aims
54
aims Structure prediction tries to build models of 3D structures of proteins that could be useful for understanding structure-function relationships.
-
Upload
eagan-lancaster -
Category
Documents
-
view
25 -
download
0
description
aims. Structure prediction tries to build models of 3D structures of proteins that could be useful for understanding structure-function relationships. Possible scenarios. 1. No homology 1D predictions. Sequence motifs. Limited functional prediction. Ab-initio prediction - PowerPoint PPT Presentation
Transcript of aims

aims
Structure prediction tries to build models of 3D structures of proteins that could be useful for understanding structure-function relationships.

Possible scenarios
1. No homology1D predictions. Sequence motifs. Limited functional prediction. Ab-
initio prediction
2. Homology exist but cannot be recognized easily (psi-blast, threading)
Low resolution fold predictions are possible. No functional information.
3. Homology can be recognized using sequence comparison tools or protein family databases (blast, clustal, pfam,...).
Structural and functional predictions are feasible

1D prediction
Prediction is based on averaging aminoacid properties
AGGCFHIKLAAGIHLLVILVVKLGFSTRDEEASS
Average over a window


1D prediction. Properties
Secondary structure propensitites Hydrophobicity Accesibility ...
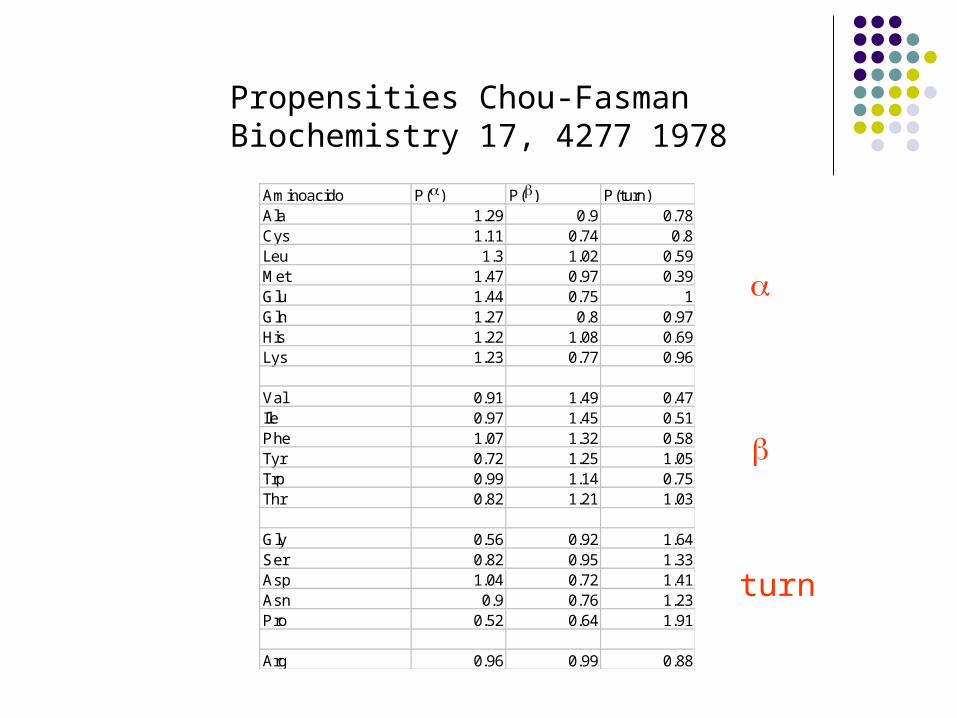
Aminoacido P() P() P(turn)Ala 1.29 0.9 0.78Cys 1.11 0.74 0.8Leu 1.3 1.02 0.59Met 1.47 0.97 0.39Glu 1.44 0.75 1Gln 1.27 0.8 0.97His 1.22 1.08 0.69Lys 1.23 0.77 0.96
Val 0.91 1.49 0.47Ile 0.97 1.45 0.51Phe 1.07 1.32 0.58Tyr 0.72 1.25 1.05Trp 0.99 1.14 0.75Thr 0.82 1.21 1.03
Gly 0.56 0.92 1.64Ser 0.82 0.95 1.33Asp 1.04 0.72 1.41Asn 0.9 0.76 1.23Pro 0.52 0.64 1.91
Arg 0.96 0.99 0.88
Propensities Chou-FasmanBiochemistry 17, 4277 1978
turn

