
Appendix 2 Clinical Laboratory Tests · Web viewmethods to have low user dependency remove the...
48
Common Protocol Template Patient Library v005 Common Protocol Template (CPT) Patient Library v005 Section in CPT Library Content 5.1 Inclusion Criteria: Sex Instructions and content, including decision tree for contraception, barriers, and pregnancy testing requirements 5.2. Exclusion Criteria Instructions and suggested and common text 7.1. Discontinuation of Study Intervention Common text for Liver Chemistry Stopping Criteria and QTc Stopping Criteria 10.2 Appendix 2 Clinical Laboratory Tests Instructions and text for pregnancy testing 10.4 Appendix 4 Contraceptive and Barrier Guidance and Collection of Pregnancy Information Instructions and text for contraceptive use Appendix 6: Liver Safety: Suggested Actions and Followup- Assessments [and Study Intervention Rechallenge Guidelines] Instructions and text for liver safety 5.1 Inclusion Criteria Sex 4. © 2018 TransCelerate BioPharma 1
Transcript of Appendix 2 Clinical Laboratory Tests · Web viewmethods to have low user dependency remove the...
Common Protocol Template Patient Library v005
Common Protocol Template (CPT) Patient Library v005
Section in CPT Library Content
5.1 Inclusion Criteria: Sex Instructions and content, including decision tree for contraception, barriers, and pregnancy testing requirements
5.2. Exclusion Criteria Instructions and suggested and common text
7.1. Discontinuation of Study Intervention Common text for Liver Chemistry Stopping Criteria and QTc Stopping Criteria
10.2 Appendix 2 Clinical Laboratory Tests Instructions and text for pregnancy testing
10.4 Appendix 4 Contraceptive and Barrier Guidance and Collection of Pregnancy Information
Instructions and text for contraceptive use
Appendix 6: Liver Safety: Suggested Actions and Followup- Assessments [and Study Intervention Rechallenge Guidelines]
Instructions and text for liver safety
5.1 Inclusion CriteriaSex
4.
© 2018 TransCelerate BioPharma 1

Common Protocol Template Patient Library v005
© 2018 TransCelerate BioPharma 2

Common Protocol Template Patient Library v005
<Start of common text>
a. Male Participants:
Male participants are eligible to participate if they agree to the following during the intervention period and for at least [X days/weeks, corresponding to time needed to eliminate study intervention(s) (e.g., 5 terminal half-lives) plus an additional 90 days (a spermatogenesis cycle) after the last dose of study intervention]:
Refrain from donating sperm
PLUS either: Be abstinent from heterosexual [or homosexual] intercourse as their preferred and usual
lifestyle (abstinent on a long term and persistent basis) and agree to remain abstinent
OR
Must agree to use contraception/barrier as detailed below o Agree to use a male condom
1.) 2.) when having sexual intercourse with a woman of childbearing potential who is not currently pregnant
o [Agree to use male condom when engaging in any activity that allows for passage of ejaculate to another person]
<End of common text>
<Start of common text>
a. Male Participants:
Male participants are eligible to participate if they agree to the following during the intervention period and for at least [X days/weeks, corresponding to time needed to eliminate study intervention(s) (e.g., 5 terminal half-lives) after the last dose of study intervention)]:
Refrain from donating sperm
Plus either: Be abstinent from heterosexual [or homosexual] intercourse as their preferred and usual
lifestyle (abstinent on a long term and persistent basis) and agree to remain abstinent
OR
Must agree to use contraception/barrier as detailed belowo Agree to use a male condom [and should also be advised of the benefit for a female
partner to use a highly effective method of contraception as a condom may break or leak] when having sexual intercourse with a woman of childbearing potential who is not currently pregnant
o [Agree to use male condom when engaging in any activity that allows for passage of ejaculate to another person]
© 2018 TransCelerate BioPharma 3

Common Protocol Template Patient Library v005
<End of common text>
<Start of common text>
a. Female Participants:
A female participant is eligible to participate if she is not pregnant or breastfeeding, and at least one of the following conditions applies:
o Is not a woman of childbearing potential (WOCBP)
OR
o Is a WOCBP and using a contraceptive method that is highly effective (with a failure rate of <1% per year), [preferably] with low user dependency, as described in Appendix [4] during the intervention period and for at least [X days/weeks, corresponding to the time needed to eliminate any study intervention(s) (e.g., 5 terminal half-lives) plus 30 days (a menstrual cycle)] after the last dose of study intervention [and agrees not to donate eggs (ova, oocytes) for the purpose of reproduction during this period.]. The investigator should evaluate the effectiveness of the contraceptive method in relationship to the first dose of study intervention.
oA WOCBP must have a negative highly sensitive pregnancy test ([urine or serum] as required by local regulations) within [24 hours] before the first dose of study intervention.
o [If a urine test cannot be confirmed as negative (e.g., an ambiguous result), a serum pregnancy test is required. In such cases, the participant must be excluded from participation if the serum pregnancy result is positive.]
Additional requirements for pregnancy testing during and after study intervention are located in Appendix [2].
The investigator is responsible for review of medical history, menstrual history, and recent sexual activity to decrease the risk for inclusion of a woman with an early undetected pregnancy
<End of common text>
<Start of common text>
A female participant is eligible to participate if she is not pregnant or breastfeeding, and at least one of the following conditions applies:
o Is not a woman of childbearing potential (WOCBP)
OR
o Is a WOCBP and using a contraceptive method that is highly effective (with a failure rate of <1% per year), [preferably] with low user dependency, as described in Appendix [4] during the intervention period and for at least [X days/weeks, corresponding to the time needed to eliminate any study intervention(s) (e.g., 5 terminal half-lives)] after the
© 2018 TransCelerate BioPharma 4

Common Protocol Template Patient Library v005
last dose of study intervention [and agrees not to donate eggs (ova, oocytes) for the purpose of reproduction during the study and for a period of (insert timeframe)]. The investigator should evaluate the effectiveness of the contraceptive method in relationship to the first dose of study intervention.
oA WOCBP must have a negative highly sensitive pregnancy test ([urine or serum] as required by local regulations) within [24 hours] before the first dose of study intervention.
o [If a urine test cannot be confirmed as negative (e.g., an ambiguous result), a serum pregnancy test is required. In such cases, the participant must be excluded from participation if the serum pregnancy result is positive].
Additional requirements for pregnancy testing during and after study intervention are located in Appendix [2].
The investigator is responsible for review of medical history, menstrual history, and recent sexual activity to decrease the risk for inclusion of a woman with an early undetected pregnancy
<End of common text>
<Start of common text>
a. Female Participants:
A female participant is eligible to participate if she is not pregnant or breastfeeding, and at least one of the following conditions applies:
o Is not a woman of childbearing potential (WOCBP)
OR
o Is a WOCBP and using a contraceptive method that is highly effective, with a failure rate of <1%, as described in Appendix [4] during the intervention period and for at least [X days/weeks, corresponding to the time needed to eliminate any study intervention(s) (e.g., 5 terminal half-lives)] after the last dose of study intervention. The investigator should evaluate the effectiveness of the contraceptive method in relationship to the first dose of study intervention.
o A WOCBP must have a negative highly sensitive [Appendix 2] pregnancy test (urine or serum as required by local regulations) within [specify timeframe] before the first dose of study intervention.
o [If a urine test cannot be confirmed as negative (e.g., an ambiguous result), a serum pregnancy test is required. In such cases, the participant must be excluded from participation if the serum pregnancy result is positive].
Additional requirements for pregnancy testing during and after study intervention are located in Appendix [2].
© 2018 TransCelerate BioPharma 5

