
Application of the LFER in Organic Electrochemistry ...
20
CROATICA CHEMICA ACTA CCACAA 58 (2) 107-126 (1985) CCA-1563 YU ISSN 0011-1643 UDC 541.13 Author's review Application of the LFER in Organic Electrochemistry: Jnterpretation of the Hammett Reaction Constant (e) for Processes of Reversible Electroreduction in Aprotic Solvents Marek K. Kalinowski and Tadeusz M. Krygowski Department of Chemistry, University of Warsaw, 02093 Warsaw, ul. Pasteura 1, Poland Received November 11, 1983 The paper is an overview of the application of the concept of linear free energy relationships (LFER), most notably through the Hammett equation, to investigations of reversible organic elec- trochemical reactions in aprotic solvents. In sequence the following topics are covered: the effect of solvent properties on the equili- brium, ion pairing effects, the direct application of Hammett's equation on polarographic data, entropic and enthalpic contribu- tions to substituent effects, and the quantum chemical interpreta- tation of the reaction rate constant in aprotic media. The applicat- ion of polarographic techniques is advocated as more sensitive and easier to interpret than the techniques of homogeneous kinetics. In organic chemistry a number of empirical models for describing relations between structure and chemical and physico-chemical properties has emerged. The most successful and most intensively studied are those based on linear free energy relationships+" (LFER), with the Hammett equatiorr'." as the most prominent example. Originally this equation was used only for the interpretation of substituent effects in chemical equilibrium and kinetics of meta- and para-substituted derivatives of benzene. In traditional form it reads 19 Kx -lg KR = (! (J (1) where Kx and KR stand for either equilibrium or rate constants for substituted and unsubstituted species, respectively. In the late'40 and later applications of eq (1) were extended to various physicochemical properties and to much more complex substituted systems than mentioned above", Since Hammett's proposal, evidence has been accumulated demonstrating dependence of the reaction constant e on solvents. This indicates the import- ance of environmental factors contributing to the transmission of substituent effects through the aromatic moiety; some of these factors will be discussed here. In the past, several suggestions were made to elucidate the solvent in- fluence on e-values. Hammett himself? suggested the following approximate formulae
Transcript of Application of the LFER in Organic Electrochemistry ...

CROATICA CHEMICA ACTA CCACAA 58 (2) 107-126 (1985)
CCA-1563YU ISSN 0011-1643
UDC 541.13Author's review
Application of the LFER in Organic Electrochemistry:Jnterpretation of the Hammett Reaction Constant (e) for Processes
of Reversible Electroreduction in Aprotic SolventsMarek K. Kalinowski and Tadeusz M. Krygowski
Department of Chemistry, University of Warsaw, 02093 Warsaw, ul. Pasteura 1,Poland
Received November 11, 1983
The paper is an overview of the application of the conceptof linear free energy relationships (LFER), most notably throughthe Hammett equation, to investigations of reversible organic elec-trochemical reactions in aprotic solvents. In sequence the followingtopics are covered: the effect of solvent properties on the equili-brium, ion pairing effects, the direct application of Hammett'sequation on polarographic data, entropic and enthalpic contribu-tions to substituent effects, and the quantum chemical interpreta-tation of the reaction rate constant in aprotic media. The applicat-ion of polarographic techniques is advocated as more sensitive andeasier to interpret than the techniques of homogeneous kinetics.
In organic chemistry a number of empirical models for describing relationsbetween structure and chemical and physico-chemical properties has emerged.The most successful and most intensively studied are those based on linearfree energy relationships+" (LFER), with the Hammett equatiorr'." as themost prominent example. Originally this equation was used only for theinterpretation of substituent effects in chemical equilibrium and kinetics ofmeta- and para-substituted derivatives of benzene. In traditional form it reads
19 Kx -lg KR = (! (J (1)
where Kx and KR stand for either equilibrium or rate constants for substitutedand unsubstituted species, respectively. In the late'40 and later applicationsof eq (1) were extended to various physicochemical properties and to muchmore complex substituted systems than mentioned above",
Since Hammett's proposal, evidence has been accumulated demonstratingdependence of the reaction constant e on solvents. This indicates the import-ance of environmental factors contributing to the transmission of substituenteffects through the aromatic moiety; some of these factors will be discussedhere.
In the past, several suggestions were made to elucidate the solvent in-fluence on e-values. Hammett himself? suggested the following approximateformulae

108 M. K. KALINOWSKI AND T. M. KRYGOWSKI
where R is the gas constant, T is temperature, e is the dielectric constant ofthe solvent and d is the distance from the substituent to the reaction site.BI and B2 are constants, depending on purely electrostatic interactions betweenthe reacting system and the medium, and susceptibility of the reaction tothe changes in charge density at the reaction site, respectively. This relationhas some validity but there are many exceptions", Another approximate inter-pretation was given by Grunwald and Gutbezahl? who suggested a line ardependence of e-values for the dissociation of aniline ions on the YO para-meter of solvents'". Many other solvent parameters have subsequently beenused in attempts to interpret the variation of reaction constant with solventl1-16.Evidently the reaction constant depends as well on a type of reaction: it isvery interesting to note that electroreduction of substituted salicylaldehydeanils and electrooxidation of corresponding free radical anions in acetonitrileyields in considerably different e-values17.
Now let us consider applications of the Hammett equation in electro-analysis. First attempts at correlating polarographic data - as the mostconvenient experimental technique of measurernents'" - with substituentconstants, 0, were published in the early 50-tiesl9-23.
Then rapid development of the electroanalytical methods'" and growinginterest in electrochemical properties of organic substances led to numerousresults, which then have been reviewed by Zuman= in the late'60-s. Initiallyelectrochemical processes were studied in aqueous and in water-containingsolutions, but to simplify the electrode reactions aprotic solvents began tobe commonly used.
In general, the shift of the half wave potential, <5 Elt225, with respect tothe value of the unsubstituted compound, to that of the substituted moleculefor a reversible system
(3)
is given(511 GO
----nF
(4)
(where F is the Faraday of charge).
In other words, the half-wave potential
the logarithm of the equilibrium constant,
For an irreversible system'"
for this system is proportional tokb
K = -k ' of reaction (3).f
RT --Elt2 = --- In 0.87 kf Y tI D-l
no;F(5)
and, consequently,
(6)
where tI is drop time, D is diffusion coefficient of depolarizer, a denotestransfer coefficient and no; stands for the number of electrons transferred inthe limiting potential step.

