
and Campylobacter fetus and N-glycans as targets for ...
254
N-linked glycosylation in Campylobacter jejuni and Campylobacter fetus and N-glycans as targets for antibody-based detection A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Biology, Medicine and Health 2017 Danielle Weaver School of Biological Sciences / Division of Infection, Immunity and Respiratory Medicine
Transcript of and Campylobacter fetus and N-glycans as targets for ...

N-linked glycosylation in Campylobacter jejuni
and Campylobacter fetus and N-glycans as
targets for antibody-based detection
A thesis submitted to the University of Manchester for the degree of Doctor
of Philosophy in the Faculty of Biology, Medicine and Health
2017
Danielle Weaver
School of Biological Sciences / Division of Infection,
Immunity and Respiratory Medicine

2
Table of Contents
List of Figures ....................................................................................................................... 8
List of Tables ...................................................................................................................... 11
Abbreviations ..................................................................................................................... 12
Abstract ............................................................................................................................... 15
Declaration ......................................................................................................................... 16
Copyright Statement ........................................................................................................... 16
Acknowledgements ............................................................................................................ 17
Chapter 1. Introduction ....................................................................................................... 18
1.1. The Genus Campylobacter .......................................................................................... 19
1.1.1 C. jejuni and C. coli are the most common Campylobacter pathogens ................ 19
1.1.2. Campylobacter fetus: a veterinary and human pathogen. .................................... 20
1.1.3. Emerging Campylobacter pathogens. .................................................................. 21
1.1.4. Overview of Campylobacter in the farming industry. .......................................... 22
1.2. Features of Protein glycosylation. ............................................................................... 23
1.2.1. N-linked protein glycosylation in Eukaryotes and Archaea ................................. 24
1.3. O-linked protein glycosylation in Bacteria .................................................................. 25
1.4. Overview of N-glycosylation in Bacteria .................................................................... 26
1.5. N-linked glycosylation in Campylobacter ................................................................... 27
1.5.1. Campylobacter jejuni N-glycosylation. ................................................................ 27
1.5.1.1. The C. jejuni N-linked glycosylation pathway .................................................. 28
1.5.1.2 The Campylobacter oligosaccharyltransferase, PglB ......................................... 30
1.5.2. The role of N-glycosylation in C. jejuni ............................................................... 31
1.5.3. N-glycosylation in other Campylobacter species. ................................................ 32
1.5.3.1. N-linked glycosylation in C. fetus ................................................................. 35
1.5.4. N-linked glycosylation in closely related genera, Helicobacter and Wolinella ... 36
1.5.5. N-linked glycosylation in hydrothermal vent Bacteria ........................................ 36
1.6. Campylobacter detection/identification ...................................................................... 37

3
1.6.1. Culture and phenotypic testing ............................................................................. 37
1.6.2. Molecular methods for detecting Campylobacter in poultry ............................... 38
1.6.3. Methods of detecting Campylobacter fetus in cattle. ........................................... 43
1.6.4. Emerging methods for Campylobacter detection ................................................. 44
1.6.5. N-linked glycans as targets for antibody-based detection. ................................... 45
1.7 Aims ............................................................................................................................. 47
Chapter 2. Materials and Methods. ..................................................................................... 48
2.1 Bacterial strains and plasmids ................................................................................. 49
2.2 Isolation of Campylobacter from retail chicken ...................................................... 49
2.2.1 Culture and isolation of Campylobacter chicken meat isolates ....................... 49
2.2.2 Verification and Molecular typing of isolates .................................................. 49
2.3 Whole cell lysate preparation for SDS-PAGE ........................................................ 50
2.4 SDS-PAGE and western blotting ............................................................................ 50
2.5 Dot blot assay .......................................................................................................... 51
2.6 Flow cytometry ........................................................................................................ 51
2.7. Polymerase Chain Reaction ..................................................................................... 52
2.8. DNA agarose gel electrophoresis ............................................................................ 54
2.9. DNA restriction digestion and ligation .................................................................... 55
2.10. E. coli competent cell preparation and heat shock transformation ...................... 55
2.11. C. fetus conjugation ............................................................................................. 55
2.12. Protein expression in E. coli BL21 (DE3) ........................................................... 56
2.13. Nickel affinity chromatography ........................................................................... 56
2.14. Ion exchange chromatography ............................................................................. 57
2.15. Post-purification protein treatment ...................................................................... 57
2.16. Mass Spectrometry .............................................................................................. 57
2.16.1. Sample preparation and MALDI-MS analysis ............................................. 57
2.16.2. LC-MS/MS ................................................................................................... 58
2.17. Bioinformatics ..................................................................................................... 59

4
Chapter 3. Characterisation of an antiserum raised against a Campylobacter jejuni N-
linked glycoprotein ............................................................................................................. 60
3.1. Introduction ............................................................................................................. 61
3.1.1. Overview of C. jejuni NGRP antiserum production process ........................... 61
3.1.2. Project aims ...................................................................................................... 61
3.2. Characterisation of CjNgp reactivity against Campylobacter whole cell lysate
extracts. ............................................................................................................................... 63
3.2.1. The reactivity of CjNgp antiserum against C. jejuni N-linked glycan. ............ 63
3.2.2. CjNgp reactivity with a variety of C. jejuni strains ......................................... 65
3.2.3. CjNgp reactivity with recent Campylobacter chicken meat isolates ............... 67
3.2.4. Campylobacter species specificity of CjNgp. .................................................. 69
3.3. CjNgp binding untreated cells of Campylobacter. .................................................. 71
3.3.1. CjNgp binding to untreated C. jejuni cells is not dependent on N-linked
glycosylation. .................................................................................................................. 71
3.3.2. Reactivity against the NGRP protein influences the ability of CjNgp to bind
untreated C. jejuni cells. ................................................................................................. 73
3.3.3. CjNgp binding to untreated cells of Campylobacter species ........................... 75
3.3.4. CjNgp labels whole cells of C. jejuni 11168H ................................................. 77
3.4. Discussion ................................................................................................................ 79
Chapter 4. Development and characterisation of an antiserum raised against a
Campylobacter fetus N-linked glycoprotein ...................................................................... 83
4.1 Introduction ............................................................................................................. 84
4.2 Production of a recombinant Campylobacter fetus N-linked glycoprotein for use as
immunogen. ........................................................................................................................ 86
4.2.1 Using a C. fetus chromosomal integration vector to integrate an N-glycoprotein
was not a suitable approach. ........................................................................................... 86
4.2.2 Production of a C. fetus conjugative vector encoding an N-glycoprotein. ...... 88
4.2.3 Production of N-linked glycoprotein immunogen in C. fetus. ......................... 90
4.2.4 Purification of the N-linked glycoprotein immunogen from C. fetus. ............. 92

5
4.3 Characterisation of C. fetus NGRP antiserum (CfNgp) reactivity with
Campylobacter whole cell lysate extracts. ......................................................................... 95
4.3.1 CfNgp reactivity with C. fetus fetus NCTC 10842. ......................................... 95
4.3.2 CfNgp reactivity with numerous C. fetus strains. ............................................ 97
4.3.3 Campylobacter species specificity of CfNgp. .................................................. 99
4.3.4 C. jejuni N-linked glycan structural specificity of CfNgp. ............................ 101
4.4 CfNgp binding to Campylobacter cells. ................................................................ 103
4.4.1 CfNgp binding untreated cells of C. fetus. ..................................................... 103
4.4.2 The influence of pglB on CfNgp C. fetus cell binding. .................................. 104
4.4.3 CfNgp binding to untreated cells of Campylobacter species. ........................ 106
4.5 Discussion .............................................................................................................. 108
Chapter 5. Towards engineering the C. fetus N-linked glycan using glycocompetent E.
coli containing a hybrid C. jejuni-C. fetus system ............................................................ 115
5.1 Introduction ................................................................................................................ 116
5.1.1 Comparison of the C. fetus and C. jejuni pgl loci ............................................... 116
5.2 Strategy to investigate the role of C. fetus glycosyltransferases in N-glycan assembly
by integrating them into an E. coli system. ...................................................................... 118
5.2.1 An uncharacterised predicted glycosyltransferase gene lies within C. fetus pgl
locus .............................................................................................................................. 118
5.2.2 Model of a hybrid C. jejuni/C. fetus glycosylation machinery. ........................... 120
5.3 Identification of initial C. fetus glycosyltransferase to act on C. jejuni pglH::kn
trisaccharide. ..................................................................................................................... 122
5.3.1 Cloning strategy ................................................................................................... 122
5.3.1.1 Cloning pglH1 into pETNGRP ............................................................... 122
5.3.1.2 Cloning pglH2 into pETNGRP ............................................................... 125
5.3.1.3 Cloning Cf1389 into pETNGRP ............................................................. 127
5.3.1.4 Cloning CjpglH into pETNGRP ............................................................. 129
5.3.2 Only CfpglH1 decreased the mobility of NGRP glycoforms in the ppglpglH::kn
background. .................................................................................................................. 131

6
5.4 Identification of second C. fetus glycosyltransferase to act on C. jejuni pglH::kn
trisaccharide. ..................................................................................................................... 133
5.4.1 Cloning strategy ................................................................................................... 133
5.4.1.1 Construction of pNH1H2 ............................................................................. 133
5.4.1.2 Construction of pNH1-1389 ......................................................................... 133
5.4.2 C. fetus pglH2 decreased NGRP glycoform mobility in the ppglpglH::kn CfpglH1
background. .................................................................................................................. 135
5.5 Structural analysis of N-linked glycoproteins produced from hybrid systems. ......... 137
5.5.1 Purification of NGRP from C. jejuni/C. fetus hybrid N-linked glycosylation
systems in E. coli .......................................................................................................... 137
5.5.2. MALDI-TOF MS analysis of purified NGRP glycoforms ................................. 139
5.5.2 Production and purification of modified NGRP in hybrid systems. ................... 143
5.5.3 MS analysis of purified NGRP3. ......................................................................... 146
5.5.3.1 The sequon-containing tryptic peptide of unglycosylated NGRP3 was
detected by MALDI TOF Mass spectrometry. ......................................................... 146
5.5.3.2 MALDI-TOF MS analysis of glycosylated NGRP3 using CHCA matrix. .. 149
5.5.3.3 MALDI-TOF MS analysis of glycosylated NGRP3 using DHB matrix. ..... 151
5.5.3.4 LC-MS/MS analysis of glycosylated NGRP3 .............................................. 153
5.6 Construction and analysis of hybrid pgl system containing C. fetus pglH genes and
predicted glycosyltransferase cf1389. .............................................................................. 159
5.6.1 Cloning strategy ................................................................................................... 159
5.6.2 Addition of cf1389 to a ppglpglH::kn CfpglH1H2 background resulted in three
apparent forms of NGRP3 ............................................................................................ 160
5.6.3 MALDI-TOF MS analysis of glycosylated NGRP3 produced in the presence of C.
fetus pglH genes and cf1389. ........................................................................................ 162
5.7 Discussion ................................................................................................................... 164
Chapter 6. C. fetus N-glycoproteome prediction and the conservation of N-glycoproteins
between Campylobacter species ....................................................................................... 168
6.1 Introduction ................................................................................................................ 169
6.2 C. fetus N-glycoproteome prediction and validation .................................................. 172

7
6.2.1 The predicted C. fetus fetus 82-40 N-glycoproteome .......................................... 172
6.2.2.2 Validation of a newly predicted N-glycoprotein, Cf0445 ............................ 180
6.3 Analysis of the C. fetus predicted N-glycoproteome .................................................. 182
6.3.1 Subcellular locations of predicted C. fetus N-glycoproteins. .............................. 182
6.3.2 Sequon distribution in C. fetus predicted N-glycoproteins. ................................ 185
6.3.3 Amino acid composition of sequons in C. fetus predicted N-glycoproteins. ...... 187
6.4 Predicted conservation of N-glycoproteins within the Campylobacter genus ........... 189
6.4.1 Predicted conservation of N-glycoproteins between C. jejuni and C. fetus ........ 189
6.4.2.1 N-glycosylation of components of the CmeABC multidrug efflux system is
predicted to be conserved in several Campylobacter species .................................. 195
6.5 Discussion ................................................................................................................... 197
Chapter 7. Conclusion and future work ............................................................................ 202
7.1 N-linked glycoprotein glycans as targets for antibody-based detection/identification
.......................................................................................................................................... 203
7.2 Developing glycocompetent E. coli producing the C. fetus N-linked glycan. ........... 206
7.3 The C. fetus predicted N-glycoproteome and putative conservation of N-glycoproteins
amongst Campylobacter species. ..................................................................................... 208
7.4 Final conclusion .......................................................................................................... 211
References ........................................................................................................................ 213
Appendix .......................................................................................................................... 252
Final word count: 50,869

8
List of Figures
Figure 1.1. Campylobacter jejuni Pgl pathway. 29
Figure 1.2. N-linked glycan structures produced by Campylobacter species,
Helicobacter pullorum and Wolinella succinogenes. 34
Figure 3.1. N-linked glycan structural specificity of CjNgp antiserum. 64
Figure 3.2. Reactivity of CjNgp antiserum with diverse C. jejuni reference strains. 66
Figure 3.3. Reactivity of CjNgp antiserum with Campylobacter chicken meat
isolates. 68
Figure 3.4. Campylobacter species specificity of CjNgp antiserum. 70
Figure 3.5. CjNgp C. jejuni cell binding in dot blot assay. 72
Figure 3.6. CjNgp cell binding upon pre-incubation with BL21 ± NGRP lysates. 74
Figure 3.7. CjNgp binding to cells of Campylobacter species. 76
Figure 3.8. Flow cytometric analysis of CjNgp antiserum labelling of fluorescent
C. jejuni 11168gfp4. 78
Figure 4.1 Chromosomal integration of NGRP into C. fetus did not produce
sufficient protein yield. 87
Figure 4.2. Construction of pG1-N and conjugation into C. fetus. 89
Figure 4.3. Production of glycosylated NGRP in C. fetus 91
Figure 4.4. Nickel affinity chromatography purification of glycosylated NGRP
from C. fetus. 94
Figure 4.5. Initial characteriseration of CfNgp C. fetus reactivity. 96
Figure 4.6. CfNgp antiserum C.fetus strain coverage. 98
Figure 4.7. Campylobacter species specificity of CfNgp antiserum 100
Figure 4.8. C. jejuni N-linked glycan structural specificity of CfNgp antiserum. 102
Figure 4.9. CfNgp C. fetus fetus and C. fetus venerealis cell binding in dot blot
assay. 103
Figure 4.10. The pglB-independency of CfNgp C. fetus cell binding. 105
Figure 4.11. CfNgp binding to cells of Campylobacter species. 107

9
Figure 5.1. Comparison of the pgl loci and N-glycan structures of C. jejuni and
C. fetus 119
Figure 5.2. Plan to investigate predicted glycosyltransferases in C. fetus N-linked
glycosylation by creating hybrid C. jejuni/C fetus pgl system in E. coli. 121
Figure 5.3. Construction of pNH1. 124
Figure 5.4. Construction of pNH2. 126
Figure 5.5. Construction of pN1389. 128
Figure 5.6. Construction of pNCjH. 130
Figure 5.7. Identification of C. fetus PglH1 glycosyltransferase activity using a
C. jejuni pglH::kn N-glycan structure. 132
Figure 5.8. Construction of pNH1H2 and pNH1-1389. 134
Figure 5.9. Further construction of a hybrid C. jejuni/C. fetus N-glycan through
C. fetus PglH2 activity. 136
Figure 5.10. Purification of NGRP from hybrid C. jejuni/C. fetus pgl systems
expressed in E. coli BL21. 138
Figure 5.11. MALDI-TOF mass spectrometry analysis of NGRP expressed in
hybrid C. jejuni/C. fetus pgl systems. 142
Figure 5.12. Amino acid sequence of NGRP3, a modified version of NGRP
optimised for glycopeptide analysis by mass spectrometry. 143
Figure 5.13. Purification of NGRP3 from hybrid C. jejuni/C. fetus pgl systems
expressed in E. coli BL21. 145
Figure 5.14. MALDI-TOF Mass spectrometry analysis of unglycosylated NGRP3. 148
Figure 5.15. MALDI-TOF Mass spectrometry analysis of glycosylated NGRP3
using CHCA matrix. 150
Figure 5.16. MALDI-TOF Mass spectrometry analysis of glycosylated NGRP3
using DHB matrix. 152
Figure 5.17. LC-MS/MS analysis of unglycosylated NGRP3 and NGRP3 modified
with C. jejuni pglH::kn N-glycan. 154
Figure 5.18. LC-MS/MS analysis of NGRP3. 156
Figure 5.19. Two possible mechanisms of N-glycan assembly in the hybrid
C. jejuni/C. fetus systems. 158
Figure 5.20. Production and purification of NGRP3 from E. coli BL21 161

10
ppglpglH::kn pX12-N3.
Figure 5.21. MALDI-TOF Mass spectrometry analysis NGRP3 produced in
C. jejuni/C.fetus hybrid system containing three predicted transferases from
C. fetus.
163
Figure 6.1 Manual curation Bioinformatics workflow for prediction of
N-glycoproteomes in Campylobacter. 171
Figure 6.2 Decision tree for subcellular localisation assignment of proteins during
manual curation of results from bioinformatics N-glycoprotein prediction pipeline. 173
Figure 6.3. Validation of PglB-dependent modification of Cf0781 in C. fetus. 179
Figure 6.4. Validation of PglB-dependent modification of Cf0445 in C. fetus. 181
Figure 6.5. Subcellular localisation predictions of C. fetus proteins. 184
Figure 6.6. Distribution of N-glycosylation sequons in C. jejuni and C. fetus
experimentally identified and predicted N-glycoproteins. 186
Figure 6.7. Logo representations of amino acid abundance in N-glycosylation
sequons of C. jejuni and C. fetus known and predicted N-glycoproteins. 188
Figure 6.8 Schematic of N-glycoprotein conservation analysis results. 190

11
List of Tables
Table 2.1. Primer sequences. 52
Table 5.1. Predicted tryptic digest of NGRP when expressed from pET22(+)-
derived plasmids. 139
Table 5.2. Predicted m/z of potential tryptic glycopeptides from NGRP. 141
Table 5.3. Predicted tryptic digest of NGRP3 when expressed from pET22(+)-
derived plasmids. 147
Table 5.4. Predicted m/z of potential tryptic glycopeptides from NGRP3. 149
Table 6.1. Complete predicted C. fetus N-glycoproteome. 174
Table 6.2. Identification of N-glycosylation sequon-containing C.fetus
homologues of known C. jejuni N-glycoproteins. 191
Table 6.3 Putative conservation of experimentally identified C. jejuni N-
glycoproteins in Campylobacter species. 196
Appendix Table 1. Bacterial strains used. 252
Appendix Table 2. Plasmids used. 254

12
Abbreviations
Anti-WC 11168 Anti-whole cell C. jejuni 11168H antiserum
Asn/N Asparagine
Asp/D Aspartate
ATCC American Type Culture Collection
Bis-tris Bis-(2-hydroxy-ethyl)-amino-tris(hydroxymethyl)-methane
BLAST Basic Local Alignment Search Tool®
BLASTP Protein Basic Local Alignment Search Tool®
BSA Bovine serum albumin
BVC Bovine Venereal Campylobacteriosis
CCDA Charcoal cefoperazone deoxycholate agar
CDP Cytidine diphosphate
CDT Cytolethal distending toxin
CELLO SubCELlular LOcalisation predictor
CFU Colony forming units
CHCA α-cyano-4-hydroxycinnamic acid
cPCR Colony PCR
CV Column volume
C. f fetus C. fetus subspecies fetus
C. f ven C. fetus subspecies venerealis
C. hyo hyo C. hyointestinalis subspecies hyointestinalis
DHB 2,5-dihydroxybenzoic acid
DiNAcBac Di-N-acetylbacillosamine
DNA Deoxyribonucleic acid
EDTA Ethylenediaminetetraacetic acid
EFSA European Food Safety Authority
ER Endoplasmic reticulum
FA Formic acid
fOS Free oligosaccharide
Gal Galactose
GalNAc N-acetylgalactosamine

13
GBS Guillain Barré Syndrome
gDNA Genomic DNA
GDP Guanosine diphosphate
GFP Green Fluorescent Protein
Glc Glucose
GlcNAc N-acetylglucosamine
Glu/E Glutamic acid
GT Glycosyltransferase
Hex Hexose
HexNAc N-acetylhexosamine
His Histidine
IPTG Isopropyl β-D-1-thiogalactopyranoside
LB Lysogeny broth
LC-MS/MS Liquid chromatography-tandem mass spectrometry
LDS Lithium dodecyl sulfate
LLO Lipid-linked oligosaccharide
LPS Lipopolysaccharide
MALDI LIFT-
TOF/TOF MS
Matrix-assisted laser desorption/ionisation LIFT-time-of-
flight/time-of-flight mass spectrometry
Man Mannose
MOPS 3-(N-morpholino)propanesulfonic acid
NCBI National Centre for Biotechnology Information
NCTC National Collection of Type Cultures
OD600 Optical density at 600 nm
OIE World Organisation for Animal Health
OPG Osmoregulated periplasmic glucan
OST Oligosaccharyltransferase
P Proline
PBS Phosphate buffered saline
PBS-T Phosphate-buffered saline - Tween
PCR Polymerase Chain Reaction
RBS Ribosome binding site
R-PE R-Phycoerythrin

14
SDS-PAGE Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Ser/S Serine
SNP Single nucleotide polymorphism
SOC Super optimal broth
SpI Signal peptide I
SpII Signal peptide II (lipoprotein)
TAE Tris base, acetic acid and EDTA
TFA Trifluoroacetic acid
Thr/T Threonine
TM Transmembrane
Tris Tris(hydroxymethyl)aminomethane
T4SS Type IV secretion system
UDP Uridine diphosphate
UndP Undecaprenyl phosphate
VBNC Viable but non-culturable

15
Abstract
N-linked glycosylation in Campylobacter jejuni and Campylobacter fetus and N-
glycans as targets for antibody-based detection
Danielle Weaver, Doctor of Philosophy at the University of Manchester, 2017.
Campylobacter spp., especially C. jejuni and C. coli, are the leading cause of bacterial
gastroenteritis in Europe. There is a recognised need to develop detection tools which can
be performed on farms to facilitate reducing the presence of Campylobacter in poultry. A
similar application could be beneficial for detection of C. fetus, a veterinary pathogen
which causes significant economic loss in the cattle industry. Campylobacter species
perform protein N-linked glycosylation and in C. jejuni at least 150 proteins, many of
which are surface-exposed, may be modified. Therefore, the first portion of this thesis
investigated the feasibility of using N-linked glycans as targets for antibody-based
detection of Campylobacter species. To do this, a His-tagged N-glycoprotein was
expressed and purified from C. fetus and used as immunogen to raise an antiserum termed
CfNgp. The Campylobacter N-glycan reactivity of this antiserum was characterised and it
was shown to react with N-glycoproteins and cells of C. fetus and other emerging
Campylobacter species such as C. concisus. Immunoblotting techniques and flow
cytometry were used to characterise an antiserum (CjNgp) raised against a C. jejuni N-
linked glycoprotein and demonstrated that it can specifically detect cells of C. jejuni,
C. coli and other emerging Campylobacter species found in poulty. This thesis also
describes the investigation of the relatively uncharacterised C. fetus N-linked
glycosylation system. Functional analysis of C. fetus predicted glycosyltransferases was
acheived by developing glycocompetent E. coli containing a hybrid C. jejuni/C. fetus pgl
system. The N-glycan structures biosynthesised were analysed using mass spectrometry
and this novel approach discovered the activity of two C. fetus glycosyltransferase
enzymes. Finally, this work used a bioinformatics pipeline to produce a C. fetus predicted
N-linked glycoproteome and experimentally verified a newly identified N-linked
glycoprotein. This pipeline was also applied to investigate the putative conservation of N-
linked glycoproteins throughout the Campylobacter genus and highlighted ‘core’ N-linked
glycoproteins which are key targets for experimental investigation. Overall, this work
demonstrates that Campylobacter N-linked glycans are attractive targets for antibody-
based detection, expands our knowledge of C. fetus N-linked glycosylation and contributes
to the broader understanding of this intriguing aspect of Campylobacter biology.

16
Declaration
No portion of the work referred to in this thesis has been submitted in support of an
application for another degree or qualification of this or any other university or other
institute of learning
Copyright Statement
I. The author of this thesis (including any appendices and/or schedules to this thesis)
owns certain copyright or related rights in it (the “Copyright”) and she has given
The University of Manchester certain rights to use such Copyright, including for
administrative purposes.
II. Copies of this thesis, either in full or in extracts and whether in hard or electronic
copy, may be made only in accordance with the Copyright, Designs and Patents
Act 1988 (as amended) and regulations issued under it or, where appropriate, in
accordance Presentation of Theses Policy You are required to submit your thesis
electronically Page 11 of 25 with licensing agreements which the University has
from time to time. This page must form part of any such copies made.
III. The ownership of certain Copyright, patents, designs, trademarks and other
intellectual property (the “Intellectual Property”) and any reproductions of
copyright works in the thesis, for example graphs and tables (“Reproductions”),
which may be described in this thesis, may not be owned by the author and may be
owned by third parties. Such Intellectual Property and Reproductions cannot and
must not be made available for use without the prior written permission of the
owner(s) of the relevant Intellectual Property and/or Reproductions.
IV. Further information on the conditions under which disclosure, publication and
commercialisation of this thesis, the Copyright and any Intellectual Property and/or
Reproductions described in it may take place is available in the University IP
Policy (http://documents.manchester.ac.uk/DocuInfo.aspx?DocID=24420), in any
relevant Thesis restriction declarations deposited in the University Library, The
University Library’s regulations
(http://www.library.manchester.ac.uk/about/regulations/) and in The University’s
policy on Presentation of Theses.

17
Acknowledgements
I would like to thank my supervisor, Dr. Dennis Linton, for providing his time and
guidance during my PhD. I am also very grateful to Dr. Adrian Jervis for passing on his
wisdom at the beginning and for continued help and advice upon leaving the group. I
would like to extend my gratitude to the Society for Applied Microbiology for funding this
PhD.
I would like to acknowledge all those in the Microbiology lab for the technical assistance
and conversation along the way. In particular, thanks to Ange for all her help and advice. I
would also like to thank Nader for all the laughs and out-of-hours discussions.
Finally, I would like to thank my friends and family for all the good times. I am
particularly thankful to Elena for supporting me and providing positivity when mine had
run dry. Finally, a special thanks to Mam and Dad for all the support and encouragement
throughout.

18
Chapter 1. Introduction

19
1.1. The Genus Campylobacter
1.1.1 C. jejuni and C. coli are the most common Campylobacter pathogens
Campylobacter is a genus of Gram-negative Epsilon-proteobacteria with a low GC content
and a curved or helical rod morphology (Lastovica et al., 2014). All but two species,
C. gracilis and C. hominis, are motile (Vandamme et al., 1995; Lawson et al., 2001). The
majority are microaerobic, but some prefer anaerobic conditions and require hydrogen or
formate for growth (Kaakoush et al., 2015). The genus contains a variety of pathogens
able to cause gastroenteritis in humans, and the occurrence of campylobacteriosis appears
to have risen in the recent decade (Kaakoush et al., 2015). In particular, C. jejuni and
C. coli are the most common agents of campylobacteriosis. Together with C. upsaliensis
and C. lari these form the so-called ‘thermotolerant’ Campylobacter group. All
Campylobacter species grow between 30 and 42 °C, but this group grow best at 42° C and
are commonly found in birds (Lastovica et al., 2014). The presence of these organisms in
poultry is the main reservoir for human infection (EFSA, 2010). In the UK over 60% of
retail chicken meat is positive for Campylobacter, and over 10% is heavily contaminated
(> 1000 colony forming units per gram) (PHE, 2017). In the European Union
Campylobacter is the most common bacterial cause of gastroenteritis, with more than
200,000 cases in 2015 (EFSA, 2016).
As the major human pathogen of the genus, C. jejuni is the best characterised species.
Infrequently, Campylobacter infections can also lead to Guillain Barré Syndrome (GBS)
which causes temporary paralysis that can last many months, and even be fatal (van Doorn
et al., 2008). This is a result of molecular mimicry, as certain variants of C. jejuni lipo-
oligosaccharide (LOS) found on its surface can induce an autoimmune response against
human neuronal gangliosides (Young et al., 2007). C. jejuni LOS and its capsular
polysaccharide are structurally variable which is thought to help evade the host immune
response. In addition, C. jejuni has a variety of features which aid its adherence to and
invasion of host cells. These include adhesins such as CadF and JlpA, the flagellar type III
secretion system and the mitosis-blocking cytotoxin, cytolethal distending toxin or CDT
(Jin et al., 2001; Konkel et al., 2004, 1997; Pickett and Whitehouse, 1999).
There have been growing concerns of drug resistance within Campylobacter species, in
particular against the cornerstone treatments, fluoroquinolone and macrolide antibiotics.
Indeed, the World Health Organisation has recently designated fluoroquinolone-resistant
Campylobacter spp. as high priority in regards to the need for novel antibiotics (WHO,

20
2017). The expansion of macrolide resistance is particularly concerning in C. coli, where
rates of resistant human isolates were over three times higher in 2014 compared to 2011
(NARMS, 2016). Of particular note is CmeABC, an efflux pump that provides protection
against bile salts but also contributes to drug resistance (Lin et al., 2002). In addition to
developing intrinsic mutations which can provide resistance, it can also work in
conjunction with other mutations such as in the DNA gyrase gene, gyrA, to mediate
enhanced antibiotic resistance (Luangtongkum et al., 2009; Luo et al., 2003). This system
has been found to play a role in drug resistance of several Campylobacter species (Guo et
al., 2010).
1.1.2. Campylobacter fetus: a veterinary and human pathogen.
Campylobacter fetus consists of three subspecies, C. fetus subsp. fetus, C. fetus subsp.
venerealis and C. fetus subsp. testudinum (Patrick et al., 2013; Veron and Chatelain,
1973). C. fetus subsp. fetus causes disease in sheep, cattle and, less frequently, humans.
Veterinary infections often result in abortion (Campero et al., 2005), whereas human
infections can involve a variety of outcomes ranging from diarrhoea to systemic disease
(Wagenaar et al., 2014). Most C. fetus subsp. fetus human infections involve
immunocompromised, pregnant or elderly individuals. In contrast, C. fetus subsp.
venerealis is an extremely rare cause of human vaginosis (Holst et al., 1987), but has a
specific host niche within the reproductive tract of cattle, where it can cause abortion and
temporary infertility (Mshelia et al., 2010). This disease, named Bovine Venereal
Campylobacteriosis (BVC), is a considerable burden on the cattle industry as it can cause
infertility for up to 8 months (Mshelia et al., 2007). The most recently described member
of the species, C. fetus testudinum, is primarily associated with reptiles but can also infect
humans (Choi et al., 2016; Fitzgerald et al., 2014; Patrick et al., 2013). Although, like C.
fetus fetus, C. fetus testudinum can cause systemic infection in the immunocompromised
or elderly, these instances have been described much less frequently in the literature.
C. fetus harbour many virulence factors also found in C. jejuni. For example, it contains
CDT, the invasion protein CiaB and adhesins such and CadF and PEB1 (Ali et al., 2012).
In contrast to C. jejuni, C. fetus does not have a capsular polysaccharide. However, a
striking and distinct aspect of C. fetus biology is the crystalline array of S-layer proteins
on its surface. S layers are a common feature in Archaea but are less common in Bacteria,
making C. fetus relatively unusual. In C. fetus, the S layer is subject to antigenic variation
due to alternating expression of two S layer protein homologs, SapA and SapB (Garcia et

21
al., 1995), contributing to immune evasion and serum resistance (Blaser et al., 1988;
Thompson, 2002).
Comparative genomics recently identified considerable conservation of virulence factors
between C. fetus subspecies, including the S layer proteins, CadF, CiaB and CdtABC
(Gilbert et al., 2016). However, there are differences proposed to explain the distinct host
niches of each subspecies (Gilbert et al., 2016). For example, the most defining feature of
C. fetus venerealis which separates it from the other subspecies is the existence of a
pathogenicity island (PAI) encoding a type IV secretion system (T4SS) (Gorkiewicz et al.,
2010).
1.1.3. Emerging Campylobacter pathogens.
Several Campylobacter species are said to be ‘emerging’ pathogens as their incidence in
disease is becoming more appreciated due to recent advances in detection and
identification techniques (Man, 2011). These species include C. lari, C. upsaliensis, C.
concisus and C. hyointestinalis and likely remain underrecognised as causes of human
disease (Kaakoush et al., 2015). For example, a study of paediatric diarrhoeic stools
reported the isolation of Campylobacter species other than C. jejuni and C. coli from 48%
of samples (Allos and Lastovica, 2008). In addition, many of these emerging
Campylobacter species harbour cytolethal distending toxin (CDT) genes (Man, 2011).
C. lari has been isolated from a variety of organisms including mammals, birds and even
molluscs (Man, 2011). In humans, C. lari can cause enteritis (Simor and Wilcox, 1987)
and more infrequently, bacteraemia (Martinot et al., 2001; Werno et al., 2002). Similarly,
C. upsaliensis can cause gastroenteritis (Goossens et al., 1990) and rare cases of
bacteraemia and abortion have been documented (Gurgan and Diker, 1994; Patton et al.,
1989). Unlike other thermotolerant Campylobacter species which commonly reside in
birds, this species is primarily found in dogs and cats (Baker et al., 1999).
C. ureolyticus, recently reclassified from the genus Bacteroides (Vandamme et al., 2010),
is another emerging Campylobacter pathogen. Its incidence in human infection appears to
be increasing and O’Donovan et al. (2014) argue that it may now represent the second
most common Campylobacter species causing gastroenteritis, surpassing C. coli.
Interestingly, cattle appear to be the reservoir for this species, with transmission via
contaminated milk (Koziel et al., 2012).
C. concisus was first identified from the human oral cavity (Tanner et al., 1981) and has
since been isolated from dogs and cats (Chaban et al., 2010; Petersen et al., 2007). This

22
species can cause gastroenteritis often with milder symptoms that last longer than
infections caused by C. jejuni/C. coli (Kaakoush et al., 2015; Nielsen et al., 2012). It has
also been linked with inflammatory bowel disease and periodontitis; however, its role in
these diseases is still contested (Kaakoush and Mitchell, 2012).
C. hyointestinalis consists of two subspecies, C. hyointestinalis subsp. hyointestinalis and
C. hyointestinalis subsp. lawsonii, which are found in the gastrointestinal tracts of cattle
and swine, respectively (Gebhart et al., 1985; Miller et al., 2016; On et al., 1995). There
are rare cases of C. hyointestinalis enteritis in humans (Edmonds et al., 1987) thought to
be transmitted from infected swine, where it also causes enteritis (Gorkiewicz et al., 2002).
Furthermore, C. hyointestinalis subsp. hyointestinalis can cause potentially fatal enteritis
in calves (Diker et al., 1990).
1.1.4. Overview of Campylobacter in the farming industry.
Campylobacter are usually transmitted to humans through consumption of contaminated
meat. Therefore, to reduce human infection, the problem needs to be addressed at the farm
level. Campylobacter spp. are widely distributed in the environment, having been
identified in wild birds, domestic animals, freshwater and sewage (Jones, 2001). As a
result, biosecurity measures are often implemented on farms to reduce livestock exposure.
In broiler chickens, maternal antibodies seem to provide some protection as colonisation
does not usually occur until chicks are at least 2 weeks old (Sahin et al., 2003). However,
once acquired, the colonisation often persists throughout its lifetime (Lee and Newell,
2006). It is traditionally thought that this colonisation is asymptomatic and thus
Campylobacter is often viewed as a chicken gut commensal (Hermans et al., 2012).
However, recent data suggest that C. jejuni colonisation induces a host inflammatory
response and, in certain chicken breeds, can cause chronic inflammation and diarrhoea
(Humphrey et al., 2014). Further studies have also identified that colonisation can impair
nutrient uptake and growth in commercial chickens (Awad et al., 2015a, 2015b).
The majority of Campylobacter transmission through a broiler flock occurs horizontally,
although there is some evidence suggesting vertical transfer can occur (Newell and
Fearnley, 2003). Transmission is mostly via the faecal-oral route, particularly due to
contaminated water. Once colonisation is established in one individual, Campylobacter
can spread rapidly throughout an entire flock within a few days (Lee and Newell, 2006).
However, the often asymptomatic nature of Campylobacter colonisation in poultry means
contamination of a flock can go unnoticed without active monitoring. Therefore, regular

23
surveillance of Campylobacter infection should be undertaken. Methods implemented for
identifying these organisms are outlined in section 1.6.
At the farm level, preventative measures include enhanced biosecurity, chicken
vaccination, chlorinating drinking water and competitive exclusion (e.g. probiotics)
(Umaraw et al., 2017). Antibiotics have traditionally been used as both preventative and
treatment measures. However, as discussed, the emergence of antibiotic resistance within
Campylobacter is concerning and hence alternative measures are being sought (Johnson et
al., 2017). For example, both bacteriocin and bacteriophage therapy can successfully
reduce the load of Campylobacter in broilers (Johnson et al., 2017; Umaraw et al., 2017).
Another option to prevent Campylobacter infection in humans is to eliminate the organism
during carcass processing, where contamination of meat with cecal contents often occurs.
These methods of treatment include multi-stage scalding of carcasses, chilling, irradiation
and freezing the meat (Umaraw et al., 2017).
C. fetus infection of cattle causes significant economic losses in agriculture, with reports
of up to a 66% reduction in gross profit in the initial year of infection (McMillen et al.,
2006). As this organism can also cause infections in humans, an infected herd also
presents a human health risk. Bulls can act as asymptomatic carriers (Samuelson and
Winter, 1966), meaning infected animals are difficult to identify. Thus, herds should be
routinely surveyed for infection. Tools used to detect and identify C. fetus from samples
will be discussed in section 1.6. Prevention and control measures for C. fetus include
increased biosecurity, artificial insemination, isolation of infected individuals and
vaccination of bulls (Mshelia et al., 2007). All members of contaminated herds are often
vaccinated to reduce the duration of infection, however, cows can still remain infected for
more than one mating season (OIE, 2008). Although antibiotic treatment is successful for
bulls, it usually does not fully eliminate C. fetus from the reproductive tract of cows
(Mshelia et al., 2007). Due to this, C. fetus infection often results in culling to prevent
further infection spread and hence incurs further cost (Truyers et al., 2014).
1.2. Features of Protein glycosylation.
A key aspect of Campylobacter biology is the extensive protein glycosylation systems.
The following sections outline the general features of protein glycosylation within all three
Domains of life, with a particular focus on N-linked glycosylation in Bacteria. Protein

24
glycosylation describes the enzymatic covalent linkage of carbohydrates onto proteins. It
is predicted that over 70% of eukaryotic proteins are glycosylated (Dell et al., 2010). The
diversity of glycan structures identified in eukaryotes is astounding, and a single
glycosylation site can harbour one of a variety of complex structures resulting in vast
microheterogeneity (Moremen et al., 2012). As a result, protein glycosylation serves many
functions, such as protein folding and cell adhesion, which can in turn have implications in
immunity, inflammation and cancer (Moremen et al., 2012). The two main types of protein
glycosylation are N-linked and O-linked. The former involves transfer of a glycan onto a
nitrogen atom within an asparagine (Asn or N) residue. The latter corresponds to addition
of a glycan onto an oxygen atom of the amino acid residues, serine (Ser) and threonine
(Thr) (Dell et al., 2010). In animals, well characterised O-glycosylated proteins include
mucins and collagen, each modified with distinct glycan structures. However, N-linked
glycosylation is the most predominant type of glycosylation, as around 90% of eukaryotic
glycoproteins are modified with N-glycans (Apweiler et al., 1999).
1.2.1. N-linked protein glycosylation in Eukaryotes and Archaea
In eukaryotes, N-glycan structures are first assembled at the cytosolic face of the
endoplasmic reticulum (ER) membrane onto a lipid carrier (dolichol phosphate in
eukaryotes), forming the lipid-linked oligosaccharide (LLO) precursor (Burda and Aebi,
1999). Several glycosyltransferase enzymes encoded by the asparagine-linked
glycosylation (ALG) pathway genes act sequentially to construct this LLO. At the
cytoplasmic face, a glycan structure consisting of two N-acetylglucosamine (GlcNAc)
residues and five mannose (Man) residues is formed by glycosyltransferase enzymes
which utilise soluble nucleotide-activated sugar donors (Breitling and Aebi, 2013). This
intermediate is then flipped across the membrane and additional ALG glycosyltransferases
elaborate this structure within the lumen of the ER to give GlcNAc2 Man9Glc3. Finally, the
oligosaccharyltransferase (OST) complex transfers the constructed glycan en bloc from
the precursor to target polypeptides containing the acceptor tripeptide sequon, N-X-S/T
(where X ≠ P) (Moremen et al., 2012). The OST is a hetero-oligomeric enzyme consisting
of up to nine subunits in higher eukaryotes, although it is the STT3 protein which is
catalytically active (Kelleher et al., 2003; Kelleher and Gilmore, 2005). There are two
isoforms of this subunit, STT3A and STT3B. The former is involved in co-translational N-
glycosylation, where the OST complex associates with the SEC61 translocon and modifies
polypeptides as they are entering the rough ER (Shrimal et al., 2015). An OST complex
containing STT3B then acts on any remaining unmodified N-glycosylation sites in a co-

25
translational or post-translocational manner (Ruiz-Canada et al., 2009; Shrimal et al.,
2015). Although all N-glycoproteins are first modified with an identical N-glycan
structure, this is then extensively remodelled in the Golgi apparatus (Moremen et al.,
2012). The N-glycans can be trimmed and further elaborated, often with residues such as
fucose and sialic acid, resulting in a variety of structures that can be complex and highly
branched.
N-glycosylation was considered a unique feature of eukaryotes until the S layer protein of
an archaea, Halobacterium salinarum, was identified to contain an N-linked glycan
(Mescher et al., 1974). The most characterised archaeal N-glycosylation pathway is now
that of Haloferax volcanii, which is encoded by Agl (archaeal glycosylation) genes (Jarrell
et al., 2014). This N-glycosylation process begins at the cytoplasmic face of the cell
membrane, where sugar residues are transferred to a dolichol-phosphate or dolichol-
pyrophosphate lipid carrier to form LLO (Schwarz and Aebi, 2011). An unusual feature of
this process is that two distinct types of LLO are formed, one containing the first four
residues of the N-glycan and the other carrying a mannose residue which will be the
terminal sugar (Eichler, 2013). Once these LLOs have been flipped across the membrane,
in a similar manner as the eukaryotic pathway, the OST transfers the tetrasaccharide to
asparagine residues within N-X-S/T (X ≠ P) motifs. The archaeal OSTs are a single
enzyme rather than a complex, and are homologous to the eukaryotic STT3 catalytic
subunit (Magidovich and Eichler, 2009). The final residue of the N-glycan is transferred
by the AglS enzyme (Cohen-Rosenzweig et al., 2012). Overall, archaeal N-glycan
pathways and the structures they construct are more diverse than analogous eukaryotic
systems (Schwarz and Aebi, 2011). For example, in Archaea at least twenty types of sugar
residues have been identified in N-linked glycans (Eichler, 2013), with uronic acid
residues appearing as a distinct feature. Furthermore, considering the limited number of
organisms studied so far, this diversity will likely expand considerably in the future
(Eichler, 2013).
1.3. O-linked protein glycosylation in Bacteria
O-linked glycosylation was first identified in Bacteria when a trisaccharide was identified
on the Neisseria meningitidis pilin protein (Stimson et al., 1995). Pilin O-glycosylation
also occurs in Neisseria gonorrhoeae and numerous pilin glycosylation (pgl) genes
encoding glycosyltransferases and sugar biosynthesis enzymes are now known (Aas et al.,

26
2007; Power et al., 2003). It was later demonstrated that Neisseria pilin modification was
performed by a general O-linked glycosylation pathway and at least twelve O-
glycoproteins has been identified (Ku et al., 2009; Vik et al., 2009). Analogous to
eukaryotic N-glycosylation, these pathways consist of OSTs, PglL and PglO in N.
meningitidis and N. gonorrhoeae, respectively, which mediate the block transfer of glycan
onto Ser or Thr residues (Aas et al., 2007; Dell et al., 2010; Faridmoayer et al., 2007). The
O-glycan structure of N. meningitidis consists of two galactose (Gal) residues and either
2,4-diacetamido-2,4,6-trideoxyhexose (DATDH) or 2-glyceramido-4-acetamido-2,4,6-
trideoxyhexose (GATDH) as the reducing end sugar (Chamot-Rooke et al., 2007).
Distinct O-linked glycosylation systems where sugar residues are sequentially added to
flagellar proteins exist in many Bacteria, including Campylobacter (Logan, 2006). C.
jejuni modifies the flagellin protein, FlaA, with as many as 19 O-glycans (Thibault et al.,
2001). These O-glycans are composed mainly of pseudaminic acid, legionaminic acid and
their derivatives and such modification is required for flagellar assembly and motility
(Goon et al., 2003; Logan et al., 2009; Thibault et al., 2001). Genetic loci encoding the O-
glycosylation system display considerable interstrain variation and can contain upto 50
genes (Mcnally et al., 2006). Many of these genes contain homopolymeric tracts which
likely alter glycan structure due to phase variation, as demonstrated for Cj1295 (Hitchen et
al., 2010). However, this complex system and its numerous glycosyltransferases remain
relatively uncharacterised.
1.4. Overview of N-glycosylation in Bacteria
In 2002, the discovery of an N-linked glycosylation system in C. jejuni (Wacker et al.,
2002; Young et al., 2002) showed that this process exists in all three domains of life.
General similarities can be drawn between the N-glycosylation systems of Bacteria and
Eukaryotes. In Bacteria, N-glycan structures are constructed as LLO precursors at the
cytoplasmic face of the inner membrane, flipped into the periplasm and a homologue of
the eukaryotic STT3 subunit then catalyses transfer of N-glycan structures en bloc onto
target sequences. In contrast to the eukaryotic system, Bacterial N-glycan structures
transferred by an OST can differ in a species specific manner. However, the extensive
downstream processing applied to eukaryotic N-glycans is not a feature observed in
Bacteria (Dell et al., 2010).

27
A striking aspect of Bacterial N-glycosylation is that, unlike in eukaryotes and archaea
where the process is universal (Dell et al., 2010), it only occurs in some species. Thus far,
N-glycosylation has been identified mainly in Gram-negative Bacteria, and only within
some species of the epsilonproteobacteria (Campylobacter, Helicobacter, Wolinella),
deltaproteobacteria (Desulfovibrio) and gammaproteobacteria (Haemophilus influenzae)
(Dell et al., 2010). However, the Gammaproteobacterial pathway is unique as it occurs in
the cytoplasm and does not utilise an LLO and hence likely evolved separately (Nothaft
and Szymanski, 2013). This pathway was identified in Haemophilus influenzae where an
adhesin, HMW1, was found to be modified with mono or di-hexose N-glycans (thought to
contain glucose and galactose residues) at up to 31 sites (Grass et al., 2010; Gross et al.,
2008).
1.5. N-linked glycosylation in Campylobacter
1.5.1. Campylobacter jejuni N-glycosylation.
The N-glycosylation locus of C. jejuni was first described as the wla locus and thought to
be involved in LPS biosynthesis (Fry et al., 1998). However, it was soon recognised to
encode a protein glycosylation system and thus renamed the pgl locus (Szymanski et al.,
1999). The original LPS biosynthesis association was likely due to the presence of an
upstream gene, waaC, which can indeed modify LOS (Klena et al., 1998). In addition, the
pgl locus contains a bifunctional UDP-GlcNAc/ UDP-glucose 4-epimerase, gne (also
known as galE), which provides sugar donor substrates for the LOS, capsular
polysaccharide and protein glycosylation pathways (Bernatchez et al., 2005; Fry et al.,
2000).
Soybean Agglutinin (SBA) lectin affinity purification was used to identify the first
putative N-glycoproteins of C. jejuni, PEB3 and CgpA, and as SBA is an N-
acetylgalactosamine (GalNAc) -specific lectin this suggested the N-glycan structure
contained this sugar (Linton et al., 2002). The 16 kb pgl locus can be functionally
transferred into E. coli allowing recombinant N-glycoprotein production (Wacker et al.,
2002). Using such ‘glycocompetent E. coli’ and tandem mass spectrometry (MS), the N-
glycan structure produced by the C. jejuni pgl pathway was identified as a heptasaccharide
consisting of five N-acetyl-hexosamine (HexNAc) residues, a 2,4-diacetamido-2,4,6-
trideoxyhexose (DATDH) and a branching hexose (Hex) (Wacker et al., 2002). Nuclear

28
magnetic resonance (NMR) analysis of SBA-purified C. jejuni N-glycoproteins confirmed
the N-linked glycan structure as GalNAc-α1,4-GalNAc-α1,4-[Glcβ1,3-]GalNAc- α1,4-
GalNAc-α1,4-GalNAc- β1,3-Bacillosamine-β1 (Young et al., 2002). Bacillosamine refers
to 2,4-diacetamido-2,4,6-trideoxy-β-D-glucopyranose and is correctly known as Di-N-
acetylbacillosamine (DiNAcBac). This heptasaccharide structure is conserved among
members of this species and over 60 C. jejuni N-glycoproteins have been experimentally
identified (Scott et al., 2014, 2011; Young et al., 2002). However, bioformatics analyses
suggest that approximately 150 C. jejuni proteins may be N-glycosylated (Frost, 2015;
Scott et al., 2011).
1.5.1.1. The C. jejuni N-linked glycosylation pathway
The C. jejuni N-glycosylation process has been studied extensively and, using data
acquired from in vitro assays and pgl gene mutagenesis both in C. jejuni and the
glycocompetent E. coli system, the role of Pgl enzymes is now well understood (Fig. 1.1).
Biosynthesis of the C. jejuni N-linked glycan first occurs in the cytoplasm, where the
structure is assembled onto undecaprenyl pyrophosphate (UndP) to create the LLO
precursor. Pgl glycosyltransferases mediate this process and although soluble, they are
relatively hydrophobic and likely interact with the cell membrane (Glover et al., 2005).
All Pgl glycosyltransferases utilise nucleotide sugars as donors and thus the pgl locus
encodes enzymes responsible for their biosynthesis. A three stage reaction catalysed by
PglDEF creates UDP-DiNAcBac (Olivier et al., 2006), providing substrate for PglC to
transfer the first residue of the N-glycan, DiNAcBac, to the undecaprenyl-phosphate
(UndP) precursor (Glover et al., 2006; Linton et al., 2005). PglAJH then act sequentially
to transfer the five GalNAc residues, respectively, with PglH adding the final three
residues (Glover et al., 2005; Troutman and Imperiali, 2009). PglAJH all utilise UDP-
GalNAc as a donor substrate which is provided by the bifunctional epimerase, Gne
(Bernatchez et al., 2005). Finally, transfer of the β 1,3-linked branching glucose residue is
catalysed by the glucosyltransferase PglI (Glover et al., 2005; Kelly et al., 2006; Linton et
al., 2005). In vitro assays using purified C. jejuni PglH found it was a polymerase with a
single active site and that its affinity for the N-glycan increases with increasing GalNAc
residues (Troutman and Imperiali, 2009). It is thought that upon PglH transferring three
GalNAc residues the reaction is inhibited until PglI competes for the product and transfers
the final branching residue, blocking further PglH binding (Troutman and Imperiali,
2009). The complete N-glycan precursor is then flipped across the inner membrane into
the periplasm by an ABC transporter, PglK (Kelly et al., 2006). The inner membrane

29
protein OST, PglB, uses this LLO as substrate and transfers the heptasaccharide structure
onto target asparagine residues in protein acceptor sequences (Wacker et al., 2002). The
acceptor sequon in C. jejuni is D/E-X-N-X-S/T (where X ≠ P), which is an extended
version of the eukaryotic equivalent (Kowarik et al., 2006b).
C. jejuni also produces free oligosaccharide (fOS) structurally identical to its N-glycan in
a PglB-dependent manner (Liu et al., 2006; Nothaft et al., 2009). This hydrolytic activity
of PglB requires the WWDYG motif known to be essential for N-linked protein
glycosylation (Nothaft et al., 2009). A reduction in free oligosaccharide release due to
altered PglB activity was observed in response to high salt concentrations or sucrose
(Nothaft et al., 2009). In contrast, the extent of N-linked glycosylation appeared
unchanged under these conditions. This indicated that fOS may be similar to
osmoregulated periplasmic glucans (OPGs) known in other proteobacterial subsets
(Nothaft et al., 2009). OPGs are found in alpha, beta and gamma proteobacteria and can be

30
cyclic or linear glucose polymers (Bohin, 2000). An absence of OPGs can cause
pleiotropic effects and are associated with the virulence of many pathogens (Bontemps-
Gallo and Lacroix, 2015). OPGs have been suggested to be involved in osmoadaptation,
cell signalling, cell envelope integrity and cell division (Bontemps-Gallo et al., 2017).
However, the function of fOS in C. jejuni remains unclear. Nevertheless, it appears to be a
prominent feature in C. jejuni as it accounts for around 2.5 % of its dry cell weight
(Dwivedi et al., 2013).
1.5.1.2 The Campylobacter oligosaccharyltransferase, PglB
Campylobacter PglB enzymes are encoded on the genomes of all but one Campylobacter
species, Campylobacter canadensis (Nothaft et al., 2012). They are members of the
glycosyltransferase superfamily GT-C, which include glycosyltransferases that harbour
between 8 and 13 transmembranes helixes and contain a modified DxD catalytic motif
(Liu and Mushegian, 2003). The X-ray structure of C.lari PglB with an acceptor peptide
was solved and demonstrated that its periplasmic and transmembrane domains contribute
to acceptor sequon binding and catalysis (Lizak et al., 2011). This work also identified
amino acid residues key for catalysis. Although the full X-ray structure of C. jejuni PglB
is unavailable, mutagenesis studies have similarly revealed the importance of the
corresponding amino acids for binding and catalysis (Nothaft and Szymanski, 2013). The
substrate specificity of C. jejuni PglB is relatively relaxed and it is able to transfer
truncated C. jejuni N-glycan structures to protein (Linton et al., 2005). Furthermore, PglB-
mediated transfer of diverse E. coli- and Pseudomonas aeruginosa-derived O-antigen
structures onto protein was observed in E. coli (Feldman et al., 2005).
The N-glycosylation sequon that PglB modifies is D/E-X-N-X-S/T (where X ≠ P)
(Kowarik et al., 2006b) and in vitro assays suggest that DQNAT results in optimal N-
glycosylation (Chen et al., 2007). The requirement for the extended sequon in comparison
to the eukaryotic counterpart, N-X-S/T, was structurally explained when Lizak et al.
(2011) identified that residue R331 of C. lari PglB forms a stabilising salt bridge with the
-2 Asp. This residue is conserved in Bacteria (Lizak et al., 2011) and the equivalent
residue (R328) in C. jejuni PglB has since been implicated in restricting the sequon
specificity also (Ollis et al., 2014). However, using glycopeptide enrichment and tandem
MS, rare instances of atypical sequons have been identified in N-glycoproteins from C.
jejuni 11168O (the ‘original’ 11168 isolate) (Scott et al., 2014). Three examples of non-
canonical sequons were found and had leucine or glutamine at the -2 position or alanine at
the +2 position.

