene (Scheme 1.23), the phenylseleno radical
produced by irradiation of diphenyl diselenide adds preferentially to the alkyne, giving
a vinyl radical which adds preferentially to the electron-rich alkene, the resulting
intermediate adding to the electron-poor alkene, followed by cyclisation. Reaction of
the carbon radical (1.46) with diphenyl diselenide is faster than polymerisation and
allows formation of cyclopentanes in up to 76% yield. With the use of diphenyl
disulfide, the radical (1.46) is not trapped as readily and polymerised products
predominate.[112]

Chapter 1
25
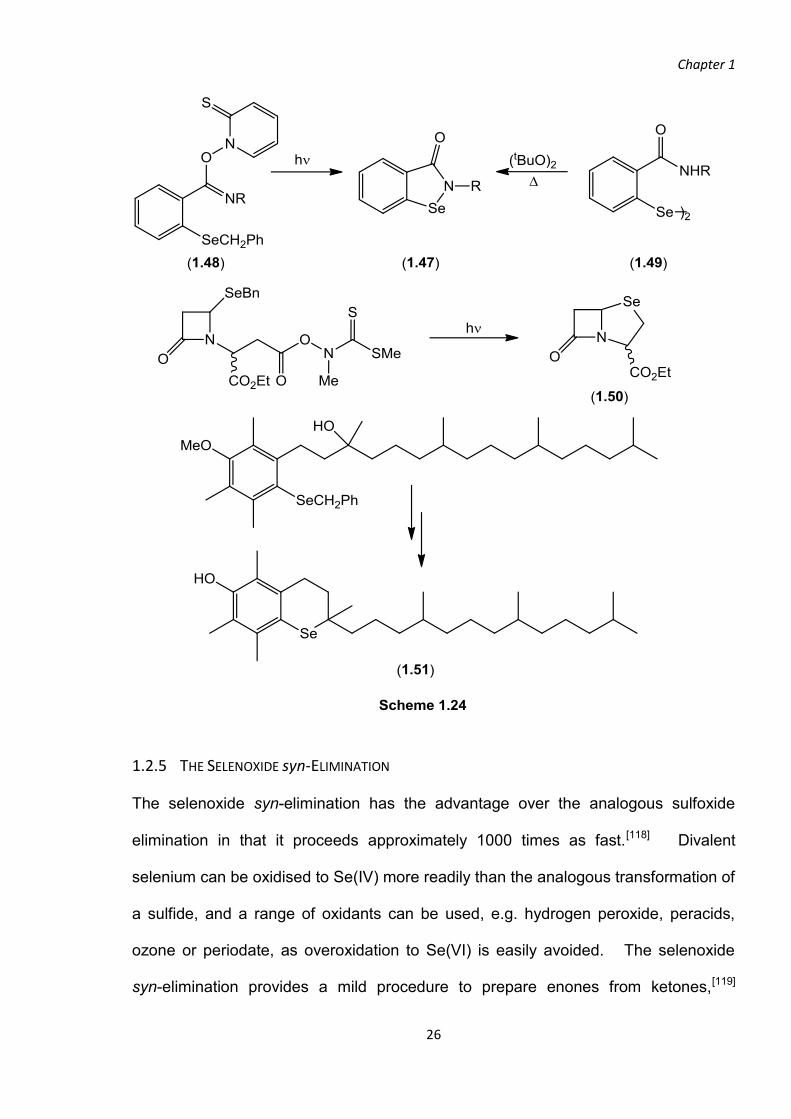
Homolytic substitution at selenium is an established path to selenium-containing
heterocycles.[113-114] The anti-inflammatory compound ebselen and its analogues[115]
(1.47) were prepared by the reaction of the diselenides (1.49) with t-butyl peroxide or
by irradiation of the PTOC imidate esters (1.48). The selenacycles of the selenium
analogues, (1.50) and (1.51), of the -lactamase inhibitor, sulbactam[116] and
tocopherol[26, 117] were also constructed via homolytic substitution (Scheme 1.24).
Chapter 1
26
1.2.5 THE SELENOXIDE syn-ELIMINATION The selenoxide syn-elimination has the advantage over the analogous sulfoxide
elimination in that it proceeds approximately 1000 times as fast.[118] Divalent
selenium can be oxidised to Se(IV) more readily than the analogous transformation of
a sulfide, and a range of oxidants can be used, e.g. hydrogen peroxide, peracids,
ozone or periodate, as overoxidation to Se(VI) is easily avoided. The selenoxide
syn-elimination provides a mild procedure to prepare enones from ketones,[119]
Chapter 1
27
acetylenes from vinyl selenoxides,[120] and allylic alcohols from epoxides after ring-
opening with a selenolate anion.[36] Protic solvents retard the elimination reaction by
hydrogen-bonding to the selenoxide oxygen, while an electron withdrawing
substituent on selenium will increase the reaction rate[121] and the use of a non-
nucleophilic base inhibits the re-addition of ‘RSeOH’ to the double bond.[121] If there
is a -hydroxyl substituent, the syn-elimination generally occurs regioselectively away
from the oxygen, giving the allylic rather than the vinylic product.[36, 122] If the
selenoxide is allylic, a [2,3]-sigmatropic rearrangement can occur faster than the syn-
elimination to give an allylic selenenate which hydrolyses to an allylic alcohol[36, 123]
(Scheme 1.25).
Unlike optically active sulfoxides, which are stable and separable, optically active
selenoxides are configurationally labile and racemise easily via the hydrate, a
process facilitated by acid catalysis.[124-125] Optically active selenoxides have been
prepared by enantioselective oxidation[126-128] by kinetic resolution,[125, 129] by
deracemisation[130] or by preparing diastereomeric selenoxides by oxidation of a
selenide possessing a chiral substituent,[37, 63, 131] and by resolution of stabilised
selenoxides with an optically active column.[132-134]

Chapter 1
28
Selenoxides (1.52) were kinetically resolved with camphor sulfonamide under
anhydrous conditions.[125] The formation of a dihydrate was sterically inhibited by the
bulky 2,4,6-triisopropylphenyl group, rendering the selenoxides stable with a half-life
of 30 hours in the presence of water (Scheme
1.26).[125] The 2,4,6-triisopropylphenyl group
also contributed kinetic stability to selenoxide
(1.53) which was further stabilized to
racemisation via intramolecular coordination to
the amino group of the 8-dimethylamino-1-
naphthyl substituent.[132]
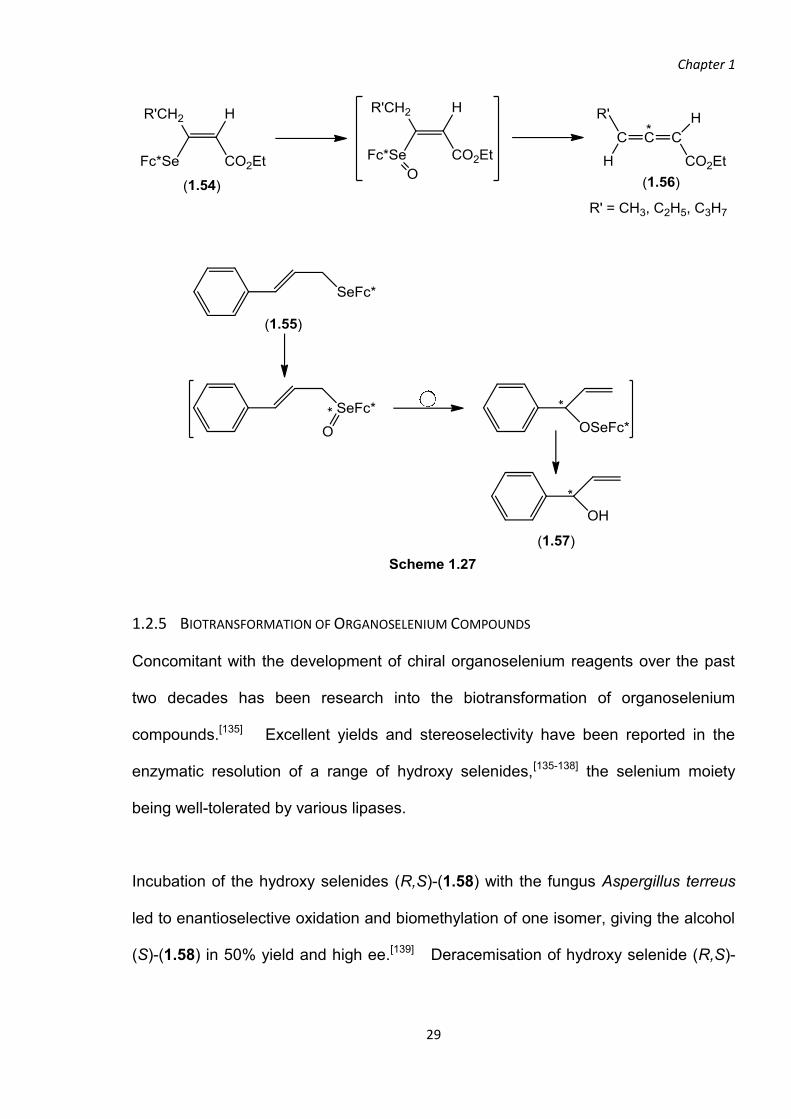
Stabilisation of the selenoxides (1.54) and (1.55) was attributed to steric and/or
electronic effects[63] but not coordination of Se to N of the chiral ferrocenyl substituent
as x-ray analysis showed no evidence of such an interaction.[131] Syn-elimination
and 2,3-sigmatropic rearrangement occurred with almost no loss of optical purity
furnishing chiral allenecarboxylic esters (1.56) in up to 89% ee[63, 131] and the allylic
alcohol (1.57) in up to 89% ee,[131] respectively (Scheme 1.27).
Chapter 1
29
1.2.5 BIOTRANSFORMATION OF ORGANOSELENIUM COMPOUNDS Concomitant with the development of chiral organoselenium reagents over the past
two decades has been research into the biotransformation of organoselenium
compounds.[135] Excellent yields and stereoselectivity have been reported in the
enzymatic resolution of a range of hydroxy selenides,[135-138] the selenium moiety
being well-tolerated by various lipases.
Incubation of the hydroxy selenides (R,S)-(1.58) with the fungus Aspergillus terreus
led to enantioselective oxidation and biomethylation of one isomer, giving the alcohol
(S)-(1.58) in 50% yield and high ee.[139] Deracemisation of hydroxy selenide (R,S)-

Chapter 1
30
(1.59), also catalysed by A. terreus, afforded the (R)-isomer in 98% yield and 99%
ee[140] (Scheme 1.28).
Selenium-containing chiral amines such as (1.60) were resolved by dynamic kinetic
resolution, giving the amides (1.61) in 74% yield and 99% ee.[141-142] Racemisation
of the amines was catalysed using Pd-BaSO4 with the acylation step catalysed by
Candida antarctica lipase B (CAL-B)[141] (Scheme 1.29).

Chapter 1
31
meta- or para-Organoselenoacetophenones, (1.62) for example, can be reduced to
chiral alcohols (1.63) in high yield and ee after incubation with whole fungal cells[143]
or fresh carrot[144] (Scheme 1.30).
Through the many contributions to the development of organoselenium chemistry
over the last four decades, selenium-mediated transformations now occupy an
established and significant place in organic synthesis.

Chapter 2
32
2 THE AMIDOSELENATION OF ALKENES
2.1 INTRODUCTION
The amidoselenation of alkenes was first described by Toshimitsu et al.[145] in 1981.
In the literature procedure, [145] an alkene is treated with phenylselenenyl halide, a
nitrile and aqueous triflic acid to give a -amidoalkyl phenyl selenide. The
mechanism of this reaction was proposed[145] to be a variation of that of the Ritter
reaction[146] in which amides are formed from a nitrile and a carbonium ion under
strongly acidic conditions.[147] The seleniranium ion (2.1) which is initially formed is
in equilibrium with the haloselenide adduct[145] (2.2). Attack by nitrogen on the
seleniranium ion (2.1) gives an imidoyl halide (2.3) which is then hydrolysed to yield
the amide (2.4) (Scheme 2.1).
In previous work of our research group,[148] the amidoselenation of cyclohexene and
cyclopentene was investigated using two equivalents of phenylselenenyl bromide in
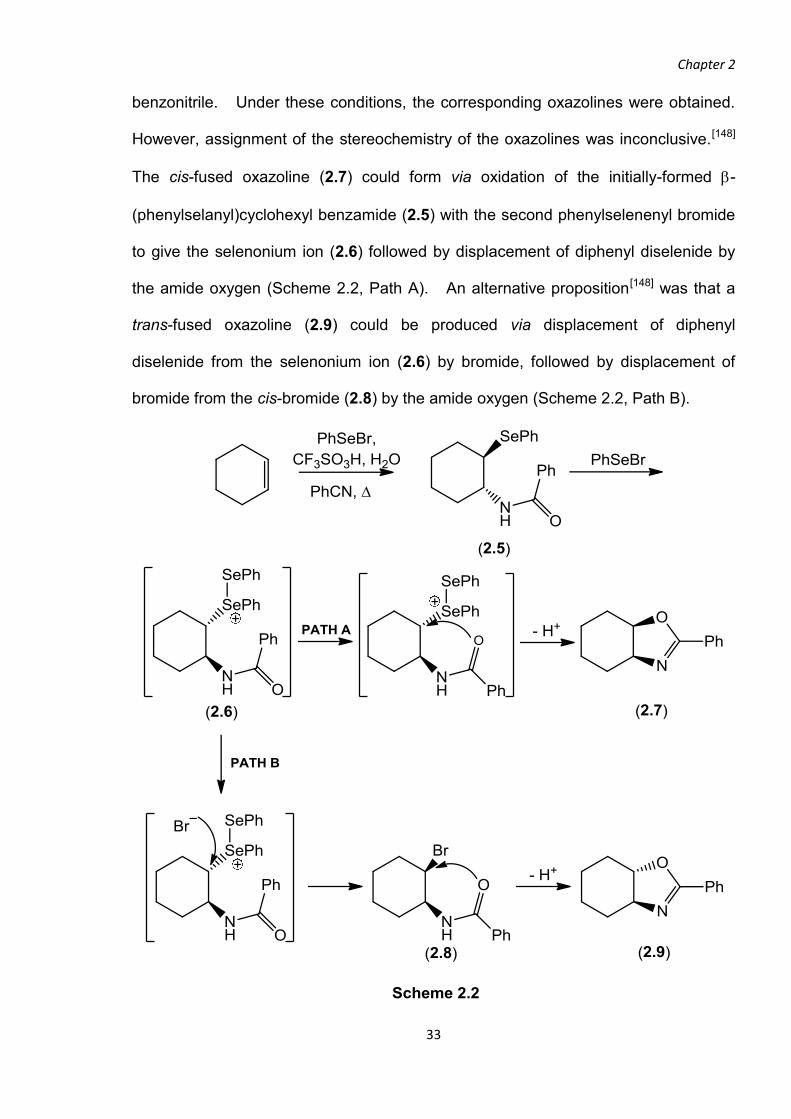
Chapter 2
33
benzonitrile. Under these conditions, the corresponding oxazolines were obtained.
However, assignment of the stereochemistry of the oxazolines was inconclusive.[148]
The cis-fused oxazoline (2.7) could form via oxidation of the initially-formed -
(phenylselanyl)cyclohexyl benzamide (2.5) with the second phenylselenenyl bromide
to give the selenonium ion (2.6) followed by displacement of diphenyl diselenide by
the amide oxygen (Scheme 2.2, Path A). An alternative proposition[148] was that a
trans-fused oxazoline (2.9) could be produced via displacement of diphenyl
diselenide from the selenonium ion (2.6) by bromide, followed by displacement of
bromide from the cis-bromide (2.8) by the amide oxygen (Scheme 2.2, Path B).

Chapter 2
34
Both the cis- and the trans-oxazolines, (2.7) and (2.9), are known compounds.[149-150]
The initial aim of the present work was to investigate whether a trans-oxazoline (2.9)
could be formed in the manner described above.
2.2 INVESTIGATION OF THE FORMATION OF THE trans-OXAZOLINE (2.9)
When cyclohexene was heated with two equivalents of phenylselenenyl bromide and
aqueous triflic acid in benzonitrile at a range of temperatures from 100 to 160°C, only
the cis-oxazoline[149] (2.7) was obtained, in yields of 5-30%. At the higher reaction
temperatures of 140-160°C which, it was proposed,[148] might allow for strain in the
transition state to the trans-oxazoline to be overcome, the yield of the cis-oxazoline
(2.7) was lowest.
The reaction could be investigated by treatment of the ‘intermediate’ amido selenide
with one equivalent of phenylselenenyl bromide. For this it was necessary to
prepare 2-(phenylselanyl)cyclohexyl benzamide (2.5).
The amido selenide[145] (2.5) was obtained in 66% yield from cyclohexene in
benzonitrile heated to 90°C, according to a variation[148] of the literature
amidoselenation procedure.[145] Treatment of 2-(phenylselanyl)cyclohexyl
benzamide (2.5) with phenylselenenyl bromide in benzonitrile at 115°C afforded the
cis-oxazoline (2.7) as a minor product (3%) along with trans-2-

Chapter 2
35
bromocyclohexylbenzamide (2.10, 31%) and cis-2-bromocyclohexylbenzamide (2.8,
10%) (Scheme 2.4). Stereochemistry of the bromide isomers was assigned based
on the ring methine proton coupling constants: a trans-diaxial coupling constant of
10.5 Hz for the trans-bromide (2.10) and a coupling constant of 3.0 Hz for the cis-
isomer (2.8). While the two ring methine protons of the trans-bromide (2.10) occur at
similar frequencies, 4.14 and 4.02, the analogous signals of the cis-bromide (2.8)
are more differentiated. The CHBr proton of (2.8) resonates at 4.49, 0.69 ppm
downfield from the CHN proton signal, a multiplet centred at 4.1, so-assigned to
account for coupling to the NH proton. In a model of the cis-bromide (2.8) in which
steric interactions are minimised, the molecule adopts a conformation with the bulky
amide group equatorial and the bromine axial. Deshielding of the equatorial CHBr
proton can therefore be attributed to the deshielding cone of the cyclohexane ring
carbons as well as the aromatic amide group. Mass spectra of both bromides
provided evidence of the bromine substituent with two weak molecular ions of similar
intensity occurring at m/z 281 and 283. In both spectra, fragmentation led to the
expected peaks at m/z 202, 122 and 105 due to loss of bromine from the molecular
ion, the protonated benzamide ion and the benzoyl cation, C6H5C=O+, respectively.
Spectral data for the cis-bromide (2.8) compared well with that of cis-2-
bromocyclohexylacetamide.[151]
Treatment of 2-(phenylselanyl)cyclohexyl benzamide (2.5) with phenylselenenyl
bromide in dichloromethane at room temperature gave a 3:1 mixture of the cis-
oxazoline (2.7) and the cis-bromide (2.8). When the reaction was conducted in
refluxing acetonitrile the cis-oxazoline (2.7) was again the main product along with
the trans-bromide (2.10) and the cis-bromide (2.8).

Chapter 2
36
With addition of tetraethylammonium bromide to the reaction mixture, the cis-bromide
(2.8) could be made to predominate. At room temperature in dichloromethane an
approximately 60% yield of (2.8) was thus obtained as 75% of the product along with
the cis-oxazoline (2.7) and unreacted amido selenide (2.5) in minor amounts. In
refluxing acetonitrile with addition of tetraethylammonium bromide the cis-bromide
(2.8) also made up over 50% of the product which also included the cis-oxazoline
(2.7) and trans-bromide (2.10). These observations are consistent with reported
results[152] from a procedure in which the selenide is oxidised with molecular chlorine
rather than phenylselenenyl bromide and from a previous study[151] in which
molecular bromine was used as the oxidant. At room temperature and in refluxing
acetonitrile it appears that displacement of diphenyl diselenide by bromide ion to give
the cis-bromide (2.8) competes with displacement by the amide oxygen to give the
cis-oxazoline (2.7). Conducting the reaction in benzonitrile at 115°C may have
provided sufficient energy for the ring-opening of the oxazoline (2.7) by bromide ion
to give the trans-bromide (2.10).

Chapter 2
37
For the purpose of verifying the stereochemistry of the oxazoline products of these
reactions, it was decided to prepare the cis-oxazolines via the established procedure
of Toshimitsu et al.[153] in which excess m-CPBA (2.5-5 equivalents) followed by
potassium hydroxide (7.5-11 equivalents) are added to a solution of the selenide at
room temperature in an alcohol solvent. These conditions were employed by
Toshimitsu et al.[153] in the cyclisation of the 2-pyridylselenoamide (2.11) to the 2-
methyloxazoline (2.12) (Scheme 2.5) and in the generation of three- to six-membered
N-tosyl nitrogen-containing heterocycles (2.13) from N-{(phenylseleno)alkyl}-p-
toluenesulfonamides (2.14) and the pyrrolidine (2.15) from the amidoselenide (2.16)
(Scheme 2.5).

Chapter 2
38
Following the procedure developed by Toshimitsu et al.,[153] the oxidation of 2-
(phenylselanyl)cyclohexyl benzamide (2.5) with 4 equivalents of m-CPBA in
isopropanol followed by addition of 7.5 equivalents of potassium hydroxide, gave only
a 12% yield of the expected cis-oxazoline (2.7) and, unexpectedly, the N-
benzoylaziridine[154] (2.17) which was isolated in a yield of 85% (Scheme 2.6). The
symmetry of this aziridine[154] (2.17) was reflected in the 13C NMR spectrum in which
there appeared only three alkyl signals, at 37.02, 23.87 and 19.93, and in the 1H
NMR spectrum in which the ring methine protons appeared as a narrow multiplet at
2.75. The mass spectrum of (2.17) showed a strong peak at m/z 202 due to M+H
and fragments at m/z 105 and m/z 96 due to the benzoyl cation and loss of the
benzoyl group from the molecular ion respectively. From these investigations it was
concluded from that the trans-oxazoline (2.9) could not be prepared from the reaction
of cyclohexene with two equivalents of phenylselenenyl halide.
Conditions effecting the cyclisation of amides to oxazolines are various and well-
established.[150, 155-162] However, N-acylaziridines are only obtained from amides
Chapter 2
39
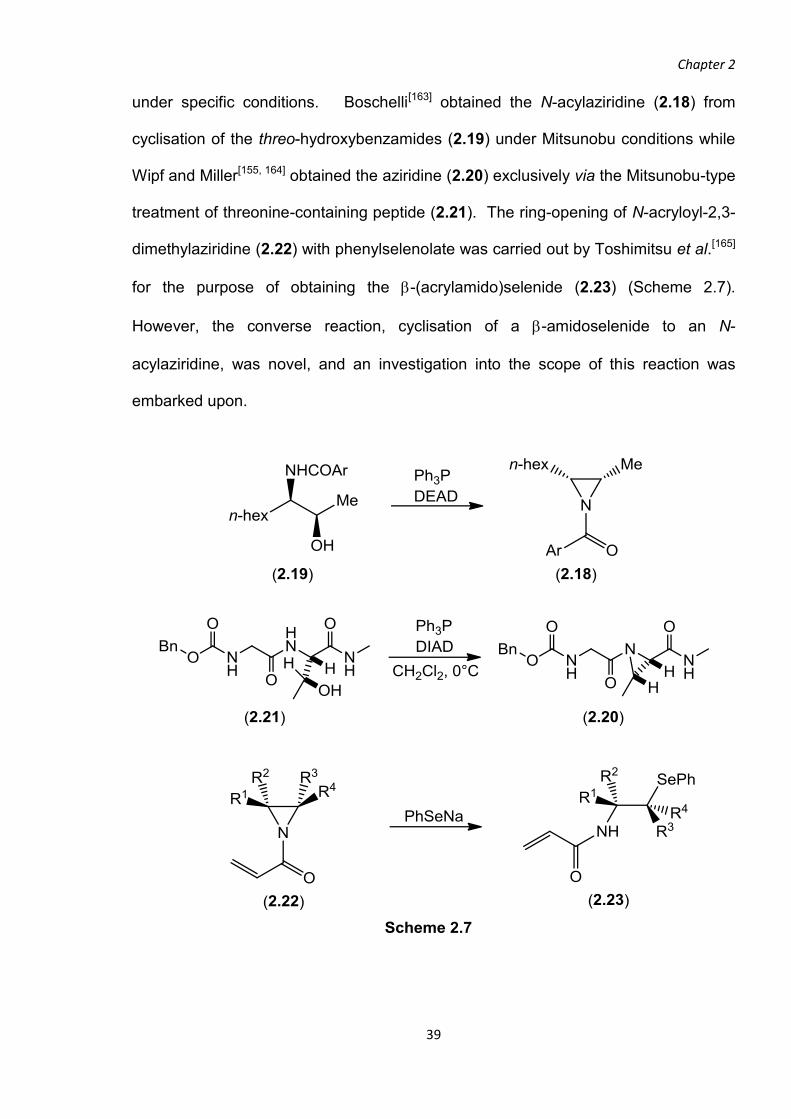
under specific conditions. Boschelli[163] obtained the N-acylaziridine (2.18) from
cyclisation of the threo-hydroxybenzamides (2.19) under Mitsunobu conditions while
Wipf and Miller[155, 164] obtained the aziridine (2.20) exclusively via the Mitsunobu-type
treatment of threonine-containing peptide (2.21). The ring-opening of N-acryloyl-2,3-
dimethylaziridine (2.22) with phenylselenolate was carried out by Toshimitsu et al.[165]
for the purpose of obtaining the -(acrylamido)selenide (2.23) (Scheme 2.7).
However, the converse reaction, cyclisation of a -amidoselenide to an N-
acylaziridine, was novel, and an investigation into the scope of this reaction was
embarked upon.

Chapter 2
40
2.3 ONE-POT PREPARATION OF -AMIDO SELENIDES
Investigation of this aziridine-forming reaction necessitated the preparation of a range
of -amido selenide substrates. Following the straightforward preparation of 2-
(phenylselanyl)cyclohexyl benzamide (2.5), using a variation of the literature
procedure[145] developed by Cooper,[148] benzamidoselenation was attempted with the
alkenes cyclopentene, cycloheptene, 1-octene and trans-2-hexene using the same
procedure.
Unlike the reaction with cyclohexene, yields of the -amido selenides (2.24) and
(2.27) derived from cyclopentene and cycloheptene were poor (16% and 2%
respectively), although comparable with the yields of the same compounds obtained

Chapter 2
41
by Cooper.[148] Both the cyclopentyl and cycloheptyl derivatives were characterised
by 1H NMR data showing the three diagnostic signals of the methine ring and NH
protons in the regions expected.[145] Mass spectra in both cases showed a strong
molecular ion and a selenium-containing fragment due to loss of benzamide as well
as a fragment at m/z 188 (2.24) and m/z 216 (2.27) due to loss of C6H5Se from the
parent molecule. Concomitant with amidoselenation of the 5-, 6- and 7-membered
cycloalkenes was the formation of the corresponding cis-oxazolines (2.25, 32%),
(2.7, 6%) and (2.28, 24%). The Ritter products, N-cyclopentylbenzamide[154] (2.26,
4%) and N-cycloheptylbenzamide[166-167] (2.29, 20%) were also produced from the
reactions with cyclopentene and cycloheptene respectively (Scheme 2.8). An
additional by-product from the reaction with cycloheptene was the syn-elimination
product, N-(cyclohept-2-en-1-yl)benzamide (2.30, 2%). N-(cyclohept-2-en-1-
yl)benzamide (2.30) was characterised in the 1H NMR spectrum by the appearance
of two alkene proton peaks at 5.88 and 5.64, an NH proton signal at 6.23 and a
fourth downfield signal at 4.82 due to the allylic CHN proton. The alkene protons
had a coupling constant of 12.3 Hz, within the range expected for alkene protons of a
cycloheptene ring.[168] In the 13C NMR spectrum the alkene carbons appeared at
128.81 and 127.11 while the mass spectrum showed a strong molecular ion at m/z
215.
Following the literature amidoselenation procedure,[145] acetamidoselenation of
cyclohexene afforded an 88% yield of -(phenylselanyl)cyclohexyl acetamide[145]
(2.31) (Scheme 2.9).

Chapter 2
42
Amidoselenation of 1-octene in benzonitrile gave a mixture of the Markovnikov and
anti-Markovnikov products in 59% yield from which the Markovnikov product (2.32)
was isolated by crystallisation. The Markovnikov compound (2.32) was
distinguishable from its regioisomer (2.33) in the 1H NMR spectrum by two doublets
of doublets at 3.29 and 3.22 attributed to the diastereotopic CH2Se protons
coupled to the neighbouring CHN proton (4.8 and 5.4 Hz) and with a geminal
coupling constant of 12.8 Hz. The 1H NMR signals of the diastereotopic CH2N
protons of the regioisomer (2.33) were well differentiated at 3.80 and 3.51 and
appeared as two doublets of doublets of doublets, coupled to the NH proton as well
as the CHSe proton and with a geminal coupling constant of 13.5 Hz. These signals
were downfield from the CHSe multiplet which appeared at 3.41.
Benzamidoselenation of trans-2-hexene gave a poor yield (12%) of the -amido
selenides as a mixture of the Markovnikov and anti-Markovnikov products (2.34) and
(2.35) in a ratio of 47:53 along with the oxazolines (2.36) and (2.37) in a ratio of 52:48
(Scheme 2.10). The slight predominance of the anti-Markovnikov amido selenide
(2.34) over its regioisomer (2.35) could be due to the more facile transformation of
the adduct (2.34) into the oxazoline (2.36) by displacement of PhSeSePh from the
less-hindered carbon.

Chapter 2
43
The two amido selenide isomers (2.34) and (2.35) were distinguishable
spectroscopically by the pattern of their methine signals in the 1H NMR spectrum.
The CHSe signal of the Markovnikov isomer (2.34), appeared as a clearly defined
doublet of quartets at 3.65 while a more complex signal approximating a doublet of
doublets of triplets at 4.33 was attributable to the CHN proton. The CHN signal of
the anti-Markovnikov isomer (2.35) appeared as a twelve-line signal at 4.47,
interpreted as a doublet of doublets of quartets, coupled to the CHSe, NH and methyl
protons, with the CHSe protons resonating as a less complex doublet of doublets of

Chapter 2
44
doublets at 3.56. In the 13C NMR spectra of the Markovnikov isomer (2.34), the
CHN signal resonated at 53.88, downfield from the CHSe signal at 47.51 due to
its more electronegative amide substituent. In contrast, in the 13C spectrum of
(2.35), the signal for the C3 CHSe carbon appeared at 54.97, identifiable by its
distinct CSe satellites, while the signal of the C2 CHN carbon appeared upfield at
48.66, the effect of the position at C3 in the carbon chain overriding the effect of the
more electronegative substituent at C2.
The two oxazoline regioisomers (2.36) and (2.37) were distinguishable in the 1H NMR
spectrum by the doublet of quartets at carbon 2 which occurred at 4.39 due to the
CHO proton in isomer (2.36) and at 3.91 due to the CHN proton in isomer (2.37).
The mass spectra of the two oxazolines also reflected the differences in their
structures: the mass spectrum of isomer (2.36) showed a base peak at m/z 44 which
was attributed to the acetaldehyde fragment, and a prominent peak at m/z 160 due to
loss of C3H7, whereas isomer (2.37) fragmented to give a base peak at m/z 131 due
to loss of C3H7CHO.
The Ritter products (2.26) and (2.29), which had not been reported previously from
the amidoselenation reaction,[145, 148] may have arisen from the addition of H+ and
benzonitrile to the double bond. The ability for (2.26) to be formed in this manner
was confirmed by reacting cyclopentene with aqueous trifluoromethanesulfonic acid

Chapter 2
45
in benzonitrile, giving N-cyclopentylbenzamide[154] (2.26) in 25% yield. Similarly,
cyclohexene gave N-cyclohexylbenzamide[154] (2.38), in 55% yield (Scheme 2.11).

Chapter 2
46
It has been shown that in the addition of a selenenyl halide to an alkene, the alkene
and the haloselenide adduct (2.2) are in equilibrium and can interconvert via the
seleniranium ion (2.1, Scheme 2.1).[48, 60] The direction of the equilibrium is
influenced by the nature of the alkene, the counterion, and the reaction
temperature.[48] It is therefore reasonable to propose that in the amidoselenation
reaction with cyclopentene and cycloheptene, the equilibrium between the alkene
and the seleniranium ion may lie more toward the alkene, than in the reaction with
cyclohexene. The unconsumed cyclopentene (or cycloheptene) is then free to
undergo the Ritter reaction to give the N-cycloalkyl amide (2.26) (or (2.29)) (Scheme
2.12). The unconsumed selenium reagent is available to react with the amido
selenide (2.24) (or (2.27)) to give a phenylselenonium intermediate (2.39) (or (2.40))
which then cyclises to the cis-oxazoline (2.7) (or (2.28)) with loss of diphenyl
diselenide. The syn-elimination product (2.30) from the reaction with cycloheptene
may have been generated by elimination of H+ and diphenyl diselenide from the
selenonium intermediate (2.40).
2.4 ALTERNATIVE SOLVENTS FOR THE AMIDOSELENATION REACTION
In order to verify the stereochemistry of the oxazolines and amidoselenides by X-ray
crystal determination, the preparation of a p-bromobenzamido selenide derived from
p-bromobenzonitrile was undertaken. The amidoselenation reaction could not be
conducted with the nitrile as solvent using the solid p-bromobenzonitrile, and
therefore, a non-nitrile solvent was required. Since previous reactions had been
carried out in refluxing acetonitrile, initial consideration was given to solvents with a
boiling point of at least 82°C, namely dimethylacetamide and toluene. Trial reactions
were conducted using either cyclohexene or cyclopentene and four to five
equivalents of benzonitrile (Table 2.1).

Chapter 2
47
TABLE 2.1
BENZAMIDOSELENATION IN NON-NITRILE SOLVENTSa
alkene solvent
reaction temp (°C)
% isolated yield
amido selenide
hydroxy selenide
N-cycloalkyl benzamide
cis-oxazoline
cyclohexene
dimethyl acetamide 90-95 - 41 - -
toluene 96-115 7 - 17 15
dichloromethane 39.5 67 1 - -
dichloromethane r.t. 90b - - -
chloroform 62 10 20 - -
cyclopentene benzonitrile r.t.-55c 55 - - -
dichloromethane 39.5 10 - 37 - a 5 eq.benzonitrile in solvent specified, 1 eq. TfOH, 5 eq. H2O b Product not isolated c r.t. for 6 days then 55°C for 12 h with additional TfOH
Attempted amidoselenation of cyclohexene in dimethylacetamide at 90–95°C gave
only 2-(phenylseleno)cyclohexanol[169] (2.41) (Scheme 2.13). Transformation
beyond the hydroxy selenide stage requires protonation of the hydroxyl group which
is lost on formation of the seleniranium ion. The dimethylacetamide may have
‘sequestered’ the acid, preventing protonation of the hydroxyl group.
From amidoselenation of cyclohexene in toluene at 96-115°C, N-
cyclohexylbenzamide (2.38, 17%), the cis-oxazoline (2.7, 15%) and the -amido
selenide (2.5, 7%) were obtained.

Chapter 2
48
In refluxing chloroform, a mixture of the hydroxy selenide (2.41) and the -amido
selenide (2.5) was produced, in low yield in a ratio of 2:1. Unsuitability of these non-
polar solvents could be attributed to poor solvation of the charged seleniranium
intermediate and, particularly with toluene, low availability of water to react with the
seleniranium ion as a result of an inhomogeneous reaction mixture.
The reaction with cyclohexene in refluxing dichloromethane was reasonably
successful, giving the -amido selenide (2.5) in 67% yield. However, this success
could not be replicated with cyclopentene, from which a mixture of the -amido
selenide (2.24) and the Ritter product (2.26) were produced, demonstrating
competition between the Ritter reaction with cyclopentene, H+, and benzonitrile and
formation of the seleniranium ion/haloselenide adduct.
At room temperature, the reaction of cyclohexene in dichloromethane gave a 90%
yield of the -amido selenide (2.5) before purification, which indicated that a higher
reaction temperature was not necessary for, and might hinder, the amidoselenation
reaction.
The reaction with cyclopentene in benzonitrile at room temperature gave a 55% yield
of the -amido selenide (2.24). This reaction was monitored by TLC over three

Chapter 2
49
days, after which, TLC analysis showed there to be a mixture of the -amido selenide
(2.24) and the hydroxy selenide[169] (2.42). Further trifluoromethanesulfonic acid was
added and the mixture reacted for a further 3 days and finally heated to 55°C for 12
hours. The subsequent conversion of the hydroxy selenide (2.42) to amido selenide
(2.24) suggested that isolation of the alcohol before treating it with
trifluoromethanesulfonic acid and the nitrile might be a cleaner route to -amido
selenides (Scheme 2.14).
2.5 TWO-STEP PREPARATION OF -AMIDO SELENIDES
Amidoselenation via hydroxyselenation using chloroacetonitrile or bromoproprionitrile
in reagent quantity in dichloromethane at room temperature has been reported by
Toshimitsu et al.[41, 165] Hydroxyselenation[169] of cyclohexene with phenylselenenyl
chloride in an acetonitrile-water mixture gave 2-(phenylseleno)cyclohexanol (2.41) in
high yield. The reaction of the alcohol (2.41) with benzonitrile and
trifluoromethanesulfonic acid in dichloromethane at room temperature for 48 hours
gave the -amido selenide (2.5) in excellent yield with the overall yield higher than for
the one-step amidoselenation of cyclohexene.

Chapter 2
50
This two-step procedure (Scheme 2.15) gave good overall yields of the -benzamido
selenides (2.24) and (2.27) derived from cyclopentene and cycloheptene via the
hydroxy selenides (2.42) and (2.43) and avoided the complication of the oxazoline,
Ritter and syn-elimination by-products. Also using this procedure, a good yield
(77%) of -acetamidocycloheptyl phenyl selenide (2.44) was afforded compared with
the literature yield of 55% for the one-step procedure.[145]
The two-step process was successful in the reaction of 2-(phenylseleno)cyclohexanol
(2.41) using the solid nitrile, p-bromobenzonitrile, with the preparation of 2-
(phenylselanyl)cyclohexyl p-bromobenzamide (2.45) proceeding in 87% yield
(Scheme 2.16). Table 2.2 summarises the results of the one- and two-step
procedures.

Chapter 2
51
TABLE 2.2
PERCENTAGE YIELDS FOR 1-STEP AND 2-STEP AMIDO SELENIDE PREPARATION
alkene nitrile
1-step amido selen- ation
2-step amidoselenation
hydroxy selenide
amido selenide
Yield over two steps
cyclopentene benzonitrile 16 77 76 59
cyclohexene
acetonitrile 88 - - -
benzonitrile 66 89 93 83
p-bromo-benzonitrile - 89 87 77
cycloheptene acetonitrile - 91 85 77
benzonitrile 2 91 57 52
cyclooctene benzonitrile - 69 45 31
trans-2-hexene benzonitrile 12 85 97 82
1-octene benzonitrile 59 71 82 58
p-bromo- benzonitrile - 71 43 31
One-step amidoselenation of cyclooctene was not attempted after the success of the
two-step procedure. Hydroxyselenation to give 2-(phenylseleno)cyclooctanol (2.46)
proceeded in 69% yield; however, only a 45% yield of 2-(phenylselanyl)cyclooctyl
benzamide (2.47) was obtained in the amidoselenation step. Low yields were also
reported by Toshimitsu et al.[169] for the reaction of 2-phenylselenocyclooctanol with
acrylonitrile (39%) and chloroacetonitrile (24%).
Hydroxyselenation of 1-octene gave a mixture of the Markovnikov and anti-
Markovnikov products (2.48) and (2.49) in 71% yield in a ratio of 85:15. The two

Chapter 2
52
hydroxy selenides were isolated by chromatography. Under electrospray conditions,
the high resolution mass spectrum of each isomer showed the expected mass for an
M-OH fragment. In the 1H NMR spectrum of the Markovnikov isomer (2.48), the
methine CHO proton resonated as a multiplet centered at 3.65, with two doublets of
doublets, at 3.15 and 2.89, due to the diastereotopic methylene protons under
selenium. In the 1H NMR spectrum of the anti-Markovnikov compound (2.49), the
signals of the diastereotopic protons under oxygen appeared as two doublets of
doublets, at 3.56 and 3.45, downfield, as expected, from the multiplet at 3.16
due to the proton under selenium.

Chapter 2
53
The amidoselenation step was carried out on the mixture of the hydroxy selenide
regioisomers, (2.48) and (2.49), as the reaction proceeds via a selenonium ion
intermediate, eliminating any advantage conferred by starting with a single
regioisomer. The reaction of the mixture of hydroxy selenides with benzonitrile gave
a mixture of the benzamido selenides (2.32) and (2.33) in a ratio of 95:5. The
Markovnikov adduct (2.32) was isolated from the mixture in 82% yield by
recrystallisation (Scheme 2.17).
Amidoselenation of a mixture of the hydroxy selenides (2.48) and (2.49) derived from
1-octene with three equivalents of p-bromobenzonitrile in dichloromethane gave a
51% yield of a mixture of the Markovnikov and anti-Markovnikov p-bromobenzamido
selenides (2.50) and (2.51), with the Markovnikov isomer predominating (Scheme
2.18) along with a small amount of the oxazoline (2.52) derived from amido selenide
(2.50). 80% of the nitrile was recovered, giving a theoretical yield of 60%, indicating
that the reaction was not very efficient. The 1H NMR spectrum of the Markovnikov
amido selenide (2.50) closely resembled that of the bromine-free compound (2.32) in
showing a three-proton system, with the signals due to the diastereotopic protons
under selenium appearing as two clean strongly-coupled doublets of doublets at
3.30 and 3.20 with vicinal coupling to the multiplet at 4.39 due to the CHN proton.
The anti-Markovnikov isomer (2.51) was not obtained pure but was assigned from its
three-proton system of two distinct signals, at 3.83 and 3.44 due to the CH2N
protons and its CHSe multiplet at 3.38.

Chapter 2
54
Hydroxyselenation of trans-2-hexene gave an 85% yield of the Markovnikov and anti-
Markovnikov products (2.53) and (2.54) in a ratio of 55:45 (Scheme 2.19).
Chromatography partially separated the two alcohols, making it possible to
distinguish the NMR signals of the individual isomers. The doublet of quartets at C2
was diagnostic, and appeared at 3.44 for the CHSe signal of the Markovnikov
isomer (2.53), and at 3.85 for the CHO signal of the anti-Markovnikov isomer (2.54).
The C3 proton signals appeared as doublets of doublets of doublets at 3.62 for the
CHO proton of isomer (2.53) and at 3.37 for the CHSe proton of isomer (2.54). In
the 13C NMR spectrum, carbon-selenium coupling was evident in the CSe signal of
the Markovnikov isomer, at 47.40, and of the anti-Markovnikov isomer, at 57.27.
Further reaction of the mixture of (2.53) and (2.54) gave the amido selenides as a

Chapter 2
55
mixture of the Markovnikov and anti-Markovnikov products (2.34) and (2.35) in a ratio
of 53:47 in 97% yield.
For characterisation purposes, a mixture of the two regioisomers (2.34) and (2.35)
was subjected to chromatography in order to purify them but also in an attempt at
their separation. Although full separation was not achieved, fractions were obtained
that were enriched in one or other regioisomer. From the first-eluted enriched
fraction were obtained large transparent crystals. A crystal structure
determination[170] showed that this material, the anti-Markovnikov isomer (2.35), was
chiral with absolute configuration (2S,3R)-2-(benzamido)-3-(phenylseleno)hexane.
Only two stereoisomers of each regioisomer would be expected as the selenonium
ion intermediate would constrain the stereochemistry of C2 relative to C3. The
racemate appears to have crystallised as a conglomerate,[171] resolving
spontaneously into enantiomorphous crystals. This is a phenomenon which has only

Chapter 2
56
been observed in 5 to 10% of organic racemates[171] and potentially allows for the
mechanical separation of enantiomers.
These explorations of the amidoselenation reaction led to the conclusion that the two-
step procedure with isolation of the hydroxy selenide intermediate was superior to the
one-step procedure, giving higher yields in most cases and simpler product mixtures.
2.6 PREPARATION OF THE trans-OXAZOLINE (2.9)
In 1950 Johnson and Schubert[150] reported the preparation of the trans-oxazoline
(2.9) by the treatment of trans-2-aminocyclohexanol hydrochloride with ethyl
iminobenzoate (2.55) (Scheme 2.20). The identity of the trans-fused product was
verified from the melting point of the known product of its hydrolysis, trans-2-
benzoyloxycyclohexylamine hydrochloride (2.56).
In order to obtain spectral data for the trans-oxazoline (2.9) to distinguish it from the
cis-oxazoline (2.7) and to verify that it had not been produced in the amidoselenation

Chapter 2
57
reaction, the trans-oxazoline (2.9) was prepared, following the literature
procedure,[150] from ethyl iminobenzoate (2.55) and commercially available trans-2-
aminocyclohexanol hydrochloride.
Ethyl iminobenzoate hydrochloride (2.57) was prepared according to the procedure of
MacKenzie et al.[172] in 89% yield from benzonitrile, ethanol and hydrogen chloride.
Deprotonation[173] gave ethyl iminobenzoate (2.55) in 87% yield after Kugelrohr
distillation (Scheme 2.21). From the reaction of ethyl iminobenzoate (2.55) with
trans-2-aminocyclohexanol hydrochloride[150] the trans-oxazoline (2.9) was obtained
(35%). Recrystallisation gave colourless crystals which melted at 78-79.5°C,
comparing well with the literature[150] melting point of 73-77°C for the trans-oxazoline,
and differentiating it from compound (2.7), with its melting point of 42-45°C, in accord
with the literature[150] value of 46-48°C for the cis-oxazoline.
Comparison of the 1H and 13C NMR spectra of the trans- and cis-oxazolines, (2.9)
and (2.7), shows distinct differences. The 1H NMR signals of the CHN and CHO
protons of the trans-oxazoline (2.9) are approximately 0.9 ppm upfield from the
analogous cis-oxazoline signals. In order for the fused ring system to accommodate
the trans-geometry, the trans-oxazoline CHN and CHO protons would occur in the
axial position and would not be affected by the C-C deshielding cone of the
cyclohexyl ring, whereas the cis-oxazoline CHN and CHO protons are more likely to

Chapter 2
58
found within the deshielding cones of the cyclohexyl C-C bonds. The 1H NMR
signals of the trans-oxazoline CHN and CHO protons appear as doublets of doublets
of doublets with coupling constants of 13.8, 11.7 and 3.6 Hz and 13.8, 11.7 and 3.3
Hz respectively. The two sets of trans-diaxial coupling constants is in contrast with
the typical cis coupling constant of 8.1 Hz exhibited by the CHN and CHO protons of
the cis-oxazoline (2.7).
In the 13C NMR spectrum, the CHN and CHO and other alkyl signals of the trans-
oxazoline (2.9) are downfield in comparison with the cis-oxazoline signals.
The assignment of cis-stereochemistry to the product obtained from the reaction of
two equivalents of phenylselenenyl bromide, nitrile and cyclohexene is therefore
strongly supported by these results and spectral data.

