
Application of Wolff Rearrangerment in Arndt-Eister
12
[220] H. Druckrej, R. Preussmann, and S. Irankoric, Ann. N. Y. Acad. Sci. 163, 676 (1969). [221] See also ref. [39b]. [222] N. P. Sen. Toxic Constituents of Animal Foodstuffs. Academic Press, New York 1974, Vol. 5, p. 131. [223] K. Hujnsr Getreide, Mehl. Brot 27, 249 (1973). [224] H. Druckre!,. Transplacental Carcinogenesis. International Agency for Research on Cancer, Lyon 1973, p. 45, and further literature cited therein. [225] If oxygen is passed into solutions of lithionitrosamines at low tempera- ture. or if peroxides [di-tert-butyl peroxide, bis(trimethylsilyl) peroxide] are added, then vigorous evolution of gas rapidly sets in on warming or on attempted evaporation. The structure of the decomposition products, which we are currently examining, should reveal whether the a-hydroxy nitrosamine derivatives postulated in Scheme 6 are actually formed. [226] M. Okada er a/., Gann 63, 391, 637 (1972): 65, 13, 69 (1974). 12271 L. Blarrmann and R. Prrussmann. Z. Krebsforsch. 79, 3 (1973). [228] F. W Krugrr Pr a/., Z. Krebsforsch. 76, 147 (1971); 79, 90 (1973); 80, 189 ( 1973). [229] R. Schornral, Brit. J. Cancer 28, 436 (1973), further references cited therein. [230] D. Daibur, Med. Dissertation, Universitat Freiburg 1966. [231] G. Eisenbrand and R. Prrussmann, Arzneim.-Forsch. 20, 1513 (1970). [232] The maximum working place concentrations of dimethylnitrosamines have been set at zero in the USA: Chem. Eng. News 51, No. 2, p. 4 (1973). The Wolff Rearrangement of a-Diazo Carbonyl Compounds By Herbert Meier and Klaus-Peter Zeller[*] The readily accessible u-diazo carbonyl compounds are distinguished by their high reactivity, which opens up a variety of preparative applications under modified conditions. Wolff rearrange- ments of these compounds, induced thermally, photochemically, or catalytically, afford ketenes. Free and complexed carbenes, 1,3-dipoles, 1,3-diradicals, and the antiaromatic oxirenes have been considered as intermediates or transition states. The present progress report attempts to integrate preparative and theoretical aspects. 1. Introduction a-Diazo carbonyl compounds (1) contain the CO-CN2 group, which is capable of resonance, as characteristic struc- tural unit. The C< bond can be part of a carbon chain or of a cyclic system. If it belongs to an aromatic ring then an inner dia- zonium phenoxide ("0-quinone diazide") (2) is present. Key positions in the attached groups may also be occupied by heteroatoms. In this context, the u-diazo carboxylic acid derivatives (I), R'=OR, NH2, NHR, NR2, etc., warrant spe- cial attention. [*I Univ.-Doz. Dr. H. Meier and Dr. K. P. Zeller [**] [**I European Science Exchange Program Fellow. 74 Tiibingen I, Auf der Morgenstelle (Germany) Apart from the CO stretching vibration, a simple analytical feature of the open-chain u-diazo carbonyl structure is the diazo band lying between 2090 and 2190cm-' (usually at 2130cm-') in the IR spectrum. Mutual interaction between the diazo and the carbonyl group lowers the CO frequency and raises the N2 frequency. u-Diazo carbonyl compounds are particularly reactive sub- stances. Reactions involving loss of the N2 group are generally induced thermally, photochemically, catalytically, or by (Lewis) acids. Whereas the decomposition by acids is applic- able to all diazoalkanes, in the other three processes one observes a rearrangement that is specific for a-diazo carbonyl compounds and is known after its discoverer as the Wolff rearrangement". 21. The preparative value of the Wolff rearran- gement is due to the ready accessibility of the a-diazo carbonyl compounds[31 and to the wide range of reactions of the result- ing ketenes (4). 32 Angew. Chem. intmnat. Edit. J Vol. 14 (1975) No. I
-
Upload
tran-duy-quynh -
Category
Documents
-
view
32 -
download
2
Transcript of Application of Wolff Rearrangerment in Arndt-Eister

[220] H. Druckre j , R. Preussmann, and S . Irankoric, Ann. N. Y. Acad. Sci. 163, 676 (1969). [221] See also ref. [39b]. [222] N. P. Sen. Toxic Constituents of Animal Foodstuffs. Academic Press, New York 1974, Vol. 5, p. 131. [223] K. Hujnsr Getreide, Mehl. Brot 27, 249 (1973). [224] H. Druckre!, . Transplacental Carcinogenesis. International Agency for Research on Cancer, Lyon 1973, p. 45, and further literature cited therein. [225] If oxygen is passed into solutions of lithionitrosamines at low tempera- ture. or if peroxides [di-tert-butyl peroxide, bis(trimethylsilyl) peroxide] are added, then vigorous evolution of gas rapidly sets in on warming or on attempted evaporation. The structure of the decomposition products, which
we are currently examining, should reveal whether the a-hydroxy nitrosamine derivatives postulated in Scheme 6 are actually formed. [226] M. Okada er a / . , Gann 63, 391, 637 (1972): 65, 13, 69 (1974). 12271 L. Blarrmann and R. Prrussmann. Z. Krebsforsch. 79, 3 (1973). [228] F . W K r u g r r P r a/., Z . Krebsforsch. 76, 147 (1971); 79, 90 (1973); 80, 189 ( 1973). [229] R. Schornral, Brit. J . Cancer 28, 436 (1973), further references cited therein. [230] D. Daibur, Med. Dissertation, Universitat Freiburg 1966. [231] G. Eisenbrand and R. Prrussmann, Arzneim.-Forsch. 20, 1513 (1970). [232] The maximum working place concentrations of dimethylnitrosamines have been set at zero in the USA: Chem. Eng. News 5 1 , No. 2, p. 4 (1973).
The Wolff Rearrangement of a-Diazo Carbonyl Compounds
By Herbert Meier and Klaus-Peter Zeller[*]
The readily accessible u-diazo carbonyl compounds are distinguished by their high reactivity, which opens up a variety of preparative applications under modified conditions. Wolff rearrange- ments of these compounds, induced thermally, photochemically, or catalytically, afford ketenes. Free and complexed carbenes, 1,3-dipoles, 1,3-diradicals, and the antiaromatic oxirenes have been considered as intermediates or transition states. The present progress report attempts to integrate preparative and theoretical aspects.
1. Introduction
a-Diazo carbonyl compounds ( 1 ) contain the CO-CN2 group, which is capable of resonance, as characteristic struc- tural unit.
The C< bond can be part of a carbon chain or of a cyclic system. If it belongs to an aromatic ring then an inner dia- zonium phenoxide ("0-quinone diazide") (2) is present.
Key positions in the attached groups may also be occupied by heteroatoms. In this context, the u-diazo carboxylic acid derivatives ( I ) , R'=OR, NH2, NHR, NR2, etc., warrant spe- cial attention.
[*I Univ.-Doz. Dr. H. Meier and Dr. K. P. Zeller [**]
[**I European Science Exchange Program Fellow. 74 Tiibingen I , Auf der Morgenstelle (Germany)
Apart from the C O stretching vibration, a simple analytical feature of the open-chain u-diazo carbonyl structure is the diazo band lying between 2090 and 2190cm-' (usually at 2130cm-') in the IR spectrum. Mutual interaction between the diazo and the carbonyl group lowers the CO frequency and raises the N2 frequency. u-Diazo carbonyl compounds are particularly reactive sub- stances. Reactions involving loss of the N2 group are generally induced thermally, photochemically, catalytically, or by (Lewis) acids. Whereas the decomposition by acids is applic-
able to all diazoalkanes, in the other three processes one observes a rearrangement that is specific for a-diazo carbonyl compounds and is known after its discoverer as the Wolff
rearrangement". 21. The preparative value of the Wolff rearran- gement is due to the ready accessibility of the a-diazo carbonyl compounds[31 and to the wide range of reactions of the result- ing ketenes ( 4 ) .
32 Angew. Chem. intmnat. Edit . J Vol. 14 (1975) No. I

2. Methods of Conducting the Reaction
2.1. Thermolysis
Thermal energy can induce a-diazo carbonyl compounds to lose nitrogen and undergo Wolff rearrangement. The decom- position temperatures range between room temperature and 750°C in gas phase pyrolysisL4! Although generally greater than that of common diazoalkanes, the thermal stability of a-diazo carbonyl compounds can be reduced by electronic effects[5* 6 l and even more effectively by steric effects[']. Thus the diazo ketone ( 5 ) , whose -CO-CN2- is twisted owing to ring strain, decomposes on formation at room tempera- ture['].
The tendency to undergo Wolff rearrangement often shows a marked initial increase at the expense of other competing reactions as the thermolysis temperature is raised. Reaction in boiling aniline or boiling benzyl alcohol with addition oftertiary bases suchascollidine, isoquinoline, or N,N-dimethyl- aniline has become a proven preparative method.
sary. Decreasing wavelength (increasing energy) of the radiation used leads to increasing amounts of by-products. However, the lowest energy excited singlet state S 1 of diazo ketones, generated by irradiation at the wavelength of a low-intensity forbidden transition in the near UV or visible range, displays only modest reactivity (cf. Section 6). In the presence of triplet sensitizers, Wolff rearrangement is greatly reduced or completely suppressed in favor of other carbene reactions.