HAMMETT CONSTANT 109
If (ntt)x = (ntt)H and ax = aH then:
<5El/2= !1Gi' -!1 Gt ~··o!1G.ej (7)
Thus, the Hammett equation when applied to reversible as well as irre-versible red-ox systems'"
(8)
belongs to the family of LFER - equations'". The formal rate constant, kf'involved in reaction (5) which is often looked upon as the rate constant ofthe surface heterogeneous process, may not be quantitatively completelyequivalent to the rate constant of a homogeneous chemical reaction. It isimpossible to predict theoretically whether the heterogeneous and homogene-ous rate constants are affected by substituent in the same way. Howeverthousands of reaction series fit eq. (8) well and hence support a convictionthat heterogeneous rate constants in electrochemical reactions are influencedin a very similar way to the rate constant for homogeneous processes. Thusthe half wave potentials can be treated as a measure of the reactivity oforganic substances. For this reason electro analytical techniques offer animportant and powerful tool in extending our knowledge about reactivity oforganic molecules.
The original Hammett equation has been modified and generalized bymany workers'v'. An important step forward was made by Streitwieser'? whoextended it to polynuclear aromatic systems. Then this idea was associatedwith the Brown and Okamoto'" substituent constants a+ and Eaborn's'" rateconstants for deteritriation of benzenoid hydrocarbons in trifluoroacetic acidleading to a new, more general, set of position constants29,30. This equationcalled the Hammett-Streitwieser equation has the same form as the originalHammett equation (1) with a replaced by a/: r denotes here a position in apolynuclear benzenoid hydrocarbon, whereas superscript + recalls connectionof this scale of constants with the electrophilic reaction carried out to estimatethem. By definition a/ describes the Bri:insted basicity of position r of anaromatic hydrocarbon with benzene taken as reference. In principle the or"'scale is associated with localization energy'", L/, and the concept of gene-ralized substituents--". The applicability of the Hammett-Streitwieser equationis very wide - as far as chemical and physicochemical properties of benzenoidhydrocarbons and their monosubstituted derivatives are concerned30,S4-37. Dueto the similarity of the basic ideas of the Hammett and the Hammett-Streit-wieser equations, the interpretation of reaction constants is very similar tothat described for Hammett's e.
ELECTROREDUCTION OF ORGANIC COMPOUNDS IN APROTIC MEDIA
Electroreduction of organic compounds can be studied either in the absenceof proton donors in formally aprotic solvents or in the presence of wateror other Bri:insted acids. Studies under these conditions offer some advantagesboth from the practical point of view (preparative electrosynthesis, electro-chemical polymerization) and from certain theoretical aspects.
Most organic compounds in aprotic conditions are reduced electrochemi-cally in two one-electron steps. The first step is, generally, not accompanied

110 M. K. KALINOWSKI AND T. M. KRYGOWSKI
by a protonation antecedent or subsequent to the electron transfer. Thereforethe half-wave potential of the first wave:
(9)
is a direct measure of the difference between the standard free enthalpiesof the parent molecule, R, and the radical anion formed (reagents in solvatedform are marked by subscript solv). Radical anions, R-'-, and particularlydianions, R2-, are strong Lewis bases which are even able to extract protonsfrom residual water, or from other organic molecules, including the mole-cules of the compound that undergoes electroreduction. As a result, the overallprocesses involved in the second wave are frequently irreversible. Their inve-stigation is rather difficult and successful in only a few cases'" .For this reasonthe reaction
(10)
will not be considered here.
1. The Effect of SoIvent on EquiIibrium (9)
One of the major problems of organic electrochemistry is the descriptionof the medium effect on the characteristics of electrochemical processes. Fromthe theoretical viewpoint+", the energetics of process (9) can be compared withthe gas phase electron affinity, EA, of the depolarizer and with the differencein free solvation enthalpies of the reactant and the corresponding radicalanion, L'1L'1G~olv' The half -wave potential is given by
(11)
where (Ge10)Hg is the free enthalpy of the electron in an electrode. In practiceit is not easy to estimate the real L'1L'1G~olv values, but the influence of solventproperties on redox potentials of organic compounds can be considered interms of empirical equations based on LFER.
In general, the solvent effect on /'!.. G~Olv for charged species is composedof contributions due to non-specific electrostatic interactions, and specificinteractions between solute and solvent treated as the Lewis acid and base.Thus, the Koppel and Palm+' empirical equation in the following form canbe used'":
/i-I n2-1o Q = aA + ,8B + y--- + 1] --- + const2 e + 1 n2 + 2
(12)
where Q is the measured physicochemical property, whereas n stands for re-fractive index for a given solvent. However, most solvents applied in organicelectrochemistry are those with large values of dielectric permittivity (e> 10)
and hence the non-specific contributions (dependent on ~ or e -1 )may bee 2e + 1
neglected"; In this case the application of the two-parameter equation describ-ing solvent effect in terms of amphoteric Lewis acid-base properties ofsolvent44,45 is particularly advantageous. This equation may be written in ageneral form as below
o E1J2 = aA + ,8B + const (13)

HAMMETT CONSTANT 111
where A and B stand for the Lewis acidity and basicity solvent parameters,respectively. For reaction series in which solute-solvent interactionsare ofone mechanism only (or predominantly) e. g. solutes chiefly interact as Lewisacids or bases, eq. (13) reduces to line ar regression with solvent explanatoryparameters B or A, respectively. Accordingly, the solvent effect on EI/2o ofone-electron, reversible electroreductiorr'" values of quinones-" and phenazinesš?(free radical anions are formed during electrochemical processes in solutionscontaining 0.1 M (C2Hs)4NCl04)may be expressed by equation (14)
o EI/2°= a AN + const (14)
where AN is the acceptor numberš? i. e. the solvent parameter describing itsLewis acidity, while a defines the sensitivity of reaction (9) to the solvationeffect.
Interpretation of eq. (14) is very simple based on the donor-acceptorconcept for solvent-solute interaction'". Accordingly, the stronger a solventas a Lewis acid, the stronger is the solvation of the radical anion, and hence,EI/Zo is shifted towards amore positive potential. It should be pointed outthat similar line ar correlations between the logarithm of standard rate eon-stants of reversible electroreduction of p-dicyanobenzene and anthracene andAN were also obtained in arecent study'",
Another situation arises in the case of electrooxidation of phenothiazine(PHN). As shown in ref.49, the formal potential of the PHN+·/PHN systemmeasured voltammetrically in 9 solvents containing 0.1 M NaCl04 is linearlydependent on solvent basicity, expressed by Gutmann's donor number+', DN.In this case eq. (15) is fulfilled.
o EI// = tJ DN + const (15)
Obviously, the reason is that the oxidized form (free radical cation, whichis a Lewis acid) is much more strongly affected by inter action s with solventmolecules than its parent molecule.
From a general point of view it should be noted here that in some redoxsystems the solvent has to be treated as a Lewis amphoteric species-š. Sucha situation has been described and discussed in the electrochemistry of para-fuchsineš-, dibiphenylene-ethenešš and diphenylpicrylhydrazylš"; in all thesecases eq. (13) was successfully used to interpret experimental results.2. Ion Pair Effects
In many cases reaction (9) is more complex and the electron transferprocess is accompanied by ion association and should be described by ageneral scheme for the first step of polarographic reduction as (16)
Rsolv + e E1/2 •••••••• R~olv••••••••
(16)

