
Application of Broad-Spectrum Resequencing …jvi.asm.org/content/84/18/9557.full.pdf... Lyssavirus...
18
JOURNAL OF VIROLOGY, Sept. 2010, p. 9557–9574 Vol. 84, No. 18 0022-538X/10/$12.00 doi:10.1128/JVI.00771-10 Copyright © 2010, American Society for Microbiology. All Rights Reserved. Application of Broad-Spectrum Resequencing Microarray for Genotyping Rhabdoviruses Laurent Dacheux, 1 ‡ Nicolas Berthet, 2 ‡ Gabriel Dissard, 3 Edward C. Holmes, 4 Olivier Delmas, 1 Florence Larrous, 1 Ghislaine Guigon, 3 Philip Dickinson, 5 Ousmane Faye, 6 Amadou A. Sall, 6 Iain G. Old, 7 Katherine Kong, 5 Giulia C. Kennedy, 5 Jean-Claude Manuguerra, 8 Stewart T. Cole, 9 Vale ´rie Caro, 3 Antoine Gessain, 2 and Herve ´ Bourhy 1 * Institut Pasteur, Lyssavirus Dynamics and Host Adaptation Unit, Paris, France 1 ; Institut Pasteur, Epidemiology and Pathophysiology Oncogenic Virus Unit, CNRS URA3015, Paris, France 2 ; Institut Pasteur, Genotyping of Pathogens and Public Health Technological Platform, Paris, France 3 ; Center for Infectious Disease Dynamics, Department of Biology, The Pennsylvania State University, University Park, Pennsylvania 4 ; Affymetrix, Santa Clara, California 5 ; Institut Pasteur de Dakar, Arbovirology Laboratory, Dakar, Senegal 6 ; Institut Pasteur, European Office, Paris, France 7 ; Institut Pasteur, Laboratory for Urgent Responses to Biological Threats, Paris, France 8 ; and Institut Pasteur, Bacterial Molecular Genetics Unit, Paris, France 9 Received 12 April 2010/Accepted 29 June 2010 The rapid and accurate identification of pathogens is critical in the control of infectious disease. To this end, we analyzed the capacity for viral detection and identification of a newly described high-density resequencing microarray (RMA), termed PathogenID, which was designed for multiple pathogen detection using database similarity searching. We focused on one of the largest and most diverse viral families described to date, the family Rhabdoviridae. We demonstrate that this approach has the potential to identify both known and related viruses for which precise sequence information is unavailable. In particular, we demonstrate that a strategy based on consensus sequence determination for analysis of RMA output data enabled successful detection of viruses exhibiting up to 26% nucleotide divergence with the closest sequence tiled on the array. Using clinical specimens obtained from rabid patients and animals, this method also shows a high species level concordance with standard reference assays, indicating that it is amenable for the development of diagnostic assays. Finally, 12 animal rhabdoviruses which were currently unclassified, unassigned, or assigned as tentative species within the family Rhabdoviridae were successfully detected. These new data allowed an unprecedented phylogenetic analysis of 106 rhabdoviruses and further suggest that the principles and methodology developed here may be used for the broad-spectrum surveillance and the broader-scale investigation of biodiversity in the viral world. The ability to simultaneously screen for a large panel of pathogens in clinical samples, especially viruses, will represent a major development in the diagnosis of infectious diseases and in surveillance programs for emerging pathogens. Cur- rently, most diagnostic methods are based on species-specific viral nucleic acid amplification. Although rapid and extremely sensitive, these methods are suboptimal when testing for a large number of known pathogens, when viral sequence diver- gence is high, when new but related viruses are anticipated, or when no clear viral etiologic agent is suspected. To overcome these technical difficulties, newer technologies have been em- ployed, especially microarrays dedicated to pathogen detec- tion. Indeed, DNA microarrays have been shown to be a pow- erful platform for the highly multiplexed differential diagnosis of infectious diseases. For example, pathogen microarrays can be simultaneously used to screen various viral or bacterial families and have been successfully used in the detection of microbial agents from different clinical samples (10–12, 19, 32, 35, 41, 42, 48). The “classical” DNA microarrays developed so far are based on the use of long-oligonucleotide pathogen-specific probes (50 nucleotides [nt]). Although powerful in terms of sensi- tivity, these diagnostic tools have the disadvantage of de- creased specificity, making it necessary to target multiple markers, and rely on hybridization patterns for pathogen iden- tification, leading to unquantifiable errors (4). Moreover, these methods lack comprehensive information about the pathogen at the single-nucleotide level, which could represent a major problem when the sequences in question show a high degree of similarity (21). The microarray-based pathogen resequencing assay represents a promising alternative tool with which to overcome these limitations. This method identifies each spe- cific pathogen and is capable of resequencing, or “fingerprint- ing,” multiple pathogens in a single test. Indeed, this technol- ogy uses tiled sets of 10 5 to 10 6 probes of 25mers, which contain one perfectly matched and three mismatched probes per base for both strands of the target genes (16). This tech- nology also offers the potential for a single test that detects and discriminates between a target pathogen and its closest phylo- genetic neighbors, which expands the repertoire of identifiable organisms far beyond those that are initially included in the array. Successful results have been obtained using this tech- * Corresponding author. Mailing address: Unite ´ Dynamique des lyssavirus et adaptation a ` l’ho ˆte, Institut Pasteur, 25 rue du Docteur Roux, 75724 Paris Cedex 15, France. Phone: 33 1 45 68 87 85. Fax: 33 1 40 61 30 20. E-mail: [email protected]. ‡ Contributed equally to this work. Published ahead of print on 7 July 2010. 9557 on June 1, 2018 by guest http://jvi.asm.org/ Downloaded from
Transcript of Application of Broad-Spectrum Resequencing …jvi.asm.org/content/84/18/9557.full.pdf... Lyssavirus...

JOURNAL OF VIROLOGY, Sept. 2010, p. 9557–9574 Vol. 84, No. 180022-538X/10/$12.00 doi:10.1128/JVI.00771-10Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Application of Broad-Spectrum Resequencing Microarray forGenotyping Rhabdoviruses�
Laurent Dacheux,1‡ Nicolas Berthet,2‡ Gabriel Dissard,3 Edward C. Holmes,4 Olivier Delmas,1Florence Larrous,1 Ghislaine Guigon,3 Philip Dickinson,5 Ousmane Faye,6 Amadou A. Sall,6
Iain G. Old,7 Katherine Kong,5 Giulia C. Kennedy,5 Jean-Claude Manuguerra,8Stewart T. Cole,9 Valerie Caro,3 Antoine Gessain,2 and Herve Bourhy1*
Institut Pasteur, Lyssavirus Dynamics and Host Adaptation Unit, Paris, France1; Institut Pasteur, Epidemiology andPathophysiology Oncogenic Virus Unit, CNRS URA3015, Paris, France2; Institut Pasteur, Genotyping of
Pathogens and Public Health Technological Platform, Paris, France3; Center for Infectious Disease Dynamics,Department of Biology, The Pennsylvania State University, University Park, Pennsylvania4; Affymetrix,
Santa Clara, California5; Institut Pasteur de Dakar, Arbovirology Laboratory, Dakar, Senegal6;Institut Pasteur, European Office, Paris, France7; Institut Pasteur, Laboratory for
Urgent Responses to Biological Threats, Paris, France8; and Institut Pasteur,Bacterial Molecular Genetics Unit, Paris, France9
Received 12 April 2010/Accepted 29 June 2010
The rapid and accurate identification of pathogens is critical in the control of infectious disease. To this end,we analyzed the capacity for viral detection and identification of a newly described high-density resequencingmicroarray (RMA), termed PathogenID, which was designed for multiple pathogen detection using databasesimilarity searching. We focused on one of the largest and most diverse viral families described to date, thefamily Rhabdoviridae. We demonstrate that this approach has the potential to identify both known and relatedviruses for which precise sequence information is unavailable. In particular, we demonstrate that a strategybased on consensus sequence determination for analysis of RMA output data enabled successful detection ofviruses exhibiting up to 26% nucleotide divergence with the closest sequence tiled on the array. Using clinicalspecimens obtained from rabid patients and animals, this method also shows a high species level concordancewith standard reference assays, indicating that it is amenable for the development of diagnostic assays. Finally,12 animal rhabdoviruses which were currently unclassified, unassigned, or assigned as tentative species withinthe family Rhabdoviridae were successfully detected. These new data allowed an unprecedented phylogeneticanalysis of 106 rhabdoviruses and further suggest that the principles and methodology developed here may beused for the broad-spectrum surveillance and the broader-scale investigation of biodiversity in the viral world.
The ability to simultaneously screen for a large panel ofpathogens in clinical samples, especially viruses, will representa major development in the diagnosis of infectious diseasesand in surveillance programs for emerging pathogens. Cur-rently, most diagnostic methods are based on species-specificviral nucleic acid amplification. Although rapid and extremelysensitive, these methods are suboptimal when testing for alarge number of known pathogens, when viral sequence diver-gence is high, when new but related viruses are anticipated, orwhen no clear viral etiologic agent is suspected. To overcomethese technical difficulties, newer technologies have been em-ployed, especially microarrays dedicated to pathogen detec-tion. Indeed, DNA microarrays have been shown to be a pow-erful platform for the highly multiplexed differential diagnosisof infectious diseases. For example, pathogen microarrays canbe simultaneously used to screen various viral or bacterialfamilies and have been successfully used in the detection of
microbial agents from different clinical samples (10–12, 19, 32,35, 41, 42, 48).
The “classical” DNA microarrays developed so far are basedon the use of long-oligonucleotide pathogen-specific probes(�50 nucleotides [nt]). Although powerful in terms of sensi-tivity, these diagnostic tools have the disadvantage of de-creased specificity, making it necessary to target multiplemarkers, and rely on hybridization patterns for pathogen iden-tification, leading to unquantifiable errors (4). Moreover, thesemethods lack comprehensive information about the pathogenat the single-nucleotide level, which could represent a majorproblem when the sequences in question show a high degree ofsimilarity (21). The microarray-based pathogen resequencingassay represents a promising alternative tool with which toovercome these limitations. This method identifies each spe-cific pathogen and is capable of resequencing, or “fingerprint-ing,” multiple pathogens in a single test. Indeed, this technol-ogy uses tiled sets of 105 to 106 probes of 25mers, whichcontain one perfectly matched and three mismatched probesper base for both strands of the target genes (16). This tech-nology also offers the potential for a single test that detects anddiscriminates between a target pathogen and its closest phylo-genetic neighbors, which expands the repertoire of identifiableorganisms far beyond those that are initially included in thearray. Successful results have been obtained using this tech-
* Corresponding author. Mailing address: Unite Dynamique deslyssavirus et adaptation a l’hote, Institut Pasteur, 25 rue du DocteurRoux, 75724 Paris Cedex 15, France. Phone: 33 1 45 68 87 85. Fax: 331 40 61 30 20. E-mail: [email protected].
‡ Contributed equally to this work.� Published ahead of print on 7 July 2010.
9557
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

nology, especially for the detection of broad-spectrum respira-tory tract pathogens using respiratory pathogen microarrays (2,25, 26) or the detection of a broad range of biothreat agents (1,23, 36, 45). The amplification step, which is more often limitingfor this technology, has also benefited from recent develop-ments. Phi29 polymerase-based amplification methods provideamplified DNA with minimal changes in sequence and relativeabundance for many biomedical applications (3, 31, 40). Theamplification factor varied from 106 to 109, and it was alsodemonstrated that coamplification occurred when viral RNAwas mixed with bacterial DNA (3). This whole-transcriptomeamplification (WTA) approach can also be successfully appliedto viral genomic RNA of all sizes. Amplifying viral RNA byWTA provides considerably better sensitivity and accuracy ofdetection than random reverse transcription (RT)-PCR in thecontext of resequencing microarrays (RMAs) (3).
The rhabdoviruses are single-stranded, negative-sense RNAgenome viruses classified into six genera, three of which—Vesiculovirus, Lyssavirus, and Ephemerovirus—include arthro-pod-borne agents that infect birds, reptiles, and mammals, aswell as a variety of non-vector-borne mammalian or fish viruses(International Committee on Taxonomy of Viruses database[ICTVdb]) (reviewed in reference 7). These rhabdoviruses arethe etiological agents of human diseases, such as rabies, thatcause serious public health problems. Some rhabdoviruses alsocause important economic losses in livestock. The three othersgenera include Nucleorhabdovirus and Cytorhabdovirus, whichare arthropod-borne viruses infecting plants, and Novirhab-dovirus, which comprises fish viruses. Other than the well-characterized rhabdoviruses that are known to be importantfor agriculture and public health, there is also a constantlygrowing list of rhabdoviruses, isolated from a variety of verte-brate and invertebrate hosts, that are partially characterizedand are still waiting for definitive genus or species assignment.Considering the large spectrum of potential animal reservoirsof these viruses compared to the few identified virus species, itis highly likely that the number of uncharacterized rhabdovi-ruses is immense.
Unclassified or unassigned viruses have been tentativelyidentified as members of the family Rhabdoviridae by electronmicroscopy, based on their bullet-shaped morphology—a char-acteristic trait of members of this family—or using their anti-genic relationships based on serological tests (9, 38). Genesequencing and phylogenetic relationships have then been pro-gressively applied to complete this initial virus taxonomy (6, 22,27). Importantly, a strongly conserved domain in the rhabdovi-rus genome, within the polymerase gene, is a useful target forthe exploration of the distant evolutionary relationships amongthese diverse viruses (6). This region corresponds to block IIIof the viral polymerase, a region predicted to be essential forRNA polymerase function, as it is highly conserved amongmost of the RNA-dependent RNA polymerases (14, 33, 46). Adirect application using this sequence region was recently de-scribed for lyssavirus RNA detection in human rabies diagnosis(13). Taking advantage of these characteristics, this polymer-ase region was also used to design probes for high-densityRMAs, also called PathogenID arrays (Affymetrix), which areoptimized for the detection and sequence determination ofseveral RNA viruses, particularly rhabdoviruses (1).
In the present study, PathogenID microarrays containing
probes for the detection of up to 126 viruses were tested usinga consensus sequence determination strategy for the analysis ofoutput RMA data. We demonstrate that this approach has thepotential to identify, in experimentally infected and clinicalspecimens, known but also phylogenetically related rhabdovi-ruses for which precise sequence information was not avail-able.
MATERIALS AND METHODS
Design of the PathogenID microarray for rhabdovirus detection. Two gener-ations of PathogenID arrays were used in this study: PathogenID v1.0, containingprobes for the detection of 42 viruses (including 3 prototype rhabdoviruses), 50bacteria, and 619 toxin or antibiotic resistance genes (previously described inreference 1), and PathogenID v2.0, which is able to detect 126 viruses (including30 different rhabdoviruses), 124 bacteria, 673 toxin or antibiotic resistance genes,and two human genes as controls. These arrays include prototype sequences ofall of the species (or genotypes) of the genus Lyssavirus, of the other majorgenera defined in the family Rhabdoviridae, such as Ephemerovirus and Vesicu-lovirus, and of 13 rhabdoviruses awaiting classification or tentatively classifiedamong minor groups such as the Le Dantec and Hark Park groups (6). For all ofthe selected probes tiled on the two versions of the PathogenID array, the sameconserved region of the viral polymerase gene was used (block III). However, thesize of the target region tiled on the array was longer in the second version (upto 937 nt in length for some sequences, compared to roughly 500 nt in the firstversion) (Tables 1 and 2).