31
It was first thought that C. jejuni PglB modified folded proteins and could only modify N-
glycosylation sequons present at exposed flexible loops (Kowarik et al. 2006). However,
more recent crystal structures of C. jejuni N-glycoproteins demonstrates that N-
glycosylation sequons can lie within alpha-helices or structured turns (Kawai et al., 2012;
Rangarajan et al., 2007; Silverman and Imperiali, 2016). Furthermore, in vivo analyses by
Silverman & Imperiali (2016) suggest that N-linked glycosylation is coupled with Sec
pathway-mediated protein translocation and hence N-glycoproteins can be modified prior
to complete folding. This is reminiscent of the eukaryotic system, where the OST complex
can couple with the SEC61 translocon resulting in co-translational N-glycosylation of
nascent polypeptides as they enter the ER (Shrimal et al., 2015). C. lari PglB itself is an
N-glycoprotein, harbouring two glycosylation sites which are modified in glycocompetent
E. coli (Lizak et al., 2011). The PglB of C. jejuni also contains an experimentally
identified N-glycosylation sequon (Scott et al., 2011).
1.5.2. The role of N-glycosylation in C. jejuni
In eukaryotes, N-glycosylation has numerous functions and is intricately involved in
protein folding, quality control, targeting and secretion (Helenius and Aebi, 2001;
Moremen et al., 2012). In C. jejuni, the specific function(s) of N-linked glycosylation are
unknown, however, loss of this modification has pleiotropic effects and impairs virulence
(Nothaft and Szymanski, 2013). For example, pglH and pglB mutants have reduced ability
to adhere to and invade human epithelial cells in vitro and colonise the gastrointestinal
tracts of chickens in vivo (Jones et al., 2004; Karlyshev et al., 2004; Szymanski et al.,
2002). Furthermore, mutation of pglE or pglF, which are involved in the biosynthesis of
DiNABac residue, was also shown to reduce colonisation in chickens (Hendrixson and
DiRita, 2004). However, the mechanism through which N-linked glycosylation impairs C.
jejuni virulence is unclear.
There are limited studies regarding the function conferred by N-linked glycosylation of
individual C. jejuni N-glycoproteins and most studies have failed to identify a requirement
for this modification. Thus far, influence of N-glycosylation on protein function has only
been demonstrated for VirB10 (Larsen et al., 2004), a protein component of the plasmid-
encoded type IV Secretion System (T4SS) of C. jejuni 81-176. Larsen et al. (2004)
demonstrated that N-glycosylation of VirB10 significantly contributes to natural
competence as loss of VirB10 or its N-glycosylation conferred similar reductions in
natural transformation efficiency. In contrast, abolishing N-glycosylation of a zinc
transporter system component, ZnuA, did not affect its function (Davis et al., 2009). In

32
addition, N-glycosylation did not influence the antigenicity or abundance of a surface-
exposed lipoprotein, JlpA (Scott et al., 2009). Futhermore, N-glycan modification was not
vital for the activity of two mechanosensitive channel N-glycoproteins, Cj1025 and
Cj0263 (Kakuda et al., 2012). Finally, C. jejuni expressing a non-glycosylated version of
Cj1496c, a periplasmic N-glycoprotein which contributes to adherence and invasion in
vitro and chick colonisation, did not give a similar phenotype to the gene knockout strain
(Kakuda and DiRita, 2006).
As discussed, current experimental evidence does not suggest that N-glycosylation is
required for the function of specific proteins. Instead, N-linked glycosylation may have a
broader function. For example, Alemka et al. (2013) demonstrated that N-glycosylation
may protect proteins from degradation by chicken gut proteases. In this study, chicken
cecal contents reduced the fitness or growth of a C. jejuni pglB mutant compared to the
wildtype and this phenotype could be partially complemented by addition of protease
inhibitors (Alemka et al., 2013). Another study has found that an Fc fusion protein
containing the extracellular region of the human Macrophage Galactose-type lectin (MGL)
receptor can bind the terminal GalNAc residues of C. jejuni N-glycoproteins (van Sorge et
al., 2009). Furthermore, the authors found that, in comparison to wildtype cells, pglA,
pglH or pglJ mutants induced higher production of interleukin 6 by human dendritic cells
in vitro, suggesting that Campylobacter N-glycans may dampen the host immune response
(van Sorge et al., 2009).
1.5.3. N-glycosylation in other Campylobacter species.
N-linked glycosylation is found in all Campylobacter species excluding C. canadensis and
the general features of the C. jejuni pgl locus are usually conserved in similarly organised
loci (Jervis et al., 2012; Nothaft et al., 2012). However, in many instances, distinct
predicted glycosyltransferase and sugar biosynthesis genes are also present. In line with
this, the N-glycans produced by Campylobacter species are structurally diverse (Jervis et
al., 2012; Nothaft et al., 2012)(Fig. 1.2). However, there is some conservation of N-glycan
structure among certain species. In addition, Nothaft et al. (2012) found that eighteen
Campylobacter species produce fOS that is structurally identical to their corresponding N-
glycans. Interestingly, DiNAcBac has been identified as the reducing end sugar in every
Campylobacter N-glycan analysed thus far. Furthermore, DiNAcBac is unique to a subset
of bacterial pathogens including Neisseria gonorrhoeae and Acinetobacter baumannii,
prompting suggestion of a putative role in virulence (Morrison and Imperiali, 2014). In

33
line with this, Schutter et al. (2017) have recently described inhibitors active against the
PglD enzyme.
A C. jejuni N-glycan reactive antiserum, hR6 (Amber and Aebi, unpublished), reacts with
all the thermotolerant Campylobacter species, C. jejuni, C. coli, C. upsaliensis and C. lari
(Jervis et al., 2012; Nothaft et al., 2012). Furthermore, members of this group contain pgl
loci most closely resembling that of C. jejuni. Unsurprisingly, C. coli, C. upsaliensis and
C. helveticus are now known to produce an identical heptasaccharide N-glycan as C.
jejuni (Jervis et al., 2012; Nothaft et al., 2012). However, C. lari lacks a pglI homologue
and thus it produces a hexsaccharide N-glycan without the branching glucose found in the
N-glycan of the other thermotolerant species.
The hR6 antiserum is not reactive with Campylobacter species more distantly related to C.
jejuni, such as C. fetus and C. concisus, and the pgl loci of these species are more variable
(Jervis et al., 2012). For example, some Campylobacter species including C. concisus
have two pglB homologues, referred to as pglB1 and pglB2 (Jervis et al., 2012). In C.
concisus, insertional mutagenesis of these homologues has indicated that pglB1 is
responsible for the majority of N-glycosylation activity but the role of pglB2 remains
unclear (Frost, 2015). The variation in Campylobacter pgl pathways means that the N-
glycan structures identified thus far exhibit much diversity, with some species known to
produce more than one N-glycan structure. Notably, four distinct N-glycan structures have
been found in the human gut commensal bacterium, C. hominis (Nothaft et al., 2012).
There have been conflicting reports regarding the glycan structure produced by the pgl
pathway of C. concisus. Nothaft et al. (2012) reported a fOS structure including a 234 Da
residue from C. concisus 13826. In contrast, Jervis et al. (2012) identified in vitro–
generated glycopeptides harbouring a 217 Da residue-containing N-glycan using
membrane preparations from C. concisus NCTC 11485. Nothaft et al. (2012) proposed
that these differences may be due to intraspecies diversity, which is broadly known as a
feature of C. concisus (Deshpande et al., 2013). C. ureolyticus, an emerging human
pathogen, has been demonstrated to produce an identical N-linked glycan to C. concisus
13826 (Nothaft et al., 2012). However, the pgl locus of C. concisus contains more
predicted glycosyltransferase genes than in C. ureolyticus which suggests that more than
one N-glycan structure might be produced by C. concisus.

34

35
1.5.3.1. N-linked glycosylation in C. fetus
C. fetus contains homologues of all C. jejuni pgl genes excluding pglI. In addition, there
are two additional open reading frames that putatively encode a glycosyltransferase and a
sugar biosynthesis enzyme (Jervis et al., 2012). The pgl locus of C. hyointestinalis is
similar to that of C. fetus and identical N-glycan structures have been identified in both
species (Jervis et al., 2012; Nothaft et al., 2012). Using an in vitro N-glycosylation assay
and MALDI LIFT- TOF/TOF MS, Jervis et al (2012) observed the production of a
hexasaccharide N-glycan which appeared similar to that of C. jejuni but lacked a terminal
HexNAc residue. However, Nothaft et al. (2012) combined LC MS/MS and NMR to
analyse C. fetus fOS and identified that, in contrast to C. jejuni, C. fetus produce two
hexasaccharide fOS structures which contain GlcNAc sugars and differ at the non-
reducing end. These structures, referred to here as type 1 and type 2, were α-GlcNAc-6-[
β-Glc-3]-α-GlcNAc-4-α-GlcNAc-4-α-GalNAc-3-α,β-diNAcBac and α-GlcNAc-6-[β-
GlcNAc-3] -α-GlcNAc-4-α-GlcNAc-4-α-GalNAc-3-α,β-diNAcBac, respectively. MS
analysis of C. fetus N-glycopeptides confirmed that two types of N-glycan are transferred
to protein: HexNAc-[HexNAc]-HexNAc3-diNAcBac and HexNAc-[Hex]-HexNAc3-
diNAcBac. These structures most likely contain identical sugar residues as the type 1 and
type 2 fOS structures, respectively. Furthermore the glycoforms were found at a ratio of
~4:1, with the type 2 GlcNAc branch-containing structure predominant both in fOS and N-
glycopeptides (Nothaft et al., 2012). The authors suggested that having two N-glycan
structures may provide protection from the host immune system (Nothaft et al., 2012),
which is thought to be the case for O-linked glycosylation in Neisseria (Borud et al.,
2014).
Excluding N-glycan structural analysis, there are limited data regarding the C. fetus pgl
pathway. However, analysis of C. fetus glycopeptides identified 32 unique N-
glycosylations sites consisting of the C. jejuni-type sequon, D/E-X-N-X-S/T (where X ≠
P) (Nothaft et al., 2012). These N-glycopeptides correspond to 25 N-glycoproteins.
Furthermore, Nothaft et al. (2012) used C. fetus fOS to generate polyclonal antisera,
termed GRPII-1 and GRPII-2, reactive against the type 1 and type 2 structures,
respectively. Immunoblotting demonstrated these antisera to react with numerous
presumed N-glycoproteins in C. fetus cell lysates, suggesting a similar extent of protein N-
glycosylation in C. fetus as in C. jejuni.

36
1.5.4. N-linked glycosylation in closely related genera, Helicobacter and Wolinella
N-glycosylation systems similar to those of Campylobacter are now known in other
epsilonproteobacteria, such as Helicobacter and Wolinella succinogenes. PglB othologues
have been identified in three Helicobacter species, H. pullorum, H. canadensis and H.
winghamensis, of which H. pullorum is most characterised (Jervis et al., 2010). H.
pullorum was first identified from poultry and humans with gastroenteritis (Stanley et al.,
1994). This species is considered an emerging enterohepatic pathogen (Javed et al., 2017)
and has also been associated with Crohn’s disease, chronic gallbladder inflammation and
bactaeremia (Apostolov et al., 2005; Bohr et al., 2004; Tee et al., 2001). However, its
pathogenicity is debated as Ceelen et al. (2005) found that H. pullorum prevalence in
human stool did not vary between healthy individuals and those with gastrointestinal
disease. Interestingly, H. pullorum contains two pglB orthologues, pglB1 and pglB2,
which are not found within an ordered locus (Jervis et al., 2010). H. pullorum PglB1 could
N-glycosylate two of four sequons present in the C. jejuni N-glycoprotein, Cj0114, in
glycocompetent E. coli (Jervis et al., 2010). Furthermore, using an in vitro assay, the N-
glycan structure transferred by PglB1 was identified as a pentasaccharide containing
unknown 216 and 217 Da sugar residues and lacking the common reducing end sugar,
DiNAcBac (Jervis et al., 2010). However, the role of PglB2 remains unclear and, unlike
pglB1, attempts to inactivate pglB2 in H. pullorum failed, suggesting that it may be
essential (Jervis et al., 2010).
W. succinogenes is a non-pathogenic species originally isolated from a bovine rumen
(Tanner et al., 1981; Wolin et al., 1961). This species contains a pgl cluster similarly
ordered as in C. jejuni but with additional predicted glycosyltransferases present (Baar et
al., 2003). As with C. jejuni, a single pglB gene is present and the reducing end sugar of its
N-glycan is DiNAcBac (Dell et al., 2010). However, the N-glycan is a hexasaccharide
which, similarly to H. pullorum, contains a 216 Da sugar and an uncharacterised 232 Da
sugar (Dell et al., 2010).
1.5.5. N-linked glycosylation in hydrothermal vent Bacteria
Most recently, pglB homologues were identified in proteobacteria which dwell in
hydrothermal vents and limited research has begun to gain insight into the activity of these
enzymes (Mills et al., 2016). Deferribacter desulfuricans, Nitratiruptor tergarcus and
Sulfurovum lithotrophicum contain functional PglB homologues which can, to some
extent, complement C. jejuni PglB activity in glycocompetent E. coli (Mills et al., 2016).
However, the PglB enzymes of these thermophilic Bacteria displayed stricter specificity

37
for target proteins compared to C. jejuni PglB. The OSTases of N. tergarcus and S.
lithotrophicum had a similar requirement for a negatively charged residue at the -2
position of an acceptor sequon. The D. desulfuricans PglB displayed low activity in E. coli
and its requirements could not be studied further, however, it lacks the R331 residue
implicated in C. jejuni PglB sequon specificity and thus likely does not require the
extended sequon (Mills et al., 2016). Such an observation was made for the PglB enzyme
encoded by Desulfovibrio desulfuricans which, like Deferribacter, is a sulfate-reducing
member of the delta-proteobacteria (Ielmini and Feldman, 2011). The N-glycan structures
synthesised by the Pgl systems of these deltaproteobacterial species are unknown.
Nevertheless, the discovery of C. jejuni PglB homologues in these species further
highlights the question of why N-glycosylation exists in such an ecologically diverse
subset of Bacteria. Mills et al. (2016) suggested that in these deep sea vent Bacteria N-
glycosylation may provide thermostability and protection against high osmolarity.
1.6. Campylobacter detection/identification
1.6.1. Culture and phenotypic testing
Isolation and identification of Campylobacter from samples is a particularly time-
consuming process which involves enrichment, solid microbiological culture isolation of
the organism and then further testing of Campylobacter-like colonies (Oyarzabal and
Fernández, 2016). Samples with relatively low numbers of Campylobacter cells, such as
food or environmental samples, are first enriched by incubating in specialised broth such
as Bolton or Preston for at least 24 hours (Kim et al., 2009). Enriched samples are then
plated onto solid culture media. For the identification of Campylobacter in a clinical
sample, Public Health England guidelines suggest microaerobic incubation on
Campylobacter selective media such as charcoal cefoperazone deoxycholate agar (CCDA)
for 48 hours at 42 °C (PHE, 2014). Cells from a Campylobacter-like colony are then
examined microscopically to confirm a curved or ‘S’ shaped rod. This is followed by
Gram staining and an oxidase test to confirm the identity as Campylobacter. Therefore,
confirming a Campylobacter species is present using this method takes at least a few days.
In order to speciate the identified organism, PHE recommend further biochemical testing
(either manually or using commercial kits), MALDI-TOF MS and/or PCR.

38
The well-established protocol described above is optimal for the isolation of C. jejuni and
C. coli from samples. However, other Campylobacter species can have different growth
requirements and isolation is sub-optimal using such procedures. For example, a German
hospital identified a “pseudo-outbreak” of C. concisus upon introducing hydrogen into the
culture environment (Casanova et al., 2015). Furthermore, C. fetus and C. upsaliensis are
susceptible to the concentrations of cefoperazone present in CCDA selective media
(Kulkarni et al., 2002; Schulze et al., 2006). Therefore, modified methods are required to
isolate all possible Campylobacter species from a sample.
1.6.2. Molecular methods for detecting Campylobacter in poultry
As traditional culture methods for the detection and identification of Campylobacter spp.
have limitations, molecular methods of identification are often adopted. Such techniques
have been applied for detecting Campylobacter spp. from a variety of samples, including
human stool, poultry faeces and meat. Molecular methods either target DNA or proteins.
DNA-based technologies adapted for Campylobacter detection include polymerase chain
reaction (PCR) and loop-mediated isothermal amplification (LAMP) (Dong et al., 2014;
Gharst et al., 2013). Techniques targeting proteins utilise antibodies and include latex
agglutination tests (LATs) and enzyme immunoassays (EIAs) (Gharst et al., 2013). A
recent project undertaken by the Department for Environment, Food and Rural Affairs
(DEFRA) (code: OZ0621) concluded that there is a significant need for a rapid on-farm
test to assist Campylobacter control strategies. The available methods for detection of
Campylobacter spp. will be discussed in this context.
Several distinct PCR-based methods have been implemented for Campylobacter detection
and identification, including single reaction PCR, multiplex PCR and quantitative PCR.
Commonly used target sequences of Campylobacter include ribosomal DNA and the
flagellin genes, flaA and flaB (Kirk and Rowe, 1994; Linton et al., 1997; Oyofo et al.,
1992). Muliplex PCR (mPCR) uses more than one primer pair per reaction which can, for
example, allow for simultaneous identification of different Campylobacter species. Kamei
et al. (2016) described an mPCR which targets the CDT and can identify and differentiate
between C. jejuni, C. coli, C. upsaliensis, C. lari, C. fetus and C. hyointestinalis in pure
culture. Quantitative PCR (qPCR) (also known as Real Time PCR) involves coupling
fluorescent detection techniques with conventional PCR to allow amplification to be
monitored in ‘real time’ (Heid et al., 1996). This method is faster than traditional PCR as
it negates the need for further processing following the reaction, such as gel
electrophoresis (Jensen et al., 2005). De Boer et al. (2015) combined three qPCR assays to

39
detect Campylobacter spp. in 926 broiler faeces samples and found that 83% of samples
were positive in comparison to the 65% detected by traditional culture methods. PCR
based assays can offer such higher sensitivity due to detecting viable but non-culturable
(VBNC) or dead Campylobacter cells (Gharst et al., 2013). However, the sensitivity can
decrease significantly when using complex sample matrices, thus it is often advisable to
enrich samples and extract DNA before analysis (Gharst et al., 2013). For example, a 24
hour enrichment step followed by DNA extraction is recommended when using the
commercially available qPCR assay, iQ-Check® Campylobacter (BIO-RAD). Another
limitation of PCR methods is that they are often less effective for the detection of the so-
called ‘emerging’ Campylobacter species as they are usually optimised for the
identification of C. jejuni and C. coli (E.g. The iQ-Check® assay cannot detect C.
upsaliensis). Thus, these techniques are not promising tools for on-site detection of
Campylobacter species in poultry.
Loop-mediated isothermal amplification (LAMP) is a highly sensitive method in which a
gene can be amplified using a single 60-65 °C step (Notomi et al., 2000). Furthermore, a
positive result can be seen by the naked eye due to the production of a turbid solution
(Hara-Kudo et al., 2005). Dong et al. (2014) applied a LAMP assay for the detection of C.
jejuni from enriched cattle faeces and found that the assay was less sensitive to inhibitors
found in the enrichment broth in comparison to PCR. However, the reported sensitivity
was 84 %. Therefore, although such methods are relatively rapid, they still require
significant sample preparation and in practice do not offer high sensitivity. Thus, these
methods are not highly suited to point-of-care testing in a farm environment.
In general, DNA-based methods tend to have higher sensitivity than antibody-based tools
(Platts-Mills et al., 2014). However, the former usually requires specialised equipment and
hence samples need transporting to a laboratory for analysis. As a result, this incurs further
cost and time. In contrast, antibody-based methods or immunological methods are simpler
and produce rapid results. Therefore, antibody-based methods are appropriate for
analysing samples on-site.
Several types of immunological assays for the detection of Campylobacter spp. have been
described in the literature. Such techniques can utilise polyclonal or monoclonal
antibodies. Polyclonal antibodies describe a set of antibodies which although all reactive
against one antigen, have varying specificities and affinities (Boenisch, 2009). In contrast,
monoclonal antibodies are those produced from a single B-cell clone and all bind identical
epitopes. Polyclonal antiserum is relatively simple to produce by immunizing animals,

40
such as rabbits, with an antigen and later harvesting the sera from its blood. The
production of monoclonal antibodies is more complex and time consuming, as it requires
isolation of an animals B cells, fusion with a myeloma cell line (to confer immortality) and
subsequent screening to identify the desired antibody-producing cell line (Boenisch,
2009).
Latex agglutination tests (LATs) are well established antibody-based tests that have long
been used to confirm pathogen identification from clinical samples. The principle involves
coupling antibodies (commonly polyclonal) to latex so that the formations of antigen-
antibody complexes are made visible to the naked eye. The application of this method for
Campylobacter identification was first reported by Hodinka and Gilligan (1988). More
recently, Miller et al. (2008) reviewed three such LAT assays marketed for Campylobacter
detection: Microgen M46 Campylobacter (Microgen Bioproducts Ltd.), CAMPY (jcl)
(Scimedx Corporation) and Dryspot Campylobacter (Oxoid). However, the Microgen and
CAMPY (jcl) assays gave false positives when analysing Acinetobacter baumanii, an
organism commonly co-isolated with Campylobacter (Oyarzabal et al., 2005). In addition,
none of the assays were able to identify all eight C. upsaliensis strains tested. Furthermore,
LATs are prone to non-specific agglutination. For example, Hazeleger et al. (1992) found
commercial LAT assays to give unacceptable amounts of non-specific agglutination when
testing against enriched stool samples. The authors also reported that the limit of detection
could vary considerably in a strain- and species-dependent manner, varying from 7 x 105 –
5 x 108 CFU/ml. Hence, it has been suggested that these assays are useful for confirming
culture-based identification but not for independent use (Gharst et al., 2013).
More robust antibody-based detection methods are enzyme immunoassays (EIAs), which
involve coupling antibodies to enzymes to afford a colour change as a result of the
antibody-antigen interaction. These include enzyme-linked immunosorbent assays
(ELISAs) in microplate formats and lateral flow immunoassays. The latter are modified
EIAs contained within a plastic casing commonly referred to as a ‘dipstick’ (Oyarzabal
and Battie, 2012). Lateral flow assays (LFAs) often contain antibodies labelled with
colloidal gold nanoparticles, which upon aggregating on the test line produce a visible
colour change (Sajid et al., 2015). Although ELISA formats allow for quantification,
LFAs produce faster results and are simpler to use (Granato et al., 2010).
There are commercial automated ELISA systems, VIDAS® and miniVIDAS®
(bioMérieux), marketed for the identification of Campylobacter from food samples.
However, the sensitivity of these devices relies on 48 hour enrichment of samples (Liu et

41
al., 2009). Furthermore, they are not portable and hence not applicable to on-farm testing.
Non-automated commercial ELISA formats also exist for the detection of Campylobacter,
including RIDASCREEN® Campylobacter (R-BioPharm AG), Premier® CAMPY
(Meridian Bioscience) and ProSpecT™ Campylobacter (Remel). However, these assays
can suffer from poor sensitivity and considerable false positive results are often reported
(Oyarzabal and Battie, 2012; Tissari and Rautelin, 2007). For example, studies have
reported ProSpecT to have a sensitivity and specificity as low as 62% and 84%,
respectively, when analysing stool samples (Hindiyeh et al., 2000; Tribble et al., 2008).
Furthermore, Giltner et al. (2013) found that Premier® CAMPY (Meridian Biosciences)
gave considerable false positives, and warned that although it was a fast alternative to
routine identification procedures, all positive results should be subsequently confirmed
with culture. In addition, these EIAs are in microtiter plate format which would not be
ideal for on-site detection at farms.
Lateral flow assays are the most applicable type of immunoassay for point-of-care testing,
providing rapid results alongside being extremely user-friendly. Commercially available
LFAs for the detection of Campylobacter include RIDA®QUICK, ImmunoCard STAT!,
Xpect, SinglePath and NH Immunochromato. These assays are marketed for analysis of
clinical stool samples or food samples. Although most suggest pre-enrichment of samples
some assays, such as ImmunoCard STAT and RIDAQUICK, are intended to analyse
patient stool samples directly, requiring only dilution of sample with reagents provided.
However, a recent study conducted by the CDC ruled that available immunoassays were
not appropriate for standalone Campylobacter spp. identification from stool due to
unacceptably low sensitivities and positive predictive values (PPVs; the probability that a
positive result will be a true positive) (Fitzgerald et al., 2016). Specifically, this work
analysed two microplate format assays, Premier and ProSpecT, and two LFAs,
ImmunoCard STAT and Xpect. Nevertheless, Oyarzabal and Battie (2012) argue that
optimisation of such assays may allow for considerable improvement. Indeed, many of the
commercial assays utilise anti-Campylobacter polyclonal antisera for which no further
details are available. As the targets of these sera are not publicly known, it can be argued
that more focused research onto appropriate target antigens may be necessary to create
more efficacious assays. Furthermore, the use of culture methods as a reference from
which to calculate sensitivity and specificity of other assays has been raised into question.
For example, VBNC cells of Campylobacter may be detected by non-culture-based tests
and due to comparison with a reference culture method, this result may be construed as a
false positive. In evidence of this, when analysing available EIAs, Granato et al., (2010)

42
found that upon using RT-PCR to further analyse contradictory results between EIAs and
culture, the PPV of the EIAs improved (i.e. less false positives were apparent).
Furthermore, Gómez-Camarasa et al. (2014) found that the PPV of RIDAQUICK changed
from 77% to 94% when including PCR in the reference method. This assay performed
well when analysing stool sample with minimal preparation, further suggesting that LFAs
are promising for on-site analysis at farms. However, analysis of the species specificity of
this assay has not been published.
Although there are numerous assays described for the detection and identification of
Campylobacter species, manufacturers usually recommend an enrichment step be included
to concentrate samples prior to analysis (Gharst et al., 2013). The only available assay
targeted for Campylobacter detection directly from poultry samples is SinglePath Direct
Campylobacter poultry kit (Merck). Merck claims this test can be performed in less than 2
hours and provides a sensitivity and specificity of 96% and 100%, respectively, when
analysing chicken caecal droppings (n = 120). The limit of detection is reported at 107
CFU/ml, which allows for differentiation between ‘Campylobacter high and low risk
flocks’. Targeting flocks with a high burden may be a feasible option for reducing chicken
meat-associated human Campylobacteriosis, as a risk assessment study found that
incidence could be 30 times lower if the level of Campylobacter on chicken carcasses was
reduced by 2 log (Rosenquist et al., 2003). However, this product is no longer available to
purchase. Wadl et al. (2009) reported the development of an LFA capable of detecting 7.2
Log CFU/g C. jejuni cells in chicken faeces. As chickens are often reported to shed up to
107-10
8 CFU/g of
C. jejuni cells in their faeces, this test seemed feasible. However, in
reality the assay did not perform well, only identifying 3 (17%) of the 18 infected flocks
tested.
The Food Standards Agency (FSA) recently published results of a project which aimed to
identify an appropriate method for detecting Campylobacter-infected flocks on-site
(Madden et al., 2016). The project analysed commercially available applications including
two lateral flow devices (SinglePath and ImmunoCard STAT! CAMPY), a LAMP
(Loopamp) kit and a qPCR (Mericon Campylobacter) kit. It was established that the LFDs
did not have sufficient sensitivity and specificity whereas the LAMP kit produced false
positives with Arcobacter spp., bacteria commonly found in chickens. This study
concluded that the qPCR kit was most suitable overall for rapidly detecting
Campylobacter infected-flocks. However, it was not able to be performed on-site and
hence farmers were not informed of the results until after at least 36 hours. Therefore,
there remains a need for rapid detection methods which can be performed on-site for the

43
detection of Campylobacter. Furthermore, ease-of-use would be a significant advantage,
meaning farmers can complete the test unaided. Hence, the development of more sensitive
and specific LFAs would be ideal.
Overall, EIAs for the identification of Campylobacter tend to suffer from poor sensitivity
and often give a high rate of false positives. Furthermore, the majority available are
marketed for the identification of C. jejuni and C. coli from clinical specimens and/or
enriched food samples. Therefore, there is still a requirement for tools which are optimised
to provide rapid results for Campylobacter monitoring on-site and also able to detect the
more ‘emerging’ pathogens within the genus.
1.6.3. Methods of detecting Campylobacter fetus in cattle.
Traditionally, C. fetus infection was identified solely using culture methods which,
contrary to those used for other Campylobacter species, includes incubation at 37 °C and
5% hydrogen if possible (Vandamme, 2000). The defining biochemical tests to
differentiate between C. fetus fetus and C. fetus venerealis are the glycine tolerance and
hydrogen sulphide production tests, for which only the former is positive. However, the
validity of this has been called into question as a variant of C. fetus venerealis termed
biovar intermedius (Veron and Chatelain, 1973) can be positive in these assays (van der
Graaf-van Bloois et al., 2014). Furthermore, such features often do not parallel virulence
capabilities, for example, strains typed as C. fetus fetus based on phenotypic and
genotyping methods can display virulence features on the accessory genome (van der
Graaf-van Bloois et al., 2014). Therefore, the authors argued that C. fetus diagnostics,
particularly the methods used for subspecies differentiation, need considerable reviewing.
As described in section 1.6.2., there are several commercially available Campylobacter
detection tests. However, they are mostly optimised for the detection of the so-called
thermotolerant species. For example, the manufacturer of the DrySpot kit (Thermo
Scientific) advices that results can be variable with C. fetus. More recently, C. fetus
identification time has been reduced by screening samples using an ELISA and
subsequently confirming positives using culture (OIE, 2017). This assay utilises
monoclonal antibodies targeting C. fetus LPS (Brooks et al., 2002) and has a reported
100% sensitivity (Brooks et al., 2004; Devenish et al., 2005). However, samples require
incubation for at least four days prior to analysis and the procedure itself takes a further
two days (Devenish et al., 2005; OIE, 2017). An immunofluorescence test (Mellick et al.,
1965) using these monoclonal antibodies can be applied directly to genital fluid, however,
it requires several steps and an immunofluorescence microscope (OIE, 2017).

44
Several DNA-based methods have been reported for the identification of C. fetus. Efforts
have mainly focused on developing methods which can differentiate between C. fetus fetus
and C. fetus venerealis, as the latter is more concerning to the farming industry. For
example, the identification of a C. fetus venerealis-infected bull at an artificial
insemination facility can result in temporary suspension of activities and recent semen
samples to be discarded (van der Graaf-van Bloois et al., 2013). However, subspecies
differentiation of C. fetus is notoriously difficult, and often requires more complex
methods such as amplified fragment length polymorphism (Wagenaar et al., 2001) and
multilocus sequence typing (van Bergen et al., 2005). Multiple authors have reported PCR
assays for such purpose, although only those which target nahE of C. fetus venerealis can
do so reliably (OIE, 2017). However, as discussed above, the clinical relevance of such
subspecies differentiation has been challenged (van der Graaf-van Bloois et al., 2014).
Available DNA-based assays for C. fetus identification were reviewed by van der Graaf-
Bloois et al. (2013). Although some assays gave 100% sensitivity and specificity, all were
applied to pure cultures or extracted DNA. For example, these authors reported the PCR
method described by Hum et al. (1997) to give 100% sensitivity, however, Schmidt et al.
(2010) found the sensitivity to be 85.7% when analysing spiked preputial scrapings.
Furthermore, although there have been reports of using qPCR assays to detect C. fetus in
preputial samples (Chaban et al., 2012), these procedures still require sample
transportation to a laboratory and some sample preparation. Thus, such assays are most
appropriate for typing samples following culture identification in a laboratory, not direct
detection. Therefore, no methods available for detecting C. fetus are practical for on-site
detection. Development of such assays could provide farmers with a simple method to
monitor C. fetus infection, and thus rapidly identify an infected herd. In addition, this
could improve C. fetus detection in developing countries which is of particular difficulty
due to a lack of resources for technical molecular diagnostic methods (Mshelia et al.,
2010).
1.6.4. Emerging methods for Campylobacter detection
In the past decade, more novel methods such as biosensors and microarrays have been
developed for the detection of Campylobacter spp. Such methodologies allow for
detection of multiple pathogens simultaneously. For example, Suo et al. (2010) described a
DNA-based microarray capable of detecting C. jejuni, E. coli O157:H7, Salmonella
enterica and Listeria monocytogenes from fresh meat samples. This methodology
essentially allows the results of numerous qPCR assays to be visualised on a single glass

45
slide, thus bypassing the need for agarose gel electrophoresis. There are also similar
platforms which automate qPCR assays and are available commercially, such as
Verigene® Enteric Pathogens test and BioFire FilmArray™. These methods can provide
extensive analysis of a sample with high sensitivity (Huang et al., 2016), however, they
are relatively complex and hence are most suited to diagnostic laboratories and not field
analysis.
Biosensors generally utilise DNA or antibodies as detecting agents. The interaction of the
sensing biomolecule with an analyte is detected by a transducer which converts this signal
into an electrical output (Borisov and Wolfbeis, 2008). The method by which the
transducer detects an interaction defines the type of biosensor as optical, electrochemical
or mass-sensitive (Yang et al., 2013). Surface plasmon resonance devices are a particularly
popular form of optical biosensor which offer fast, highly sensitive and label-free analysis
without the need for numerous reagents (Skottrup et al., 2008; Yang et al., 2013).
Furthermore, commercial platforms are increasingly accessible. For example,
Gnanaprasaka et al. (2011) developed a DNA-based biosensor able to detect C. jejuni
using the Spreeta™ SPR platform. A comprehensive review of biosensors described for
the detection of Campylobacter spp. by Yang et al. (2013) found that C. jejuni was often
the sole target. Furthermore, although these biosensors have high sensitivity for C. jejuni,
it often reduces when analysing other species. For example, an immunosensor described
by Sapsford et al. (2004) could detect as little as 9.7 x 102
cells of C. jejuni but only 7.8 x
105
or more C. coli cells. In addition, there are no reports in the literature of biosensors
optimised for the detection of C. fetus. The development of antibodies capable of detecting
the common and emerging Campylobacter species with equal sensitivity could be a merit
to these methodologies. Furthermore, as the technology expands and devices become
increasingly portable these approaches will likely be readily applicable to on-farm testing.
1.6.5. N-linked glycans as targets for antibody-based detection.
As discussed, there is a need for fast and simple detection methods for pathogenic
Campylobacter species found in poultry and also the veterinary pathogen, C. fetus. In
particular, detection tests which could be performed on-site at farms are desirable.
Antibody-based detection methods such as lateral flow devices or immunosensors are
suitable technologies. For detection of Campylobacter species, a surface-exposed target
antigen which is a dominant and constant feature would be ideal.

46
The C. jejuni cell surface displays an abundance of carbohydrates, including the LOS,
capsular polysaccharide and O-glycosylated flagella (Karlyshev et al., 2005). These
features display interstrain diversity and phase-variable expression and are therefore
limited as targets for detection/identification of Campylobacter species. However, the C.
jejuni N-linked glycosylation pathway is not phase variable and produces only a single N-
glycan structure (Linton et al., 2005). At least 150 proteins may be N-glycosylated in C.
jejuni, including outer membrane proteins which could be surface exposed. The C. jejuni
N-glycan structure is conserved in closely related Campylobacter species such as C. coli
but not in more distantly related species such as C. fetus (Jervis et al., 2012; Nothaft et al.,
2012). Therefore, C. jejuni N-linked glycans might be considered promising targets for
antibody-based detection.
The N-glycan-reactive antiserum, hR6, is often used in Campylobacter N-glycosylation
research as it reacts with the two or three terminal GalNAc residues of the C. jejuni
heptasaccharide (Amber and Aebi, unpublished). As a result, hR6 is reactive with N-
glycoproteins of all Campylobacter species producing the C. jejuni type N-glycan and also
C. lari (Jervis et al., 2012; Nothaft et al., 2012). This antiserum does not react with other
Campylobacter species that produce N-glycans which vary at the non-reducing end in
comparison to C. jejuni, such as C. fetus and C. concisus. Furthermore, fluorescent
microscopy has demonstrated that hR6 can label cells of C. jejuni, suggesting that many
N-glycoproteins are surface exposed (Alemka et al., 2013). Therefore, C. jejuni N-glycan-
reactive antiserum could be used for specific, antibody-based detection of C. jejuni, C. coli
and other emerging species.
Detection assays for the human and veterinary pathogen, C. fetus, are limited. The cell
surface of C. fetus is unusual amongst Campylobacter species in that it contains a
paracrystalline layer of antigenically-variable surface layer proteins (SLPs) (Garcia et al.,
1995; Thompson, 2002). C. fetus do not produce capsular polysaccharide but do harbour
putative loci for synthesis of variable LOS and O-glycosylated flagellin (Gilbert et al.,
2016). Although C. fetus produce two N-linked glycan structures, they do not appear to be
subject to variable expression. Furthermore, antisera raised against each of the C. fetus
fOS structures are reactive with numerous C. fetus N-glycoproteins but not those of other
species such as C. jejuni, C. coli and C. concisus (Nothaft et al., 2012). Furthermore,
fluorescent microscopy revealed that these antisera can label cells of C. fetus fetus and C.
fetus venerealis (Nothaft et al., 2012). Therefore, similar to in C. jejuni, C. fetus N-linked

47
glycans are favourable targets for antibody-based detection as they appear a conserved
feature of C. fetus cells.
1.7 Aims
There are two overall aims for this research. The first is the investigation into
Campylobacter N-linked glycans as targets for antibody-based detection/identification of
Campylobacter species (Chapters 3 and 4). The second describes functional
characterisation of N-linked glycosylation in C. fetus (Chapters 5 and 6).
As discussed, it would useful to develop assays for the on-site detection of Campylobacter
human pathogens in poultry and the veterinary pathogen, C. fetus, in cattle. Antibody-
based detection methods are simple and rapid and therefore applicable for this purpose.
Furthermore, Campylobacter N-linked glycans could be attractive targets for antibody-
based detection/identification. To this end, this project aimed to characterise two antisera
raised against the N-glycans of C. jejuni and C. fetus (Chapters 3 and 4, respectively), and
evaluate the potential of these antisera to be used to detect Campylobacter species. An
antiserum, CjNgp, against a C. jejuni N-glycoprotein was already in production (Jervis &
Linton, unpublished) and this antiserum was characterised here. However, a C. fetus N-
glycan-reactive antiserum was unavailable and was newly developed as part of the work
described in this thesis.
Secondly, as the C. fetus N-linked glycosylation system is relatively uncharacterised, this
project aimed to analyse this further. Glycocompetent E. coli containing the C. jejuni pgl
locus is a powerful tool used to study C. jejuni N-glycosylation. Such a system expressing
the C. fetus N-glycan would be similarly useful for understanding N-glycosylation system
of this species. Therefore, this project aimed to develop glycocompetent E. coli producing
the C. fetus N-glycan structures (Chapter 5). In addition, using a bioinformatics tool, it
aimed to predict the C. fetus N-glycoproteome and assess conservation of predicted N-
glycoproteins more broadly amongst Campylobacter species (Chapter 6).

48
Chapter 2. Materials and Methods.

49
2.1 Bacterial strains and plasmids
Bacterial strains used are listed in Appendix Table 1. All Campylobacter strains were
grown on blood agar (BA) plates containing Columbia blood agar base (Oxoid Ltd.)
supplemented with 5% defibrinated horse blood (TCS Biosciences) and containing
selective antibiotics as required. Most Campylobacter strains were grown in a VA500
workstation (Don Whitley Ltd., UK) in a microaerobic atmosphere (85% N2, 10% CO2,
and 5% O2). However, C. concisus, C. lanienae, C. sputorum and C. hyointestinalis
require a microaerobic hydrogen-containing environment generated in a 2.5 litre gas
chamber (Oxoid Ltd.) using a CampyGen™ atmosphere generation system sachet and 10
ml of a 1% sodium borohydride solution. Most Campylobacter species were incubated at
42°C, with C. fetus and C. concisus incubated at 37 °C. E. coli strains were grown in
lysogeny broth (LB) or LB agar (Oxoid Ltd.) containing antibiotics as required and
incubated at 37 °C. Antibiotics were purchased from Sigma and used at the following
concentrations: 50 µg/ml kanamycin, 100 µg/ml ampicillin and 34 µg/ml chloramphenicol.
For Campylobacter, chloramphenicol was used at 17 µg/ml.
2.2 Isolation of Campylobacter from retail chicken
2.2.1 Culture and isolation of Campylobacter chicken meat isolates
Chicken meat was purchased from a variety of supermarkets during 2014 and 2015.
Packaging and tools used to handle meat were sterilised using 100% ethanol, and samples
were obtained aseptically. Sample-inoculated swabs or ~ 1 inch2
skin pieces were
incubated in Bolton broth (Oxoid Ltd.) with Bolton selective supplement (Fluka
Analytical) for 48 hours at 42°C in a VA500 workstation-generated microaerobic
atmosphere. Samples were plated onto Blood Free Campylobacter selectivity agar base
(Oxoid Ltd.) with CCDA selective supplement (Fluka Analytical) and incubated for a
further 48 hours, before single Campylobacter-like colonies were picked and streaked onto
Columbia blood agar base (Oxoid Ltd.) supplemented with 5% defibrinated horse blood
(TCS Biosciences).
2.2.2 Verification and Molecular typing of isolates
Isolates were confirmed as Campylobacter using PCR with genus-specific 16S rDNA
primers, C412F and C1288 (892 and 893 in Table 2.1), as described by Linton et al.
(1996). Species-specific PCR was performed using primer pairs HIP400F/HIP1134R for
C. jejuni and CC18F and CC519R for C. coli (Linton et al., 1997). To ensure strains were
distinct, isolates were typed by sequencing the hypervariable region of flaB using primers

50
1246 and 1247 (Table 2.1). Amplicons were sequenced on an Applied Biosystems 3730
DNA analyzer (Life Technologies). Isolates (DW1-10) are described in Appendix Table 1.
2.3 Whole cell lysate preparation for SDS-PAGE
Whole cell lysates were prepared using BugBuster™ Protein Extraction Reagent
(Novagen) or incubating cells suspended in sample buffer at 95 °C for 10 minutes. Cells
were harvested from agar plates and suspended in 0.6 ml PBS. Absorbance at 600 nm
(OD600) was measured using an Ultrospec 2100 pro UV/visible spectrophotometer
(Amersham Biosciences). The remaining 0.5 ml cell suspension was centrifuged at 16000
g for 3 minutes and the supernatant removed. Whole cell lysates were produced by
resuspending this pellet in 50-100 µl BugBuster™ Protein Extraction Reagent (Novagen)
with 0.5 µl DNaseI (Qiagen) and incubating for 20 minutes at room temperature. Cell
lysate solutions were centrifuged at 16000 g for 3 minutes and the supernatant transferred
to a clean Eppendorf. NuPAGE™ LDS sample buffer (4X) (Thermofisher Scientific) and
water were added to the supernatants to give a final volume of 10 µl per optical density at
OD600 of 0.1. The samples were then incubated at 95°C for 5 minutes. Alternatively, for
samples created without the use of BugBuster™ Protein Extraction Reagent, a cell
suspension was made to a final volume of 10 µl per optical density at OD600 of 0.1 using
NuPAGE™ LDS sample buffer (4X), incubated at 95°C for 10 minutes and centrifuged at
16000 g at 16162 xg for 20 minutes. The supernatant was transferred to a fresh Eppendorf
and stored at 4 °C.
2.4 SDS-PAGE and western blotting
Samples were loaded onto NuPage Bis-tris precast 12% gels (Life Technologies) and
electrophoresed at 200 V for 50 minutes in NuPage MOPS buffer using an XCell
SureLock™ Mini-Cell Electrophoresis System (Life Technologies) and BIORAD
powerpac300. Alternatively, samples were run on hand-cast gels containing 12% ProtoGel
Acrylamide (National Diagnostics) in SDS-PAGE running buffer (containing 0.3% (w/v)
Tris, 1.4% (w/v) glycine and 0.1% (w/v) SDS). Hand-cast gels were electrophoresed at
160 V for 80 minutes using the apparatus described above. Precision plus All Blue
Standards (Bio-Rad) was used as a molecular weight marker. Gels were stained with
InstantBlue (Expedeon) coomassie stain for at least 20 minutes at room temperature and
washed in water prior to imaging.
For western blotting, SDS-PAGE gels were transferred to a nitrocellulose membrane in an
XCell II™ Blot Module (Life Technologies) for 1 hour at 30V in Transfer buffer

51
containing 0.24 % (w/v) Tris and 1.1 % (w/v) glycine. Membranes were blocked with 5 %
(w/v) powdered milk (Marvel) or 5 % BSA in PBS with 0.1 % (w/v) Tween (PBS-T) at
4°C overnight or 30 minutes at room temperature on a rocker. Membranes were probed
with primary antibodies in PBS-T with either 3 % (w/v) milk or BSA at room temperature
for one hour. Antibody dilutions were as follows: 1/2000 (CjNgp), 1/1000 (CfNgp) or
1/1000 (Anti-HexaHis). Membranes were washed three times in PBS-T. IRDye® 800CW
Goat anti-Rabbit IgG secondary antibodies (LI-COR Biosciences) or IRDye® 680RD
Donkey anti-Mouse IgG secondary antibodies (LI-COR Biosciences) were added at a
dilution of 1/10000 in 3 % milk or 3 % BSA-PBS-T and incubated at room temperature
for one hour. Membranes were washed three times with PBS-T. Membranes were then
stored in PBS until visualised using an ODYSSEY Infrared Imaging System (LI-COR
Biosciences) and associated Image Studio™ software.
2.5 Dot blot assay
The dot blot assay protocol was modified from Qian et al. (2008). Cells were grown for 24
hours on blood agar plates, harvested, washed in 600 µl PBS and resuspended at various
OD600 values. Cells numbers were estimated based on an OD600 of 1 being equal to around
1 x 109
C. jejuni cells per ml (Qian et al., 2008). Cell suspensions (1 µl) were pipetted onto
a nitrocellulose membrane, left to air-dry for 15 minutes and the membrane blocked in
PBS with 0.1% Tween and 5% BSA for 30 minutes. Following a brief wash in PBS-T,
primary and secondary antibodies were incubated in 3% BSA-PBS-T for 1 hour and the
membrane washed with PBS-T three times following incubations. Dot blots were stored in
PBS and visualised using an ODYSSEY (LI-COR Biosciences) Infrared scanner and
associated software.
2.6 Flow cytometry
Cells were harvested from overnight blood agar plates, suspended into PBS and
centrifuged at 12470 g for 3 minutes. Cells were washed again in PBS, resuspended at an
OD600 of 0.5 in PBS with 0.05 % (w/v) Tween and 1 % (w/v) BSA and incubated at room
temperature for 30 minutes on an end-over-end rocker. Pimary antibody was added at a
dilution of 1/250 and incubated for an hour at room temperature. Cells were washed once
in PBS-T-BSA, R-phycoerythrin Goat Anti-Rabbit IgG secondary antibodies (Life
Technologies) added at 1/250 in PBS-T-BSA and incubated for 45 minutes at room
temperature in the dark. Cells were washed with PBS-T-BSA and resuspended in PBS.
Data was aquired on a Canto Flow Cytometer using BDFACSDIVA software. Ungated

52
data analysis was performed using Flowjo version 9 (Tree Star) and event counts were set
to 10,000.
2.7. Polymerase Chain Reaction
Genomic DNA was extracted using an Archive Pure DNA cell/tissue kit according to
manufacturer’s instructions (5PRIME) and PCR performed in a Techne thermal cycler
(Bibby Scientific Ltd.). Primers (Table 2.1) were purchased from Eurofins MWG Operon
and used at a concentration of 1 pmol µl-1
. For colony PCR, MyTaq Red Mix (Bioline)
was used as the PCR master mix. Target DNA was included at a concentration of 1 ng µl-
1. Reactions were 30 cycles of: 94°C for 30 seconds, annealing temperature at 5 degrees
below the melting temperature for 30 seconds and elongation at 72°C for 30 seconds kb-1
.
Upon completion, samples were held at 4°C. For PCR for use in molecular cloning,
CloneAmp HiFi PCR premix (Takara Bio USA) was used and reaction parameters were as
suggested by the manufacturer. PCR products were purified using QIAquick PCR
purification kit (Qiagen) and amplicons sequenced on an Applied Biosystems 3730 DNA
analyzer (Life Technologies) using between 300 and 500 ng of DNA and 4 pmol of primer
in 10 µl of milliQ water.
Table 2.1. Primer sequences.
Name (number) Sequence Reference
(if applicable)
T7 universal F (80)
()promoter
GGATGACACTTTTCGGAGC N/A
T7 universal R (82) CATTGTAGCACGTGTGTC N/A
Cm200Rout (546) CTTGGAAAGGAACACCGCCG
N/A
Cj0114NpET22-R1 (790) TCTTTGCGTTCACAAGCAGC Jervis & Linton,
unpublished C412F (892) ACCTGATTGCGCAAGGATG Linton et al., 1996
C1288 (893) GCGACTGGATCCATGTAATTAGTTT
CTTAATCG
Linton et al., 1996
NGRP-F (1184) GTAACTAGATCTGCGAAATTAAGA
CAAAGTG
Jervis & Linton,
unpublished
FlaB_Cjc-L (1426) GCTAAGAGATCTTCAATTTGCTGCT
TCTTTTAT
Korczak et al., 2009
CjflaBR (1247) TACATTGGATCCGCTATTTTCATGC
AAAATTTCC
This work
rrlfetusF (1268) TAAGTGCAAGTGGTAGGAG This work
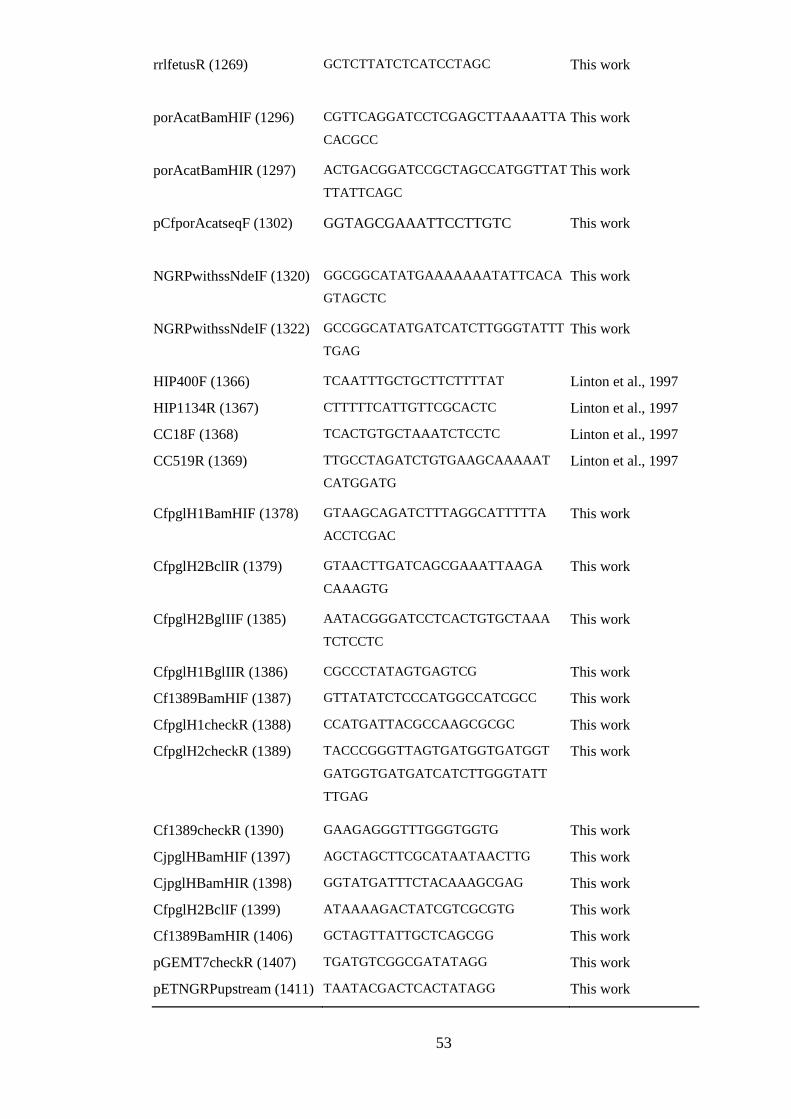
53
rrlfetusR (1269) GCTCTTATCTCATCCTAGC This work
porAcatBamHIF (1296) CGTTCAGGATCCTCGAGCTTAAAATTA
CACGCC
This work
porAcatBamHIR (1297) ACTGACGGATCCGCTAGCCATGGTTAT
TTATTCAGC
This work
pCfporAcatseqF (1302)
GGTAGCGAAATTCCTTGTC
This work
NGRPwithssNdeIF (1320) GGCGGCATATGAAAAAAATATTCACA
GTAGCTC
This work
NGRPwithssNdeIF (1322) GCCGGCATATGATCATCTTGGGTATTT
TGAG
This work
HIP400F (1366) TCAATTTGCTGCTTCTTTTAT Linton et al., 1997
HIP1134R (1367) CTTTTTCATTGTTCGCACTC Linton et al., 1997
CC18F (1368) TCACTGTGCTAAATCTCCTC Linton et al., 1997
CC519R (1369) TTGCCTAGATCTGTGAAGCAAAAAT
CATGGATG
Linton et al., 1997
CfpglH1BamHIF (1378) GTAAGCAGATCTTTAGGCATTTTTA
ACCTCGAC
This work
CfpglH2BclIR (1379) GTAACTTGATCAGCGAAATTAAGA
CAAAGTG
This work
CfpglH2BglIIF (1385) AATACGGGATCCTCACTGTGCTAAA
TCTCCTC
This work
CfpglH1BglIIR (1386) CGCCCTATAGTGAGTCG This work
Cf1389BamHIF (1387) GTTATATCTCCCATGGCCATCGCC This work
CfpglH1checkR (1388) CCATGATTACGCCAAGCGCGC This work
CfpglH2checkR (1389) TACCCGGGTTAGTGATGGTGATGGT
GATGGTGATGATCATCTTGGGTATT
TTGAG
This work
Cf1389checkR (1390) GAAGAGGGTTTGGGTGGTG This work
CjpglHBamHIF (1397) AGCTAGCTTCGCATAATAACTTG This work
CjpglHBamHIR (1398) GGTATGATTTCTACAAAGCGAG This work
CfpglH2BclIF (1399) ATAAAAGACTATCGTCGCGTG This work
Cf1389BamHIR (1406) GCTAGTTATTGCTCAGCGG This work
pGEMT7checkR (1407) TGATGTCGGCGATATAGG This work
pETNGRPupstream (1411) TAATACGACTCACTATAGG This work

54
CjpglHcheckR (1424) GATAGCTGATCACTTTTTCATTGTT
CGCACTC
This work
pETNpglcheck R (1425) TTAGGCATTTTTAACCTCGGC This work
M13 universal F (1432) GCGGCTGTGATAGTTTAGTCG N/A
Cf1389BclIF (1439) AGAACGGTTAATCCACTTGC This work
pRYGG1/2_seq_F (1445) GCTCGCTGATCACTATTTTCATGCA
AAATTTCC This work
CfH2midcheck_R (1450) CTCAGACGTAAAATTAAAGCCG This work
NGRPSmaI_H8_R (1484) TTGTTTCTCGAGATCATCTTGGGTAT
TTTGAG This work
CfpglH1_F_out (1485) ATGGCCATGGGAGATATAACAAGC
AATTC This work
Cf1389_mid_F (1486) TTCAGGCTTACTTGCGTCT This work
CfpglH1midR (1558) CGTCACGGATCCATGAATATAGGC
AAGGGTATAGC This work
Cf0445BamHI_F (1566) CGGCGCGGATCCATGAAAAAAATA
CTTAGCCTTATGTTC This work
Cf0781BamHI_F (1567)
TACCCGGGTTAGTGATGGTGATGGT
GATGGTGATGTTCACTTACGTCAATCC
CAA
This work
Cf0781SmaI_R (1572)
TACCCGGGTTAGTGATGGTGATGGT
GATGGTGATGCTTAAATAAAGAAAT
TTCTTC
This work
Cf0445SmaI_R (1573) GTAAAACGACGGCCAGT This work
2.8. DNA agarose gel electrophoresis
DNA electrophoresis was performed in a Mini-Sub Cell GT cell (Bio-Rad) in 1% agarose
gels containing 0.1% ethidium bromide in TAE (40 mM Tris-acetate, 1 mM EDTA, pH 8)
buffer. Gels were exposed to 110 V for one hour using a Consort EV265 electrophoresis
power supply. Samples were prepared using 6X loading buffer (30% (w/v) glycerol,
0.25% (w/v) bromophenol blue, 0.25% (w/v) xylene cyanol FF). Hyperladder 1 kb
(Bioline) was used as the DNA molecular weight marker. Gels were visualised using a
Uvipro Silver geldoc (UVITEC).

