
Appendix: Automated Methods for Structure Comparison Basic problem: how are any two given structures...
18
Appendix: Automated Methods for Structure Comparison • Basic problem: how are any two given structures to be automatically compared in a meaningful way? • How are distant relationships to be recognized? program method DALI distance matrix comparison (basis for FSSP structural classification) SSAP dynamic programming (used in CATH to classify topologies) VAST convert secondary structures to vectors and align vectors
-
date post
21-Dec-2015 -
Category
Documents
-
view
212 -
download
0
Transcript of Appendix: Automated Methods for Structure Comparison Basic problem: how are any two given structures...

Appendix: Automated Methods for Structure Comparison
• Basic problem: how are any two given structures to be automatically compared in a meaningful way?
• How are distant relationships to be recognized?
program method
DALI distance matrix comparison (basis
for FSSP structural classification)
SSAP dynamic programming (used in CATH
to classify topologies)
VAST convert secondary structures to vectors
and align vectors

Structure comparison is pretty easy when two proteins are very similar
• when two proteins are so similar that the sequences can be reliably aligned, say >35% identical, structure comparison can proceed from the seq. alignment:
1. Align the sequences
sequence 1: YIREV-GKL
sequence 2: YITQVRNKA
2. Superpose the structures to minimize the RMSD for equivalent residue pairs in the alignment
note: thesestructures do notcorrespond to the sequences above

it is harder when the proteins are very
different... • if one cannot align the sequence reliably, how does one
establish which residues, if any, play equivalent structural roles in the two proteins?
• the answer is to attempt to align the structures directly in such a way that structural equivalencies in the two proteins are revealed
• we will discuss how the distance-matrix based algorithm of DALI solves this problem

Distance Matrices•2D representation of 3D structure•plot sequence against itself•identify pairs of residues which are close in space to each other•usually distance between C-alpha carbons is used•identify closeness between residues as dark parts of the matrix

