
and Non-fuel-stimulated Insulin Secretion*DS
13
Adipose Triglyceride Lipase Is Implicated in Fuel- and Non-fuel-stimulated Insulin Secretion * □ S Received for publication, February 25, 2009, and in revised form, April 9, 2009 Published, JBC Papers in Press, April 22, 2009, DOI 10.1074/jbc.M109.006650 Marie-Line Peyot ‡ , Claudiane Guay ‡1 , Martin G. Latour ‡ , Julien Lamontagne ‡2 , Roxane Lussier ‡ , Marco Pineda ‡ , Neil B. Ruderman § , Guenter Haemmerle ¶ , Rudolf Zechner ¶ ,E ´ rik Joly ‡ , S. R. Murthy Madiraju ‡ , Vincent Poitout ‡ ** 3 , and Marc Prentki ‡4 From the ‡ Molecular Nutrition Unit and the Montreal Diabetes Research Center, Centre de Recherche du Centre Hospitalier de l’Universite ´ de Montre ´al, and Departments of Nutrition and **Medicine, University of Montreal, Montreal, Quebec H1W 4A4, Canada, the § Departments of Medicine and Physiology and Biophysics, Boston University School of Medicine and Diabetes Unit, Section of Endocrinology, Boston University School of Medicine and Boston Medical Center, Boston, Massachusetts 02118, and the ¶ Institute of Molecular Biosciences, Karl-Franzens-University, Graz 8010, Austria Reduced lipolysis in hormone-sensitive lipase-deficient mice is associated with impaired glucose-stimulated insulin secretion (GSIS), suggesting that endogenous -cell lipid stores provide signaling molecules for insulin release. Measurements of lipol- ysis and triglyceride (TG) lipase activity in islets from HSL / mice indicated the presence of other TG lipase(s) in the -cell. Using real time-quantitative PCR, adipose triglyceride lipase (ATGL) was found to be the most abundant TG lipase in rat islets and INS832/13 cells. To assess its role in insulin secretion, ATGL expression was decreased in INS832/13 cells (ATGL-knock- down (KD)) by small hairpin RNA. ATGL-KD increased the ester- ification of free fatty acid (FFA) into TG. ATGL-KD cells showed decreased glucose- or Gln Leu-induced insulin release, as well as reduced response to KCl or palmitate at high, but not low, glucose. The K ATP -independent/amplification pathway of GSIS was con- siderably reduced in ATGL-KD cells. ATGL / mice were hypoin- sulinemic and hypoglycemic and showed decreased plasma TG and FFAs. A hyperglycemic clamp revealed increased insulin sensitivity and decreased GSIS and arginine-induced insulin secretion in ATGL / mice. Accordingly, isolated islets from ATGL / mice showed reduced insulin secretion in response to glucose, glucose palmitate, and KCl. Islet TG content and FFA esterification into TG were increased by 2-fold in ATGL / islets, but glucose usage and oxidation were unaltered. The results demonstrate the impor- tance of ATGL and intracellular lipid signaling for fuel- and non- fuel-induced insulin secretion. Free fatty acids (FFA) 5 and other lipid molecules are impor- tant for proper glucose-stimulated insulin secretion (GSIS) by -cells. Thus, deprivation of fatty acids (FA) in vivo (1) dimin- ishes GSIS, whereas a short term exposure to FFA enhances it (1–3). In contrast, a sustained provision of FA, particularly in the presence of high glucose in vitro, is detrimental to -cells in that it reduces insulin gene expression (4) and secretion (5) and induces -cell apoptosis (6). The FA supply to the -cells can be from exogenous sources, such as plasma FFAs and lipoproteins, or endogenous sources, such as intracellular triglyceride (TG) stores. Studies from our laboratory (7–10) and others (11, 12) support the concept that the hydrolysis of endogenous TG plays an important role in fuel-induced insulin secretion because TG depletion with leptin (13) or inhibition of TG lipol- ysis by lipase inhibitors such as 3,5-dimethylpyrazole (7) or orl- istat (11, 12) markedly curtail GSIS in rat islets. Furthermore, mice with -cell-specific knock-out of hormone-sensitive lipase (HSL), which hydrolyzes both TG and diacylglycerol (DAG), show defective first phase GSIS in vivo and in vitro (14). Lipolysis is an integral part of an essential metabolic pathway, the TG/FFA cycle, in which FFA esterification onto a glycerol backbone leading to the synthesis of TG is followed by its hydrolysis with the release of the FFA that can then be re-ester- ified. Intracellular TG/FFA cycling is known to occur in adipose tissue of rats and humans (15, 16) and also in liver and skeletal muscle (17). It is generally described as a “futile cycle” as it leads to the net hydrolysis of ATP with the generation of heat (18). However, several studies have shown that this cycle has impor- tant functions in the cell. For instance, in brown adipose tissue, it contributes to overall thermogenesis (17, 19). In islets from the normoglycemic, hyperinsulinemic, obese Zucker fatty rat, increased GSIS is associated with increased glucose-stimulated lipolysis and FA esterification, indicating enhanced TG/FFA * This work was supported, in whole or in part, by a National Institutes of Health grant (to N. R., V. P., and M. P.). This work was also supported by grants from the Canadian Diabetes Association and the Canadian Institute of Health Research (to M. P.). □ S The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1. 1 Supported by graduate studentships from the Fonds de Recherche en Sante ´ du Que ´ bec and Programme de Biologie Mole ´ culaire de l’Universite ´ de Montre ´al. 2 Supported by graduate studentships from the Fonds de Recherche en Sante ´ du Que ´ bec. 3 Recipient of a Canadian Chair in Diabetes and Pancreatic Beta-cell Function. 4 Recipient of a Canadian Chair in Diabetes and Metabolism. To whom corre- spondence should be addressed: Montreal Diabetes Research Center, Cen- tre de Recherche du Centre Hospitalier de l’Universite ´ de Montre ´al, Technopo ˆ le Angus (Rm. 401), 2901, Rachel East St., Montreal, Quebec H1W 4A4, Canada. Tel.: 514-890-8000 (Ext. 23642); Fax: 514-412-7648; E-mail: [email protected]. 5 The abbreviations used are: FFA, free fatty acid; ATGL, adipose triglyceride lipase; BSA, bovine serum albumin; d-BSA, defatted BSA; DAG, diacylglyc- erol; FA, fatty acid; GSIS, glucose-stimulated insulin secretion; HSL, hor- mone-sensitive lipase; KD, knockdown; KRBH, Krebs-Ringer bicarbonate buffer containing HEPES; MAG, monoacylglycerol; NEFA, nonesterified fatty acid; shRNA, small hairpin RNA; PBS, phosphate-buffered saline; PL, phospholipid; TG, triglyceride; TGL, triglyceride lipase; WAT, white adipose tissue; WT, wild type; ANOVA, analysis of variance; RT, reverse transcription; HGC, hyperglycemic clamp; IPGTT, intraperitoneal glucose tolerance test; Rh123, rhodamine 123; AUC, area under the curve. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 284, NO. 25, pp. 16848 –16859, June 19, 2009 © 2009 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. 16848 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009 by guest on February 19, 2018 http://www.jbc.org/ Downloaded from
Transcript of and Non-fuel-stimulated Insulin Secretion*DS

Adipose Triglyceride Lipase Is Implicated in Fuel- andNon-fuel-stimulated Insulin Secretion*□S
Received for publication, February 25, 2009, and in revised form, April 9, 2009 Published, JBC Papers in Press, April 22, 2009, DOI 10.1074/jbc.M109.006650
Marie-Line Peyot‡, Claudiane Guay‡1, Martin G. Latour‡, Julien Lamontagne‡2, Roxane Lussier‡, Marco Pineda‡,Neil B. Ruderman§, Guenter Haemmerle¶, Rudolf Zechner¶, Erik Joly‡, S. R. Murthy Madiraju‡, Vincent Poitout‡�**3,and Marc Prentki‡�4
From the ‡Molecular Nutrition Unit and the Montreal Diabetes Research Center, Centre de Recherche du Centre Hospitalier del’Universite de Montreal, and Departments of �Nutrition and **Medicine, University of Montreal, Montreal, Quebec H1W 4A4, Canada,the §Departments of Medicine and Physiology and Biophysics, Boston University School of Medicine and Diabetes Unit, Section ofEndocrinology, Boston University School of Medicine and Boston Medical Center, Boston, Massachusetts 02118, and the ¶Instituteof Molecular Biosciences, Karl-Franzens-University, Graz 8010, Austria
Reduced lipolysis in hormone-sensitive lipase-deficient miceis associatedwith impaired glucose-stimulated insulin secretion(GSIS), suggesting that endogenous �-cell lipid stores providesignaling molecules for insulin release. Measurements of lipol-ysis and triglyceride (TG) lipase activity in islets from HSL�/�
mice indicated the presence of other TG lipase(s) in the �-cell.Using real time-quantitative PCR, adipose triglyceride lipase(ATGL) was found to be the most abundant TG lipase in ratislets and INS832/13 cells. To assess its role in insulin secretion,ATGLexpressionwasdecreased in INS832/13cells (ATGL-knock-down (KD)) by small hairpin RNA. ATGL-KD increased the ester-ification of free fatty acid (FFA) into TG. ATGL-KD cells showeddecreased glucose- orGln�Leu-induced insulin release, aswell asreduced response toKCl or palmitate at high, but not low, glucose.The KATP-independent/amplification pathway of GSIS was con-siderably reduced inATGL-KDcells.ATGL�/�micewerehypoin-sulinemicandhypoglycemicandshoweddecreasedplasmaTGandFFAs.Ahyperglycemicclamprevealed increased insulinsensitivityand decreased GSIS and arginine-induced insulin secretion inATGL�/� mice. Accordingly, isolated islets from ATGL�/� miceshowedreduced insulin secretion inresponse toglucose, glucose�
palmitate, and KCl. Islet TG content and FFA esterification intoTGwere increased by 2-fold in ATGL�/� islets, but glucose usageand oxidationwere unaltered. The results demonstrate the impor-tance of ATGL and intracellular lipid signaling for fuel- and non-fuel-induced insulin secretion.