55
2.9. DNA restriction digestion and ligation
Plasmids were extracted using QIAprep Spin miniprep kit (Qiagen) and PCR products
were purified using QIAquick PCR purification kit (Qiagen). DNA concentrations were
measured using a NanoDrop 1000 spectrophotometer (Thermo Scientific). Restriction
enzymes used were either purchased from Roche or NEB. Most restriction digests were
performed for one hour at 37°C, whereas SmaI enzyme was incubated at 25 °C. T4 DNA
Ligase (NEB) and 10X Ligation buffer were purchased from Roche. Ligation reactions
were performed at 4°C overnight.
2.10. E. coli competent cell preparation and heat shock
transformation
All reagents, pipette tips and falcon tubes were pre-chilled and centrifugation was
performed at 4 °C. One ml of an overnight culture was used to inoculate 100 ml pre-
warmed LB broth. Cells were grown at 37°C with shaking until an OD600 of 0.6-0.7,
placed on ice for 15 minutes, transferred to 50 ml falcon tubes, centrifuged at 3500 rpm
for 10 minutes in a Heraeus™ Megafuge 40R centrifuge (Thermo Scientific) and the
supernatant discarded. Cells were washed with 25 ml cold 0.1M CaCl2, centrifuged as
above and the supernatant removed. Cells were resuspended in 2 ml cold 0.1M CaCl2 and
incubated on ice for at least two hours. Following this, glycerol was added to give a final
concentration of 15% and the suspension aliquoted into 100 µl fractions. Cells were snap-
frozen in liquid nitrogen and stored at -80°C.
For purchased competent E. coli XL-1 Blue cells (Agilent), transformations were carried
out according to manufacturer’s instructions. For laboratory-prepared cells, 50 µl of cells
were aliquoted into pre-chilled tubes, 1.7 µl of β-mercaptoethanol added and incubated for
ten minutes on ice. One µl of DNA was added, cells incubated for 30 minutes on ice, 42°C
for 45 seconds and then incubated on ice for two minutes. 900 µl of preheated SOC
medium (20 g tryptone, 5 g yeast extract, 0.5 g NaCl per 1L) was added and tubes
incubated at 37 °C for one hour with shaking. 200 µl of cells were plated onto four LB
plates containing appropriate antibiotics and incubated overnight at 37 °C.
2.11. C. fetus conjugation
C. fetus conjugation was carried out according to the protocol of Kienesberger et al.
(2007), with slight modification based on personal communication (Kienesberger, 2015).
All C. fetus incubation steps and the mating step were carried out under microaerobic

56
conditions. C. fetus cells were incubated on blood agar plates supplemented with 5%
defibrinated horse blood (with selection if necessary) for 24 hours at 37 °C. The donor
strain, E. coli S17-1 λpir pG1-N, was in cultured in 5 ml LB with chloramphenicol
overnight at 37 °C and 0.5 ml of this overnight culture was subsequently used to inoculate
3 ml of LB with chloramphenicol and incubated for 3 hours at 37 °C. The OD600 of this
culture was measured and ~107
cells (based on OD600 of 0.1 equating to 5 x 107
cells/ml)
were resuspended in 20 µl of sterile PBS. C. fetus cells were harvested from the 24 hour
incubation plate and suspended in sterile PBS. Based on an OD600 of 0.1 equating to 5 x
108
cells, 109 C. fetus cells were centrifuged and the 20 µl of prepared E. coli cells were
used to resuspend this pellet. This mating mixture was spotted onto a nitrocellulose filter
(25 mm, 0.22 µm pore size)(Millipore), placed in the centre of a BA plate and incubated
microaerobically for 3 hours at 37 °C to allow plasmid mobilisation. Cells were harvested
from the filter in a sterile 20 ml universal tube using 1 ml sterile PBS (prewarmed to 37
°C), vortexed and plated onto four BA plates containing Nalidixic acid (75 µg/ml)
(ACROS Organics) and chloramphenicol (30 µg/ml) to select for C. fetus and the
transferred plasmid, respectively. When using C. fetus pglB::kn, these plates also included
kanamycin. Plates were incubated for 8-14 days at 37 °C.
2.12. Protein expression in E. coli BL21 (DE3)
Protein expression from E. coli was achieved using plasmids derived from pET22 b (+)
(Novagen). E. coli BL21 containing the appropriate expression vector was incubated in 5
ml LB broth (with selective antibiotics) overnight (16 hours) at 37 °C. This culture was
then used to inoculate 250 ml LB broth (with relevant antibiotics) and incubated at 37 °C
until the culture reached an OD600 of 0.6. At this point, 1 mM IPTG (Thermo Fisher
Scientific) was added and the culture incubated for either 2 hours at 37 °C or overnight at
16 °C to allow for protein expression. To confirm the desired protein had been produced, a
whole cell lysate sample was prepared and analysed using SDS-PAGE and western
blotting as described above.
2.13. Nickel affinity chromatography
Following protein induction in E. coli or 24 hour incubation of C. fetus, cells were
centrifuged at 8000 rpm in a SORVALL® RC5B Plus centrifuge for 20 minutes at 4 °C.
Cell pellets were weighed and cells resuspended to a final volume of 5 ml/g in Nickel
binding buffer: 20 mM sodium phosphate (pH 7.4), 0.5 M NaCl, 20 mM imidazole
containing Complete™ mini EDTA-free protease inhibitor cocktail and DNase (1/1000).
Cells were sonicated on ice at 30% setting for 5 second pulses with 10 second intervals.

57
Sonication was carried out for at least 4 minutes. To remove cell debris, the sonicated
solution was centrifuged at 16,000 rpm in a SORVALL® RC5B Plus centrifuge for 30
minutes at 4 °C. The supernatant was removed and filtered using a 0.35 µm pore size
syringe filter (Millex).
All buffers were filtered and degassed prior to use. Proteins were purified using a 5 ml
HisTrap FF column (GE Healthcare). The column was first washed with at least 25 ml of
water and equilibrated with 5 ml of 20 mM imidazole binding buffer (solution as above).
Sample was applied and the flow-through collected. The protein-bound column was
washed with 75 ml of 50 mM imidazole wash buffer and sample eluted with 10 ml of 500
mM imidazole elution buffer. Eluate was collected in 1 ml fractions (E1-10).
2.14. Ion exchange chromatography
Nickel affinity chromatography eluate was dialysed against 20 mM Tris pH 7.6 0.01 M
NaCl. Briefly, sample was applied to a HiTrapQ HP 1 ml column (GE healthcare) on an
ÄKTA HPLC system and a gradient from 20 mM Tris pH 7.6 0.01 M NaCl to 100% 20
mM Tris pH 7.6 1 M NaCl over 20 ml applied.
2.15. Post-purification protein treatment
The appropriate nickel affinity chromatography eluate fractions (as determined by SDS-
PAGE and coomassie-staining) were pooled and dialysed against 30 mM Tris pH 8.0, 30
mM NaCl overnight at 4 °C. Protein solutions were concentrated using Vivaspin
ultrafiltration columns (Sartorius) and centrifuged at 7000 rpm until sufficiently
concentrated.
For preparation of the glycosylated NGRP purified from C. fetus for use as an
immunogen, the dialysed purification eluate was concentrated to ~ 2 ml, a sample ran
alongside BSA standards and coomassie-stained (Fig. 4.4C). The total amount of NGRP
glycoforms present was estimated as around 1 mg using densitometry within Image Studio
software (LiCor biosciences). Aliquots of this preparation were lyophilised and sent to
Antibody Production Services Ltd (Life Science Group Ltd.) for production of polyclonal
rabbit antisera using their standard 77 day immunisation protocol.
2.16. Mass Spectrometry
2.16.1. Sample preparation and MALDI-MS analysis
Samples were separated using SDS-PAGE and proteins visualised with coomassie stain.
Following destaining in water overnight, protein bands of interest were excised using a

58
sterile scalpel, sliced into ~ 1 mm3
pieces and transferred to a sterile eppendorf. Gel pieces
were incubated in 20 µl of 50 mM ammonium bicarbonate (AMBIC) pH 8.4 at room
temperature for 5 minutes and the supernatant removed. This step was repeated until the
coomassie stain was no longer visible. Destained gel pieces were dried for 15 minutes in a
vacuum centrifuge. Dried gel pieces were rehydrated in 20 µl of a working solution of
trypsin (25 ng/µl in 50 mM AMBIC pH 8.4) (Promega) and incubated at room
temperature for 15 minutes. If the gel pieces swelled above the liquid level, 20-30 µl of 50
mM AMBIC pH 8.4 was added to submerge them. The in-gel tryptic digests were then
incubated overnight at 37 °C. Following incubation, the supernatant was transferred to a
sterile Eppendorf. To stop the reaction, the gel pieces were incubated in 50 µl of
Trifluoroacetic acid (TFA) optima™ LC-MS grade (Fisher Scientific) for 10 minutes at 37
°C. To elute peptides, the pieces were incubated in acetonitrile (ACN) for 15 minutes at 37
°C. The tryptic peptide-containing supernatant was removed and combined with the
hydrophilic peptides initially extracted. This TFA and ACN step was repeated once. The
tryptic peptide solution was then dried in a vacuum centrifuge for upto two hours.
Finally, peptides were concentrated and purified using C18 ZipTip® pipette tips (Merck).
Samples were acidified using 10 µl of 0.1% trifluoroacetic acid (TFA). ZipTips were
equilibrated using two cycles of 50% ACN followed by five cycles of 0.1% TFA. Samples
were bound to the tip using five cycles. Samples were washed with two cycles of 0.1%
TFA and eluted in 4-10 µl in 50% ACN, 0.1 % FA. Samples were either analysed using α-
cyano-4-hydroxycinnamic acid (CHCA) (Sigma) or 2,5-Dihydroxybenzoic acid (DHB)
(Sigma) as the matrix. CHCA was prepared as a 1 mg/ml solution in 80% ACN, mixed 1:1
with sample solution. Two microlitres of this solution was applied to a ground steel
MALDI target plate and left to air dry. DHB was prepared at 20 mg/ml in 0.1 % TFA and
mixed 1:1 with the sample solution. Half a microlitre of this solution was pipetted onto a
ground steel MALDI target plate and left to air dry. MALDI-TOF MS was performed on a
Bruker Ultraflex II mass spectrometer in positive-ion relfection mode and at 40-50% laser
power. Data were analysed using FlexAnalysis version 3.0 software (Bruker Daltonics).
2.16.2. LC-MS/MS
Digested samples were produced as previous and analysed by LC-MS/MS using an
UltiMate® 3000 Rapid Separation LC (RSLC, Dionex Corporation) coupled to a Q
Exactive™ HF Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (Thermo Fisher
Scientific) with Higher-energy Collisional Dissociation (HCD) fragmentation.

59
Samples were separated using a multistep gradient from 95% A (0.1% formic acid in
water) and 5% B (0.1% formic acid in ACN) to 7% B at 1 min, 18% B at 35 min, 27% B
in 43 min and 60% B at 44 min at 300 nL min-1
, using a 75 mm x 250 μm i.d. 1.7 M
CSH C18, analytical column (Waters).
Raw data files were converted to .mgf files using Mascot Daemon software (Matrix
Science) and data analysed using mMass v 5.5 (Niedermeyer and Strohalm, 2012). Files
were scanned for precursor ions at predicted m/z ratios. Fragmentation spectra were
manually analysed for the presence of glycan-derived species such as m/z 204.09, which
corresponds to a HexNAc residue and is characteristic of glycopeptide fragmentation
spectra obtained using HCD (Zhao et al., 2011). In addition, spectra were scanned for
disaccharide oxonium ions such as m/z 366 (HexHexNAc), as they can help elucidate
glycan structure (Toghi Eshghi et al., 2016).
2.17. Bioinformatics
The molecular weight of proteins was predicted using the compute pI/Mw tool of the
ExPASy bioinformatics resource portal (Swiss Institute of Bioinformatics). The following
programs were downloaded locally on a computer running a Linux operating system to
facilitate use of the N-glycoprotein prediction Perl script described by Frost (2015): NCBI
BLAST version (v) 2.3.0+ (Camacho et al., 2009), Pftools v 2.3 (ExPASy), PSORTb v
3.0.4 (Yu et al., 2006), LipoP v 1.0 (Juncker et al., 2003) and TMHMM v 2.0b (Krogh et
al., 2001). Proteomes, excluding protein sequences derived from pseudogenes, were
extracted in FASTA format from genomes using Artemis Genome Browser (Rutherford et
al., 2000). These proteomes were then fed to the perlscript. The tab delimited text files
produced by the perlscript were copied to Microsoft Excel for manual curation. Manual
analysis utilised CELLO v 2.5 (Yu et al., 2006) and SignalP 4.1 Server (Petersen et al.,
2011). Sequence logos were created using WebLogo 3 (Crooks et al., 2004). Predicted N-
glycoprotein comparisons utilised the online standard protein BLAST® (BLASTP)
program and query sequences were searched against the revelant organism within the non-
redundant protein sequence database. The total percent identity between two proteins was
determined using the BLAST® global alignment tool.

60
Chapter 3. Characterisation of an
antiserum raised against a
Campylobacter jejuni N-linked
glycoprotein

61
3.1. Introduction
3.1.1. Overview of C. jejuni NGRP antiserum production process
At the start of this project, an antiserum raised against a C. jejuni N-linked glycoprotein
antiserum was in production (Jervis and Linton, unpublished). This antiserum was raised
against a truncated form of a C. jejuni glycoprotein, Cj0114 that has four N-linked
glycosylation sites, two in an N-terminal coiled-coil domain and two in a flexible linker
region. Its C-terminal tetratricopeptide repeat (TPR) domain is not N-glycosylated and
hence was removed to give the truncated form, termed NGRP (N-Glycosylation Reporter
Protein). Due to being small and heavily N-glycosylated this recombinant protein made an
ideal candidate to use for the production of a C. jejuni N-linked glycoprotein antiserum.
Production of NGRP glycosylated with the C.jejuni heptasaccharide was achieved using
glycocompetent E. coli (Wacker et al., 2002). NGRP was cloned into pET22b(+) to give
pNGRP which allowed expression of His6-tagged NGRP directed to the periplasm. This
plasmid was transformed into E. coli BL21 (DE3) cells containing the plasmid ppgl
(encoding the functional pgl locus of C. jejuni) (Wacker et al., 2002). Glycosylated His6-
tagged NGRP was purified by Nickel-affinity chromatography and used as an immunogen
for production of rabbit polyclonal antiserum (Antibody Production Services Ltd.). The
final antiserum produced was named CjNgp (C. jejuni N-glycoprotein).
3.1.2. Project aims
There is a considerable need for detection methods that could be used on-site to detect
Campylobacter in poultry. Antibody-based detection/identification methods are
particularly suitable, as they are generally rapid and simple to use. As discussed in chapter
1, N-linked glycans are promising targets for antibody-based detection or identification of
Campylobacters. More specifically, an identical N-glycan structure is produced by C.
jejuni, C. coli and C. upsaliensis, pathogenic species which are found on chicken meat.
Therefore, antibodies targeting this N-glycan structure have the potential to be developed
into an assay able to detect several important Campylobacter pathogens. Indeed,
previously described antisera reactive against the C. jejuni heptasaccharide have been
shown to react similarly well with cell lysates of C. coli and C upsaliensis (Jervis et al.,
2012; Nothaft et al., 2012). In addition, these antisera are able to react with cell lysates of

62
the hexasaccharide N-glycan-producing C. lari, another Campylobacter pathogen that
contaminates chicken meat. In conclusion, it appears that antibodies against the C. jejuni
heptasaccharide N-glycan could be used to create a test able to detect at least four species
of Campylobacter which are a human infection risk in association with chicken meat. This
project aimed to to assess the feasibility of CjNgp to be used in an antibody-based method
for identification/detection of important Campylobacter pathogens found in poultry. To do
this, the following aspects of antiserum CjNgp were characterised:
C. jejuni N-linked glycan reactivity and specific N-glycan structural specificity.
C. jejuni strain coverage and reactivity against recent chicken meat isolates.
Campylobacter species specificity.
Binding untreated cells of Campylobacter species.

63
3.2. Characterisation of CjNgp reactivity against Campylobacter
whole cell lysate extracts.
3.2.1. The reactivity of CjNgp antiserum against C. jejuni N-linked glycan.
In order to assess if CjNgp was reactive towards the C. jejuni N-linked heptasaccharide, it
was used to probe western blots containing C. jejuni wildtype and a range of pgl mutant
whole cell lysates (Fig. 3.1B). Whole cell lysates were separated by SDS-PAGE and then
either stained with coomassie (Fig. 3.1A), or western blotted for probing with antiserum
(Fig. 3.1B). With a C. jejuni lysate, CjNgp reacted with many proteins (Fig. 3.1B). In
particular, a strong reactive band at around 42 kDa was visible. When probed against a C.
jejuni pglB mutant only a single reactive band at around 35 kDa was visible. The
oligosaccharyltransferase PglB is responsible for transferring the N-linked glycan structure
onto target proteins, thus a mutant lacks N-linked glycoproteins (Wacker et al., 2002).
Therefore, all but a single CjNgp-reactive band in a C. jejuni lysate were pglB-dependent.
Thus, CjNgp is reactive towards the N-linked glycan produced by C. jejuni.
Deletion of C. jejuni pgl glycosyltransferase genes results in transfer of truncated glycans
onto target proteins (Linton et al., 2005) and N-linked glycan structures produced by each
strain are presented (Fig. 3.1C). As with a wildtype lysate, CjNgp exhibited reactivity with
numerous bands within a pglI mutant lysate, but with reduced intensity (Fig. 3.1B). In
contrast to its particularly strong reactivity with an ~ 42 kDa band in a wildtype lysate,
with a pglI mutant it reacted with two distinct bands at around 42 kDa and 41 kDa in size.
Reactivity with lysates from pglHJA mutants was reduced to a similar level to that
observed with the pglB mutant. Importantly, it appeared that the single reactive band
remaining in the pglB and pglHJA mutants decreased in size as glycan structure was
truncated, suggesting it corresponds to a glycoprotein. This is likely Cj0114, the native N-
glycoprotein from which NGRP was constructed, as its apparent unglycosylated size (35
kDa) in C. jejuni pglB::kn correlates with what is predicted (35.4 kDa). Therefore, CjNgp
is reactive with this native protein, but its N-glycan reactivity was likely abolished upon
reduction to tri-, di- and monosaccharides. These data confirm that CjNgp has significant
reactivity against the C. jejuni N-linked glycan, the majority of which is directed towards
the terminal four sugars (GalNAc3Glc) of the heptasaccharide structure. However, the
antiserum also appears to react with the N-glycoprotein Cj0114 regardless of N-glycan
modification.

64

65
3.2.2. CjNgp reactivity with a variety of C. jejuni strains
In order to assess if CjNgp has consistently high reactivity with N-glycoproteins of a range
of C. jejuni strains, it was used to probe western blots containing whole cell lysates of an
additional 11 strains. This strain set included Penner reference strains and clinical strains
from patients with Miller-Fischer syndrome (F1-F3) and Guillian Barré Syndrome (G2)
(Appendix Table 1). Whole cell lysates were separated by SDS-PAGE and then either
stained with coomassie (Fig. 3.2A), or western blotted for probing with antiserum (Fig.
3.2B). As previously, CjNgp reacted with a multitude of bands in a C. jejuni 11168H
whole cell lysate (Fig. 3.2B). A C. jejuni 11168H pglB mutant lysate served as a negative
control. In contrast to the result seen with the previous pglB::kn sample, CjNgp did not
react with a 35 kDa band in this instance. This could be due to a difference in extraction
method (BugBuster® Protein Extraction Reagent) that fails to solubilise Cj0114 and/or
possibly due to reduced stability of this protein when not N-glycosylated.
CjNgp strongly reacted with whole cell lysates of each of the strains tested and although
glycoprotein profiles were very similar, subtle differences were observed. Again, a strong
band at around 42 kDa in size is seen with all the strains tested, though the mobility and
intensity of the band does vary between strains. Therefore, the CjNgp antiserum reacted
with numerous glycoproteins in a variety of C. jejuni strains. Hence, CjNgp antiserum has
the potential to detect a broad range of C. jejuni strains if integrated into an antibody-
based test.

66

67
3.2.3. CjNgp reactivity with recent Campylobacter chicken meat isolates
To verify that the CjNgp antiserum reacts with currently circulating strains of
Campylobacter, strains were isolated from retail chicken meat. Isolates were identified
using Campylobacter-specific PCR (Linton et al., 1996) followed by C. jejuni and C. coli
species-specific PCR assays (Linton et al., 1997). Strains were typed by sequencing of the
flaB variable region (See section 2.2). Isolation experiments carried out in 2014 and 2015
from two supermarkets resulted in 10 distinct strains, 6 C. jejuni and 4 C. coli (Appendix
Table 1).
Whole cell lysates were separated by SDS-PAGE and then either stained with coomassie
(Fig. 3.3A), or western blotted for probing with antiserum CjNgp (Fig. 3.3B). Many
immunoreactive bands were evident with each isolate. The pattern of reactivity with each
C. jejuni isolate was very similar to that observed with C. jejuni 11168H. As for the C.
coli isolates, there were a few differences in the reactive band profile however the overall
level of reactivity was just as marked. Overall, CjNgp reacted very strongly towards whole
cell lysates of Campylobacter recently found to be infecting retail chicken meat, whether
they were C. jejuni or C. coli. These data suggest CjNgp is appropriate for detecting
Campylobacter strains recently contaminating chicken flocks.

68

69
3.2.4. Campylobacter species specificity of CjNgp.
To investigate the species specificity of the CjNgp antiserum, it was used to probe whole
cell lysates of Campylobacter species which encompass five N-linked glycan structural
groups described (Jervis et al., 2012). Whole cell lysates were separated by SDS-PAGE
and then either stained with coomassie (Fig. 3.4B), or western blotted for probing with
antiserum CjNgp (Fig. 3.4A).
As seen previously, CjNgp reacted with C. jejuni 11168H but not the corresponding pglB
knockout (Fig. 3.4A). As expected given the results of the chicken isolates reactivity,
CjNgp reacted to a similarly strong degree with C. coli. Alongside these, CjNgp also gave
numerous reactive bands with C. upsaliensis and C. helveticus, species which produce an
identical N-linked heptasaccharide structure. With C. lari, CjNgp reacted with several
bands but at a lower intensity. Compared with the C. jejuni glycan structure, C. lari
produces a linear hexasaccharide lacking the glucose sugar (Schwarz et al., 2011),
suggesting that CjNgp binds this residue within C. jejuni N-glycan. CjNgp did not react
with whole cell lysates of any of the four remaining species tested (C. concisus, C. fetus,
C. hyointestinalis and C lanienae) (Fig. 3.4A). These four species all produce distinct N-
glycans that vary more considerably in structure compared to the C. jejuni group and C.
lari. These results confirm that CjNgp is highly reactive towards the N-linked glycan
structure produced by C. jejuni, C. coli, C. upsaliensis and C. helveticus and, to a lesser
extent, the linear hexasaccharide structure produced by C. lari. However, CjNgp does not
bind to C. concisus, C. fetus, C. hyointestinalis or C. lanienae N-linked glycans. These
data indicate CjNgp could specfically detect both the common and so-called ‘emerging’
Campylobacter pathogens found in poultry, but not cross react with other Campylobacter
species.

70

71
3.3. CjNgp binding untreated cells of Campylobacter.
3.3.1. CjNgp binding to untreated C. jejuni cells is not dependent on N-linked
glycosylation.
Many assays for detecting/identifying Campylobacter require samples to be pre-treated,
for example cells are lysed or DNA extracted, before analysis. A reagent that can directly
bind untreated cells would allow preparation time to be reduced. Hence, for CjNgp to be a
successful reagent in an antibody-based Campylobacter detection system, it would be
useful if it could bind untreated Campylobacter cells. To investigate this, CjNgp was used
to probe dot blots containing untreated cells of C. jejuni. Furthermore, to assess if
antibody reactivity was against N-glycan, C. jejuni pgl mutants were also included in the
experiment. Although a pglB mutant does not produce N-linked glycoproteins, the glycan
structure is still in the cell in its precursor form, lipid-linked oligosaccharide (LLO) (Jervis
et al., 2012). This lipid-linked precursor migrates very rapidly in SDS-PAGE of cell
lysates so that it does not interfere with antibody-protein interactions, making the strain a
suitable control in 3.2. However, it is possible that dot blots containing untreated cells may
present LLO in this strain. Therefore, it was thought that a pglB mutant may not be the
most appropriate negative control. To circumvent this potential issue, the pglA mutant,
which produces a severely truncated N-glycan, was also included. As shown in 3.2.1.,
CjNgp does not react with the N-glycan structure present in this mutant. Dot blots
performed with no primary antibody present or using pre-immune serum gave no
reactivity (not shown). As a positive control, anti- whole cell C. jejuni 11168 (anti-WC
11168) antiserum was included (Fig. 3.5B) (Linton, unpublished). Dot blots were
performed similarly to the protocol described by Qian et al. (2008). Briefly, the dot blot
procedure involved harvesting cells from agar plates following 24 hours growth, washing
in 1ml PBS, and resuspending at various OD600 values. Cell numbers are estimated based
on OD600 of 1 equating to around 1 x 109 C. jejuni cells (Qian et al., 2008). One µl of each
cell suspension was pipetted onto nitrocellulose membranes and air dried for 15 minutes.
Membranes were then blocked, probed and washed as in a western blotting procedure (see
methods).
When used in a dot blot assay, anti-WC 11168 detected as little as 104
cells of C. jejuni
wildtype, pglB::kn and pglA::kn strains (Fig. 3.5B). Although this limit of detection was
consistent with each strain, the observed reactivity was lower with the pglB mutant in
comparison to the wildtype and pglA mutant. When CjNgp was used to probe a dot blot, it

72
could detect 105
cells and above (Fig. 3.5A). Using CjNgp, slightly more reactivity was
seen with the wildtype strain in comparison to each knockout. These data show that CjNgp
is able to bind to untreated cells of C. jejuni in a dot blot assay format. However, the
antiserum does not perform as well as anti-WC 11168 antiserum. Furthermore, cells
lacking the pglB or pglA genes were still bound by CjNgp to an extent not far below that
observed with the wildtype. This suggests that the cell binding observed is not due to N-
linked glycan reactivity within the antiserum. As CjNgp displays reactivity against Cj0114
(the native progenitor of NGRP) (Section 3.2.1.), reactivity against this protein may be
responsible for C. jejuni cell binding.

73
3.3.2. Reactivity against the NGRP protein influences the ability of CjNgp to bind
untreated C. jejuni cells.
Section 3.3.1 demonstrated that the ability of CjNgp to detect untreated cells of C. jejuni
was not dependent on N-linked glycosylation. Using western blotting, the antiserum was
inferred to react with the Cj0114 protein, the basis for production of the N-glycan carrier
immunogen, NGRP (See section 3.2.1.). Therefore, the NGRP reactivity within the
antiserum may be influencing its ability to bind untreated C. jejuni cells. To investigate
this, dot blots were performed using CjNgp that had been pre-treated to adsorb out
reactivity against unglycosylated NGRP. To achieve this, CjNgp was pre-incubated with a
membrane containing lysates of E. coli BL21 expressing NGRP prior to use in a dot blot
assay. To account for the loss of antibodies due to non-specific binding, pre-incubation
with a blank membrane was performed as a control. Also, to ensure no loss of antibody
binding was due to adsorption of E.coli-reactive antibodies, CjNgp pre-treated with a
membrane containing BL21 lysates was also included. Following each pre-incubation,
CjNgp was used to probe dot blots containing untreated cells of C. jejuni wildtype, C.
jejuni pglB::kn and C. jejuni pglA::kn, as previously (Fig. 3.6). In addition a spot of
purified NGRP was included to assess NGRP reactivity present.
When pre-incubated with a blank membrane, CjNgp weakly reacted with as few as 105
cells of all strains tested (Fig. 3.6A). As seen previously, CjNgp reacted similarly with C.
jejuni cells with and without functional pglB or pglA genes, giving a strong signal with
each of the strains when 107
cells were present. CjNgp gave a very strong reaction with
purified NGRP. Upon pre-incubation with BL21 lysates, no change in cell binding was
observed (Fig. 3.6B). In addition, a similarly strong signal was seen with purified NGRP.
When CjNgp was pre-incubated with BL21 NGRP lysates, a marked change in cell
binding was observed (Fig. 3.6C). This pre-adsorbed CjNgp could only reasonably detect
5 x 105
C. jejuni wildtype cells and the overall intensity of reactivity was considerably
lower. When probing C. jejuni pglB::kn cells, a signal was only identifiable with 107
cells.
With C. jejuni pglA::kn, there was no signal at all using CjNgp. However, even following
this BL21 NGRP pre-incubation, some slight NGRP reactivity still remained.
In summary, removing most of the NGRP-reactive antibodies from CjNgp resulted in a
reduction in its ability to bind C. jejuni cells. In this assay, with wildtype C. jejuni cells, its
limit of detection appeared to shift from 105 cells to 5 x 10
5 cells upon removing the
majority of NGRP-reactive antibodies. Moreover it could no longer detect C. jejuni cells
lacking pglA and only gave a sufficient signal with a pglB mutant when 107
cells were

74
present. Therefore, these data suggest that CjNgp contains a large proportion of antibodies
reactive with the NGRP protein rather than the heptasaccharide N-glycan. Furthermore,
these data suggest that these NGRP-reactive antibodies considerably contribute to, but are
not fully responsible for, the ability of CjNgp to bind untreated cells.

75
3.3.3. CjNgp binding to untreated cells of Campylobacter species
As described in previous sections, CjNgp can bind to untreated cells of C. jejuni,
suggesting it would be feasible to integrate this antiserum into a C. jejuni detection tool.
Moreover, there are several other Campylobacter species found alongside C. jejuni in
poultry that are also known to cause human disease. A tool able to detect such
Campylobacter species would be useful. This work has previously demonstrated that
CjNgp is highly reactive with lysates of disease-causing Campylobacter species found on
poultry, but not with lysates of other Campylobacter pathogens, including the veterinary
pathogen, C. fetus venerealis (section 3.2.4). To investigate if this pattern is maintained
with regards to untreated cell binding, CjNgp was used to probe dot blot assays containing
five Campylobacter species. Again, anti-WC 11168 was used for comparison.
In this dot blot assay anti-WC 11168 could detect 5 x 105
cells of each of the five species
used (Fig. 3.7B). However, this antiserum reacted with C. fetus fetus considerably less in
comparison to the remaining four species (C. jejuni, C. coli, C, upsaliensis and C. lari).
When CjNgp was used, it could detect 5 x 105
cells and above of C. jejuni, C. coli, C,
upsaliensis and C. lari (Fig. 3.7A). However, it gave the strongest reaction with C. jejuni
and C. coli, then C. upsaliensis then C. lari. CjNgp was not able to bind upto 8 x 105
cells
of C. fetus.
In conclusion, when testing against untreated cells, the Campylobacter species specificity
of CjNgp was similar to the observation made when using cell lysates. This observation is
that CjNgp is most reactive towards Campylobacter species producing the N-linked
heptasaccharide structure, gives lower reactivity with C. lari and harbours no reactivity for
C. fetus. Here it was also shown that, as previously found with C. jejuni cells in section
3.3., CjNgp has a lower affinity for the Campylobacter species tested here in comparison
to the anti-WC 11168 serum. However, in contrast to the anti-WC 11168 serum, CjNgp
does not cross react with untreated cells of C. fetus.

76

77
3.3.4. CjNgp labels whole cells of C. jejuni 11168H
Previous analysis using dot blot assays demonstrated that CjNgp can bind untreated cells.
However, cells may have lysed using this method as it involves drying the cells onto a
membrane. As N-linked glycoproteins are either periplasmic or membrane proteins, a
considerable percentage could be surface-exposed and accessible for antibody binding
when cells are whole. Therefore, to assess if CjNgp is able to label whole cells of C. jejuni
flow cytometric analysis was used. To allow for cell detection, the green fluorescent
protein (GFP)-producing strain C. jejuni 11168Hgfp4 developed by Jervis et al. (2015)
was utilised. The FITC-channel was used to detect GFP (Fig. 3.8 x-axes). Antibody
labelling of cells was detected using R-phycoerthrin (R-PE) goat anti-rabbit IgG secondary
antibody (Fig. 3.8, y-axes). Cells not incubated with antiserum were included to rule out
any staining resulting from the secondary antibody (Fig. 3.8A). As a positive control, anti-
whole cell C. jejuni 11168 (anti-WC 11168) antiserum was included (Fig. 3.8B) (Linton,
unpublished). Finally, pre-immune rabbit serum served to investigate if non-specific
binding occurred with normal rabbit serum alone (Fig. 3.8D).
GFP production was unexpectedly low in all samples (Fig.3.8 x-axes), however, the
analysis still gave a useful indication of antibody labelling. As seen in figure 3.8A, C.
jejuni cells not treated with serum displayed very minimal R-PE labelling. When
incubated with anti-WC 11168, the cells exhibited an obvious shift in R-PE staining, with
the majority of the population giving a high signal (Fig. 3.8B). In contrast, pre-immune
rabbit antiserum did not confer significant R-PE staining of C. jejuni (Fig. 3.8D). Finally,
a large majority of the population exhibited R-PE staining upon treatment with CjNgp
antiserum (Fig. 3.8C). However, this shift was seen with fewer of the population and the
signal achieved was not as intense in comparison to with the anti-WC 11168 antiserum.
Therefore, CjNgp appears capable of labelling whole cells of C. jejuni 11168, but not to
the same as extent as an anti-whole cell C. jejuni 11168 antiserum.

78

79
3.4. Discussion
This Chapter described the characterisation of an antiserum, CjNgp, raised against a C.
jejuni N-glycoprotein, NGRP, expressed in glycocompetent E. coli. CjNgp antiserum was
assessed with regards to its Campylobacter reactivity; its N-linked glycan structural
specificity, strain coverage, species specificity and the ability to bind Campylobacter cells.
The overall aim was to preliminarily assess the feasibility of integrating the antiserum into
a Campylobacter detection/identification test.
The CjNgp antiserum was very reactive with a C. jejuni 11168H lysate and gave only a
single band of reactivity upon deletion of pglB, demonstrating its reactivity against the N-
linked glycan structure. The ~ 35 kDa CjNgp-reactive band observed with a pglB mutant
likely corresponds to Cj0114, the native protein from which NGRP was developed. A
band putatively corresponding to this protein was observed within each C. jejuni pgl
mutant lysate tested – with its mobility changing dependent on N-glycan length.
Therefore, CjNgp harbours reactivity against this protein with and without N-linked
glycan modification. CjNgp N-glycan reactivity was influenced by loss of the branching
glucose sugar upon deletion of pglI. Further truncation of the N-glycan suggested the
antiserum is most reactive with the terminal four residues (GalNAc3Glc). An antiserum,
termed hR6, specific for the C. jejuni N-glycan already exists in the literature (Amber and
Aebi, unpublished). In contrast to these data, Nothaft et al. (2009) demonstrated that hR6
reacts equally well with C. jejuni wildtype and pglI::kn lysates, suggesting its reactivity is
independent of the branching glucose sugar. Instead its reactivity is mostly dependent on
the terminal three GalNAc residues, having very minimal reactivity against a pglH::kn
lysate and no reactivity if the N-glycan is further truncated. This difference is not
surprising as these are polyclonal antisera and is likely due to varying antibody responses
in the immunized rabbits.
The pgl locus is conserved within C. jejuni, however, prediction and/or experimental
identification of C. jejuni N-glycoproteins has only been performed in a few strains (Ding
et al., 2009; Scott et al., 2011). Therefore the extent of variation in the size of the C. jejuni
N-linked glycoproteome among different strains is undetermined. The amount of N-linked
glycosylation that takes place within C. coli is even less understood. CjNgp reacted with a
multitude of bands within the lysates of 11 C. jejuni reference strains and clinical isolates,
demonstrating that an equally large N-glycoproteome is present in several members of this
species. Furthermore, CjNgp displayed consistently high reactivity within C. jejuni and C.
coli strains recently isolated from retail chicken meat. These observations provide further

80
evidence that the N-linked glycan structure produced by C. jejuni and C. coli is well
conserved and that N-glycosylation remains a predominant protein modification among
strains of these species. Furthermore, it suggests this N-glycan-reactive antiserum could
create a detection tool suitable for a variety of C. jejuni and C. coli strains, including those
recently contaminating poultry.
The Campylobacter species specificity of CjNgp was assessed by probing western blots of
whole cell lysates of a variety of species. CjNgp was very reactive towards the three
Campylobacter species (C. coli, C. upsaliensis and C. helveticus) known to produce the C.
jejuni-type heptasaccharide N-glycan structure. This is in agreement with the findings of
Jervis et al. (2012) and Nothaft et al. (2012) when using hR6 antiserum, and provides
further evidence that C. jejuni N-glycan-reactive antisera are promising tools for detecting
these Campylobacter pathogens. However, in contrast to the characterised reactivity of
hR6, CjNgp was considerably less reactive with C. lari. As C. lari produces a linear N-
glycan identical to that of a C. jejuni pglI mutant, this observation further proves the
importance of this branching glucose residue for CjNgp reactivity. These data demonstrate
a drawback of using polyclonal sera for detection tools, as variability in rabbit responses
may result in such differences in reactivity. However, these data suggest the CjNgp
antiserum could be promising for the broad detection of Campylobacter species found in
chickens, including the common C. jejuni and C. coli and those considered to be emerging
pathogens (C. upsaliensis and C. lari) (Man, 2011). Such broad reactivity against
pathogenic Campylobacter species found in poultry is favourable as methods for
Campylobacter detection are often restricted to C. jejuni and C. coli. For example, Miller
et al. (2008) found that three commercially available Campylobacter LAT assays were
unable to identify any of the eight C. upsaliensis strains tested. Furthermore, these data
suggest that a detection tool using CjNgp could be similarly sensitive when detecting C.
jejuni, C. coli and C. upsaliensis. This would be another advantage over currently
available methods of detection because, for example as seen with an immunosensor
developed by Sapsford et al. (2004), they can have lower sensitivity for Campylobacter
species other than C. jejuni.
CjNgp did not react with cell lysates of any other Campylobacter species tested, each of
which are known to produce N-glycan structures distinct from that of C. jejuni (Jervis et
al., 2012; Nothaft et al. 2012). The hR6 antiserum described elsewhere also displays an
absence of reactivity with these species (Jervis et al., 2012; Nothaft et al., 2012). Therefore
CjNgp has the potential to be used to identify common Campylobacter human

81
gastrointestinal pathogens without cross reacting with other species such as C. fetus, C.
hyointestinalis, C. concisus or C. lanienae.
To integrate CjNgp into a rapid detection/identification tool it would be advantageous for
it bind to Campylobacter cells without pre-treatment steps. Dot blot assays showed that
CjNgp could bind untreated cells of C. jejuni wildtype, pglB::kn and pglA::kn strains to a
similar extent, suggesting N-glycosylation was not important for cell binding. Adsorbing
NGRP-reactive antibodies from the antiserum demonstrated that NGRP reactivity
considerably influenced the ability of CjNgp to bind C. jejuni cells. However, the
remaining antibodies within CjNgp were able to bind C. jejuni wildtype cells but not C.
jejuni pglA::kn cells, suggesting that these antibodies were N-glycan-reactive. These
antibodies were also able to bind C. jejuni pglB::kn cells, but to a reduced extent. This
reduction in binding as opposed to complete loss is likely due to the presence of LLO
within these pglB::kn cells. Therefore, NGRP reactivity considerably influences the ability
of CjNgp to bind untreated cells of C. jejuni, even though it is a periplasmic protein. The
reason this protein is readily accessible in a dot blot assay may be due to surface damage
of the cells during preparation. Nevertheless, it appears that the N-glycan reactive
antibodies within CjNgp are able to bind untreated C. jejuni cells, and thus C. jejuni N-
glycan remains a potential target for developing a rapid antibody-based detection assay.
Dot blot assays also demonstrated CjNgp can bind cells of C. coli, C. upsaliensis and C.
lari but not C. fetus. In contrast to the similar reactivity against whole cell lysates of C.
jejuni, C. coli and C. upsaliensis, CjNgp was more reactive with C. jejuni cells than those
of the other species. It should be noted that NGRP-reactive antibodies may have bound to
these cells as they contain Cj0114 homologues but that such antibodies would likely be
more reactive with Cj0114 and thus have higher affinity for C. jejuni. However, as N-
glycan-reactive antibodies present within CjNgp can bind untreated cells of C. jejuni, it is
envisaged that purification of the N-glycan-reactive antibodies from this antiserum would
still provide a reagent able to similarly bind C. jejuni, C. coli, C. upsaliensis and C. lari
cells. Furthermore, in contrast to anti-WC 11168, CjNgp does not cross react with C. fetus
cells. Therefore, an advantage of CjNgp over an anti-whole cell serum is that it is specific
for the common gastrointestinal Campylobacter pathogens found in poultry.
Flow cytometric analysis demonstrated that CjNgp could bind whole cells of C. jejuni,
suggesting that N-glycans may be significantly surface-exposed. Surface localisation of N-
glycan in C. jejuni has previously been shown with hR6 antiserum using fluorescent
microscopy (Alemka et al., 2013). However, flow cytometric analysis here indicated that

82
an N-glycan reactive antiserum may not label C. jejuni cells as efficiently as an anti-whole
cell 11168 antiserum. Nevertheless, CjNgp still has the advantage of specificity for the
common Campylobacter human pathogens found on chicken meat. Furthermore, future
work to purify and concentrate N-glycan reactive antibodies from the antiserum could
allow enhanced Campylobacter sensitivity (See Chapter 7).
This chapter has demonstrated that CjNgp is an antiserum reactive against the C. jejuni N-
linked glycan structure, more specifically the four terminal sugar residues (GalNAc3Glc).
This reactivity results in the ability to react with a multitude of C. jejuni and C. coli N-
linked glycoproteins from numerous strains, including reference strains, clinical isolates
and recent retail chicken meat contaminants. In addition CjNgp reacts significantly with C.
upsaliensis, C. helveticus and, to a lesser extent, C. lari N-glycoproteins. Importantly, it
does not cross react with the N-linked glycoproteins produced by other species of
Campylobacter such as C. fetus and C. consisus. Furthermore, this antiserum is able to
bind untreated cells of C. jejuni, C. coli, C.upsaliensis and C. lari. A portion of this
binding activity is due to presence of protein-reactive antibodies present in the serum.
However, upon removing these antibodies, CjNgp still exhibits the ability to bind to
untreated cells of C. jejuni. Furthermore, CjNgp was demonstrated to label whole cells of
C. jejuni. Therefore, CjNgp has been shown to be a promising candidate for development
of a tool capable of detecting harmful poultry meat contaminants such as C. jejuni, C. coli,
C.upsaliensis and C. lari.

83
Chapter 4. Development and
characterisation of an antiserum raised
against a Campylobacter fetus N-linked
glycoprotein

84
4.1 Introduction
C. fetus consists of three subspecies, C. fetus fetus (C. f fetus), C. fetus venerealis (C. f
ven) (Veron and Chatelain, 1973) and the more recently proposed C. fetus testudinum
(Patrick et al., 2013). C. f fetus causes sporadic abortion in sheep and cattle (van Bergen et
al., 2005). However, it is also an opportunistic human pathogen capable of systemic
infections (Wagenaar et al., 2014). C.f ven is the causative agent of Bovine Genital
Campylobacteriosis (BCG), which is a significant burden to the cattle industry (Mshelia et
al., 2010). C. fetus testudinum is associated with reptiles but has also proven to be an
opportunistic human pathogen (Patrick et al., 2013).
Traditional culture and isolation of C. fetus is known to be difficult and solely relying on
this method can allow infected herds to remain undiagnosed (Truyers et al., 2014).
Although numerous molecular assays have been developed, most are better suited to
research than routine diagnostics (van der Graaf-van Bloois et al., 2013). Furthermore,
these methods require laboratories with sufficient equipment, a factor which is limiting for
developing countries (Mshelia et al., 2010). Instead, simple detection methods that can be
used in the field are desirable. Antibody-based detection is suitable for such on-farm
detection as it can be quick, easy and require very little sample preparation. Current
immunological methods utilised for diagnosis of bovine genital campylobacteriosis tend to
have poor sensitivity and can be misleading due to reliance on the varying humoral
responses of cattle (Truyers et al., 2014). For this reason it has been suggested that several
methods should be used in parallel to obtain accurate results (Truyers et al., 2014).
Therefore, improved tests are needed. In addition, such assays could be applicable to
detecting C. fetus in human samples as there are currently none available.
As described in 3.1, Campylobacter N-linked glycans are promising target candidates for
antibody-based detection tools. C. fetus produce N-glycan structures distinct from that of
all other Campylobacter species except for C. hyointestinalis (Jervis et al., 2012; Nothaft
et al., 2012). C. hyointestinalis consists of two subspecies, C. hyointestinalis lawsonii and
C. hyointestinalis hyoinestinalis (C. hyo hyo) (On et al., 1995). The former is associated
with swine whereas the latter is isolated from bovine intestinal tracts (Miller et al., 2016).
Therefore, as C. hyo hyo is not specifically found in the reproductive tract of cattle it
should not pose a serious cross contamination risk when screening for C. fetus. However,
C. hyo hyo can lead to fatal enteritis in calves (Diker et al., 1990) and detection of this
species in significantly contaminated preputial/vaginal samples could also be favourable.
Additionally, C. hyointestinalis has been associated with human gastrointestinal illness

85
and even bacteraemia (Gorkiewicz et al., 2002; Pasternak et al., 1984). Hence, C. fetus N-
glycans could be utilised to develop a test suitable for detecting this species, and possibly
C. hyointestinalis, in agricultural animals and/or human samples. Antisera, termed GRPII-
1 and GRPII-2, raised against each of the two reported C. fetus free oligosaccharide
structures display considerable reactivity with numerous proteins in C. fetus fetus and C.
fetus venerealis .(Nothaft et al., 2012). Although promising, each antiserum displayed
varying characteristics and further research into the potential for C. fetus N-glycans to be
used as detection targets was warranted. The study described here aimed to develop a C.
fetus N-linked glycan-reactive polyclonal antiserum to allow for further research.
Firstly, due to the ease and success of using NGRP as the glycoprotein immunogen when
raising the CjNgp antiserum (see Chapter 3), it was chosen again here. Previous
production of glycosylated NGRP was undertaken in glycocompetent E. coli expressing
the C. jejuni pgl locus (Wacker et al., 2002). However, no such E. coli system exists for
producing N-glycoproteins with the C. fetus N-glycan structure. To develop an equivalent
system for C. fetus N-glycan production in E. coli would have meant laborious cloning
and screening with no guarantee of a correctly functioning end-product. Instead, this
project aimed to integrate NGRP into C. fetus to allow for glycosylation via the native pgl
system.
Following successful development of a C. fetus N-glycan-reactive antiserum, this project
aimed to characterise the serum to assess its feasibility to be used for C. fetus
identification or detection purposes. Similarly as in Chapter 2, the following features of
CfNgp were characterised:
C. fetus N-glycan reactivity.
C. fetus subspecies and strain coverage.
Campylobacter species specificity.
C. jejuni N-linked glycan structural specificity.
Ability to bind cells of Campylobacter species.

86
4.2 Production of a recombinant Campylobacter fetus N-linked
glycoprotein for use as immunogen.
4.2.1 Using a C. fetus chromosomal integration vector to integrate an N-
glycoprotein was not a suitable approach.
Genetic manipulation of C. fetus is difficult and available tools are relatively limited. Gene
integration within the 23S rRNA gene, rrl, on the C. jejuni genome has been demonstrated
(Karlyshev and Wren, 2005). These vectors exploit the fact that C. jejuni harbours three
identical copies of ribosomal RNA genes. As C. fetus also has multiple, identical copies of
ribosomal rRNA genes, this study aimed to similarly develop a suicide vector which could
allow integration of an N-glycoprotein gene into the 23S rRNA gene of C. fetus.
Briefly, a 1.6 kb region of the C. fetus rrl gene was amplified from C. fetus fetus 10482
genomic DNA (gDNA) using primers 1268 and 1269 (Table 2.1) and ligated to pGEMT-
easy vector (Promega) to give pGEM23SCf. Jervis et al. (2015) previously developed a
suicide vector, pCJC4, for high-level protein expression in C. jejuni which included a
construct containing the C. jejuni porA promoter and a chloramphenicol resistance cassette
(cat). This construct was utilised here and was amplified from pCJC4 using primers 1296
and 1297 (Table 2.1) which add BamHI sites to each end. This construct was inserted
within the centre of the rrl region of pGEM23SCf to give plasmid pCfporAcat. In chapter
3, using glycocompetent E. coli, NGRP was directed to the periplasm using the pelB
signal sequence of pET22b(+) to allow modification by the C. jejuni N-linked
glycosylation machinery (Jervis and Linton, unpublished). To produce glycosylated
NGRP in C. fetus a different approach was required. NGRP was amplified along with the
native Cj0114 signal peptide (Fig. 4.1A) using primers 1320 and 1322 (Table 2.1) which
add a C-terminal Hexa-Histag and an NdeI site to each end. This PCR product was cloned
between the porA promoter and the cat cassette within pCfporAcat to give plasmid pCfN
(Fig. 4.1B).
Successful transformation of pCfN into C. fetus was challenging to attain. Transformants
were checked by PCR which amplified both the porA promoter and the sequence of NGRP
using primers 546 and 1302 (Table 2.1) (not shown). Western blotting whole cell lysates
of positive clones demonstrated two weak anti-His(6)-reactive bands around 28 kDa and
31 kDa that were not present in wildtype C. fetus (Fig. 4.1C). Transformation of pCfN into
C. fetus pglB::kn was unsuccessful and therefore it could not be confirmed that these anti-

87
his(6)-reactive proteins were N-glycosylated versions of NGRP. Furthermore, the poor
yield of protein was not suitable for producing sufficient immunogen to raise an
antiserum. Therefore, an alternative approach was used to integrate an N-glycoprotein in
C. fetus.

88
4.2.2 Production of a C. fetus conjugative vector encoding an N-glycoprotein.
As discussed above, integrating an N-glycoprotein-encoding gene onto the chromosome of
C. fetus was challenging and unsuccessful for producing a modified N-glycoprotein in C.
fetus in this case. However, Kienesberger et al. (2007) have described a conjugative
plasmid transmission method for introducing exogenous DNA into C. fetus. This was
achieved using E. coli – C. fetus shuttle vectors which utilise the C. fetus sapA promoter to
drive expression of downstream genes (Kienesberger et al., 2007). One such vector,
pRYGG1 (Appendix Table 2), was used to express the target glycoprotein, NGRP, in C.
fetus.
Similarly to in section 4.2.1, NGRP was amplified along with the native Cj0114 signal
peptide from C. jejuni 11168H gDNA using primers 1427 and 1484 (Table 2.1). This
primer pair introduced a 5’ BamHI site and a 3’ polyhistidine (His8) tag-encoding
sequence followed by a SmaI site. The resulting 600 bp product (not shown) was digested
with BamHI/SmaI and ligated into SmaI and BamHI-digested pRYGG1 to give pG1-N
(Fig. 4.2D). Following transformation, colonies were checked by PCR using primer pair
1445 and 1484 (Fig. 4.2A) and NGRP insert sequenced using primers 1445 and 1432
(indicated in Fig. 4.2D). To allow for conjugative transfer, pG1-N was transformed into
electro-competent E.coli S17-1 λpir cells. The predicted size of NGRP with a His8-tag is
21.8 kDa. Production of an ~ 22 kDa His-tagged protein in an E.coli S17-1 λpir lysate was
confirmed by western blotting (Fig. 4.2B). These donor cells were subsequently used to
introduce pG1-N into both wildtype C. fetus fetus and the corresponding pglB::kn mutant
(Jervis et al., 2012) via conjugation and strains were verified by colony PCR using primers
1445 and 1484 (Fig. 4.2C).