2.3. Catalysis
The decomposition temperature of a-diazo carbonyl com- pounds can be drastically lowered by metal (ion) catalysts Ag, Age, Cu, Cue, Cu2@, Pt, Pd, Rh, etc.'Most frequently use is made of freshly precipitated Ag(1) oxide in the presence of alkaline reagents. With the exception of CuI, copper cata- lysts not only exert a n activating effect on nitrogen elimination but also stabilize the resulting carbene (see Section 6.2) by complex formation[31. The same applies to rhodium[12' and palladium catalysts" 'I. As a consequence, Wolff rearrange- ment no longer occurs or becomes very difficult. Catalysts are sometimes also used during photolysis of a-diazo carbonyl compounds[141 (Table 1).
Table 1. Catalysts for Wolff rearrangement of 3-diazo ketones.
Catalyst Ref.
2.2. Photolysis
The photochemical variant of the Wolff rearrangement discov- ered by Homer[*] is frequently superior to other methods. It also succeeds when the thermal or catalytic approach merely results in a C-H insertion, as with diazocamphor (8jr9 - ' *I.
AgLWa,S,O, AgzOINa 2S1O~iNazCOz AgNOz C6Hi-C02Ag/tertiary base CFA-COZAg colloidal Ag from AglO + HlCO CUO Cu I/acetonitrile CuIacetonitrile platinum
&O (8) N Z
3. Secondary Reactions of Ketene Products
The ketenes ( 4 ) are generally not isolated after a Wolff re- arrangement ; instead their reaction products are obtained.
3.1. Addition of Nucleophiles
The limit of the photochemical method is reached when the desired reaction product is itself photolabile under the irradia- tion conditions, e. g. with 2-diazo-1,3-indandione ( I I ) . The choice of wavelength should be guided by the common photochemical principle: as long as possible, as short as neces-
If the Wolff rearrangement is conducted in the presence of nucleophilic reagents whose proton activity is not excessively high, then the ketene intermediates yield carboxylic acids ( 1 4 ) or their derivatives['. 2 2 , Z2a].
This synthesis represents the principal application of the Wolff rearrangement, particularly in connection with the homologi- zation of carboxylic acids'151. Remarkably, the photochemical
+CHIOH H
( I 2 ) . 45-50% aCooCH3 0
Angew. Chem. infernaf. E d i f . 1 Vol. 14 ( 1 9 7 5 ) 1 No. I 33

X = OH, OR, NHz, NHR, NHOR, NR2, S R
Wolff rearrangement of 2-diazo 1,3-diketones in the presence of water leads to P-keto carboxylic acids which otherwise prove to be unisolable[" ' 9 I s "1 (cf. Section 4.5).
3.2. Intermolecular and Intramolecular Cycloadditions
While the relatively stable ketenes resulting from decom- position of corresponding cr-diazo ketones in inert solvents are isolable, neither aldo ketenes nor their dimers can be prepared. In some cases the more labile aldo ketenes react with intact cr-diazo ketone to give 3-buten-4-olides (15) [23-261 or pyrazoles ( 1 6 ) [ 2 7 1 .
occur preferentially at the ketene stage in the photochemical reaction[371 but at the carbene stage in the catalytic p r o ~ e s s l ~ ~ !
H COCHN, H C0-C-H
H CH=C=O 6 b;& On thermolysis of I-diazo-2-0x0- ( I 7) and 2-diazo-l-oxo-1,2- dihydronaphthalene (28), Yates and Robb obtained the same 2-methylene-l,3-dioxole derivative ( I 9)lz81.
Nearly all o-quinone diazides behave in this manner on ther- molysis[28a1. The resulting ketene reacts with unrearranged primary fragments (carbenes), yielding compounds of type (21).
P
The ketenes formed "in situ" in the Wolff rearrangement have frequently been subjected to cycloadditions with C=C, C=N, and N=N double bonds or with o - q u i n o n e ~ [ ~ ~ - ~ ~ ~ . The trap- ping of an unstable aminoketene to give the a-amino p-lactam (23) should be cited as an The scope of this cycloaddition is limited above all by reaction of the starting compound itself or its primary fragments (car- benes) prior to rearrangement. The case of the diazo ketone (24) is interesting because intramolecular cycloadditions
4. Substrates Capable of Rearrangement
4.1. Open-Chain a-Diazo Ketones
Whereas little is known about the Wolff rearrangement of a-diazo aldehydes[39- 4'1, numerous publications have appeared on the fragmentation of open-chain a-diazo ketonesF3! In principle, alkyl, aryl, and aralkyl groups can migrate. The rerr-butyl group in (27)'421 and the triphenyl- methyl group in (30)[431 qualify as borderline cases.
H3C 0
While the tert-butyl group of (27) invariably displays only a limited proclivity to migrate, the triphenylmethyl group undergoes clean photochemical reaction. In the catalytic pro- cess, however, anomalous attack at the ortho position of a phenyl ring occurs in compound (30). 9-Diazoacetylfluorene behaves similarly to triphenyldiazoacetylmethane (30)[431. The presence of additional functional groups, particularly in cr position to the CO group, can interfere with the Wolff rearrangement or lead to secondary reactions. Studies on the influence of, for example, h a 1 0 [ ~ ~ - ' ~ ] , substituted hy- d r o ~ y l [ ~ ~ * "1,freeandsubstitutedamino~56 ~ 641,cyclopropyl[651,
34 Angew. Chem. internat. Edi t . J Vol. 14 f 1 9 7 5 ) J No. I

and cyclopropenyl g r o ~ p s [ ~ ' - ~ ~ - ~ 'I h ave been reported. Incor- poration of double or triple bonds into the @-diazo carbonyl compounds, especially in the ctp position to the CO group[781, suppresses the Wolff rearrangement either completely or par-
occur with the @$-unsaturated diazo carbonyl compounds (33)[791 and (34)[*'l.
tialIy[34. 37. 38. 7 2 - 7 8 1 . Th e Wolff rearrangement is known to
4.2. Cyclic a-Diazo Ketones
Apart from the Arndt-Eistert homologization["- 821, ring contraction is the main application of the Wolff rearrange- ment. Above all the photochemical variant proves suitable
cyclics[85. 8 7 , i o n - 1031, paracyclophanes[104- 1061 , ster- ). The formation of the oxetane (36) can be con-
sidered as an example["3,
for producing strained systems (bicyclics[". 83-991 3 POlY-
aids[ I07 - 1 I21
R' R Z 0
O g N 2 R' R2 @OOH
A, hv, cat.
"* R R2
2-Diazo-1 -0xoacenaphthene is a negative example-it fails to undergo ring contraction[' ' 'I.
4.3. o-Quinone Diazides
As originally demonstrated by 0. Siis["", '17,117a1, the Wolff rearrangement can also be implemented with o-quinone di- azides. In this case the rearrangement involves ring contraction, thus providing access to the cyclopentadiene series (Sus reac- tion). This reaction is exploited in a photocopying process (Diazotype, offset process)" 1 6 - ' ' * I in which an increase in pH promotes coupling of rearrangement products with unreacted o-quinone diazide, e.g. (38) with (37), to yield the azo dye (39).
( 3 7 )
I SO3H SO3H S O a
( 3 8 ) (39)
Coupling reactions can be avoided by working in acid media. Thus irradiation of derivatives of l-diazo-2-oxo-1,2-dihydro- naphthalene and 2-diazo-l-oxo-1,2-dihydronaphthalene leads to the corresponding derivatives of 1-indenecarboxylic acid[l16. 1 1 9 , 1 2 0 1 [see formula ( I 9) in Section 3.21. Application to the pyridine and quinoline series["'- l2 '1 permits synthesis of pyrrole- and indolecarboxylic acids. Photochemical trans- formation of 3-diazo-2-0~0-2,3-dihydropyridine to 2-pyrrole- carboxylic acid is a remarkable reaction because it involves migration of an sp2 nitrogen[12"! Whereas (40)[122* '231,
( 4 1 ) [ ' 2n1, and (42)" 241 undergo smooth Wolff rearrangement,
only 2 % ring contraction occurs with (43)[12'], presumably owing to an increase of ca. 50kcal/mol in strain energy['"!
( 4 3 ) w (44 ) 2 %
Thermal decomposition of o-quinone diazides in inert solvents leads to 1,3-dioxoles, only part of the keto carbene being subject to Wolff rearrangement (cf. Section 3.2).
4.4. Derivatives of a-Diazo Carboxylic Acids
The first example of Wolff rearrangement of a functional group bound to a heteroatom was observed in the photolysis of 0-diazoacetylserine (45) in water['26'.
hu N~=CH-CO-O-CH~<H-CO~~ -
I H20 145) NH3@
H O ~ C -C H~-O-CH,-CH-C 0.p I
( 4 6 ) m3@
Nowadays a series of examples is known for the migration of substituted hydroxyl[I2'- 1331, mercapto['341, and amino groups['27. 135-1371, which are all usually induced photochem- ically. Formation of dimethyl methoxymalonate from
,COzCH3 hv /C0-0CH3 CD,OD ,COOCH3 N2=C, - :c, D3CO-C?
