
Affinity Chromatography (AC) - Hebrew University of...
93
1 Affinity Chromatography (AC)
Transcript of Affinity Chromatography (AC) - Hebrew University of...

1
Affinity Chromatography
(AC)

2
Affinity Chromatography (AC)
• Principles of AC
• Main stages in Chromatography
• How to prepare Affinity gel - Ligand Immobilization - Spacer arms – Coupling methods – Coupling tips
• Types of AC
• Elution Conditions
• Affinity Tags and Fusion Proteins: Chelating, Strep-tag, GST, MBP, etc
• Cleavage sites – Proteases
• Parameters for development optimization (Troubleshooting): binding, washing, elution
• Examples
• Binding equilibrium, competitive elution, kinetics
• Future Considerations

3
What is affinity chromatography?
Affinity chromatography is a technique of liquid chromatography which
separates molecules through biospecific interactions.
The molecule to be purified is specifically and reversibly adsorbed to a
specific ligand
The ligand is immobilized to an insoluble support (“matrix”): resin, “chip”,
Elisa plate, membrane, western, etc
Introduction of a “spacer arm” between the ligand and the matrix to
improve binding
Elution of the bound target molecule: a) non specific or b) specific elution
method

4
What is it used for?
Monoclonal and polyclonal antibodies
Fusion proteins
Enzymes
DNA-binding proteins . . . . . .
ANY protein where we have a binding partner!!

5
Sample: 600 ml mouse monoclonal IgG 2a Gel: rProtein A Sepharose Fast Flow Elution: 20 mM Sodium Citrate, pH 4.0 Result: 83 mg Mab, recovery 95%
AU
2.0
1.5
1.0
0.5
0.0
0 200 400 600 Volume (ml)
Starting material
Eluate
94
43
30
SDS-PAGE reduced
Purifying monoclonal antibodies

6
Designing and preparing an affinity gel
Choosing the matrix
Designing the ligand - Spacer arms
Coupling methods

7
Ligand Immobilization
Matrix
+ Activating
agent +
Ligand
Activated matrix
Immobilised ligand

8
Designing the ligand
Essential ligand properties: interacts selectively and
reversibly with the target
Carries groups which can couple it to the matrix without
losing its binding activity
Available in a pure form

9
Steric considerations & spacer arms
Small ligand (<1,000)
Risk of steric interference with
binding between matrix and
target molecule
Often need spacer arm
but watch out for
adsorption to the spacer! Spacer arm

10
Design of spacer arms
Alkyl chain Real risk of unspecific interactions between spacer and target molecule
Hydrophilic chain Risk of unspecific interactions greatly reduced No coupling reaction will use 100% of the available binding sites. Using a gel with pre-attached spacer arms will leave some uncoupled spacers, increasing the possibility of non-specific interactions. The unused spacer arms should not be too much of a problem if they are hydrophilic. If available, this problem may be eliminated by using a ligand with a pre-attached spacer arm.
OH
O
O
H
H

11
Choosing a coupling group
• Ligands are coupled to the matrix via reactive groups
Amino -NH2
Hydroxyl -OH
Aldehyde -CHO
Thiol -SH
Carboxyl -COOH
• The group coupled must not be too near the site for binding the target

12
Useful Coupling Methods
Coupling chemistry Kinds of group coupled Ligand
N-hydroxysuccinimide -NH2 Protein ,Peptide. Sugar.Polynucleotide.Cofactor
CNBr -NH2 Protein , Sugar.Polynucleotide.Cofactor
Carbodiimide -NH2 Small ligands. Non polar ligand in organic
via hydrophilic spacer arm solvents. Derivatized material
Epoxide -SH-NH2-OH Sugars. Small ligands
via hydrophilic spacer arm
Thiol exchange -SH Protein ,Peptide. Polynucleotide
Mild Conditions Low MW thiol containig ligand

13
Useful Coupling Methods
Sepharose Cyanogen bromide
Cyanate ester formed
C N
Br+O
Gel HO
Gel C N + H Br
Cyanogen bromide activation
+ Lig NH2
OGel C N Gel
OC
NH
NH
Lig
CNBr-activated Sepharose
Protein ligand N-substituted isourea
Cyanogen bromide coupling
Reaction at pH 8. Isourea may be cleaved by nucleophilic attack at high pH.
Multiple bonds formed with protein ligands. No spacer arm present.
WARNING!: Cyanogen bromide is dangerous. Consult safety data before start working

14
A general protocol for ligand coupling
Prepare the ligand solution in coupling buffer
Prepare the activated matrix
Mix the ligand solution and the activated gel in coupling buffer until
coupling is complete
Check total ligand before and after coupling: ligand/ml resin
Block any remaining active groups
Wash the coupled gel alternately at high and low pH to remove adsorbed
ligand
Transfer to storage buffer

15
Tips: Preparing the activated matrix - couple buffer
If the activated matrix is freeze-dried, allow it to swell before washing
Always use the recommended swelling and washing solutions!
The swelling, washing and equilibration are best performed on a sintered
glass filter. Facilitates quickly removal of excess liquid from the media
Couple buffer: make sure there are no components, e.g. amines, that can
couple in place of the ligand, e.g. Tris buffer has a primary amine
To suppress ionic adsorption, add NaCl, 0.5 M.
pH is important. Always use the recommended pH.

16
Tips: Coupling reaction, washing and storage
Ligands which are poorly soluble in water, may usually be added in an organic solvent.
The ligand should be available at a high level of purity. Any contaminants present in the
ligand sample will likely be coupled to the matrix.
Mix the activated gel and the ligand solution by swirling or end-over-end mixing - Avoid
magnetic stirrers as these can crush the gel and produce fines!!
Take samples of ligand before and after reaction: to calculate ligand per ml resin
Time and temperature are in the standard protocol. Most coupling reactions are
performed at room temperature
Block any remaining active groups with a small neutral ligand (Tris buffer for amino
reactions)
Wash the coupled gel at alternating high and low pH to remove adsorbed substances.
Keep in storage buffer (Na Azide , Thimerosol 0.02% or ethanol if possible)

17
7
20
2408
18647
A. Tobin et al. (1996) J. Biol. Chem. 271, 3907-3916
• 10 mg homogenous protein
G Protein Receptor Kinase
Technique
Ppt
HIC
AIEX
CIEX
AC
Pig brain homogenate
RESOURCE Q
RESOURCE S
HiTrap Heparin
Purification factor
Comment
Ammonium sulfate precipitation
Butyl Sepharose Fast Flow

18
Placental extract in 1.5% Triton X-100
Membrane Protein
Technique
Blue Sepharose
DEAE Sephacel
SP Sepharose
Muc2 Sepharose
Mono S
AC
AIEX
CIEX
AC
CIEX
Purification factor
Comment
• Main purification step
• Final polishing and purity check, 20 mg obtained
T. White et al. (1995) J. Biol. Chem. 270, 24156-24165
3
4
6
242
1442
• Concentration and capture

19
AC
DNA-binding Protein
Technique
DNA-1 Sepharose
DNA-2 Sepharose
Mono S
CIEX
AC
AC
CIEX
Purification factor
Comment
• General AC step for DNA-binding proteins
• Removal step, non-specific DNA binding activity removed
• Main purification step
• Final polishing, 20 mg obtained
J. Berthelsen et al. (1996) J. Biol. Chem. 271, 3822-3830
5
8
9
2447
4943
HeLa cell nuclear extract
SP Sepharose High Performance
Heparin Sepharose Fast Flow