Virus strains and biological samples analyzed. Detailed descriptions of all ofthe prototype and field virus strains used in this study and their sources are listedin Tables 1 and 2. Briefly, 16 and 31 different viruses were tested using Patho-genID v1.0 (15 lyssaviruses and 1 vesiculovirus) and PathogenID v2.0 (14 lyssa-viruses, 1 vesiculovirus, and 12 unassigned and 4 tentative species of animalrhabdoviruses according to ICTVdb), respectively. Samples tested included invitro-infected cells, a synthetic nucleotide target (when the corresponding virusstrain was not available), brain biopsy specimens obtained from experimentallyinfected mice, and biological specimens from various animals (bat, cat, dog, andfox brains) and humans (brain, saliva, and skin biopsy specimens).
Extraction and amplification of viral RNA. RNA extraction from biologicalsamples was processed with TRI Reagent (Molecular Research Center) accord-ing to the manufacturer’s recommendations. After extraction, viral RNAs werereverse transcribed and then amplified using the whole-transcriptome amplifica-tion (WTA) protocol (QuantiTect Whole Transcriptome kit; Qiagen) as de-scribed previously (3).
Microarrays assay. All of the amplification products obtained from viral RNAwere quantified by Quantit BR (Invitrogen) according to the manufacturer’sinstructions or by the NanoDrop ND-1000 spectrophotometer instrument(Thermo Scientific). A recommended amount of target DNA was fragmentedand labeled according to GeneChip Resequencing Assay manual (Affymetrix).The microarray hybridization process was carried out according to the protocolrecommended by the manufacturer (Affymetrix). All of the details and param-eter settings for the data analysis (essentially conversion of raw image filesobtained from scanning of the microarrays into FASTA files containing thesequences of base calls made for each tiled region of the microarray) have beendescribed previously (1). The base call rate refers to the percentage of base callsgenerated from the full-length tiled sequence.
Data analysis. In the first approach, resequencing data obtained by the Patho-genID v1.0 microarray were manually submitted to the NCBI nr/nt database forBLASTN query. The default BLAST options were modified. The word size wasset to 7 nt. The expected threshold was increased from its default value of 10 to100,000 to reduce the filtering of short sequences and sequences rich in unde-termined calls, which can assist correct taxonomic identification. To avoid false-negative results induced by high numbers of undetermined nucleotides in thesequences, the “low complexity level filter” (�F) was also turned off. BLASTsorts the resulting hits according to their bit scores so that the sequence that isthe most similar to the entry sequence appears first. Identification of the virusstrains tested was considered successful only when the best hit was unique andcorresponded to the expected species or isolate (according to the nucleotidesequences of these viruses already available in the NCBI nr/nt database).
In the second approach, an automatic bioinformatics-based analysis of RMAdata provided by PathogenID v2.0 was developed, including a consensus se-quence determination strategy completed with a systematic BLAST strategy. Thegeneral workflow of this strategy is represented in Fig. 1. A Perl script reads the
9558 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

input data, which consist of one FASTA file per sample that contains all of thesequences read by the GSEQ software from the hybridization. A modified ver-sion of the filtering process described by Malanoski et al. (29) is applied to thesequences. The retained sequences contain stretches of nucleotides that areascertained according to the following algorithm. Briefly, sequences that do notcontain subsequences fulfilling specific parameters (minimum nucleotide length[m] and maximum undetermined nucleotide content [N]) defined by the user arediscarded. These parameters differ from those described in the original filteringprocess, where m was fixed to 20 and N was a value depending on m, leading tothe filtering out of all short subsequences, even with a high base call rate. Forsubsequence determination, the program starts from the first base call of thesequence considered and searches for the first m base window area that scoresthe elongation threshold defined by the user, which represents another differencefrom the filtering process described by Malanoski et al., where this elongationthreshold was fixed at 60% (29). The subsequence is extended by one base (m �1) if the percentage of N remains inferior to the elongation threshold. When thisthreshold is exceeded, the elongation is stopped and the subsequence is con-served. This process is reiterated until the end of the sequence is reached togenerate as many informative sequences as possible. All of our analyses wereperformed with the following filtering parameters: m � 12, N � 10, and elon-gation threshold � 10%.
A systematic BLAST strategy to search for sequence homologues was thenperformed with the filtered sequences containing subsequences. These se-
quences individually undergo a BLAST analysis based on a local viral andbacterial database (sequences obtained after filtering from the NCBI nr/nt da-tabase, updated and used for BLAST queries in December 2009), and thetaxonomies of the best BLAST hits are retrieved (Fig. 1A). The default BLASToptions were modified as previously described. When several hits obtain thehighest bit score, the script automatically retrieves the taxonomies of the 10 firstBLAST hits. The final taxonomic identification of each virus strain tested wasdone by the user as follows: (i) identification at the species or isolate level whena unique best hit corresponds to the expected species or isolate, (ii) identificationat the genus level (if available) when multiple best viral hits exist and correspondto different species within the same genus of the family Rhabdoviridae, (iii)identification at the family level when multiple best viral hits exist and corre-spond to different rhabdoviruses genera, or (iv) negative or inaccurate identifi-cation when a BLAST query is not possible or when multiple best hits correspondto other viral families, respectively.
For the consensus sequence determination strategy, resequencing data ob-tained from rhabdoviral tiled sequences are filtered as previously described andthen submitted to a multiple alignment with CLUSTAL W (39), from which aconsensus sequence is determined (Fig. 1B). For each sequence in the alignment,if a called base has undetermined calls on both sides, it is replaced by anundetermined call. If different calls appear in the sequences for a given position,the majority base call is added to the consensus. The positions that contain anundetermined call or a gap are not considered in the majority base call compu-
TABLE 1. Description of virus species belonging to the family Rhabdoviridae used for selection of tiled sequences and for validation of thePathogenID v1.0 microarray
Genus and speciesa
(abbreviation) Strain Host species/vector Origin
Yr offirst
isolation
Tiledregionb (nt)
Length(nt)
Biological sampletested
GenBankaccession no.
Origin of tiled sequencesLyssavirus Rabies virus
(RABV)PV Vaccine 7452–7953 502 NC_001542
Vesiculovirus Vesicularstomatitis Indianavirus (VSIV)
VSVLMS 7453–7953 497 K02378
Ephemerovirus Bovineephemeral fevervirus (BEFV)
BB7721 Bos taurus Australia 1968 7454–7952 498 NC_002526
Rhabdoviruses testedLyssavirus species
Rabies virus (RABV) 8764THA Human Thailand 1983 Human brain EU293111Rabies virus (RABV) 9147FRA Red fox France 1991 Fox brain EU293115Rabies virus (RABV) 93128MAR Fixed strain Morocco ? Mouse brain GU815994Rabies virus (RABV) 9811CHI Dog China 1998 Mouse brain GU815995Rabies virus (RABV) 0435AFG Dog Afghanistan 2004 Mouse brain GU815996Rabies virus (RABV) 9001FRA Dog bitten by
batFrench Guiana 1990 Mouse brain EU293113
Rabies virus (RABV) 9026CI Dog Ivory Coast 1990 Mouse brain GU815997Rabies virus (RABV) 9105USA Fox USA 1991 Fox brain GU815998Rabies virus (RABV) 9233GAB Dog Gabon 1992 Dog brain GU815999Rabies virus (RABV) 93127FRA Fixed strain France ? Mouse brain GU816000Rabies virus (RABV) 9503TCH Fixed strain
(Vnukovo,SAD)
Czechoslovakia ? Mouse brain GU816001
Rabies virus (RABV) 9737POL Raccoon dog Poland 1997 Mouse brain GU816002Rabies virus (RABV) Challenge virus
strain(CVS_IP13)
Fixed strain Mouse brain GU816003
Rabies virus (RABV) ERA Fixed strain Mouse brain GU816005Rabies virus (RABV) LEP Fixed strain Chicken embryo
fibroblastsGU816004
Vesiculovirus Vesicularstomatitis Indianavirus (VSIV)
Orsay(0503FRA)
Fixed strain BSR cellsc GU816006
a Classifications and names of viruses correspond to approved virus taxonomy according to ICTVdb. Names in italics are those of validated virus species.b Position according to the reference Pasteur virus genome (NC_001542) after alignment of all of the tiled sequences with the reference sequence.c Clone of the baby hamster kidney cell line BHK-21.
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9559
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

TA
BL
E2.
Des
crip
tions
ofvi
rus
spec
ies
belo
ngin
gto
the
fam
ilyR
habd
oviri
dae
used
for
sele
ctio
nof
tiled
sequ
ence
san
dfo
rva
lidat
ion
ofth
ePa
thog
enID
v2.0
mic
roar
ray
Gen
usor
grou
paan
dsp
ecie
s(a
bbre
viat
ion)
aU
A/T
S/U
Cb
Stra
inH
ost
spec
ies/
vect
oror
sour
ceT
iled
regi
on(n
t)c
Len
gth
(nt)
Bio
logi
cal
sam
ple
test
edO
rigi
nof
sam
ple
Yr
offir
stis
olat
ion
Gen
Ban
kac
cess
ion
no.
Ori
gin
oftil
edse
quen
ces
Lys
savi
rus
Gen
otyp
e1,
Rab
ies
viru
s(R
AB
V)
PVV
acci
ne70
40–7
977
937
NC
_001
542
Gen
otyp
e2,
Lag
osba
tvi
rus
(LB
V)
8619
NG
AB
at,E
idol
onhe
lvum
7040
–797
793
7N
iger
ia19
56E
U29
3110
Gen
otyp
e3,
Mok
ola
viru
s(M
OK
V)
MO
KV
Cat
7040
–797
793
7Z
imba
bwe
1981
NC
_006
429
Gen
otyp
e4,
Duv
enha
gevi
rus
(DU
VV
)94
286S
AB
at,M
inop
teru
ssp
ecie
s70
40–7
977
937
Sout
hA
fric
a19
81E
U29
3120
Gen
otyp
e5,
Eur
opea
nba
tly
ssav
irus
1(E
BL
V-1
)89
18F
RA
Bat
,Ept
esic
usse
rotin
us70
40–7
977
937
Fra
nce
1989
EU
2931
12G
enot
ype
6,E
urop
ean
bat
lyss
aviru
s2
(EB
LV
-2)
9018
HO
LB
at,M
yotis
dasy
cnem
e70
40–7
977
937
Net
herl
ands
1986
EU
2931
14G
enot
ype
7,A
ustr
alia
nba
tly
ssav
irus
(AB
LV
)A
BL
hH
uman
7040
–797
793
7A
ustr
alia
1986
AF
4180
14
Ves
icul
oviru
sC
hand
ipur
avi
rus
(CH
PV)
I65
3514
Hum
an70
40–7
981
935
Indi
a19
65A
J810
083
Isfa
han
viru
s(I
SFV
)91
026-
167
Phl
ebot
omus
papa
tasi
7040
–798
193
5Ir
an19
75A
J810
084
Ves
icul
arst
omat
itis
New
Jers
eyvi
rus
(VSN
JV)
VSV
NJ-
OB
osta
urus
,equ
ine/
Cul
exni
grip
alpu
s,C
ulic
oide
ssp
ecie
s,M
anso
nia
indu
bita
ns
7040
–798
193
5U
nite
dSt
ates
1949
AY
0748
04
Ves
icul
arst
omat
itis
Indi
ana
viru
s(V
SIV
)V
SVL
MS
?70
40–7
981
935
??
K02
378
Peri
net
viru
s(P
ER
V)
TS
Ar
Mg
802
Cul
exan
tenn
atus
7089
–750
240
5M
adag
asca
r19
78A
Y85
4652
Spri
ngvi
rem
iaof
carp
viru
s(S
VC
V)
TS
VR
-139
0C
yprin
usca
rpio
7040
–798
193
5Y
ugos
lavi
a19
71U
1810
1
Eph
emer
oviru
sA
dela
ide
Riv
ervi
rus
(AR
V)
DPP
61B
osta
urus
7089
–750
240
8A
ustr
alia
1981
AY
8546
35B
ovin
eep
hem
eral
feve
rvi
rus
(BE
FV
)B
B77
21B
osta
urus
7089
–750
240
8A
ustr
alia
1968
AY
8546
42K
imbe
rley
viru
s(K
IMV
)T
SC
S36
8B
osta
urus
7089
–750
240
8A
ustr
alia
1980
AY
8546
37K
oton
kan
viru
sd(K
OT
V)
UA
IbA
r233
80C
ulic
oide
ssp
ecie
s70
89–7
502
408
Nig
eria
1967
AY
8546
38
Oth
erdi
mar
habo
dvir
uses
d
Alm
piw
argr
oup
Alm
piw
arvi
rus
(AL
MV
)U
AM
RM
4059
Abl
epha
rus
bout
onii
virg
atus
7089
–750
241
1A
ustr
alia
1966
AY
8546
45H
umpt
ydo
ovi
rus
(HD
OO
V)
UA
CS
79L
asio
hele
asp
ecie
s70
89–7
502
411
Aus
tral
ia19
75A
Y85
4643
Oak
-Val
evi
rus
(OV
RV
)U
AC
S13
42C
ulex
spec
ies
7089
–750
240
8A
ustr
alia
1981
AY
8546
70H
art
Park
grou
pF
land
ers
viru
s(F
LA
NV
)U
A61
-748
4C
ulis
eta
mel
anur
a70
89–7
502
410
Uni
ted
Stat
es19
61A
F52
3199
Nga
inga
nvi
rus
(NG
AV
)U
AN
RM
1455
6C
ulic
oide
sbr
evita
rsis
7089
–750
240
8A
ustr
alia
1970
AY
8546
49Pa
rry
Cre
ekvi
rus
(PC
RV
)U
AO
R18
9C
ulex
annu
liros
tris
7089
–750
240
8A
ustr
alia
1972
AY
8546
47W
onga
belv
irus
(WO
NV
)U
AC
S26
4C
ulic
oide
sau
stro
palp
alis
7089
–750
240
8A
ustr
alia
1979
AY
8546
48L
eD
ante
can
dK
ern
Can
yon
grou
pF
ukuo
kavi
rus
(FU
KV
)U
AF
UK
-11
Cul
icoi
des
punc
tatu
s70
89–7
502
408
Japa
n19
82A
Y85
4651
Le
Dan
tec
viru
s(L
DV
)U
AD
akH
D76
3H
uman
7089
–750
240
8Se
nega
l19
65A
Y85
4650
Tib
roga
rgan
grou
p,T
ibro
garg
anvi
rus
(TIB
V)
UA
CS
132
Cul
icoi
des
brev
itars
is70
89–7
502
408
Aus
tral
ia19
76A
Y85
4646
Oth
eran
imal
rhab
dovi
ruse
sT
upai
arh
abdo
viru
s(T
UPV
)U
AT
RV
1591
Tup
aia
bela
nger
i70
89–7
502
408
Tha
iland
?N
C_0
0702
0Si
gma
viru
s(S
IGM
AV
)U
A23
4HR
CD
roso
phila
mel
anog
aste
r62
20–6
642
408
??
X91
062
Sea
trou
trh
abdo
viru
s(S
TR
V)
UC
28/9
7Sa
lmo
trut
tatr
utta
7108
–757
641
5Sw
eden
1996
AF
4349
92
Rha
bdov
irus
este
sted
Lys
savi
rus
Gen
otyp
e1
Rab
ies
viru
s(R
AB
V)
9312
7FR
AF
ixed
stra
inM
ouse
brai
nF
ranc
e?