112 M. K. KALINOWSKI AND T. M. KRYGOWSKI.,'il i
where El/20 and E1t~ stand for half-wave potentials describing reversiblereduction without ion pairing and followed by it, respectively, while M+denotes the cation of the supporting electrolyte. For reaction scheme (16) amodified Nernst formula may be written which describes a potential shiftdue to ion pairing processes:C.
where
(18)
and
K Tas[R~] [M+]2
[R~ .•• (M+>z](18a)
Ii tripIe ions R~ ... (M"), are not formed and Kas [M+] » 1 eq. (17) is reducedto the formš?
(j Eipf = RT In {K [M+]}1/2 F as (19)
Ii small amounts of water are present in aprotic solvents used in expe-riments, reaction scheme (16) may be still more cornplex. Even if the processremains reversible it is necessary to take into account the formation of sepa-rated ion pairs like e. g. (R-r- IH20/M+)58. These effects are, however, subtleand in most cases do not affect too much the observed El/2 - values. Foranother stoichiometry of ion association intervening in reaction (16) see ref.59
In the early paper of Holleck and Becher'" the relation between E1ti fora fixed depolarizer and the nature of the cations M+ in a given solvent wasobserved. This reaction has subsequently been described quantitatively bythe expression'"
• Eipf Eipf _ E1/2 _ I/. rhu 112- 1/2- o =:».• 'P (20)
where lA- is a regression coefficient (slope) while I('[J stands for the ionic potent-ial of the cation'", defined as charge of the cation divided by its radius. Whenthe experimental cation radius r is replaced by its corrected value (r + x)following the Latimer, Pitzer and Slansky'" modification of the Born theoryof ion solvation, the fit of eq. (20) becomes considerably better=. This allowsns to conclude that a decisive factor in the medium effect described by eq.. (20)is the solvation of the cation. The stronger Lewis acid it is (the higher itsionic potential) the greater is the free energy of solvation and the lessfavorable is the association between cations and radical anions'", On theother hand it is clear that eq. (20) belongs to the wide family of LFER equat-ions: it relates changes in free energy of ion pairing, estimated by (jE~~Jandthe free energy of solvation of cations described by the Born-type approach(I('[J - value). The value of the correlation term, x, can be approximatelyestimated by the empirical relation (21)65
(DN - 9.7) (x - 0.62) = 0.84 (21)

1,30
1,25
1,20
HAMMETT CONSTANT
o 1,0 2,0
113J2"1-
Mg
1,10
1.05
1,00
o
0,95
Figure 1. Variation in half-wave potential of 1 mM fluorenone (A) and 1 mM nitro-benzene (B) with ionic potential rf> of the supporting electrolyte cations inN,N-dime-methylformamide, Concentrations of electrolytes were 0.1 M and 0.05 M for theperchlorates of monovalent and divalent cations, respectively. A refers to the Ieft,
B - to the right-hand potential scale. Figure taken from ref. 61.
Figure 1. shows applications of eq. (20) to the first electroreduction stepof nitrobenzene and fluorenone in N,N-dimethylformamide. As it can be seen,the half-wave potential of nitrobenzene, determined at constant metal ionconcentration, is a linear function of oi{! , which indicates that a nitrobenzeneradical anion undergoes an ion pairing process even with tetraalkylammoniumcations. Experiments with fluorenone give a plot with two linear sections.The first horizontal section (oi{! smaller than 1) corresponds to the formationof free radical anions which do not form ion pairs with such large cations.For srnaIler counter ions a descending branch is observed, i. e. eq. (20) isfollowed.
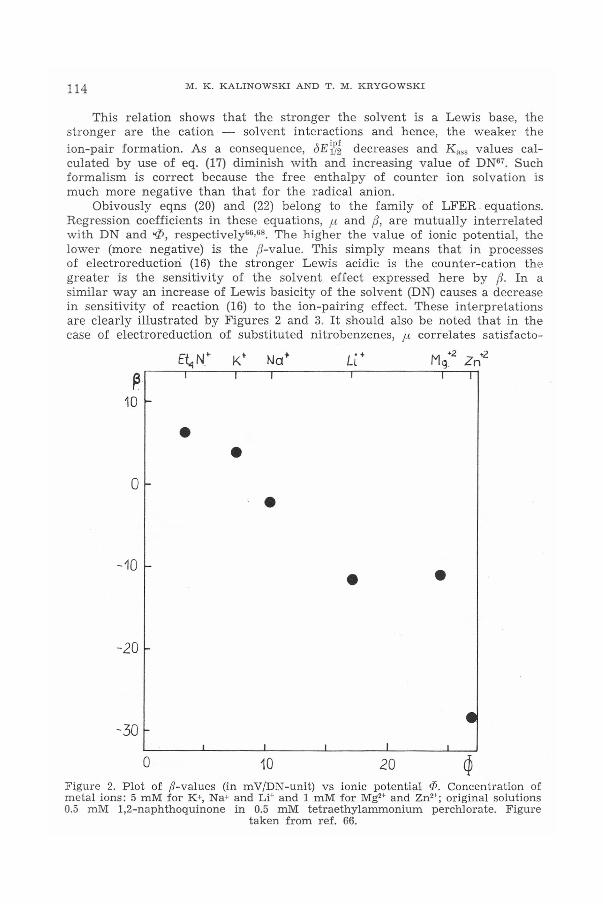
Another factor influencing E~~~ - values is directly due to solvent effects.In sufficiently polar solvents containing a given kind of cation, the mediumeffect on reversible electroreduction may be expected expressed by theempirical equation'" (Figure 2).
J E~}~= fJ DN + const (22)
114 M. K. KALINOWSKI AND T. M. KRYGOWSKI
This relation shows that the stronger the solvent is a Lewis base, thestronger are the cation - solvent interactions and hence, the weaker theion-pair formation. As a consequence, oE~~~decreases and Kass values cal-culated by use of eq. (17) diminish with and increasing value of DNG7. Suchformalism is correct because the free enthalpy of counter ion solvation ismuch more negative than that for the radical anion.
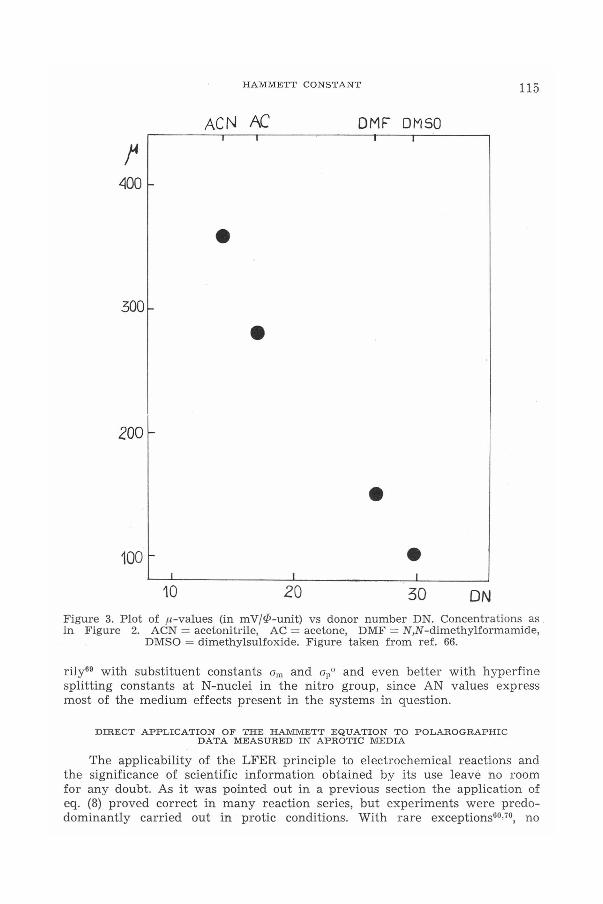
Obivously eqns (20) and (22) belong to the family of LFER equations.Regression coefficients in these equations, ,u and tJ, are mutually interrelatedwith DN and ..p, respectively'".", The higher the value of ionic potential, thelower (more negative) is the tJ-value. This simply means that in processesof electroreduction (16) the stronger Lewis acidic is the counter-cation thegreater is the sensitivity of the solvent effect expressed here by tJ. In asimilar way an increase of Lewis basicity of the solvent (DN) causes a decreasein sensitivity of reaction (16) to the ion-pairing effect. These interpretationsare clearly illustrated by Figures 2 and 3. It should also be noted that in thecase of electroreduction of substituted nitrobenzenes, fl correlates satisfacto-
E~N" Kt Na+ U· M +2 ZntZ9
~I I I I I I
10 -• •
O •-10
-20 I-
-30
•
I I
•
o 10 20
•Figure 2. Plot of ji'-values (in mV!DN-unit) vs ionic potential P. Concentration ofmetal ions: 5 mM for K+,Na+and Li+and 1 mM for Mg2+and Zn2+;original solutions0.5 mM 1,2-naphthoquinone in 0.5 mM tetraethylammonium perchlorate. Figure
taken from ref. 66.
HAMMETT CONSTANT 115
ACN AC DMF OMSO
300
•
I I I
400 -
•200 f-
••
10 20 30 ONFigure 3. Plot of /-l-values (in mV/P-unit) vs donor number DN. Concentrations asin Figure 2. ACN = acetonitrile, AC = acetone, DMF = N,N-dimethylformamide,
DMSO = dimethylsulfoxide. Figure taken from ref. 66.
rily69 with substituent constants Om and opo and even better with hyperfinesplitting constants at N-nuclei in the nitro group, since AN values expressmost of the medium effects present in the systems in question.
DIRECT APPLICATION OF THE HAMMETT EQUATION TO POLAROGRAPHICDATA MEASURED IN APROTIC MEDIA
The applicability of the LFER principle to electrochemical reactions andthe significance of scientific information obtained by its use Ieave no roomfor any doubt. As it was pointed out in a previous section the application ofeq. (8) proved correct in many reaction series, but experiments were predo-dominantly carried out in protic conditions. With rare exceptionsv''.?'', no

