
AntitumorEffectsandMechanismsofAZD4547on FGFR2...
12
Cancer Biology and Signal Transduction Antitumor Effects and Mechanisms of AZD4547 on FGFR2-Deregulated Endometrial Cancer Cells Yeonui Kwak 1 , Hanna Cho 1 , Wooyoung Hur 2 , and Taebo Sim 1,2 Abstract Uncontrolled activation of FGFRs induces the progression of various cancers. It was recently reported that FGFR2-activating mutants are implicated in about 12% of endometrial carcinomas. AZD4547, a potent pan-FGFR inhibitor, is currently being eval- uated in clinical trials for several FGFR-driven cancers. However, AZD4547 has not been examined yet against FGFR2 mutant– driven endometrial cancers. Thus, we evaluated the activity of AZD4547 against four different endometrial cancer cells, includ- ing AN3-CA, MFE296, MFE280, and HEC1A, where all but HEC1A cells express distinctive FGFR2 mutations. We found that AZD4547 exhibits potent antiproliferative activity (EC 50 ¼ 31 nmol/L) against AN3-CA cells harboring FGFR2-K310R/N550K mutant. Analysis using a phospho-kinase array revealed that AZD4547 blocks FGFR2 downstream signaling, such as p38, ERK1/2, JNK, p70S6K, and PLCg . Moreover, oral administration of AZD4547 (30 mg/kg, every day) remarkably delayed tumor growth in a mouse xenograft model of AN3-CA cells. Unbiased reporter gene assay showed that AZD4547 antagonizes the aFGF- induced activation of several transcription factors, including EGR1, ELK-1/SRF, AP-1, and NFkB. Genome-wide transcriptome analysis revealed that AZD4547 perturbs a number of transcrip- tions, and EGR1 was identified as one of the major targets of AZD4547. The significance of the FGFR2–EGR1 axis in endome- trial cancer progression has not been reported. In addition, using kinome-wide inhibition profiling analysis, we first identified potential new target kinases of AZD4547, including MAP4K3, MAP4K5, IRR, RET, and FLT3. Our study demonstrated that AZD4547 exhibits its therapeutic activity against endometrial cancer cells by perturbing various regulatory mechanisms related to FGFR signaling. Mol Cancer Ther; 14(10); 2292–302. Ó2015 AACR. Introduction Fibroblast growth factor receptors (FGFR), consisting of four genes named FGFR1–FGFR4, are one of the subfamilies of recep- tor tyrosine kinases (RTK; ref. 1). Eighteen different FGF ligands bind to the distinct FGFRs or their alternative splicing isoforms through formation of ternary complexes with heparan sulfate proteoglycans (2). FGFRs regulate important physiologic process- es, including cell growth, and their dysregulated activities are implicated in various human diseases. For example, FGFR1 over- expression resulting from 8p12 amplification induces carcino- genesis in the breast (3) and is implicated in 22% of squamous cell lung cancers (4). In about 15% to 20% of patients, multiple myeloma is caused by overexpression of FGFR3 within strong IgH enhancers as a result of a t(4;14)(p16.3;q32.3) chromosomal translocation (5). In 60% to 70% of non–muscle-invasive super- ficial bladder cancers, gain-of-function FGFR3 mutations, such as S249C/Y373C, in the extracellular region and mutations of K650 in the kinase domain are indicative of a high risk for tumor recurrence (6). In addition, a FGFR3–ETV6-fused gene generated by a t(4;12)(p16;p13) chromosomal translocation causes periph- eral T-cell lymphoma (7). FGFR1–TACC1 and FGFR3–TACC3- fused genes are associated with glioblastoma multiforme (8). Overexpression of FGFR4 has been shown to be associated with 7% to 8% of rhabdomyosarcoma, which is the most common soft-tissue sarcoma in children (9, 10). Moreover, FGFR1–3 mutations found in cancers are also involved in various skeletal disorders, such as Apert syndrome and Crouzon syndrome (11–13). About 10% to 12% of endometrial cancers are associ- ated with constitutively active FGFR2 mutants (14, 15), where S252W mutation in the extracellular domain increases the bind- ing affinity with FGF, and N550K mutant in the kinase domain loses the autoinhibitory conformation (13, 16). Despite muta- tions of both the PI3K pathway and FGFR2, FGFR2 inhibition alone using siRNA FGFR2 knockdown or small-molecule inhibi- tors blocks the growth of endometrial cancers harboring PTEN/ FGFR2 mutations, while it shows no effect in HEC1A endometrial cancers expressing wild-type FGFR2 along with PTEN/KRAS muta- tions (17, 18). Owing to their prominent roles in cancers, FGFRs have become significant targets for inhibitor development (19). This effort has led to the discovery of several FGFR inhibitors, including dovi- tinib, BGJ398, ponatinib, LY2874455, and AZD4547, all of which have entered clinical trials as potential anticancer drugs. As a frontrunner in the group of pan-FGFR inhibitors, AZD4547 was demonstrated to promote favorable therapeutic outcomes against a variety of FGFR-deregulated cancer models, including glioblas- toma, non–small cell lung cancer, gastric cancer, and multiple myeloma (20–23). However, little is known about the efficacy of 1 KU-KIST Graduate School of Converging Science and Technology, Korea University, Seongbuk-gu, Seoul, Republic of Korea. 2 Chemical Kinomics Research Center, Korea Institute of Science and Technology, Seongbuk-gu, Seoul, Republic of Korea. Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/). Corresponding Author: Taebo Sim, Korea Institute of Science and Technology, Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 136-791, Republic of Korea (South). Phone: 822-958-6437; Fax: 822-958-5189; E-mail: [email protected], [email protected] doi: 10.1158/1535-7163.MCT-15-0032 Ó2015 American Association for Cancer Research. Molecular Cancer Therapeutics Mol Cancer Ther; 14(10) October 2015 2292 on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032
Transcript of AntitumorEffectsandMechanismsofAZD4547on FGFR2...

Cancer Biology and Signal Transduction
AntitumorEffectsandMechanismsofAZD4547onFGFR2-Deregulated Endometrial Cancer CellsYeonui Kwak1, Hanna Cho1,Wooyoung Hur2, and Taebo Sim1,2
Abstract
Uncontrolled activation of FGFRs induces the progression ofvarious cancers. It was recently reported that FGFR2-activatingmutants are implicated in about 12% of endometrial carcinomas.AZD4547, a potent pan-FGFR inhibitor, is currently being eval-uated in clinical trials for several FGFR-driven cancers. However,AZD4547 has not been examined yet against FGFR2 mutant–driven endometrial cancers. Thus, we evaluated the activity ofAZD4547 against four different endometrial cancer cells, includ-ing AN3-CA, MFE296, MFE280, and HEC1A, where all butHEC1A cells express distinctive FGFR2 mutations. We found thatAZD4547 exhibits potent antiproliferative activity (EC50 ¼ 31nmol/L) against AN3-CA cells harboring FGFR2-K310R/N550Kmutant. Analysis using a phospho-kinase array revealed thatAZD4547 blocks FGFR2 downstream signaling, such as p38,ERK1/2, JNK, p70S6K, and PLCg . Moreover, oral administration
of AZD4547 (30 mg/kg, every day) remarkably delayed tumorgrowth in a mouse xenograft model of AN3-CA cells. Unbiasedreporter gene assay showed that AZD4547 antagonizes the aFGF-induced activation of several transcription factors, includingEGR1, ELK-1/SRF, AP-1, and NFkB. Genome-wide transcriptomeanalysis revealed that AZD4547 perturbs a number of transcrip-tions, and EGR1 was identified as one of the major targets ofAZD4547. The significance of the FGFR2–EGR1 axis in endome-trial cancer progression has not been reported. In addition, usingkinome-wide inhibition profiling analysis, we first identifiedpotential new target kinases of AZD4547, including MAP4K3,MAP4K5, IRR, RET, and FLT3. Our study demonstrated thatAZD4547 exhibits its therapeutic activity against endometrialcancer cells by perturbing various regulatory mechanisms relatedto FGFR signaling. Mol Cancer Ther; 14(10); 2292–302. �2015 AACR.
IntroductionFibroblast growth factor receptors (FGFR), consisting of four
genes named FGFR1–FGFR4, are one of the subfamilies of recep-tor tyrosine kinases (RTK; ref. 1). Eighteen different FGF ligandsbind to the distinct FGFRs or their alternative splicing isoformsthrough formation of ternary complexes with heparan sulfateproteoglycans (2). FGFRs regulate important physiologic process-es, including cell growth, and their dysregulated activities areimplicated in various human diseases. For example, FGFR1 over-expression resulting from 8p12 amplification induces carcino-genesis in thebreast (3) and is implicated in22%of squamous celllung cancers (4). In about 15% to 20% of patients, multiplemyeloma is caused by overexpression of FGFR3within strong IgHenhancers as a result of a t(4;14)(p16.3;q32.3) chromosomaltranslocation (5). In 60% to 70% of non–muscle-invasive super-ficial bladder cancers, gain-of-function FGFR3mutations, such asS249C/Y373C, in the extracellular region and mutations of K650
in the kinase domain are indicative of a high risk for tumorrecurrence (6). In addition, a FGFR3–ETV6-fused gene generatedby a t(4;12)(p16;p13) chromosomal translocation causes periph-eral T-cell lymphoma (7). FGFR1–TACC1 and FGFR3–TACC3-fused genes are associated with glioblastoma multiforme (8).Overexpression of FGFR4 has been shown to be associated with7% to 8% of rhabdomyosarcoma, which is the most commonsoft-tissue sarcoma in children (9, 10). Moreover, FGFR1–3mutations found in cancers are also involved in various skeletaldisorders, such as Apert syndrome and Crouzon syndrome(11–13). About 10% to 12% of endometrial cancers are associ-ated with constitutively active FGFR2 mutants (14, 15), whereS252W mutation in the extracellular domain increases the bind-ing affinity with FGF, and N550K mutant in the kinase domainloses the autoinhibitory conformation (13, 16). Despite muta-tions of both the PI3K pathway and FGFR2, FGFR2 inhibitionalone using siRNA FGFR2 knockdown or small-molecule inhibi-tors blocks the growth of endometrial cancers harboring PTEN/FGFR2mutations, while it shows no effect inHEC1A endometrialcancers expressingwild-type FGFR2alongwithPTEN/KRASmuta-tions (17, 18).