GU
8160
00R
abie
svi
rus
(RA
BV
)87
64T
HA
Hum
anH
uman
brai
nT
haila
nd19
83E
U29
3111
Rab
ies
viru
s(R
AB
V)
0833
9FR
AH
uman
(pro
babl
yco
ntam
inat
edby
bat)
Hum
ansa
liva
Fra
nce
(Fre
nch
Gui
ana)
2008
GU
8160
07
Rab
ies
viru
sh(R
AB
V)
0702
9SE
NH
uman
Skin
biop
sySe
nega
l20
06G
enot
ype
2,L
agos
bat
viru
s(L
BV
)86
19N
GA
Bat
,Eid
olon
helv
umM
ouse
brai
nN
iger
ia19
56E
U29
3110
Gen
otyp
e3,
Mok
ola
viru
s(M
OK
V)
8610
0CA
MSh
rew
Mou
sebr
ain
Cam
eroo
n19
81N
C_0
0642
9G
enot
ype
4,D
uven
hage
viru
s(D
UV
V)
8613
2SA
Hum
anM
ouse
brai
nSo
uth
Afr
ica
1971
EU
2931
19
9560 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
Gen
otyp
e5
Eur
opea
nba
tly
ssav
irus
1su
btyp
ea
(EB
LV
-1a)
0724
0FR
AC
at(c
onta
min
ated
byba
t)C
atbr
ain
Fra
nce
2007
EU
6265
52
Eur
opea
nba
tly
ssav
irus
1su
btyp
ea
(EB
LV
-1b)
0834
1FR
AB
at,E
ptes
icus
sero
tinus
Bat
brai
nF
ranc
e20
08G
U81
6009
Eur
opea
nba
tly
ssav
irus
1su
btyp
eb
(EB
LV
-1b)
8918
FR
AB
at,E
ptes
icus
sero
tinus
Mou
sebr
ain
Fra
nce
1989
EU
2931
12
Gen
otyp
e6,
Eur
opea
nba
tly
ssav
irus
2(E
BL
V-2
)90
18H
OL
Bat
,Myo
tisda
sycn
eme
Mou
sebr
ain
Hol
land
1986
EU
2931
14G
enot
ype
7,A
ustr
alia
nba
tly
ssav
irus
(AB
LV
)98
10A
US
Bat
Mou
sebr
ain
Aus
tral
ia?
GU
8160
08G
enot
ype
8(t
enta
tive
spec
ies)
,Dak
arba
tly
ssav
irus
(DB
LV
)U
C04
06SE
N(A
nD42
443)
Bat
,Eid
olon
helv
umM
ouse
brai
nSe
nega
l19
85E
U29
3108
Not
assi
gned
,Wes
tC
auca
sian
bat
viru
s(W
CB
V)
UC
Bat
,Myo
tissc
hrei
bers
iiPl
asm
ide
Rus
sia
2002
EF
6142
58
Ves
icul
oviru
sV
esic
ular
stom
atiti
sIn
dian
avi
rus
(VSI
V)
Ors
ay(0
503F
RA
)?
BSR
cells
f?
?G
U81
6006
Bot
eke
viru
s(B
TK
V)
TS
Dak
ArB
1077
(041
7RC
A)
Coq
uille
ttidi
am
acul
ipen
nis
Mou
sebr
ain
Cen
tral
Afr
ican
Rep
ublic
1968
GU
8160
14
Juro
navi
rus
(JU
RV
)T
SB
eAr
4057
8(0
414B
RE
)H
emag
ogus
speg
azzi
nii
Mou
sebr
ain
Bra
zil
1962
GU
8160
24Po
rton
’svi
rus
(PO
RV
)T
S16
43(0
416M
AL
)M
anso
nia
unifo
rmis
Mou
sebr
ain
Mal
aysi
a(S
araw
ak)
?G
U81
6013
Eph
emer
oviru
sK
oton
kan
viru
sd(K
OT
V)
UA
IbA
r233
80(9
145N
IG)
Cul
icoi
des
spec
ies
Mou
sebr
ain
Nig
eria
1967
AY
8546
38K
imbe
rley
viru
s(K
IMV
)T
SC
S36
8B
osta
urus
Mou
sebr
ain
Aus
tral
ia19
80A
Y85
4637
Oth
eran
imal
rhab
odvi
ruse
sH
art
Park
grou
pK
ames
evi
rus
(KA
MV
)U
AM
P61
86(0
8343
OU
G)
Cul
exan
nulio
risM
ouse
brai
nU
gand
a19
67G
U81
6011
Mos
suri
lvir
us(M
OSV
)U
ASA
Ar
1995
(041
8MO
Z)
Cul
exsi
tiens
Mou
sebr
ain
Moz
ambi
que
1959
GU
8160
12
Kol
ongo
and
Sand
jimba
grou
p,Sa
ndjim
bavi
rus
(SJA
V)
UA
Dak
AnB
373d
(072
44R
CA
)A
croc
epha
lus
scho
enob
aenu
sM
ouse
brai
nC
entr
alA
fric
anR
epub
lic19
70G
U81
6019
Le
Dan
tec
and
Ker
nC
anyo
ngr
oup
Keu
ralib
avi
rus
(KE
UV
)U
AD
akA
nD53
14(9
715S
EN
,042
0SE
N)
Tat
era
kem
piM
ouse
brai
nSe
nega
l19
68G
U81
6021
Nko
lbis
son
viru
s(N
KO
V)
UA
Ar
Y31
/65
(042
5CA
M)
Ere
tmap
odite
sle
ucop
ous
Mou
sebr
ain
Ivor
yC
oast
,C
amer
oon
1965
GU
8160
22
Ung
roup
edG
arba
viru
sg(G
AR
V)
UA
Dak
AnB
439a
(042
2RC
A)
Cor
ytho
rnis
cris
tata
Mou
sebr
ain
Cen
tral
Afr
ican
Rep
ublic
1970
GU
8160
18
Nas
oule
viru
sg(N
ASV
)U
AD
akA
nB42
89a
(041
0RC
A)
And
ropa
dus
vire
nsM
ouse
brai
nC
entr
alA
fric
anR
epub
lic19
73G
U81
6017
Oua
ngo
viru
sg(O
UA
V)
UA
Dak
AnB
1582
a(9
718R
CA
)P
loce
usm
elan
ocep
halu
sM
ouse
brai
nC
entr
alA
fric
anR
epub
lic19
70G
U81
6015
Bim
bovi
rusg
(BB
OV
)U
AD
akA
nB10
54d
(971
6RC
A)
Eup
lect
esaf
erM
ouse
brai
nC
entr
alA
fric
anR
epub
lic19
70G
U81
6016
Ban
gora
nvi
rus
(BG
NV
)U
AD
akA
rB20
53(0
424R
CA
)C
ulex
perf
uscu
sM
ouse
brai
nC
entr
alA
fric
anR
epub
lic19
69G
U81
6010
Gos
sas
viru
sh(G
OSV
)U
AD
akA
nD40
1(0
8344
SEN
)T
adar
ida
spec
ies
Mou
sebr
ain
Sene
gal
1964
NA
i
aU
nles
sst
ated
othe
rwis
e,th
ecl
assi
ficat
ions
and
nam
esof
viru
ses
corr
espo
ndto
appr
oved
viru
sta
xono
my
acco
rdin
gto
ICT
Vdb
.Nam
esin
italic
are
thos
eof
valid
ated
viru
ssp
ecie
s.b
UA
,una
ssig
ned;
TS,
tent
ativ
esp
ecie
s;U
C,u
ncla
ssifi
ed(n
otfo
und
inIC
TV
db).
cPo
sitio
nac
cord
ing
toth
ere
fere
nce
Past
eur
viru
sge
nom
e(N
C_0
0154
2)af
ter
alig
nmen
tofa
llof
the
tiled
sequ
ence
sw
ithth
ere
fere
nce
sequ
ence
(exc
eptf
ortil
edse
quen
ces
from
ungr
oupe
drh
abdo
viru
ses
TU
PVan
dSI
GM
AV
,whi
chw
ere
alig
ned
inde
pend
ently
with
the
refe
renc
ese
quen
ce).
dT
axon
omic
alcl
assi
ficat
ion
acco
rdin
gto
refe
renc
e6.
eA
977-
ntfr
agm
ent
ofth
epo
lym
eras
ege
ne(f
rom
nt70
20to
nt79
97,a
ccor
ding
toth
ere
fere
nce
Past
eur
viru
sge
nom
e�N
C_0
0154
2�)
was
synt
hesi
zed
invi
tro
and
then
clon
edin
topl
asm
idpC
R2.
1(O
pero
n).
fC
lone
ofth
eba
byha
mst
erki
dney
cell
line
BH
K-2
1.g
Not
dete
cted
usin
gPa
thog
enID
v2.0
mic
roar
ray
but
ampl
ified
byPC
Ror
nest
edPC
Rus
ing
cons
ensu
sor
spec
ific
prim
ers.
hN
otde
tect
edus
ing
Path
ogen
IDv2
.0m
icro
arra
yor
ampl
ified
byPC
Ror
nest
edPC
Rus
ing
cons
ensu
sor
spec
ific
prim
ers.
iN
A,n
otap
plic
able
.
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9561
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
tation. If multiple base calls tie for the majority, an undetermined call appears atthis position in the consensus sequence. This procedure generally increases thelength and accuracy of the query sequence for subsequent analysis. Homologysearching of the consensus sequences is performed with BLAST using the pa-rameters previously described, and the taxonomy of the best hit is retrieved as forthe systematic homology searching approach. We tested if the resulting consen-sus sequences had higher identification accuracy than any individual sequence orcould be used to design PCR primers for a characterization of a potential novelisolate.
Sequencing confirmation. Conventional sequencing was undertaken after thePCR amplification of viral targets directly from biological samples (after RNAextraction and RT) or from 10- to 100-fold water-diluted WTA products. Primerdesign was first based on consensus sequences obtained using the consensussequence determination strategy previously described and/or on rhabdovirusnucleotide sequences available in GenBank. Depending on the results obtainedand the virus strain tested, the primer design, the set of primers used, and thePCR conditions used for partial polymerase gene amplification were then ad-justed (list of primers and the PCR conditions are available on request from thecorresponding author). All PCR products were obtained using the proofreadingDNA polymerase ExtTaq (Takara). Sequence assembly and consensus sequenceswere obtained using Sequencher 4.7 (Gene Codes).
Phylogenetic analysis. The data set of 15 newly sequenced rhabdoviruses fromthis study (including the Sandjimba and Kolongo viruses previously only identi-fied on the basis of partial nucleoprotein gene sequences, as well as Piry virus, forwhich the nucleotide sequences of different genes were available) was comparedwith the corresponding block III polymerase amino acid sequences of 91 otherrhabdoviruses collected from GenBank (see Table 6). DNA translation wasperformed with BioEdit software (17), and sequence alignment was performedusing the CLUSTAL W program (39) and then checked for accuracy by eye. Thisresulted in a final alignment of 106 sequences 160 amino acid residues in length.Phylogenetic analysis of these sequences was then undertaken using the Bayesianmethod available in the MRBAYES package (18). This analysis utilized theWAG model of amino acid replacement with a gamma distribution of among-siterate variation. Chains were run for 10 million generations (with a 10% burn in),at which point all of the parameter estimates had converged. The level of supportfor each node is provided by Bayesian posterior probability (BPP) values.
Nucleotide sequence accession numbers. The GenBank accession numbers forthe sequences newly acquired are designated GU815994 to GU816024 and areindicated in Tables 1, 2 and 6.
RESULTS
Identification of lyssaviruses based on two successive Patho-genID microarray generations using a systematic BLASTstrategy. To test whether PathogenID microarrays, and specif-ically the prototype tiled regions, could be used for the iden-
tification of a broad number of viral variants without relying onpredetermined hybridization patterns, representative animalviruses from the family Rhabdoviridae (including unassigned ortentatively classified rhabdoviruses according to ICTVdb) werestudied. The capability of these RMAs to identify and discrim-inate between near phylogenetic neighbors was first testedusing one sequence of the genus Lyssavirus (strain PV, geno-type or species 1) tiled on the first generation of the Patho-genID microarray (Table 1). It was possible to use BLAST to
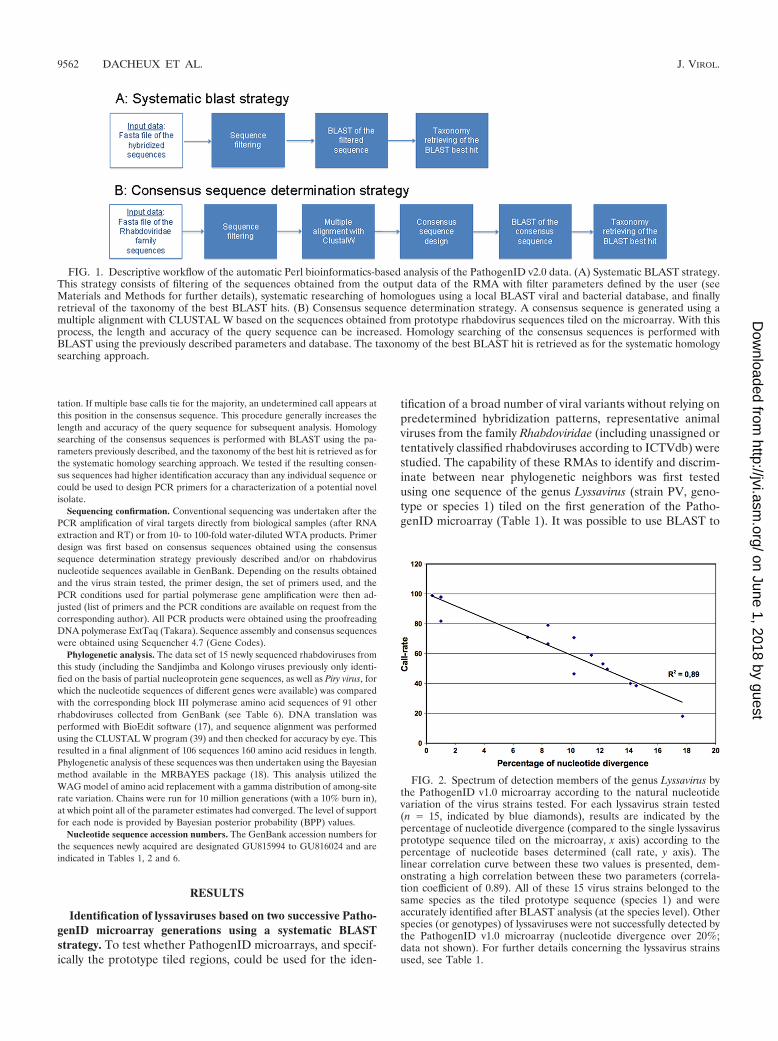
FIG. 1. Descriptive workflow of the automatic Perl bioinformatics-based analysis of the PathogenID v2.0 data. (A) Systematic BLAST strategy.This strategy consists of filtering of the sequences obtained from the output data of the RMA with filter parameters defined by the user (seeMaterials and Methods for further details), systematic researching of homologues using a local BLAST viral and bacterial database, and finallyretrieval of the taxonomy of the best BLAST hits. (B) Consensus sequence determination strategy. A consensus sequence is generated using amultiple alignment with CLUSTAL W based on the sequences obtained from prototype rhabdovirus sequences tiled on the microarray. With thisprocess, the length and accuracy of the query sequence can be increased. Homology searching of the consensus sequences is performed withBLAST using the previously described parameters and database. The taxonomy of the best BLAST hit is retrieved as for the systematic homologysearching approach.
