
Amino acid disorders. Phenylketonuria (PKU) Enzyme defect: phenylalanine hydroxylase (12th...
57
Amino acid disorders
-
Upload
peter-goodwin -
Category
Documents
-
view
217 -
download
1
Transcript of Amino acid disorders. Phenylketonuria (PKU) Enzyme defect: phenylalanine hydroxylase (12th...

Amino acid disorders

4-OH-fenilpirüvik asit
Homogentisik asit
Malleoloasitik asit
Fümarilasetoasetik asit
Süksinil- aseton
Fümarik asit + asetoasetik asit
Fenilalanin hidroksilaz
4-OH fenilpirü- vik asit dioksijenaz
Tirozin aminotrasferaz
Homogentisik
asit oksidaz
İzomeraz
Fümarilasetoasetat
hidrolaz
Fenilpirüvat
Parahidroksifenilasetat
Fenillaktat
Fenilasetat
Fenilasetilglütamin
ALKAPTONÜRİ
FENİLKETONÜRİ
NTBC (-)
Tirozin hidroksilaz + BH4, folik asit, niasin, demir)
ALBİNİZM
FENİLALANİN TİROZİN
Delta-amino- Levülinik asit (d-ALA)
Porfobilinojen
d-ALA Dehidraz
Dihidroksifenilalanin (DOPA)
Triptofan
5-hidroksi-
triptofan (5-HT)
Triptofan
dihidroksilaz
BH4
Serotonin
Norepinefrin
Melanin
Metionin Adenozil- metionin
Metionin adenoziltransferaz
Melanositik tirozin hidroksilaz
Tetrahidrobiyopterin (BH4)
TETRAHİDROBİOPTERİN YETERSİZLİĞİ
Tiroksin
TİROZİNEMİ TİP III HAWKİNSİNÜRİ YENİDOĞANIN GEÇİCİ HİPERTİROZİNEMİSİ
TİROZİNEMİ TİP II
TİROZİNEMİ TİP I
Epinefrin
Dopamin
B6 vit(+)
Homovalinik asit
C vit(+)
C vit(+)
C vit(+)
B6 vit(+)

Phenylketonuria (PKU)
• Enzyme defect: phenylalanine hydroxylase (12th chromosome): more than 400 mutations
• Incidence: Average 1:10,000 (Highest incidence in Turkey, 1: 4,000)

Phenylketonuria (PKU): Variants
1. Classical phenylketonuria (complete or near complete enzyme deficiency): phenylalanine levels above 20 mg/dL (<1200 mmol/L) require diet therapy
2. Atypical phenylketonuria (partial enzyme deficiency): (enzyme activity %1-5) require partial diet therapy
3. Benign phenylketonuria. phenylalanine levels below: 10 mg/dL (<600 mmol/L) no clinical findings, not requiring diet therapy
3. Malign phenylketonuria: Tetrahydrobiopterin (BH4=cofactor of phenylalanine hydroxylase): Severe neurologic findings, does not respond diet therapy. Dopamine and setotonin may be helpful.

Phenylketonuria (PKU): Clinical findings
• Severe brain damage, progressive motor-mental retardation
• Spasticity
• Paralysis
• Convulsions
• Self-mutilation
• Light colored skin and eye (yellow hair, blue eyes; tyrosine deficiency)
• Mouse-like odor in urine and sweat.


Phenylketonuria: Diagnosis
• High phenylalanine (N: <2mg/dL) and low tyrosine (N: <2mg/dL) levels,
• Ferric chloride test gives green color in urine (not reliable).
• Neonatal screening: Guthrie-card (taken between 3rd and 7th days of life)

Phenylketonuria:Therapy• Phenylalanine restricted diet, supplementation of
tyrosine, essential amino acids and trace elements.
Goals of the therapy:• 0-10 years: phenylalanine values: 0.7-4 mg/dL• 11-16 years: phenylalanine values: <15 mg/dL• 16+ years: phenylalanine values: <20 mg/dL• Pregnant mothers with PKU: phenylalanine
values < 7mg/dL
Prognosis: with immediate and efficient treatment, normal development and intelligence