20
Advantages and Disadvantages of affinity chromatography
Easy to achieve otherwise difficult
separations
Often high purity in one step
(but not always)
Fast separations (depends of the
kinetics of binding and elution)
Can be use to remove specific
contaminants
Economics (scale-up)
Buffer / elution limitations
Sometimes needs harsh or
denaturizing elution conditions
Do not resolve all the problems

21
The main stages in affinity chromatography
Equilibration Of gel and sample to binding conditions
Sample application
Wash contaminants
Elution of the target
Re-equilibration Of the gel to binding conditions Or keep in storage buffer (20% Ethanol or 0.02%NaAzide in Buffer)

22
Columns and equipment
Column volume: according to amount of target and gel capacity
Avoid using excess of resin: to avoid low affinity binding of
impurities
Column length: not usually critical, short and wide
Equipment: no special demands
Equilibrate column before applying sample and follow standard
protocol
Tips

23
Sample preparation
Adjust pH, buffer salts and additives to promote binding
Filter or centrifuge to remove particles
Make sure that components known to interfere with binding
are absent
Consider alternative capture steps before affinity purification
(cost of resin, crude impurities or interfering substances,
concentrate sup, etc)
Tips

24
Sample application
Binding buffer: usually neutral pH, and high salt concentration when is
possible (0.3-0.5M NaCl) to avoid hydrophobic interactions of non specific
proteins to the resin. Use additives only if necessary.
For batch binding, incubation of around 1.5 hours at 4°C is enough. For
extremely slow binding kinetics, incubate overnight at 4°C.
Loading directly on column
- Strong affinity and fast binding: High flow rate
- Weak affinity and/or slow binding: Low flow rate
Tips

25
Elution and re equilibration
Follow standard protocol
Optimize alternative wash strategies to increase final purity
If eluting at extreme pH: collect the target protein in a small
amount of concentrated buffer at a neutralizing pH
Re-equilibrate column immediately with binding or storage
buffer
Regeneration: use buffers that do not harm the ligand
Tips

26
Column Regeneration
Wash with basic and acidic buffers.
– 10 col. vol. 0.1 M Tris.HCl, 0.5 M NaCl, pH 8.5
– 10 col. vol. 0.1 M NaAc, 0.5 M NaCl, pH 4.5
– Use chaotropic agents or detergents if needed
Re-equilibrate with 10 column volumes starting buffer. Use more if needed
Ni columns can be washed with 0.5N NaOH, neutralized, destriped with
100mM neutral EDTA, wash, charge with 100mM Ni SO4, wash and store
with 20% Ethanol
Tips

27
Column Storage
To maximize the useful lifetime of an affinity resin, choose a
storage buffer that does not harm the ligand
Typical buffers are 20% ethanol solution or binding buffer
containing a bacteriostatic agent (e.g. 0.02% Thimerosal or
0.02% sodium azide)
Many of the commercial resins can be used hundreds of times
Tips

28
Affinity Chromatography (AC)
• Principles of AC
• Main stages in Chromatography
• How to prepare Affinity gel - Ligand Immobilization - Spacer arms – Coupling methods – Coupling tips
• Types of AC
• Elution Conditions
• Affinity Tags and Fusion Proteins: Chelating, Strep-tag, GST, MBP, etc
• Cleavage sites – Proteases
• Parameters for development optimization (Troubleshooting): binding, washing, elution
• Examples
• Binding equilibrium, competitive elution, kinetics
• Future Considerations

29
Type of Affinities
Mono-specific ligands
• Specific for a single substance
Antigen antibody
Hormone receptor
• Usually home-made gels
• Elution scheme must be worked
out for each case:
• Often general elution
• Little help from the literature
Group-specific ligands
• Specific for a group of structurally
or functionally similar substances:
Lectins glycoproteins
Protein G IgG antibodies
Dye-stuffs enzymes
• Often ready-made gels
• Known elution schemes
• Standard tested elution protocols
• Often competitive elution

30
Group-specific ligands - 1
Ligand Specificity
Protein A Fc region of IgG
Protein G Fc region of IgG
Concanavalin A Glucopyranosyl & Mannopyranosyl groups
Peanut Lectin Terminal galactose group
Cibacron Blue Broad range of enzymes, serum albumin
Procion Red NADP+ dependent enzymes
Lysine Plasminogen, ribosomal RNA

31
Group-specific ligands - 2
Ligand Specificity
Arginine Serine proteases
Benzamidine Serine proteases
Calmodulin Proteins regulated by calmodulin
Heparin Coagulation factors, lipoproteins, lipases,
hormones, steroid receptors, protein synthesis
factors, Nucleic acid-binding enzymes
Metal chelate resins Proteins and peptides which contain
(Co, Ni, Zn, Cu, Fe) 6-12 Histidines

32
Designing the elution scheme
General elution conditions
– Low pH, salt, GuHCl, Urea etc.
– Often harsh conditions
Specific eluents
– Competing free ligand
– Competing binding substances
– Sometimes expensive compounds (like peptides)

33
Reconstituting
buffer
+
Changing buffer conditions
Usually decrease pH or increase ionic strength
decrease polarity adding up to 10 % dioxane or up to 50 % ethylene glycol
Denaturing buffer
Usually extremes of pH or chaotropic agents
General elution conditions
+
There are no guaranteed general elution methods in affinity chromatography
Low pH, e.g. glycine HCl, pH 2.8, is the closest approach

34
General elution conditions • pH • 100 mM glycine•HCl, pH 2.5-3.0 • 100 mM citric acid, pH 3.0 • 50-100 mM triethylamine or triethanolamine, pH 11.5 • 150 mM ammonium hydroxide, pH 10.5
• Ionic strength and/or chaotropic effects • 3.5-4.0 M magnesium chloride, pH 7.0 in 10 mM Tris • 5 M lithium chloride in 10 mM phosphate buffer, pH 7.2 • 3.0 M Potassium chloride • 2.5 M sodium iodide, pH 7.5 • 0.2-3.0 sodium thiocyanate
• Denaturing • 2-6 M guanidine•HCl • 2-8 M urea • 1% deoxycholate • 1 % SDS
• Organic • 10% dioxane • 50% ethylene glycol, pH 8-11.5 (also chaotropic)
• Competitor • >0.1 M counter ligand or analog
Elution conditions are intended to break
the ionic, hydrophobic and hydrogen bonds
that hold the ligand and the target together
Successful eluting conditions will be
dependent upon the specific ligand-target
interaction that is occurring.
Ideally, an elution condition effectively
releases the target without causing
permanent damage, but all eluting
conditions result in some loss of functionality
Empirical evidence is needed to determine
which elution condition is the best.

35
Elution at Low pH IgG antibodies on rProtein A Sepharose
Column: HiTrap rProtein A
Binding: 20 mM sodium phosphate, pH 7.0
Elution: 0.1 M glycine HCl, pH 3
Note: Collect IgG into a small volume of strong buffer, pH 7, to
preserve its antibody activity.