COzCH3 - N 2 CO43CH3 COOCH3 (49 )
,COOCD3 4 (47 )
,0CH3 CDiOD o=c =c, - HsCO-C,D COzCH3 COOCH3
( 4 8 ) (50)
dimethyl diazomalonate (47) involves, for example, about 30% of an intramolecular methoxy group migration since irradiation of (47) in perdeuteriomethanol affords an 80% overall yield of the insertion product (49) and the rearrange- ment product (50) in the ratio 9:4[I3O1.
4.5. 2Diazo 1,SDicarbonyl Compounds
On rearrangement of unsymmetrically substituted[ 1381 2-diazo 1,3-dicarbonyl compounds (51 ), both R ' and R 2 can migrate, since a diacylcarbene (52) is present after loss of N2. Studies on compounds ( 5 1 ) (see Table 2) therefore provide an indica- tion of the relative migration tendencies of various groups (cf. Section 6).
Angew. Chem. internat. Edit. / Vol. 14 f1975) 1 No. I 35

R'-C-C-C-R~ II II II 5. Competing Reactions
5.1. [1.2]-Hydrogen Shifts
wDiazo ketones of type (60) very frequently react via a [ 1.21-H shift at the carbene stage, especially under catalytic
(53) (52) (54 ) conditions, to form cr,P-unsaturated ketones (61)[16'- l 7 'I.
(51) 0 N 2 0
l - N 2 - R 2 R2\
,c =o I1 II o=c,'
R' -R' c =c =o
R'
o=c =c= - R'-C<~-C-R~ - R2 0 0
Table 2. Wolff rearrangement of unsymmetrically substituted open chain 2-diazo 1.3-dicarbonyl compounds of type ( 5 1 ) .
R' R2 Mode of f53) + (541 (531 : f 5 4 ) Ref. reactions yield ["4]
hv hv
A hv Ag@-cat. A A hv A hv A hv A A A hv hv hv A A A A hv hv A hv
84 82
> 90 > 90
74
64 74 53 21 15 -
-
44 57 78 88 -
- -
7 20-28 28 14-18 16 -25
49 51 [151a] 13 87 [151.
151a] 0 100 [152] 0 100 [152] 0 100 [I531
62 38 [152] 60 40 [I541 4 96 [I521
76 24 [152] 25 75 [I521 0 100 [I521 0 I00 [I521
80 20 [I541
0 100 [155] 0 100 [I551 0 100 [150] 0 100 [I501 0 100 [156] 0 100 [156] 0 100 11561 0 100 [79] 0 100 [79]
40 60 [155] 0 100 [I371
98 2 [154]
0 100 r1371
Cyclic 2-diazo 1,3-dicarbonyl compounds can also be included in these investigations. The arrows in formulas (55) ' ' 571,
(56)[l5'I and ( 5 7 ) [ ' 5 9 1 indicate the centers of selective migra- tory aptitude.
R'-C-C-CH,-R v A, hu, cat. R'-C-C<H,R II II - Nz I I 0 N2 0
k (601
R'-C<II=CH-R
(61)
N2 A [1.2]-methyl shift is the principal reaction route of 4-diazo- 2,2,5,5-tetramethyl-3-hexanone (27)f421 (cf. Section 4.1).
(55) O (57)
4.6. Other Diazo Carbonyl Compounds 5.2. Hydrogen Abstraction
Thermally['601 or photochemically induced[L61. 1621 W olff rearrangement of 1 -diazo 2,3-dicarbonyl compounds requires migration of an electron-impoverished carbonyl carbon to a carbenoid center, thus imparting not only preparative but also theoretical interest to the reaction. Final confirmation of the Wolff rearrangement of 1,3-bisdiazo 2-carbonyl compounds ( 5 8 ) [ ' 6 3 - 1661 is still outstanding.
Saturation of the transient carbene by H abstraction from the solvent occasionally accompanies the Wolff rearrangement. In ethyl cr-diazotrifluoroacetate (62)I5'' it becomes the major reaction:
F&<-C-COOC2H5 + C2H50H 5 F&-C-CH2-COOC2H5 II II - N2 II 0 N2 (62) (63)
+ HsC-CHO
In the photochemical process, the lowest triplet state (of the diazo ketone) is responsible for H abstraction['401. The extent of this competing reaction depends upon the wavelength and solvent [ l 4 O 1 .
36 Angew. Chem. internat. Edi t . J Vol. 14 ( 1 9 7 5 ) J No. I
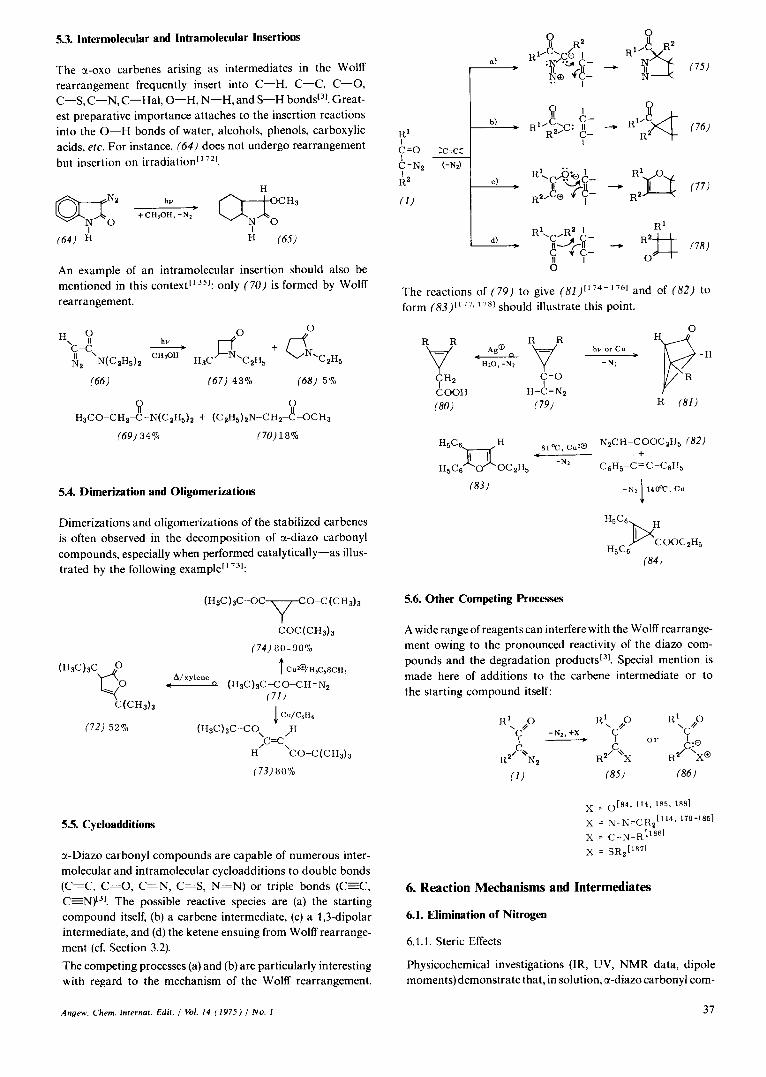
5.3. Intermolecular and Intramolecular Insertions
The ct-0x0 carbenes arising as intermediates in the Wolff rearrangement frequently insert into C-H, C-C, C-0, C-S, C-N, C-Hal, 0-H, N-H, and S-H bonds[31. Great- est preparative importance attaches to the insertion reactions into the 0-H bonds of water, alcohols, phenols, carboxylic acids, etc. For instance, (64) does not undergo rearrangement but insertion on irradiation" 721.
An example of an intramolecular insertion should also be mentioned in this context[I3']: only (70) is formed by Wolff rearrangement.
(66) (67) 43% (68) 5 %
Q R H3CO-CHZ-C-N( CzH,), + (C 2H5) ~ N - C H Z - C ~ C H ~
(69) 34% (70)18%
5.4. Dimerization and Oiigomerizations
Dimerizations and oligomerizations of the stabilized carbenes is often observed in the decomposition of a-diazo carbonyl compounds, especially when performed catalytically-as illus- trated by the following example[' 731:
(72) 5 2 % (H3C)3C-C0, 21 ,c=c
H 'C 0-C (C H3) 3
(73) 80%
5.5. Cycloadditions
a-Diazo carbonyl compounds are capable of numerous inter- molecular and intramolecular cycloadditions to double bonds (C=C, C=O, C=N, C=S, N=N) or triple bonds (CEC, CEN)[31. The possible reactive species are (a) the starting compound itself, (b) a carbene intermediate, (c) a 1,3-dipolar intermediate, and (d) the ketene ensuing from Wolff rearrange- ment (cf. Section 3.2). The competing processes (a) and (b) are particularly interesting with regard to the mechanism of the Wolff rearrangement.