89

90
4.2.3 Production of N-linked glycoprotein immunogen in C. fetus.
Whole cell lysates of C. fetus pG1-N and corresponding pglB::kn strain were subjected to
SDS-PAGE and coomassie staining to visualise NGRP production (Fig. 4.3A). E. coli
S17-1 λpir pG1-N was included as a reference. In addition, plasmid-free versions of each
strain were included as negative controls.
Consistent with Figure 4.1C, an ~ 22 kDa band was visible in the coomassie-stained cell
lysate of E. coli S17-1 λpir only when it harboured pG1-N (Fig. 4.3A). Up to four
glycoforms of NGRP (predicted ~ 23.2, 24.4, 25.6 and 26.8 kDa) were expected in C.
fetus pG1-N, but were not evident. Neither was unglycosylated NGRP (~ 22 kDa)
observed in C. fetus pglB::kn pG1-N.
To detect NGRP expression in these C. fetus strains, whole cell lysates were western
blotted and probed with anti-His6 antibodies (Fig. 4.3B). Similar to the observation in
4.2.2, an anti-His6 reactive band at around 22 kDa was observed with E. coli S17-1 λpir
pG1-N (Fig. 4.3B, lane 2). However, at least three anti-His6-reactive bands between 27
and 31 kDa in size were visible in a C. fetus pG1-N lysate (Fig. 4.3B, lane 3). Moreover,
in the N-glycosylation-deficient pglB mutant, a single anti-His6-reactive band was
observed at around 25 kDa (Fig. 4.3B, lane 4). Importantly, no anti-His6-reactive bands
were evident in C. fetus strains lacking pG1-N (Fig. 4.3B, lanes 5 and 6).
Although the presence of multiple anti-His6-reactive forms reducing to a single higher
mobility band upon pglB deletion indicates N-linked glycan modification, the mobility of
this form (Fig. 4.3B, lane 4) was less than predicted and of that observed in E. coli (Fig.
4.3B, lane 2). To ensure the observed proteins were NGRP-derivatives, the NGRP-reactive
CjNgp antiserum was also used to probe C. fetus lysates (Fig. 4.3C). CjNgp antiserum
reacts with NGRP and C. jejuni N-linked glycan but not with C. fetus N-linked glycan
(See 3.2.4). Hence, this antiserum would react with NGRP produced in C. fetus but would
not bind any C. fetus N-glycan. Each band reactive with anti-His6 antibodies was also
bound by CjNgp, suggesting these His-reactive bands are forms of NGRP. In addition, an
unexpected band reactive with CjNgp alone was observed at around 42 kDa. Therefore,
these data suggest that, although the mobility of unglycosylated NGRP was less than
observed in E. coli, NGRP was expressed and N-glycosylated in C. fetus. However, a
larger CjNgp-reactive protein was also observed. One explanation for this band is an
NGRP-derivative without an accessible His-tag. Nevertheless, as this unexplained band
was not anti-His6-reactive, it was not deemed problematic as glycosylated NGRP was to
be purified by its His-tag using Nickel affinity chromatography.

91

92
4.2.4 Purification of the N-linked glycoprotein immunogen from C. fetus.
Although some discrepancies were observed in the apparent size of NGRP produced in C.
fetus pG1-N, glycoforms of His-tagged NGRP were evident and Nickel-affinity
chromatography was employed to purify this glycosylated NGRP (gNGRP). Filtered cell
lysate was prepared from material harvested from 200 plates of C. fetus pG1-N. Lysate
was applied to a 5 ml HisTrap FF column, washed with 75 ml of 50 mM imidazole wash
buffer and eluted with 10 ml 500 mM imidazole elution buffer, with samples collected in 1
ml fractions (E1-10). Elution fractions 5 and 6 were separated by SDS-PAGE and
coomassie stained (Fig. 4.4A). Lysate, lysate flow through, wash flow through and elution
fraction 5 were separated by SDS-PAGE, western blotted and probed with anti-His6 and
CjNgp (Fig. 4.4B).
Elution fractions 5 and 6 contained bands between 27 and 31 kDa in size (Fig, 4.4A) that
were reactive with anti-His6 and CjNgp (Fig. 4.4B), confirming the presence of His-
tagged NGRP forms. These protein bands were analysed by mass spectrometry and the
results searched against C. jejuni and C. fetus protein sequences within Swiss-Prot and
TrEMBL databases. C. jejuni Cj0114 (YbgF), the native progenitor of NGRP, was the first
protein hit, confirming these proteins are NGRP. However, glycosylated NGRP had only
been partially purified, as other proteins were evident in the eluate (Fig. 4.4A). Three
distinct bands at 42, 44, and 46 kDa were CjNgp-reactive but not anti-His6 reactive (Fig.
4.4B), although they had been enriched by Nickel-affinity purification. Furthermore, the
pattern of these CjNgp-reactive bands was not identical to that observed with coomassie-
staining, as a stronger band was evident at around 46 kDa when coomassie-stained. To
identify this ~ 46 kDa protein the band was excised, subjected to mass spectrometry and
the results searched against C. jejuni and C. fetus protein sequences within Swiss-Prot and
TrEMBL databases. The protein with the most peptide matches (56) was a C. fetus
glutamate dehydrogenase (CFF8240-1557) with a theoretical size of 49.8 kDa and the
second hit was C. jejuni Cj0114 (9 peptide matches), the native protein from which NGRP
was constructed. This suggested that the significant band at ~ 46 kDa corresponded to
CFF8240-1557 but the three CjNgp-reactive proteins were derived from NGRP. As these
forms were not anti-His6-reactive, the possibility of these proteins being translational
read-through products was explored by analysing the codons downstream of NGRP in
pG1-N. However, this is unlikely as it would only give a ~ 25 kDa protein (Maximum size
being ~ 29.8 kDa if four N-glycan sites modified.).

93
Attempts to further purify gNGRP using ion exchange chromatography were unsuccessful
(not shown). An absorbance peak at the end of the salt concentration gradient
corresponded to fractions containing similar protein bands as prior to the chromatography
and thus no separation was observed. Due to poor yield and time constraints, further
rounds of purification were not undertaken. Instead, nickel affinity chromatography
elution fractions were pooled and dialysed to remove imidazole and reduce salt levels. The
preparation was concentrated to ~ 2 ml and a sample ran alongside BSA standards and
coomassie-stained (Fig. 4.4C). The total amount of NGRP glycoforms present was
estimated to be around 1 mg using densitometry within Image Studio software (LiCor
biosciences). Aliquots of the preparation were lyophilised and sent to Antibody Production
Services Ltd (Life Science Group Ltd.) for production of polyclonal rabbit antisera using
their standard 77 day immunisation protocol.

94

95
4.3 Characterisation of C. fetus NGRP antiserum (CfNgp)
reactivity with Campylobacter whole cell lysate extracts.
4.3.1 CfNgp reactivity with C. fetus fetus NCTC 10842.
As described in section 4.2, a recombinant N-glycoprotein was produced and partially
purified from C. fetus NCTC 10842 and used as an immunogen to develop a C. fetus N-
glycan-reactive polyclonal antiserum. The resulting antiserum was named CfNgp (C. fetus
N-glycoprotein). To initially identify if this antiserum contained any C. fetus N-glycan
reativity a test bleed sample (day 49) was used to probe a western blot containing whole
cell lysates of C. fetus 10842 wildtype and pglB::kn strains (Fig. 4.6C). The observed
reactivity was compared to that of the pre-immune serum (Day 0) (Fig. 4.5B).
Pre-immune serum gave numerous faint reactive bands identical between wildtype and
pglB knockout (Fig. 4.5B). Day 49 CfNgp serum displayed considerably higher reactivity
with the C. fetus cell lysates (Fig. 4.5C). When probing C. fetus wildtype lysate, CfNgp
resulted in many reactive bands. In contrast, with C. fetus pglB::kn lysate, CfNgp gave
only one strong reactive band at around 49 kDa. In summary, immunization with
glycosylated NGRP resulted in an antiserum reactive against numerous C.fetus proteins in
a pglB-dependent manner, indicating they were N-linked glycoproteins. The antiserum
also contained reactivity against a single pglB-independent band, likely the contaminating
C. fetus glutamate dehydrogenase (predicted 49.8 kDa) identified in the partially purified
NGRP preparation used for immunization. These data indicate the potential of CfNgp for
detecting C. fetus N-linked glycoproteins. In the following sections the antiserum was
used in its final form (day 77 terminal bleed) and employed at the most appropriate
dilution (1/1000), as decided by probing blots similar to Figure 4.5C with CfNgp at a
variety of dilutions (not shown).

96

97
4.3.2 CfNgp reactivity with numerous C. fetus strains.
Previous N-glycan structural characterisation work in C. fetus has identified two distinct
structures in both C. fetus fetus 82-40 and C. fetus venerealis, α-GlcNAc-6-[ β-Glc-3]-α-
GlcNAc-4-α-GalNAc-3-α,β-diNAcBac and α-GlcNAc-6-[β-GlcNAc-3]-α-GlcNAc-4-α-
GalNAc-3-α,β-diNAcBac, with the latter being most predominant (Nothaft et al., 2012).
However, only a HexNAc-[Hex]-HexNAc3-diNAcBac N-glycan species was identified in
the strain used in this thesis, C. fetus fetus 10842 (Jervis et al., 2012). To ensure CfNgp
antiserum is equally reactive with strains of C. fetus other than in which the immunogen
was produced, it was used to probe western blots of cell lysates from a variety of C. fetus
strains (Fig. 4.6B). This included C. fetus 10842 wildtype and pglB::kn, C. fetus veneralis
10354 as well as 18 C. fetus human isolates obtained from Public Health England (PHE).
PHE isolates were collected from a variety of human samples (including blood, tissue and
faeces) between 2004 and 2015 (Table 2.1). Although only two have been subtyped as C.
fetus fetus, it is likely that most or all are also this subspecies as C. fetus venerealis is very
rarely found in humans (Wagenaar et al., 2014). All samples were seperated by SDS-
PAGE and coomassie stained to confirm protein level consistency (Fig. 4.6A, isolates 9-
18 are not shown).
As in the previous section, CfNgp gave numerous immunoreactive bands with wildtype
C.ff 10842, all except one of which were pglB-dependent (Fig. 4.6B). With the C. f ven
lysate, CfNgp exhibited similar reactivity, with a notable band at less than 15 kDa. With
all the C. fetus human isolates, CfNgp gave a near identical pattern to that observed with
C. f fetus 10842 (Fig. 4.6B, isolates 9-18 not shown). Therefore, CfNgp reacts similarly
well with cell lysates of numerous C. fetus strains, encompassing both subspecies.

98

99
4.3.3 Campylobacter species specificity of CfNgp.
Previously a C. jejuni N-linked glycan-reactive antiserum was shown to specifically react
with species that produce structurally related N-linked glycan (section 3.2.4). This
antiserum does not react with species of Campylobacter such as C. fetus and C. concisus
which produce a variety of less structurally related N-glycan structures. This feature
makes the antiserum a promising reagent for creating a detection tool specific for an
important set of Campylobacter pathogens. To a similar end, the species specificity of
CfNgp was determined by probing western blots of whole cell lysates of a variety of
Campylobacter species (Fig. 4.7B).
CfNgp produced numerous immunoreactive bands with wildtype C. fetus lysate and only a
single ~ 49 kDa band with a pglB mutant (Fig. 4.7B). CfNgp also reacted with all the
remaining Campylobacter species tested, which could be divided into a higher and a lower
reactivity group. With the higher reactivity group (consisting of C. hyointestinalis, C.
concisus, C. lanienae and C. sputorum) the immunoreactivity observed was similar to that
observed with C. fetus. This is unsurprising for C. hyointestinalis as it produces identical
N-glycan structures to C. fetus (Jervis et al., 2012; Nothaft et al., 2012). However, this
level of reactivity was not expected with C. concisus, C. lanienae and C. sputorum as they
produce related but structurally distinct N-glycan structures.
The lower reactivity group (C. jejuni, C. coli, C. upsaliensis, C. helveticus and C. lari)
produced fewer immunoreactive bands with reduced intensity (Fig. 4.7B). These five
species are reactive with the C.jejuni N-glycan antiserum, CjNgp (Fig. 3.4, section 3.2.4),
and produce either the canonical C. jejuni-type N-glycan heptasaccharide or the C. lari
hexasaccharide structure lacking a branching glucose (Jervis et al., 2012; Nothaft et al.,
2012). Therefore, although CfNgp reacts with numerous N-glycoproteins produced by C.
fetus and C. hyointestinalis, it is not specific for their N-glycan structures as it also reacts
similarly with cell lysates of C. consisus, C. sputorum and C. lanienae. CfNgp also reacts
with whole cell lysates of C. jejuni, C. coli, C. upsaliensis, C. helveticus and C. lari,
however, the observed reactivity was of lower intensity. This suggests that the antiserum
contains antibodies against diverse N-glycan structures and this was investigated further
using C. jejuni pgl mutants that produce a variety of N-glycan structures.

100

101
4.3.4 C. jejuni N-linked glycan structural specificity of CfNgp.
Section 4.3.3 demonstrated the broad reactivity of CfNgp against Campylobacter species,
with a higher and a lower reactivity group identified. Species within the higher reactivity
group produce N-glycans more structurally related to C. fetus than those in the lower
reactivity group. To prove the immunoreactive bands observed with species in the lower
reactivity group were N-glycoproteins, CfNgp was used to probe a western blot of C.
jejuni wildtype and pglB::kn lysates. To further elucidate the C. jejuni N-glycan structural
specificity of CfNgp, lysates of C. jejuni pgl mutants producing truncated N-glycans were
also tested (Fig. 4.8B). C. fetus fetus wildtype and pglB::kn lysates were included as a
positive and negative control, respectively.
As seen previously, CfNgp reacted with a number of proteins in a C. jejuni cell lysate (Fig.
4.8B). Upon deletion of pglB, only one reactive band remained, with the rest being pglB-
dependent and hence a result of N-glycan reactivity. This remaining reactive protein (~ 35
kDa) is likely Cj0114, the protein from which NGRP was constructed, as glycosylated
NGRP was used as immunogen and CfNgp undoubtedly contains antibodies against the
protein. When the branching glucose residue was absent from the C. jejuni N-glycan (pglI
mutant), CfNgp produced a similar number of reactive bands. Interestingly, in a few cases
this truncation appeared to increase the intensity of the reactivity. Upon further loss of the
three terminal GalNAc residues (pglH mutant), CfNgp was reactive with fewer proteins.
Further truncation in pglJ and pglA mutants resulted in only a single reactive protein, at
around 37 kDa and 36 kDa, respectively. These remaining reactive bands correspond to
Cj0114 harbouring the respective truncated N-glycan structures, hence antibodies within
CfNgp are likely reactive against the protein and not the truncated N-glycan structures.
In conclusion, the majority of CfNgp reactivity with a C. jejuni cell lysate is pglB-
dependent, with the exception of the reactivity against Cj0114. Furthermore, CfNgp C.
jejuni N-glycan reactivity is independent of the branching glucose, the presence of which
appears to reduce the level of reactivity in some cases. Instead, the majority of the CfNgp
C. jejuni N-glycan reactivity is directed towards the four terminal GalNAc residues.

102

103
4.4 CfNgp binding to Campylobacter cells.
4.4.1 CfNgp binding untreated cells of C. fetus.
Bacterial detection tools often require pre-treatment of cells, for example, a cell lysis or
DNA extraction step. However, if a reagent can effectively bind untreated cells the
preparation time can be reduced, allowing faster detection. Therefore it would be useful if
CfNgp could react with untreated cells of C. fetus. To test this, CfNgp was used to probe
dot blots containing untreated cells of C. fetus fetus and C. fetus venerealis (Fig. 4.9A).
Polyclonal serum raised against cells of C. jejuni 11168H, anti-WC 11168, was also used
for comparison as it is known to react with C. fetus cells (See section 3.3.4) (Fig. 4.9B).
Similar blots performed without primary antibody or probed with pre-immune serum gave
no reactivity (not shown). Dot blots were performed similarly to the protocol described by
Qian et al. (2008), as explained in section 3.3.1.
In this dot blot assay, CfNgp could detect 103
or more cells of C. fetus fetus (Fig. 4.9A).
CfNgp had a lower affinity for cells of C. fetus venerealis, requiring a least 104
cells for
detection. In contrast, anti-WC 11168 was only able to detect as few as 10
5 cells of either
subspecies (Fig. 4.9B). Therefore, CfNgp can bind untreated cells of both C. fetus fetus
and C. fetus venerealis.

104
4.4.2 The influence of pglB on CfNgp C. fetus cell binding.
Section 4.4.1. demonstrated that CfNgp is able to bind untreated cells of C. fetus. To
investigate if this immunoreactivity is N-glycoprotein-dependent, the antiserum was used
to probe dot blots containing untreated cells of C. fetus wildtype and pglB::kn (Fig.
4.10B). The respective pre-immune serum was also tested (Fig. 4.10A). As discussed in
3.3.1., LLO is found within Campylobacter pglB mutants and hence may be bound by
anti-N-glycan antibodies if presented in a dot blot assay. In 3.3.1, this issue was taken into
account and a C. jejuni pglA mutant also included in the experiment. However, it should
be noted that a C. fetus pglA mutant was not available here. Hence, this experiment served
only to assess if cell binding was due to N-glycoprotein reactivity, not overall N-glycan
reactivity.
Due to the immunogen being only partially purified, there is some reactivity against C.
fetus proteins within the antiserum, as evidenced by mild reactivity with a pglB::kn lysate
upon western blotting (see section 4.3.1). Therefore, the influence of such reactivity on the
ability of CfNgp to bind cells was also investigated. To achieve this, some of this protein
reactivity was adsorbed from the antiserum by pre-incubating CfNgp with a membrane
containing C. fetus pglB::kn cell lysates before use in a dot blot (Fig. 4.10D). To account
for potential loss of reactivity due to non-specific binding to a membrane, CfNgp was also
pre-incubated with an empty membrane prior to dot blotting (Fig. 4.10C).
When probed with the rabbit pre-immune serum, no cells were detected (Fig. 4.10A).
Upon probing with CfNgp, as little as 103
cells of C fetus wt and pglB::kn were detectable,
with no difference in reactivity between each strain (Fig. 4.10B). Upon pre-incubation
with a blank membrane, the binding ability of CfNgp was unchanged (Fig. 4.10C).
However upon pre-incubation with a membrane containing C. fetus pglB::kn lysates, the
cell binding ability of CfNgp was reduced (Fig. 4.10D). Instead, the antiserum only gave a
significant signal when at least 104
cells of either strain were present. Furthermore,
although the limit of detection did not differ, this treated CfNgp appeared to bind wildtype
cells with a slightly stronger affinity than cells of the pglB mutant.
In conclusion, absorption of some C. fetus protein cross-reactivity reduced the ability of
CfNgp to bind untreated C. fetus cells. This suggests that unglycosylated protein reactivity
within CfNgp influences its ability to detect cells of C. fetus. Furthermore, although a
marginally higher affinity for wildtype cells indicated some cell binding was due to N-
glycoprotein-reactivity, it appeared the majority of cell binding activity within CfNgp was
not influenced by PglB activity. However, in contrast to a western blot, where the LLO

105
(likely abundant in a pglB::kn mutant) is not present, the antiserum may have had access
to this component in the untreated cells displayed by this dot blot method. Therefore, it is
likely that some proportion of the binding observed with C. fetus pglB::kn is due to the
availability of LLO and not C. fetus proteins. Nevertheless, these data suggest that both N-
linked glycan reactivity and protein reactivity contribute to the ability of CfNgp to bind C.
fetus cells.

106
4.4.3 CfNgp binding to untreated cells of Campylobacter species.
Previous experiments found CfNgp to be reactive against cell lysates of several
Campylobacter species, even those which produce N-glycan structures different from the
C. fetus N-glycan. This reactivity could be divided into two groups, one of high reactivity
and one of low reactivity. The high reactivity group contained C. fetus, C. hyointestinalis,
C. consisus, and C. lanienae. The low reactivity group consisted of C. jejuni, C. coli, C.
upsaliensis, C. helveticus and C. lari. Section 4.4.1. showed that CfNgp can bind to
untreated cells of C. fetus, which belongs to the high reactivity group. Here it was
investigated if CfNgp is able to bind untreated cells of species from the low reactivity
group in a similar dot blot assay. Cells of Campylobacter species from this ‘low reactivity’
group (excluding C. helveticus) were spotted onto nitrocellulose and probed with CfNgp
(Fig. 4.11A). The cell number spotted ranged from 5 x 103
to 104
cells, as this includes the
lower range at which CfNgp was previously observed to detect C. fetus cells. Again, anti-
WC 11168 was used for comparison purposes (Fig. 4.11B).
As expected, CfNgp detected between 5 x 103
and 104
cells of C. fetus fetus (Fig. 4.11A).
CfNgp was also able to detect C. jejuni, C. coli and C. lari at these cell numbers.
However, the observed signal was at a lower intensity. Furthermore, C. upsaliensis was
very weakly detected by CfNgp at these cell numbers. In contrast, anti-WC 11168 was
unable to detect C. fetus at any cell number used here (Fig. 4.11B). The remaining four
species were detectable by anti-WC 11168 at all cell numbers, with C. jejuni giving the
strongest signal. In conclusion, although CfNgp is able to easily detect between 5 x 103
and 104
C. fetus cells, the antiserum can also detect species from the ‘low reactivity’ group
within this cell number range. However, the reaction intensity is discriminatory between
C. fetus and those species from the ‘low reactivity’ group, as observed when analysing the
antiserum using western blotting.

107

108
4.5 Discussion
Rapid antibody-based tools such as lateral flow devices would be advantageous for
C. fetus detection in cattle and possibly identification from human samples also. C. fetus
N-linked glycan has the potential to be a useful target for detecting this species. Indeed,
antisera raised against BSA conjugates of each of the two C. fetus fOS structures can label
C. fetus fetus cells using fluorescence microscopy (Nothaft et al., 2012). This study aimed
to produce a C. fetus N-glycan-reactive antiserum and characterise its potential to be used
for C. fetus detection purposes. A recombinant glycoprotein (NGRP) was expressed and
glycosylated in the native C. fetus and purified. This purified N-glycoprotein was used to
raise polyclonal antiserum termed CfNgp. The Campylobacter reactivity of CfNgp was
assessed including its C. fetus strain coverage, Campylobacter species specificity, and
ability to bind untreated cells of Campylobacter.
This study described the first documented integration of a recombinant N-glycoprotein
into C. fetus. A His-tagged N-glycoprotein, NGRP, was first introduced using a newly
developed chromosomal integration vector which utilised a strong C. jejuni promoter.
However, this approach proved to be unsuitable for producing N-glycoprotein immunogen
for two reasons. Firstly, it was difficult to successfully gain C. fetus transformants and
therefore was achieved in wildtype C. fetus but not the N-glycosylation-negative control
strain C. fetus pglB::kn. Secondly, the wildtype C. fetus transformants did not produce
sufficient protein yield as anti-His(6)-reactive bands were barely visible when western
blotting whole cell lysates. In agreement with this, Kienesberger et al. (2007) have shown
that C. fetus require endogenous promoter elements to drive significant gene expression
and thus developed suitable conjugative vectors for this species. Therefore His-tagged
NGRP was integrated into one such vector and successfully conjugated into C. fetus
wildtype and pglB::kn strains. This approach resulted in the successful production of
readily-detectable forms of His-tagged NGRP in C. fetus.
Interestingly, the apparent size of NGRP was larger in C. fetus compared to what was
expected and that observed in the E. coli donor strain. As the conjugated plasmids were
not re-isolated from C. fetus and sequenced it is possible that a mutation in the NGRP
sequence created a larger sized protein. Alternatively, it could have been due to additional
post-translational modification in C. fetus, but this was beyond the scope of this study.
Nevertheless, NGRP appeared to be glycosylated by PglB at several sites, as expected.
Although detectable by western blotting, glycoprotein expression was not as high as
anticipated. Hence, purifying sufficient quantities of glycoprotein immunogen was

109
relatively laborious. NGRP was successfully extracted from C. fetus cell lysates using
Nickel affinity chromatography; however, several additional proteins were evident in the
partially purified preparation. Three proteins at around 42, 44, and 46 kDa in size were
reactive with CjNgp antiserum, indicating they could be NGRP-derivatives. A similar
phenomenon of a CjNgp-reactive band at around 40 kDa has been observed when
expressing NGRP using a pET22(+)b vector in E. coli BL21 (not shown). The closest
characterised homologue of Cj0114, the native protein from which NGRP was
constructed, is E. coli YbgF (21% identity). YbgF contains an N-terminal coiled coil
domain (Krachler et al., 2010). As NGRP includes the N-terminal long α-helical region of
Cj0114, it could be speculated that these observed bands may relate to NGRP forming a
dimeric coiled-coil. Indeed, bioinformatics analysis on NGRP using COILS/PCOILS tool
gives a probability of 1 for a predicted coiled coil at its N-terminus. Therefore, these lower
mobility forms could have been dimers of NGRP glycoforms. However, there was an
abundance of protein at around 46 kDa when coomassie staining in comparison to western
blotting, suggesting that another CjNgp-unreactive protein was present. This was
identified as CFF8240-1557, a C. fetus predicted glutamate dehydrogenase, using mass
spectrometry. This protein is not rich in Histidine residues (1.98%), but a SWISS-MODEL
prediction suggests the protein forms a homo-hexamer with each subunit having 3 His
residues in a surface exposed alpha helix. This may explain why the protein was extracted
using Nickel affinity chromatography. A brief attempt to further purify gNGRP using ion-
exchange chromatography was unsuccessful and, due to time constraints, a partially
purified NGRP preparation was used as antigen for raising rabbit polyclonal antiserum,
termed CfNgp.
Initial characterisation of CfNgp at day 49 found it reacted with numerous N-
glycoproteins in C. fetus 10842, the strain from which the antigen was derived. In C. fetus
pglB::kn CfNgp reacted with a few proteins, in particular a strong reaction was observed
with an ~ 49 kDa protein. This is likely the glutamate dehydrogenase which contaminated
the NGRP purification preparation. Upon completion of the antiserum and when CfNgp
was used at its most appropriate dilution, reactivity against this glutamate dehydrogenase
remained. However, other proteins within C. fetus pglB::kn no longer gave a significant
reaction with the antiserum, suggesting that the majority of reactivity was now against C.
fetus N-glycoproteins. Therefore, CfNgp reacted with numerous C. fetus fetus N-
glycoproteins and also reacted similarly well with N-glycoproteins of the causative agent
of BVC, C. fetus venerealis.

110
The only other reported sera reactive against C. fetus N-glycans are GRPII-1 and GRPII-2
developed by Nothaft et al. (2012). These polyclonal sera were raised against free
oligosaccharide (fOS)-BSA conjugates of each distinct fOS structure extracted from C.
fetus fetus 82-40. GRPII-1 was raised against HexNAc-[Hex]-HexNAc3-diNAcBac-BSA
(type 1) and GRPII-2 against HexNAc-[HexNAc]-HexNAc3-diNAcBac-BSA (type 2)
(Nothaft et al., 2012). In similarity with CfNgp, GRPII-1 reacts equally with cell lysates of
C. fetus fetus and C.fetus venerealis. In contrast, GRPII-2 is more reactive with a C. fetus
fetus cell lysate. Nothaft et al. (2012) reported that type 2 glycopeptides are around 3.5
times more abundant than type 1 glycopeptides in C. fetus fetus 82-40. However, Jervis et
al. (2012) only identified the type 1 glycan structure when analysing C. fetus fetus 10842
N-glycosylation in vitro. It may be that, in contrast, this strain produces more type 1 N-
glycan than type 2 N-glycan. This could explain why the reactivity pattern of CfNgp bears
more similarity to GRPII-1 than GRPII-2, if NGRP was modified with more type 1 than
type 2 N-glycan. Alternatively, as the exact enzymes responsible for creating these
strucures are unknown and the complete genome sequence of this strain is not available, it
is possible that C. fetus 10842 may only produce the type 1 N-glycan.
Furthermore, CfNgp reacted similarly with 18 C. fetus clinical isolates as it did with C.
fetus fetus 10842. These data suggest that C. fetus strains recently infecting humans have
an unchanged N-glycan structure and similar N-glycoproteome size in comparison to type
strains isolated decades ago and since maintained in laboratories. This finding is
encouraging in regard to the potential for using C. fetus N-glycans as detection targets.
The Campylobacter species specificity of CfNgp was investigated and, as expected based
on their identical reported glycan structures (Jervis et al., 2012; Nothaft et al., 2012),
CfNgp reacted with C. hyointestinalis hyointestinalis to a similar extent as with C. fetus.
This is in agreement with the behaviour of GRPII-1, but not GRPII-2 as it reacts
minimally with C. hyo hyo (as with C. f ven) (Nothaft et al., 2012). Further species tested
all produce N-glycan structures distinct from that of C. fetus, however, CfNgp reacted
similarly well with C. concisus, C. lanienae and C. sputorum sputorum also. As this
reactivity was not proven to be due to N-glycan reactivity, it cannot be ruled out that
CfNgp harbours some reactivity against native proteins of these species, possibly due to
homology with C. fetus proteins which contaminated the antigen preparation. However,
the broad extent and pattern of reactivity suggests the majority is N-glycoprotein
reactivity.

111
The observations made here are somewhat similar to that of Nothaft et al. (2012) who
reported that their GRPII sera reacted weakly with C. lanienae and C. sputorum. Notably
GRPII-2 demonstrated considerably less reactivity with C. lanienae and C. sputorum
sputorum than GRPII-1. However, with CfNgp we observed equivalent reactivity with C.
fetus and these species. C. sputorum sputorum produces a heptasaccharide structure which
is similar to the C. fetus type 2 N-glycan except that it has an additional terminal residue
which is a glucose (Jervis et al., 2012; Nothaft et al., 2012). Furthermore, an identical N-
glycan structure was found in C. lanienae. Nothaft et al. (2012) proposed that the
branching GlcNAc of these species is similar configured as the GlcNAc at the
nonreducing end of the C. fetus N-glycan and that this causes the weak reactivity of GRPII
sera with these species. The apparent higher reactivity CfNgp has for these species in
comparison to GRPII sera may be due to antibodies with such reactivity being more
dominant in CfNgp serum. Another explanation may be that CfNgp bears reactivity
against the reducing end of the N-glycan, which is identical in these Campylobacter
species. In addition, these epitopes could encompass the amino acid sequon of N-
glycoproteins, explaining why this observation is more pronounced with CfNgp than
GRPII-1, as GRPII-1 was raised against a fOS-BSA conjugate and not a traditional N-
linked glycoprotein. The considerable cross-reactivity that CfNgp displays against C.
lanienae and C. sputorum could pose an issue if it was used to detect C. fetus in cattle. For
example, C. sputorum is found in cattle but does not cause disease (On et al., 1998).
Therefore this cross-reactivity could cause an inappropriate positive result.
In contrast to the considerable reactivity CfNgp displayed with C. concisus 13826, GRPII
sera do not react with this strain (Nothaft et al., 2012). Interestingly, N-glycan structural
data produced by Nothaft et al. (2012) regarding C. concisus 13826 conflicts findings from
this laboratory (Frost & Linton, unpublished). The striking difference observed between
the C. concisus reactivity of GRPII sera and CfNgp here may be due to differences evident
between the version of the strain used by Nothaft et al. (2012) and what was utilised in this
study. Alternatively, C. concisus 13826 may produce more than one N-glycan structure, a
phenomenon known in C. hominis and C. fetus (Nothaft et al., 2012). However, to result in
this conflicting reactivity between sera, the expression of these N-glycans would need to
vary in some manner. Lastly, the theory discussed above regarding reactivity towards the
reducing end of the N-glycan structure could also explain some of the reactivity of CfNgp
against the C. concisus N-glycan. This is unlikely to be the sole factor because if this was
true, CfNgp would presumably be equally reactive with Campylobacter species such as C.
jejuni as they produce N-glycans which are identical at the reducing end. However, this is

112
not the case (see below). Nevertheless, as C. ureolyticus produces an identical N-glycan to
C. concisus (Nothaft et al., 2012), this suggests that CfNgp may also react with this
emerging Campylobacter species.
Furthermore, CfNgp displayed weak reactivity with Campylobacter species which
produce the C. jejuni N-glycan structure (C. jejuni/coli/upsaliensis/helveticus) and also C.
lari. In contrast, the GRP-II sera harbour no reactivity for these species (Nothaft et al.,
2012). Again, this could be due to CfNgp being reactive with the identical reducing end
portion of these N-glycan structures. Further elucidation of the specific nature of this
reactivity was achieved by analysing C. jejuni pgl mutants. This experiment demonstrated
that CfNgp does not react with the branching glucose residue; in fact its absence appeared
to increase reactivity with some proteins. Instead, CfNgp reactivity with C. jejuni
glycoproteins depended on the presence of the four terminal GalNAc residues. At least
two GalNAc residues appeared to be required for reactivity with C. jejuni glycoproteins to
be evident. These data appear to conflict the theory that CfNgp could harbour reactivity
against residues at the reducing end of the N-glycan. However, Kelly et al. (2006) reported
that no N-glycan is detectable via NMR and lectin blotting in C. jejuni pglHJA knockout
mutants. This suggests that the reduction in reactivity seen with these mutants could be
influenced by N-glycoprotein depletion and does not prove that the serum is unreactive
with the reducing end. Together, these data suggest that CfNgp contains antibodies
reactive against several N-glycan structural epitopes which in turn confers broad reactivity
against Campylobacter species.
The dot method was used as a preliminary investigation into the ability of CfNgp to bind
untreated cells of Campylobacter. This assay demonstrated that CfNgp was able to detect
as few as 103 cells of C. fetus fetus and 10
4 of C. fetus ven. Previously Nothaft et al. (2012)
reported GRPII sera were able to label C. fetus cells using fluorescence microscopy.
However, no difference in the ability to label each C. fetus subspecies was obvious in their
study. The variation in the limit of detection between each subspecies seen here may be
apparent due to a dot blot assay allowing for more sensitive assessment of cell binding
ability. The variation itself may be explained by differing amounts of surface accessible
N-glycans between each subspecies. In addition, some C. fetus fetus-specific antibodies
may exist within CfNgp due to C. fetus fetus proteins contaminating the antigen
preparation. Although minimal reactivity is seen with a C. fetus pglB::kn cell lysate,
components of the cell which are not available when probing western blots cannot be
accounted for (e.g. LLO/insoluble proteins) and may be exposed during dot blotting.
Furthermore, surface accessible proteins that are mildly reactive during western blotting

113
conditions may impact cell surface binding more significantly than expected due to correct
folding providing additional conformational epitopes.
Although Nothaft et al. (2012) reported cell labelling of C. fetus with GRPII sera, the
authors did not verify that this result was solely due to N-glycoprotein reactivity. Here,
CfNgp was able to detect cells of C. fetus pglB::kn to the same extent as the wildtype
strain. However, if some C. fetus protein-reactive antibodies were adsorbed from the
serum its cell detection limit increased to 104
cells and the displayed reactivity was of a
higher intensity with the wildtype strain. This suggests that protein reactivity within the
antiserum is responsible for some portion of the cell binding observed. However, this
study did not ensure that all non-glycan reactivity within the antiserum was removed so it
does not elucidate to what extent the influence is. Nevertheless, as removing some of this
reactivity allowed for a difference in the ability to bind the wildtype and pglB::kn strain to
be visible, it proves that the N-glycan reactivity does contribute to the ability to bind
untreated cells. Notably, C. fetus pglB::kn contains N-linked glycan precursor, LLO,
which could result in N-glycan-reactive antibodies binding to this strain. LLO may have
been exposed if cell surface damage occurred during preparation and/or cells did not
remain intact upon membrane drying prior to the dot blot assay. Therefore, anti-N-glycan
antibodies within CfNgp may contribute to cell binding to a higher extent than is
suggested by the results reported here. Hence, further investigation into the ability of N-
glycan reactive antibodies to bind to C. fetus cells is warranted. A C. fetus pglA::kn mutant
would be a more appropriate control for investigating this, however, such a mutant is
currently unavailable.
CfNgp was also able to bind cells of C. jejuni-type N-glycan producing species and C.
lari, albeit at a lower intensity, in a dot blot assay. This is in contrast to the GRPII antisera
produced by Nothaft et al. (2012) which were unable to label C. jejuni cells under
fluorescence microscopy. Therefore it seems that the cross reactivity CfNgp displayed
with cell lysates of these Campylobacter species still persists when probing untreated
cells. These results suggest that if CfNgp were integrated into a detection test, it could
potentially give positive results when faced with the so-called ‘thermophilic’
Campylobacter species such as C. jejuni and C. lari. Although there have been cases of C.
jejuni causing abortion in cattle and sheep (Larson et al., 1992; Mannering et al., 2006),
these species do not usually reside in the reproductive tracts of ruminants. However, they
are often found in the intestinal tracts of healthy animals (Stanley and Jones, 2003) and
this could pose a slight cross-contamination risk.

114
In conclusion, a recombinant N-glycoprotein produced in C. fetus fetus was successfully
used to raise an antiserum, CfNgp, reactive with N-linked glycoproteins produced by a
variety of C. fetus fetus strains and C. fetus venerealis. In contrast to previously reported
sera reactive against C. fetus N-linked glycan, CfNgp also reacts similarly with the N-
linked glycans of other Campylobacter species, such as C. concisus. This antiserum can
also bind to untreated cells of many Campylobacter species. Although protein-reactive
antibodies contributed to this ability to bind cells, adsorbing some of this reactivity
demonstrated that N-glycan-reactive antibodies were also able to bind C. fetus cells.
Therefore, these data suggest that CfNgp could be used to develop a rapid test which can
detect several Campylobacter species, with particular emphasis on C. fetus and rarer,
‘emerging’ pathogenic Campylobacter species such as C. concisus and possibly C.
ureolyticus.

115
Chapter 5. Towards engineering the
C. fetus N-linked glycan using
glycocompetent E. coli containing a
hybrid C. jejuni-C. fetus system

116
5.1 Introduction
Chapter 4 described the development of an antiserum reactive against N-glycans of several
Campylobacter species including C. fetus and C. concisus. Such an antiserum could be
used in an immunoassay for detecting a variety of Campylobacter species. However,
production and purification of the N-linked glycoprotein immunogen from C. fetus was
time consuming and laborious with a low yield. This method would not be suitable for
larger scale production of C. fetus glycoprotein if further antisera were made. In Chapter 3,
to obtain immunogen for raising antiserum CjNgp, high-yield production of C. jejuni N-
linked glycoprotein was achieved with relative ease using the glycocompetent E. coli
system developed by Wacker et al. (2002). The development of an analogous C. fetus N-
linked glycosylation system functioning in E. coli would similarly be useful for larger
scale production of C. fetus N-glycoprotein immunogen for production of CfNgp antisera.
It would also be useful for further research into the C. fetus N-linked glycosylation process
itself.
5.1.1 Comparison of the C. fetus and C. jejuni pgl loci
To reconstitute the C. fetus N-glycosylation system in E. coli, the appropriate genes need
identifying. Although the C. fetus pgl locus has not been fully characterised, the function
of some genes are inferred from homology with the C. jejuni pgl locus (Fig. 5.1A).
Similarities between the C. jejuni and C. fetus pgl loci reflect the similarities in their N-
glycan structures, with identical sugar residues (DiNAcBacGalNAc2) at the reducing end
(Fig, 5.1B) (Jervis et al., 2012; Nothaft et al., 2012). Both species contain pglK and pglB,
the former encoding the flippase mediating the transit of the LLO precursor across the
inner membrane (Kelly et al., 2006) and the latter the oligosaccharyltransferase which
transfers the assembled N-glycan onto target proteins (Wacker et al., 2002). The C. fetus
pgl locus contains homologues of C. jejuni genes encoding sugar biosynthesis enzymes
responsible for production of UDP-DiNAcBac (pglDEF) (Olivier et al., 2006) and UDP-
GalNAc (gne also known as galE) (Bernatchez et al., 2005), sugar-nucleotide donors used
in the Pgl systems of both species. C. fetus also contains orthologues of C. jejuni genes
encoding the glycosyltransferases, pglCAJ, which mediate the transfer of DiNAcBac and
the two subsequent GalNAc residues (Fig, 5.1B) (Glover et al., 2006, 2005; Linton et al.,
2005) found in each N-glycan structure.

117
The non-reducing end of the C. jejuni and C. fetus N-glycans vary in structure and hence
their pgl loci begin to differ with regards to the genes responsible for constructing this
portion. C. jejuni pglH encodes the glycosyltransferase which acts after PglJ, adding a
further three GalNAc residues sequentially (Troutman and Imperiali, 2009). However, the
C. fetus N-glycan does not contain these additional GalNAc residues, but rather two
GlcNAc residues (Nothaft et al., 2012). The C. fetus pgl locus harbours two homologues
of C. jejuni pglH, namely cf1385 and cf1386, and referred to as pglH2 and pglH1,
respectively. The products of pglH2 and pglH1 share 38% and 39% identity, respectively,
with C. jejuni PglH and 48% identity with one another. As with C. jejuni pglH, C. fetus
pglH1 and pglH2 are members of Glycosyltransferase Family 4 of the CAZy
(carbohydrate active enzymes) database. PglH1, PglH2 and C. jejuni PglH contain a GT1
amsD-like domain (with a putative nucleotide diphosphate-binding pocket) and a
Glycosyltransferase group 1 domain, placing them within the Glycosyltransferase GTB-
type superfamily. It is likely that one or both of PglH1H2 are responsible for transfer of at
least two GlcNAc residues onto the DiNAcBacGalNAc2-UndP precursor.
In C. jejuni, the final sugar transferred to the N-glycan precursor is the branching glucose
residue added by PglI (Glover et al., 2005; Linton et al., 2005). Although C. fetus
produces N-glycan with a branching glucose residue, it does not have a pglI homologue.
Furthermore, there is no obvious candidate within the C. fetus pgl locus for the addition of
the alternative branching residue, GlcNAc. However, it is possible that either C. fetus
PglH1 or PglH2 could be responsible for transfer of this residue. Therefore, based on
homology with C. jejuni pgl genes alone, there are no genes predicted to encode the
glycosyltransferases responsible for adding these branching sugars (Glc or GlcNAc).

118
5.2 Strategy to investigate the role of C. fetus glycosyltransferases
in N-glycan assembly by integrating them into an E. coli system.
5.2.1 An uncharacterised predicted glycosyltransferase gene lies within C. fetus pgl
locus
Three uncharacterised genes, cf1389-cf1391, are situated between pglK and galE within
the C. fetus 82-40 pgl locus. Of these, cf1389 is annotated as a predicted
glycosyltransferase matching to Pfam protein family PF00535, a diverse family which
transfer sugar from UDP-Glc, UDP-GalNAc, GDP-Mannose or CDP-abequose to a
variety of substrates. The gene product is, like C. jejuni PglI, characterised as a
Glycosyltransferase 2 family protein within the CAZy (carbohydrate active enzymes)
database. C. fetus cf1389 is not a homologue of C. jejuni pglI, and alignment using
BLASTP gives 31% identity over only 17% of PglI. However, in C. fetus the glucose
residue is linked to a GlcNAc, not a GalNAc as in C. jejuni, and hence a considerably
different enzyme may be responsible for its addition. Therefore, it is possible that cf1389
encodes a glycosyltransferase responsible for adding either of the branching sugars (Glc or
GlcNAc) to the C. fetus N-linked glycan.

119

120
5.2.2 Model of a hybrid C. jejuni/C. fetus glycosylation machinery.
As discussed, the first three residues of the C. fetus N-glycan (DiNAcBacGalNAc2) are
identical to those in C. jejuni. Therefore, using a truncated N-glycan consisting of these
residues could provide a base onto which the remaining residues of C. fetus N-glycan
could be transferred. Such a system would provide an alternative to cloning the entire pgl
locus of C. fetus into E. coli to create C. fetus N-linked glycoprotein. Furthermore,
glycosyltransferases which build the remainder of the C. fetus N-glycan could be
investigated by introducing gene candidates to the system individually and in various
combinations. Such a truncated trisaccharide can be produced in E. coli using
ppglpglH::kn (Linton et al., 2005). C. jejuni PglB does not have strict substrate specificity,
as it can transfer truncated C. jejuni N-glycan structures (Linton et al., 2005) and even
diverse O-antigen structures produced by E. coli and Pseudomonas aeruginosa onto
protein (Feldman et al., 2005). Hence it would likely be able to transfer the C. fetus N-
glycan to a target protein. Therefore, a plan to engineer the full C. fetus N-glycan in E. coli
by introducing C. fetus predicted transferases to the available C. jejuni pglH::kn system
was devised (Fig. 5.2).

121

122
5.3 Identification of initial C. fetus glycosyltransferase to act on C.
jejuni pglH::kn trisaccharide.
5.3.1 Cloning strategy
The strategy devised included using the available ppglpglH::kn plasmid which, when
expressed in E. coli BL21 with a target protein, results in modification with a truncated
trisaccharide version of the C. jejuni N-linked glycan structure (Linton et al., 2005).
Previously this laboratory successfully used a C. jejuni glycoprotein, Cj0114, as a
glycosylation reporter protein in H. pullorum (Jervis et al., 2010). Since then, a truncated
version, termed NGRP (N-glycosylation reporter protein) (See 3.1.1), was constructed
(Jervis & Linton, unpublished). His-tagged NGRP was introduced using pETNGRP (see
Appendix Table 2) and used as a target protein for N-glycosylation.
To avoid requiring a third plasmid, each C. fetus glycosyltransferase gene candidate was
introduced onto the backbone of pETNGRP. A BglII restriction site upstream of the T7
promoter (Fig. 5.3A) was used for insertion of each gene of interest to avoid the predicted
glycosyltransferases being over-expressed upon IPTG induction. The ability to combine
BglII-digested DNA with compatible ends created by BamHI and BclI was also utilised.
The open reading frame of each gene and 80-100 bp upstream of the start codon was
cloned to incorporate the ribosomal start site and any potential native promoter elements.
Notably, the complete genome sequence of the C. fetus strain used here, NCTC 10842,
was not available hence the genome of C. fetus 82-40 was used as a template. To identify
if any of the C. fetus predicted glycosyltransferases could transfer sugar onto the C. jejuni
pglH::kn N-glycan, each gene was individually introduced into pETNGRP. To provide a
positive control, C. jejuni PglH activity was complemented in this system by re-
introducing a functional copy of pglH at the BglII site of pETNGRP. Construction of these
vectors is described in the following sections.
5.3.1.1 Cloning pglH1 into pETNGRP
pglH1 was amplified from C. fetus fetus 10842 genomic DNA (gDNA) using primers
1378 and 1386 (Table 2.1), introducing a 5’ BamHI and a 3’ BglII site. Agarose gel
electrophoresis confirmed production of the predicted 1.2 kb product (Fig. 5.3B). This was
digested with BamHI and BglII and ligated to BglII-digested pETNGRP. Following
transformation, ligation of pglH1 was demonstrated by colony PCR (cPCR) using primers
82 and 1378 (indicated in Fig. 5.3C) to amplify pglH1 along with ngrp (results not

123
shown). Purified plasmids from positive clones were subjected to further PCR to validate
the size of pglH1 using primers 1388 and 1411 (Fig. 5.3C). PCR products around 1.2 kb in
size were produced from each plasmid, as expected (Fig. 5.3D). Plasmids were sequenced
using primers 1411 and 1425. This revealed two basepair mismatches, -51G>T and
(769C>T) in each of the three plasmids. The former did not appear to interrupt any
obvious promoter elements and the latter was a silent mutation. As these mismatches were
identical in all three clones and did not appear likely to interrupt gene expression or
function they were assumed to be single nucleotide polymorphisms (SNPs) in the C. fetus
strain being used. Furthermore, the sequence of NGRP was confirmed as correct via
sequencing using a universal T7 promoter primer (Table 2.1, number 80). This plasmid
was designated pNH1.

124

125
5.3.1.2 Cloning pglH2 into pETNGRP
pglH2 was amplified from C. fetus fetus 10842 gDNA using primer pair 1379/1399 (Table
2.1), which introduced a BclI site at each end (Fig. 5.4A). This product was digested with
BclI and ligated to BglII-digested pETNGRP. Following transformation, colonies were
screened by PCR using primers 1389 and 1411 (Fig. 5.4B). Colonies 5 and 8 gave PCR
products around 1.2 kb in size, as expected (Fig. 5.4C). Plasmids extracted from these
clones were sequenced with primers 1411 and 1425. Each plasmid had a 655A>C
mutation within pglH2, translating to a K219Q substitution in the protein product.
Repeating the cloning process created another plasmid (clone 15) with this mutation.
Hence it was presumed a C. fetus fetus 10842 SNP. A further PCR check was included
using primers 82 and 1411, in order to also cover the NGRP region of each plasmid (Fig.
5.4B). A predicted product of 1.97 kb was expected from this reaction. Although each
clone produced a PCR product ~ 2 kb in size, clones 5 and 8 each produced an additional
unexpected product, either larger or smaller in size, respectively (Fig. 5.4D). Therefore,
plasmid extracted from clone 15 was chosen and termed pNH2.

126

127
5.3.1.3 Cloning Cf1389 into pETNGRP
Cf1389 was amplified from C. fetus fetus 10842 gDNA using primers 1387 and 1406
(Table 2.1), adding a BamHI site to each end of the PCR product. An ~ 1.1 kb PCR
product, as predicted, was observed (Fig. 5.5A). Following BamHI digestion, the product
was ligated with BglII-digested pETNGRP. Following transformation, positive clones
were identified by PCR using primers 1390 and 1411 (Fig. 5.5B). Clones 1 and 3 gave
PCR products at ~ 1.2 kb (Fig. 5.5C). Plasmids were purified and sequenced using primers
1411 and 1425. One base mismatch, 783A>G, was found in each clone. However, this
mutation was silent as the codon continued to encode a glycine residue (GGA>GGG). In
addition, the NGRP sequence within each plasmid was confirmed by sequencing using
primer 80. The plasmid, termed pN1389, purified from clone 1 was used in subsequent
work.
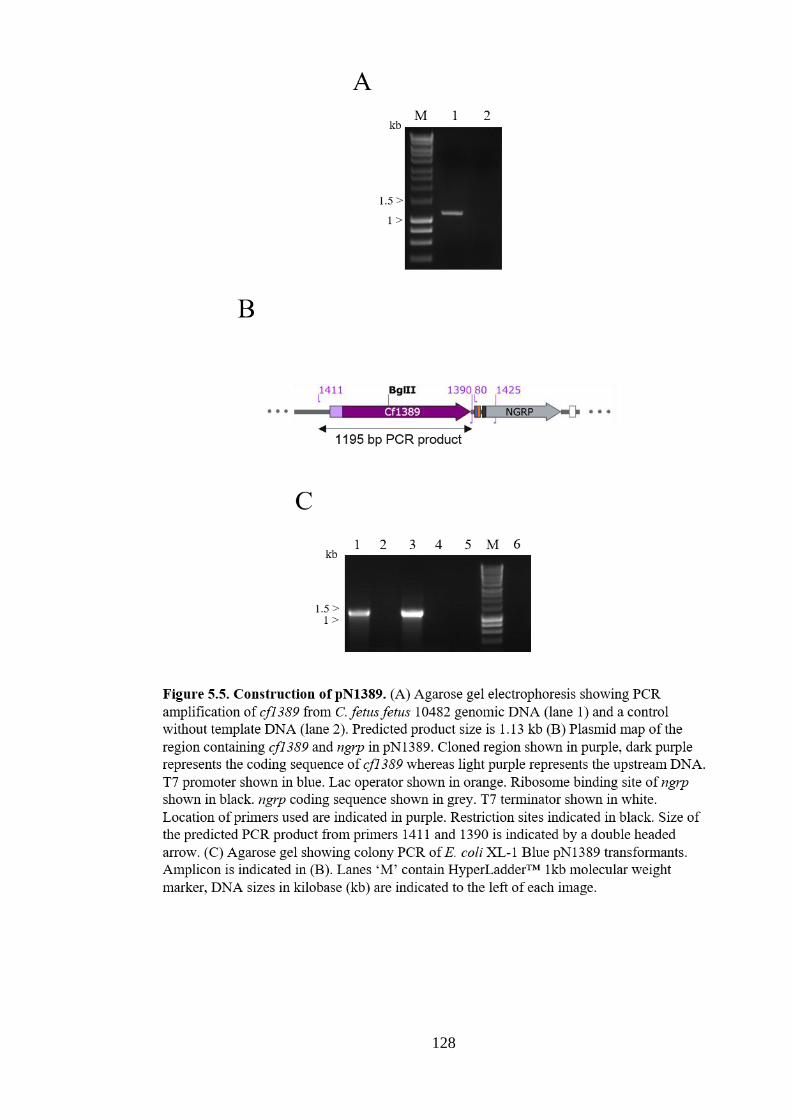
128

129
5.3.1.4 Cloning CjpglH into pETNGRP
To provide a positive control, the C. jejuni glycosyltransferase gene, pglH, was similarly
cloned into pETNGRP. This is predicted to genetically complement the pglH insertional
knockout and restore heptasaccharide N-glycan production. pglH was amplified from C.
jejuni 11168H gDNA using primer pair 1397/1398 (Table 2.1), which add a BamHI site to
each end of the PCR product. Production of an ~1.2 kb PCR product was demonstrated via
agarose gel electrophoresis (predicted product 1.24 kb)(Fig. 5.6A) and following BamHI
digestion, this product was ligated with BglII-digested pETNGRP. Transformatants were
were identified by PCR using primers 1411 and 1424 (Fig. 5.6B). PCR products of ~ 1.3
kb were confirmed from clones 3 and 7 (Fig. 5.6C). Plasmid from clone 3 was sequenced
using primers 1411 and 1425 with no mismatches found. Furthermore the plasmid was
sequenced using primer 80 to check the sequence of NGRP and this plasmid was termed
pNCjH.