Chapter 3
59
3 CYCLISATION OF -AMIDOALKYL PHENYL SELENIDES
3.1 INITIAL ATTEMPTS TO OPTIMISE THE FORMATION OF N-ACYLAZIRIDINES
The conditions used in the oxidation of 2-(phenylselanyl)cyclohexyl benzamide (2.5),
which unexpectedly generated the N-acylaziridine (2.17), provided the starting point
for the investigation of our new method for the generation of
N-acylaziridines. The -(phenylselanyl)alkyl amides, (2.24),
(2.5), (2.27), (2.31), (2.45) and (2.32) were used as the
substrates for the initial investigation. According to the
literature procedure,[153] the -amido selenide was dissolved in isopropanol and
treated with at least three equivalents of m-CPBA, The use of an excess of m-CPBA
as oxidant[174-176] and an alcohol as solvent[174-176] have been shown to be effective
conditions for the oxidation of a selenide to a selenone. An excess of oxidant has
also been shown to facilitate the oxidation of a selenoxide to a selenone, so as to
avoid the selenoxide syn-elimination as a side-reaction.[175]

Chapter 3
60
Using these general conditions,[153] and with variation of some parameters, a clean
reaction giving the aziridine exclusively was not achieved from any of the starting -
amido selenides. Using 10.8 equivalents of hydroxide and 4 equivalents of peracid,
a 95:5 ratio of aziridine (2.17) to oxazoline (2.7) was obtained from 2-

Chapter 3
61
(phenylselanyl)cyclohexyl benzamide (2.5). Increasing the amount of base to 13.5
equivalents did not increase the proportion of aziridine (2.17) in the product.
The reaction of 2-(phenylselanyl)cyclopentyl benzamide (2.24) using 10.5 equivalents
of hydroxide and 3.9 equivalents of peracid gave a 60:40 ratio of aziridine (3.1) to
oxazoline (2.25). With 13.4 equivalents of base the product ratio decreased to 45:55
(Scheme 3.1). Using ethanol in place of isopropanol as solvent also favoured the
oxazoline (2.25), giving a 30:70 ratio of aziridine (3.1) to oxazoline (2.25). The
bridgehead CHN protons of known[154] aziridine (3.1) appeared as a singlet-like peak
at 3.19. Unlike the bridgehead proton signals for the other fused aziridines
prepared (vide infra), the complete coalescence of this signal suggests more rapid
pyramidal inversion at nitrogen (Scheme 3.1).
Increasing the quantity of base from 8 to 10 equivalents in the reaction of 2-
(phenylselanyl)cycloheptyl benzamide (2.27) led to a small increase from 15:85 to
25:75 in the ratio of aziridine (3.2) to oxazoline (2.28) produced (Scheme 3.1).
Lowering the reaction temperature to 0°C decreased the ratio of aziridine (3.2) to
oxazoline (2.28) to 15:85. Carrying out the reaction at 37°C also appeared to favour
the oxazoline, giving a 10:80:10 mixture of aziridine (3.2), oxazoline (2.28) and the
syn-elimination product, N-(cyclohept-2-en-1-yl)benzamide (2.30). In the 1H NMR
spectrum of the aziridine (3.2) the signals of the bridgehead CHN protons appeared
as a narrow multiplet centred at 2.72. The symmetry of the molecule was again
apparent in the 13C spectrum which showed only four alkyl and four aromatic signals
and the carbonyl carbon at 180.0. The oxazoline (2.28) was characterised in the
1H NMR spectrum by a doublet of doublets of doublets at 4.86 and a doublet of

Chapter 3
62
triplets at 4.42, assigned to the CHO and CHN protons. An M+H peak was
apparent in an electrospray high resolution mass spectrum, while in the 13C NMR
spectrum there appeared seven alkyl signals, one at 83.21 and another at 69.83
due to the CHO and CHN carbons respectively.
The bromine substituent of 2-(phenylselanyl)cyclohexyl p-bromobenzamide (2.45)
might be expected to facilitate aziridine-formation by rendering the amide proton
more acidic and thus more easy to deprotonate. However, cyclisation of (2.45) with
4.1 equivalents of peracid and 8.5 equivalents of hydroxide resulted in a mixture of
the aziridine (3.3) and oxazoline (3.4) in a ratio of 60:40 (Scheme 3.1). The NMR
spectra of the bromine-substituted aziridine (3.3) and oxazoline (3.4) differed from the
spectra of the bromine-free analogues (2.17) and (2.7) only in displaying pairs of
aromatic proton signals integrating to two hydrogens and in the aromatic 13C peaks in
which deshielding of the two substituted carbons was evident. In the mass spectrum
of the aziridine (3.3) the bromine substituent was indicated by a molecular ion at m/z
281 and a peak of almost equal intensity at m/z 279. Loss of bromine gave a small
peak at m/z 200. Peaks due to the p-bromobenzoyl cation appeared at m/z 185 and
183 with loss of C=O from this cation giving peaks at m/z 157 and 155. The IR
spectrum showed a C-Br stretch at 1304 cm-1 and an absorption of medium intensity
at 849 cm-1 attributable to the C-H bend of a disubstituted benzene ring.

Chapter 3
63
In previous work of our research group[148] the oxidation of 2-
(phenylselanyl)cyclohexyl acetamide (2.31) under acidic conditions, in which the
number of equivalents of peracid exceeded that of hydroxide, had given the ring-
contracted amide (3.6) and the lactone (3.5). The current study also obtained a
mixture of (3.5), (3.6) and starting material from the reaction of the acetamide (2.31)
with 3.2 equivalents of m-CPBA and 2 equivalents of hydroxide (Scheme 3.2).
1,2-alkyl shifts similar to the contraction to give (3.6) have been reported[176] following
oxidation of cyclic methoxyselenides (3.8) and (3.10) to give the ring-contracted
acetals (3.9) and (3.11) (Scheme 3.3).
Cyclisation of the acyclic amido selenide (2.32) produced no aziridine; from the
reactions both with zero and with 7.8 equivalents of potassium hydroxide, the
oxazoline (3.12) was the sole product (Scheme 3.4). The mass spectrum of (3.12)
showed an M+H peak at m/z 232, fragmentation of the alkyl chain and a peak due to
the benzoyl cation at m/z 105. Six alkyl signals appeared in the 13C NMR spectrum,
with two signals at 72.52 and 66.80 due to the carbons under oxygen and
nitrogen respectively and a signal at 163.29 of the O-C=N carbon. In the 1H NMR

Chapter 3
64
spectrum two distinct signals due to the diastereotopic methylene protons appeared
at 4.48 and 4.03 and flanked a multiplet centred at 4.27 due to the proton under
nitrogen.
Results of these reactions using potassium hydroxide as base could not be replicated
consistently, possibly due to the difficulty in obtaining dry carbonate-free powdered
hydroxide. Precedents for the N-alkylation of amides indicate that N-alkylation will
only occur reliably in preference to O-alkylation if the amide is first deprotonated
using a strong base in an inert solvent.[177] Sodium hydride was therefore
substituted for potassium hydroxide; addition of sodium hydride to isopropanol would
generate the stronger base, isopropoxide ion.
Use of sodium hydride as base gave more consistent results, however, the conditions
which would generate aziridines as the sole products remained elusive except for the
cyclohexene derivatives, 2-(phenylselanyl)cyclohexyl benzamide (2.5) and 2-
(phenylselanyl)cyclohexyl p-bromobenzamide (2.45) from which the aziridines (2.17)
and (3.3) were cleanly produced in the reactions using 3-4.5 equivalents of peracid
and ten and eight equivalents of sodium hydride respectively.

Chapter 3
65
FIGURE 3.1
OXIDATION OF -AMIDO SELENIDES (2.24), (2.5) AND (2.27):
PROPORTION OF AZIRIDINE IN PRODUCT VERSUS EXCESS OF BASEa (KOH OR NaH) USED
a equivalents base minus equivalents m-CPBA
Binary mixtures of the aziridine (3.1) and oxazoline (2.25) were obtained in the
oxidation of the 2-(phenylselanyl)cyclopentyl benzamide (2.24) using sodium hydride
as base. A reaction profile using 3.9 equivalents of m-CPBA and incrementally
increasing the quantity of sodium hydride from two to ten equivalents, confirmed the
dependence of aziridine-formation on the basicity of the reaction medium (Figure
3.1). An excess of about 2.5 equivalents of NaH was optimal, giving the aziridine
(3.1) as just over 50% of the product; with a greater excess of base, the yield of
aziridine (3.1) declined. For the same reaction but with KOH as the base the NaH
data are translated to the right along the x-axis, the weaker hydroxide base being
required in greater excess for an equivalent result. Figure 3.1 also illustrates the
significant difference in response to the same reaction conditions of three
cycloalkylamidoselenides which differ only in the number of ring carbons.

Chapter 3
66
Using 6 to 10 equivalents of sodium hydride as base and about 4 equivalents of m-
CPBA, a predominance of the aziridine (3.2) was achieved consistently in the
oxidation of 2-(phenylselanyl)cycloheptyl benzamide (2.27). Ratios of aziridine (3.2)
to oxazoline (2.28) in the crude product were of the order of 3-4 to 1. However, loss
of product occurred upon purification by chromatography with concomitant generation
of N-(cyclohept-2-en-1-yl)benzamide (2.30), through -elimination of the aziridine[178]
(3.2) (Scheme 3.5).
The acyclic amido selenide, 1-(phenylselanyl)-2-octyl benzamide (2.32), was reacted
with 8.6 equivalents of sodium hydride and 3.9 equivalents of m-CPBA. Though
creating a more strongly basic medium than previous reactions using potassium
hydroxide or no base at all, these conditions again produced no aziridine, only the
oxazoline (3.12), in 87% isolated yield.
The hindered base potassium tert-butoxide proved to be similar in effect to sodium
hydride in the reaction of 2-(phenylselanyl)cyclohexyl benzamide (2.5) giving the
aziridine (2.17) as the sole product and in the reaction of 2-(phenylselanyl)cyclopentyl
benzamide (2.24), which using this base gave a 51:49 mixture of the aziridine (3.1)
and oxazoline (2.25), essentially identical to the result using sodium hydride.

Chapter 3
67
3.2 CYCLISATION OF -AMIDO SELENIDES AT LOW TEMPERATURE
With the resurgence of interest in organoselenium chemistry in the 1970s came new
approaches to overcoming the well-known[179] difficulties in generating selenones.
Shimizu, Ando and Kuwajima[174] reported that m-CPBA was an effective oxidant for
the conversion of vinyl selenides to selenones and that methanol or t-butanol as
solvent were preferable to dichloromethane in facilitating the oxidation of the
selenoxide. The work of Krief et al.[175] showed that use of a peracid in
dichloromethane at 20°C was effective in the generation of a range of dialkyl and
alkyl phenyl selenones. Krief et al.[175] also noted that potassium permanganate as
the oxidant provided the advantage that the by-products of oxidation were inorganic
and could be removed in the aqueous layer. Uemura et al.[176] found methanol to be
preferable to dichloromethane and other alcohols as with this solvent the rate of
oxidation of the selenoxide to the selenone increased. Toshimitsu et al.[153]
recommended ethanol or 2-propanol rather than methanol to avoid formation of
methyl meta-chlorobenzoate by esterification of m-CBA. Other oxidants such as
peroxides and periodate have been found to be ineffective in the oxidation of
selenium beyond the +IV oxidation state in organic compounds.[175-176]
The conditions developed by Toshimitsu et al.[153] were those followed up to this point
of the present investigation. Variation of these conditions above room temperature
offered no improvement in the yield of aziridine. There appears to be no reference in
the literature to attempts to produce a selenone below 0°C except by Paetzold and
Bochman[180] who reported the preparation of dialkyl selenones by ozonisation of the
corresponding selenoxides at -10°C. However, the indication[180] that generation of a
selenone at temperatures below 0°C may be possible was encouraging, and

Chapter 3
68
therefore an investigation into the possibility of the preparation of aziridines at low
temperature was undertaken.
Thus, 2-(phenylselanyl)cyclopentyl benzamide (2.24) was treated with 3.2
equivalents of m-CPBA and 6 equivalents of potassium tert-butoxide in
tetrahydrofuran at –6°C, giving the surprising result of a 73% yield of the aziridine
(3.1) with minor amounts of the oxazoline (2.25) and syn-elimination product, N-
(cyclopent-2-ene-1-yl)benzamide[154] (3.13) (Scheme 3.6). When the reaction was
carried out at –60°C, cyclisation proceeded cleanly to the aziridine (3.1) which was
isolated in a yield of 75%.
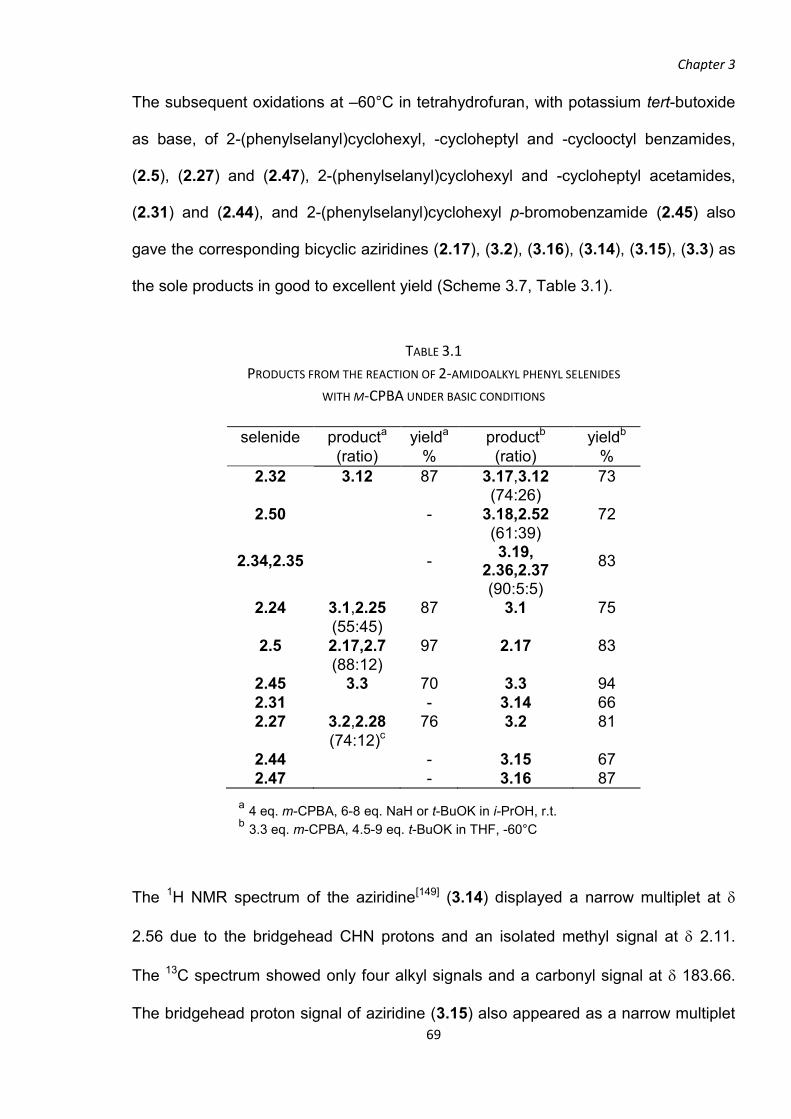
Chapter 3
69
The subsequent oxidations at –60°C in tetrahydrofuran, with potassium tert-butoxide
as base, of 2-(phenylselanyl)cyclohexyl, -cycloheptyl and -cyclooctyl benzamides,
(2.5), (2.27) and (2.47), 2-(phenylselanyl)cyclohexyl and -cycloheptyl acetamides,
(2.31) and (2.44), and 2-(phenylselanyl)cyclohexyl p-bromobenzamide (2.45) also
gave the corresponding bicyclic aziridines (2.17), (3.2), (3.16), (3.14), (3.15), (3.3) as
the sole products in good to excellent yield (Scheme 3.7, Table 3.1).
TABLE 3.1
PRODUCTS FROM THE REACTION OF 2-AMIDOALKYL PHENYL SELENIDES
WITH M-CPBA UNDER BASIC CONDITIONS
selenide producta yielda productb yieldb
(ratio) % (ratio) % 2.32 3.12 87 3.17,3.12 73
(74:26) 2.50 - 3.18,2.52 72
(61:39)
2.34,2.35 - 3.19, 2.36,2.37 83
(90:5:5) 2.24 3.1,2.25 87 3.1 75
(55:45) 2.5 2.17,2.7 97 2.17 83
(88:12) 2.45 3.3 70 3.3 94 2.31 - 3.14 66 2.27 3.2,2.28 76 3.2 81
(74:12)c 2.44 - 3.15 67 2.47 - 3.16 87
a 4 eq. m-CPBA, 6-8 eq. NaH or t-BuOK in i-PrOH, r.t. b 3.3 eq. m-CPBA, 4.5-9 eq. t-BuOK in THF, -60°C
The 1H NMR spectrum of the aziridine[149] (3.14) displayed a narrow multiplet at
2.56 due to the bridgehead CHN protons and an isolated methyl signal at 2.11.
The 13C spectrum showed only four alkyl signals and a carbonyl signal at 183.66.
The bridgehead proton signal of aziridine (3.15) also appeared as a narrow multiplet

Chapter 3
70
centred at 2.61 in the 1H NMR spectrum. The mass spectrum showing a very
weak molecular ion at m/z 153 and the base peak being at m/z 110, due to loss of
the acetyl group.
In the 1H NMR spectrum of the aziridine[154] (3.16), the bridgehead proton signals
occurred as a narrow multiplet at 2.52. This compound (3.16) exhibited a simple
13C spectrum showing four aryl and four alkyl signals and the carbonyl carbon signal
at 179.71. The mass spectrum showed a weak molecular ion at m/z 229, a peak
at m/z 201 due to loss of ethylene with a base peak at m/z 124 corresponding to loss
of the benzoyl group.
In contrast to the results from reactions conducted at room temperature on 1-
(phenylselanyl)-2-octyl benzamide (2.32), in which only the oxazoline (3.12) was
produced, the low temperature cyclisation of this amidoselenide gave a 3:1 mixture of
the aziridine (3.17) and oxazoline (3.12). The reaction of 1-(phenylselanyl)-2-octyl p-
bromobenzamide (2.50) under the low temperature conditions also gave a 3:1
mixture of the aziridine (3.18) to oxazoline (2.52) (Scheme 3.8). The bromine-
substituted aziridine (3.18) and oxazoline (2.52) were again only distinguishable from
the unsubstituted aziridine (3.17) and oxazoline (3.12) in their NMR spectra by the

Chapter 3
71
pattern of aromatic proton and carbon signals.
The 1H NMR spectra of (3.18) and (2.52) showed
two sets of aromatic protons, each integrating to
two hydrogens, while in the 13C spectrum the
substituted aromatic carbon signals were shifted
upfield as expected for a bromine-substituted
benzene ring. The diastereotopic ring protons of
aziridines (3.17) and (3.18), with signals at 2.49 and 2.19 for (3.17) and 2.50
and 2.19 for (3.18), are coupled to the vicinal proton, Hc, with coupling constants of
3.6 Hz and 6 Hz respectively. Given an HaCCHc, dihedral angle approaching 120°
and a small HbCCHc dihedral angle, the 3Jac value would be expected to be smaller
than the 3Jbc value, in accord with the Karplus correlation.[181-182] Therefore, a trans-
and cis-relationship to Hc were assigned to the proton (Ha) with signal at 2.49 (
2.50) and the proton (Hb) with signal at 2.19 respectively.

Chapter 3
72
Cyclisation of an approximately 50:50 mixture of 2-(phenylselanyl)-3-hexyl
benzamide (2.34) and 3-(phenylselanyl)-2-hexyl benzamide (2.35) under the low
temperature oxidation conditions gave a mixture that was 90% aziridine (3.19) and
5% of each of the two oxazolines, (2.36) and (2.37), and an isolated yield of aziridine
(3.19) of 69% (Scheme 3.9).
As a result of the stereoselectivity of the amidoselenation of trans-2-hexene and the
cyclisation reaction, aziridine (3.19) was formed as a pair of enantiomers with
configuration (R,S) and (S,R). This aziridine was isolated as a colourless oil. The
13C NMR spectrum showed six clean alkyl and four aryl signals and the carbonyl
signal at 177.92. In the 1H NMR spectrum the C2 CHN proton appeared as a
doublet of quartets at 2.59 coupled to the methyl group with a coupling constant of
5.7 Hz, the methyl group signal appearing as a doublet at 1.19. The C3 CHN
proton appeared as a doublet of doublets of doublets at 2.44 with a coupling
constant of 3.3 Hz to the C2 CHN proton. The mass spectrum showed a moderately
strong molecular ion at m/z 203 with fragmentation of the alkyl chain giving peaks at
m/z 188, 174 and 160. The familiar peak for the benzoyl cation appeared at m/z 105
and a peak at m/z 98 was attributed to loss of the benzoyl group from the molecular
ion.
While there is evidence that the N-acylaziridine nitrogen in crystalline samples has
considerable pyramidal character,[183] there is nevertheless sufficient sp2 character in
such compounds in solution to render the barrier to inversion of nitrogen very low.[184]
Early NMR studies[185-186] of the barrier to inversion of N-acylaziridines were unable to
observe any decoalescence at low temperature. No decoalescence of the aziridine

Chapter 3
73
ring proton signal of N-acetylaziridine was observed at –160°C on a 60MHz
spectrometer.[186] Boggs and Gerig[185] observed no decoalescence in N-
benzoylaziridine at –155°C and attributed this to the significant contribution of the
structure in which the lone-pair electrons are delocalised, so that the NCO system is
in the same plane as the three-membered ring, thereby lowering the barrier to
inversion.
Of the N-acylaziridines prepared in the present study, only the cyclopentene
derivative (3.1) displayed a singlet for the bridgehead protons. The analogous
protons of the other fused aziridines appeared as narrow multiplets in 1H spectra
recorded at room temperature on a 300MHz spectrometer, suggesting that
thermodynamic data of such compounds may now be obtainable.
In order to determine that the intermediate was the selenone and not the selenoxide,
the low-temperature reaction was carried out with only sufficient m-CPBA to oxidise
the selenide to the selenoxide. Under these conditions, 2-(phenylselanyl)cyclopentyl

Chapter 3
74
benzamide (2.24) and 2-(phenylselanyl)cycloheptyl benzamide (2.27) were expected
to be transformed into N-(cyclopent-2-ene-1-yl)benzamide[154] (3.13) and N-
(cyclohept-2-ene-1-yl)benzamide (2.30), via the syn-elimination of the respective
selenoxides. These starting materials, (2.24) and (2.27), were chosen because the
selenoxide syn-elimination of five- and seven-membered ring selenoxides is known to
be facile;[121, 145] six-membered ring selenoxides are known to be very slow to
undergo the syn-elimination.[121, 145] The products from this treatment of the five-
membered ring amido selenide (2.24) were the expected N-(cyclopent-2-ene-1-
yl)benzamide (3.13) along with the starting amido selenide (2.24) and the aziridine
(3.1) in a ratio of 55:30:15. The slight excess of peracid would account for the
aziridine (3.1) produced, via the selenone. Similar treatment of the seven-
membered ring amido selenide (2.27) with 1.05 equivalents of m-CPBA gave N-
(cyclohept-2-ene-1-yl)benzamide (2.30 ,58%) along with some starting amido
selenide (2.27, 13%) (Scheme 3.10). The starting material in these reactions was
presumably regenerated from the selenoxide under the reductive workup conditions.
To determine that the low temperature, and not the solvent or base, was the main
factor influencing the direction of the reaction, 2-(phenylselanyl)cycloheptyl
benzamide (2.27) was oxidised in tetrahydrofuran with 3.2-3.3 equivalents of m-
CPBA and 6 equivalents of potassium tert-butoxide as base at –15°C and at 0°C.

Chapter 3
75
Mixtures of the aziridine (3.2) and N-(cyclohept-2-ene-1-yl)benzamide (2.30) were
produced, the latter more favoured with the higher temperature (Scheme 3.11). The
syn-elimination product (2.30) was not observed in reactions carried out at –60°C,
suggesting that at higher temperatures the syn-elimination pathway is competitive
with oxidation of the selenoxide. At room temperature and at 0°C in isopropanol, the
syn-elimination product (2.30) only occurred in trace quantities, probably as a result
of the inhibitory effects of (i) strong hydrogen bonds between the solvent and the
selenoxide[121] and (ii) formation of the selenoxide hydrate as a result of water
present in the reaction mixture.[121]
Aziridines are known to undergo isomerization to oxazolines with acid-catalysis[187] or
in the presence of a nucleophile.[178] To determine if the reaction conditions were
affecting the product ratios, the aziridine (2.17) was (i) refluxed with silica in
dichloromethane and (ii) stirred with m-CPBA in ethanol. No change occurred at all
when the aziridine (2.17) was refluxed with silica in dichloromethane. A minor
amount of isomerization to the cis-oxazoline (2.7) was observed after treatment of the
aziridine (2.17) with m-CPBA. However, the amount was not sufficient to account for
the oxazoline formed from oxidation of the amido selenide (2.5).
2-(Phenylselanyl)cyclohexyl benzamide (2.5) was treated with 10 equivalents of
potassium hydroxide in 2-propanol to confirm that base alone could not induce
cyclisation. After stirring for 5 hours, a 1H NMR spectrum showed no new product
and after work up, 80% of the starting material was recovered.

Chapter 3
76
3.3 SUMMARY OF RESULTS FROM THE CYCLISATION OF -AMIDO SELENIDES
The results of this study of the reaction of a range of -amido selenides with an
oxidising agent under basic conditions, showed that three products could arise – the
aziridine, oxazoline and the selenoxide syn-elimination product – in combinations
depending on the reaction conditions.
Conditions favouring the aziridine were a strongly basic medium, a non-polar, aprotic
solvent and low temperature. The requirement for strongly basic conditions is
consistent with the general principle that N-alkylation of amides requires prior
deprotonation of the amide with a strong base[177] while a non-polar, aprotic solvent
would be expected to enhance the nucleophilicity of the amide anion. With
cyclisation to the aziridine occurring at low temperature, the aziridine would appear to
be the kinetic product under strongly basic conditions, deprotonation of the amide
lowering the activation energy of the transition state.
The syn-elimination reaction of susceptible substrates, e.g. 2-benzamidocycloheptyl
and 2-benzamidocyclopentyl phenyl selenides, was inhibited under the conditions
which favoured the aziridine. Under non-polar, aprotic conditions the selenoxide
oxygen would not be solvated, which would facilitate the elimination reaction.
However, the low temperature may not provide sufficient energy to overcome the
syn-elimination activation energy.
Cyclisation to the oxazoline was favoured at a higher temperature, in a protic solvent
and a weakly basic reaction medium. A higher temperature appears to be
necessary to overcome the activation energy of the transition state to the oxazoline.

Chapter 3
77
Although the higher temperature would allow the selenoxide syn-elimination to
compete with cyclisation to the oxazoline, this effect would be countered by a protic
solvent which would inhibit the elimination reaction. In the formation of oxazolines
from N-2-bromoethylbenzamides, Heine[156] found a 2- to 4-fold rate acceleration
where the aromatic ring was para-substituted with electron-withdrawing groups and
concluded that deprotonation occurred either prior to, or concomitantly with,
cyclisation to the oxazoline. Cyclisation of the acyclic amido selenides (2.32) and
(2.50) to the oxazolines with no base and with an excess of base showed that the
oxazolines could form either from the neutral amide or the amide anion, which is
consistent with Heine’s[156] conclusion.
While the cyclic amido selenides cyclised exclusively to aziridines under the low-
temperature conditions, mixtures of aziridine and oxazoline resulted from similar
treatment of the acyclic amido selenides (2.32), (2.50) and (2.34) and (2.35). This
suggests that deprotonation of the amide may be necessary for the internal N-
alkylation of these acyclic amidoselenides, but does not determine the course of the
cyclisation. 1-(Phenylselanyl)-2-octyl benzamide (2.32) and 1-(phenylselanyl)-2-
octyl p-bromobenzamide (2.50) gave the same product ratio of aziridine to oxazoline,
indicating that any stabilisation of the amide anion due to the p-bromo group did not
affect the direction of cyclisation.
3.4 FACTORS DETERMINING THE FORMATION OF 3- VERSUS 5-MEMBERED RINGS
Although there may be no specific data for the ring strain of the aziridines and
oxazolines of the present study, nevertheless it is apparent by comparison of the ring
strain of cyclopropane (27.5 kcal/mol[188]), aziridine (26.7 kcal/mol[189], 27.7
kcal/mol[189]), cyclopentane (6.2 kcal/mol[188]) and cyclopentene (4.1 kcal/mol[188]) that

Chapter 3
78
the aziridines would be considerably more strained than the corresponding
oxazolines. As well as having less bond angle strain than a three-membered
ring,[188] and a double bond, which would reduce torsional strain,[188] the oxazoline
ring has two heteroatoms which would also be expected to reduce the non-bonded
interactions.[190] An acyl substituent on nitrogen would be expected to increase the
angle strain through greater sp2 character of the aziridine nitrogen.
Despite their high ring strain, three-membered rings often display surprisingly high
rates of formation in comparison to larger, less strained rings.[191] One explanation
for this phenomenon is that there is less loss of entropy upon formation of a three-
membered ring compared with that of a larger ring. Illuminati and Mandolini[192]
demonstrated that the loss of entropy upon formation of a three-membered lactone
was low compared with less strained five- or six-membered lactones (although this
interpretation has been disputed[193]). The fast rate of formation of the three-
membered, compared with the five-membered, lactones was therefore attributed to
compensation of the ring strain through reduced loss of entropy upon formation of the
transition state.[192]
Differences in loss of entropy upon cyclisation could explain the differences in the
results for the acyclic amido selenides compared with the relatively rigid
cycloalkylamido selenides of the present study. The acyclic amido selenides
possess a greater number of degrees of freedom than the cycloalkyl substrates.
The difference in the loss of entropy upon cyclisation of the acyclic amido selenides
to the three- versus the five-membered ring would therefore be relatively less than
the same comparison for the cyclic amido selenides.

Chapter 3
79
3.5 OCCURRANCE AND UTILITY OF N-ACYLAZIRIDINES
Aziridines are useful intermediates in organic synthesis and can be regarded as
‘spring-loaded rings’[194] which readily undergo ring-opening reactions with C, O, N, S,
Se and halogen nucleophiles.[195-197] The ring-opening of aziridines generally
proceeds more readily if the ring is activated by incorporation of an electron-
withdrawing group on the ring nitrogen.[195] Activation of an aziridine ring with an N-
acyl group can accelerate the rate of ring-opening by up to 1018, through stabilisation
of the amide anion and increased ring strain due to the sp2 character of the
nitrogen.[198]
Unlike the epoxide ring, the aziridine ring is not particularly common in natural
products. Some notable aziridine-containing natural compounds are the mitomycins
and azinomycins, some of which were discovered and characterised in the late
1950s.[199] These compounds possess antitumor and antibiotic properties, with the
aziridine ring playing a key role in their mode of action through DNA alkylation.[199]
Desymmetrization of meso-aziridines is an effective and reliable way to obtain
enantiomerically pure -substituted amines. In recent years a number of catalysts
have been developed which enable the catalytic desymmetrization of meso-aziridines
in high yield and ee.[200] Some examples of the catalysts which can be used in the
ring opening of N-acylaziridines are the chiral guanidine (3.20), for ring-opening with
thiols,[201] VAPOL-hydrogen phosphate (3.21), for azide[202] and selenium[203]
nucleophiles and the chiral dimeric yttrium complex (3.22), which catalyses ring
opening with azide and cyanide nucleophiles.[204] Ring-opening of meso-N-
acylaziridines with malonates under the catalysis of the heterobimetallic La(O-

Chapter 3
80
iPr)3/Yb(Otf)3/Schiff base (3.23) produces -amino acids in up to 99% yield and
>99.5% ee.[205]
Lewis acid catalysed ring-opening of an N-acylaziridine is the penultimate step in a
number of syntheses[206-209] of the anti-influenza drug Tamiflu (3.24), while catalytic
desymmetrization of a meso-N-acylaziridine under the catalysis of the yttrium
complex with the chiral ligand (3.25) is a key step in the Tamiflu synthesis developed
by Shibasaki and co-workers (Scheme 3.12).[206-207]

Chapter 3
81
N-acylaziridines are usually prepared by acylation of the corresponding N-H
aziridine.[178, 183, 185, 210-211] Methods of preparing N-H aziridines encompass addition
of a nitrene or equivalent to a double bond, addition of a carbene or equivalent to an
imine or cyclisation of an aminoalcohol or equivalent.[212] Methods involving addition
reactions are often low in stereocontrol and require harsh conditions.[212] In contrast,
the formation of -amidoselenides has been shown to be trans-stereospecific,[145]
while cyclisation of tosylamino phenyl selenides to N-tosyl-pyrrolidines[213] and -
alkylamino phenyl selenides to aziridines[214] has been shown to proceed with
inversion. The generation of N-acylaziridines via cyclisation of -amido selenides
therefore provides an alternative approach to the preparation of these useful
synthons.

Chapter 3
82
Recent advances in the development of chiral electrophilic selenium reagents have
led to the preparation of heterocycles in high optical purity.[215] Tiecco et al.[216] have
prepared diastereomeric 2-amidoalkyl camphorselenides which were separated and
cyclised to the enantiopure oxazolines via activation of the seleno-moiety with
phenylselenenyl triflate. These developments suggest that there is potential for the
methodology developed in the present study to be extended to the preparation of
chiral N-acylaziridines.

Chapter 4
83
4 AMIDOSELENATION VIA ADDITION OF ‘PHENYLSELENENYL
PERCHLORATE’
4.1 INTRODUCTION
In the one-pot amidoselenation reaction of alkenes with phenylselenenyl halide and
nitrile,[145] the halide ion remaining after the formation of the seleniranium ion can
lead to undesired side-reactions. For example, Toshimitsu et al.[165] found that the
one-pot preparation of 1-(acrylamido)-2-(phenylseleno)cyclohexane (4.1) from
cyclohexene and phenylselenenyl chloride in acrylonitrile was complicated by the
further reaction of the desired product (4.1) with hydrogen chloride which added to
the carbon-carbon double bond, giving the chloroamide (4.2) (Scheme 4.1).
Conducting the amidoselenation reaction in two steps with isolation of the
hydroxyselenide intermediate avoided this side-reaction and gave the acrylamido
selenide (4.1) in high yield.[165]
In the addition of a selenenyl halide to a double bond, the presence of the halide ion
can drive the equilibrium between the alkene and the seleniranium ion to the left,
resulting in lower yields.[217] The formation of N-cycloalkyl amides in the
amidoselenation reaction (Chapter 2) was attributed to the reversal of the formation
of the seleniranium ion, facilitated by the presence of the chloride ion (Scheme 4.2).

Chapter 4
84
Conducting the reaction in two steps with isolation of the hydroxy selenide avoided
this side-reaction also.
It was of interest to prepare -amido selenides from their essential components -
alkene, nitrile, selenenyl halide and hydroxide ion - without the use of strong acid, via
a path which does not involve the hydroxy selenide intermediate. Sequestration of
the halide ion after addition of a selenenyl halide to a double bond has led to the
formation of stable seleniranium salts, such as the seleniranium
hexafluorophosphates and hexafluoroantimonates (4.3) prepared by Schmid and
Garratt[56] (Scheme 4.3), and can increase the reactivity of the seleniranium ion as
well as increase product yield.[66, 218]

Chapter 4
85
Through addition of silver tetrafluoroborate to a mixture of alkene, phenylselenenyl
chloride and ethyl carbamate, sequestration of the chloride nucleophile allowed the
reaction of the seleniranium ion (4.4) with the weakly nucleophilic carbamate, leading
to the formation of 2-(phenylseleno)alkylcarbamates (4.5, Scheme 4.4).[57]
In a reaction analogous to the amidoselenation of alkenes, Hassner et al.[219]
prepared -bromoalkyl amides (4.6) via attack on a bromonium ion (4.7) by nitrile
followed by hydrolysis (Scheme 4.5). To avoid competition between the bromide ion
(produced in the formation of the bromonium ion (4.7)) and the nitrile in nucleophilic
attack upon (4.7), silver perchlorate was added to sequester the bromide ion.
Introduction of hydroxide to the mixture containing the nitrilium ion (4.8) led to the
successful bromoamidation of a range of alkenes.[219] Addition of azide instead of
hydroxide gave the -bromotetrazoles (4.9) (Scheme 4.5).

Chapter 4
86
4.2 PREPARATION OF -(PHENYLSELANYL)CYCLOHEXYL AMIDES
These precedents[56-57, 219] for the formation and reactions of seleniranium and
nitrilium ions suggested that conducting the amidoselenation reaction with
sequestration of the halide ion might be a way to prepare a -amido selenide and by-
pass the formation of the hydroxy selenide intermediate. Addition of silver
perchlorate to a solution of cyclohexene, phenylselenenyl bromide and nitrile would
remove the bromide ion from solution and expose the seleniranium ion (4.10) to
attack by nitrile (Scheme 4.6). An equilibrium mixture of the seleniranium and
nitrilium ions (4.10) and (4.11) would result; addition of hydroxide to this mixture
would lead to either or both the hydroxy selenide (2.41) by reaction with (4.10) or the
acetamide (2.31) by reaction with (4.11) (Scheme 4.6). The direction of this

Chapter 4
87
equilibrium would depend on the nucleophilicity of the nitrile and relative stabilities of
the nitrilium and seleniranium ions.
In the first attempt at this reaction, silver perchlorate was added to a dry
dichloromethane solution of cyclohexene, acetonitrile and phenylselenenyl chloride,
giving an immediate precipitate of silver chloride. Slow introduction of aqueous
sodium hydroxide to the stirred suspension gave only the hydroxy selenide (2.41), in
56% yield. In the second attempt, using phenylselenenyl bromide instead of the
chloride, 1.5 equivalents of water in acetonitrile was slowly added after precipitation
of the silver salt, affording 2-(phenylselanyl)cyclohexyl acetamide (2.31) in 57%
isolated yield, a result comparable with the 60% yield of -bromocyclohexyl
acetamide (4.9, R=Me) obtained by Hassner et al.[219] The hydroxy selenide (2.41)
was also isolated in 15% yield under the latter conditions (Scheme 4.6). The greater
success with addition of water rather than hydroxide to the mixture of the nitrilium and
seleniranium ions (4.10) and (4.11) could be due to an irreversible reaction of (4.10)

Chapter 4
88
with hydroxide to give the hydroxy selenide (2.41) unlike its
reaction with water which would be reversible.
Using benzonitrile in place of acetonitrile, introduction of a
mixture of water in nitrile to the reaction mixture was not
possible for the reason of immiscibility. Slow addition of
water to the seleniranium ion-nitrilium ion equilibrium generated from cyclohexene,
phenylselenenyl bromide and silver perchlorate gave only a 33% yield of 2-
(phenylselanyl)cyclohexyl benzamide (2.5). The lower yield may be a reflection of
the lower nucleophilicity of benzonitrile compared with acetonitrile.[220-221]
4.3 ALTERNATIVES TO THE -AMIDO SUBSTITUENT
Addition of azide anion to the mixture containing the nitrilium ion could potentially
give the phenylseleno- analogues of the -bromotetrazoles (4.9) prepared by
Hassner et al.[219] With the aim of preparing such a phenylselenotetrazole, sodium
azide was introduced to a mixture of cyclohexene, phenylselenenyl bromide,
acetonitrile and silver perchlorate in dichloromethane, giving a complex mixture from
which 2-(phenylselanyl)cyclohexyl acetamide (2.31) was isolated in 24% yield along
with a product whose spectral data were consistent with the tetrazole (4.12) (Scheme
4.7).

Chapter 4
89
A mass corresponding to the expected MH+ mass for the tetrazole (4.12) was
obtained in a high resolution mass spectrum. A peak at m/z 322, attributable to the
molecular ion of the tetrazole (4.12), was shown in a low resolution
spectrum along with a selenium-containing fragment at m/z 238
consistent with the loss of 1H-5-methyl-tetrazole (4.13) from the
molecular ion.
The cyclohexyl methine peaks in the 1H NMR spectrum of the tetrazole (4.12)
occurred as doublets of doublets of doublets at 4.16 and 3.67 with a trans-diaxial
coupling constant of 11.4 Hz. Compared with the analogous acetamide (2.31), these
peaks were downfield, due to the electron-withdrawing nature of the tetrazole ring.
The methyl singlet appeared at 2.57, deshielded by the tetrazole ring current, while
in the 13C NMR spectrum, a carbon signal corresponding to the amidine carbon, N-
C=N, occurred at 150.99. These 1H and 13C NMR signals were consistent with the
spectra of other 1,5-disubstituted tetrazoles.[222-223]
A second attempt at this reaction
gave a viscous brown oil from
which trituration and
recrystallisation led to the isolation
of a product with spectral
characteristics similar but not
identical to those of the tetrazole (4.12). In the 1H NMR spectrum, the cyclohexyl
methine protons resonated at 4.53 and 3.55 with a trans-diaxial coupling constant
of 11.7 Hz. These protons each integrated to 2 hydrogens compared with the 3

Chapter 4
90
hydrogens of the methyl singlet, suggesting a double addition. X-ray analysis[224] of
the crystals showed the product to be the ‘meso’ tetrazolium perchlorate (4.14). The
downfield shifts, compared with the tetrazole, of both the CHN proton signal and the
methyl singlet at 3.51 could therefore be attributed to further deshielding by the
positively charged tetrazolium ring.
Over weeks in solution, decomposition of an NMR sample of the tetrazolium salt
(4.14) occurred to give the tetrazole (4.12), as indicated by NMR analysis.
The tetrazolium salt (4.14) had resulted from a reaction mixture which was slightly
deficient in azide which suggests stoichiometry as the reason for the double addition.
Yields of both the tetrazole (4.12) and tetrazolium perchlorate (4.14) were poor (14%
and 9% respectively), probably at least partly due to the difficulty in their isolation.
Using a modification of the carbamatoselenation procedure of Francisco et al.,[57]
namely substituting silver perchlorate for the silver tetrafluoroborate specified in the
literature procedure, 2-(phenylselanyl)cyclohexyl carbamate (4.15) was obtained in
82% yield (Scheme 4.7). The ethoxy group was evident in the 1H NMR spectrum,
the quartet appearing at 4.20 and the triplet at 1.25. The peak due to the CHSe
proton occurred at 3.06 as a doublet of doublets of doublets with a trans-diaxial
coupling constant of 10.8 Hz. The signal due to the CHN proton was a less well-
defined multiplet at 3.50. The mass spectrum gave a molecular ion at m/z 327 and
further fragments at m/z 281, 238 and 170 corresponding to loss of CH3CH2O.,
NH2CO2CH2CH3 and C6H5Se. respectively from the molecular ion.

Chapter 4
91
This examination of the amidoselenation reaction of cyclohexene, with sequestration
of the halide ion, showed these conditions to be capable of producing reasonable
yields of -amido selenides. The cost and hygroscopicity of the silver reagent limit
the utility of the method, although it could be useful if the particular target were a -
phenylselenoalkyl tetrazole. Further work on this reaction could involve exploring
the effect of low temperature on the stability of, or equilibrium between, the nitrilium
and seleniranium ions.

Chapter 5
92
5 CLOSER EXAMINATION OF A SELENOXIDE AND A SELENONE
5.1 PREPARATION OF N-[2-(PHENYLSELENINYL)CYCLOHEXYL]BENZAMIDE
AND N-[2-(PHENYLSELENONYL)CYCLOHEXYL]BENZAMIDE
In the cyclisation of -amidoselenides, the intermediate selenoxides and selenones
were not observed among the reaction products. It was expected that isolation of
these oxidised intermediates would be difficult due to the thermal instability of
selenoxides[1] and the vulnerability of the selenonyl group to
nucleophiles.[175]
However, it was possible that the selenoxide (5.1) might be
relatively stable for two reasons. Firstly, cyclohexyl
selenoxides are more reluctant than other cycloalkyl
selenoxides to undergo the syn-elimination reaction. For example, the half-life of
phenylseleninyl cyclohexane (5.2) is 364 times that of phenylseleninyl cyclopentane
(5.3) (Scheme 5.1).[121] This has been attributed, at least partly, to unfavourable
dihedral angles in the transition state.[121]

Chapter 5
93
Secondly, the selenoxide (5.1) could be stabilised by an intramolecular hydrogen
bond between the NH hydrogen and the SeO oxygen. Spectroscopic evidence has
been cited[145] for the existence of such a bond in 2-acetylamido- and 2-(n-
propyl)amido- cyclohexyl phenyl selenoxides, (5.4) and (5.5), which do not readily
undergo the elimination reaction to yield allyl amides. These selenoxides exhibit
deshielding of the NH proton in the NMR spectrum, and in the IR spectrum, show an
increase in the carbonyl stretching frequency and a lowering of the NH stretching
frequency in comparison with the selenide.[145] An intramolecular hydrogen bond is
also believed to be the reason for the unusual stability of the 2-
(phenylseleninyl)cyclohexanols (5.6)[225] and (5.7).[226]
Indeed, in the oxidation of 2-(phenylselanyl)cyclohexyl benzamide (2.5) as described
in Chapter 3, the syn-elimination product was not observed, despite the occurrence
of the respective syn-elimination product from the oxidation of both 2-
(phenylselanyl)cyclopentyl benzamide (2.24) and 2-(phenylselanyl)cycloheptyl
benzamide (2.27). It was therefore of interest to prepare the selenoxide (5.1) to see
if it exhibited the same hydrogen-bonding properties as other cyclohexyl selenoxides
and whether an intramolecular hydrogen bond would be present in the crystalline
state.

Chapter 5
94
2-(Phenylseleninyl)cyclohexyl benzamide (5.1) was obtained in 96% yield by
oxidation of the amido selenide (2.5) with 1.1 equivalents of m-CPBA in
dichloromethane at room temperature. While the amido selenide (2.5) has two
asymmetric carbons, the trans-addition of the amidoselenation reaction constrains
the number of stereoisomers to one pair of enantiomers.[145] Three chirality centres
are present in the selenoxide - the two methine ring carbons and the selenium atom –
so that four stereoisomers of the selenoxide, R,R,SSe-(5.1), S,S,SSe-(5.1), R,R,RSe-
(5.1), S,S,RSe-(5.1) would be expected. However, inversion at selenium can
transform one stereoisomer into a diastereomer with the same configuration at the
ring carbons, but the opposite configuration at selenium. The 1H NMR spectrum of
2-(phenylseleninyl)cyclohexyl benzamide (5.1) showed a single, poorly-resolved peak
due to the methine protons, indicating rapid inversion at selenium between
diastereomeric isomers.[124] Recrystallisation of the selenoxide by slow infusion of
ethyl acetate into a methanol solution gave colourless needles which X-ray analysis
revealed to be crystals of R,R,SSe-(5.1) with the unit cell comprised of two
conformational isomers, both having configuration S at selenium and configuration R
at both the CHN and CHSe carbons.[227] The enantiomer, S,S,RSe-(5.1), with
configuration R at selenium and configuration S at both chiral carbons was
presumably also present in the crystal. SeO-HN intermolecular hydrogen bonds
were exhibited in the crystal but no intramolecular hydrogen bonds were evident.[227]
The crystalline selenoxide enantiomers R,R,SSe-(5.1) and S,S,RSe-(5.1), are
assumed to be the thermodynamically-favoured isomers, inversion at selenium via
the hydrate being facilitated by the methanol and/or water present in the
recrystallisation medium.[124, 228]

Chapter 5
95
In a 1H NMR spectrum of a solution of these crystals, the CHN peak appeared as a
doublet of doublets of doublets of doublets at 3.60 with coupling constant of 4.8 Hz
to the NH proton which resonated at 8.10 and with a trans-diaxial coupling of 11.1
Hz to the CHSe peak which appeared as a doublet of doublets of doublets at 3.39.
Over several hours, inversion at selenium occurred in the NMR sample of (5.1), to
give a mixture of all four stereoisomers, R,R,SSe-(5.1), S,S,SSe-(5.1), R,R,RSe-(5.1)
and S,S,RSe-(5.1). Well-defined peaks due to the CHN and CHSe methine protons
of the isomers R,R,RSe-(5.1) and S,S,RSe-(5.1) appeared at 4.04 and 3.14
respectively. Assuming an intramolecular hydrogen bond analogous with the
structures of (5.4) through (5.7),[145, 225-226] then the CHN proton at 3.60 of isomers
R,R,SSe-(5.1) and S,S,SSe-(5.1) is shielded relative to the corresponding proton in the
selenide (2.5) ( 3.96) due to its 1,3 relationship to the benzene ring of the selenium
moiety. The CHSe proton at 3.39 is deshielded compared with the analogous
proton in the selenide at 3.15 due to its trans-1,2 position with respect to this
benzene ring as well as the electron-withdrawing effect of the selenoxide group.
A model of the isomers R,R,RSe-(5.1) and S,S,RSe-(5.1) incorporating an
intramolecular hydrogen bond shows that the CHSe proton (at 3.14) is now cis
relative to the benzene ring and hence more shielded than in the selenide, while the

Chapter 5
96
CHN proton is unaffected and resonates at a similar frequency to the corresponding
proton in the selenide.
A 77Se NMR spectrum of a mixture of the four selenoxide stereoisomers (5.1)
exhibited resonances at 872.8 and 843.7, in the region expected for
selenoxides.[229] Duddeck et al.[230] reported two peaks in the 77Se NMR spectra of
monosubstituted cyclohexyl phenyl selenides and selenoxides and attributed these to
the equatorial and axial conformers. However, as compound (5.1) contained two
large vicinal groups, it can be assumed that the conformer with both substituents
equatorial would be of lower energy with a high barrier to inversion. It is therefore
proposed that the two sets of signals in the 77Se and 1H NMR spectra are due to the
two enantiomeric pairs of selenoxides R,R,SSe-(5.1), S,S,SSe-(5.1) and R,R,RSe-(5.1),
S,S,RSe-(5.1).
The crystalline selenoxide isomers (5.1) exhibited a strong absorption at 814 cm-1 in
the IR spectrum (KBr disc), characteristic of the selenoxide SeO stretch[231] and in the
mass spectrum, a molecular ion at m/z 375. The selenoxide crystals were stable
indefinitely at room temperature.
Oxidation of 2-(phenylselanyl)cyclohexyl benzamide (2.5) with 3 equivalents of m-
CPBA in tetrahydrofuran gave a white precipitate which was collected and washed
with cold tetrahydrofuran to remove m-CBA and excess m-CPBA. A 1H NMR
spectrum of this product showed two cyclohexyl methine signals at 4.16 and 3.97
coupled to each other with a trans-diaxial coupling constant of 11.4 Hz, the signal at
3.97 also being coupled (6.6 Hz) to the NH signal at 7.31. The substantial
electron-withdrawing effect of the selenonyl moiety resulted in a downfield shift of the

Chapter 5
97
CHSe signal of 0.77 and 1.02 ppm compared with the selenoxide isomers (5.1) and a
shift of 1.01 ppm compared with the selenide (2.5). Consequently, and in contrast
to the selenide and selenoxide, the CHSe 1H resonance of the selenone (5.8) was
further downfield than the CHN signal. A high resolution mass spectrum obtained
under electrospray conditions showed a satisfactory MH+ peak, while a low resolution
mass spectrum showed an MH+ peak at m/z 392 and a peak at m/z 376 due to loss
of oxygen from the MH+ ion. In the IR spectrum, the product (5.8) exhibited two
strong absorptions at 935 cm-1 and 879 cm-1, characteristic of the selenone
asymmetric and symmetric O=Se=O stretches respectively.[231]
A solution of the product (5.8) in tetrahydrofuran, with a few drops of dichloromethane
added to aid dissolution, was washed with 30% sodium hydroxide solution, resulting
in a mixture of the aziridine (2.17) and cis-oxazoline (2.7) in a ratio of 7:3 (Scheme
5.2). The high proportion of aziridine produced was further support that in the
isolated compound the selenium atom was oxidised to the +VI oxidation state since
treatment of the selenoxide (5.1) with base would be expected to give either no
reaction or the syn-elimination product. If the product (5.8) was further along the path
to an oxazoline, then treatment with base would be expected to give predominantly
the cis-oxazoline (2.7).