FIG. 2. Spectrum of detection members of the genus Lyssavirus bythe PathogenID v1.0 microarray according to the natural nucleotidevariation of the virus strains tested. For each lyssavirus strain tested(n � 15, indicated by blue diamonds), results are indicated by thepercentage of nucleotide divergence (compared to the single lyssavirusprototype sequence tiled on the microarray, x axis) according to thepercentage of nucleotide bases determined (call rate, y axis). Thelinear correlation curve between these two values is presented, dem-onstrating a high correlation between these two parameters (correla-tion coefficient of 0.89). All of these 15 virus strains belonged to thesame species as the tiled prototype sequence (species 1) and wereaccurately identified after BLAST analysis (at the species level). Otherspecies (or genotypes) of lyssaviruses were not successfully detected bythe PathogenID v1.0 microarray (nucleotide divergence over 20%;data not shown). For further details concerning the lyssavirus strainsused, see Table 1.
9562 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
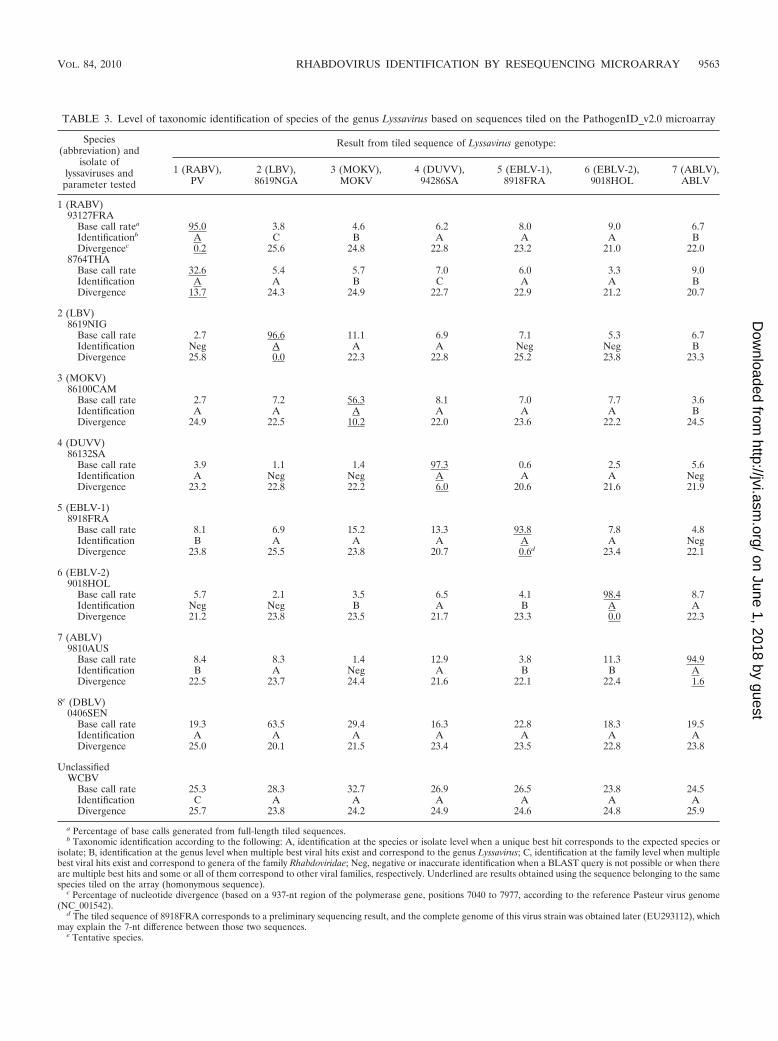
TABLE 3. Level of taxonomic identification of species of the genus Lyssavirus based on sequences tiled on the PathogenID_v2.0 microarray
Species(abbreviation) and
isolate oflyssaviruses and
parameter tested
Result from tiled sequence of Lyssavirus genotype:
1 (RABV),PV
2 (LBV),8619NGA
3 (MOKV),MOKV
4 (DUVV),94286SA
5 (EBLV-1),8918FRA
6 (EBLV-2),9018HOL
7 (ABLV),ABLV
1 (RABV)93127FRA
Base call ratea 95.0 3.8 4.6 6.2 8.0 9.0 6.7Identificationb A C B A A A BDivergencec 0.2 25.6 24.8 22.8 23.2 21.0 22.0
8764THABase call rate 32.6 5.4 5.7 7.0 6.0 3.3 9.0Identification A A B C A A BDivergence 13.7 24.3 24.9 22.7 22.9 21.2 20.7
2 (LBV)8619NIG
Base call rate 2.7 96.6 11.1 6.9 7.1 5.3 6.7Identification Neg A A A Neg Neg BDivergence 25.8 0.0 22.3 22.8 25.2 23.8 23.3
3 (MOKV)86100CAM
Base call rate 2.7 7.2 56.3 8.1 7.0 7.7 3.6Identification A A A A A A BDivergence 24.9 22.5 10.2 22.0 23.6 22.2 24.5
4 (DUVV)86132SA
Base call rate 3.9 1.1 1.4 97.3 0.6 2.5 5.6Identification A Neg Neg A A A NegDivergence 23.2 22.8 22.2 6.0 20.6 21.6 21.9
5 (EBLV-1)8918FRA
Base call rate 8.1 6.9 15.2 13.3 93.8 7.8 4.8Identification B A A A A A NegDivergence 23.8 25.5 23.8 20.7 0.6d 23.4 22.1
6 (EBLV-2)9018HOL
Base call rate 5.7 2.1 3.5 6.5 4.1 98.4 8.7Identification Neg Neg B A B A ADivergence 21.2 23.8 23.5 21.7 23.3 0.0 22.3
7 (ABLV)9810AUS
Base call rate 8.4 8.3 1.4 12.9 3.8 11.3 94.9Identification B A Neg A B B ADivergence 22.5 23.7 24.4 21.6 22.1 22.4 1.6
8e (DBLV)0406SEN
Base call rate 19.3 63.5 29.4 16.3 22.8 18.3 19.5Identification A A A A A A ADivergence 25.0 20.1 21.5 23.4 23.5 22.8 23.8
UnclassifiedWCBV
Base call rate 25.3 28.3 32.7 26.9 26.5 23.8 24.5Identification C A A A A A ADivergence 25.7 23.8 24.2 24.9 24.6 24.8 25.9
a Percentage of base calls generated from full-length tiled sequences.b Taxonomic identification according to the following: A, identification at the species or isolate level when a unique best hit corresponds to the expected species or
isolate; B, identification at the genus level when multiple best viral hits exist and correspond to the genus Lyssavirus; C, identification at the family level when multiplebest viral hits exist and correspond to genera of the family Rhabdoviridae; Neg, negative or inaccurate identification when a BLAST query is not possible or when thereare multiple best hits and some or all of them correspond to other viral families, respectively. Underlined are results obtained using the sequence belonging to the samespecies tiled on the array (homonymous sequence).
c Percentage of nucleotide divergence (based on a 937-nt region of the polymerase gene, positions 7040 to 7977, according to the reference Pasteur virus genome(NC_001542).
d The tiled sequence of 8918FRA corresponds to a preliminary sequencing result, and the complete genome of this virus strain was obtained later (EU293112), whichmay explain the 7-nt difference between those two sequences.
e Tentative species.
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9563
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

successfully identify virus strains with approximately 18% nu-cleotide divergence compared to the prototype (Fig. 2). Thehybridization of 15 virus strains representative of the geneticdiversity found in this species indicated that a single tiledsequence was able to detect all of the variant strains belongingto the same species.
In addition, we evaluated the spectrum of detection of thesecond generation of the PathogenID microarray, which in-cluded one prototype sequence representative of each of theseven described species in the genus Lyssavirus (Table 2). Allof the isolates tested led to the correct species identificationusing a systematic BLAST strategy when hybridizing a targetbelonging to the same species that is tiled on the array (Table3). Moreover, all of the tested isolates of a known genotypewere also recognized by heterospecific tiled sequences (Table3). We also investigated the capacity of this RMA to detectmore distantly related viruses not yet classified into a species.Isolates 0406SEN and WCBV, which have been proposed torepresent new species of the genus Lyssavirus (5, 15), weresurprisingly recognized by almost all of the seven species se-quences tiled on the PathogenID v2.0 microarray (Table 3).This recognition indicates that each sequence tiled on the arrayhas the ability to identify strains that are more than 18%divergent, and up to 25.9% in some cases (Table 3). Thisanalysis also reveals that information on a strain hybridized onPathogenID v2.0 can be obtained from distinct species or iso-lates tiled on the array. Evaluation of the spectrum of detec-tion of this RMA was further extended to two other genera ofthe family Rhabdoviridae—Ephemerovirus and Vesiculovirus(Table 4). Here again, successful identification was achievedusing homospecific sequences tiled on the array, confirmingthe reliability of the identification.
In both experiments (Tables 3 and 4), low base call rate
values were obtained for several combinations of hybridizedand tiled sequences. These values were sufficient for viral iden-tification by BLAST, despite the presence of sequence reads asshort as 14 nt. This indicates that most of these short sequencescorresponded to highly conserved sequence domains. The ac-curacy of these short sequences was checked by comparisonwith those obtained by classical sequencing (data not shown).
Identification of lyssaviruses based on the consensus se-quence determination strategy. A bioinformatic workflow wasdeveloped to gather stretches of sequence reads obtained withmore or less distantly related sequences tiled on PathogenIDv2.0. The aim of this strategy was to enlarge the length of thesequence determined in order to improve the sensitivity of theBLAST analysis compared to previously described methodol-ogies (29). All of the sequence reads obtained from prototypesequences of the genus Lyssavirus (at least 12 nt long with nomore than one undetermined base, whether or not they ini-tially led to a positive BLAST identification) were used togenerate a contiguous sequence. When overlapping fragmentswere identified, a consensus sequence was generated to re-move ambiguous or undetermined base calls. The methodol-ogy used to obtain consensus sequences confirmed the speciesidentification after BLAST analysis in the case of the sevenlyssavirus nucleotide sequences used for hybridization (Table5). Moreover, these consensus sequences were found to bemore powerful in identifying unclassified or new species oflyssaviruses not tiled on the RMA than resequencing datacollected individually from each tiled sequences, as shown forstrains 0406SEN and WCBV. In both cases, an increase in thebase call rate was observed using this consensus sequencestrategy, from 63.5% (best base call rate obtained from indi-vidual prototype sequences) to 75.9% for strain 0406SEN andfrom 32.7% to 60.9% for WCBV (Tables 3 and 5). Once again,
TABLE 4. Levels of taxonomic identification of virus species among the genera Vesiculovirus and Ephemerovirus based on Vesiculovirus andEphemerovirus sequences tiled on the PathogenID_v2.0 microarray
Rhabdovirus genus and species(isolate) and parameter tested
Result from specific rhabdovirus sequence tiled
Vesiculovirus Ephemerovirus LyssavirusRABV(PV)CHPV ISFV PERV SVCV VSIV VSNJV ARV BEFV KIMV KOTV
Vesiculovirus VSIV (0503FRA)Base call ratea 1.2 4.1 1.0 1.0 98.6 2.9 0 0 0 0 0Scoreb Neg Neg Neg Neg A A Neg Neg Neg Neg Neg
EphemerovirusKIMVc (CS 368)
Base call rate 1.9 1.1 0 0 0.3 0.3 9.4 7.3 70.6 9.1 1.4Score Neg Neg Neg Neg Neg Neg Neg Neg A Neg Neg
KOTVd (Ib Ar23380, 9145NIG)Base call rate 6.6 3.8 5.7 3.2 3.7 7.2 8.8 5.2 3.4 100 2.1Score Neg Neg Neg Neg Neg C Neg Neg Neg A Neg
Lyssavirus RABV (93127FRA)Base call rate 0.3 1.2 2.6 1.5 0 0 0.1 0 0.1 2.3 95.0Score Neg Neg Neg Neg Neg Neg Neg Neg Neg Neg A
a Percentage of base calls generated from full-length tiled sequences.b Taxonomic identification according to the following: A, identification at the species or isolate level when a unique best hit corresponds to the expected species or
isolate; C, identification at the family level when multiple best viral hits exist and correspond to genera of the family Rhabdoviridae; Neg, negative or inaccurateidentification when a BLAST query is not possible or when multiple best hits exist and some or all of them correspond to other viral families. Underlined are resultsobtained using the sequence belonging to the same species or isolate tiled on the array (homonymous sequence).
c TS, tentative species according to ICTVdb.d Taxonomic classification according to reference 6.
9564 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

this increase in nucleotide base determination was associatedwith a relatively high accuracy (91.8% and 97.3% concordancebetween the consensus sequences and the reference sequencesof isolates 0406SEN and WCBV, respectively (Table 5). Tofurther demonstrate the ability of this strategy to detect andidentify novel virus species, consensus sequences were gener-ated based only on six of the seven prototype tiled sequences(excluding the homospecific sequence of the same species tiledon the array). All of the strains of the seven species tested wereaccurately and specifically identified using this restricted ap-proach (Table 5). These results indicate that the consensussequences obtained could improve the detection of a noveldomain(s) not identified using only the closest prototype se-quence tiled on the RMA.
Assessment of clinical specimens. A total of 17 brain biopsysamples originating from experimentally infected mice and var-ious clinical samples (n � 8) obtained from the National Ref-erence Centre for Rabies at the Institut Pasteur were tested forlyssavirus detection and identification using the two versions ofthe PathogenID microarray (Tables 1 and 2). These specimenswere previously collected from humans and animals with clin-ically documented encephalitis and suspected of having rabies.They were used to compare RMA results with conventionalmethods of diagnosis, including the RT-heminested PCR (RT-hnPCR) technique for the intra vitam diagnosis of rabies inhumans (13), the fluorescent-antibody test, the rabies tissueculture inoculation test, and the enzyme-linked immunosor-bent assay for the postmortem diagnosis of humans and ani-mals (8, 47). Among the eight clinical samples, most were brainbiopsy specimens collected from different rabid mammals, in-cluding a bat, a cat, a dog, and two foxes, and from a human.The two other samples comprised a saliva specimen and a skinbiopsy sample collected from two different rabid human pa-tients (Tables 1 and 2). Except for the skin biopsy case, whichwas not recognized, this comparison demonstrated a completeconcordance between our method and conventional methodsfor all of the samples tested. Hence, the accuracy of the se-quences provided with PathogenID microarray was close tothat obtained using classical sequencing (data not shown). Thefailure to detect lyssaviruses in the skin biopsy samples wasprobably due to insufficient sensitivity of the current RMAmethod, as viral RNA was only weakly detected after RT-hnPCR.
In sum, these results demonstrated that the newly developedamplification process by WTA coupled to hybridization to thePathogenID microarray allowed the detection of a large rangeof viral variants from various complex biological samples, in-cluding clinical samples (Tables 1 and 2).