Some programs (www.expasy.org)
BCM PSSP - Baylor College of Medicine Prof - Cascaded Multiple Classifiers for Secondary Structure Prediction GOR I (Garnier et al, 1978) [At PBIL or at SBDS] GOR II (Gibrat et al, 1987) GOR IV (Garnier et al, 1996) HNN - Hierarchical Neural Network method (Guermeur, 1997) Jpred - A consensus method for protein secondary structure prediction at
University of Dundee nnPredict - University of California at San Francisco (UCSF) PredictProtein - PHDsec, PHDacc, PHDhtm, PHDtopology, PHDthreader,
MaxHom, EvalSec from Columbia University PSA - BioMolecular Engineering Research Center (BMERC) / Boston PSIpred - Various protein structure prediction methods at Brunel University SOPM (Geourjon and Deléage, 1994) SOPMA (Geourjon and Deléage, 1995) AGADIR - An algorithm to predict the helical content of peptides

1D Prediction
Original methods: 1 sequence and uniform parameters (25-30%)
Original improvements: Parameters specific from protein classes
Present methods use sequence profiles obtained from multiple alignments and neural networks to extract parameters (70-75%, 98% for transmembrane helix)


PredictProtein (PHD)
1. Building of a multiple alignment using Swissprot, prosite, and domain databases
2. 1D prediction from the generated profile using neural networks
3. Fold recognition
4. Confidence evaluation
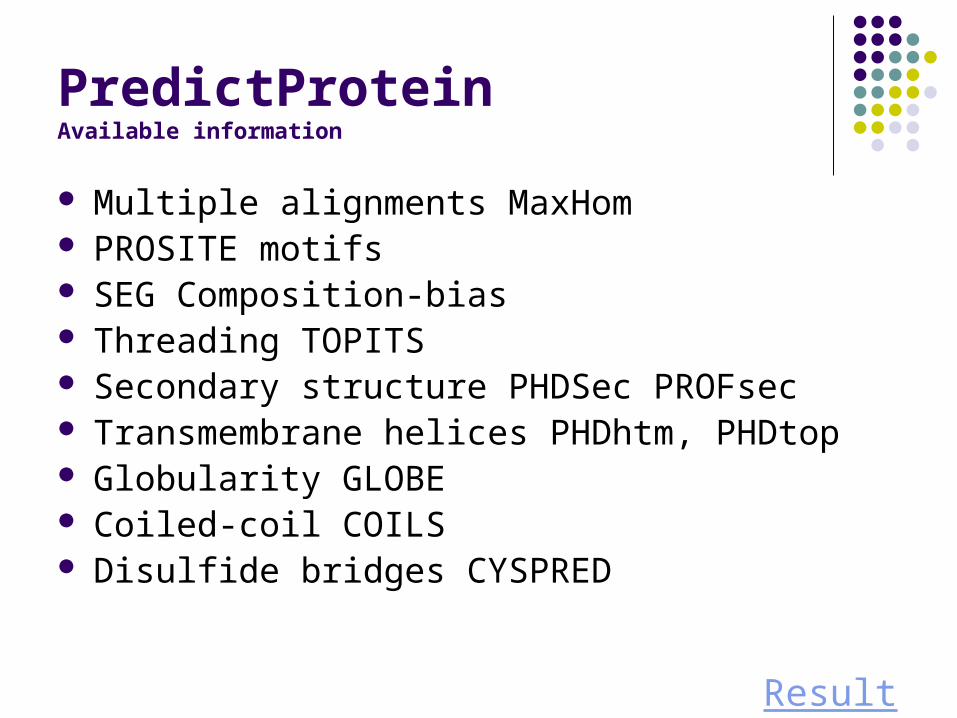
PredictProteinAvailable information
Multiple alignments MaxHom PROSITE motifs SEG Composition-bias Threading TOPITS Secondary structure PHDSec PROFsec Transmembrane helices PHDhtm, PHDtop Globularity GLOBE Coiled-coil COILS Disulfide bridges CYSPRED
Result

PredictProteinAvailable information
Signal peptides SignalP O-glycosilation NetOglyc Chloroplast import signal CloroP Consensus secondary struc. JPRED Transmembrane TMHMM, TOPPRED SwissModel

Methods for remote homology
Homology can be recognized using PSI-Blast
Fold prediction is possible using threading methods
Acurate 3D prediction is not possible: No structure-function relationship can be inferred from models