Chapter 5
98
Recrystallisation of the selenone (5.8) from tetrahydrofuran/hexane gave fine
colourless needles which were stable under nitrogen at –15°C, but decomposed
when stored at room temperature.
5.2 HYDROGEN-BONDING IN THE SELENIDE (2.5), SELENOXIDE (5.1) AND SELENONE (5.9)
Spectroscopic indicators of the extent of hydrogen bonding in the three compounds -
selenide (2.5), selenoxide (5.1) and selenone (5.8) - are shown in Table 5.1. 1H
NMR spectra were recorded as 0.005 M solutions in deuterochloroform; IR spectra
of KBr discs of the oxidised species and nujol mulls of the selenide and selenoxide
were compared with spectra of dilute solutions in chloroform and/or dichloromethane.
TABLE 5.1
HYDROGEN BOND INDICATORS IN THE SELENIDE (2.5), SELENOXIDE(5.1) AND SELENONE (5.8)
R
NMRA
(ppm) IR (solid)
(cm-1) IR (solution)B
(cm-1)
NH NH CO SeO SeO2 NH CO SeO SeO2
SePh 6.13 nujol 3319
s br 1631 CHCl3 3691 1655
Se(O)Ph 8.07
KBr 3230
s br 1655 814
CH2Cl2
3431
3257 w br
1661 826 809
nujol 3223 s br 1654 814
Se(O2)Ph 7.19 KBr 3309
s br 1657 935
879
CHCl3 3688 1664 933 881
CH2Cl2 3684 1666 935 880
A 1H NMR, 0.005M in CDCl3 B 0.001M in CHCl3, 0.002M in CH2Cl2

Chapter 5
99
Expected intermolecular hydrogen bonding in the solid selenide (2.5) is indicated by
a difference of 24 cm-1 between the frequency of the selenide carbonyl stretch in the
solid state (1631 cm-1) and in dilute solution (1655 cm-1) along with a strong
intermolecular NH stretch at 3319 cm-1 in the spectrum of the solid which becomes a
discrete free NH stretch at 3691cm-1 in the spectrum of the solution. The NH signal
at 6.13 in the 1H NMR spectrum of the selenide (2.5) is consistent with the absence
of intramolecular hydrogen bonding.
The X-ray structure of the selenoxide (5.1) shows an intermolecular hydrogen bond
between the Se=O oxygen and the amide hydrogen. If this is the case, then the
SeO stretch at 814 cm-1 in both the KBr disc and nujol mull infrared spectra of the
selenoxide could be interpreted as characteristic of the intermolecularly hydrogen
bonded SeO. The solution infrared spectrum of the selenoxide (5.1) appeared to
show two SeO stretches, at 826 and 809 cm-1. These two bands could represent the
free SeO and intramolecularly hydrogen bonded SeO stretches respectively. The
sharp (3431 cm-1) and broad (3257 cm-1) NH stretches in the dichloromethane
solution of the selenoxide are consistent with this interpretation. Intramolecular
hydrogen bonding in the solution of the selenoxide (5.1) is also supported by the
downfield resonance ( 8.07) of the NH proton in the 1H NMR spectrum.
Some intramolecular hydrogen bonding in the selenone (5.8) is suggested by the
somewhat downfield signal of the NH proton at 7.19. However, this is in contrast
with the sharp NH stretches and the absence of any broad NH stretch in the solution
infrared spectra, which suggest that no intramolecular hydrogen bonding of the NH
hydrogen is occurring in the selenone in dilute solution. The absence of any

Chapter 5
100
significant difference in the frequencies of the SeO symmetric and asymmetric
stretches in the spectrum of the solid compared with the analogous stretches in the
chloroform and dichloromethane solution spectra also do not support the involvement
of the SeO2 group in hydrogen bonding. The less polar selenone selenium-oxygen
bond would be expected to result in weaker hydrogen bonding to the group if it
occurred.[232-233] The effect this would have on the Se=O stretching frequencies
may therefore not be significant.
The slight increase in the carbonyl stretching frequencies of the selenoxide (5.1) and
selenone (5.8) on going from the solid state to the solution is perhaps indicative of
some bonding, other than hydrogen bonding, to the carbonyl group in the solid.
In the dilute chloroform solution infrared spectra of (5.8), evidence of intramolecular
hydrogen bonding between the NH proton and an oxygen on selenium may be
masked by hydrogen bonding between the Se-O oxygen and solvent,[233] and
therefore, this data provides no conclusive evidence regarding such intramolecular
hydrogen bonding. Unfortunately, the selenoxide was not soluble in the less polar,
non-hydrogen-bonding solvent carbon tetrachloride, which may have provided more
conclusive evidence of intramolecular hydrogen bonding.[233] Dichloromethane, less
polar and less hydrogen-bonding than chloroform, was chosen as a compromise.
The solid IR spectra of the KBr discs of the selenoxide (5.1) and selenone (5.8) may
also not be a valid reflection of the bonding in the crystal due to mechanochemical
changes which may occur in the preparation of a KBr disc.[234]

Chapter 5
101
5.3 NMR-SCALE OXIDATION OF N-[2-(PHENYLSELANYL)CYCLOHEXYL]BENZAMIDE (2.5)
It was of interest to follow the course of the oxidation of the selenide using NMR to
observe the transformation of the selenide to the selenoxide and then to the
selenone, and to observe the subsequent decomposition of the selenone. 2-
(Phenylselanyl)cyclohexyl benzamide (2.5) was chosen as the subject of NMR
studies of the oxidation reaction due to the stability of its selenoxide (5.1).
The oxidation reaction was observed by 1H NMR at room temperature in two
experiments: one in methylene chloride-d2 (CD2Cl2) and the other in tetrahydrofuran-
d8 (THF-d8) containing a small amount CD2Cl2 to aid dissolution. The experiments
were carried out at approximately the same concentration as the preparatory-scale
oxidation reactions.
FIGURE 5.1
OXIDATION OF 2-(PHENYLSELANYL)CYCLOHEXYL BENZAMIDE IN CD2CL2:
PROPORTION (%) OF COMPOUNDS (5.1), (5.8) AND (5.9) IN PRODUCT VERSUS REACTION TIME (t)

Chapter 5
102
In CD2Cl2, the oxidation reaction was followed by 1H NMR with six spectra recorded
at approximately six minute intervals and then one spectrum recorded at 90 minutes’
reaction time (Figure 5.1).
Spectrum 1, at approximately 6 minutes, showed that all of the starting material had
been consumed, most of the product being the selenoxide together with a small
amount of the selenone. After 12 minutes, there appeared two broad signals at
5.05 and 4.51, apparently due to a product of decomposition of the selenone (5.8).
The subsequent spectra showed a steady diminution of the selenoxide signals, an
increase in the concentration of the selenone which peaked at 24 minutes’ reaction
time before decreasing, and finally, predominance of the decomposition product.
The two diagnostic signals of the decomposition product were shifted further
downfield with each spectrum and finally appeared as a multiplet at 5.48 and a
doublet of doublets of doublets at 4.80. This product was assigned the structure of
the oxazolinium ion (5.9). As confirmation of this assignment, a 1H NMR sample of
the cis-oxazoline (2.7) in CDCl3 was shaken with two drops of concentrated HCl.
Protonation of the oxazoline resulted in a downfield shift of the methine CHN and
CHO protons to 5.46 and 4.79 respectively, almost identical with the signals of the
product obtained in the NMR-scale oxidation of the selenide (Scheme 5.3). Slight
differences in chemical shift could be attributed to a different solvent (CDCl3 versus
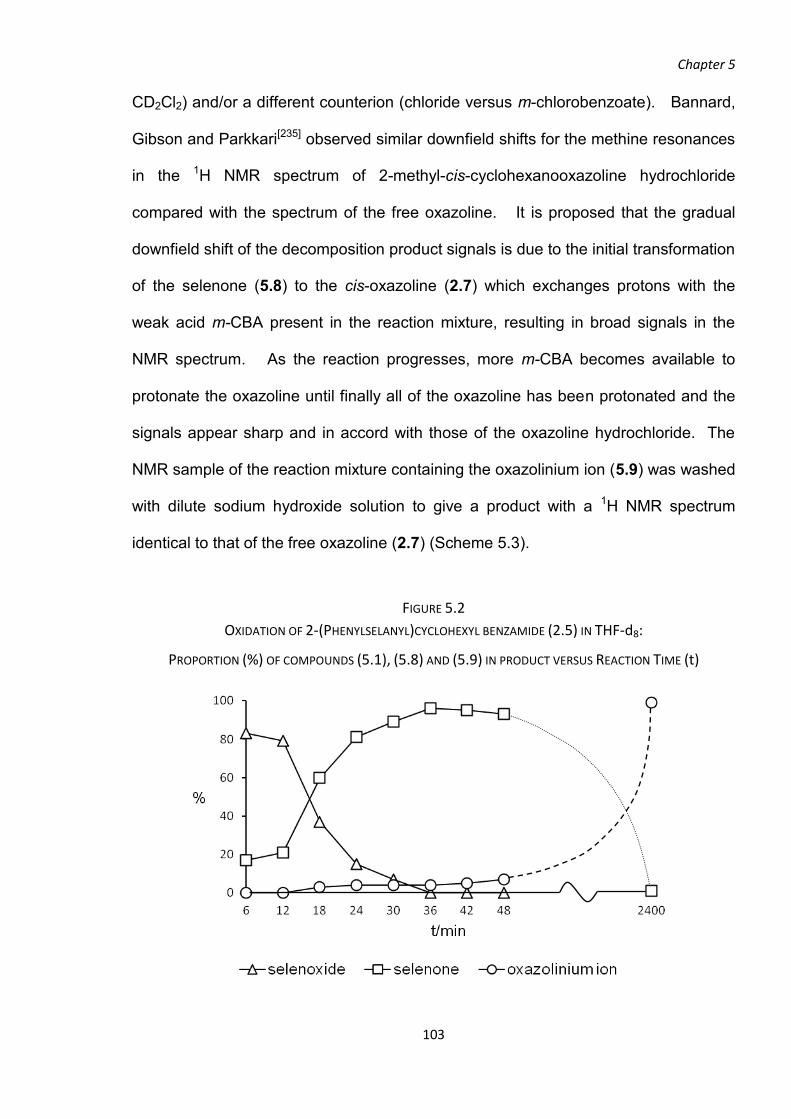
Chapter 5
103
CD2Cl2) and/or a different counterion (chloride versus m-chlorobenzoate). Bannard,
Gibson and Parkkari[235] observed similar downfield shifts for the methine resonances
in the 1H NMR spectrum of 2-methyl-cis-cyclohexanooxazoline hydrochloride
compared with the spectrum of the free oxazoline. It is proposed that the gradual
downfield shift of the decomposition product signals is due to the initial transformation
of the selenone (5.8) to the cis-oxazoline (2.7) which exchanges protons with the
weak acid m-CBA present in the reaction mixture, resulting in broad signals in the
NMR spectrum. As the reaction progresses, more m-CBA becomes available to
protonate the oxazoline until finally all of the oxazoline has been protonated and the
signals appear sharp and in accord with those of the oxazoline hydrochloride. The
NMR sample of the reaction mixture containing the oxazolinium ion (5.9) was washed
with dilute sodium hydroxide solution to give a product with a 1H NMR spectrum
identical to that of the free oxazoline (2.7) (Scheme 5.3).
FIGURE 5.2
OXIDATION OF 2-(PHENYLSELANYL)CYCLOHEXYL BENZAMIDE (2.5) IN THF-d8:
PROPORTION (%) OF COMPOUNDS (5.1), (5.8) AND (5.9) IN PRODUCT VERSUS REACTION TIME (t)

Chapter 5
104
The oxidation of the selenide was followed by 1H NMR in THF-d8, with a small
amount of CD2Cl2 added to aid dissolution. Nine spectra were recorded, eight at
intervals of approximately 6 minutes, then one after 40 hours’ reaction time (Figure
5.2).
After 6 minutes all of the selenide (2.5) had been consumed and the spectrum
showed a mixture of the selenoxide (5.1) and the selenone (5.8) in a ratio of
approximately 4 to 1. After 18 minutes, the selenone was the predominant product
and weak signals due to the oxazolinium ion were evident. After 36 minutes the
selenoxides (5.1) had been consumed and the selenone (5.8) and oxazolinium (5.9)
ion made up the product in a ratio of approximately 95 to 5. The transformation of
selenone (5.8) to oxazolinium ion (5.9) is slow in tetrahydrofuran and even after 48
minutes the selenone (5.8) still made up approximately 90% of the product.
Inversion at selenium is also inhibited in this solvent as predominantly one selenoxide
isomer was observed by NMR. A spectrum recorded after 40 hours’ reaction time
showed the oxazolinium ion (5.9) along with other unidentified minor products. Basic
workup of the sample gave the cis-oxazoline (2.7) along with small amounts of
unidentified products.
Due to the parameters involved with 77Se NMR, namely the high concentration and
lengthy time required to attain a spectrum, it was not possible to follow the oxidation
reaction using this technique. However, a reaction mixture of the selenide (2.5) and
4.8 equivalents of m-CPBA in THF at approximately sixteen times the preparatory
concentration was prepared in an NMR tube. A spectrum was recorded at –60°C;
after 570 transients a signal at 1010 attributed to the selenone[229] (5.8) and two

Chapter 5
105
selenoxide signals,[229] at 859.5 and 843.9, were observed. This spectrum was
an average of the reaction up to that point and therefore the relative concentration of
the products could not be determined. The difference in -value of one of the
selenoxide peaks compared with the selenoxide spectrum conducted in CDCl3 may
be due to solvent, temperature or concentration effects.
At the higher concentration required to run the 77Se NMR experiment, the selenone
(5.8) precipitated out of solution as a white solid. A 1H NMR spectrum of the
precipitate in deuterochloroform showed two well-defined peaks at 4.15 and 3.96,
identical with those of the isolated selenone (5.9), together with a minor amount of
the cis-oxazoline (2.7). The cis-oxazoline (2.7) may have formed in the original
reaction mixture or during collection and dissolution of the precipitate. A spectrum
recorded of this deuterochloroform sample after 18 hours showed that the selenone
(5.8) had been cleanly and completely transformed into the cis-oxazoline (2.7).
2-(Phenylseleninyl)cyclohexyl benzamide (5.1) was found to be as stable as
structurally-similar previously-reported cyclohexyl selenoxides.[145, 225-226] However,
contrary to proposals in the literature,[145, 226] X-ray analysis showed hydrogen
bonding in the crystalline selenoxide (5.1) to be intermolecular rather than
intramolecular. NMR and IR data suggest that an intramolecular hydrogen bond
may be important for stability of the selenoxide in solution.
2-(Phenylselenonyl)cyclohexyl benzamide (5.8) was found to be a surprisingly stable
compound both as a solid and in solution in tetrahydrofuran. Comparison of the

Chapter 5
106
course of the reaction in CD2Cl2 and THF-d8 showed that the selenone (5.8) forms
more rapidly in tetrahydrofuran and decomposes much more slowly in this solvent.
Monitoring of the oxidation reaction by 1H NMR in the absence of base suggested
that, in both polar and non-polar solvent, the selenone (5.8) decomposed to the cis-
oxazoline (2.7) which was then protonated by acid present in the reaction mixture to
give the oxazolinium ion (5.9). This is supported by the observation that isolation of
the selenone (5.8) from m-CBA and dissolution in deuterochloroform resulted in
cyclisation to the cis-oxazoline (2.7) with no intermediate or further transformation to
the oxazolinium ion (5.9).
Results from the current work have clearly shown that base is not necessary in the
cyclisation of -selenonyl amide to the oxazoline, although Heine[156] has reported
that base is involved in the rate-determining step in the cyclisation of -bromo amides
to oxazolines. Heine[156] suggested that deprotonation of a -bromo amide may
occur concomitantly with cyclisation to the oxazoline. If deprotonation occurs
concomitantly with cyclisation of the selenone (5.8) to the cis-oxazoline (2.7) then an
intramolecular hydrogen bond could facilitate the transformation (Scheme 5.4).

Chapter 6
107
6 PREPARATION AND CYCLISATION OF -HYDROXY SELENIDES
6.1 INTRODUCTION
Although the parent oxetane has been known since 1878,[236] there are relatively few
synthetic approaches to oxetanes. The Paterno-Büchi reaction[237-239] affords
oxetanes from the photocycloaddition of a carbonyl compound and an alkene
(equation 1). The reaction can proceed with regio- and facial selectivity depending
on the choice of the carbonyl and alkene substrates.[237-239] Enantiomerically pure
oxetanes have been prepared from the photocycloaddition of enantiomerically pure
silyl enol ethers to aromatic aldehydes[240-241] (equation 2).
The intramolecular Williamson ether synthesis furnishes oxetanes from the base-
induced cyclisation of 1,3-halohydrins and related substrates (equation 3). The
cyclisation of optically active substrates can provide access to optically active
oxetanes. For example, optically pure 2,2-substituted oxetanes were prepared via
optically active 1,3-chlorohydrin intermediates, generated by enantioselective
reduction of -halogenoketones[242] (equation 4).

Chapter 6
108
Biggs[243] developed a method of generating oxetane by thermal decomposition of the
tributyltin derivative of a 1,3-bromoacetate, itself derived from corresponding 1,3-diol
(equation 5). This method avoids the use of strong base required for cyclisation of -
substituted alcohols and provided oxetane in a yield of 40%.

Chapter 6
109
Using the methylene transfer reagent, dimethyloxosulfonium methylide, a ketone can
be transformed to an oxetane via the corresponding epoxide[244] (equation 6). Using
this sulfur ylide in concert with a chiral heterobimetallic catalyst, enantioselectivity
was amplified over the two steps, giving 2,2-disubstituted oxetanes in up to >99.5%
ee[245] (equation 7).
Oxetanes have been prepared from selenium-containing precursors by
selenocyclisation of unsaturated substrates. For example,
phenylselenoetherification of 2,4-dimethyl-1,4-pentadiene using N-
phenylselenenylsuccinimide gave bis(phenylseleno)-oxetane[246] (equation 8);
selenocyclisation of a 2-ene-1,5-diol with N-phenylselenenylphthalimide, provided the
two oxetanes[247] (equation 9). Displacement of the selenonyl group affords 3-
alkoxyoxetanes after conjugate addition of an alkoxide to a 3-
hydroxyvinylselenone[174, 248] (equation 10).

Chapter 6
110
6.2 ATTEMPTED ONE-POT PREPARATION OF 2-PHENYLOXETANE
It was thus of interest to investigate the utility of the selenonyl group and in particular,
our methodology for aziridine-formation from -amidoselenides, for the preparation of
oxetanes.
In theory, an oxetane could simply be formed by cyclisation of a -hydroxyselenone in
a one-pot preparation involving the ring-opening of an epoxide with an -metallo-alkyl
selenone to give an alkoxyselenone which might then cyclise in situ with loss of the
selenonyl moiety (Scheme 6.1).
The deprotonation of methyl phenyl selenone and addition of the anion to an
aldehyde in the expectation of forming a -hydroxyselenone was explored by
Saez.[249] Using LDA or LiHMDS as the base, Saez unexpectedly produced an
epoxide as a result of in situ displacement of the selenonyl group by the alkoxide ion
(Scheme 6.2).

Chapter 6
111
With a view to adapting this method of Saez[249] for the preparation of an oxetane
from the reaction of an -lithio selenone with an epoxide, methyl phenyl selenide[74]
(6.1) was prepared in 81% isolated yield by sodium hydride reduction of diphenyl
diselenide[75] and treatment of the resulting sodium phenylselenolate with methyl
iodide (Scheme 6.3).[74] Oxidation with m-CPBA gave methyl phenyl selenone[249]
(6.2) in 41% recrystallised yield. Although an attempt to replicate Saez’ result by
metallation of methyl phenyl selenone with LDA and reaction with benzaldehyde did
not result in the production of styrene oxide, when the reaction was conducted using
potassium tert-butoxide as the base, styrene oxide was obtained in low yield. The
subsequent attempt to prepare 2-phenyloxetane (6.3) by deprotonation of methyl
phenyl selenone with potassium tert-butoxide and reaction of the anion with styrene
oxide was, however, unsuccessful; methyl phenyl selenone and styrene oxide were
recovered from the reaction (Scheme 6.3).
While the attack of -lithio selenoxides on aldehydes and ketones has been
reported[250] in the production of -hydroxy selenides, there are no reports of the

Chapter 6
112
opening of epoxides with -lithio selenoxides to give -hydroxy selenoxides, which
would be useful intermediates on the path to oxetanes. Adapting a procedure
described by Reich[250] for the reaction of -lithio selenoxides with aldehydes and
ketones, oxidation of methyl phenyl selenide (6.1) to the selenoxide (6.4) with m-
CPBA, in situ deprotonation with LDA and introduction of styrene oxide to the mixture
at –78°C gave no reaction, with recovery of the styrene oxide (Scheme 6.4).
6.3 PREPARATION AND ATTEMPTED CYCLISATION OF 3-PHENYL-3-PHENYLSELENOPROPANOL
An alternative approach to the preparation of -hydroxy selenides could be the
addition of phenyl selenol to an ,-unsaturated aldehyde or ketone, such as
cinnamaldehyde, followed by reduction of the carbonyl group.
Sodium phenylselenolate was prepared by sodium hydride reduction of diphenyl
diselenide.[75] Introduction of cinnamaldehyde to the sodium phenylselenolate
suspension gave 3-phenyl-3-phenylselenopropanal (6.5) in approximately 80% crude
yield. Chromatography resulted in the decomposition of some of this product as was
evident from the appearance of a yellow band of diphenyl diselenide during elution of
the propanal (6.5) (Scheme 6.5). Decomposition of the propanal (6.5) during
chromatography had been observed previously in our research group[251-252] and
could occur via the elimination of phenyl selenol followed by its oxidation to diphenyl
diselenide. The isolated yield of 3-phenyl-3-phenylselenopropanal (6.5) was

Chapter 6
113
approximately 51%, contaminated with cinnamaldehyde and diphenyl diselenide.
The mass spectrum of 3-phenyl-3-phenylselenopropanal (6.5) showed a selenium-
containing molecular ion at m/z 290 and in the 1H NMR spectrum, two doublets of
doublets of doublets at 3.27 and 3.12 with a geminal coupling constant of 17.4 Hz
attributed to the diastereotopic methylene protons. These signals also showed
coupling to the benzylic proton whose signal appeared at 4.81, and to a triplet at
9.69 due to the aldehydic proton.
Sodium borohydride reduction of the impure aldehyde gave the -hydroxy selenide,
3-phenyl-3-phenylseleno-1-propanol (6.6), in 56% isolated yield (Scheme 6.5).
Assignment of this structure was supported by a molecular ion at m/z 292 in the
mass spectrum with fragments due to loss of C6H5Se at m/z 135 and further loss of
water at m/z 117. In the 1H NMR spectrum, two distinct doublets of triplets at 3.72
and 3.59 with a geminal coupling constant of 10.8 Hz were attributed to the
diastereotopic protons under oxygen. Signals due to the second diastereotopic
methylene protons, vicinal to the benzylic proton, appeared as a doublet of doublets

Chapter 6
114
of doublets at 2.29 with coupling constants of 6.0 and 6.3 Hz to the vicinal
methylene protons and of 7.8 Hz to the benzylic proton, the triplet due to which
appeared at 4.44. Also produced was cinnamyl alcohol, (6.7) through reduction of
the cinnamaldehyde present in the starting material. A third product exhibited two
triplets at 3.65 and 2.69 and a multiplet at 1.87 in the 1H NMR spectrum,
consistent with its being 3-phenyl-1-propanol (6.8) (Scheme 6.5). Further reduction
of the cinnamaldehyde double bond to give this product (6.8) is plausible as sodium
borohydride reduction of a double bond is known to occur where the double bond is
conjugated with a carbonyl group and is especially facile in a cinnamyl system.[256-257]
Attempted cyclisation of 3-phenyl-3-phenylseleno-1-propanol (6.6) under the
conditions developed for the preparation of aziridines, namely, oxidation with m-
CPBA at -78°C in THF solution, addition of 5 equivalents of potassium tert-butoxide
and warming to room temperature, gave a mixture, the 1H NMR spectrum of which
showed no indication of the expected triplet at ~ 5.9 of 2-phenyloxetane[258-259] (6.3)
(Scheme 6.6). Previous workers[251-252] in this research group had made attempts to
cyclise 3-phenyl-3-phenylseleno-1-propanol (6.6) to the oxetane; a range of reaction
conditions had been explored, including oxidation with (i) m-CPBA, (ii) H2O2 with acid
catalysis, (iii) oxone in a medium buffered at pH 11 and (iv) oxone in a medium
buffered at pH 8. However, no evidence had been observed for the formation of the
oxetane under any of the conditions used. For this reason, further attempts to
cyclise 3-phenyl-3-phenylseleno-1-propanol (6.6) were not pursued in the present
work. Competition between elimination and oxidation of the seleninyl group,
competition between the displacement and elimination of the selenonyl group, and

Chapter 6
115
hindrance of the secondary carbon by the bulky phenyl and phenylseleno groups
could all contribute to the reluctance of this molecule to cyclise to an oxetane.
6.3 PREPARATION AND CYCLISATION OF -HYDROXY SELENIDES BEARING A PRIMARY
SELENIUM MOIETY
Efforts were directed toward the possibility that a -hydroxy selenide bearing a
primary seleno group and a secondary hydroxyl group would be a more viable
oxetane-precursor. These -hydroxy selenides could be prepared by the ring-
opening of an epoxide with phenylselenomethyllithium.[86]
Bis(phenylseleno)methane (6.9) was prepared in 94% isolated yield by sodium
hydride reduction of diphenyl diselenide[75] and the reaction of the resulting sodium
phenylselenolate with methylene iodide.[74, 260] Selenium-metal exchange was
effected by treatment of the selenoacetal with n-butyllithium,[86] giving
phenylselenomethyllithium (6.10). Addition of HMPA and a solution of styrene oxide
in THF to the mixture gave the -hydroxy selenide, 1-phenyl-3-phenylseleno-1-
propanol[261] (6.11, 44%), along with the expected by-product n-butyl phenyl selenide
(6.12, 62%). HMPA is added to this reaction mixture to inhibit the decomposition of
phenylselenomethyllithium (6.10) to lithium phenylselenolate.[86, 89] However, also

Chapter 6
116
obtained was an 18% yield of 1-phenyl-2-phenylselenoethanol[136] (6.13), resulting
from the reaction of styrene oxide with lithium phenylselenolate (Scheme 6.7).
The mass spectrum of 1-phenyl-3-phenylseleno-1-propanol[261] (6.11) showed a
strong molecular ion at m/z 292 with a selenium-containing fragment at m/z 185
attributed to PhSeCHCH2+
, and a signal at m/z 107 attributed to C6H5CH2O+. In the
1H NMR spectrum, two doublets of doublets of triplets, at 2.18 and 2.0, with a
geminal coupling constant of 14.1 Hz, were assigned to the diastereotopic methylene
protons at C2. These protons were coupled to the benzylic proton whose signal
appeared at 4.83, and to the methylene protons under selenium, which resonated
as a triplet at 2.98.
An attempt to cyclise 1-phenyl-3-phenylseleno-1-propanol (6.11) to 2-phenyloxetane
(6.3), under the conditions which produced the aziridine from a -amido selenide,
gave a complex mixture whose 1H NMR spectrum showed no evidence of the
oxetane[258-259]. A second attempt at this reaction, varying the method by carrying
out the oxidation at ambient temperature and then cooling the mixture to –78°C

Chapter 6
117
before addition of base, gave predominantly 1-phenyl-1,3-propanediol[262-263] (6.14),
which was isolated in 23% yield (Scheme 6.8).
Efforts were directed to determine whether the conditions described by Kuwajima,
Shimizu and Ando[174, 248] for the preparation of 3-methoxyoxetanes (equation 10)
would be generally conducive to the cyclisation of -hydroxy selenides to oxetanes.
This procedure[174, 248] uses methanol as the solvent, which has been shown to
optimise the rate of oxidation of a number of selenoxides to the respective
selenones.[176] Thus, 1-phenyl-3-phenylseleno-1-propanol (6.11) was oxidised with
three equivalents of m-CPBA in methanol at room temperature for 30 minutes.
Aqueous sodium hydroxide was added and the reaction allowed to continue for 18
hours, resulting in a mixture of 2-phenyloxetane[258-259] (6.3) and 3-methoxy-1-phenyl-
1-propanol[262] (6.15) in a ratio of 2:1, along with 1-phenyl-1,3-propanediol[262-263]
(6.14). Chromatography of the mixture isolated 2-phenyloxetane[258-259] (6.3, 20%)
and 3-methoxy-1-phenyl-1-propanol[262] (6.15, 12%) (Scheme 6.8). The five ring
protons of 2-phenyloxetane (6.3) appeared as five distinct signals in the 1H NMR
spectrum: a triplet at 5.82, due to the -proton, and two pairs of signals - at 4.84
and 4.67, and at 3.03 and 2.67 - due to the - and - diastereotopic methylene
protons respectively.
The -hydroxy selenide (6.11) was oxidised with 2.5 equivalents of m-CPBA in
methanol but without the addition of hydroxide; 3-methoxy-1-phenyl-1-propanol[262]
(6.15) was obtained in 41% yield as the predominant product, with no evidence of the
oxetane in 1H NMR spectrum, indicating that the cyclisation reaction could not
proceed in the absence of base.

Chapter 6
118
A further two -hydroxy selenides, 1-phenylseleno-3-undecanol (6.17) and 4-
phenylseleno-1-phenyl-2-butanol (6.18), were prepared by the ring-opening of an
epoxide with phenylselenomethyllithium (6.10). 1-Phenylseleno-3-undecanol (6.17)
was isolated in 24% yield along with the -hydroxy selenide,1-phenylseleno-2-
decanol[264] (6.19, 14%) from the reaction of the phenylselenomethyllithium (6.10)
and 1,2-epoxydecane (Scheme 6.9). The expected eleven alkyl signals were
evident in the 13C NMR spectrum of the -hydroxy selenide (6.17). Both the low and
high resolution mass spectra showed a molecular ion, and a prominent fragment due
to loss of OH. In the 1H NMR spectrum, the CHO proton appeared as a multiplet
centered at 3.72, while signals due to the diastereotopic methylene protons under
selenium were almost coincident and appeared as a multiplet centered at ~ 3.01
which was not further elucidated. The signals due to the diastereotopic methylene
protons at C2 were also almost coincident and appeared as a complex multiplet
centered at 2.83.

Chapter 6
119
The -hydroxy selenide[264] (6.19) exhibited a strong molecular ion at m/z 314 with a
peak at m/z 297 due to loss of OH and a prominent selenium-containing peak at m/z
172 assigned to PhSeCH3+. The 13C spectrum showed ten alkyl carbons while the
1H NMR spectrum gave three diagnostic signals: two distinct doublets of doublets at
3.15 and 2.88 due to the diastereotopic protons under selenium, coupled to the
CHO proton which resonated at 3.67.
The oxidation of 1-phenylseleno-3-undecanol (6.17) under the conditions described
by Shimizu, Ando and Kuwajima[174, 248] gave a mixture of 2-octyloxetane[265] (6.20)
and 1-methoxy-3-undecanol (6.21), each in a yield of about 30%, estimated from the
1H NMR integrations in a spectrum of the crude product (Scheme 6.10). Purification
by chromatography resulted in very small yields, perhaps due to the volatility of both
compounds. The oxetane (6.20) exhibited five distinct alkyl signals in the 1H NMR
spectrum, corresponding to the five ring protons. Two doublets of doublets of
doublets, at 4.66 and 4.50, with a geminal coupling of 5.7 Hz, were attributed to
the diastereotopic -methylene ring protons. Signals due to the diastereotopic -
methylene ring protons appeared at 2.64 and 2.35 with a germinal coupling
constant of 10.8 Hz, and were coupled to the -methine proton which resonated at
4.82. Two poorly-resolved multiplets, at 1.78 and 1.66, corresponded to the
diastereotopic methylene protons of the alkyl chain.

Chapter 6
120
In the 1H NMR spectrum of 1-methoxy-3-undecanol (6.21), two doublets of doublets
of doublets at 3.78 and 3.63, with a germinal coupling constant of 9.3 Hz, were
assigned to the diastereotopic methylene protons at C1. The neighbouring
diastereotopic methylene protons at C2 appeared as a multiplet at 1.73. A
multiplet at 3.78 and a singlet at 3.36 were assigned to the methine CHO and the
methoxy protons respectively. A D2O shake resulted in greater resolution of the
CHOH multiplet at 3.78, supporting the assignment of this structure rather than of
the isomeric 3-methoxy-1-undecanol. Three of the twelve signals in the 13C
spectrum of (6.21) resonated downfield ( 71.83, 71.57 and 58.90) corresponding to
the three carbons attached to oxygen. A high resolution mass spectrum showed a
molecular ion, with the base peak at m/z 185.1901, due to loss of OH.
The ring-opening of 2-benzyloxirane with phenylselenomethyllithium (6.10) gave the
-hydroxy selenide, 4-phenylseleno-1-phenyl-2-butanol (6.18, 13%), along with the -
hydroxy selenide, 3-phenylseleno-1-phenyl-2-propanol[266] (6.22, 2%). Also
produced was 4,4-bis(phenylseleno)-1-phenyl-2-butanol (6.23, 14%) (Scheme 6.11).
In the mass spectrum, the -hydroxy selenide (6.18) gave a molecular ion at m/z 306
and a fragment at m/z 213 due to loss of C7H9. In the 1H NMR spectrum, the
diastereotopic methylene protons under selenium resonated as two doublets of
triplets at 3.15 and 3.05, with a geminal coupling constant of 12.3 Hz. Signals of

Chapter 6
121
the benzylic diastereotopic methylene protons appeared as doublets of doublets at
2.85 and 2.72 with a geminal coupling constant of 13.5 Hz. A poorly resolved
multiplet at 4.02 was assigned to the CHO proton. The third pair of diastereotopic
methylene protons at C2 resonated as a complex multiplet at 1.95. The -hydroxy
selenide (6.22) was characterised by three alkyl signals in the 13C NMR spectrum
and three corresponding sets of alkyl signals in the 1H NMR spectrum. A 16-line
signal centred at 3.93 in the 1H NMR spectrum was assigned to the CHO proton.
This signal was coupled to the diastereotopic methylene protons under selenium
which resonated as two doublets of doublets at 3.13 and 2.93, and to a doublet of
doublets at 2.93 which was attributed to the benzylic diastereotopic methylene
protons The mass spectrum of the selenoacetal (6.23) showed a molecular ion at
m/z 462 and fragment at m/z 187 due to loss of the phenylseleno group and water.
In the 13C NMR spectrum, C-Se coupling was apparent for two aromatic signals, at
134.83 and 134.33. The 1H NMR spectrum showed a one-hydrogen multiplet at
4.25 which was assigned to the CHO proton, and a doublet of doublets at 4.71,
assigned to the proton under the two selenium atoms. The benzylic diastereotopic
methylene protons resonated as two doublets of doublets at 2.70 and 2.66 while
two doublets of doublets of doublets, at 2.14 and 2.05, were attributed to the
remaining diastereotopic methylene protons at C2. Strong bands at 1069, 1022 and
1000 cm-1 in the infrared spectrum of the selenoacetal (6.23) were consistent with
similar absorptions in the infrared spectrum of bis(phenylseleno)methane (6.9), and
also the expected absorptions of an acetal or ketal,[267] and could be attributed to C-
Se stretching.

Chapter 6
122
-Hydroxyalkylselenoacetals analogous with compound (6.23) have arisen in the
work of Krief et al.[86] from the reactions of bis(phenylseleno)methane and 1,1-
bis(phenylseleno)ethane with alkehydes and ketones. Krief et al.[86] proposed that
these products are generated via the metallation of the selenoacetal by the
selenoalkyllithium (Scheme 6.12).
The reaction[174, 248] of 1-phenyl-4-phenylseleno-2-butanol (6.18) with m-CPBA in
methanol followed by addition of aqueous sodium hydroxide gave a complex mixture
from which 2-benzyloxetane[268] (6.24) and 4-methoxy-1-phenyl-2-butanol (6.25) were
isolated in yields of 10% and 15% respectively (Scheme 6.13). 2-Benzyloxetane[268]

Chapter 6
123
(6.24) was characterised by four alkyl carbon signals in the 13C NMR spectrum and
seven distinct alkyl signals in the 1H NMR spectrum, each integrating to one
hydrogen. The apparent quintet due to the CHO proton resonated at 5.04, while
the signals of the CH2O protons appeared as distinct doublets of doublets of doublets
at 4.65 and 4.48 with a geminal coupling constant of 5.7 Hz. Signals due to the
benzylic methylene protons appeared as two strongly coupled doublets of doublets at
3.09 and 2.98, with a geminal coupling constant of 13.8 Hz. Two distinct, but
more complex, signals at 2.63 and 2.44 were assigned to the remaining
diastereotopic methylene protons.
4-Methoxy-1-phenyl-2-butanol (6.25) gave an M+H peak in the mass spectrum, with
prominent fragment at m/z 162 due to loss of water, and a fragment at m/z 131 due
to an additional loss of OCH3. The 13C spectrum of (6.25) showed five alkyl signals
while five sets of alkyl protons were also apparent in the 1H NMR spectrum. A three-
hydrogen singlet at 3.34 was evidence of an methoxy group. The CHO methine
proton appeared as a poorly resolved pentuplet, while the diastereotopic methylene
protons - to the methoxy group appeared as two distinct doublets of doublets of
doublets at 3.62 and 3.52. Two doublets of doublets at 2.81 and 2.76 were
assigned to the benzylic diastereotopic methylene protons. The remaining multiplet
centred at 1.73 was assigned to the remaining methylene protons.

Chapter 6
124
These preliminary investigations demonstrated that oxetanes may be prepared from
-hydroxy selenides via a variation of the literature procedure.[174, 248] Future work
would involve optimising the yield by exploring the parameters of reaction time and
temperature and the use of a less nucleophilic solvent.
6.4 OXETANES IN NATURAL PRODUCTS AND DRUG DESIGN
In natural product investigations, the discovery of an oxetane ring is often regarded
as unusual or unique in a family of compounds. Some recent examples include the
limonoid (6.26), isolated from the leaves and twigs of Melia toosendan,[269] the
sesquiterpene dimer (6.27), containing a hemiacetal oxetane, isolated from the
leaves of Xylopia aromatica,[270] the macrolactin (6.28), isolated from the fermentation
broth of a marine Bacillus sp. and found to exhibit antibacterial activity,[271] and the
herbicidal and bacteriocidal Oxetin (6.29), obtained from the culture filtrate of a
Streptomyces sp.[272] The diterpenoid (6.30), isolated from the leaves and twigs of
Trigonostemon chinensis,[273] and Mitrephorone A (6.31), isolated from Mitrephora
glabra,[274] both display anticancer activity.[273-274] Oxetanocin A (6.32), isolated from
the fermentation broth of Bacillus megaterium, attracted considerable interest as it
was shown to inhibit the in vitro replication of HIV.[275-278]

Chapter 6
125

Chapter 6
126
The oxetane moiety is not unusual among the taxane compounds found in the Yew
trees of the genus Taxus. Of the approximately 550 taxanes isolated from species
of this genus, more than one-quarter contain an oxetane ring.[279] Taxol (6.33),
which was isolated from T. brevifolia in the late 1960s, is currently used in cancer
chemotherapy. The role of the oxetane ring in the bioactivity of Taxol is believed to
be two-fold: firstly, in contributing to the conformational rigidity of the four-ring
scaffold and secondly, as a hydrogen-bond acceptor in the tubulin protein binding
pocket.[280]
Despite having similar ring strain to oxiranes, oxetanes are not quite as susceptible
toward acid-catalysed ring-opening,[281-282] and in the absence of acid catalysis, are
considerably less reactive than oxiranes toward nucleophiles.[282-284] 3-Substituted
oxetanes are particularly resistant to nucleophilic attack, as substitution results in
lower ring-strain[285] and ring-opening would lead to unfavourable non-bonding
interactions. Oxetanes have the highest affinity for hydrogen-bonding among the
common cyclic ethers,[286] and comparable or greater hydrogen-bonding ability than
carbonyl compounds, with the exception of amides.[287-289]

Chapter 6
127
These physicochemical properties make the oxetane unit an attractive component for
incorporation into a drug molecule.[290-292] An oxetane moiety can be incorporated
into a drug to ‘block’ a reactive methylene group or to introduce conformational
constraint, without increasing the lipophilicity of the molecule.[290-292] The oxetane
unit is thus a structural alternative to a gem-dimethyl group which has the
disadvantage of increasing the lipophilicity of the drug, thereby exposing it to
enzymatic degradation.[290, 292] The oxetane unit also provides the polarity and
comparable hydrogen bonding ability of a carbonyl group, but without the carbonyl
group’s inherent reactivity.[290, 292] The oxetane moiety can thus be regarded as a
‘bioisostere’ of both a gem-dimethyl group and a carbonyl group.[293]
Morpholine (6.34) is incorporated
into a number of drugs in order to
increase their aqueous solubility
but has the disadvantage that it is
susceptible to oxidative metabolic
attack.[294] The spirocyclic oxetane (6.35) has been proposed as a viable substitute
for morpholine, and has been shown to be stable at pH 1-10 and resistant to
oxidative degradation.[294]
Recent studies in medicinal chemistry suggest that oxetanes will find extensive
application in drug design in the future.[290-292] As synthetic approaches to oxetanes
are few, further exploration of the use of the selenonyl group in the synthesis of
oxetanes, with the mild conditions required for its displacement, could therefore be a
worthwhile endeavour.

Experimental 7.1
128
7 EXPERIMENTAL
7.1 GENERAL EXPERIMENTAL
All solvents were redistilled prior to use. Tetrahydrofuran (THF) was distilled from
sodium wire and sodium benzophenone under a nitrogen atmosphere immediately
prior to use. Sodium hydride was used as a 60% dispersion in oil. meta-
Chloroperbenzoic acid was recrystallised from dichloromethane and was 80% pure
as determined by iodometric titration. Other reagents were purified according to
standard procedures.[295]
Flash chromatography was carried out with Merck Kieselgel 60 (230-400 mesh).
Thin layer chromatography (TLC) was performed on MERCK aluminium-backed silica
gel 60 F254 plates. TLC plates were visualised with UV light at 254 nm or using an
ammonium molybdate dip.
1H and 13C NMR spectra were obtained using either Varian Gemini 2000
Spectrometers (1H: 199.954, 13C: 50.283 MHz and 1H: 300.145, 13C: 75.479 MHz) or
a Varian INOVA Spectrometer (1H: 599.842, 13C: 150.842 MHz). Unless otherwise
stated, spectra were recorded as solutions in deuterochloroform at 25°C. Chemical
shifts () are reported in parts per million (ppm), relative to an internal standard of
tetramethylsilane (0 ppm) for 1H spectra, an internal standard of chloroform (77.0
ppm) for 13C spectra and an external standard of diphenyl diselenide (463 ppm) for
77Se spectra. Hydrogen multiplicities are abbreviated as s (singlet), d (doublet), t
(triplet), q (quartet), qn (quintuplet), m (multiplet).

Experimental 7.1
129
Infrared spectra were recorded on an ATI Mattson Genesis FT IR spectrometer or a
Perkin-Elmer 1720X FT IR spectrometer or a Perkin-Elmer Spectrum 100 ATR FT IR
spectrometer. Liquids were recorded as liquid films, solids as nujol mulls between
sodium chloride plates or as dispersions in pressed potassium bromide discs.
Solution spectra were obtained using a 0.5 mm path-length solution cell with sodium
chloride windows.
Electron impact (EI) mass spectra were recorded with a VG ZAB 2HF mass
spectrometer operating at 70 eV or a Shimadzu mass spectrometer at the University
of Adelaide. Electrospray (ESI) mass spectra were recorded with a Finnigan LCQ
mass spectrometer at the University of Adelaide. Electron impact high resolution
mass spectra (EI HRMS) were recorded on a Kratos Concept ISQ mass
spectrometer at the University of Tasmania. Electrospray high resolution mass
spectra (ESI HRMS) were recorded with an LTQ Orbitrap XL ETD spectrometer at
the University of Adelaide. Elemental analyses were performed at the University of
Otago, New Zealand.
Melting points were determined using a Kofler hot stage apparatus fitted with a
Reichert microscope and are uncorrected.
X-ray crystal structures were determined by Dr Edward Tiekink at the University of
Adelaide or by Professor Allan White at the University of Western Australia.
Unless otherwise stated, ratios of products were estimated from the integration of
peaks in the 1H NMR spectrum. The peaks used for -amidoselenides, -

Experimental 7.1
130
bromoamides, aziridines and cis-oxazolines were the protons under the two vicinal
substituents of the ring or chain, and for the syn-elimination products, the two alkene
protons and the proton under the amide group.
Data is described for each compound in the Experimental for the chapter in which it
first appears. Upon each occurrence thereafter the page containing the data is cited.
In subsequent preparations of the same compound, the compound was identified by
its accordance with spectra of a previous sample(s).

Experimental 7.2
131
7.2 WORK DESCRIBED IN CHAPTER 2
Amidoselenation of cyclohexene with 2 equivalents phenylselenenyl bromide
Procedure 7.2A: To a solution of cyclohexene in benzonitrile was added
phenylselenenyl bromide followed by aqueous TfOH. The mixture was stirred at the
specified bath temperature for 1 h then allowed to cool to r.t. Saturated aqueous
NaHCO3 (10 mL) was added and the products were extracted with CHCl3 (2 x 25
mL). The combined organic layers were washed with saturated aqueous NaCl (10
mL), dried (MgSO4), and the solvent evaporated at reduced pressure.
(i) reaction at 100°C
Following Procedure 7.2A, a mixture of cyclohexene (92 mg, 1.1 mmol) and
phenylselenenyl bromide (465 mg, 1.97 mmol) in benzonitrile (5.5 mL), TfOH (0.09
mL, 1 mmol) and water (0.09 mL, 5 mmol) was stirred at 97-107°C.
Chromatography (CHCl3/hexane 15:85 to remove diphenyl diselenide then
EtOAc/hexane 50:50) gave a fraction (93 mg) which contained the cis-oxazoline[149-
150] (2.7, data: page 137) and the selenide (2.5, data: page 137) in a ratio of 80:20 as
estimated from integration of peaks in the 1H NMR spectrum.
(ii) reaction at 120°C
Following Procedure 7.2A, a mixture of cyclohexene (120 mg, 1.46 mmol) and
phenylselenenyl bromide (706 mg, 2.99 mmol) in benzonitrile (8 mL), TfOH (0.13 mL,
1.5 mmol) and water (0.13 mL, 7.2 mmol) was stirred at 120°C. Chromatography
(CHCl3/hexane 20:80 to remove diphenyl diselenide, then EtOAc/hexane 50:50) gave
the cis-oxazoline[149-150] (2.7, data: page 137) as a pale brown gum (88 mg, 30%).