Owing to their prominent roles in cancers, FGFRs have becomesignificant targets for inhibitor development (19). This effort hasled to the discovery of several FGFR inhibitors, including dovi-tinib, BGJ398, ponatinib, LY2874455, andAZD4547, all ofwhichhave entered clinical trials as potential anticancer drugs. As afrontrunner in the group of pan-FGFR inhibitors, AZD4547 wasdemonstrated to promote favorable therapeutic outcomes againsta variety of FGFR-deregulated cancer models, including glioblas-toma, non–small cell lung cancer, gastric cancer, and multiplemyeloma (20–23). However, little is known about the efficacy of
1KU-KIST Graduate School of Converging Science and Technology,Korea University, Seongbuk-gu, Seoul, Republic of Korea. 2ChemicalKinomics ResearchCenter, Korea Institute of Science and Technology,Seongbuk-gu, Seoul, Republic of Korea.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Taebo Sim, Korea Institute of Science and Technology,Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 136-791, Republic of Korea (South).Phone: 822-958-6437; Fax: 822-958-5189; E-mail: [email protected],[email protected]
doi: 10.1158/1535-7163.MCT-15-0032
�2015 American Association for Cancer Research.
MolecularCancerTherapeutics
Mol Cancer Ther; 14(10) October 20152292
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032

AZD4547 against FGFR2 mutant–driven endometrial cancer.Here, we investigated a detailed anticancer mechanism ofAZD4547against FGFR2mutant–expressing endometrial cancers.
Materials and MethodsReagents
Inhibitors, including AZD4547, BGJ398, sorafenib, dasatinib,and cabozantinib, were purchased from Selleck Chemicals.Recombinant human acidic FGF (aFGF) was from R&D Systems(#231-BC). Heparin sulfate (#H3149), collagen (#C7661), andiodonitrotetrazolium chloride (#I8377) were from SigmaAldrich. Antibodies for FGFR2, actin, phospho-GSK3b (Ser9),caspase-3, and cleaved PARP were purchased from Santa CruzBiotechnology. Other primary antibodies were purchased fromCell Signaling Technology: phospho-FGFR (Tyr653/654, #3471),phospho-FRS2a (Tyr436, #3861), phospho-ERK1/2 (Thr202/Tyr204, #4370), phospho-AKT (Thr308, #4056; Ser473,#4058), phospho-JNK (Thr183/Tyr185, #4668), phospho-p38(Thr180/Tyr182, #4511), phospho-RET (Tyr905, #3221), phos-pho-FLT3 (Tyr591, #3474), phospho-DDR1 (Tyr792, #11994),phospho-FMS (Tyr723, #3155), phospho-PLK1 (Thr210, #9062),phosphor PLCg (Tyr783, #2821), phospho-STAT1 (Tyr701,#9167), phospho-STAT3 (Ser727, #9134), phospho-p70S6K(Thr421/Ser424, #9204), phospho-CREB (Ser133, #9198), phos-pho-IkB (Ser32/36, #9246), FGFR1 (#9740), FLT3(#3462), RET(#3220), AKT (#4685), ERK (#4695), HSP60 (#4870), and EGR1(#4153). Antibodies for immunohistochemistry were anti-Ki67(Leica) anti-phospho-S6 (S235/236; Cell Signaling Technology,#2211) anti-cleaved caspase-3 (Cell Signaling Technology;#9661).
Cell cultureMFE280, MFE296, and MOLM-14 cells were purchased from
DSMZ. TT, MV4-11, U2OS, AN3-CA, and HEC1A cells werepurchased from the ATCC. Ishikawa cells were purchased fromSigma-Aldrich. Every cell linewas usedwithin 6months after theirDNA profiles were confirmed with short tandem repeat (STR)analysis by the Korean Cell Line Bank. AN3-CA (endometrium),Ishikawa (endometrium), andHEK293T cells weremaintained inDMEM supplemented with 10% FBS and antibiotics (100 U/mLpenicillin and 100 mg/mL streptomycin). HEC1A cells were cul-tured in DMEM/F12 media supplemented with 10% FBS andantibiotics. MFE280 (endometrium) and MFE296 (endometri-um) cells were maintained in a RPMI1640/MEM (1:1) supple-mented with 20% FBS and antibiotics. TT cells (medullary thy-roid) were cultured in RPMI1640 media supplemented with 2mmol/L glutamine, 15% FBS, and antibiotics. U2OS (osteosar-coma), MV4-11 (acute myeloid leukemia), and MOLM-14 (acutemyeloid leukemia) cells were grown in RPMI1640 supplementedwith 10% FBS and antibiotics. Parental Ba/F3 cells were grown inhigh-glucose RPMI1640 supplemented with 10% FBS and anti-biotics in the presence of IL3,whereas transformedBa/F3 cell lineswere grown in the same media without IL3.
Kinome profilingKinome profiling was performed by Reaction Biology Corp.
AZD4547 (1 mmol/L) was tested against 336 recombinant humankinases at 10 mmol/L ATP in duplicate mode. The extent to whichkinase activity is inhibited by AZD4547 was indicated as percent-age relative to DMSO-treated kinase activity (100%).
Establishment of Tel-fused tyrosine kinase–transformedBa/F3 cell lines
Ba/F3 cells lines transformed with TEL-fused human FLT3 orRET were established as described (24).
Immunoblot analysisCells were incubated overnight in the base media containing
0.5% FBS, treated primarily with compound for 2 hours, andsubsequently stimulated by a mixture of 10 ng/mL aFGF and 10mg/mL heparin sulfate for 20 minutes, unless otherwise specified.
Proliferation assayCells were plated at 5,000 cells/well in 96-well plates.
Each compound was added to wells at 10 points of 3-foldserial dilution (0–50 mmol/L). For adherent cells, cells weretreated with compounds one day after cell seeding. After 72-hourexposure, cell viability was measured using an MTT assay kit(Promega). Each assay was performed in duplicate mode threeindependent times. Cell viability of compound-treated wells wasnormalized relative to 0.5% DMSO-treated wells (100%). GI50values were calculated using Prism 5.0 software (GraphPad).
Apoptosis and cell-cycle analysisFor apoptosis assay, cells were treated with AZD4547 and
BGJ398 for 48hours. Allfloating andattached cellswere harvestedby trypsinization andwashedwith PBS (DPBS pH7.4). Cells werestained using an Annexin V-FLUOS staining Kit (Roche, # 11 988549 001). For cell-cycle analysis, cells were treated with com-pounds for 24 hours and harvested as above and fixed in 70%ethanol at �20�C overnight. Cells were harvested by centrifuga-tion at 500 � g, washed with cold DPBS, then suspended inpropidium iodide/RNase solution (Cell Signaling Technology,#4087) and incubated for 30 minutes in a dark condition beforeflow cytometer analysis (BD Biosciences).
Soft-agar assayOn the 0.5% bottom agar, cells in the complete media contain-
ing 0.3%agarwas plated at a density of 2,000 cells in 6-well plates.The plates were incubated for 3 weeks at 37�C and 5% CO2.Compounds diluted in culturemedia were added on the top agar.Media were refreshed twice a week. Spheroids were stained usingiodonitrotetrazolium chloride (Sigma Aldrich) for 24 hours. Theentire area of eachwell was photographedwithoutmagnification,and colonies in each well were counted using ImageJ software.
Phospho-kinase array analysisAN3-CA cells serum-starved media (DMEM containing 0.5%
FBS) overnight were treated with either DMSO or AZD4547 (300nmol/L) for 2 hours. Cells were stimulated with 10 ng/mL aFGFand 10 mg/mL heparin sulfate for 20 minutes. Cells were thenlysed and subjected to the analysis using Human-PhosphokinaseArray Kit (R&D Systems) using the manufacturer's protocol.
Reporter gene assayReporter gene assays were conducted using a Cignal 45-Path-
way Reporter Assay Kit (SABiosciences) following the manufac-turer's protocol. Eighteen hours after transfection, media in eachplate were changed to Opti-MEM containing DMSO, DMSO plusaFGF (10ng/mL), or AZD4547 (1mmol/L) plus aFGF (10ng/mL).After 24hours, Dual-Glo luciferase assay reagents (Promega)were
Therapeutic Effects of AZD4547 on Endometrial Cancers
www.aacrjournals.org Mol Cancer Ther; 14(10) October 2015 2293
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032

added. Luminescence values were normalized by dividing fireflyemission by Renilla emission as relative luminescence units(RLU). RLU values related to DMSO controls were representedas fold changes.
Microarray analysisAN3-CA cells were treated in three different conditions for 18
hours; (i)DMSO(0.5%)only, (ii) cotreatmentof aFGF(10ng/mL)andDMSO (0.5%), and (iii) cotreatment of aFGF (10 ng/mL) andAZD4547 (300 nmol/L) in duplicate mode and then lysed with 1mL of TRIzol reagent (Life Technologies). Total mRNAs wereisolated using RNeasy Mini Kit (Qiagen) and were subjected toGeneChip PrimeViewTMHumanGene Expression Array (Affyme-trix). Biotinylated cRNA was prepared according to the standardAffymetrix protocol from 500 ng total RNA (Expression AnalysisTechnicalManual, 2001,Affymetrix). After fragmentation, 12mgofbiotinylated cRNA were hybridized for 16 hours at 45�C onGeneChip Human Genome Array. GeneChips were washed andstained in the Affymetrix Fluidics Station 450. GeneChips werescannedusing theAffymetrixGeneChipScanner30007G.Thedatawere analyzed using Robust Multiarray Analysis (RMA) withAffymetrix default analysis settings and global scaling as thenormalizationmethod. The trimmedmean target intensity of eacharray was arbitrarily set to 100. The normalized and log trans-formed intensity values were then analyzed using GeneSpring GX12.6 (Agilent Technologies). Fold change filters included therequirement that the genes be present in at least 150% of controlsfor upregulated genes and lower than 66% of controls for down-regulated genes. Hierarchical clustering data were clustered intogroups that behave similarly across experiments using GeneSpringGX12.6.1 (Agilent Technologies). Thismicroarray datawas depos-ited in GEO public repository (GEO accession no. GSE61481).