Common Protocol Template Patient Library v005
The investigator is responsible for review of medical history, menstrual history, and recent sexual activity to decrease the risk for inclusion of a woman with an early undetected pregnancy
<End of common text>
<Start of common text>
b. Female Participants:
o A female participant is eligible to participate if she is not pregnant or breastfeeding, and at least one of the following conditions applies:
o Is not a woman of childbearing potential (WOCBP)
OR
o Is a WOCBP and using an acceptable contraceptive method as described in Appendix [4] during the intervention period (at a minimum until after the last dose of study intervention). The investigator should evaluate the effectiveness of the contraceptive method in relationship to the first dose of study intervention.
o A WOCBP must have a negative highly sensitive (Appendix [2]) pregnancy test (urine or serum as required by local regulations) within [specify timeframe] before the first dose of study intervention.
o [If a urine test cannot be confirmed as negative (e.g., an ambiguous result), a serum pregnancy test is required. In such cases, the participant must be excluded from participation if the serum pregnancy result is positive].
Additional requirements for pregnancy testing during and after study intervention are located in Appendix [2].
The investigator is responsible for review of medical history, menstrual history, and recent sexual activity to decrease the risk for inclusion of a woman with an early undetected pregnancy
5.2 Exclusion Criteria
MEDICAL CONDITIONS
1. Symptomatic herpes zoster within 3 months prior to screening2. Evidence of active or latent tuberculosis (TB) as documented by
medical history and examination, chest X-rays (posterior anterior and lateral), and TB testing: either a positive tuberculin skin test (TST; defined as a skin induration <5 mm at 48 to 72 hours, regardless of Bacillus Calmette-Guerin (BCG) or other vaccination history) or a positive (not indeterminate) QuantiFERON®-TB Gold test. NOTE: The choice to perform a TST or a QuantiFERON-TB Gold test will be made by the investigator according to local licensing and standard of care. The QuantiFERON-TB Gold test can only be used in countries where it is licensed, and
© 2018 TransCelerate BioPharma 6

Common Protocol Template Patient Library v005
the use of this test is dependent on previous treatment(s). This test may not be suitable if previous treatment(s) produced significant immunosuppression.
3. Significant allergies to humanized monoclonal antibodies
4. Clinically significant multiple or severe drug allergies, intolerance to topical corticosteroids, or severe post-treatment hypersensitivity reactions (including, but not limited to, erythema multiforme major, linear immunoglobulin A [IgA] dermatosis, toxic epidermal necrolysis, and exfoliative dermatitis)
5. Lymphoma, leukemia, or any malignancy within the past 5 years except for basal cell or squamous epithelial carcinomas of the skin that have been resected with no evidence of metastatic disease for 3 years
6. Breast cancer within the past 10 years
7. Abnormalities in lumbar spine previously known or determined by a screening lumbar X-ray (if conducted)
8. History of clinically significant back pain, back pathology, and/or back injury (e.g., degenerative disease, spinal deformity, or spinal surgery) that may predispose participant to complications or technical difficulty with lumbar puncture
9. Evidence or history of significant active bleeding or coagulation disorder or use of non-steroidal anti-inflammatory drugs or other drugs that affect coagulation or platelet function within 14 days prior to lumbar catheter insertion
10. Allergy to lidocaine (Xylocaine®) or its derivatives
11. Medical or surgical conditions for which lumbar puncture is contraindicated
<Start of common text for Phase I1studies>
12. Alanine transaminase (ALT) >1.5x upper limit of normal (ULN)
13. Bilirubin >1.5xULN (isolated bilirubin >1.5xULN is acceptable if bilirubin is fractionated and direct bilirubin <35%)
14. Current or chronic history of liver disease, or known hepatic or biliary abnormalities (with the exception of Gilbert's syndrome or asymptomatic gallstones)
<End of common text for Phase 1 studies>
<Start of common text for Phase 2 studies>
12. Alanine transaminase (ALT) >2.0x upper limit of normal (ULN)
13. Bilirubin >1.5xULN (isolated bilirubin >1.5xULN is acceptable if bilirubin is fractionated and direct bilirubin <35%)
14. Current or chronic history of liver disease, or known hepatic or biliary abnormalities (with the exception of Gilbert's syndrome or asymptomatic gallstones)
<End of common text for Phase 2 studies>
© 2018 TransCelerate BioPharma 7

Common Protocol Template Patient Library v005
<Start of common text for Phase 3 studies>
12. Alanine transaminase (ALT) >2x upper limit of normal (ULN)
13. Bilirubin >1.5xULN (isolated bilirubin >1.5xULN is acceptable if bilirubin is fractionated and direct bilirubin <35%)
14. Current unstable liver or biliary disease per investigator assessment defined by the presence of ascites, encephalopathy, coagulopathy, hypoalbuminaemia, esophageal or gastric varices, persistent jaundice, or cirrhosis. NOTE : Stable chronic liver disease (including Gilbert’s syndrome, asymptomatic gallstones, and chronic stable hepatitis B or C -e.g., presence of hepatitis B surface antigen [HBsAg] or positive hepatitis C antibody test result at screening or within 3 months prior to starting study intervention) is acceptable if the participant otherwise meets entry criteria
<End of common text for Phase 3 studies><Start of common text for Phase 1--4 studies in Patients>
<End of common text>
PRIOR/CONCOMITANT THERAPY
16. [Past or] intended use of over-the-counter or prescription medication [including herbal medications] within [X] days prior to dosing. [Specific medications listed in Section 6.5 may be allowed.]
17. Live vaccine(s) within 1 month prior to screening, or plans to receive such vaccines during the study
18. Treatment with biologic agents (such as monoclonal antibodies including marketed drugs) within 3 months or 5 half-lives (whichever is longer) prior to dosing.
PRIOR/CONCURRENT CLINICAL TRIAL EXPERIENCE
19. Participation in the study would result in loss of blood or blood products in excess of [X] mL within [X]
20. Exposure to more than [X] new chemical entities within 12 months prior to the first dosing day
21. Current enrollment or past participation within the last [X] days before [signing of consent] in any other clinical study involving an investigational study treatment or any other type of medical research
22 .Current enrollment or past participation within the last [X] days before [signing of consent] in this clinical study.
DIAGNOSTIC ASSESSMENTS
23 Positive pre-study drug/alcohol screen
© 2018 TransCelerate BioPharma 8

Common Protocol Template Patient Library v005
24 Positive human immunodeficiency virus (HIV) antibody test
<Start of common text for Phase 1 or 2 studies>
For studies of immunosuppressive agents
NOTE:
For potent immunosuppressive agents
25 Presence of Hepatitis B surface antigen (HBsAg) at screening or within 3 months prior to first dose of study intervention.
26 Positive Hepatitis C antibody test result at screening or within 3 months prior to starting study intervention. NOTE: Participants with positive Hepatitis C antibody due to prior resolved disease can be enrolled, only if a confirmatory negative Hepatitis C RNA test is obtained.
27 Positive Hepatitis C RNA test result at screening or within 3 months prior to first dose of study intervention. NOTE: Test is optional and participants with negative Hepatitis C antibody test are not required to also undergo Hepatitis C RNA testing
<End of common text for Phase 1 or 2 studies>
OTHER EXCLUSIONS
28 Regular alcohol consumption within [X] months prior to the study defined as: For [X sites] : an average weekly intake of > [X] units for males or > [X] units for females. One unit is equivalent to 8 g of alcohol: a half-pint (~240 mL) of beer, 1 glass (125 mL) of wine or 1 (25 mL) measure of spirits
30 Sensitivity to heparin or heparin-induced thrombocytopenia31 Sensitivity to any of the study interventions, or components thereof, or drug or other
allergy that, in the opinion of the investigator [or Medical Monitor], contraindicates participation in the study.
© 2018 TransCelerate BioPharma 9