Experimental 7.2
132
(iii) reaction at 150°C
Following Procedure 7.2A, a mixture of cyclohexene (92 mg, 1.1 mmol) and
phenylselenenyl bromide (499 mg, 2.12 mmol) in benzonitrile (6 mL), TfOH (0.09 mL,
1 mmol) and water (0.09 mL, 5 mmol) was stirred at 146-151°C. Chromatography
(CHCl3/hexane 15:85 to remove diphenyl diselenide then EtOAc/hexane 30:70) gave
the cis-oxazoline[149-150] (2.7, data: page 137) as a brown gum (33 mg, 15%).
(iv) reaction at 160°C
Following Procedure 7.2A, a mixture of cyclohexene (128 mg, 1.56 mmol) and
phenylselenenyl bromide (769 mg, 3.26 mmol) in benzonitrile (6 mL), TfOH (0.14 ml,
1.6 mmol) and water (0.14 mL, 8.0 mmol) was stirred at 160°C. Chromatography
(EtOAc/hexane, gradient of 25:75 to 40:60) gave diphenyl diselenide followed by the
cis- oxazoline[149-150] (2.7, data: page 137) as a brown oil (15 mg, 5%).
Reaction of trans-2-benzamidocyclohexyl phenyl selenide (2.5) with
phenylselenenyl bromide
(i) reaction in benzonitrile at 115°C
A mixture of the amido selenide (2.5, 161 mg, 0.448 mmol) and phenylselenenyl
bromide (120 mg, 0.509 mmol) in benzonitrile (5.5 mL) was stirred at a bath
temperature of 115°C for 4 h. The mixture was allowed to cool and the solvent was
evaporated at reduced pressure. Chromatography (EtOAc/hexane 40:60) gave
trans-2-bromocyclohexyl benzamide (2.10) which crystallised from the eluting solvent
as white crystals (39 mg, 31%), m.p.152-154°C. Found: C, 55.17; H, 5.40; N, 5.03.
C13H16NOBr requires C, 55.33; H, 5.72; N, 4.96%. max: 3228, 3080, 1635, 1574,
1342, 1194, 706 cm-1. 1H NMR: 7.81-7.77, m, 2H, ArH; 7.54 - 7.41, m, 3H, ArH;
6.20, d, J 6.0 Hz, 1H, NH; 4.14, ddt, J 3.6, 7.8, 10.5 Hz, 1H, CHN; 4.02, ddd, J 4.2,

Experimental 7.2
133
10.5, 11.1 Hz, 1H, CHBr; 2.48 - 2.32, m, 2H; 2.04-1.91, m, 1H; 1.81-1.76, m, 2H;
1.58-1.29, m, 3H. 13C NMR: 167.06, C=O; 134.85, 131.46, 128.59, 126.96, all Ar;
55.83, CHN; 55.25, CHBr; 37.17, 33.29, 26.55, 24.35. MS: m/z 281 (M+, 79Br), 202
(M+-Br), 122 (C6H5CONH3+), 105 (C6H5CO+), 77 (C6H5
+). Further elution gave cis-2-
bromocyclohexyl benzamide (2.8, 13 mg, 10%). Recrystallisation from EtOAc gave
(2.8) as white crystals, m.p. 150.5-152°C. EI HRMS: 281.0409 C13H16NO79Br
requires 281.0416. max: 3324, 2943, 1633, 1525, 1489, 1447, 1313, 1296, 1282,
1263, 1244, 1103, 824, 799, 718, 691, 657 cm-1. 1H NMR: 7.80-7.76, m, 2H, ArH;
7.54-7.42, m, 3H, ArH; 6.33, d, J 6.9 Hz, 1H, NH; 4.79, m, 1H, CHBr; 4.16-4.07, m,
1H, CHN; 2.25-2.18, m, 1H; 2.06-1.94, m, 1H; 1.86-1.67, m, 4H; 1.61-1.43, m, 2H.
13C NMR: 166.63, C=O; 134.58, 131.85, 128.85, 127.17, all Ar; 61.39, CHBr; 50.89,
CHN; 34.01; 28.06; 24.79; 19.99. MS: m/z 281 (M+, 79Br), 202 (M+-Br), 122
(C6H5CONH3+), 105 (C6H5CO+), 77 (C6H5
+). Further elution gave the cis-
oxazoline[149] (2.7, 3 mg, 3%, data: page 137).
(ii) reaction in CH2Cl2 at r.t.
To a solution of the amido selenide (2.5, 71 mg, 0.20 mmol) in CH2Cl2 (5 mL) was
added phenylselenenyl bromide (57 mg, 0.24 mmol) and the mixture was stirred at
r.t. for 48 h. An aliquot of the mixture was taken and the solvent evaporated at
reduced pressure. 1H NMR analysis showed a substantial amount of unreacted
amido selenide (2.5) and therefore, a further portion of phenylselenenyl bromide (26
mg, 0.11 mmol) was added and the mixture was stirred a further 44 h. The mixture
was diluted with CH2Cl2 then washed with saturated aqueous NaHCO3 and saturated
aqueous NaCl and dried (MgSO4), and the solvent removed at reduced pressure to
give a pale brown oil (114 mg). 1H NMR analysis showed this product to be a

Experimental 7.2
134
mixture of the cis-oxazoline[149] (2.7, data: page 137), the cis-bromide (2.8, data:
page 133) and the amido selenide (2.5) in a ratio of 75:20:5.
(iii) reaction in refluxing CH3CN
A solution of the amido selenide (2.5, 72 mg, 0.20 mmol) and phenylselenenyl
bromide (74 mg, 0.31 mmol) in CH3CN (8 mL) was refluxed for 3 h, then cooled and
diluted with CH2Cl2 (25 mL). The mixture was washed with saturated aqueous
NaHCO3 (10 mL) and saturated aqueous NaCl (10 mL) and dried (Na2SO4) and the
solvent evaporated at reduced pressure to give a yellow solid (110 mg). 1H NMR
analysis showed the product to be a mixture of cis-oxazoline[149] (2.7, data: page
137), the trans-bromide (2.10, data: page 132) and the cis-bromide (2.8, data: page
133) in a ratio of 60:25:15.
(iv) in CH2Cl2 with addition of Et4NBr
To a solution of the amido selenide (2.5, 68 mg, 0.19 mmol) in CH2Cl2 (5 mL) was
added phenylselenenyl bromide (73 mg, 0.31 mmol) followed by Et4NBr (66 mg, 0.31
mmol) and the mixture was stirred at r.t. for 4 d. The mixture was diluted with
CH2Cl2, washed with saturated aqueous NaHCO3 and saturated aqueous NaCl and
dried (MgSO4) and the solvent evaporated at reduced pressure to give a yellow solid
(93 mg). 1H NMR analysis showed the product to be a mixture of the cis-bromide
(2.8), the cis-oxazoline[149] (2.7, data: page 137) and the amido selenide (2.5) in a
ratio of 75:15:10. Chromatography (EtOAc/hexane, gradient of 5:95 to 15:85) gave
a fraction (34 mg) containing the cis-bromide (2.8) and the amido selenide (2.5) in a
ratio of 90:10. Recrystallisation of this mixture from EtOAc gave the cis-bromide
(2.8, data: page 133) as white crystals, m.p. 150.5-152°C. Further elution gave the
cis-oxazoline[149] (2.7, 4 mg, 10%).

Experimental 7.2
135
(v) in refluxing CH3CN with addition of Et4NBr
A solution of the amido selenide (2.5, 71 mg, 0.20 mmol), phenylselenenyl bromide
(69 mg, 0.29 mmol) and Et4NBr (63 mg, 0.30 mmol) in CH3CN (8 mL) was refluxed
for 3 h, then cooled and diluted with CH2Cl2 (20 mL). The mixture was washed with
saturated aqueous NaHCO3 (10 mL) and saturated aqueous NaCl (10 mL) and dried
(Na2SO4) and the solvent evaporated at reduced pressure to give a yellow solid (108
mg). 1H NMR analysis showed the product to be a mixture of the cis-bromide (2.8,
data: page 133), the cis-oxazoline[149] (2.7, data: page 137), the trans-bromide (2.10,
data: page 132) and the amido selenide (2.5) in a ratio of 50:20:15:15.
Oxidation of 2-amidoalkyl phenyl selenide with m-CPBA and with KOH as base
To a stirred solution of the amido selenide (2.5, 151 mg, 0.421 mmol) in i-PrOH (20
mL) was added powdered KOH (178 mg, 3.17 mmol) followed by m-CPBA (362 mg,
80%, 1.68 mmol) and the mixture was stirred at r.t. for 1 h. Aqueous Na2S2O3 (0.5
M, 15 mL) and saturated aqueous NaHCO3 (10 mL) were added and the products
were extracted with CHCl3 (2 x 25 mL). The combined organic extracts were dried
(Na2SO4) and the solvent evaporated at reduced pressure. Chromatography
(EtOAc/hexane, gradient of 10:90 to 30:70) gave 7-benzoyl-7-
azabicyclo[4.1.0]heptane[154] (2.17) as a white solid (72 mg, 85%), which crystallised
from the eluting solvent as white crystals, m.p. 79.5-80.5°C (lit.[154] m.p. 77°C).
max(Nujol) 3059, 3008, 2951, 1716, 1666, 1630, 1599, 1577, 1547, 1450, 1410,
1336, 1311, 1294, 1263, 1230, 1186, 1119, 1072, 754, 737, 708, 627 cm-1. 1H
NMR: 8.00-7.97, m, 2H, ArH; 7.56-7.51, m, 1H, ArH; 7.47-7.41, m, 2H, ArH; 2.76-
2.75, m, 2H, CHN; 2.12-2.03, m, 2H; 1.95-1.85, m, 2H; 1.62-1.50, m, 2H; 1.41-1.29,
m, 2H. 13C NMR: 180.25, C=O; 133.77, 132.45, 129.11, 128.35, all Ar; 37.02,

Experimental 7.2
136
23.87, 19.93. MS: m/z 202 (M++H), 105 (PhCO+), 96 (M+-PhCO), 77 (C6H5+).
Further elution gave the cis-oxazoline[149] (2.7, data: page 137) as a pale yellow gum
(10 mg, 12%)
One-Step Amidoselenation[145, 148]
Procedure 7.2B: To a solution of the alkene in nitrile was added phenylselenenyl
chloride followed by a solution of TfOH in water. The mixture was stirred at the
temperature and for the time specified, then cooled to r.t. Saturated aqueous
NaHCO3 (10 mL) was added and the products were extracted with CHCl3 (2 x 25
mL). The combined organic extracts were washed with saturated aqueous NaCl (10
mL), dried (MgSO4), and the solvent was evaporated at reduced pressure. The
crude product was purified by chromatography.
(a) trans-2-(Phenylselanyl)cyclohexyl acetamide (2.31)
Following Procedure 7.2B, a mixture of cyclohexene (0.506 mL, 5.00 mmol) and
phenylselenenyl chloride (958 mg, 5.00 mmol) in CH3CN (28 mL) and TfOH (0.44
mL, 5.0 mmol) in water (0.45 mL, 25 mmol) was refluxed for 2 h. Chromatography
(gradient of EtOAc/hexane 60:40 to EtOAc/hexane/MeOH 60:30:10) gave 2-
(phenylseleno)cyclohexanol[169] (2.41) as a brown oil (7 mg, 1%). max (neat) 3429,
3070, 3055, 2931, 2856, 1577, 1477, 1446, 1437, 1381, 1356, 1271, 1255, 1186,
1113, 1065, 1036, 1022, 957, 741, 694 cm-1. 1H NMR: 7.61-7.58, m, 2H, ArH;
7.34-7.26, m, 3H, ArH; 3.33, dt, J 4.2, 9.9 Hz, 1H, CHO; 2.94, s, 1H, OH; 2.90, ddd,
3.9, 9.9, 12.1 Hz, 1H, CHSe; 2.21-2.12, m, 2H; 1.76-1.70, m, 1H; 1.65-1.60, m, 1H;
1.44-1.17, m, 4H; 13C NMR (200 MHz): 136.08, 129.01, 128.11, 126.72, all Ar;
72.24, CHO; 53.48, CHSe; 33.82, 33.31, 26.75, 24.36. 77Se NMR: 333.43. MS:
m/z 256 (M+), 239, (M+-OH), 158 (C6H5SeH+), 99 (M+-C6H5Se), 81 (C6H9+). Further

Experimental 7.2
137
elution gave the title compound[145] which crystallised from the eluting solvent as fine
white needles (1.298 g, 88%), m.p. 153-155°C (lit.[145] m.p. 149-150°C). EI HRMS:
297.0628 C14H19NOSe requires 297.0633. max (Nujol) 3307, 1643, 1543, 1317,
1178, 1113, 976, 744, 692, 598 cm-1. 1H NMR: 7.59-7.30, m, 2H, ArH; 7.30-7.26,
m, 3H, ArH; 5.43, d, J 7.5 Hz, 1H, NH; 3.81, ddt, J 4.2, 7.5, 11.1 Hz, 1H, CHN; 3.01,
dt, J 3.9, 11.1 Hz, CHSe; 2.21-2.10, m, 2H; 1.90, s, 3H, CH3; 1.70-1.47, m, 3H; 1.42-
1.11, m, 3H. 13C NMR: 169.20, C=O; 135.36, 129.02, 128.27, 127.75, all Ar;
53.32, CHN; 47.94, CHSe; 34.05, 33.81, 26.65, 24.57, 23.40. MS: m/z 297 (M+),
238 (M+-NH2COCH3), 157 (C6H5Se+), 140 (M+-C6H5Se), 98 (C6H12N+), 81 (C6H9+).
(b) trans-2-(Phenylselanyl)cyclohexyl benzamide (2.5)
Following Procedure 7.2B, a mixture of cyclohexene (239 mg, 2.91 mmol) and
phenylselenenyl chloride (617 mg, 3.22 mmol) in benzonitrile (15 mL) and TfOH (0.26
mL, 2.9 mmol) in water (0.26 mL, 14 mmol) was stirred at a bath temperature of
100°C for 1 h. Chromatography (CHCl3/hexane 15:85 to remove diphenyl
diselenide, then EtOAc/hexane 50:50) gave the title compound[145] which crystallised
from the eluting solvent as colourless needles (691 mg, 66%), m.p. 143-144°C (lit.[145]
m.p. 133-134°C). max (Nujol) 3319, 1631, 1577, 1539, 1327, 1178, 741, 694, 665
cm-1. 1H NMR: 7.69-7.66, m, 2H, ArH; 7.58-7.54, m, 2H, ArH; 7.51-7.50, m, 1H,
ArH; 7.49-7.38, m, 2H, ArH; 7.25-7.21, m, 3H, ArH; 6.16, d, J 7.5 Hz, 1H, NH; 3.96,
ddt, J 3.6, 7.5, 11.1 Hz, 1H, CHN; 3.15, dt, J 3.9, 11.1 Hz, 1H, CHSe; 2.39-2.33, m,
1H; 2.26-2.19, m, 1H; 1.76-1.68, m, 1H; 1.64-1.55, m, 1H; 1.50-1.20, m, 4H. 13C
NMR: 166.69, C=O; 135.41, 134.89, 131.25, 129.08, 128.46, 128.07, 127.77,
126.89, all Ar; 53.91, CHN; 48.06, CHSe; 34.04, 26.80, 24.61. MS: m/z 359 (M+),
238 (M+-C6H5CONH2), 202 (M+-C6H5Se), 158 (C6H5SeH+), 122 (C6H5CONH3+), 105
(C6H5CO+), 81 (C6H9+), 77 (C6H5
+). Further elution gave cis-3a,4,5,6,7,7a-

Experimental 7.2
138
hexahydro-2-phenylbenzoxazole[149] (2.7) as a pale yellow gum (37 mg, 6%) which
was recrystallised from hexane to give colourless crystals, m.p. 42-45°C (lit.[149] m.p.
47-48°C). max 3313, 3274, 1720, 1637, 1577, 1543, 1346, 1275, 1176, 1151, 1111,
1066, 1026, 976, 903, 775, 696 cm-1. 1H NMR: 7.99-7.95, m, 2H, ArH; 7.51-7.37,
m, 3H, ArH; 4.68, dt, J 5.1, 8.1 Hz, 1H, CHO; 4.13, ddd, J 6.0, 6.6, 8.1 Hz, 1H, CHN;
1.97-1.78, m, 2H; 1.69-1.38, m, 4H. 13C NMR: 164.27, C=N; 131.16, 128.24,
126.92, all Ar; 78.86, CHO; 63.54, CHN; 27.66, 26.22, 19.78, 19.07. MS: m/z 202
(MH+), 122 (C6H5CONH3+), 105 (C6H5CO+), 77 (C6H5
+).
(c) trans-2-(Phenylselanyl)cyclopentyl benzamide (2.24)
Following Procedure 7.2B, a mixture of cyclopentene (342 mg, 5.02 mmol) and
phenylselenenyl chloride (1.18g, 3.22 mmol) in benzonitrile (15 mL) and TfOH (0.45
mL, 5.1 mmol) in water (0.45 mL, 25 mmol) was stirred at a bath temperature of 100-
105°C for 1 h. Chromatography (EtOAc/hexane 40:60) gave the title compound
which crystallised from the eluting solvent as colourless needles (275 mg, 16%),
m.p.121.5-123.5°C. C18H19NOSe requires: C 62.79, H 5.56, N 4.07. Found: C 62.89,
H 5.51, N 4.14. EI HRMS: 345.0634 C18H19NOSe requires 345.0633. max (Nujol)
3273, 2854, 1631, 1549, 1358, 1323, 1296, 1221, 1182, 1070, 737, 692 cm-1. 1H
NMR: 7.63-7.59, m, 4H, ArH; 7.47-7.44, m, 1H, ArH; 7.40-7.35, m, 2H, ArH; 7.27-
7.22, m, 3H, ArH; 6.10, br d, J 6.0 Hz, 1H, NH; 4.31, qn, J 7.5 Hz, 1H, CHN; 3.44, dt,
J 7.5, 8.1 Hz, 1H, CHSe; 2.35-2.22, m, 2H; 1.81-1.72, m, 3H; 1.55-1.48, m, 1H. 13C
NMR: 167.31, C=O; 134.98, 134.59, 131.38, 129.12, 128.73, 128.45, 127.70,
126.88, all Ar; 58.06, CHN; 46.82, CHSe; 31.74, 31.56, 22.06. MS: m/z 345 (M+),
224 (M+-C6H5CONH2), 188 (M+-C6H5Se), 105 (C6H5CO+), 77 (C6H5+). Further elution
gave a fraction (99 mg) containing a mixture of trans-2-(phenylselanyl)cyclopentyl
benzamide (2.24) and N-cyclopentylbenzamide[154] (2.26) in a ratio of 1:1, as

Experimental 7.2
139
estimated from integrations in the 1H NMR spectrum (data for (2.26): page 143).
Further elution gave cis-4,5,6,6a-tetrahydro-2-phenyl-3aH-cyclopentoxazole[149]
(2.25) (296 mg, 32%) as a brown oil. max 3410, 3282, 3060, 2960, 2870, 1649,
1579, 1495, 1450, 1354, 1323, 1296, 1257, 1201, 1095, 1065, 1024, 947, 781, 696
cm-1. 1H NMR: 7.94-7.90, m, 2H, ArH; 7.49-7.37, m, 3H, ArH; 5.11, dd, J 5.7, 7.2
Hz, 1H, CHO; 4.73, t, J 7.2 Hz, 1H, CHN; 2.12-1.99, m, 2H; 1.81-1.25, m, 4H. 13C
NMR: 163.81, C=N; 131.09, 128.23, 127.84, all Ar; 84.70, CHO; 71.80, CHN;
34.64, 33.85, 22.12. MS: m/z 187 (M+), 158 (M-C2H5+), 130 (M-C3H5O+), 104
(C6H5CNH+), 77 (C6H5+).
(d) trans-N-2-(Phenylselanyl)cycloheptyl benzamide (2.27)
Following Procedure 7.2B, a mixture of cycloheptene (491 mg, 5.11 mmol) and
phenylselenenyl chloride (1.196 g, 6.24 mmol) in benzonitrile (19 mL) and TfOH (0.45
mL, 5.1 mmol) in water (0.45 mL, 25 mmol) was stirred at a bath temperature of 96-
110°C for 20 h. Chromatography (EtOAc/hexane 25:75) gave a fraction (145 mg)
containing the title compound (2.27; data: page 152), the cis-oxazoline, cis-
4,5,6,7,8,8a-hexahydro-2-phenyl-3aH-cycloheptoxazole (2.28), N-cycloheptyl
benzamide[166-167] (2.29), and the syn-elimination product, N-(cyclohept-2-en-1-
yl)benzamide (2.30; data: page 180), in a ratio of 20:25:40:15. Simultaneous
equations translate these ratios into the following approximate yields, respectively:
2%: 3%: 5%: 2%. Further elution gave a fraction from which N-
cycloheptylbenzamide[166-167] (2.29) was isolated as a pale brown solid (163 mg,
15%) by trituration and crystallization from CH2Cl2/hexane, m.p. 126-130°C (lit.[167]
m.p. 127-129°C). 1H NMR: 7.76-7.30, m, 2H, ArH; 7.51 -7.39, m, 3H, ArH; 6.08, br
d, J 9.6 Hz, 1H, NH; 4.19-4.15, m, 1H, CHN; 2.08-2.00, m, 2H; 1.67-1.48, m, 10H.
13C NMR: 166.28, C=O; 136.20, 131.10, 128.44, 126.74, all Ar; 50.83, CHN; 35.17,

Experimental 7.2
140
28.02, 24.15. MS: m/z 217 (M+), 121 (C6H5CONH2+), 105 (C6H5CO+). The mother
liquor contained mainly the cis-oxazoline, cis-4,5,6,7,8,8a-hexahydro-2-phenyl-3aH-
cycloheptoxazole (2.28) (222 mg, 21%). ESI HRMS: 216.13793 C14H17NO+H
requires 216.13883. max 3240, 2925, 2857, 1637, 1625, 1576, 1555, 1489, 1461,
1445, 1327, 1269, 1075, 1054, 888, 803 cm-1. 1H NMR: 7.97-7.93, m, 2H, ArH;
7.50 - 7.37, m, 3H, ArH; 4.86, ddd, J 6.0, 6.9, 10.2 Hz, 1H, CHO; 4.42, ddd, J 3.6,
9.6, 10.2 Hz, 1H, CHN; 2.08 - 1.36, m, 10H. 13C NMR: 162.31, C=N; 131.04,
128.49, 128.21, 128.16, all Ar; 83.21, CHO; 69.83, CHN; 31.57, 30.91, 30.88, 26.06,
24.33. MS: m/z 215 (M+), 105 (C6H5CO+).
(e) 1-(Phenylselanyl)-2-octyl benzamide (2.32)
Following Procedure 7.2B, a mixture of 1-octene (0.471 mL, 3.00 mmol) and
phenylselenenyl chloride (578 mg, 3.02 mmol) in benzonitrile (18 mL) and TfOH
(0.270 mL, 3.05 mmol) in water (0.27 mL, 15 mmol) was stirred at a bath temperature
of 105°C for 2 h. Chromatography (CH2Cl2/hexane 15:85 then a gradient of
EtOAc/hexane 5:95 to 50:50) gave slightly impure 1-(phenylselanyl)-2-octyl
benzamide (2.32) as an orange oil (692 mg, 59%). Recrystallisation from
CH2Cl2/hexane gave the title compound as fine white needles, m.p. 69.5-71.5°C. EI
HRMS: 389.1258 C21H27NOSe requires 389.1259. max (KBr) 3296, 3059, 2953,
2927, 2850, 1628, 1603, 1577, 1537, 1491, 1477, 1466, 1437, 1421, 1412, 1375,
1350, 1321, 1302, 1281, 1072, 1022, 733, 698, 688, 667 cm-1. 1H NMR: 7.59-7.53,
m, 4H, ArH; 7.50-7.44, m, 1H, ArH; 7.40-7.34, m, 2H, ArH; 7.27-7.24, m, 1H, ArH;
7.23-7.19, m, 2H, ArH; 6.14, d, J 8.7 Hz, 1H, NH; 4.43-4.37, m, 1H, CHN; 3.29, dd, J
5.4, 12.9 Hz, 1H, CHaHbSe; 3.22, dd, J 4.8, 12.9 Hz, 1H, CHaHbSe; 1.71-1-61, m, 2H;
1.36-1.24, m, 8H; 0.86, t, J 6.9 Hz, 3H, CH3. 13C NMR: 166.84, C=O; 132.70,
131.29, 130.09, 129.24, 128.51, 128.42, 127.06, 126.80, all Ar; 49.58, CHN; 34.55;

Experimental 7.2
141
33.88, CHSe; 31.64, 29.04, 26.01, 22.52, 13.97. MS: m/z 389 (M+), 268 (M-
C6H5CONH+), 232 (M-C6H5Se+), 127 (C8H17N+), 105 (C6H5CO+), 77 (C6H5+). The
mother liquor contained a mixture of the title compound (2.32) and a second
compound which was identified as the regioisomer, 2-(phenylselanyl)-1-octyl
benzamide (2.33), on the basis of the following signals: 1H NMR: 6.69, br s, 1H,
NH; 3.80, ddd, J 4.2, 6.3, 13.5 Hz, 1H, CHaHbNH; 3.51, ddd, J 5.1, 8.1, 13.5 Hz, 1H,
CHaHbNH; 3.41, m, 1H, CHSe.
(f) 2-(Phenylselanyl)-3-hexyl benzamide (2.34) and 3-(Phenylselanyl)-2-hexyl benzamide (2.35)
Following Procedure 7.2B, a mixture of trans-2-hexene (0.629 mL, 5.00 mmol) and
phenylselenenyl chloride (994 mg, 5.19 mmol) in benzonitrile (20 mL) and TfOH (0.44
mL, 5.0 mmol) in water (0.44 mL, 24 mmol) was stirred at a bath temperature of 90°C
for 1.5 hours. Chromatography (CH2Cl2/hexane 20:80 to remove diphenyldiselenide
then a gradient of Et2O/hexane 5:95 to 50:50) gave a fraction that was predominantly
trans-4-methyl-2-phenyl-5-propyl-4,5-dihydro-1,3-oxazole (2.37) as a yellow oil (56
mg, 6%). max (neat) 3062, 3032, 2960, 2929, 2872, 1720, 1649, 1603, 1579, 1533,
1495, 1450, 1375, 1340, 1319, 1296, 1273, 1244, 1113, 1082, 1065, 1053, 1026,
877, 781, 696 cm-1. 1H NMR: 7.95-7.92, m, 2H, ArH; 7.49-7.37, m, 3H, ArH; 4.39,
qn, J 6.3 Hz, 1H, CHO; 3.77, q, J 6.3 Hz, 1H, CHN; 1.73-1.66, m, 2H; 1.58-1.39, m,
5H; 1.41, d, J 6.3 Hz, 3H, CH(O)CH3; 0.98, t, J 7.2 Hz, 3H, CH2CH3. 13C NMR:
162.40, C=N; 130.96, 128.23, 128.12, 128.08, all Ar; 81.14, CHO; 73.36, CHN;
37.78, 20.92, 18.87, 14.01. MS: m/z 203 (M+), 160 (M+-C3H7), 130 (M+-CH3CHO-
C2H5), 105 (C6H5CO+), 77 (C6H5+), 44 (CH3CHO+). Further elution gave a fraction
(A) containing a mixture of the two oxazoline isomers (2.36) and (2.37) and the title
compounds (2.34) and (2.35) followed by a fraction (B) containing a mixture of the

Experimental 7.2
142
title compounds and the oxazoline (2.36). Fraction (A) was further chromatographed
(EtOH/CH2Cl2 1:99) to give a mixture of the title compounds as a yellow oil (76 mg,
4%). max (Nujol) 3371, 1637 cm-1. 2-(Phenylselanyl)-3-hexyl benzamide (2.34) 1H
NMR: 7.60-7.57, m, 2H, ArH; 7.55-7.52, m, 2H, ArH; 7.48-7.45, m, 1H, ArH; 7.41-
7.35, m, 2H, ArH; 7.22-7.17, m, 3H, ArH; 6.23, d, J 9.3 Hz, 1H, NH; 4.33, ddt, J 3.6,
9.3, 9.9 Hz, 1H, CHN; 3.65, dq, J 3.6, 7.2, Hz, 1H, CHSe; 1.75-1.36, m, 4H; 1.57, d, J
7.2 Hz, 3H, CHCH3; 0.95, t, J 7.2 Hz, 3H, CH2CH3. 13C NMR: 166.94, C=O; 134.54,
134.13, 131.35, 129.52, 129.22, 128.47, 127.48, 126.80, all Ar; 53.88, CHN; 47.51,
CHSe; 33.24, 20.09, 19.52, 13.97. 3-(Phenylselanyl)-2-hexyl benzamide (2.35) 1H
NMR: 7.54-7.50, m, 2H, ArH; 7.48-7.41, m, 3H, ArH; 7.37-7.29, m, 2H, ArH; 7.19-
7.15, m, 3H, ArH; 6.48, d, J 8.7 Hz, 1H, NH; 4.47, ddq, J 3.3, 6.6, 8.7 Hz, 1H, CHN;
3.56, ddd, J 3.3, 6.9, 7.5 Hz, 1H, CHSe; 1.79-1.64, m, 2H; 1.63-1.46, m, 2H; 1.26, d,
J 6.6 Hz, 3H, CHCH3; 0.95, t, J 7.2 Hz, 3H, CH2CH3. 13C NMR: 166.25, C=O;
136.89, 133.55, 131.26, 129.76, 129.33, 128.37, 127.27, 126.75, all Ar; 54.97, CHSe;
48.66, CHN; 36.62, 21.44, 15.52, 13.78. Further elution gave a fraction which was
mainly the oxazoline (2.37) as a yellow oil (60 mg, 6%) followed by a fraction which
was mainly trans-5-methyl-2-phenyl-4-propyl-4,5-dihydro-1,3-oxazole (2.36) as a
yellow oil (40 mg, 4%). max (neat) 3060, 3032, 2960, 2931, 2872, 1720, 1643,
1603, 1579, 1537, 1493, 1450, 1375, 1342, 1321, 1302, 1269, 1176, 1157, 1113,
1078, 1063, 1026, 953, 928, 891, 837, 781, 741, 696 cm-1. 1H NMR: 7.95-7.93, m,
2H, ArH; 7.49-7.37, m, 3H, ArH; 4.19, dt, J 5.1, 7.2 Hz, 1H, CHO; 3.91, dq, J 6.9, 6.9
Hz, 1H, CHN; 1.95-1.44, m, 4H; 1.34, d, J 6.9 Hz, 3H, CH(N)CH3; 0.99, t, J 7.2 Hz,
3H, CH2CH3. 13C NMR: 162.64, C=N; 131.01, 128.27, 128.14, 128.08, all Ar;
86.62, CHO; 67.21, CHN; 36.95, 21.44, 18.45, 13.87. MS: m/z 203 (M+), 188 (M+–
CH3), 174 (M+-C2H5), 160 (M+-C3H7), 131 (M+-C3H7CHO), 104 (C6H5CNH+), 103

Experimental 7.2
143
(C6H5CN+), 77 (C6H5+). Chromatography (acetone/CH2Cl2 5:95) of fraction (B) gave
a mixture of the title compounds as a yellow oil (141 mg, 8%) followed by a fraction
which was mainly the oxazoline (2.36) as a yellow oil (93 mg, 9%).
N-Cyclohexylbenzamide (2.38)
To a stirred solution of cyclohexene (205 mg, 2.49 mmol) in benzonitrile (10 mL) was
added a solution of TfOH (0.22 mL, 2.5 mmol) in water (0.22 mL, 12 mmol) and the
mixture was stirred at a bath temperature of 120°C for 1 h. Saturated aqueous
NaHCO3 (10 mL) was added and the mixture was extracted with CHCl3. The
combined organic extracts were washed with saturated aqueous NaCl (10 mL), dried
(MgSO4), and the solvent removed under reduced pressure. Chromatography
(EtOAc/hexane 50:50) gave the title compound[296-297] as a white solid (280 mg,
55%), m.p. 154-156°C (lit.[297] m.p. 152–154°C). max (Nujol) 3330, 3236, 3074,
1639, 1562, 11331, 700 cm-1. 1H NMR(200 MHz): 7.78-7.73, m, 2H, ArH; 7.50-
7.38, m, 3H, ArH; 5.95, m, 1H, NH; 3.99, m, 1H, CHN; 2.07-2.00, m, 2H; 1.79-1.14,
m, 8H. 13C NMR: 166.58, C=O; 135.16, 131.19, 128.49, 126.78, all Ar; 48.65,
CHN; 33.25, 25.59, 24.89. MS: m/z 203 (M+), 122 (C6H5CONH3+), 105 (C6H5CO+),
77 (C6H5+).
N-Cyclopentylbenzamide (2.26)
To a solution of cyclopentene (207 mg, 3.04 mmol) in benzonitrile (10 mL) was added
a solution of TfOH (0.26 mL, 2.9 mmol) in water (0.26 mL, 14 mmol) and the mixture
was stirred at a bath temperature of 100°C for 2 h. Saturated aqueous NaHCO3 (10
mL) was added and the products extracted with CHCl3. The combined organic
extracts were washed with saturated aqueous NaCl (10 mL), dried (MgSO4), and the

Experimental 7.2
144
solvent removed under reduced pressure. Chromatography (EtOAc/hexane 50:50)
gave the title compound[154, 167] as a white solid (146 mg, 25%), m.p. 162-164°C
(lit.[167] m.p. 156-157°C). max (Nujol) 3290, 1628, 1545, 1315, 1184, 1076, 1028,
931, 890, 804, 696 cm-1
. 1H NMR: 7.77-7.32, m, 2H, ArH; 7.50-7.27, m, 3H, ArH;
6.26, br s, 1H, NH; 4.39, dqn, J 6.9, 6.9 Hz, 1H, CHN; 2.13-2.02, m, 2H; 1.76-1.57,
m, 4H; 1.55-1.43, m, 2H. 13C NMR: 167.12, C=O; 134.93, 131.12, 128.39, 126.79
all Ar; 51.64, 33.13, 23.77. MS: m/z 189 (M+), 122 (C6H5CONH3+), 105 (C6H5CO+),
77 (C6H5+).
Amidoselenation in non-nitrile solvents
(a) Reaction of cyclohexene
(i) in dimethyl acetamide
To a solution of phenylselenenyl chloride (284 mg, 1.48 mmol) and benzonitrile (770
mg, 7.47 mmol) in dimethyl acetamide (7 mL) was added cyclohexene (134 mg, 1.63
mmol) followed by TfOH (0.13 mL, 1.5 mmol) in water (0.13 mL, 7.2 mmol). The
mixture was stirred at a bath temperature of 90-95°C for 1 h, then cooled, diluted with
saturated aqueous NaHCO3 (10 mL), extracted with CHCl3 (2 x 30 mL), washed with
saturated aqueous NaCl (10 mL) and dried (NaSO4). Evaporation of the solvent at
reduced pressure and chromatography (CHCl3/hexane 15:85 to remove diphenyl
diselenide, then EtOAc/hexane 20:80) gave 2-(phenylseleno)cyclohexanol (2.41) as
a red oil (172 mg, 41%, data: page 136).
(ii) in toluene
To a solution of cyclohexene (97 mg, 1.2 mmol) and benzonitrile (513 mg, 4.97
mmol) in toluene (1 mL) was added phenylselenenyl chloride (228 mg, 1.19 mmol) in
toluene (5.5 mL) followed by TfOH (0.09 mL, 1 mmol) in water (0.09 mL, 5 mmol).

Experimental 7.2
145
The mixture was stirred at a bath temperature of 96-115°C for 80 min and then
allowed to cool to room temperature. Saturated aqueous NaHCO3 (10 mL) was
added and the products were extracted with CHCl3 (2 x 25 mL). The combined
organic extracts were washed with saturated aqueous NaCl (10 mL), dried (MgSO4),
and the solvent evaporated at reduced pressure. Chromatography of the residue
gave a fraction (70 mg) containing 2-(phenylselanyl)cyclohexyl benzamide (2.5) and
N-cyclohexylbenzamide[296-297] (2.38) in a ratio of 1:2.5. Simultaneous equations
translate these ratios into the approximate yields, respectively: 7% and 17%.
Further elution gave the cis-oxazoline (2.7, 37 mg, 15%, data: page 137).
(iii) in CHCl3 at reflux
A solution of phenylselenenyl chloride (280 mg, 1.46 mmol) in CHCl3 (4.5 mL)
followed by benzonitrile (0.75 mL, 7.3 mmol) were added to a solution of cyclohexene
(148 mL, 1.46 mmol) in CHCl3 (4 mL) at 0°C. The solution was warmed to r.t. and
stirred for 30 min. A solution of TfOH (0.13 mL, 1.5 mmol) in water (0.13 mL, 7.2
mmol) was added and the mixture was refluxed for 2h. Saturated aqueous NaHCO3
(15 mL) was added and the aqueous layer extracted with CHCl3 (3 x 20 mL). The
combined organic layers were washed with saturated aqueous NaCl (15 mL) and
dried (MgSO4) and the solvent removed at reduced pressure to give a brown oil.
Chromatography (CHCl3/hexane 15:85 then a gradient of EtOAc/hexane 5:95 to
50:50) gave 2-(phenylseleno)cyclohexanol (2.41) as a red oil (74 mg, 20%, data:
page 136). Further elution gave a fraction which was predominantly 2-
(phenylselanyl)cyclohexyl benzamide (2.5) as a pale brown oil (50 mg, 10%), data:
page 137).

Experimental 7.2
146
(iv) in CH2Cl2 at reflux
A solution of phenylselenenyl chloride (281 mg, 1.47 mmol) in CH2Cl2 (4.5 mL)
followed by benzonitrile (0.75 mL, 7.3 mmol) were added to a solution of cyclohexene
(0.148 mL, 1.46 mmol) in CH2Cl2 (4 mL) at 0°C. The solution was warmed to r.t. and
a solution of TfOH (0.13 mL, 1.5 mmol) in water (0.13 mL, 7.2 mmol) was added and
the mixture was refluxed for 2h. Saturated aqueous NaHCO3 (15 mL) was added
and the aqueous layer extracted with CHCl3 (3 x 15 mL). The combined organic
layers were washed with saturated aqueous NaCl (15 mL) and dried (MgSO4) and
the solvent removed at reduced pressure to give a yellow solid. Chromatography
(CHCl3/hexane 15:85 then a gradient of EtOAc/hexane 5:95 to 60:40) gave 2-
(phenylseleno)cyclohexanol (2.41) as a brown liquid (5 mg, 1%, data: page 136).
Further elution gave 2-(phenylselanyl)cyclohexyl benzamide (2.5) as a white solid
(349 mg, 67%, data: page 137).
(v) in CH2Cl2 at r.t.
Cyclohexene (0.034 mL, 0.34 mmol) was added to a solution of phenylselenenyl
chloride (70 mg, 0.37 mmol) in CH2Cl2 (1 mL). Benzonitrile (0.17 mL, 1.7 mmol) and
TfOH (0.05 mL, 0.57 mmol) in water (0.015 mL, 0.83 mmol) were added and the
mixture was stirred at r.t. for 24 h. After 24 h TLC analysis showed that the hydroxy
selenide had formed but that the amido selenide was still only a minor product and 1
drop TfOH was added. Stirring was continued for a further 3 d at which point TLC
analysis showed the main product to be the amido selenide. Saturated aqueous
NaHCO3 (10 mL) was added and the aqueous layer extracted with CH2Cl2 (2 x 20
mL). The combined organic layers were washed with saturated aqueous NaCl (10
mL) and dried (MgSO4) and the solvent removed at reduced pressure to give a

Experimental 7.2
147
yellow oil which 1H NMR analysis showed to be predominantly 2-
(phenylselanyl)cyclohexyl benzamide (2.5) (113 mg, 90%, data: page 137).
(b) Reaction of cyclopentene
(i) in CH2Cl2 at reflux
A solution of phenylselenenyl chloride (290 mg, 1.51 mmol) in CH2Cl2 (3 mL)
followed by benzonitrile (0.75 mL, 7.3 mmol) were added to a solution of
cyclopentene (0.13 mL, 1.5 mmol) in CH2Cl2 (5.5 mL) at 0°C. The solution was
warmed to r.t. and a solution of TfOH (0.13 mL, 1.5 mmol) in water (0.03 mL, 1.7
mmol) was added and the mixture was refluxed for 2h. Saturated aqueous NaHCO3
(15 mL) was added and the aqueous layer extracted with CHCl3 (3 x 20 mL). The
combined organic layers were washed with saturated aqueous NaCl (15 mL) and
dried (MgSO4), and the solvent removed at reduced pressure to give a yellow solid.
Chromatography (EtOAc/hexane 25:75 to 50:50) gave a fraction (65 mg) containing a
mixture of 2-(phenylselanyl)cyclopentyl benzamide (2.24) and N-cyclopentyl
benzamide (2.26) in a ratio of 2:1. Further elution gave N-cyclopentyl benzamide
(2.26) (90 mg, 32%, data: page 143).
(ii) in benzonitrile at r.t. then 55°C
Cyclopentene (0.09 mL, 1.0 mmol) and water (0.09 mL, 5.0 mmol) were added to a
solution of phenylselenenyl chloride (197 mg, 1.02 mmol) in benzonitrile (3 mL). The
solution was stirred at r.t. for 3 d. TLC analysis showed the formation of the hydroxy
selenide and further TfOH (0.09 mL, 1.02 mmol) was added and the mixture was
stirred a further 3 d. TLC analysis showed that the reaction had not gone to
completion and a further 3 drops TfOH were added and the mixture stirred for 5 h at
a bath temperature of 55°C. TLC analysis showed that the reaction was still not
complete and a further 3 drops TfOH was added and the mixture was stirred 7 h at

Experimental 7.2
148
bath temperature 55°C. The mixture was cooled and saturated aqueous NaHCO3
(10 mL) was added and the aqueous layer extracted with CH2Cl2 (3 x 15 mL). The
combined organic layers were dried (MgSO4) and the solvent removed at reduced
pressure to give a yellow solid. Chromatography (CHCl3/hexane 15:85 then a
gradient of EtOAc/hexane 5:95 to 30:70) gave 2-(phenylselanyl)cyclopentyl
benzamide (2.24) (192 mg, 55%, data: page 138).
Hydroxyselenation
Procedure 7.2C:[169] Phenylselenenyl chloride was added to a solution of the
alkene in acetonitrile followed by water and the solution was stirred at room
temperature for 48 h. Saturated aqueous NaHCO3 (15 mL) was added and the
products were extracted with CH2Cl2 (3 x 20 mL). The combined organic layers
were washed with saturated aqueous NaCl (15 mL) and dried (Na2SO4) and the
solvent removed under reduced pressure. The crude product was purified by
column chromatography (CHCl3/hexane 15:85 to remove diphenyl diselenide
followed by a gradient of EtOAc/hexane 5:95 to 50:50).
(a) trans-2-(Phenylseleno)cyclopentanol (2.42)
Following Procedure 7.2C, a mixture of cyclopentene (0.44 mL, 5.0 mmol),
phenylselenenyl chloride (960 mg, 5.01 mmol) and water (3 mL) in acetonitrile (15
mL) gave the title compound as a pale yellow oil (927 mg, 77 %). max (neat) 3377,
3070, 3057, 2958, 2870, 1579, 1477, 1437, 1336, 1302, 1196, 1120, 1093, 1070,
1034, 978, 843, 737, 692, 669 cm-1. 1H NMR: 7.59-7.55, m, 2H, ArH; 7.30-7.25, m,
3H, ArH; 4.15, dd, J 4.8, 11.3 Hz, 1H, CHO; 3.40, ddd, J 1.3, 5.7, 11.3 Hz, 1H, CHSe;
2.30-2.23, m, 1H; 2.10, br s, 1H, OH; 2.10-2.01, m, 1H; 1.86-1.56, m, 4H; 13C NMR:
134.09, 129.33, 129.02, 127.37, all Ar; 78.95, CHO; 49.70, JCSe 264 Hz, CHSe;

Experimental 7.2
149
32.83, 31.07, 22.00. 77Se NMR: 349.68; MS: m/z 242 (M+), 225 (M+-OH), 158
(C6H5SeH+), 85 (M+-C6H5Se), 77 (C6H5+), 67 (C5H7
+).
(b) trans-2-(Phenylseleno)cyclohexanol (2.41)
Following Procedure 7.2C, a mixture of cyclohexene (0.75 mL, 7.4 mmol),
phenylselenenyl chloride (1.42 g, 7.42 mmol) and water (5 mL) in acetonitrile (22 mL)
gave the title compound as a pale yellow oil (1.68 g, 89%, data: page 136)
(c) trans-2-(Phenylseleno)cycloheptanol (2.43)
Following Procedure 7.2C, a mixture of cycloheptene (0.93 mL, 8.0 mmol),
phenylselenenyl chloride (1.54 g, 8.04 mmol) and water (4.8 mL) in acetonitrile (24
mL) gave the title compound as a pale yellow oil (1.96 g, 91%). max (neat) 3433,
3070, 3055, 2926, 2858, 1577, 1477, 1456, 1437, 1385, 1265, 1211, 1072, 1022,
999, 739, 692, 671 cm-1. 1H NMR: 7.60-7.58, m, 2H, ArH; 7.33-7.25, m, 3H, ArH;
3.58, ddt, J 1.5, 3.6, 9.6 Hz, 1H, CHO; 3.10, dt, J 3.3, 9.6 Hz, 1H, CHSe; 2.82, br s,
1H, OH; 2.25-2.17, m, 1H; 2.04-1.95, m, 1H; 1.73-1.54, m, 5H; 1.52-1.37, m, 3H. 13C
NMR: 135.27, 135.21, 128.94, 127.82, all Ar; 74.97, CHO; 56.08, JCSe 239 Hz,
CHSe; 33.56, 32.51, 27.19, 26.48, 21.67. MS: m/z 270 (M+), 158 (C6H5SeH+), 113
(M+-C6H5Se), 95 (C7H11+), 78 (C6H6
+), 77 (C6H5+), 67 (C5H7
+), 55 (C4H7+).
(d) trans-2-(Phenylseleno)cyclooctanol (2.46)
Following Procedure 7.2C, a mixture of cyclooctene (0.977 mL, 7.52 mmol),
phenylselenenyl chloride (1.44 g, 7.52 mmol) and water (4.5 mL) in acetonitrile (23
mL) gave the title compound as a pale yellow oil (1.46 g, 69%). max(neat) 3454,
3070, 3055, 2924, 2854, 1577, 1475, 1464, 1439, 1387, 1354, 1329, 1302, 1271,
1228, 1113, 1072, 1041, 1022, 999, 968, 739, 692, 669 cm-1. 1H NMR: 7.61-7.58,
m, 2H, ArH; 7.33-7.27, m, 3H, ArH; 3.70, ddd, J 3.0, 5.7, 9.9 Hz, 1H, CHO; 3.32, ddd,
J 2.7, 8.7, 9.9 Hz, 1H, CHSe; 2.90, br s, 1H, OH; 2.30-2.22, m, 1H; 1.93-1.85, m, 2H;

Experimental 7.2
150
1.82-1.40, m, 9H. 13C NMR: 135.39, 135.33, 129.04, 127.91, all Ar; 73.67, CHO;
55.38, JCSe 236 Hz, CHSe; 31.95, 31.70, 26.78, 26.73, 25.32, 23.58. 77Se NMR:
368.22. MS: m/z 284 (M+), 158 (C6H5SeH+), 127 (M+-C6H5Se), 109 (C8H13+).
(e) R,S- and S,R-2-(Phenylseleno)-3-hexanol (2.53) and R,S- and S,R-3-(Phenylseleno)-2-hexanol (2.54) Following Procedure 7.2C, a mixture of trans-2-hexene (0.629 mL, 5.00 mmol),
phenylselenenyl chloride (993 mg, 5.18 mmol) and water (3 mL) in acetonitrile (15
mL) gave a mixture of the title compounds, the Markovnikov and anti-Markovnikov
hydroxy selenides (2.53) and (2.54), in a ratio of 55:45 as a yellow oil (1.214 g).
Chromatography (CH2Cl2/hexane 15:85 to remove diphenyldiselenide then a gradient
of EtOAc/hexane 5:95 to 25:75) gave a fraction containing the Markovnikov and anti-
Markovnikov isomers (2.53) and (2.54) in a ratio of 66:34 as a yellow oil (868 mg,
67%). Further elution gave a fraction containing (2.53) and (2.54) in a ratio of 17:83
as a colourless oil (227 mg, 18%). max (neat mixture of (2.53) and (2.54)) 3438
cm-1, br, OH str. MS (mixture of (2.53) and (2.54)): m/z 258 (M+), 213 (M+-C2H5O,
(2.54)), 186 (M+-C4H8O, (2.53)), 158 (C6H5SeH+), 101 (M+-C6H5Se), 78 (C6H6+), 77
(C6H5+), 55 (C4H7
+) 45 (C2H5O+), 43 (C3H7+). NMR data of compound (2.53): 1H
NMR: 7.60-7.54, m, 2H, ArH; 7.30-7.25, m, 3H, ArH; 3.62, ddd, J 3.0, 3.6, 8.7 Hz,
1H, CHO; 3.44, dq, J 3.0, 7.2 Hz, 1H, CHSe; 2.27, br s, 1H, OH; 1.74-1.59, m, 2H;
1.55-1.26, m, 2H; 1.39, d, J 7.2 Hz, 3H, CH(Se)CH3; 0.88, t, J 6.9 Hz, 3H, CH2CH3.
13C NMR: 134.64, 129.08, 127.66, 127.46, all Ar; 72.45, CHO; 47.40, JCSe 239 Hz,
CHSe; 35.76, 19.29, 14.90, 13.85. NMR data of compound (2.54): 1H NMR: 7.61-
7.55, m, 2H, ArH; 7.29-7.23, m, 3H, ArH; 3.85, dq, J 3.6, 6.3 Hz, 1H, CHO; 3.27, ddd,
J 3.6, 4.5, 9.3 Hz, 1H, CHSe; 2.48, br s, 1H, OH; 1.74-1.59, m, 2H; 1.55-1.26, m, 2H;
1.20, d, J 6.3 Hz, 3H, CH(O)CH3; 0.92, t, J 6.9 Hz, 3H, CH2CH3. 13C NMR: 134.26,

Experimental 7.2
151
129.52, 128.96, 127.31, all Ar; 68.84, CHO; 57.27, JCSe 252 Hz, CHSe; 33.21, 21.45,
19.72, 13.71.
(f) 1-(Phenylseleno)-2-octanol (2.48) and 2-(phenylseleno)-1-octanol (2.49)
Following Procedure 7.2C, a mixture of 1-octene (0.81 mL, 5.2 mmol),
phenylselenenyl chloride (1.00 g, 5.23 mmol) and water (3 mL) in acetonitrile (15 mL)
gave a mixture of the title compounds, the Markovnikov and anti-Markovnikov
hydroxy selenides (2.48) and (2.49), in a ratio of 85:15 as a yellow oil.
Chromatography (EtOAc/hexane 15:85) gave 1-(phenylseleno)-2-octanol (2.48) as a
yellow oil (291 mg, 51%). ESI HRMS: 269.07989 C14H22OSe-OH requires
269.08084. max (neat) 3402, 2954, 2927, 2856, 1579, 1477, 1466, 1437, 1072,
1022, 737, 690 cm-1. 1H NMR: 7.55-7.51, m, 2H, ArH; 7.28-7.24, m, 3H, ArH;
3.69-3.62, m, 1H, CHO; 3.15, dd, J 3.6, 12.6 Hz, 1H, CHaHbSe; 2.89, dd, J 8.7, 12.6
Hz, 1H, CHaHbSe; 2.43, br s, 1H, OH; 1.56-1.26, m, 10H; 0.87, t, J 6.8 Hz, 3H, CH3.
13C NMR: 132.91, 129.37, 129.12, 127.16, all Ar; 69.81, CHO; 37.16, CHSe; 36.53,
31.67, 29.17, 25.70, 22.51, 14.01. MS: m/z 286 (M+), 201 (M+-C6H13), 183 (M+-
C6H13-H2O), 172 (C6H5SeCH3+), 157 (C6H5Se+), 129 (M+-C6H5Se), 77 (C6H5
+), 69
(C5H9+), 55 (C4H7
+). Further elution gave a fraction containing both regioisomers,
(2.48) and (2.49), as a yellow oil (750 mg, 20%) followed by 2-(phenylseleno)-1-
octanol (2.49) as a yellow oil (6 mg, 0.4%). ESI HRMS: 269.07993 C14H22OSe-OH
requires 269.08084. 1H NMR: 7.51-7.45, m, 2H, ArH; 7.25-7.17, m, 3H, ArH; 3.56,
dd, J 5.1, 11.4 Hz, 1H, CHaHbO; 3.45, dd, J 6.6, 11.4, 1H, CHaHbO; 3.16, m, 1H,
CHSe; 1.90, br s, 1H, OH; 1.61-1.19, m, 10H; 0.81, t, J 6.9 Hz, 3H, CH3. 13C NMR:
135.43, 133.17, 129.08, 127.94, all Ar; 64.22, CHO; 50.63, CHSe; 31.66, 31.58,
28.99, 27.76, 22.58, 14.05. MS: m/z 286 (M+), 255 (M-CH2OH), 201 (M+-C6H13), 183
(M+-C6H13-H2O), 156 (C6H4Se+), 129 (M+-C6H5Se), 111 (C8H15+).