130

131
5.3.2 Only CfpglH1 decreased the mobility of NGRP glycoforms in the
ppglpglH::kn background.
To investigate if C. fetus glycosyltransferases are able to transfer sugar (s) to the C. jejuni
pglH::kn truncated N-glycan structure, pNH1, pNH2 and pN1389 were individually
transformed into competent cells of E. coli BL21 (λDE3) ppglpglH::kn. For reference,
NGRP glycosylated with the full C. jejuni heptasaccharide N-linked glycan was produced
by transforming pETNGRP and ppgl into E. coli BL21 (λDE3). To create the truncated N-
glycan structure, pETNGRP was introduced into E. coli BL21 (λDE3) ppglpglH::kn. As a
positive control, pETNCjH was introduced into E. coli BL21 (λDE3) ppglpglH::kn to
complement PglH activity and reconstitute the C. jejuni heptasaccharide N-glycan
structure. Presence of the appropriate plasmids was confirmed by PCR (not shown).
NGRP production was induced by addition of 1 mM IPTG and following incubation at
25°C overnight whole cell lysates were prepared. A western blot of whole cell lysates was
probed with anti-His6 antibodies (Fig. 5.7). E. coli ppgl pETNGRP produced three
detectable forms of His-tagged NGRP. The major forms migrated at around 23 and 26
kDa, corresponding to unglycosylated NGRP (21.6 kDa predicted size) and NGRP with a
single glycosylation site modified (referred to as g1, labelled with asterisk), respectively.
The least dominant form migrated at 25 kDa and likely corresponds to unmodified NGRP
with it its signal peptide present. Upon inactivation of the C. jejuni pglH gene on ppgl, the
g1 form now migrated at around 24 kDa. This g1 form represents modification with a
truncated trisaccharide N-glycan, as C. jejuni PglH catalyses transfer of the fourth, fifth
and sixth sugar residues of the C. jejuni heptasaccharide (Troutman and Imperiali, 2009).
Introduction of C. fetus pglH2 or cf1389 along with ppglpglH::kn did not alter the
migration of g1 NGRP forms. However, adding C. fetus pglH1 into the system caused the
electrophoretic mobility of the g1 form to decrease, increasing in apparent size by ~ 1
kDa. Another His-tagged glycoform of NGRP was visible at around 27 kDa, which
correlates with two N-glycosylation site being modified (g2). Finally, upon re-
introduction of C. jejuni pglH into the system (on pNCjH), the g1 form returned to its
wildtype apparent size of around 26 kDa. Furthermore, the g2 form was also visible in this
strain.
In summary, introducing the C. fetus predicted glycosyltransferase gene, pglH1, decreased
the electrophoretic mobility of NGRP glycoforms produced by E. coli containing
ppglpglH::kn. Such a shift is consistent with addition of glycan residue (s). In contrast,
addition of genes pglH2 and cf1389 did not influence the apparent electrophoretic mobility

132
of NGRP glycoforms in this system. These data suggest that the predicted
glycosyltransferase, PglH1, is able to utilise the truncated C. jejuni pglH::kn N-glycan
structure as an acceptor substrate. This would indicate that PglH1 is a glycosyltransferase
responsible for addition of the fourth sugar residue of the C. fetus hexasaccharide
structure.

133
5.4 Identification of second C. fetus glycosyltransferase to act on C.
jejuni pglH::kn trisaccharide.
5.4.1 Cloning strategy
The reduced electrophoretic mobility of NGRP glycoforms due to C. fetus pglH1 allowed
investigation into the identity of the glycosyltransferase able to transfer sugar(s) to this
new N-glycan structure. Thus, plasmids containing pglH1 and also one of the two
glycosyltransferase genes, pglH2 and cf1389, were constructed.
5.4.1.1 Construction of pNH1H2
As described in 5.2.3, C. fetus pglH1 and pglH2 are overlapping genes. Therefore, they
were amplified together with primers 1378 and 1379 (Table 2.1), adding a 5’ BamHI and a
3’ BclI site. An ~ 2.2 kb PCR product was confirmed via agarose gel electrophoresis (Fig.
5.8A), digested with BamHI and BclI, ligated with BglII-digested pETNGRP and the
ligation electroporated into E. coli XL-1 Blue. Transformants were identified by
production of an ~ 2.3 kb cPCR product from primers 1411 and 1389 (not shown).
Plasmid was extracted from clone 11 and sequenced using primers 1411, 1425 and 1450.
Sequences were identical to those found previously when individually cloning pglH1 and
pglH2 (See 5.3.1), confirming that these are strain-specific SNPs. The plasmid was termed
pNH1H2 (Fig. 5.8B).
5.4.1.2 Construction of pNH1-1389
To create pNH1-1389, cf1389 was cloned into pNH1 at the BglII site downstream of
pglH1. Primers 1439 and 1406 (Table 2.1) were used to amplify cf1389 with a 5’ BclI and
a 3’ BamHI site. An ~ 1.1 kb PCR product was confirmed by gel electrophoresis (1.13 kb
predicted) (Fig. 5.8C) and, following digestion, was ligated into BglII-digested pNH1 and
the ligation electroporated into E. coli XL-1 Blue. Transformants were identified by the
production of an ~ 2.3 kb cPCR product from primers 1411 and 1390 (not shown).
Plasmid DNA was extracted from clones 15 and 28 and sequenced using primers 1425 and
1485. The basepair mismatch (783A>G), a silent mutation also found when cloning
cf1389 into pETNGRP, was detected, confirming a strain-specific SNP. The plasmid was
termed pNH1-1389 (Fig. 5.8D).

134

135
5.4.2 C. fetus pglH2 decreased NGRP glycoform mobility in the ppglpglH::kn
CfpglH1 background.
To investigate if either of the C. fetus predicted glycosyltransferase genes, pglH2 or
cf1389, could transfer sugar (s) to the N-glycan created by E. coli BL21 (λDE3)
ppglpglH::kn pNH1, the plasmids described in 5.4.1 were each transformed into E. coli
BL21 (λDE3) ppglpglH::kn. Clones positive for pNH1H2 and pNH1-1389 were identified
by PCR using primer pairs 1411/1450 and 1411/1390, respectively. NGRP production was
induced in each strain as previously (section 5.3.2.), and a western blot of whole cell
lysates of these induced strains along with induced E. coli BL21 (λDE3) ppglpglH::kn
containing pNH1 or pNCjH was probed with anti-His6 antibodies (Fig. 5.9).
As previously, all strains produced unglycosylated NGRP around 23 kDa (Fig. 5.9) and E.
coli BL21 (λDE3) ppglpglH::kn pNH1 produced g1 and g2 forms at ~ 25 kDa and ~27
kDa, respectively. Notably, g1 and g2 forms with pNH1H2 had reduced electrophoretic
mobility, whilst glycosylated forms produced with pNH1-1389 did not. With pNCjH,
bands corresponding to g1 and g2 forms modified with full size C. jejuni N-glycan were
evident ( ~26.5 kDa and ~ 29 kDa). Therefore, adding cf1389 into E. coli ppglpglH::kn
pNH1 system had no detectable effect on NGRP glycoforms, but pglH2 caused a
reduction in electrophoretic mobility consistent with addition of sugar residue (s). These
data suggest that the pglH2-encoded predicted glycosyltransferase is able to transfer sugar
residue (s) to the N-glycan structure following the action of PglH1. Therefore, this would
indicate that PglH2 is a glycosyltransferase which transfers the fifth sugar residue onto the
C. fetus N-glycan structure.

136

137
5.5 Structural analysis of N-linked glycoproteins produced from
hybrid systems.
5.5.1 Purification of NGRP from C. jejuni/C. fetus hybrid N-linked glycosylation
systems in E. coli
His-tagged NGRP was purified from E. coli BL21 (λDE3) containing ppglpglH::kn and
pETNGRP, pNH1 or pNH1H2 using nickel affinity chromatography. Cells were induced
with 1 mM IPTG overnight at 16 °C, sonicated and applied to a 5 ml HisTrap FF column.
Cell lysate, lysate flow-through and eluate were separated by SDS-PAGE and stained with
coomassie (Fig. 5.10). The unglycosylated and g1 forms of NGRP were apparent in the
induced cell lysates of each strain (Fig. 5.10, indicated by asterisks), and absent from the
column flow-through. Consistent with western blotting in 5.3.2 and 5.4.2, the mobility of
the g1 form decreased upon addition of C. fetus pglH1 then pglH2 into the system (pNH1
then pNH1H2). The unglycosylated and g1 forms of NGRP purified from E. coli BL21
(λDE3) ppglpglH::kn pETNGRP were not distinct bands due to their similar mobility and
the high concentration of each protein in the eluate (Fig. 5.10, left). Glycosylated forms
were apparent in eluate from E. coli containing pNH1 and pNH1H2, with g1 and g2 forms
evident at around 25 and 27 kDa with pNH1 and around 25.5 and 27.5 kDa with pNH1H2
(Fig. 5.10, pNH1 – middle, pNH1H2 – right). Therefore, NGRP was purified from the
hybrid systems created in 5.3 and the observation of glycoforms with reduced
electrophoretic mobility was further evidenced. These preparations were analysed by mass
spectrometry to structurally characterise the attached N-glycans.
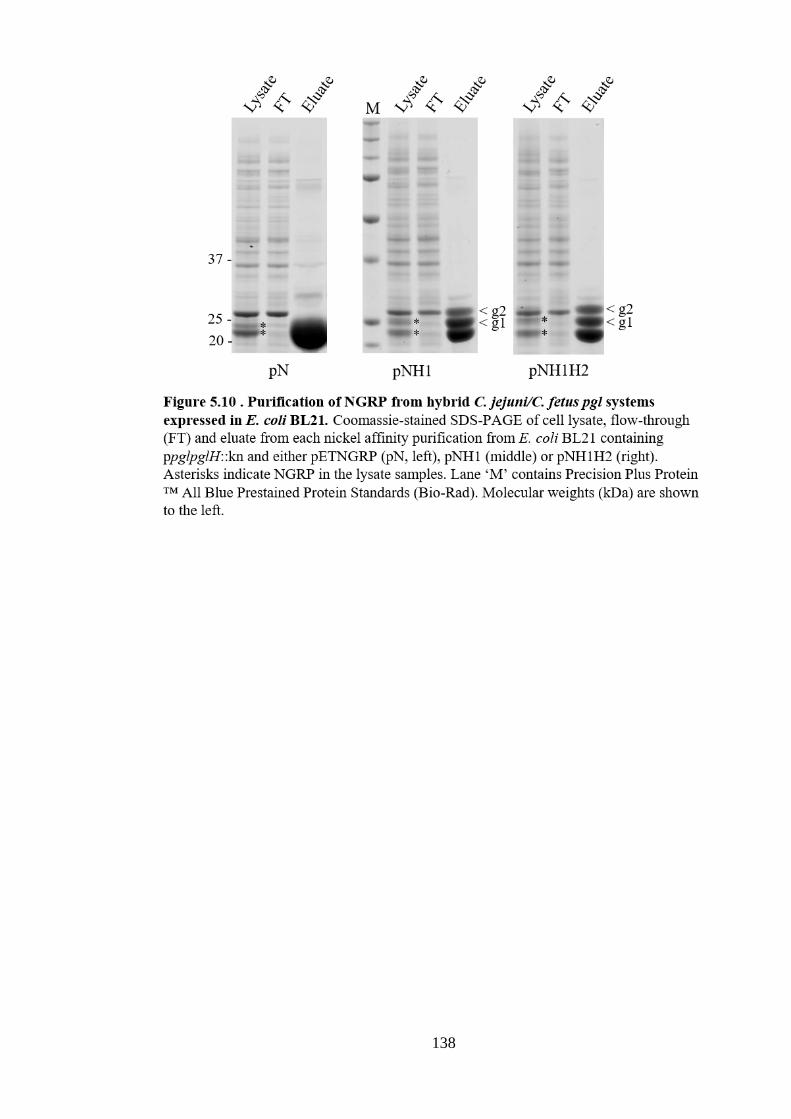
138

139
5.5.2. MALDI-TOF MS analysis of purified NGRP glycoforms
To structurally characterise the N-glycans produced by the hybrid C. jejuni/C. fetus pgl
systems, the dialysed eluates described in 5.5.1 were separated by SDS-PAGE and bands
were excised, digested with trypsin and subjected to MALDI-TOF MS using α-cyano-4-
hydroxycinnamic acid (CHCA) matrix. The g2 forms of NGRP purified from E. coli BL21
(λDE3) containing ppglpglH::kn and pNH1 or pNH1H2 were excised for analysed. As
only the g1 form of NGRP purified from E. coli BL21 (λDE3) ppglpglH::kn pETNGRP
was available, and was not fully distinguishable from the unglycosylated band, the upper
limit of this band was excised for analysis.
Each sample gave peaks corresponding to at least seven of the ten predicted tryptic
peptides of NGRP (Table 5.1 peptides 2-4, 6-9), confirming the identity of this protein. A
further peak corresponds to a predicted tryptic peptide with m/z 1885.9662 arising due to
one missed cleavage site (residues 21-36).

140
Three predicted tryptic peptides from NGRP (Table 5.1 peptides 1, 2 and 7) contain N-
glycosylation sequons, and the predicted m/z values of these peptides with possible N-
glycan structures attached are displayed in Table 5.2. The largest sequon-containing
peptide of predicted m/z 4059.8411 (Table 5.2, peptide 1) contains two sequons and, as
glycan modification can significantly reduce ionisation efficiency (Song et al., 2013),
detecting such a large glycopeptide seemed the least likely. As expected, no peaks
corresponded to this sequon-containing peptide modified with any of the predicted N-
glycan structures in the spectra. The two remaining potential glycopeptides (Table 5.2,
peptides 2 and 7) each have a single sequon and smaller predicted m/z values of 2478.1743
and 1216.6168, and hence were more likely to have higher detectability. Unfortunately, no
species with such mass to charge ratios were found in any NGRP preparation. Instead,
peaks with m/z values corresponding to these peptides without glycan were evident (Fig.
5.11, indicated with arrows). One explanation for these findings could be that the two
sequons within the largest tryptic peptide (4059.8411) were preferentially modified,
creating large glycopeptides which could not be detected (smallest predicted m/z;
5329.1587 – modified with truncated N-glycan produced by ppglpglH::kn). Therefore,
MALDI MS analysis of NGRP, with its four N-glycan sites, did not yield structural N-
glycan data. In light of this issue, further MALDI-TOF MS analysis was undertaken using
a modified form of NGRP.

141

142

143
5.5.3 Production and purification of modified NGRP in hybrid systems.
Due to difficulties in observing glycopeptide ions when analysing trypsin-digested NGRP
using MALDI-TOF MS, a different protein was integrated into the hybrid C. jejuni/C.
fetus pgl systems to allow N-glycan structural analysis. A modified version of NGRP,
termed NGRP3, is a glycosylation reporter protein created to be more amenable for mass
spectrometry (MS) analysis (Jervis & Linton, unpublished). To achieve this, three of the
four N-glycan sequons were inactivated by site directed mutagenesis and the single
remaining sequon is located in the flexible linker region that appears to be more readily
modified (Jervis et al. 2010 and section 5.5.2). In addition, residues surrounding this
remaining sequon were altered to create a smaller tryptic peptide which has a higher
positive charge, to enhance its ionisation efficiency during mass spectrometry (Fig. 5.12).
To replace NGRP in the hybrid systems developed in 5.3 and 5.4, the original NGRP
sequence was first digested out of pNH1/pNH1H2 using XhoI and NcoI. NGRP3 was
amplified from pNGRP3 using primers 790 and 1184 (Table 2.1), adding a 5’ NcoI and 3’
XhoI. Following digestion with XhoI and NcoI, the product was ligated with digested
pNH1 and pNH1H2 to give pH1-N3 and pH1H2-N3, respectively. Following
transformation, the presence of NGRP3 was identified by PCR using primers 790 and
1184. Extracted plasmids were sequenced using primer 82 to check the NGRP3 sequence.
Primers 1411 and 1425 were used to sequence the pglH1 region of pH1-N3. The pglH1-
pglH2 region of pH1H2-N3 was sequenced using primers 1388, 1389, 1411, 1425 and
1559.

144
Plasmids pNGRP3, pH1-N3 and pH1H2-N3 were transformed into E. coli BL21 (λDE3)
ppglpglH::kn. Positive clones were identified by PCR using primers 790 and 1184 for
pNGRP3 and primers 1388 and 1411 for pH1-N3 or pH1H2-N3. To produce
unglycosylated protein, pNGRP3 was also transformed into E. coli BL21 (λDE3). NGRP3
production was induced in each strain using 1 mM IPTG at 16°C overnight. Whole cell
lysates were separated by SDS-PAGE and coomassie-stained, with unglycosylated
NGRP3 present as an ~ 21 kDa protein (predicted 21.78 kDa) (Fig. 5.13A). A g1 form of
NGRP3 was evident in each strain carrying C.fetus pgl genes, at around 23 and 23.5 kDa
with pglH1 and pglH1H2, respectively.
NGRP3 was purified from lysates of each IPTG-induced strain using Nickel affinity
chromatography. Eluates were dialysed against 30 mM Tris 0.15 M NaCl pH 7.5. Bands
corresponding to unglycosylated NGRP at ~ 20 kDa were evident in each eluate when
diluted (1/10) and separated by SDS-PAGE (Fig. 5.13B). In the presence of ppglpglH::kn,
a g1 form of NGRP at around 21 kDa was also purified. Upon addition of pH1-N3 or
pH1H3-N3 the g1 forms purified were around 22 kDa or 22.5 kDa, respectively.
Therefore, forms of NGRP3 glycosylated with varying N-glycan structures similar to
those observed with the hybrid C. jejuni/C. fetus pgl systems in sections 5.3 and 5.4 were
purified. The structures of these N-glycans were analysed by mass spectrometry.

145

146
5.5.4 MS analysis of purified NGRP3.
5.5.4.1 The sequon-containing tryptic peptide of unglycosylated NGRP3 was detected by
MALDI TOF Mass spectrometry.
NGRP3-containing eluates (as described in 5.5.3) were separated by SDS-PAGE, protein
bands excised and tryptic peptides prepared for MALDI-TOF MS analysis. To confirm
that the sequon-containing tryptic peptide of NGRP3 (Table 5.3 peptide 8) could be
detected when unmodified, unglycosylated NGRP3 produced in E. coli BL21
ppglpglH::kn pH1-N3 was excised and analysed. Two matrices, CHCA and 2, 5-
dihydroxybenzoic acid (DHB), were used to assess which was most appropriate for
detection of this peptide. The spectra using each matrix are compared in Figure 5.14.
Better coverage of NGRP3 was achieved using DHB (Fig. 5.14B), with 7 peaks matching
possible tryptic peptides (Table 5.3, peptides 1-4, 6, 8 and 9) compared to 3 matches with
CHCA (peptides 2,4 and 8). A further three peaks (m/z 1885, 2718 and 3217) were
identified in each spectrum that correspond to masses of possible tryptic peptides arising
due to a missed cleavage site. The sequon-containing tryptic peptide (predicted m/z
1167.55) was identified at m/z 1167.32 and 1167.38 using CHCA and DHB, respectively.
However, the peak identified using DHB was of very low intensity (Fig 5.14B, indicated
by grey arrow). Therefore, the sequon-containing tryptic peptide from NGRP3 was readily
detectable by MALDI-TOF MS when using CHCA as the sample matrix, but did not give
a significant peak with DHB matrix.

147

148

149
5.5.4.2 MALDI-TOF MS analysis of glycosylated NGRP3 using CHCA matrix.
As CHCA allowed improved identification of the sequon-containing tryptic peptide from
unglycosylated NGRP in comparison to DHB, this matrix was chosen for analysing
NGRP3 glycoforms using MALDI-TOF mass spectrometry. NGRP3-containing eluates
were separated by SDS-PAGE and coomassie stained (as described in 5.5.3), and the g1
forms of NGRP3 excised for mass spectrometry analysis.
At least 7 peaks corresponded to predicted NGRP3 tryptic peptides in each sample
analysed (Fig. 5.15, Peptides 1-4, 6, 9 and 10 in Table 5.3), confirming the identity of
NGRP3. A peak with a m/z value corresponding to tryptic peptide 7 (Table 5.3) was
produced by NGRP3 from pH1H2-N3. In addition, peaks corresponding to the masses of
tryptic peptides arising from missed cleavages were identified in all samples (m/z 1886
from all samples and m/z 2134 from pN3 and pH1-N3). Peaks corresponding to the
sequon-containing tryptic peptide, HHTDSNSTIR (Table 5.3 peptide 8), were not
observed.
The m/z values of remaining peaks were cross referenced against the predicted m/z values
of tryptic peptide 8 (HHTDSNSTIR) modified with the expected N-glycan structures
(Table 5.4). No peaks from pN3 or pN3-H1 had m/z values similar to any of the predicted
glycopeptides. Therefore, no structural data for the attached N-glycans were obtained
using MALDI-TOF MS with CHCA matrix.

150

151
5.5.4.3 MALDI-TOF MS analysis of glycosylated NGRP3 using DHB matrix.
CHCA matrix allowed identification of the sequon-containing tryptic peptide from
unglycosylated NGRP3 (Section 5.5.4.1) but did not when this peptide was N-
glycosylated (Section 5.5.4.2). Although with DHB the peak intensity for this unmodified
peptide was low, this matrix is commonly used for glycan analysis (Morelle et al., 2006)
and Mills et al. (2016) successfully identified glycopeptides from a similar NGRP variant
using this matrix. Hence, the glycoforms of NGRP3 analysed above were reanalysed using
DHB matrix (Fig. 5.16). The mass range of the detector was selected between m/z 1700
and 2500.
In the m/z range tested, only unmodified tryptic peptides 1-5 were detectable. Peaks
corresponding to peptides 1-4 were each found from pH1-N3 and pH1H2-N3 and those
corresponding to 1,3 and 4 from pN3 (Table 5.3). Additionally, each NGRP glycoform
gave two peaks corresponding to tryptic peptides with one miscleavage (m/z 1885 and
2134). Of the remaining peaks, one at m/z 2207.906 evident in pH1H2-N3 (Fig. 5.16,
bottom panel) could correspond to the sequon-containing tryptic peptide with a
DiNAcBac-HexNAc4 modification (Table 5.4, m/z 2207.87). Unfortunately, this peak was
of low intensity and tandem MS fragmentation was unsuccessful.
In summary, MALDI-TOF mass spectrometry analysis of NGRP3 glycoforms mainly
resulted in identification of peaks corresponding to unmodified tryptic peptides. However,
using DHB matrix, an ion with at m/z 2207 was identified in glycosylated NGRP3
produced from pH1H2-N3. This species could be a glycopeptide containing a DiNAcBac-
HexNAc4 modification. This would suggest that the addition of both C. fetus pglH
homologs into a C. jejuni pglH::kn system results in the transfer of a further two HexNAc
residues onto the N-linked glycan structure. However, tandem MS data could not be
obtained from this species and hence the structure was not confirmed. To elucidate if this
species is such a glycopeptide, more sensitive mass spectrometry analysis was undertaken.
In addition, further analysis was warranted to confirm the structure of N-glycan present in
glycosylated NGRP3 expressed from pN3 and pH1-N3.

152

153
5.5.4.4 LC-MS/MS analysis of glycosylated NGRP3
To investigate the N-glycan structures created by the E. coli hybrid pgl systems described
in this chapter, purified NGRP3 preparations were analysed by LC-MS/MS. Analysis was
performed on the Q Exactive™ HF Hybrid Quadrupole-Orbitrap™ Mass Spectrometer
with Higher-energy Collisional Dissociation (HCD) fragmentation. MS/MS scan events
with precursor ions at the predicted m/z values were identified using mMass v5.5
(Niedermeyer and Strohalm, 2012) and scanned for a HexNAc oxonium ion (m/z 204.09),
which is characteristic of glycopeptide fragmentation spectra obtained using HCD (Zhao
et al., 2011). Marker ions at m/z 126, 138, 144, 168 and 186 can also be found,
representing further fragmented HexNAc residues (Zhao et al., 2011). Disaccharide
oxonium ions such as m/z 366 (HexHexNAc) can also be identified, which can help
elucidate glycan structure (Toghi Eshghi et al., 2016). Identification of these ions was used
to confirm scans corresponded to glycopeptides and are labelled on each spectrum (Fig.
5.17).
The presence of the unmodified sequon-containing tryptic peptide, 145
HHTDSNSTIR154
,
as a doubly charged species at m/z 584.21 was confirmed in NGRP3 produced in the
absence of Campylobacter pgl genes. Fragmentation of this parent ion resolved peaks
corresponding to 7 y-ions and 1 b-ion (Fig 5.17A). From glycosylated NGRP3 produced
in the presence of ppglpglH::kn, a precursor ion with m/z 901.41 corresponding to
145HHTDSNSTIR
154 with DiNAcBac-HexNAc2 in a doubly charged state was confirmed.
Fragmentation resolved peaks representing the peptide with partial N-glycans attached,
including DiNAcBac-HexNAc and DiNAcBac (Fig 5.17B).

154

155
A precursor ion of m/z 722.99 was identified from glycosylated NGRP3 produced in the
presence of ppglpglH::kn and pH1-N3 (5.18A). This m/z corresponds to
145HHTDSNSTIR
154 with DiNAcBac-HexNAc3-Hex in a triply charged state. This
confirms previous data suggesting that PglH1 modifies the trisaccharide produced by the
C. jejuni pglH::kn locus and identifies the transferred residues as a HexNAc and a Hex.
Fragmentation yielded ions with m/z values 1395 and 1598 corresponding to the peptide
with truncated glycan structures of DiNAcBac and DiNAcBac-HexNAc, respectively (Fig
5.18A). Further ions corresponded to y-ions with DiNAcBac, DiNAcBac-HexNAc or
DiNAcBac-HexNAc2 modification. Although no ions containing DiNAcBacHexNAc3
were identified, the marker ion of m/z 366.14 (HexNAcHex) was detected. Therefore, the
lack of a species containing DiNAcBacHexNAc3 is likely due to the first fragmentation
event removing the terminal HexNAc and Hex residues in conjunction.
Analysis of glycosylated NGRP3 produced in the presence of ppglpglH::kn and pH1H2-
N3 also yielded a precursor ion of m/z 722.99 (5.18B, upper spectrum). Fragmentation of
this ion yielded a peak, m/z 1778, corresponding to the m/z of a b7 ion with a DiNAcBac-
HexNAc3-Hex modification. A peak at an m/z of a b2 peptide ion was also evident.
However, another precursor ion of m/z 736.66 representing 145
HHTDSNSTIR154
with
DiNAcBac-HexNAc4 in a triply charged state (Fig 5.18B, lower spectrum) was identified
from glycosylated NGRP3 produced in this system. Notably, precursor ions at this m/z
were over three times more abundant than m/z 722.99 ions (112 and 29 scan events,
respectively). Fragmentation of a m/z 736.66 parent ion produced species corresponding to
the peptide with partial N-glycan structures of DiNAcBac-HexNAc3, DiNAcBac-
HexNAc2, DiNAcBac-HexNAc and DiNAcBac attached (Fig.5.18B, lower spectrum). In
addition, several species with m/z values corresponding to y-ions with either DiNAcBac or
DiNAcBac-HexNAc were identified. Ions corresponding to the unmodified peptide and its
b2, b3 and b4 ions were also observed. Lastly, an ion of m/z 407.17 was identified which
likely corresponds to two HexNAc residues. These data suggest that two pentasaccharide
N-glycan structures were present, the DiNAcBac-HexNAc3-Hex structure first produced
by introduction of PglH1 alone and another structure, DiNAcBac-HexNAc4, produced
only when PglH2 is also present.

156

157
Therefore, LC-MS/MS analysis confirmed the production of NGRP3 modified with a
DiNAcBac-HexNAc2 glycan from E. coli BL21 ppglpglH::kn pETNGRP. These data
provided evidence that upon addition of C. fetus pglH1 into the system, a further HexNAc
and Hex residue were transferred to the N-glycan structure giving DiNAcBac-HexNAc3-
Hex. Upon introduction of C. fetus pglH2 in conjunction with pglH1, an N-glycan
structure consisting of DiNAcBac-HexNAc4 was also present. Therefore, these data
suggest that C. fetus PglH1 may be responsible for adding the first GlcNAc residue and
the branching glucose within the C. fetus N-linked glycan. Secondly, these data suggest
that PglH2 may transfer the alternative branching GlcNAc residue (See Fig. 5.19, pathway
to the left). The latter structure is the more dominant structure in C. fetus (Nothaft et al.,
2012), which may explain the abundance of this ion in comparison to the other. However,
it should be noted that as the C. jejuni PglI enzyme was present in the system, it is possible
that C. fetus PglH1 attached a GlcNAc residue which was then acted upon by C. jejuni
PglI, transferring a branching glucose residue (Fig. 5.19, pathway to the right). Such
interference would be similar to its native role of transferring a branching glucose residue
to the third GalNAc residue of the C. jejuni N-glycan (Kelly et al., 2006). Nevertheless,
these data suggest that pglH1H2 can mediate the construction of a partial C. fetus N-linked
glycan using a C. jejuni pglH::kn trisaccharide N-glycan as a substrate. To engineer the
full N-glycan structure, the predicted transferase gene cf1389 was introduced into this
pglH1H2-containing system.

158

159
5.6 Construction and analysis of hybrid pgl system containing C.
fetus pglH genes and predicted glycosyltransferase cf1389.
5.6.1 Cloning strategy
This chapter has demonstrated a role for C. fetus pglH homologues in building the C. fetus
N-glycan, adding two of the three GlcNAc residues and possibly the branching Glc. To
investigate if the predicted glycosyltransferase gene cf1389 is involved in creating the
remainder of the C. fetus N-glycan structure, a plan to introduce this gene along with
pglH1 and pglH2 was devised. Briefly, this plan included cloning the three genes together
into pGEM®-T-easy vector, amplifying the construct with BclI ends, then digesting and
ligating it with BglII-digested pETNGRP. Genes pglH1 and pglH2 were amplified as in
5.4.1.1. The product was A-tailed using MyTaq™ Red Mix (Bioline) and ligated into
pGEM®-T-Easy, creating pGEMH1H2. Positive clones were identified by cPCR using
primers 80 and 81. The sequence of pglH1H2 was checked by sequencing using primers
80, 81 and 1450. Cf1389 was amplified as in 5.3.1.3, digested with BamHI and ligated
with BamH1-digested pGEMH1H2 to give pGEMX12. Following transformation, positive
clones were identified by cPCR using primer 80 and 1388. Further cPCR to confirm that
pglH1H2 and cf1389 were in the same orientation was performed using primers 80 and
1390 (1188 bp predicted). The presence of all three correctly sized genes was confirmed
by PCR using primers 1379 and 1439. Cf1389 was sequenced using primers 80 and 1486.
The pglH1H2-cf1389 region was amplified from pGEMX12 using primers 1379 and 1439
(Table 2.1), adding BclI sites to each end. This ~3.3 kb product was digested with BclI
and ligated into BglII-digested pETNGRP. Positive transformants were identified by
cPCR using primers 1390 and 1411 (Table 2.1). Plasmid was purified and the sequence of
the C. fetus genes confirmed by sequencing using primers 1388, 1411, 1425, 1485 and
1486. To offer improved performance during mass spectrometry analysis NGRP was
replaced with NGRP3 as with vectors in 5.5.2, creating pX12-N3. C. fetus genes were
sequenced using primers 1388, 1389, 1411, 1425, 1485 and 1558. Following
transformation of pX12-N3 into E. coli BL21 (λDE3) ppglpglH::kn, positive clones were
identified by PCR using primers 1390 and 1411 (not shown).

160
5.6.2 Addition of cf1389 to a ppglpglH::kn CfpglH1H2 background resulted in
three apparent forms of NGRP3
NGRP3 production was induced in E. coli BL21 (λDE3) ppglpglH::kn pX12-N3 using 1
mM IPTG at 16°C overnight. Notably, the yield of biomass was lower compared with
previous strains used in this chapter (~ 6.7 g pellet from 1 L induced culture, in contrast to
between 10 and 16g pellets obtained per 1 L culture). Although NGRP3 production was
not obvious in the coomassie-stained lysate of this induced strain (not shown), his6-tagged
NGRP3 was purified using Nickel affinity chromatography. The sample was eluted in 10
mL elution buffer and 1 mL fractions collected. Fractions 4-8 were combined, dialysed
against 30 mM Tris 0.15 M NaCl pH 7.5 and separated by SDS-PAGE (Fig. 5.18A).
Proteins were observed at around 19.5 kDa and 21.5 kDa, likely corresponding to
unglycosylated NGRP3 and NGRP3- DiNAcBac-HexNAc4, respectively. In addition a
considerable amount of a protein at around 23 kDa was purified, suggesting the possible
presence of another form of NGRP3.
A western blot of this eluate and that from E. coli BL21 (λDE3) ppglpglH::kn pH1H2-N3
was probed with anti-his6 antibodies (Fig. 5.20B). Due to the low biomass yield from
pX12-N3, approximately 20 times more eluate sample was loaded onto the gel in
comparison with the eluate from pH1H2-N3. In both strains unglycosylated NGRP3 was
confirmed at around 21 kDa (predicted 21.78 kDa) and a g1 form of NGRP3 at around
23.5 kDa. However, the 23 kDa protein observed by coomassie-staining was not reactive,
suggesting this protein was not a form of NGRP3 and rather another protein contaminating
the preparation.
In summary, the addition of cf1389 into the hybrid glycosylation system resulted in lower
biomass and protein yield upon induction. Nevertheless, production of a g1 form of
NGRP3 similarly sized to that seen previously was identified. To further study the forms
of NGRP3 produced in this system, they were analysed by mass spectrometry.

161

162
5.6.3 MALDI-TOF MS analysis of glycosylated NGRP3 produced in the presence
of C. fetus pglH genes and cf1389.
To investigate the N-glycan structure of glycosylated NGRP3 purified from E. coli BL21
(λDE3) ppglpglH::kn pX12-N3, the g1 band was excised and analysed by MALDI-TOF
mass spectrometry using CHCA. Peaks with m/z ratios corresponding to 9 tryptic peptides
of NGRP3 (one with 1 missed cleavage site) were identified (Fig. 5.21). This included a
relatively small peak at m/z 1167.793, corresponding to the unmodified sequon-containing
tryptic peptide, indicating the presence of unglycosylated NGRP3. However, another small
peak with an m/z of 2208.393 was produced, suggesting the presence of the tryptic peptide
modified with DiNAcBac-HexNAc4 (as found with pH1H2-N3).
Therefore, these data suggest that both unglycosylated NGRP3 and NGRP3 modified with
DiNAcBac-HexNAc4 were produced. The peak corresponding to the unmodified sequon-
containing tryptic peptide may be due to cross contamination from the ~ 21.5 kDa
unglycosylated form of NGRP3 evident in the preparation (Fig. 5.21). Alternatively, it
could be due to unglycosylated NGRP3 with its signal peptide attached which, if present,
may have been indistinguishable from glycosylated NGRP3 by coomassie staining and
western blotting. If this is true, another tryptic peptide with a predicted m/z of 3735.9077
would be possible, however, this was above the detectable mass range during analysis so
would not have been identified. Nevertheless, these data suggest that introduction of
cf1389 into the hybrid pgl system did not change the N-glycan structure created and hence
this gene does not appear to play a role in C. fetus N-linked glycosylation.
163

164
5.7 Discussion
This Chapter investigated the role of predicted C. fetus transferase genes, cf1389, pglH1
and pglH2, in assembling residues at the non-reducing end of the N-linked glycan
structure. These genes were integrated into an E. coli system which produces an N-
glycoprotein reporter protein, NGRP, modified with a truncated C. jejuni N-glycan to
identify if they were able to add sugar residues onto this structure.
Each candidate gene was singly introduced into the E. coli ppglpglH::kn system to identify
the first-acting glycosyltransferase gene. Western blotting revealed that addition of
CfpglH1 reduced the electrophoretic mobility of NGRP, indicating an increase in its
molecular weight, likely due to transfer of a larger N-glycan structure. Upon introducing
cfpglH1 in combination with either cfpglH2 or cf1389, a further shift in electrophoretic
mobility was observed with cfpglH2 only. Therefore, it appeared that the gene products of
cfpglH1 and cfpglH2 were able to act sequentially on a C. jejuni pglH::kn N-glycan
structure.
The various N-glycosylated NGRP products were purified using nickel affinity
chromatography and the doubly N-glycosylated (g2) form was analysed using MALDI-
TOF MS. However, none of the three possible tryptic glycopeptides were resolved. Instead
two of these possible tryptic peptides were observed in an unmodified state. The
remaining possible tryptic glycopeptide contains two N-glycosylation sequons, and if fully
modified, even with the truncated C. jejuni pglH::kn glycan, would give a peptide with an
m/z of 5329. Hence, this peptide was likely not resolved due to creating an ion with such a
large m/z. Therefore, it may be that the g2 form of NGRP is predominantly modified at the
two sites, N150 and N156, which reside within a single tryptic peptide. In support of this,
site-directed mutagenesis of the native precursor of NGRP, Cj0114, has previously
identified that these sites are preferentially modified in H. pullorum, as these are predicted
to reside in a flexible linker region within the protein (Jervis et al., 2010).
To alleviate the issues regarding MS analysis of NGRP, an alternative version of this
protein, NGRP3, was integrated into the systems developed. This version has been altered
to create a novel sequon-containing tryptic peptide that is small and positively charged
(Jervis & Linton, unpublished). NGRP3 was similarly modified by the hybrid N-
glycosylation systems and analysed by MALDI MS. Two different sample matrices were
tested, CHCA and DHB, the latter often applied for glycopeptide analysis. No possible
glycopeptides were identified from the systems containing CfpglH1 alone with either

165
matrix. However, using DHB, a possible glycopeptide was identified from gNGRP3
produced in the presence of cfpglH1H2, although attempts to further fragment this ion
failed. Nevertheless, the size of the peptide indicated an N-glycan structure consisting of
DiNAcBacHexNAc4. Therefore, these data suggested that cfpglH1H2 work in sequence to
transfer the first two GalNAc residues of the C. fetus N-glycan structure.
To gain further insight into the N-glycan structures produced by the hybrid systems,
gNGRP3 samples were analysed using LC-MS/MS. Confirming the MALDI MS findings,
glycopeptides containing an N-glycan structure of DiNAcBacHexNAc4 were identified
from NGRP3 produced in the presence of cfpglH1H2. However, when cfpglH1 alone was
present, glycopeptides containing a DiNAcBacHexNAc3Hex N-glycan were identified. In
addition, this structure was also found when pglH1H2 were present, albeit at a lower
abundance in comparison to the DiNAcBacHexNAc4-containing glycopeptides. Two
possible hypotheses were formed (Fig. 5.19) regarding the mechanism behind these
observed N-glycan structures. In each case, C. fetus PglH1 adds a GlcNAc residue onto
the C. jejuni pglH::kn trisaccharide. Following this, either C. fetus PglH1 or C. jejuni PglI
transfers a branching glucose to this intermediate tetrasaccharide to form the
DiNAcBacHexNAc3Hex N-glycan identified in the pglH1-containing system. The
DiNAcBacHexNAc4 N-glycan identified upon introducing C. fetus pglH2 into this system
is then a result of PglH2 transferring either the branching or linear GlcNAc residue onto
the tetrasaccharide intermediate (created by the PglH1-mediated addition of a GlcNAc).
C. jejuni PglI transfers the branching Glc onto GalNAc within the C. jejuni N-glycan
structure (Linton et al., 2005). Although C. fetus PglH1 is hypothesised to add a GlcNAc
in this system, and hence should not provide a substrate for C. jejuni PglI, this was not
verified. Therefore, it is plausible that interference from C. jejuni PglI occurred. As this
reaction would not be optimal for this enzyme, it can be expected that the Hex-containing
structure would be less abundant than that created by PglH2 activity. Indeed, the Hex-
containing structure appeared over three times less abundant than the DiNAcBacHexNAc4
N-glycan. However, in C. fetus the GlcNAc branch-containing structure is 4 times more
abundant than the Glc-branching structure (Nothaft et al., 2012). Therefore, the relative
abundancies of the N-glycan structures observed do not favour either hypothesis. If
CfPglH1 does catalyse the addition a GlcNAc and a Glc residue onto two different
acceptor substrates, GalNAc and GlcNAc, this would be a striking finding. Although
bacterial glycosyltransferases are broadly known to have more relaxed acceptor
specificities than their eukaryotic counterparts (Johnson, 1999), such relaxed specificity is

166
not typical for the GTs of the ε-proteobacteria Pgl systems. It has been demonstrated that,
if a GlcNAc is transferred in place of DiNAbac (by an E. coli GT), this can be utilised as
an acceptor substrate by C. jejuni PglA. However, this happens at a lower efficiency and is
only observed in the E. coli host system not in vivo (Linton et al., 2005). Furthermore,
although there are examples of bacterial GTs which can utilise more than one sugar donor
(Naegeli et al., 2014), this is not a representative feature of Pgl systems. For example,
although C. jejuni PglH transfers multiple sugar residues onto the N-linked glycan
structure, each residue is a GalNAc and hence the enzyme has specificity for only a single
sugar donor. Therefore, the suggested role of C. fetus PglH1 would be an unusual feature
of N-linked glycosylation in ε-proteobacteria.
As introducing cfpglH1 and H2 did not build the full C. fetus N-glycan hexasaccharide
structure, it was hypothesised that the predicted glycosyltansferase, cf1389, may be
responsible for transfer of the final GlcNAc residue. However, introducing this gene into
the hybrid C. jejuni/C. fetus pgl system containing cfH1H2 caused a reduction in both
biomass yield and N-glycosylation efficiency. Furthermore, the N-glycosylation that was
identified appeared to include the DiNAcBacHexNAc4, as found in the system without
cf1389 present. Therefore, it seemed that the product of cf1389 did not contribute to N-
glycosylation in this system, and even inhibited the process. This prompted further
bioinformatics analysis into this gene and the two uncharacterised genes downstream, all
of which are located within the C. fetus pgl locus in C. fetus fetus 82-40.
As these genes are not found in the C. fetus 04/554 pgl locus, it was hypothesised that this
region may result from integration of bacteriophage DNA into the genome. Indeed,
PHASTER (PHAge Search Tool Enhanced Release) web server, a tool for identifying
prophage DNA (Arndt et al., 2016), found this region to be probable prophage DNA
within the C. fetus 82-40 genome. Genes cf1389 and cf1390 matched sequences from
Chronobacter phage ENT47670 (Genbank accession HQ201308), identified from
Chronobacter sakazakii. The former matched with a bactoprenol glucosyl transferase,
which could explain the negative impact introduction of this gene had on the C. fetus
engineered system created in this chapter. Bactoprenol glucosyl transferases catalyse the
addition of glucose to undecaprenyl-pyrophosphate (also known as UndPP/Bactoprenol)
(Allison and Verma, 2000). Hence, if Cf1389 transfers a glucose/GlcNAc residue onto
UndP, it could interfere with the N-linked glycosylation system by utilising the UndP
substrate needed to create LLO precursor. Furthermore, this could have a far-reaching
effect in E. coli, inhibiting other systems which require an UndP substrate, such as

167
peptidoglycan and lipopolysaccharide biosynthesis (Heijenoort, 2001; Wang and Quinn,
2010). Such interference may only be significant in the E. coli system due to a potentially
higher copy number of Cf1389 than when expressed in vivo.
Cf1390 matched an uncharacterised hypothetical protein of Chronobacter phage
ENT47670. Performing protein BLASTP searches of Cf1390 identified a possible
Glucos_trans_II domain, placing it within the GtrII superfamily. The canonical protein of
this superfamily is glucosyl transferase II (gtrII) from Shigella phage SfII, which mediates
transfer of a glucose residue onto a rhamnose present within the O-antigen repeat unit
(Mavris et al., 1997). Furthermore, performing a BLASTP search with the predicted
product of cf1391 found the protein to have a GtrA-like domain. GtrA is a Shigella phage
SfX protein thought to mediate transport of UndP-Glc across the inner membrane.
Therefore, it appears that cf1389-91 constitute a horizontally-acquired LPS O-antigen
modification locus termed GtrABC, as documented in Shigella and Salmonella (Allison &
Verma, 2000; Davies et al., 2013). In line with this, the more recent availability of
complete genomes of C. hyointestinalis (Miller, Yee, & Chapman, 2016) reveal there are
no homologues of cf1389-1391 in these species. Therefore, as these C. hyointestinalis
produce identical N-glycan structures to C. fetus, this suggests that none of these genes
play a role in the N-glycosylation pathway.
In conclusion, glycosyltransferase activity of C. fetus PglH1 and PglH2 was demonstrated
using the C. jejuni pglH::kn trisaccharide as a substrate. These data revealed that CfPglH1
likely mediates transfer of one GlcNAc residue and is followed by the activity of CfPglH2,
which may add a further GlcNAc residue. Furthermore, CfPglH1 may also transfer a
branching glucose residue. However, this transfer could have been due to interference
from the C. jejuni PglI enzyme. Nevertheless, these enzymes were not sufficient to
construct the entire non-reducing end of the C. fetus N-glycan, as the N-glycan structures
produced by the pglH1H2-containing system each lacked a GlcNAc residue. Investigation
into another potential glycosyltransferase within the C. fetus pgl locus found this protein to
not be involved in the N-glycosylation process, instead likely forming part of an O-antigen
serotype conversion prophage locus. Therefore, the glycosyltransferase responsible for
adding the final GlcNAc residue of the C. fetus N-glycan structure remains unknown.
Furthermore, the identity of the enzyme responsible for transferring the branching glucose
residue to the C. fetus N-glycan remains unclear.

168
Chapter 6. C. fetus N-glycoproteome
prediction and the conservation of N-
glycoproteins between Campylobacter
species

169
6.1 Introduction
The C. fetus Pgl system has not been studied extensively and the extent of N-glycan
protein modification in comparison to C. jejuni is not known. In C. jejuni there are over 60
experimentally identified N-glycoproteins (Scott et al., 2014, 2011; Young et al., 2002). In
C. fetus, Nothaft et al. (2012) identified 65 N-glycopeptides using LC-MS/MS,
corresponding to 32 distinct N-glycosylation sites from 25 proteins. Numerous reactive
bands were evident upon probing C. fetus whole cell lysates with antiserum raised against
C. fetus free oligosaccharides (Nothaft et al., 2012), which suggests there are many more
N-glycoproteins. This is also evident from the extent of reactivity observed with an
antiserum raised against a C. fetus N-linked glycoprotein (See Chapter 4). Although
glycopeptide enrichment coupled with LC-MS/MS is a powerful tool for discovering
glycopeptides, there are limitations as many N-glycoproteins may be insoluble, or
glycosylation sites may reside within large tryptic peptides which cannot be resolved
easily. In addition, glycoproteins which are less abundant can be missed. In line with this,
an N-glycoproteome prediction pipeline suggests there are at least 148 potential N-linked
glycoproteins in C. jejuni NCTC 11168 (Frost, 2015). Therefore, it is likely that C. fetus
N-glycosylates more proteins than have been experimentally identified thus far, and that
the N-glycoproteome prediction pipeline could reveal more potential N-glycoproteins.
As C. jejuni is known to N-glycosylate numerous proteins, many studies have tried to
elucidate the function of this modification. For example, Alemka et al. (2013)
demonstrated that N-glycosylation influences the growth of C. jejuni in the presence of
chicken caecal contents ex vivo, likely due to N-glycan modification providing protection
from the action of chicken proteases. Other evidence also suggests that Campylobacter N-
glycosylation is broadly important for bacterial fitness, cell attachment, colonisation and
survival in the host (Hendrixson and DiRita, 2004; Szymanski et al., 2002; van Sorge et
al., 2009). However, studies on the influence of N-glycosylation on individual proteins are
limited and largely do not suggest that modification alters protein activity. An influence of
N-glycosylation on the function of virB10, a type IV secretion system component, has
been demonstrated (Larsen et al., 2004). However, studies on proteins such as ZnuA and
JlpA suggest that N-glycan modification does not significantly affect protein function
(Davis et al., 2009; Kakuda and DiRita, 2006; Scott et al., 2009). Therefore, it can be
argued that N-glycosylation does not afford advantages to specific target proteins, but
rather the abundance of the modification itself provides an overall advantage, such as

170
increased resistance to proteases, sufficient for continued selection. If this is correct, there
is likely low level conservation of the identity of N-glycosylation target proteins between
different Campylobacter species, provided that each species has a significant number of
modified proteins overall. As experimental data regarding the N-glycoproteins of
Campylobacter species other than C. jejuni are relatively sparse, bioinformatics analysis
could also be utilised to putatively identify the extent of N-glycoprotein conservation
among C. jejuni and these species.
This chapter describes the prediction of the C. fetus fetus 82-40 N-glycoproteome using
the previously developed bioinformatics pipeline (Frost, 2015). This prediction pipeline is
a perl script which inputs protein sequences into PSORTb (Yu et al., 2006), LipoP
(Juncker et al., 2003) and TMHMM (Krogh et al., 2001) to predict subcellular localisation
and then identifies any D/E-X-N-S/T sequons in the sequences, generating an output file
which can be manually curated to produce a list of predicted N-glycoproteins (Fig. 6.1).
PglB-mediated modification of a newly predicted N-glycoprotein was confirmed
experimentally in C. fetus. Lastly, the conservation of N-glycoproteins among
Campylobacter species including C. fetus was investigated.

171

172
6.2 C. fetus N-glycoproteome prediction and validation
6.2.1 The predicted C. fetus fetus 82-40 N-glycoproteome
Protein sequences, extracted from the C. fetus fetus 82-40 genome using Artemis
(Rutherford et al., 2000), were fed into the N-glycoprotein prediction pipeline (Fig. 6.1)
and the output file manually curated as described in Figure 6.2. Proteins not confidently (<
9) assigned a subcellular location by PSORTb but with predicted transmembrane (TM)
regions or signal peptide cleavage sites by the other programs were assigned as ‘unknown
extracytoplasmic’ and still considered a potential target for PglB. Although the predicted
N-glycoproteome of C. jejuni 11168 NCTC was previously established by Frost (2015)
using this pipeline, the process was repeated to allow for accurate comparison with the
newly generated C. fetus data. Furthermore, the protein sequences extracted from the C.
jejuni 11168 genome by Frost (2015) included the products of pseudogenes; such
sequences were excluded from this analysis. Four experimentally identified C. jejuni N-
glycoproteins (Cj0494, Cj1345c, Cj1496c and Cj1032) had been missed by the prediction
pipeline as they were predicted as cytoplasmic. These proteins were added to give the total
predicted C. jejuni N-glycoproteome consisting of 158 proteins.
Of the total 1719 C. fetus proteins, 308 (18%) contained N-glycosylation sequon
sequences. Of these 308, 129 were identified as possible N-glycoproteins based on
subcellular localisation predictions (Table 6.1). This included 20 of the 25 N-glycoproteins
previously experimentally identified by Nothaft et al. (2012). The remaining five proteins
(CFF8240_0198, 0217, 0365, 0372 and 1782) were not identified as they were classified
as ‘unknown’ by PSORTb and contained no predicted transmembrane domains or signal
peptide cleavage sites according to TMHMM and LipoP, respectively (Table 6.1, shown
without colour). Subsequent analysis of these proteins using an alternative localisation
tool, CELLO (Yu et al., 2006), predicted that all but Cf0372 were extracytoplasmic.
However, SignalP 4.1 (Petersen et al., 2011), a server which predicts signal peptide (SP)
cleavage sites, suggested that Cf0372 does harbour a SP cleavage site. Therefore, although
LipoP can identify SpI or SpII (lipoprotein) signal peptide cleavage sites, it appears that
SignalP may be more accurate for identifying SpI cleavage sites. This is likely due to the
tool being optimised for specific identification of SpI cleavage sites and possibly because
it is recently updated (version 4.1 is currently available) whereas there is only a single
version of LipoP released in 2003. These findings indicate that the prediction pipeline
could be improved if more accurate protein localisation prediction tools were included.

173
Upon combining the experimental data set of Nothaft et al. (2012) with this prediction
pipeline result, the predicted total C. fetus N-glycoproteome consists of 134 proteins,
making up 7.8% of its total proteome and containing 219 sequons in total. These figures
are similar to that found in C. jejuni, which has a predicted total N-glycoproteome of 158
proteins, constitutes 9.7% of its total proteome and contains 185 sequons. Therefore, it
appears that the extent of N-glycosylation is as significant in C. fetus as it is in C. jejuni.