RT-PCRTotal RNAs were extracted from cells treated with the indi-
cated condition using TRIzol reagent (Invitrogen) according tothe manufacturer's instruction. Total RNAs (2 mg) was used tosynthesize cDNA with M-MLV reverse transcriptase (Promega).Equal amount of cDNA was amplified with PCR for 30 cycles.The exact size of amplified PCR products was assessed byelectrophoresis in 3% agarose gels and visualized by Eco-stardye (Biofect #ES301-1000). Primer sequences are listed inSupplementary Table S1.
Gene set enrichment analysisWe utilized 509 genesets publicly available in molecular sig-
nature database (MSigDB) as described (25). These gene sets arecategorized by transcription factor–binding motif in their pro-moter regions. For each gene set, we calculated enrichment scoreto evaluate the distribution of members of a given gene setthroughout total gene list. Enrichment score (ES) means themaximum distance form X axis in the enrichment plot, and highES reflects that a gene set is overrepresented at the top of the entirelist. Variation in gene set size was adjusted in normalized ES(NES). False discovery rate (FDR) reflects the probability of falsepositive interpretation of the gene set with NES.
Xenograft modelAnimal experiments were approved by the Institutional Animal
Care and Use Committee of Korea University. Tumor xenografts
were established by subcutaneous injection into the right flankof 5-week-old female balb/c nude mice (Orient Bio Inc.) withAN3-CA cells (5� 106 cells/0.2 mL)mixed withMatrigel (BectonDickenson) to 1:1.When the tumor volume reached 100mm3 onaverage, the tumor-bearing mice (n ¼ 21) were sorted randomlyinto 3 treatment groups: vehicle (n¼ 7), 10mg/kg AZD4547 (n¼7), 30mg/kg AZD4547 (n¼ 7). The final concentration of vehiclewas 5%NMP, 6% solutol, 20% PEG400, 69%distilled water, and1 N HCl. Mice were orally gavaged once daily for 15 days. Tumorvolume and body weight were measured twice a week. Theformula used to calculate tumor volumes is [longest length �(length vertical to the longest length)2/2] mm3. Body weightswere measured once a week.
ImmunohistochemistryTumors were removed from subcutaneously xenografted
mice 4 hours after the final oral administration. Each tumor wastransferred to cassettes, followed by fixation in 10% formalinsolution overnight at 4�C. Each formalin-fixed tumor was washedin flowing water for 20 minutes. The tissue cassettes were trans-ferred to a solution of 70% ethanol. Tissues were embedded inparaffin and sliced. Paraffin-embedded tissue slices were stainedusing the following antibodies; Ki67 antibody (1:200), anti-phospho FGFR (1:50), anti-EGR1 (1:50), anti-phospho ERK1/2(1:100), anti-phospho S6 (1:200), and anti-cleaved caspase 3(1:100). The staining intensity was quantitated using the thresh-old function of ImageJ software.
ResultsAZD4547 displays antiproliferative effects against FGFR2mutant–expressing endometrial cancer cells
We measured the kinase-inhibitory activities of AZD4547against WT FGFR1–4 and their mutants (Table 1). AZD4547profoundly inhibited FGFR1–3 with the IC50 values of 2, 1, and7 nmol/L, respectively, and it showed a lower potency on FGFR4(IC50¼ 56 nmol/L). The kinase-inhibitory potencies of AZD4547were also assessed against the three gatekeeper mutants (V561M-FGFR1, V550E/L-FGFR4) and five activating mutants, includingN550H-FGFR2, K650E/M-FGFR3, G697C-FGFR3, and N535K-FGFR4. N550H-FGFR2 (IC50¼ 8 nmol/L), K650E-FGFR3 (IC50¼21 nmol/L), K650M-FGFR3 (IC50 ¼ 82 nmol/L), and G697C-FGFR3 (IC50 ¼ 3 nmol/L), were all potently inhibited byAZD4547, but gatekeeper mutants (V561M-FGFR1, V550M/E-FGFR4) and N535K-FGFR4 were much less sensitive to AZD4547(IC50 > 600 nmol/L).
On the basis of the potent kinase-inhibitory activity against theN550H-FGFR2 mutant (IC50 ¼ 8 nmol/L), we measured theantiproliferative activity of AZD4547 against endometrial cancercell lines harboring FGFR2-activating mutants (Table 1). We alsoincluded HEC1A endometrial cancers expressing normal FGFR2.AZD4547 strongly suppressed the proliferation of AN3-CA cellsharboring the FGFR2 doublemutant K310R/N550Kwith theGI50value of 31 nmol/L. MFE296 (GI50 ¼ 730 nmol/L) cells andMFE280 (GI50¼ 218 nmol/L) cells, harboringN550K-FGFR2 andS252W-FGFR2, respectively,were about 7- to 20-fold less sensitiveto AZD4547. On the other hand, endometrial cancer cells withother FGFR2 aberration such as Ishikawa (FGFR2 overexpression)and HEC1A (normal FGFR2) cells were resistant to AZD4547.This implicates that AZD4547 was highly effective to FGFR2-activating mutant-addicted endometrial cancers.
Kwak et al.
Mol Cancer Ther; 14(10) October 2015 Molecular Cancer Therapeutics2294
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032
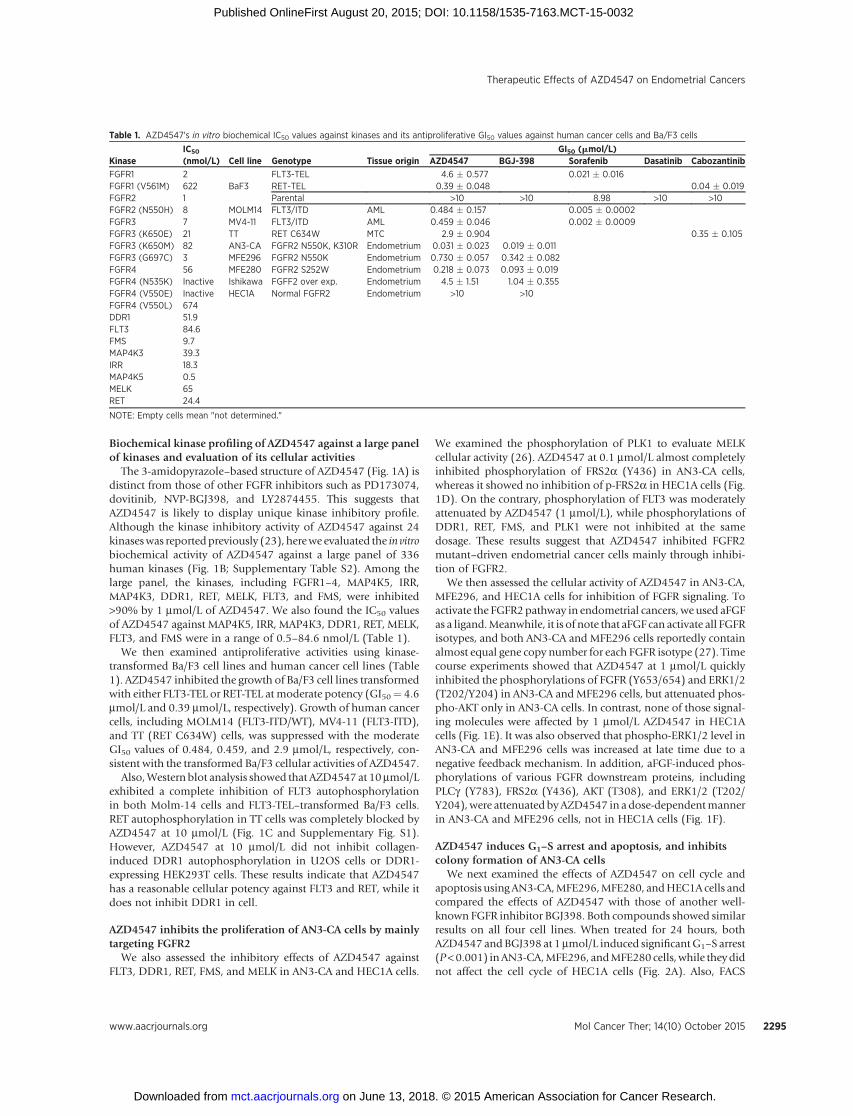
Biochemical kinase profiling of AZD4547 against a large panelof kinases and evaluation of its cellular activities
The 3-amidopyrazole–based structure of AZD4547 (Fig. 1A) isdistinct from those of other FGFR inhibitors such as PD173074,dovitinib, NVP-BGJ398, and LY2874455. This suggests thatAZD4547 is likely to display unique kinase inhibitory profile.Although the kinase inhibitory activity of AZD4547 against 24kinaseswas reportedpreviously (23), herewe evaluated the in vitrobiochemical activity of AZD4547 against a large panel of 336human kinases (Fig. 1B; Supplementary Table S2). Among thelarge panel, the kinases, including FGFR1–4, MAP4K5, IRR,MAP4K3, DDR1, RET, MELK, FLT3, and FMS, were inhibited>90% by 1 mmol/L of AZD4547. We also found the IC50 valuesof AZD4547 against MAP4K5, IRR, MAP4K3, DDR1, RET, MELK,FLT3, and FMS were in a range of 0.5–84.6 nmol/L (Table 1).
We then examined antiproliferative activities using kinase-transformed Ba/F3 cell lines and human cancer cell lines (Table1). AZD4547 inhibited the growth of Ba/F3 cell lines transformedwith either FLT3-TEL or RET-TEL at moderate potency (GI50¼ 4.6mmol/L and 0.39 mmol/L, respectively). Growth of human cancercells, including MOLM14 (FLT3-ITD/WT), MV4-11 (FLT3-ITD),and TT (RET C634W) cells, was suppressed with the moderateGI50 values of 0.484, 0.459, and 2.9 mmol/L, respectively, con-sistent with the transformed Ba/F3 cellular activities of AZD4547.