Maternal PKU= phenylketonuric fetopathy
• Normal phenylalanine levels
• Microcephaly
• Cardiac defects
• Motor-mental retardation
• No therapy
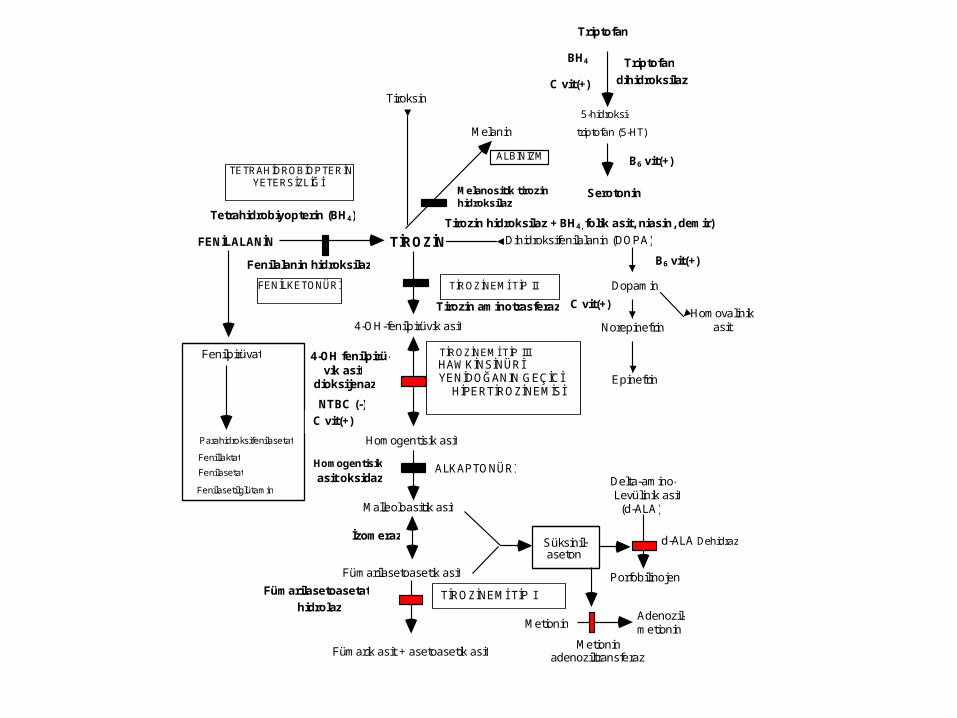
4-OH-fenilpirüvik asit
Homogentisik asit
Malleoloasitik asit
Fümarilasetoasetik asit
Süksinil- aseton
Fümarik asit + asetoasetik asit
Fenilalanin hidroksilaz
4-OH fenilpirü- vik asit dioksijenaz
Tirozin aminotrasferaz
Homogentisik
asit oksidaz
İzomeraz
Fümarilasetoasetat
hidrolaz
Fenilpirüvat
Parahidroksifenilasetat
Fenillaktat
Fenilasetat
Fenilasetilglütamin
ALKAPTONÜRİ
FENİLKETONÜRİ
NTBC (-)
Tirozin hidroksilaz + BH4, folik asit, niasin, demir)
ALBİNİZM
FENİLALANİN TİROZİN
Delta-amino- Levülinik asit (d-ALA)
Porfobilinojen
d-ALA Dehidraz
Dihidroksifenilalanin (DOPA)
Triptofan
5-hidroksi-
triptofan (5-HT)
Triptofan
dihidroksilaz
BH4
Serotonin
Norepinefrin
Melanin
Metionin Adenozil- metionin
Metionin adenoziltransferaz
Melanositik tirozin hidroksilaz
Tetrahidrobiyopterin (BH4)
TETRAHİDROBİOPTERİN YETERSİZLİĞİ
Tiroksin
TİROZİNEMİ TİP III HAWKİNSİNÜRİ YENİDOĞANIN GEÇİCİ HİPERTİROZİNEMİSİ
TİROZİNEMİ TİP II
TİROZİNEMİ TİP I
Epinefrin
Dopamin
B6 vit(+)
Homovalinik asit
C vit(+)
C vit(+)
C vit(+)
B6 vit(+)