36
Elution at High salt concentration DNA-binding proteins on Heparin Sepharose
Column: HiTrap Heparin
Binding: 20 mM Tris-HCl, pH 8, 0.5 M NaCl
Elution: Binding buffer + 1 to 2 M NaCl
37
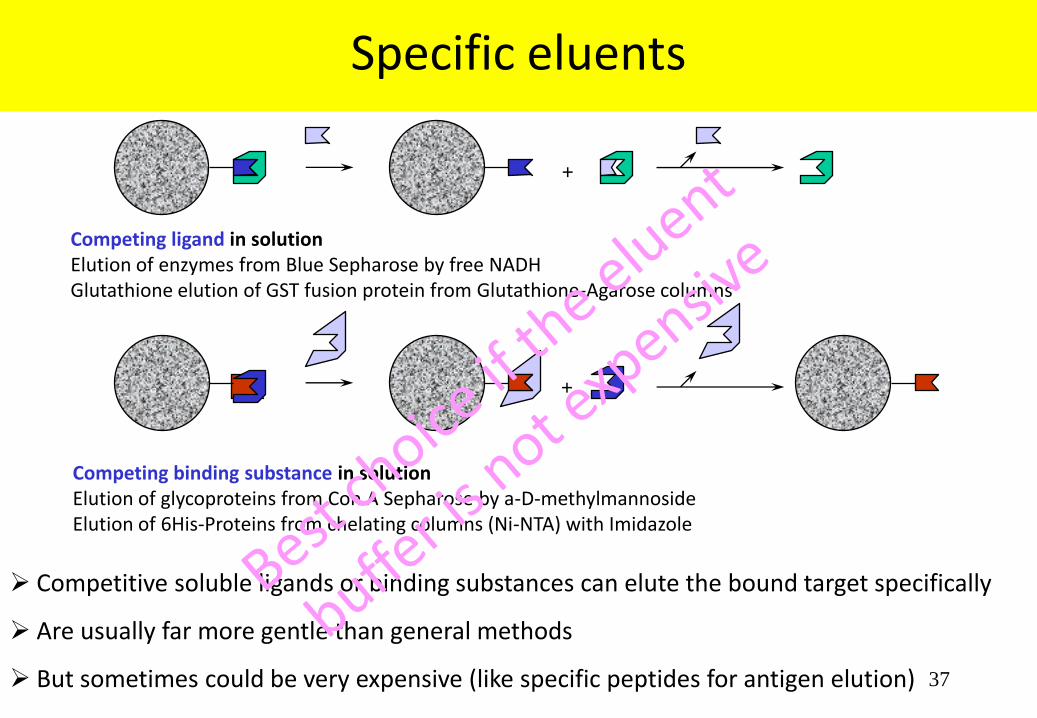
Competing binding substance in solution Elution of glycoproteins from Con A Sepharose by a-D-methylmannoside Elution of 6His-Proteins from chelating columns (Ni-NTA) with Imidazole
Specific eluents
+
Competing ligand in solution Elution of enzymes from Blue Sepharose by free NADH Glutathione elution of GST fusion protein from Glutathione-Agarose columns
+
Competitive soluble ligands or binding substances can elute the bound target specifically
Are usually far more gentle than general methods
But sometimes could be very expensive (like specific peptides for antigen elution)

38
Lectins
Lectin Specificity
Concanavalin A α-mannose, α-glucose
Glycine Max (soybean) N-Acetyl Galactosamine
Ulex Europaeus I α-L-fucose
Wheat germ lectin N-Acetyl Glucosamine & NeuNAc
Peanut lectin α-mannose
Ricinus Communis, RCA 120 ß-Galactose
Arachis hypogaea ß-Gal(1->3)GalNAc
Bandeira Simplicifolia, BS-I α-Gal & α Gal NAc
Maackia amurensis Sialic acid
Lymulus polyphemus NeuNAc
Lectins are proteins
which bind well-defined
sugar residues in
polysaccharides,
glycoproteins, etc
Elution with competing
free binding substance:
sugar

39
Dye-stuffs
Dye-stuff Sites recognised
Cibacron Blue 3G-A NADH-binding and similar
Procion Red NADPH-binding and similar
Many dye-stuffs mimic binding sites of natural biologically active molecules
Elution with High Salt
The structural analogies are not exact.
Related structures are also recognized.
Blue Sepharose also binds serum albumin.

40
Affinity Chromatography (AC)
• Principles of AC
• Main stages in Chromatography
• How to prepare Affinity gel - Ligand Immobilization - Spacer arms – Coupling methods – Coupling tips
• Types of AC
• Elution Conditions
• Affinity Tags and Fusion Proteins
• Chelating, Strep-tag, GST, MBP, etc
• Cleavage sites – Proteases
• Parameters for development optimization (Troubleshooting): binding, washing, elution
• Examples
• Binding equilibrium, competitive elution, kinetics
• Future Considerations

41
Affinity Tags - I
Small stretches of amino acids added to the N-terminal or C-terminal end
of a protein with a high affinity for a specific biological or chemical ligand
Allow purification and detection of the expressed protein
Enables different proteins to be purified using a common method (HTPS)
Most popular: His-tag, comprised of six histidine that provide specific
binding to metal chelate resins
Polyarginine or polyaspartic acid tags can be used to alter binding of a
protein on ion-exchange resins

42
Affinity Tags - II
The Strep-tag II (8 amino acids, WSHPQFEK), which binds with high
selectivity to Strep-Tactin.
The tag may be placed at either end of the protein or in a region with
appropriate surface exposure to allow binding or recognition.
To enable removal of the tag, a linker region is typically included between
the tag and the native protein sequence.
The linker contributes to increased accessibility of the affinity tag and is
often required for effective endoprotease cleavage.

43
Fusion proteins Solubility-enhancement tags (SETs)
Use to overcome some of the problems of bacterial expression: protein aggregation, poor
expression levels and difficult purification.
The most popular fusion systems employ maltose-binding protein (MBP), glutathion S-
tranferase (GST), thioredoxin (TRX), SUMO and NusA. These genes are well expressed and
the proteins are highly soluble and provide specific characteristics to aid purification.
Typically, the gene of interest is inserted “in frame” at the 3’ end of the carrier protein, in
place of the termination codon.
To enable removal of the fusion protein, a linker region is typically included between the
fusion protein and the native protein sequence.
Specific recognition of the linked regions with specific proteases: TEV, Prescission, SUMO
protease, Enterokinase

44
Advantages / disadvantages of fusion proteins
In several cases
facilitate protein purification
facilitate protein refolding /
increase solubility
improve protein yield (high
expression)
prevent proteolysis
But in other cases
Target is not soluble after cleavage
change in protein conformation
lower protein yields
alteration in biological activity
undesired flexibility in structural studies
Toxicity
Extra work if you need to cleave

45
IMAC: Immobilized metal affinity chromatography Chelating-Chromatography for poly-His fusion proteins
The most widely used.
It’s small in size. Less immunogenically active
It does not need to be removed for downstream applications
Availability of a large number of expression vectors
Tag may be placed at either the N or C terminus
A protease cleavage site allows the tag to be removed after purification
The interaction of the tag with the Ni2+column does not depend of the tag structure,
making it possible to purify otherwise insoluble proteins using denaturating conditions
(8 M urea or 6 M GuHCl )
Avoid use of chelating: EDTA, DTT, etc. (cell chelating molecules in low expression)