Experimental 7.2
152
Amidoselenation of -hydroxy selenides
Procedure 7.2D:[165] To a solution of the hydroxy selenide in CH2Cl2 was added the
nitrile followed by a solution of TfOH in water. The mixture was stirred for
approximately 48 h as specified, treated with saturated aqueous NaHCO3 (15 mL),
and the products were extracted with CH2Cl2 (3 x 20 mL). The combined organic
layers were washed with saturated aqueous NaCl (15 mL), dried (Na2SO4), and the
solvent removed under reduced pressure. Chromatography (CHCl3/hexane 15:85 to
remove diphenyl diselenide followed by a gradient of EtOAc/hexane 5:95 to 50:50)
gave the 2-amidoalkyl phenyl selenide.
(a) trans-2-(phenylselanyl)cyclopentyl benzamide (2.24)
Following Procedure 7.2D, but with a reaction time of 41 h, the reaction of trans-2-
(phenylseleno)cyclopentanol (2.42) (667 mg, 2.77 mmol), benzonitrile (2.0 mL, 20
mmol), TfOH (0.26 mL, 2.9 mmol) and water (0.2 mL, 10 mmol) in CH2Cl2 (2.6 mL)
gave the title compound as a white solid (726 mg, 76%, data: page 138).
(b) trans-2-(Phenylselanyl)cyclohexyl benzamide (2.5)
Following Procedure 7.2D, but with a reaction time of 22 h, the reaction of trans-2-
(phenylseleno)cyclohexanol (2.41) (4.12 g, 16.2 mmol), benzonitrile (13 mL, 130
mmol), TfOH (1.43 mL, 16.2 mmol) and water (0.29 mL, 16 mmol) in CH2Cl2 (11 mL)
gave the title compound as a white solid (5.382 g, 93%, data: page 137).
(c) trans-N-2-(Phenylselanyl)cycloheptyl benzamide (2.27)
Following Procedure 7.2D the reaction of trans-2-(phenylseleno)cycloheptanol (2.43)
(1.91 g, 7.09 mmol), benzonitrile (7.0 mL, 75 mmol), TfOH (0.63 mL, 7.1 mmol) and
water (0.13 mL, 7.2 mmol) in CH2Cl2 (5 mL) gave the title compound as a white solid
which, after chromatography, crystallised from the eluting solvent as colourless
crystals (1.498 g, 57%), m.p. 148-150.5°C. EI HRMS: 373.0947 C20H23NOSe

Experimental 7.2
153
requires 373.0947. max (Nujol) 3311, 1631, 1577, 1529, 1323, 1186, 737, 694 cm-1.
1H NMR: 7.71-7.68, m, 2H, ArH; 7.62-7.58, m, 3H, ArH; 7.56-7.38, m, 2H, ArH;
7.30-7.23, m, 3H, ArH; 6.29, br d, J 7.5 Hz, 1H, NH; 4.22, ddd, J 3.3, 7.5, 9.6 Hz, 1H,
CHN; 3.39, ddd, J 3.3, 8.4, 9.6 Hz, 1H, CHSe; 2.19-2.06, m, 2H; 1.94-1.46, m, 8H.
13C NMR: 166.45, C=O; 134.88, 134.73, 131.27, 129.30, 129.11, 128.46, 127.58,
126.86, all Ar; 56.05, CHN; 50.50, CHSe; 33.64, 33.08, 27.79, 26.35, 23.66. MS: m/z
371 (M+-H2), 251 (M-H-C6H5CONH2+), 216 (M-C6H5Se+), 158 (C6H5SeH+), 122
(C6H5CONH3+), 105 (C6H5CO+), 77 (C6H5
+)
(d) trans-N-2-(phenylselanyl)cyclooctyl benzamide (2.47)
To a solution of trans-2-(phenylseleno)cyclooctanol (2.46) (906 mg, 3.20 mmol) in
benzonitrile (4 mL, 40 mmol), cooled to 0°C, was added dropwise a solution of TfOH
(0.32 mL, 3.6 mmol) in water (0.064 mL, 3.6 mmol). The mixture was allowed to
warm to r.t. and stirred for 49 h followed by work-up according to Procedure 7.2D.
Evaporation of the CH2Cl2 under reduced pressure gave a yellow benzonitrile
solution from which crystallised a white solid which was collected, washed with cold
Et2O and recrystallised from acetonitrile to give the title compound as white needles
(293 mg, 24%), m.p. 133-136°C. The remaining benzonitrile was distilled from the
mother liquor at low pressure and the residue chromatographed (a gradient of
EtOAc/hexane 12:88 to 50:50) to give the title compound which crystallised from the
eluting solvent as white needles (258 mg, 21%). EI HRMS: 387.1099 C21H25NOSe
requires 387.1102. max (Nujol) 3327, 1631, 1577, 1531, 1323, 1232, 1161, 1078,
741, 719, 694, 663, 606 cm-1. 1H NMR: 7.67-7.63, m, 2H, ArH; 7.55-7.36, m, 5H,
ArH; 7.26-7.21, m, 3H, ArH; 6.26, d, J 7.2 Hz, 1H, NH; 4.33, ddt, J 3.0, 7.2, 10.8 Hz,
1H, CHN; 3.53, ddd, J 2.7, 7.2, 10.8 Hz, 1H, CHSe; 2.27- 2.22, m, 1H; 2.02-1.50, m,
13H. 13C NMR: 166.48, C=O; 134.78, 134.73, 131.22, 129.59, 129.15, 128.42,

Experimental 7.2
154
127.57, 126.86, all Ar; 55.00, CHN; 50.01, CHSe; 32.56, 31.17, 26.51, 26.22, 25.73,
25.21. MS: m/z 387 (M+), 266 (M+-C6H5CONH2), 230 (M+-C6H5Se), 157 (C6H5Se+),
122 (C6H5CONH3+), 109 (C8H13
+), 105 (C6H5CO+), 77 (C6H5+).
(e) trans-N-[2-(phenyselanyl)cyclohexyl]acetamide[145] (2.44)
To a solution of 2-(phenylseleno)cycloheptanol (2.43) (915 mg, 3.40 mmol) in CH3CN
(20 mL) was added a solution of TfOH (0.3 mL, 3 mmol) in water (0.06 mL, 3 mmol)
and the solution was stirred at r.t. for 50 h. The mixture was diluted with CH2Cl2 (30
mL) and worked up according to Procedure 7.2D to give a yellow solid (1.16 g).
Chromatography (gradient of EtOAc/hexane 25:75 to 100:0) gave the title compound
as a pale yellow solid (902 mg, 85%) which was recrystallised from ethyl acetate to
afford white crystals, m.p. 113.5-115°C (lit.[298] m.p. 107-108°C). EI HRMS:
311.0787 C15H21NOSe requires 311.0789. max (Nujol) 3298, 3070, 1635, 1539,
1315, 1184, 953, 742, 694, 604 cm-1. 1H NMR: 7.58-7.55, m, 2H, ArH; 7.30-7.26,
m, 3H, ArH; 5.68, d, J 7.2 Hz, 1H, NH; 4.06, ddt, J 3.6, 7.8, 9.3 Hz, 1H, CHN; 3.24,
ddd, J 3.3, 8.7, 9.3 Hz, 1H, CHSe; 2.12-2.04, m, 1H; 1.90, s, 3H, CH3; 1.97-1.43, m,
10H. 13C NMR: 169.30, C=O; 134.92, 129.85, 129.35, 127.82, all Ar; 55.87, CHN;
50.67, CHSe; 33.95, 32.94, 28.20, 26.70, 23.83, 23.70. MS: m/z 311 (M+), 252 (M+-
NHCOCH3-H), 157 (C6H5Se+), 154 (M+-C6H5Se), 112 (C7H14N+), 95 (C7H11+), 77
(C6H5+).
(f) trans-N-2-(phenylselanyl)cyclohexyl p-bromobenzamide (2.45)
Following Procedure 7.2D the reaction of trans-2-(phenylseleno)cyclohexanol (2.41)
(58 mg, 0.23 mmol), 4-bromobenzonitrile (123 mg, 0.676 mmol), TfOH (0.02 mL, 0.2
mmol) and water (0.01 mL, 0.6 mmol) ) in CH2Cl2 (1 mL) and chromatography
(CHCl3/hexane 15:85 then a gradient of EtOAc/hexane 5:95 to 100:0) gave 4-
bromobenzonitrile as a white solid (67 mg, 54% recovery) followed by the title

Experimental 7.2
155
compound as a white solid which, after chromatography, crystallised from the eluting
solvent as colourless crystals (86 mg, 87%), m.p. 154-157.5°C. max (Nujol) 3338,
1633, 1591, 1537, 1327, 1186, 1012, 839, 757, 742, 692 cm-1. 1H NMR: 7.55-7.47,
m, 4H, ArH; 7.29-7.21, m, 5H, ArH; 6.06, br d, J 7.2 Hz, 1H, NH; 3.95, dddd, J 3.6,
7.2, 10.5, 11.1 Hz, 1H, CHN; 3.14, dt, J 3.9, 11.1 Hz, CHSe; 2.36-2.32, m, 1H; 2.26-
2.22, m, 1H; 1.76-1.56, m, 3H; 1.49-1.19, m, 3H. 13C NMR: 165.70, C=O; 135.25,
135.52, 131.65, 129.17, 128.53, 128.07, 127.83, 125.95, all Ar; 54.27, 47.94, 34.08,
33.96, 26.81, 24.61. MS: m/z 438 (M+-H, 81Br), 436 (M+-H, 79Br), 282 (M+-C6H5Se,
81Br), 280 (M+-C6H5Se, 79Br), 238 (M+-BrC6H4CONH2), 202 (BrC6H4CONH3+, 81Br),
200 (BrC6H4CONH3+, 79Br), 185 (BrC6H4CO+, 81Br), 183 (BrC6H4CO+, 79Br), 157
(C6H5Se+), 104 (C6H4CO+), 81 (C6H9+).
(g) S,R- and R,S-2-(phenylselanyl)-3-hexyl benzamide (2.34) and S,R- and R,S-3-
(phenylselanyl)-2-hexyl benzamide (2.35)
To a solution of a 17:83 mixture of 2-(phenylseleno)-3-hexanol (2.53) and 3-
(phenylseleno)-2-hexanol (2.54) (226 mg, 0.879 mmol) in benzonitrile (3 mL) was
added a solution of TfOH (0.08 mL, 0.9 mmol) in water (0.02 mL, 1 mmol) and the
mixture was stirred at r.t. for 48 h. The mixture was diluted with CH2Cl2 and worked
up according to Procedure 7.2D to give a yellow oil (662 mg). Chromatography
(CH2Cl2/hexane 15:85 then a gradient of EtOAc/hexane 5:95 to 30:70) gave a 53:47
mixture of the title compounds as a pale yellow solid (306 mg, 97%). Further
chromatography (gradient of Et2O/hexane 10:90 to 50:50) gave fractions enriched in
one or other isomer. Solvent was evaporated slowly from the first amido selenide-
containing fraction, giving 2-benzamido-3-(phenylseleno)hexane (2.35) as colourless
plates, m.p. 76-78.5°C. ESI HRMS: 362.10136 C19H23NOSe+H requires 362.10176.
max: 3321, 2938, 1633, 1578, 1523, 1489, 1447, 1296, 1076, 1022, 800, 736, 719,

Experimental 7.2
156
690 cm-1. MS: m/z 361 (M+), 240 (M+-C6H5CONH2), 204 (M-C6H5Se+). The
structure of (2.35) was confirmed by an X-ray structure determination.[170]
(h) 1-(phenylselanyl)-2-octyl benzamide (2.32) and 2-(phenylselanyl)-1-octyl benzamide (2.33) To a solution of a mixture of 1-(phenylseleno)-2-octanol (2.48) and 2-(phenylseleno)-
1-octanol (2.49) (801 mg, 2.80 mmol) in benzonitrile (3 mL) was added a solution of
TfOH (0.25 mL, 2.8 mmol) in water (0.05 mL, 3 mmol) and the solution was stirred at
r.t. for 48 h. The mixture was diluted with CH2Cl2 and worked up according to
Procedure 7.2D to give a yellow oil (1.377 g). Chromatography (CH2Cl2/hexane
15:85 then a gradient of EtOAc/hexane 5:95 to 30:70) gave a mixture of the title
compounds (2.32) and (2.33) in a ratio of 9:1 as pale pink solid (1.119 g, 103%).
Recrystallisation from dichloromethane/hexane afforded the title compound (2.32) as
a pale yellow solid (892 mg, 82%, data: page 140).
(i) N-[1-(phenylselanyl)-2-octyl] p-bromobenzamide (2.50) and N-[2-(phenylselanyl)-1-octyl] p-bromobenzamide (2.51) Following Procedure 7.2D the reaction of a mixture of 1-(phenylseleno)-2-octanol
(2.48) and 2-(phenylseleno)-1-octanol (2.49) (360 mg, 1.26 mmol), p-
bromobenzonitrile (734 mg, 4.03 mmol), TfOH (0.120 mL, 1.36 mmol) and water
(0.03 mL, 2 mmol) in dichloromethane (10 mL) and chromatography (gradient of
CH2Cl2/hexane 50:50 to 100:0) gave p-bromobenzonitrile as a colourless solid (589
mg, 80% recovery). Further elution gave a mixture of the title compounds (2.50) and
(2.51) and the oxazoline (2.52) (data: page 176) in a ratio of 2.5:0.1:0.25 as a yellow
solid (125 mg, 21%) followed by a fraction containing a mixture of the title
compounds (2.50) and (2.51) in a ratio of 2.5:1 as a yellow solid (178 mg, 30%)
which was recrystallised from EtOAc to give the title compound, N-[1-(phenylselanyl)-
2-octyl] p-bromobenzamide (2.50), as colourless needles, m.p. 103–105.5°C. ESI

Experimental 7.2
157
HRMS: 468.04291 C21H26NOSeBr+H requires 468.04358. max (Nujol) 3346, 2953,
1630, 1591, 1525, 1414, 1296, 1070, 1012, 843, 758, 735, 692 cm-1. 1H NMR:
7.55-7.47, m, 4H, ArH; 7.40-7.37, m, 2H, ArH; 7.23-7.19, m, 3H, ArH; 6.06, d, J 8.4
Hz, 1H, NH; 4.44-4.33, m, 1H, CHN; 3.29, dd, J 4.5, 13.2 Hz, CHaHbSe; 3.20, dd, J,
5.1, 13.2 Hz, CHaHbSe; 1.70-1.60, m, 4H; 1.30-1.24, m, 6H; 0.86, t, J 6.6 Hz, 3H,
CH3. 13C NMR: 165.86, C=O; 133.27, 132.62, 131.63, 129.96, 129.34, 128.42,
127.16, 125.99, all Ar; 49.68, 34.45, 33.66, 31.65, 29.03, 26.01, 22.54, 14.04. MS:
m/z 469 (M+, 81Br), 467 (M+, 79Br), 312 (M+-C6H5Se, 81Br), 310 (M+-C6H5Se, 79Br), 268
(M+-BrC6H4CONH2), 185 (BrC6H4CO+, 81Br), 183 (BrC6H4CO+, 79Br), 157 (C6H5Se+),
104 (C6H4CO+), 91 (C7H7+). N-[2-(phenylselanyl)-1-octyl] p-bromobenzamide (2.51)
was identified from the following 1H NMR signals in a spectrum of the mixture of
(2.50) and (2.51): 6.55, br s, 1H, NH; 3.83, ddd, J 3.6, 6.6, 13.5 Hz, 1H, CHaHbNH;
3.44, ddd, J 4.8, 8.4, 13.5 Hz, CHaHbNH; 3.41-3.35, m, 1H, CHSe.
Preparation of trans-3a,4,5,6,7,7a-hexahydro-2-phenylbenzoxazole (2.9)
Ethyl iminobenzoate hydrochloride (2.57)[172, 299]
Benzonitrile (7.5 ml, 73 mmol) and absolute ethanol (5.0 ml, 86 mmol) were placed in
a quick-fit test tube and the solution was cooled to 0°C. Hydrogen chloride was
bubbled through the solution for about 1.5 h. The reaction mixture was securely
stoppered and kept at 5°C. After 4 d the mixture was almost completely crystalline.
After a further 15 d the crystals were collected, washed once with dry ether, and the
residual solvent evaporated under reduced pressure over KOH to give the title
compound[299] (12.07 g, 89%) which was stored over KOH and not purified further.
1H NMR: 8.41-8.37, m, 2H, ArH; 7.75-7.68, m, 1H, ArH; 7.61-7.54, m, 2H, ArH;
4.94, q, J 6.6 Hz, 2H, CH2; 1.63, t, J 6.6 Hz, CH3.

Experimental 7.2
158
Ethyl iminobenzoate (2.55)[173]
Ethyl iminobenzoate hydrochloride (2.57, 10.011 g, 53.9 mmol) was added in
portions to a stirred mixture of aqueous KOH (42 ml, 2M, 84 mmol) and CH2Cl2 (105
ml) cooled externally with ice. The layers were separated and the organic layer was
washed with water (2 x 30 ml) and dried (MgSO4) and the solvent was evaporated
under reduced pressure. Kugelrohr distillation (b.p. 40°C/0.1mm, lit.[299] b.p. 56-
60°C/0.6mm) gave the title compound[299] (6.986 g, 87%) as a colourless oil. max
(neat): 3332, 3299, 3061, 2983, 2939, 2900, 1635, 1579, 1478, 1449, 1399, 1373,
1331, 1298, 1169, 1078, 1029, 1020, 999, 873, 828, 783, 696, 677 cm-1. 1H NMR:
7.76-7.74, m, 2H, ArH; 7.47-7.38, m, 3H, ArH; 4.33, q, J 7.2 Hz, 2H, CH2; 1.43, t, J
7.2 Hz, 3H, CH3. 13C NMR: 167.76, C=N; 132.97, 130.70, 128.35, 126.61, all Ar;
61.72, CH2; 14.15, CH3. MS: m/z 149 (M+), 122 (C6H5CONH3+), 121 (C6H5CONH2
+),
105 (C6H5CO+), 77 (C6H5+).
trans-3a,4,5,6,7,7a-Hexahydro-2-phenylbenzoxazole (2.9)[150]
d,l-trans-2-Aminocyclohexanol (683 mg, 4.50 mmol) was added to a solution of ethyl
iminobenzoate (2.55, 838 mg, 5.62 mmol) in dry ethylene dichloride (45 ml) and the
mixture was refluxed for 24 h. After cooling, the mixture was filtered to remove
suspended salt and the filtrate was concentrated under reduced pressure. Kugelrohr
distillation (130°C/15 mm) recovered ethyl iminobenzoate as a colourless liquid,
leaving a pale brown solid containing the oxazoline, imine and benzamide in a ratio of
35:40:25 together with small amounts of unidentified products. Chromatography
(Et2O/hexane 70:30) gave the oxazoline contaminated with imine as a white solid
(319mg, 35%). Further evaporation of imine under reduced pressure gave the trans-
oxazoline (2.9) as a white solid, m.p. 64-67°C. Recrystallisation from MeOH/Et2O

Experimental 7.2
159
gave the title compound as white crystals, m.p. 78-79.5°C [lit.[150] m.p. 73-77°C].
max (KBr) 2934, 2859, 1622, 1600, 1575, 1492, 1448, 1358, 1334, 1318, 1291, 1255,
1230, 1145, 1103, 1086, 1067, 1048, 1013, 927, 890, 870, 779, 697, 553 cm-1. 1H
NMR: 8.00-7.97, m, 2H, ArH; 7.51-7.38, m, 3H, ArH; 3.76, ddd, J 3.6, 11.7, 13.8
Hz, 1H, CHO; 3.25, ddd, J 3.3, 11.7, 13.8 Hz, 1H, CHN, 2.45-2.38, m, 2H; 1.97-1.84,
m, 2H; 1.78, ddd, J 4.2, 12.0, 24.0 Hz, 1H; 1.54, ddd, J 3.3, 12.0, 23.7 Hz, 1H; 1.43-
1.34, m, 2H. 13C NMR: 165.96, C=N; 131.36, 131.11, 128.29, 128.02, all Ar;
87.02, CHO; 71.49, CHN; 30.50, 29.65, 25.06, 24.34. MS: m/z 201 (M+), 172 (M+-
CHO), 158 (M+-CH2CO), 130 (M+-C3H7CO), 117 (M+-C5H8O), 105 (C6H5CO+), 104
(C6H5CNH+), 77 (C6H5+).

Experimental 7.3
160
7.3 WORK DESCRIBED IN CHAPTER 3
Oxidation of 2-amidoalkyl phenyl selenide with KOH as base
Procedure 7.3A: To a stirred solution of the selenide in i-PrOH was added powdered
KOH followed by m-CPBA and the suspension was stirred at r.t. for 1-2 h. Aqueous
Na2S2O3 (0.5 M, 15 mL) and saturated aqueous NaHCO3 (10 mL) were added and
the products were extracted with CHCl3 (2 x 25 mL). The combined organic extracts
were dried (MgSO4) and the solvent evaporated at reduced pressure.
(a) Reaction of trans-2-(phenylselanyl)cyclohexyl benzamide (2.5)
(i) with 4 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.5, 100 mg, 0.280
mmol) with KOH (63 mg, 1.1 mmol) and m-CPBA (194 mg, 0.899 mmol) in i-PrOH
(20 mL) followed by chromatography (EtOAc/hexane 25:75 to 80:20) gave the
aziridine[154] (2.17, trace, data: page 135). Further elution gave the amido selenide
(2.5) as a colourless oil (13 mg, 13%). Further elution gave the cis-oxazoline[149]
(2.7, data: page 137) as a pale yellow gum (28 mg, 50%).
(ii) with 10.8 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.5, 60 mg, 0.17
mmol) with KOH (102 mg, 1.81 mmol) and m-CPBA (147 mg, 0.681 mmol) in i-PrOH
(11 ml) gave a semi-solid (31 mg, 93%), being a mixture of the aziridine[154] (2.17,
data: page 135) and the cis-oxazoline[149] (2.7, data: page 137) in a ratio of 95:5.
(iii) with 13.5 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.5, 60 mg, 0.17
mmol) with potassium hydroxide (129 mg, 2.30 mmol) and m-CPBA (144 mg, 0.668
mmol) in i-PrOH (11 mL) gave a mixture (23 mg, 68%) of the aziridine[154] (2.17,
data: page 135) and the cis-oxazoline[149] (2.7, data: page 137) in a ratio of 95:5.

Experimental 7.3
161
(b) Reaction of trans-2-(phenylselanyl)cyclopentyl benzamide (2.24)
(i) with 4 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.24, 81 mg, 0.23
mmol) with KOH (52 mg, 0.93 mmol) and m-CPBA (203 mg, 0.941 mmol) in i-PrOH
(15 mL) followed by chromatography (EtOAc/hexane 25:75 to 50:50) gave the
aziridine[154] (3.1,1 mg, 2%, data: page 166) as a brown oil. Further elution gave the
amido selenide (2.24, 10 mg, 13%, data: page 138) as a white solid, then the cis-
oxazoline (2.25, data: page 139) as a brown oil (23 mg, 52%).
(ii) with 7.5 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.24, 60 mg, 0.18
mmol) with KOH (74 mg, 1.3 mmol) and m-CPBA (151 mg, 0.700 mmol) in i-PrOH
(11 mL) gave a mixture of the aziridine[154] (3.1, data: page 166) and the cis-
oxazoline (2.25) in a ratio of 25:75. Chromatography (EtOAc/hexane 45:55) gave
the aziridine[154] (3.1, 4 mg, 12%, data: page 166). Further elution gave the cis-
oxazoline (2.25, 10 mg, 31%, data: page 139).
(iii) with 10.5 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.24, 60 mg, 0.18
mmol) with KOH (106 mg, 1.88 mmol) and m-CPBA (151 mg, 0.700 mmol) in i-PrOH
(11 ml) gave a mixture (28 mg, 87%) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 60:40.
(iv) with 13.4 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.24, 60 mg, 0.18
mmol) with KOH (136 mg, 2.42 mmol) and m-CPBA (151 mg, 0.700 mmol) in i-PrOH
(11 mL) gave a mixture (31 mg, 96%) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 45:55.

Experimental 7.3
162
(v) in ethanol with 11 equivalents of potassium hydroxide
Following Procedure 7.3A but with EtOH rather than i-PrOH as solvent, the reaction
of the amido selenide (2.24, 60 mg, 0.17 mmol) with KOH (103 mg, 1.84 mmol) and
m-CPBA (212 mg, 0.983 mmol) in EtOH (9.5 mL) gave a mixture (31 mg, 96%) of the
aziridine[154] (3.1, data: page 166) and the cis-oxazoline (2.25, data: page 139) in a
ratio of 30:70.
(c) Reaction of 1-(phenylselanyl)-2-octyl benzamide (2.32)
(i) with 0 equivalents of potassium hydroxide
Following Procedure 7.3A but with no KOH, the reaction of the amido selenide (2.32,
101 mg, 0.260 mmol) and m-CPBA (102 mg, 0.473 mmol) in i-PrOH (10 mL) followed
by chromatography (EtOAc/hexane 15:85 to 30:70) gave 4-(n-hexyl)-2-phenyl-4,5-
dihydro-oxazole (3.12) as a pale yellow oil (27 mg, 45%). ESI HRMS: 232.16936
C15H21NO+H requires 232.16959. max 2955, 2926, 2856, 1650, 1450, 1356, 1270,
1080, 1061, 1025, 970, 779 cm-1. 1H NMR: 7.96-7.93, m, 2H, ArH; 7.47-7.37, m,
3H, ArH; 4.48, dd, J 9.3, 8.1 Hz, 1H, CHHO; 4.27, m, 1H, CHN; 4.03, dd, J 8.1, 7.8
Hz, CHHO; 1.79-1.71, m, 1H; 1.57-1.29, m, 8H; 0.89, t, J 6.9 Hz, 3H, CH3. 13C
NMR: 163.29, C=N; 131.07, 128.27, 128.18, 127.97, all Ar; 72.52, CHO; 66.80,
CHN; 35.94, 31.69, 29.24, 25.82, 22.53, 13.97. MS: m/z 232 (M++H), 202 (M+-
C2H5), 188 (M+-C3H7), 174 (M+-C4H9), 161 (M+-C5H10), 146 (M+-C6H13), 122
(C6H5CONH2+), 105 (C6H5CO+), 91 (C7H7
+), 77 (C6H5+). Further elution gave the
selenide (2.32, trace, data: page 140)).
(ii) with 7.8 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.32, 120 mg, 0.310
mmol) with KOH (136 mg, 2.42 mmol) and m-CPBA (265 mg, 1.23 mmol) in i-PrOH

Experimental 7.3
163
(12 mL) gave a yellow oil whose 1H NMR spectrum showed to be a mixture with the
oxazoline (3.12, data: page 162) as the predominant product.
(d) Reaction of trans-2-(phenylselanyl)cyclohexyl acetamide (2.31)
Following Procedure 7.3A, the reaction of the acetamide (2.31, 100 mg, 0.338 mmol)
with KOH (38 mg, 0.67 mmol) and m-CPBA (234 mg, 1.08 mmol) in i-PrOH (10 mL)
followed by chromatography (EtOAc/hexane 60:40) gave 6-acetamidehexano-6-
lactone (3.5, 10 mg, 17%) as a pale yellow oil. 1H NMR: 5.33, dd, J 3.6, 6.3 Hz,
1H, CH(O)N; 2.65, m, 1H, CHaHbC(O)O; 2.55, m, 1H, CHaHbC(O)O; 2.09, s, CH3;
2.08-1.55, m, 6H. Further elution gave N-(1-isopropoxy-1-cyclopentyl)acetamide
(3.6) as a pale yellow oil (6 mg, 9%). 1H NMR: 5.71, d, J 8.4 Hz, 1H, NH; 5.06, dd,
J 8.4, 9.3 Hz, 1H, CH(O)N; 3.78, sept, J 6.3 Hz, 1H, CH(CH3)2; 2.01, s, 3H,
C(O)CH3; 2.07-1.96, m, 1H, CHCH(O)N; 1.81-1.67, m, 2H; 1.63-1.50, m, 4H; 1.48-
1.25, m, 2H; 1.15, d, J 6.3 Hz, 3H, CH(CH3)CH3; 1.12, d, J 6.3 Hz, 3H, CH(CH3)CH3.
(e) Reaction of 2-(phenylselanyl)cyclohexyl p-bromobenzamide (2.45)
Following Procedure 7.3A but stirring for 16 h at r.t., the reaction of the selenide
(2.45, 80 mg, 0.18 mmol) with KOH (87 mg, 1.6 mmol) and m-CPBA (159 mg, 0.737
mmol) in i-PrOH (10 mL) gave a yellow solid (64 mg) as a mixture of the cis-
oxazoline (3.4, data: page 170) and the aziridine (3.3, data: page 173) in a ratio of
60:40.
(f) Reaction of trans-N-2-(phenylselanyl)cycloheptyl benzamide (2.27)
(i) with 6 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.27, 149 mg, 0.400
mmol) with KOH (136 mg, 2.42 mmol) and m-CPBA (278 mg, 1.29 mmol) in i-PrOH
(14 mL) gave a yellow solid (98 mg) which 1H NMR analysis showed to be a mixture

Experimental 7.3
164
of the aziridine (3.2, data: page 168), the cis-oxazoline (2.28, data: page 140) and the
syn-elimination product (2.30, data: page 180) in a ratio of 10:50:40.
(ii) with 8 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.27) (100 mg, 0.269
mmol) with KOH (121 mg, 2.16 mmol) and m-CPBA (187 mg, 0.867 mmol) in i-PrOH
(15 mL) gave a yellow solid (92 mg) which 1H NMR analysis showed to be a mixture
of the aziridine (3.2, data: page 168) and the cis-oxazoline (2.28, data: page 140) in a
ratio of 15:85 along with a trace of the syn-elimination product (2.30, data: page 180).
(iii) with 10 equivalents of potassium hydroxide
Following Procedure 7.3A, the reaction of the amido selenide (2.27) (150 mg, 0.403
mmol) with KOH (224 mg, 3.99 mmol) and m-CPBA (278 mg, 1.29 mmol) in i-PrOH
(14 mL) gave a pale brown oil (66 mg, 76%) which 1H NMR analysis showed to be a
mixture of the aziridine (3.2, data: page 168) and the cis-oxazoline (2.28, data: page
140) in a ratio of 25:75.
(iv) with 8 equivalents of potassium hydroxide at 0°C
Following Procedure 7.3A but carrying out the reaction at 0°C, the reaction of the
amido selenide (2.27) (150 mg, 0.403 mmol) with KOH (181 mg, 3.22 mmol) and m-
CPBA (280 mg, 1.30 mmol) in i-PrOH (15 mL) gave a mixture of the aziridine (3.2,
data: page 168) and the cis-oxazoline (2.28, data: page 140) in a ratio of 15:85.
(v) with 9 equivalents of potassium hydroxide at 37°C
Following Procedure 7.3A but carrying out the reaction at 37°C, the reaction of the
amido selenide (2.27) (52 mg, 0.14 mmol) with KOH (71 mg, 1.3 mmol) and m-CPBA
(121 mg, 0.561 mmol) in i-PrOH (7.5 mL) gave a pale yellow oil (31 mg) which 1H
NMR analysis showed to be a mixture of the aziridine (3.2, data: page 168), the cis-

Experimental 7.3
165
oxazoline (2.28, data: page 140) and the syn-elimination product (2.30, data: page
180) in a ratio of 10:80:10.
Oxidation of 2-amidoalkyl phenyl selenide with NaH as base
Procedure 7.3B: NaH (60% suspension in oil) was added with stirring to dry i-PrOH
under a N2 atmosphere. To the resulting i-PrONa/i-PrOH mixture was added the
selenide and stirring was continued until the selenide had dissolved. A solution of
m-CPBA in i-PrOH was added and the resulting mixture was stirred a further 1.5-2h.
Aqueous Na2S2O3 (0.5 M, 15 mL) and saturated aqueous NaHCO3 (10 mL) were
added and the products were extracted with CHCl3 (2 x 25 mL). The combined
organic extracts were washed with saturated aqueous NaCl, dried (MgSO4) and the
solvent evaporated at reduced pressure.
(a) Reaction of trans-2-(phenylselanyl)cyclopentyl benzamide (2.24)
(i) with 2 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.24, 74 mg, 0.22
mmol) with NaH (18 mg, 60%, 0.45 mmol) and m-CPBA (186 mg, 0.862 mmol) in i-
PrOH (12 mL) gave a mixture (54 mg) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 2:98.
(ii) with 4 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.24, 74 mg, 0.22
mmol) with NaH (35 mg, 60%, 0.88 mmol) and m-CPBA (185 mg, 0.858 mmol) in i-
PrOH (12 mL) gave a mixture (69 mg) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 25:75.

Experimental 7.3
166
(iii) with 6 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.24, 71 mg, 0.21
mmol) with NaH (49 mg, 60%, 1.2 mmol) and m-CPBA (179 mg, 0.830 mmol) in i-
PrOH (12.5 mL) gave a mixture (63 mg) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 40:60.
(iv) with 8 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.24, 71 mg, 0.21
mmol) with NaH (65 mg, 60%, 1.6 mmol) and m-CPBA (176 mg, 0.816 mmol) in i-
PrOH (12.5 mL) gave a mixture (66 mg) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 55:45.
(v) with 10 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.24, 75 mg, 0.22
mmol) with NaH (88 mg, 60%, 2.2 mmol) and m-CPBA (186 mg, 0.862 mmol) in i-
PrOH (12.5 mL) gave a mixture (70 mg) of the aziridine[154] (3.1, data: page 166) and
the cis-oxazoline (2.25, data: page 139) in a ratio of 50:50.
(vi) with 8 equivalents of sodium hydride at 0°C
Following Procedure 7.3B but with the flask placed in ice, the reaction of the amido
selenide (2.24, 75 mg, 0.22 mmol) with NaH (70 mg, 60%, 1.7 mmol) and m-CPBA
(183 mg, 0.848 mmol) in i-PrOH (12 mL) gave a mixture (66 mg) of the aziridine[154]
(3.1) and the cis-oxazoline (2.25) in a ratio of 45:55 as estimated from integrations of
1H NMR signals. Chromatography (EtOAc/hexane 25:75) gave 6-benzoyl-6-
azabicyclo[3.1.0]hexane[154] (3.1) as a colourless oil (10 mg, 24%). max (KBr) 3035,
2966, 2956, 2924, 2850, 1664, 1643, 1595, 1577, 1450, 1433, 1414, 1390, 1348,
1319, 1288, 1221, 1107, 1076, 1012, 941, 808, 733, 694 cm-1. 1H NMR: 7.99-
7.95, m, 2H, ArH; 7.55-7.50, m, 1H, ArH; 7.46-7.40, m, 2H, ArH; 3.19, s, 2H, CHN;

Experimental 7.3
167
2.13, dd, J 12.6, 8.0 Hz, 2H; 1.71-1.62, m, 3H; 1.43-1.35, m, 1H. 13C NMR:
178.06, C=O; 133.76, 132.29, 128.78, 128.30, all Ar; 43.68, CHN; 27.01, 19.58 MS:
m/z 187 (M+), 105 (C6H5CO+), 77 (C6H5+), 55 (C4H7
+). Further elution gave the cis-
oxazoline (2.25, data: page 139) as a pale yellow oil (14 mg, 34%).
(b) Reaction of trans-2-(phenylselanyl)cyclohexyl benzamide (2.5)
(i) with 4 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.5, 100 mg, 0.279
mmol) with NaH (49 mg, 60%, 1.2 mmol) and m-CPBA (194 mg, 0.899 mmol) in i-
PrOH (16 mL) gave a mixture (76 mg) containing the aziridine[154] (2.17, data: page
135) and the cis-oxazoline[149] (2.7, data: page 137) in a ratio of 20:80.
(ii) with 6 equivalents of sodium hydride
Following Procedure 7.3B, reaction of the amido selenide (2.5, 100 mg, 0.279 mmol)
with NaH (72 mg, 60%, 1.8 mmol) and m-CPBA (193 mg, 0.895 mmol) in i-PrOH (15
mL) gave a mixture (36 mg) containing the aziridine[154] (2.17, data: page 135) and
the cis-oxazoline (2.7, data: page 137) in a ratio of 80:20.
(iii) with 10 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.5, 79 mg, 0.22
mmol) with NaH (88 mg, 60%, 2.2 mmol) and m-CPBA (204 mg, 0.946 mmol) in i-
PrOH (12 mL) gave a mixture (66 mg) of the aziridine[154] (2.17) in an approximate
ratio of 90:10 with other products including the amido selenide (2.5).
Chromatography (EtOAc/hexane 15:85) gave the aziridine[154] (2.17, data: page 135)
as a white solid (26 mg, 59%). Further elution gave a mixture (3.3 mg) containing
the amido selenide (2.5) and the cis-oxazoline (2.7, data: page 137) in a ratio of 15:1.

Experimental 7.3
168
(c) Reaction of trans-N-2-(phenylselanyl)cycloheptyl benzamide (2.27)
(i) with 6 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.27) (83 mg, 0.22
mmol) with NaH (54 mg, 60%, 1.4 mmol) and m-CPBA (180 mg, 0.834 mmol) in i-
PrOH (12.5 mL) gave a mixture (36 mg) containing the aziridine (3.2), the cis-
oxazoline (2.28) and the syn-elimination product (2.30) in a ratio of 70:25:5.
Chromatography (EtOAc/hexane gradient of 15:85 to 50:50) gave the 8-benzoyl-8-
azabicyclo[5.1.0]octane (3.2) as a pale yellow oil (16 mg, 35%). Crystallisation from
CH2Cl2/hexane gave white, star-like crystals, m.p. 104.5-106.5°C. ESI HRMS:
216.13884 C14H17NO+H requires 216.13829. max 3301, 2924, 1672, 1544, 1450,
1428, 1313, 1296, 1259, 1175, 1133, 1091, 1071, 1022, 740, 668 cm-1. 1H NMR:
7.98-7.95, m, 2H, ArH; 7.56-7.51, m, 1H, ArH; 7.47-7.42, m, 2H, ArH; 2.73-2.71, m,
2H, CHN; 2.12-1.93, m, 4H; 1.71-1.57, m, 5H; 1.30-1.26, m, 1H. 13C NMR:
179.99, C=O; 133.54, 132.32, 128.93, 128.27 all Ar; 41.83, CHN; 31.40, 29.05,
25.40. MS: m/z 215 (M+), 110 (M+-C6H5CO), 105 (C6H5CO+), 77 (C6H5+).
Further elution gave a mixture (8 mg, 17%) of the cis-oxazoline (2.28, data: page
140) and the syn-elimination product (2.30, data: page 180) in a ratio of 55:45.
(ii) with 8 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.27) (82 mg, 0.22
mmol) with NaH (72 mg, 60%, 1.8 mmol) and m-CPBA (185 mg, 0.858 mmol) in i-
PrOH (12.5 mL) gave a mixture (74 mg) containing the aziridine (3.2), the cis-
oxazoline (2.28) and the syn-elimination product (2.30) in a ratio of 75:15:10 as
estimated from integrations of 1H NMR signals. Chromatography (EtOAc/hexane
15:85) gave the aziridine (3.2, data: page 168) as a colourless oil (25 mg, 53%).

Experimental 7.3
169
Further elution gave a mixture (15 mg) containing the cis-oxazoline (2.28, data: page
140) and the syn-elimination product (2.30, data: page 180) in a ratio of 40:60.
(iii) with 10 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.27) (79 mg, 0.21
mmol) with NaH (88 mg, 60%, 2.2 mmol) and m-CPBA (187 mg, 0.867 mmol) in i-
PrOH (12 mL) gave a mixture (86 mg) containing the aziridine (3.2), the cis-oxazoline
(2.28), the syn-elimination product (2.30) and the amido selenide (2.27) in a ratio of
70:20:10:10 as estimated from integrations of 1H NMR signals. Chromatography
(CHCl3/hexane to remove diphenyl diselenide then EtOAc/hexane 5:95 to 25:75)
gave the aziridine (3.2, data: page 168) as a pale yellow oil (21 mg, 46%). Further
elution gave a mixture (13 mg) containing the cis-oxazoline (2.28, data: page 140)
the syn-elimination product (2.30, data: page 180) and the selenide (2.27, data: page
152) in a ratio of 45:35:20.
(d) Reaction of 1-(phenylselanyl)-2-octyl benzamide (2.32)
(i) with 8.6 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.32, 85 mg, 0.22
mmol) with NaH (75 mg, 60%, 1.9 mmol) and m-CPBA (186 mg, 0.862 mmol) in i-
PrOH (12 mL) gave a pale yellow oil (72 mg). Chromatography (EtOAc/hexane
15:85) gave the oxazoline (3.12, 44 mg, 87%, data: page 162) as a pale yellow oil.
(e) Reaction of 2-(phenylselanyl)cyclohexyl p-bromobenzamide (2.45)
(i) with 4 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.45, 94 mg, 0.22
mmol) with NaH (38 mg, 60%, 0.95 mmol) and m-CPBA (185 mg, 0.858 mmol) in i-
PrOH (12 mL) gave a pale yellow solid containing the aziridine (3.3) and cis-
oxazoline (3.4, data: page 170) in a ratio of 1:3 as estimated from integrations of 1H

Experimental 7.3
170
NMR signals. Chromatography (EtOAc/hexane gradient of 15:85 to 45:55) gave the
aziridine (3.3, data: page 173) as a white solid (12 mg, 20%). Further elution gave
cis-3a,4,5,6,7,7a-hexahydro-2-(4’-bromophenyl)benzoxazole (3.4) as a pale yellow
solid (40 mg, 67%). Recrystallisation from EtOAc/hexane gave white crystals, m.p.
46-47°C. ESI HRMS: 298.04329 C13H14NOBr+H3O requires 298.04426. max 2940,
1645, 1591, 1486, 1401, 1346, 1264, 1071, 1011, 979, 916, 832, 728, 673 cm-1. 1H
NMR: 7.85-7.80, m, 2H, ArH; 7.57-7.52, m, 2H, ArH; 4.69, dt, J 5.4, 8.1 Hz, 1H,
CHO; 4.12, dt, J 6.3, 8.1 Hz, 1H, CHN; 1.93-1.83, m, 2H; 1.64-1.51, m, 2H; 1.46-
1.37, m, 2H; 1.27-1.19, m, 2H. 13C NMR: 163.48, C=O; 131.53, 131.52, 129.62,
125.81, all Ar; 79.09, CHO; 63.62, CHN; 27.65, 26.20, 19.79, 19.06.
(ii) with 6 equivalents of sodium hydride
Following Procedure 7.3B, the reaction of the amido selenide (2.45, 82 mg, 0.19
mmol) with NaH (48 mg, 60%, 1.2 mmol) and m-CPBA (158 mg, 0.732 mmol) in i-
PrOH (12.5 mL) gave the aziridine (3.3) in a clean reaction. Chromatography
(CHCl3/hexane 15:85 to remove diphenyl diselenide then EtOAc/hexane 15:85 to
25:75) gave the aziridine (3.3, data: page 173) as a colourless solid (37 mg, 70%).
Oxidation of 2-amidoalkyl phenyl selenide with t-BuOK as base
Procedure 7.3C: To dry i-PrOH under a N2 atmosphere was added with stirring t-
BuOK and stirring was continued until the salt had dissolved. The selenide was
added and the mixture was stirred until the solid had dissolved. m-CPBA was added
and the resulting mixture was stirred a further 1.5 h. Aqueous Na2S2O3 (0.5 M, 15
mL) and saturated aqueous NaHCO3 (10 mL) were added and the products were
extracted with CHCl3 (3 x 20 mL). The combined organic extracts were washed with
saturated aqueous NaCl (10 mL), dried (MgSO4) and the solvent was evaporated at
reduced pressure.