$=o :c=c: I
The reactions of (79) to give (81)11'4-1761 and of (82) to form (83)["'* 1 7 * ] should illustrate this point.
p 2
COOH c =o I
H-C=Nz (801 (79)
hv or Cu
- Ni
5.6. Other Competing Processes
A wide range of reagents can interferewith the Wolff rearrange- ment owing to the pronounced reactivity of the diazo com- pounds and the degradation products[31. Special mention is made here of additions to the carbene intermediate or to the starting compound itself:
6. Reaction Mechanisms and Intermediates
6.1. Elimination of Nitrogen
6.1.1. Steric Effects
Physicochemical investigations (IR, UV, NMR data, dipole moments) demonstrate that, in solution, a-diazo carbonyl com-
Angew. Chem. internat. Edi t . f Vol. 14 (1975) f No. 1 37

pounds generally exist in Z / E equilibrium about the C-C bond having partial double bond characterf189- 1931 (AG f98 = 15 kcal/mol).
In view of a concerted mechanism of the Wolff rearrangement starting from the Z-conformation, the two rotamers have occasionally been assigned differing reactivities" 891. The be- havior of compounds ( 5 ) ( Z , labile) and (27) (E , 3% Wolff rearrangement) fits this concept well, but still does not prove its validity. Steric hindrance has been proposed as an alterna- tive explanation for the inertness of (27), and the lack of delocalization energy for the lability of ( 5 ) . Resonance of the coplanar CO-CN2 group normally leads to a gain of about 0.7 p in delocalization energy['94! However, resonance is impossible in the twisted heptacycle (5) since the CO and C N 2 are mutually orthogonal. Kinetic studies['95- 1 9 ~ 1
and the finding that substituents have different effects on nitrogen e l i m i n a t i ~ n ~ ' ~ ~ . 2o01 and on migratory aptitude con- traindicate a concerted mechanism, regardless of the manner in which the reaction is conducted.
6.1.2. Electronic Effects
For the case of substituted azibenzils (87), electronic effects occurring on thermal nitrogen elimination can be correlated with the Hammett o constants['96!
Electron-withdrawing groups RZ decelerate the decomposi- tion, while their electron-repelling counterparts have an acceler- ating action. The effect of groups R' is exactly opposite, but less significant[196* l9'1.
In the photochemical variant, N 2 elimination can be accom- plished both by direct irradiation in the entire absorption range of the a-diazo carbonyl compound and on triplet sensi- tization. The apparent integral quantum yield is always lower than 1[2001. Values of 0.6, 0.5, and 0.07[2011 are obtained for diazoacetophenone at 250,294, and 325 nm. The longest wave- length forbidden transition['931, which leads to the lowest energy singlet excited state S I , is therefore not very effective. S undergoes mainly radiationless deactivation. Under suitable conditions additional photochromic valence isomerization to diazirines ( 8 9 ) is possible[201* 'OZ1.
R 1881
I
R 1891
Numerous quantum yield measurements have been performed on substituted diazoacetophenones[2001. Electron-withdraw-
ing and electron-repelling para substituents lower the quantum yield. No uniform trend could be established for electronic effects in the catalytic variant['98. 1991.
6.1.3. Medium Effects
Elimination of nitrogen is independent of both the rearrange- ment and also the presence of nucleophilic reagents['95* 1961. It obeys a first-order rate law[1961. This disproves theaddition- protonation mechanism discussed by E i ~ t e r t [ ~ ~ ~ ] , which assumes participation of the nucleophile prior to nitrogen elimination. There remain certain medium e K e c t ~ [ ' ~ ~ - 19'. 2041
arising from polarity changes and from hydrogen bonding to the carbonyl oxygen or the diazo carbon. The presence of such aggregates in mixed systems of aprotic and protic solvents has also been detected by UV spectroscopy[2051. The resulting barrier to resonance facilitates thermal elimination of N,. However, the absence of H/D isotope e f fec t~~"~~demon- strates that complete H @ transfer does not take place in the rate-determining step. Whereas monomolecular decomposition can be assumed in the thermal and photochemical processes, transient participa- tion of the metal or metal ion occurs in the catalytic variant. According to a hypothesis put forward by Huisgenf2061, the electrophilic Ag@ ion adds to the diazo carbon and thus effects nitrogen elimination and migration of the group. Indeed, reaction of diazomethyl ketones (90) with silver(1) oxide in tetrahydrofuran at - 15°C affords silver-substituted diazo ketones (91 )['071.
Newman and Beal[171, who have developed a homogeneous medium for the AgO catalyzed rearrangement (methanol in the presence of silver benzoate and triethylamine) assume a radical chain process to occur under these conditions. Trieth- ylamine has a kind of initiating effect as proton transfer agent and Age likewise for electron In recent kinetic studies['981 a complex between the deprotonated diazo ketone and Ag[N(C2H5)3]? has been proposed as crucial interme- diate.
6.2. Primary Fragments
The singlet carbene (a) , the 1,3-diradical ( b ) , the 1,3-dipole (c), and the two-spin systems ( d ) and ( e ) are reasonable possibilities for the ground state of the primary fragments left after elimination of nitrogen.
In the plane of the rs bond skeleton there is a p orbital of the oxygen and an sp2 or p orbital of the "diazo carbon" to be populated; in a plane at right angles there are three
38 Angew. Chem. internat. Edit. 1 Vol. 14 f 1979) 1 No. I

p orbitals comprising an ally1 system. ( a ) and (c) are singlet states. A scalar and dipolar spin coupling can be assumed in ( b ) , and dipolar spin couplings in ( d ) and (e). This leads to singlet and triplet splitting, so that a total of five singlet and three triplet states will result. If the sp2/p alternative or antibonding levels are included, then the number of possibil- ities increases considerably. The energetic proximity of these states makes any theoretical decision difficult. In the thermal case [ e . g . (92), (94 ) ] an indication of the nature of the primary fragments is provided by trapping- and side reactions (cf. Section 5).
Thus a singlet carbene and 1,3-dipole participation may be assumed. However, in the numerous 1,3-dipolar cycloadditions with C=C, C=O, C=S, C=N, C z , and C E N it is often doubtful whether the primary fragments or the original a-diazo carbonyl compound itself is involved in the reaction. With primary fragments showing a low tendency to undergo rearrangement and additional stabilization by complex formation with copper, trapping reactions, yielding e . g . (97) are much more
C H ? - C H3
In the Ag'-catalyzed Wolff rearrangement the free keto car- bene is apparently present after elimination of nitrogen. The reaction course during direct photolysis was followed by absorption spectrometry (flash photolysis)[" 'I and by ESR spectroscopy['13~ ' 14] . A t 77K, azibenzil affords a triplet car- bene exhibiting hardly any Wolff rearrangement ( < 3 %). Marked deviation from an angle of 180" can be deduced from the ratio of its zero field parameters (D/hc=0.3815, E/hc=0.0489 cm I ) . (The triplet carbene disappears in favor of an oxygen radical when the temperature is r a i ~ e d [ ~ l ~ I . ) The Wolff rearrangement of azibenzil increases up to a yield of 85 -90% at room In the triplet-sensitized decomposition of a-diazo carbonyl compounds, Wolff re- arrangement occurs at most as a minor reaction['52*215-217! The major processes are H abstraction and insertions. The most obvious conclusion to be drawn from all these observations is that the photochemical Wolff rearrangement takes place in a singlet state of the primary fragment. Since the relative migratory aptitudes of individual groups differ from those in the thermal variant (cf. Sections 4.7 and 6.4), the process must involve a singlet excited state. For small
singlet-triplet splitting, an intersystem crossing of low activa- tion energy in both directions can be assumed so that reactions of the triplet state are also observed on direct photolysis, and the Wolff rearrangement is found to occur in sensitized photolysis. The quantum yield for the transition S1-T' increases with the temperature and the energy of the illuminat- ing light[140!
n
F3CyOyCH3 + H,C-CN
hu -
(101) ROOC
Photochemically generated primary fragments, e. g. from (98), can also be trapped in a kind of carbene reaction or a 1,3-dipo- Iar reaction[' l 8 -" '1.
6.3. The Oxirene Intermediate
The oxirenes (102), antiaromatic 4rr electron systems, formed by valence isomerization of the primary fragments should possess similar energies to the a-0x0 carbenes["' - 2241. The true role of the oxirenes in decomposition of a-diazo carbonyl compounds long remained unclear due to a lack of experimen- tal ~ o r k [ ~ ~ ~ - ~ ~ ~ l . C onclusive proof of the intermediate appearance of oxirenes during the photochemical Wolff rear- rangement has been obtained with the aid of three independent methods employing isotopic labeling.
11021
The extent of oxirene"] formation depends upon the condi- tions, e. g . on theconcentration of water[2281. The carbene-oxir- ene equilibrium makes it difficult to reach any conclusion regarding the migratory aptitude of the groups R ' and R2. In all systems investigated so far[2291, however, the initial carbene formed after nitrogen elimination undergoes reaction. Oxirene participation could be ruled out on sensitized photo- lysis, on catalysis, and on thermolysis in 2311 .
Proof of oxirene formation that is completely independent of the Wolff rearrangement was obtained with unsymmetri- cal a-diazo dialkyl ketones['701 and a number of special aryl- substituted a-diazo ketonedZ3' 'I.
['I No oxirene could be detected on photolysis of a-diazohornoadarnant- anone in the presence of water [228a].
Angew. Chem. internat. Edit. / Vol. 14 ( 1 9 7 5 ) / No. 1 39

0
[ 1 . 2 1 - H - s h i f t 1 0
etc.