Also,Western blot analysis showed that AZD4547 at 10mmol/Lexhibited a complete inhibition of FLT3 autophosphorylationin both Molm-14 cells and FLT3-TEL–transformed Ba/F3 cells.RET autophosphorylation in TT cells was completely blocked byAZD4547 at 10 mmol/L (Fig. 1C and Supplementary Fig. S1).However, AZD4547 at 10 mmol/L did not inhibit collagen-induced DDR1 autophosphorylation in U2OS cells or DDR1-expressing HEK293T cells. These results indicate that AZD4547has a reasonable cellular potency against FLT3 and RET, while itdoes not inhibit DDR1 in cell.
AZD4547 inhibits the proliferation of AN3-CA cells by mainlytargeting FGFR2
We also assessed the inhibitory effects of AZD4547 againstFLT3, DDR1, RET, FMS, and MELK in AN3-CA and HEC1A cells.
We examined the phosphorylation of PLK1 to evaluate MELKcellular activity (26). AZD4547 at 0.1 mmol/L almost completelyinhibited phosphorylation of FRS2a (Y436) in AN3-CA cells,whereas it showed no inhibition of p-FRS2a in HEC1A cells (Fig.1D). On the contrary, phosphorylation of FLT3 was moderatelyattenuated by AZD4547 (1 mmol/L), while phosphorylations ofDDR1, RET, FMS, and PLK1 were not inhibited at the samedosage. These results suggest that AZD4547 inhibited FGFR2mutant–driven endometrial cancer cells mainly through inhibi-tion of FGFR2.
We then assessed the cellular activity of AZD4547 in AN3-CA,MFE296, and HEC1A cells for inhibition of FGFR signaling. Toactivate the FGFR2 pathway in endometrial cancers, we used aFGFas a ligand.Meanwhile, it is of note that aFGF can activate all FGFRisotypes, and both AN3-CA and MFE296 cells reportedly containalmost equal gene copy number for each FGFR isotype (27). Timecourse experiments showed that AZD4547 at 1 mmol/L quicklyinhibited the phosphorylations of FGFR (Y653/654) and ERK1/2(T202/Y204) in AN3-CA and MFE296 cells, but attenuated phos-pho-AKT only in AN3-CA cells. In contrast, none of those signal-ing molecules were affected by 1 mmol/L AZD4547 in HEC1Acells (Fig. 1E). It was also observed that phospho-ERK1/2 level inAN3-CA and MFE296 cells was increased at late time due to anegative feedback mechanism. In addition, aFGF-induced phos-phorylations of various FGFR downstream proteins, includingPLCg (Y783), FRS2a (Y436), AKT (T308), and ERK1/2 (T202/Y204),were attenuated byAZD4547 in a dose-dependentmannerin AN3-CA and MFE296 cells, not in HEC1A cells (Fig. 1F).
AZD4547 induces G1–S arrest and apoptosis, and inhibitscolony formation of AN3-CA cells
We next examined the effects of AZD4547 on cell cycle andapoptosis usingAN3-CA,MFE296,MFE280, andHEC1A cells andcompared the effects of AZD4547 with those of another well-known FGFR inhibitor BGJ398. Both compounds showed similarresults on all four cell lines. When treated for 24 hours, bothAZD4547 andBGJ398 at 1mmol/L induced significantG1–S arrest(P<0.001) inAN3-CA,MFE296, andMFE280 cells,while theydidnot affect the cell cycle of HEC1A cells (Fig. 2A). Also, FACS
Table 1. AZD4547's in vitro biochemical IC50 values against kinases and its antiproliferative GI50 values against human cancer cells and Ba/F3 cells
KinaseIC50
(nmol/L) Cell line Genotype Tissue originGI50 (mmol/L)
AZD4547 BGJ-398 Sorafenib Dasatinib Cabozantinib
FGFR1 2 FLT3-TEL 4.6 � 0.577 0.021 � 0.016FGFR1 (V561M) 622 BaF3 RET-TEL 0.39 � 0.048 0.04 � 0.019FGFR2 1 Parental >10 >10 8.98 >10 >10FGFR2 (N550H) 8 MOLM14 FLT3/ITD AML 0.484 � 0.157 0.005 � 0.0002FGFR3 7 MV4-11 FLT3/ITD AML 0.459 � 0.046 0.002 � 0.0009FGFR3 (K650E) 21 TT RET C634W MTC 2.9 � 0.904 0.35 � 0.105FGFR3 (K650M) 82 AN3-CA FGFR2 N550K, K310R Endometrium 0.031 � 0.023 0.019 � 0.011FGFR3 (G697C) 3 MFE296 FGFR2 N550K Endometrium 0.730 � 0.057 0.342 � 0.082FGFR4 56 MFE280 FGFR2 S252W Endometrium 0.218 � 0.073 0.093 � 0.019FGFR4 (N535K) Inactive Ishikawa FGFF2 over exp. Endometrium 4.5 � 1.51 1.04 � 0.355FGFR4 (V550E) Inactive HEC1A Normal FGFR2 Endometrium >10 >10FGFR4 (V550L) 674DDR1 51.9FLT3 84.6FMS 9.7MAP4K3 39.3IRR 18.3MAP4K5 0.5MELK 65RET 24.4
NOTE: Empty cells mean "not determined."
Therapeutic Effects of AZD4547 on Endometrial Cancers
www.aacrjournals.org Mol Cancer Ther; 14(10) October 2015 2295
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032

analysis showed that treatment of either compound for 48 hoursinduced apoptosis in AN3-CA and MFE296 cells, whereas it didnot induce apoptosis in HEC1A cells (Fig. 2B). Upregulation ofcleaved PARP was observed in AN3-CA and MFE280 cells (Fig.2C). We also observed enhancement of p53 transcription activityby AZD4547 (1 mmol/L) in AN3-CA cells, suggesting thatAZD4547 promoted apoptosis and cell-cycle arrest through acti-vation of p53 (Fig. 2D). Furthermore, we evaluated the effect ofAZD4547 on anchorage-independent growth of the three celllines using a soft-agar assay (Fig. 2E). Treatment with AZD4547(0.3 mmol/L or 1 mmol/L) for 3 weeks almost completely blockedthe formation of the colonies of AN3-CA andMFE296 cells, whileit did not block the colony formation of HEC1A cells harboringnormal FGFR2.
AZD4547 inhibits multiple signalings, including JNK andERK1/2, in endometrial cancers
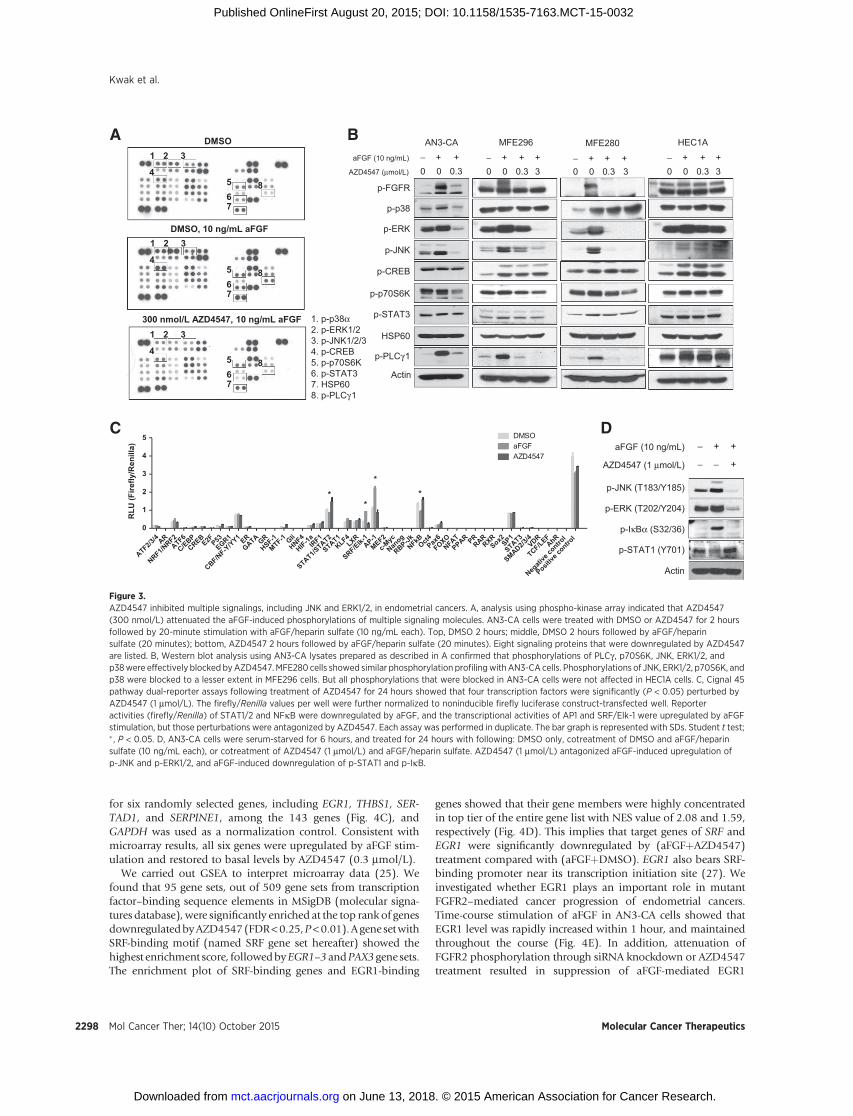
We then used a phospho-kinase array to find which signalingpathways are blocked by AZD4547 in AN3-CA cells. AZD4547 at300 nmol/L attenuated the aFGF-induced phosphorylations of
p38 (T180/Y182), ERK1/2 (T202/Y204, T185, Y187), JNK (T183/Y185, T221/Y223), CREB (S133), p70S6K (T421/S424), STAT3(Y705), and PLCg (Y783; Fig. 3A). We performed Western blotanalysis usingAN3-CA cells and confirmed that phosphorylationsof PLCg , p70S6K, JNK, ERK1/2, and p38 were blocked byAZD4547 (Fig. 3B).We repeated the same analysis usingMFE296,MFE280, andHEC-1A cells aswell. AZD4547 caused similar levelsof inhibition of phosphorylations in MFE280 cells and AN3-CAcells. Phosphorylations of JNK, ERK1/2, p70S6K, and p38 wereblocked to a lesser extent in MFE296 cells. But all phosphoryla-tions that were blocked in AN3-CA cells were not affected inHEC1A cells. Also, AZD4547 at 3 mmol/L slightly inhibitedphospho-GSK3b (Ser 9) in AN3-CA and MFE296 cells (Supple-mentary Fig. S2). This suggests that the activity of AZD4547mightbe related mainly with its ability to block MAPK pathways.