Free fatty acids (FFA)5 and other lipid molecules are impor-tant for proper glucose-stimulated insulin secretion (GSIS) by�-cells. Thus, deprivation of fatty acids (FA) in vivo (1) dimin-ishes GSIS, whereas a short term exposure to FFA enhances it(1–3). In contrast, a sustained provision of FA, particularly inthe presence of high glucose in vitro, is detrimental to �-cells inthat it reduces insulin gene expression (4) and secretion (5) andinduces�-cell apoptosis (6). The FA supply to the�-cells can befrom exogenous sources, such as plasma FFAs and lipoproteins,or endogenous sources, such as intracellular triglyceride (TG)stores. Studies from our laboratory (7–10) and others (11, 12)support the concept that the hydrolysis of endogenous TGplays an important role in fuel-induced insulin secretionbecause TG depletion with leptin (13) or inhibition of TG lipol-ysis by lipase inhibitors such as 3,5-dimethylpyrazole (7) or orl-istat (11, 12) markedly curtail GSIS in rat islets. Furthermore,mice with �-cell-specific knock-out of hormone-sensitivelipase (HSL), which hydrolyzes both TG and diacylglycerol(DAG), show defective first phase GSIS in vivo and in vitro (14).Lipolysis is an integral part of an essentialmetabolic pathway,
the TG/FFA cycle, in which FFA esterification onto a glycerolbackbone leading to the synthesis of TG is followed by itshydrolysis with the release of the FFA that can then be re-ester-ified. Intracellular TG/FFA cycling is known to occur in adiposetissue of rats and humans (15, 16) and also in liver and skeletalmuscle (17). It is generally described as a “futile cycle” as it leadsto the net hydrolysis of ATP with the generation of heat (18).However, several studies have shown that this cycle has impor-tant functions in the cell. For instance, in brown adipose tissue,it contributes to overall thermogenesis (17, 19). In islets fromthe normoglycemic, hyperinsulinemic, obese Zucker fatty rat,increased GSIS is associated with increased glucose-stimulatedlipolysis and FA esterification, indicating enhanced TG/FFA
* This work was supported, in whole or in part, by a National Institutes ofHealth grant (to N. R., V. P., and M. P.). This work was also supported bygrants from the Canadian Diabetes Association and the Canadian Instituteof Health Research (to M. P.).
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Table 1.
1 Supported by graduate studentships from the Fonds de Recherche en Santedu Quebec and Programme de Biologie Moleculaire de l’Universite deMontreal.
2 Supported by graduate studentships from the Fonds de Recherche en Santedu Quebec.
3 Recipient of a Canadian Chair in Diabetes and Pancreatic Beta-cell Function.4 Recipient of a Canadian Chair in Diabetes and Metabolism. To whom corre-
spondence should be addressed: Montreal Diabetes Research Center, Cen-tre de Recherche du Centre Hospitalier de l’Universite de Montreal,Technopole Angus (Rm. 401), 2901, Rachel East St., Montreal, Quebec H1W4A4, Canada. Tel.: 514-890-8000 (Ext. 23642); Fax: 514-412-7648; E-mail:[email protected].
5 The abbreviations used are: FFA, free fatty acid; ATGL, adipose triglyceridelipase; BSA, bovine serum albumin; d-BSA, defatted BSA; DAG, diacylglyc-erol; FA, fatty acid; GSIS, glucose-stimulated insulin secretion; HSL, hor-mone-sensitive lipase; KD, knockdown; KRBH, Krebs-Ringer bicarbonatebuffer containing HEPES; MAG, monoacylglycerol; NEFA, nonesterifiedfatty acid; shRNA, small hairpin RNA; PBS, phosphate-buffered saline; PL,phospholipid; TG, triglyceride; TGL, triglyceride lipase; WAT, white adiposetissue; WT, wild type; ANOVA, analysis of variance; RT, reverse transcription;HGC, hyperglycemic clamp; IPGTT, intraperitoneal glucose tolerance test;Rh123, rhodamine 123; AUC, area under the curve.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 284, NO. 25, pp. 16848 –16859, June 19, 2009© 2009 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
16848 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

cycling (10). Stimulation of lipolysis by glucose has also beenobserved in isolated islets from normal rats (12) and HSL�/�
mice (8) indicating the presence of glucose-responsive TG/FFAcycling in pancreatic �-cells.The identity of the key lipases involved in the TG/FFA cycle
in pancreatic islets is uncertain. HSL is expressed in islets (20),is up-regulated by long term treatment with elevated glucose(21), and is associated with insulin secretory granules (22). Inaddition, our earlier results suggested that elevated HSLexpression correlates with augmented TG/FFA cycling in isletsof Zucker fatty rats (10). However, it appears that other lipasesmay contribute to lipolysis and the regulation of GSIS in islettissue. Thus, results from studies using HSL�/� mice showedunalteredGSIS (8, 23), except in fastedmalemice (8, 9) inwhichlipolysis was decreased but not abolished. Furthermore,HSL�/�mice show residual TG lipase activity (8) indicating thepresence of other TG lipases.Recently, adipocyte triglyceride lipase (ATGL; also known as
Desnutrin, TTS-2, iPLA2-�, and PNPLA2) (24–26) was foundto account for most if not all of the residual lipolysis in HSL�/�
mice (26, 27). Two homologues of ATGL, Adiponutrin andGS2, have been described in adipocytes (24). All three enzymescontain a patatin-like domain with broad lipid acyl-hydrolaseactivity. However, it is not known if adiponutrin and GS2 areactually TG hydrolases. An additional lipase, TG hydrolase orcarboxylesterase-3, has been identified in rat adipose tissue (28,29). Although the hydrolysis of TG is catalyzed by all theselipases, HSL can hydrolyze both TG and DAG, the latter beinga better substrate (30).In this study, we observed that besides HSL, ATGL (31), adi-
ponutrin, and GS2 are expressed in rat islets and INS832/13cells, withATGLbeing themost abundant.We then focused onthe role of ATGL in fuel-stimulated insulin secretion in twomodels, INS832/13 �-cells in which ATGL expression wasreduced by RNA interference-knockdown (ATGL-KD) andATGL�/� mice.
EXPERIMENTAL PROCEDURES
Cell Culture—Rat insulinoma INS832/13 cells (32) (passages54–63) were cultured at 11.1 mM glucose in RPMI 1640medium supplemented with 10% (w/v) fetal bovine serum, 10mMHEPES, 2mMglutamine, 1mM sodiumpyruvate, and 50�M�-mercaptoethanol (complete RPMI) at 37 °C in a humidifiedatmosphere (5% CO2, 95% air). Cells were seeded at 4 � 106cells 2 days before transfection to reach a 60–70% confluence atthe day of transfection.Animals—10-Week-old overnight fasted male ATGL�/�
mice (33) backcrossed to theC57BL/6 strain formore than ninegenerations were used. Control mice used in this study wereC57BL/6 wild type littermates. The mice are not from theC57BL/6J background and therefore do not harbor a mutationin the nicotinamide nucleotide transhydrogenease gene (34).Wistar rats (200–250 g) were obtained fromCharles River Lab-oratories (St. Constant, Quebec, Canada). Mice and rats werehoused under control temperature (23 °C) and light conditions(12-h light/dark cycle) with free access to water and standarddiet (11% fat by energy). Serum insulin, glucose, nonesterifiedFFA, and TGweremeasured in overnight fasted pentobarbital-
anesthetizedmice (8). Themeasurements ofwhole body fat andlean mass were done in fed mice by quantitative magnetic res-onance (EchoMRI, Echo Medical Systems). All procedureswere approved by the Institutional Committee for the Protec-tion of Animals at the Centre de Recherche du Centre Hospi-talier de l’Universite de Montreal.Islet Isolation and Culture—Fed or overnight fasted male
Wistar rats and male ATGL�/� mice and their wild type con-trols were anesthetized with sodium pentobarbital (Somnotol,MTC Pharmaceuticals, Hamilton, Ontario, Canada) and killedby exsanguination. Pancreatic rat ormice islets were isolated bycollagenase digestion of the pancreas according to the methodof Gotoh et al. (35). After digestion and washing, islets wereseparated from digested exocrine tissue by Histopaque gradi-ent, after which rat islets were hand-picked for immediateextraction of RNA or protein preparation forWestern blotting.Mouse islets were handpicked and kept before experiments for2 h in culture in complete RPMI medium containing 2.8 mMglucose without �-mercaptoethanol at 37 °C in a humidifiedatmosphere containing 5% CO2 (8).Short Hairpin RNA-mediated ATGL Gene Suppression—
Short hairpin (sh) RNA-mediated gene suppression was per-formed as described previously (36). In brief, five 19-nucleotideshRNAs against rat ATGL (GenBankTM accession numberNM_001108509) with a 9-nucleotide loop were synthesized,annealed, and ligated to the PmeI and XbaI sites of pDLDU6(37). Efficiency of the various shRNA constructs on ATGLmRNA levels was verified in INS832/13 cells, and themost effi-cient shRNA was chosen for subsequent experiments. Thesense target sequence of shRNA-ATGL chosen is 5�-AAAGACCAT CCG TGG TTG TCT-3� (beginning at nucleotide 371 ofATGL sequence). The sense sequence with no known rat genehomology for scrambled control shRNA is 5�-CTG AGC ATTCAT TGG TCG C-3� (scrATGL). The pDLDU6 empty-vector(referred to as Mock) was also used as control.Cell Transfection—shRNA-pDLDU6 constructs were intro-
duced into INS832/13 cells by nucleofection (36) at a concen-tration of 5 �g of DNA for 6.9 � 106 cells. After transfection,cells were seeded in 12-well plates with 3 � 105 cells for mRNApreparation and insulin secretion assays; 6-well plates with 6 �105 cells for immunoblot analysis, TG hydrolase activity, TGcontent, fatty acid (FA) esterification, and lipolysis measure-ments; 48-well plates with 6 � 104 cells for mitochondrialmembrane potential determinations; 96-well plates with 2 �104 cells for the assessment of ATP content; and in 25-cm2
flasks with 1.5 � 106 cells for glucose oxidation determina-tion. Experiments with shRNA-ATGL were performed 96 hpost-transfection.Real Time Quantitative PCR Analysis—Total RNA was
extracted from rat adipocytes using theRNeasy lipid tissueminikit (Qiagen, Mississauga, Ontario, Canada) and from rat andmouse islets and cells using the RNeasy mini kit (Qiagen) withRNase-free DNase (Qiagen). RNA (3 �g) was reverse-tran-scribed to cDNA usingMoloneymurine leukemia virus reversetranscriptase (Invitrogen) and hexamers as described previ-ously (38). The primers (IDT, Coralville, IA) used are listed insupplemental Table 1. Gene expression was determined by thestandard curve method and normalized to the expression of
ATGL and Insulin Secretion
JUNE 19, 2009 • VOLUME 284 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 16849
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

cyclophilin or �-actin. Real time PCR analysis was performedusing the Rotor-Gene R3000 (Corbett Research, Mortlake,New South Wales, Australia) and the LCR Faststart DNAMasterPLUS SYBR Green reagent (Roche Applied Science).The number of mRNA molecules of the different lipases per�g of total RNA was evaluated employing a correspondingstandard curve. For the determination of the effect of glu-cose on ATGL mRNA expression, INS832/13 cells at70–80% confluence were employed after washing twice withPBS and culturing in complete RPMI medium at 3, 11, or 16mM glucose for 24 h.Triglyceride Lipase Activity—Transfected cells were washed
twice in PBS and centrifuged for 5 min at 200 � g. Cell homo-genates were then prepared in 0.2 ml of homogenization buffer(0.25 mM sucrose, 1 mM EDTA, 10 mM Tris, pH 7.0, 20 �g/mlleupeptin, 2 �g/ml antipain, and 1 �g/ml pepstatin A). Triglyc-eride lipase activity assay was performed on the cell homoge-nates as described previously (8).Western Blotting Analysis—Transfected cells collected by
trypsinization and isolated rat islets were washed three timeswith PBS. Total protein extracts from islets and cells wereobtained as described previously (36).Inguinal fat pads were surgically removed from fed rats, pul-
verized under liquid nitrogen, and extracted with the use ofice-cold lysis buffer (20 mM Tris-HCl, pH 7.2, containing 150mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% (v/v) Triton X-100,0.1% SDS, and protease inhibitors) for 30 min at 4 °C. Insolublematerial was removed by centrifugation at 10,000� g at 4 °C for10 min. The supernatant was collected and the protein assayedwith a BSA protein assay kit (Pierce). Protein extracts fromcells, islets, and fat (15 �g/lane) were separated on 10% SDS-PAGE and electroblotted to nitrocellulose membrane (What-man, Hanestrabe, Dassel, Germany). Membranes were blockedwith 5% (w/v) nonfat dry skimmedmilk in Tris-buffered saline,pH 7.5, with 0.1% (v/v) Tween (TBS-T) for 30 min at 37 °C andincubated with rabbit anti-ATGL antibody (1/200 dilution inTBS-T, Cayman Chemical Co., Ann Arbor, MI) overnight at4 °C. Blots were washed with TBS-T, blocked 15 min at 37 °Cwith TBS-T, 5% milk, and exposed to horseradish peroxidase-conjugated goat anti-rabbit IgG (1/10,000 dilution in TBT-T,Bio-Rad) for 1 h at room temperature. Membranes werewashed again and developed by enhanced chemiluminescenceusing a standard kit (SuperSignalWest Pico chemiluminescentsubstrate, Pierce). Band intensity was measured by densitome-try and analyzed using image analysis Genesnap software fromthe G-Box (PerkinElmer Life Sciences). For the normalizationof ATGL protein levels, membranes were incubated with anti-actin antibody (1/500 dilution in TBS-T).Triglyceride Content—TG content was measured in trans-
fected cells and mice islets as described (9).Insulin Secretion and Insulin Content Measurements—
Transfected cells (96 h post-transfection) were washed withPBS and cultured for 2 h in complete RPMI medium at 1 mMglucose. They were then preincubated for 40 min at 37 °C in 1ml of Krebs-Ringer bicarbonate (KRBH) buffer containing 10mM HEPES, 1 mM glucose, and 0.5% defatted BSA (d-BSA),after which they were incubated for 45 min in KRBH, 0.5%d-BSA plus different test agents as indicated in figure legends.