174
Table 6.1. Complete predicted C. fetus N-glycoproteome. Colour indicates predicted
location assignment based on prediction pipeline described in Figure 6.1: Blue; inner
membrane. Green; periplasmic. Red; Outer membrane. Purple; extracellular. Grey; unknown
but extracytoplasmic. White; predicted as cytoplasmic. Bold indicates experimental
identification by Nothaft et al. (2012).
Locus
Tag
No. of
sequons Sequon sequence (s) Predicted location
Cf0010 2 EENSS, DQNGS Extracellular
Cf0014 1 DTNSS Extracytoplasmic
Cf0018 2 DINLS, DINSS Inner Membrane
Cf0026 1 DFNRS Extracellular
Cf0065 1 EINAT Inner Membrane
Cf0067 3 DKNST, DDNGT, DDNKT Extracytoplasmic
Cf0092 1 EINAS Extracellular
Cf0107 3 DANAT, DTNDT, DNNIT Periplasmic
Cf0117 1 EQNAS Outer Membrane
Cf0118 1 EANST Inner Membrane
Cf0119 2 DYNGT, ENNST Inner Membrane
Cf0121
(ccoG) 1 DVNDT Inner Membrane
Cf0134 1 DDNFT Extracytoplasmic
Cf0136 1 DDNKS Extracytoplasmic
Cf0138 1 DINSS Inner Membrane
Cf0151 1 DKNQS Inner Membrane
Cf0191 1 ENNTT Extracytoplasmic
Cf0198 2 DINST, DINGS Cytoplasmic
Cf0217 2 DANYS, DLNDT Cytoplasmic
Cf0243 1 DENST Inner Membrane
Cf0272 1 ELNAT Inner Membrane
Cf0290 7 DKNNS, ESNMT, DENVT, DSNLS, DENES,
DINLS, DKNTT Extracytoplasmic
Cf0301 2 DLNLT, DYNTT Extracytoplasmic
Cf0312 11
DANLS, EANIT, EFNFS, DTNSS, DANAS,
DVNYS, DNNIS, DINIT, DINGT, DTNIT,
ETNES
Outer Membrane
Cf0358 2 EGNLT, DLNNS Extracytoplasmic
Cf0365 1 DNNET Cytoplasmic
Cf0367 2 DINKT, DKNGT Inner Membrane
Cf0368 2 EDNIS, DNNFT Inner Membrane
Cf0372 2 DVNRT, DNNSS Cytoplasmic
Cf0384 2 DTNAT, DLNLT Extracytoplasmic
Cf0398 1 DENVS Extracytoplasmic
Cf0401 1 DGNIS Inner Membrane
Cf0404 1 DVNNT Inner Membrane
Cf0431 4 EDNIT, DHNIT, DSNFS, DKNIS Inner Membrane
Cf0440 1 ERNLS Periplasmic
Cf0443 1 DHNTT Extracytoplasmic
Cf0444 3 DKNET, EQNAS, DNNET Extracytoplasmic
Cf0445 1 DENLT Periplasmic
Cf0449 1 DVNTT Extracytoplasmic
Cf0459 1 DLNLT Inner Membrane

175
Cf0461 1 DNNVT Extracellular
Cf0490 3 DINET,EINFT,DGNDT Extracellular
Cf0502 1 DANYS Extracytoplasmic
Cf0505 2 ENNES, DINGT Extracytoplasmic
Cf0511 1 EINIS Inner Membrane
Cf0517 2 DINST, DLNST Outer Membrane
Cf0529 1 DINFS Inner Membrane
Cf0536 1 ELNLT Inner Membrane
Cf0537 1 DWNSS Inner Membrane
Cf0553 2 DINQT, EQNSS Inner Membrane
Cf0557 1 EKNSS Inner Membrane
Cf0569 2 DVNDT, ESNAT Inner Membrane
Cf0572 3 EQNAS, DINGT, DDNSS Extracytoplasmic
Cf0583 1 ESNTS Inner Membrane
Cf0607 4 DGNIT, DQNET, ESNVS, EYNST Outer Membrane
Cf0625 2 DTNGT, DINGS Inner Membrane
Cf0636 2 EQNRT, DKNIT Extracytoplasmic
Cf0682 1 DFNLS Inner Membrane
Cf0684 1 DDNNT Inner Membrane
Cf0699 6 ELNLT, DLNYT, DFNIS, DANAS, DVNVT,
DENKT Outer Membrane
Cf0705
(flgC) 1 ELNKS Periplasmic
Cf0708 6 DSNVT, DTNET, DENIT, DANAS, DLNKT,
DNNIT Extracytoplasmic
Cf0723 1 DINFS Inner Membrane
Cf0725
(int) 2 EKNAS, ENNMS Inner Membrane
Cf0728
(secD) 1 DSNLT Inner Membrane
Cf0735 2 DINGS, DNNTT Extracytoplasmic
Cf0738 2 EKNVT, EDNNS Extracellular
Cf0745 1 ENNNT Outer Membrane
Cf0748 1 DNNDS Inner Membrane
Cf0758 1 EQNAT Extracytoplasmic
Cf0760 1 DFNQT Outer Membrane
Cf0781 1 DINGT Periplasmic
Cf0786
(pyrD) 1 DKNAT Inner Membrane
Cf0787 1 EKNES Inner Membrane
Cf0799 1 DFNIT Inner Membrane
Cf0800 2 DRNLS, DLNTT Periplasmic
Cf0802
(cutF) 3 DSNGS, DINQT, DDNET Outer Membrane
Cf0825 1 DDNNS Inner Membrane
Cf0845
(murG) 1 EHNIS Inner Membrane
Cf0855 1 DINSS Inner Membrane
Cf0872 2 EHNDT, DRNIT Inner Membrane
Cf0875
(ppk) 1 EKNSS Inner Membrane
Cf0898 1 DNNSS Inner Membrane
Cf0944 2 DQNFT, DGNST Inner Membrane
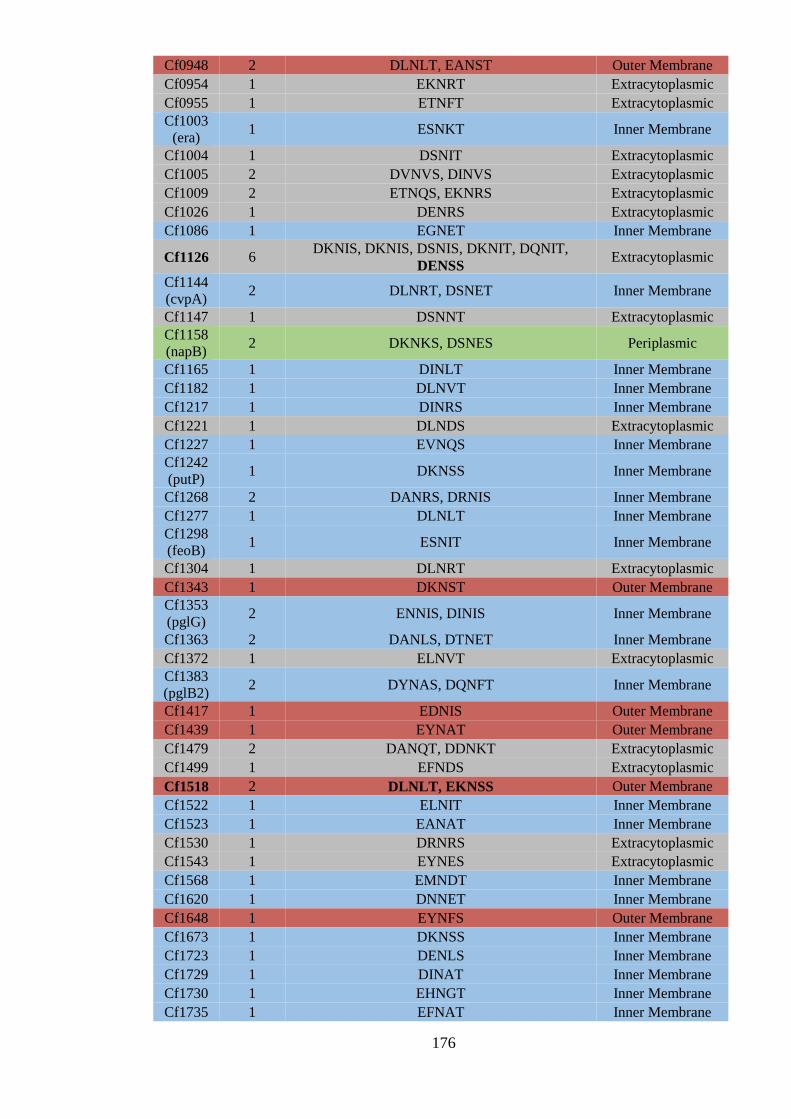
176
Cf0948 2 DLNLT, EANST Outer Membrane
Cf0954 1 EKNRT Extracytoplasmic
Cf0955 1 ETNFT Extracytoplasmic
Cf1003
(era) 1 ESNKT Inner Membrane
Cf1004 1 DSNIT Extracytoplasmic
Cf1005 2 DVNVS, DINVS Extracytoplasmic
Cf1009 2 ETNQS, EKNRS Extracytoplasmic
Cf1026 1 DENRS Extracytoplasmic
Cf1086 1 EGNET Inner Membrane
Cf1126 6 DKNIS, DKNIS, DSNIS, DKNIT, DQNIT,
DENSS Extracytoplasmic
Cf1144
(cvpA) 2 DLNRT, DSNET Inner Membrane
Cf1147 1 DSNNT Extracytoplasmic
Cf1158
(napB) 2 DKNKS, DSNES Periplasmic
Cf1165 1 DINLT Inner Membrane
Cf1182 1 DLNVT Inner Membrane
Cf1217 1 DINRS Inner Membrane
Cf1221 1 DLNDS Extracytoplasmic
Cf1227 1 EVNQS Inner Membrane
Cf1242
(putP) 1 DKNSS Inner Membrane
Cf1268 2 DANRS, DRNIS Inner Membrane
Cf1277 1 DLNLT Inner Membrane
Cf1298
(feoB) 1 ESNIT Inner Membrane
Cf1304 1 DLNRT Extracytoplasmic
Cf1343 1 DKNST Outer Membrane
Cf1353
(pglG) 2 ENNIS, DINIS Inner Membrane
Cf1363 2 DANLS, DTNET Inner Membrane
Cf1372 1 ELNVT Extracytoplasmic
Cf1383
(pglB2) 2 DYNAS, DQNFT Inner Membrane
Cf1417 1 EDNIS Outer Membrane
Cf1439 1 EYNAT Outer Membrane
Cf1479 2 DANQT, DDNKT Extracytoplasmic
Cf1499 1 EFNDS Extracytoplasmic
Cf1518 2 DLNLT, EKNSS Outer Membrane
Cf1522 1 ELNIT Inner Membrane
Cf1523 1 EANAT Inner Membrane
Cf1530 1 DRNRS Extracytoplasmic
Cf1543 1 EYNES Extracytoplasmic
Cf1568 1 EMNDT Inner Membrane
Cf1620 1 DNNET Inner Membrane
Cf1648 1 EYNFS Outer Membrane
Cf1673 1 DKNSS Inner Membrane
Cf1723 1 DENLS Inner Membrane
Cf1729 1 DINAT Inner Membrane
Cf1730 1 EHNGT Inner Membrane
Cf1735 1 EFNAT Inner Membrane

177
Cf1737 1 DKNAS Inner Membrane
Cf1750 1 DFNDS Extracytoplasmic
Cf1756
(nhaA) 1 DFNLS Inner Membrane
Cf1782 3 EYNQT, DSNST, DVNIS Cytoplasmic
Cf1804 1 ETNKT Inner Membrane

178
6.2.2. Validation of predicted C.fetus N-glycoproteins
Numerous N-glycoproteins have been predicted in C. fetus using the bioinformatics
pipeline described above with 20 being previously identified experimentally by Nothaft et
al. (2012). However, a predicted extracytoplasmic protein containing an N-glycosylation is
not necessarily an N-glycoprotein in vivo. For example, the sequon could reside within a
buried structural feature and hence not be accessible (Silverman and Imperiali, 2016). To
demonstrate that N-glycoproteins predicted using the bioinformatics pipeline were
modified in vivo, His-tagged versions of predicted proteins were introduced into C. fetus
wildtype and pglB::kn strains. Although the prediction pipeline utilised the genome of C.
fetus fetus 82-40, this strain was not available and thus C. fetus fetus 10842 was utilised
instead.
6.2.2.1 Proof of principle validation of experimentally identified C. fetus N-glycoprotein,
Cf0781
As a proof of principle, a His-tagged version of a known C. fetus N-glycoprotein, Cf0781
(Gene ID: CFF8240_0781), was introduced into C. fetus wildtype and pglB::kn strains.
This protein was chosen as it was predicted to be periplasmic and contained only a single,
experimentally confirmed sequon (Nothaft et al., 2012). cf0781 was amplified from C.
fetus 10842 gDNA using primer pair 1567/1572 (Table 2.1), adding a 5’ BamHI site and
3’ Octa-His tag and SmaI site. An ~ 0.8 kb product was confirmed using agarose gel
electrophoresis (not shown). The digested product was ligated to BamHI- and SmaI-
digested pRYGG1 to give p0781 and transformed into E. coli XL-1 Blue. The sequence of
the octa-his-tagged version of Cf0781 in p0781 is shown in Figure 6.3A. Positive clones
were confirmed by cPCR using primers 1445 and 1572. The sequence of cf0781 within
p0781 was checked using primers 1445 and 1432. Plasmid p0781 was then transformed
into E. coli S17 λpir cells in preparation for conjugation into C. fetus wildtype and
pglB::kn strains. Primers 1432 and 1445 were also used to identify positive E. coli S17
clones and then C. fetus transformants. To confirm production of this protein, a western
blot containing whole cell lysates of C. fetus wildtype and pglB::kn strains with and
without p0781 was probed with anti-His(6) antibodies (Fig. 6.3B).
No His-reactive proteins were detected in the absence of p0781 (Fig. 6.3B). Upon
introduction of the plasmid into wildtype C. fetus, an anti-His6 reactive protein was
detected at around 27 kDa. This is consistent with the predicted size, ~ 27.4 kDa, of
(8)His-tagged Cf0781 with a single N-glycan modification. In the presence of p0781 but
without pglB, an anti-His6 reactive protein was observed at around 26 kDa, which

179
correlates with the predicted size of 26.4 kDa for unglycosylated Cf0781. These data
confirm that Cf0781 is modified in a C. fetus PglB dependent manner.

180
6.2.2.2 Validation of a newly predicted N-glycoprotein, Cf0445
A His(8)-tagged version of a protein, Cf0445, predicted to be an N-glycoprotein using the
bioinformatics approach described in section 6.2 was introduced into C. fetus. Cf0445 is a
relatively small predicted periplasmic protein containing a cytochrome c553 domain and a
single predicted N-glycosylation sequon.
cf0445 (Gene ID: CFF8240_0445) was amplified from C. fetus 10842 gDNA using primer
pair 1566/1573 (Table 2.1), adding a 5’ BamHI site and 3’ Octa-His tag and SmaI site. An
~ 0.5 kb product was confirmed using agarose gel electrophoresis (not shown). The
digested product was ligated to BamHI- and SmaI-digested pRYGG1 to give p0445 and
transformed into E. coli XL-1 Blue. The sequence of the octa-his-tagged version of
Cf0445 in p0445 is shown in Figure 6.4A. Positive clones were confirmed by PCR using
primers 1445 and 1573. The sequence of cf0445 was confirmed using primers 1445 and
1432. Verified plasmid was transformed into E. coli S17 λpir cells in preparation for
conjugation into C. fetus wildtype and pglB::kn. All subsequent cPCR used to confirm
positive clones utilised primers 1432 and 1445. To confirm production of the protein, a
western blot containing whole cell lysates of C. fetus wildtype and pglB::kn strains with
and without p0445 was probed with anti-His(6) antibodies (Fig. 6.4B).
No reactivity was identified in C. fetus cells in the absence of p0445 (Fig. 6.4B). In the
presence of p0445, an anti-His(6)-reactive protein was produced, with apparent sizes of
around 19 and 17.5 kDa in wildtype and pglB::kn C. fetus cells, respectively. These likely
correspond to unmodified (predicted 17.1 kDa) and g1 form (predicted 18.3 kDa) of His-
tagged Cf0445. Therefore, these data demonstrate that Cf0445 is indeed modified in a C.
fetus PglB-dependent manner.

181

182
6.3 Analysis of the C. fetus predicted N-glycoproteome
6.3.1 Subcellular locations of predicted C. fetus N-glycoproteins.
Following the methodology outlined in Figure 6.2, N-glycoproteins were assigned a
predicted subcellular localisation. This bioinformatics pipeline previously found that the
majority of C. jejuni experimentally identified and predicted N-glycoproteins were
designated as inner membrane proteins (53.2%), followed by the outer membrane (9.1%),
periplasmic (6.5%) and extracellular (0.6%) (Frost, 2015). Similar analyses were
performed on C. fetus proteins to identify the predicted subcellular localisations of N-
glycoproteins experimentally identified by Nothaft et al. (2012), the predicted total N-
glycoproteome (described in 6.2.1) and the total set of C. fetus proteins predicted to
extracytoplasmic (Fig. 6.5).
Analysis of the 25 experimentally identified C. fetus N-glycoproteins revealed that the
largest proportion, consisting of nine proteins, was those predicted to be extracytoplasmic
but not confidently assigned a specific location (Fig. 6.5A, ‘unknown extracytoplasmic’).
In addition, five proteins had no indication of residing outside the cytoplasm and thus
were predicted as cytoplasmic (Fig. 6.5A). The remaining eleven proteins consisted of
four inner membrane, four outer membrane and three periplasmic proteins. No proteins
were predicted to be extracellular.
In comparison, analysis of the C. fetus predicted total N-glycoproteome (those predicted
and identified experimentally) (Fig. 6.5B) found that the largest proportion was predicted
to be inner membrane proteins (49.3%) and the smallest fraction was predicted to be
extracellular (4.5%). There were a considerable number (26.9%) of proteins assigned as
‘unknown extracytoplasmic’ due to contradictions between the three prediction tools
included in the pipeline. For example, 7 were categorised as cytoplasmic by PSORTb but
had a signal peptide and/or transmembrane (TM) region(s) predicted by LipoP and
TMMHM, respectively. The remainder of the proteins assigned as ‘unknown
extracytoplasmic’ were unclassifiable using PSORTb, but also displayed either a predicted
signal peptide or TM region. Furthermore, as above, there were five proteins (accounting
for 3.7% in this instance) assigned as cytoplasmic due to having no indication of being
extractyoplasmic.
These data reveal that outer membrane N-glycoproteins may be twice as abundant as
periplasmic N-glycoproteins in C. fetus, accounting for 10.4% and 5.2% of the predicted

183
total N-glycoproteome, respectively (Fig. 6.5B). This finding is not simply a result of their
relative abundance within the extracytoplasmic pool of proteins (Fig. 6.5C: 6.4% and
5.1% for OM and periplasmic, respectively). These observations are similar to what has
been observed in C. jejuni. Although Scott et al. (2011) found that, of the experimentally
identified N-glycoproteins, more were predicted to be periplasmic than outer membrane
proteins, combining these N-glycoproteins with those predicted by the bioinformatics
pipeline suggested there are potentially more outer membrane (8.9%) than periplasmic
(6.3%). The lower abundance of membrane proteins found experimentally may be because
membrane proteins are more challenging to extract and solubilise prior to mass
spectrometry analysis (Scott et al., 2011). In summary, the localisation of N-glycoproteins
in C. fetus appears to be similar to that of C. jejuni with predominantly inner membrane
proteins followed by outer membrane then periplasmic.

184

185
6.3.2 Sequon distribution in C. fetus predicted N-glycoproteins.
Previous analysis of sequon frequency within individual N-glycoproteins found that 85%
of C. jejuni 11168 predicted N-glycoproteins contain a single sequon (Frost, 2015) (Fig.
6.6A). Similarly to this, using the predicted total C. jejuni N-glycoproteome established
here, it was found that 72% of proteins harboured only a single sequon and 17% contained
two sequons (Fig. 6.6A). The most extensively glycosylated predicted N-glycoprotein
identified, Cj0633, contained eight sequons and Scott et al. (2014) have experimentally
identified two of these sequons in C. jejuni. Furthermore, eight glycoforms are observed
by western blotting when Cj0633 is expressed in glycocompetent E. coli (Frost, 2015). To
determine the distribution of sequons within experimentally identified and predicted C.
fetus N-glycoproteins, the frequency of sequons per protein was analysed.
Analysis of the 33 C. fetus N-glycosylation sequons experimentally identified by Nothaft
et al. (2012) demonstrated that 76% of N-glycoproteins contained a single modified
sequon and 20% contained two sequons. The highest number of modified sequons per
protein was four, which was evident in only a single protein. Similarly, examination of the
C. fetus predicted total N-glycoproteome found the majority of N-glycoproteins contained
one (64%) or two (25%) sequons (Fig. 6.6B). However, the remaining proteins had 3, 4, 6,
7 or 11 sequons. The protein with eleven potential N-glycosylation sequons,
CFF8240_0312, was experimentally determined as an N-glycoprotein by Nothaft et al.
(2012); however, the glycopeptides corresponded to only one of the possible eleven
sequons. This protein is predicted to reside in the outer membrane, and hence it could be
that the possible sites are within TM helices and not accessible for modification. However,
the TMHMM server predicts only a single TM helix encompassing residues 7 and 26, with
the remainder of the protein following this residing outside the cell. This suggests that as
no sequons lie within the TM region they still have the potential to be modified (Table
6.1).
In conclusion, the predicted N-glyoproteome of C. fetus has revealed that its N-
glycoproteins may be more extensively N-glycosylated than indicated by experimental
data. The pattern of sequon distribution was similar to that of C. jejuni. Strikingly, one
predicted N-glycoprotein, Cf0312, contained 11 possible sequons and is potentially the
most highly N-glycosylated protein identified in Campylobacter thus far.

186

187
6.3.3 Amino acid composition of sequons in C. fetus predicted N-glycoproteins.
The extended sequon, D/E-X-N-X-S/T (X ≠ P), is required for C. jejuni N-glycosylation
(Kowarik et al. 2006) and other Campylobacter species including C. fetus also appear to
follow this rule (Nothaft et al., 2012). Chen et al. (2007) proposed that DQNAT is the
optimum acceptor sequence for C. jejuni PglB using an in vitro assay. In agreement with
this, in vitro analysis of C. lari PglB found Thr to be favoured over Ser at the +2 position,
giving a higher rate of glycosylation (Gerber et al., 2013). Furthermore, analysis of N-
glycosylation sequons in all predicted and verified C. jejuni N-glycoproteins demonstrated
that Asp (D) is most common in the -2 position. However, Ser (S) is more abundant at the
+2 position (Frost, 2015). The amino acid composition of sequons within the C. jejuni
predicted total N-glycoproteome is shown in Figure 6.7A. To identify the most abundant
amino acids in C. fetus N-glycosylation sequons, the distribution of residues in both
predicted and known C. fetus N-glycoproteins was analysed.
Analysing sequon sequences from C. fetus N-glycopeptides identified by Nothaft et al.
(2012) revealed overrepresentation of Asp (94%) and Thr (78%) at the -2 and +2
positions, respectively (not shown). This trend is also observed when analysing sequons of
the predicted total N-glycoproteome, although the relative abundancies are not as marked,
with 70% Asp and 55% Thr at the -2 and +2 positions, respectively (Fig. 6.7B). Therefore,
these data suggest that, in agreement with in vitro Campylobacter PglB substrate
specificity studies, C. fetus N-glycosylation sequons preferentially consist of D-X-N-X-T
sequences.

188

189
6.4 Predicted conservation of N-glycoproteins within the
Campylobacter genus
6.4.1 Predicted conservation of N-glycoproteins between C. jejuni and C. fetus
Although many N-glycoproteins from C. jejuni and C. fetus have been experimentally
identified, the extent of conservation of N-glycosylation targets among these species is
unknown. As discussed in section 6.1, some evidence suggests that N-glycosylation is not
required for the function of specific target proteins and rather the modification may afford
a general advantage, such as improved resistance against proteolytic enzymes (Alemka et
al., 2013). Understanding the conservation of N-glycoproteins among Campylobacter
species may provide suggestive evidence of proteins which require N-glycosylation. To
this end, a sample of the core N-glycoproteins found in C. jejuni and C. fetus were
identified using a combination of the bioinformatics pipeline described above and the
protein BLAST® search tool (NCBI) (BLASTP).
Previous work identified the core N-glycoproteome of C. jejuni using experimentally
validated and predicted N-glycoproteins from four C. jejuni strains (Frost, 2015). This list
included 109 N-glycoproteins, 65 of which have been experimentally identified as N-
glycoproteins (Frost, 2015; Scott et al., 2014, 2011; Young et al., 2002). To identify if
these experimentally identified N-glycoproteins were conserved in C. fetus, the protein
sequences were queried against C. fetus fetus 82-40 using BLASTP to identify
homologues. Matches with an E value of 0.01 or higher were not designated as
homologues. Results from the N-glycoprotein prediction pipeline were then used to
identify if the homologues contained N-glycosylation sequons (Table 6.2). Seventeen of
the C. jejuni experimentally identified N-glycoproteins had no homologues in C. fetus
(Fig. 6.8). Of the 48 C. fetus proteins with homologues among the C. jejuni N-
glycoproteins, 34 contained potential N-glycosylation sequons, 10 of which have been
experimentally identified as sites of N-glycosylation in C. fetus (Nothaft et al. 2012, Table
6.2 experimentally identified in bold). Notably, every C. fetus protein homologue of C.
jejuni outer membrane proteins contained sequons. In contrast only two of the five C. fetus
homologues of C. jejuni periplasmic N-glycoproteins contained sequons. Therefore,
overall, 31 of the C. jejuni known N-glycoproteins did not have N-glycosylation sequon-
containing homologues in C. fetus.

190
Four of the C. fetus proteins identified in this comparison, Cf0214, Cf0318, Cf0928 and
Cf1178, were not within the predicted total C. fetus N-glycoproteome established in
section 6.2.1 as there was no indication of extracytoplasmic localisation. However, further
analysis of these proteins using CELLO predicted all except Cf0928 as outer membrane.
Cf0928 was predicted as either extracellular or outer membrane with similar probabilities.
Therefore, based on homology with known C. jejuni N-glycoproteins, this comparison
revealed four sequon-containing C. fetus proteins which may be N-glycosylated and hence
could be included in its predicted N-glycoproteome. This would increase the C. fetus
predicted total N-glycoproteome to 138 proteins.
In summary, of the 65 experimentally identified C. jejuni N-glycoproteins, 34 had
homologues which also contained N-glycosylation sequons in C. fetus, suggesting a
moderate level of conservation between the N-glycosylation protein targets of these
species. If excluding C. jejuni N-glycoproteins which had no significant protein
homologue in C. fetus, as many as 71% of N-glycoproteins were predicted to be modified
in both species. Furthermore, more C. jejuni outer membrane N-glycoproteins than
periplasmic N-glycoproteins were predicted to be conserved as N-glycoproteins in C.
fetus.

191
Table 6.2. Identification of N-glycosylation sequon-containing C.fetus homologues
of known C. jejuni N-glycoproteins. Colour represents subcellular location
assignment; Blue; inner membrane. Green; periplasmic. Red; Outer membrane. Purple;
extracellular. Grey; unknown extracytoplasmic. Light grey; assigned as cytoplasmic.
‘No homol’ indicates no homologous protein was identified in C. fetus. Bold indicates
the protein has been experimentally identified as an N-glycoprotein.
Locus Tag
(Name) # Sequon(s)
Sequon
position(s)
Identity
(coverage)
Locus
Tag
(Name)
# Sequon(s)
Sequon
position
(s)
Cj0011c 1 EANFT 49-53 50 (93) Cf0510 0 - -
Cj0017c
(dsbI) 2
EINKT DNNFS
3-7
290-294 45 (99) Cf1646 0 - -
Cj0081
(cydA) 2
DNNES
ENNDT
283-287
351-355 no homol
Cj0089 1 DFNKS 73-77 no homol
Cj0114 4
ENNFT
DANLS
DSNST
ENNNT
99-103
153-157
171-175
177-181
41 (99) Cf1518 2 DLNLT
EKNSS
104-108
153-157
Cj0131 1 DDNTS 73-77 52(93) Cf1499 1 EFNDS 83-87
Cj0143c
(znuA) 1 EQNTS 26-30 44 (98) Cf1731 0 - -
Cj0152c 6
EQNNT
ETNRT DKNIS
ENNIS
ENNTT
DFNIS
126-130
163-167
182-186
188-192
193-197
250-254
32 (91) Cf0290 7
DKNNS
ESNMT
DENVT
DSNLS
DENES
DINLS
DKNTT
101-105
124-128
136-140
141-145
146-150
220-224
242-246
Cj0158 2 EKNIS
DKNHS
24-28
119-123 no homol
Cj0168c 1 DVNQT 26-30 no homol
Cj0176c 1 DLNKT 29-33 no homol
Cj0182 3 DSNST
ENNAT
EKNIS
58-62
70-74
394-398
61(84) Cf0147 0 - -
Cj0200c 1 DNNKT 33-37 no homol
Cj0235c
(secG) 2
ENNNT
DVNSS
87-91
118-122 63 (71) Cf1783 0 - -
Cj0238 2 DANIS
DENSS
24-28
56-60 42 (94) Cf1737 1 DKNAS 45-49
Cj0256
(eptC) 1 ENNHT 213-217 48 (96) Cf1723 1 DENLS 474-478
Cj0277
(mreC) 1 DQNST 91-95 44 (92) Cf0318 1 DKNST 75-79
Cj0289c
(peb3) 1 DFNVS 88-92 no homol
Cj0313 2 DLNLS
DGNIT
173-177
196-200 47 (100) Cf0272 1 ELNAT 207-211
Cj0365c
(cmeC) 2
EANYS
ENNSS
30-34
47-51 36 (96) Cf0117 1 EQNAS 354-358
Cj0366c
(cmeB) 2
DRNVS DRNAS
634-638
653-657 61 (99) Cf0118 1 EANST 638-642
Cj0367c
(cmeA) 2
DFNRS DNNNS
121-125
271-275 45 (95) Cf0119 2
DYNGT
ENNST
247-251
273-277
Cj0371 1 DLNGT 75-79 53 (93) Cf1659 0 - -

192
Cj0397c 1 DFNNT 105-109 37 (98) Cf1147 1 DSNNT 104-108
Cj0399 1 DLNNT 179-183 45 (100) Cf1144
(cvpA) 2
DLNRT
DSNET
160-164
180-184
Cj0454c 1 ENNKS 91-95 32 (90) Cf1372 1 ELNVT 121-125
Cj0494 1 DNNIT 26-30 no homol
Cj0508 1 DANLS 312-316 60 (97)
Cf1032 0 - -
Cj0511 1 DQNIS 67-71 66 (99) Cf0944 2 DQNFT
DGNST
64-68
145-149
Cj0515 3 DFNIS ELNAT
DFNAS
103-107
207-211
234-238
38 (90) Cf0948 2 DLNLT
EANST
96-100
130-134
Cj0530 5
DGNLS
DFNAS DFNIT
DFNAS
DSNKT
156-160
180-184
389-393
519-523
617-621
28 (77) Cf0928 8
DYNIS
DSNLT
DINDT
DLNIT
DANSS
DLNRT
DGNFT
DINMT
2-6
164-168
260-264
272-276
391-395
416-420
478-482
578-582
Cj0587 1 DNNLS 282-286 no homol
Cj0592c 4
DINQS
ENNES
ENNQS
DVNMT
97-101
103-107
127-131
137-141
no homol
Cj0599 3 EANIT
DLNST
DNNIT
97-101
109-113
168-172
50 (99) Cf0517 2 DINST
DLNST
111-115
157-161
Cj0608 2 DLNLT DANNT
35-39
412-416 36 (97) Cf0760 1 DFNQT 38-42
Cj0610c 5
DENLS DENTS
DANIS
ENNRS
EENAS
82-86
98-102
113-117
296-300
331-335
no homol
Cj0633 8
DNNKS
DLNTS
EQNLS
EQNLS
EQNQS
DTNLT
DQNLT
DKNHS
73-77
90-94
97-101
104-108
110-114
123-127
129-133
180-184
39 (94) Cf1126 6
DKNIS
DKNIS
DSNIS
DKNIT
DQNIT
DENSS
69-73
82-86
97-101
104-108
133-137
141-145
Cj0648 3 ESNTS
DFNLS
EGNVT
49-53
82-86
103-107
31 (95) Cf1187 2 ELNST
DDNIT
41-45
90-94
Cj0652
(pbpC) 3
DRNGT
DLNAS
ENNNT
55-59
99-103
467-471
54 (99) Cf1182 1 DLNVT 96-100
Cj0694 3 DFNKT
DQNIS DQNSS
132-136
306-310
426-430
42 (99) Cf0569 2 DVNDT
ESNAT
239-243
297-301
Cj0734c
(hisJ) 1 ESNAS 27-31 48 (89) Cf1232 0 - -
Cj0776c 3
DENQS
ENNQS
DTNTS
87-91
103-107
111-115
no homol
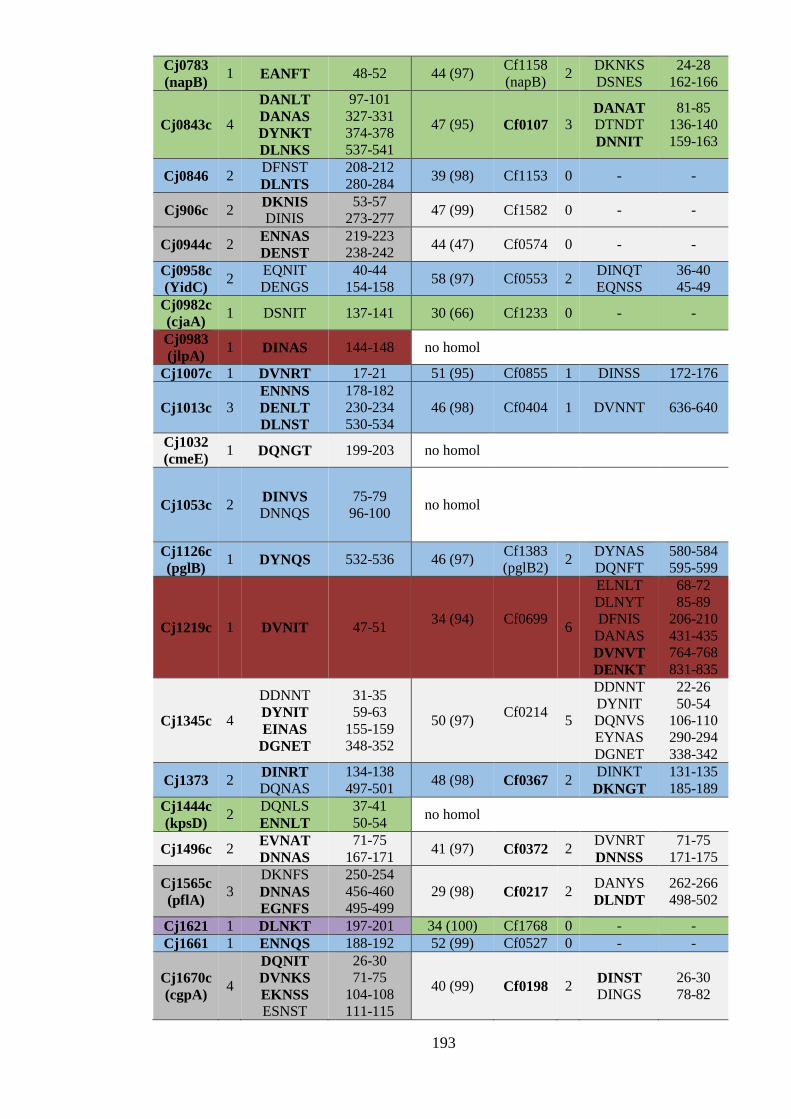
193
Cj0783
(napB) 1 EANFT 48-52 44 (97)
Cf1158
(napB) 2
DKNKS
DSNES
24-28
162-166
Cj0843c 4
DANLT
DANAS
DYNKT
DLNKS
97-101
327-331
374-378
537-541
47 (95) Cf0107 3 DANAT
DTNDT
DNNIT
81-85
136-140
159-163
Cj0846 2 DFNST
DLNTS
208-212
280-284 39 (98) Cf1153 0 - -
Cj906c 2 DKNIS
DINIS
53-57
273-277 47 (99) Cf1582 0 - -
Cj0944c 2 ENNAS
DENST
219-223
238-242 44 (47) Cf0574 0 - -
Cj0958c
(YidC) 2
EQNIT
DENGS
40-44
154-158 58 (97) Cf0553 2
DINQT
EQNSS
36-40
45-49
Cj0982c
(cjaA) 1 DSNIT 137-141 30 (66) Cf1233 0 - -
Cj0983
(jlpA) 1 DINAS 144-148 no homol
Cj1007c 1 DVNRT 17-21 51 (95) Cf0855 1 DINSS 172-176
Cj1013c 3 ENNNS
DENLT
DLNST
178-182
230-234
530-534
46 (98) Cf0404 1 DVNNT 636-640
Cj1032
(cmeE) 1 DQNGT 199-203 no homol
Cj1053c 2 DINVS DNNQS
75-79
96-100 no homol
Cj1126c
(pglB) 1 DYNQS 532-536 46 (97)
Cf1383
(pglB2) 2
DYNAS
DQNFT
580-584
595-599
Cj1219c 1 DVNIT 47-51 34 (94)
Cf0699
6
ELNLT
DLNYT
DFNIS
DANAS
DVNVT
DENKT
68-72
85-89
206-210
431-435
764-768
831-835
Cj1345c 4
DDNNT
DYNIT
EINAS
DGNET
31-35
59-63
155-159
348-352
50 (97) Cf0214
5
DDNNT
DYNIT
DQNVS
EYNAS
DGNET
22-26
50-54
106-110
290-294
338-342
Cj1373 2 DINRT DQNAS
134-138
497-501 48 (98) Cf0367 2
DINKT
DKNGT
131-135
185-189
Cj1444c
(kpsD) 2
DQNLS
ENNLT
37-41
50-54 no homol
Cj1496c 2 EVNAT
DNNAS
71-75
167-171 41 (97) Cf0372 2
DVNRT
DNNSS
71-75
171-175
Cj1565c
(pflA) 3
DKNFS
DNNAS
EGNFS
250-254
456-460
495-499
29 (98) Cf0217 2 DANYS
DLNDT
262-266
498-502
Cj1621 1 DLNKT 197-201 34 (100) Cf1768 0 - -
Cj1661 1 ENNQS 188-192 52 (99) Cf0527 0 - -
Cj1670c
(cgpA) 4
DQNIT
DVNKS
EKNSS ESNST
26-30
71-75
104-108
111-115
40 (99) Cf0198 2 DINST
DINGS
26-30
78-82

194
6.4.2 Predicted conservation of N-glycoproteins among further Campylobacter species
Section 6.4.1 identified 34 experimentally identified C. jejuni N-glycoproteins which also
have predicted N-glycoprotein homologues in C. fetus. To identify if these putatively
conserved N-glycoproteins were also found in other Campylobacter species, the same
methodology was applied to species closely related to C. jejuni and C. fetus: C. coli
RM4661, C. lari RM2100, C. hyointestinalis LMG 9260 and C. concisus 13826. The
findings are displayed in Table 6.3 and the percent identity between each C. jejuni known
N-glycoprotein and its homologue is shown. Precent identity was found using BLAST®
global alignment tool.
Nineteen of the thirty-four C. jejuni N-glycoproteins had homologues containing N-
sequons in all Campylobacter species analysed (Table 6.3). As previously, this conserved
set contained mostly predicted inner membrane proteins. Three were predicted as outer
membrane proteins and none were predicted to reside in the periplasm. One N-
glycoprotein (Cj0843c) did not have a homologue in C. coli but in all other species there
were N-sequon-containing homologues. The remaining fourteen proteins had homologues
in all species tested, but N-sequons were not identified in every homologue. Eleven
proteins lacked an N-sequon-containing homologue in a single species whereas three
proteins lacked N-sequon-containing homologues in two species.
No overall pattern in the presence/absence of N-sequons was immediately apparent and
the conservation of sequons did not appear to correlate with percentage identity. For
example, homologues of Cj0530 had low percentage identity (as low as 23%) but
contained sequons whereas the C. coli homologue of NapB has high (84%) identity but
lacks a sequon. Therefore, it appeared that the observed conservation of N-glycosylation
sequon occurrence was independent of amino acid sequence identity.
In conclusion, there appears to be moderate conservation of proteins predicted to be N-
glycosylated between different Campylobacter species. Nineteen C. jejuni N-
glycoproteins had putative N-glycoprotein homologues in the five Campylobacter species
tested. This suggests that these proteins may be ‘core’ Campylobacter N-glycoproteins
and make promising candidates for experimentally addressing if N-glycosylation can
influence protein function.

195
6.4.2.1 N-glycosylation of components of the CmeABC multidrug efflux system is
predicted to be conserved in several Campylobacter species
The C. jejuni CmeABC multidrug efflux system (Lin et al., 2002) is a known colonisation
factor in chickens (Hermans et al., 2011). Mutation of the CmeABC locus or its regulator,
CmeR, can increase the susceptibility of C. jejuni to bile salts and a variety of
antimicrobials (Lin et al., 2002). Moreover, a CmeB mutant is unable to colonise chickens
(Lin et al., 2003). All three components have been experimentally identified as N-
glycoproteins in C. jejuni (Scott et al., 2011; Wacker et al., 2002). CmeA (also known as
AcrA) is the most extensively studied and is often used as a ‘model’ N-glycoprotein
(Nothaft et al., 2010). It has been reported that abolishing CmeA N-glycan modification
confers reductions in antimicrobial resistance and chicken colonisation similar to a cmeA
knockout, suggesting modification significantly influences its function (Nothaft and
Szymanski, 2013). In addition, Alemka et al. (2013) described data which suggests N-
glycosylation of CmeA provides resistance from proteolysis. However, both these data
sets remain unpublished.
N-glycosylation sequon-containing homologues of CmeA and CmeC were identified in C.
fetus, C. coli, C. lari, C. concisus and C. hyointestinalis. However, N-sequon-containing
homologues of CmeB were only identified in C. coli and C. lari, not C. concisus or C.
hyointestinalis. Further analysis of the predicted sequons present in the identified
homologues revealed that in other Campylobacter species, components were often
predicted to contain only a single sequon. However, although each C. jejuni component
has two predicted N-glycosylation motifs, only one sequon has been experimentally
identified from each component. This may explain why only one motif is conserved per
protein in other species.
In conclusion, N-glycan modification is a putatively conserved feature of the CmeABC
system in Campylobacter species, with CmeA and CmeC being predicted N-glycoproteins
in the further five species studied. CmeB is conserved as a sequon-containing predicted N-
glycoprotein in three of the five species studied. Therefore, this bioinformatics analysis
further suggests that N-glycosylation may be important for the CmeABC multidrug efflux
system.

196
Table 6.3 Putative conservation of experimentally identified C. jejuni N-
glycoproteins in Campylobacter species. Background colour of C. jejuni N-
glycoproteins represents subcellular location assignment; Blue; inner membrane.
Green; periplasmic. Red; Outer membrane. Purple; extracellular. Grey; unknown
extracytoplasmic. Light grey; assigned as cytoplasmic. No colour indicates that
the homologue is also a predicted N-glycoprotein. Dark grey background
indicates that the homologue does not contain a sequon. The percentage identity is
shown for each homologue (identified using BLAST® global alignment). A cross
indicates there is no significant homologue. Bold and underlined indicates the
homologue has been experimentally identified as an N-glycoprotein in C. fetus
(Nothaft et al., 2012).
Predicted N-glycoprotein homologue in Campylobacter
species
C. fetus C. coli C. lari C. hyo C. concisus
C. je
jun
i p
rote
in n
ame
CmeB 60 98 75 59 59
NapB 44 84 65 45 49
Cj0131 50 99 60 50 50
Cj0114 40 75 52 40 36
EptC 47 75 51 46 22
MreC 41 75 54 44 44
Cj0313 47 78 54 46 47
Cj0399 45 81 59 44 41
Cj0454c 29 59 31 27 26
Cj0515 35 64 45 35 34
Cj0843c 45 54 43 41
Cj1013c 46 79 63 46 47
PglB 45 81 56 45 47
Cj1219c 33 72 43 32 31
Cj1373 47 83 60 45 46
Cj0152c 29 62 38 31 30
Cj0238 41 84 53 42 39
CmeC 35 88 54 37 33
CmeA 43 88 56 43 43
Cj0397c 37 65 43 40 39
Cj0511 65 89 67 64 64
Cj0530 24 69 39 30 31
Cj0599 49 91 63 49 42
Cj0608 35 79 49 33 33
Cj0633 36 62 45 38 30
Cj0648 30 75 42 29 32
PbpC 54 82 60 54 54
Cj0694 42 73 48 42 40
Cj0958c 56 81 68 56 56
Cj1007c 49 83 56 50 45
Cj1345c 48 91 66 49 46
Cj1496c 36 88 59 38 38
PflA 28 80 47 31 31
CgpA 39 78 43 37 28

197
6.5 Discussion
This chapter described the in silico prediction of the total C. fetus N-glycoproteome and
investigated the conservation of N-linked glycosylation sequon occurence among proteins
of several Campylobacter species. By combining an available experimental data set
(Nothaft et al., 2012) and that of a bioinformatics pipeline, a total C. fetus predicted N-
glycoproteome containing 134 proteins was identified. However, the bioinformatics
pipeline only predicted 80% of the experimentally identified C. fetus N-glycoproteins,
highlighting flaws in its ability to correctly predict the subcellular location of proteins.
Further analysis of these N-glycoproteins not identified by the pipeline using online
prediction servers, CELLO and SignalP, identified them as extracytoplasmic. Therefore, it
would improve sensitivity of the pipeline if these tools were integrated in the future.
Nevertheless, these data suggest that N-glycan modification is a significant feature of C.
fetus, occurring to a similar extent as in C. jejuni, which has 148 predicted N-
glycoproteins (Frost, 2015). These figures correspond to 7.8 % and 9.5 % of total proteins
predicted in C. fetus and C. jejuni, respectively. These results correlate well with the
results of probing C. fetus lysates with C. fetus N-glycan-reactive antibodies, which
previously indicated the presence of a multitude of N-glycoproteins (Nothaft et al. 2012,
Chapter 4). Therefore, this bioinformatics analysis further suggests that Campylobacter
species N-glycosylate a considerable number of proteins. However, despite significant
advances in understanding the mechanism of N-linked glycosylation in Campylobacter,
the question as to why Campylobacter proteins are N-glycosylated remains unanswered.
Furthermore, whether N-glycan modification is required for the function of specific
proteins or serves a more general role remains unclear. Only limited experimental data
exists surrounding these questions and further investigation to gain a more holistic view of
Campylobacter N-glycosylation may provide insight into these questions. Towards this
end, data gained using the N-glycoprotein prediction pipeline was analysed in further
detail.
Further analysis of the C. fetus predicted N-glycoproteome found that most were predicted
to reside in the inner membrane, which is also true for predicted C. jejuni N-glycoproteins
(Frost, 2015). Predicted outer membrane N-glycoproteins were twice as abundant as
predicted periplasmic N-glycoproteins, although total numbers of predicted outer
membrane and periplasmic proteins are more similar. Although predicted outer membrane
N-glycoproteins were also more (1.4X) abundant than periplasmic in C. jejuni (Frost,

198
2015), the difference is more marked in C. fetus. As N-glycosylation has been reported to
confer resistance against chicken gut protease activity (Alemka et al., 2013), this relatively
higher abundance of predicted outer membrane N-glycoproteins may reflect a preferential
requirement for surface-exposed proteins to be modified with N-glycan to provide such
protection.
The distribution of sequons among N-glycoproteins was also investigated. Using the
experimentally identified N-glycosylation sequons identified from C. fetus, sequon
frequency per protein was between 1 and 4, with 76% of the identified N-glycoproteins
harbouring a single sequon. Analysis of N-glycosylation motifs identified in the C. fetus
predicted N-glycoproteome found that although most (64%) proteins were still predicted
to contain a single site, a larger proportion (25%) was predicted to be modified at two
sites. Furthermore, proteins with between 1 and 11 potential modification sites were
identified. Structural prediction of the N-glycoprotein with the most predicted sequons
(Cj0312) suggested the 11 sequons reside outside the cell. Importantly, one of these
sequons was experimentally identified in a glycopeptide by Nothaft et al. (2012),
providing evidence that it is indeed an N-glycoprotein. Extensive N-glycosylation has
been documented in a C. jejuni protein, Cj0633, which has 8 modification sites that can be
modified in the available E. coli Pgl system (Frost, 2015). In the future, it would be
interesting to confirm if Cf0312 is indeed N-glycosylated to such a high extent, and if so,
would make it the most extensively N-glycosylated protein reported in ε-proteobacteria
thus far. It would also be the N-glycoprotein containing the second-most modification
sites in Bacteria overall, with HMW1 of Haemophilus influenzae having 31 sites modified
with mono or di-hexoses (Gross et al., 2008). Furthermore, it may be that such high level
N-glycosylation protects this C. fetus protein from degradation by host gut proteases, as
suggested in C. jejuni (Alemka et al., 2013).
The amino acid composition of sequons from experimentally identified C. fetus N-
glycoproteins was analysed, finding that the most common motif was D-X-N-X-T. This
motif was also most abundant in the sequons of the total C. fetus predicted N-
glycoproteome. This is in agreement with in vitro assays investigating the activity of C.
jejuni and C. lari PglB enzymes, which found that D and T at the -2 and +2 positions,
respectively, confer the highest efficiency of glycosylation (Gerber et al., 2013; Kowarik
et al., 2006b). However, this finding is in contrast to the composition of sequons in C.
jejuni, where Ser is more prominent at the +2 position when analysing experimentally
identified N-glycoproteins or the total predicted set (Frost, 2015). These findings suggest
that D-X-N-X-T may be the preferred N-glycosylation sequon in C. fetus. However, as

199
these data rely mostly on predicted and not proven N-glycosylation sequons, they must be
taken with caution and further experimental validation is required. Futhermore, it may be
that the most efficient N-glycosylation sequons are not the most abundant.
Although the bioinformatics pipeline predicted numerous N-glycoproteins in C. fetus, the
presence of an extended D/E-X-N-X-S/T sequon is not sufficient for glycosylation.
Although it was originally thought that N-glycosylation sites had to reside within flexible
loops in order to be modified in C. jejuni (Kowarik et al. 2006), more recent evidence
suggests that this is not the case. The crystal structures of C. jejuni N-glycoproteins JlpA
and PEB3 revealed N-glycosylation sites present in an alpha-helix and structured turn,
respectively (Kawai et al., 2012; Rangarajan et al., 2007). Silverman & Imperiali (2016)
have since demonstrated that N-linked glycosylation is coupled with protein translocation
mediated by the Sec pathway and hence N-glycoproteins can be modified prior to
complete folding. Nevertheless, although it appears that PglB can modify regions within a
variety of protein structures, if sequons reside within buried structural features they may
not be modified (Silverman and Imperiali, 2016). Therefore, some of the predicted N-
glycosylation sequons identified in this chapter may not be accessible. Nevertheless, His-
tagged versions of two predicted N-glycoproteins identified by the workflow were shown
to be modified by PglB in C. fetus. Glycopeptides from the first protein, Cf0718, had been
previously identified by Nothaft et al. (2012) and hence this served as a proof of principle
to demonstrate that N-glycosylation could be successfully validated using this approach.
Secondly, PglB-mediated modification of a previously unknown predicted N-glycoprotein,
Cf0445, was demonstrated. This provides some evidence towards the accuracy of the N-
glycoprotein predictions of the bioinformatics pipeline; however, more extensive
experimental validation/identification is required.
Limited experimental evidence has demonstrated that N-glycosylation is not required for
the function of individual proteins in C. jejuni. Some evidence suggests that N-
glycosylation instead serves a more general role and provides protection from chicken gut
proteases (Alemka et al., 2013). However, it could be that particular proteins have a
stronger requirement for N-glycosylation to provide such protection than others. For
example, N-glycosylation may be particularly advantageous for surface-exposed proteins.
Nevertheless, it can be argued that if a protein requires N-glycosylation, whether it is for
protein function or protection, there would be an evolutionary pressure to maintain this
modification. Therefore, understanding the conservation of N-glycoproteins among
Campylobacter species may provide suggestive evidence of proteins which require N-
glycosylation to function and contribute to uncovering the role of Campylobacter N-linked

200
glycosylation. Towards this end, the conservation of sequon occurrence for experimentally
validated C. jejuni N-glycoproteins which appear to be core N-glycoproteins for this
species was analysed. Thirty-four of sixty-five experimentally identified C. jejuni N-
glycoproteins were predicted to be N-glycosylated in C. fetus, and ten of these were
previously experimentally identified as C. fetus N-glycoproteins (Nothaft et al., 2012).
Interestingly, more outer membrane proteins were conserved as predicted N-glycosylation
targets than periplasmic proteins. This finding may reflect the need for certain surface-
exposed proteins to be N-glycosylated to provide protection from protease activity. Some
of the C. fetus homologues identified were not characterised as predicted N-glycoproteins
by the bioinformatics workflow but did contain N-glycosylation sequons. This issue was
alleviated by manual analysis using CELLO which predicted the proteins were
extracytoplasmic, further highlighting that improvements to the localisation predictive
ability of the bioinformatics workflow could be made. Therefore, this analysis also
identified another four C. fetus predicted N-glycoproteins, increasing the total predicted N-
glycoproteome to contain 138 proteins. The conserved C. jejuni proteins cover a range of
functions, including motility (PflA) (Yao et al., 1994), drug efflux (CmeABC)(Lin et al.,
2002), post-translational modification (EptC) (Scott et al., 2012), nitrate reduction (NapB)
(Pittman et al., 2007) and cell shape determination (MreC) (Divakaruni et al., 2007).
These data suggested that the targets of N-glycan modification may be moderately
conserved in Campylobacter and warranted investigation into conservation among other
species.
Further analysis identified that of the 34 C. jejuni N-glycoproteins with homologues
predicted to be N-glycosylated in C. fetus, 19 of these were also predicted to be N-
glycoproteins in another four Campylobacter species (C. coli, C. lari, C. hyointestinalis
and C. concisus). The conservation of N-glycosylation sequons did not appear to strictly
correlate with high percentage identity homologues which suggested N-glycan
modification may be required for the function of certain proteins. However, it may be that
N-glycosylation serves a general role, such as protection from proteases (Alemka et al.,
2013), and that certain proteins have a stronger requirement for this. For example, more
surface exposed proteins may benefit from such protection more than proteins present
inside the cell. In agreement with this, there were generally more predicted outer
membrane N-glycoproteins than periplasmic and more outer membrane than periplasmic
N-glycoproteins were putatively conserved among species. However, an argument against
this theory is that the majority of N-glycoproteins are not surface exposed as most are

201
predicted to be inner membrane proteins. Therefore, it is clear that further experimental
study into the role of N-glycosylation in protein function is warranted.
Interestingly, this analysis found that ZnuA and JlpA are not conserved as N-glycoproteins
throughout these Campylobacter species. This is consistent with experimental evidence
suggesting that N-glycosylation is not required for their function in C. jejuni (Davis et al.,
2009; Scott et al., 2009). Therefore, it can be speculated that these proteins are not
representative in such studies, and that further analysis of specific conserved N-
glycoproteins needs to be undertaken. For example, the conserved proteins included
CmeA and CmeC from the tripartite drug efflux system, CmeABC (Lin et al., 2002), and
PflA (Yao et al., 1994), a protein associated with motility in Campylobacter. It would be
interesting to identify if N-glycosylation is essential for the function of these
Campylobacter proteins in particular.
In summary, the complete C. fetus predicted N-glycoproteome containing 138 candidate
targets was identified. The general features of this N-glycoproteome bear similarity with
the predicted N-glycoproteome of C. jejuni, containing largely inner membrane proteins
followed by outer membrane and periplasmic proteins. The analysis revealed an N-
glycoprotein with 11 potential N-glycosylation sites which, if fully modified, would make
it the most extensively N-glycosylated protein in ε-proteobacteria to date. Two of the
predicted N-glycoproteins were demonstrated to be modified by C. fetus PglB in vivo,
providing limited evidence of the robustness of this predictive approach. Lastly,
conservation of predicted N-glycoproteins across Campylobacter species was investigated.
Of the 65 experimentally confirmed C. jejuni N-glycoproteins thought to be part of the
‘core’set for the species, 34 had homologues predicted to be N-glycosylated in C. fetus.
Further analysis revealed that 19 are also predicted to be N-glycoproteins in C. coli, C.
lari, C. hyointestinalis and C. concisus, suggesting some conservation of N-glycoproteins
between several Campylobacter pathogens covering broad host niches. Interestingly, this
set did not include N-glycoproteins for which N-glycan modification is not thought to be
responsible for function. Instead, it may be that the putatively conserved N-glycoproteins
identified here have a requirement for N-glycosylation. These findings inform the wider
question of what is/are the function(s) of the N-glycosylation of so many Campylobacter
proteins and identify conserved N-glycoproteins for which further study is attractive.