Tyrosinemia Type I
Enzyme defect: Fumarylacetoacetate hydroxylase
Clinical findings
• Acute infantile form: Severe liver failure, vomiting, bleeds, sepsis, hypoglycemia, renal tubulopathy (Fanconi syndrome)
• Chronic form: Hepatomegaly, cirrhosis, growth retardation, rickets, hematoma, tubulopathy, neuropathy, and abdominal pain (due to porphyrines)

Tyrosinemia Type I: Diagnosis
• High succinylacetone levels (diagnostic). tyrosine levels: normal or slightly elevated.
• Methionine: high
• Delta-aminolevulinic acid: high (colic)
• Alfa-feto protein: very high (marker of hepatocellular carcinoma)

Tyrosinemia Type I: Therapy
• NTBC 1 mg/kg: blocks the accumulation of toxic metabolites (succinylacetone); beware tyrosine elevation and give tyrosine-restricted diet
• If this therapy fails consider liver transplantation.

Tyrosinemia Type I: Complications
• Renal failure• Hepatocellular carcinoma
(monitor alfa-feto protein), check periodically liver ultrasongraphy and biopsy.
Prognosis: Relatively good under NTBC treatment.

4-OH-fenilpirüvik asit
Homogentisik asit
Malleoloasitik asit
Fümarilasetoasetik asit
Süksinil- aseton
Fümarik asit + asetoasetik asit
Fenilalanin hidroksilaz
4-OH fenilpirü- vik asit dioksijenaz
Tirozin aminotrasferaz
Homogentisik
asit oksidaz
İzomeraz
Fümarilasetoasetat
hidrolaz
Fenilpirüvat
Parahidroksifenilasetat
Fenillaktat
Fenilasetat
Fenilasetilglütamin
ALKAPTONÜRİ
FENİLKETONÜRİ
NTBC (-)
Tirozin hidroksilaz + BH4, folik asit, niasin, demir)
ALBİNİZM
FENİLALANİN TİROZİN
Delta-amino- Levülinik asit (d-ALA)
Porfobilinojen
d-ALA Dehidraz
Dihidroksifenilalanin (DOPA)
Triptofan
5-hidroksi-
triptofan (5-HT)
Triptofan
dihidroksilaz
BH4
Serotonin
Norepinefrin
Melanin
Metionin Adenozil- metionin
Metionin adenoziltransferaz
Melanositik tirozin hidroksilaz
Tetrahidrobiyopterin (BH4)
TETRAHİDROBİOPTERİN YETERSİZLİĞİ
Tiroksin
TİROZİNEMİ TİP III HAWKİNSİNÜRİ YENİDOĞANIN GEÇİCİ HİPERTİROZİNEMİSİ
TİROZİNEMİ TİP II
TİROZİNEMİ TİP I
Epinefrin
Dopamin
B6 vit(+)
Homovalinik asit
C vit(+)
C vit(+)
C vit(+)
B6 vit(+)

Tyrosinemia Type II
• Enzyme defect: Cytosolic tyrosine aminotransferase
• Clinical findings: Painful corneal lesions (lacrimation, photophobia, scars), mild mental retardation
• Diagnosis: High tyrosine and phenylalanine levels
• Therapy: Tyrosine and phenylalanine-restricted diet