At the end of the incubation, media were kept for insulin meas-urement by radioimmunoassay using a human insulin standard(Linco Research, St. Charles, MO). Total insulin content wasmeasured following acid/ethanol (0.2 mM HCl in 75% ethanol)extraction of cells, and protein cellular content was determinedby using a BCA protein assay kit.For insulin secretion in islets, freshly isolated islets that were
cultured as described above in RPMI medium for only 2 h forrecovery from isolation were distributed in 12-well plates (10islets/well) and preincubated for 45min at 37 °C in KRBH, 0.5%d-BSA, and 2.8mMglucose. Theywere then incubated for 1 h inKRBH, 0.5% d-BSA, and 2.8, 8.3, or 16.7 mM glucose, in thepresence or absence of 0.4mM palmitate, or 35mMKCl. Insulinin themedia at the end of the incubation and insulin contents ofislets were quantified as indicated above.IntraperitonealGlucose Tolerance Test—Intraperitoneal glu-
cose tolerance tests (IPGTT)were performed in consciousmicein themorning after a 16-h fast. A 15% glucose solution (1.5 g ofglucose/kg of body weight) was administrated intraperitone-ally. Tail blood samples (�70 �l) were collected into hepa-rinized tubes at 0, 15, 30, 60, and 120 min for measurement ofglycemia and insulinemia.Hyperglycemic Clamp—One-step hyperglycemic clamps
(HGC) were performed on conscious mice. A 20% dextrosesolution (Baxter Healthcare Corp.) was infused through thejugular vein to clamp plasma glucose at�300mg/dl for 60min.Glucose infusion rate of the exogenous glucose infusion wasadjusted based on instantaneous glycemia assessments using ahandheld glucometer (Accu-Check, Roche Applied Science).At time 60 min of the clamp, an arginine bolus injection wasperformed (intravenously, 1 mmol/kg; Sandoz Canada Inc.) toassess the maximal insulin response. Blood samples were col-lected from the tail vessels, rapidly spun to separate erythro-cytes, and kept frozen until insulin determination by enzyme-linked immunosorbent assay (insulin mouse ultrasensitiveelectroimmunoassay, ALPCODiagnostics, Salem,NH) at times0, 5, 15, 30, 60, 61, and 70 min.Glucose Oxidation in INS Cells—Transfected INS832/13
cells, cultured and preincubated as described under insulinsecretion experiments, were incubated for 2 h in 2 ml ofKRBH, 0.5% d-BSA with 1 mM glucose plus 0.1 �Ci/ml[U-14C]glucose (290 mCi/mmol; Amersham Biosciences) or10 mM glucose plus 0.2 �Ci/ml of [U-14C]glucose. At thebeginning of the incubation, 25-cm2 flasks were sealed with astopper containing a piece of Whatman GF/B paper soakedin 5% KOH. At the end of the incubation, perchloric acid wasinjected into each flasks, and the liberated CO2 was trappedinto Whatman paper. The trapped 14CO2 was measured byliquid scintillation counting.Islets Glucose Metabolism—Groups of 20 freshly isolated
islets, cultured and preincubated as described for insulin secre-tion, were incubated at 37 °C for 2 h in KRBH, 0.25% d-BSAcontaining 0.5 �Ci of D-[5-3H]glucose (16 Ci/mmol) and 1�Ci/ml D-[U-14C]glucose (250mCi/mmol), 2.8 or 16.7mM glu-cose. Incubation was stopped by the addition of citrate/NaOHbuffer (400 mM, pH 4.9) containing antimycin-A (10 �M), rote-none (10 �M), and potassium cyanide (5 mM) as described pre-viously (39). Glucose oxidationwasmeasured by the generation
ATGL and Insulin Secretion
16850 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

of potassium hydroxide-trapped 14CO2 after 60 min of incuba-tion at room temperature. Glucose utilization was determinedby measuring the amount of 3H2O.Fatty Acid Esterification andOxidation—For fatty acid ester-
ification determination in INS832/13 cells, transfected cellscultured and preincubated as for insulin secretion experiments,were incubated for 45 min in 2 ml of KRBH containing 0.1�Ci/ml [14C]palmitate (57.5 mCi/mmol; Amersham Bio-sciences), 0.2 mM palmitate, 0.5% d-BSA, 1 mM carnitine, and 1or 10 mM glucose. For FA esterification and oxidation in miceislets, batches of 50 freshly isolated islets cultured as for insulinsecretion experiments were preincubated for 45 min in 1 ml ofKRBH containing 0.1 mM (FA oxidation) or 0.2 mM palmitate,0.25% d-BSA (FA esterification) and 2.8mM glucose. They werethen incubated for 2 h in 0.5 ml of KRBH, 0.25% d-BSA, 0.1 mM(oxidation) or 0.2mMpalmitate (esterification), 1mM carnitine,2 �Ci/ml [9,10-3H]palmitate (51 Ci/mmol, Amersham Bio-sciences), and 2.8 or 16.7 mM glucose. At the end of the incuba-
tion, cells or islets were collected forFA esterification determination,washed in cold PBS, and resus-pended in 3ml of Folch reagent (40).Total lipids were extracted and sep-arated by thin layer chromatogra-phy using a solvent for neutral lipids(petroleum ether/ether/acetic acid;70/30/1) to measure the incorpora-tion of labeled palmitate into com-plex lipids as described previously(10). For FA oxidation, after 2 h,incubation media were transferredinto Eppendorf tubes for separationof 3H2O from labeled fatty acids asdescribed previously (41).Lipolysis Measurement—Trans-
fected INS832/13 cells cultured andpreincubated as for insulin secre-tion experiments were incubatedfor 2 h in 1ml of KRBH, 0.5% d-BSAand 1 or 10 mM glucose. Batches of60 freshly isolated islets, cultured asfor insulin secretion experiments,were incubated for 3 h in 0.2 ml ofKRBH, 0.07% d-BSA at 2.8 or 16.7mM glucose. At the end of the incu-bation, media were kept to measureglycerol release as an index of lipol-ysis. An enzymatic luminescencedetection method, based on thereduction of NAD� to NADH in aseries of enzymatic reactions (8),was used to measure glycerol in themedium.ATP Content and Mitochondrial
Membrane Potential—TransfectedINS832/13 cells cultured as describedfor insulin secretion experimentswere preincubated for 1 h in
KRBH, 0.07% d-BSA at 1 mM glucose, after which they wereincubated for 10 min in KRBH, 0.07% d-BSA at 1 or 10 mMglucose. At the end of the incubation, ATP was extractedfrom the cells and assayed using an ATP bioluminescenceassay (ATPlite kit, PerkinElmer Life Sciences) according tothe manufacturer’s instructions.For mitochondrial membrane potential determination,
transfected cells were loaded with rhodamine 123 (Rh123,Invitrogen) for 20 min. They were then washed and incubatedin KRBH, 0.07% d-BSA at 1 mM glucose for 30 min, after whichthey were washed and incubated in the same solution for 10min. At the end of the incubation, basal fluorescence (Rh123excited at 485 nm and the emitted fluorescence monitored at530 nm)wasmeasured on a FLUOstarmicroplate reader (BMGLabtech, Offenburg, Germany). Then 1 or 10 mM glucose withor without 0.3 mM fluoro-carbonyl cyanide phenylhydrazone, amitochondrial membrane uncoupler, was added, and 10 minlater Rh123 fluorescence was recorded.
FIGURE 1. ATGL expression relative to other triglyceride lipases in adipose tissue, islets, and INS832/13cells and its regulation by the dietary state. ATGL, adiponutrin (ADPN), GS2, and HSL mRNA levels weredetermined by real time RT-PCR in adipose tissue (A) and islets (B) from overnight fasted and fed rats, and inINS832/13 cells cultured in complete RPMI medium at 11.1 mM glucose (C). The effect of glucose on ATGLexpression was studied in INS832/13 exposed for 24 h at 3, 11, or 16 mM glucose (G) in complete RPMI mediumand normalized to cyclophilin (D). Immunoblot analysis of ATGL (E) in adipose tissue (AT) (adipose tissue fromtwo different rats, 1st and 2nd lanes), rat islets (two different rats, 3rd and 4th lanes), and INS832/13 cells (twodifferent passages, 5th and 6th lanes) is shown. The data are expressed as means � S.E. of four rats (A and B) orfour different passages for INS832/13 cells (C and D). *, p � 0.05; **, p � 0.01; ***, p � 0.001 versus nutritionalstate, by unpaired two-tailed Student’s t test. NB, number.
ATGL and Insulin Secretion
JUNE 19, 2009 • VOLUME 284 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 16851
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from
Statistical Analysis—All results are expressed as means �S.E. Statistical significance was calculated with the Student’s ttest or, formultiple comparisons, one-way or two-ways analysisof variance (ANOVA) with Bonferroni post hoc testing as indi-cated. A p value of �0.05 was considered significant.