AZD4547 perturbs various transcription factors, includingSRF/Elk-1 and AP-1
Next, we performed 45 pathway dual-reporter gene assaysto investigate which transcription factors were affected by
A
Percent control
336 assays tested32 interactions mappedS-score (35) = 0.11S-score (5) = 0.02
C
p-FLT3
Molm-14
FLT3
AZD4547
mol/L
Sorafenib
U2OS
AZD4547
mol/L
Dasatinib+collagen
TTAZD4547
mol/L
Cabozantinib
p-DDR1
Actin
p-RET
RET
D
p-RET (Y905)
p-DDR1 (Y792)
p-FLT3 (Y591)
p-FMS (Y723)
Actin
AZD4547 (μmol/L)
p-FRS2 (Y436)
p-PLK1 (T210)
AN3-CA HEC1A
p-ERK1/2 (T202/Y204)
p-FGFR(Y653/654)
AKT
p-AKT (S473)
p-AKT (T308)
ERK
FGFR2
Time (h) 0 63 12 241HEC1AMFE296AN3-CA
E
0 63 12 241 0 63 12 241
AZD4547 (nmol/L)
p-AKT(T308)
p-FGFR(Y653/654)
p-FRS2(Y436)
p-ERK1/2(T202/Y204)
p-PLC (Y783)
p-AKT(S473)
AKT
FGFR2
aFGF aFGF aFGF
HEC1AMFE296AN3-CAF
B
3,00
03,
000
3,00
0
0
0.1
0.1–1
1–55–10
10–35>35
Figure 1.In vitro and cellular activities of AZD4547 against FGFRs and other kinases. A, chemical structure of AZD4547. B, kinome-wide inhibition profiling of AZD4547at 1 mmol/L. Kinases showing >75% inhibition were mapped on kinome tree. The sizes of dots indicate the remaining activity of each kinase after the inhibitionby AZD4547. C, AZD4547 moderately inhibited the autophosphorylations of FLT3 (Molm-14 cells) and RET (TT cells), but not that of DDR1 in U2OS cells. Forinduction of DDR1 phosphorylation, serum-starved U2OS cells were pretreated with compounds for 2 hours before stimulation with collagen (10 ng/mL) for1 hour. D, AZD4547 potently suppressed phosphorylation of FRS2a, whereas it showedmild inhibition of p-FLT3 and no inhibition of p-RET, p-DDR1, p-FMS, or p-PLK1in both AN3-CA and HEC1A cells. E, a time-course experiment showed that AZD4547 (1 mmol/L) quickly downregulated p-FGFR and p-ERK1/2 in AN3-CA andMFE296 cells, but downregulated p-AKT only in AN3-CA cells, whereas it showed no effect in HEC1A cells. Phospho-ERK1/2 level in AN3-CA and MFE296 cells wasincreased at late time due to a negative feedback mechanism. F, AZD4547 blocked p-FGFR2 and its downstream signalings in a dose-dependent manner inendometrial cancer cells harboring FGFR2 mutants (AN3-CA, MFE296), not in HEC1A cells harboring normal FGFR2.
Kwak et al.
Mol Cancer Ther; 14(10) October 2015 Molecular Cancer Therapeutics2296
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032

AZD4547 in AN3-CA cells. Among 45 transcription factors,four transcription factors were significantly (P < 0.05) per-turbed by AZD4547 (Fig. 3C). Reporter activities (firefly/Renilla) of STAT1/2 and NFkB were observed to be down-regulated by aFGF, and antagonized by AZD4547. In contrast,the transcriptional activity of AP1 and SRF/Elk-1 were upre-gulated by aFGF stimulation, and AZD4547 blocked the upre-gulation of AP1 and SRF/Elk-1. The perturbations of these fourtranscription factors were verified by measuring the phospholevels of the transcription factors or their upstream regulators(Fig. 3D). Western blot analysis showed that phosphorylationsof JNK, ERK1/2, and IkBa, which are well-known upstreamregulators of AP-1, Elk-1, and NFkB, respectively, wereenhanced by aFGF, and were antagonized by AZD4547. Like-wise, phosphorylation of STAT1 was downregulated by aFGFstimulation, and was antagonized by AZD4547.
AZD4547 changes the transcriptions of a number of genes,including EGR1
In addition, we carried out a genome-wide transcriptomeanalysis using AN3-CA cells. AN3-CA cells were treated withvehicle (DMSO only), with vehicle and aFGF (DMSOþaFGF),or with AZD4547 and aFGF (AZD4547þaFGF). RNAs were har-vested at 18 hours after treatment and analyzed using AffymetixPrimeview arrays (Fig. 4A). We identified that 143 probes wereupregulated more than 2-fold by aFGF stimulation relative toDMSO only (P < 0.01), and that 240 probes were significantlydownregulated less than 0.5-fold after cotreatment of aFGF andAZD4547 relative to (aFGFþDMSO) treatment (P < 0.01; Sup-plementary Table S3 and S4). Thus, we assumed that all 143 genesoverlapped in both probe sets would be the FGFR downstreamtarget genes (Fig. 4B; Supplementary Table S5). To validate themicroarray results, reverse transcription (RT)-PCR was performed
A
B CA
N3-
CA
HE
C1A
MFE
296
1 10.3 0.3 BGJ398 (μmol/L)AZD4547 (μmol/L)
0 100 200 300 400 500
DMSO0.3 mmol/L AZD4547
1 mmol/L AZD45470.3 mmol/L BGJ398
1 mmol/L BGJ398
******
******
Number of colonies
0 200 400 600 800 1,000
DMSO0.3 mmol/L AZD4547
1 mmol/L AZD45470.3 mmol/L BGJ398
1 mmol/L BGJ398
Number of colonies
0 200 400 600
DMSO0.3 mmol/L AZD4547
1 mmol/L AZD45470.3 mmol/L BGJ398 1 mmol/L BGJ398
******
***
***
Number of colonies
E
AZD4547 (1 μmol/L)
PARP1/2
Actin
AN3-CA MFE296 HEC1A MFE280
− 48 72 h− 48 72 − 48 72 − 48 72
++−+−−
aFGF (10 ng/mL)
AZD4547 (1 μmol/L)
D DMSO
100
50
0
100
50
0
100
50
0
100
50
0
Pop
ulat
ion
(%)
Pop
ulat
ion
(%)
Pop
ulat
ion
(%)
Pop
ulat
ion
(%)
AN3-CA
AN3-CA
MFE296
MFE296
MFE280HEC1A
HEC1A
Sub-G1G0–G1
G2–MS
Sub-G1G0–G1
G2–MS
Sub-G1G0–G1
G2–MS
Sub-G1G0–G1
G2–MS
DMSO
AZD4547
BGJ398
DMSO
AZD4547
BGJ398
DMSO
AZD4547
BGJ398
DMSO
AZD4547
BGJ398
DMSO
AZD4547
BGJ398
DMSO
AZD4547
BGJ398
DMSO
AZD4547
BGJ398
Apo
ptos
is (%
)
Apo
ptos
is (%
)
Apo
ptos
is (%
)
25
20
15
10
5
0
25
20
15
10
5
0
25
20
15
10
5
0
RLU
4
3
2
1
0
Figure 2.AZD4547 induces G1–S arrest and apoptosis mediated through p53, and inhibits colony formation of AN3-CA cells. A, treatment of AZD4547 orBGJ398 (1 mmol/L) for 24 hours induced significant G1–S arrest in AN3CA, MFE296, and MFE280 cells, while they did not affect the cell cycle of HEC1A cells.B, treatment of AZD4547 or BGJ398 (1 mmol/L) for 48 hours induced apoptosis in AN3-CA cells, whereas it induced almost no apoptosis in MFE296and HEC1A cells. Each graph in A and B presents the means and the SDs of three independent experiments that were run on different days. One-way ANOVA;� , P < 0.05; ��, P < 0.01. C, treatment of AZD4547 or BGJ398 (1 mmol/L) for 48 hours increased the level of cleaved PARP in AN3-CA and MFE280cells, but not in MFE296 and HEC1A cells. D, treatment of AZD4547 (1 mmol/L) for 24 hours enhanced p53 transcription activity. Assays were done induplicate, and average and SD are depicted. Student t test; � , P < 0.05. E, AZD4547 suppresses anchorage-independent growth of AN3-CA and MFE296cells, but not in HEC1A cells. Assays were performed three independent times, and bar graph represents the average (n ¼ 3) and SD. One-wayANOVA; ��� , P < 0.001.
Therapeutic Effects of AZD4547 on Endometrial Cancers
www.aacrjournals.org Mol Cancer Ther; 14(10) October 2015 2297
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032
for six randomly selected genes, including EGR1, THBS1, SER-TAD1, and SERPINE1, among the 143 genes (Fig. 4C), andGAPDH was used as a normalization control. Consistent withmicroarray results, all six genes were upregulated by aFGF stim-ulation and restored to basal levels by AZD4547 (0.3 mmol/L).