(103)
h v : 57% A ( 1 6 5 "C) : 64%
cat . : 1 0 0 %
1 [ I . Z ] - H - s h i f t
etc. 0
43% 36%
0 or,
(104)
The trapping reaction yielding (105 ) strongly suggests that the oxirenes are true intermediates, and not merely transition statesI2 '1.
hv - -N,
0
6.4. Rearrangement of the Primary Fragments
The examples cited in Section 4 demonstrate that hydrogen, alkyl, aryl, substituted hydroxyl, mercapto, and amino groups can all migrate. Thus H, C(sp3), C(sp2), C(sp), N(sp3), N(sp2), 0 , or S represent migrating centers. Apart from this absolute migratory disposition, the relative migratory aptitudes of indi- vidual groups established primarily with 2-diazo 1,3-dicar- bony1 compounds (Section 4.5) also warrant interest. The following series is generally valid for the thermal reac- tion[152. 1 5 5 . 156, 1 3 7 1 .
H C,H5 C H 3 > NRI z OR
In the photochemical variant, phenyl and methyl change places. The electronic effects are similar in both cases[1521:
The mesityl and above all the 2,4,6-tribromophenyl group migrate far more readily than purely electronic effects of methyl and bromo substituents would suggest" 541. This pronounced steric effect is most probably due to restriction of resonance in the keto carbene intermediate (106).
All these findings show that a high electronegativity at the migrating center and an electron-poor R I-CO bond tend to favor the relative migratory aptitude of the group R ' (in accord with classical ideas of an anionotropic migration). In contrast, a partial double bonding character of this bond arising from participation of the C 0 group in resonance of R ' have an unfavorable effect, especially in the excited state. The group R2 also influences the migratory disposition of
Regardless of the way in which the reaction is conducted, the migrating groups are not severed entirely from the molecu- lar framework, thus largely or completely conserving all stereo- chemical information[234- 2421. In particular all intermolecular mechanisms can therefore be discounted.
R 112331,
7. Conclusion
The principles of the above mechanistic and preparative inves- tigations on the Wolff rearrangement show how fresh aspects have repeatedly come to light during the course of work on a 70-year old reaction. A considerable extension of this topic has recently been provided by electron-impact induced Wolff r e a r ~ - a n g e m e n t [ ~ ~ ~ - ~ ~ ~ ] , and by thermal and photolytic rearrangements of related systems such as a-diazo sul- f o n e ~ [ ~ ~ ' ] , a-diazo phosphane 2491, 0-0x0 sulfonium y l i d e ~ [ ~ ~ ~ ] and their S - o ~ i d e s ' ~ ~ ' ~ , 1 , 2 , 3 - t r i a ~ o l e s ~ ~ ~ ~ ~ , 1,2,3- thiadiaz01es '~~~- 2 5 h 1 , and 1,2,3-selenadia~oles~~~~~ 259!
Received: July 18, 1974 [A 36 IE] German version: Angew. Chem. <97,43 (1975)
[ I ] L. WolPY, Liebigs Ann. Chem. 325, 129 (1902): 333, I (1904): 394, 23 ( I 9 12).
[2] Work independent of Wow was published almost simultaneously by G. SrhrBtrr, Ber. Deut. Chem. Ges. 42, 2346 (1909); 49, 2704 (1916).
[3] Recent surveys of general syntheses and reactions of z-diazo carbonyl compounds: B. Eisfrrr. M. Rrgirz, G . Heck. and H . Schwa11 in Houben-Weyl- Muller: Methoden der Organischen Chemie. 4th Edit., Thieme, Stuttgart 1968, Vol. 1014. pp. 473N.: F . We!.gand and H . J . Besfmann, Angew. Chem. 72, 535 (1960): W Ried and H . M m g l e r . Fortschr. Chem. Forsch. 5, 1 (1965): W K i r m s r : Carbene Chemistry. 2nd Edit., Academic Press, New York 1971; W! J . Baron, M . R . De Camp, M . E. Hendrick, M . Jones, J r . , R . H . Lecin, and M . B. Sohn in M . Jones, Jr . and R. A . Moss: Carbenes. Wiley, New York 1973, Vol. I , pp. 2m.: L. L. Rodina and I . K. Korohifsyna, Russ. Chem. Rev. 36, 260 (1967); A. Musfafa, Advan. Photochem. 2, 113 (1964). [4] M . P . Caua and R . J . Spangler, J. Amer. Chem. SOC. 89, 4550 (1967). [5] W Jugelt and D. Schmidt, Tetrahedron 25,969 (1969).
[6] A. Mrlzer and E . F . Jenny , Tetrahedron Lett. 1968, 4503.
[7] A. [ I P Groor. J . A. Borrma, J . dr Valk. and H . Wvnhrrg, J. Org. Chem. 33, 4025 (1968).
[ 8 ] L. Horner and E. Spietschka, Chem. Ber. 85, 225 (1952).
[9] J . Bredt and W H o l z , J. Prakt. Chem. [2] 95, 133 (1917). [lo] L. Hornrr and E. Spierschka, Chem. Ber. 88, 934 ( I 955).
[ l t ] T Gibson and W F . Erman, J . Org. Chem. 31, 3028 (1966). 1121 R . Paulissen el a/. , Tetrahedron Lett. 1973, 2233.
[13] N . Yoshimura, S:J. Marahashi, and J. Morifani . J. Organometal. Chem. 52, C 58 (1973).
[ I41 U . R . Gharak ef a/ . , J. C. S. Chem. Comm. 1973, 548. [ I51 F . Arndr and B. E i s f e r f , Ber. Deut. Chem. Ges. 68, 200 (1935): cf. also ref. [S l ] .
[ I61 A. Burger and S . Acokian, J. Org. Chem. 5, 606 (1940).
[ I71 M. S. Newman and P . F . B e d , J. Amer. Chem. SOC. 72, 5163 (1950). [ I81 R . Casanooa and T Rrichsrrin. Helv. Chim. Acta 32, 649 (1949).
1191 P. Yares and J . Crawford . J. Amer. Chem. SOC. 88. 1562 (1966).
[20] P. Yorrs and J . Fuggrr, Chem. Ind. (London) 1957, 151 I . 1211 L. WofL Liebigs Ann. Chem. 394, 23 (1912). [22] F. Wrygand and H . J . Besfmann, Chem. Ber. 92, 528 (1959). [22a] M . T W! Hearn and H . D. Ward, Austral.-J. Chem. 1969, 1731. R = CH3, B r
40 Angew. Chrm. infernat. Edit . J Vo1. 14 11975) 1 No. I

[23] K. B. Wibrrg and 7: W Hirrron, J. Amer. Chem. SOC. 76, 5367 (1954). [24] J . Quinrana, M. Torres, and F . Serrurosa, Tetrahedron 29, 2065 (1973). [25] P . Yarvs and 7: J . Clark, Tetrahedron Lett. 1961, 435. [26] J. E. Baldwin, Tetrahedron 20, 2933 (1964). [27] M. Tukrhayashi and 7: Ibara, Bull. Chem. SOC. Jap. 41, 1700 (1968). 1281 P . Yarrs and E. W! Robh, J. Amer. Chem. SOC. 79, 5760 (1957).
[28a] 19: Rird and H. M m g l e r , Fortschr. Chem. Forsch. 5, 1 (1965); R. Clinging, F . M. Dean, and G . H. Mitchell, Tetrahedron 30. 4065 (1974).
[29] W Kirmse and L. Horner, Chem. Ber. 89, 2759 (1956). [30] L. Hornrr and E. Spierschka, Chem. Ber. 89, 2765 (1956). [31] L. Horner. E. Spirrschka, and A . Gross, Liebigs Ann. Chem. 573, 17
[32] W Ried and W Radr, Liebigs Ann. Chem. 676, 110 (1964); 688, 170 I 1965).
1331 W! Fischer and E. Fahr, Tetrahedron Lett. 1966, 5245. [34] P . Yares and A . G . Fallis, Tetrahedron Lett. 1968, 2493. 1351 S. .Ma.sarnunr and K . Fukumoro, Tetrahedron Lett. 1965, 4647. [36] E. Muller and P . Heinrich, Chem.-Ztg. 95, 567 (1971): 96, 112 (1972). [37] P . K. Freeman and D . G. Kuper, Chem. Ind. (London) 1965,424. 1381 J . Meiniuald and G . H. Wahl, Chem. Ind. (London) 1965.424. 1391 A . Kranrz, J . C. S. Chem. Comm. 1973, 670. [40] Z . Arnold, Chem. Commun. 1967, 299. [41] J. Krrtyrra, Z . Janoiikk. and Z . Arnold, Collect. Czech. Chem. Commun. 35, 3618 (1970). [42] M. S. Nrwman and A. Arkrl, J. Org. Chem. 24, 385 (1959). [43] A . L. Wilds cf a / . , Tetrahedron Lett. 1965, 4841 ; J. Amer. Chem. SOC. 84. 1503 (1962); 1. Org. Chem. 34, 2401 (1969). 1441 J . MichalsLy and I . Rakovo, J . Prakt. Chem. [4] 8, 181 (1959). [45] J . Mic,halsky, M. Holik. and A. P o d p e r o w , Monatsh. Chem. 90, 814
[46] A. Rordig and H. Lunk, Chem. Ber. 87, 971 (1954). [47] A . Rordig and R . Maier , Chem. Ber. 86, 1467 (1953). [48] F . J. Buckle, F . L. Pafrison, and B. C. Saunders, J . Chem. SOC. 1949, 1471, 2774. [49] F. Brown and W K . R. Musgraoe, J . Chem. SOC. 1953. 2087. [50] J . D. Parker ef a/. , J . Org. Chem. 23, 1166(1958).