Threading
Unknown sequence is “folded” in a number of known structures
Scoring functions evaluate the fitting between sequence and structure according to statistical functions and sequence comparison

ATTWV....PRKSCTATTWV....PRKSCT
..........
10.510.5 5.2>> ..........
SELECTED HITSELECTED HIT

ATTWV....PRKSCTATTWV....PRKSCT SequenceSequenceHHHHH....CCBBBBHHHHH....CCBBBB Pred. Sec. Struc.Pred. Sec. Struc.eeebb....eeebebeeebb....eeebeb Pred. accesibilityPred. accesibility
..........
SequenceSequence GGTV....ATTW ........... ATTVL....FFRKGGTV....ATTW ........... ATTVL....FFRKObs SS Obs SS BBBB....CCHH ........... HHHB.....CBCB BBBB....CCHH ........... HHHB.....CBCB Obs Acc. Obs Acc. EEBE.....BBEB ........... BBEBB....EBBEEEBE.....BBEB ........... BBEBB....EBBE

Technical aspectsTechnical aspects
Alignment:Alignment: Dynamic programming (Needleman & Dynamic programming (Needleman & Wunsch, 1970)Wunsch, 1970)
Scoring FunctionScoring Function::
wwseqseq.P.Pseqseq + w + wstrstr . (P . (PSSSS + P + PACAC))
PPseqseq: Dayhoff matrix, P: Dayhoff matrix, PSSSS y P y PACAC: probability model on : probability model on
pred. SS and ACpred. SS and AC

Threading accurancyThreading accurancy
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
% ACIERTOS
5 10 15 20 25
% IDENTIDAD SECUENCIAS



Comparative modelling
Good for homology >30%
Accurancy is very high for homology > 60%

Remainder
The model must be USEFUL Only the “interesting” regions of the protein need
to be modelled

Expected accurancy
Strongly dependent on the quality of the sequence alignment
Strongly dependent on the identity with “template” structures. Very good structures if identity > 60-70%.
Quality of the model is better in the backbone than side chains
Quality of the model is better in conserved regions


Steps
1. Choose templates: Proteins with experimental 3D structure with significant homology (BLAST, PFAM, PDB)
2. Building multiple alignment of templates. Alignment quality is critical for accurancy.
Always use structure-based alignment. Reduce redundancies

Steps
1. Alignment of template structures
2. Alignment of unknown sequence against template alignment
• Structural alignment may not concide with evolution-based alignment.
• Gaps must be chosen to minimize structure distortion


Steps
1. Alignment of template structures
2. Alignment of unknown sequence against template alignment
3. Build structure of conserved regions (SCR)• Coordinates come from either a single structure or
averages.• Side chains are adapted to the original or placed in
standard conformations
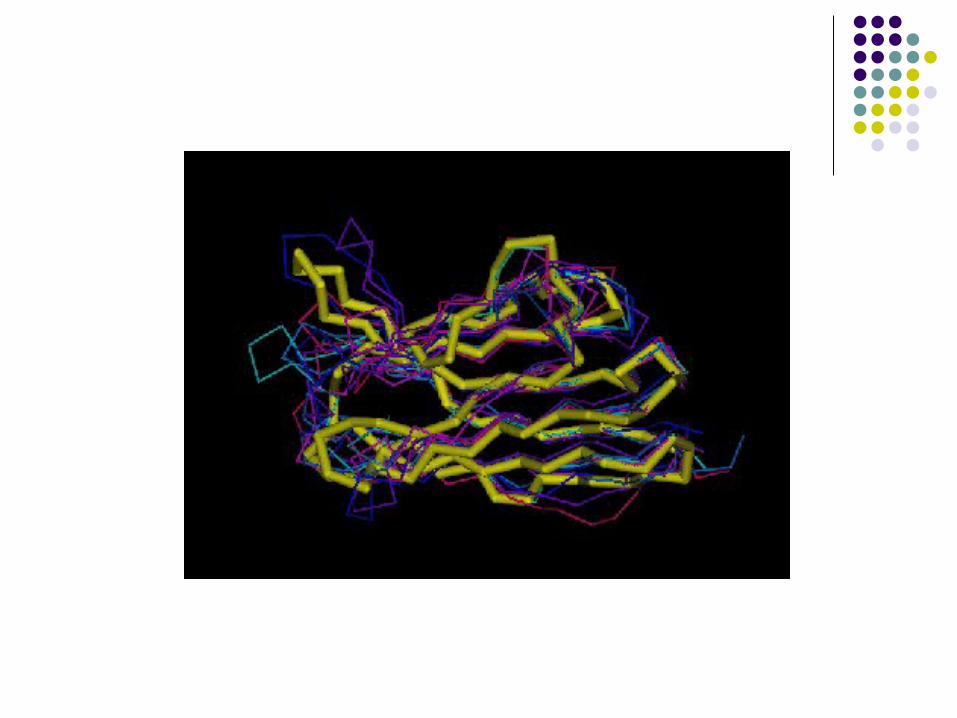