202
Chapter 7. Conclusion and future work

203
7.1 N-linked glycoprotein glycans as targets for antibody-based
detection/identification
One focus of this thesis was assessing Campylobacter N-linked glycans as targets for
antibody-based detection of Campylobacter species. Chapter 3 described the
characterisation of an antiserum, CjNgp, raised against a recombinant C. jejuni N-
glycoprotein, NGRP. This antiserum reacted with numerous N-glycoproteins in many C.
jejuni strains, including strains recently isolated from chicken meat. CjNgp also reacted
with the N-glycoproteins of C. coli, C. upsaliensis, C. helveticus and, to a lesser extent, C.
lari. CjNgp could also detect untreated cells of these Campylobacter species. The
antiserum contained antibodies reactive with both the carrier protein, NGRP, and the
associated N-linked glycan, but it was demonstrated that both types of antibodies
interacted with C. jejuni cells. Furthermore, flow cytometry demonstrated that CjNgp
labelled whole cells of C. jejuni, although to a lesser extent that an anti-whole cell C.
jejuni antiserum. One advantage of CjNgp over the whole cell antiserum was that it does
not cross react with C. fetus. Therefore, CjNgp shows promise as a tool for detecting both
the common human Campylobacter pathogens (C. jejuni and C. coli) and also emerging
pathogens found in poultry (C. upsaliensis and C. lari) without cross-reacting with other
Campylobacter species such as C. fetus and C. hyointestinalis. However, these species are
not found in chicken and would not usually pose a cross-reactivity risk. Instead, these
species can be found in cattle and swine and therefore this suggests that a detection test
using CjNgp could also be used to identify Campylobacter species such as C. jejuni and C.
coli from cattle and pigs without producing false-positives with species such as C. fetus
and C. hyointestinalis, respectively. For example, although poultry is the main reservoir
for human campylobacteriosis (EFSA, 2010), one study found that over 70% of beef liver
products are infected with Campylobacter (Noormohamed and Fakhr, 2013). Therefore,
such a tool for detecting C. jejuni, C. coli and emerging Campylobacter pathogens from
other farmed animals could also be useful.
Chapter 4 described the development and characterisation of a novel antiserum, CfNgp,
raised against a recominant N-glycoprotein containing C. fetus N-linked glycan. This
involved expression of the recombinant N-glycoprotein to be used as immunogen, NGRP,
in C. fetus and subsequent purification. The resulting polyclonal antiserum was reactive
with N-linked glycoproteins of C. fetus fetus, C. fetus venerealis and a variety of C. fetus
clinical isolates. CfNgp displayed broad reactivity with Campylobacter species,

204
particularly with C. hyointestinalis, C. consisus, C. sputorum and C. lanienae. This
antiserum was also able to bind Campylobacter cells. Protein-reactive antibodies within
the antiserum partly contributed to this ability. Overall, these data suggest that CfNgp
could be utilised to detect C. fetus and also less common Campylobacter pathogens, such
as C. hyointestinalis and C. concisus. Notably, CfNgp was similarly reactive with C. fetus
and C. sputorum. This cross-reactivity poses a significant disadvantage to using this
antiserum for detecting C. fetus in cattle as C. sputorum is found in cattle but does not
cause disease in these animals. However, species such as C. sputorum and C. concisus can
cause gastroenteritis in humans (Lindblom et al., 1995) and therefore it appears this
antiserum could be better suited for detection of Campylobacter species from human
samples. Furthermore, Campylobacter species including C. concisus and C. ureolyticus
are particularly difficult to isolate and culture in the laboratory and such a detection test
could improve detection of these species. However, as the previously described C. fetus N-
glycan-reactive GRP-II antisera display very little or no reactivity against species such as
C. concisus and C. sputorum (Nothaft et al., 2012), it can be argued that, overall, C. fetus
N-linked glycan is still a potential target for antibody-based detection of C. fetus in cattle.
This research instead highlights that N-glycan-reactive antibodies with stricter specificity
would be favourable for such a purpose.
One significant feature of both antisera is the presence of protein-reactive antibodies,
particularly those reactive with NGRP and its native progenitor, Cj0114. These antibodies
influence the ability of CjNgp and CfNgp to bind cells of C. jejuni and C. fetus,
respectively. There are homologues of Cj0114 in C. coli, C. upsaliensis, C. lari and
C. fetus. In light of this, the CjNgp cell binding reactivity observed with these species
could be markedly influenced by NGRP reactivity within the antiserum. Similarly, such
reactivity within CfNgp may influence the observed cell binding of C. jejuni and others
tested here. Therefore, such protein reactivity is a potential issue which needs to be
addressed further. It would be interesting to purify the N-glycan-reactive antibodies from
these antisera and analyse if their Campylobacter specificity is altered. Furthermore,
purification and concentration of N-glycan-reactive antibodies may make the reagents
more reactive towards target Campylobacter cells. For example, carrying this out with
CjNgp could produce a reagent which is more effective at labelling C. jejuni cells than the
anti-whole cell C. jejuni 11168 antiserum. Furthermore, the sensitivity of this reagent may
be more consistent when detecting cells of C. jejuni, C coli and C. upsaliensis. This would
provide an advantage over currently available detection methods, as they can have reduced
sensitivity for species other than C. jejuni (Sapsford et al., 2004). Antibody purification

205
could be achieved using an affinity chromatography column containing an N-glycoprotein
other than NGRP/Cj0114. Alternatively a column containing the fOS produced by each
species could be developed. Purification of fOS could be undertaken according to the
protocol of Nothaft et al. (2012), which was successfully used to create fOS-BSA
conjugate immunogens for producing the C. fetus N-glycan-reactive GRPII sera. Such
affinity chromatography columns would retain N-glycan-reactive antibodies, which could
be eluted from the column upon removing the non-N-glycan-reactive antibodies and other
serum components. These approaches would generate high-titre reagents specific for the
respective N-linked glycan structures and may allow more sensitive detection of the
corresponding Campylobacter species.
Such purified anti-N-glycan antibodies could subsequently be integrated into detection
platforms such as LFDs or biosensors and their performance assessed in a more ‘real
world’ applications. Such analysis could include determining the sensitivity and
specificity of the platform. In particular, the occurrence of cross-reactivity with bacterial
species often resident within poultry caeca and human faeces would be necessary for
CjNgp and CfNgp, respectively. For example, Clostridium, Bacteroides, and
Gallibacterium species are often found in poultry caeca (Pauwels et al., 2015; Stanley et
al., 2015). In line with this, the compatibility of such assays when directly testing
poultry/human samples could be investigated.
As NGRP-reactivity was a significant feature of the antisera described here, it may be
more appropriate to use an alternative N-glycoprotein antigen when producing such
antisera in the future. For example, Nothaft et al. (2016) have developed a small
‘GlycoTag’ from a C. jejuni N-glycoprotein which is N-glycosylated at nine sites and was
readily expressed and purified from E. coli. Such a small protein may result in less
protein-reactive antibodies in the antiserum. Alternatively, an N-glycoprotein unique to
the respective target Campylobacter species could be used so that any resulting protein
reactivity should not cause cross-reactivity with other species. This could be particularly
useful for further development of C. fetus N-glycan-reactive antisera so that the cross-
reactivity with C. jejuni and other species is reduced. Furthermore, the development of
glycocompetent E. coli producing the C. fetus N-linked glycan would provide a more
effective method of producing and purifying large quantities of C. fetus N-glycoprotein
(See below) and an alternative carrier protein, for example GlycoTag, could be introduced
into such a system. Another option would be to use fOS-BSA conjugates as immunogen,
which was successfully utilised by Nothaft et al. (2012) to develop the GRP-II sera. As
described in section 4, these antisera displayed stricter Campylobacter species specificity

206
than CfNgp as they did not react with C. concisus. However, it must be noted that
polyclonal antisera can display batch-to-batch variation and such specificity may not be
reproducible. In conclusion, this study has highlighted the limitations of using polyclonal
antisera and it can be argued that monoclonal antibodies would be more suitable in the
future. However, the production of monoclonal antibodies is more time-consuming and
expensive than raising polyclonal sera and these findings demonstrate that the latter are a
feasible tool to preliminarily assess potential target antigens.
Nevertheless, in their current form, these antisera will be useful for N-glycosylation
research. CjNgp can be used similarly as the previously available hR6 antiserum, and
assist investigation into the N-linked glycosyation systems of C. coli, C. lari and others.
Due to its broad reactivity against Campylobacter species, CfNgp will prove a valuable
tool for Campylobacter N-glycan research into the less established systems in species such
as C. fetus and C. concisus. For example, C. concisus harbours two pglB enzymes and this
antiserum could aid the study of their activities. In addition, CfNgp has the potential to be
used for N-glycoprotein pulldown experiments to identify N-glycoproteins in multiple
Campylobacter species.
7.2 Developing glycocompetent E. coli producing the C. fetus N-
linked glycan.
Chapter 5 described a strategy toward developing glycocompetent E. coli able to
synthesise the C. fetus N-glycan structures. By exploiting the known structural features of
the C. jejuni and C. fetus N-linked glycan, this approach used part of the C. jejuni Pgl
system to produce a trisaccharide N-glycan which could be used as a basal substrate by
predicted C. fetus glycosyltransferases. Introducing C. fetus pglH1 and pglH2 into this
system, respectively, demonstrated that these genes encoded enzymes able to sequentially
modify this truncated N-glycan. Structural analysis revealed that PglH1 and PglH2 could
each add a HexNAc sugar, likely GlcNAc residues given the structure of C. fetus N-
glycan. Although this approach of combining two N-linked glycosylation pathways was
partially successful, it was limited by unexpected interactions with the components of the
two systems. Upon introduction of PglH1, a Hex sugar was also transferred along with the
presumed GlcNAc, however, the enzyme responsible for this transfer is to be determined
as C. jejuni PglI may have interfered with the system. Another predicted

207
glycosyltransferase, encoded by Cf1389, did not contribute to further N-glycan
biosynthesis and rather appeared to cause deleterious effects in the glycocompetent E. coli.
Subsequent genome analysis suggests that this glycosyltransferase is instead part of a
prophage O-antigen modification locus. Nevertheless, this work successfully developed a
hybrid C. jejuni/C. fetus N-glycosylation system in E. coli and provided strong evidence
that C.fetus PglH1 and PglH2 are active glycosyltransferases that act sequentially to add
GlcNAc residues to the N-linked glycan structure.
The main limitation of this work is the likely interference by the C. jejuni PglI enzyme.
Therefore, future work would require C. jejuni PglI to be removed from the system and
could be achieved by insertional mutagenesis of pglI within ppglpglH::kn. Re-analysis of
the N-glycan structures produced in the absence of C. jejuni pglI would identify if the Hex
residue in question was still transferred upon introduction of C. fetus pglH1. If so, it would
seem that C. fetus PglH1 was responsible for the transfer of this residue. However, if the
N-glycan structure did not contain a Hex residue in the absence of C. jejuni pglI it would
suggest that the presence of this residue was due to interference from the C. jejuni
pathway. In this case, it could be concluded that C. fetus PglH1 is responsible for the
transfer of the HexNAc residue, likely a GlcNAc, only. Such activity would better reflect
its homology with C. jejuni PglH enzyme which transfers linear GalNAc residues to the
C. jejuni N-glycan. NMR could then be used to characterise the specific sugars and
glycosidic linkages present to confirm that the HexNAc residues transferred by C. fetus
glycosyltransferases are GlcNAc residues, as predicted.
Another limitation of this approach was the difficulty in obtaining N-glycan structural data
using MALDI-TOF-MS. Although using an improved version of NGRP, NGRP3,
somewhat improved the outcome of the MALDI-TOF-MS analysis, it was not sufficient.
Therefore, LC-MS/MS anaylsis was undertaken to alleviate these issues. However, LC-
MS/MS is a more costly technology and produces large quantities of spectra which require
time-consuming data analysis. In contrast, MALDI-TOF-MS is a cheaper and simple
alternative. Another variant of NGRP, modified to produce a further positively charged N-
glycan sequon-containing tryptic peptide, was successfully used by Mills et al. (2016) to
gain N-glycan structural data using MALDI-TOF-MS. Introducing this NGRP variant into
the experimental strategy described here could therefore improve the applicability of this
approach.
Finally, to identify the glycosyltransferase(s) responsible for transferring the remaining
C. fetus N-linked glycan residues, predicted glycosyltransferase genes common to C. fetus

208
and C. hyointestinalis could be introduced into the system. Such investigation is now
possible with the recent availability of complete C. hyointestinalis genome sequences
(Miller et al., 2016). Development of the full C. fetus N-glycan structures in E. coli would
allow more rapid and high-yield production of N-glycoprotein, which could readily
provide antigen for antibody purification (see above). Furthermore, as the majority of
Campylobacter N-linked glycan structures are structurally identical at the reducing end,
this work provides an experimental framework which could be adapted for investigating
glycosyltransferase enzymes of other Campylobacter species.
7.3 The C. fetus predicted N-glycoproteome and putative
conservation of N-glycoproteins amongst Campylobacter species.
Chapter 6 described prediction of the C. fetus N-glycoproteome using a bioinformatics
pipeline which analyses the predicted subcellular locations of proteins and the occurrence
of N-glycosylation sequons. Combining experimental data and the results of the N-
glycoprotein prediction pipeline, it was found that there are at least 134 possible N-linked
glycoproteins in C. fetus. PglB modification of two predicted N-glycoproteins, Cf0718 and
Cf0445, the latter of which was previously unknown as an N-glycoprotein, was validated
in C. fetus. However, the bioinformatics pipeline did not predict some previously
experimentally identified N-glycoproteins due to incorrect assignment of their subcellular
location, whereas manual analysis using another localisation prediction tool, CELLO,
identified these proteins as extracytoplasmic. Therefore, it appears the subcellular location
predictive ability of the pipeline could be improved if this tool was integrated.
Analysis of the C. fetus predicted N-glycoproteome found that it generally had similar
features to that previously found with C. jejuni (Frost, 2015; Scott et al., 2014). For
example, investigating the predicted subcellular localisations of C. fetus predicted N-
glycoproteins found that most N-glycoproteins are likely inner membrane proteins, as
found in C. jejuni (Frost, 2015; Scott et al., 2014). Similar to what was previously found
with the C. jejuni predicted N-glycoproteome (Frost, 2015), there were more predicted
outer membrane N-glycoproteins than periplasmic, further suggesting that N-glycosylation
may be particularly important for surface exposed proteins. This could be due to N-glycan
modification increasing resistance to proteolytic degradation by extracellular proteases, as
suggested for C. jejuni by Alemka et al. (2013). Like in C. jejuni, most C. fetus predicted

209
N-glycoproteins contain only one or two sequons. However, there were instances where
more sequons were evident, with one predicted N-glycoprotein identified containing 11
sequons making this potentially the most N-glycosylated Campylobacter protein.
Although C. fetus N-glycoproteins experimentally identified by Nothaft et al. (2013)
contain the extended sequon, D/E-X-N-X-S/T (X ≠ P), known to be a requirement for N-
linked glycosylation in C. jejuni, the acceptor sequon specificity of C. fetus PglB has not
been studied in detail. This prediction pipeline found that, in agreement with
Campylobacter PglB in vitro assays (Chen et al., 2007; Gerber et al., 2013), the amino
acid composition of sequons in C. fetus predicted N-glycoproteins most commonly
contained aspartate and threonine at the -2 and +2 positions, respectively. However, the
abundance of these amino acid residues does not mean that D-X-N-X-T are the most
efficiently modified sequons in vivo. The acceptor sequon specificity of C. fetus PglB
could be investigated further by validating several of the predicted N-glycoproteins
identified by the pipeline in vivo, as was done with Cf0718 and Cf0445 here. Site-directed
mutagenesis of the asparagine residues within predicted sequons would confirm the sites
of N-glycosylation and identify if any are preferentially modified. Together, such analyses
may begin to reveal the structural preferences and acceptor sequon specificity of the
C. fetus PglB enzyme, which is relatively uncharacterised. Furthermore, this methodology
could be used to investigate if Cf0312 is modified at the eleven N-glycan sequons
predicted and therefore demonstrate if it is the most N-glycosylated Campylobacter
protein identified thus far.
One flaw of this prediction pipeline is that it gives no indication of where the identified
sequons reside within protein structures. Experimental evidence suggests that sequons
which lie within buried structural features may not be modified (Silverman and Imperiali,
2016) and thus presence of a sequon is not sufficient for N-glycosylation. Therefore, it
would be useful to integrate structural predictions into the pipeline to identify N-sequons
which are found in buried structural regions and thus not likely to be modified in vivo.
Detailed protein structure modelling of all predicted N-glycoproteins would be a large and
complex task bioinformatically, however, PredictProtein (Yachdav et al., 2014) is an
online tool which gives a simple output of protein structural prediction and solvent
accessibility. Such a tool may be appropriate for integration into the N-glycoprotein
prediction pipeline. Including such analysis would undoubtedly reduce the size and alter
the overall features of the predicted N-glycoproteome, although the extent of this is
unknown. A subset of these predicted N-glycoproteins could then be validated in vivo to
provide evidence of the accuracy of the pipeline.

210
The second part of Chapter 6 investigated the conservation of a set of experimentally
identified C. jejuni N-glycoproteins thought to be core N-glycoproteins of this species.
The role of N-linked glycosylation in Campylobacter is unclear and limited experimental
evidence has demonstrated that N-glycosylation is not required for the function of
individual proteins in C. jejuni (Davis et al., 2009; Kakuda et al., 2012; Kakuda and
DiRita, 2006; Scott et al., 2009). Some evidence suggests that N-glycosylation instead
serves a more general role and provides protection from chicken gut proteases (Alemka et
al., 2013). Understanding the conservation of N-glycoproteins among Campylobacter
species may help identify proteins which require N-glycosylation for their function and/or
protection, as they will likely be conserved as N-glycosylation targets. Such N-
glycoproteins would provide promising candidates for experimentally studying the role of
N-glycosylation and this investigation served to identify a portion of these N-
glycoproteins. Fourty-eight of the sixty-five C. jejuni N-glycoproteins studied had C. fetus
homologues. Thirty-four of these contained sequons and thus are predicted to be N-
glycosylated in C. fetus. Notably, ten of these predicted C. fetus N-glycoproteins were
experimentally identified by Nothaft et al. (2012). More outer membrane proteins than
periplasmic proteins were predicted to be N-glycoproteins in both species, suggesting that
surface exposure may be a stronger drive for the conservation of N-glycosylation. It can be
speculated that such conservation is due the proposed protection of N-glycan against
protease-mediated degradation, as suggested by Alemka et al. (2013). Analysis among a
further four Campylobacter species, C. coli, C. lari, C.hyointestinalis and C. concisus,
revealed that nineteen of the thirty-four C. jejuni N-glycoproteins also had predicted N-
glycoprotein homologues in these species. Therefore, there appeared to be some
conservation of the proteins targeted for N-glycan modification throughout the
Campylobacter genus.
Interestingly, N-glycoproteins for which experimental research identified N-glycosylation
was dispensible for protein function are those which are not predicted to be conserved as
N-glycoproteins among Campylobacter species. It may be that there is no evolutionary
pressure driving conservation of N-glycan modification of these proteins because it does
not confer a significant advantage. Therefore, in contrast, it can be argued that N-
glycoproteins which are putatively conserved as N-glycoproteins among Campylobacter
species may be so because N-glycan modification does contribute to protein function
and/or protection. Therefore, this work identified promising protein candidates for
experimentally researching the role of N-glycans. In particular, a few N-glycoproteins
identified have known phenotypes (eg. Antibiotic resistance and motility for CmeABC

211
and PflA, respectively), and thus it would be experimentally tractable to abolish N-
glycosylation of these proteins and identify if it is required for function.
An issue with this method is that when analysing the conservation of N-glycoproteins
among Campylobacter species, the number and location of conserved N-sequons was not
analysed. If N-sequons in an N-glycoprotein are similarly located in its homologue, this
may further suggest that N-glycosylation is important for the function of those proteins.
To investigate this, protein sequence alignments and protein structure predictions for the
conserved set of N-glycoproteins could be created. Such analysis may reveal the specific
N-glycosylation sites that are conserved in several species and therefore pinpoint which
sequons to investigate experimentally.
Overall, this bioinformatics prediction approach revealed a large number of possible
C. fetus N-glycoproteins and identified a subset of the possible ‘core’ N-glycoproteins
among Campylobacter species. Although this approach has its drawbacks, predicting N-
glycoproteins complements experimental methods of identification which, at present,
remain limiting. Furthermore, this analysis highlighted areas of improvement to the
pipeline which would provide an increasingly powerful tool for investigating N-linked
glycosylation in Campylobacter species. In the future, this valuable tool could be further
adapted to contribute to the understanding of N-linked glycosylation in other Bacterial
species.
7.4 Final conclusion
This thesis provided evidence that Campylobacter N-linked glycans are attractive targets
for antibody-based detection of Campylobacter species. A C. jejuni N-glycan-reactive
antiserum, CjNgp, showed promise for specific detection of Campylobacter pathogens
found in poultry, including C. jejuni, C. coli and C. lari. Furthermore, this antiserum could
be useful for detecting these organisms in other farmed animals without cross-reacting
with Campylobacter species that may be resident. An antiserum, CfNgp, was raised
against a recombinant C. fetus N-glycoprotein and shown to be able to detect the
veterinary and human pathogen, C. fetus and emerging human pathogens such as
C. concisus and C. ureolyticus. As its broad reactivity against Campylobacter N-linked
glycans could potentially cause cross-reactivity when sampling cattle, it was concluded
that this antiserum may be better suited for detection of such Campylobacter species from

212
human samples. Limitations of these antisera, in their neat form, include protein-cross
reactivity and low sensitivity. Nevertheless, this work demonstrated that, with further
improvement, N-glycan-reactive antibodies are suitable for developing Campylobacter
detection assays that are rapid and simple to perform. Such assays would be valuable for
detecting Campylobacter pathogens in agricultural and/or human samples and could be an
asset to Campylobacter surveillance programs. In addition, the N-glycan-reactive antisera
described here will also be a useful tool for experimental research into Campylobacter N-
linked glycosylation systems.
Secondly, this thesis developed glycocompetent E. coli containing a hybrid C. jejuni/C.
fetus Pgl system. This novel approach successfully demonstrated the activity of two
C. fetus glycosyltransferases, PglH1 and PglH2. Although this experimental system
proved useful and adaptable, it was hindered by possible interference from the C. jejuni
pathway. However, removal of a single C. jejuni enzyme would alleviate this issue and
provide a robust system for continued investigation into C. fetus glycosyltransferases. This
approach would also be a valid method to investigate glycosyltransferases in other
Campylobacter species. In particular, it provides a favourable alternative to newly cloning
entire pgl loci into E. coli or constructing insertional pgl gene knockouts in
Campylobacter, both of which are relatively difficult and laborious tasks.
In conclusion, this thesis has identified the C. fetus predicted N-glycoproteome and
revealed similarities with that of C. jejuni. Furthermore, investigation into the putative
conservation of N-glycosylation protein targets revealed considerable conservation among
six Campylobacter species and identified key candidates for experimental study into the
function of Campylobacter N-linked glycosylation. This study identified limitations within
the bioinformatics pipeline which could inform subsequent work and move towards
developing a more robust N-glycoprotein prediction tool. In the future, this bioinformatics
analysis could further advance the understanding of N-glycosylation throughout the
Campylobacter genus. Futhermore, it has the potential to be adapted for predicting N-
glycoproteins in other bacterial species, for example, the deep-sea vent Bacteria which
were recently identified to contain N-linked glycosylation machinery (Mills et al., 2016).
In conclusion, this work has demonstrated that prediction of Campylobacter N-
glycoproteins is informative and complements experimental N-linked glycosylation
research.

213
References
Aas, F.E., Vik, A., Vedde, J., Koomey, M., Egge-Jacobsen, W., 2007. Neisseria
gonorrhoeae O-linked pilin glycosylation: functional analyses define both the
biosynthetic pathway and glycan structure. Mol. Microbiol. 65, 607–24.
doi:10.1111/j.1365-2958.2007.05806.x
Alemka, A., Nothaft, H., Zheng, J., Szymanski, C.M., 2013. N-glycosylation of
Campylobacter jejuni surface proteins promotes bacterial fitness. Infect. Immun. 81,
1674–1682. doi:10.1128/IAI.01370-12
Ali, A., Soares, S.C., Santos, A.R., Guimarães, L.C., Barbosa, E., Almeida, S.S., Abreu,
V. a C., Carneiro, A.R., Ramos, R.T.J., Bakhtiar, S.M., Hassan, S.S., Ussery, D.W.,
On, S., Silva, A., Schneider, M.P., Lage, A.P., Miyoshi, A., Azevedo, V., 2012.
Campylobacter fetus subspecies: Comparative genomics and prediction of potential
virulence targets. Gene 508, 145–156. doi:10.1016/j.gene.2012.07.070
Allison, G.E., Verma, N.K., 2000. Serotype-converting bacteriophages and O-antigen
modification in Shigella flexneri. Trends Microbiol. 8, 17–23. doi:10.1016/S0966-
842X(99)01646-7
Allos, B.M., Lastovica, A.J., 2008. Clinical Significance of Campylobacter and Related
species other than Campylobacter jejuni and Campylobacter coli, in: Nachamkin, I.,
Szymanski, C.M., Blaser, M.J. (Eds.), Campylobacter, Third Edition. American
Society of Microbiology, Washington, pp. 123–149.
doi:10.1128/9781555815554.ch7
Apostolov, E., Al-Soud, W.A., Nilsson, I., Kornilovska, I., Usenko, V., Lyzogubov, V.,
Gaydar, Y., Wadström, T., Ljungh, A., 2005. Helicobacter pylori and other
Helicobacter species in gallbladder and liver of patients with chronic cholecystitis
detected by immunological and molecular methods. Scand. J. Gastroenterol. 40, 96–

214
102.
Apweiler, R., Hermjakob, H., Sharon, N., 1999. On the frequency of protein
glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim.
Biophys. Acta 1473, 4–8.
Arndt, D., Grant, J.R., Marcu, A., Sajed, T., Pon, A., Liang, Y., Wishart, D.S., 2016.
PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids
Res. 44, 1–6. doi:10.1093/nar/gkw387
Awad, W.A., Molnár, A., Aschenbach, J.R., Ghareeb, K., Khayal, B., Hess, C., Liebhart,
D., Dublecz, K., Hess, M., 2015a. Campylobacter infection in chickens modulates the
intestinal epithelial barrier function. Innate Immun. 21, 151–160.
doi:10.1177/1753425914521648
Awad, W.A., Smorodchenko, A., Hess, C., Aschenbach, J.R., Molnár, A., Dublecz, K.,
Khayal, B., Pohl, E.E., Hess, M., 2015b. Increased intracellular calcium level and
impaired nutrient absorption are important pathogenicity traits in the chicken
intestinal epithelium during Campylobacter jejuni colonization. Appl. Microbiol.
Biotechnol. 99, 6431–41. doi:10.1007/s00253-015-6543-z
Baar, C., Eppinger, M., Raddatz, G., Simon, J., Lanz, C., Klimmek, O., Nandakumar, R.,
Gross, R., Rosinus, A., Keller, H., Jagtap, P., Linke, B., Meyer, F., Lederer, H.,
Schuster, S.C., 2003. Complete genome sequence and analysis of Wolinella
succinogenes. Proc. Natl. Acad. Sci. U. S. A. 100, 11690.
doi:10.1073/pnas.1932838100
Baker, J., Barton, M.D., Lanser, J., 1999. Campylobacter species in cats and dogs in South
Australia. Aust. Vet. J. 77, 662–6.

215
Benjamin, J., Leaper, S., Owen, R.J., Skirrow, M.B., 1983. Description of Campylobacter
laridis, a new species comprising the nalidixic acid resistant
thermophilicCampylobacter (NARTC) group. Curr. Microbiol. 8, 231–238.
doi:10.1007/BF01579552
Bernatchez, S., Szymanski, C.M., Ishiyama, N., Li, J.J., Jarrell, H.C., Lau, P.C., Berghuis,
A.M., Young, N.M., Wakarchuk, W.W., 2005. Single bifunctional UDP-GlcNAc/Glc
4-epimerase supports the synthesis of three cell surface glycoconjugates in
Campylobacter jejuni. J. Biol. Chem. 280, 4792–4802.
Blaser, M.J., Smith, P.F., Repine, J.E., Joiner, K.A., 1988. Pathogenesis of Campylobacter
fetus infections. Failure of encapsulated Campylobacter fetus to bind C3b explains
serum and phagocytosis resistance. J. Clin. Invest. 81, 1434–1444.
doi:10.1172/JCI113474
Bohin, J.-P., 2000. Osmoregulated periplasmic glucans in Proteobacteria. FEMS
Microbiol. Lett. 186, 11–19. doi:10.1111/j.1574-6968.2000.tb09075.x
Bohr, U.R.M., Glasbrenner, B., Primus, A., Zagoura, A., Wex, T., Malfertheiner, P., 2004.
Identification of enterohepatic Helicobacter species in patients suffering from
inflammatory bowel disease. J. Clin. Microbiol. 42, 2766–8.
doi:10.1128/JCM.42.6.2766-2768.2004
Bontemps-Gallo, S., Bohin, J.-P., Lacroix, J.-M., 2017. Osmoregulated Periplasmic
Glucans. EcoSal Plus 7. doi:10.1128/ecosalplus.ESP-0001-2017
Bontemps-Gallo, S., Lacroix, J.-M., 2015. New insights into the biological role of the
osmoregulated periplasmic glucans in pathogenic and symbiotic bacteria. Environ.
Microbiol. Rep. 7, 690–697. doi:10.1111/1758-2229.12325

216
Borisov, S.M., Wolfbeis, O.S., 2008. Optical biosensors. Chem. Rev. 108, 423–461.
doi:10.1021/cr068105t
Borud, B., Anonsen, J.H., Viburiene, R., Cohen, E.H., Samuelsen, A.B.C., Koomey, M.,
2014. Extended glycan diversity in a bacterial protein glycosylation system linked to
allelic polymorphisms and minimal genetic alterations in a glycosyltransferase gene.
Mol. Microbiol. 94, 688–699. doi:10.1111/mmi.12789
Breitling, J., Aebi, M., 2013. N-linked protein glycosylation in the endoplasmic reticulum.
Cold Spring Harb. Perspect. Biol. 5, a013359. doi:10.1101/cshperspect.a013359
Brooks, B.W., Devenish, J., Lutze-Wallace, C.L., Milnes, D., Robertson, R.H., Berlie-
Surujballi, G., 2004. Evaluation of a monoclonal antibody-based enzyme-linked
immunosorbent assay for detection of Campylobacter fetus in bovine preputial
washing and vaginal mucus samples. Vet. Microbiol. 103, 77–84.
doi:10.1016/j.vetmic.2004.07.008
Brooks, B.W., Robertson, R.H., Lutze-Wallace, C.L., Pfahler, W., 2002. Monoclonal
antibodies specific for Campylobacter fetus lipopolysaccharides. Vet. Microbiol. 87,
37–49. doi:10.1016/s0378-1135(02)00026-3
Burda, P., Aebi, M., 1999. The dolichol pathway of N-linked glycosylation. Biochim.
Biophys. Acta - Gen. Subj. 1426, 239–257. doi:10.1016/S0304-4165(98)00127-5
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., Madden,
T.L., 2009. BLAST+: architecture and applications. BMC Bioinformatics 10, 421.
doi:10.1186/1471-2105-10-421
Campero, C.M., Anderson, M.L., Walker, R.L., Blanchard, P.C., Barbano, L., Chiu, P.,
Martínez, A., Combessies, G., Bardon, J.C., Cordeviola, J., 2005.

217
Immunohistochemical identification of Campylobacter fetus in natural cases of
bovine and ovine abortions. J. Vet. Med. Ser. B Infect. Dis. Vet. Public Heal. 52,
138–141. doi:10.1111/j.1439-0450.2005.00834.x
Casanova, C., Schweiger, A., Von Steiger, N., Droz, S., Marschall, J., 2015.
Campylobacter concisus pseudo-outbreak caused by improved culture conditions. J.
Clin. Microbiol. 53, 660–662. doi:10.1128/JCM.02608-14
Ceelen, L., Decostere, A., Verschraegen, G., Ducatelle, R., Haesebrouck, F., 2005.
Prevalence of Helicobacter pullorum among patients with gastrointestinal disease and
clinically healthy persons. J. Clin. Microbiol. 43, 2984–6.
doi:10.1128/JCM.43.6.2984-2986.2005
Chaban, B., Chu, S., Hendrick, S., Waldner, C., Hill, J.E., 2012. Evaluation of a
Campylobacter fetus subspecies venerealis real-time quantitative polymerase chain
reaction for direct analysis of bovine preputial samples. Can. J. Vet. Res. 76, 166–
173.
Chaban, B., Ngeleka, M., Hill, J.E., 2010. Detection and quantification of 14
Campylobacter species in pet dogs reveals an increase in species richness in feces of
diarrheic animals. BMC Microbiol. 10, 73. doi:10.1186/1471-2180-10-73
Chamot-Rooke, J., Rousseau, B., Lanternier, F., Mikaty, G., Mairey, E., Malosse, C.,
Bouchoux, G., Pelicic, V., Camoin, L., Nassif, X., Dumenil, G., 2007. Alternative
Neisseria spp. type IV pilin glycosylation with a glyceramido acetamido
trideoxyhexose residue. Proc. Natl. Acad. Sci. 104, 14783–14788.
doi:10.1073/pnas.0705335104
Chen, M.M., Glover, K.J., Imperiali, B., 2007. From peptide to protein: comparative
analysis of the substrate specificity of N-linked glycosylation in C. jejuni.
Biochemistry 46, 5579–5585. doi:10.1021/bi602633n

218
Choi, H.S., Shin, S.U., Bae, E.H., Ma, S.K., Kim, S.W., 2016. Infectious Spondylitis in a
Patient with Chronic Kidney Disease: Identification of Campylobacter fetus subsp.
testudinum by 16S Ribosomal RNA Sequencing. Jpn. J. Infect. Dis. 69, 517–519.
doi:10.7883/yoken.JJID.2015.461
Cohen-Rosenzweig, C., Yurist-Doutsch, S., Eichler, J., 2012. AglS, a novel component of
the Haloferax volcanii N-glycosylation pathway, is a dolichol phosphate-mannose
mannosyltransferase. J. Bacteriol. 194, 6909–16. doi:10.1128/JB.01716-12
Crooks, G.E., Hon, G., Chandonia, J.-M., Brenner, S.E., 2004. WebLogo: A Sequence
Logo Generator. Genome Res. 14, 1188–1190. doi:10.1101/gr.849004
Davies, M.R., Broadbent, S.E., Harris, S.R., Thomson, N.R., van der Woude, M.W., 2013.
Horizontally acquired glycosyltransferase operons drive Salmonellae
lipopolysaccharide diversity. PLoS Genet. 9. doi:10.1371/journal.pgen.1003568
Davis, L.M., Kakuda, T., DiRita, V.J., 2009. A Campylobacter jejuni znuA orthologue is
essential for growth in low-zinc environments and chick colonization. J. Bacteriol.
191, 1631–1640. doi:10.1128/JB.01394-08
De Boer, P., Rahaoui, H., Leer, R.J., Montijn, R.C., Van Der Vossen, J.M.B.M., 2015.
Real-time PCR detection of Campylobacter spp.: A comparison to classic culturing
and enrichment. doi:10.1016/j.fm.2015.05.006
de Lorenzo, V., Timmis, K.N., 1994. Analysis and construction of stable phenotypes in
gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods
Enzymol. 235, 386–405.
De Schutter, J.W., Morrison, J.P., Morrison, M.J., Ciulli, A., Imperiali, B., 2017.
Targeting Bacillosamine Biosynthesis in Bacterial Pathogens: Development of

219
Inhibitors to a Bacterial Amino-Sugar Acetyltransferase from Campylobacter jejuni.
J. Med. Chem. 60, 2099–2118. doi:10.1021/acs.jmedchem.6b01869
Dell, A., Galadari, A., Sastre, F., Hitchen, P., 2010. Similarities and differences in the
glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010,
148178. doi:10.1155/2010/148178
Deshpande, N.P., Kaakoush, N.O., Wilkins, M.R., Mitchell, H.M., 2013. Comparative
genomics of Campylobacter concisus isolates reveals genetic diversity and provides
insights into disease association. BMC Genomics 14, 585. doi:10.1186/1471-2164-
14-585
Devenish, J., Brooks, B., Perry, K., Milnes, D., Burke, T., McCabe, D., Duff, S., Lutze-
Wallace, C.L., 2005. Validation of a monoclonal antibody-based capture enzyme-
linked immunosorbent assay for detection of Campylobacter fetus. Clin. Diagn. Lab.
Immunol. 12, 1261–1268. doi:10.1128/cdli.12.11.1261-1268.2005
Diker, K.S., Diker, S., Ozlem, M.B., 1990. Bovine diarrhea associated with
Campylobacter hyointestinalis. Zentralbl. Veterinarmed. B 37, 158–60.
Ding, W., Nothaft, H., Szymanski, C.M., Kelly, J., 2009. Identification and Quantification
of Glycoproteins Using Ion-Pairing Normal-phase Liquid Chromatography and Mass
Spectrometry. Mol. Cell. Proteomics 8, 2170–2185. doi:10.1074/mcp.M900088-
MCP200
Divakaruni, A. V., Baida, C., White, C.L., Gober, J.W., 2007. The cell shape proteins
MreB and MreC control cell morphogenesis by positioning cell wall synthetic
complexes. Mol. Microbiol. 66, 174–188. doi:10.1111/j.1365-2958.2007.05910.x
Dong, H.-J., Cho, A.-R., Hahn, T.-W., Cho, S., 2014. Development of a loop-mediated

220
isothermal amplification assay for rapid, sensitive detection of Campylobacter jejuni
in cattle farm samples. J. Food Prot. 77, 1593–1598. doi:10.4315/0362-028X.JFP-14-
056
Dwivedi, R., Nothaft, H., Reiz, B., Whittal, R.M., Szymanski, C.M., 2013. Generation of
free oligosaccharides from bacterial protein N-linked glycosylation systems.
Biopolymers 99, 772–783. doi:10.1002/bip.22296
Edmonds, P., Patton, C.M., Griffin, P.M., Barrett, T.J., Schmid, G.P., Baker, C.N.,
Lambert, M. a, Brenner, D.J., 1987. Campylobacter hyointestinalis associated with
human gastrointestinal disease in the United States. J Clin Microbiol 25, 685–691.
EFSA, 2016. The European Union summary report on trends and sources of zoonoses,
zoonotic agents and food-borne outbreaks in 2015. EFSA J. 14.
doi:10.2903/j.efsa.2016.4634
EFSA, 2010. Scientific Opinion on Quantification of the risk posed by broiler meat to
human campylobacteriosis in the EU. EFSA J. 8, 1–89.
doi:10.2903/j.efsa.2010.1437.Available
Eichler, J., 2013. Extreme sweetness: protein glycosylation in archaea. Nat. Rev.
Microbiol. 11, 151–156. doi:10.1038/nrmicro2957
Faridmoayer, A., Fentabil, M.A., Mills, D.C., Klassen, J.S., Feldman, M.F., 2007.
Functional characterization of bacterial oligosaccharyltransferases involved in O-
linked protein glycosylation. J. Bacteriol. 189, 8088–98. doi:10.1128/JB.01318-07
Feldman, M.F., Wacker, M., Hernandez, M., Hitchen, P.G., Marolda, C.L., Kowarik, M.,
Morris, H.R., Dell, A., Valvano, M.A., Aebi, M., 2005. Engineering N-linked protein
glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia

221
coli. Proc. Natl. Acad. Sci. U. S. A. 102, 3016–3021. doi:10.1073/pnas.0500044102
Fitzgerald, C., Patrick, M., Gonzalez, A., Akin, J., Polage, C.R., Wymore, K., Gillim-
Ross, L., Xavier, K., Sadlowski, J., Monahan, J., Hurd, S., Dahlberg, S., Jerris, R.,
Watson, R., Santovenia, M., Mitchell, D., Harrison, C., Tobin-D’Angelo, M.,
DeMartino, M., Pentella, M., Razeq, J., Leonard, C., Jung, C., Achong-Bowe, R.,
Evans, Y., Jain, D., Juni, B., Leano, F., Robinson, T., Smith, K., Gittelman, R.M.,
Garrigan, C., Nachamkin, I., 2016. Multicenter Evaluation of Clinical Diagnostic
Methods for Detection and Isolation of Campylobacter spp. from Stool. J. Clin.
Microbiol. 54, 1209–1215. doi:10.1128/JCM.01925-15
Fitzgerald, C., Tu, Z. c., Patrick, M., Stiles, T., Lawson, A.J., Santovenia, M., Gilbert,
M.J., van Bergen, M., Joyce, K., Pruckler, J., Stroika, S., Duim, B., Miller, W.G.,
Loparev, V., Sinnige, J.C., Fields, P.I., Tauxe, R. V., Blaser, M.J., Wagenaar, J.A.,
2014. Campylobacter fetus subsp. testudinum subsp. nov., isolated from humans and
reptiles. Int. J. Syst. Evol. Microbiol. 64, 2944–2948. doi:10.1099/ijs.0.057778-0
Fouts, D.E., Mongodin, E.F., Mandrell, R.E., Miller, W.G., Rasko, D.A., Ravel, J.,
Brinkac, L.M., DeBoy, R.T., Parker, C.T., Daugherty, S.C., Dodson, R.J., Durkin,
A.S., Madupu, R., Sullivan, S.A., Shetty, J.U., Ayodeji, M.A., Shvartsbeyn, A.,
Schatz, M.C., Badger, J.H., Fraser, C.M., Nelson, K.E., 2005. Major structural
differences and novel potential virulence mechanisms from the genomes of multiple
Campylobacter species. Plos Biol. 3, 72–85. doi:10.1371/journal.pbio.0030015
Frost, H.M., 2015. N-linked protein glycosylation in Campylobacter and Helicobacter
species. Ph.D. Univ. Manchester.
Fry, B.N., Feng, S., Chen, Y.Y., Newell, D.G., Coloe, P.J., Korolik, V., 2000. The galE
gene of Campylobacter jejuni is involved in lipopolysaccharide synthesis and
virulence. Infect. Immun. 68, 2594–2601. doi:10.1128/IAI.68.5.2594-2601.2000

222
Fry, B.N., Korolik, V., Brinke, J.A., Pennings, M.T.T., Zalm, R., Teunis, B.J.J., Coloe,
P.J., Zeijst, B.A.M. Van Der, 1998. The lipopolysaccharide biosynthesis locus of
Campylobacter jejuni 81116.
Garcia, M.M., Lutze-Wallace, C.L., Denes, A.S., Eaglesome, M.D., Holst, E., Blaser,
M.J., 1995. Protein shift and antigenic variation in the S-layer of Campylobacter fetus
subsp. venerealis during bovine infection accompanied by genomic rearrangement of
sapA homologs. J. Bacteriol. 177, 1976–80.
Gebhart, C.J., Edmonds, P., Ward, G.E., Kurtz, H.J., Brenner, D.J., 1985. “Campylobacter
hyointestinalis” sp. nov.: a new species of Campylobacter found in the intestines of
pigs and other animals. J. Clin. Microbiol. 21, 715–720.
Gerber, S., Lizak, C., Michaud, G., Bucher, M., Darbre, T., Aebi, M., Reymond, J.L.,
Locher, K.P., 2013. Mechanism of bacterial oligosaccharyltransferase: In vitro
quantification of sequon binding and catalysis. J. Biol. Chem. 288, 8849–8861.
doi:10.1074/jbc.M112.445940
Gharst, G., Oyarzabal, O. a, Hussain, S.K., 2013. Review of current methodologies to
isolate and identify Campylobacter spp. from foods. J. Microbiol. Methods 95, 84–
92. doi:10.1016/j.mimet.2013.07.014
Gilbert, M.J., Miller, W.G., Yee, E., Zomer, A.L., van der Graaf-van Bloois, L.,
Fitzgerald, C., Forbes, K.J., Méric, G., Sheppard, S.K., Wagenaar, J. a, Duim, B.,
2016. Comparative genomics of Campylobacter fetus from reptiles and mammals
reveals divergent evolution in host-associated lineages. Genome Biol. Evol. 8, 2006–
19. doi:10.1093/gbe/evw146
Giltner, C.L., Saeki, S., Bobenchik, A.M., Humphries, R.M., 2013. Rapid detection of
Campylobacter antigen by enzyme immunoassay leads to increased positivity rates. J.
Clin. Microbiol. 51, 618–620. doi:10.1128/JCM.02565-12

223
Glover, K.J., Weerapana, E., Chen, M.M., Imperiali, B., 2006. Direct biochemical
evidence for the utilization of UDP-bacillosamine by PglC, an essential glycosyl-1-
phosphate transferase in the Campylobacter jejuni N-linked glycosylation pathway.
Biochemistry 45, 5343–5350. doi:10.1021/bi0602056
Glover, K.J., Weerapana, E., Imperiali, B., 2005. In vitro assembly of the
undecaprenylpyrophosphate-linked, heptasaccharide for prokaryotic N-linked
glycosylation. Proc. Natl. Acad. Sci. U. S. A. 102, 14255–14259.
doi:10.1073/pnas.0507311102
Gnanaprakasa, T.J., Oyarzabal, O.A., Olsen, E. V, Pedrosa, V.A., Simonian, A.L., 2011.
Tethered DNA scaffolds on optical sensor platforms for detection of hipO gene from
Campylobacter jejuni. Sensors and Actuators B-Chemical 156, 304–311.
doi:10.1016/j.snb.2011.04.037
Gómez-Camarasa, C., Gutiérrez-Fernández, J., Rodríguez-Granger, J.M., Sampedro-
Martínez, A., Sorlózano-Puerto, A., Navarro-Marí, J.M., 2014. Evaluation of the
rapid RIDAQUICK Campylobacter® test in a general hospital. Diagn. Microbiol.
Infect. Dis. 78, 101–104. doi:10.1016/j.diagmicrobio.2013.11.009
Goon, S., Kelly, J.F., Logan, S.M., Ewing, C.P., Guerry, P., 2003. Pseudaminic acid, the
major modification on Campylobacter flagellin, is synthesized via the Cj1293 gene.
Mol. Microbiol. 50, 659–71.
Goossens, H., Vlaes, L., De Boeck, M., Pot, B., Kersters, K., Levy, J., De Mol, P., Butzler,
J.P., Vandamme, P., 1990. Is Campylobacter upsaliensis an unrecognised cause of
human diarrhoea? Lancet. 335, 584–6.
Gorkiewicz, G., Feierl, G., Zechner, R., Zechner, E.L., 2002. Transmission of
Campylobacter hyointestinalis from a pig to a human. J. Clin. Microbiol. 40, 2601–5.
doi:10.1128/JCM.40.7.2601

224
Gorkiewicz, G., Kienesberger, S., Schober, C., Scheicher, S.R., Guelly, C., Zechner, R.,
Zechner, E.L., 2010. A genomic island defines subspecies-specific virulence features
of the host-adapted pathogen Campylobacter fetus subsp venerealis. J. Bacteriol. 192,
502–517. doi:10.1128/jb.00803-09
Granato, P. a, Chen, L., Holiday, I., Rawling, R. a, Novak-Weekley, S.M., Quinlan, T.,
Musser, K. a, 2010. Comparison of premier CAMPY enzyme immunoassay (EIA),
ProSpecT Campylobacter EIA, and ImmunoCard STAT! CAMPY tests with culture
for laboratory diagnosis of Campylobacter enteric infections. J. Clin. Microbiol. 48,
4022–7. doi:10.1128/JCM.00486-10
Grass, S., Lichti, C.F., Townsend, R.R., Gross, J., St. Geme, J.W., 2010. The Haemophilus
influenzae HMW1C protein is a glycosyltransferase that transfers hexose residues to
asparagine sites in the HMW1 adhesin. PLoS Pathog. 6, e1000919.
doi:10.1371/journal.ppat.1000919
Gross, J., Grass, S., Davis, A.E., Gilmore-Erdmann, P., Townsend, R.R., St. Geme, J.W.,
2008. The Haemophilus influenzae HMW1 adhesin is a glycoprotein with an unusual
N-linked carbohydrate modification. J. Biol. Chem. 283, 26010–26015.
doi:10.1074/jbc.M801819200
Guo, B., Lin, J., Reynolds, D.L., Zhang, Q., 2010. Contribution of the multidrug efflux
transporter CmeABC to antibiotic resistance in different Campylobacter species.
Foodborne Pathog. Dis. 7, 77–83. doi:10.1089/fpd.2009.0354
Gurgan, T., Diker, K.S., 1994. Abortion associated with Campylobacter upsaliensis. J.
Clin. Microbiol. 32, 3093–4.
Hara-Kudo, Y., Yoshino, M., Kojima, T., Ikedo, M., 2005. Loop-mediated isothermal
amplification for the rapid detection of Salmonella. FEMS Microbiol. Lett. 253, 155–
161. doi:10.1016/j.femsle.2005.09.032

225
Hazeleger, W.C., Beumer, R.R., Rombouts, F.M., 1992. The use of latex agglutination
tests for determining Campylobacter species. Lett. Appl. Microbiol. 14, 181–184.
doi:10.1111/j.1472-765X.1992.tb00679.x
Heid, C.A., Stevens, J., Livak, K.J., Williams, P.M., 1996. Real time quantitative PCR.
Genome Res. 6, 986–94. doi:10.1101/GR.6.10.986
Helenius, A., Aebi, M., 2001. Intracellular functions of N-linked glycans. Science 291,
2364–9. doi:10.1126/science.291.5512.2364
Hendrixson, D.R., DiRita, V.J., 2004. Identification of Campylobacter jejuni genes
involved in commensal colonization of the chick gastrointestinal tract. Mol.
Microbiol. 52, 471–484. doi:10.1111/j.1365-2958.2004.03988.x
Hermans, D., Pasmans, F., Heyndrickx, M., Van Immerseel, F., Martel, A., Van Deun, K.,
Haesebrouck, F., 2012. A tolerogenic mucosal immune response leads to persistent
Campylobacter jejuni colonization in the chicken gut. Crit. Rev. Microbiol. 38, 17–
29. doi:10.3109/1040841X.2011.615298
Hermans, D., Van Deun, K., Martel, A., Van Immerseel, F., Messens, W., Heyndrickx,
M., Haesebrouck, F., Pasmans, F., 2011. Colonization factors of Campylobacter
jejuni in the chicken gut. Vet. Res. 42, 82. doi:10.1186/1297-9716-42-82
Hindiyeh, M., Jense, S., Hohmann, S., Benett, H., Edwards, C., Aldeen, W., Croft, A.,
Daly, J., Mottice, S., Carroll, K.C., 2000. Rapid detection of Campylobacter jejuni in
stool specimens by an enzyme immunoassay and surveillance for Campylobacter
upsaliensis in the greater Salt Lake City area. J. Clin. Microbiol. 38, 3076–3079.
Hitchen, P., Brzostek, J., Panico, M., Butler, J.A., Morris, H.R., Dell, A., Linton, D., 2010.
Modification of the Campylobacter jejuni flagellin glycan by the product of the

226
Cj1295 homopolymeric-tract-containing gene. Microbiology 156, 1953–1962.
doi:10.1099/mic.0.038091-0
Hodinka, R.L., Gilligan, P.H., 1988. Evaluation of the campyslide agglutination test for
confirmatory identification of selected Campylobacter species. J. Clin. Microbiol. 26,
47–9.
Holst, E., Wathne, B., Hovelius, B., Mårdh, P.A., 1987. Bacterial vaginosis:
microbiological and clinical findings. Eur. J. Clin. Microbiol. 6, 536–41.
Huang, R.S.P., Johnson, C.L., Pritchard, L., Hepler, R., Ton, T.T., Dunn, J.J., 2016.
Performance of the Verigene® enteric pathogens test, Biofire FilmArrayTM
gastrointestinal panel and Luminex xTAG® gastrointestinal pathogen panel for
detection of common enteric pathogens. Diagn. Microbiol. Infect. Dis. 86, 336–339.
doi:10.1016/j.diagmicrobio.2016.09.013
Hum, S., Quinn, K., Brunner, J., On, S., 1997. Evaluation of a PCR assay for identification
and differentiation of Campylobacter fetus subspecies. Aust. Vet. J. 75, 827–831.
doi:10.1111/j.1751-0813.1997.tb15665.x
Humphrey, S., Chaloner, G., Kemmett, K., Davidson, N., Williams, N., Kipar, A.,
Humphrey, T., Wigley, P., 2014. Campylobacter jejuni is not merely a commensal in
commercial broiler chickens and affects bird welfare. MBio 5, e01364-14.
doi:10.1128/mBio.01364-14
Ielmini, M. V., Feldman, M.F., 2011. Desulfovibrio desulfuricans PglB homolog
possesses oligosaccharyltransferase activity with relaxed glycan specificity and
distinct protein acceptor sequence requirements. Glycobiology 21, 734–742.
doi:10.1093/glycob/cwq192

227
Jarrell, K.F., Ding, Y., Meyer, B.H., Albers, S.-V., Kaminski, L., Eichler, J., 2014. N-
linked glycosylation in Archaea: a structural, functional, and genetic analysis.
Microbiol. Mol. Biol. Rev. 78, 304–41. doi:10.1128/MMBR.00052-13
Javed, S., Gul, F., Javed, K., Bokhari, H., 2017. Helicobacter pullorum: An emerging
zoonotic pathogen. Front. Microbiol. 8, 604. doi:10.3389/fmicb.2017.00604
Jensen, a N., Andersen, M.T., Dalsgaard, a, Baggesen, D.L., Nielsen, E.M., 2005.
Development of real-time PCR and hybridization methods for detection and
identification of thermophilic Campylobacter spp. in pig faecal samples. J. Appl.
Microbiol. 99, 292–300. doi:10.1111/j.1365-2672.2005.02616.x
Jervis, A.J., Butler, J.A., Wren, B.W., Linton, D., 2015. Chromosomal integration vectors
allowing flexible expression of foreign genes in Campylobacter jejuni. BMC
Microbiol. 15, 230. doi:10.1186/s12866-015-0559-5
Jervis, A.J., Butler, J. a., Lawson, A.J., Langdon, R., Wren, B.W., Linton, D., 2012.
Characterization of the structurally diverse N-linked glycans of Campylobacter
species. J. Bacteriol. 194, 2355–2362. doi:10.1128/JB.00042-12
Jervis, A.J., Langdon, R., Hitchen, P., Lawson, A.J., Wood, A., Fothergill, J.L., Morris,
H.R., Dell, A., Wren, B., Linton, D., 2010. Characterization of N-Linked Protein
Glycosylation in Helicobacter pullorum. J. Bacteriol. 192, 5228–5236.
doi:10.1128/jb.00211-10
Jin, S., Joe, A., Lynett, J., Hani, E.K., Sherman, P., Chan, V.L., 2001. JlpA, a novel
surface-exposed lipoprotein specific to Campylobacter jejuni, mediates adherence to
host epithelial cells. Mol. Microbiol. 39, 1225–36.
Johnson, K.F., 1999. Synthesis of oligosaccharides by bacterial enzymes. Glycoconj. J. 16,