RESULTS
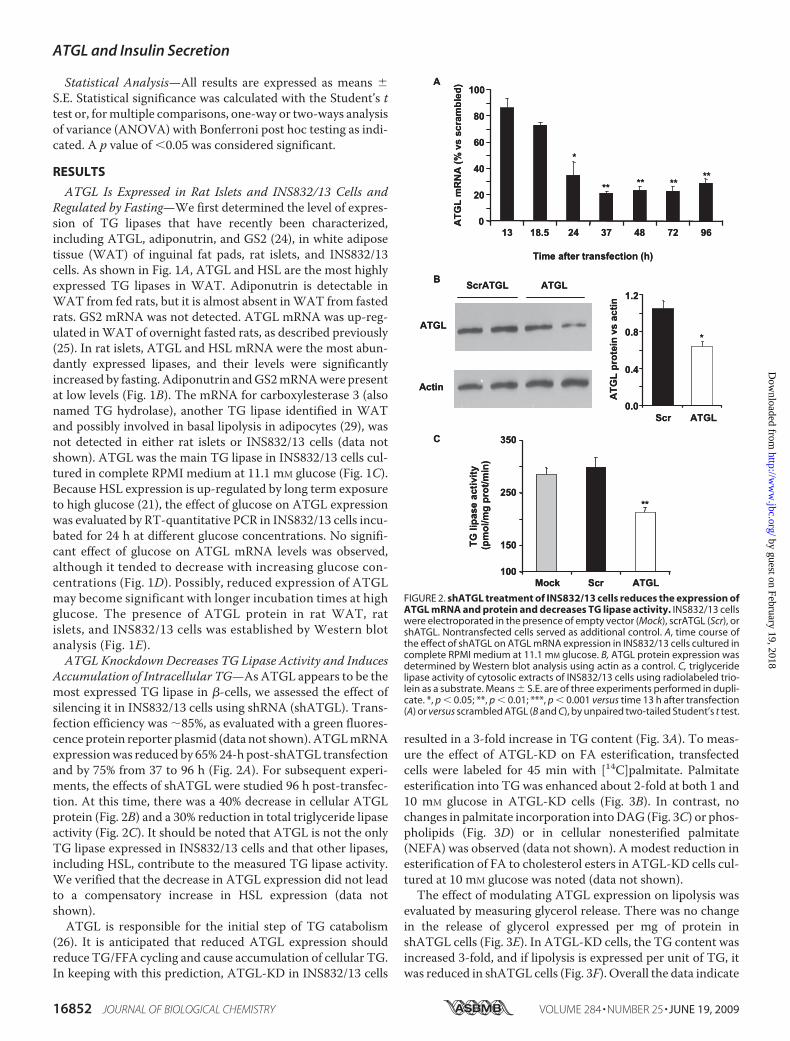
ATGL Is Expressed in Rat Islets and INS832/13 Cells andRegulated by Fasting—We first determined the level of expres-sion of TG lipases that have recently been characterized,including ATGL, adiponutrin, and GS2 (24), in white adiposetissue (WAT) of inguinal fat pads, rat islets, and INS832/13cells. As shown in Fig. 1A, ATGL and HSL are the most highlyexpressed TG lipases in WAT. Adiponutrin is detectable inWAT from fed rats, but it is almost absent inWAT from fastedrats. GS2 mRNA was not detected. ATGL mRNA was up-reg-ulated inWAT of overnight fasted rats, as described previously(25). In rat islets, ATGL and HSL mRNA were the most abun-dantly expressed lipases, and their levels were significantlyincreased by fasting. Adiponutrin andGS2mRNAwere presentat low levels (Fig. 1B). The mRNA for carboxylesterase 3 (alsonamed TG hydrolase), another TG lipase identified in WATand possibly involved in basal lipolysis in adipocytes (29), wasnot detected in either rat islets or INS832/13 cells (data notshown). ATGL was the main TG lipase in INS832/13 cells cul-tured in complete RPMI medium at 11.1 mM glucose (Fig. 1C).Because HSL expression is up-regulated by long term exposureto high glucose (21), the effect of glucose on ATGL expressionwas evaluated by RT-quantitative PCR in INS832/13 cells incu-bated for 24 h at different glucose concentrations. No signifi-cant effect of glucose on ATGL mRNA levels was observed,although it tended to decrease with increasing glucose con-centrations (Fig. 1D). Possibly, reduced expression of ATGLmay become significant with longer incubation times at highglucose. The presence of ATGL protein in rat WAT, ratislets, and INS832/13 cells was established by Western blotanalysis (Fig. 1E).ATGL Knockdown Decreases TG Lipase Activity and Induces
Accumulation of Intracellular TG—AsATGL appears to be themost expressed TG lipase in �-cells, we assessed the effect ofsilencing it in INS832/13 cells using shRNA (shATGL). Trans-fection efficiency was �85%, as evaluated with a green fluores-cence protein reporter plasmid (data not shown).ATGLmRNAexpressionwas reduced by 65%24-h post-shATGL transfectionand by 75% from 37 to 96 h (Fig. 2A). For subsequent experi-ments, the effects of shATGL were studied 96 h post-transfec-tion. At this time, there was a 40% decrease in cellular ATGLprotein (Fig. 2B) and a 30% reduction in total triglyceride lipaseactivity (Fig. 2C). It should be noted that ATGL is not the onlyTG lipase expressed in INS832/13 cells and that other lipases,including HSL, contribute to the measured TG lipase activity.We verified that the decrease in ATGL expression did not leadto a compensatory increase in HSL expression (data notshown).ATGL is responsible for the initial step of TG catabolism
(26). It is anticipated that reduced ATGL expression shouldreduce TG/FFA cycling and cause accumulation of cellular TG.In keeping with this prediction, ATGL-KD in INS832/13 cells
resulted in a 3-fold increase in TG content (Fig. 3A). To meas-ure the effect of ATGL-KD on FA esterification, transfectedcells were labeled for 45 min with [14C]palmitate. Palmitateesterification into TG was enhanced about 2-fold at both 1 and10 mM glucose in ATGL-KD cells (Fig. 3B). In contrast, nochanges in palmitate incorporation intoDAG (Fig. 3C) or phos-pholipids (Fig. 3D) or in cellular nonesterified palmitate(NEFA) was observed (data not shown). A modest reduction inesterification of FA to cholesterol esters in ATGL-KD cells cul-tured at 10 mM glucose was noted (data not shown).
The effect of modulating ATGL expression on lipolysis wasevaluated by measuring glycerol release. There was no changein the release of glycerol expressed per mg of protein inshATGL cells (Fig. 3E). In ATGL-KD cells, the TG content wasincreased 3-fold, and if lipolysis is expressed per unit of TG, itwas reduced in shATGL cells (Fig. 3F). Overall the data indicate
FIGURE 2. shATGL treatment of INS832/13 cells reduces the expression ofATGL mRNA and protein and decreases TG lipase activity. INS832/13 cellswere electroporated in the presence of empty vector (Mock), scrATGL (Scr), orshATGL. Nontransfected cells served as additional control. A, time course ofthe effect of shATGL on ATGL mRNA expression in INS832/13 cells cultured incomplete RPMI medium at 11.1 mM glucose. B, ATGL protein expression wasdetermined by Western blot analysis using actin as a control. C, triglyceridelipase activity of cytosolic extracts of INS832/13 cells using radiolabeled trio-lein as a substrate. Means � S.E. are of three experiments performed in dupli-cate. *, p � 0.05; **, p � 0.01; ***, p � 0.001 versus time 13 h after transfection(A) or versus scrambled ATGL (B and C), by unpaired two-tailed Student’s t test.
ATGL and Insulin Secretion
16852 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

that TG metabolism of INS832/13 cells is affected uponreduced expression of ATGL.Knockdown of ATGL Expression Does Not Affect Glucose and
EnergyMetabolism—Various parameters of glucose and energymetabolism were determined to assess the potential toxicity ofthe constructs or of reducingATGL expression. ATGL-KDhadno effect on glucose oxidation at low or high glucose concen-trations in INS832/13 cells (Fig. 4A). A very modest reductionin ATP content was observed at both low and high glucose inATGL-KD cells; however, there was no difference in the frac-tional increase in ATP content from 1 to 10 mM glucosebetween scrATGL and shRNA-ATGL cells (Fig. 4B). Insulingranules contain a large amount of ATP (42). BecauseATGL-KD causes a modest reduction in the insulin content ofINS832/13 cells (see below), the slight reduction in ATP con-tent is possibly due to a reduced number of secretory granules.Mitochondrial membrane potential was measured inINS832/13 cells using rhodamine 123, a fluorescent lipophiliccation. In scrATGL- and shATGL-transfected cells, 10mM glu-cose caused mitochondrial membrane potential hyper-polar-ization to the same extent, indicated by a fall in the intensity ofRh123 fluorescence, and membrane potential at basal glucosewas similar (Fig. 4C). These results indicate that ATGL-KDdoes not affect glucose and energy metabolism in INS832/13cells.
Reduction in ATGL Expression Lowers Fuel-induced InsulinSecretion—The effect of ATGL-KD on insulin secretion wasmeasured in cells in response to glucose, amino acids (leucineplus glutamine), GLP-1, and palmitate. scrATGL-transfectedcells had the same response to different secretagogues asmock-transfected cells (not shown for Fig. 5A; see Fig. 5B). Basal insu-lin release at 1 mM glucose was the same for scrATGL andshATGL cells. The response to KCl, GLP-1, and palmitate atlow glucose was not affected by ATGL-KD (Fig. 5, A and B). Incontrast, reduced ATGL expression decreased the effect ofamino acids on insulin release by 60% and GSIS both in theabsence and presence of palmitate by 50–55% (Fig. 5A). Total
FIGURE 3. Knockdown of ATGL expression in INS832/13 cells increases FAesterification into TG and causes TG deposition. Cellular TG content wasmeasured 96 h post-transfection with scrATGL, shATGL, or under mock ornontransfected (NT) conditions (A). Means � S.E. are of 15 different wells infour separate experiments. B–D, cells were incubated for 45 min in KRBHcontaining [1-14C]palmitate at 1 and 10 mM glucose (G). B–D show palmitateesterification into TG (B), DAG (C), and PL (D). Means � S.E. of three independ-ent experiments are done in triplicate. Glycerol release is shown in E and F andexpressed per protein content (E) or per TG content (F). Data representmeans � S.E. of five independent experiments performed in duplicate ortriplicate. *, p � 0.05; **, p � 0.01; ***, p � 0.001 versus scrambled ATGL for thesame glucose concentration, one way-ANOVA, Bonferroni post hoc test.