We carried out GSEA to interpret microarray data (25). Wefound that 95 gene sets, out of 509 gene sets from transcriptionfactor–binding sequence elements in MSigDB (molecular signa-tures database), were significantly enriched at the top rank of genesdownregulatedbyAZD4547(FDR<0.25,P<0.01).Agene setwithSRF-binding motif (named SRF gene set hereafter) showed thehighest enrichment score, followedbyEGR1–3 andPAX3 gene sets.The enrichment plot of SRF-binding genes and EGR1-binding
genes showed that their gene members were highly concentratedin top tier of the entire gene list with NES value of 2.08 and 1.59,respectively (Fig. 4D). This implies that target genes of SRF andEGR1 were significantly downregulated by (aFGFþAZD4547)treatment compared with (aFGFþDMSO). EGR1 also bears SRF-binding promoter near its transcription initiation site (27). Weinvestigated whether EGR1 plays an important role in mutantFGFR2–mediated cancer progression of endometrial cancers.Time-course stimulation of aFGF in AN3-CA cells showed thatEGR1 level was rapidly increased within 1 hour, and maintainedthroughout the course (Fig. 4E). In addition, attenuation ofFGFR2 phosphorylation through siRNA knockdown or AZD4547treatment resulted in suppression of aFGF-mediated EGR1
DMSO1 2 3
4
DMSO, 10 ng/mL aFGF 1 2 3
4
300 nmol/L AZD4547, 10 ng/mL aFGF 1 2 3
4
AaFGF (10 ng/mL)
AZD4547 (μmol/L)
− + +0 0 0.3
AN3-CA MFE296− + +0 0 0.3
+3
B
ATF2/3/4 AR
NRF1/NRF2
ATF6
C/EBPCREB
E2FP53EGR1
CBF/NF-Y
/YY1 ERGATA GR
HSF-1MTF-1 Gli
HNF4
HIF-1aIRF1
STAT1/STAT2
STAT1KLF4
LXR
SRF/Elk-1AP-1
MEF2c-M
yc
Nanog
RBP-JkNFkBOct4Pax
6FOXO
NFATPPAR PR
RARRXR
Sox2SP1
STAT3
SMAD2/3/4VDR
TCF/LEFAhR
Negati
ve co
ntrol
Positive
contro
l 0
1
2
3
4
5 DMSOaFGFAZD4547
*
** *
RLU
(Fire
fly/R
enill
a)
C
1. p-p38α2. p-ERK1/23. p-JNK1/2/34. p-CREB5. p-p70S6K6. p-STAT37. HSP608. p-PLCγ1
p-FGFR
p-p38
p-ERK
p-JNK
p-CREB
p-STAT3
HSP60
p-PLCγ1
p-p70S6K
Actin
56
8
7
56
8
7
56
8
7
aFGF (10 ng/mL)
AZD4547 (1 μmol/L) +
+−
− −
+
p-JNK (T183/Y185)
p-ERK (T202/Y204)
p-STAT1 (Y701)
Actin
D
p-IκBα (S32/36)
MFE280
− + +0 0 0.3
+3
HEC1A− + +0 0 0.3
+3
Figure 3.AZD4547 inhibited multiple signalings, including JNK and ERK1/2, in endometrial cancers. A, analysis using phospho-kinase array indicated that AZD4547(300 nmol/L) attenuated the aFGF-induced phosphorylations of multiple signaling molecules. AN3-CA cells were treated with DMSO or AZD4547 for 2 hoursfollowed by 20-minute stimulation with aFGF/heparin sulfate (10 ng/mL each). Top, DMSO 2 hours; middle, DMSO 2 hours followed by aFGF/heparinsulfate (20 minutes); bottom, AZD4547 2 hours followed by aFGF/heparin sulfate (20 minutes). Eight signaling proteins that were downregulated by AZD4547are listed. B, Western blot analysis using AN3-CA lysates prepared as described in A confirmed that phosphorylations of PLCg , p70S6K, JNK, ERK1/2, andp38were effectively blocked byAZD4547.MFE280 cells showed similar phosphorylation profilingwithAN3-CA cells. Phosphorylations of JNK, ERK1/2, p70S6K, andp38 were blocked to a lesser extent in MFE296 cells. But all phosphorylations that were blocked in AN3-CA cells were not affected in HEC1A cells. C, Cignal 45pathway dual-reporter assays following treatment of AZD4547 for 24 hours showed that four transcription factors were significantly (P < 0.05) perturbed byAZD4547 (1 mmol/L). The firefly/Renilla values per well were further normalized to noninducible firefly luciferase construct-transfected well. Reporteractivities (firefly/Renilla) of STAT1/2 and NFkB were downregulated by aFGF, and the transcriptional activities of AP1 and SRF/Elk-1 were upregulated by aFGFstimulation, but those perturbations were antagonized by AZD4547. Each assay was performed in duplicate. The bar graph is represented with SDs. Student t test;� , P < 0.05. D, AN3-CA cells were serum-starved for 6 hours, and treated for 24 hours with following: DMSO only, cotreatment of DMSO and aFGF/heparinsulfate (10 ng/mL each), or cotreatment of AZD4547 (1 mmol/L) and aFGF/heparin sulfate. AZD4547 (1 mmol/L) antagonized aFGF-induced upregulation ofp-JNK and p-ERK1/2, and aFGF-induced downregulation of p-STAT1 and p-IkB.
Kwak et al.
Mol Cancer Ther; 14(10) October 2015 Molecular Cancer Therapeutics2298
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032
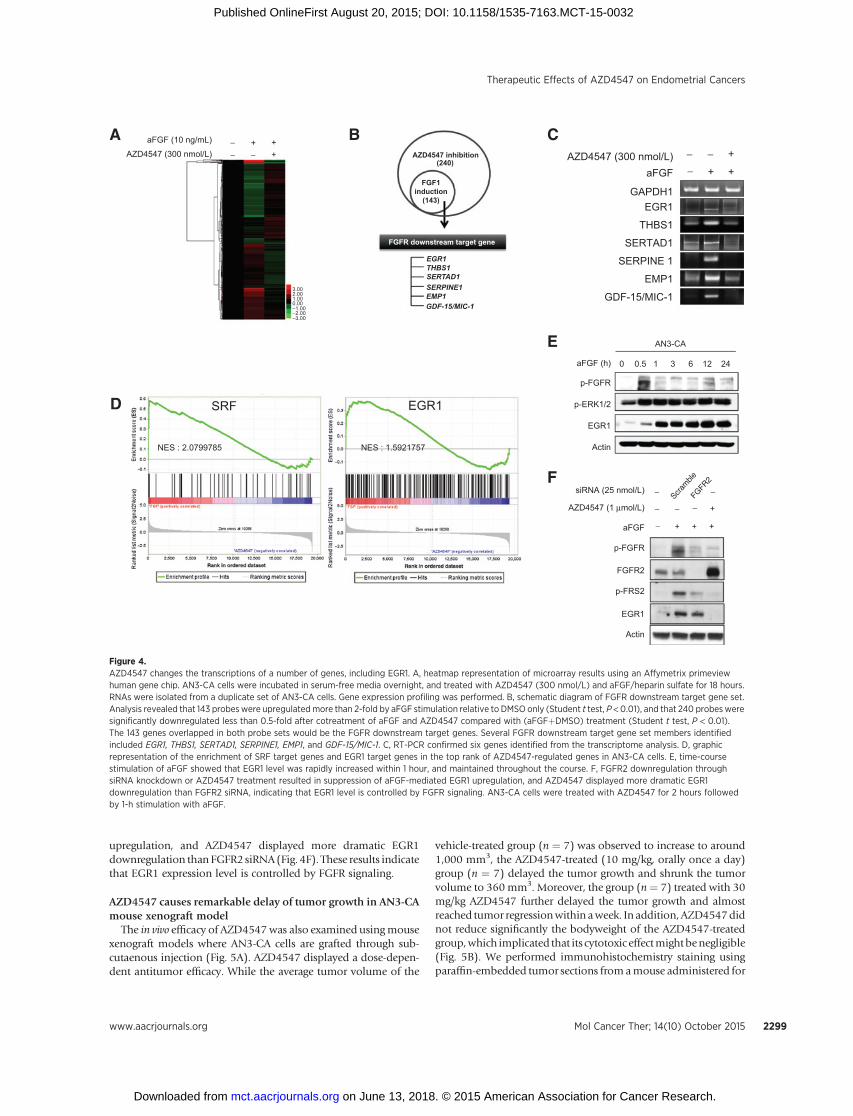
upregulation, and AZD4547 displayed more dramatic EGR1downregulation than FGFR2 siRNA(Fig. 4F). These results indicatethat EGR1 expression level is controlled by FGFR signaling.