[Sl] F. We!.gond, U: Schrwnkr, and H . J. Brsrmann, Angew. Chem. 70, 506 ( I 958). [S2] B. L. Dyatk in and E. P . Mocholina, Izv. Akad. Nauk SSSR, Ser. Khim. 1965, 1035. 1531 F. W'j.ganrl and K . K o c h , Angew. Chem. 73, 531 (1961). [54] J. H. Looker and L. L. Braun, J. Org. Chem. 23, 1062 (1958). [55] F . W Bachelor and G . A. Miano, Tetrahedron Lett. 1967, 4733. 1561 K. Balenovie. I! Thaller, and L. Filipooic, Helv. Chim. Acta 34, 744 I I95 I ). 1571 K. Balenovie and J . Dcornik, J. Chem. SOC. 1954, 2976. [58] K. Balmovic, D. Fles, and I . Jambresic, Croat. Chem. Acta 28. 303 I 1956). [59] K. Balenocic, 1. Jambresic, and I . Ranogajec, Croat. Chem. Acta 29, 87 (1957). [60] K. Balenouic, Chem. Ind. (London) 1955, 1673. [61] F . W<,rgand, Chem. Ber. 91. 1037 (1958). [62] F . Wc.,vgand, P . Klinke, and I . Eigen, Chem. Ber. 90, 1896 (1957). [63] D . Fles and M . Markoaac-Prpic, Croat. Chem. Acta 28, 73 (1956); 29, 79 ( I 957).
[64] K. Balenocic and N . Srimac, Croat. Chem. Acta 29, 153 (1957). [6S] W C. Agosra er a/ . , Tetrahedron Lett. 1969, 4517.
[66] S. Masamune and M. Karo, J. Amer. Chem. SOC. 87, 4190 (1965).
[67] S. Masamune. J. Amer. Chem. SOC. 86, 735 (1964).
[68] W von E. Doering and M. Pomeranrz, Tetrahedron Lett. 1964, 961.
[69] S. Masamune and N . 7: Casfellricci, Proc. Chem. SOC. 1964, 298. [70] S. Masamune and K . Fukumoro, Tetrahedron Lett. 1965, 4647.
[71] A. S. Monahan, J . Org. Chem. 33, 1441 (1968).
[72] W ron E. Doering r f a/., Tetrahedron 23, 3943 ( I 967).
[73] W L'OII E. Doering, E. 7: Fossel. and R. L. Keye, Tetrahedron 21, 25 (1965). [74] M. M. F a n r i a n d C. D. Gursche, J . Org. Chem. 31, 1390 (1961). [7S] K. M o r i and M . Marsui, Tetrahedron Lett. 1969. 2729.
[76] A . A . Plente and M. 7: Bogerr, J. Org. Chem. 6. 669 (1941). [77] R. Grrwe and A. Bokranz. Chem. Ber. 88, 49 (1955).
(1951).
I 1 959).
1781 J . H. Woriz and S. N . Buco, J. Org. Chem. 20, 210 (1955).
[79] A. Roedig, E. Fahr. and H. Aman, Chem. Ber. 97, 77 (1964). [XO] R . Srlsarajan and J . H . B o y - , J. Org. Chem. 36, 1679 (1971).
[81] B. Eisrerr in W Foersr: Neuere Methoden der praparativen organischen Chemie. 4th Edit. Verlag Chemie, Weinheim 1963, Vol. I , p. 359.
[SZ] W E. Bachmann and W S. Srruue, Org. React. I. 38 (1942). [83] K. B. Wibrrg, B. R. Lowry, and 7: H. Colbj, J . Amer. Chem. SOC. 83, 3988 (1961). [84] L. Hornrr, M! Kirmse, and K . Murh, Chem. Ber. 91, 430 (1958). [SS] L. Horner, K . Murh, and H. G. Schmelzer, Chem. Ber. 92. 2953 (1959).
[86] P . R. Brook and B. I! Brophy, Tetrahedron Lett. 1969, 4187. [87] M. P . Caiq R. L. Lirle, and D. R. Napier. J. Amer. Chem. SOC. 80, 2257 ( I 958). [SS] J. Afrinwald, A. Lewis, and P. G. Gassmaii, J. Amer. Chem. SOC. 82, 2649 (1960): 84, 977 (1962). [89] J . Meinwald and J . K . Crandall, J. Amer. Chem. SOC. 88, 1292 (1966). [90] J . Meinwald and P . G . Gassman, J. Amer. Chem. SOC. 82. 2857 (1960). [91] J. Mrinwald t'r a/ . , J. Org. Chem. 33. 99 (1968). [92] J . Meinivald er a/., J. Org. Chem. 29, 3469 (1964). [93] K. B. Wibery and R . W Ubersax, Tetrahedron Lett. 1968, 3063.
[94] 7: Gibson and W F . Erman, J. Org. Chem. 31. 3028 (1966). [95] K. B. Wiberg and B. A. Hess. J. Org. Chem. 31. 2250 (1966). [96] K. B. Wiberg and A. dr Mrijerr . Tetrahedron Lett. 1969, 519. [97] A. J . Ashe, Tetrahedron Lett. 1969, 519.
[98] L. Horner and E. Spierschka, Chem. Ber. 88. 934 (1955). [99] A . 7: Blomquisr and C. Borromlry, Liebigs Ann. Chem. 653, 67 (1962). [loo] 7: Tanida and E Hara, J. Amer. Chem. SOC. 88, 4289 (1966); 9 / , 1170(1969). [ I O I ] L. Hornrr and D . W Basron. Chem. Ber. 98, 1252 (1965). [I021 W G. Dauben and D. L. Whalen, Tetrahedron Lett. 1966, 3743. [ 1031 S. Huneck, Chem. Ber. 98, I837 (1965). [I041 N . L. Allinger rr a/. . J. Amer. Chem. SOC. 85, 1171 (1963). [IOS] M. G. Nrwron, 7: J . Walter, and N . L. Allingrr. J. Amer. Chem. SOC. 95. 5652 (1973). [I061 N. L. Allingrr and 7: J . Walrrr, J. Amer. Chem. SOC. 94. 9267 (1972). [I071 A. Hassner, A. W Coulrer. and W S. Seese. Tetrahedron Lett. l962. 759. [I081 J . L. Mareos, 0. Chao, and H . Flows, Tetrahedron 19. 1051 (1963). [I091 S . Huneck, Tetrahedron Lett. 1963. 375. [ I lo] G . Mullrr, C. Huynh, and J . Morhirri, Bull. SOC. Chim. Fr. 1962, 296. [ I 1 I ] M. P . Cura and E. M o r o z , J. Amer. Chem. SOC. 84, I I5 ( I 962). [ I 121 J . Meinwold, G. G . Curtis, and P . G. Gassman, J . Amer. Chem. SOC. 84, I I6 ( I 962). [I131 I. K. Korobitsyna and L. L. Rodina, Zh. Org. Khim. I , 932 (1965). [114] I. K . Korobirsyna, L. L. Rodina,and L. M. Srashkom. Khim. Geterotsikl. Soed. 2, 843 (1966): Zh. Obshch. Khim. 33, 3109 (1963). [ I IS] W Rird and H . Lohwasser. Liebigs Ann. Chem. 683, I I8 I 1965). 11161 0. Sus, Liebigs Ann. Chem. 556, 65, 85 (1944): 557. 237 (1947): 579. 133 (1953). [ I 17) 0. Siis, Z. Wiss. Photogr.. Photophysik, Photochem. 50, 11. 476 (1955). [ I 17a] Cf. also P . A. S. Smirh and W! L. Burr,, J. Org. Chem. 26. 27 (1961). [ I I X ] J . rle Jonge and R. Dijksrra, Rec. Trav. Chim. Pays-Bas 67, 328 (1948); 69. 1448 (1950). [ I 191 0. Sus er a/. , Liebigs Ann. Chem. 583. 150 ( I 953). [I201 0. Sus and K . Moller, Liebigs Ann. Chem. 593. 91 (1955). [I211 Cf. also 0. Siis and K . MBller, Liebigs Ann. Chem. 599, 233 (1956). [I221 0. Sus and R. Dierrich, Liebigs Ann. Chem. 6l7, 20 (1958): Angew. Chem. 70, 25 (1958). [I231 €3. M . Trosr and P . L. Kinsoti, Tetrahedron Lett. 1973, 2675. [I241 L. H o r n w er a / . . Liebigs Ann. Chem. 661, 44 (1963). 11251 B. M. Post and P. L. Kinon, J . Amer. Chem. SOC. 92, 2591 (1970). [I261 J . Shafer, P . Baronowsky, R. Laumw, F . Finn, and F. H. Wesrheimrr. J . Biol. Chem. 241, 421 (1966). [I271 H. Chairnorich, R. J . Vauqhon. and F. H. Westhermer, J. Amer. Chem. SOC. 90, 4088 ( 1968). [I281 0. P. Srrausz, 7: Do Minh. and H. E. Gunning, J. Amer. Chem. SOC. 90. 1660 (1968); 91. 1261 (1969): cf. also X9, 6785 (1967). [I291 G . 0. Schenck and A. Rirrrr, Tetrahedron Lett. 1968. 3189. 11301 K.-P. Zeller, Chem. Ztg. 97, 37 (1973); cf. also [139]. [I311 7: Do M i d i and 0. P . Srrausz, J. Amer. Chem. SOC. YZ, 1766 (1970).