Distance matrices
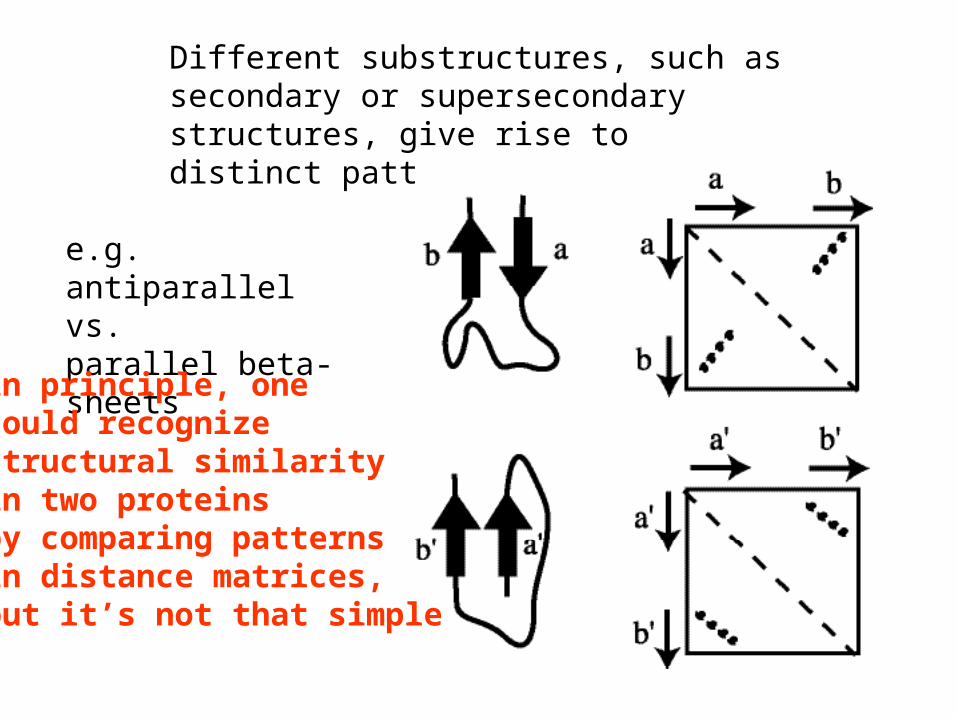
Different substructures, such as secondary or supersecondary structures, give rise to distinct patterns in the matrix
e.g. antiparallel vs.parallel beta-sheets
in principle, onecould recognizestructural similarityin two proteinsby comparing patternsin distance matrices,but it’s not that simple
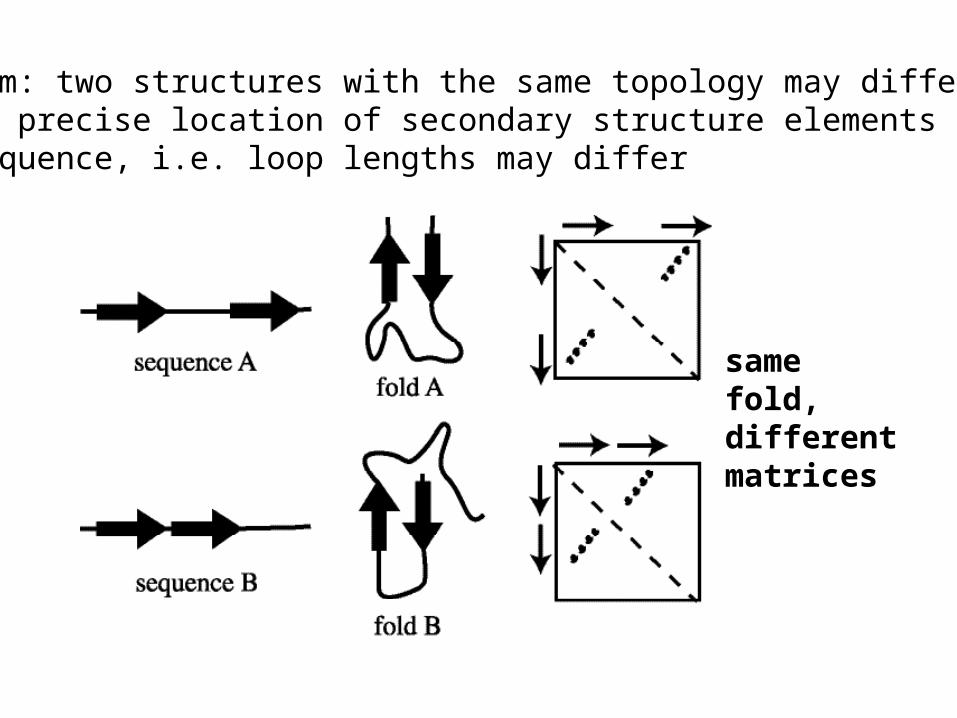
Problem: two structures with the same topology may differin the precise location of secondary structure elements alongthe sequence, i.e. loop lengths may differ
samefold,differentmatrices
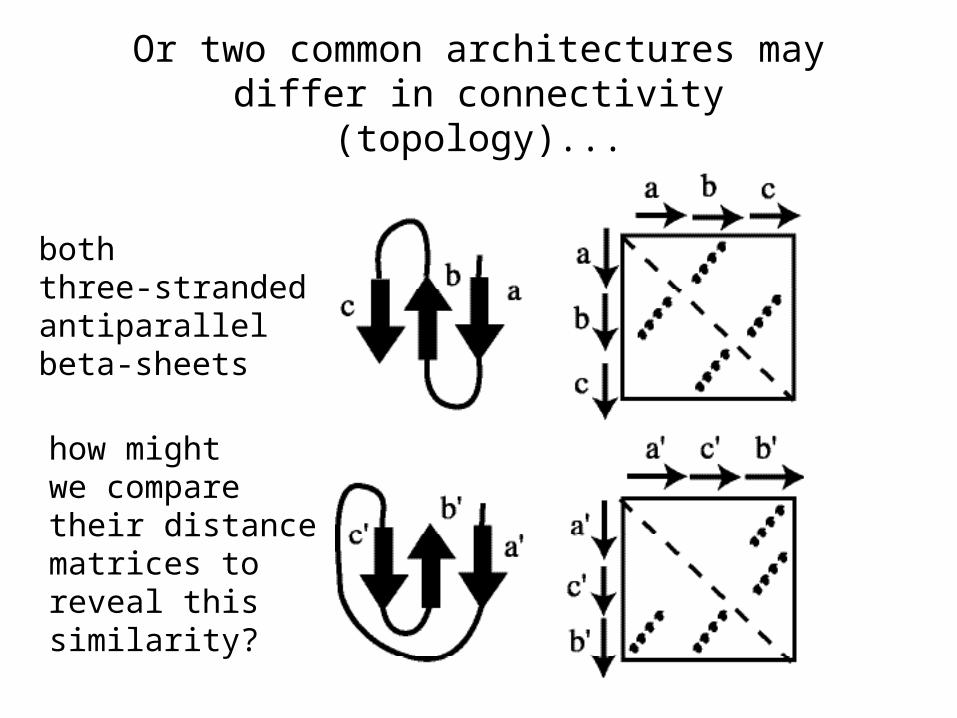
Or two common architectures may differ in connectivity (topology)...
boththree-strandedantiparallelbeta-sheets
how mightwe comparetheir distancematrices to reveal thissimilarity?