Common Protocol Template Patient Library v005
7.1 Discontinuation of Study Intervention Liver Chemistry Stopping Criteria <Start of common text>
Study intervention will be discontinued for a participant if liver chemistry stopping criteria are met.
<Start of common text for Phase 1 studies>
Phase 1 Liver Chemistry Stopping Algorithm
Continue Study Treatment
Discontinue Study Treatment
*INR value not applicable to subjects on anticoagulants
ALT ≥3xULN
No
Yes
Must refer to Liver Safety Required Actions and Follow up Assessments section in the Appendix
Report as an SAE if possible Hy’s Law case: ALT≥3xULN and Bilirubin≥2xULN (>35% direct) or INR>1.5, if measured*
Abbreviations: ALT = alanine transaminase; INR = international normalized ratio; SAE = serious adverse event; ULN = upper limit of normal.
Liver Safety: Suggested Actions and Follow-up Assessments can be found in Appendix 6 .
<End of common text for Phase 1 studies>
© 2018 TransCelerate BioPharma 10

Common Protocol Template Patient Library v005
<Start of common text for Phase 2 studies>
Phase 2 Liver Chemistry Stopping Criteria and Increased Monitoring Algorithm
Continue Study Treatment
Discontinue Study Treatment
PlusBilirubin≥2xULN (>35%
direct) or plus
INR>1.5, if measured*PossibleHy’s Law
ALT≥3xULN ALT≥5xULN
ALT≥3xULNPlus
Symptoms of liver injury
or hypersensitivity
ALT≥3xULNbut able to monitor
weekly for 4 weeks
No
Yes
YesYes Yes
No No No
No
Yes
ALT≥3xULNpersist for4 weeks or stopping criteria
met
Yes
No
*INR value not applicable to subjects on anticoagulants
Yes
If subject to be monitored weekly must refer to Liver Safety Required Actions and Follow up Assessments section in the Appendix
Must refer to Liver Safety Required Actions and Follow up Assessments section in the Appendix
Report as an SAE if possible Hy’s Law case: ALT≥3xULN and Bilirubin≥2xULN (>35% direct) or INR>1.5, if measured*
Abbreviations: ALT = alanine transaminase; INR = international normalized ratio; SAE = serious adverse event; ULN = upper limit of normal.
Liver Safety: Suggested Actions and Follow-up Assessments can be found in Appendix [6] .
<End of common text for Phase 2 studies>
© 2018 TransCelerate BioPharma 11

Common Protocol Template Patient Library v005
<Start of common text for Phase 3-4 studies>
Phase 3-4 Liver Chemistry Stopping Criteria and Increased Monitoring Algorithm
Continue Study Treatment
Discontinue Study Treatment
Plus Bilirubin≥2xULN (>35%
direct) or plusINR>1.5, if measured*Possible Hy’s Law
ALT≥3xULNALT
≥8xULN
PlusSymptoms of
liver injuryor
hypersensitivity
No
Yes
YesYes
No No No
See algorithm for continued therapy with
increased liver chemistry monitoring
Yes
*INR value not applicable to subjects on anticoagulants
ALT ≥3xULN
but <8xULN
Yes
Must refer to Liver Safety Required Actions and Follow up Assessments section in the Appendix
Report as an SAE if possible Hy’s Law case: ALT≥3xULN and Bilirubin≥2xULN (>35% direct) or INR>1.5, if measured*
Abbreviations: ALT = alanine transaminase; INR = international normalized ratio; SAE = serious adverse event; ULN = upper limit of normal.
Liver Safety: Suggested Actions and Follow-up Assessments can be found in Appendix [6].
© 2018 TransCelerate BioPharma 12

Common Protocol Template Patient Library v005
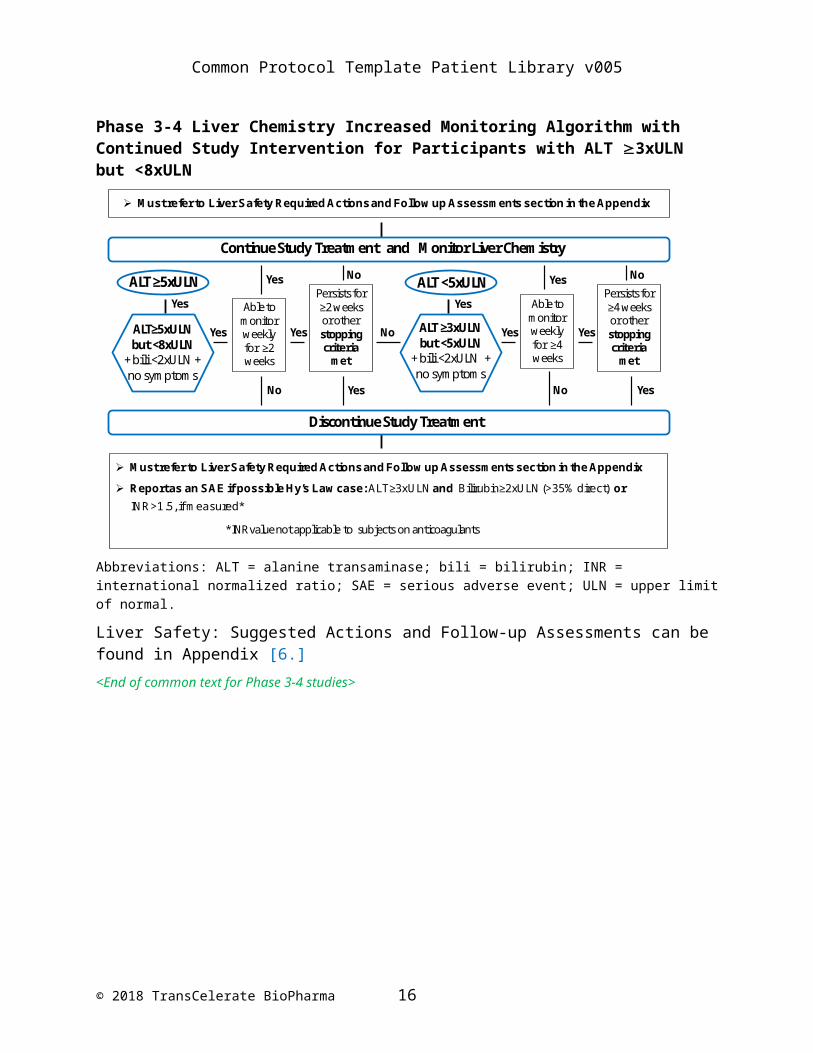
Phase 3-4 Liver Chemistry Increased Monitoring Algorithm with Continued Study Intervention for Participants with ALT 3xULN but <8xULN
Continue Study Treatment and Monitor Liver Chemistry
Discontinue Study Treatment
ALT≥5xULN but <8xULN
+ bili <2xULN +no symptoms
No
Must refer to Liver Safety Required Actions and Follow up Assessments section in the Appendix
Yes
ALT ≥3xULN but <5xULN
+ bili <2xULN +no symptoms
Able to monitor weekly for ≥2 weeks
Persists for ≥2 weeks or other stopping criteria
met
No
YesYes
Yes
NoNo
Able to monitor weekly for ≥4 weeks
Persists for ≥4 weeks or other stopping criteria
met
NoYes Yes Yes Yes
ALT ≥5xULN ALT <5xULN Yes Yes
*INR value not applicable to subjects on anticoagulants
Must refer to Liver Safety Required Actions and Follow up Assessments section in the Appendix
Report as an SAE if possible Hy’s Law case: ALT≥3xULN and Bilirubin≥2xULN (>35% direct) or INR>1.5, if measured*
Abbreviations: ALT = alanine transaminase; bili = bilirubin; INR = international normalized ratio; SAE = serious adverse event; ULN = upper limit of normal.
Liver Safety: Suggested Actions and Follow-up Assessments can be found in Appendix [6.]
<End of common text for Phase 3-4 studies>
© 2018 TransCelerate BioPharma 13