116 M. K. KALINOWSKI AND T. M. KRYGOWSKI
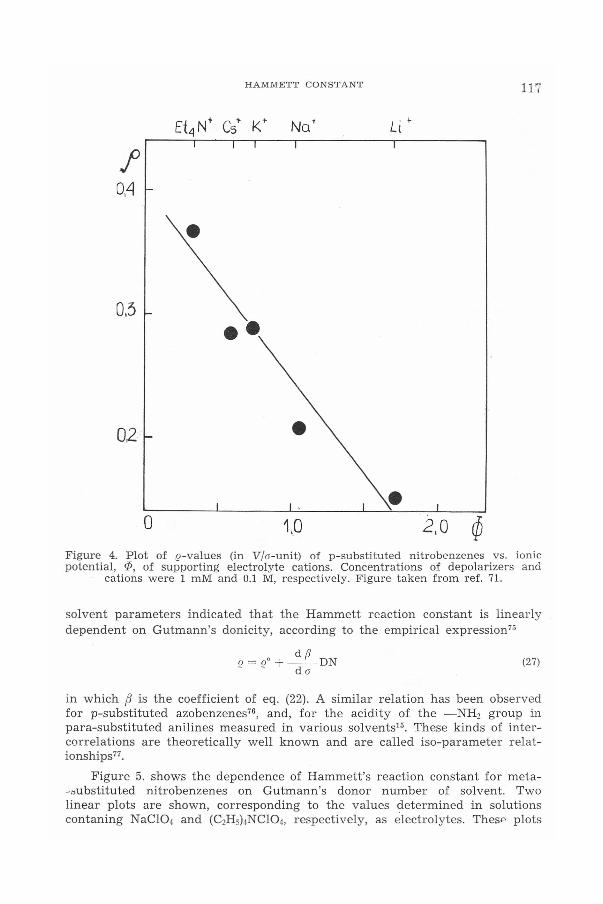
attempts have been made to correlate e values with the fundamental cha-racteristics of aprotic media, such as solvent properties and the kind of sup-porting electrolyte. This seems rather incomprehensible because as early as1962 Holleck and Becher?'' demonstrated indirectly that for substituted nitro-benzenes in N,N-dimethylformamide and acetonitrile EI/2 depends on bothof these factors. Based on their results Zuman?" found a linear correlationbetween half-wave potentials of the unsubstituted nitrobenzenes in solutionscontaining a given cation (Et4N+, Cs+,Na+ and Li") and the values of e obtainedfor a series of substituted nitrobenzenes in the same supporting electrolyte.Then, growing interest in the ion pairing phenomena in organic electrochemi-stry led to amore quantitative description of experimental results. At thetime, the influence of the supporting electrolyte cations on the e constantwas investigated in the case of the polarographic reduction of para-sub-stituted nitrobenzenes and benzaldehydes in dimethylformamide solutions. Itwas found'" that the formation of ion pairs between corresponding free radicalanions and cations of the supporting electrolyte influences the value of thereaction constant according to the following formula
12 = 12° + (d;.</da) ep (23)
where (J denotes the reaction constant in the presence of a given cation withwhich ion association occurs, (JD signifies the reaction constant measured inthe absence of ion association, while f-t and O<P are the parameters of eq. (20).The plot of the reaction constant e versus t[> for p-substituted nitrobenzenesis presented in Figure 4.
Relation (23) was also used to compare the effects of supporting electro-lyte cations on the reaction constant of para-substituted azobenzenes andtheir heterocyclic aza-analogs'". Its meaning may be simply justified on thebasis of eqns (8) and (20). Formally, the reaction constant (J for the seriesconforming to both of these equations may be presented as follows:
do Eipf1/2
da(24)
After substituting eq. (20) in relation (24) we have?"
12= d (EI/2° + ;.< ep) = d EI/2° + ~~ epda da da
(25)
which is obviously equivalent to eq. (23) since
d EI/2°/d o = 12° (26)
As it was previously pointed out, f-t describes the sensitivity of a givenradical anion toward ion association effects. Therefore, it is dependent on theproperties of the solvent used67,69. On the other hand, it is evident that the econstant should also be influenced by the kind of solvent. For one electronreversible electroreduction of m-substituted nitrobenzenes such a dependencehas actually been observed .The correlation of corresponding e valu es with
fO,LJ
0,3
0.2,
HAMMETT CONSTANT
Li"
117
Figure 4. Plot of ,Q-values (in V/O'-unit) of p-substituted nitrobenzenes vs. ionicpotential, ep, of supporting electrolyte cations. Concentrations of depolarizers and
cations were 1 mM and 0.1 M, respectively. Figure taken from ref. 71.
-.-
solvent parameters indicated that the Hammett reaction constant is linear1ydependent on Gutmann's donicity, according to the empirical expression "
o 1.0 2,0
(27)
in which fJ is the coefficient of eq. (22). A similar relation has been observedfor p-substituted azobenzenes'", and, for the acidity of the -NH2 group inpara-substituted anilines measured in various solvents'". These kinds of inter-correlations are theoretically well known and are called iso-parameter relat-ionships?",
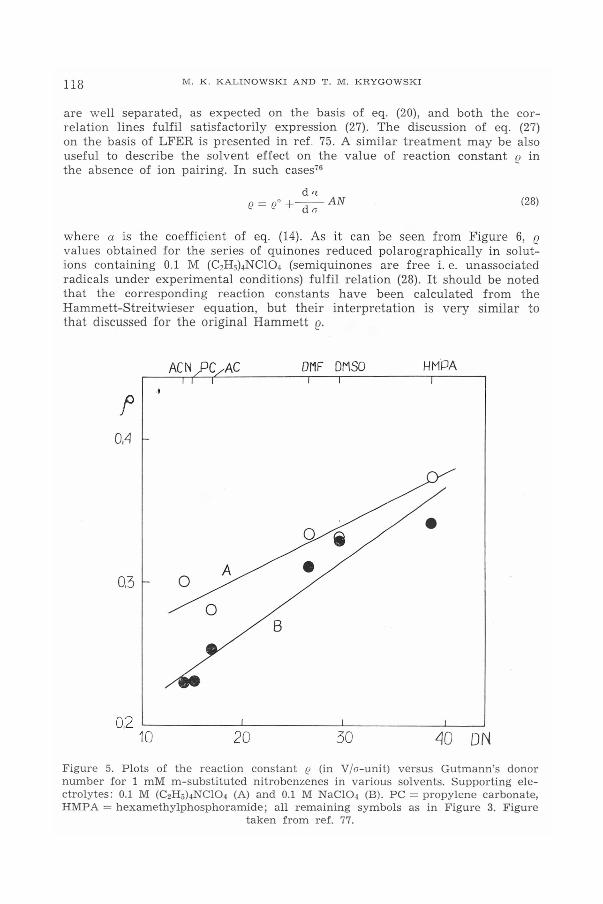
Figure 5. shows the dependence of Hammett's reaction constant for meta--substituted nitrobenzenes on Gutmann's donor number of solvent. Twolinear plots are shown, corresponding to the values determined in solutionscontaning NaCI04 and (C2Hs)4NCI04,respectively, as electrolytes, 'I'hes= plots
d(3o = nO +--DN~" d o
118 M. K. KALINOWSKI AND T. M. KRYGOWSKI
are well separated, as expected on the basis of eq. (20), and both the cor-relation lines fulfil satisfactorily expression (27). The discussion of eq. (27)on the basis of LFER is presented in ref. 75. A similar treatment may be alsouseful to describe the solvent effect on the value of reaction constant (! inthe absence of ion pairing. In such cases?"
d r(
n = 0° +--AN'" c; do (28)
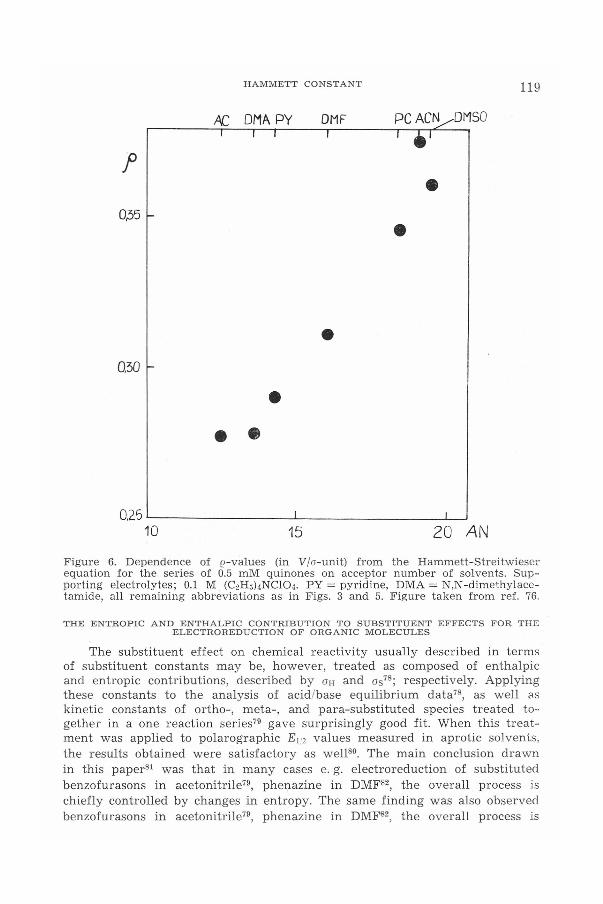
where a is the coefficient of eq. (14). As it can be seen from Figure 6, ()valu es obtained for the series of quinones reduced polarographically in solut-ions containing 0.1 M (C2Hs)4NCl04(semiquinones are free i. e. un associatedradicals under experimental conditions) fulfil relation (28). It should be notedthat the corresponding reaction constants have been calculated from theHammett-Streitwieser equation, but their interpretation is very similar tothat discussed for the original Hammett (2.
O,LJ
0,210
ACN C AC OMF DMSO
LJO ONFigure 5. Plots of the reaction constant f2 (in V/o-unit) versus Gutmann's donornumber for 1 mM m-substituted nitrobenzenes in various solvents. Supporting ele-ctrolytes: 0.1 M (C2H5)4NCI04(A) and 0.1 M NaCI04 (B). PC = propylene carbonate,HMPA = hexamethylphosphoramide; all remaining symbols as in Figure 3. Figure
taken from ref. 77.
r
B
·f
•0,3
20 30
r0,35 -
0.30 -
HAMMETT CONSTANT 119
,AC DMA PY, "
OMF, PCAC~DMSO
, .'•
••
• •
•
0.25 '------ L-' -L-...l'
10 15 20 ANFigure 6. Dependence of e-values (in Vla-unit) from the Hammett-Streitwieserequation for the series of 0.5 mM quinones on acceptor number of solvents. Sup-porting electrolytes; 0.1 M (C2H5)4NCI04.PY = pyridine, DMA= N,N-dimethylace-tamide, all rernaining abbreviations as in Figs. 3 and 5. Figure taken from ref. 76.
THE ENTROPIC AND ENTHALPIC CONTRIBUTION TO SUBSTITUENT EFFECTS FOR THEELECTROREDUCTION OF ORGANIC MOLECULES
The substituent effect on chemical reactivity usually described in termsof substituent constants may be, however, treated as composed of enthalpicand entropic contributions, described by OH and 0578; respectively. Applyingthese constants to the analysis of acid/base equilibrium data", as well askinetic constants of ortho-, meta-, and para-substituted species treated to-gether in a one reaction series'" gave surprisingly good fit. When this treat-ment was applied to polarographic E':2 values measured in aprotic solvents,the results obtained were satisfactory as well'". The main conclusion drawnin this paper'" was that in many cases e. g. electroreduction of substitutedbenzofurasons in acetonitrtle?", phenazine in DMF82, the over all process ischiefly controlled by changes in entropy. The same finding was also observedbenzofurasons in acetonitrile", phenazine in DMF82, the overall process is