FIGURE 4. Decreased ATGL expression in INS832/13 cells does not affectglucose and mitochondrial metabolism. Nontransfected cells (NT) orINS832/13 cells were subjected to electroporation in the presence of eithercontrol (mock and scrATGL), or shATGL plasmids were cultured for 96 h priorto experiments. A, glucose (G) oxidation was measured in cells incubated for2 h in KRBH at 1 or 10 mM glucose with [U-14C]glucose. Means � S.E. are ofnine separate determinations in three independent experiments. B, total ATPcontent was determined in cells incubated for 10 min in KRBH at 1 or 10 mM
glucose. Means � S.E. are of 18 separate determinations in three independ-ent experiments. C, mitochondrial membrane potential was monitored asrhodamine 123 fluorescence. After dye loading, basal fluorescence was deter-mined in cells cultured at 1 mM glucose, and then fluorescence was recordedfor 10 min at 10 mM glucose. Means � S.E. are of 12 separate determinationsin two independent experiments. *, p � 0.05 versus scrambled ATGL for thesame glucose concentration; ***, p � 0.001 versus 1 mM glucose for the samegroup; one way-ANOVA, Bonferroni post hoc test.
ATGL and Insulin Secretion
JUNE 19, 2009 • VOLUME 284 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 16853
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

insulin content was decreased by 35% in shATGL-transfectedcells (4.13� 0.15 and 2.73� 0.11 ng of insulin/�g of protein forscrATGL and shATGL, respectively, n � 7).To ascertain whether the reduction of GSIS in ATGL-KD
cells involves KATP-independent/amplification mechanism(s)(43, 44), cells were incubated in the presence of 35 mM KCl �diazoxide, a KATP channel opener. shATGL treatment ofINS832/13 cells barely affected KCl-induced insulin secretion
at low (1 mM) glucose (Fig. 5, A andB). However, in the presence of ele-vated KCl with or without diazox-ide, GSIS was markedly curtailed inshATGL cells as compared withmock- or scrATGL-transfectedcells (Fig. 5B). Thus, the activity ofthe “amplification” arm of GSIS, asevaluated by the difference in insu-lin secretion between 1 and 10 mMglucose in cells incubated with highKCl without or with diazoxide (45),was reduced by 60–75% inshATGL-treated cells (Fig. 5C).These results indicate that thedefect of GSIS in ATGL-KD cells islargely due to altered KATP-inde-pendent/amplification of insulinsecretion.ATGL�/� Mice Are Hypoinsu-
linemic and Hypoglycemic andShow Decreased Plasma TG andFFA Levels—To confirm the impor-tance of ATGL in the regulation ofinsulin secretion, ATGL�/� micewere used. Body weight and bloodchemistry of ATGL�/� mice fol-lowing an overnight fast are shownin Table 1. Fasted ATGL�/� micewere slightly heavier thanATGL�/� mice. Circulating insulinlevels were reduced by 70% inATGL�/� mice, consistent with the40% reduction in serum insulinreported in fed ATGL�/� mice on amixed genetic background (50%
C57BL/6 and 50% 129/Ola) (33). As expected, plasma TG andFFA were reduced (about 40%), and glucose levels were signif-icantly (25%) decreased in ATGL�/� mice. Whole body fatmass was increased by 2-fold in ATGL�/� mice, as reportedbefore on mice with a mixed genetic background (33).Reduced First Phase GSIS in Vivo in Insulino-sensitive Glu-
cose-normotolerant ATGL�/� Mice—HGC were performed in10-week-old overnight fasted conscious male mice to investi-gate the consequence of ATGL deficiency on insulin secretionin vivo. Conscious ATGL�/� mice had lower basal glycemia(105 � 4 versus 145 � 7 mg/dl) (Fig. 6A) and insulinemia(51.7 � 7.7 versus 123.3 � 13.6 pmol/liter) (Fig. 6B) thanATGL�/� mice. During the clamp, the glucose infusion ratewas adjusted to maintain blood glucose at � 300 mg/dl (Fig.6A). Insulin secretion in response to hyperglycemia wasreduced in ATGL�/� mice (Fig. 6B). Calculation of the areaunder the curve (AUC) for the first 15 min of the clamp, sub-tracting basal insulinemia, indicated that first phase GSIS wasreduced by 72% inATGL�/�mice (Fig. 6D). However, theAUCfor the second phase GSIS (15–60 min of the clamp) ofATGL�/� mice was not significantly different from that ofATGL�/� mice (Fig. 6E). Consistent with reduced first phase
FIGURE 5. ATGL knockdown in INS832/13 cells reduces fuel-induced insulin secretion. A, insulin releasewas measured in mock, scrATGL, and shATGL cells incubated as indicated for 45 min at 1 or 10 mM glucose with0.5% d-BSA in the presence or absence of 0.25 mM palmitate (Pal), 10 nM GLP-1, 35 mM KCl, or 5 mM glutamineplus 5 mM leucine. B, effect of ATGL knockdown on the KATP-independent/amplification pathway(s) of insulinsecretion was determined in cells incubated at 1 or 10 mM glucose (G) in the presence or absence of 35 mM KClwith or without 0.25 mM diazoxide (Dz). C shows the differences in insulin release between 1 and 10 mM glucosefrom data shown in B. Means � S.E. are of 9 –15 separate determinations from 3 to 5 independent experiments.*, p � 0.05; ***, p � 0.001 versus scrambled ATGL for the same incubation condition; one way-ANOVA, Bonfer-roni post hoc test.
TABLE 1Plasma insulin, glucose, FFA, TG levels and body weight in 10-week-oldovernight fasted male ATGL�/� and ATGL�/� mice
ATGL�/� ATGL�/�
Insulin (pmol/liter) 111 � 18 (n � 35) 31 � 7a (n � 31)Glucose (mg/dl) 167 � 5 (n � 40) 128 � 7a (n � 36)FFA (mmol/liter) 0.32 � 0.03 (n � 23) 0.20 � 0.01a (n � 22)TG (mmol/liter) 0.46 � 0.02 (n � 24) 0.28 � 0.03a (n � 20)BW (g) 23.6 � 0.3 (n � 85) 24.6 � 0.3b (n � 66)BW (g)c 26.0 � 0.7 (n � 10) 27.3 � 0.8 (n � 7)Body fat (g)c 2.5 � 0.3 (n � 10) 4.9 � 0.2a (n � 7)Lean massc 20.5 � 0.9 (n � 10) 19.4 � 0.7 (n � 7)
a p � 0.001 versus ATGL�/� by unpaired, two-tailed Student’s t test.b p � 0.05.c Bodyweight (BW), body fat, and leanmass in 9-week-old fedmalemice are shown.Means � S.E.
ATGL and Insulin Secretion
16854 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

GSIS, insulin secretion in response to an arginine bolus wasreduced by 50% in ATGL�/� mice (Fig. 6C). The M/I index ofinsulin sensitivity (46) was increased in ATGL�/� mice (2.42�0.21 versus 1.47 � 0.13 �mol�kg�1�min�1 glucose infusion rateper pmol/liter insulin) indicating that ATGL�/� mice aremoresensitive to insulin than wild type mice (Fig. 6F). In ATGL�/�
mice, glucose tolerance was unchanged in comparison withATGL�/� mice (Fig. 6G). Normal glucose tolerance inATGL�/� mice was maintained despite lower plasma insulinlevels (Fig. 6H). These data confirm that ATGL�/� mice showimproved insulin sensitivity in association with reduced insulinsecretion, allowing unchanged glucose tolerance.Impaired Insulin Secretion in Isolated Islets from ATGL�/�
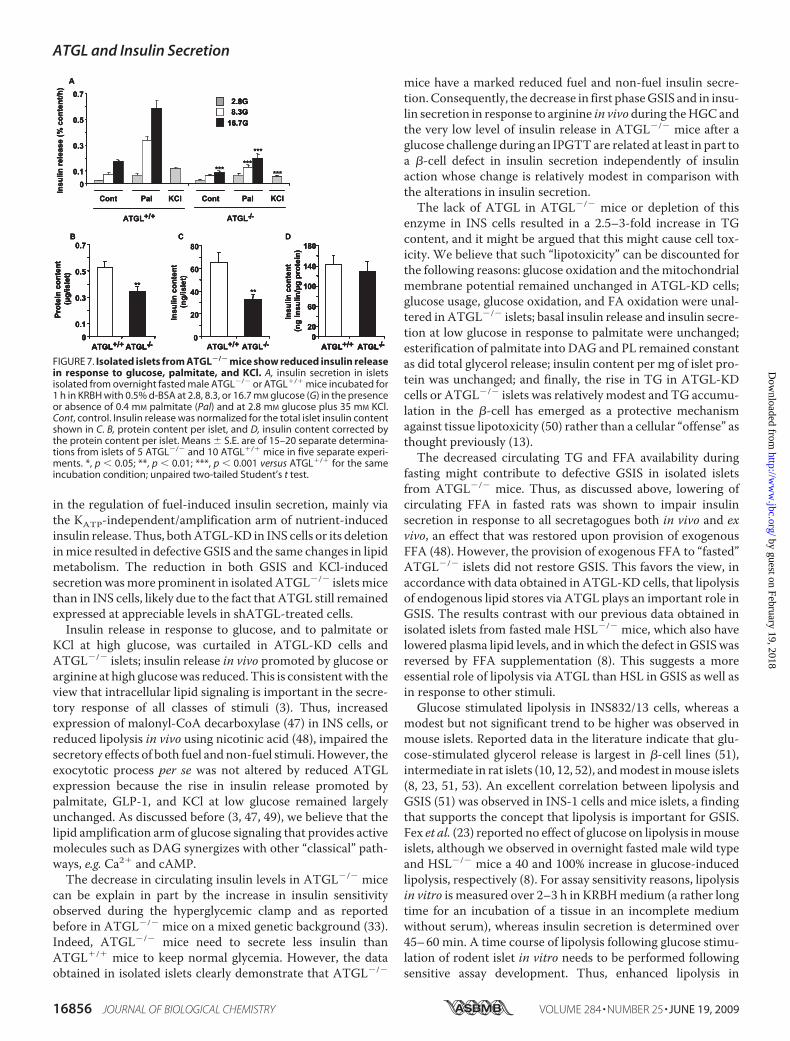
Mice—Insulin release was measured in isolated islets fromovernight-fasted ATGL�/� and ATGL�/� male mice. InATGL�/� mice, glucose increased insulin secretion by 3- and7-fold at 8.3 and 16.7 mM glucose, respectively, in comparisonwith the value at 2.8mM glucose (Fig. 7A). Exogenous palmitate
induced a robust enhancement ofinsulin secretion in control islets atall glucose concentrations, and thisenhancement was curtailed inATGL�/� islets. Fig. 7A shows thatGSIS either in the absence or pres-ence of palmitate was dramaticallyreduced in islets deficient in ATGL,even when the data are expressedper total islet insulin content. Insu-lin secretion in response to a depo-larizing concentration of KCl wasalso reduced by 50% in ATGL�/�
islets. At low glucose, palmitatecause a 2–3-fold increase in insulinrelease in ATGL�/� islets, and thissecretory response remained unal-tered in ATGL�/� islets (Fig. 7A).Altogether the data indicate thatATGL�/� islets show a markedreduction in insulin secretion inresponse to all classes of tested stim-uli (glucose, palmitate in the pres-ence of elevated glucose, and KCl).Total insulin content was re-
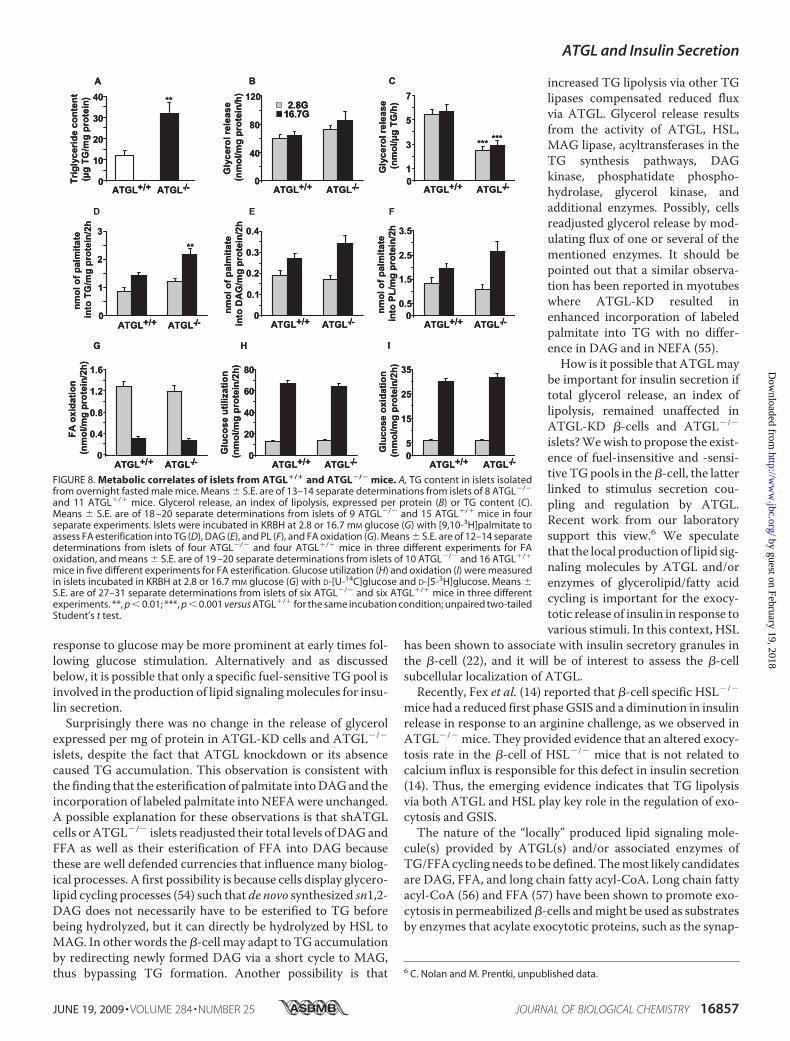
duced by 50% in ATGL�/� islets(Fig. 7C), largely due to the factthat ATGL�/� islets were smaller,as indicated by their protein con-tent that was reduced by �35%(Fig. 7B). However, islet insulincontent corrected for protein con-tent per islet was similar in bothislet groups (Fig. 7D).Metabolic Correlates in ATGL�/�
Islets—As observed in shATGL-KDcells, the deletion of ATGL led to a2.6-fold increase in the islet TGcon-tent of ATGL�/� mice (Fig. 8A).Similar to shATGL-KD cells, lack of