AZD4547 causes remarkable delay of tumor growth in AN3-CAmouse xenograft model
The in vivo efficacy of AZD4547was also examined usingmousexenograft models where AN3-CA cells are grafted through sub-cutaenous injection (Fig. 5A). AZD4547 displayed a dose-depen-dent antitumor efficacy. While the average tumor volume of the
vehicle-treated group (n ¼ 7) was observed to increase to around1,000 mm3, the AZD4547-treated (10 mg/kg, orally once a day)group (n ¼ 7) delayed the tumor growth and shrunk the tumorvolume to 360mm3. Moreover, the group (n¼ 7) treated with 30mg/kg AZD4547 further delayed the tumor growth and almostreached tumor regressionwithin aweek. In addition,AZD4547didnot reduce significantly the bodyweight of the AZD4547-treatedgroup,which implicated that its cytotoxic effectmightbenegligible(Fig. 5B). We performed immunohistochemistry staining usingparaffin-embedded tumor sections fromamouse administered for
A aFGF (10 ng/mL)
AZD4547 (300 nmol/L) ++−
− −+
B
aFGFAZD4547 (300 nmol/L)
−−
+
−
+
+
GAPDH1
THBS1
SERPINE 1
EMP1
GDF-15/MIC-1
SERTAD1
EGR1
C
SRF EGR1D
NES : 2.0799785 NES : 1.5921757
AN3-CA
0 0.5 1 3 6 12 24aFGF (h)
EGR1
Actin
p-ERK1/2
p-FGFR
E
p-FGFR
p-FRS2
Actin
aFGF
AZD4547 (1 μmol/L)
−
−
+
−
+
+
siRNA (25 nmol/L)
−
+
− −
FGFR2
EGR1
F
Scram
bleFGFR2
EGR1THBS1SERTAD1SERPINE1EMP1GDF-15/MIC-1
AZD4547 inhibition
FGF1induction
(143)
(240)
FGFR downstream target gene
3.002.001.000.00−1.00−2.00−3.00
Figure 4.AZD4547 changes the transcriptions of a number of genes, including EGR1. A, heatmap representation of microarray results using an Affymetrix primeviewhuman gene chip. AN3-CA cells were incubated in serum-free media overnight, and treated with AZD4547 (300 nmol/L) and aFGF/heparin sulfate for 18 hours.RNAs were isolated from a duplicate set of AN3-CA cells. Gene expression profiling was performed. B, schematic diagram of FGFR downstream target gene set.Analysis revealed that 143 probeswere upregulatedmore than 2-fold by aFGF stimulation relative to DMSO only (Student t test, P < 0.01), and that 240 probesweresignificantly downregulated less than 0.5-fold after cotreatment of aFGF and AZD4547 compared with (aFGFþDMSO) treatment (Student t test, P < 0.01).The 143 genes overlapped in both probe sets would be the FGFR downstream target genes. Several FGFR downstream target gene set members identifiedincluded EGR1, THBS1, SERTAD1, SERPINE1, EMP1, and GDF-15/MIC-1. C, RT-PCR confirmed six genes identified from the transcriptome analysis. D, graphicrepresentation of the enrichment of SRF target genes and EGR1 target genes in the top rank of AZD4547-regulated genes in AN3-CA cells. E, time-coursestimulation of aFGF showed that EGR1 level was rapidly increased within 1 hour, and maintained throughout the course. F, FGFR2 downregulation throughsiRNA knockdown or AZD4547 treatment resulted in suppression of aFGF-mediated EGR1 upregulation, and AZD4547 displayed more dramatic EGR1downregulation than FGFR2 siRNA, indicating that EGR1 level is controlled by FGFR signaling. AN3-CA cells were treated with AZD4547 for 2 hours followedby 1-h stimulation with aFGF.
Therapeutic Effects of AZD4547 on Endometrial Cancers
www.aacrjournals.org Mol Cancer Ther; 14(10) October 2015 2299
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032
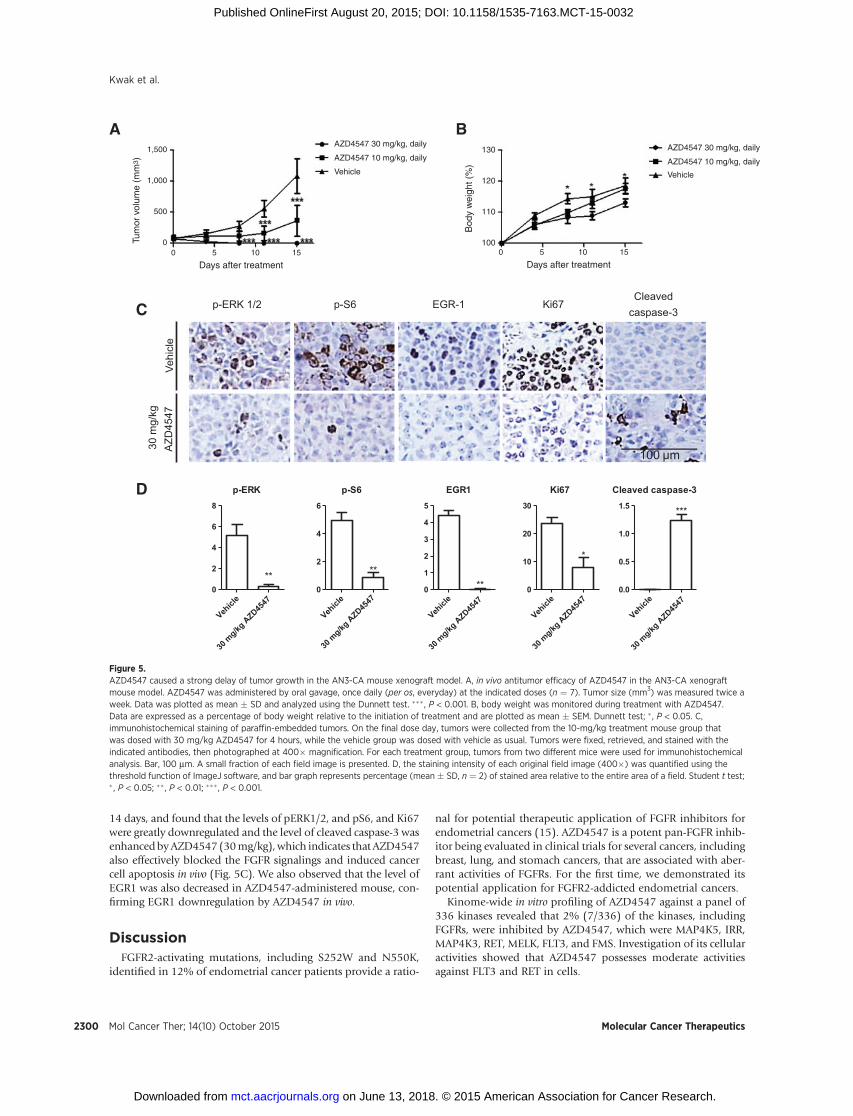
14 days, and found that the levels of pERK1/2, and pS6, and Ki67were greatly downregulated and the level of cleaved caspase-3 wasenhancedbyAZD4547 (30mg/kg),which indicates that AZD4547also effectively blocked the FGFR signalings and induced cancercell apoptosis in vivo (Fig. 5C). We also observed that the level ofEGR1 was also decreased in AZD4547-administered mouse, con-firming EGR1 downregulation by AZD4547 in vivo.
DiscussionFGFR2-activating mutations, including S252W and N550K,
identified in 12% of endometrial cancer patients provide a ratio-
nal for potential therapeutic application of FGFR inhibitors forendometrial cancers (15). AZD4547 is a potent pan-FGFR inhib-itor being evaluated in clinical trials for several cancers, includingbreast, lung, and stomach cancers, that are associated with aber-rant activities of FGFRs. For the first time, we demonstrated itspotential application for FGFR2-addicted endometrial cancers.
Kinome-wide in vitro profiling of AZD4547 against a panel of336 kinases revealed that 2% (7/336) of the kinases, includingFGFRs, were inhibited by AZD4547, which were MAP4K5, IRR,MAP4K3, RET, MELK, FLT3, and FMS. Investigation of its cellularactivities showed that AZD4547 possesses moderate activitiesagainst FLT3 and RET in cells.
p-S6
Vehicl
e
30 m
g/kg A
ZD4547
0
2
4
6
**
p-ERK
Vehicl
e
30 m
g/kg A
ZD4547
0
2
4
6
8
**
Ki67
Vehicl
e
30 m
g/kg A
ZD4547
0
10
20
30
*
Cleaved caspase-3
Vehicl
e
30 m
g/kg A
ZD4547
0.0
0.5
1.0
1.5 ***
EGR1
Vehicl
e
30 m
g/kg A
ZD4547
0
1
2
3
4
5
**
A B
100 μm
D
1,500
1,000
500
00 5 10 15
Days after treatment0 5 10 15
Days after treatment
Tum
or v
olum
e (m
m3 )
Bod
y w
eigh
t (%
)
AZD4547 30 mg/kg, daily
AZD4547 10 mg/kg, daily
Vehicle
AZD4547 30 mg/kg, daily
AZD4547 10 mg/kg, daily
Vehicle
130
120
110
100
EGR-1Cleaved
caspase-3p-ERK 1/2 Ki67p-S6C
Vehi
cle
30 m
g/kg
AZD
4547
Figure 5.AZD4547 caused a strong delay of tumor growth in the AN3-CA mouse xenograft model. A, in vivo antitumor efficacy of AZD4547 in the AN3-CA xenograftmouse model. AZD4547 was administered by oral gavage, once daily (per os, everyday) at the indicated doses (n ¼ 7). Tumor size (mm3) was measured twice aweek. Data was plotted as mean � SD and analyzed using the Dunnett test. ��� , P < 0.001. B, body weight was monitored during treatment with AZD4547.Data are expressed as a percentage of body weight relative to the initiation of treatment and are plotted as mean � SEM. Dunnett test; � , P < 0.05. C,immunohistochemical staining of paraffin-embedded tumors. On the final dose day, tumors were collected from the 10-mg/kg treatment mouse group thatwas dosed with 30 mg/kg AZD4547 for 4 hours, while the vehicle group was dosed with vehicle as usual. Tumors were fixed, retrieved, and stained with theindicated antibodies, then photographed at 400� magnification. For each treatment group, tumors from two different mice were used for immunohistochemicalanalysis. Bar, 100 mm. A small fraction of each field image is presented. D, the staining intensity of each original field image (400�) was quantified using thethreshold function of ImageJ software, and bar graph represents percentage (mean � SD, n ¼ 2) of stained area relative to the entire area of a field. Student t test;� , P < 0.05; �� , P < 0.01; ��� , P < 0.001.
Kwak et al.
Mol Cancer Ther; 14(10) October 2015 Molecular Cancer Therapeutics2300
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032

We found that AZD4547 was most effective to AN3-CA cellsexpressing FGFR2 N550K/K310R mutation. AZD4547 effectivelyblocked autophosphorylation of FGFRs and its subsequent down-stream signaling, inducing cell-cycle arrest at G1–S transition andapoptosis in AN3-CA cells, whereas it had no significant activityon HEC1A harboring normal FGFR2. In addition, like otherFGFR-targeted inhibitors such as PD173074, dovitinib, andBGJ-398, AZD4547 showed weaker activity against MFE296(FGFR2 N550K) and MFE280 (FGFR2 S252W) cells than againstAN3-CA cells (15, 18, 28).
