
Application of Hidden Markov Model for Sequence Analysis and Use for Predicting Protein Localization...
28
Application of Hidden Markov Model for Sequence Analysis and Use for Predicting Protein Localization By: Manchikalapati Myerow Shivananda
-
date post
21-Dec-2015 -
Category
Documents
-
view
219 -
download
0
Transcript of Application of Hidden Markov Model for Sequence Analysis and Use for Predicting Protein Localization...

Application of Hidden Markov Model for Sequence Analysis and Use for Predicting Protein Localization
By: Manchikalapati Myerow Shivananda Monday, April 14, 2003

Mathematical Modeling
Mathematical Modeling in biology and chemistry
Using probabilistic models Bayes Theorem and Maximum
Likelihood Theorem Ex: HMM

What is Markov Chain ? A directed graph with a collection of
states with transition probabilities. Models a random process with finite
states.
Markov Assumption – The chain is memory less and current state probability depends on previous state. This allows us to predict behavior.

Hidden Markov Model Hidden Markhov Model
A probabilistic model that is composed of states which are not observable events.
A statistical model that describes a probability distribution over a number of possible sequences.
HMM has the following components: States Symbol emission probabilities State transition probabilities
Why Hidden? Only the symbol sequence that a hidden state emits is observable.
Protein Modeling using HMM.

What is Hidden? in the Markov Model
Observed sequence is a probabilistic function of underlying Markov chain
In HMMs the state sequence is not uniquely determined by the observed symbol sequence, but must be inferred probabilistically from it.

Definition of Profile
A profile is a description of the consensus of a multiple sequence alignment.
Alignment Methods
Position Specific Scoring System
Position Independent(Pairwise alignment)Scoring SystemEx: BLAST, FASTA

Profile HMM Is a linear state machine consisting of a series of
nodes, each of which corresponds roughly to a position (column) in the alignment from which it was built.
The HMM will have a set of positions which would correspond to the columns in a multiple alignment and each column can have one of the three states: Insert, Delete and Match.
Profile HMMs can be used to do sensitive database searching using statistical descriptions of a sequence family's consensus.

Profile HMM vs Std Profiles Profile HMMs have a
formal probabilistic basis and have a consistent theory behind gap and insertion scores.
Profile HMMs apply a statistical method to estimate the true frequency of a residue at a given position in the alignment from its observed frequency.
In general, producing good profile HMMs requires less skill and manual intervention than producing good standard profiles.
Standard profile methods use heuristic methods.
Standard profiles use the observed frequency itself to assign the score for that residue.

Three Algorithms of HMM The Viterbi algorithm: get the most
probable state sequence. The Forward/Backward algorithm: score
an observation sequence against a model. Expectation/Maximization: get the
parameters of the model from the data.
For all HMM applications, the algorithms are fairly standard. Only the design of the model are different.

Application of HMM Gene finding Chromosome identification Protein applications include
Database searching Homology detection
Ex:One could take a single sequence of interest, and query it against the model to determine if it contained certain domains of interest.

HMM and its basic elements
1)Match States(M1,M2..)2)Delete State(D1,D2…)3)Insert States(I0,I1…)4) Begin State5)End State6)Emmision Probabilities7) Transition Probabilites8) Parameters

Problems “DEFINE” HMM Architecture
Problem at hand (given below)defines architecture(to the left)
Finding Ungapped Motifs - BLOCKS
Finding Multiple MotifsMETA-MEME
Finding Protein Familes ProfileHMMs(Krogh)
HMMER2 architecture is used in SAM,HMMER.

HMM Profile alignment flow chart in Pfam

Three Important Questions that HMM should answer
Scoring1Q) How likely is a given sequence
coming from the model? Alignment2Q)What is the optimal path for
generating a given sequence Training 3Q) Given a set of sequences how can
you learn about the HMM parameters

Q1)How likely is the given Seq (ACCY) coming from the model
Answer Forward Algorithm
Prob(A in state I0) = 0.4*0.3=0.12
Prob(C in state I1) = 0.05*0.06*0.5 = 0.015
Prob(C in state M1) = 0.46*0.01= 0.005
Prob(C in state M2) = (0.005*0.97) +(0.015*0.46)= .012
Prob(Y in state I3) = .012*0.015*0.73*0.01 = 1.31x10-7
Prob(Y in state M3) = .012*0.97*0.2 = 0.002