46
Immobilized metal ion affinity chromatography (IMAC) – for poly-His fusion proteins
The basis of the purification is the interaction of the imidazole moiety of the poly His with
the metal (Nickel in most cases).
The metal is immobilized to a support through complex formation with a chelate that is
covalently attached to the support
In some resins we can use
Co , Cu , Zi or Fe instead of Ni
and obtain different results
Ni-NTA, Co resins, etc

47
IMAC Binding Conditions:
20 mM phosphate or TrisHCl buffers
+ (0.5 M NaCl) or low imidazol
concentrations (10-20mM) to avoid non-
specific binding of contaminants to the resin.
Buffers may include 8 M urea or 6 M GuHCl
when purifying inclusion bodies solubilised
proteins.
Elution Conditions:
Usually Imidazol (Histidine analoge) or
low pH (~4.5)
Alternatives: EDTA, Histidine

48
Strep-tag /Strep-Tactin system According to Expression and purification of proteins using Strep-tag and/or 6xHistidine-tag. A comprehensive manual
from IBA GmbH
The Strep-tag II is a short peptide (8 mino acids, WSHPQFEK), which binds with high
selectivity to Strep-Tactin, an engineered streptavidin.
This technology allows one-step purification of almost any recombinant protein under
physiological conditions, thus preserving its bioactivity (elution with 2.5mM desthiobiotin).
The Strep-tag system can be used to purify functional Strep-tag II proteins from any
expression system including baculovirus, mammalian cells, yeast, and bacteria.
The Strep-tag/Strep-Tactin interaction is compatible with a variety of reagents (detergents,
reducing agents, etc.) making the system attractive for purifying metallo- and membrane
proteins, large proteins and protein complexes.

49
Strep-tag/
Strep-Tactin system
According to Expression and purification of
proteins using Strep-tag and/or 6xHistidine-tag.
A comprehensive manual
from IBA GmbH

50
Strep/6 x Histidine system (double-tag) IBA and QIAGEN
Useful for full length recombinant proteins purification at high purity under
standardized and non-denaturative conditions (Imidazol and desthiobiotin
elution)
Especially useful to eliminate “difficult to resolve” protease cleavage
fragments of target protein.
Recombinant proteins that carry 6xHistidine-tag at the N-terminus and
Strep tag II at the C-terminus (or vice versa).
Efficiently expressed in E. coli, yeast, insect, or mammalian cells.
Recommendation: use IMAC as first capture step. No buffer exchange is
required for the second purification step: Strep-Tactin resin

51
A panel of commonly used affinity tags selected for purification of recombinant fusion proteins and their
associated characteristics
Preparative Purification of Recombinant Proteins: Current Status and Future Trends Mayank Saraswat et al. Hindawi Publishing Corporation - BioMed Research International Volume 2013, Article ID 312709,
http://dx.doi.org/10.1155/2013/312709

52
Gluthathione S-transferase or GST-fusion proteins
• Column: Glutathione Sepharose 4B, pre-packed
• Binding: PBS (+ 1% Triton X-100)
• Elution: 5-10 mM Glutathione, 50 mM Tris.HCl, pH 8.0
r-Protein
Cleavage site
Specific ligand Matrix Affinity tag Target protein
GST: 26kDa cytoplasmic protein. Binds specifically to glutathione
Highly use in immunoprecipitation
Less use for increasing solubility
Commercial vectors containing either factor Xa, Prescision or a thrombin
recognition site to allow cleavage and removal of the GST.
Useful for dimerization

53
Maltose-binding protein pMAL ™
MBP: a 43kDa secreted protein from E. coli,
binds specifically to maltose
Purification: Maltose or Amylose agarose
columns and Dextrin Seph (most recommended)
Can be secreted to the periplasm
Or expressed without the signal peptide in the
cytoplasm.
Very effective for solubility enhancement
TEV protease cleavage site
Sometimes additional 6His in N-terminal
Use sometimes to aid crystallization of target
protein

54
The Mechanism of Solubility Enhancement by MBP Sreejith Raran-Kurussi and David S. Waugh PLOS ONE (2012) 7 (11): e49589 - doi:10.1371
Recently, MBP has also been used to maintain proteins that contain
disulfide-bonds in a soluble state in the E. coli cytoplasm so that
they could be acted upon by appropriate redox enzymes that were
co-expressed in the same cellular compartment
MBP serves as a passive participant in the folding process; passenger proteins either fold spontaneously or with the
assistance of chaperones.
Chaperones and/or chaperonins seem to come into play after a passenger protein has been rendered soluble by MBP.
MBP serves primarily as a ‘‘holdase’’, keeping the
incompletely folded passenger protein from forming
insoluble aggregates until either spontaneous or
chaperone-mediated folding can occur.
A third class of passenger proteins is unable to fold
and exists in an incompletely folded state and
typically precipitate after they are cleaved

11/19/2014 55
SUMO (small ubiquitin-like
modifier)
Increased expression
Increased solubility
Acidic protein ~10kDa
Both the tag and the protease have 6xHis tags
SUMO Protease leaves no unwanted residues on the N-terminus
SUMO Protease extremely efficiently (1:500)

56
Thioredoxin
Thioredoxin: 12 kDa intracellular E. coli protein. Very soluble, and highly over-expressed
Periplasmic or cytoplasmatic expression
Most popular to increase disulfide bonds
6His Tag can be add to the N terminal of TRX for metal-chelator purification
Since TRX is thermostable, and some heat-stable fusion proteins can be purified by thermal
denaturation of contaminants by thermoosmotic shock (osmotic shock coupled with heat-
treatment) According to Q.-R. Guo et al. / Protein Expression and Puriffication 49 (2006) 32–38