ATGL did not alter islet lipolysis when expressed per mg ofprotein (Fig. 8B). However, when normalized for TG content,lipolysis was reduced by 50% inATGL�/� islets (Fig. 8C). Therewas no change in HSL transcript expression in islets fromATGL�/� mice (data not shown). Islets from ATGL�/� miceshowed a 55% significant increase in FA esterification into TGat 16.7mM glucose compared with ATGL�/� islets (Fig. 8D). Incontrast, FA esterification into DAG (Fig. 8E) and PL (Fig. 8F)was not significantly different in islets from both genotypes. FAoxidation (Fig. 8G), glucose utilization (Fig. 8H), and glucoseoxidation (Fig. 8I) were similar at low and high glucose in iso-lated islets fromATGL�/� and ATGL�/� mice, indicating thatATGL deficiency does not induce metabolic toxicity of islettissue.
DISCUSSION
The results show that ATGL is expressed in rat islets andINS832/13 cells, and that this key lipolytic enzyme plays a role
FIGURE 6. ATGL�/� mice are insulino-sensitive and glucose-normotolerant and have a defect in glucose-and arginine-stimulated insulin secretion in vivo. A hyperglycemic clamp was performed in overnightfasted male wild type (ATGL�/�) and ATGL KO (ATGL�/�) mice. A, glucose levels during the clamp. B, insulinlevels during the clamp and C, in response to an arginine (Arg) bolus (1 mmol/kg). D, first phase insulin secretionin response to elevated glucose (Gluc) expressed as AUC from 0 to 15 min. E, second phase insulin secretion(AUC) from 15– 45 min. F, M/I index of insulin sensitivity calculated by dividing the glucose infusion rate (M)during the last 30 min of the clamp by circulating insulin levels (I) during the same period. M/I index isexpressed as �mol�kg�1�min�1 glucose infusion per pmol/liter insulin. G and H, glycemia (G) and insulinemia(H) during an IPGTT in overnight fasted male ATGL�/� and ATGL�/� mice. Mean � S.E. are of 9 and 5 animalsper group for HGC and IPGTT, respectively. *, p � 0.05; **, p � 0.01; ***, p � 0.001 versus ATGL�/� for the sametime; two-way ANOVA, Bonferroni post hoc test for A–C and G and H, and unpaired two-tailed Student’s t testfor D–F.
ATGL and Insulin Secretion
JUNE 19, 2009 • VOLUME 284 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 16855
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from
in the regulation of fuel-induced insulin secretion, mainly viathe KATP-independent/amplification arm of nutrient-inducedinsulin release. Thus, bothATGL-KD in INS cells or its deletioninmice resulted in defective GSIS and the same changes in lipidmetabolism. The reduction in both GSIS and KCl-inducedsecretionwasmore prominent in isolated ATGL�/� isletsmicethan in INS cells, likely due to the fact that ATGL still remainedexpressed at appreciable levels in shATGL-treated cells.Insulin release in response to glucose, and to palmitate or
KCl at high glucose, was curtailed in ATGL-KD cells andATGL�/� islets; insulin release in vivo promoted by glucose orarginine at high glucosewas reduced. This is consistentwith theview that intracellular lipid signaling is important in the secre-tory response of all classes of stimuli (3). Thus, increasedexpression of malonyl-CoA decarboxylase (47) in INS cells, orreduced lipolysis in vivo using nicotinic acid (48), impaired thesecretory effects of both fuel and non-fuel stimuli. However, theexocytotic process per se was not altered by reduced ATGLexpression because the rise in insulin release promoted bypalmitate, GLP-1, and KCl at low glucose remained largelyunchanged. As discussed before (3, 47, 49), we believe that thelipid amplification arm of glucose signaling that provides activemolecules such as DAG synergizes with other “classical” path-ways, e.g. Ca2� and cAMP.
The decrease in circulating insulin levels in ATGL�/� micecan be explain in part by the increase in insulin sensitivityobserved during the hyperglycemic clamp and as reportedbefore in ATGL�/� mice on a mixed genetic background (33).Indeed, ATGL�/� mice need to secrete less insulin thanATGL�/� mice to keep normal glycemia. However, the dataobtained in isolated islets clearly demonstrate that ATGL�/�
mice have a marked reduced fuel and non-fuel insulin secre-tion.Consequently, the decrease in first phaseGSIS and in insu-lin secretion in response to arginine in vivo during theHGCandthe very low level of insulin release in ATGL�/� mice after aglucose challenge during an IPGTTare related at least in part toa �-cell defect in insulin secretion independently of insulinaction whose change is relatively modest in comparison withthe alterations in insulin secretion.The lack of ATGL in ATGL�/� mice or depletion of this
enzyme in INS cells resulted in a 2.5–3-fold increase in TGcontent, and it might be argued that this might cause cell tox-icity. We believe that such “lipotoxicity” can be discounted forthe following reasons: glucose oxidation and themitochondrialmembrane potential remained unchanged in ATGL-KD cells;glucose usage, glucose oxidation, and FA oxidation were unal-tered in ATGL�/� islets; basal insulin release and insulin secre-tion at low glucose in response to palmitate were unchanged;esterification of palmitate into DAG and PL remained constantas did total glycerol release; insulin content per mg of islet pro-tein was unchanged; and finally, the rise in TG in ATGL-KDcells or ATGL�/� islets was relatively modest and TG accumu-lation in the �-cell has emerged as a protective mechanismagainst tissue lipotoxicity (50) rather than a cellular “offense” asthought previously (13).The decreased circulating TG and FFA availability during
fasting might contribute to defective GSIS in isolated isletsfrom ATGL�/� mice. Thus, as discussed above, lowering ofcirculating FFA in fasted rats was shown to impair insulinsecretion in response to all secretagogues both in vivo and exvivo, an effect that was restored upon provision of exogenousFFA (48). However, the provision of exogenous FFA to “fasted”ATGL�/� islets did not restore GSIS. This favors the view, inaccordance with data obtained in ATGL-KD cells, that lipolysisof endogenous lipid stores via ATGL plays an important role inGSIS. The results contrast with our previous data obtained inisolated islets from fasted male HSL�/� mice, which also havelowered plasma lipid levels, and in which the defect inGSISwasreversed by FFA supplementation (8). This suggests a moreessential role of lipolysis via ATGL than HSL in GSIS as well asin response to other stimuli.Glucose stimulated lipolysis in INS832/13 cells, whereas a
modest but not significant trend to be higher was observed inmouse islets. Reported data in the literature indicate that glu-cose-stimulated glycerol release is largest in �-cell lines (51),intermediate in rat islets (10, 12, 52), andmodest inmouse islets(8, 23, 51, 53). An excellent correlation between lipolysis andGSIS (51) was observed in INS-1 cells and mice islets, a findingthat supports the concept that lipolysis is important for GSIS.Fex et al. (23) reported no effect of glucose on lipolysis inmouseislets, although we observed in overnight fasted male wild typeand HSL�/� mice a 40 and 100% increase in glucose-inducedlipolysis, respectively (8). For assay sensitivity reasons, lipolysisin vitro is measured over 2–3 h in KRBHmedium (a rather longtime for an incubation of a tissue in an incomplete mediumwithout serum), whereas insulin secretion is determined over45–60 min. A time course of lipolysis following glucose stimu-lation of rodent islet in vitro needs to be performed followingsensitive assay development. Thus, enhanced lipolysis in
FIGURE 7. Isolated islets from ATGL�/� mice show reduced insulin releasein response to glucose, palmitate, and KCl. A, insulin secretion in isletsisolated from overnight fasted male ATGL�/� or ATGL�/� mice incubated for1 h in KRBH with 0.5% d-BSA at 2.8, 8.3, or 16.7 mM glucose (G) in the presenceor absence of 0.4 mM palmitate (Pal) and at 2.8 mM glucose plus 35 mM KCl.Cont, control. Insulin release was normalized for the total islet insulin contentshown in C. B, protein content per islet, and D, insulin content corrected bythe protein content per islet. Means � S.E. are of 15–20 separate determina-tions from islets of 5 ATGL�/� and 10 ATGL�/� mice in five separate experi-ments. *, p � 0.05; **, p � 0.01; ***, p � 0.001 versus ATGL�/� for the sameincubation condition; unpaired two-tailed Student’s t test.