120 M. K. KALINOWSKI AND T. M. KRYGOWSKI
in line83 with the most recent studies on iso-kinetic temperature=, which wasdescribed as an estimated blend of entropy and enhalpy changes in thechanges of free energy, which is, usually indirectly measured in chemical orphysicochemical measurements. It was clearly shown'" that for reaction series,for which the absolute value of the isokinetic temperature is lower than thetemperature of the experiment, the dominant contribution to free energychanges comes from T!1 S-terms. This finding has important consequencesin the theory of substituent effects'",
It should be pointed out that the effect of the temperature on the valuesof reversible electrode potentials has been investigated to a very limitedextent. The temperature dependence of the potentials proper for electro-reduction of aromatic hydrocarbons in acetonitrile was found to vary in amanner predictable from the distribution of charges in free radical anions'".It was also demonstrated'" that in the case of the electroreduction of sub-stituted nitrobenzenes the entropy of the formation of radical anions isinfluenced both by steric and electronic effects of the alkyl substituents. Theearlier work of Andrieux and Saveant indicateds? that the entropy and ent-halpy factors of successive electroreduction of aromatic dinitro compoundsare dependent on the nature of solvent and supporting electrolyte cation.Parker's recent review'" gives additional data on the entropy f'actor in electro-chemical processes.
QUANTUM CHEMICAL INTERPRETATION OF THE REACTION CONSTANT
Quantum chemistry has several times been used to interpret the Hammettequationš"?'. In the case of polarographic data for reversible electroreductionsuch an attempt was made90,92 taking into account the well-known possibilityof describing physicochemical properties of n-electron systems in terms of thesimple HMO theory'", In the case of standard reduction potential (approxi-mated by E1/2-values) the appropriate quantity is the energy of the lowestvacant molecular orbital'", ELVMO, as estimated by HMO theory. Since, ingeneral, reaction constant is defined by a simple derivative expression
(29)
where Q is the measured property depending on the nature of the substituent,for our case one may write
dELVMae =-d"-u- (30)
In order to complete the quantum chemical description of (), one must beable to interpret o with in the context of HMO theory. Such an interpretationwas given'" by relating o to the so called Hiickel effective inductive para-meter. The definition of this parameter is based on the following assumptions:(i) changes in n-electron structure and energy caused by a substituent in aposition para to the reaction centre can be described by a simple correctionto the Coulomb integral of the carbon atom at the substituent position: thatis, the Coulomb integral at becomes at + d at (ii) the corrections d at dependlinearly on the Hammett constants Op. The first of these assumptions was