Etapas
1. Alignment of template structures
2. Alignment of unknown sequence against template alignment
3. Build structure of conserved regions (SCR)
4. Build of unconserved regions (“loops” usually)
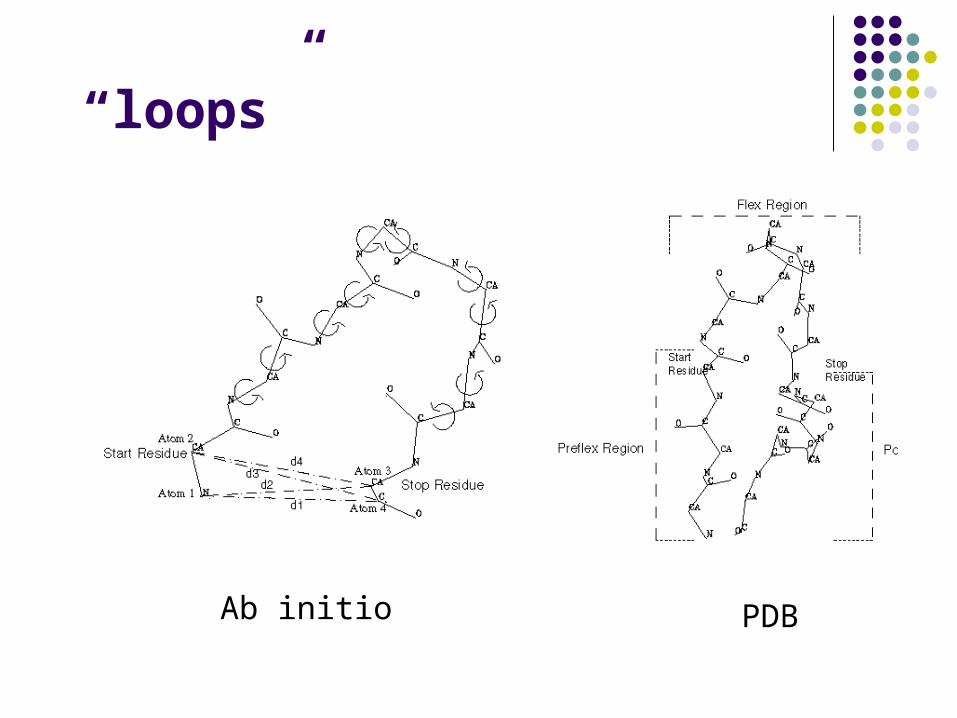
Ab initio PDB
“loops”

“loops”Chosen manually or energy-based

Optimization
1. Optimize side chain conformation1. Energy minimization restricted to standard conformers
and VdW energy
2. Optimize everything• Global energy minimization with restrains• Molecular dynamics


Quality test
No energy differences between a correct or wrong model
The structure must by “chemically correct” to use it in quantitative predictions

Analysis software
PROCHECK WHATCHECK Suite Biotech PROSA

Sources of information
300 best structures in PDB
Molecular geometry from CSD database
Theoretical data (Ramachandran, etc.)

Procheck
Covalent geometry Planarity Dihedral angels Quirality Non-bonded interactions Satisfied/unsatisfies Hydrogen-bonds Disulfide bonds



Whatcheck

Prediction software
SwissModel (automatic) http://www.expasy.org/swissmod/
SwissModel Repository http://swissmodel.expasy.org/repository/
3D-JIGSAW (M.Stenberg) http://www.bmm.icnet.uk/servers/3djigsaw/
Modeller (A.Sali) http://salilab.org/modeller/modeller.html
MODBASE (A. Sali) http://alto.compbio.ucsf.edu/modbase-cgi/index.cgi



Resultspdbv

Final test
The model must justify experimental data (i.e. differences between unknown sequence and templates) and be useful to understand function.