Common Protocol Template Patient Library v005
QTc Stopping Criteria<Start of common text for Phase 1-4 studies in patients>
QTc >500 msec OR Uncorrected QT >600 msec
[Change from baseline of QTc >60 msec]
For participants with underlying bundle branch block, follow the discontinuation criteria listed below:
Baseline QTc with Bundle Branch Block Discontinuation QTc Threshold with Bundle Branch Block
< 450 msec > 500 msec
450 to 480 msec ≥ 530 msec
<End of common text for Phase 1-4 studies in patients>
© 2018 TransCelerate BioPharma 14

Common Protocol Template Patient Library v005
10.2 Appendix 2 Clinical Laboratory Tests <Start of common text>
Pregnancy testing (urine or serum as required by local regulations) should be conducted at monthly intervals during intervention
Pregnancy testing (urine or serum as required by local regulations) should be conducted at the end of relevant systemic exposure plus an additional 30 days and correspond with the time frame for female participant contraception in Section 5.1, Inclusion Criteria
<End of common text>
<Start of common text>
Pregnancy testing (urine or serum as required by local regulations) should be conducted at monthly intervals during intervention
Pregnancy testing (urine or serum as required by local regulations) should be conducted at the end of relevant systemic exposure and correspond with the time frame for female participant contraception in Section 5.1, Inclusion Criteria
<End of common text>
<Start of common text>
Pregnancy testing (urine or serum as required by local regulations) should be conducted at [specify intervals based upon mechanism of action, study design etc. X] during intervention
Pregnancy testing (urine or serum as required by local regulations) should be conducted at the end of relevant systemic exposure.
<End of common text>
<Start of common text>
Additional serum or urine pregnancy tests may be performed, as determined necessary by the investigator or required by local regulation, to establish the absence of pregnancy at any time during the subject's participation in the study.
<End of common text>
© 2018 TransCelerate BioPharma 15
Common Protocol Template Patient Library v005
10.4Appendix 4 Contraceptive and Barrier Guidance and Collection of Pregnancy Information
Contraception Guidance:
© 2018 TransCelerate BioPharma 16

Common Protocol Template Patient Library v005
<Start of common text>
CONTRACEPTIVESa ALLOWED DURING THE STUDY INCLUDE:Highly Effective Methodsb That Have Low User Dependency Implantable progestogen-only hormone contraception associated with inhibition of ovulationc
Intrauterine device (IUD) Intrauterine hormone-releasing system (IUS)c
Bilateral tubal occlusion Vasectomized partner (Vasectomized partner is a highly effective contraceptive method provided that the partner is the sole sexual
partner of the woman of childbearing potential and the absence of sperm has been confirmed. If not, an additional highly effective method of contraception should be used. Spermatogenesis cycle is approximately 90 days.)
Highly Effective Methodsb That Are User Dependent Combined (estrogen- and progestogen-containing ) hormonal contraception associated with inhibition of
ovulationc
o oralo intravaginalo transdermalo injectable
Progestogen-only hormone contraception associated with inhibition of ovulationc
o oralo injectable
Sexual abstinence(Sexual abstinence is considered a highly effective method only if defined as refraining from heterosexual intercourse during the entire period of risk associated with the study intervention. The reliability of sexual abstinence needs to be evaluated in relation to the duration of the study and the preferred and usual lifestyle of the participant.)
a) Contraceptive use by men or women should be consistent with local regulations regarding the use of contraceptive methods for those participating in clinical studies.
b) Failure rate of <1% per year when used consistently and correctly. Typical use failure rates differ from those when used consistently and correctly.
c.) If hormonal contraception is prohibited, delete this footnote. If hormonal contraception efficacy is potentially decreased due to interaction with study intervention(s) add the following footnote:[Male condoms must be used in addition to hormonal contraception] If locally required, in accordance with Clinical Trial Facilitation Group (CTFG) guidelines, acceptable contraceptive methods are limited to those which inhibit ovulation as the primary mode of action.
Note: Periodic abstinence (calendar, symptothermal, post-ovulation methods), withdrawal (coitus interruptus), spermicides only, and lactational amenorrhoea method (LAM) are not acceptable methods of contraception for this study. Male condom and female condom should not be used together (due to risk of failure with friction)
<End of common text>
© 2018 TransCelerate BioPharma 17

Common Protocol Template Patient Library v005
<Start of common text>
CONTRACEPTIVESa ALLOWED DURING THE STUDY INCLUDE:Highly Effective Methodsb That Have Low User Dependency Failure rate of <1% per year when used consistently and correctly. Implantable progestogen-only hormone contraception associated with inhibition of ovulationc
Intrauterine device (IUD) Intrauterine hormone-releasing system (IUS)c
Bilateral tubal occlusion Vasectomized partner
(Vasectomized partner is a highly effective contraceptive method provided that the partner is the sole sexual partner of the woman of childbearing potential and the absence of sperm has been confirmed. If not, an additional highly effective method of contraception should be used. Spermatogenesis cycle is approximately 90 days.)
Highly Effective Methods b That Are User Dependent Failure rate of <1% per year when used consistently and correctly.Combined (estrogen- and progestogen-containing ) hormonal contraception associated with inhibition of ovulationc
oral intravaginal transdermal injectable
Progestogen-only hormone contraception associated with inhibition of ovulationc
oral injectable
Sexual abstinence(Sexual abstinence is considered a highly effective method only if defined as refraining from heterosexual intercourse during the entire period of risk associated with the study intervention. The reliability of sexual abstinence needs to be evaluated in relation to the duration of the study and the preferred and usual lifestyle of the participant.)
a) Contraceptive use by men or women should be consistent with local regulations regarding the use of contraceptive methods for those participating in clinical studies.
b) Failure rate of <1% per year when used consistently and correctly. Typical use failure rates differ from those when used consistently and correctly.
c) If hormonal contraception is prohibited, delete this footnote. If hormonal contraception efficacy is potentially decreased due to interaction with study intervention add the following footnote: [Male condoms must be used in addition to hormonal contraception]. If locally required, in accordance with Clinical Trial Facilitation Group (CTFG) guidelines, acceptable contraceptive methods are limited to those which inhibit ovulation as the primary mode of action.
Note: Periodic abstinence (calendar, symptothermal, post-ovulation methods), withdrawal (coitus interruptus), spermicides only, and lactational amenorrhoea method (LAM) are not acceptable methods of contraception. Male condom and female condom should not be used together (due to risk of failure with friction)
<End of common text>
© 2018 TransCelerate BioPharma 18