4-OH-fenilpirüvik asit
Homogentisik asit
Malleoloasitik asit
Fümarilasetoasetik asit
Süksinil- aseton
Fümarik asit + asetoasetik asit
Fenilalanin hidroksilaz
4-OH fenilpirü- vik asit dioksijenaz
Tirozin aminotrasferaz
Homogentisik
asit oksidaz
İzomeraz
Fümarilasetoasetat
hidrolaz
Fenilpirüvat
Parahidroksifenilasetat
Fenillaktat
Fenilasetat
Fenilasetilglütamin
ALKAPTONÜRİ
FENİLKETONÜRİ
NTBC (-)
Tirozin hidroksilaz + BH4, folik asit, niasin, demir)
ALBİNİZM
FENİLALANİN TİROZİN
Delta-amino- Levülinik asit (d-ALA)
Porfobilinojen
d-ALA Dehidraz
Dihidroksifenilalanin (DOPA)
Triptofan
5-hidroksi-
triptofan (5-HT)
Triptofan
dihidroksilaz
BH4
Serotonin
Norepinefrin
Melanin
Metionin Adenozil- metionin
Metionin adenoziltransferaz
Melanositik tirozin hidroksilaz
Tetrahidrobiyopterin (BH4)
TETRAHİDROBİOPTERİN YETERSİZLİĞİ
Tiroksin
TİROZİNEMİ TİP III HAWKİNSİNÜRİ YENİDOĞANIN GEÇİCİ HİPERTİROZİNEMİSİ
TİROZİNEMİ TİP II
TİROZİNEMİ TİP I
Epinefrin
Dopamin
B6 vit(+)
Homovalinik asit
C vit(+)
C vit(+)
C vit(+)
B6 vit(+)

Alcaptonuria• Enzyme defect: Homogentisate
oxygenase
• Clinical findings: black discoloration in urine at acid pH; mild arthritis in adults
• Diagnosis: High homogentisic acid levels in urine
• Therapy: Protein-restricted diet? NTBC?
• Prognosis: Relatively good without treatment

Methionine metabolism

CLASSICAL HOMOCYSTINURIA
• Enzyme defect: Cystationine-ß-synthase
• Mechanism: Accumulation of homocysteine (collagen disorder)
• Clinical findings: Progressive disease, usually starting with school age. Marfan-like appearance (archnodactyly), progressive myopia (the earliest finding), lens dislocation, epilepsy, mental retardation, osteoporosis, thromboembolism !!!

Marfan syndrom

HOMOCYSTINURIA
• Diagnosis: High methionine, high homocysteine (N: 0-3.5 µmol/L) and low cysteine levels. Positive nitroprusside test in fresh urine
• Therapy: Pyridoxine (Vit. B6): 50-1000 mg/day + folic acid 10 mg/day.
• If this fails diet + betaine (100 mg/kg) up to 3X3 g
• Goal: Keep homocysteine <30µmol/L.

MILD HYPERHOMOCYSTEINEMIACauses
• Methylene tetrahydrofolate reductase (MTHFR) polymorphism, thermolabile variant, homozygosity, up to 5% in Europeans, 60% in Asiasns
• Heterozygosity for cystationine-ß-synthase
• Endogenous and exogenous disorders of folic acid metabolism
• Vitamin B12 deficiency

MILD HYPERHOMOCYSTEINEMIA
Clinical findings:• Premature vascular disease in the 3rd
and 4th decade (infarctions, thrombosis embolies)
Maternal hyperhomocysteinemia: congenital defects
• Neural tube defects• Cardiac output defects• Renal defects• Pyloric stenosis?


Maple syrup urine disease
Enzyme: Branched-chain alfa-ketoacid dehydrogenase complex
Incidence: 1:200,000, autosomal recessive
Clinical findings
• Severe form: Progressive encephalopathy, cerebral edema, lethargy, coma after the 3rd day of life, “çemen” odor in urine and sweat
• Mild form: Developmental retardation, recurrent ketoacidotic decompensation

Diagnosis:• “Çemen” odor in urine and sweat, positive DNPH
test in urine (non-spesific), • Aminoacid analysis: high valine, leucine,
isoleucine and alloisoleucine (diagnostic) levels.
Therapy: • Acute: Detoxification (dialysis, exchange
transfusion)Augmentation of anabolism : Glucose + insulin
• Chronic: Diet (monitor leucine level) ± vitamin B1 (thiamin): 5 mg/kg/day

Disorders of amino acid transport

Methionine Malabsorption
• Methionine malabsorption in renal tubules and intestines.
• Clinical findings: White hair, convulsions,, diarrhea, edema , mental retardation, odor (like beer).
• Therapy: Diet deficient in methionine.