57
Fusion Tag Comparison Study LifeSensors (http://www.lifesensors.com/r_and_d/protein_expression.php3)
SUMO (Small Ubiquitin-like MOdifier) and NusA fusion tags dramatically outperform
glutathiones transferase (GST), maltose binding protein (MBP), thioredoxin (TRX), and
ubiquitin (Ub). The protein target tested in this study is GDF-8, a growth/differentiation factor.
UN: un-induced, IN: induced, S: soluble, IB: inclusion body.
UN IN S IB UN IN S IB UN IN S IB UN IN S IB UN IN S IB UN IN S IB UN IN S IB
GDF8 ALONE Ub-GDF8 SUMO-GDF8 MBP-GDF8 GST-GDF8 TRX-GDF8 NUS A-GDF8
58
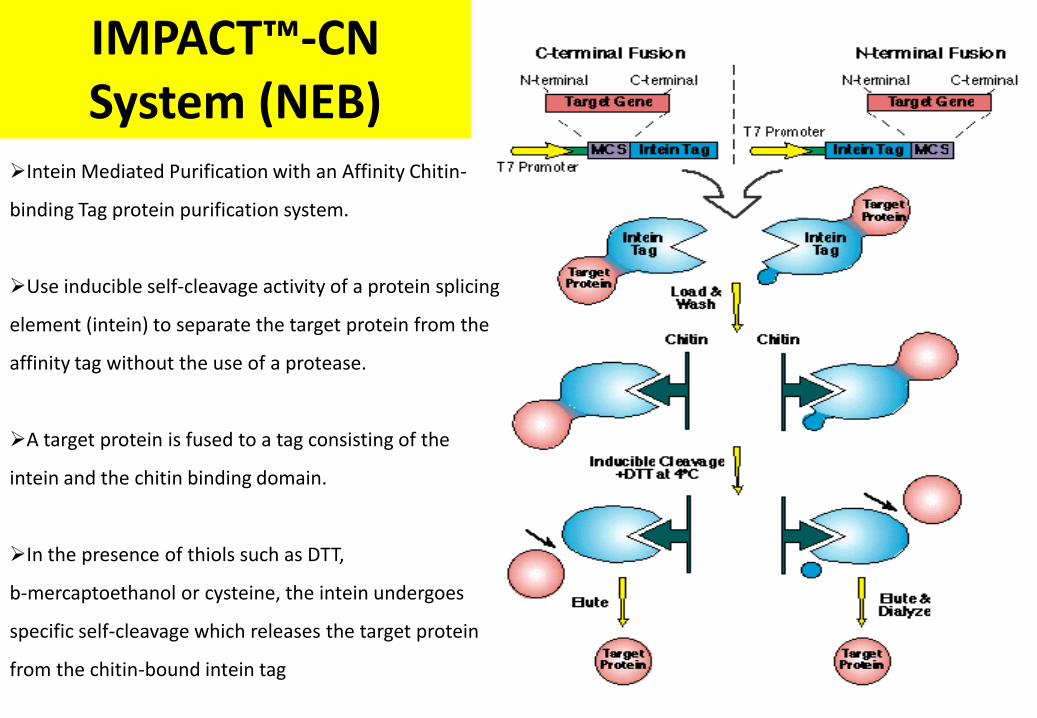
IMPACT™-CN System (NEB)
Intein Mediated Purification with an Affinity Chitin-
binding Tag protein purification system.
Use inducible self-cleavage activity of a protein splicing
element (intein) to separate the target protein from the
affinity tag without the use of a protease.
A target protein is fused to a tag consisting of the
intein and the chitin binding domain.
In the presence of thiols such as DTT,
b-mercaptoethanol or cysteine, the intein undergoes
specific self-cleavage which releases the target protein
from the chitin-bound intein tag

59
PinPoint™ Xa from Promega Production and purification of fusion proteins that are biotinylated
in vivo
In vivo Biotinylated Affinity Tags: biotinylation reaction in E. coli through
biotin ligase holo-enzyme.
Fusion purification tag with a single biotin specifically on one Lys residue
The system use a monomeric avidin (Soft Release Avidin Resin): allows
protein elution with a non-denaturing 5mM biotin buffer.
(Avidin-biotin interactions are very and requires denaturing conditions)
Tag cleavage with Factor Xa

60
Comparison of Affinity tag technologies According to J.J. Lichty et al. / Protein Expression and Puriffication 41 (2005) 98–105
mg10Tag Size(aa) Resin Eluting agent Source Capacity Cost Cost/
MBP 396 Amylose Maltose Biolabs 3mg/ml $105/10ml $12
HIS 6 Talon Imidazole Clontech 5–14mg/ml $220/25ml $18
Ni–NTA Imidazole Qiagen 5–10mg/ml $257/25ml $21
GST 218 GSH–Sepharose Glutathione Amersham 10 mg/ml $396/25ml $36
CBP 28 Calmodulin EGTA Stratagene 2mg/ml $227/10ml $114
Strep II 8 Strep-Tactin Desthiobiotin IBA 50–100 nmol/ml $1100/25ml $293
FLAG 8 Anti-FLAG M2 FLAG peptide Sigma 0.6mg/ml $1568/25ml $1045
HPC 12 Anti-Prot.C Ab EDTA Roche 2–10 nmol/ml $299/1ml $4983

61
Fusion protein cleavage methods
Chemicals cleavage is effective but often requires extreme conditions (low
pH or high temp.), and is often non-specific (cyanogen bromide,
hydroxylamine, etc).
Enzymatic digestion is the method of choice for soluble fusion protein
cleavage. Reaction is carried out under relative mild conditions.
Commonly used endo-proteases : thrombin, factor Xa, enterokinase
Best sellers: TEV, SUMO and rhinovirus 3C protease (Prescission)
Exoprotease: TAGZyme, carboxypeptidase A (C-terminal tag)
Intein cleavage by reduction agents such as DTT, or low pH

62
Purification of Fusion Proteins
Desorption
Proteolytic cleavage
Linker sequence
Gel
Ligand
r-Protein Affinity
tail + L
r-Protein Affinity
tail
Gel L
r-Protein Affinity tail
Proteolytic cleavage
r-Protein Affinity
tail +

63
Cleavage Sites • Thrombin Amersham-Biosciences, Novagen, SIGMA, Roche
Leu-Val-Pro-Arg▼Gly-Ser Protease Capture: Benzamidine-Agarose
Secondary cleavage sites. Biotinylated form available for removal with immobilized streptavidin.
• Factor Xa Amersham-Biosciences, NEB, Roche
Ile-Glu/Asp-Gly-Arg▼ Protease Capture: Benzamidine-Agarose
Will not cleave if followed by proline and arginine. Secondary cleavage sites following Gly-Arg sequences.
• Enterokinase NEB, Novagen, Roche
Asp-Asp-Asp-Asp-Lys▼ Protease Capture: Trypsin Inhibitor-Agarose
The site will not cleave if followed by a proline residue. Secondary cleavage sites at other basic residues, depending on conformation of protein substrate. Active from pH 4.5 to 9.5 and between 4°C and 45°C
• TEV protease Invitrogen – Life Technologies
Glu-Asn-Leu-Tyr-Phe-Gln▼Gly Protease Capture: Ni-NTA (6His recomb. TEV)
Seven-residue recognition site, making it a highly site-specific protease. Active over a wide range of temperatures. Protease available as a His-tag fusion protein, allowing for protease removal after recombinant protein cleavage.
• PreScission Amersham-Biosciences
Leu-Glu-Val-Leu-Phe-Gln▼Gly-Pro
Genetically engineered form of human rhinovirus 3C protease with a GST fusion tag, allowing for facile cleavage and purification of GST-tagged proteins along with protease removal after recombinant protein cleavage. Enables low-temperature cleavage of fusion proteins containing the eight residue recognition sequence.
• TAGZyme Quiagen: His-tag removal by Exoproteolytic Digestion
Protease Capture: Ni-NTA (6His recomb. enzyme)
The TAGZyme System is an efficient and specific solution for the complete removal of small N-terminal His tags and other amino acid tags by the use of exopeptidases. These recombinant enzymes contain a C-terminal His tag and can therefore be bound to Ni-NTA matrices.
• Intein Site (dithiothreitol cleavage) NEB
DTT elimination by dialysis
Uses self-cleavable affinity tags. Even after cleavage un-natural termini are present on the protein of interest.