Common Protocol Template Patient Library v005
<Start of common text>
CONTRACEPTIVESa ALLOWED DURING THE STUDY INCLUDE:Highly Effective Methodsb That Have Low User Dependency Failure rate of <1% per year when used consistently and correctly. Implantable progestogen-only hormone contraception associated with inhibition of ovulationc
Intrauterine device (IUD) Intrauterine hormone-releasing system (IUS)c
Bilateral tubal occlusion Vasectomized partner
(Vasectomized partner is a highly effective contraceptive method provided that the partner is the sole sexual partner of the woman of childbearing potential and the absence of sperm has been confirmed. If not, an additional highly effective method of contraception should be used. Spermatogenesis cycle is approximately 90 days.)
Highly Effective Methodsb That Are User Dependent Failure rate of <1% per year when used consistently and correctly.Combined (estrogen- and progestogen-containing ) hormonal contraception associated with inhibition of ovulationc
oral intravaginal transdermal injectableProgestogen-only hormone contraception associated with inhibition of ovulationc
oral injectableSexual abstinence(Sexual abstinence is considered a highly effective method only if defined as refraining from heterosexual intercourse during the entire period of risk associated with the study intervention. The reliability of sexual abstinence needs to be evaluated in relation to the duration of the study and the preferred and usual lifestyle of the participant.)
a) Contraceptive use by men or women should be consistent with local regulations regarding the use of contraceptive methods for those participating in clinical studies.
b) Failure rate of <1% per year when used consistently and correctly. Typical use failure rates differ from those when used consistently and correctly.
c.) If hormonal contraception is prohibited, delete this footnote. If hormonal contraception efficacy is potentially decreased due to interaction with study intervention add the following footnote: [Male condoms must be used in addition to hormonal contraception]. If locally required, in accordance with Clinical Trial Facilitation Group (CTFG) guidelines, acceptable contraceptive methods are limited to those which inhibit ovulation as the primary mode of action.
Note: Periodic abstinence (calendar, symptothermal, post-ovulation methods), withdrawal (coitus interruptus), spermicides only, and lactational amenorrhoea method (LAM) are not acceptable methods of contraception. Male condom and female condom should not be used together (due to risk of failure with friction)
<End of common text>
© 2018 TransCelerate BioPharma 19

Common Protocol Template Patient Library v005
<Start of common text>
CONTRACEPTIVESa ALLOWED DURING THE STUDY INCLUDE:Highly Effective Methodsb That Have Low User Dependency Failure rate of <1% per year when used consistently and correctly. Implantable progestogen-only hormone contraception associated with inhibition of ovulationb
Intrauterine device (IUD) Intrauterine hormone-releasing system (IUS) b
Bilateral tubal occlusion Vasectomized partner
(Vasectomized partner is a highly effective contraceptive method provided that the partner is the sole sexual partner of the woman of childbearing potential and the absence of sperm has been confirmed. If not, an additional highly effective method of contraception should be used. Spermatogenesis cycle is approximately 90 days.)
Highly Effective Methodsb That Are User Dependent Failure rate of <1% per year when used consistently and correctly.Combined (estrogen- and progestogen-containing ) hormonal contraception associated with inhibition of ovulationc
oral intravaginal transdermal injectable
Progestogen-only hormone contraception associated with inhibition of ovulationc
oral injectable
Sexual abstinenceSexual abstinence is considered a highly effective method only if defined as refraining from heterosexual intercourse during the entire period of risk associated with the study intervention. The reliability of sexual abstinence needs to be evaluated in relation to the duration of the study and the preferred and usual lifestyle of the participant.)ACCEPTABLE METHODSd
Progestogen-only oral hormonal contraception where inhibition of ovulation is not the primary mode of action
Male or female condom with or without spermicidee
Cervical cap, diaphragm, or sponge with spermicide A combination of male condom with either cervical cap, diaphragm, or sponge with spermicide (double-
barrier methods)c
a) Contraceptive use by men or women should be consistent with local regulations regarding the use of contraceptive methods for those participating in clinical studies.
b) Failure rate of <1% per year when used consistently and correctly. Typical use failure rates differ from those when used consistently and correctly.
c.) If hormonal contraception is prohibited, delete this footnote. If hormonal contraception efficacy is potentially decreased due to interaction with study intervention add the following footnote: [Male condoms must be used in addition to hormonal contraception]. If locally required, in accordance with Clinical Trial Facilitation Group (CTFG) guidelines, acceptable contraceptive methods are limited to those which inhibit ovulation as the primary mode of action.
d) Considered effective, but not highly effective - failure rate of ≥1% per year. Periodic abstinence (calendar, symptothermal, post-ovulation methods), withdrawal (coitus interruptus), spermicides only, and lactational amenorrhoea method (LAM) are not acceptable methods of contraception.
© 2018 TransCelerate BioPharma 20

Common Protocol Template Patient Library v005
e) Male condom and female condom should not be used together (due to risk of failure with friction).
<End of common text>
© 2018 TransCelerate BioPharma 21

Common Protocol Template Patient Library v005
Appendix 6: Liver Safety: Suggested Actions and Follow-up Assessments [and Study Intervention Rechallenge Guidelines]
<Start of common text for Phase I studies >
Phase 1 liver chemistry stopping criteria are designed to assure participant safety and to evaluate liver event etiology.
Phase 1 Liver Chemistry Stopping Criteria and Follow-Up Assessments
Liver Chemistry Stopping Criteria – Liver Stopping Event
ALT-absoluteALT≥3xULNIf ALT ≥3xULN AND bilirubin 2xULN (>35% direct bilirubin) OR international normalized ratio (INR) >1.5, report as a serious adverse event (SAE).1,2
See additional actions and follow-up assessments listed below
Suggested Actions and Follow up Assessments
Actions Follow Up Assessments
Immediately discontinue study intervention. Report the event to the [sponsor] within 24
hours Complete the liver event case report form
(CRF), and complete a SAE data collection tool if the event also met the criteria for an SAE.2
Perform liver chemistry follow-up assessments. Monitor the participant until liver chemistry
test abnormalities resolve, stabilize, or return to baseline (see MONITORING).
Do not restart/rechallenge participant with study intervention unless allowed per protocol and [sponsor] approval is granted.
If restart/rechallenge is either not allowed per protocol or not granted, permanently discontinue study intervention. The participant may continue in the study for any protocol-specified follow-up assessments
MONITORING:If ALT 3xULN AND bilirubin 2xULN or INR >1.5: Repeat liver chemistry tests (include ALT,
aspartate transaminase [AST], alkaline phosphatase, bilirubin and INR) and perform
Viral hepatitis serology3
Obtain INR and recheck with each liver chemistry assessment until the transaminases values show downward trend
Obtain blood sample for pharmacokinetic (PK) analysis [insert time interval recommended by clinical pharmacokinetics representative] after the most recent dose4
Serum creatine phosphokinase (CPK) and lactate dehydrogenase (LDH)
Fractionate bilirubin, if total bilirubin 2xULN
Obtain complete blood count with differential to assess eosinophilia
Record the appearance or worsening of clinical symptoms of liver injury, or hypersensitivity, on the adverse event (AE) CRF
Record use of concomitant medications (including acetaminophen, herbal remedies, and
© 2018 TransCelerate BioPharma 22