Q2)What is the optimal path for generating a given seq(ACCY)
Answer: Viterbi Algorithim1. The probability that the amino acid A was generated by
state I0 is computed and entered as the first element of the matrix.
2. The probabilities that C is emitted in state M1 (multiplied by the probability of the most likely transition to state M1 from state I0) and in state I1 (multiplied by the most likely transition to state I1 from state I0) are entered into the matrix element indexed by C and I1/M1.
3. The maximum probability, max(I1, M1), is calculated.4. A pointer is set from the winner back to state I0.5. Steps 2-4 are repeated until the matrix is filled. Prob(A in state I0) = 0.4*0.3=0.12 Prob(C in state I1) = 0.05*0.06*0.5 = .015 Prob(C in state M1) = 0.46*0.01 = 0.005 Prob(C in state M2) = 0.46*0.5 = 0.23 Prob(Y in state I3) = 0.015*0.73*0.01 = .0001 Prob(Y in state M3) = 0.97*0.23 = 0.22 The most likely path through the model can now be
found by following the back-pointers.

3Q)Given a set of sequences how do you learn about HMM params
The Learning Task given:– a model– a set of sequences (the
training set) do:– find the most likely
parameters to explain the training sequences
the goal is find a model that generalizes well to sequences we haven’t seen before
Answer: Baum-Welch(Forward Backward) Algorithm
initialize parameters of model
iterate until convergence– calculate the expected
number of times each transition or emission is used
– adjust the parameters to maximize the likelihood of these expected values

HMMER in the Workflow

Tripartite structure of signal peptide
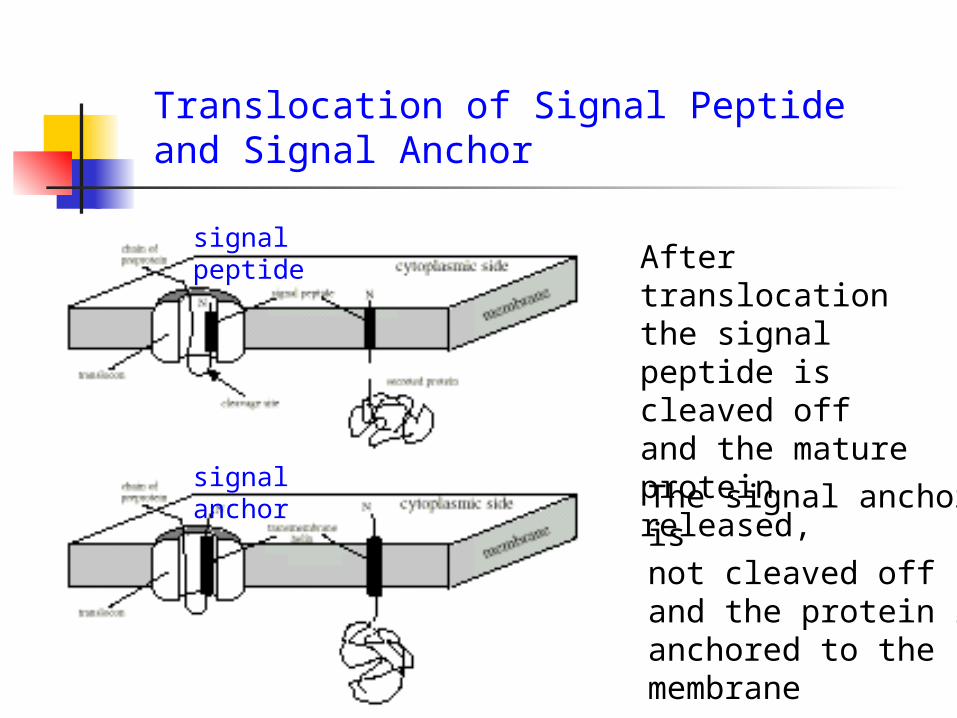
Translocation of Signal Peptide and Signal Anchor
After translocation the signal peptide is cleaved off and the mature protein released,
signal peptide
signal anchorThe signal anchor is not cleaved off and the protein is anchored to the membrane

(Nielsen, H and Krogh A. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc. Sixth Int. Conf on Intelligent Systems for Molecular Biology, 122-130. AAAI Press, 1998.)
Model not based on Multiple sequence alignment (profile)
Compare model to neural network in eukaryotes and prokaryotes
Two HMM Models for Signal PeptidesFirst Model:

The model used for signal peptides. The states in a shaded box are tied to each other.
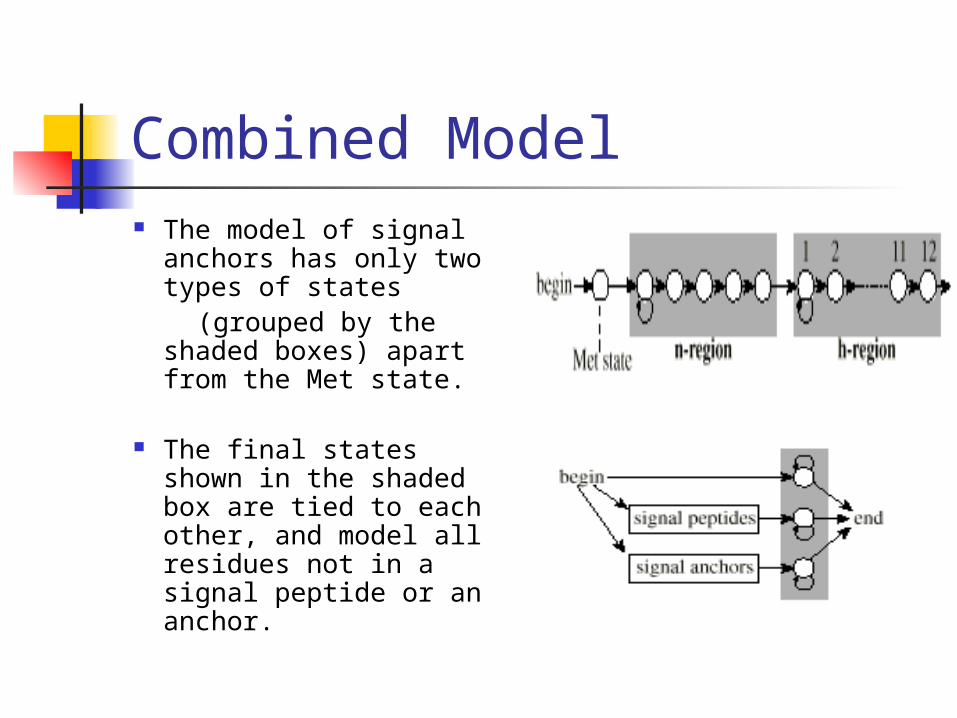
Combined Model The model of signal
anchors has only two types of states
(grouped by the shaded boxes) apart from the Met state.
The final states shown in the shaded box are tied to each other, and model all residues not in a signal peptide or an anchor.

Hidden Markov model (HMM) vs. neural network (NN) Cleavage site location: percentage of signal peptide
sequences where the cleavage site was placed correctly
Discrimination values: correlation coefficients (Mathews 1975).
Protein types: signal peptides (sig) cytoplasmic or nuclear—proteins (non-sec), and signal anchors (anc).
NN simple= S-score NN combined= Y-score

Second model for Signal Peptide
Barash S, Wang W, and Shi Y. Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem and Biophys Res Com 294, 835-842, 2002.
Profile HMM method using HMMER software

Steps for Model Building with HMMER N-terminal region of 416 non-redundant
human secreted proteins
Training in hmmalign: all start Met aligned in first column, 406/416 cleavage sites aligned
Build model with MLL estimation (random model= Swiss Prot 34)
Evaluate alignment model: 416/416 start Met, 406/416 cleavage site, 416/416 h-region
Re-estimate HMM with maximum discrimination method

Model Validation Used hmmemit program to generate
artificial sequences of variable bit scores
In vitro validation using secretion test plasmid constructs: using secretory alkP with native signalP replaced by HMM signal peptides, the signal strengths correlate with the bit scores (transcription or translation effect?)
Ranked signal strengths of known natural human secretory proteins: above average serum proteins such as albumin were found to have high bit scores

Conclusion HMM and its applicability to sequence analysis
has been discussed
Two different HMM architectures for modeling the signal peptide have been shown
Both are able to perform the task of separating secreted proteins from cytoplasmic and nuclear proteins with excellent discrimination
Discrimination of signal peptides from signal anchors is a little less clean
Multiple modeling strategies may be beneficial depending on the nature of the query and available data for training


