HAMMETT CONSTANT 121verified some time age and is described by Streitweiser-" as a method ofinterpreting the half-wave potentials of poly-methyl derivati ves of naphtha-lene. The second was derived empirically from IR investigations of substituenteffects on the carbonyl stretching frequency of 16 derivatives of anthraqui-none'". It has been successfully applied to the interpretation of the polaro-graphic and spectral properties of anthraquinones=-", the polarographic pro-perties of substituted nitrobenzene, azobenzene, and azopyridine'", and theNMR properties of substituted thiophenes'".
From the above empirical considerations, one may replace d o by thed at and equation (30) then reads as
(31)
Furthermore, on the basis of the simple HMO perturbation theory, dE;/dat ,.== C2it. where Ci,t is the coefficient at the t-position for the i-th molecularorbital of the unsubstituted molecule. Thus, one may define liHMO in thecase of a reversible electroreduction reaction as
- C2(lHMO - LVlIIO, t (32)
According to the simple interpretation of the HMO model C ~VMO, t is thespin density at the t-position in the corresponding anion radical of theunsubstituted molecule. The spin density, in turn, is normally assumed tobe proportional to the hyperfine splitting constant of the hydrogen atom atthe same position as determined from the ESR spectrum'". 'I'hus, the aboveanalysis, points to the conclusion that liHMO is directly proportional to thehyperfine splitting constant of the hydrogen atom at the substituent positionin the parent molecule.
The valu es of li determined from the variation in half-wave potentialwith substituent for eight parent molecules are plotted in Figure 7 againstAH,t, the hyperfine splitting constant at the substituent position. The correlat-ion obtained may be considered satisfactory and is qualitative support of themodel presented for li,
CONCLUDING REMARKS
In conclusion, we wish to point out that polarography seems to be a verypowerful technique in the field of LFER investigations. Measurements of thehalf-wave potentials are usually easy and fast procedures, often much fasterthan determination of rate or equilibrium constants. The elucidation of ele-ctrochemical processes is also usually simpler than the elucidation of themechanism of a homogeneous reaction. The deviations from the mechanismoperating in the whole series can sometimes be detected more easily in pola-rography than in homogeneous kinetics: one ought to compare the chara-cteristics and slopes of registered waves. Generally these deviations can hecaused by (i) steric effects for a given substance, (ii) specific effects of substi-tuent in the given molecule and in the particular environment, as e. g. ortho--effects, field effects etc, which occur in a particular reaction series but whichare not incorporated in the o-substituent constant, and (iii) specific effects

]22 M. K. KALINOWSKI AND T. M. KRYGOWSKI
0,2
06
0,6
2.0 6.0 AHFigure 7. Plot of experimental Hammett reaction constant f2 determined from pola-rographic data for reduction reactions against the e.s.r. hyperfine splitting constantfor the proton at the substituent position in the parent molecule, AH., for the follo-wing compounds: 1, stilbene; 2, c-substituted anthraquinone; 3, azobenzene; 4, ji'-sub-stituted anthraquinone; 5, benzophenone; 6, nitrobenzene in acetonitrile; 7, nitro-benzenne in dimethylformamide; 8, benzaidehyde. Position t refers to para for allmolecules except anthraquinone where it refers to a or ji' as noted. In all casesmeasurememts of AH.! and (I were carried out in the same solvent. AH values are
given in gauss (1 gauss = 10-4 T).
which may operate in polarographic measurements, such as adsorption phe-nomena. The identification of these effects is usually not difficult.
It seems proper to add that the accuracy of polarographic measurementsis usually quits high and hence attempts to use these measurements forestimation »new« Hammett substituent constants98,99. Under common experi-mental conditions a difference of 0.01 V in half-wave potentials is well repro-duced. Under carefully controlled conditions the preci sion of measurementsincreases even tenfold. This means that for (j = 0.3 V/a-unit, the differencein substituent constants of 0.01 a-units is significant, which is simply notattainable by non-electroanalytical methods. Polarographic Q-values are, asarule, in the range of 0.1-0.6 V/a-unit. It should be pointed out that Q-valuesdetermined from kinetics and equilibria of homogeneous reactions and thosefrom polarographic measurements are on different scales. In order to transformthe polarographic (j-values into the usual scale of magnitude for (j, the pola-

HAMMETT CONSTANT 123rographic e-values have to be multiplied by factor nF/2.3 RT = 16.91 n, wherethe symbols have the same meaning as in eqns 4-6. For this reason, pola-rography seems to be a useful method for the determination of not onlyreaction constants but also of accurate values of substituent constants usingproperly chosen secondary standards. Even in those cases where very smalldifferences in measured values are important, as e. g. in the proofs of hyper-conjugation, polarography may be very serviceable.
In this account we have summarized some studies on the environmentaleffects affecting the value of the reaction constant. In our opinion, this isonly the first step in more systhematic investigations in this field. We suggestin particular assume that the effect of temperature changes should be exa-min ed, since these changes influence the equilibria between different typesof ion pairs (tight, solvent separated etc). Obviously, polarographic measure-ments in non-aqueous media are connected with difficulties involved in thechanges of the potential of the reference electrode and in the problems withliquid-junction potentials. These difficulties, however, can be circumventedby measuring the changes in the difference (EJ/2)x - (EJ/2)H and/er by anapplication of internal standards reducing the role of liquid junction effects.
AcknowLedgments. -- The Authors wish to thank C. D. Johnson (Norwieh, DK),John Shorter (Hull, DK) N. Trinajstić (Zagreb) and Romuald 1. Zalewski (Poznan,Poland) for reading the draft of this review and parti.cularly for their useful com-ments. This paper was sponsored by MR 1-11 projeet.
REFERENCES
1. Advances in Linear Free Energy ReLationships, N. B. Ch a p man and J.S hor ter eds, Plenum Press, London 1972.
2. Corretation Anatysis in Chemistry - Recent Advances, N. B. Ch a p man andJ. S hor ter eds., Plenum Press, London 1978.
3. J. S hor ter, Corretation AnaLysis of Organic Reactivity, Res. Studies Press,Liv. of J. Wiley and Sons, Chichester 1982.
4. C. D. J o h n s o n, The Hammett Equation, University Press, Cambridge 1973.5. L. P. Ham m e t t, PhysicaL Organic Chemistry, McGraw-Hill, New York,
1940;2nd Ed. MeGraw-Hill New York 1970.6. H. H. J a f f e, Chem. Revs. 53 (1953)191.7. L. P. Ham m e t t, J. Amer. Chem. Soc., 59 (1937)96.8. O. Ex ner, ref. (1) p. 26.9. E. G r u n w a l d and B. Gu t bez a h l, J. Am. Chem. Soc., 75 (1953)559.
10. For details ef. J. E. Le f f 1er and E. G r u n w a l d, Rates and Equitibria ofOrganie Reactions, J. Wiley, New York 1963p. 289 ff.
11. A. B u c k l e y, N. B. Ch a p man, M. R. J. Da e k, J. S hor ter, and H.M. Wall, J. Chem. Soc. C (1968)631.
12. A. P. Ste fan i, J. Am. Chem. Soc., 90 (1968)1964.13. T. Mat s u i and N. Tok ura, BuLt. Chem. Soc. Japan, 44 (1971)756.14. T. Mat s u i and L. G. He p 1e r, Can. J. Chem., 54 (1976)1296.15. G. La u n a s, B. Wo j tko w i a k, and T. M. Kr y g ow s k i, Can. J. Chem.
57 (1979)3065.16.W. H. AsI a m, N. B. Ch a p man, M. Ch art on, and J. S hor ter, J.
Chem. Res., Synop., (1981)188.17. J. E. Kud e r, H. W. G i b s o n, and D. WY ehi e k, J. Org. Chem., 40 (1975)
875.18. For details of eleetroanalytieal teehniques ef. Z. Gal u s, Fundamentals of
ELectrochemical AnaLysis, Ellis Harwood Ltd, Chiehester, 1976.19. F. S. Seh u 1t z, Iowa State Coll. J. Sci., 26 (1952)280.20. R. W. B oe km a n and D. E. P e ars o n, J. Am. Chem. Soc., 74 (1952)4128.21. E. I m o t o and R. Mo to y ama, BuU. Chem. Soc. Japan, 55 (1952)384.