The results from phospho-kinase array experiment using AN3-CA cells revealed that AZD4547 perturbed mainly ERK1/2 andJNK signalings. Also our reporter gene assays showed that theactivities of SRF/Elk-1 and AP-1, which are effector transcriptionfactors of ERK1/2 and JNK, respectively, were downregulated byAZD4547. This is consistent with the report that downregulationof ERK1/2 activity is associated with therapeutic effects of FGFRinhibitor (PD173074) in endometrial cancer cells harboringmutants of PTEN and FGFR2 (17). Our results also demonstratedthat the anticancer activity ofAZD4547might bemediatedmainlythrough inhibition of the MAPK pathway.
GSEA analysis from our gene transcription profiling suggestedthat AZD4547 suppressed cancer progression of AN3-CA cells viaEGR1 downregulation. Acidic FGF is a well-known strong activa-tor of EGR1 (29), and EGR1 gene was identified to have severalfunctional elements, including serum response elements (SREs),AP-1, cAMP response elements (CRE), and SP-1, at the 30 end of itspromoter (30). Nonetheless, the significance of FGFR2-EGR1 axisin endometrial cancer progression has not been reported yet.EGR1 expression was dependent on FGFR2 activity, becausesiRNA knockdown of FGFR2 perturbed aFGF-induced EGR1upregulation. In addition, an in vivo xenograft model of AN3-CAshowed that EGR1 was effectively downregulated in AZD4547-administered tumors. Our study suggests that EGR1 downregula-tion is related to AZD4547's efficacy against endometrial cancers.
We also showed that AZD4547 suppressed anchorage-inde-pendent growth of AN3-CA cells and induced significant tumorregression in the AN3-CAmouse xenograft model in conjunctionwith a significant decrease in proliferation markers, including
p-ERK1/2 and Ki67, in tumors. The efficacy of AZD4547might beassociated with the findings from unbiased reporter gene assaysand transcriptome analysis using AN3-CA cells. AZD4547 per-turbed the activities of a number of transcription factors, such asEGR-1 and SP1, which are related to proliferation as well asupregulating p53 activity.
We here report the detailed mechanism by which AZD4547expresses its anticancer effect against FGFR2 mutant–dependentendometrial cancer cells. Our study demonstrates that AZD4547exhibits its therapeutic activity against endometrial cancer cellsby perturbing various regulatory mechanisms related to FGFRsignaling.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: Y. Kwak, T. SimAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Kwak, H. ChoAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): Y. Kwak, W. Hur, T. SimWriting, review, and/or revision of the manuscript: Y. Kwak, W. Hur, T. SimAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): Y. Kwak, T. SimStudy supervision: T. Sim
Grant SupportThis work was supported by the Korea Institute of Science and Technology
(KIST), the Creative/Challenging Research Program (2011-0028676) of theNational Research Foundation of Korea (NRF), a grant (D33400) of the KoreaBasic Science Institute, and a grant from the Creative Fusion Research Programthrough the Creative Allied Project funded by the National Research Council ofScience and Technology (CAP-12-1-KIST; to T. Sim).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received January 12, 2015; revised July 14, 2015; accepted August 3, 2015;published OnlineFirst August 20, 2015.
References1. Johnson DE, Williams LT. Structural and functional diversity in the FGF
receptor multigene family. Adv Cancer Res 1993;60:1–41.2. Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon
A, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals adual role for heparin in FGFR binding and dimerization. Mol Cell2000;6:743–50.
3. Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A,et al. FGFR1 emerges as a potential therapeutic target for lobular breastcarcinomas. Clin Cancer Res 2006;12:6652–62.
4. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al.Frequent and focal FGFR1 amplification associates with therapeuticallytractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med2010;2:62ra93.
5. Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM, Bergsagel PL. Thet(4;14) translocation in myeloma dysregulates both FGFR3 and a novelgene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood 1998;92:3025–34.
6. van Rhijn BW, Montironi R, Zwarthoff EC, Jobsis AC, van der Kwast TH.Frequent FGFR3 mutations in urothelial papilloma. J Pathol 2002;198:245–51.
7. Yagasaki F,WakaoD, Yokoyama Y, Uchida Y,Murohashi I, KayanoH, et al.Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-celllymphoma with a t(4;12)(p16;p13) chromosomal translocation. CancerRes 2001;61:8371–4.
8. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al.Transforming fusions of FGFR and TACC genes in human glioblastoma.Science 2012;337:1231–5.
9. Taylor JGt, Cheuk AT, Tsang PS, Chung JY, Song YK, Desai K, et al.Identification of FGFR4-activating mutations in human rhabdomyosarco-mas that promote metastasis in xenotransplanted models. J Clin Invest2009;119:3395–407.
10. De Giovanni C, Landuzzi L, Nicoletti G, Lollini PL, Nanni P. Molecularand cellular biology of rhabdomyosarcoma. Future Oncol 2009;5:1449–75.
11. Ibrahimi OA, Chiu ES, McCarthy JG, Mohammadi M. Understandingthe molecular basis of Apert syndrome. Plastic Reconstr Surg 2005;115:264–70.
12. Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S.Mutations in the fibroblast growth factor receptor 2 gene cause Crouzonsyndrome. Nat Genet 1994;8:98–103.
Therapeutic Effects of AZD4547 on Endometrial Cancers
www.aacrjournals.org Mol Cancer Ther; 14(10) October 2015 2301
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032

13. Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factorreceptor 2 ligand-binding specificity in Apert syndrome. Proc Natl Acad SciU S A 2000;97:14536–41.
14. Byron SA, Gartside M, Powell MA, Wellens CL, Gao F, Mutch DG, et al.FGFR2 point mutations in 466 endometrioid endometrial tumors: rela-tionship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopatho-logical features. PLoS ONE 2012;7:e30801.
15. Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, et al.Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc NatlAcad Sci U S A 2008;105:8713–7.
16. Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, et al. A molecularbrake in the kinase hinge region regulates the activity of receptor tyrosinekinases. Mol Cell 2007;27:717–30.
17. Byron SA, Gartside MG, Wellens CL, Mallon MA, Keenan JB, Powell MA,et al. Inhibition of activated fibroblast growth factor receptor 2 in endo-metrial cancer cells induces cell death despite PTEN abrogation. Cancer Res2008;68:6902–7.
18. Konecny GE, Kolarova T, O'Brien NA, Winterhoff B, Yang G, Qi J, et al.Activity of the fibroblast growth factor receptor inhibitors dovitinib(TKI258) andNVP-BGJ398 inhuman endometrial cancer cells.Mol CancerTher 2013;12:632–42.
19. Katoh M, Nakagama H. FGF receptors: Cancer biology and therapeutics.Med Res Rev 2014;34:280–300.
20. Liu L, Ye TH,HanYP, SongH,ZhangYK, Xia Y, et al. Reductions inmyeloid-derived suppressor cells and lungmetastases usingAZD4547 treatment of ametastatic murine breast tumor model. Cell Physiol Biochem 2014;33:633–45.
21. Xie L, Su X, Zhang L, Yin X, Tang L, Zhang X, et al. FGFR2 gene amplificationin gastric cancer predicts sensitivity to the selective FGFR inhibitorAZD4547. Clin Cancer Res 2013;19:2572–83.
22. Zhang J, Zhang L, Su X, Li M, Xie L, Malchers F, et al. Translating thetherapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung
cancer through the use of patient-derived tumor xenograft models. ClinCancer Res 2012;18:6658–67.
23. Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al.AZD4547: an orally bioavailable, potent, and selective inhibitor of thefibroblast growth factor receptor tyrosine kinase family. Cancer Res2012;72:2045–56.
24. Melnick JS, Janes J, Kim S, Chang JY, Sipes DG, Gunderson D, et al. Anefficient rapid system for profiling the cellular activities of molecularlibraries. Proc Natl Acad Sci U S A 2006;103:3153–8.
25. SubramanianA, TamayoP,Mootha VK,Mukherjee S, Ebert BL,GilletteMA,et al. Gene set enrichment analysis: a knowledge-based approach forinterpreting genome-wide expression profiles. Proc Natl Acad Sci U S A2005;102:15545–50.
26. Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, et al.MELK-dependent FOXM1 phosphorylation is essential for proliferation ofglioma stem cells. Stem Cells 2013;31:1051–63.
27. Bai A, Meetze K, Vo NY, Kollipara S, Mazsa EK, WinstonWM, et al. GP369,an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity againsthuman cancers driven by activated FGFR2 signaling. Cancer Res 2010;70:7630–9.
28. Byron SA, Chen H, Wortmann A, Loch D, Gartside MG, Dehkhoda F, et al.The N550K/H mutations in FGFR2 confer differential resistance toPD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neopla-sia 2013;15:975–88.
29. Benz AH, Shajari M, Peruzki N, Dehghani F, Maronde E. Earlygrowth response-1 induction by fibroblast growth factor-1 viaincrease of mitogen-activated protein kinase and inhibition of pro-tein kinase B in hippocampal neurons. Br J Pharmacol 2010;160:1621–30.
30. Pagel JI, Deindl E. Early growth response 1-a transcription factor in thecrossfire of signal transduction cascades. Indian J Biochem Biophys2011;48:226–35.
Mol Cancer Ther; 14(10) October 2015 Molecular Cancer Therapeutics2302
Kwak et al.
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032
2015;14:2292-2302. Published OnlineFirst August 20, 2015.Mol Cancer Ther Yeonui Kwak, Hanna Cho, Wooyoung Hur, et al. FGFR2-Deregulated Endometrial Cancer CellsAntitumor Effects and Mechanisms of AZD4547 on
Updated version
10.1158/1535-7163.MCT-15-0032doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2015/08/20/1535-7163.MCT-15-0032.DC1
Access the most recent supplemental material at:
Cited articles
http://mct.aacrjournals.org/content/14/10/2292.full#ref-list-1
This article cites 30 articles, 15 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/14/10/2292.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/14/10/2292To request permission to re-use all or part of this article, use this link
on June 13, 2018. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 20, 2015; DOI: 10.1158/1535-7163.MCT-15-0032


