HARTNUP DISEASE• Defect: Intestinal and renal tubular reabsorption
defect of the neutral amino acids (alanine, valine, threonine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, histidine, glycine; tryptophan deficiency leads nicotinic acid and serotonine deficiency.
• Clinical finding: Photodermatitis, cerebellar ataxia; often asymptomatic
• Diagnosis: High levels of neutral amino acids in urine low levels of neutral amino acids in plasma.
• Therapy: Nicotinamide 40-300 mg/day, sun protection

LYSINURIC PROTEIN INTOLERANCE
• Defect: Intestinal and renal tubular reabsorption defect of the dibasic amino acids (lysine, arginine and ornithine) lead blockage of urea cycle; lysine deficiency
• Clinical findings: Intestinal protein intolerance, failure to thrive, osteoporosis, and hyperammonemia with progressive encephalopathy
• Diagnosis: Hyperammonemia, low lysine, arginine and ornithine in plasma, high LDH levels.
• Therapy: Citrulline substitution, protein restriction

CYSTINURIA
• Defect: Renal tubular reabsorption defect of the dibasic amino acids (lysine, arginine, ornithine and cystine)
• Clinical findings: Neprolithiasis (cystine crystallizes above 1250 µmol/L at pH 7.5)
• Diagnosis: Positive nitroprusside test in urine, increased levels of acids lysine, arginine, ornithine and cystine in urine, plasma levels are generally normal.
• Therapy: High (>5L) fluid intake, alkalisation of the urine (urinary infections!). Consider penisillamine (1-2 g/day), mercaptopropionylglycine or captopril in selected cases.

ORGANIC ACIDEMIAS
Pahogenesis • Mitochondrial accumulation of related CoA-
metabolites
Clinical findings
Acute neonatal form• Lethargy * Coma• Feeding problems *
Hypotonia/hypertonia• Myoclonic jerks * Cerebral edema• Dehydration * Unusual odor

ORGANIC ACIDEMIAS: Forms
Acute intermittent form• Recurrent episodes of acidotic coma• Ataxia• Focal neurologic signs
Chronic progressive form• Failure to thrive, Anorexia • Chronic vomiting• Hypotonia• Developmental retardation

ORGANIC ACIDEMIAS
Laboratory findings• Acidosis (increased anion gap)• Hyperammonemia• Hyperlactatemia
Diagnosis• Organic acids in urine (GC-MS)• Enzyme and DNA studies

ORGANIC ACIDEMIAS:Therapy
Acute• Remove toxins: dialysis, hemofiltration and
exchange transfusion• Interrupt catabolic state• Stop protein intake• Give carnitine (100-300 mg/kg)
Long term• Protein restricted diet (special formulas if
available)• Carnitine• Vitamins (Vit. B12, Vit. B1, Vit. B2, biotin)

Features of some organic acidemias
Izovaleric acidemia Ketoacidosis, dehydration, neutropenia, thromboscytopenia, hyperammonemia, sweety feet odor
Propionic acidemia Motor-mental retardation, ketoacidosis, dehydration, neutropenia, thromboscytopenia, hyperammonemia, hipoglycemia
Methylmalonic acidemia Motor-mental retardation, ketoacidosis, neutropenia, thromboscytopenia, hyperammonemia, hypoglycemia, response to vit B12 (+)

Biotinidase deficiencyBiotin (complex)
Biotin (free)
piruvate carboxylase propionyl
CoA carboxylase
beta-methylcrotonyl CoA carboxylase
asetyl CoA carboxylase
Biotinidase

Biotinidase deficiencyIncidenseWorld. 1:60,000 Turkey: 1:10,000
Clinical and laboratory findings• Severe metabolic acidosis • Alopecia • Seborrheic skin eruptions• Refractory convulsions
Therapy 5-10 mg/day biotin (life long).