Application of the RMA strategy to characterize new rhab-doviruses. Broad-spectrum detection was demonstrated usingthe consensus sequences-based analysis strategy among virusesof the family Rhabdoviridae, and the more distantly relatedviruses examined included many viruses that are not yet clas-sified as species. Accordingly, 17 different rhabdoviruses weretested by using brain samples from experimentally infectedmice (n � 16) or infected cell suspension. These viruses in-cluded four strains belonging to the genus Vesiculovirus,with Vesicular stomatitis Indiana virus (VSIV) and Boteke(BOTK), Jurona (JURV), and Porton’s (PORV) viruses, thelatter three of which are currently classified as tentative
TABLE 5. Identification of species of the genus Lyssavirus based onLyssavirus sequences tiled on the PathogenID_v2.0 microarray and
using the consensus sequence determination strategy
Lyssavirus species(abbreviation), isolate, and
parameter tested
Result obtained with analysisstrategy of use of:
Prototypesequence
only
Consensus sequence
Including alltiled
sequences
Excludingprototypesequence
1 (RABV)93127FRA
Base call ratea 95.0 96.3 32.7BLAST scoreb 791 801 38Accuracyc 100 99.9 95.9
8764THABase call rate 32.6 47.4 26.7BLAST score 46 64 31Accuracy 94.8 99.1 98.4
2 (LBV), 8619NIGBase call rate 96.6 96.4 28.1BLAST score 816 814 39Accuracy 99.9 99.9 97.7
3 (MOKV), 86100CAMBase call rate 56.3 67.4 28.4BLAST score 66 112 64Accuracy 98.2 99.8 98.5
4 (DUVV), 86132SABase call rate 97.3 97.3 18.1BLAST score 843 833 20Accuracy 99.9 99.8 96.4
5 (EBLV-1), 8918FRABase call rate 93.8 96.0 41.1BLAST score 757 807 83Accuracy 100 100 97.9
6 (EBLV-2), 9018HOLBase call rate 98.4 98.8 26.8BLAST score 871 879 44Accuracy 100 99.9 99.6
7 (ABLV), ABLVBase call rate 94.9 95.6 29.7BLAST score 749 741 40Accuracy 100 99.9 94.5
8e (DBLV), 0406SENBase call rate NAd 75.9 NABLAST score NA 82 NAAccuracy NA 91.8 NA
?, WCBVBase call rate NA 60.9 NABLAST score NA 56 NAAccuracy NA 97.3 NA
a Percentage of base calls generated from full-length tiled sequences.b BLAST score (bit score) obtained after BLAST query on a local viral and
bacterial database using the consensus sequence determination strategy with m(minimum nucleotide length) � 12 and N (maximum undetermined nucleotidecontent) � 10. Default BLAST parameters, except for the minimum word length(7 nt), the expect threshold (increased from the default of 10 to 100,000), and thelow complexity level filter (�F) turned off. All of the BLAST scores indicatecorrect identification at the species or isolate level (i.e., unique best hit corre-sponds to the expected species or isolate).
c Percentage of nucleotides correctly identified, compared to the sequence ob-tained after classical sequencing of the corresponding Lyssavirus species tested.
d NA, not applicable.e Tentative species.
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9565
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

TA
BL
E6.
Des
crip
tions
and
final
clas
sific
atio
nsof
rhab
dovi
ruse
sus
edfo
rph
ylog
enet
ican
alys
is
Gen
usan
dna
mea
(spe
cies
)U
A/T
S/U
Cb
Abb
revi
atio
nSt
rain
Prin
cipa
lhos
tsp
ecie
s/ve
ctor
cSa
mpl
eor
igin
Yr
offir
stis
olat
ion
Gen
Ban
kac
cess
ion
no.
Lys
savi
rus
Rab
ies
viru
s(1
)R
AB
V90
01F
RA
Dog
bitt
enby
bat
Fre
nch
Gui
ana
1990
EU
2931
13R
abie
svi
rus
(1)
RA
BV
9147
FR
AF
oxF
ranc
e19
91E
U29
3115
Rab
ies
viru
s(1
)R
AB
V87
43T
HA
Hum
anT
haila
nd19
83E
U29
3121
Rab
ies
viru
s(1
)R
AB
V97
04A
RG
Bat
,Tad
arid
abr
asili
ensi
sA
rgen
tina
1997
EU
2931
16R
abie
svi
rus
(1)
RA
BV
9706
CH
IV
acci
neA
GC
hina
AY
8546
63R
abie
svi
rus
(1)
RA
BV
9702
IND
Hum
anIn
dia
1997
AY
8546
65L
agos
bat
viru
s(2
)L
BV
8619
NG
AB
at,E
idol
onhe
lvum
Nig
eria
1956
EU
2931
10M
okol
avi
rus
(3)
MO
KV
MO
KV
Cat
Zim
babw
e19
81N
C_0
0642
9M
okol
avi
rus
(3)
MO
KV
8610
0CA
MSh
rew
Cam
eroo
n19
74E
U29
3117
Mok
ola
viru
s(3
)M
OK
V86
101R
CA
Rod
ent
Rep
ublic
ofC
entr
alA
fric
a19
81E
U29
3118
Duv
enha
gevi
rus
(4)
DU
VV
9428
6SA
Bat
,Min
iopt
erus
spec
ies
Sout
hA
fric
a19
81E
U29
3120
Duv
enha
gevi
rus
(4)
DU
VV
8613
2SA
Hum
anSo
uth
Afr
ica
1971
EU
2931
19E
urop
ean
bat
lyss
aviru
s1
(5)
EB
LV
-189
18F
RA
Bat
,Ept
esic
usse
rotin
usF
ranc
e19
89E
U29
3112
Eur
opea
nba
tly
ssav
irus
1(5
)E
BL
V-1
0812
0FR
AB
at,E
ptes
icus
sero
tinus
Fra
nce
2008
EU
6265
51E
urop
ean
bat
lyss
aviru
s2
(6)
EB
LV
-290
18H
OL
Bat
,Myo
tisda
sycn
eme
Net
herl
ands
1986
EU
2931
14E
urop
ean
bat
lyss
aviru
s2
(6)
EB
LV
-293
37SW
IB
at,M
yotis
daub
ento
nii
Switz
erla
nd19
93A
Y85
4657
Aus
tral
ian
bat
lyss
aviru
s(7
)A
BL
VA
BL
hH
uman
Aus
tral
ia19
86A
F41
8014
Aus
tral
ian
bat
lyss
aviru
s(7
)A
BL
VA
BL
b(S
6-12
56)
Bat
,Sac
cola
imus
spec
ies
Aus
tral
ia19
96N
C_0
0324
3D
akar
bat
lyss
avir
us(8
�pro
pose
d�)
UC
DB
LV
0406
SEN
(AnD
4244
3)B
at,E
idol
onhe
lvum
Sene
gal
1985
EU
2931
08
Dak
arba
tly
ssav
irus
(8�p
ropo
sed�
)U
CD
BL
VK
E13
1B
at,E
idol
onhe
lvum
Ken
ya20
07E
U25
9198
Irku
tvi
rus
UC
IRK
VB
at,M
urin
ale
ucog
aste
rR
ussi
a20
02E
F61
4260
Oze
rnoe
viru
sU
CH
uman
Rus
sia
2007
FJ9
0510
5A
rava
nvi
rus
UC
AR
AV
Bat
,Myo
tisbl
ythi
Kyr
gyzs
tan
1991
EF
6142
59K
huja
ndvi
rus
UC
KH
UV
Bat
,Myo
tism
ysta
cinu
sT
ajik
ista
n20
01E
F61
4261
Wes
tC
auca
sian
bat
viru
sU
CW
CB
VB
at,M
inio
pter
ussc
hrei
bers
iiR
ussi
a20
02E
F61
4258
Ves
icul
oviru
sC
hand
ipur
avi
rus
CH
PVI
6535
14H
uman
;do
mes
tican
imal
sd;h
edge
hog,
Ate
lerix
spec
ies;
dipt
eran
,Phl
ebot
omus
spec
ies
Indi
a19
65A
J810
083
Coc
alvi
rus
CO
CV
TR
VL
4023
3L
ives
tock
,equ
ine,
bovi
ne;m
ites,
Gig
anto
lael
aps
spec
ies
Tri
nida
dan
dT
obag
o,T
rini
dad
1961
EU
3736
57
Isfa
han
viru
sIS
FV
9102
6-16
7D
ipte
ran,
Phl
ebot
omus
papa
tasi
Iran
1975
AJ8
1008
4P
iryvi
rus
PIR
YV
BeA
n24
232
(041
3BR
E)
Hum
an;o
poss
um,P
hila
nder
opos
sum
Bra
zil
1960
GU
8160
23V
esic
ular
stom
atiti
sN
ewJe
rsey
viru
sV
SNJV
VSV
NJ-
OSe
vera
lliv
esto
cksp
ecie
s,in
clud
ing
Bos
taur
usan
deq
uine
s;se
vera
ldip
tera
nsp
ecie
s,in
clud
ing
Cul
exni
grip
alpu
s,C
ulic
oide
ssp
ecie
s,an
dM
anso
nia
indu
bita
ns
Uta
h19
49A
Y07
4804
Ves
icul
arst
omat
itis
New
Jers
eyvi
rus
VSN
JVV
SVN
J-H
Seve
rall
ives
tock
spec
ies,
incl
udin
gSu
ssc
rofa
;se
vera
ldip
tera
nsp
ecie
s,in
clud
ing
Cul
exni
grip
alpu
s,C
ulic
oide
ssp
ecie
s,an
dM
anso
nia
indu
bita
ns
Geo
rgia
1952
AY
0748
03
Ves
icul
arst
omat
itis
Indi
ana
viru
sV
SIV
Mud
d-Su
mm
ers
(MS)
Bov
ine,
Bos
taur
usIn
dian
a19
25E
U84
9003
Ves
icul
arst
omat
itis
Indi
ana
viru
sV
SIV
85C
LB
Bov
ine
Col
ombi
a19
85A
F47
3865
Ves
icul
arst
omat
itis
Indi
ana
viru
sV
SIV
98C
OE
Equ
ine
Col
orad
o19
98A
F47
3864
Ves
icul
arst
omat
itis
Ala
goas
viru
sV
SAV
Indi
ana
3E
quin
eliv
esto
ck(m
ule)
,Bos
taur
us;d
ipte
rans
,P
hleb
otom
ussp
ecie
sB
razi
l19
64E
U37
3658
Juro
navi
ruse
TS
JUR
VB
eAr
4057
8(0
414B
RE
)D
ipte
ran,
Hem
agog
ussp
egaz
zini
iB
razi
l19
62G
U81
6024
Peri
net
viru
sT
SPE
RV
Ar
Mg
802
Dip
tera
ns,A
noph
eles
cous
tani
,Cul
exan
tenn
atus
,Cul
exgr
.pip
iens
,Man
soni
aun
iform
is,P
hleb
otom
usbe
rent
ensi
s
Mad
agas
car
1978
AY
8546
52
Pike
fry
rhab
dovi
rus
TS
PFR
VF
4F
ish,
Eso
xlu
cius
Net
herl
ands
1972
FJ8
7282
7
9566 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Scop
htha
lmus
max
imus
rhab
dovi
rus
UC
SMR
VQ
Z-2
005
Fis
h,Sc
opht
halm
usm
axim
usC
hina
?A
Y89
5167
Spri
ngvi
rem
iaof
carp
viru
sT
SSV
CV
Fija
n_ce
ll(V
R-1
390,
isol
ated
from
fat
head
min
now
cells
)
Fis
h,C
ypri
nus
carp
ioY
ugos
lavi
a19
71A
J318
079
Spri
ngvi
rem
iaof
carp
viru
sT
SSV
CV
Fija
n_tis
sue
(VR
-139
0,is
olat
edfr
omtis
sues
ofdi
seas
edco
mm
onca
rp)
Fis
h,C
ypri
nus
carp
ioY
ugos
lavi
a19
71U
1810
1
Spri
ngvi
rem
iaof
carp
viru
sT
SSV
CV
BJ0
505-
2F
ish,
Cyp
rinu
sca
rpio
Chi
na20
05E
U17
7782
Eph
emer
oviru
sA
dela
ide
Riv
ervi
rus
AR
VD
PP61
Bov
ine,
Bos
taur
usA
ustr
alia
1981
AY
8546
35B
errim
ahvi
rus
BR
MV
DPP
63B
ovin
e,B
osta
urus
Aus
tral
ia19
81A
Y85
4636
Bov
ine
ephe
mer
alfe
ver
viru
sB
EF
VC
s19
33B
ovin
e,B
osta
urus
Aus
tral
ia19
73A
Y85
4641
Bov
ine
ephe
mer
alfe
ver
viru
sB
EF
VC
s42
Dip
tera
n,A
noph
eles
banc
rofti
Aus
tral
ia19
75A
Y85
4639
Bov
ine
ephe
mer
alfe
ver
viru
sB
EF
VB
B77
21B
ovin
e,B
osta
urus
Aus
tral
ia19
68N
C_0
0252
6K
imbe
rley
viru
sT
SK
IMV
CS
368
Bov
ine,
Bos
taur
usA
ustr
alia
1980
AY
8546
37K
oton
kan
viru
sU
AK
OT
VIb
Ar
2338
0D
ipte
ran,
Cul
icoi
des
spec
ies
Nig
eria
1967
AY
8546
38A
lmpi
war
grou
pA
lmpi
war
viru
sU
AA
LM
VM
RM
4059
Mam
mal
s,d
bovi
ne,e
quin
e,ov
ine,
kang
aroo
,ba
ndic
oot,
hum
an;b
irds
;dliz
ard,
Abl
epha
rus
bout
onii
virg
atus
and
othe
rsk
inks
d
Aus
tral
ia19
66A
Y85
4645
Cha
rlev
ille
viru
sU
AC
HV
VC
h98
24H
uman
;ddi
pter
an,P
hleb
otom
usan
dL
asio
hele
asp
ecie
sA
ustr
alia
1969
AY
8546
44
Cha
rlev
ille
viru
sU
AC
HV
VC
h98
47H
uman
;ddi
pter
an,P
hleb
otom
usan
dL
asio
hele
asp
ecie
sA
ustr
alia
1969
AY
8546
72
Hum
pty
doo
viru
sU
AH
DO
OV
CS
79D
ipte
rans
,Las
iohe
lea
spec
ies,
Cul
icoi
des
mar
ksi
Aus
tral
ia19
75A
Y85
4643
Har
tPa
rkgr
oup
Ban
gora
nvi
ruse
UA
BG
NV
Dak
ArB
2053
(042
4RC
A)
Bir
d,T
urdu
slib
onya
nus;
dipt
eran
,Cul
expe
rfus
cus
Cen
tral
Afr
ican
Rep
ublic
1969
GU
8160
10
Fla
nder
svi
rus
UA
FL
AN
V61
-748
4B
irds
,Sei
urus
auro
capi
llus,
Age
laiu
sph
oeni
ceus
;dip
tera
ns,C
ulis
eta
mel
anur
a,C
ulex
spec
ies
New
Yor
k19
61A
F52
3199
Kam
ese
viru
seU
AK
AM
VM
P61
86(0
8343
OU
G)
Dip
tera
ns,A
edes
afric
anus
,Cul
exsp
ecie
s,in
clud
ing
Cul
exan
nulio
ris
Uga
nda
1967
GU
8160
11
Mos
suri
lvir
use
UA
MO
SVSA
Ar
1995
(041
8MO
Z)
Bir
ds,A
ndro
padu
svi
rens
,Col
iusp
asse
rm
acro
urus
;dip
tera
ns,A
edes
abno
rmal
is,
Cul
exsp
ecie
s,in
clud
ing
Cul
exsi
tiens
Moz
ambi
que
1959
GU
8160
12
Nga
inga
nvi
rus
UA
NG
AV
MR
M14
556
Mam
mal
s,d
wal
labi
es,k
anga
roos
,bov
ines
;di
pter
an,C
ulic
oide
sbr
evita
rsis
Aus
tral
ia19
70A
Y85
4649
Parr
yC
reek
viru
sU
APC
RV
OR
189
Dip
tera
n,C
ulex
annu
liros
tris
Aus
tral
ia19
72A
Y85
4647
Port
on’s
viru
seT
S(V
SV)
POR
V16
43(0
416M
AL
)D
ipte
ran,
Man
soni
aun
iform
isM
alay
sia
(Sar
awak
)?