DALI algorithm
• not useful to compare entire matrices
• instead, chop distance matrices into all possible submatrices of 6x6 amino acids
• compare this set of submatrices for pattern similarities rather than comparing entire matrix
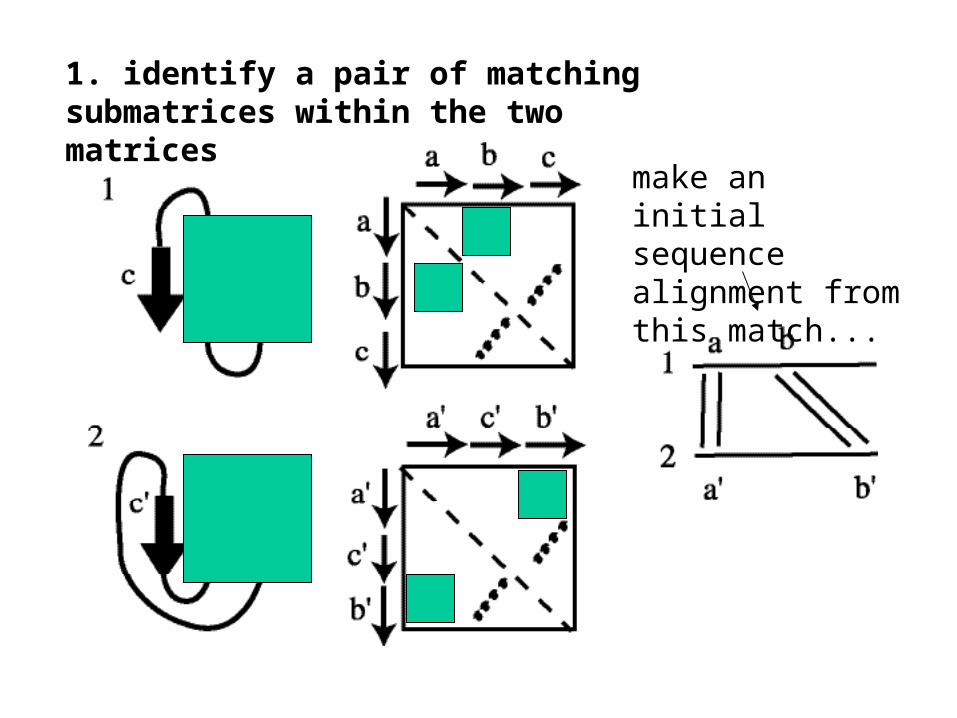
1. identify a pair of matching submatrices within the two matrices
make an initialsequence alignment from this match...

2. Identify a second pair which overlaps the first(contains one common structural element)

3. Combine overlapping pairs
overall alignmentof structurallyequivalent sequenceregions

4. Rearrange and “collapse” the matrixaccording to the aligned regions of the sequence
now the commonstructural elementsare aligned as arethe structurallyequivalent residuesin the sequence!

All together now...

The Power of DALI
• DALI is quite powerful because it can recognize architectural similarities even when topologies are different.
• It is also flexible because it can be made more topologically restrictive (i.e. no swapping of segments in chain allowed) to focus on closer relationships

FSSP uses DALI alignments to classify structures
all PDB entries
representative set of structures
representative set of domains
group domains into fold types
(clusters of similar structures)
and make set of representatives of each fold
eliminate similar sequences
divide into domains
align domains with DALI!
8320
947
1484
540

Judging DALI alignments• Z-score: how much better than average is the alignment,
i.e. how many standard deviations from the mean of a distribution of alignments of random pairs of proteins.
>16 very close, 8-16 pretty close, <8 not so close.
• RMSD: root mean square deviation of alpha carbons for the matching portion of the structures.
• LALI: length of alignment (recognizably matching portion of the structures)
• LSEQ2: total length of the sequence being matched.
• %IDE: % sequence identity between the two sequences

if you go into FSSP, and search for a particular structure, you’ll get an output of its best DALI alignments with other structures
STRID2 Z RMSD LALI LSEQ2 %IDE PROTEIN
1plc 24.4 0.0 99 99 100 Plastocyanin (cu2+, ph 6.0)
2pcy 23.4 0.2 99 99 100 Apo-plastocyanin (pH 6.0)
1bqk 12.1 2.0 89 124 29 pseudoazurin
1aac 11.0 1.9 84 104 24 amicyanin
1ibzA 9.1 2.5 83 111 19 nitrosocyanin
1qhqA 8.3 2.4 87 139 29 auracyanin
1rcy 8.2 2.5 90 151 17 rusticyanin biological_unit
1qniA 7.7 2.2 78 572 19 nitrous-oxide reductase
1kcw 7.1 2.4 81 1017 17 ceruloplasmin biological_unit
2cuaA 7.0 2.2 80 122 15 cua fragment
1nwpA 6.7 3.1 85 128 24 azurin


