Common Protocol Template Patient Library v005
liver event follow-up assessments within 24 hours.
Monitor participant twice weekly until liver chemistry test abnormalities resolve, stabilize, or return to baseline.
A specialist or hepatology consultation is recommended.
If ALT ≥3xULN AND bilirubin <2xULN and INR ≤1.5: Repeat liver chemistry tests (include ALT,
AST, alkaline phosphatase, bilirubin and INR) and perform liver chemistry follow-up assessments within 24 to 72 hours.
Monitor participants weekly until liver chemistry abnormalities resolve, stabilize, or return to baseline.
other over-the-counter medications) on the concomitant medications CRF.
Record alcohol use on the liver event alcohol intake CRF.
If ALT ≥3xULN AND bilirubin 2xULN or INR >1.5: Anti-nuclear antibody, anti-smooth
muscle antibody, Type 1 anti-liver kidney microsomal antibodies, and quantitative total immunoglobulin G (IgG) or gamma globulins.
Serum acetaminophen adduct high performance liquid chromatography (HPLC) assay (quantifies potential acetaminophen contribution to liver injury in participants with definite or likely acetaminophen use in the preceding week [James, 2009].) NOTE: Not required in China.
Liver imaging (ultrasound, magnetic resonance, or computerized tomography) and/or liver biopsy to evaluate liver disease; complete liver imaging and/or liver biopsy CRFs.
1. Serum bilirubin fractionation should be performed if testing is available. If serum bilirubin fractionation is not immediately available, discontinue study intervention if ALT 3xULN and bilirubin 2xULN. Additionally, if serum bilirubin fractionation testing is unavailable, record the absence/presence of detectable urinary bilirubin on dipstick which is indicative of direct bilirubin elevations suggesting liver injury.
2. All events of ALT 3xULN and bilirubin 2xULN (>35% direct bilirubin) or ALT 3xULN and INR >1.5 may indicate severe liver injury (possible ‘Hy’s Law’) and must be reported as an SAE (excluding studies of hepatic impairment or cirrhosis). The INR stated threshold value will not apply to participants receiving anticoagulants.
3. Hepatitis A immunoglobulin M (IgM) antibody; HBsAg and HBcAb; hepatitis C RNA; cytomegalovirus IgM antibody; Epstein-Barr viral capsid antigen IgM antibody (or if unavailable, heterophile antibody or monospot testing); and hepatitis E IgM antibody.
4. PK sample may not be required for participants known to be receiving placebo or non-comparator interventions. Record the date/time of the PK blood sample draw and the date/time of the last dose of study intervention prior to the PK blood sample draw on the CRF. If the date or time of the last dose is unclear, provide the participant’s best approximation. If the date/time of the last dose cannot be approximated OR a PK sample cannot be collected in the time period indicated above, do not obtain a PK sample. Instructions for sample handling and shipping are in the [Study Reference Manual].
© 2018 TransCelerate BioPharma 23

Common Protocol Template Patient Library v005
References: James LP, Letzig L, Simpson PM, Capparelli E, Roberts DW, Hinson JA,et al. Pharmacokinetics of Acetaminophen-Adduct in Adults with Acetaminophen Overdose and Acute Liver Failure. Drug Metab Dispos 2009; 37:1779-1784.
<End of common text for Phase 1 studies >
© 2018 TransCelerate BioPharma 24

Common Protocol Template Patient Library v005
<Start of common wording for Phase 2 studies>
Phase 2 liver chemistry stopping criteria are designed to assure participant safety and to evaluate liver event etiology.
Phase 2 Liver Chemistry Stopping Criteria and Follow-Up Assessments
Liver Chemistry Stopping Criteria
ALT-absolute ALT 5xULN
ALT Increase ALT 3xULN persists for 4 weeks
Bilirubin1, 2 ALT 3xULN and bilirubin 2xULN (>35% direct bilirubin)
INR2 ALT 3xULN and international normalized ratio (INR) >1.5, if INR measured
Cannot Monitor
ALT 3xULN and cannot be monitored weekly for 4 weeks
Symptomatic3 ALT 3xULN associated with symptoms (new or worsening) believed to be related to liver injury or hypersensitivity
Suggested Actions and Follow-up Assessments
Actions Follow-Up Assessments
Immediately discontinue study intervention. Report the event to the [sponsor] within 24
hours. Complete the liver event case report form
(CRF), and complete a serious adverse event (SAE) data collection tool if the event also met the criteria for an SAE.2
Perform liver chemistry follow-up assessments.
Monitor the participant until liver chemistry test abnormalities resolve, stabilize, or return to baseline (see MONITORING).
Do not restart/rechallenge participant with study intervention unless allowed per protocol and [sponsor] approval is granted
If restart/rechallenge not allowed per protocol or not granted, permanently discontinue study intervention and continue participant in the study for any protocol specified follow up assessments
Viral hepatitis serology4
Obtain INR and recheck with each liver chemistry assessment until the transaminases values show downward trend
Obtain blood sample for pharmacokinetic (PK) analysis [insert time interval recommended by clinical pharmacokinetics representative] after the most recent dose5.
Serum creatine phosphokinase (CPK) and lactate dehydrogenase (LDH)
Fractionate bilirubin, if total bilirubin 2xULN
Obtain complete blood count with differential to assess eosinophilia
Record the appearance or worsening of clinical symptoms of liver injury, or hypersensitivity, on the adverse event (AE) report form
Record use of concomitant
© 2018 TransCelerate BioPharma 25

Common Protocol Template Patient Library v005
MONITORING:If ALT 3xULN AND bilirubin 2xULN or INR >1.5: Repeat liver chemistry tests (include ALT,
aspartate transaminase [AST], alkaline phosphatase, bilirubin and INR) and perform liver event follow-up assessments within 24 hours.
Monitor participant twice weekly until liver chemistry test abnormalities resolve, stabilize, or return to baseline.
A specialist or hepatology consultation is recommended.
If ALT 3xULN AND bilirubin <2xULN and INR 1.5: Repeat liver chemistry tests (include ALT,
AST, alkaline phosphatase, bilirubin and INR) and perform liver chemistry follow-up assessments within 24 to 72 hours.
Monitor participants weekly until liver chemistry abnormalities resolve, stabilize, or return to baseline.
medications (including acetaminophen, herbal remedies, and other over-the-counter medications) on the concomitant medications CRF.
Record alcohol use on the liver event alcohol intake CRF
If ALT 3xULN AND bilirubin 2xULN or INR >1.5: Anti-nuclear antibody, anti-smooth
muscle antibody, Type 1 anti-liver kidney microsomal antibodies, and quantitative total immunoglobulin G (IgG) or gamma globulins.
Serum acetaminophen adduct high performance liquid chromatography (HPLC) assay (quantifies potential acetaminophen contribution to liver injury in participants with definite or likely acetaminophen use in the preceding week [James, 2009].) NOTE: Not required in China.
Liver imaging (ultrasound, magnetic resonance, or computerized tomography) and/or liver biopsy to evaluate liver disease; complete liver
1. Serum bilirubin fractionation should be performed if testing is available. If serum bilirubin fractionation is not immediately available, discontinue study intervention if ALT 3xULN and bilirubin 2xULN. Additionally, if serum bilirubin fractionation testing is unavailable, record the absence/presence of detectable urinary bilirubin on dipstick which is indicative of direct bilirubin elevations suggesting liver injury.
2. All events of ALT 3xULN and bilirubin 2xULN (>35% direct bilirubin) or ALT 3xULN and INR >1.5 may indicate severe liver injury (possible ‘Hy’s Law’) and must be reported as an SAE (excluding studies of hepatic impairment or cirrhosis). The INRstated threshold value will not apply to participants receiving anticoagulants.
3. New or worsening symptoms believed to be related to liver injury (such as fatigue, nausea, vomiting, right upper quadrant pain or tenderness, or jaundice) or hypersensitivity (such as fever, rash or eosinophilia).
4. Includes:Hepatitis A immunoglobulin M (IgM) antibody; HBsAg and HBcAb; hepatitis C RNA; cytomegalovirus IgM antibody; Epstein-Barr viral capsid antigen IgM antibody (or if unavailable, heterophile antibody or monospot testing); and hepatitis E IgM antibody.
5. PK sample may not be required for participants known to be receiving placebo or non-comparator interventions. Record the date/time of the PK blood sample draw and the date/time of the last dose of study intervention prior to the blood sample draw on the CRF. If the date or time of the last dose is unclear, provide the participant’s best approximation. If the date/time of the last dose cannot be approximated OR a PK sample cannot be collected in the time period indicated above, do not obtain a PK sample. Instructions for sample handling and shipping are in the [Study Reference Manual].
© 2018 TransCelerate BioPharma 26