GU
8160
13W
onga
belv
irus
UA
WO
NV
CS
264
Sea
bird
s;d
dipt
eran
,Cul
icoi
des
aust
ropa
lpal
isA
ustr
alia
1979
AY
8546
48
Le
Dan
tec
grou
pF
ukuo
kavi
rus
UA
(Ker
nC
anyo
nG
roup
)F
UK
VF
UK
-11
Bov
ine;
dipt
eran
,Cul
icoi
des
punc
tatu
s,C
ulex
trita
enio
rhyn
chus
Japa
n19
82A
Y85
4651
Keu
ralib
avi
ruse
UA
KE
UV
Dak
AnD
5314
(971
5SE
N,0
420S
EN
)R
oden
ts,T
ater
asp
ecie
s,in
clud
ing
Tat
era
kem
pi,T
ater
illus
spec
ies
Sene
gal
1968
GU
8160
21
Le
Dan
tec
viru
sU
AL
DV
Dak
HD
763
Hum
anSe
nega
l19
65A
Y85
4650
Nko
lbis
son
viru
seU
A(K
ern
Can
yon
Gro
up)
NK
OV
Ar
YM
31/6
5(0
425C
AM
)D
ipte
ran,
Aed
essp
ecie
s,E
retm
apod
ites
spec
ies,
incl
udin
gE
retm
apod
ites
leuc
opou
s,C
ulex
tele
silla
Cam
eroo
n19
65G
U81
6022
Con
tinue
don
follo
win
gpa
ge
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9567
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

TA
BL
E6—
Con
tinue
d
Gen
usan
dna
mea
(spe
cies
)U
A/T
S/U
Cb
Abb
revi
atio
nSt
rain
Prin
cipa
lhos
tsp
ecie
s/ve
ctor
cSa
mpl
eor
igin
Yr
offir
stis
olat
ion
Gen
Ban
kac
cess
ion
no.
Mou
ssa
grou
pM
ouss
avi
rus
UC
MO
USV
C23
Dip
tera
n,C
ulex
dece
nsIv
ory
Coa
st20
04F
J985
748
Mou
ssa
viru
sU
CM
OU
SVD
24D
ipte
ran,
Cul
exsp
ecie
sIv
ory
Coa
st20
04F
J985
749
Sand
jimba
grou
pB
imbo
viru
seU
AB
BO
VD
akA
nB10
54d
(971
6RC
A)
Bir
d,E
uple
ctes
afer
Cen
tral
Afr
ican
Rep
ublic
1970
GU
8160
16
Bot
eke
viru
seT
S(V
SV)
BT
KV
Dak
ArB
1077
(041
7RC
A)
Dip
tera
n,C
oqui
lletti
dia
mac
ulip
enni
sC
entr
alA
fric
anR
epub
lic19
68G
U81
6014
Gar
bavi
ruse
UA
GA
RV
Dak
AnB
439a
(042
2RC
A)
Bir
ds,C
oryt
horn
iscr
ista
ta,N
ecta
rina
pulc
hella
Cen
tral
Afr
ican
Rep
ublic
1970
GU
8160
18
Kol
ongo
viru
sU
AK
OL
VD
akA
nB10
94d
(971
7RC
A)
Bir
ds,E
uple
ctes
afer
,Plo
ceus
cucu
llatu
sC
entr
alA
fric
anR
epub
lic19
70G
U81
6020
Nas
oule
viru
seU
AN
ASV
Dak
AnB
4289
a(0
410R
CA
)B
ird,
And
ropa
dus
vire
nsC
entr
alA
fric
anR
epub
lic19
73G
U81
6017
Oak
-Val
evi
rus
UA
OV
RV
CS
1342
Fer
ralp
igs;
ddi
pter
an,A
edes
vigi
lax,
Cul
exsp
ecie
s,in
clud
ing
(Cul
exed
war
dsi)
Aus
tral
ia19
81A
Y85
4670
Oua
ngo
viru
seU
AO
UA
VD
akA
nB15
82a
(971
8RC
A)
Bir
d,P
loce
usm
elan
ocep
halu
sC
entr
alA
fric
anR
epub
lic19
70G
U81
6015
Sand
jimba
viru
sU
ASJ
AV
Dak
AnB
373d
(072
44R
CA
)B
ird,
Acr
ocep
halu
ssc
hoen
obae
nus
Cen
tral
Afr
ican
Rep
ublic
1970
GU
8160
19
Sigm
agr
oup
Dro
soph
ilaaf
finis
sigm
avi
rus
UC
DA
ffSV
10D
ipte
rian
,Dro
soph
ilaaf
finis
New
Con
nect
icut
2007
GQ
4109
80D
roso
phila
mel
anog
aste
rsi
gma
viru
sU
ASI
GM
AV
(DM
elSV
)A
P30
Dip
teri
an,D
roso
phila
mel
anog
aste
rF
lori
da20
05N
C_0
1313
5
Dro
soph
ilam
elan
ogas
ter
sigm
avi
rus
UA
SIG
MA
V(D
Mel
SV)
HA
P23
Dip
teri
an,D
roso
phila
mel
anog
aste
rF
ranc
e?
GQ
3752
58
Dro
soph
ilaob
scur
asi
gma
viru
sU
CD
Obs
SV10
AD
ipte
rian
,Dro
soph
ilaob
scur
aU
nite
dK
ingd
om20
07G
Q41
0979
Sini
star
grou
pSi
nipe
rca
chua
tsir
habd
ovir
usU
CSC
RV
Fis
h,Si
nipe
rca
chua
tsi
Chi
na?
NC
_008
514
Star
ryflo
unde
rrh
abdo
viru
sU
CSF
RV
Fis
h,P
latic
hthy
sst
ella
tus
Was
hing
ton
2000
AY
4506
44
Tib
roga
rgan
grou
pT
ibro
garg
anvi
rus
UA
TIB
VC
S13
2B
ovin
es,d
wat
erbu
ffalo
es,c
attle
;dip
tera
n,C
ulic
oide
sbr
evita
rsis
Aus
tral
ia19
76A
Y85
4646
Tup
aia
viru
sT
S(V
SV)
TU
PVT
RV
1591
Tre
esh
rew
,Tup
aia
bela
nger
iT
haila
nd?
NC
_007
020
Nov
irhab
dovi
rus
Hira
me
rhab
dovi
rus
HIR
RV
CA
9703
Fis
h,in
clud
ing
cult
ured
Kor
ean
flou
nder
s,P
aral
icht
hys
oliv
aceu
s,P
leco
glos
sus
altiv
elis
,M
ilio
mac
roce
phal
us,a
ndSe
bast
esin
erm
is
Kor
ea19
97N
C_0
0509
3
Infe
ctio
ushe
mat
opoi
etic
necr
osis
viru
sIH
NV
HV
7601
AB
2316
60
Infe
ctio
ushe
mat
opoi
etic
necr
osis
viru
sIH
NV
WR
AC
Fis
h,in
clud
ing
salm
onid
Onc
orhy
nchu
sts
chaw
ytsc
haId
aho
NC
_001
652
Snak
ehea
drh
abdo
viru
sSH
RV
Fis
h,in
clud
ing
Oph
icep
halu
sst
riat
us,C
laria
sbr
atac
hus,
and
Oxy
eleo
tism
arm
orat
usT
haila
ndN
C_0
0090
3
Vira
lhem
orrh
agic
sept
icem
iavi
rus
VH
SVK
RR
V98
22F
ish,
Japa
nese
flou
nder
Japa
nA
B17
9621
Vira
lhem
orrh
agic
sept
icem
iavi
rus
VH
SV07
-71
Fis
h,O
ncor
hync
hus
myk
iss
Fra
nce
AJ2
3339
6V
iralh
emor
rhag
icse
ptic
emia
viru
sV
HSV
JF00
Ehi
1F
ish,
Par
alic
hthy
sol
ivac
eus
Japa
n20
00A
B49
0792
Vira
lhem
orrh
agic
sept
icem
iavi
rus
VH
SV14
-58
Fis
h,O
ncor
hync
hus
myk
iss
Fra
nce
AF
1438
63
Nuc
leor
habd
oviru
sM
aize
mos
aic
viru
sM
MV
Plan
ts(h
ost)
,Gra
min
ae,i
nclu
ding
Zea
may
s;he
mip
tera
ns(v
ecto
r),D
elph
acid
aeU
nite
dSt
ates
NC
_005
975
9568 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Ric
eye
llow
stun
tvi
rus
RY
SVPl
ant
(hos
t),O
ryza
sativ
a;ho
mop
tera
ns(v
ecto
r),C
icad
ellid
aeN
C_0
0374
6
Sonc
hus
yello
wne
tvi
rus
SYN
VPl
ants
(hos
t),A
ster
acea
e,in
clud
ing
Sonc
hus
oler
aceu
s;he
mip
tera
ns(v
ecto
r),A
phid
idae
NC
_001
615
Iran
ian
mai
zem
osai
cnu
cleo
rhab
dovi
rus
UC
IMM
NV
Plan
ts(h
ost)
,Gra
min
ae,i
nclu
ding
Zea
may
s;he
mip
tera
ns(v
ecto
r),D
elph
acid
aeIr
anN
C_0
1154
2
Mai
zefin
est
reak
viru
sU
CM
FSV
Plan
ts(h
ost)
,Gra
min
ae,i
nclu
ding
Zea
may
s;ho
mop
tera
ns(v
ecto
r),C
icad
ellid
aeG
eorg
ia19
99N
C_0
0597
4
Orc
hid
fleck
viru
sfU
CO
FV
SoPl
ants
(hos
t),O
rchi
dace
ae,i
nclu
ding
Cym
bidi
umsp
ecie
s;ac
arid
(vec
tor)
,B
revi
palp
usca
lifor
nicu
s
Japa
nN
C_0
0960
9
Tar
ove
inch
loro
sis
viru
sU
CT
aVC
VPl
ant
(hos
t),C
oloc
asia
escu
lent
aF
ijiIs
land
sN
C_0
0694
2
Cyt
orha
bdov
irus
Bar
ley
yello
wst
riate
mos
aic
BY
SMV
Zan
jan-
1Pl
ants
(hos
t),G
ram
inae
,inc
ludi
ngT
ritic
umsp
ecie
s;he
mip
tera
ns(v
ecto
r),D
elph
acid
aeIr
anF
J665
628
Let
tuce
necr
otic
yello
ws
viru
sL
NY
V31
8Se
vera
l(ho
st)
plan
tfa
mili
esan
dsp
ecie
s,in
clud
ing
Alli
umsa
tivum
and
Lac
tuca
sativ
a;he
mip
tera
ns(v
ecto
r),A
phid
idae
Aus
tral
iaN
C_0
0764
2
Nor
ther
nce
real
mos
aic
viru
sN
CM
VPl
ants
(hos
t),G
ram
inae
,inc
ludi
ngH
orde
umvu
lgar
e;he
mip
tera
ns(v
ecto
r),D
elph
acid
aeJa
pan
NC
_002
251
Stra
wbe
rry
crin
kle
viru
sSC
VH
B-A
1Pl
ant
(hos
t),F
raga
ria
spec
ies;
hem
ipte
rans
(vec
tor)
,Aph
idid
aeA
Y33
1389
Stra
wbe
rry
crin
kle
viru
sSC
V37
-2Pl
ant
(hos
t),F
raga
ria
spec
ies;
hem
ipte
rans
(vec
tor)
,Aph
idid
aeA
Y33
1388
Stra
wbe
rry
crin
kle
viru
sSC
V37
-1Pl
ant
(hos
t),F
raga
ria
spec
ies;
hem
ipte
rans
(vec
tor)
,Aph
idid
aeA
Y33
1387
Let
tuce
yello
wm
ottle
viru
sU
CL
YM
oVPl
ant
(hos
t),L
actu
casa
tiva
Fra
nce
1998
NC
_011
532
Taa
stru
pgr
oup,
Taa
stru
pvi
rus
UC
TV
Hem
ipte
ran
(pot
entia
lvec
tor)
,Psa
mm
otet
tixal
ienu
sF
ranc
e19
96A
Y42
3355
aN
ames
ofvi
ruse
sin
italic
sco
rres
pond
toap
prov
edsp
ecie
sac
cord
ing
toth
eIn
tern
atio
nalC
omm
ittee
onT
axon
omy
ofV
irus
esda
taba
se.
bU
A,u
nass
igne
d;T
S,te
ntat
ive;
UC
,unc
lass
ified
(not
foun
din
ICT
Vdb
).c
Inbo
ldis
the
host
spec
ies
from
whi
chth
evi
rus
was
first
isol
ated
,if
that
info
rmat
ion
isav
aila
ble.
dSe
rolo
gica
ldet
ectio
non
ly.
eF
irst
iden
tifica
tion
base
don
nucl
eic
acid
dete
rmin
atio
nan
dcl
assi
ficat
ion
base
don
phyl
ogen
ican
alys
is(t
his
stud
y).
fA
lso
tent
ativ
ely
clas
sifie
din
toth
ene
wge
nus
Dic
horh
abdo
viru
sac
cord
ing
toits
unus
ualb
ipar
tite
geno
me
(20)
.