64
Incubation of HLT-435aaCtermBP2 with TEV-protease
Shajar Rotem, Assaf Friedler
Protein µl 5 5 5 5 5 5 5 5 5 5
TEV-pr. µl 0 0.2 1 2.5 5 0 0.2 1 2.5 5
Temp ºC 4 4 4 4 4 22 22 22 22 22
HTL-435aaCtermBP2
435aaCtermBP2
TEV-protease
HTL

65
Disadvantage of Proteolytic Cleavage
Spurious, non-specific proteolytic cleavage of the fusion protein
Non-covalent forces maintain proteins connected after cleavage: separation problems
Some proteases need elevated temperatures (25-37°C) for efficient cleavage: can denature
or cause aggregation of the fusion protein
Incomplete cleavage, which reduces the yield and/or introduces heterogeneity to the
purified protein
Buffer and additive restriction (mainly detergents)
$ and time: The need for additional steps to separate the cleaved fusion protein from the
fusion tag, remove the protease, and exchange buffer or desalt
Increase purity (negative column or other chromatographic procedure)
Targets are mainly C-terminal of the solubility protein (exoproteases as carboxypeptidase A)

67
Self-cleaving elastin-like polypeptide tag Purification of recombinant protein without the need for affinity
chromatography or proteolytic tag removal
Simple bioseparations using self-cleaving elastin-like polypeptide tags. Banki M. et al. - Nature Methods 2, 659 - 662 (2005) Self-cleaving ELP tags consisting of repeating pentapeptides of VPGXG (X = any amino acid) fused to a controllable self-cleaving intein
ELPs can be designed to undergo a reversible transition from soluble to insoluble upon temperature upshift
The reversibility of the precipitation is distinct from simple irreversible aggregation or misfolding, and can be used by several groups to purify ELPs and protein products fused to ELPs
Intein-fused affinity tags get inducible self-cleave under mild conditions (room temperature and slightly acidic pH)
PROBLEMS: Slow growth.
Expression level
Optimization will be required for new, uncharacterized products on a case-by-case basis

68
Affinity Chromatography (AC)
• Principles of AC
• Main stages in Chromatography
• How to prepare Affinity gel - Ligand Immobilization - Spacer arms – Coupling methods – Coupling tips
• Types of AC
• Elution Conditions: Affinity Tags and Fusion Proteins
• Chelating, Strep-tag, GST, MBP, etc
• Cleavage sites – Proteases
• Parameters for development optimization (Troubleshooting): binding, washing, elution
• Examples
• Binding equilibrium, competitive elution, kinetics
• Future Considerations

69
Parameters for optimization during binding and washings- I
If protein does not bind to the resin, there are several options to choose
Check the quality of the resin (use an His-tag control protein) or check for the presence of reagents that avoid binding
Partial binding: use more resin, or bind for longer time (BUT: the longer the duration of purification, the greater the risk of protein degradation). Use unbound material for a new purification.
Try next time additives as glycerol, detergents or more NaCl (soluble aggregates??)
Tag is inaccessible: try purification under denaturating conditions (for poly-His fusion proteins)
Check by western-blot if the tag has been degraded; if this is the case, try to work all the time at 4°C and use more protease inhibitors during lysis
Construct a new vector with the tag in the opposite end of the protein.

70
Parameters for optimization during binding and washings- II
If multiple proteins bands are seen in the elution try:
Protein degradation (you can check previously with western blot) try to work all the time at 4°C and
use protease inhibitors during lysis.
Use more stringent competitive conditions during binding and washing (example: use low Imidazole concentrations during binding and washings to increase competition for the same sites on the resin)
Decrease resin volume (allows higher competition between fusion protein and contaminants for
the same sites on the resin)
If contaminants are associated with the tagged protein, try to disrupt the non-
specific interaction by adding to the wash buffer before elution additives as ß-ME, glycerol up to 50%, detergents as Triton X-100, NP40 or Tween 20 up to 2% or increase ionic strength up to 1.5M NaCl or KCl.
Increase the washing step volume.
Consider an additional purification step before or after purification.
Consider the use of a pre-column of beads without ligand to adsorb proteins that bound to
beads non-specifically

71
Parameters for optimization during elution
Optimize resolution
Adjust gradient slope/shape, or step elution with increasing competitor concentration
Include additives in buffers
Adjust flow rate
Adjust column volume
Bead size and quality of the resin
If the protein slightly elutes or does not elute from the column
Use higher competitor concentration (Examle: up to 1M Imidazol for chelating columns),
or additives
Reduce elution flow-rate
Change elution conditions, consider elution under denaturating conditions

72
Parameters for optimization during protease cleavage
and purification
Cleavage depends of target/protease ratio, volume, temperature, time, buffer, etc
If possible try to cut at low temperature
Cleavage can be done inside dialysis bags under dialysis to prepare protein to next step
If necessary add additives in buffers to avoid aggregation of the target after cleavage
High aggregation can affect cleavage
Some targets slightly bound to the IMAC resin in the negative step: increase competition
(Imidazol, etc)
Cleavage is OK, but proteins elute together because of other interactions. Try to reduce
these interactions (high salt, detergents, etc)
Alternatives to negative affinity: IEX, HIC, GF

73
Lysis of 10ml culture
Bound and washed (in the presence of 10, 20, 30 mM imidazol) to nickel resin
Elution was with 300 mM imidazol
stop of ARNO-(His tag) cc17 pDest Optimization during binding
Each well contains 10 micro liter of the desired step (from 40 microliters )+ 5 micro liter sample buffer.
Each of the marker proteins is 2 micro-grams.
20kDa
14 kDa
6.5 kDA
His6-cc
Tamar Shultz
Altschuler lab
Elution 300 mM Imidazole
Ste
p 1
Ste
p 2
Ste
p 2
Ste
p 2
Ste
p 2
Ste
p 1
Ste
p 1
Ste
p 1
0 10 20 30
Elution marker
Binding and washing- x mM imidazol

11/19/2014 74
P38 CAPTURE -Optimization
Incubate each fraction (batch binding) 60min 4°C with 150µl Ni-NTA (equilibrated with different [Imidazol]).
Spin 3min 3000rpm 4°C. Discharge unbound material.
Wash resin 6x1ml washing buffer + different [Imidazol].
Elution 3x300µl elution buffer (250mM Imidazol). Run 12%PAGE-SDS of each elution.
Ni-NTA equilibration (mM Imidazol) 10 10 10 20 20 20 30 30 30 40 40 40
[Imidazol] for binding (mM Imidazol) 10 10 10 20 20 20 30 30 30 40 40 40
[Imidazol] for washing (mM Imidazol) 10 10 10 20 20 20 30 30 30 40 40 40
[Imidazol] for elution (mM Imidazol) 250 250 250 250 250 250 250 250 250 250 250 250
Elution number 1 2 3 1 2 3 MW 1 2 3 1 2 3
Cells lysis from 250ml culture.
Spin 30min 20000g 4°C.
Divide supernatant in 4 fractions,
add to each one different [Imidazol].