124 M. K. KALINOWSKI AND T. M. KRYGOWSKI
22. S. K o i d o and R. Mo t o y ama, J. ELectrochem. Soc. Japan, 20 (1952) 314.23. P. Z uma n, Chem. Listy, 47 (1953) 1234.24. P. Z uma n, Substituent Effects in Organic Polarography, Plenum Press, New
York, 1967.25. b is the so-called chemical operator introduced by Le f f 1e r and G r u n W a 1d,
ref. 10 p. 22. It may describe variation in substituent, solvent or generallymedium, realized in a given reaction series.
26. equation (10) is known as the polarographic Hammett equation. Obviously itmay be also applied in other e1ectroana1ytica1 techniques as eg. cyclic voltam-metry, chronopotentiometry etc.
27. A. Str e i t w i ser Jr., MolecuLar OrbitaL Theory, J. Wiley, New York, 1960.28. H. V. B r o w n and Y. Oka m o t o, J. Am. Chem. Soc., 79 (1957) 1913.29. R. B ake r, C. E a b o r n, and R. Ta y lo r, J. Chem. Soc. Perkin II, (1972) 97.30. T. M. Kr y g o w s k i, Tetrahedron, 28 (1972) 4981; the term position constant
recalls the fact that reactivity of reaction site depends in benzenoid hydrocarbonson position of attack and further on position to which the reaction site is atta-ched to the aromatic moiety. This problem is closely related to the idea ofgeneralized substi tuentt-" .
31. W. G. W h ela n d, J. Am. Chem. Soc., 64 (1942) 900 see as well ref. 27.32. T. M. Kr y g o w s k i, BuH. Acad. Sci. Polon. Ser. Sci. Chim., 19 (1971) 49.33. T. M. Kr y g o w s k i, BuLL Acad. Sci. Polon. Ser. Sci. Chim., 19 (1971) 61.34. T. M. Kr y g o w s k i, M. Ste n c el, and Z. Gal u s, J. ELectroanal. Chem..,
39 (1972) 359.35. T. M. Kr y go w s k i, BuH. Acad. Sci. Polon. Ser. Sci. Chim., 19 (1971) 433.36. B. K ami e il s k i and T. M. Kr y g o w s k i, Tetr. Letiers, (1972) 681.37. T. M. Kr y g o w s k i and J. Kr u s z e w s k i, Bull. Acad. Polon. Sci. Ser. Sci.
Chim. 22 (1974) 1059.38. Par k e r and co-workers!" have found that organic compounds can accept
reversibly a second electro n but only if the solution is perfectly purified fromelectrophilic contaminations. This is usually realized by addition of suspendedneutral alumina. This finding is wery important for the interpretation of themechanisms of e1ectroreduction of organic compounds in aprotic conditions.
39. O. Ham m eri c h and V. D. Par k er, Electrochim. Acta 18 (1973) 537.40. G. J. H o i j t i n k and J. van Sc h o ote n, Rec. Trav. Chim. Pays-Bas 71
(1952) 1089.41. I. A. K o P p e 1 and V. A. Pal m, ref. 1 p. 203 ff.42. For a wide and detailed review on using empirical (and theoretical as well)
models of solvent effects on chemical reactivity and physicochemical propertiescf. Ch. Rei c h ard t, SoLvent Effects in Organic Chemistry, Verlag Chemie,Weinheim 1979.
43. T. M. Kr y g o w s k i, E. Mi 1c zar e k, and P. K. W r o n a, J. Chem. Soc.Per kin II, (1980) 1563.
44. T. M. Kr y g o w s k i and W. R. F a W cet t, J. Amer. Chem. Soc. 97 (1975)2143.
45. R. W. F a W cet t and T. M. Kr y g o w s k i, Can. J. Chem. 54 (1976) 3283.46. The potentials were measured as referred to the potential of the ferrocene/ferri-
cinium redox couple. This internal standard has been recommended in ref.47 toelim ina te the liquid junction potential.
47. H. Str e h 1o w, The Chemistry of Non-Aqueous SoLvents. J. J. La g o w s k iEd., vol. (1) p. 129, Academic Press, New York 1966.
48. J. S. J a w o r s k i, E. L e s n i e w s k a and M. K. K <oi 1i n o w s k i, J. ELectro-anal. Chem. 105 (1979) 329.
49. B. Pad u s z e k and M. K. Kal i n o w s k i, Electrochim. Acta 28 (1983) 639.50. W. Mayer, V. G u t man n and W. G e r g e r, Monats. Chem. 106 (1975)
1235.51. V. G u t man n - The Donor-Acceptor Approach to MolecuLar Interactions,
Plenum Press, New York, 1978.52. W. R. F a w cet t and J. S. Ja w o r s k i, J. Phys. Chem. 87 (1983) 2972.53. V. G u t man n and E. W Yc h era, J. Inorg. Nucl. Chem. Lett. 2 (1966) 257.54. M. K. Kal i n o w s k i and I. S z e 1q ž ek, PoLish J. Chem. 52 (1978) 1853.55. E. Wagner, T. Paroi, and M. K. Ka1inowski, Can. J. Chem. 59 (1981)
2957.