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9569
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from
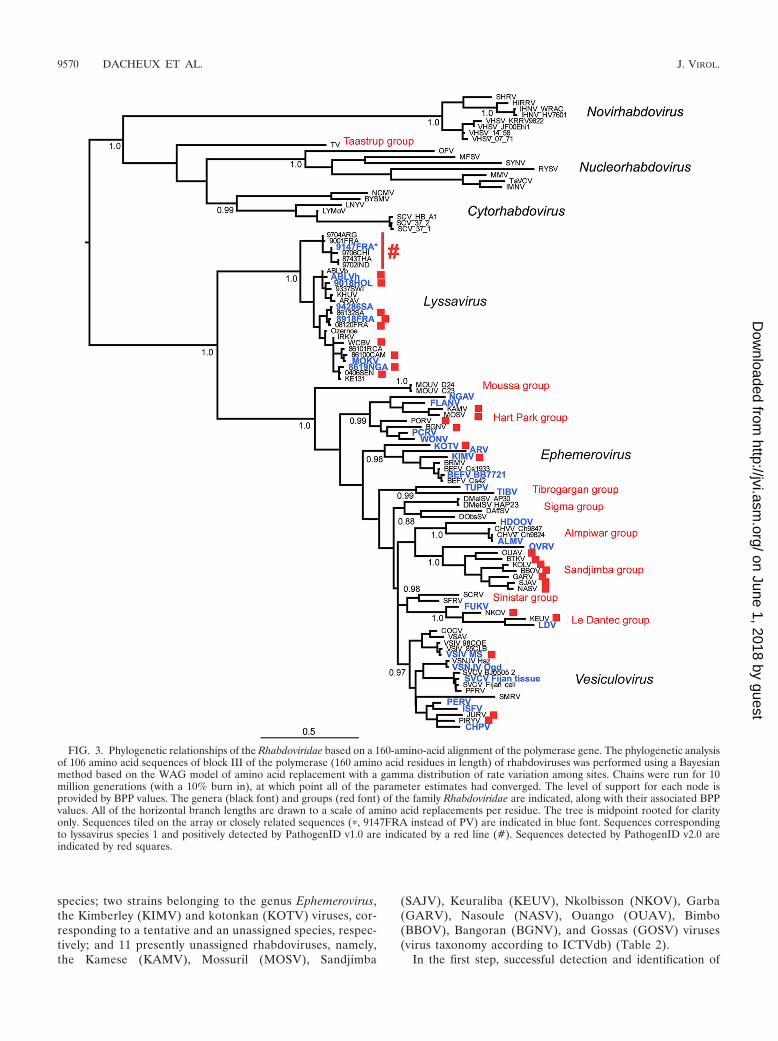
species; two strains belonging to the genus Ephemerovirus,the Kimberley (KIMV) and kotonkan (KOTV) viruses, cor-responding to a tentative and an unassigned species, respec-tively; and 11 presently unassigned rhabdoviruses, namely,the Kamese (KAMV), Mossuril (MOSV), Sandjimba
(SAJV), Keuraliba (KEUV), Nkolbisson (NKOV), Garba(GARV), Nasoule (NASV), Ouango (OUAV), Bimbo(BBOV), Bangoran (BGNV), and Gossas (GOSV) viruses(virus taxonomy according to ICTVdb) (Table 2).
In the first step, successful detection and identification of
FIG. 3. Phylogenetic relationships of the Rhabdoviridae based on a 160-amino-acid alignment of the polymerase gene. The phylogenetic analysisof 106 amino acid sequences of block III of the polymerase (160 amino acid residues in length) of rhabdoviruses was performed using a Bayesianmethod based on the WAG model of amino acid replacement with a gamma distribution of rate variation among sites. Chains were run for 10million generations (with a 10% burn in), at which point all of the parameter estimates had converged. The level of support for each node isprovided by BPP values. The genera (black font) and groups (red font) of the family Rhabdoviridae are indicated, along with their associated BPPvalues. All of the horizontal branch lengths are drawn to a scale of amino acid replacements per residue. The tree is midpoint rooted for clarityonly. Sequences tiled on the array or closely related sequences (�, 9147FRA instead of PV) are indicated in blue font. Sequences correspondingto lyssavirus species 1 and positively detected by PathogenID v1.0 are indicated by a red line (#). Sequences detected by PathogenID v2.0 areindicated by red squares.
9570 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

these viruses using the PathogenID v2.0 microarray was ob-tained for 12 (70.5%) out of 17 viruses; accurate taxonomicpositioning—that is, within the family Rhabdoviridae—was alsoachieved, and for some, the corresponding genus (when avail-able) was also matched accurately (data not shown). In thesecond step, specific and consensus primers were designedbased on the stretches of sequences identified by the microar-ray using the consensus sequence determination strategy andthen subsequently used for PCR and classical sequencing ofthe amplified target nucleotide sequences. For four (GARV,NASV, OUAV, and BBOV) of the five rhabdoviruses notdetected by the microarray, a region of 1,000 nt of the poly-merase gene encompassing that tiled on the array was success-fully amplified by PCR and sequenced using the primers de-scribed above. The only exception was the GOSV isolate,which remained undetected by either the microarray or PCR.Further, two other rhabdoviruses not previously tested with thePathogenID v2.0 microarray—Kolongo virus (KOLV, an un-classified species) and Piry virus (PIRYV, a vesiculovirus)—were also amplified and sequenced using these primers.
All of the newly sequenced nucleotide regions of the poly-merase gene were further translated into protein sequencesand aligned with 88 sequences of animal or plant rhabdovi-ruses obtained from GenBank, producing a total data set of106 sequences 160 amino acid residues in length. A Bayesianphylogenetic analysis of these sequences tentatively distin-guished 15 groups of viruses based on their strongly supportedmonophyly (Table 6 and Fig. 3). The members of the sixgenera—Ephemerovirus, Lyssavirus, Vesiculovirus, Cytorhab-dovirus, Nucleorhabdovirus, and Novirhabdovirus—fall intowell-supported monophyletic groups (BPP value, �0.97) (Fig.3). Interestingly, this analysis suggested the existence of at leastnine more groups of currently unclassified rhabdoviruses,which reflect important biological characteristics of the virusesin question. Five of these groups have been proposed previ-ously and were further supported by our analysis (data avail-able at the CRORA database website [http://www.pasteur.fr/recherche/banques/CRORA/]) (6, 27; reviewed in reference7). The first group, tentatively named the Hart Park group,contains the previously described Parry Creek (PCRV),Wongabel (WONV), Flanders (FLANV), and Ngaingan(NGAV) viruses added to the newly identified viruses BGNV,KAMV, MOSV, and PORV. This group has a large distribu-tion that encompasses Africa, Australia, Malaysia, and theUnited States. These viruses have a wide host range, as theyhave been found to infect dipterans, birds, and mammals. Thesecond group is the Almpiwar group, containing four mem-bers—two strains of Charleville (CHVV) virus, i.e., CHVV_Ch9824 and CHVV_Ch9847—and the Almpiwar (ALMV) andHumpty doo (HDOOV) viruses. Viruses of this group wereisolated in Australia and are associated with infections ofdipterans and lizards but also birds and mammals, includinghumans. Another group, herein referred to as the Le Dantecgroup, was also seen to form a distinct cluster with Le Dantecvirus (LDV), Fukuoka virus (FUKV), and the two newly mo-lecularly identified viruses KEUV and NKOV. Members wereisolated in Japan and Africa, where they were shown toinfect dipterans and mammals, including humans. Thefourth group has been tentatively named the Tibrogargangroup and includes the Tupaia (TUPV) and Tibrogargan
(TIBV) viruses. These viruses were isolated in SoutheastAsia, Australia, and New Guinea from dipterans and mam-mals. Finally, we observed the Sigma group as previouslydescribed (27). It includes Drosophila affinis (DAffSV), Dro-sophila obscura (DObsSV), and two strains of Drosophilamelanogaster (SIGMAV_AP30 and SIGMAV_HAP23)sigma viruses, infecting Drosophila flies which were found inthe United States and Europe.
In addition, four other tentative groups of viruses are newlydescribed in this study. The Sandjimba group includes the firstmolecularly classified viruses BBOV, BTKV, NASV, GARV,and OUAV and the previously described Oak-Vale virus(OVRV), SJAV, and KOLV (identification of the latter twobased only on a limited region of the nucleoprotein gene).These viruses were isolated from birds and dipterans from theCentral African Republic and Australia (data available at http://www.pasteur.fr/recherche/banques/CRORA/) (6, 9). Interest-ingly, all of the African members of this group clusteredclosely, whereas the sole Australian virus was more divergent,suggesting a potential geographical segregation. Second, theSinistar group includes the Siniperca chuatsi rhabdovirus(SCRV) isolated from mandarin fish in China (37) and thestarry flounder rhabdovirus (SFRV) from starry flounder inthe United States (30). These two viruses appear to be moreclosely related to the Le Dantec group than to viruses in thegenus Vesiculovirus, in which several other fish rhabdovirusesare classified. The third one is the Moussa group, including twoisolates of Moussa virus (MOUV_D24 and MOUV_C23) col-lected from mosquitoes in Ivory Coast (34). Finally, a phylo-genetic analysis suggests the presence of another group withinthe plant rhabdoviruses: the Taastrup group, which comprisesthe single isolate Taastrup virus (TV) isolated from leafhop-pers (Psammotettix alienus) originally collected in France (28).All of these groups were strongly supported by the Bayesiananalysis (BPP value, �0.98), with the exception of the Sigmagroup, which exhibits a BPP value of 0.88.
In addition, classification of some uncharacterized rhab-doviruses from our phylogenetic analysis diverged from thatpreviously suggested by serology (according to ICTVdb) andwill probably need further investigation to determine theirprecise taxonomic positions within the family Rhabdoviridae(Table 6) (9, 38). In particular, PORV and BTKV, previ-ously identified as vesiculoviruses, were included within theHart Park and Sandjimba groups, respectively, and NKOVwas classified into the Le Dantec group instead of the KernCanyon group. Moreover, in contrast to a previous phylo-genetic study (22), TUPV was found to be more closelyrelated to TIBV than to any other isolates in the Sandjimbagroup. Finally, our study confirmed the previous serology-based classification of JURV and the recently identifiedScophthalmus maximus rhabdovirus (SMRV) within the Ve-siculovirus genus (38, 49).
DISCUSSION
We have analyzed the capacity of viral detection and iden-tification of two versions of a newly described RMA, termedPathogenID, which was designed specifically for multiplepathogen detection using database similarity searching (1). Toevaluate this microarray, we focused on one of the largest and
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9571
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

most diverse viral families described to date, the Rhabdoviridae(ICTVdb, reviewed in reference 7). All of the virus strainstested (except WCBV) were extracted from biological samplesand amplified using a nonspecific and unbiased WTA step aspreviously described (3). Rhabdovirus-targeted sequenceswere selected among blocks of conservation within the poly-merase gene (6). This region was chosen so as to encompass asufficient number of homologous but also polymorphic sites.The key advantage of this RMA strategy is that it does notrequire a specific match between the samples tested and tiledsequences; indeed, mismatches add value as they allow precisetyping of the unknown genetic resequenced element. In ourcase, the conserved nature of the target region of the polymer-ase gene (block III) and the capability of detection of the RMAallows a precise taxonomic identification (i.e., family, genus,species) and also provides key information on phylogeneticrelationships for some unclassified, unassigned, or tentativespecies of rhabdoviruses. For example, results obtained by thePathogenID v1.0 microarray evaluation demonstrated thatmost of the intraspecies nucleotide diversity found in the genusLyssavirus can be covered by a single prototype sequence tiledon the microarray. Using the second version of PathogenIDwhich included one prototype sequence of each of the sevenspecies recognized thus far within the genus Lyssavirus, weextended the spectrum of detection of the RMA to potentiallyall of the known or unknown lyssaviruses (i.e., positive detec-tion of virus isolates presenting up to 25.9% nucleotide diver-gence with the tiled sequence considered), which is greaterthan that previously reported (24–26, 43, 44).
This study also indicates that accurate viral identificationmay still be possible even when only shorter sequences areobtained from individual tiled prototype sequences. Indeed,taken individually, these short stretches of nucleotide sequencecould not give positive results during the initial BLAST query.However, when used in the consensus sequence determinationstrategy employed here, they improved the identification ofvirus strains distantly related with that tiled on the RMA. Forexample, we were able to test and detect rhabdoviruses basedon sequence data obtained with tiled sequences that originatedfrom other viral genera.
The strategy developed here also allowed the potential de-tection of genetically diverse rhabdoviruses previously identi-fied or unknown by using a limited number of sequences tiledon the microarray. Using the PathogenID v2.0 microarray, wewere able to identify 30 rhabdoviruses in total. This included 12viruses currently unclassified, unassigned, or assigned as ten-tative species within the family Rhabdoviridae (according toICTVdb). Moreover, the consensus sequence-based analysis ofRMA results was shown to be accurate compared to sequencesobtained through classical sequencing (Table 5 and data notshown). Sequence data provided by the PathogenID v2.0 mi-croarray were also extremely helpful in the design of specificprimers to further sequence the targeted region of the viralpolymerase gene of some other rhabdoviruses. Finally, thisapproach allowed us to undertake the largest phylogeneticanalysis of the family Rhabdoviridae (Table 6 and Fig. 3), eventhough it is important to note that the list of viruses andpotential taxa described here is still incomplete and more vi-ruses will clearly be characterized in the near future. Despitethese phylogenetic divisions, all of the viruses included in these
proposed groups are closely related to vesiculoviruses andephemeroviruses and were found to infect a large spectrum ofanimals, included dipterans and mammals (and previously re-ferred to as the dimarhabdovirus supergroup (6) but also liz-ards (Almpiwar group), birds (especially the Sandjimba groupbut also with Hart Park group), and fish (Sinistar group) (Ta-ble 6).
Although promising, inadequate sequence selection for thedesign of the RMA, and consequently a lack of coverage of theviral sequence space, represents an important limitation. Aproper selection of blocks of conserved sequence across taxo-nomic subdivisions in the viral world could be similarly definedand targeted by the RMA assay, and in doing so improve thedetection power of this tool and therein greatly aid in theidentification of members of the family Rhabdoviridae or evenother viral families. The results presented here validated theusefulness of the design methodology. It emphasizes the gainin identification using a consensus sequence strategy determi-nation compared to a systematic BLAST strategy (29). Indeed,this strategy allows us to use and accurately analyze the RMAoutput data, even if only short subsequences with a high basecall rate are obtained. It provides an informative alternative tocurrent molecular methods, such as classical or multiplex PCR,for the rapid identification of viral pathogens. It is currentlybeing applied to assist in a new generation of RMA aimed atthe detection and identification of genetically diverse and un-known viral pathogens and more broadly of any virus presentin a clinical specimen. In contrast to conventional microarrays,it is not limited by the requirement of prior knowledge of theidentities of viruses present in biological samples and it is notrestricted to the detection of a limited number of candidateviruses. As such, this strategy has a great potential for beingimplemented as a high-throughput platform to identify moredivergent viral organisms. This technology could be especiallyuseful in clinical diagnosis or in surveillance programs fordetecting uncharacterized viral pathogens or highly variablevirus strains in the same taxonomic genus or family, which isfrequently the case for RNA viruses (2). The potential appli-cations of such a methodology therefore appear to be numer-ous: differential diagnostics for illnesses with multiple potentialcauses (for example, central nervous diseases like encephalitisand meningitis), tracking of emergent pathogens, the distinc-tion of biological threats from harmless phylogenetic neigh-bors, and the broader-scale investigation of biodiversity in theviral world.
ACKNOWLEDGMENTS
This work was supported by grant UC1 AI062613 (G. C. Kennedy)from the U.S. National Institute of Allergy and Infectious Diseases,National Institute of Health; the Programme Transversal de Recher-che (PTR DEVA 246) from the Institut Pasteur, Paris, France; theEuropean Commission, through the VIZIER Integrated Project(LSHG-CT-2004-511966); and the Institut Pasteur International Net-work Actions Concertees InterPasteuriennes (2003/687). We thank thesponsorship of the Total-Institut Pasteur for financial support.
We are grateful to D. Blondel, H. Zeller, and the CRORA databasefor having provided some of the rhabdovirus isolates tested in thisstudy. We are also grateful to the technical staff of the Genotyping ofPathogens and Public Health Technological Platform for their pa-tience and their excellent work in the sequencing of the differentrhabdoviruses.