11/19/2014 75 100gr cells (6L culture). Cell disruption with Mountain Goulin in 900ml lysis buffer. Batch binding to 15ml Ni-NTA 90min 4°C in
the presence of 20mM Imidazol. Wash with 20mM Imidazol buffer and elution with 5cv gradient 20-250mM Imidazol.
Pool P38
61 OD280nm
P38 CAPTURE - Affinity Chromatography
Unb Wash 4 5 6 7 MW 8 9 10 11 12
11/19/2014 76
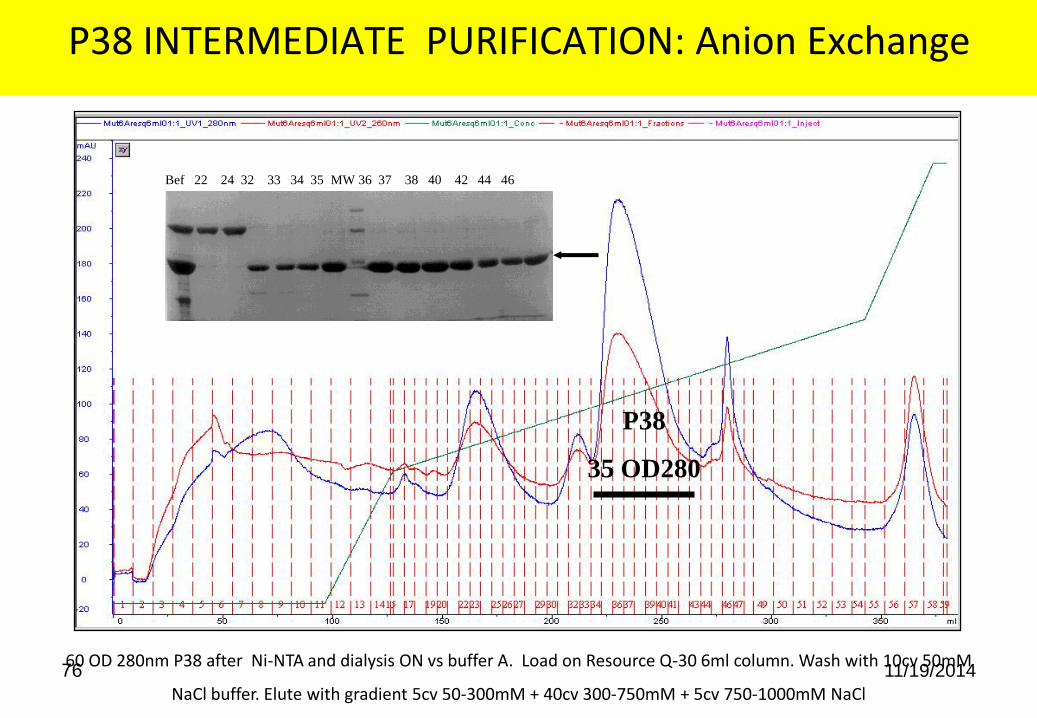
P38 INTERMEDIATE PURIFICATION: Anion Exchange
60 OD 280nm P38 after Ni-NTA and dialysis ON vs buffer A. Load on Resource Q-30 6ml column. Wash with 10cv 50mM
NaCl buffer. Elute with gradient 5cv 50-300mM + 40cv 300-750mM + 5cv 750-1000mM NaCl
P38
35 OD280
Bef 22 24 32 33 34 35 MW 36 37 38 40 42 44 46
11/19/2014 77
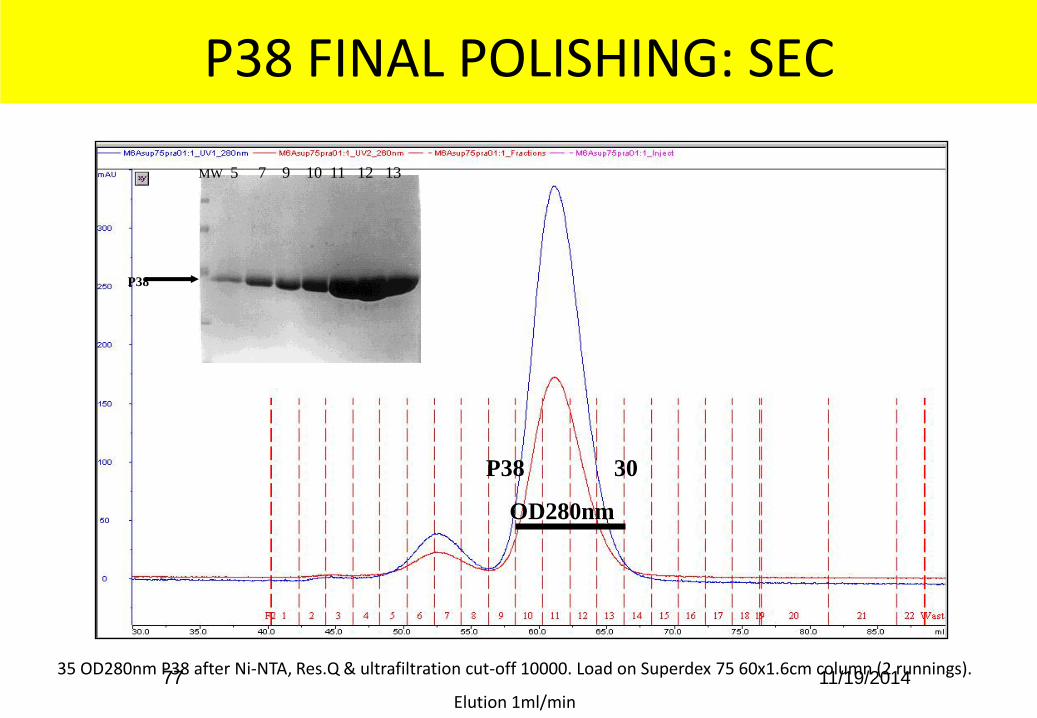
P38 FINAL POLISHING: SEC
35 OD280nm P38 after Ni-NTA, Res.Q & ultrafiltration cut-off 10000. Load on Superdex 75 60x1.6cm column (2 runnings).
Elution 1ml/min
P38 30
OD280nm
MW 5 7 9 10 11 12 13
P38

78
AbrBHiTrapNiHP1mlb001:1_UV1_280nm AbrBHiTrapNiHP1mlb001:1_UV2_260nm AbrBHiTrapNiHP1mlb001:1_Conc AbrBHiTrapNiHP1mlb001:1_Fractions AbrBHiTrapNiHP1mlb001:1_Inject AbrBHiTrapNiHP1mlb001:1_Logbook
0
100
200
300
400
mAU
0.0 10.0 20.0 30.0 40.0 50.0 60.0 70.0 ml
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
Load + 35 cv 0%B + 8cv 4%B + 8cv 10%B + 5cv 10-100%B + 10cv 100%B (500mM Imidazol)
IMAC purification: Low Imidazol step washing before elution Case study: AbrB- Collaboration with A.Kaplan & D. Schatz
Sol 1 3 5 9 10 12 13 14 15 16 17 19 20
34
26
17
10
AbrB

11/19/2014 79
HLT-p53CT- Affinity start with pellet of 1.5L culture
Ni-Sepharose FF 14ml Ronen Gabizon – Assaf Friedler Group
HLTp53CTNiNTA16ml004:1_UV1_280nm HLTp53CTNiNTA16ml004:1_UV2_260nm HLTp53CTNiNTA16ml004:1_Conc HLTp53CTNiNTA16ml004:1_Fractions HLTp53CTNiNTA16ml004:1_Inject HLTp53CTNiNTA16ml004:1_Logbook
0
500
1000
1500
2000
2500
mAU
200 250 300 350 400 450 ml
F4 Waste 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
Load + 10cv 0%B + 3cv 8%B + 4cv 15%B + 4cv 100%B
POOL 17-22: 3.5OD x 35ml ~ 276mg