HAMMETT CONSTANT 12556. M. K. Ka1inowski and J. K1imkiewicz, Monats. Chem. 114 (1983)
1035.57. M. E. P e ove r, J. D. Da vie s, J. ELectroanaL Chem. 6 (1963) 43.58. M. L i P s z taj n, T. M. Kr y g o w s k i, and Z. Gal u s, J. ELectroanaL Chem.
49 (1973) 17.59. A. Las i a and M. K. Kal i n o w s k i, J. ELectroanaL Chem. 36 (1972) 511.60. L. H o Il e c k and D. B e c h e r, J. ELectroanaL Chem. 4 (1962) 321.61. M. K. Kal i n o w s k i, Chem. Phys. Lett. 8 (1970) 55.62. G. H. Car tle d g e, J. Am. Chem. Soc. 50 (1982) 2855.63. W. M. Lat ime r, K. S. P i t z e r, and C. M. Sl a n sky, J. Chem. Phys.
7 (1939) 108.64. T. M. Kr y g o w s k i, M. L i p s z taj n, and Z. Gal u s, J. ELectroanaL Chem.
42 (1973) 261.65. N. T ana k a, ELectrochim. Acta 21 (1976) 701.66. T. M. Kr y g o w s k i, J. ElectroanaL Chem. 35 (1972) 436.67. E. Le S n i e w s k a - L ada and M. K. Kal in o w s k i, Electrochim. Acta 28
(1983) 1415.68. M. K. Kal i n o w s k i and B. Te n der end e - G u m i il s ka, J. ELectroanaL
Chem. 55 (1974) 277.69. M. L i p s z taj n, T. M. Kr y go w s k i, M. S mol a r z, and W. Sz e 1 clg o w-
s k a, PoLish. J. Chem. 52 (1978) 1069.70. P. Z uma n, The ELudidation of Organic ELectrode Processes, Academic Press,
New York 1969.71. M. K. Kal i n o w s k i, Chem. Phys. Lett. 8 (1972) 378.72. H. Kr y sz c z y il s k a and M. K. Kal i n o w s k i, Rocz. Chem. 45 (1971) 174.73. Relation (25) may be considered as an example of the additivity rule within
Ham m e t t' s treatment. In the general case, if the reaction proceeds in twosteps (I and II) fulfilling the Hammett equation one should treat the overallreaction constant as the sum eJ + elI. Consequently, dE1i2o/da in eq. (25) is thereaction constant for electroreduction not influenced by ion pairing whereasdfl/da is equivalent to the reaction constant of the ion association process. Itshould be noted that a similar analysis has been done by Kargin and Ivanova".In the very interesting paper by these authors an attempt was made to calculatethe effect of the subsequent protonation reaction on the e values for substitutedbenzophenones in dimethylformamide with a thousandfold excess of phenol. Asa result, the protonation reaction constant of the substituted benzophenone anionradicals was estimated.
74. YU. M. K arg i n and V. Kh. Iva n ova, Reaktisionnaya Sposobnost Organ.Soed. 7 (1970) 255.
75. M. K. Kal i n o w s k i and B. O s i e c k a, Roczn. Chem. 50 (1976) 299.76. J. S. J a w o r s k i, E. Le s n i e w s k a, - L ada, and M. K. Kal i n o w s k i,
PoLish J. Chem. 54 (1980) 1313.77. V. A. Pal m, Osnovy koLitchestvennoy teorii organitheskikh soedinenii, 2nd
edition, Izd. Khimiya, Leningrad, 1972.78. for details of the idea of T. M. Kr y g o w s k i and W. R. F a w cet t, Can. J.
Chem. 53 (1975) 3622.79. T. M. Kr y g o w s k i and W. R. F a w cet t, J. Chem. Soc., Perkin II, (1977)
2033.80. W. R. F a w cet t and T. M. Kr y g o w s k i, J. ELectroanaL Chem. 77 (1977)
47.81. W. R. F a w cet t, P. A. For t e, R. O. L o u t f s, and J. M. P rok i p cak,
Can. J. Chem. 50 (1972) 263.82. A. 1. B rod sky, L. L. Go r die n k o, and L. S. D e g t i ari e v, ELectro-
chim. Acta 13 (1965) 1095.!l3. T. M. Kr y g o w s k i and J. G u i Il eme, J. Chem. Soc., Perkin II, (1982) 531.84. For details about the meaning and definition and estimation of isokinetic tem-
perature see O. Ex ner, Progr. Phys. Org. Chem. 10 (1973) 411.85. M. Sva a n and V. D. Par k e r, Acta Chem. Scand. B, 35 (1981) 559.86. M. Sva a n and V. D. Par k e r, Acta Chem. Scand. B, 36 (1982) 357.87. C. P. And rie u x and J. M. Save a n t, J. ELectroanaL Chem. 57 (1974) 27.88. V. D. Par k e r, Phys. Org. Chem. 19 (1983) 131.

126 M. K. KALINOWSKI AND T. M. KRYGOWSKI
89. S. Eh r e n s o n, Progr. Phys. Org. Chem., A. Str e i t w i e ser and R. W.Ta f t, Edts., vol. 2 p. 165 (1969) and references cited therein.
90. T. M. Kr y go w s k i and A. Per j e s s s, BuU. Acad. Polon. Sci., Ser. Sci.Chim. 22 (1974) 437.
91. R. P o nec, CoU. CzechosL Chem. Commun. 45 (1980) 1646.92. W. R. F a W cet t and T. M. Kr y g o W s k i, unpublished, 1975.83. W. K e m ula and T. M. Kr y g o W s k i, BuLL Acad. PoL Sci., Ser. Sci. ChiJn.
15 (1967) 479.94. W. K e m ula and T. M. Kr y g o w s k i, Tetr. Letters, (1968) 8135.95. T. M. Kr y go W s k i and P. Tom a s i k, BuLL Acad. PoL Sci. Ser. Sci. Chim.
18 (1970) 303.96. B. Ka m i e il s k i and T. M. Kr y g o W s k i, Tetr. Letters (1971) 103.97. H. M. Mc C o n n e l I, J. Chem. Phys. 24 (1956) 764.98. Yu. M. Ka r g i n, T. A. P o d k o v y r i n a. G. A. K h m u tova, and T. G.
Man n a f o v, Zh. Obshch. Khim. 46 (1976) 2459.99. Yu. M. Ka r g i n, V. Z. Lat y p ova, O. G. Y ako v lev a, A. A. Va f i n a,
A. N. U s t y u g o v and A. V. Il y a s o v, Zh. Obshch. Khim. 52 (1982) 655.
SAŽETAK
Primjena linearnih relacija slobodne energije u organskoj elektrokemiji.Interpretacija Hammettove reakcijske konstante na procese reverzibiIne
elektroredukcije u aprottčnim otapalima .
Marek K. KaLinowski i Ttuieusz M. Krygowski
Dan je pregled primjena linearnih relacija slobodne energije, poglavito pri-mjena Hammettove jednadžbe, na istraživanja reverzibilnih organskih elektro-kemijskih reakcija u aprotičnim otapalima. U pregledu teorijskih i praktičkihrezultata posebno su izabrana slijedeća poglavlja: o utjecaju svojstava otapala naravnoteže, o utjecaju stvaranja ionskih parova, o direktno] primjeni Hammettovejednadžbe na polarografske podatke, o entropijskim i entalpijskim doprinosimasupstituenata, te o kvantno-kemijskim interpretacijama reakcijskih konstanti uaprotičnim otapalima. Polarografskim tehnikama pridaje se posebno značenje uproučavanju organsko-kemtiskih reakcija u aprotičnim otapalima stoga što su oneosjetljivije i prikladnije za interpretaciju rezultata nego tehnike homogena reakcij-ske kinetike.


