9572 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

REFERENCES
1. Berthet, N., P. Dickinson, I. Filliol, A. K. Reinhardt, C. Batejat, T. Vallaeys,K. A. Kong, C. Davies, W. Lee, S. Zhang, Y. Turpaz, B. Heym, G. Coralie, L.Dacheux, A. M. Burguiere, H. Bourhy, I. G. Old, J. M. Manuguerra, S. T.Cole, and G. C. Kennedy. 2007. Massively parallel pathogen identificationusing high-density microarrays. Microb. Biotechnol. 1:79–86.
2. Berthet, N., I. Leclercq, A. Dublineau, S. Shigematsu, A. M. Burguiere, C.Filippone, A. Gessain, and J. C. Manuguerra. 2010. High-density rese-quencing DNA microarrays in public health emergencies. Nat. Biotech-nol. 28:25–27.
3. Berthet, N., A. K. Reinhardt, I. Leclercq, S. van Ooyen, C. Batejat, P.Dickinson, R. Stamboliyska, I. G. Old, K. A. Kong, L. Dacheux, H. Bourhy,G. C. Kennedy, C. Korfhage, S. T. Cole, and J. C. Manuguerra. 2008. Phi29polymerase based random amplification of viral RNA as an alternative torandom RT-PCR. BMC Mol. Biol. 9:77.
4. Bodrossy, L., and A. Sessitsch. 2004. Oligonucleotide microarrays in micro-bial diagnostics. Curr. Opin. Microbiol. 7:245–254.
5. Botvinkin, A. D., E. M. Poleschuk, I. V. Kuzmin, T. I. Borisova, S. V.Gazaryan, P. Yager, and C. E. Rupprecht. 2003. Novel lyssaviruses isolatedfrom bats in Russia. Emerg. Infect. Dis. 9:1623–1625.
6. Bourhy, H., J. A. Cowley, F. Larrous, E. C. Holmes, and P. J. Walker. 2005.Phylogenetic relationships among rhabdoviruses inferred using the L poly-merase gene. J. Gen. Virol. 86:2849–2858.
7. Bourhy, H., A. Gubala, R. P. Weir, and D. Boyle. 2008. Animal rhabdovi-ruses, p. 111–121. In B. W. J. Mahy and M. H. V. Van Regenmortel (ed.),Encyclopedia of virology, vol. 1. Elsevier, Oxford, United Kingdom.
8. Bourhy, H., P. E. Rollin, J. Vincent, and P. Sureau. 1989. Comparative fieldevaluation of the fluorescent-antibody test, virus isolation from tissue cul-ture, and enzyme immunodiagnosis for rapid laboratory diagnosis of rabies.J. Clin. Microbiol. 27:519–523.
9. Calisher, C. H., N. Karabatsos, H. Zeller, J. P. Digoutte, R. B. Tesh, R. E.Shope, A. P. Travassos da Rosa, and T. D. St. George. 1989. Antigenicrelationships among rhabdoviruses from vertebrates and hematophagousarthropods. Intervirology 30:241–257.
10. Chiu, C. Y., A. A. Alizadeh, S. Rouskin, J. D. Merker, E. Yeh, S. Yagi, D.Schnurr, B. K. Patterson, D. Ganem, and J. L. DeRisi. 2007. Diagnosis of acritical respiratory illness caused by human metapneumovirus by use of apan-virus microarray. J. Clin. Microbiol. 45:2340–2343.
11. Chiu, C. Y., A. L. Greninger, K. Kanada, T. Kwok, K. F. Fischer, C. Runckel,J. K. Louie, C. A. Glaser, S. Yagi, D. P. Schnurr, T. D. Haggerty, J. Parson-net, D. Ganem, and J. L. DeRisi. 2008. Identification of cardioviruses relatedto Theiler’s murine encephalomyelitis virus in human infections. Proc. Natl.Acad. Sci. U. S. A. 105:14124–14129.
12. Chiu, C. Y., A. Urisman, T. L. Greenhow, S. Rouskin, S. Yagi, D. Schnurr,C. Wright, W. L. Drew, D. Wang, P. S. Weintrub, J. L. Derisi, and D. Ganem.2008. Utility of DNA microarrays for detection of viruses in acute respiratorytract infections in children. J. Pediatr. 153:76–83.
13. Dacheux, L., J. M. Reynes, P. Buchy, O. Sivuth, B. M. Diop, D. Rousset, C.Rathat, N. Jolly, J. B. Dufourcq, C. Nareth, S. Diop, C. Iehle, R. Rajerison,C. Sadorge, and H. Bourhy. 2008. A reliable diagnosis of human rabies basedon analysis of skin biopsy specimens. Clin. Infect. Dis. 47:1410–1417.
14. Delarue, M., O. Poch, N. Tordo, D. Moras, and P. Argos. 1990. An attemptto unify the structure of polymerases. Protein Eng. 3:461–467.
15. Delmas, O., E. C. Holmes, C. Talbi, F. Larrous, L. Dacheux, C. Bouchier,and H. Bourhy. 2008. Genomic diversity and evolution of the lyssaviruses.PLoS One 3:e2057.
16. Hacia, J. G. 1999. Resequencing and mutational analysis using oligonucle-otide microarrays. Nat. Genet. 21:42–47.
17. Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignmenteditor and analysis program for Windows 95/98/NT. Nucleic Acids Symp.Ser. 41:95–98.
18. Huelsenbeck, J. P., and F. Ronquist. 2001. MRBAYES: Bayesian inferenceof phylogenetic trees. Bioinformatics 17:754–755.
19. Kistler, A., P. C. Avila, S. Rouskin, D. Wang, T. Ward, S. Yagi, D. Schnurr,D. Ganem, J. L. DeRisi, and H. A. Boushey. 2007. Pan-viral screening ofrespiratory tract infections in adults with and without asthma reveals unex-pected human coronavirus and human rhinovirus diversity. J. Infect. Dis.196:817–825.
20. Kondo, H., T. Maeda, Y. Shirako, and T. Tamada. 2006. Orchid fleck virusis a rhabdovirus with an unusual bipartite genome. J. Gen. Virol. 87:2413–2421.
21. Kothapalli, R., S. J. Yoder, S. Mane, and T. P. Loughran, Jr. 2002. Microar-ray results: how accurate are they? BMC Bioinformatics 3:22.
22. Kuzmin, I. V., G. J. Hughes, and C. E. Rupprecht. 2006. Phylogeneticrelationships of seven previously unclassified viruses within the familyRhabdoviridae using partial nucleoprotein gene sequences. J. Gen. Virol.87:2323–2331.
23. Leski, T. A., B. Lin, A. P. Malanoski, Z. Wang, N. C. Long, C. E. Meador, B.Barrows, S. Ibrahim, J. P. Hardick, M. Aitichou, J. M. Schnur, C. Tibbetts,and D. A. Stenger. 2009. Testing and validation of high density resequencingmicroarray for broad range biothreat agents detection. PLoS One 4:e6569.
24. Lin, B., K. M. Blaney, A. P. Malanoski, A. G. Ligler, J. M. Schnur, D.Metzgar, K. L. Russell, and D. A. Stenger. 2007. Using a resequencingmicroarray as a multiple respiratory pathogen detection assay. J. Clin. Mi-crobiol. 45:443–452.
25. Lin, B., A. P. Malanoski, Z. Wang, K. M. Blaney, A. G. Ligler, R. K. Rowley,E. H. Hanson, E. von Rosenvinge, F. S. Ligler, A. W. Kusterbeck, D. Metz-gar, C. P. Barrozo, K. L. Russell, C. Tibbetts, J. M. Schnur, and D. A.Stenger. 2007. Application of broad-spectrum, sequence-based pathogenidentification in an urban population. PLoS One 2:e419.
26. Lin, B., Z. Wang, G. J. Vora, J. A. Thornton, J. M. Schnur, D. C. Thach,K. M. Blaney, A. G. Ligler, A. P. Malanoski, J. Santiago, E. A. Walter, B. K.Agan, D. Metzgar, D. Seto, L. T. Daum, R. Kruzelock, R. K. Rowley, E. H.Hanson, C. Tibbetts, and D. A. Stenger. 2006. Broad-spectrum respiratorytract pathogen identification using resequencing DNA microarrays. GenomeRes. 16:527–535.
27. Longdon, B., D. J. Obbard, and F. M. Jiggins. 2010. Sigma viruses fromthree species of Drosophila form a major new clade in the rhabdovirusphylogeny. Proc. Biol. Sci. 277:35–44.
28. Lundsgaard, T. 1997. Filovirus-like particles detected in the leafhopperPsammotettix alienus. Virus Res. 48:35–40.
29. Malanoski, A. P., B. Lin, Z. Wang, J. M. Schnur, and D. A. Stenger. 2006.Automated identification of multiple micro-organisms from resequencingDNA microarrays. Nucleic Acids Res. 34:5300–5311.
30. Mork, C., P. Hershberger, R. Kocan, W. Batts, and J. Winton. 2004. Isolationand characterization of a rhabdovirus from starry flounder (Platichthys stel-latus) collected from the northern portion of Puget Sound, Washington,USA. J. Gen. Virol. 85:495–505.
31. Paez, J. G., M. Lin, R. Beroukhim, J. C. Lee, X. Zhao, D. J. Richter, S.Gabriel, P. Herman, H. Sasaki, D. Altshuler, C. Li, M. Meyerson, and W. R.Sellers. 2004. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. NucleicAcids Res. 32:e71.
32. Palacios, G., P. L. Quan, O. J. Jabado, S. Conlan, D. L. Hirschberg, Y. Liu,J. Zhai, N. Renwick, J. Hui, H. Hegyi, A. Grolla, J. E. Strong, J. S. Towner,T. W. Geisbert, P. B. Jahrling, C. Buchen-Osmond, H. Ellerbrok, M. P.Sanchez-Seco, Y. Lussier, P. Formenty, M. S. Nichol, H. Feldmann, T.Briese, and W. I. Lipkin. 2007. Panmicrobial oligonucleotide array for diag-nosis of infectious diseases. Emerg. Infect. Dis. 13:73–81.
33. Poch, O., I. Sauvaget, M. Delarue, and N. Tordo. 1989. Identification of fourconserved motifs among the RNA-dependent polymerase encoding ele-ments. EMBO J. 8:3867–3874.
34. Quan, P. L., S. Junglen, A. Tashmukhamedova, S. Conlan, S. K. Hutchi-son, A. Kurth, H. Ellerbrok, M. Egholm, T. Briese, F. H. Leendertz, andW. I. Lipkin. 2010. Moussa virus: a new member of the Rhabdoviridaefamily isolated from Culex decens mosquitoes in Cote d’Ivoire. VirusRes. 147:17–24.
35. Quan, P. L., G. Palacios, O. J. Jabado, S. Conlan, D. L. Hirschberg, F. Pozo,P. J. Jack, D. Cisterna, N. Renwick, J. Hui, A. Drysdale, R. Amos-Ritchie, E.Baumeister, V. Savy, K. M. Lager, J. A. Richt, D. B. Boyle, A. Garcia-Sastre,I. Casas, P. Perez-Brena, T. Briese, and W. I. Lipkin. 2007. Detection ofrespiratory viruses and subtype identification of influenza A viruses byGreeneChipResp oligonucleotide microarray. J. Clin. Microbiol. 45:2359–2364.
36. Taitt, C. R., A. P. Malanoski, B. Lin, D. A. Stenger, F. S. Ligler, A. W.Kusterbeck, G. P. Anderson, S. E. Harmon, L. C. Shriver-Lake, S. K. Pol-lack, D. M. Lennon, F. Lobo-Menendez, Z. Wang, and J. M. Schnur. 2008.Discrimination between biothreat agents and ‘near neighbor’ species using aresequencing array. FEMS Immunol. Med. Microbiol. 54:356–364.
37. Tao, J. J., G. Z. Zhou, J. F. Gui, and Q. Y. Zhang. 2008. Genomic sequenceof mandarin fish rhabdovirus with an unusual small non-transcriptionalORF. Virus Res. 132:86–96.
38. Tesh, R. B., A. P. Travassos Da Rosa, and J. S. Travassos Da Rosa. 1983.Antigenic relationship among rhabdoviruses infecting terrestrial vertebrates.J. Gen. Virol. 64(Pt. 1):169–176.
39. Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W:improving the sensitivity of progressive multiple sequence alignment throughsequence weighting, position-specific gap penalties and weight matrix choice.Nucleic Acids Res. 22:4673–4680.
40. Vora, G. J., C. E. Meador, D. A. Stenger, and J. D. Andreadis. 2004. Nucleicacid amplification strategies for DNA microarray-based pathogen detection.Appl. Environ. Microbiol. 70:3047–3054.
41. Wang, D., L. Coscoy, M. Zylberberg, P. C. Avila, H. A. Boushey, D. Ganem,and J. L. DeRisi. 2002. Microarray-based detection and genotyping of viralpathogens. Proc. Natl. Acad. Sci. U. S. A. 99:15687–15692.
42. Wang, D., A. Urisman, Y. T. Liu, M. Springer, T. G. Ksiazek, D. D. Erdman,E. R. Mardis, M. Hickenbotham, V. Magrini, J. Eldred, J. P. Latreille, R. K.Wilson, D. Ganem, and J. L. DeRisi. 2003. Viral discovery and sequencerecovery using DNA microarrays. PLoS Biol. 1:E2.
43. Wang, Z., L. T. Daum, G. J. Vora, D. Metzgar, E. A. Walter, L. C. Canas,A. P. Malanoski, B. Lin, and D. A. Stenger. 2006. Identifying influenzaviruses with resequencing microarrays. Emerg. Infect. Dis. 12:638–646.
44. Wang, Z., A. P. Malanoski, B. Lin, C. Kidd, N. C. Long, K. M. Blaney, D. C.
VOL. 84, 2010 RHABDOVIRUS IDENTIFICATION BY RESEQUENCING MICROARRAY 9573
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

Thach, C. Tibbetts, and D. A. Stenger. 2008. Resequencing microarray probedesign for typing genetically diverse viruses: human rhinoviruses and entero-viruses. BMC Genomics 9:577.
45. Wilson, W. J., C. L. Strout, T. Z. DeSantis, J. L. Stilwell, A. V. Carrano, andG. L. Andersen. 2002. Sequence-specific identification of 18 pathogenic mi-croorganisms using microarray technology. Mol. Cell. Probes 16:119–127.
46. Xiong, Y., and T. H. Eickbush. 1990. Origin and evolution of retroelementsbased upon their reverse transcriptase sequences. EMBO J. 9:3353–3362.
47. Xu, G., P. Weber, Q. Hu, H. Xue, L. Audry, C. Li, J. Wu, and H. Bourhy.
2007. A simple sandwich ELISA (WELYSSA) for the detection of lyssavirusnucleocapsid in rabies suspected specimens using mouse monoclonal anti-bodies. Biologicals 35:297–302.
48. Yoo, S. M., J. Y. Choi, J. K. Yun, J. K. Choi, S. Y. Shin, K. Lee, J. M. Kim,and S. Y. Lee. 2010. DNA microarray-based identification of bacterial andfungal pathogens in bloodstream infections. Mol. Cell. Probes 24:44–52.
49. Zhang, Q. Y., J. J. Tao, L. Gui, G. Z. Zhou, H. M. Ruan, Z. Q. Li, and J. F.Gui. 2007. Isolation and characterization of Scophthalmus maximus rhab-dovirus. Dis. Aquat. Organ. 74:95–105.
9574 DACHEUX ET AL. J. VIROL.
on June 1, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from


