ATGL and Insulin Secretion
16856 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from
response to glucose may be more prominent at early times fol-lowing glucose stimulation. Alternatively and as discussedbelow, it is possible that only a specific fuel-sensitive TG pool isinvolved in the production of lipid signalingmolecules for insu-lin secretion.Surprisingly there was no change in the release of glycerol
expressed per mg of protein in ATGL-KD cells and ATGL�/�
islets, despite the fact that ATGL knockdown or its absencecaused TG accumulation. This observation is consistent withthe finding that the esterification of palmitate intoDAGand theincorporation of labeled palmitate into NEFAwere unchanged.A possible explanation for these observations is that shATGLcells or ATGL�/� islets readjusted their total levels of DAGandFFA as well as their esterification of FFA into DAG becausethese are well defended currencies that influence many biolog-ical processes. A first possibility is because cells display glycero-lipid cycling processes (54) such that de novo synthesized sn1,2-DAG does not necessarily have to be esterified to TG beforebeing hydrolyzed, but it can directly be hydrolyzed by HSL toMAG. In other words the�-cell may adapt to TG accumulationby redirecting newly formed DAG via a short cycle to MAG,thus bypassing TG formation. Another possibility is that
increased TG lipolysis via other TGlipases compensated reduced fluxvia ATGL. Glycerol release resultsfrom the activity of ATGL, HSL,MAG lipase, acyltransferases in theTG synthesis pathways, DAGkinase, phosphatidate phospho-hydrolase, glycerol kinase, andadditional enzymes. Possibly, cellsreadjusted glycerol release by mod-ulating flux of one or several of thementioned enzymes. It should bepointed out that a similar observa-tion has been reported in myotubeswhere ATGL-KD resulted inenhanced incorporation of labeledpalmitate into TG with no differ-ence in DAG and in NEFA (55).How is it possible that ATGLmay
be important for insulin secretion iftotal glycerol release, an index oflipolysis, remained unaffected inATGL-KD �-cells and ATGL�/�
islets?Wewish to propose the exist-ence of fuel-insensitive and -sensi-tive TG pools in the�-cell, the latterlinked to stimulus secretion cou-pling and regulation by ATGL.Recent work from our laboratorysupport this view.6 We speculatethat the local production of lipid sig-naling molecules by ATGL and/orenzymes of glycerolipid/fatty acidcycling is important for the exocy-totic release of insulin in response tovarious stimuli. In this context, HSL
has been shown to associate with insulin secretory granules inthe �-cell (22), and it will be of interest to assess the �-cellsubcellular localization of ATGL.Recently, Fex et al. (14) reported that �-cell specific HSL�/�
mice had a reduced first phaseGSIS and a diminution in insulinrelease in response to an arginine challenge, as we observed inATGL�/� mice. They provided evidence that an altered exocy-tosis rate in the �-cell of HSL�/� mice that is not related tocalcium influx is responsible for this defect in insulin secretion(14). Thus, the emerging evidence indicates that TG lipolysisvia both ATGL and HSL play key role in the regulation of exo-cytosis and GSIS.The nature of the “locally” produced lipid signaling mole-
cule(s) provided by ATGL(s) and/or associated enzymes ofTG/FFAcycling needs to be defined. Themost likely candidatesare DAG, FFA, and long chain fatty acyl-CoA. Long chain fattyacyl-CoA (56) and FFA (57) have been shown to promote exo-cytosis in permeabilized�-cells andmight be used as substratesby enzymes that acylate exocytotic proteins, such as the synap-
6 C. Nolan and M. Prentki, unpublished data.
FIGURE 8. Metabolic correlates of islets from ATGL�/� and ATGL�/� mice. A, TG content in islets isolatedfrom overnight fasted male mice. Means � S.E. are of 13–14 separate determinations from islets of 8 ATGL�/�
and 11 ATGL�/� mice. Glycerol release, an index of lipolysis, expressed per protein (B) or TG content (C).Means � S.E. are of 18 –20 separate determinations from islets of 9 ATGL�/� and 15 ATGL�/� mice in fourseparate experiments. Islets were incubated in KRBH at 2.8 or 16.7 mM glucose (G) with [9,10-3H]palmitate toassess FA esterification into TG (D), DAG (E), and PL (F), and FA oxidation (G). Means � S.E. are of 12–14 separatedeterminations from islets of four ATGL�/� and four ATGL�/� mice in three different experiments for FAoxidation, and means � S.E. are of 19 –20 separate determinations from islets of 10 ATGL�/� and 16 ATGL�/�
mice in five different experiments for FA esterification. Glucose utilization (H) and oxidation (I) were measuredin islets incubated in KRBH at 2.8 or 16.7 mM glucose (G) with D-[U-14C]glucose and D-[5-3H]glucose. Means �S.E. are of 27–31 separate determinations from islets of six ATGL�/� and six ATGL�/� mice in three differentexperiments. **, p � 0.01; ***, p � 0.001 versus ATGL�/� for the same incubation condition; unpaired two-tailedStudent’s t test.
ATGL and Insulin Secretion
JUNE 19, 2009 • VOLUME 284 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 16857
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

tosomal associated protein-25 (SNAP-25) (58) and synaptotag-min (59), to enhance their association with the plasma mem-brane. Furthermore, phorbol esters, commonly used as stableand potent DAGmimics, via protein kinase C activation, causeSNAP-25 phosphorylation and stimulation of insulin exocyto-sis (60). DAG can also act on vesicle exocytosis via its binding tothe C1 domain of the synaptic vesicle priming protein Munc13(61), and we report that GSIS is defective in Munc13-1-defi-cient islets (62).The cellular insulin content was reduced in islets of
ATGL�/� mice and in shATGL-transfected INS cells. Recentstudies have revealed a tight coupling between insulin secretionand biosynthesis. Islet cell autoantigen 512 (ICA512), an intrin-sic tyrosine phosphatase-like protein of the insulin secretorygranulemembrane (63), is cleaved (64) following granule fusionto the plasma membrane. The resulting cleaved cytosolic frag-ment of this protein (ICA512-CCF) translocates to the nucleuswhere it prolongs the activity of STATs and thus insulin genetranscription and granule biogenesis (65). Possibly, thedecreased exocytosis of insulin because of reduced ATGLexpression in the �-cell leads to an adaptive reduced insulinbiosynthesis and storage.The role of lipolysis in human islets is not known, butwehave
observed both ATGL andHSL expression in human islets (datanot shown). The possible importance of ATGL in human�-cellfunction and insulin secretion is supported by the identificationof ATGL gene polymorphisms associated with type 2 diabetes(66). Furthermore, ATGL mutations leading to a truncatedATGL protein are responsible for a neutral lipid storage diseasewith myopathy that is characterized by systemic TG accumu-lation (67, 68). Recently, a novel mutation in the ATGL geneleading to a lack of the C-terminal region of the ATGL proteinwas identified in a patientwith neutral lipid storage diseasewithmyopathy (69). Interestingly, this patient showed a decrease ininsulin secretory capacity with age. Whether ATGL partici-pates in lipolysis and insulin secretion in human islets remainsto be examined.In conclusion, the results support the concept that �-cell
lipolysis via ATGL is important for the provision of lipid-sig-naling molecules necessary for insulin secretion in response tofuel and non-fuel stimuli. Additional work is needed to conclu-sively identify these lipid signaling molecules, to understandhowATGL is regulated in the �-cell, and to determine whetherthis enzyme directly produces coupling factors (DAG and FFA)for insulin secretion or indirectly via glycerolipid/FFA cyclingor other metabolic pathway(s) of lipid metabolism.
Acknowledgments—We thankGrace Ferguson andMelanie Ethier forvaluable technical help.