Angew. Chrm. inrernar. Edir. 1 Vol. 14 11975) No. I 41

[I321 I . G. Csizmadia, J . Font, and 0. P. Srrausz, J. Amer. Chem. Soc. 90, 7360 (1968). [I331 D. G. Thornfan, R. K . Gosari, and 0. P. Srrausz, J. Amer. Chem. Soc. 92, 1768 (1970). [I341 S. S. Hixon and S. H. Hixon, J. Org. Chem. 37, 1279 (1972). 11351 R. R. Rondo, J. Amer. Chem. Soc. 92,6706 (1970). 11361 E. Miiller and P. Heinrich, Chem.-Ztg. 95, 567 (1971); 96, 112 (1972). [I371 N. 7: Buu and J . 7: Edward, Can. J. Chem. 50, 3719 (1972); cf. also 50, 3730 (1972). [ 1381 Concerning the symmetrically substituted compounds, cf., e. 9.. refs. [1,4, 130, 139-1501. [139] D. C . Richardson, M . E. Hendrick, and M . Jones, J. Amer. Chem. SOC. 93, 3790 (197 I ) . [140] N . Baumann, Helv. Chim. Acta 55, 2716 (1972). [141] H. Sferter and K . Kiehs, Chem. Ber. 98, 1181, 2099 (1965). 11421 H. Schwall, K Schmidt, and B. E i s f e r f , Chem. Ber. 102, 1731 (1969). [143] H. Vesrhambre and D. Vocelle, Can. J. Chem. 47, 1981 (1969). [144] B. Eisrerr, H. Elias, E. Kosch, and R. Wollheim, Chem. Ber. 92, 130 (1959). [145] B. Eisrerr, G. Bock, E. Kosch, and F. Spalink, Chem. Ber. 93, 1461 ( 1960). [146] B. Eisrerf, D. Greiber, and I . Caspari, Liebigs Ann. Chem. 659, 79 (1972). 11471 H. Ledon, G. Linsfrumelle, and J . Syloestre, Bull. Soc. Chim. Fr. 1973, 2065. [148] I . Arnold and J . Sanliovd, Collect. Czech. Chem. Commun. 38, 2641 (1973). [149] J . K . Korobitsjna and W A. Nikolaeu, Zh. Org. Khim. 7, 413 (1971). [ISO] L. Horner and E. Spietschka, Chem. Ber. 85, 225 (1952). [ lSl] J . K. Korobifsyna and W A. Nikolaeo, Zh. Org. Khim. 7, 413 (1971). [151a] W A. Nikolaeu, C. D. Kotok , and J . K . Korobitsyna, Zh. Org. Khim.
[152] K.-P. Zeller, H . Meier, and E. Miiller, Tetrahedron 28, 5831 (1972). [1S3] F. M . Stojanouic and Z. Arnold, Collect. Czech. Chem. Commun. 32. 2155 (1967). [I541 G. Hayes and G. Holr, J. C. S. Perkin I 1973, 1206. [155] H. Meier. unpublished. [156] H. Staudinger and H. Hirzel, Ber. Deut. Chem. Ges. 49, 2526 (1917).
[ I571 M I D. Barker, R. Gilbert, J . P. Lapoinre, H . Veschambre, and D. Vocelle, Can. J. Chem. 47,2853 (1969). [I581 B. Eistert and G. Heck, Liebigs Ann. Chem. 681, 138 (1965). [I591 G. Lowe and D. D. Ridley, J. C. S . Chem. Comm. 1973, 328; J. C. S. Perkin I 1973, 2024. [I601 H. Sraudinger, J . Becker, and H. Hirzel, Ber. Deut. Chem. Ges. 49, 2522 (1916).
[161] W Kirmse and L. Horner, Chem. Ber. 89, 2759 (1956). [162] C . W Bird and C. K . Wong, Tetrahedron Lett. 1972,4281. [163] W Kirmse, Angew. Chem. 71, 539 (1959). [164] R. Tasouac, M . Srefanouic, and A. Stojiljkouic, Tetrahedron Lett. 1967, 2729. 11651 R. F. Rorch and D. L. Fields, J . Org. Chem. 34, 1480 (1969). 11661 P. J . W h i f m a n and B. M . Trost, J. Amer. Chem. SOC. 91, 7534 (1969).
[I671 I! Franzen, Liebigs Ann. Chem. 603. 202 (1957). [168] S. Hauptmanrrand K. Hirsrhberg, J . Prakt. Chem. [4] 34, 269 (1966). [169] M . Reg i f z and J . Riirer, Chem. Ber. 101, 1263 (1968). [170] M. Reg i f z and J . Riirer, Chem. Ber. 102, 3877 (1969).
[171] S. A. Marlin and P. G. Sammes, J. C. S. Perkin I 1972, 2623. [I721 E. Voigr and H. Meier. Angew. Chem., in press. [I731 J. Quinrana, M. Torres, and F. Serrafosa, Tetrahedron 29, 2065 (1973). [174] S. Masamune, J. Amer. Chem. SOC. 86, 735 (1964). [175] S. Masamune and N . Casrelluci, Proc. Chem. SOC. 1964, 298. [176] M I oon E. Doering and M . Pomerantz, Tetrahedron Lett. 1964, 961.
[I771 R. Breslow, R. Winter, and M . Barfisre, J. Org. Chem. 24, 415 (1959). [I781 R. Breslow and D. Chipman, Chem. Ind. (London) 1960. 1105.
[ 1791 W Ried and K . Lohwasser, Liebigs Ann. Chem. 683, I18 (1965). [I801 R. Huisgen and R. Fleisrhmann, Liebigs Ann. Chem. 623, 47 (1959).
[ lSl] C. Grundmnnn, Liebigs Ann. Chem. 536, 26 (1938).
[l82] W Kirmse. L. Horner, and H . Hoffmann, Liebigs Ann. Chem. 614, 19 (1958). [1%3] H. Reimlinger, Chem. Ber. 97, 339 (1964): Angew. Chem. 74, 153 (1962); Angew. Chem. internat. Edit. 1, 157 (1962).
10. 1335 (1974).
[I841 C. Oberger and J . Anselme, J. Org. Chem. 29, 1188 (1964).
[185] M. 'Enaka, T. Nogai, and N . Takura. Chem. Lett. 1972, 1207: J. Org. Chem. 38, 1602 (1973). [ I861 E . Ciganek, J. Org. Chem. 35, 862 (1970).
[I871 W Ando ef ul., J. Amer. Chem. Soc. 91, 2786 (1969). [t88] S. A. Marlin and P. G. Sammes, J . C. S. Perkin I 1973, 2851.
11891 F. Kaplan and G. K . Meloj, J. Amer. Chem. Soc. 88, 950 (1966); Tetrahedron Lett. 1964, 2427. [190] H . Kessler and D. Rosenrhal, Tetrahedron Lett. 1973, 393.
[I911 S. Sorrisso and A. Fofani , J. C. S. Perkin I I 1973, 2142. [I921 G. Piazzo, S. Sorriso. and A. Fofani, Tetrahedron 24, 4751 (1968); J. Chem. Soc. B 1971, 805. [I931 J . G. Csizmadia er al., Tetrahedron 25, 2121 (1969).
[194] P. Schusrer and 0. E. Polansky, Monatsh. Chem. 96, 396 (1965).
[I951 W Barrz and M . Regirz, Chem. Ber. 103, 1463 (1970). [I961 W Jungelt and D. Schmidt, Tetrahedron 25, 969 (1969). [I971 A. Melzer and E. F. Jenny, Tetrahedron Lett. 1968, 4503. [I981 Y Yukawa, E %no, and 7: Ibafa. Bull. Chem. SOC. Jap. 40, 2613, 2618 (1967). [199] Y Yukawa and 7: lbata, Bull. Chem. SOC. Jap. 42, 802 (1969).
[ZOO] W Kirmes and L. Horner, Liebigs Ann. Chem. 625, 34 (1959). [201] H. Meier and E. Voigr, unpublished.
[202] G. Lowe and J . Parker, Chem. Commun. 1971, 1135: R. A. Franich. G. Lowe, and J . Porker, J. C. S . Perkin I 1972, 2034. [203] B. Eisrert, Chem. Ber. 68, 208 (1935); cf. also A. L. Wilds, N . F. Woolsey, J . uan den Berghe, and C. H . Winesrock, Tetrahedron Lett. 1965, 484 1.
[204] U . Mnzzucato, G. Canzzo, and A. Foffani, Tetrahedron Lett. 1963.1525.