Common Protocol Template Patient Library v005
Phase 2 Liver Chemistry Increased Monitoring Criteria with Continued Study Intervention
Liver Chemistry Increased Monitoring Criterion and Follow-Up
Criterion Actions
ALT 3xULN and <5xULN and bilirubin <2xULN, without symptoms believed to be related to liver injury or hypersensitivity, and who can be monitored weekly for 4 weeks
Notify the [sponsor within 24 hours of learning of the abnormality to discuss participant safety.
Participant can continue study intervention Participant must return weekly for repeat liver
chemistry tests (ALT, AST, alkaline phosphatase, bilirubin) until the abnormalities resolve, stabilize or return to baseline.
If at any time, the participant meets liver chemistry stopping criteria, proceed as described in [location].
If, after 4 weeks of monitoring, ALT <3xULN and bilirubin <2xULN, monitor participants twice monthly until liver chemistry tests resolve, stabilize, or return to baseline.
ReferencesJames LP, Letzig L, Simpson PM, Capparelli E, Roberts DW, Hinson JA, et al. Pharmacokinetics of Acetaminophen-Adduct in Adults with Acetaminophen Overdose and Acute Liver Failure. Drug Metab Dispos 2009; 37:1779-1784.
<End of common wording for Phase 2 studies>
© 2018 TransCelerate BioPharma 27

Common Protocol Template Patient Library v005
<Start of common wording for Phase 3-4studies>
Phase 3-4liver chemistry stopping criteria are designed to assure participant safety and to evaluate liver event etiology.
Phase 3-4 liver Chemistry Stopping Criteria and Follow-Up assessments
Liver Chemistry Stopping Criteria
ALT-absolute ALT 8xULN
ALT Increase ALT 5xULN but <8xULN persists for 2 weeksALT 3xULN but <5xULN persists for 4 weeks
Bilirubin1, 2 ALT 3xULN and bilirubin 2xULN (>35% direct bilirubin)
INR2 ALT 3xULN and international normalized ratio (INR) >1.5, if INR measured
Cannot Monitor
ALT 5xULN but <8xULN and cannot be monitored weekly for 2 weeksALT 3xULN but <5xULN and cannot be monitored weekly for 4 weeks
Symptomatic3 ALT 3xULN associated with symptoms (new or worsening) believed to be related to liver injury or hypersensitivity
Suggested Actions and Follow up Assessments
Actions Follow Up Assessments
Immediately discontinue study intervention.
Report the event to the [sponsor] within 24 hours.
Complete the liver event case report form (CRF), and complete a serious adverse event (SAE) data collection tool if the event also met the criteria for an SAE.2
Perform liver chemistry follow-up assessments.
Monitor the participant until liver chemistry test abnormalities resolve, stabilize, or return to baseline (see MONITORING).
Do not restart/rechallenge participant with study intervention unless allowed per protocol and [sponsor] approval is granted.
If restart/rechallenge not allowed per protocol or not granted, permanently discontinue study intervention and continue participant in the study for any protocol specified follow up assessments.
Viral hepatitis serology4
:only in those with underlying chronic hepatitis B at study entry (identified by positive hepatitis B surface antigen), quantitative hepatitis B deoxyribonucleic acid (DNA) and hepatitis delta antibody5
Obtain INR and recheck with each liver chemistry assessment until the transaminases values show downward trend
Obtain blood sample for pharmacokinetic (PK) analysis [insert time interval recommended by clinical pharmacokinetics representative] after the most recent dose5
Serum creatine phosphokinase (CPK) and lactate dehydrogenase (LDH)
Fractionate bilirubin, if total bilirubin 2xULN
© 2018 TransCelerate BioPharma 28

Common Protocol Template Patient Library v005
MONITORING:For bilirubin or INR criteria Repeat liver chemistry tests (include ALT,
aspartate transaminase [AST], alkaline phosphatase, bilirubin and INR) and perform liver event follow-up assessments within 24 hours.
Monitor participant twice weekly until liver chemistry test abnormalities resolve, stabilize, or return to baseline.
A specialist or hepatology consultation is recommended.
For all other criteria Repeat liver chemistry tests (include ALT,
AST, alkaline phosphatase, bilirubin and INR) and perform liver chemistry follow-up assessments within 24 to 72 hours.
Monitor participants weekly until liver chemistry abnormalities resolve, stabilize, or return to baseline.
Obtain complete blood count with differential to assess eosinophilia
Record the appearance or worsening of clinical symptoms of liver injury, or hypersensitivity, on the adverse event (AE) report form
Record use of concomitant medications (including acetaminophen, herbal remedies, and other over-the-counter medications) on the concomitant medications CRF.
Record alcohol use on the liver event alcohol intake CRF
For bilirubin or INR criteria: Anti-nuclear antibody, anti-smooth
muscle antibody, Type 1 anti-liver kidney microsomal antibodies, and quantitative total immunoglobulin G (IgG) or gamma globulins.
Serum acetaminophen adduct high performance liquid chromatography (HPLC) assay (quantifies potential acetaminophen contribution to liver injury in participants with definite or likely acetaminophen use in the preceding week [James, 2009]). NOTE: Not required in China.
Liver imaging (ultrasound, magnetic resonance, or computerizsed tomography) andor liver biopsy to evaluate liver disease; complete Liver Imaging and/or Liver Biopsy CRFs.
1. Serum bilirubin fractionation should be performed if testing is available. If serum bilirubin fractionation is not immediately available, discontinue study intervention if ALT 3xULN and bilirubin 2xULN. Additionally, if serum bilirubin fractionation testing is unavailable, record the absence/presence of detectable urinary bilirubin on dipstick which is indicative of direct bilirubin elevations suggesting liver injury.
2. All events of ALT 3xULN and bilirubin 2xULN (>35% direct bilirubin) or ALT 3xULN and INR >1.5 may indicate severe liver injury (possible ‘Hy’s Law’) and must be reported as an SAE (excluding studies of hepatic impairment or cirrhosis). The INR stated threshold value will not apply to participants receiving anticoagulants.
3. New or worsening symptoms believed to be related to liver injury (such as fatigue, nausea, vomiting, right upper quadrant pain or tenderness, or jaundice) or hypersensitivity (such as fever, rash or eosinophilia).
© 2018 TransCelerate BioPharma 29

Common Protocol Template Patient Library v005
4. Includes:Hepatitis A immunoglobulin M (IgM) antibody; HBsAg and HBcAb; hepatitis C RNA; cytomegalovirus IgM antibody; Epstein-Barr viral capsid antigen IgM antibody (or if unavailable, heterophile antibody or monospot testing); and hepatitis E IgM antibody.
5. If hepatitis delta antibody assay cannot be performed,, it can be replaced with a polymerase chain reaction (PCR) of hepatitis D RNA virus (where needed) [Le Gal, 2005].
6. PK sample may not be required for participants known to be receiving placebo or non-comparator interventions. Record the date/time of the PK blood sample draw and the date/time of the last dose of study intervention prior to the PK blood sample draw on the CRF. If the date or time of the last dose is unclear, provide the participant’s best approximation. If the date/time of the last dose cannot be approximated OR a PK sample cannot be collected in the time period indicated above, do not obtain a PK sample. Instructions for sample handling and shipping are in the [Study Reference Manual].
Phase 3-4Liver Chemistry Increased Monitoring Criteria with Continued Study Intervention
Liver Chemistry Increased Monitoring Criteria
Criteria Actions
ALT 5xULN and <8xULN and bilirubin <2xULN without symptoms believed to be related to liver injury or hypersensitivity, and who can be monitored weekly for 2 weeks.ORALT 3xULN and <5xULN and bilirubin <2xULN without symptoms believed to be related to liver injury or hypersensitivity, and who can be monitored weekly for 4 weeks.
Notify the [sponsor] within 24 hours of learning of the abnormality to discuss participant safety.
Participant can continue study intervention Participant must return weekly for repeat liver
chemistry tests (ALT, AST, alkaline phosphatase, bilirubin) until the abnormalities resolve, stabilize, or return to baseline.
If at any time, the participant meets liver chemistry stopping criteria, proceed as described in [location].
If ALT decreases from ALT 5xULN and <8xULN to ≥3xULN but <5xULN, continue to monitor liver chemistries weekly.
If, after 4 weeks of monitoring, ALT <3xULN and bilirubin <2xULN, monitor participants twice monthly until liver chemistry tests resolve, stabilize, or return to baseline.
ReferencesJames LP, Letzig L, Simpson PM, Capparelli E, Roberts DW, Hinson JA, et al. Pharmacokinetics of Acetaminophen-Adduct in Adults with Acetaminophen Overdose and Acute Liver Failure. Drug Metab Dispos 2009; 37:1779-1784.
Le Gal F, Gordien E, Affolabi D, Hanslik T, Alloui C, Dény P, et al. Quantification of Hepatitis Delta Virus RNA in Serum by Consensus Real-Time PCR Indicates Different Patterns of Virological Response to Interferon Therapy in Chronically Infected Patients. J Clin Microbiol. 2005;43(5):2363–2369.
© 2018 TransCelerate BioPharma 30