228
141–146. doi:10.1023/A:1026440509859
Johnson, T.J., Shank, J.M., Johnson, J.G., 2017. Current and potential treatments for
reducing Campylobacter colonization in animal hosts and disease in humans. Front.
Microbiol. 8, 1–14. doi:10.3389/fmicb.2017.00487
Jones, K., 2001. Campylobacters in water, sewage and the environment. J. Appl.
Microbiol. 90, 68S–79S. doi:10.1046/j.1365-2672.2001.01355.x
Jones, M.A., Marston, K.L., Woodall, C.A., Maskell, D.J., Linton, D., Karlyshev, A. V,
Dorrell, N., Wren, B.W., Barrow, P.A., 2004. Adaptation of Campylobacter jejuni
NCTC11168 to high-level colonization of the avian gastrointestinal tract. Infect.
Immun. 72, 3769–76. doi:10.1128/IAI.72.7.3769-3776.2004
Juncker, A.S., Willenbrock, H., von Heijne, G., Brunak, S., Nielsen, H., Krogh, A., 2003.
Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12,
1652–1662. doi:10.1110/ps.0303703
Kaakoush, N.O., Castano-Rodriguez, N., Mitchell, H.M., Man, S.M., 2015. Global
epidemiology of Campylobacter infection. Clin. Microbiol. Rev. 28, 687–720.
doi:10.1128/CMR.00006-15
Kaakoush, N.O., Mitchell, H.M., 2012. Campylobacter concisus – A new player in
intestinal disease. Front. Cell. Infect. Microbiol. 2, 1–15.
doi:10.3389/fcimb.2012.00004
Kakuda, T., DiRita, V.J., 2006. Cj1496c encodes a Campylobacter jejuni glycoprotein that
influences invasion of human epithelial cells and colonization of the chick
gastrointestinal tract. Infect. Immun. 74, 4715–4723. doi:10.1128/IAI.00033-06

229
Kakuda, T., Koide, Y., Sakamoto, A., Takai, S., 2012. Characterization of two putative
mechanosensitive channel proteins of Campylobacter jejuni involved in protection
against osmotic downshock. Vet. Microbiol. 160, 53–60.
doi:10.1016/j.vetmic.2012.04.044
Kamei, K., Kawabata, H., Asakura, M., Samosornsuk, W., Hinenoya, A., Nakagawa, S.,
Yamasaki, S., 2016. A cytolethal distending toxin gene-based multiplex PCR assay
for Campylobacter jejuni, C. fetus, C. coli, C. upsaliensis, C. hyointestinalis, and C.
lari. Jpn. J. Infect. Dis. 69, 256–258. doi:10.7883/yoken.JJID.2015.182
Karlyshev, A. V., Ketley, J.M., Wren, B.W., 2005. The Campylobacter jejuni glycome.
FEMS Microbiol. Rev. 29, 377–390. doi:10.1016/j.femsre.2005.01.003
Karlyshev, A. V., Linton, D., Gregson, N.A., Wren, B.W., 2002. A novel paralogous gene
family involved in phase-variable flagella-mediated motility in Campylobacter jejuni.
Microbiology 148, 473–480. doi:10.1099/00221287-148-2-473
Karlyshev, a V, Everest, P., Linton, D., Cawthraw, S., Newell, D.G., Wren, B.W., 2004.
The Campylobacter jejuni general glycosylation system is important for attachment
to human epithelial cells and in the colonization of chicks. Microbiology 150, 1957–
1964. doi:10.1099/mic.0.26721-0
Karlyshev, a V, Wren, B.W., 2005. Development and application of an insertional system
for gene delivery and expression in Campylobacter jejuni. Appl. Environ. Microbiol.
71, 4004–4013. doi:10.1128/AEM.71.7.4004
Kawai, F., Paek, S., Choi, K.-J., Prouty, M., Kanipes, M.I., Guerry, P., Yeo, H.-J., 2012.
Crystal structure of JlpA, a surface-exposed lipoprotein adhesin of Campylobacter
jejuni. J. Struct. Biol. 177, 583–588. doi:10.1016/j.jsb.2012.01.001

230
Kelleher, D.J., Gilmore, R., 2005. An evolving view of the eukaryotic
oligosaccharyltransferase. Glycobiology 16, 47R–62R. doi:10.1093/glycob/cwj066
Kelleher, D.J., Karaoglu, D., Mandon, E.C., Gilmore, R., 2003. Oligosaccharyltransferase
isoforms that contain different catalytic stt3 subunits have distinct enzymatic
properties. Mol. Cell 12, 101–111. doi:10.1016/S1097-2765(03)00243-0
Kelly, J., Jarrell, H., Millar, L., Tessier, L., Fiori, L.M., Lau, P.C., Allan, B., Szymanski,
C.M., 2006. Biosynthesis of the N-linked glycan in Campylobacter jejuni and
addition onto protein through block transfer. J. Bacteriol. 188, 2427–2434.
doi:10.1128/JB.188.7.2427-2434.2006
Kienesberger, S., Gorkiewicz, G., Joainig, M.M., Scheicher, S.R., Leitner, E., Zechner,
E.L., 2007. Development of experimental genetic tools for Campylobacter fetus.
Appl. Environ. Microbiol. 73, 4619–4630. doi:10.1128/AEM.02407-06
Kim, S.A., Lee, Y.M., Hwang, I.G., Kang, D.H., Woo, G.J., Rhee, M.S., 2009. Eight
enrichment broths for the isolation of Campylobacter jejuni from inoculated
suspensions and ground pork. Lett. Appl. Microbiol. 49, 620–626.
doi:10.1111/j.1472-765X.2009.02714.x
Kirk, R., Rowe, M.T., 1994. A PCR assay for the detection of Campylobacter jejuni and
Campylobacter coli in water. Lett. Appl. Microbiol. 19, 301–3.
Konkel, M.E., Garvis, S.G., Tipton, S.L., Anderson, D.E., Cieplak, W., 1997.
Identification and molecular cloning of a gene encoding a fibronectin-binding protein
(CadF) from Campylobacter jejuni. Mol. Microbiol. 24, 953–63.
Konkel, M.E., Klena, J.D., Rivera-Amill, V., Monteville, M.R., Biswas, D., Raphael, B.,
Mickelson, J., 2004. Secretion of virulence proteins from Campylobacter jejuni is

231
dependent on a functional flagellar export apparatus. J. Bacteriol. 186, 3296–3303.
doi:10.1128/JB.186.11.3296-3303.2004
Korlath, J.A., Osterholm, M.T., Judy, L.A., Forfang, J.C., Robinson, R.A., 1985. A point-
source outbreak of campylobacteriosis associated with consumption of raw milk. J.
Infect. Dis. 152, 592–6.
Kowarik, M., Numao, S., Feldman, M.F., Schulz, B.L., Callewaert, N., Kiermaier, E.,
Catrein, I., Aebi, M., 2006a. N-linked glycosylation of folded proteins by the
bacterial oligosaccharyltransferase. Science (80-. ). 314, 1148–1150.
doi:10.1126/science.1134351
Kowarik, M., Young, N.M., Numao, S., Schulz, B.L., Hug, I., Callewaert, N., Mills, D.C.,
Watson, D.C., Hernandez, M., Kelly, J.F., Wacker, M., Aebi, M., 2006b. Definition
of the bacterial N-glycosylation site consensus sequence. Embo J. 25, 1957–1966.
doi:10.1038/sj.emboj.7601087
Koziel, M., Lucey, B., Bullman, S., Corcoran, G.D., Sleator, R.D., 2012. Molecular-based
detection of the gastrointestinal pathogen Campylobacter ureolyticus in
unpasteurized milk samples from two cattle farms in Ireland. Gut Pathog. 4, 14.
doi:10.1186/1757-4749-4-14
Krachler, A.M., Sharma, A., Cauldwell, A., Papadakos, G., Kleanthous, C., 2010. TolA
modulates the oligomeric status of YbgF in the bacterial periplasm. J. Mol. Biol. 403,
270–285. doi:10.1016/j.jmb.2010.08.050
Krogh, A., Larsson, B., von Heijne, G., Sonnhammer, E.L.., 2001. Predicting
transmembrane protein topology with a hidden markov model: application to
complete genomes. J. Mol. Biol. 305, 567–580. doi:10.1006/jmbi.2000.4315

232
Ku, S.C., Schulz, B.L., Power, P.M., Jennings, M.P., 2009. The pilin O-glycosylation
pathway of pathogenic Neisseria is a general system that glycosylates AniA, an outer
membrane nitrite reductase. Biochem. Biophys. Res. Commun. 378, 84–89.
doi:10.1016/j.bbrc.2008.11.025
Kulkarni, S.P., Lever, S., Logan, J.M.J., Lawson, A.J., Stanley, J., Shafi, M.S., 2002.
Detection of Campylobacter species: a comparison of culture and polymerase chain
reaction based methods. J. Clin. Pathol. 55, 749–53.
Larsen, J.C., Szymanski, C., Guerry, P., 2004. N-linked protein glycosylation is required
for full competence in Campylobacter jejuni 81-176. J Bacteriol.186, 6508–6514.
doi:10.1128/JB.186.19.6508
Larson, D.J., Wesley, I. V., Hoffman, L.J., 1992. Use of oligodeoxynucleotide probes to
verify Campylobacter jejuni as a cause of bovine abortion. J. Vet. Diagnostic
Investig. 4, 348–351. doi:10.1177/104063879200400324
Lastovica, A.J., On, S.L.W., Zhang, L., 2014. The Family Campylobacteraceae, in: The
Prokaryotes. Springer Berlin Heidelberg, Berlin, Heidelberg, pp. 307–335.
doi:10.1007/978-3-642-39044-9_274
Lawson, A.J., Logan, J.M., On, S.L., Stanley, J., 2001. Campylobacter hominis sp. nov.,
from the human gastrointestinal tract. Int. J. Syst. Evol. Microbiol. 51, 651–660.
doi:10.1099/00207713-51-2-651
Lee, M.D., Newell, D.G., 2006. Campylobacter in Poultry: Filling an Ecological Niche.
Avian Dis. 50, 1–9. doi:10.1637/7474-111605R.1
Lin, J., Michel, L.O., Zhang, Q., 2002. CmeABC functions as a multidrug efflux system in
Campylobacter jejuni. Antimicrob. Agents Chem. 46, 2124–2131.

233
doi:10.1128/AAC.46.7.2124
Lin, J., Sahin, O., Michel, L.O., Zhang, Q., 2003. Critical role of multidrug efflux pump
CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect.
Immun. 71, 4250–4259. doi:10.1128/IAI.71.8.4250
Lindblom, G.B., Sjögren, E., Hansson-Westerberg, J., Kaijser, B., 1995. Campylobacter
upsaliensis, C. sputorum sputorum and C. concisus as common causes of diarrhoea in
Swedish children. Scand. J. Infect. Dis. 27, 187–8.
Linton, D., Allan, E., Karlyshev, A. V, Cronshaw, A.D., Wren, B.W., 2002. Identification
of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in
Campylobacter jejuni. Mol. Microbiol. 43, 497–508.
Linton, D., Dorrell, N., Hitchen, P.G., Amber, S., Karlyshev, A. V., Morris, H.R., Dell, A.,
Valvano, M. a., Aebi, M., Wren, B.W., 2005. Functional analysis of the
Campylobacter jejuni N-linked protein glycosylation pathway. Mol. Microbiol. 55,
1695–1703. doi:10.1111/j.1365-2958.2005.04519.x
Linton, D., Karlyshev, A. V, Hitchen, P.G., Morris, H.R., Dell, A., Gregson, N.A., Wren,
B.W., 2000. Multiple N-acetyl neuraminic acid synthetase (neuB) genes in
Campylobacter jejuni: identification and characterization of the gene involved in
sialylation of lipo-oligosaccharide. Mol. Microbiol. 35, 1120–1134.
doi:10.1046/j.1365-2958.2000.01780.x
Linton, D., Lawson, A.J., Owen, R.J., Stanley, J., 1997. PCR detection, identification to
species level, and fingerprinting of Campylobacter jejuni and Campylobacter coli
direct from diarrheic samples. J. Clin. Microbiol. 35, 2568–2572.
Linton, D., Owen, R.., Stanley, J., 1996. Rapid identification by PCR of the genus

234
Campylobacter and of five Campylobacter species enteropathogenic for man and
animals. Res. Microbiol. 147, 707–718. doi:10.1016/S0923-2508(97)85118-2
Liu, J., Mushegian, A., 2003. Three monophyletic superfamilies account for the majority
of the known glycosyltransferases. Protein Sci. 12, 1418–31. doi:10.1110/ps.0302103
Liu, L., Hussain, S.K., Miller, R.S., Oyarzabal, O.A., 2009. Efficacy of Mini VIDAS for
the detection of Campylobacter spp. from retail broiler meat enriched in bolton broth,
with or without the supplementation of blood. J. Food Prot. 72, 2428–2432.
Liu, X., McNally, D.J., Nothaft, H., Szymanski, C.M., Brisson, J.-R., Li, J., 2006. Mass
spectrometry-based glycomics strategy for exploring N-linked glycosylation in
Eukaryotes and Bacteria. Anal. Chem. 78, 6081–6087. doi:10.1021/ac060516m
Lizak, C., Gerber, S., Numao, S., Aebi, M., Locher, K.P., 2011. X-ray structure of a
bacterial oligosaccharyltransferase. Nature 474, 350–355. doi:10.1038/nature10151
Logan, J.M., Burnens, a, Linton, D., Lawson, a J., Stanley, J., 2000. Campylobacter
lanienae sp. nov., a new species isolated from workers in an abattoir. Int. J. Syst.
Evol. Microbiol. 50 Pt 2, 865–72.
Logan, S.M., 2006. Flagellar glycosylation - a new component of the motility repertoire?
Microbiology 152, 1249–1262. doi:10.1099/mic.0.28735-0
Logan, S.M., Hui, J.P.M., Vinogradov, E., Aubry, A.J., Melanson, J.E., Kelly, J.F.,
Nothaft, H., Soo, E.C., 2009. Identification of novel carbohydrate modifications on
Campylobacter jejuni 11168 flagellin using metabolomics-based approaches. FEBS
J. 276, 1014–1023. doi:10.1111/j.1742-4658.2008.06840.x

235
Luangtongkum, T., Jeon, B., Han, J., Plummer, P., Logue, C.M., Zhang, Q., 2009.
Antibiotic resistance in Campylobacter: emergence, transmission and persistence.
Future Microbiol. 4, 189–200. doi:10.2217/17460913.4.2.189
Luo, N., Sahin, O., Lin, J., Michel, L.O., Zhang, Q., 2003. In vivo selection of
Campylobacter isolates with high levels of fluoroquinolone resistance associated
with gyrA mutations and the function of the CmeABC efflux pump. Antimicrob.
Agents Chem. 47, 390–4. doi:10.1128/aac.47.1.390-394.2003
Madden, R.H., Hutchison, M., Young, F., Taylor, M., 2016. Development of a rapid on-
farm test for the detection of Campylobacter. FSA Project FS241049.
Magidovich, H., Eichler, J., 2009. Glycosyltransferases and oligosaccharyltransferases in
Archaea: putative components of the N -glycosylation pathway in the third domain of
life. FEMS Microbiol. Lett. 300, 122–130. doi:10.1111/j.1574-6968.2009.01775.x
Man, S.M., 2011. The clinical importance of emerging Campylobacter species. Nat. Rev.
Gastroenterol. Hepatol. 8, 669–685. doi:10.1038/nrgastro.2011.191
Mannering, S. a., West, D.M., Fenwick, S.G., Marchant, R.M., O’Connell, K., 2006.
Pulsed-field gel electrophoresis of Campylobacter jejuni sheep abortion isolates. Vet.
Microbiol. 115, 237–242. doi:10.1016/j.vetmic.2006.01.005
Martinot, M., Jaulhac, B., Moog, R., De Martino, S., Kehrli, P., Monteil, H., Piemont, Y.,
2001. Campylobacter lari bacteremia. Clin. Microbiol. Infect. 7, 96–97.
doi:10.1046/j.1469-0691.2001.00212.x
Mavris, M., Manning, P.A., Morona, R., 1997. Mechanism of bacteriophage SfII-mediated
serotype conversion in Shigella flexneri. Mol. Microbiol. 26, 939–50.
doi:10.1046/j.1365-2958.1997.6301997.x

236
McMillen, L., Fordyce, G., Doogan, V.J., Lew, A.E., 2006. Comparison of culture and a
novel 5’ Taq nuclease assay for direct detection of Campylobacter fetus subsp.
venerealis in clinical specimens from cattle. J. Clin. Microbiol. 44, 938–45.
doi:10.1128/JCM.44.3.938-945.2006
Mcnally, D.J., Hui, J.P.M., Aubry, A.J., Mui, K.K.K., Guerry, P., Brisson, J.-R., Logan,
S.M., Soo, E.C., 2006. Functional characterization of the flagellar glycosylation locus
in Campylobacter jejuni 81–176 using a focused metabolomics approach. J. Biol.
Chem. 281, 18489-18498. doi:10.1074/jbc.M603777200
Mellick, P.W., Winter, A.J., Mcentee, K., 1965. Diagnosis of vibriosis in the bull by use
of the fluorescent antibody technique. Cornell Vet. 55, 280–94.
Mescher, M.F., Stroming.Jl, Watson, S.W., 1974. Protein and carbohydrate composition
of cell envelope of Halobacterium salinarium. J. Bacteriol. 120, 945–954.
Miller, R.S., Speegle, L., Oyarzabal, O. a., Lastovica, A.J., 2008. Evaluation of three
commercial latex agglutination tests for identification of Campylobacter spp. J. Clin.
Microbiol. 46, 3546–3547. doi:10.1128/JCM.01027-08
Miller, W.G., Yee, E., Chapman, M.H., 2016. Complete genome sequences of
Campylobacter hyointestinalis subsp. hyointestinalis strain LMG 9260 and C.
hyointestinalis subsp. lawsonii strain LMG 15993. Genome Announc. 4, e00665-16.
doi:10.1128/genomeA.00665-16
Mills, D.C., Jervis, A.J., Abouelhadid, S., Yates, L.E., Cuccui, J., Linton, D., Wren, B.W.,
2016. Functional analysis of N-linking oligosaccharyl transferase enzymes encoded
by deep-sea vent proteobacteria. Glycobiology 26, 398–409.
doi:10.1093/glycob/cwv111

237
Morelle, W., Canis, K., Chirat, F., Faid, V., Michalski, J.C., 2006. The use of mass
spectrometry for the proteomic analysis of glycosylation. Proteomics 6, 3993–4015.
doi:10.1002/pmic.200600129
Moremen, K.W., Tiemeyer, M., Nairn, A. V., 2012. Vertebrate protein glycosylation:
diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462.
doi:10.1038/nrm3383
Morrison, M.J., Imperiali, B., 2014. The renaissance of bacillosamine and its derivatives:
Pathway characterization and implications in pathogenicity. Biochemistry 53, 624–
638. doi:10.1021/bi401546r
Mshelia, G.D., Amin, J.D., Woldehiwet, Z., Murray, R.D., Egwu, G.O., 2010.
Epidemiology of bovine venereal campylobacteriosis: Geographic distribution and
recent advances in molecular diagnostic techniques. Reprod. Domest. Anim. 45.
doi:10.1111/j.1439-0531.2009.01546.x
Mshelia, G.D., Singh, J., Amin, J.D., Woldehiwet, Z., Egwu, G.O., Murray, R.D., 2007.
Bovine venereal campylobacteriosis: an overview. CAB Rev. Perspect. Agric. Vet.
Sci. Nutr. Nat. Resour. 2, 14 pp. doi:10.1079/PAVSNNR20072080
Naegeli, A., Michaud, G., Schubert, M., Lin, C.W., Lizak, C., Darbre, T., Reymond, J.L.,
Aebi, M., 2014. Substrate specificity of cytoplasmic N-glycosyltransferase. J. Biol.
Chem. 289, 24521–24532. doi:10.1074/jbc.M114.579326
Newell, D.G., Fearnley, C., 2003. Sources of Campylobacter colonization in broiler
chickens. Appl. Environ. Microbiol. 69, 4343–4351. doi:10.1128/AEM.69.8.4343
Niedermeyer, T.H.J., Strohalm, M., 2012. mMass as a software tool for the annotation of
cyclic peptide tandem mass spectra. PLoS One 7, e44913.

238
doi:10.1371/journal.pone.0044913
Nielsen, H.L., Engberg, J., Ejlertsen, T., B?cker, R., Nielsen, H., 2012. Short-term and
medium-term clinical outcomes of Campylobacter concisus infection. Clin.
Microbiol. Infect. 18, E459–E465. doi:10.1111/j.1469-0691.2012.03990.x
Noormohamed, A., Fakhr, M.K., 2013. A higher prevalence rate of Campylobacter in
retail beef livers compared to other beef and pork meat cuts. Int. J. Environ. Res.
Public Health 10, 2058–68. doi:10.3390/ijerph10052058
Nothaft, H., Davis, B., Lock, Y.Y., Perez-Munoz, M.E., Vinogradov, E., Walter, J., Coros,
C., Szymanski, C.M., 2016. Engineering the Campylobacter jejuni N-glycan to create
an effective chicken vaccine. Sci. Rep. 6, 26511. doi:10.1038/srep26511
Nothaft, H., Liu, X., McNally, D.J., Li, J., Szymanski, C.M., 2009. Study of free
oligosaccharides derived from the bacterial N-glycosylation pathway. Proc. Natl.
Acad. Sci. U. S. A. 106, 15019–15024. doi:10.1073/pnas.0903078106
Nothaft, H., Liu, X., McNally, D.J., Szymanski, C.M., 2010. N-linked protein
glycosylation in a bacterial system, in: Methods in Molecular Biology (Clifton, N.J.).
pp. 227–243. doi:10.1007/978-1-60761-454-8_16
Nothaft, H., Scott, N.E., Vinogradov, E., Liu, X., Hu, R., Beadle, B., Fodor, C., Miller,
W.G., Li, J., Cordwell, S.J., Szymanski, C.M., 2012. Diversity in the protein n-
glycosylation pathways within the Campylobacter Genus. Mol. Cell. Proteomics 11,
1203–1219. doi:10.1074/mcp.M112.021519
Nothaft, H., Szymanski, C.M., 2013. Bacterial protein N-glycosylation: New perspectives
and applications. J. Biol. Chem. 288, 6912–6920. doi:10.1074/jbc.R112.417857

239
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., Hase,
T., 2000. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28,
E63.
O’Donovan, D., Corcoran, G.D., Lucey, B., Sleator, R.D., 2014. Campylobacter
ureolyticus: A portrait of the pathogen. Virulence 1–9. doi:10.4161/viru.28776
Olivier, N.B., Chen, M.M., Behr, J.R., Imperiali, B., 2006. In vitro biosynthesis of UDP-
N,N ’-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein
glycosylation system. Biochemistry 45, 13659–13669. doi:10.1021/bi061456h
Ollis, A. a, Zhang, S., Fisher, A.C., DeLisa, M.P., 2014. Engineered
oligosaccharyltransferases with greatly relaxed acceptor-site specificity. Nat. Chem.
Biol. 1–9. doi:10.1038/nchembio.1609
On, S.L., Bloch, B., Holmes, B., Hoste, B., Vandamme, P., 1995. Campylobacter
hyointestinalis subsp. lawsonii subsp. nov., isolated from the porcine stomach, and an
emended description of Campylobacter hyointestinalis. Int. J. Syst. Bacteriol. 45,
767–74. doi:10.1099/00207713-45-4-767
On, S.L.W., Atabay, H.I., Corry, J.E.L., Harrington, C.S., Vandamme, P., 1998. Emended
description of Campylobacter sputorum and revision of its infrasubspecific (biovar)
divisions, including C. sputorum biovar paraureolyticus, a urease-producing variant
from cattle and humans. Int. J. Syst. Bacteriol. 48, 195–206. doi:10.1099/00207713-
48-1-195
Oyarzabal, O.A., Fernández, H., 2016. Isolation and identification of Campylobacter spp.
in poultry, in: Campylobacter spp. and related organisms in poultry. Springer
International Publishing, Cham, pp. 19–35. doi:10.1007/978-3-319-29907-5_2

240
Oyarzabal, O.A., Macklin, K.S., Barbaree, J.M., Miller, R.S., 2005. Evaluation of agar
plates for direct enumeration of Campylobacter spp. from poultry carcass rinses.
Appl. Environ. Microbiol. 71, 3351–3354. doi:10.1128/aem.71.6.3351-2254.2005
Oyarzabal, O. a, Battie, C., 2012. Immunological methods for the detection of
Campylobacter spp. - Current applications and potential use in biosensors, in: Trends
in Immunolabelled and Related Techniques. InTech, pp 203-225.
Oyofo, B.A., Thornton, S.A., Burr, D.H., Trust, T.J., Pavlovskis, O.R., Guerryl, P., 1992.
Specific detection of Campylobacter jejuni and Campylobacter coli by using
polymerase chain reaction. J. Clin. Microbiol. 30, 2613–2619.
Palmer, S.R., Gully, P.R., White, J.M., Pearson, A.D., Suckling, W.G., Jones, D.M.,
Rawes, J.C.L., Penner, J.L., 1983. Water-borne outbreak of Campylobacter
gastroenteritis. Lancet 321, 287–290. doi:10.1016/S0140-6736(83)91698-7
Pasternak, J., Bolivar, R., Hopfer, R.L., Fainstein, V., Mills, K., Rios, A., Bodey, G.P.,
Fennell, C.L., Totten, P.A., Stamm, W.E., 1984. Bacteremia caused by
Campylobacter-like organisms in two male homosexuals. Ann. Intern. Med. 101,
339–41.
Patrick, M.E., Gilbert, M.J., Blaser, M.J., Tauxe, R. V., Wagenaar, J. a., Fitzgerald, C.,
2013. Human infections with new subspecies of Campylobacter fetus. Emerg. Infect.
Dis. 19, 1678–1680. doi:10.3201/eid1910.130883
Patton, C.M., Shaffer, N., Edmonds, P., Barrett, T.J., Lambert, M.A., Baker, C., Perlman,
D.M., And, S., Brenner3, D.J., 1989. Human disease associated with Campylobacter
upsaliensis (Catalase-negative or weakly positive Campylobacter species) in the
United States. J. Clin. Microbiol. 66–73.

241
Pauwels, J., Taminiau, B., Janssens, G.P.J., De Beenhouwer, M., Delhalle, L., Daube, G.,
Coopman, F., 2015. Cecal drop reflects the chickens’ cecal microbiome, fecal drop
does not. J. Microbiol. Methods 117, 164–170. doi:10.1016/j.mimet.2015.08.006
Penner, J.L., Hennessy, J.N., Congi, R. V., 1983. Serotyping of Campylobacter jejuni and
Campylobacter coli on the basis of thermostable antigens. Eur. J. Clin. Microbiol. 2,
378–383. doi:10.1007/BF02019474
Petersen, R.F., Harrington, C.S., Kortegaard, H.E., On, S.L.W., 2007. A PCR-DGGE
method for detection and identification of Campylobacter, Helicobacter, Arcobacter
and related Epsilobacteria and its application to saliva samples from humans and
domestic pets. J. Appl. Microbiol. 103, 2601–2615. doi:10.1111/j.1365-
2672.2007.03515.x
Petersen, T.N., Brunak, S., von Heijne, G., Nielsen, H., 2011. SignalP 4.0: discriminating
signal peptides from transmembrane regions. Nat. Methods 8, 785–6.
doi:10.1038/nmeth.1701
Pickett, C.L., Whitehouse, C.A., 1999. The cytolethal distending toxin family. Trends
Microbiol. 7, 292–297. doi:10.1016/S0966-842X(99)01537-1
Pittman, M.S., Elvers, K.T., Lee, L., Jones, M.A., Poole, R.K., Park, S.F., Kelly, D.J.,
2007. Growth of Campylobacter jejuni on nitrate and nitrite: electron transport to
NapA and NrfA via NrfH and distinct roles for NrfA and the globin Cgb in protection
against nitrosative stress. Mol. Microbiol. 63, 575–590. doi:10.1111/j.1365-
2958.2006.05532.x
Platts-Mills, J.A., Liu, J., Gratz, J., Mduma, E., Amour, C., Swai, N., Taniuchi, M.,
Begum, S., Peñataro Yori, P., Tilley, D.H., Lee, G., Shen, Z., Whary, M.T., Fox, J.G.,
McGrath, M., Kosek, M., Haque, R., Houpt, E.R., 2014. Detection of Campylobacter
in stool and determination of significance by culture, enzyme immunoassay, and PCR

242
in developing countries. J. Clin. Microbiol. 52, 1074–80. doi:10.1128/JCM.02935-13
Power, P.M., Roddam, L.F., Rutter, K., Fitzpatrick, S.Z., Srikhanta, Y.N., Jennings, M.P.,
2003. Genetic characterization of pilin glycosylation and phase variation in Neisseria
meningitidis. Mol. Microbiol. 49, 833–47.
Public Health England, 2017. A microbiological survey of Campylobacter contamination
in fresh whole UK-produced chilled chickens at retail sale. FSA Project FS102121.
Qian, H., Pang, E., Du, Q., Chang, J., Dong, J., Say, L.T., Fook, K.N., Ai, L.T., Kwang, J.,
2008. Production of a monoclonal antibody specific for the major outer membrane
protein of Campylobacter jejuni and characterization of the epitope. Appl. Environ.
Microbiol. 74, 833–839. doi:10.1128/AEM.01559-07
Rangarajan, E.S., Bhatia, S., Watson, D.C., Munger, C., Cygler, M., Matte, A., Young,
N.M., 2007. Structural context for protein N-glycosylation in bacteria: The structure
of PEB3, an adhesin from Campylobacter jejuni. Protein Sci. 16, 990–995.
doi:10.1110/ps.062737507
Rosenquist, H., Nielsen, N.L., Sommer, H.M., Nørrung, B., Christensen, B.B., 2003.
Quantitative risk assessment of human campylobacteriosis associated with
thermophilic Campylobacter species in chickens. Int. J. Food Microbiol. 83, 87–103.
doi:10.1016/S0168-1605(02)00317-3
Ruiz-Canada, C., Kelleher, D.J., Gilmore, R., 2009. Cotranslational and posttranslational
N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136,
272–83. doi:10.1016/j.cell.2008.11.047
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M.A., Barrell,
B., 2000. Artemis: sequence visualization and annotation. Bioinformatics 16, 944–5.
doi:10.1093/BIOINFORMATICS/16.10.944

243
Sahin, O., Luo, N., Huang, S., Zhang, Q., 2003. Effect of Campylobacter-specific
maternal antibodies on Campylobacter jejuni colonization in young chickens. Appl.
Environ. Microbiol. 69, 5372-5379. doi:10.1128/AEM.69.9.5372
Sajid, M., Kawde, A.-N., Daud, M., 2015. Designs, formats and applications of lateral
flow assay: A literature review. J. Saudi Chem. Soc. 19, 689–705.
doi:10.1016/j.jscs.2014.09.001
Samuelson, J.D., Winter, A.J., 1966. Bovine vibriosis: the nature of the carrier state in the
bull. J. Infect. Dis. 116, 581–92.
Sandstedt, K., Ursing, J., 1991. Description of Campylobacter upsaliensis sp. nov.
previously known as the CNW Group. Syst. Appl. Microbiol. 14, 39–45.
doi:10.1016/S0723-2020(11)80359-0
Sapsford, K.E., Rasooly, A., Taitt, C.R., Ligler, F.S., 2004. Detection of Campylobacter
and Shigella species in food samples using an array biosensor. Anal. Chem. 76, 433–
440. doi:10.1021/ac035122z
Schmidt, T., Venter, E.H., Picard, J. a, 2010. Evaluation of PCR assays for the detection of
Campylobacter fetus in bovine preputial scrapings and the identification of
subspecies in South African field isolates. J. S. Afr. Vet. Assoc. 81, 87–92.
doi:10.4102/jsava.v81i2.111
Schulze, F., Bagon, A., Müller, W., Hotzel, H., 2006. Identification of Campylobacter
fetus subspecies by phenotypic differentiation and PCR. J. Clin. Microbiol. 44, 2019–
24. doi:10.1128/JCM.02566-05
Schwarz, F., Aebi, M., 2011. Mechanisms and principles of N-linked protein
glycosylation. Curr. Opin. Struct. Biol. 21, 576–582. doi:10.1016/j.sbi.2011.08.005

244
Schwarz, F., Lizak, C., Fan, Y.Y., Fleurkens, S., Kowarik, M., Aebi, M., 2011. Relaxed
acceptor site specificity of bacterial oligosaccharyltransferase in vivo. Glycobiology
21, 45–54. doi:10.1093/glycob/cwq130
Scott, N.E., Bogema, D.R., Connolly, A.M., Falconer, L., Djordjevic, S.P., Cordwell, S.J.,
2009. Mass spectrometric characterization of the surface-associated 42 kDa
lipoprotein JlpA as a glycosylated antigen in strains of Campylobacter jejuni. J.
Proteome Res. 8, 4654–4664. doi:10.1021/pr900544x
Scott, N.E., Marzook, N.B., Cain, J. a, Solis, N., Thaysen-Andersen, M., Djordjevic, S.P.,
Packer, N.H., Larsen, M.R., Cordwell, S.J., 2014. Comparative proteomics and
glycoproteomics reveal increased N-linked glycosylation and relaxed sequon
specificity in Campylobacter jejuni NCTC11168 O. J. Proteome Res.
doi:10.1021/pr5005554
Scott, N.E., Nothaft, H., Edwards, A.V.G., Labbate, M., Djordjevic, S.P., Larsen, M.R.,
Szymanski, C.M., Cordwell, S.J., 2012. Modification of the Campylobacter jejuni N-
linked glycan by EptC protein-mediated addition of phosphoethanolamine. J. Biol.
Chem. 287, 29384–29396. doi:10.1074/jbc.M112.380212
Scott, N.E., Parker, B.L., Connolly, A.M., Paulech, J., Edwards, A.V.G., Crossett, B.,
Falconer, L., Kolarich, D., Djordjevic, S.P., Højrup, P., Packer, N.H., Larsen, M.R.,
Cordwell, S.J., 2011. Simultaneous glycan-peptide characterization using hydrophilic
interaction chromatography and parallel fragmentation by CID, higher energy
collisional dissociation, and electron transfer dissociation MS applied to the N-linked
glycoproteome of Campylobacter jejuni. Mol. Cell. Proteomics 10, M000031–
MCP201. doi:10.1074/mcp.M000031-MCP201
Shrimal, S., Cherepanova, N.A., Gilmore, R., 2015. Cotranslational and
posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin.
Cell Dev. Biol. 41, 71–78. doi:10.1016/j.semcdb.2014.11.005

245
Silverman, J.M., Imperiali, B., 2016. Bacterial N-glycosylation efficiency is dependent on
the structural context of target sequons. J. Biol. Chem. 291, 22001–22010.
doi:10.1074/jbc.M116.747121
Simor, A.E., Wilcox, L., 1987. Enteritis associated with Campylobacter laridis. J. Clin.
Microbiol. 25, 10–2.
Skottrup, P.D., Nicolaisen, M., Justesen, A.F., 2008. Towards on-site pathogen detection
using antibody-based sensors. Biosens. Bioelectron. 24, 339–348.
doi:10.1016/j.bios.2008.06.045
Song, E., Pyreddy, S., Mechref, Y., 2013. Quantification of glycopeptides by multiple
reaction monitoring LC-MS/MS. Rapid Commun. Mass Spectrom. 26, 1941–1954.
doi:10.1002/rcm.6290.Quantification
Stanley, D., Geier, M.S., Chen, H., Hughes, R.J., Moore, R.J., 2015. Comparison of fecal
and cecal microbiotas reveals qualitative similarities but quantitative differences.
BMC Microbiol. 15, 51. doi:10.1186/s12866-015-0388-6
Stanley, J., Burnens, A.P., Linton, D., On, S.L.W., Costas, M., Owen, R.J., 1992.
Campylobacter helveticus sp. nov., a new thermophilic species from domestic
animals: characterization, and cloning of a species-specific DNA probe. J. Gen.
Microbiol. 138, 2293–2303. doi:10.1099/00221287-138-11-2293
Stanley, J., Linton, D., Burnens, A.P., Dewhirst, F.E., On, S.L., Porter, A., Owen, R.J.,
Costas, M., 1994. Helicobacter pullorum sp. nov.-genotype and phenotype of a new
species isolated from poultry and from human patients with gastroenteritis.
Microbiology 140 ( Pt 1, 3441–9. doi:10.1099/13500872-140-12-3441
Stimson, E., Virji, M., Makepeace, K., Dell, A., Morris, H.R., Payne, G., Saunders, J.R.,

246
Jennings, M.P., Barker, S., Panico, M., 1995. Meningococcal pilin: a glycoprotein
substituted with digalactosyl 2,4-diacetamido-2,4,6-trideoxyhexose. Mol. Microbiol.
17, 1201–14.
Studier, F.W., Moffatt, B.A., 1986. Use of bacteriophage T7 RNA polymerase to direct
selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130.
doi:10.1016/0022-2836(86)90385-2
Suo, B., He, Y., Paoli, G., Gehring, A., Tu, S.-I., Shi, X., 2010. Development of an
oligonucleotide-based microarray to detect multiple foodborne pathogens. Mol. Cell.
Probes 24, 77–86. doi:10.1016/j.mcp.2009.10.005
Szymanski, C.M., Burr, D.H., Guerry, P., 2002. Campylobacter protein glycosylation
affects host cell interactions. Infect. Immun. 70, 2242–2244.
doi:10.1128/iai.70.4.2242-2244.2002
Szymanski, C.M., Yao, R.J., Ewing, C.P., Trust, T.J., Guerry, P., 1999. Evidence for a
system of general protein glycosylation in Campylobacter jejuni. Mol. Microbiol. 32,
1022–1030. doi:10.1046/j.1365-2958.1999.01415.x
Tanner, A.C.R., Badger, S., Lai, C.-H., Listgarten, M.A., Visconti, R.A., Socransky, S.S.,
1981. Wolinella gen. nov., Wolinella succinogenes (Vibrio succinogenes Wolin et al.)
comb. nov., and description of Bacteroides gracilis sp. nov., Wolinella recta sp. nov.,
Campylobacter concisus sp. nov., and Eikenella corrodens from humans with
periodontal disease. Int. J. Syst. Bacteriol. 31, 432–445. doi:10.1099/00207713-31-4-
432
Tee, W., Montgomery, J., Dyall‐ Smith, M., 2001. Bacteremia caused by a Helicobacter
pullorum– like organism. Clin. Infect. Dis. 33, 1789–1791. doi:10.1086/323983

247
Thibault, P., Logan, S.M., Kelly, J.F., Brisson, J.-R., Ewing, C.P., Trust, T.J., Guerry, P.,
2001. Identification of the carbohydrate moieties and glycosylation motifs in
Campylobacter jejuni flagellin. J. Biol. Chem. 276, 34862–34870.
doi:10.1074/jbc.M104529200
Thompson, S. a, 2002. Campylobacter surface-layers (S-layers) and immune evasion.
Ann. Periodontol. 7, 43–53. doi:10.1902/annals.2002.7.1.43
Tissari, P., Rautelin, H., 2007. Evaluation of an enzyme immunoassay-based stool antigen
test to detect Campylobacter jejuni and Campylobacter coli. Diagn. Microbiol. Infect.
Dis. 58, 171–175. doi:10.1016/j.diagmicrobio.2006.12.023
Toghi Eshghi, S., Yang, W., Hu, Y., Shah, P., Sun, S., Li, X., Zhang, H., 2016.
Classification of tandem mass spectra for identification of N- and O-linked
glycopeptides. Sci. Rep. 6, 37189. doi:10.1038/srep37189
Tribble, D.R., Baqar, S., Pang, L.W., Mason, C., Houng, H.-S.H., Pitarangsi, C., Lebron,
C., Armstrong, A., Sethabutr, O., Sanders, J.W., 2008. Diagnostic approach to acute
diarrheal illness in a military population on training exercises in Thailand, a region of
Campylobacter hyperendemicity. J. Clin. Microbiol. 46, 1418–25.
doi:10.1128/JCM.02168-07
Troutman, J.M., Imperiali, B., 2009. Campylobacter jejuni PglH is a single active site
processive polymerase that utilizes product inhibition to limit sequential glycosyl
transfer reactions. Biochemistry 48, 2807–2816. doi:10.1021/bi802284d
Truyers, I., Luke, T., Wilson, D., Sargison, N., 2014. Diagnosis and management of
venereal campylobacteriosis in beef cattle. BMC Vet. Res. 10, 280.
doi:10.1186/s12917-014-0280-x

248
Umaraw, P., Prajapati, A., Verma, A.K., Pathak, V., Singh, V.P., 2017. Control of
Campylobacter in poultry industry from farm to poultry processing unit: A review.
Crit. Rev. Food Sci. Nutr. 57, 659–665. doi:10.1080/10408398.2014.935847
van Bergen, M. a P., Dingle, K.E., Maiden, M.J., Newell, D.G., van Der Graaf-Van
Bloois, L., van Putten, J.P.M., Wagenaar, J. a, 2005. Clonal nature of Campylobacter
fetus as defined by multilocus sequence typing. J. Clin. Microbiol. 43, 5888–5898.
doi:10.1128/JCM.43.12.5888
van der Graaf-van Bloois, L., Miller, W.G., Yee, E., Rijnsburger, M., Wagenaar, J.A.,
Duim, B., 2014. Inconsistency of phenotypic and genomic characteristics of
Campylobacter fetus subspecies requires reevaluation of current diagnostics. J. Clin.
Microbiol. 52, 4183–8. doi:10.1128/JCM.01837-14
van der Graaf-van Bloois, L., van Bergen, M. a P., van der Wal, F.J., de Boer, A.G., Duim,
B., Schmidt, T., Wagenaar, J. a, 2013. Evaluation of molecular assays for
identification Campylobacter fetus species and subspecies and development of a C.
fetus specific real-time PCR assay. J. Microbiol. Methods 95, 93–7.
doi:10.1016/j.mimet.2013.06.005
van Doorn, P.A., Ruts, L., Jacobs, B.C., 2008. Clinical features, pathogenesis, and
treatment of Guillain-Barré syndrome. Lancet Neurol. 7, 939–950.
doi:10.1016/S1474-4422(08)70215-1
van Sorge, N.M., Bleumink, N.M.C., van Vliet, S.J., Saeland, E., van der Pol, W.-L., van
Kooyk, Y., van Putten, J.P.M., 2009. N-glycosylated proteins and distinct
lipooligosaccharide glycoforms of Campylobacter jejuni target the human C-type
lectin receptor MGL. Cell. Microbiol. 11, 1768–81. doi:10.1111/j.1462-
5822.2009.01370.x
Vandamme, P., Daneshvar, M.I., Dewhirst, F.E., Paster, B.J., Kersters, K., Goossens, H.,

249
Moss, C.W., 1995. Chemotaxonomic analyses of Bacteroides gracilis and
Bacteroides ureolyticus and reclassification of B. gracilis as Campylobacter gracilis
comb. nov. Int. J. Syst. Bacteriol. 45, 145–52. doi:10.1099/00207713-45-1-145
Vandamme, P., Debruyne, L., De Brandt, E., Falsen, E., 2010. Reclassification of
Bacteroides ureolyticus as Campylobacter ureolyticus comb. nov., and emended
description of the genus Campylobacter. Int. J. Syst. Evol. Microbiol. 60, 2016–2022.
doi:10.1099/ijs.0.017152-0
Veron, M., Chatelain, R., 1973. Taxonomic study of the Genus Campylobacter Sebald and
Veron and designation of the neotype strain for the type species, Campylobacter fetus
(Smith and Taylor) Sebald and Veron. Int. J. Syst. Bacteriol. 23, 122–134.
doi:10.1099/00207713-23-2-122
Vik, A., Aas, F.E., Anonsen, J.H., Bilsborough, S., Schneider, A., Egge-Jacobsen, W.,
Koomey, M., 2009. Broad spectrum O-linked protein glycosylation in the human
pathogen Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. 106, 4447–4452.
doi:10.1073/pnas.0809504106
Wacker, M., Linton, D., Hitchen, P.G., Nita-Lazar, M., Haslam, S.M., North, S.J., Panico,
M., Morris, H.R., Dell, A., Wren, B.W., Aebi, M., 2002. N-linked glycosylation in
Campylobacter jejuni and its functional transfer into E. coli. Science 298, 1790–
1793. doi:10.1126/science.298.5599.1790
Wadl, M., Pölzler, T., Flekna, G., Thompson, L., Slaghuis, J., Köfer, J., Hein, I., Wagner,
M., 2009. Easy-to-use rapid test for direct detection of Campylobacter spp. in
chicken feces. J. Food Prot. 72, 2483–8.
Wagenaar, J.A., van Bergen, M.A.P., Newell, D.G., Grogono-Thomas, R., Duim, B.,
2001. Comparative study using amplified fragment length polymorphism
fingerprinting, PCR genotyping, and phenotyping to differentiate Campylobacter

250
fetus strains isolated from animals. J. Clin. Microbiol. 39, 2283–2286.
doi:10.1128/jcm.39.6.2283-2286.2001
Wagenaar, J. a., Van Bergen, M. a P., Blaser, M.J., Tauxe, R. V., Newell, D.G., Van
Putten, J.P.M., 2014. Campylobacter fetus infections in humans: Exposure and
disease. Clin. Infect. Dis. 58, 1579–1586. doi:10.1093/cid/ciu085
Werno, A.M., Klena, J.D., Shaw, G.M., Murdoch, D.R., 2002. Fatal Case of
Campylobacter lari prosthetic joint infection and bacteremia in an immunocompetent
patient. J. Clin. Microbiol. 40, 1053–1055. doi:10.1128/JCM.40.3.1053–1055.2002
Wolin, M.J., Wolin, E.A., Jacobs, N.J., 1961. Cytochrome-producing anaerobic Vibrio
succinogenes, sp. n. J. Bacteriol. 81, 911–7.
Yachdav, G., Kloppmann, E., Kajan, L., Hecht, M., Goldberg, T., Hamp, T.,
Honigschmid, P., Schafferhans, A., Roos, M., Bernhofer, M., Richter, L., Ashkenazy,
H., Punta, M., Schlessinger, A., Bromberg, Y., Schneider, R., Vriend, G., Sander, C.,
Ben-Tal, N., Rost, B., 2014. PredictProtein--an open resource for online prediction of
protein structural and functional features. Nucleic Acids Res. 42, W337–W343.
doi:10.1093/nar/gku366
Yang, X., Kirsch, J., Simonian, A., 2013. Campylobacter spp. detection in the 21st
century: a review of the recent achievements in biosensor development. J. Microbiol.
Methods 95, 48–56. doi:10.1016/j.mimet.2013.06.023
Yao, R., Burr, D.H., Doig, P., Trust, T.J., Niu, H., Guerry, P., 1994. Isolation of motile
and non-motile insertional mutants of Campylobacter jejuni: The role of motility in
adherence and invasion of eukaryotic cells. Mol. Microbiol. 14, 883–893.
doi:10.1111/j.1365-2958.1994.tb01324.x

251
Young, K.T., Davis, L.M., Dirita, V.J., 2007. Campylobacter jejuni: molecular biology
and pathogenesis. Nat. Rev. Microbiol. 5, 665–679. doi:10.1038/nrmicro1718
Young, N.M., Brisson, J.R., Kelly, J., Watson, D.C., Tessier, L., Lanthier, P.H., Jarrell,
H.C., Cadotte, N., Michael, F.S., Aberg, E., Szymanski, C.M., 2002. Structure of the
N-linked glycan present on multiple glycoproteins in the gram-negative bacterium,
Campylobacter jejuni. J. Biol. Chem. 277, 42530–42539.
doi:10.1074/jbc.M206114200
Yu, C.-S., Chen, Y.-C., Lu, C.-H., Hwang, J.-K., 2006. Prediction of protein subcellular
localization. Proteins Struct. Funct. Bioinforma. 64, 643–651.
doi:10.1002/prot.21018
Zhao, P., Viner, R., Teo, C.F., Boons, G.-J., Horn, D., Wells, L., 2011. Combining high-
energy C-trap dissociation and electron transfer dissociation for protein O-GlcNAc
modification site assignment. J. Proteome Res. 10, 4088–4104.
doi:10.1021/pr2002726

252
Appendix
Table 2.1. Bacterial strains used.
Bacterial strain Reference/Source
Campylobacter jejuni 11168H Karlyshev et al. 2002
C. jejuni 11168gfp4 Jervis et al. 2015
C. jejuni 11168H pgI::kan Linton et al. 2002
C. jejuni 11168H pglA::kan Linton et al. 2002
C. jejuni 11168H pglB::kan Wacker et al. 2002
C. jejuni 11168H pglD::kan Linton et al. 2002
C. jejuni 11168H pglE::kan Linton et al. 2002
C. jejuni 11168H pglF::kan Linton et al. 2002
C. jejuni 11168H pglH::kan Linton et al. 2002
C. jejuni 11168H pglJ::kan Linton et al. 2002
C. jejuni 81116 Palmer et al. 1983
C. jejuni 81-176 Korlath et al. 1985
C. jejuni DW10 This study
C. jejuni DW2 This study
C. jejuni DW3 This study
C. jejuni DW7 This study
C. jejuni DW8 This study
C. jejuni DW9 This study
C. jejuni Guillain-Barré strain G2 (Linton et al., 2000)
C. jejuni Miller Fisher strain F1 (Linton et al., 2000)
C. jejuni Miller Fisher strain F2 (Linton et al., 2000)
C. jejuni Miller Fisher strain F3 (Linton et al., 2000)
C. jejuni Penner serotype reference strain P1 Penner et al. 1983
C. jejuni Penner serotype reference strain P10 Penner et al. 1983
C. jejuni Penner serotype reference strain P19 Penner et al. 1983
C. jejuni Penner serotype reference strain P3 Penner et al. 1983
C. jejuni Penner serotype reference strain P4 Penner et al. 1983
Campylobacter coli DW1 This study
C. coli DW4 This study
C. coli DW5 This study
C. coli DW6 This study
C. coli RM2228 Veron and Chatelain 1973
Campylobacter concisus 13826 Tanner et al. 1981
Campylobacter fetus clinical isolate 1* PHE
Campylobacter fetus clinical isolate 10# PHE
Campylobacter fetus clinical isolate 11~ PHE
Campylobacter fetus clinical isolate 12+ PHE
Campylobacter fetus clinical isolate 13# PHE
Campylobacter fetus clinical isolate 14# PHE

253
Campylobacter fetus clinical isolate 15+ PHE
Campylobacter fetus clinical isolate 16# PHE
Campylobacter fetus clinical isolate 17~
PHE
Campylobacter fetus clinical isolate 19 PHE
Campylobacter fetus clinical isolate 2+ PHE
Campylobacter fetus clinical isolate 3+ PHE
Campylobacter fetus clinical isolate 4+ PHE
Campylobacter fetus clinical isolate 5 PHE
Campylobacter fetus clinical isolate 7+
PHE
Campylobacter fetus clinical isolate 8#
PHE
Campylobacter fetus clinical isolate 9+
PHE
Campylobacter fetus subsp. fetus NCTC 10842 Veron and Chatelain 1973
Campylobacter fetus subsp. fetus NCTC 10842
pglB::kan
Jervis et al. 2012
Campylobacter fetus subsp. fetus clinical isolate 18! PHE
Campylobacter fetus subsp. fetus clinical isolate 6+ PHE
Campylobacter fetus subsp. venerealis NCTC 10354
Veron and Chatelain 1973
Campylobacter helveticus NCTC 12470 Stanley et al. 1992
Campylobacter hyointestinalis subsp. hyointestinalis
NCTC 11608
Gebhart et al. 1985
Campylobacter lanienae NCTC 13004 Logan et al. 2000
Campylobacter lari RM2100 Benjamin et al. 1983
Campylobacter sputorum subsp. sputorum NCTC 11528 Veron and Chatelain 1973
Campylobacter upsaliensis NCTC 11541 Sandstedt and Ursing 1991
Campylobacter upsaliensis RM3195 Fouts et al. 2005
E. coli BL21 (λDE3) Studier and Moffatt 1986
E. coli S17-1 λpir de Lorenzo and Timmis
1994 E. coli XL-1 Blue Agilent
PHE; Public Health England. For clinical isolates, symbol indicates site of origin:
*Tissue
+Blood
#Faeces
~Hip aspirate
!Shoulder.

254
Appendix Table 2. Plasmids used.
Plasmid Description Resistance Reference
pRYGG1 sapA108 promoter in multiple cloning
region of pRY111 Chlor
Kienesberger et al.
2007
pG1-N
pRYGG1 containing bases 1 to 561 of
cj0114 plus eight 3’ histidine codons
cloned at the BamHI and SmaI sites.
Chlor This study
pET22 (+)b
pET system vector carrying an N-
terminal pelB signal sequence and a C-
terminal His-Tag®.
Amp Novagen
pNGRP
pET22(+)b with bases 73 to 561 of
cj0114 cloned between NcoI and XhoI
sites.
Amp Jervis & Linton,
unpublished
pNH1 pNGRP with bases -105 to 1053 of C.
fetus pglH1 cloned at the BglII site Amp This study
pNH2 pNGRP with bases -77 to 1046 of C.
fetus pglH2 cloned at the BglII site Amp This study
pN1389 pNGRP with bases -98 to 1010 of C.
fetus cf1389 cloned at the BglII site Amp This study
pNCjH pNGRP with bases -136 to 1080 of C.
jejuni pglH cloned at the BglII site Amp This study
pNH1H2
pNGRP with bases -105 to 1053 and 1
to 1046 of C. fetus pglH1 and pglH2,
respectively, cloned at the BglII site
Amp This study
pNH1-1389 pNH1 with bases -98 to 1010 of C. fetus
cf1389 cloned at the BglII site Amp This study
pNX12
pNGRP with bases -105 to 1053 and 1
to 1046 of C. fetus pglH1 and pglH2,
respectively, and bases -98 to 1010 of C.
fetus cf1389 cloned at the BglII site
Amp This study
ppgl pACYC containing the 16 kb C. jejuni
pgl locus. Chlor Wacker et al. 2002
ppglpglH::kn ppgl with a kanamycin cassette inserted
within pglH.
Chlor
Kan Linton et al. 2005
p0445
pRYGG1 with the ORF of C. fetus
cf0445 plus eight 3’ histidine codons
cloned at the BamHI and SmaI sites.
Chlor This study
p0781
pRYGG1 with the ORF of C. fetus
cf0781 plus eight 3’ histidine codons
cloned at the BamHI and SmaI sites.
Chlor This study
Chlor; chloramphenicol. Amp; ampicillin. Kan; kanamycin.


