Experimental 7.3
171
(a) Reaction of trans-2-(phenylselanyl)cyclopentyl benzamide (2.24)
Following Procedure 7.3C, the reaction of the amido selenide (2.24, 78 mg, 0.23
mmol) with t-BuOK (204 mg, 1.82 mmol) and m-CPBA (195 mg, 0.904 mmol) in i-
PrOH (12 mL) gave a mixture of the aziridine (3.1) and the cis-oxazoline (2.25) in a
ratio of 51:49. Chromatography (EtOAc/hexane 25:75) gave the aziridine[154] (3.1,
data: page 166) as a pale yellow oil (18 mg, 42%). Further elution gave the cis-
oxazoline (2.25, data: page 139) as a pale yellow oil (15 mg, 35%).
(b) Reaction of trans-2-(phenylselanyl)cyclohexyl benzamide (2.5)
Following Procedure 7.3C, the reaction of the amido selenide (2.5, 80 mg, 0.22
mmol) with t-BuOK (199 mg, 1.8 mmol) and m-CPBA (199 mg, 0.923 mmol) in i-
PrOH (12 mL) gave a colourless oil (49 mg) whose 1H NMR spectrum showed it to
contain the aziridine[154] (2.17) along with traces of other products. Chromatography
(EtOAc/hexane 20:80 to 45:55) gave the aziridine[154] (2.17, data: page 135) as a
pale yellow solid (38 mg, 85%).
Oxidation of trans-2-(phenylselanyl)cyclopentyl benzamide (2.24) with t-BuOK
as base at –6°C
The amido selenide (2.24, 76 mg, 0.22 mmol) was dissolved in THF (15 mL) and the
solution was cooled in an ice-salt bath at –6°C. A solution of m-CPBA (152 mg,
80%, 0.705 mmol) in THF (10 mL) was added dropwise to the cooled solution. The
mixture was stirred for 20 min by which time the bath temperature had risen to –3°C.
t-BuOK (148 mg, 1.32 mmol) was added and the resulting suspension was stirred 2 h
by which time the bath temperature was 15°C. Aqueous Na2S2O3 (0.5 M, 15 mL)
and saturated aqueous NaHCO3 (20 mL) were added and the products were
extracted with CH2Cl2 (2 x 25 mL). The combined organic extracts were washed with

Experimental 7.3
172
saturated aqueous NaCl (10 mL), dried (MgSO4) and the solvent evaporated at
reduced pressure to give a yellow oil (44 mg). Chromatography (EtOAc/hexane
20:80 to 30:70) gave the aziridine[154] (3.1, data: page 166) as a pale yellow oil (30
mg, 73%). Further elution gave a mixture (2 mg) of the cis-oxazoline (2.25, data:
page 139) and syn-elimination product[154] (3.13, data: page 178) in a ratio of 55:45
as a pale yellow oil.
Oxidation and cyclisation of 2-amidoalkyl phenyl selenide at low temperature
Procedure 7.3E: The selenide was dissolved in dry THF (20 mL) and the flask was
placed in a dry ice/acetone bath cooled to a bath temperature of between -60°C and
-70°C. A solution of m-CPBA in dry THF (20 mL) was added dropwise to the cooled
solution and the mixture was stirred for 1h with the bath temperature below –60°C.
t-BuOK was added in one portion and the resulting mixture was stirred for a further
1h. The flask was removed from the cooling bath and allowed to warm over 0.5-1h.
Aqueous Na2S2O3 (0.5 M, 15 mL) and saturated aqueous NaHCO3 (10 mL) were
added and the aqueous phase was extracted with Et2O (30 mL). The organic extract
was washed with aqueous NaOH (10%, 10 mL) and saturated aqueous NaCl (10 mL)
and dried (MgSO4) and the solvent evaporated at reduced pressure.
(a) 7-Acetyl-7-azabicyclo[4.1.0]heptane (3.14)
Following Procedure 7.3E, the reaction of 2-(phenylselanyl)cyclohexyl acetamide
(2.31, 250 mg, 0.844 mmol), m-CPBA (594 mg, 2.75 mmol) and t-BuOK (571 mg,
5.09 mmol) gave a pale yellow oil (119 mg) containing the aziridine (3.14).
Chromatography (Et2O/CH2Cl2 gradient of 0:100 to 10:90) gave the title
compound[149] as a colourless oil (77 mg, 66%). max (KBr) 2934, 2857, 1657, 1553,
1449, 1413, 1373, 1303, 1248, 1221, 1075, 1043 cm-1. 1H NMR: 2.63-2.32, m, 2H,

Experimental 7.3
173
CHN; 2.11, s, 3H, CH3; 1.90, m, 2H; 1.85, m, 2H; 1.50-1.38, m, 2H; 1.32-1.23, m, 2H.
13C NMR: 183.66, C=O; 35.82, 23.77, 23.41, 19.81.
(b) 7-Benzoyl-7-azabicyclo[4.1.0]heptane (2.17)
Following Procedure 7.3E, the reaction of trans-2-(phenylselanyl)cyclohexyl
benzamide (2.5, 152 mg, 0.424 mmol) with m-CPBA (290 mg, 1.34 mmol) and t-
BuOK (284 mg, 75%, 1.90 mmol) gave a pale yellow liquid (88 mg) containing the
aziridine[154] (2.17, data: page 135). Chromatography (EtOAc/hexane 20:80)
afforded the title compound as a pale yellow solid (71 mg, 83%), which crystallised
from the eluting solvent to give white crystals, m.p. 79.5–80.5 °C (lit.[154] m.p. 77°C).
(c) 6-Benzoyl-6-azabicyclo[3.1.0]hexane (3.1)
Following Procedure 7.3E, the reaction of trans-2-(phenylselanyl)cyclopentyl
benzamide (2.24, 144 mg, 0.418 mmol) with m-CPBA (291 mg, 1.35 mmol) and t-
BuOK (282 mg, 75%, 1.88 mmol) gave a pale yellow oil (67 mg) containing the
aziridine[154] (3.1, data: page 166) and a trace of the cis-oxazoline (2.25, data: page
139). Chromatography (EtOAc/hexane 20:80) afforded the title compound as a
colourless oil (59 mg, 75%).
(d) 7-(4’-Bromobenzoyl)-7-azabicyclo[4.1.0]heptane (3.3)
Following Procedure 7.3E, the reaction of 2-(phenylselanyl)cyclohexyl p-
bromobenzamide (2.45, 95 mg, 0.22 mmol) with m-CPBA (151 mg, 0.700 mmol) and
t-BuOK (146 mg, 75%, 0.976 mmol) gave a pale yellow solid (82 mg) containing the
aziridine (3.3). Chromatography (CH2Cl2/hexane 15:85 to remove diphenyl
diselenide then EtOAc/hexane, gradient of 5:95 to 50:50) afforded the title compound
which crystallised from the eluting solvent as white crystals (57 mg, 94%), m.p. 110–
113°C. EI HRMS: 279.0247 C13H14NOBr requires 279.0259. max (KBr) 2947,
2933, 2860, 1662, 1587, 1568, 1481, 1441, 1412, 1396, 1371, 1344, 1304, 1252,

Experimental 7.3
174
1232, 1171, 1117, 1084, 1070, 1009, 849, 762 cm-1. 1H NMR: 7.87-7.82, m, 2H,
ArH; 7.61-7.57, m, 2H, ArH; 2.76-2.75, m, 2H, CHN; 2.10-2.00, m, 2H; 1.96-1.88, m,
2H; 1.59-1.50, m, 2H; 1.41-1.32, m, 2H. 13C NMR: 179.14, C=O; 132.53, 131.58,
130.51, 127.35, all Ar; 37.23, CHN; 23.81, 19.90. MS: m/z 281 (M+, 81Br), 279 (M+,
79Br), 278 (M+-1, 79Br), 200 (M+-Br), 185 (BrC6H4CO+, 81Br), 183 (BrC6H4CO+, 79Br),
157 (BrC6H4+, 81Br), 155 (BrC6H4
+, 79Br), 96 (C6H10N+), 69 (C5H9+), 55 (C4H7
+), 41
(C3H5+).
(e) 8-Benzoyl-8-azabicyclo[5.1.0]octane (3.2)
Following Procedure 7.3E, the reaction of trans-2-benzamidocycloheptyl phenyl
amido selenide (2.27) (81 mg, 0.22 mmol) with m-CPBA (156 mg, 0.723 mmol) and t-
BuOK (148 mg, 75%, 0.989 mmol) gave a pale yellow oil (38 mg) containing the
aziridine (3.2) and traces of other products. Chromatography (EtOAc/hexane 20:80)
afforded the title compound as a pale yellow solid (38 mg, 81%, data: page 168).
(f) 9-Benzoyl-9-azabicyclo[6.1.0]nonane (3.16)
Following Procedure 7.3E, the reaction of trans-2-(phenylselanyl)cyclooctyl
benzamide (2.47, 85 mg, 0.22 mmol) with m-CPBA (154 mg, 0.714 mmol) and t-
BuOK (149 mg, 75%, 0.998 mmol) gave a pale yellow solid (49 mg) containing the
aziridine (3.16) and traces of other products. Chromatography (EtOAc/hexane
20:80) afforded the title compound as a pale yellow solid (44 mg, 87%).
Recrystallisation from Et2O/hexane gave white crystals, m.p. 52.5-54.5°C (lit.[154]
m.p. 72.5°C) 1H NMR: 8.00-7.96, m, 2H, ArH; 7.56-7.51, m, 1H, ArH; 7.47-7.41,
m, 2H, ArH; 2.54-2.50, m, 2H, CHN; 2.34-2.28, m, 2H; 1.71-1.43, m, 10H. 13C NMR:
179.71, C=N; 133.67, 132.35, 129.02, 128.23, all Ar; 41.50, CHN; 26.70, 26.39,
26.36. MS: m/z 229 (M+), 228 (M+-H), 201 (M+-C2H4), 124 (M+-C6H5CO), 105
(C6H5CO+), 97 (C7H13+), 77 (C6H5
+).

Experimental 7.3
175
(g) 8-Acetyl-8-azabicyclo[5.1.0]octane (3.15)
Following Procedure 7.3E, the reaction of trans-2-acetamidocycloheptyl phenyl
selenide (2.44, 101 mg, 0.325 mmol) with m-CPBA (229 mg, 1.06 mmol) and t-BuOK
(211 mg, 1.88 mmol) gave a yellow oil which was chromatographed (EtOAc/hexane,
gradient 15:85 to 30:70) to afford the title compound as a colourless oil (33 mg, 67%).
EI HRMS 153.1151 C9H15NO requires 153.1154. max (neat) 2925, 2851, 1694,
1455, 1442, 1365, 1299, 1245, 1224, 733 cm-1. 1H NMR: 2.62-2.60, m, 2H, CHN;
2.10, s, 3H, CH3; 2.01-1.83, m, 4H; 1.63-1.43, m, 4H; 1.25-1.19, m, 2H. 13C NMR:
183.83, C=O; 40.80, CHN; 31.25, CH3; 28.82, 25.19, 23.39. MS: m/z 153 (M+),
110 (M+-CH3CO), 96 (C7H12+).
(h) N-benzoyl-2-methyl-3-(n-propyl)aziridine (3.19)
Following Procedure 7.3E, the reaction of a mixture of 3-(phenylselanyl)-2-hexyl
benzamide (2.35) and 2-(phenylselanyl)-3-hexyl benzamide (2.34) (100 mg, 0.277
mmol) with m-CPBA (236 mg, 1.09 mmol) and t-BuOK (248 mg, 2.21 mmol) gave
yellow oil (54 mg) containing the aziridine (3.19) and oxazolines, (2.36) and (2.37), in
a ratio of 90:5:5. Chromatography (EtOH/CH2Cl2, gradient of 0.75:99.25 to 5:95)
afforded the title compound as a colourless oil (39 mg, 69%). ESI HRMS:
204.13862 C13H17NO+H requires 204.13829. max (neat) 3062, 3030, 2962, 2931,
2873, 1668, 1601, 1581, 1531, 1491, 1450, 1383, 1340, 1323, 1271, 1225, 1174,
1151, 1122, 1095, 1070, 1026, 725, 702 cm-1. 1H NMR: 8.02-7.99, m, 2H, ArH;
7.56-7.51, m, 1H, ArH; 7.47-7.41, m, 2H, ArH; 2.59, dq, J 3.3, 5.7 Hz, 1H, CH3CHN;
2.44, ddd, J 3.3, 5.1, 7.5 Hz, 1H, CH2CHN; 1.73-1.62, m, 1H; 1.53-1.40, m, 2H; 1.32-
1.23, m, 1H; 1.19, d, J 5.7 Hz, 3H, CHCH3; 0.95, t, J 7.2 Hz, 3H, CH2CH3. 13C NMR:
177.92, C=O; 134.59, 132.27, 128.82, 128.27, all Ar; 44.26, CH3CHN; 40.52,
CH2CHN; 33.48, 20.37, 16.63, 13.69. MS: m/z 203 (M+), 188 (M+-CH3), 174 (M+-

Experimental 7.3
176
CH2CH3), 160 (M+-(CH2)2CH3), 105 (PhCO+), 98 (M+-PhCO), 77 (C6H5+), 56 (C4H8
+).
Further elution gave a fraction containing the oxazoline (2.36, data: page 142) and
unidentified products as a colourless oil (6 mg, 10%). Further elution gave the
oxazoline (2.37, data: page 141) as a colourless oil (2 mg, 4%).
(i) N-(4’-Bromobenzoyl)-2-(n-hexyl)aziridine (3.18)
Following Procedure 7.3E, the reaction of N-[2-(phenylselanyl)-1-octyl] p-
bromobenzamide (2.50, 66 mg, 0.14 mmol) with m-CPBA (98 mg, 0.45 mmol) and t-
BuOK (97 mg, 75%, 0.65 mmol) gave a pale yellow liquid which was
chromatographed (hexane/CH2Cl2, gradient of 10:90 to 0:100) to afford the title
compound as a pale yellow oil (19 mg, 44%). ESI HRMS: 310.07956
C15H20NOBr+H requires 310.08010. max 2955, 2927, 2856, 1675, 1587, 1466,
1397, 1311, 1226, 1171, 1089, 1069, 1011, 848, 764 cm-1. 1H NMR: 7.91-7.87, m,
2H, ArH; 7.61-7.58, m, 2H, ArH; 2.56-2.53, m, 1H, CHN; 2.49, d, J 6.0 Hz, 1H,
CHHN; 2.19, d, J 3.6 Hz, CHHN; 1.86-1.78, m, 1H; 1.44-1.26, m, 9H; 0.89, t, J 6.6
Hz, 3H, CH3. 13C NMR: 178.37, C=O; 132.39, 131.65, 130.55, 127.55, all Ar;
38.80, 32.07, 31.67, 31.66, 28.90, 26.42, 22.49, 13.97. MS: m/z 309 (M+), 280 (M+-
C2H5), 239 (M+-C5H10), 224 (M+-C6H13), 183 (BrC6H4CO+), 155 (BrC6H4+), 126
(C8H16N+). Further elution afforded 4-(n-hexyl)-2-(4’-bromophenyl)-4,5-dihydro-
oxazole (2.52) as a pale yellow oil (12 mg, 28%). ESI HRMS: 310.07990
C15H20NOBr+H requires 310.08010. max 2952, 2922, 2853, 1721, 1638, 1592,
1484, 1463, 1398, 1366, 1318, 1291, 1274, 1263, 1077, 1056, 975, 835, 756, 729,
677 cm-1. 1H NMR: 7.84-7.79, m, 2H, ArH; 7.55-7.52, m, 2H, ArH; 4.48, dd, J 8.1,
9.3 Hz, 1H, CHHO; 4.31-4.21, m, 1H, CHN; 4.03, dd, J 7.8, 8.1 Hz, CHHO; 1.79-
1.71, m, 1H; 1.61-1.26, m, 9H; 0.89, t, J 6.9 Hz, CH3. 13C NMR: 162.57, C=N;
131.51, 129.75, 126.91, 125.79, all Ar; 72.74, CHO; 66.91, CHN; 35.88, 31.70, 29.23,

Experimental 7.3
177
25.83, 22.54, 14.00. MS: m/z 309 (M+), 280 (M+-CHO, M+-C2H5), 239 (M+-C5H10),
224 (M+-C6H13), 183 (BrC6H4CO+), 155 (BrC6H4+).
(j) N-benzoyl-2-(n-hexyl)aziridine (3.17)
Following Procedure 7.3E, the reaction of 1-(phenylselanyl)-2-octyl benzamide (2.32,
110 mg, 0.283 mmol) with m-CPBA (238 mg, 1.10 mmol) and t-BuOK (273 mg, 2.43
mmol) gave a pale yellow liquid (51 mg) being a mixture of the title compound and
the isomeric oxazoline in a ratio of 75:25, as estimated by 1H NMR signals.
Chromatography (hexane/CH2Cl2, gradient of 10:90 to 5:95) afforded the title
compound as a pale yellow oil (36 mg, 56%). EI HRMS: 231.1623 C15H21NO
requires 231.1624. max (KBr) 2956, 2929, 2856, 1678, 1601, 1581, 1466, 1450,
1406, 1317, 1300, 1230, 723, 710 cm-1. 1H NMR: 8.05-8.01, m, 2H, ArH; 7.58–
7.52, m, 1H, ArH; 7.48–7.42, m, 2H, ArH; 2.58–2.49, m, 1H, CHN; 2.50, d, J 6.0 Hz,
1H, CHHN; 2.19, d, 3.6 Hz, 1H, CHHN; 1.89–1.81, m, 1H; 1.48–1.29, m, 9H; 0.86, t,
J 6.9 Hz, 3H, CH3. 13C NMR: 179.31, C=O; 133.52, 132.52, 129.05, 128.32, all Ar;
38.63, 32.14, 31.69, 31.58, 28.94, 26.44, 22.51, 13.99. MS: m/z 232 (M++H), 216
(M+-CH3), 202 (M+-C2H5), 188 (M+-C3H7), 174 (M+-C4H9), 161 (M+-C5H10), 146 (M+-
C6H13), 126 (M+-C6H5 CO), 105 (C6H5CO+), 77 (C6H5+). Further elution afforded the
oxazoline (3.12, data: page 162) as a pale yellow oil (11mg, 17%).
Oxidation of trans-2-(phenylselanyl)cyclopentyl benzamide (2.24)
with 1.1 equivalents of m-CPBA
Following procedure 7.3E, the reaction of the amido selenide (2.24, 77 mg, 0.22
mmol) with m-CPBA (53 mg, 0.25 mmol) and t-BuOK (80 mg, 0.71 mmol) gave a
yellow oil (33 mg) as a mixture of the aziridine[154] (3.1), the amido selenide (2.24)
and the syn-elimination product (3.13) in a ratio of 15:30:55. Chromatography

Experimental 7.3
178
(CH2Cl2/hexane 15:85 to remove diphenyl diselenide then EtOAc/hexane gradient of
5:95 to 50:50) gave the aziridine[154] (3.1, data: page 166) as a yellow solid (trace).
Further elution gave the cis-oxazoline (2.25, data: page 139) as a pale yellow oil (2
mg, 4%). Further elution gave the amido selenide (2.24, 4 mg, 5%, data: page 138)
as a pale brown solid. Further elution gave N-(cyclopent-2-en-1-yl)benzamide[154]
(3.13) as a yellow solid (12 mg, 29%). Recrystallisation from CH2Cl2/hexane gave
pale yellow crystals, m.p. 121-123°C (lit.[154] m.p. 123°C). max 3293, 3059, 2926,
2852, 1627, 1603, 1578, 1534, 1491, 1454, 1338, 1285, 1263, 1057, 916, 806 cm-1.
1H NMR: 7.77-7.75, m, 2H, ArH; 7.51-7.37, m, 3H, ArH; 6.12, br s, 1H, NH; 6.00,
ddd, J 2.1, 3.9, 5.7 Hz, 1H, CHCHN; 5.77, ddd, J 2.1, 4.2, 5.7 Hz, 1H, CH2CH:CH;
5.24-5.14, m, 1H, CHN; 2.53-2.29, m, 2H; 1.79-1.63, m, 2H. 13C NMR(600MHz):
166.84, C=O; 135.13, CH2CH; 134.74, 131.35, both Ar; 131.05, CHNCH; 128.53,
126.86, both Ar; 56.10, CHN; 31.58, 31.23.
Oxidation of 2-(phenylselanyl)cycloheptyl benzamide (2.27)
with 1.1 equivalents of m-CPBA
Following procedure 7.3E, the reaction of the amido selenide (2.27) (82 mg, 0.22
mmol) with m-CPBA (50 mg, 0.23 mmol) and t-BuOK (73 mg, 0.65 mmol) gave a
yellow solid (52 mg) as a mixture of the starting material and syn-elimination product
(2.30) in a ratio of 80:20.
Oxidation of trans-N-2-(phenylselanyl)cycloheptyl benzamide (2.27) with excess m-CPBA at –15°C
The amido selenide (2.27) (81 mg, 0.22 mmol) was dissolved in THF (12.5 mL) and
the solution was cooled in an ice-salt bath to –15°C. A solution of m-CPBA (152 mg,
0.705 mmol) in THF (10 mL) was added dropwise to the cooled solution. The

Experimental 7.3
179
mixture was stirred for 1 h, the bath temperature being maintained between –15 and
–9°C. t-BuOK (149 mg, 1.33 mmol) was added and the resulting suspension was
stirred 1 h by which time the bath temperature was 7°C. Aqueous Na2S2O3 (0.5 M,
15 mL) and saturated aqueous NaHCO3 (15 mL) were added and the aqueous phase
extracted with Et2O (3 x 20 mL). The combined organic extracts were washed with
saturated aqueous NaHCO3 (4 x 10 mL) followed by saturated aqueous NaCl (15
mL), dried (MgSO4) and the solvent evaporated at reduced pressure to give a pale
yellow liquid (43 mg) estimated to be a mixture of the aziridine (3.2) and the syn-
elimination product (2.30) in a ratio of 75:25 from integrations of 1H NMR signals.
Chromatography (EtOAc/hexane 20:80 to 50:50) gave the aziridine (3.2, data: page
168) as a yellow oil (28 mg, 59%). Further elution gave the syn-elimination product
(2.30, data: page 180) as a pale yellow solid (9 mg, 19%).
Oxidation of trans-N-2-(phenylselanyl)cycloheptyl benzamide (2.27)
with excess m-CPBA at 0°C
The amido selenide (2.27) (83 mg, 0.22 mmol) was dissolved in THF (12.5 mL) and
the solution was cooled in an ice bath to 0°C. A solution of m-CPBA (155 mg, 0.719
mmol) in THF (10 mL) was added to the cooled solution. The mixture was stirred for
1 h, the bath temperature being maintained at 0°C. t-BuOK (150 mg, 1.33 mmol)
was added and the resulting suspension was stirred 1 h by which time the bath
temperature was 4°C. The flask was removed from the cooling bath and the mixture
was stirred a further 15 min. Aqueous Na2S2O3 (0.5 M, 15 mL) and saturated
aqueous NaHCO3 (15 mL) were added and the aqueous phase extracted with Et2O
(3 x 20 mL). The combined organic extracts were washed with saturated aqueous
NaHCO3 (4 x 10 mL) followed by saturated aqueous NaCl (15 mL), dried (MgSO4)

Experimental 7.3
180
and the solvent evaporated at reduced pressure to give a pale yellow solid (43 mg)
as a mixture of the aziridine (3.2) and the syn-elimination product (2.30) in a ratio of
1:9 as estimated from integrations of 1H NMR signals. Chromatography
(EtOAc/hexane 20:80 to 45:55) gave the aziridine (3.2, data: page 168) as a pale
yellow oil (3 mg, 6%). Further elution gave N-(cyclohept-2-en-1-yl)benzamide (2.30)
as a pale yellow solid (29 mg, 61%). Recrystallisation from CH2Cl2/hexane gave
white crystals, m.p. 122-124°C. EI HRMS: found 215.1309 C14H17NO requires
215.1311. max 3291, 2927, 1628, 1602, 1578, 1541, 1491, 1332, 1309, 1278, 1261,
719, 692, 680, 663 cm-1. 1H NMR: 7.79-7.75, m, 2H, ArH; 7.53-7.40, m, 3H, ArH;
6.23, m, 1H, NH; 5.88, dddd, J 2.1, 5.4, 6.6, 12.3 Hz, 1H, CH2CHCH; 5.64, ddd, J
2.1, 2.4, 12.3 Hz, 1H, CH2CHCH; 4.82, m, 1H, CHN; 2.24-2.17, m, 2H; 1.97-1.89, m,
2H; 1.83-1.62, m, 3H; 1.49-1.39, m, 1H. 13C NMR: 166.68, C=O; 135.18, 134.78,
132.90, 131.59, all Ar; 128.81, CH2CHCH; 127.11, CH2CHCH; 51.15, CHN; 34.24,
CH2CHCH; 28.84, 27.77, 27.10. MS: m/z 215 (M+), 122(C6H5CONH3+), 105
(C6H5CO+), 94 (C7H10+); 77 (C6H5
+).
Attempted isomerisation of aziridine (2.17) to cis-oxazoline (2.7)
(i) stirring with silica
The aziridine (2.17, 32 mg, 0.16 mmol) was stirred with silica in CH2Cl2 at r.t. for 24 h
at which time TLC analysis indicated no change in the reaction mixture. The mixture
was then refluxed for 30 min at which time TLC analysis again showed no new
product. The mixture was filtered and the solvent removed at reduced pressure to
give the aziridine[154] (2.17, data: page 135).

Experimental 7.3
181
(ii) with m-CPBA
The aziridine (2.17, 5 mg, 0.025 mmol) was stirred in EtOH (3 mL) at r.t. for 1.75 h
after which time no new product was observed by TLC analysis. m-CPBA (10.9 mg,
0.06 mmol) was added and the mixture stirred for a further 1.75 h. Aqueous
Na2S2O3 (0.5 M, 5 mL) and saturated aqueous NaHCO3 were added and the mixture
was extracted with CHCl3. The combined organic layers were dried (MgSO4) and the
solvent removed at reduced pressure. 1H NMR analysis of the product showed the
aziridine[154] (2.17, data: page 135) as the predominant product along with a small
amount of the cis-oxazoline (2.7, data: page 137) and other products in minor
amounts which were not identified.
Attempt to cyclise trans-2-(phenylselanyl)cyclohexyl benzamide (2.5) by
treatment with hydroxide
The amido selenide (2.5, 30 mg, 0.08 mmol) was stirred with KOH (47 mg, 0.8 mmol)
in i-PrOH (5.5 mL) at r.t. for 5 h. The mixture was acidified with dropwise addition of
HCl (0.1 M), then extracted with CHCl3 (2 x 20 mL). The combined organic layers
were dried (MgSO4) and the solvent removed at reduced pressure to give the amido
selenide (2.5, 24 mg, 80%).

Experimental 7.4
182
7.4 WORK DESCRIBED IN CHAPTER 4
Attempted acetamidoselenation of cyclohexene
Phenylselenenyl chloride (213 mg, 1.11 mmol) was added to a solution of
cyclohexene (95 L, 0.94 mmol) in dry acetonitrile (5 mL) and the mixture was cooled
to 0°C under N2. To the resulting yellow solution was added silver perchlorate (216
mg, 1.04 mmol), giving a white precipitate. The mixture was stirred at 0°C for 10 min
and to it was added aqueous KOH (0.5 mL) and stirring was continued for a further
10 min at 0°C. The mixture was diluted with Et2O (40 mL) and decanted. The Et2O
layer was washed with water (10 mL), dried (MgSO4) and the solvent evaporated at
reduced pressure to give a yellow oil (208 mg). Chromatography (CHCl3/hexane
15:85 then a gradient of EtOAc/hexane 5:95 to 60:40) gave 2-
(phenylseleno)cyclohexanol[169] (2.41) as a pale red oil (135 mg, 56%) which was
identified by comparison of its 1H NMR spectrum with that of (2.41) prepared
previously (see page 136).
N-[2-(Phenylselanyl)cyclohexyl]acetamide (2.31)
To a solution of cyclohexene (100 L, 0.987 mmol) in acetonitrile (4 mL) under N2
was added phenylselenenyl bromide (231 mg, 0.979 mmol) and the mixture was
cooled to 0°C. To it was added silver perchlorate (221 mg. 1.07 mmol) and the
mixture was stirred for 5 min. A solution of water (2 drops) in acetonitrile (2 mL) was
added over 50 min followed by a solution of water (3 drops) in acetonitrile (0.5 mL)
added over 15 min. The mixture was diluted with CH2Cl2 (40 mL) and the CH2Cl2
layer was decanted from the precipitated silver salts, washed with water (10 mL),
dried (MgSO4) and the solvent evaporated at reduced pressure. Chromatography

Experimental 7.4
183
(CHCl3/hexane 15:85 then a gradient of EtOAc/hexane 5:95 to 75:25)) gave 2-
(phenylseleno)cyclohexanol[169] (2.41) as a red oil (39 mg, 15%) which was identified
by comparison of its 1H NMR spectrum with that of (2.41) prepared previously (see
page 136). Further elution gave the title compound[145] as a white solid (166 mg,
57%) which was identified by comparison of its 1H NMR spectrum with that of (2.31)
prepared previously (see page 136).
trans-N-[2-(Phenylselanyl)cyclohexyl]benzamide (2.5)
To a solution of cyclohexene (100 L, 0.987 mmol) in benzonitrile (4 mL) under N2
was added phenylselenenyl bromide (262 mg, 1.11 mmol) and the mixture was
cooled to 5°C. To it was added silver perchlorate (228 mg, 1.10 mmol) and the
mixture was stirred for 10 min. Water (2 drops) was added dropwise and the mixture
was stirred for a further 50 min, then diluted with CHCl3 (35 mL). The mixture was
filtered to remove the precipitated silver salts and the organic layer was washed with
water (10 mL), dried (MgSO4) and the solvent evaporated at reduced pressure.
Chromatography (CHCl3/hexane 15:85 then a gradient of EtOAc/hexane 5:95 to
50:50) gave the title compound[145] as a pale pink solid (118 mg, 33%) which was
identified by comparison of its 1H NMR spectrum with that of (2.5) prepared
previously (see page 137).
5-Methyl-1-[trans-2-(phenylselanyl)cyclohexyl]-1H-tetrazole (4.12)
Cyclohexene (101 L, 0.997 mmol) was added to a solution of phenylselenenyl
bromide (238 mg, 1.01 mmol) in acetonitrile (4 mL) under N2 and the mixture was
cooled to 0°C, and to it was added silver perchlorate (209 mg, 1.01 mmol), giving a
white precipitate. To the suspension was added, with stirring, sodium azide (64 mg,

Experimental 7.4
184
0.98 mmol) over 20 min. The mixture was stirred at 0°C for 1.5 h, then diluted with
CH2Cl2 (40 mL), and decanted, leaving a residue of white silver salts. The CH2Cl2
layer was washed with water (7 mL) and dried (MgSO4) and the solvent evaporated
under reduced pressure to give a yellow oil (251 mg). Chromatography
(EtOAc/hexane 80:20) gave the title compound as a red oil (45 mg, 14%) which was
crystallised from CH2Cl2/Et2O as colourless crystals, m.p. 97-98°C. ESI HRMS:
323.07686 C14H18N4Se+H requires 323.07694. max (CHCl3) 3013, 2944, 2862,
1667 (C=N), 1524, 1477, 1450, 1438, 1404, 1226 (C-N), 1118, 1091, 1022, 794 cm-1.
1H NMR: 7.29-7.17, m, 5H, ArH; 4.16, dt, J 4.5, 11.4 Hz, 1H, CHN; 3.67, dt, J 4.2,
11.4 Hz, 1H, CHSe; 2.58, s, 3H, CH3; 2.48-2.43, m, 1H; 2.16-1.93, m, 2H; 1.86-1.82,
m, 1H; 1.74-1.60, m, 2H; 1.55-1.20, m, 2H. 13C NMR: 150.99, C=N; 135.30,
134.81, 129.03, 128.15, all Ar; 62.69, 47.55, 34.46, 34.01, 26.32, 24.86, 9.26. MS:
m/z 322 (M+), 238 (M+-CH3CN4H), 157 (C6H5Se+). Further elution gave a 2-
(phenylselanyl)cyclohexyl acetamide [145] (2.31) as a white solid (72 mg, 24%) which
was identified by comparison of its 1H NMR spectrum with that of (2.31) prepared
previously (see page 136). Further elution gave an orange-red solid which appeared
by 1H NMR to be a complex mixture and was not further purified.
5-Methyl-1,4-di[2-(phenylselanyl)cyclohexyl]-4H-1,2,3,4-tetraazol-1-ium
perchlorate (4.14)
Phenylselenenyl bromide (295 mg, 1.16 mmol) was added to a solution of
cyclohexene (118 L, 1.16 mmol) in acetonitrile (10 mL) under N2. After a few
minutes the mixture was still orange due to unreacted phenylselenenyl bromide, and
two more drops of cyclohexene were added. The mixture immediately became a
very pale orange and was cooled to 0°C, and to it was added silver perchlorate (241

Experimental 7.4
185
mg, 1.16 mmol), and the mixture was stirred for 10 min. Sodium azide (60 mg, 0.92
mmol) was added over 5 min and the resulting mixture was stirred for 80 min then
filtered through a bed of celite and the solvent was evaporated under reduced
pressure to give a viscous orange oil. The oil was dissolved in CHCl3 (30 mL) and
washed with water (10 mL) and saturated aqueous NaCl (10 mL) and the solvent was
evaporated under reduced pressure to give a viscous brown oil. Trituration with
Et2O/CH2Cl2 and recrystallisation from CH2Cl2/hexane gave the title compound as
pale brown crystals (34mg, 9%). This structure was confirmed by an X-ray structure
determination.[224] 1H NMR: 7.39-7.20, m, 10H, ArH; 4.53, dt, J 4.2, 11.7 Hz, 2H,
CHN; 3.55, dt, J 4.2, 11.7 Hz, 2H, CHSe; 3.12, s, 3H, CH3; 2.76-2.70, m, 2H; 2.43-
2.38, m, 2H; 2.18-1.98, m, 4H; 1.85-1.71, m, 4H; 1.57-1.41, m, 4H. 13C NMR:
151.50, C=N; 135.18, 129.47, 128.74, 126.27, all Ar; 65.77, CHN; 47.09, CHSe;
34.90, 32.88, 26.23, 24.46, 10.67. MS: m/z 322 (M+-C6H10SeC6H5); 238
(C6H9SeC6H5+).
trans-Ethyl[2-(phenylselanyl)cyclohexyl]carbamate (4.15)
Following a variation of the procedure of Francisco et al.,[57] to a mixture of ethyl
carbamate (4.75 g, 53.3 mmol), cyclohexene (0.138 mL, 1.36 mmol) and silver
perchlorate (334 mg, 1.61 mmol) in dry CH2Cl2 (45 mL) under N2 and protected from
light with aluminium foil, was added dropwise with stirring a solution of
phenylselenenyl chloride (290 mg, 1.51 mmol) in dry CH2Cl2 (10 mL) over
approximately 25 min. The resultant mixture was stirred 1 h at r.t., then poured into
10% KOH solution and filtered through a bed of celite. The aqueous layer was
extracted with Et2O (40 mL) and the combined organic layers washed with water (10
mL) and saturated aqueous NaCl (10 mL), dried (Na2SO4) and concentrated at

Experimental 7.4
186
reduced pressure to give a pale brown solid (4.3 g). The product was dissolved in
CH2Cl2 and absorbed onto silica (approximately 10 g) and the solvent evaporated.
Chromatography (EtOAc/hexane 25:75) gave the title compound as a pale pink solid
(365 mg, 82%) which was recrystallised from EtOAc to give colourless needles, m.p.
94.5-96.5°C. EI HRMS: 327.0738 C15H21NO2Se requires 327.0738. max(nujol)
3330, 3068, 3052, 1685, 1533, 1475, 1311, 1230, 1041 cm-1. 1H NMR: 7.61-7.57,
m, 2H, ArH; 7.32-7.24, m, 3H, ArH; 4.77, d, J 6.0Hz, 1H, NH; 4.12, q, J 7.2 Hz, 2H,
OCH2; 3.54-3.46, m, 1H, CHN; 3.01, ddd, J 3.9, 10.8, 10.8 Hz, 1H, CHSe; 2.21-2.07,
m, 2H; 1.69-1.48, m, 3H; 1.41-1.16, m, 3H; 1.25, t, J 7.2 Hz, 3H, CH3. 13C NMR:
155.86, C(=O)O; 135.63, 128.92, 128.00, 127.76, all Ar; 60.73, OCH2; 54.26, CHN;
48.40, CHSe; 33.99, 33.73, 26.36, 24.46; 14.62, CH3. MS: m/z 327 (M+), 281 (M+-
OC2H5), 238 (M+-NH2CO2Et), 170 (M+-C6H5Se), 81 (C6H9+). Further elution gave
ethyl carbamate as colourless crystals (3.64g, 77% recovery).

Experimental 7.5
187
7.5 WORK DESCRIBED IN CHAPTER 5
N-[2-(Phenylseleninyl)cyclohexyl]benzamide (5.1)
m-CPBA (127 mg, 0.59 mmol) was added to a solution of the amido selenide (2.5)
(199 mg, 0.555 mmol) in CH2Cl2 (10 mL) and the solution was stirred at r.t. for 70
min. The solution was diluted with CH2Cl2 (10 mL) and washed with aqueous NaOH
(10%, 3 x 15 mL) and saturated aqueous NaCl (10 mL), dried (MgSO4) and the
solvent was evaporated at reduced pressure to give the title compound as a white
solid (200 mg, 96%). Recrystallisation from MeOH/EtOAc gave the pure selenoxide
(5.1) as colourless needles, m.p. 137–138.5°C. An X-ray crystal determination
confirmed the crystals to be a mixture of R,R,SSe-(5.1) and its enantiomer S,S,RSe-
(5.1).[300] max (KBr): 3411, 3230, 3051, 2935, 2858, 1655, 1603, 1577, 1533, 1491,
1443, 1321, 1294, 814 (Se=O), 741, 698 cm-1. 1H NMR: 8.10, d, J 4.8 Hz, 1H,
NH; 7.95-7.91, m, 2H, ArH; 7.58-7.43, m, 8H, ArH; 3.60, dddd, J 4.2, 4.8, 10.8, 11.1
Hz, 1H, CHN; 3.39, ddd, J 3.6, 11.1, 12.3 Hz, 1H, CHSe; 2.45-2.40, m, 1H; 1.98-
1.93, m, 1H; 1.88-1.84, m, 1H; 1.75-1.25, m, 5H. 77Se NMR: (CDCl3/CD3OD, 2:3)
887.7; (THF/CD3OD, 2:3) 872.8, 843.7; diastereomeric mixture. Mass spectrum
m/z 375 (M+), 358 (M+-OH), 254 (M+-C6H5CONH2), 216 (M+-C4H8-H2), 200 (M+-
C6H5SeO–H2), 173 (C6H5SeO+), 157 (C6H5Se+), 122 (C6H5CONH2++H), 105
(C6H5CO+), 77 (C6H5+).
The NMR sample of R,R,SSe-(5.1) and S,S,RSe-(5.1) was allowed to stand for 24 h
after which time epimerisation at selenium had occurred to give a 1:1 mixture of
R,R,SSe-(5.1) and S,S,RSe-(5.1) and their diastereomers R,R,RSe-(5.1) and S,S,SSe-
(5.1). Data for the mixture of R,R,RSe-(5.1) and S,S,SSe-(5.1): 1H NMR: 8.85, d, J
8.1 Hz, 1H, NH; 8.00-7.88, m, 2H, ArH; 7.63-7.28, m, 8H, ArH; 4.04, dddd, J 3.9, 8.1,

Experimental 7.5
188
11,7, 12.3 Hz, 1H, CHN; 3.14, ddd, J 3.9, 10.8, 11.7 Hz, 1H, CHSe; 2.19-2.15, m, 1H;
1.92-1.87, m, 1H; 1.79-1.43, m, 3H; 1.32-0.85, m, 2H; 0.73-0.60, m, 1H.
N-[2-(Phenylselenonyl)cyclohexyl]benzamide (5.8)
m-CPBA (446 mg, 2.07 mmol) was added to a solution of the amido selenide (2.5)
(248 mg, 0.692 mmol) in dry THF (20 mL) and the solution was stirred under N2 at r.t.
for 2 h. At this time a white solid had precipitated from the mixture. This was
collected by Büchner filtration and washed with cold THF. Recrystallisation of a
sample from THF/hexane gave fine colourless needles (m.p. 99-101°C) which
noticeably coloured upon standing at r.t. ESI HRMS: 392.07610 C10H21NO3Se+H
requires 392.07594. max (KBr): 3462, 3057, 2937, 2858, 1657, 1637, 1603, 1579,
1541, 1491, 1444, 1321, 1292, 1066, 935 (as, O=Se=O), 879 (s, O=Se=O), 746,
700, 687, 671 cm-1. 1H NMR: 7.86-7.81, m, 4H, ArH; 7.64-7.59, m, 1H, ArH; 7.55-
7.49, m, 3H, ArH; 7.44-7.40, m, 2H, ArH; 7.31, d, J 6.6Hz, 1H, NH; 4.16, ddd, J 3.9,
11.4, 12.6 Hz, 1H, CHSe; 3.97, dddd, J 4.2, 6.6, 11.4, 11.4 Hz, 1H, CHN; 2.59-2.55,
m, 1H; 2.44-2.40, m, 1H; 1.96-1.72, m, 2H; 1.67-1.55, m, 2H; 1.45-1.34, m, 2H. ESI
MS: m/z 391.7 (MH+), 375.8 (MH+-O), 202.1 (M+-C6H5SeO2)
Cyclisation of 2-(phenylselenonyl)cyclohexyl benzamide (5.8)
The selenone (5.8) (14 mg, 0.036 mmol) was dissolved in dry THF (5 mL) containing
a few drops of CH2Cl2 to facilitate dissolution. The solution was immediately washed
with aqueous NaOH (30%, 15 mL), the layers were separated and the aqueous layer
was extracted with CH2Cl2 (2 x 10 mL). The combined organic layers were dried
(MgSO4) and the solvent evaporated at reduced pressure to give a pale brown oil
which 1H NMR analysis showed to be a mixture of the aziridine (2.17) and the
oxazoline (2.7) in a ratio of 7:3 (5 mg, 69%).

Experimental 7.5
189
Solution spectra of dilute solutions of (2.5), (5.1) and (5.8)
Solution spectra in CHCl3 were obtained from 4 scans over the range 4000 to 600
cm-1. The spectrum of the selenoxide (5.1) in CH2Cl2 was obtained from 20 scans
per spectrum in three spectra, over the ranges 4000 to 3000, 1700 to 1600 and 900
to 800 cm-1. The spectrum of the selenone (5.8) in CH2Cl2 was obtained from 20
scans per spectrum in three spectra, over the ranges 4000 to 3000, 1700 to 1600
and 1000 to 800 cm-1.
(i) N-[2-(Phenylselanyl)cyclohexyl]benzamide (2.5) (see also Experimental 7.2)
max (0.001M in CHCl3): 3433 (sh), 3013, 1655, 1516, 1486 cm-1. 1H NMR (0.005M
in CDCl3): 7.69-7.65, m, 2H, ArH; 7.57-7.54, m, 2H, ArH; 7.51-7.37, m, 4H, ArH;
7.31-7.22, m, 2H, ArH; 6.13, d, J 7.5 Hz, 1H, NH; 3.92, dddd, J 3.6, 7.5, 10.8, 10.8
Hz, 1H, CHN; 3.15, ddd, J 3.6, 10.8, 11.7 Hz, 1H, CHSe; 2.38-2.34, m, 1H; 2.24-
2.20, m, 1H; 1.76-1.64, m, 2H; 1.49-1.25, m, 4H.
(ii) N-[2-(Phenylseleninyl)cyclohexyl]benzamide (5.1)
max (0.001M in CHCl3): 3694 (sh), 1658, 1602, 1547, 1536, 1485, 1233, 1212, 814
(Se=O) cm-1. max (0.002M in CH2Cl2): 3431 (sh, NH), 3257 (br w, NH), 1661 (C=O),
826 (Se=O), 809 (Se=O) cm-1. max (nujol): 3223 (str, br), 2953, 2921, 1653, 1532,
1462, 1377, 1320, 1291, 814 (Se=O), 740, 699 cm-1. 1H NMR (0.005M in CDCl3):
8.07, d, J 4.8 Hz, 1H, NH; 7.94-7.91, m, 2H, ArH; 7.57-7.42, m, 8H, ArH; 3.57, dddd,
J 4.2, 4.8, 10.8, 11.1 Hz, 1H, CHN; 3.36, ddd, J 3.6, 11.1, 12.3 Hz, 1H, CHSe; 2.46-
2.42, m, 1H; 2.00-1.96, m, 1H; 1.95-1.84, m, 1H; 1.75-1.71, m, 1H; 1.62-1.27, m, 4H.
(iii) N-[2-(Phenylselenonyl)cyclohexyl]benzamide (5.8)
max (0.001M in CHCl3): 3690 (sh), 1656, 1602, 1276, 1116, 936 (as O=Se=O), 888,
868 (s O=Se=O) cm-1. max (0.002M in CH2Cl2): 3684 (sh, NH), 1666 (C=O), 935
(as O=Se=O), 880 (s O=Se=O) cm-1. 1H NMR (0.005M in CDCl3): 7.85-7.81, m,

Experimental 7.5
190
4H, ArH; 7.65-7.59, m, 1H, ArH; 7.57-7.49, m, 3H, ArH; 7.46-7.41, m, 2H, ArH; 7.19,
d, J 6.6 Hz, 1H, NH; 4.16, ddd, J 3.6, 10.8, 12.6 Hz, 1H, CHSe; 3.93, dddd, J 4.5,
6.6, 10.8, 10.8 Hz; 1H, CHN; 2.59-2.55, m, 1H; 2.48-2.44, m, 1H; 2.00-1.96, m, 1H;
1.77-1.52, m, 3H; 1.43-1.37, m, 2H.
7.5.2 NMR-Scale oxidation of 2-(phenylselanyl)cyclohexyl benzamide (2.5)
(i) 1H NMR in CD2Cl2
The amido selenide (2.5) (12 mg, 0.033 mmol) and m-CPBA (29 mg, 0.13 mmol)
were dissolved in CD2Cl2 (1.5 mL). 0.7 mL of this solution was transferred to an
NMR tube. Six 1H NMR spectra of 40 transients were recorded at 6 min intervals
followed by one spectrum recorded at 90 minutes’ reaction time. In the seven
spectra, the three compounds – selenoxide (5.1), selenone (5.8) and oxazolinium salt
(5.9) – were identified from their methine proton signals which appeared in the NMR
spectra as follows:
(a) N-[2-(Phenylseleninyl)cyclohexyl]benzamide, R,R,SSe-(5.1) and S,S,RSe-(5.1)
1H NMR: 3.99-3.89, m, 1H, CHN; 3.83-3.76, m, 1H, CHSe.
(b) N-[2-(Phenylseleninyl)cyclohexyl]benzamide, R,R,RSe-(5.1) and S,S,SSe-(5.1)
1H NMR: 4.22-4.10, m, 1H, CHN; 3.62-3.54, m, 1H, CHSe.
(c) N-[2-(Phenylselenonyl)cyclohexyl]benzamide (5.8)
1H NMR: 4.39, ddd, J 4.2, 11.4, 12.9 Hz, 1H, CHSe; 4.14, dddd, J 4.2, 7.2, 11.4,
11.4 Hz, 1H, CHN.
(d) cis-3a,4,5,6,7,7a-hexahydro-2-phenylbenzoxazole m-CBA salt (5.9)
1H NMR: 5.48, m,1H, CHO; 4.80, ddd, J 6.3, 6.6, 8.4 Hz, 1H, CHN.

Experimental 7.5
191
The proportion of each of the three products - (5.1), (5.8) and (5.9) – at each stage of
the reaction was calculated from the ratios of the integration of these methine signals
(Table 7.5.1).
TABLE 7.5.1
NMR-SCALE OXIDATION OF 2-(PHENYLSELANYL)CYCLOHEXYL BENZAMIDE (2.5) IN CD2CL2
spectrum reaction
time (min)
proportion of product (%) selenoxide
(5.1) selenone
(5.8) oxazolinium ion
(5.9)
1 6 91 9 0
2 12 58 29 11
3 18 47 36 17
4 24 37 39 25
5 30 30 37 33
6 36 26 33 41
7 90 0 0 100
After 90 minutes, the NMR sample was diluted with CH2Cl2 and washed with dilute
aqueous NaHCO3 followed by dilute aqueous NaOH. The organic layer was dried
(MgSO4) and the solvent evaporated at reduced pressure to give a colourless oil
which 1H NMR analysis showed to be a mixture with the oxazoline (2.7) as the main
product.