Urea cycle defects

Carbaglu (+)

Urea cycle defects
Incidence: 1:10,000 (cumulative)
Genetics
• Ornitine transcarbabamylase deficiency (most common urea cycle defect, X-linked)
• Argininosuccinate synthase deficiency (citrullinemia, (the second most common urea cycle defect, OR)
• Carbamylphosphate synthase I deficiency (OR)
• Argininosuccinate lyase deficiency (argininosuccinic aciduria, OR)
• Arginase deficiency (argininemia, OR)

Urea cycle defects: Clinical findings
Main symptom (acute/or chronic encephalopathy) is related to high protein intake, increased catabolism, infections or stress
Neonates: * Poor feeding * Temperature lability* Lethargy * Hyperventilation (respiratory alkalosis)* Loss of reflexes * Intracranial hemorrhages * Seizures * Progressive encephalopathy
Infants and children* Failure to thrive * Episodic encephalopathy* Feeding problems * Ataxia* Nausea, vomiting * Convulsions
Adolescents and adults* Chronic neurologic symptoms * Episodic encephalopathy* Chronic psychiatric symptoms * Behavioral problems

Urea cycle defectsLaboratory findings
• Hyperammonemia (generally >400 µmol/L in urea cycle defects)
• Amino acids in serum• Organic acids in urine
Differential diagnosis• Organic acidurias: • Liver diseases: neonatal hepatitis, galactosemia,
tyrosinemia, respiratory chain defects• Transient hyperammonemia of newborn due to
patent ductus venosus.

CPS= Karbamoil fosfat sentaz OTC= Ornitin transkarbomoilaz ASA=Arjininosüksinik asit AS=Arjininosüksinat sentazAL=Arjininosüksinat liaz(sitrüllinemi)

Urea cycle defects: Acute therapy
• Stop protein intake
• Interrupt catabolic state by high calorie infusion (carbohydrate + lipid)
• Remove ammonia when >400 µmol/L by hemodiafiltration, hemofiltration, or hemodialysis, (periton dialysis is not effective)
• Give arginine 350 mg/kg in order to support urea cycle.
• Give sodium benzoate: 350mg/kg/day
• Give sodium phenylbutyrate 250mg/kg/day
• Aim for an ammonia concentration < 200µmol/L

Urea cycle defects
Chronic therapy• Restriction of protein intake (1.0-1.5
g/kg/day) +arginine +• sodium benzoate + sodium –phenylbutyrate
Prognosis• Poor if there is prolonged coma (>3 days),
and symptoms and signs of increased intracranial pressure