11/19/2014 80
HLT-p53CT- Cation Exchange after TEV protease cleavage
ON 4ºC SP-Sepharose FF 5ml
HLTp53CTHiTrapSP5mlml005:1_UV1_280nm HLTp53CTHiTrapSP5mlml005:1_UV2_260nm HLTp53CTHiTrapSP5mlml005:1_Cond HLTp53CTHiTrapSP5mlml005:1_Conc HLTp53CTHiTrapSP5mlml005:1_Fractions HLTp53CTHiTrapSP5mlml005:1_Inject HLTp53CTHiTrapSP5mlml005:1_Logbook
0
50
100
150
mAU
500 550 600 650 700 ml
F3 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44

11/19/2014 81
HLT-p53CT- GF after Cation
Exchange
Sephacryl S100 FF 500ml
HLTp53CTSephacrylS100of500ml004:1_UV1_280nm HLTp53CTSephacrylS100of500ml004:1_UV2_260nm HLTp53CTSephacrylS100of500ml004:1_Fractions HLTp53CTSephacrylS100of500ml004:1_Inject HLTp53CTSephacrylS100of500ml004:1_Logbook
0
50
100
150
mAU
60 80 100 120 140 160 180 200 220 min
F3 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 Waste
POOL 7-14

82
Binding equilibrium
At equilibrium
K
L T
LTD
KD is the equilibrium dissociation constant, [L] is the concentration of free ligand [T] is the concentration of free target [LT] is the concentration of the ligand/target complex

83
Binding equilibrium
It can be shown* that
Bound target
Total targetL
K L0
D 0
KD is the equilibrium dissociation constant, L0 is the concentration of ligand, usually 10-4 - 10-2 M
*Graves, DJ, Wu, YT Meth. Enzymol 34 (1974) 140-163
For good binding, KD should be at least two orders of magnitude less than L0, i.e. 10-6 - 10-4 M

84
Binding equilibria
L + T LT KD 10-6 - 10-4 M
Binding
L + T LT KD 10-1 - 10-2 M
Elution
We can change KD by changing pH, temperature, salt concentration etc.
Target elutes as a sharp peak

85
Elution by changing KD - Unexpected results
KD too high during binding (low affinity)
Target is retarded as a broad peak and elutes under binding conditions as a broad, low peak
Find better binding conditions or use ligand with higher affinity
KD not low enough during elution
Target elutes in a long, low 'peak'
Try different elution conditions or use ligand with lower affinity
KD too low during binding (extremely high affinity)
Difficult to elute target. Difficult or impossible to increase KD enough to elute the target
without destroying its activity
Use ligand with lower affinity

86
Competitive elution
+
Binding
Release
C + T CT
Elution +

87
Binding equilibrium for competing ligand
At equilibrium
K
C T
CTDComp
KDComp is the equilibrium dissociation constant [C] is the concentration of free competing ligand [T] is the concentration of free target [CT] is the concentration of the competing ligand/target complex

88
Competitive elution
For good elution: Increase competitor concentration to get a sharp peak if necessary
If KDComp and KD are similar then the concentrations of competing and coupled ligand should be similar
If KDComp is 10 x KD (i.e. the free competing ligand binds more weakly) then the concentration of competing
ligand will need to be 10 x higher to get effective elution
121125AntiHer2Ni30ml005:10_UV1_280nm 121125AntiHer2Ni30ml005:10_UV2_260nm 121125AntiHer2Ni30ml005:10_Conc 121125AntiHer2Ni30ml005:10_Fractions 121125AntiHer2Ni30ml005:10_Logbook
0
100
200
300
400
500
mAU
200 300 400 500 600 700 800 ml
F3 F4 1 2 3 4 5 6 7 8 9 10 11121314 15161718 192021 23 28 33 38 43 48 53 58 63 68 71 74 77 80 83 86
121014AntiHer2Ni16ml004:10_UV1_280nm 121014AntiHer2Ni16ml004:10_UV2_260nm 121014AntiHer2Ni16ml004:10_Conc 121014AntiHer2Ni16ml004:10_Fractions 121014AntiHer2Ni16ml004:10_Logbook
0
100
200
300
400
500
600
700
800
mAU
200 250 300 350 400 450 ml
F4 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 21 23 25 27 29 31 33 35 37 39 41 43 45 47 49 50
Load lysed pellet of 2L culture on 16ml Ni-SepharoseFF used many times
Load lysed pellet of 4L culture on 30ml Ni-Sepharose6B used a few times

89
Kinetics of adsorption and desorption Unexpected results
Slow binding
Some of the target elutes under binding conditions as a broad, low peak
Slow down binding. Stopped flow binding to bind the target in portions
Slow desorption
Target elutes as a long, low 'peak'
Stopped flow elution to elute the target in pulses. Change elution scheme

90
Synthetically “mimicking” and enhancing the natural molecular affinity of binding ligands toward
targeted biomolecules
Mimetic Ligand adsorbents are biologically inert, very durable, have excellent liquid flow properties,
and are resistant to most chemical treatments.
Stable in the pH range 2 - 14, can be treated with denaturants (8M urea), detergents (Triton, SDS)
and can be sterilized by autoclaving.
Capacities can exceed 50mg protein per ml of packed gel. Low Ligand Leakage
Alkali-stable adsorbents - Enables cleaning, sterilization and depyrogenation with 1M sodium
hydroxide, ensuring long operational lifetimes.
Albumin • Proteases • Blood Proteins • Oxidases • Dehydrogenases • Ligases • Kinases • Antibodies
• Nucleases • Cytokines
Custom Media Development Programs
Chemical Combinatorial Library™, + with a rational ligand design approach
Mimetic Ligand™ Affinity Chromatography www.prometic.com

91
BAC (BioAffinity Company) Capture Select®
Camelid derived single domain antibody fragments
http://www.bacbv.com/
Custom designed ligands (12-15kDa)
Produced in Saccharomyces cerevisiae, enabling high level production with minimal purification
effort as well as easy scale-up.
Stability, affinity, and selectivity
Reduction of process steps: higher yields, reduced costs
Selectivity: high purity in single step / feed stock independent
Mild elution conditions: retaining biological activity of target
Efficient clearance of HCP, DNA, virus: high selectivity in capture step

92
IgG Select
93

94
Scil Proteins: Affilin™ Techology
Novel human binding proteins for therapy and applications in chromatography.
Affilin™ molecules are small non-immunoglobulin proteins which are designed for specific
binding of proteins and small molecules. New Affilin™ molecules can be quickly selected
from libraries which are based on two different human derived scaffold proteins.
Gamma crystalline, a human structural eye lens protein and ubiquitin one of the highest
conserved proteins known today. Both human scaffolds are small, show high temperature
stability and are almost resistant to pH changes and denaturing agents.
By engineering the protein surfaces through a locally defined randomization of solvent
exposed amino acids, completely new binding sites are created. Former non-binding
proteins are thereby transformed into specific binding proteins that can be easily
produced in the cytoplasm of E. coli
![3 l] Affinity Chromatography](https://static.fdocuments.in/doc/165x107/6234c6b4f34ba75ca16e0e55/3-l-affinity-chromatography.jpg)



![Recent developments in protein ligand affinity mass spectrometry · frontal affinity chromatography (FAC) [1], size-exclusion chromatography (SEC) [2], (pulsed) ultrafiltration [3],](https://static.fdocuments.in/doc/165x107/604c1f4e3a10f26659366e36/recent-developments-in-protein-ligand-affinity-mass-spectrometry-frontal-affinity.jpg)