Experimental 7.5
192
(ii) 1H NMR in THF-d8
The amido selenide (2.5) (12 mg, 0.033 mmol) and m-CPBA (30 mg, 0.14 mmol)
were dissolved in CD2Cl2 ( ~0.3 mL) and THF-d8 (1 mL). 0.7 mL of this solution was
transferred to an transferred to an NMR tube. Eight 1H NMR spectra of 40 transients
were acquired at 6 min intervals, followed by one spectrum at 40 hours’ reaction time.
The three compounds – selenoxide (5.1), selenone (5.8) and oxazolinium salt (5.9) –
were identified from their methine proton signals which appeared in the NMR spectra
as follows:
(a) N-[2-(Phenylseleninyl)cyclohexyl]benzamide (5.1)
1H NMR: 4.06-3.93, m, 1H, CHN; 3.38-3.30, m, 1H, CHSe.
b) N-[2-(Phenylselenonyl)cyclohexyl]benzamide (5.8)
1H NMR: 4.19, ddd, J 3.9, 11.4, 12.9 Hz, 1H, CHSe; 4.05, dddd, J 4.1, 7.2, 11.4,
11.4 Hz, 1H, CHN.
(c) cis-3a,4,5,6,7,7a-hexahydro-2-phenylbenzoxazole m-CBA salt (5.9)
1H NMR: 5.41, ddd, J 4.5, 4.8, 8.4 Hz, 1H, CHO; 4.78, ddd, J 6.3, 6.6, 8.4 Hz, 1H,
CHN.
The proportion of each of the three products - (5.1), (5.8) and (5.9) – at each stage of
the reaction was calculated from the ratios of the integration of these methine signals
(Table 7.5.2).

Experimental 7.5
193
TABLE 7.5.2
NMR-SCALE OXIDATION OF 2-(PHENYLSELANYL)CYCLOHEXYL BENZAMIDE (2.5) IN d8-THF
spectrum reaction
time (min)
proportion of product (%) selenoxide
(5.1) selenone
(5.8) oxazolinium ion
(5.9)
1 6 83 17 0
2 12 79 21 0
3 18 37 60 3
4 24 15 81 4
5 30 7 89 4
6 36 0 96 4
7 42 0 95 5
8 48 0 93 7
9 2400 0 1 99
After 40 h, the NMR sample was diluted with CH2Cl2 and washed with dilute aqueous
NaOH. The organic layer was dried (MgSO4) and the solvent evaporated at reduced
pressure to give a mixture in which the oxazoline (2.7) was the predominant product.
cis-3a,4,5,6,7,7a-hexahydro-2-phenylbenzoxazole hydrochloride (5.10)
An NMR sample of the oxazoline (2.7) in CDCl3 was shaken with two drops of
concentrated HCl. The methine signals of the hydrochloride were compared with the
product (5.9) from the NMR-scale oxidations of the selenide (2.5):

Experimental 7.5
194
cis-3a,4,5,6,7,7a-hexahydro-2-phenylbenzoxazole (2.7)
1H NMR: 4.68, ddd, J 5.1, 5.7, 8.1 Hz, 1H, CHO; 4.13, ddd, J 6.0, 6.6, 8.1 Hz, 1H,
CHN.
cis-3a,4,5,6,7,7a-hexahydro-2-phenylbenzoxazolinium hydrochloride (5.10)
1H NMR: 5.46, ddd, J 4.4, 4.8, 8.8 Hz, 1H, CHO; 4.79, ddd, J 5.8, 5.8, 8.8 Hz, 1H,
CHN.
(iii) 77Se NMR in THF-d8 at low temperature
A solution of the amido selenide (2.5) (50 mg, 0.14 mmol) in dry THF (0.7 mL) was
added to a solution of m-CPBA (144 mg, 0.67 mmol) in dry THF (0.5 mL) at r.t. 0.7
mL of this solution was used to prepare an NMR sample which was placed in the
probe which had been cooled to –30°C. The probe was further cooled to –60°C. A
spectrum was acquired at this temperature over 2 hours, spectra being recorded at
intervals of 20-30 minutes. Two signals were constant for the first 80 minutes: 77Se
NMR 1010.31, 843.86 while one signal drifted from 859.16 (30 min) to 859.24
(45 min) to 859.51 (50-80 min) to 859.59 (95-120 min). The probe was warmed
to –40°C and a spectrum acquired over 30 minutes at this temperature, 77Se NMR
860.03. The probe was then warmed to above 0°C and the sample was removed.
A white solid had precipitated from the NMR sample solution. The precipitate was
collected, giving a white solid (4 mg), which 1H NMR analysis showed to be
predominantly the selenone (5.8) with the oxazoline (2.7) as the minor product. The
precipitate was dissolved in CH2Cl2 (6 mL) and washed with aqueous NaOH (20%, 6
mL). The organic layer was dried (MgSO4) and the solvent evaporated at reduced
pressure to give a 3:1 mixture of the aziridine (2.17) and oxazoline (2.7) as a
colourless oil (2 mg, 97% from 4 mg selenone). Evaporation of the filtrate gave a
white solid which was dissolved in CDCl3 containing a few drops of CD3OD to

Experimental 7.5
195
facilitate dissolution. A 1H NMR spectrum showed essentially only aromatic signals,
consistent with the spectrum of a mixture of m-CPBA and m-CBA.

Experimental 7.6
196
7.6 WORK DESCRIBED IN CHAPTER 6
Methyl phenyl selenide[74-75] (6.1)
A solution of diphenyl diselenide (1.00 g, 3.21 mmol) in THF (10 mL) was added to a
suspension of sodium hydride (249 mg, 8.30 mmol) in THF (10 mL) under N2. The
resulting mixture was refluxed for 1 h 40 min, then cooled to r.t., and to it was added
methyl iodide (0.33 mL, 5.3 mmol) and the mixture was stirred at r.t. for 41 h. The
mixture was diluted with Et2O (20 mL) and washed with half-saturated aqueous NaCl
(15 mL). The aqueous layer was extracted with Et2O (2 x 15 mL) and the combined
organic layers were washed with saturated aqueous NaCl (10 mL) and dried
(Na2SO4). Evaporation of the solvent at reduced pressure gave a yellow oil (0.9728
g) which was purified by Kugelrohr distillation (50°C/0.1 mm) to give the title
compound[74, 301] as a pale yellow oil (738 mg, 81%). 1H NMR: 7.44-7.41, m, 2H,
ArH; 7.29-7.19, m, 3H, ArH; 2.35, s, JSeH 11.1 Hz, 3H, CH3. 13C NMR: 131.79,
130.41, 128.97, 126.07, all Ar; 7.17, Jcse 253 Hz, CH3. MS: m/z 172 (M+), 157 (M+-
CH3), 91 (C7H7+), 77 (C6H5
+).
Methyl phenyl selenone[249] (6.2)
A solution of methyl phenyl selenide (413 mg, 2.41 mmol) in CH2Cl2 was cooled to
0°C and to it was added m-CPBA (1.296 g, 6.08 mmol) and the resulting mixture was
stirred for 23 h, then allowed to warm to r.t. The mixture was then cooled to 0°C and
the precipitated m-CBA was removed by filtration. The yellow filtrate was washed
with aqueous NaOH (0.75 M, 10 mL) and saturated aqueous NaCl (10 mL), and dried
(Na2SO4), and the solvent was removed at reduced pressure to give a colourless
solid (399 mg). Recrystallisation from ethyl acetate gave the title compound[249] as

Experimental 7.6
197
colourless crystals (199 mg, 41%), m.p. 110-114°C (lit.[249] m.p. 130.5-131°C). 1H
NMR: 8.04-8.01, m, 2H, ArH; 7.78-7.65, m, 3H, ArH; 3.31, s, JSeH 8.1 Hz, 3H, CH3.
13C NMR: 142.65, 134.36, 130.31, 126.49, all Ar; 44.26, CH3.
One-pot preparation of styrene oxide
(i) Reaction of benzaldehyde and -lithiomethylphenylselenone[249]
A solution of LDA in THF was prepared by the addition of n-butyllithium (140 L, 1.85
M, 0.259 mmol) to a solution of diisopropylamine (38 L, 0.27 mmol) in THF (3 mL) at
0°C under N2.[3] The LDA solution was then cooled to -78°C. A solution of methyl
phenyl selenone (51 mg, 0.25 mmol) and benzaldehyde (35 L, 0.34 mmol) in THF
(5 mL) was cooled to –78°C and to it was added the LDA solution via cannula. The
resulting mixture was stirred for 15 min at -78°C, and to it was added a solution of
acetic acid (30 L, 0.52 mmol) in THF (0.5 mL). The mixture was stirred a further 55
min, while being allowed to warm to r.t., and then was diluted with CH2Cl2 (15 mL).
The reaction mixture was washed with a mixture of aqueous HCl (10%, 2 mL) and
saturated aqueous NaCl (2 mL), then washed with saturated aqueous NaHCO3 (3
mL) followed by saturated aqueous NaCl (3 mL) and dried (Na2SO4). Evaporation of
the solvent at reduced pressure gave a yellow oil (52 mg). NMR analysis showed
the oil to be a complex mixture with none of the expected peaks for styrene oxide [302]
apparent.
(ii) Reaction of benzaldehyde and potassium phenylselenonylmethylate
A solution of methyl phenyl selenone (29 mg, 0.14 mmol) and benzaldehyde (15 L,
0.15 mmol) in THF (7 mL) was cooled to -60°C and to it was added t-BuOK (40 mg,
0.36 mmol) and the mixture was stirred for 1.5 h, then allowed to warm to r.t. The

Experimental 7.6
198
mixture was quenched with water (10 mL) and the layers were separated. The
aqueous layer was extracted with CH2Cl2 (2 x 15 mL) and the combined organic
layers were washed with saturated aqueous NaCl (10 mL) and dried (Na2SO4).
Evaporation of the solvent at reduced pressure gave a partly-solid product (4 mg)
which NMR analysis showed to be a mixture of styrene oxide[302] and methyl phenyl
selenone in a ratio of 6:1.
Attempted one-pot preparation of 2-phenyloxetane via the reaction of styrene
oxide with potassium phenylselenonylmethylate
A solution of methyl phenyl selenone (42 mg, 0.21 mmol) and styrene oxide (52 mg,
0.43 mmol) in THF (12 mL) was cooled to 0°C and to it was added t-BuOK (46 mg,
0.41 mmol). The mixture was stirred for 50 min, the flask was removed from the ice
bath, and the mixture was diluted with water (10 mL). The layers were separated,
the aqueous layer was extracted with CH2Cl2 (15 mL), and the combined organic
layers were washed with saturated aqueous NaCl (10 mL) and dried (Na2SO4).
Evaporation of the solvent at reduced pressure gave a yellow solid (21 mg) which
NMR analysis showed to be a mixture containing methyl phenyl selenone and
styrene oxide in a ratio of 2.4:1 with none of the expected peaks for 2-
phenyloxetane[302] apparent.
Attempted preparation[250] of 1-phenyl-3-phenylseleninyl-1-propanol
A solution of LDA in THF was prepared[119] by the addition of n-butyllithium (2.1 mL,
1.5 M, 3.2 mmol) to a solution of diisopropylamine (445 L, 3.18 mmol) in THF (0.6
mL) at -78°C under N2. The solution was allowed to warm to 0°C and was kept at
this temperature under N2 until needed. To a solution of methyl phenyl selenide

Experimental 7.6
199
(260 mg, 1.52 mmol) in THF (3 mL) at -70°C was added a solution of m-CPBA (277
mg, 1.61 mmol) in THF (1.5 mL). The mixture was stirred for 30 min, then cooled to
–78°C, and to it was added the LDA solution followed by styrene oxide (175 L, 1.54
mmol). Stirring was continued for 20 min, then water (0.5 mL) containing a few
drops of acetic acid was added followed by Et2O (10 mL). The layers were separated
and the organic layer was washed with half-saturated aqueous NaCl (15 mL) and the
aqueous layer was extracted with Et2O (15 mL). The combined organic layers were
washed with saturated aqueous NaCl and dried (Na2SO4) and the solvent was
evaporated at reduced pressure to give slightly impure styrene oxide[302] (196 mg,
106%) which was identified from its IR and 1H NMR spectra. max (neat): 3085, 3052,
3037, 2989, 2912, 1726, 1496, 1479, 1454, 1390, 1292, 1258, 1202, 1128, 1074,
1027, 985, 877, 812, 759, 700 cm-1. 1H NMR: 7.38-7.25, m, 5H, ArH; 3.87, dd, J
2.6, 4.1 Hz, CHO; 3.15, dd, J 4.1, 5.6 Hz, 1H, CHH; 2.81, dd, J 2.6, 5.6 Hz, 1H, CHH.
3-Phenyl-3-phenylselenopropanal (6.5)
A solution of diphenyl diselenide (500 mg, 1.60 mmol) in THF (15 mL) was added to
a suspension of NaH (128 mg, 4.27 mmol) under N2, and the mixture was refluxed for
24 h and then cooled in ice. Cinnamaldehyde ( 0.61 mL, 4.8 mmol) was added and
the mixture was stirred for 2 h. Acetic acid (0.28 mL, 4.9 mmol) was added and this
mixture was stirred for 24 h, then poured into saturated aqueous NaHCO3 (10 mL).
The layers were separated and the aqueous layer was extracted with Et2O (30 mL).
The combined organic layers were washed with saturated aqueous NaCl (10 mL),
dried (Na2SO4) and the solvent evaporated at reduced pressure to give an orange oil
(1.044 g). Chromatography (CH2Cl2/hexane 3:2) gave the title compound (approx.
50%) contaminated with diphenyl diselenide and cinnamaldehyde as a yellow oil (580

Experimental 7.6
200
mg). From integration of the signals in the 1H NMR spectrum, the ratio of the title
compound to cinnamaldehyde in the product was estimated as 4:1. Decomposition
of the selenide during chromatography was evident from the appearance of a yellow
band (diphenyl diselenide) coincident with elution of the selenide. Data for (6.5):
max(neat) 3058, 3029, 2822, 2726, 1721,1577, 1494, 1475, 1452, 1437, 1066, 1021,
1000 cm-1. 1H NMR: 9.69, t, J 1.5 Hz, CHO; 7.67-7.61, m, 2H, ArH; 7.51-7.40, m,
2H, ArH; 7.34-7.22, m, 6H, ArH; 4.81, dd, J 7.2, 8.4 Hz, CHSe; 3.27, ddd, J 1.5, 8.4,
17.4 Hz, 1H, CHaHb; 3.12, ddd, J 1.5, 7.2, 17.4 Hz, 1H, CHaHb. 13C NMR: 199.68,
C=O; 136.00, 131.50, 129.12, 128.99, 128.50, 127.67, 127.47, 127.36, all Ar; 49.22,
CH2; 40.38, CSe. MS: m/z 290 (M+), 157 (C6H5Se+), 133 (M-C6H5Se+), 105
(C6H5CH2CH2+), 77 (C6H5
+).
3-Phenyl-3-phenylseleno-1-propanol (6.6)
To a solution of impure 3-phenyl-3-phenylseleno-1-propanal (6.5) (contaminated with
cinnamaldehyde and diphenyl diselenide) (366 mg, ~0.98 mmol in (6.5)) in EtOH (15
mL) was added NaBH4 (68 mg, 1.8 mmol) in three portions over 10 min. The
mixture was stirred at r.t. under N2 for 1 h, then diluted with water (5 mL). 10%
aqueous HCl was added dropwise until no further H2 evolution was observed. The
mixture was extracted with Et2O (2 x 15 mL) and the combined organic layers were
washed with saturated aqueous NaCl, dried (Na2SO4) and the solvent evaporated at
reduced pressure to give a yellow oil (350 mg). Chromatography (Et2O/hexane
60:40) gave the title compound as a pale yellow oil (103 mg, 36%). HRMS: 292.0366
C15H16OSe requires 292.0367. max (neat): 3575, 3359, 3059, 3028, 2935, 2879,
1577, 1493, 1475, 1452, 1437, 1155, 1041, 1022, 739, 694 cm-1. 1H NMR: 7.40-
7.38, m, 2H, ArH; 7.29-7.16, m, 8H, ArH; 4.44, t, J 7.8 Hz, 1H, CHSe; 3.72, dt, J 6.0,
10.8 Hz, 1H, CHaHbO; 3.59, dt, J 6.3, 10.8 Hz, 1H, CHaHbO; 2.29, ddd, J 6.0, 6.3, 7.8

Experimental 7.6
201
Hz, 2H, CHCH2. 13C NMR: 135.49, 131.53, 129.17, 128.82, 128.39, 127.88, 127.69,
127.04, all Ar; 61.10, CHSe; 44.85, CH2O; 38.65, CHCH2. MS: m/z 292 (M+), 157
(C6H5Se+), 135 (M+-C6H5Se), 117 (M+-C6H5Se-H2O), 105 (C8H9+), 91 (C7H7
+), 77
(C6H5+). Further elution gave a fraction containing a mixture which was
chromatographed (EtOAc/hexane 45:55) to give the title compound (55 mg, 20%) as
a pale yellow oil. Further elution gave a fraction (48 mg) containing a mixture of the
title compound (6.6), cinnamyl alcohol[253-254] (6.7) and 3-phenyl-1-propanol[255] (6.8)
in a ratio of 2:5:3. Further elution gave a fraction (6 mg) containing a mixture of
cinnamyl alcohol[253-254] (6.7) and 3-phenyl-1-propanol[255] (6.8) in a ratio of 1:1. 1H
NMR: 7.41-7.19, m, 10H, ArH; 6.62, dd, J 1.5, 15.9 Hz, 1H, CCH (6.7); 6.37, dt, J
5.7, 15.9 Hz, 1H, CCHCH (6.7); 4.33, dd, J 1.5, 5.7 Hz, 2H, CH2O (6.7); 3.68, t, J 6.3
Hz, 2H, CH2O (6.8); 2.69, t, J 7.8 Hz, CCH2 (6.8); 1.90, tt, J 6.3, 7.8 Hz, CH2CH2CH2
(6.8); 1.60-1.50, br s ,2H ,OH (6.7 and 6.8).
Bis(phenylseleno)methane[75, 260] (6.9)
A solution of diphenyl diselenide (1.000 g, 3.20 mmol) in THF (10 mL) was added via
cannula to a suspension of NaH (257 mg, 8.57 mmol) in THF (10 mL) under N2. The
mixture was refluxed for 100 min, then cooled to r.t. and to it was added methylene
iodide (335 L, 4.16 mmol). The resulting mixture was stirred for 19 h, then diluted
with Et2O (20 mL) and washed with half-saturated aqueous NaCl (15 mL). The
aqueous layer was extracted with Et2O (2 x 15 mL) and the combined organic layers
were washed with saturated aqueous NaCl (10 mL) and dried (Na2SO4) and the
solvent was removed at reduced pressure to give a yellow oil (1.125 g). Kugelrohr
distillation (150-160°C/0.2mm, lit.[260] b.p. 138°C/0.1mm) gave the title compound[91,
260] as a pale yellow oil (977 mg, 94%). max (neat): 3069, 3055, 3014, 2997, 2915,

Experimental 7.6
202
2852, 1578, 1477, 1453, 1437, 1378, 1299, 1133, 1070, 1022, 999, 733, 690, 670
cm-1. 1H NMR: 7.56-7.50, m, 4H, ArH; 7.31-7.25, m, 6H, ArH; 4.22, s, 2H, CH2. 13C
NMR: 132.99, 130.79, 129.08, 127.49, all Ar; 20.97, CH2. MS: m/z 328 (M+), 171
(M+-C6H5Se), 91 (C7H7+).
Styrene oxide (6.11)[303]
To a solution of styrene (1.0447 g, 100 mmol) in CH2Cl2 (100 mL) was added pH 8
phosphate buffer solution (0.1M, 100 mL) and the solution was cooled to 0°C. To
the solution was added m-CPBA (2.459 g, 100 mmol) in small portions over 10 min.
The flask was removed from the cooling bath and the reaction mixture was stirred for
4 h, then cooled again to 0°C and to it was added m-CPBA (2.494 g, 100 mmol) in
small portions over 20 min. The flask was again removed from the cooling bath and
stirring was continued for 3 h. Saturated aqueous Na2S2O3 (15 mL) was added and
the mixture was stirred 5 min. The layers were separated and the organic layer was
washed with Na2S2O3 (15 mL), then with half-saturated aqueous NaCl (20 mL), and
dried (Na2SO4), and the solvent was evaporated at reduced pressure to give the title
compound[302] as a colourless oil (0.9728 g, 81%). max (neat): 3085, 3052, 3037,
2989, 2912, 1726, 1496, 1479, 1454, 1390, 1292, 1258, 1202, 1128, 1074, 1027,
985, 877, 812, 759, 700 cm-1. 1H NMR: 7.38-7.25, m, 5H, ArH; 3.87, dd, J 2.6, 4.1
Hz, CHO; 3.15, dd, J 4.1, 5.6 Hz, 1H, CHH; 2.81, dd, J 2.6, 5.6 Hz, 1H, CHH.
Preparation of -hydroxy selenides via the reaction of an epoxide
with -phenylselenomethyllithium[86, 88, 252]
Procedure 7.6A: A solution of bis(phenylseleno)methane (6.9) in THF was cooled
to –78°C and to it was added n-butyllithium. The solution was stirred for 1 h and to
it was added HMPA followed by a solution of the epoxide in THF. The mixture was

Experimental 7.6
203
stirred at –78°C for 2 h and then removed from the cooling bath and stirred a
further 1.5 h. The flask was placed in ice and the reaction was quenched with
dropwise addition of saturated aqueous NH4Cl (5 mL). A further portion of saturated
aqueous NH4Cl (15 mL) was added, followed by water (10 mL). The mixture was
extracted with Et2O (2 x 20 mL) and the combined organic layers were washed with
saturated aqueous NaCl (10 mL) and dried (Na2SO4), and the solvent evaporated at
reduced pressure.
1-Phenyl-3-phenylseleno-1-propanol (6.11)
Following Procedure 7.6A, a mixture of bis(phenylseleno)methane (6.9) (1.00 g, 3.07
mmol) and n-butyllithium (1.6 M in hexane, 2.5 mL, 4.0 mmol) in THF (10 mL), stirred
for 1 h, followed by addition of HMPA (0.54 mL, 3.1 mmol) and a solution of styrene
oxide (561 mg, 4.67 mmol) in THF (5 mL) and stirred for 2 h after removal from the
cooling bath, gave a pale yellow oil (1.4317 g). Chromatography
(CH2Cl2/hexane/EtOH 68:30:2) gave n-butyl phenyl selenide[304] (6.13) as a yellow oil
(406 mg, 62%). max (neat): 3071, 3058, 2959, 2928, 2871, 2859, 1579, 1476, 1463,
1437, 1378, 1296, 1258, 1199, 1073, 1022, 999, 900, 734, 690, 670 cm-1. 1H
NMR:7.50-7.46, m, 2H, ArH; 7.29-7.22, m, 3H, ArH; 2.92, t, J 7.5 Hz, 2H, CH2Se;
1.68, tq, J 7.5, 7.5 Hz, 2H, CH3CH2; 1.43, qn, J 7.5 Hz, CH3CH2CH2; 0.91, t, 7.5 Hz,
3H, CH3. 13C NMR(200 MHz): 132.37, 129.30, 128.84, 126.56, all Ar; 32.24, 27.61,
22.92, 13.52. MS: m/z 214 (M+), 158 (C6H5SeH+), 78 (C6H6+), 77 (C6H5
+). Further
elution gave 1-phenyl-2-(phenylseleno)-ethanol[136] (6.13) as a pale yellow oil (155
mg, 18%). 1H NMR:7.60-7.52, m, 2H, ArH; 7.38-7.20, m, 8H, ArH; 4.75, dd, J 3.9,
9.3 Hz, 1H, CHOH; 3.31, dd, J 3.9, 12.6 Hz, 1H, CHaHbSe; 3.14, dd, J 9.3, 12.6 Hz,
1H CHaHbSe; 2.80, br s, 1H, OH. 13C NMR(200 MHz): 142.48, 133.07, 129.21,

Experimental 7.6
204
129.06, 128.49, 127.87, 127.34, 125.77 all Ar; 72.23, CHO; 38.20, CHSe. Further
elution gave the title compound[261] as a pale yellow oil (389 mg, 44%). max (neat):
3390, 3059, 3029, 2935, 2874, 1578, 1493, 1477, 1453, 1437, 1392, 1359, 1328,
1301, 1248, 1203, 1184, 1054, 1023, 1001, 912, 896, 763, 735, 700, 670, 649 cm-1.
1H NMR: 7.49-7.46, m, 2H, ArH; 7.37-7.22, m, 8H, ArH; 4.83, dd J 5.1, 7.8 Hz, 1H,
CHO; 2.98, dd, J 7.2, 7.8 Hz, 2H, CH2Se; 2.18, ddt, J 7.2, 7.8, 14.1 Hz, 1H,
CHCHaHbCH2; 2.05, ddt, J 5.1, 7.8, 14.1 Hz, 1H, CHCHaHbCH2, 1.95, br s, 1H, OH.
13C NMR: 143.93, 132.54, 129.95, 129.07, 128.54, 127.73, 126.84, 125.80, all Ar;
73.84, CHO; 39.02, CHSe; 23.80. MS: m/z 292 (M+), 275 (M+-OH), 185 (M+-
C6H5CH2O), 157 (C6H5Se+), 134 (M+-C6H5SeH), 117 (C9H9+), 107 (C6H5CH2O+),
77(C6H5+).
1-Phenyl-4-phenylseleno-2-butanol (6.18)
Following Procedure 7.6A, a mixture of bis(phenylseleno)methane (6.9) (880 mg,
2.69 mmol) and n-butyllithium (2.45 M in hexane, 1.44 mL, 3.53 mmol) in THF (10
mL), stirred for 70 min, followed by addition of HMPA (0.47 mL, 2.7 mmol) and a
solution of 2-benzyloxirane (540 L, 4.10 mmol) in THF (5 mL) and stirred for 6 h
after removal from the cooling bath, gave a yellow oil (596 mg). Kugelrohr distillation
(25-100°C/0.2mm) gave a fraction containing a mixture of n-butyl phenyl selenide
(6.12), HMPA and 2-benzyloxirane as a colourless oil. The residue, a yellow oil
(0.5045 g), was chromatographed (CH2Cl2) to give 1-phenyl-4,4-bis(phenylseleno)-2-
butanol (6.23) as a pale yellow oil (173 mg, 14%). EI HRMS: 461.9998 C22H22OSe2
requires 462.0002. max (neat): 3555 (sharp, OH), 3435 (broad, OH), 3057, 3026,
2931, 1578, 1494, 1476, 1437, 1069, 1022, 1000, 739, 691, 669 cm-1. 1H NMR:
7.59-7.52, m, 4H, ArH; 7.47-7.19, m, 9H, ArH; 7.17-7.13, m, 2H, ArH; 4.71, dd, J 4.8,

Experimental 7.6
205
9.3 Hz, 1H, CH(Se)Se; 4.25, m, 1H, CHO; 2.70, dd, J 5.4, 13.5 Hz, 1H, CCHaHb;
2.66, dd, J 7.5, 13.5 Hz, 1H, CCHaHb; 2.14, ddd, J 4.8, 9.0, 14.7 Hz, 1H,
CHCHaHbCH; 2.05, ddd, J 3.3, 9.3, 14.7 Hz, 1H, CHCHaHbCH; 1.75, d, J 3.0 Hz, 1H,
OH. 13C NMR(200 MHz): 137.78, 134.83, JCSe 36.4 Hz; 134.33, JCSe 38.0 Hz;
130.39, 129.31, 129.26, 129.00, 128.93, 128.49, 128.04, 127.84, 126.46, all Ar;
71.00, 43.81, 43.57, 39.78. MS: m/z 462 (M+); 287 (M+-C6H5Se-H2O); 185
(C6H5SeCH2CH2+); 147 (M+-C6H5Se- C6H5Se-H); 129 (C10H9
+); 103 (C8H7+); 91
(C7H7+); 77 (C6H5
+). Further elution gave a fraction containing a mixture as a yellow
oil (192 mg) which was chromatographed (hexane/CH2Cl2/EtOH 30:69:1) to give 1-
phenyl-3-phenylseleno-2-propanol[266] (6.22) as a yellow oil (19 mg, 2%). ESI
HRMS: 275.0333 C12H26O2-OH requires 275.0339. max (neat): 3426 (br, OH), 3060,
3028, 2923, 2849, 1671, 1650, 1579, 1478, 1451, 1437, 1428, 1314, 1297, 1260,
1071, 1022, 736, 691 cm-1. 1H NMR: 7.53-7.44, m, 2H, ArH; 7.34-7.15, m, 8H,
ArH; 3.98-3.88, m, 1H, CHO; 3.13, dd, J 4.0, 12.8 Hz, 1H, CHaHbSe; 2.93, dd, J 8.0,
12.8 Hz, 1H, CHaHbSe; 2.87, d, J 6.8 Hz, 2H, C6H5CH2; 2.35, br s, OH. 13C NMR:
137.79, 132.86, 129.38, 129.20, 128.53, 127.24, 126.60, all Ar; 71.03, CO; 42.90,
35.88. One of the aromatic carbon signals did not appear in the spectrum. MS: m/z
292 (M+); 201 (M+-C7H7); 183 (M+-C7H7-H2O); 157 (C6H5Se+); 117 (M+-C6H5Se-H2O).
Further elution gave the title compound as a pale yellow oil (109 mg, 13%). 1H
NMR: 7.56-7.50, m, 2H, ArH; 7.39-7.19, m, 8H, ArH; 4.05-3.98, m, 1H, CHO; 3.15,
dt, J 7.2, 12.3 Hz, 1H, CHaHbSe; 3.05, dt, J 7.5, 12.3 Hz, 1H, CHaHbSe; 2.85, dd, J
4.2, 13.5 Hz, 1H, CCHaHbCH; 2.72, dd, J 8.1, 13.5 Hz, 1H, CCHaHbCH; 1.99-1.92, m,
2H, CHCH2CH2Se; 1.64, br s, 1H, OH. MS: m/z 306 (M+); 213 (M+-C7H7Se-H2); 157
(C6H5Se+); 91 (C7H7+).

Experimental 7.6
206
1-Phenylseleno-3-undecanol (6.17)
Following Procedure 7.6A, a mixture of bis(phenylseleno)methane (6.9) (864 mg,
2.65 mmol) and n-butyllithium (2.5 M in hexane, 1.27 mL, 3.18 mmol), stirred for 45
min, followed by addition of 1,2-epoxydecane (740 L, 3.98 mmol) and HMPA (0.47
mL, 2.7 mmol), and stirred for 2 h after removal from cooling bath, gave a yellow oil
(1.421 g). Kugelrohr distillation (25-100°C/0.2 mm) gave a fraction containing a
mixture of n-butyl phenyl selenide, 1,2-epoxydecane and HMPA as a colourless oil.
The residue was chromatographed ((hexane/CH2Cl2/EtOH, 30:69:1) to give 1-
phenylseleno-2-decanol[264] (6.19) as a yellow oil (119 mg, 14%). max (neat): 3373,
2953, 2926, 2854, 1579, 1477, 1466, 1437, 1408, 1385, 1072, 1022, 1001, 737, 690
cm-1. 1H NMR: 7.56-7.50, m, 2H, ArH; 7.32-7.24, m, 3H, ArH; 3.66, dddd, J 3.3,
5.1, 6.9, 8.7 Hz, 1H, CHO; 3.15, dd, J 3.3, 12.6 Hz, 1H, CHaHbSe; 2.88, dd, J 8.7,
12.6 Hz, 1H, CHaHbSe; 2.36, br s, 1H, OH; 1.56-1.44, m, 2H, CH2CHO; 1.43-1.25, m,
12H; 0.87, t, J 6.6 Hz, 3H, CH3. 13C NMR: 132.99, 129.15, 129.05, 127.21, all Ar;
69.87, CH2O; 37.25; 36.59, CH2Se; 31.80; 29.53; 29.44; 29.18; 25.75; 22.60; 14.04.
MS: m/z 314 (M+), 297 (M+-OH), 172 (C6H5SeCH3+). Further elution gave the title
compound as a pale yellow solid (210 mg, 24%) which was recrystallised from
hexane to give colourless plates, m.p. 47-49.5°C. ESI HRMS: 329.1373
C17H28OSe+H requires 329.1378. max (neat): 3392, 2954, 2918, 2899, 2872, 2848,
1579, 1477, 1470, 1437, 1402, 1342, 1257, 1078, 1055, 1034, 1022, 899, 729, 690
cm-1. 1H NMR: 7.55-7.48, m, 2H, ArH; 7.32-7.26, m, 3H, ArH; 3.75-3.69, m, 1H,
CHO; 3.11-2.94, m, 2H, CH2Se; 1.88-1.78, m, 2H, CHCH2CH2Se; 1.55-1.26, m, 16H;
0.88, t, J 6.6 Hz, 3H, CH3. 13C NMR: 132.58, 132.55, 129.02, 126.78, all Ar; 71.55,
CH2O; 37.42, CH2Se; 31.82; 29.56; 29.50; 29.19; 25.52; 25.27; 24.12; 22.60; 14.02.

Experimental 7.6
207
MS: m/z 328 (M+); 311 (M+-OH); 185 (C6H5CH2CH2+); 171 (M+-C6H5; C6H5SeCH2
+);
158 (M+-C6H5SeCH2; C6H5SeH+); 141 (C9H17O+); 57 (C3H5O+); 43 (C2H3O+).
Cyclisation of -hydroxy selenides
Attempted cyclisation of 1-Phenyl-3-phenylseleno-1-propanol (6.11)
(i) in THF at low temperature
Following Procedure 7.3E, the reaction of 1-phenyl-3-phenylseleno-1-propanol (6.11)
(62 mg, 0.21 mmol), m-CPBA (207 mg, 70%, 0.84 mmol) and t-BuOK (202 mg, 1.80
mmol) gave a negligible yield of a pale yellow oil. NMR analysis showed the product
to be a complex mixture which was not purified further.
(ii) in THF at r.t.
To a solution of 1-phenyl-3-phenylseleno-1-propanol (6.11) (40 mg, 0.14 mmol) in
THF (15 mL) was added m-CPBA (100 mg, 70%, 0.41 mmol) and the mixture was
stirred at r.t. for 3 d. The mixture was then cooled to -78°C and to it was added t-
BuOK (140 mg, 1.25 mmol) and the mixture was removed from the cooling bath and
stirred for 2.5 h. The reaction was quenched with aqueous Na2S2O3 (0.5 M, 8 mL)
and the mixture was diluted with Et2O (15 mL). The layers were separated and the
organic layer was washed with aqueous NaOH (10%, 10 mL) followed by saturated
aqueous NaCl (10 mL), and dried (Na2SO4). Evaporation of the solvent at reduced
pressure gave a pale yellow oil (20 mg). Chromatography (EtOAc/hexane 60:40 to
80:20) gave 1-phenyl-1,3-propanediol[262-263] (6.14) as a colourless oil (5 mg, 23%).
1H NMR: 7.39-7.34, m, 3H, ArH; 7.33-7.27, m, 2H, ArH; 4.97, dd, J 3.6, 8.7 Hz, 1H,
CHO; 3.87, dd, J 5.1, 5.7 Hz, 2H, CH2O; 2.87, br s, 1H, OH; 2.42, br s, 1H, OH;
2.07-1.90, m, 2H, CCHaHbC. 13C NMR: 144.29, 128.50, 127.58, 125.61, all Ar;

Experimental 7.6
208
74.33, CHOH; 61.44, CH2OH; 40.50. Further elution gave a fraction which NMR
analysis showed to be complex mixture which was not purified further.
2-Phenyloxetane[169] (6.3)
A solution of 1-phenyl-3-phenylseleno-1-propanol (6.11) (102 mg, 0.350 mmol) and
m-CPBA (150 mg, 0.87 mmol) in methanol (3 mL) was stirred at r.t. for 30 min.
Aqueous NaOH (1 M, 1.75 mL, 1.75 mmol) was added and the solution was stirred a
further 18 h. Half-saturated aqueous NaCl (10 mL) was added and the aqueous
layer was extracted with Et2O (2 x 20 mL). The combined organic layers were
washed with saturated aqueous NaCl (10 mL) and dried (Na2SO4), and the solvent
was evaporated at reduced pressure to give a colourless oil (26 mg).
Chromatography (hexane/EtOAc, 9:1 to 7:3) gave the title compound[258-259] as a
colourless oil (9 mg, 20%). 1H NMR: 7.47-7.27, m, 5H, ArH; 5.82, t, J 7.5 Hz, 1H,
CHO; 4.84, ddd, J 5.7, 7.8, 8.1 Hz, CHaHbO; 4.67, ddd, J 5.4, 5.7, 9.3 Hz, 1H,
CHaHbO; 3.03, dddd, J 5.4, 7.5, 8.1, 11.1 Hz, 1H, CHCHaHb; 2.67, dddd, J 7.5, 7.8,
9.3, 11.1 Hz, 1H, CHCHaHb. Further elution gave 3-methoxy-1-phenyl-1-
propanol[262] (6.15) as a colourless oil (7 mg, 12%). 1H NMR: 7.39-7.33, m, 3H,
ArH; 7.29-7.25, m, 2H, ArH; 4.92, dd, J 3.9, 8.1 Hz, 1H, CHO; 3.62-3.53, m, 2H,
CHaHbO; 3.38, s, 3H, CH3; 3.30, br s, 1H, OH; 2.08-1.93, m, 2H, CCHaHbC. 13C
NMR: 144.39, 128.33, 127.26, 125.65, all Ar; 73.56, COH; 71.18, CH3; 58.89,
CH2OH; 38.53. Further elution gave 1-phenyl-1,3-propanediol[262-263] (6.14) as a
brown oil (trace). 1H NMR: 7.39-7.34, m, 3H, ArH; 7.33-7.27, m, 2H, ArH; 4.97,
dd, J 3.6, 8.7 Hz, 1H, CHO; 3.87, t, J 5.4 Hz, 2H, CH2O; 2.87, br s, 1H, OH; 2.42, br
s, 1H, OH; 2.07-1.90, m, 2H, CCHaHbC. 13C NMR: 144.29, 128.50, 127.58, 125.61,
all Ar; 74.33, CHOH; 61.44, CH2OH; 40.50.

Experimental 7.6
209
3-Methoxy-1-phenyl-1-propanol[262] (6.15)
A solution of 1-phenyl-3-phenylseleno-1-propanol (6.11) (55 mg, 0.19 mmol) and m-
CPBA (83 mg, 0.48 mmol) in methanol (5 mL) was stirred at r.t. for 4 d after which
the solvent was evaporated to give a pale yellow solid. The product was dissolved in
Et2O (15 mL) and washed with aqueous NaOH (10%, 8 mL) followed by saturated
aqueous NaCl (8 mL) and dried (Na2SO4). Evaporation of the solvent at reduced
pressure gave a yellow oil (35 mg) which was chromatographed (hexane/EtOAc,
75:25) to give the title compound, 3-methoxy-1-phenyl-1-propanol,[262] (6.15) as a
pale yellow oil (13 mg, 41%) whose 1H NMR spectrum was in accord with the
spectrum of (6.15) previously obtained.
2-Benzyloxetane[174] (6.24)
A solution of 1-phenyl-4-phenylseleno-2-butanol (6.18) (91 mg, 0.30 mmol) and m-
CPBA (129 mg, 0.748 mmol) in methanol (3 mL) was stirred at r.t. for 1 h. Aqueous
NaOH (1 M, 1.5 mL, 1.5 mmol) was added and the solution was stirred a further 17 h.
Half-saturated aqueous NaCl (10 mL) was added and the mixture was extracted with
Et2O (2 x 20 mL). The combined organic layers were washed with saturated
aqueous NaCl (10 mL) and dried (Na2SO4), and the solvent was evaporated at
reduced pressure to give a colourless oil (42 mg). Chromatography (hexane/EtOAc
3:1) gave the title compound[268] as a colourless oil (4.6 mg, 10%). 1H NMR: 7.33-
7.20, m, 5H, ArH; 5.04, dddd, J 6.3, 6.6, 6.9, 7.2 Hz, 1H, CHO; 4.65, ddd, J 5.7, 7.5,
8.1 Hz, 1H, CHaHbO; 4.48, ddd, J 5.7, 6.0, 9.0 Hz, 1H, CHaHbO; 3.09, dd, J 6.3, 13.8
Hz, 1H, C6H5CHaHb; 2.98, dd, J 6.6, 13.8 Hz, 1H, C6H5CHaHb; 2.63, dddd, J 6.0, 7.2,
8.4, 10.8 Hz, 1H, CHCHaHbCH2; 2.44, dddd, 6.9, 7.5, 9.0, 10.8 Hz, 1H,
CHCHaHbCH2. 13C NMR: 137.04, 129.19, 128.36, 126.38, all Ar; 82.61, CHO;

Experimental 7.6
210
67.89, CH2O; 44.00, C6H5CH2; 27.13. MS: m/z 148.1 (M+), 131.1 (M+-OH), 117.1
(M+-CH2OH), 105.1, (M+-C2H3O), 91.1 (C7H7+), 77 (C6H6
+), 65.1 (C5H5+), 57.0
(C3H5O+), 43.0 (C2H3O+). Further elution gave 1-phenyl-4-methoxy-2-butanol (6.25)
as a pale yellow oil (8 mg, 15%). max (KBr): 3438 (br, OH), 2924, 1496, 1454, 1385,
1339, 1191, 1118, 1086, 1029, 746, 701 cm-1. 1H NMR: 7.33-7.19, m, 5H, ArH;
4.02, qn, J 6.9 Hz, 1H, CHO; 3.62, ddd, J 5.1, 5.4, 9.3 Hz; 1H, CHaHbOMe; 3.52, ddd,
J 6.6, 7.5, 9.3 Hz, 1H, CHaHbOMe; 3.34, s, 3H, OCH3; 2.87, br s, 1H, OH; 2.81, dd, J
7.2, 13.5 Hz, 1H, CHaHbCH; 2.76, dd, J 6.0, 13.5 Hz, 1H, CCHaHbCH; 1.76-1.70, m,
2H, CHCH2CH2. 13C NMR: 138.55, 129.40, 128.39, 126.29, all Ar; 72.11, CHO;
71.39, CH2O; 58.82, OCH3; 43.99, C6H5CH2; 35.67. MS: m/z 181 (M+), 162 (M+-
H2O), 147 (M+-H2O-CH3), 131 (M+-H2O-OCH3), 117 (C6H5CHCHCH2+), 103 (C8H7
+),
91 (C7H7+), 89 (C4H9O2
+), 45 (CH2OCH3+).
2-Octyloxetane[174] (6.20)
A solution of 1-phenylseleno-3-undecanol (6.17) (103 mg, 0.315 mmol) and m-CPBA
(137 mg, 0.794 mmol) in methanol (3 mL) was stirred at r.t. for 30 min. Aqueous
NaOH (1 M, 1.6 mL, 1.6 mmol) was added and the mixture was stirred a further 43 h.
Half-saturated aqueous NaCl (10 mL) was added and the mixture was extracted with
Et2O (2 x 20 mL). The combined organic layers were washed with saturated
aqueous NaCl (10 mL) and dried (Na2SO4), and the solvent was evaporated at
reduced pressure to give a colourless oil (42 mg). 1H NMR analysis showed the
product to be predominantly a mixture of 2-octyloxetane (6.20) and 1-methoxy-3-
undecanol (6.21) in a ratio of 1:1. Chromatography (hexane/EtOAc 9:1) gave the
title compound[265] as a colourless oil (trace). ESI HRMS: 171.1743 C11H22O+H
requires 171.1749. 1H NMR: 4.82, qn, J 6.9 Hz, 1H, CHO; 4.66, ddd, J 5.7, 7.5,

Experimental 7.6
211
8.4 Hz, 1H, CHaHbO; 4.50, ddd, J 5.7, 5.7, 9.0 Hz, 1H, CHaHbO; 2.64, dddd, J 5.7,
6.9, 8.4, 10.8 Hz, 1H, CHCHaHbCH2O; 2.35, dddd, J 6.9, 7.5, 9.0, 10.8 Hz, 1H,
CHCHaHbCH2O; 1.83-1.76, m, 1H, CH2CH2CHaHbCHO; 1.68-1.58, m, 1H,
CH2CH2CHaHbCHO; 1.4-1.1, m, 12H; 0.88, t, 6.9 Hz, 3H, CH3. Further elution gave
a fraction containing a complex mixture (12 mg). Further elution gave 1-methoxy-3-
undecanol (6.21) as a pale yellow oil (trace). ESI HRMS: 203.2006 C12H26O2+H
requires 203.2006. max (neat): 3433 (broad, OH), 2924, 2854, 1460, 1380, 1187,
1118, 1029, 965, 721 cm-1. 1H NMR: 3.78, m, 1H, CHO; 3.63, ddd, J 5.1, 5.1, 9.3
Hz, 1H, CHaHbO; 3.55, ddd, J 5.4, 7.2, 9.3 Hz, 1H, CHaHbO; 3.36, s, 3H, OCH3;
1.73-1.63, m, 2H, CHCHaHbCH2O; 1.62-1.26, m, 15H; 0.88, t, J 6.6 Hz, 3H, CH2CH3.
13C NMR: 71.83, CH2O; 71.57, CHO; 58.90, CH3O; 37.49, 36.28, 31.88, 29.69,
29.58, 29.27, 25.60, 22.66, 14.10, CH(OH)CH2CH2O. MS: m/z 185 (M+-H), 169 (M+-
H2O), 155 (M+-CH3O), 141 (C10H21), 124 (M+-C2H5O-OH).

References
212
REFERENCES [1] M. E. Weeks, in Discovery of the Elements, Fourth ed., Journal of Chemical
Education, Easton, PA, 1939, pp. 110-121.
[2] W. C. Cooper, R. A. Zingaro, Von Nostrand Reinhold, New York, 1974, p. 835.
[3] E. Gerlach, P. Grosse, in Springer Series in Solid State Sciences, Vol. 13,
Springer-Verlag, Berlin; New York, 1979, p. 281.
[4] D. M. Pai, B. E. Springett, Rev. Mod. Phys. 1993, 65, 163.
[5] K.-Y. Law, Chem. Rev. 1993, 93, 449.
[6] T. Rezanka, K. Sigler, Phytochemistry 2008, 69, 585.
[7] M. Stadlober, M. Sager, K. J. Irgolic, Food Chem. 2001, 73, 357.
[8] A. P. Vonderheide, K. Wrobel, S. S. Kannamkumarath, C. B'Hymer, M.
Montes-Bayon, C. Ponce de Leon, J. A. Caruso, J. Agric. Food Chem. 2002,
50, 5722.
[9] C. Ip, M. Birringer, E. Block, M. Kotrebai, J. F. Tyson, P. C. Uden, D. J. Lisk, J.
Agric. Food Chem. 2000, 48, 2062.
[10] G. N. Schrauzer, J. Nutr. 2000, 130, 1653.
[11] T. C. Stadtman, Annu. Rev. Biochem. 1996, 65, 83.
[12] K. El-Bayoumy, R. Sinha, J. T. Pinto, R. S. Rivlin, J. Nutr. 2006, 136, 864S.
[13] Y. Dong, D. Lisk, E. Block, C. Ip, Cancer Res. 2001, 61, 2923.
[14] K. Schwarz, C. M. Foltz, J. Am. Chem. Soc. 1957, 70, 3292.
[15] L. D. Koller, J. H. Exon, Can. J. Vet. Res. 1986, 50, 297.
[16] G. V. Kryukov, S. Castellano, S. V. Novoselov, A. V. Lobanov, O. Zehtab, R.
Guigo, V. N. Gladyshev, Science 2003, 300, 1439.
[17] P. C. Raich, J. Lu, H. J. Thompson, G. F. Combs, Cancer Invest. 2001, 19,
540.

References
213
[18] P. D. Whanger, Br. J. Nutr. 2004, 91, 11.
[19] G. N. Schrauzer, P. F. Surai, Clin. Rev. Biotech. 2009, 29, 2.
[20] D. L. Hatfield, V. N. Gladyshev, Mol. Interventions 2009, 9, 18.
[21] C. Ip, H. E. Ganther, Carcinogenesis 1992, 13, 1167.
[22] M. J. Parnham, Exp. Opin. Invest. Drugs 1996, 5, 861.
[23] G. Mugesh, W.-W. du Mont, H. Sies, Chem. Rev. 2001, 101, 2125.
[24] K. P. Bhabak, G. Mugesh, Chem. Eur. J. 2007, 13, 4594.
[25] V. P. Singh, H. B. Singh, R. J. Butcher, Eur. J. Inorg. Chem. 2010, 637.
[26] D. Shanks, R. Amorati, M. G. Fumo, G. F. Pedulli, L. Valgimigli, L. Engman, J.
Org. Chem. 2006, 71, 1033.
[27] S. Shabaan, L. A. Ba, M. Abbas, T. Burkholz, A. Denkert, A. Gohr, L. A.
Wessjohann, F. Sasse, W. Weber, C. Jacob, Chem. Commun. 2009, 4702.
[28] R. L. Grange, J. Ziogas, J. A. Angus, C. H. Schiesser, Tetrahedron Lett. 2007,
48, 6301.
[29] H. R. P. Naik, H. S. B. Naik, T. R. R. Naik, H. R. Naika, K. Gouthamchandra,
R. Mahmood, B. M. K. Ahamed, Eur. J. Med. Chem. 2009, 44, 981.
[30] G. R. Waitkins, C. W. Clark, Chem. Rev. 1945, 235.
[31] K. B. Sharpless, K. M. Gordon, R. F. Lauer, D. W. Patrick, S. P. Singer, M. W.
Young, Chem. Scr. 1975, 8A, 9.
[32] H. L. Riley, J. F. Morley, N. A. C. Friend, J. Chem. Soc. 1932, 1875.
[33] H. L. Riley, N. A. C. Friend, J. Chem. Soc. 1932, 2342.
[34] K. B. Sharpless, K. M. Gordon, J. Am. Chem. Soc. 1976, 98, 300.
[35] K. B. Sharpless, R. F. Lauer, J. Am. Chem. Soc. 1972, 94, 7154.
[36] K. B. Sharpless, R. F. Lauer, J. Am. Chem. Soc. 1973, 95, 2697.