REFERENCES1. Stein, D. T., Esser, V., Stevenson, B. E., Lane, K. E., Whiteside, J. H.,
Daniels, M. B., Chen, S., and McGarry, J. D. (1996) J. Clin. Invest. 97,2728–2735
2. Stein, D. T., Stevenson, B. E., Chester, M. W., Basit, M., Daniels, M. B.,Turley, S. D., and McGarry, J. D. (1997) J. Clin. Invest. 100, 398–403
3. Nolan, C. J., Madiraju, M. S., Delghingaro-Augusto, V., Peyot, M. L., andPrentki, M. (2006) Diabetes 55, Suppl. 2, S16–23
4. Poitout, V., Hagman, D., Stein, R., Artner, I., Robertson, R. P., and Har-mon, J. S. (2006) J. Nutr. 136, 873–876
5. Prentki, M., Joly, E., El-Assaad, W., and Roduit, R. (2002) Diabetes 51,Suppl. 3, S405–413
6. El-Assaad,W., Buteau, J., Peyot,M. L., Nolan, C., Roduit, R., Hardy, S., Joly,E., Dbaibo, G., Rosenberg, L., and Prentki, M. (2003) Endocrinology 144,4154–4163
7. Masiello, P., Novelli, M., Bombara, M., Fierabracci, V., Vittorini, S.,Prentki, M., and Bergamini, E. (2002)Metabolism 51, 110–114
8. Peyot, M. L., Nolan, C. J., Soni, K., Joly, E., Lussier, R., Corkey, B. E.,Wang,S. P., Mitchell, G. A., and Prentki, M. (2004) Diabetes 53, 1733–1742
9. Roduit, R., Masiello, P.,Wang, S. P., Li, H., Mitchell, G. A., and Prentki, M.(2001) Diabetes 50, 1970–1975
10. Nolan, C. J., Leahy, J. L., Delghingaro-Augusto, V., Moibi, J., Soni, K.,Peyot, M. L., Fortier, M., Guay, C., Lamontagne, J., Barbeau, A., Przybyt-kowski, E., Joly, E., Masiello, P., Wang, S., Mitchell, G. A., and Prentki, M.(2006) Diabetologia 49, 2120–2130
11. Yaney, G. C., Civelek, V. N., Richard, A. M., Dillon, J. S., Deeney, J. T.,Hamilton, J. A., Korchak, H. M., Tornheim, K., Corkey, B. E., and Boyd,A. E., 3rd (2001) Diabetes 50, 56–62
12. Mulder, H., Yang, S., Winzell, M. S., Holm, C., and Ahren, B. (2004) Dia-betes 53, 122–128
13. Koyama, K., Chen, G., Wang, M. Y., Lee, Y., Shimabukuro, M., Newgard,C. B., and Unger, R. H. (1997) Diabetes 46, 1276–1280
14. Fex,M., Haemmerle, G.,Wierup,N., Dekker-Nitert,M., Rehn,M., Ristow,M., Zechner, R., Sundler, F., Holm, C., Eliasson, L., and Mulder, H. (2009)Diabetologia 52, 271–280
15. Jensen, M. D., Ekberg, K., and Landau, B. R. (2001) Am. J. Physiol. Endo-crinol. Metab. 281, E789–793
16. Vaughan, M. (1962) J. Biol. Chem. 237, 3354–335817. Reshef, L., Olswang, Y., Cassuto, H., Blum, B., Croniger, C. M., Kalhan,
S. C., Tilghman, S. M., and Hanson, R. W. (2003) J. Biol. Chem. 278,30413–30416
18. Newsholme, E. A., and Crabtree, B. (1976) Biochem. Soc. Symp. 61–10919. Hahn, P., and Novak, M. (1975) J. Lipid Res. 16, 79–9120. Mulder, H., Holst, L. S., Svensson, H., Degerman, E., Sundler, F., Ahren, B.,
Rorsman, P., and Holm, C. (1999) Diabetes 48, 228–23221. Winzell, M. S., Svensson, H., Arner, P., Ahren, B., and Holm, C. (2001)
Diabetes 50, 2225–223022. Lindvall, H., Nevsten, P., Strom, K.,Wallenberg, R., Sundler, F., Langin, D.,
Winzell, M. S., and Holm, C. (2004) J. Biol. Chem. 279, 3828–383623. Fex, M., Olofsson, C. S., Fransson, U., Bacos, K., Lindvall, H., Sorhede-
Winzell, M., Rorsman, P., Holm, C., andMulder, H. (2004) Endocrinology145, 3746–3753
24. Jenkins, C. M., Mancuso, D. J., Yan,W., Sims, H. F., Gibson, B., and Gross,R. W. (2004) J. Biol. Chem. 279, 48968–48975
25. Villena, J. A., Roy, S., Sarkadi-Nagy, E., Kim, K. H., and Sul, H. S. (2004)J. Biol. Chem. 279, 47066–47075
26. Zimmermann, R., Strauss, J. G., Haemmerle, G., Schoiswohl, G., Birner-Gruenberger, R., Riederer, M., Lass, A., Neuberger, G., Eisenhaber, F.,Hermetter, A., and Zechner, R. (2004) Science 306, 1383–1386
27. Schweiger,M., Schreiber, R., Haemmerle, G., Lass, A., Fledelius, C., Jacob-sen, P., Tornqvist, H., Zechner, R., and Zimmermann, R. (2006) J. Biol.Chem. 281, 40236–40241
28. Soni, K. G., Lehner, R., Metalnikov, P., O’Donnell, P., Semache, M., Gao,W., Ashman, K., Pshezhetsky, A. V., and Mitchell, G. A. (2004) J. Biol.Chem. 279, 40683–40689
29. Wei, E., Gao, W., and Lehner, R. (2007) J. Biol. Chem. 282, 8027–803530. Haemmerle, G., Zimmermann, R., Hayn, M., Theussl, C., Waeg, G., Wag-
ner, E., Sattler, W., Magin, T. M., Wagner, E. F., and Zechner, R. (2002)J. Biol. Chem. 277, 4806–4815
31. Fex, M., Lucas, S., Winsell, M. S., Ahren, B., Holm, C., and Mulder, H.(2006) Diabetes 55, Suppl. 2, S24–31
32. Hohmeier, H. E.,Mulder, H., Chen, G., Henkel-Rieger, R., Prentki,M., andNewgard, C. B. (2000) Diabetes 49, 424–430
33. Haemmerle, G., Lass, A., Zimmermann, R., Gorkiewicz, G., Meyer, C.,Rozman, J., Heldmaier, G., Maier, R., Theussl, C., Eder, S., Kratky, D.,Wagner, E. F., Klingenspor,M., Hoefler, G., andZechner, R. (2006) Science
ATGL and Insulin Secretion
16858 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 25 • JUNE 19, 2009
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

312, 734–73734. Freeman, H. C., Hugill, A., Dear, N. T., Ashcroft, F. M., and Cox, R. D.
(2006) Diabetes 55, 2153–215635. Gotoh, M., Maki, T., Satomi, S., Porter, J., Bonner-Weir, S., O’Hara, C. J.,
and Monaco, A. P. (1987) Transplantation 43, 725–73036. Guay, C.,Madiraju, S. R., Aumais, A., Joly, E., and Prentki,M. (2007) J. Biol.
Chem. 282, 35657–3566537. Brun, T., Duhamel, D. L., Hu He, K. H., Wollheim, C. B., and Gauthier,
B. R. (2007) Oncogene 26, 4261–427138. Roduit, R., Morin, J., Masse, F., Segall, L., Roche, E., Newgard, C. B., Assi-
macopoulos-Jeannet, F., and Prentki, M. (2000) J. Biol. Chem. 275,35799–35806
39. Massa, M. L., Borelli, M. I., Del Zotto, H., and Gagliardino, J. J. (2001) J.Endocrinol. 171, 551–556
40. Segall, L., Lameloise, N., Assimacopoulos-Jeannet, F., Roche, E., Corkey,P., Thumelin, S., Corkey, B. E., and Prentki, M. (1999) Am. J. Physiol. 277,E521–E528
41. Saddik, M., and Lopaschuk, G. D. (1991) J. Biol. Chem. 266, 8162–817042. Hutton, J. C., and Peshavaria, M. (1983) Biochem. J. 210, 235–24243. Henquin, J. C. (2000) Diabetes 49, 1751–176044. Straub, S. G., and Sharp, G. W. (2002) Diabetes Metab. Res. Rev. 18,
451–46345. Straub, S. G., James, R. F., Dunne, M. J., and Sharp, G. W. (1998) Diabetes
47, 758–76346. DeFronzo, R. A., Tobin, J. D., and Andres, R. (1979) Am. J. Physiol. 237,
E214–E22347. Roduit, R., Nolan, C., Alarcon, C., Moore, P., Barbeau, A., Delghingaro-
Augusto, V., Przybykowski, E., Morin, J., Masse, F., Massie, B., Ruderman,N., Rhodes, C., Poitout, V., and Prentki,M. (2004)Diabetes 53, 1007–1019
48. Dobbins, R. L., Chester, M. W., Stevenson, B. E., Daniels, M. B., Stein,D. T., and McGarry, J. D. (1998) J. Clin. Invest. 101, 2370–2376
49. Nolan, C. J., and Prentki, M. (2008) Trends Endocrinol. Metab. 19,285–291
50. Cnop, M., Hannaert, J. C., Hoorens, A., Eizirik, D. L., and Pipeleers, D. G.(2001) Diabetes 50, 1771–1777
51. Winzell, M. S., Strom, K., Holm, C., and Ahren, B. (2006) Nutr. Metab.Cardiovasc. Dis. 16, Suppl. 1, S11–16
52. Delghingaro-Augusto, V., Nolan, C. J., Gupta, D., Jetton, T. L., Latour,M. G., Peshavaria, M., Madiraju, S. R., Joly, E., Peyot, M. L., Prentki, M.,
and Leahy, J. (2009) Diabetologia 52, 1122–113253. Sorhede Winzell, M., and Ahren, B. (2004) Horm. Metab. Res. 36,
795–80354. Prentki, M., and Madiraju, S. R. (2008) Endocr. Rev. 29, 647–67655. Watt, M. J., van Denderen, B. J., Castelli, L. A., Bruce, C. R., Hoy, A. J.,
Kraegen, E.W., Macaulay, L., and Kemp, B. E. (2008)Mol. Endocrinol. 22,1200–1212
56. Deeney, J. T., Gromada, J., Høy,M., Olsen, H. L., Rhodes, C. J., Prentki,M.,Berggren, P. O., and Corkey, B. E. (2000) J. Biol. Chem. 275, 9363–9368
57. Olofsson, C. S., Salehi, A., Holm, C., and Rorsman, P. (2004) J. Physiol. 557,935–948
58. Gonzalo, S., and Linder, M. E. (1998)Mol. Biol. Cell 9, 585–59759. Chapman, E. R., Blasi, J., An, S., Brose, N., Johnston, P. A., Sudhof, T. C.,
and Jahn, R. (1996) Biochem. Biophys. Res. Commun. 225, 326–33260. Shu, Y., Liu, X., Yang, Y., Takahashi,M., andGillis, K. D. (2008) J. Neurosci.
28, 21–3061. Rhee, J. S., Betz, A., Pyott, S., Reim, K., Varoqueaux, F., Augustin, I., Hesse,
D., Sudhof, T. C., Takahashi,M., Rosenmund, C., and Brose, N. (2002)Cell108, 121–133
62. Kwan, E. P., Xie, L., Sheu, L., Nolan, C. J., Prentki, M., Betz, A., Brose, N.,and Gaisano, H. Y. (2006) Diabetes 55, 1421–1429
63. Solimena, M., Dirkx, R., Jr., Hermel, J. M., Pleasic-Williams, S., Shapiro,J. A., Caron, L., and Rabin, D. U. (1996) EMBO J. 15, 2102–2114
64. Trajkovski, M., Mziaut, H., Altkruger, A., Ouwendijk, J., Knoch, K. P.,Muller, S., and Solimena, M. (2004) J. Cell Biol. 167, 1063–1074
65. Mziaut, H., Trajkovski, M., Kersting, S., Ehninger, A., Altkruger, A., Le-maitre, R. P., Schmidt, D., Saeger, H. D., Lee,M. S., Drechsel, D.N.,Muller,S., and Solimena, M. (2006) Nat. Cell Biol. 8, 435–445
66. Schoenborn, V., Heid, I. M., Vollmert, C., Lingenhel, A., Adams, T. D.,Hopkins, P. N., Illig, T., Zimmermann, R., Zechner, R., Hunt, S. C., andKronenberg, F. (2006) Diabetes 55, 1270–1275
67. Akiyama, M., Sakai, K., Ogawa, M., McMillan, J. R., Sawamura, D., andShimizu, H. (2007)Muscle Nerve 36, 856–859
68. Fischer, J., Negre-Salvayre, A., and Salvayre, R. (2007) Med. Sci. 23,575–578
69. Kobayashi, K., Inoguchi, T., Maeda, Y., Nakashima, N., Kuwano, A., Eto,E., Ueno, N., Sasaki, S., Sawada, F., Fujii, M., Matoba, Y., Sumiyoshi, S.,Kawate, H., and Takayanagi, R. (2008) J. Clin. Endocrinol. Metab. 93,2877–2884
ATGL and Insulin Secretion
JUNE 19, 2009 • VOLUME 284 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 16859
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from

Joly, S. R. Murthy Madiraju, Vincent Poitout and Marc PrentkiLussier, Marco Pineda, Neil B. Ruderman, Guenter Haemmerle, Rudolf Zechner, Érik
Marie-Line Peyot, Claudiane Guay, Martin G. Latour, Julien Lamontagne, RoxaneSecretion
Adipose Triglyceride Lipase Is Implicated in Fuel- and Non-fuel-stimulated Insulin
doi: 10.1074/jbc.M109.006650 originally published online April 22, 20092009, 284:16848-16859.J. Biol. Chem.
10.1074/jbc.M109.006650Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2009/06/17/M109.006650.DC1
http://www.jbc.org/content/284/25/16848.full.html#ref-list-1
This article cites 68 references, 39 of which can be accessed free at
by guest on February 19, 2018http://w
ww
.jbc.org/D
ownloaded from


