Defects of Fatty acid oxidation

Fatty acid (plasma)
Asetil CoA
Fatty acid (mitochondria)
Carnitine Carnitine enzymes
Beta-oxidation(acyl CoA
dehydrogenases)
131 ATP
Keton bodies
HMG CoA- liaseHMG CoA- synthase
3-ketothiolase (tioforase)
Krebs cycle
Fatty acid oxidation
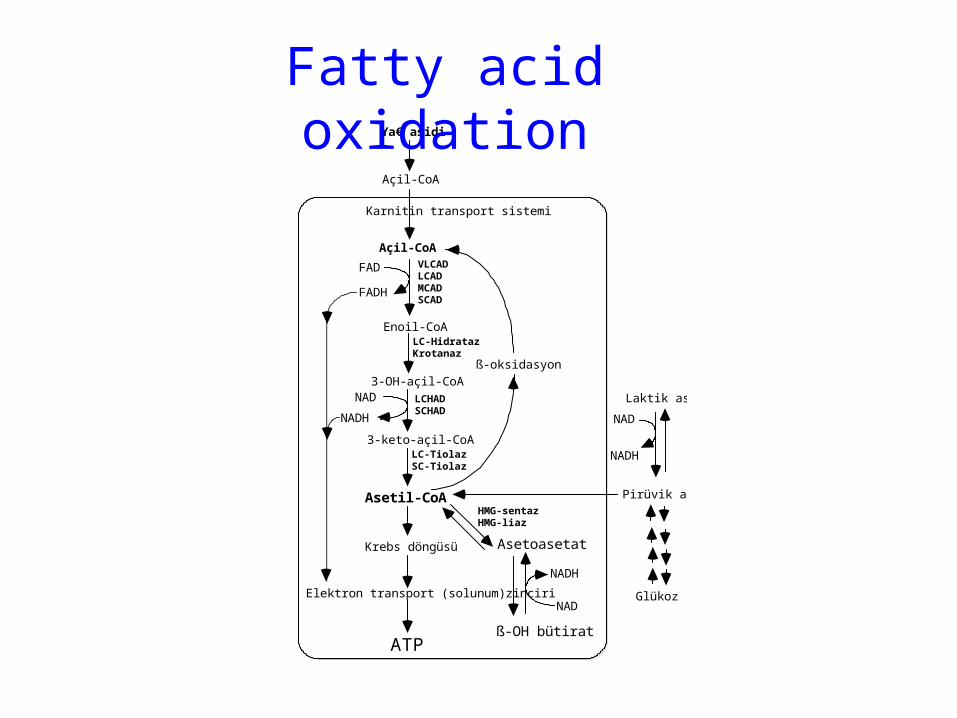
Ya€ asidi
Açil-CoA
Açil-CoA
ß-oksidasyon
Asetil-CoA Pirüvik asit
Laktik asit
Krebs döngüsü
Elektron transport (solunum)zinciri
ATP
Karnitin transport sistemi
Enoil-CoA
VLCAD LCAD MCAD SCAD
3-OH-açil-CoA
LC-Hidrataz Krotanaz
LCHAD SCHAD
3-keto-açil-CoALC-Tiolaz SC-Tiolaz
Asetoasetat
ß-OH bütirat
NAD
NADH
NAD
NADH
NAD
NADH
FAD
FADH
Glükoz
HMG-sentaz HMG-liaz
Fatty acid oxidation

• Disorders of fatty acid oxidation
• During prolonged fasting mitochonrial oxidation of fatty acids provides up to 80% of the total energy requirement.

Fatty acid oxidation: Etiology
Carnitine transporter deficiency
Defects of carnitine cycle• Carnitine palmitoyltransferase I (CPTI) deficiency• Carnitine translocase deficiency• Carnitine palmitoyltransferase II (CPTII) deficiency
ß-oxidation defects• Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency• Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency• Short-chain acyl-CoA dehydrogenase (SCAD) deficiency• Long-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency• Medium-chain hydroxyacyl-CoA dehydrogenase (LCHAD)
deficiency• Short-chain hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency

Fatty acid oxidation: Pathogenesis
• Insufficient energy production during fasting
• Deficiency of mitochondrial free CoA due to accumulation of toxic intermediary products

Clinical findings (Reyelike syndrome)
• Life-threatening hypoketotic hypoglycemic coma during catabolic states (prolonged fasting, infections, operations)
• Liver failure
• Skeletal myopathy, cardiomyopathy

Fatty acid oxidation: Laboratory findings
• Ketones: low, ammonia: high, glucose: low to normal, liver enzymes: high
• Total carnitine: low (high in CPTI deficiency)
• Acyl carnitine/total carnitine: Low
• Dicarboxilic acids in urine (GS-MS)
• Acylcarnitine profile (Diagnostic)
• Enzyme studies (Fibroblasts, lymphocytes)

Fatty acid oxidation: therapy
Acute therapy • High dose glucose (7-10 mg/kg/min),
no lipids (!)• Carnitine (100mg/kg): not in carnitine
cylce defects, and in LCHAD deficiency
Chronic therapy• Avoid prolonged fasting, careful
monitoring during catabolic states


