Common Protocol Template Patient Library v005
<End of common text for Phase 3-4studies>
© 2018 TransCelerate BioPharma 31

Common Protocol Template Patient Library v005
Liver Safety: Study Intervention Rechallenge Guidelines
<Start of common text if study intervention restart/rechallenge is allowed>
A participant who met liver chemistry stopping criteria cannot restart study intervention unless all of the following conditions are met:
[Sponsor] approval is granted (as described below) Institutional Review Board/Independent Ethics Committee (IRB/IEC) approval is
obtained Separate ICF for intervention restart/rechallenge is signed by the participant
If [sponsor] approval to restart/rechallenge the participant with study intervention is not granted, then the participant must permanently discontinue study intervention and may continue in the study for protocol-specified follow-up assessments.
Rechallenge Following Liver Chemistry Events that are Possibly Related to Study Intervention
Following study intervention-induced liver injury, rechallenge is associated with 13% mortality across all study interventions in prospective studies.1 Clinical outcomes vary with nearly 50% fatality with halothane readministered within one month of the initial injury. However, some interventions seldom result in recurrent liver injury or fatality.
Risk factors for a fatal rechallenge outcome include:
o Hypersensitivity with initial liver injury (e.g., fever, rash, eosinophilia) 1
o Jaundice or bilirubin >2xULN with initial liver injury (direct bilirubin >35% of total)
o Ongoing severe liver injury defined by ALT 3xULN AND bilirubin 2xULN (direct bilirubin >35% of total) OR INR>1.5
o Serious adverse event or fatality previously observed with rechallenges2,3
o Evidence of intervention-related preclinical liability (e.g., reactive metabolites, mitochondrial impairment)3
Rechallenge refers to resuming study intervention following drug-induced liver injury (DILI). Because of the risks associated with rechallenge after DILI, this should only be considered if there is compelling evidence of benefit from a critical or life-saving medicine, there is no alternative approved medicine available, and a benefit/risk assessment of rechallenge is considered to be favorable.
Approval by the [sponsor] for rechallenge with study intervention can be considered when:
o The Principal Investigator (PI) requests consideration of rechallenge with study intervention for a participant who is receiving compelling benefit with study intervention that exceeds risk and for whom no effective alternative therapy is available.
© 2018 TransCelerate BioPharma 32

Common Protocol Template Patient Library v005
o IRB/IEC approval for rechallenge with study intervention has been obtained.
If the rechallenge is approved by the [sponsor] in writing:
The participant must be provided with a clear description of the possible benefits and risks of study intervention administration including the possibility of recurrent, more severe liver injury or death.
The participant must provide signed informed consent specifically for the rechallenge with study intervention. Documentation of informed consent must be recorded in the study file.
Study intervention must be administered at the dose specified by the [sponsor].
Participants approved by the [sponsor] for rechallenge with study intervention must return to the clinic twice a week for liver chemistry tests until stable liver chemistry tests have been demonstrated and then standard laboratory monitoring may resume as per protocol.
If the participant meets protocol-defined liver chemistry stopping criteria after study intervention rechallenge, study intervention should be permanently discontinued.
The [sponsor] and the IRB/IEC, must be informed of the outcome for the participant following study intervention rechallenge.
The [sponsor] must be notified of any adverse events.
AND/OR
Restart Following Transient Resolving Liver Chemistry Events Not Related to Study Intervention
Restart refers to resuming study intervention following liver chemistry events for which there are clear underlying causes (other than DILI) (e.g., biliary obstruction, pancreatic events, hypotension, acute viral hepatitis). Furthermore, there should be no evidence of alcoholic hepatitis or hypersensitivity.
Approval by the [sponsor] for study intervention restart can be considered when:o The investigator requests consideration for study intervention restart if liver
chemistry events have a clear underlying cause (e.g., biliary obstruction, pancreatic events, hypotension, acute viral hepatitis) and liver chemistry tests have improved to normal or are within 1.5 x baseline and ALT <3xULN.
o Possible DILI has been excluded by the investigator and the study team. This includes the absence of markers of hypersensitivity (otherwise unexplained fever, rash, eosinophilia). Where a study intervention has an identified genetic marker associated with liver injury (e.g., lapatinib, abacavir, amoxicillin/clavulanate), the presence of the marker should be excluded. If study intervention-related liver injury cannot be excluded, the guidance on rechallenge in the previous part of this Appendix will apply.
o There is no evidence of alcoholic hepatitis.o IRB/IEC approval of study intervention restart has been obtained.
© 2018 TransCelerate BioPharma 33

Common Protocol Template Patient Library v005
If restart of study intervention is approved by the [sponsor] in writing:
The participant must be provided with a clear description of the possible benefits and risks of study intervention administration including the possibility of recurrent, more severe liver injury or death.
The participant must provide signed informed consent specifically for the restart of study intervention. Documentation of informed consent must be recorded in the study file.
Study intervention must be administered at the dose specified by the [sponsor].
Participants approved by the [sponsor] for restart of study intervention must return to the clinic twice a week for liver chemistry tests until stable liver chemistry tests have been demonstrated and then standard laboratory monitoring may resume as per protocol.
If the participant meets protocol-defined liver chemistry stopping criteria after study intervention restart, study intervention should be permanently discontinued.
The [sponsor], and the IRB/IEC, must be informed of the outcome for the participant following study intervention restart.
The [sponsor] must be notified of any adverse events.
References:1. Andrade RJ, Robles M, Lucena MI. Rechallenge in drug-induced liver injury: the
attractive hazard. Expert Opin Drug Saf. 2009;8:709-714.
2. Papay JI, Clines D, Rafi R, Yuen N, Britt SD, Walsh JS, et al. Drug-induced liver injury following positive drug rechallenge. Regul Tox Pharm. 2009;54:84-90.
3. Hunt, CM. Mitochondrial and immunoallergic injury increase risk of positive drug rechallenge after drug-induced liver injury: A systematic review. Hepatol. 2010;52:2216-2222.
<End of common text >
© 2018 TransCelerate BioPharma 34
![Introduction to Dependency Grammar [0.2cm] and Dependency ...ufal.mff.cuni.cz/~bejcek/parseme/prague/Nivre1.pdf · Introduction to Dependency Grammar and Dependency Parsing Joakim](https://static.fdocuments.in/doc/165x107/5b14bded7f8b9a201a8b9282/introduction-to-dependency-grammar-02cm-and-dependency-ufalmffcuniczbejcekparsemeprague.jpg)

