[2OS] E. Fahr, Chem. Ber. 92, 398 (1959). [206] R. Huisgen, Angew. Chem. 67, 439 (1955). 12071 U . Schallkopfand W Richer, Chem. Ber. 102,488 (1969). 12081 0. Tsuge, 1. Shinkai, and M . Koga, J. Org. Chem. 36, 745 (1971).
[209] W Ried and H . Mengler, Liebigs Ann. Chem. 678, 113 (1964). [210] W Ried and H. Mengler, Fortschr. Chem. Forsch. 5, 27 (1965). and references concerning the stereospecificity of olefin addition cited therein: cf. especially G. Rinsuh, R. Huisgen, and H. Kiinig, Chem. Ber. 97. 2893 ( 1964).
12111 R. Huisgen, G. Binsch, and L. Ghosez, Chem. Ber. 97, 2628 (1964). [212] K. Nakamura, S. Udagawa, and K . Hondo, Chem. Lett. 1972, 763. [213] A. M . Trozzolo and S. P. Fahrenholrz, Abstr. 151st Meeting Amer. Chem. SOC. 1966, K 23; A. M . Trozzolo, Accounts Chem. Res. I . 329 (1968). [214] H. Meier, IV IUPAC Symp. Photochemistry 1972. 163. [215] A. Padwa and R. Lagton, Tetrahedron Lett. 1965, 2167. [216] D. 0. Cowan er a/., J. Org. Chem. 29, 1922 (1964). [217] M. Jones and W Ando, J. Amer. Chem. Soc. 90, 2200 (1968). [218] M . Jones, J. Amer. Chem. Soc. 90, 2200 (1968). [219] F . Wejgand er a/., Angew. Chem. 73, 409 (1961). 12201 E. J . Moriconi and J . J . Murray, J . Org. Chem. 29, 3577 (1964). [221] H. Dworschok and F . Weygand, Chem. Ber. 101, 289, 302 (1968).
[222] M. J. S. Dewar and C. A. Ramsden, J. C. S. Chem. Comm. 1973, 688. 12231 A. C. Hopkinson, J . C. S . Perkin I I 1973, 794. [224] J . G. Csizmodia ef a/., J. Amer. Chem. Soc. 95. 133 (1973). [225] C . Hugget, R . 7: Arnold, and 7: S. Taylor. J. Amer. Chem. Soc. 64, 3043 (1942).
[226] K Franzen, Liebigs Abb. Chem. 614. 31 (1958). [227] 1. G. Csizmadia, J . Font, and 0. P. Sfrausz , J. Amer. Chem. Soc. 90, 7360 (1968). [228] K.-P. Zeller, H . Meier, H . Kolshorn, and E. Miiller, Chem. Ber. 105, 1875 (1972); cf. also ref. [231]. [228a] Z . Mnjerski and C. S. Redoanly. Chem. Commun. 1972, 694. 12291 D . E. Thorn fon , R. K . Gosaoi, and 0. P. Srrausz, J. Amer. Chem. SOC. 92, 1768 ( 1 970). [230] G. Frafer and 0. P. Srrausz, J . Amer. Chem. Soc. 92, 6654 (1970). 12311 J . Fenwick, G. Frarer, K . Ogi, and 0. P. Srrausz, J . Amer. Chem. SOC. 95. 124 11973).
[232] P. Heinrich and H . Meier, cf . also ref. [214].
[232a] K. G. Nogat, Dissertation. Technische Universitat Hannover 1972.
[233] F . M. Dean and A. Robertson, J. Chem. SOC. 1948, 1674.
[234] J . F . Lane and F . S. Wallis, I. Org. Chem. 6, 443 (1941).
42 Angew. Chem. internat. Edit. / Val. 14 11975) / Nu. 1

[235] K . J. Say and W Bergmann. J. Amer. Chem. SOC. 77, 1910 (1955). [236] N. A. Preobrashenski, A. M . Poljakowa, and W A. Preobrashenski, Ber. Deut. Chem. Ges. 68, 850 (1935). [237] J. F . Lane and E . S. Wallis, J. Org. Chem. 6, 443 (1941). [238] J. F . Lane and E. S . Wallis, J. Amer. Chem. SOC. 63, 1674 (1947). 12391 K . E. Wiberg and 7: W Hurton, J. Amer. Chem. SOC. 78, 1640 (1956). [240] C . D. Gursrhe, J. Amer. Chem. SOC. 70,4150 (1948). [241] F . l! Brurcher and D. D . Rosenfeld, J . Org. Chem. 29, 3154 (1964). [242] J. F. Lane et a/., J. Org. Chem. 5 , 276 (1940). 12431 K . - P . Zeller, H. Meier, and E. Miiller, Liebigs Ann. Chem. 749, 178 (I97 I ) .
[244] K.-P. Zeller, H. Meier, and 6. Miiller, Tetrahedron 28, 5831 (1972). [245] P . Kinson and E. M. Trusr, Tetrahedron Lett. 1964, 1075. 12461 D. C. de Jong, R . Y. Fossen, L. R. Dussold, and M. P. Cova, Org. Mass Spectrom. 3, 31 (1970). [247] R. J. Mulder, A. M . van Leusen, and J . Stating, Tetrahedron Lett. 1967, 3057.
12481 M. Regirr rr al., Tetrahedron Lett. 1968, 3171.
[249] W Jugrlr and D. Schmidt, Tetrahedron 25, 5569 (1969).
[250] B. M. Posr, J . Amer. Chem. SOC. 88, 1587 (1966); 89, 138 (1967).
12511 E. J. Core!. and M . Chaykocsk!, J . Amer. Chem. SOC. 86, 1640 (1964).
12521 E. M. Burgess, R . Carirhers, and L. McCullaugh, J . Amer. Chem. SOC. 90, 1923 (1968).
[253] W Kirmse and L. Horner, Liebigs Ann. Chem. 614, 4 (1958). [254] K . - P . Zeller, H. Meier, and E . Mullrr, Tetrahedron Lett. 1971, 537.
[255] P . Krauss, K . - P . Zeller, H . Meier , and E . Miiller, Tetrahedron 27. 5953.
[256] K . - P . Zeller, H. Mrier, and E. Miiller, Liebigs Ann. Chem. 766, 32 (1972).
[257] H. Meier and I . M e n d , Tetrahedron Lett. 1972, 445
[258] H . Meier and E. Vulgr, Tetrahedron 28, 187 (1972).
[259] H . Mrier and I . Menzel, J. C. S. Chem. Comm. 1971, 1059.
Isomerization Polymerization of Lactams
By H. K. R e i m s c h u e s s e l [ * ]
Isomerization polymerization affords a macromolecule whose repeating unit is derived from an iso- mer of the original monomer. Thus hexa- and heptacyclic P-carboxymethyllactams yield polymers containing glutarimide units, whereas penta- and hexacyclic P-carboxylactams furnish polymers containing succinimide units. Some dimethyl derivatives of P-carboxylactams isomerize or rearrange, and 5-oxopyrrolidine-3-acetic acid does not react at all. These observations can be explained in terms of a bicyclic intermediate.
1. Introduction
Isomerization polymerization is feasible with substituted lac- tams containing a carboxylic group capable of interacting with the amide function[']. Whereas the ordinary ring opening polymerization of lactams yields polyamides, isomerization polymerization affords polyimides. Both processes are charac- terized by competition between intramolecular cyclization and intermolecular polymerization. In the ordinary ring opening polymerization of lactams.the former reaction is part of a polymer-monomer equilibrium, and the product of cyclization is the particular lactam itself. The chemical structure of the repeating unit of the corresponding polymer molecule is in this case identical with that of the opened lactam ring. Of course, this also applies to any cyclic oligomers formed during polymerization. A rather different situation, however, charac- terizes isomerization polymerization for which no polymer- monomer equilibrium is indicated. The structures of both the propagating species entailed in this polymerization and the product of any cyclization differ from that of the starting lactam. Furthermore, no structural identity exists in this case between the lactam and the repeating unit of the polymer molecule.
['I Dr. H. K. Reirnschuessel Chemical Research Center, Allied Chemical Corporation Morristown, N. J. 07960 (USA)
Whether polymerization or cyclization is the dominating reac- tion depends for either process upon thermodynamic and kinetic factors, and upon the total molecular strain energy of the particular ring structure. This energy is directly related to the sum of all molecular deformations which are usually expressed in terms of: 1 ) bond stretching or bond compression (a function of the bond length, related to the motion of the bonded nuclei along the internuclear line); 2) bond angle bending (related to the radial scissoring motion of the bond angle--also known as angle strain or Baeyer strain); 3 ) bond torsion (refers to the rotational motion around the bond axis, related to the dihedral angle-also known as tor- sional strain, Pitzer strain, bond opposition force);
4) nonbonded interactions (refers to the interaction between substituents on neighboring carbon atoms and depends on the nuclear distance). The extent to which each type of strain contributes to the total molecular strain depends upon the ring size. In the ordinary ring opening polymerization of lactams it appears that bond angle bending (2)dominates in case of the four-mem- bered rings, whereas torsional forces ( 3 ) prevail in the five-, six-, and seven-membered rings, and both torsional forces and nonbonded interactions (4) seem to characterize the larger
Angew. Chem. infernal. Edit. / Vol. 14 (1975) / No. I 43


