References
214
[37] D. N. Jones, D. Mundy, R. D. Whitehouse, J. Chem. Soc., Chem. Commun.
1970, 86.
[38] K. B. Sharpless, M. W. Young, R. F. Lauer, Tetrahedron Lett. 1973, 22, 1979.
[39] G. Hölzle, W. Jenny, Helv. Chim. Acta 1958, 41, 593.
[40] J. N. Denis, J. Vicens, A. Krief, Tetrahedron Lett. 1979, 2697.
[41] A. Toshimitsu, T. Aoai, H. Owada, S. Uemura, M. Okano, J. Chem. Soc.,
Chem. Commun. 1980, 412.
[42] A. Hassner, A. S. Amarasekara, Tetrahedron Lett. 1987, 28, 5185.
[43] L. Engman, J. Org. Chem. 1989, 54, 884.
[44] S. Raucher, Tetrahedron Lett. 1977, 44, 3909.
[45] D. Liotta, G. Zima, Tetrahedron Lett. 1978, 50, 4977.
[46] G. H. Schmid, D. G. Garratt, Tetrahedron 1978, 34, 2869.
[47] T.-Y. Luh, W.-H. So, K. S. Cheung, S. W. Tam, J. Org. Chem. 1985, 50, 3051.
[48] S. E. Denmark, M. G. Edwards, J. Org. Chem. 2006, 71, 7293.
[49] D. Liotta, Acc. Chem. Res. 1984, 17, 28.
[50] D. Liotta, G. Zima, J. Org. Chem. 1980, 45, 2551.
[51] M. A. Cooper, A. D. Ward, Tetrahedron 2004, 60, 7963.
[52] M. A. Cooper, A. D. Ward, Tetrahedron Lett. 1995, 36, 2327.
[53] D. Liotta, G. Zima, M. Saindane, J. Org. Chem. 1982, 47, 1258.
[54] A. Toshimitsu, K. Terao, S. Uemura, J. Chem. Soc., Perkin Trans. 1 (1972-
1999) 1987, 1059.
[55] D. L. J. Clive, J. Chem. Soc., Chem. Commun. 1974, 100.
[56] G. H. Schmid, D. G. Garratt, Tetrahedron Lett. 1975, 3991.
[57] C. G. Francisco, R. Hernandez, E. I. Leon, J. A. Salazar, E. Suaraz, J. Chem.
Soc., Perkin Trans. 1 (1972-1999) 1990, 2417.

References
215
[58] A. Toshimitsu, G. Hayashi, K. Terao, S. Uemura, J. Chem. Soc., Perkin Trans.
1 (1972-1999) 1988, 2113.
[59] F. D'Onofrio, L. Parlanti, G. Piancatelli, Tetrahedron Lett. 1995, 36, 1929.
[60] T. Wirth, G. Fragale, M. Spichty, J. Am. Chem. Soc. 1998, 120, 3376.
[61] S. Tomoda, M. Iwaoka, Chem. Lett. 1988, 1895.
[62] R. Deziel, E. Malenfant, C. Thibault, Tetrahedron Lett. 1998, 39, 5493.
[63] Y. Nishibayashi, J. D. Singh, S. Uemura, Tetrahedron Lett. 1994, 35, 3115.
[64] K. Fujita, K. Murata, M. Iwaoka, S. Tomoda, Tetrahedron 1997, 53, 2029.
[65] T. Wirth, Angew. Chem., Int. Ed. Engl. 1995, 34, 1726.
[66] T. Wirth, Tetrahedron 1999, 55, 1.
[67] T. Wirth, G. Fragale, Synthesis 1998, 162.
[68] Y. Okamoto, Y. Nishibayashi, S. Uemura, A. Toshimitsu, Angew. Chem., Int.
Ed. 2005, 44, 3588.
[69] T. Wirth, K. J. Kulicke, G. Fragale, J. Org. Chem. 1996, 61, 2686.
[70] M. Miyashita, M. Hoshino, A. Yoshikoshi, Tetrahedron Lett. 1988, 29, 347.
[71] D. L. J. Clive, Tetrahedron 1978, 34, 1049.
[72] R. M. Scarborough Jr., A. B. Smith III, Tetrahedron Lett. 1977, 4361.
[73] D. Liotta, W. Markiewicz, H. Santiesteban, Tetrahedron Lett. 1977, 4365.
[74] A. Krief, M. Trabelsi, W. Dumont, Synthesis 1992, 933.
[75] P. Dowd, P. Kennedy, Synth. Commun. 1981, 11, 935.
[76] D. Liotta, H. Santiesteban, Tetrahedron Lett. 1977, 50, 4369.
[77] K. Haraguchi, H. Tanaka, T. Miyasaka, Synthesis 1989, 434.
[78] Y. Nishiyama, T. Asano, Y. Kishimoto, K. Itoh, Y. Ishii, Tetrahedron Lett. 1998,
39, 8685.
[79] M. J. Dabdoub, T. M. Cassol, C. F. Batista, Tetrahedron Lett. 1996, 37, 9005.

References
216
[80] C. Santi, S. Santoro, L. Testaferri, M. Tiecco, Synlett 2008, 1471.
[81] A. L. Braga, R. S. Schwab, E. E. Alberto, S. M. Salman, J. Vargas,
Tetrahedron Lett. 2009, 50, 2309.
[82] A. Krief, M. Derock, D. Lacroix, Synlett 2005, 2832.
[83] M. Yang, C. Zhu, F. Yuan, Y. Huang, Y. Pan, Org. Lett. 2005, 7, 1927.
[84] A. M. El-Nahas, P. Von Ragué Schleyer, J. Comput. Chem. 1994, 15, 596.
[85] J.-M. Lehn, G. Wipff, Helv. Chim. Acta 1977, 60, 1239.
[86] A. Krief, W. Dumont, M. Clarembeau, G. Bernard, E. Badaoui, Tetrahedron
1989, 45, 2005.
[87] D. Labar, W. Dumont, L. Hevesi, A. Krief, Tetrahedron Lett. 1978, 13, 1145.
[88] M. Sevrin, A. Krief, Tetrahedron Lett. 1978, 187.
[89] M. Sevrin, A. Krief, Tetrahedron Lett. 1980, 21, 585.
[90] D. Labar, A. Krief, J. Chem. Soc., Chem. Commun. 1982, 564.
[91] H. J. Reich, F. Chow, S. K. Shah, J. Am. Chem. Soc. 1979, 101, 6638.
[92] X. Huang, W. Xu, Tetrahedron Lett. 2002, 43, 5495.
[93] W. M. Xu, E. Tang, X. Huang, Synthesis 2004, 13, 2094.
[94] R. W. Hoffman, W. Klute, Chem. Eur. J. 1996, 2, 694.
[95] R. W. Hoffman, W. Klute, R. K. Dress, A. Wenzel, J. Chem. Soc., Perkin
Trans. 2 (1972-1999) 1995, 1721.
[96] W. Klute, R. K. Dress, R. W. Hoffman, J. Chem. Soc., Perkin Trans. 2 (1972-
1999) 1993, 1409.
[97] S. Nakamura, T. Ogura, L. Wang, T. Toru, Tetrahedron Lett. 2004, 45, 2399.
[98] M. Sevrin, D. Van Ende, A. Krief, Tetrahedron Lett. 1976, 30, 2643.
[99] D. L. J. Clive, G. Chittattu, C. K. Wong, J. Chem. Soc., Chem. Commun. 1978,
41.

References
217
[100] D. L. J. Clive, G. J. Chittattu, V. Farina, W. A. Kiel, S. M. Menchen, C. G.
Russell, A. Singh, C. K. Wong, N. J. Curtis, J. Am. Chem. Soc. 1980, 102,
4438.
[101] D. L. Boger, R. J. Mathvink, J. Am. Chem. Soc. 1990, 112, 4003.
[102] D. L. Boger, R. J. Mathvink, J. Am. Chem. Soc. 1990, 112, 4008.
[103] J. C. Scaiano, P. Schmid, K. U. Ingold, J. Organomet. Chem. 1976, 121, C4.
[104] A. L. J. Beckwith, P. E. Pigou, Aust. J. Chem. 1986, 39, 77.
[105] A. Stojanovic, P. Renaud, Helv. Chim. Acta 1998, 81, 353.
[106] V. H. Rawal, S. P. Singh, C. Dufour, C. Michoud, J. Org. Chem. 1993, 58,
7718.
[107] A. Stojanovic, P. Renaud, Tetrahedron Lett. 1996, 37, 9199.
[108] A. Ouchi, S. Liu, Z. Li, S. A. Kumar, T. Suzuki, T. Hyugano, H. Kitahara, J.
Org. Chem. 2007, 72, 8700.
[109] T. Hyugano, S. Liu, A. Ouchi, J. Org. Chem. 2008, 73, 8861.
[110] J. H. Byers, G. C. Lane, J. Org. Chem. 1993, 58, 3355.
[111] P. Renaud, J.-P. Vionnet, J. Org. Chem. 1993, 58, 5895.
[112] K. Tsuchii, M. Doi, T. Hirao, A. Ogawa, Angew. Chem., Int. Ed. 2003, 42,
3490.
[113] J. E. Lyons, C. H. Schiesser, K. Sutej, J. Org. Chem. 1993, 58, 5632.
[114] C. H. Schiesser, K. Sutej, J. Chem. Soc., Chem. Commun. 1992, 57.
[115] M. C. Fong, C. H. Schiesser, J. Org. Chem. 1997, 62, 3103.
[116] M. W. Carland, R. L. Martin, C. H. Schiesser, Org. Biomol. Chem. 2004, 2,
2612.
[117] N. Al-Maharik, L. Engman, J. Malmström, C. H. Schiesser, J. Org. Chem.
2001, 66, 6286.

References
218
[118] H. J. Reich, Acc. Chem. Res. 1979, 22.
[119] H. J. Reich, J. M. Renga, I. L. Reich, J. Am. Chem. Soc. 1975, 97, 5434.
[120] H. J. Reich, W. W. Willis Jr., J. Am. Chem. Soc. 1980, 102, 5967.
[121] H. J. Reich, S. Wollowitz, J. E. Trend, F. Chow, D. F. Wendelborn, J. Org.
Chem. 1978, 43, 1697.
[122] H. J. Reich, S. K. Shah, J. Am. Chem. Soc. 1975, 97, 3250.
[123] H. J. Reich, K. E. Yelm, S. Wollowitz, J. Am. Chem. Soc. 1983, 105, 2503.
[124] M. Oki, H. Iwamura, Tetrahedron Lett. 1966, 25, 2917.
[125] F. A. Davis, J. M. Billmers, O. D. Stringer, Tetrahedron Lett. 1983, 24, 3191.
[126] F. A. Davis, R. T. Reddy, J. Org. Chem. 1992, 57, 2599.
[127] N. Komatsu, Y. Nishibayashi, T. Sugita, S. Uemura, J. Chem. Soc., Chem.
Commun. 1992, 46.
[128] A. Krief, C. Colaux, W. Dumont, Tetrahedron Lett. 1997, 38, 3315.
[129] M. Kobayashi, H. Ohkubo, T. Shimizu, Bull. Chem. Soc. Jpn. 1986, 59, 503.
[130] F. Toda, K. Mori, J. Chem. Soc., Chem. Commun. 1986, 1357.
[131] Y. Nishibayashi, J. D. Singh, S. Fukuzawa, S. Uemura, J. Org. Chem. 1995,
60, 4114.
[132] H. Taka, A. Matsumoto, T. Shimizu, N. Kamigata, Heteroat. Chem. 2001, 12,
227.
[133] T. Shimizu, M. Enomoto, H. Taka, N. Kamigata, J. Org. Chem. 1999, 64, 8242.
[134] T. Soma, T. Shimizu, K. Hirabayashi, N. Kamigata, Heteroat. Chem. 2007, 18,
301.
[135] J. V. Comasseto, R. A. Gariani, Tetrahedron 2009, 65, 8447.
[136] M. Gruttadauria, P. Lo Meo, S. Riela, F. D'Anna, R. Noto, Tetrahedron:
Asymmetry 2006, 17, 2713.

References
219
[137] C. E. Costa, G. C. Clososki, H. B. Barchesi, S. P. Zanotto, M. G. Nascimento,
J. V. Comasseto, Tetrahedron: Asymmetry 2004, 15, 3945.
[138] A. T. Omori, L. F. Assis, L. H. Andrade, J. V. Comasseto, A. L. M. Porto,
Tetrahedron: Asymmetry 2007, 18, 1048.
[139] C. E. Da Costa, J. V. Comasseto, I. H.-S. Crusius, L. H. Andrade, A. L. M.
Porto, J. Mol. Catal. B: Enzym. 2007, 45, 135.
[140] L. F. Assis, E. Kagohara, A. T. Omori, J. V. Comasseto, L. H. Andrade, A. L.
M. Porto, Food Technol. Biotechnol. 2007, 45, 415.
[141] L. H. Andrade, A. V. Silva, E. C. Pedrozo, Tetrahedron Lett. 2009, 50, 4331.
[142] L. H. Andrade, A. V. Silva, Tetrahedron: Asymmetry 2008, 19, 1175.
[143] L. H. Andrade, A. T. Omori, A. L. M. Porto, J. V. Comasseto, J. Mol. Catal. B:
Enzym. 2004, 29, 47.
[144] J. V. Comasseto, A. T. Omori, A. L. M. Porto, L. H. Andrade, Tetrahedron Lett.
2004, 45, 473.
[145] A. Toshimitsu, T. Aoai, H. Owada, S. Uemura, M. Okano, J. Org. Chem. 1981,
46, 4727.
[146] J. J. Ritter, P. P. Minieri, J. Am. Chem. Soc. 1948, 70, 4045.
[147] H. C. Brown, J. T. Kurek, J. Am. Chem. Soc. 1969, 91, 5647.
[148] M. A. Cooper, Ph.D. thesis, University of Adelaide 1994.
[149] N. X. Hu, Y. Aso, T. Otsubo, F. Ogura, J. Chem. Soc., Perkin Trans. 1 (1972-
1999) 1989, 1775.
[150] W. S. Johnson, E. N. Schubert, J. Am. Chem. Soc. 1950, 72, 2187.
[151] A. M. Morella, Ph.D. thesis, University of Adelaide 1985.
[152] A. M. Morella, A. D. Ward, Tetrahedron Lett. 1985, 26, 2899.

References
220
[153] A. Toshimitsu, C. Hirosawa, S. Tanimoto, S. Uemura, Tetrahedron Lett. 1992,
33, 4017.
[154] Z. da Zhang, R. Scheffold, Helv. Chim. Acta 1993, 76, 2602.
[155] P. Wipf, C. P. Miller, Tetrahedron Lett. 1992, 33, 907.
[156] H. W. Heine, J. Am. Chem. Soc. 1956, 78, 3708.
[157] G. E. McCasland, D. A. Smith, J. Am. Chem. Soc. 1950, 72, 2190.
[158] D. Müller, G. Umbricht, B. Weber, A. Pfalz, Helv. Chim. Acta 1991, 74, 232.
[159] N. Galleoti, C. Montagne, J. Poncet, P. Jouin, Tetrahedron Lett. 1992, 33,
2807.
[160] A. B. Smith III, B. A. Salvatore, K. G. Hull, J.-W. Duan, Tetrahedron Lett. 1991,
32, 4859.
[161] K. Nakajima, H. Kawai, M. Takai, K. Okawa, Bull. Chem. Soc. Jpn. 1977, 50,
917.
[162] D. M. Roush, M. M. Patel, Synth. Commun. 1985, 15, 675.
[163] D. H. Boschelli, Synth. Commun. 1988, 18, 1391.
[164] P. Wipf, C. P. Miller, Tetrahedron Lett. 1992, 33, 6267.
[165] A. Toshimitsu, G. Hayashi, K. Terao, S. Uemura, J. Chem. Soc., Perkin Trans.
1 (1972-1999) 1986, 343.
[166] R. A. Johnson, M. E. Herr, H. C. Murray, W. C. Krueger, L. M. Pschigoda, D.
L. Duchamp, J. Org. Chem. 1992, 57, 7212.
[167] V. R. Biela, I. Hahnemann, H. Panovsky, W. Pritzkow, J. Prakt. Chem. 1966,
4, 282.
[168] H. Günther, NMR Spectroscopy, 2nd ed., John Wiley & Sons Ltd, Chichester,
1995.

References
221
[169] A. Toshimitsu, T. Aoai, H. Owada, S. Uemura, M. Okano, Tetrahedron 1985,
41, 5301.
[170] A. H. White, personal communication, 2004.
[171] J. Jacques, A. Collet, S. Wilen, Enantiomers, Racemates, and Resolutions,
Wiley, New York, New York, 1981.
[172] C. A. Mackenzie, G. A. Schmidt, L. R. Webb, J. Am. Chem. Soc. 1951, 73,
4990.
[173] M. A. Perez, C. A. Dorado, J. L. Soto, Synthesis 1983, 483.
[174] M. Shimizu, R. A., I. Kuwajima, J. Org. Chem. 1984, 49, 1230.
[175] A. Krief, W. Dumont, J. N. Denis, G. Evrard, B. Norberg, J. Chem. Soc.,
Chem. Commun. 1985, 569.
[176] S. Uemura, S. Fukuzawa, J. Chem. Soc., Perkin Trans. 1 (1972-1999) 1985,
471.
[177] B. C. Challis, J. A. Challis, in Comprehensive Organic Chemistry, Vol. 2 (Eds.:
D. H. R. Barton, W. D. Ollis), Pergamon Press, 1979, p. 1011.
[178] H. W. Heine, M. S. Kaplan, J. Am. Chem. Soc. 1967, 89, 3069.
[179] G. Ayrey, D. Barnard, D. T. Woodbridge, J. Am. Chem. Soc. 1962, 2089.
[180] R. Paetzold, G. Bochmann, Z. Anorg. Allgemeine Ch. 1968, 360, 293.
[181] M. Karplus, J. Am. Chem. Soc. 1963, 85, 2870.
[182] A. Navarro-Vazquez, J. C. Cobas, F. J. Sardina, J. Chem. Inf. Comput. Sci.
2004, 44, 1680.
[183] D. Ferraris, W. J. Drury III, C. Cox, T. Lectka, J. Org. Chem. 1998, 63, 4568.
[184] M. Oki, Applications of Dynamic NMR Spectroscopy to Organic Chemistry,
Vol. 4, 1st ed., VCH Publishers, Deerfield Beach, 1985.
[185] G. R. Boggs, J. T. Gerig, J. Org. Chem. 1969, 34, 1484.

References
222
[186] F. A. L. Anet, J. M. Osyany, J. Am. Chem. Soc. 1967, 89, 352.
[187] A. R. Meyer, D. L. Temple, D. Haidukewych, E. Mihelich, J. Org. Chem. 1974,
39, 2787.
[188] K. B. Wiberg, Angew. Chem., Int. Ed. Engl. 1986, 25, 312.
[189] T. Dudev, C. Lim, J. Am. Chem. Soc. 1998, 120, 4450.
[190] E. L. Eliel, S. H. Wilen, M. P. Doyle, Basic Organic Stereochemistry, 1st ed.,
Wiley-Interscience, New York, 2001.
[191] A. C. Knipe, C. J. M. Stirling, J. Chem. Soc. 1968, 67.
[192] G. Illuminati, L. Mandolini, Acc. Chem. Res. 1981, 14, 95.
[193] F. Lightstone, T. Bruice, Bioorg. Chem. 1998, 26, 193.
[194] H. C. Kolb, M. G. Finn, K. B. Sharpless, Angew. Chem., Int. Ed. 2001, 40,
2004.
[195] X. E. Hu, Tetrahedron 2004, 60, 2701.
[196] J. Wu, X.-L. Hou, L.-X. Dai, J. Org. Chem. 2000, 65, 1344.
[197] P. J. J. A. Buijnsters, M. C. Feiters, R. J. M. Nolte, N. A. J. M. Sommerdijk,
Chem. Commun. 2001, 269.
[198] H. Banks, J. Org. Chem. 2006, 71, 8089.
[199] P. A. S. Lowden, in Aziridines and Epoxides in Organic Synthesis, 1st ed.
(Ed.: A. K. Yudin), WILEY-VCH, Weinheim, 2006, pp. 399-442.
[200] C. Schneider, Angew. Chem., Int. Ed. 2009, 48, 2082.
[201] Y. Zhang, C. W. Kee, R. Lee, X. Fu, J. Y.-T. Soh, E. M. F. Loh, K.-W. Huang,
C.-H. Tan, Chem. Commun. 2011, 3897.
[202] E. B. Rowland, G. B. Rowland, E. Rivera-Otero, J. C. Antilla, J. Am. Chem.
Soc. 2007, 129, 12084.

References
223
[203] M. Senatore, A. Lattanzi, S. Santoro, C. Santi, G. Della Sala, Org. Biomol.
Chem. 2011, 9, 6205.
[204] B. Wu, J. C. Gallucci, J. R. Parquette, T. V. RajanBabu, Angew. Chem., Int.
Ed. 2009, 48, 1126.
[205] Y. Xu, L. Lin, M. Kanai, S. Matsunaga, M. Shibasaki, J. Am. Chem. Soc. 2011,
133, 5891.
[206] Y. Fukuta, T. Mita, N. Fukuda, M. Kanai, M. Shibasaki, J. Am. Chem. Soc.
2006, 128, 6312.
[207] T. Mita, N. Fukuda, F. X. Roca, M. Kanai, M. Shibasaki, Org. Lett. 2006, 9,
259.
[208] Y.-Y. Yeung, S. Hong, E. J. Corey, J. Am. Chem. Soc. 2006, 128, 6310.
[209] K. M. Bromfield, H. Graden, D. P. Hagberg, T. Olssen, N. Kann, Chem.
Commun. 2007, 3183.
[210] G. S. Bates, M. A. Varelas, Can. J. Chem. 1980, 58, 2562.
[211] T. A. Foglia, L. M. Gregory, G. Maerker, J. Org. Chem. 1970, 35, 1970.
[212] J. B. Sweeney, in Aziridines and Epoxides in Organic Synthesis, 1st ed. (Ed.:
A. K. Yudin), Wiley-VCH, Weinheim, 2006, pp. 117-144.
[213] A. Toshimitsu, H. Fuji, Chem. Lett. 1992, 2017.
[214] S. Boivin, F. Outurquin, C. Paulmier, Tetrahedron Lett. 2000, 41, 663.
[215] D. M. Browne, T. Wirth, Curr. Org. Chem. 2006, 10, 1893.
[216] M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli, A.
Temperini, Eur. J. Org. Chem. 2000, 3451.
[217] M. Gruttadauria, C. Aprile, R. Noto, Tetrahedron Lett. 2002, 43, 1669.
[218] S. Fukuzawa, K. Takahashi, H. Kato, H. Yamazaki, J. Org. Chem. 1997, 62,
7711.

References
224
[219] A. Hassner, L. A. Levy, R. Gault, Tetrahedron Lett. 1966, 27, 3119.
[220] M. Berthelot, M. Helbert, C. Laurence, J.-Y. Le Questel, J. Phys. Org. Chem.
1993, 6, 302.
[221] J.-Y. Le Questel, M. Berthelot, C. Laurence, J. Chem. Soc., Perkin Trans. 2
(1972-1999) 1997, 2711.
[222] J. H. Markgraf, W. T. Bachmann, D. P. Hollis, J. Org. Chem. 1965, 30, 3472.
[223] R. N. Butler, V. C. Garvin, J. Chem. Soc., Perkin Trans. 1 (1972-1999) 1980,
390.
[224] A. D. Ward, V. R. Ward, E. R. T. Tiekink, Z. Kristallogr. NCS 2001, 216, 551.
[225] M. R. Detty, J. Org. Chem. 1980, 45, 274.
[226] R. W. Rickards, W. P. Watson, Aust. J. Chem. 1980, 33, 451.
[227] E. R. T. Tiekink, personal communication, 2000.
[228] T. Takahashi, N. Kurose, S. Kawanami, Y. Arai, T. Koizumi, J. Org. Chem.
1994, 59, 3262.
[229] W. Nakanishi, Y. Ikeda, H. Iwamura, Org. Magn. Res. 1982, 20, 117.
[230] H. Duddeck, P. Wagner, A. Biallass, Magn. Reson. Chem. 1991, 29, 248.
[231] R. Paetzold, G. Bochmann, Spectrochim. Acta 1970, 26A, 391.
[232] E. V. Dikarev, R. Y. Becker, E. Block, Z. Shan, R. C. Haltiwanger, M. A.
Petrukhina, Inorg. Chem. 2003, 42, 7098.
[233] D. Barnard, J. M. Fabian, H. P. Koch, J. Chem. Soc. 1949, 2442.
[234] J. F. Fernandez-Bertran, Pure Appl. Chem. 1999, 71, 581.
[235] R. A. B. Bannard, N. C. C. Gibson, J. H. Parkkari, Can. J. Chem. 1971, 49,
2065.
[236] M. Reboul, Ann. Chim. 1878, 14, 496.

References
225
[237] J. A. Porco, S. L. Schreiber, in Comprehensive Organic Synthesis, Vol. 5
(Eds.: B. M. Trost, I. Fleming), Pergamon Press, Oxford, 1991, pp. 151-192.
[238] J. D. Coyle, H. A. J. Carless, J. Chem. Soc. Rev. 1972, 1, 465.
[239] T. Bach, Synthesis 1998, 683.
[240] T. Bach, K. Jödicke, K. Kather, J. Hecht, Angew. Chem., Int. Ed. Engl. 1995,
34, 2271.
[241] T. Bach, K. Jödicke, K. Kather, R. Fröhlich, J. Am. Chem. Soc. 1997, 119,
2437.
[242] K. Soai, S. Niwa, T. Yamanoi, H. Hikima, M. Ishizaki, J. Chem. Soc., Chem.
Commun. 1986, 1018.
[243] J. Biggs, Tetrahedron Lett. 1975, 48, 4285.
[244] K. Okuma, Y. Tanaka, S. Kaji, H. Ohta, J. Org. Chem. 1983, 48, 5133.
[245] T. Sone, G. Lu, S. Matsunaga, M. Shibasaki, Angew. Chem., Int. Ed. 2009, 48,
1677.
[246] K. C. Nicolaou, D. A. Claremon, W. E. Barnett, S. P. Seitz, J. Am. Chem. Soc.
1979, 101, 3704.
[247] P. Van de Weghe, S. Bourg, J. Eustache, Tetrahedron 2003, 59, 7365.
[248] M. Shimizu, I. Kuwajima, J. Org. Chem. 1980, 45, 4063.
[249] D. J. Saez, Ph.D. thesis, The University of Wisconsin - Madison 1984.
[250] H. J. Reich, S. K. Shah, F. Chow, J. Am. Chem. Soc. 1979, 101, 6648.
[251] M. Jevric, Honours thesis, University of Adelaide 1996.
[252] I. Scharfbillig, personal communication, 2000.
[253] National Institute of Advanced Industrial Science and Technology
http://riodb01.ibase.aist.go.jp/sdbs, accessed May 27, 2008.

References
226
[254] National Institute of Advanced Industrial Science and Technology
http://riodb01.ibase.aist.go.jp/sdbs, accessed May 29, 2008.
[255] National Institute of Advanced Industrial Science and Technology
http://riodb01.ibase.aist.go.jp/sdbs, accessed May 26, 2008.
[256] H. C. Brown, H. M. Hess, J. Org. Chem. 1969, 34, 2206.
[257] A. R. Meyer, J. Chem. Educ. 1981, 58, 628.
[258] M. Fujiwara, K. Hitomi, A. Baba, H. Matsuda, Synthesis 1990, 106.
[259] P. Picard, D. Leclercq, J.-P. Bats, J. Moulines, Synthesis 1981, 550.
[260] D. Seebach, N. Peleties, Chem. Ber. 1972, 105, 511.
[261] T. Iwama, S. Tsujiyama, H. Kinoshita, K. Kanematsu, Y. Tsurukami, H.
Iwamura, S. Watanabe, T. Kataoka, Chem. Pharm. Bull. 1999, 47, 956.
[262] J. Yun, S. L. Buchwald, J. Am. Chem. Soc. 1999, 121, 5640.
[263] J. S. Yadav, M. Sridhar Reddy, P. Purushothama Rao, A. R. Prasad,
Synthesis 2006, 4005.
[264] S. Uemura, K. Ohe, N. Sugita, J. Chem. Soc., Perkin Trans. 1 (1972-1999)
1990, 1697.
[265] M. Chini, P. Crotti, L. Favero, F. Macchia, Tetrahedron Lett. 1994, 35, 761.
[266] S.-R. Sheng, H.-R. Luo, Z.-Z. Huang, W.-K. Sun, X.-L. Liu, Synth. Commun.
2007, 37, 2693.
[267] R. M. Silverstein, G. C. Bassler, T. C. Morrill, Spectrometric Identification of
Organic Compounds, Fifth ed., John Wiley & Sons, Inc., 1991.
[268] W. Kirmse, R. Lelgemann, K. Friedrich, Chem. Ber. 1991, 124, 1853.
[269] S.-H. Dong, C.-R. Zhang, X.-F. He, H.-B. Liu, Y. Wu, J.-M. Yue, J. Nat. Prod.
2010, 73, 1344.

References
227
[270] D. Martins, E. Osshiro, N. F. Roque, V. Marks, H. E. Gottlieb, Phytochemistry
1998, 48, 677.
[271] M. A. M. Mondol, F. S. Tareq, J. H. Kim, M. Lee, H.-S. Lee, Y.-J. Lee, J. S.
Lee, H. J. Shin, J. Nat. Prod. 2011, 74, 2582.
[272] S. Omura, M. Murata, N. Imamura, Y. Iwai, H. Tanaka, A. Furusaki, T.
Matsumoto, J. Antibiot. 1984, 37, 1324.
[273] H.-D. Chen, S.-P. Yang, X.-F. He, H.-B. Liu, J. Ding, J.-M. Yue, Tetrahedron
2010, 66, 5065.
[274] C. Li, D. Lee, T. N. Graf, S. S. Phifer, Y. Nakanishi, J. P. Burgess, S. Riswan,
F. M. Setyowati, A. M. Saribi, D. D. Soejarto, N. R. Farnsworth, J. O.
Falkinham III, D. J. Kroll, A. D. Kinghorn, M. C. Wani, N. H. Oberlies, Org. Lett.
2005, 7, 5709.
[275] N. Shimida, S. Hasegawa, T. Harada, T. Tomisawa, A. Fujii, T. Takita, J.
Antibiot. 1986, 39, 1623.
[276] H. Hoshino, N. Shimizu, N. Shimada, T. Takita, T. Takeuchi, J. Antibiot. 1987,
40, 1077.
[277] D. W. Norbeck, J. B. Kramer, J. Am. Chem. Soc. 1988, 110, 7217.
[278] E. Ichikawa, K. Kato, Synthesis 2002, 1.
[279] Y.-F. Wang, Q.-W. Shi, M. Dong, H. Kiyota, Y.-C. Gu, B. Cong, Chem. Rev.
2011, 111, 7652.
[280] M. Wang, B. Cornett, J. Nettles, D. C. Liotta, J. P. Snyder, J. Org. Chem.
2000, 65, 1059.
[281] F. A. Long, J. G. Pritchard, F. E. Stafford, J. Am. Chem. Soc. 1957, 79, 2362.
[282] J. G. Pritchard, F. A. Long, J. Am. Chem. Soc. 1958, 80, 4162.
[283] S. Searles, J. Am. Chem. Soc. 1951, 73, 4515.

References
228
[284] H. D. Banks, Org. Biomol. Chem. 2009, 7, 4496.
[285] B. Ringner, S. Sunner, H. Watanabe, Acta Chem. Scand. 1971, 25, 141.
[286] M. Berthelot, F. Besseau, C. Laurence, Eur. J. Org. Chem. 1998, 925.
[287] F. Besseau, M. Luçon, C. Laurence, M. Bethelot, J. Chem. Soc., Perkin Trans.
2 (1972-1999) 1998, 101.
[288] F. Besseau, C. Laurence, M. Bertholet, J. Chem. Soc., Perkin Trans. 2 (1972-
1999) 1994, 485.
[289] J.-Y. Le Questel, C. Laurence, A. Lachkar, M. Helbert, M. Bertholet, J. Chem.
Soc., Perkin Trans. 2 (1972-1999) 1992, 2091.
[290] G. Wuitschik, E. M. Carreira, B. Wagner, H. Fischer, I. Parrilla, F. Schuler, M.
Rogers-Evans, K. Müller, J. Med. Chem. 2010, 53, 3227.
[291] G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler,
L. Polonchuk, E. M. Carreira, Angew. Chem., Int. Ed. 2006, 45, 7736.
[292] J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller, E. M. Carreira,
Angew. Chem., Int. Ed. 2010, 49, 9052.
[293] N. A. Meanwell, J. Med. Chem. 2011, 54, 2529.
[294] G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel,
H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B.
Schweizer, K. Müller, E. M. Carreira, Angew. Chem., Int. Ed. 2008, 47, 4512.
[295] D. D. Perrin, W. L. F. Armarego, Purification of Laboratory Chemicals,
Pergamon Press, 1988.
[296] R. M. Lawrence, S. A. Biller, O. M. Fryszman, M. A. Poss, Synthesis 1997,
553.
[297] N. Auzeil, M. Largeron, M.-B. Fleury, J. Chem. Soc., Perkin Trans. 2 (1972-
1999) 1999, 1703.

References
229
[298] A. Toshimitsu, T. Aoai, S. Uemura, M. Okano, J. Chem. Soc., Chem.
Commun. 1980, 1041.
[299] R. Kupfer, M. Nagel, E.-U. Wurthwein, R. Allmann, Chem. Ber. 1985, 118,
3089.
[300] A. D. Ward, V. R. Ward, E. R. T. Tiekink, Z. Kristallogr. NCS 2001, 216, 555.
[301] G. Llabres, M. Baiwir, Can. J. Chem. 1978, 56, 2008.
[302] National Institute of Advanced Industrial Science and Technology
http://riodb01.ibase.aist.go.jp/sdbs, accessed October 1, 2012.
[303] M. Imuta, H. Ziffer, J. Org. Chem. 1979, 44, 1351.
[304] Y. Okamoto, T. Yano, J. Organomet. Chem. 1971, 29, 99.

230
PUBLICATIONS
Synthesis of N-acylaziridines from -amido selenides. Virginia R. Ward,
Matthew A. Cooper, A. David Ward, Journal of the Chemical Society, Perkin
Transactions 1, 2001, 944-945.
The Synthesis of N-Benzoyl Aziridines from -Benzamidoalkyl Phenyl
Selenides. Virginia R. Ward, Matthew A. Cooper and A. David Ward, Phosphorus,
Sulfur and Silicon, 2001, 172, 195-201.
Crystal Structure of N1-[2-(phenylseleninyl)cyclohexyl]benzamide,
C19H21NO2Se. Ward, A. D.; Ward, V. R.; Tiekink, E. R. T., Zeitschrift für
Kristallographie – New Crystal Structures, 2001, 216(4) 555-557.
Crystal Structure of 5-methyl-1,4-di[2-(phenylselanyl)cyclohexyl]-4H-1,2,3,4-
tetraazol-1-ium perchlorate, [(C6H5SeC6H10)2CH3CN4]ClO4. Ward, A. D.;
Ward, V. R.; Tiekink, E. R. T., Zeitschrift für Kristallographie – New Crystal
Structures, 2001, 216(4) 551-552.

PERKIN 1lncorporati ng Acta Chemi ca Scandi navica
W6*s'{e t&a*
{*Kv&p##rwffir&*s *f *fux* &sa#&affi.{
An internation"l journal
of organic and
bioorganic chemistry
ruffiffiffi^w?.&w
RSO(www.rsc.org lperkin 1 ROYAL SOCIETY OF CHEMISTRY

Synthesis of l/-acylaziridines from F-amido selenides
Virginia R. Ward,* Matthew A. Cooper and A. David Ward
Department of Chemistry, Adelaide (Jniversity, Adelaide, Australia 5005
Received 15th March 2001, Accepted 16th March 2001First published as an Advance Article on the web 30th March 2001
MCPBA under
ErnPnzand 8 with
Product D Yield'(ratio) (%)
6a,7a (74 :26:) 73
6b,7b (61 : 39) 726c,7c (83 : I l)" 83
rlo==CZ.-lCz
The low temperature oxidation of p-amido selenides withMCPBA affords the corresponding B-amido selenones.In situ treatment of the selenones with I(OtBu givesN-acylaziridines in good to excellent yield.
Aziridines are valuable compounds due to the regio- and stereo-controlled ring-opening reactions which are central to theirchemistry.t ,A/-Acylaziridines are of particular value in such
reactions as substitution at the nitrogen atom with an electron-withdrawing group enhances the susceptibility of the aziridinering to open.l-3
.n/-Acylaziridines are usually prepared by acylation of theunsubstituted aziridine.ou The alternative approach, via cyclis-ation of B-substituted amides, often forms oxazolines,t-tt as a
result of ring-closure by oxygen rather than by nitrogen, andonly rarely produces an aziridine.a Krook and Millert2 have
shown that cyclisation of the mesylate I can be directed to give
the oxazoline 2 under weakly basic conditions (potassiumbicarbonate in hot dichloroethane) and the aziridine 3 and
Table I Products lrom the reaction of 5basic conditions
Product o
Selenide (ratio)Yield "(%,)
5a 7a5b5c8a8b8c8d8e
8f8g
87
9a,10a (il : a9) 179b 859c 70
9e,10e (14 :12)' 55
9a9b9c9d
7583
9466
9e 8l9f 6799 87
o 4 eq. MCPBA, 6-8 eq. NaH or tBuOK in iPrOH, RT. t 3.3 eq.
MCPBA,4.5-9 eq. tBuOK in THtr, -60 "C. " Some of the correspond-ing elimination product was also lormed.
o
,.A*H
! ,oMsi
,rNHoBzo'
1
oAr4
N
R1
oA*T]
R2 'R3
7
Phse. NHcoRl f;o*tHLni tRr R24n3
B-lactam 4 under strongly basic conditions (potassium tert-butoxide (tBuOK) in tetrahydrofuran (THF)), thus demonstrat-ing that cyclisation of amides to aziridines requires generationof the amide anion prior to alkylation, ?s does //-alkylationof amides in general.t3 B-Hydroxy amides of threo-stereo-chemistry, such as threonine-containing peptides, have been
found to give aziridines under Mitsunobu conditions in whichthe reduced diisopropyl azodicarboxylate anion is believed toact as the base. t4-17 The same treatment of allo-threoninederivatives, however, leads to oxazolines.15
Toshimitsuls cyclized B-amido selenide 5a to the oxazolineTain 84o/o yield through its oxidation to the selenone with MCPBAin methanol in the absence of base. We report herein that thecyclisation of B-amido selenides under strongly basic con-ditions at low temperature can be directed predominantly toaziridine formation and that where the alkyl group is cyclic,azindines are formed as the exclusive products.
Initially the cycloalkyl phenyl selenides were oxidised underconditions similar to those used by Toshimitsu,rs with an excess
of MCPBA in isopropanol (propan -2-ol) in the presence ofpotassium hydroxide (KOH). Thus the reaction of selenide 8b
using 1.5 equivalents of KOH and 3 equivalents of MCPBAgave the oxazoline in94'Yuyield. However, withT .5 equivalents ofbase the aziridine was afforded in 73% yield. Investigation of
944 J. Chem. Soc., Perkin Trans. 1,2001 ,944-945
a Rl = C6H5i R2 = H,H; R3 = CoHrg
b R1 = pBr-C6Ha; R2 - H,H; R3 = CoHrs
c approx. 1:1 mixture of
R1 = CsHs; R2 = CHr' R3 = C3H7 and
R1 = C6H5i R2 = C3H7i R3 = CHs
the oxidation of other cyclic benzamido selenides confirmedthat neutral or acidic conditions favoured the oxazoline with an
excess of base giving the azindine as the predominant product.The use of sodium hydride (NaH) or tBuOK instead of KOHimproved the ratio of aziridine to oxazoline, presumably due togeneration of the stronger base, isopropoxide ion (Table 1, con-ditions a). However, except with the cyclohexanebenzamides 8b
and 8c, we were unable to effect a clean transformation to theazindines. Oxidation of acyclic selenoamide 5a under these
conditions gave the oxazoline in 8J'Y,, yield, a replication ofToshimitsu's result. r8
The work of Krook and Miller tt suggested that cyclisation tothe aziridine might be more favoured by the use of an aproticsolvent such as THF at a lower temperature. We were unawareof any precedent for the generation of selenones at temper-atures below zero degrees; neither did we know of any reportsof the generation of selenones with MCPBA in solvents otherthan alcohols or dichloromethane. Indeed, we have found theoxidation of other selenides to be 50 to 60 times slower in THFthan in alcohols and we expected the reaction at low tem-perature in THF to be very slow, if it proceeded at all. We were
therefore surprised to find that oxidation of the cyclic amidoselenides for one hour at -60'C in THF followed by additionof tBuOK and allowing the mixture to warm to ca.0 'C over 1
hour, afforded the aziridines as the exclusive products, often inexcellent yield (Table 1, conditions b).le The acyclic compounds
DOI: 10.1039/b102468j
o
*A"frE/ \ srAN--H 7l*oazo< A'NHOBz
34
This journal is O The Royal Society of Chemistry 2001

SePh
,,,NHCOR
a n=3,R=CoHsb n=4,R=CoHsc n=4,R-pBr-CoH+d n=4,R=CHse n=5,R=COHsI n=5,R=CHgI 11 = 6, R = COHS
9
,t]-o..(cQA,(h*
N-COR
2 tr. A. Davis, H. Liu and G. V. Reddy, Tetrahedron Lett., 1996, 37 ,
s473.3 J. Wu, X. L. Hou and L. X. Dai, J. Org. Chem., 2000, 65,
1344.4 H. M. I. Osborne and J. Sweeney, Tetrahedron: Asymmetry,799J,8,
r 693.5 G. Bates and M. Varelas, Can. J. Chent, 1980, 58,2562.6 K. Okawa, K. Nakajima, T. Tanaka and Y. Kawana, Chem. Lett.,
1915,591.I J. A. Frump, Chem. Rev., 1911,71,483.8 P. Wipl and C. P. Miller, Tetrahedron Lett., 1992,33,901 .
9 K. Nakajim?, H. Kawai, M. Takai and K. Okawa, Bull. Chem. Soc.
Jpn., 19J7 , 50,917 .
10 D. M. Roush and M. M. Patel, S)'nth. Commun, 1985, 15,675.1 1 N. Galeotti, C. Montagne, J. Poncet and P. Jouin, Tetrahedron
Lett., 1992,33,2801 .
12 M. A. Krook and M. J. Miller, J. Org. Chem., 1985,50, 1126.13 B. C. Challis and J. A. Challis, Comprehensive Organic Chemistry,
ed. D. H. R. Barton and W. D. Ollis, Pergamon Press, Oxlord,7919,vo1.2, pp. 1011-1015.
14 D. Boschelli, Synth. Commun.,1988, 18, 1391.
1 5 P. Wipf and C. P. Miller , Tetrahedron Lett., 1992,33, 6267 .
16 A. K. Bose, B. P Sahu and M. S. Manhas, I Org. Chem., 1981,46,,1229.
ll Y. Nakagawa, T. Tsuno, K. Nakajima, M. Iwai, H. Kawai andK. Okawa, Bull. Chem. Soc. Jpn.,1972,45, 1162.
18 A. Toshimitsu, C. Hirosawa, S. Tanimoto and S. tlemura, Tetra-hedron Lett., 1992,33, 4017 .
19In a typical procedure, to a solution ol the selenide 8d (250 mg,
0.84 mmol) in tetrahydroluran (20 ml) cooled to -60 "C was addeddropwise, with stirring, a solution of MCPBA (594 nlg, 80Y,,,
2.7 5 mmol) in tetrahydrofuran (20 ml) and the mixture was stirred at
-60 "C for t h. Potassium tert-butoxide (571 mg, 5.1 mmol) was
added and the resulting mixture stirred for a lurther I h. Aqueoussodium thiosullate (0.5 M, I 5 ml) and saturated aq. sodiumbicarbonate (10 ml) were added and the aqueous phase extractedwith diethyl ether (30 ml). The organic extract was washed with 10%,
aq. sodium hydroxide (10 ml) and saturated aq. sodium chloride(10 ml) and dried (MgSOo) and the solvent evaporated at reducedpressure. Chromatography using a gradient of 0 to 10'2, diethyl etherin dichloromethane as eluent gave the aziridine 9d as a clear liquid(77 mg,66%,,).
20 A. Krief, W. Dumont, J. N. Denis, G. Evrard and B. Norberg,J. Chent Soc., Chem. Commurz., 1985,569.
2l A. Toshimitsu, T. Aoai, H. Owada, S. Llemura and M. Okano,J. Chent. Soc., Chem. Contntun, 1980, 412.
22 A. Toshimitsu, G. Hayashi, K. Terao and S. Uen-rura, J. Chem. Soc.,
Perkin Trans. l, 1986, 343.23 M. Hayashi, K.Ono, H. Hoshimi and N. Oguni, Telraltedron, 1996,
52,l81l .
24 Z. da Zhang and R" Scheffold, Helv" Chim. Acta, 1993, 76,2602.
10
GNHcoc6H511
5a-c also predominantly formed the corresponding aziridines6a-c under these conditions.
The oxidation of 8e with 1 equivalent of MCPBA (sufficientto give the selenoxide) and 3 equivalents of tBuOK with otherparameters constant gave the selenoxide syn-elimination productll (5S%) and starting material (13'/,,). This confirmed thatthe intermediate was the selenone and not the selenoxide. Inaddition, the ttSe NMR spectrum of a mixture of 8b andMCPBA in THF at -60 "C showed a peak atld l0l0, consistentwith the presence of a selenone.2o
When the reaction was conducted on 8e at higher temper-atures (- l5 oC, 0'C) aziridine formation decreased with a con-comitant increase in the s-t,rz-elimination product 11. At bothtemperatures only traces of oxazoline were observed. These
results indicate that although it may have little effect on themode of cyclisation, the low temperature is necessary to ensure
that the selenoxide is sufficiently long-lived to enable its furtheroxidation to the selenone.
The B-amido selenides were prepared via establishedprocedures in two steps from the corresponding alkenes,27'22
with overall yields of aziridine from the starting alkene at least
comparable to, and in one case a six-fold improvement oil,yields reported using other method s.23'24 Thus our method-ology represents an efficient and mild alternative route to//-acylaziridines.
References
1 D. Tanner, Angew. Chem.,Int. Ed" Eng\.,1994,33,599.
Chem. Soc., Perkin Trans. 1,2001 ,944-945 945

Ward, V.R., Cooper, M.A. and Ward, A.D. (2001) The Synthesis of N-Benzoyl Aziridines from β-Benzamidoalkyl Phenyl Selenides. Phosphorus, Sulfur, and Silicon and the Related Elements, v. 172 (1), pp. 195-201,
May 2001
NOTE: This publication is included in the print copy of the thesis
held in the University of Adelaide Library.
It is also available online to authorised users at:
http://dx.doi.org/10.1080/10426500108046651

A.D. Ward, V.R. Ward and E.R.T. Tiekink (2001) Crystal structure of 5-methyl-1,4 di[2-phenylselanyl)cyclohexyl]-4H-1,2,3,4-tetraazol-1-ium perchlorate, [(C6H5SeC6H10)2CH3CN4]CIO4 Zeitschrift fur Kristallographie - New Crystal Structures, v. 216, pp. 551-552, 2001
A NOTE:
This publication is included in the print copy of the thesis held in the University of Adelaide Library.

A.D. Ward, V.R. Ward and E.R.T. Tiekink (2001) Crystal structure of N1-[2-(phenylseleninyl)cyclohexyl]benzamide, C19H21NO2Se Zeitschrift fur Kristallographie - New Crystal Structures, v. 216, pp. 555-557, 2001
A NOTE:
This publication is included in the print copy of the thesis held in the University of Adelaide Library.


















