
ADVANCES IN COMPUTER AIDED DRUG DESIGNujpsr.com/sites/default/files/articles/UJPSRMN-05.pdf · F...
6
REVIEW ARTICLE Department of Pharmaceutical Chemistry Kalita et.al / UJPSR / 1 (2), 2015, 17-22 e ISSN: 2454-3764 Print ISSN: 2454-3756 ADVANCES IN COMPUTER AIDED DRUG DESIGN Abstract Key words In the early ages of drug design and discovery system, very less information was available with respect to the structure of ligands and the targets. With advancement in the technological and information system now it is possible to simulate the drug-target interactions inside the computer chips. Computer-aided drug design (CADD) is a modern, rationale and diverse discipline where various aspects of basic and applied knowledge are used to design new drugs. There are different strategies, protocols and algorithms included under computer aided drug design. It is the researcher who can choose the perfect Strategy for CADD depending on the extent of structural and other information available regarding the targets (enzyme/receptor) and the ligands. In the present review we reported a brief history of CADD together with its progress in the last few years. CADD, Structure Based Drug Design, Molecular Modelling, Docking. ARTICLE INFO: Article history: Received: 01 August 2015 Received in revised form: 09 September 2015 Accepted: 1 November 2015 Available online: 10 November 2015 Jun Moni Kalita Department of Pharmaceutical Chemistry Himalayan Pharmacy Institute Majhitar, E-Sikkim-737136, INDIA E-mail: [email protected] Phone: +919508980893 and its commercialization is a tedious and time consuming process and moreover the cost has increased drastically during the past thirty-four years. In 1962, the total cost was $4 million, which became $350 million in 1996. It has been observed that only small amount of drugs which has been synthesized will be examined in clinics and among them few will be marketed. In 1950, it has been estimated that about 7000 compounds are isolated or synthesized and then tested for therapeutic activity for each one that became a pharmaceutical product. The challenge is becoming more difficult when 10,000 compounds had to be evaluated in 1979, and this number could be as high as 20,000 today [1, 2]. So, due to this reason now the entire world's major pharmaceutical and biotechnology The discovery of a drug, its development INTRODUCTION companies started using computational design tools. These tools are used to replace the crude mechanical models by displaying of structure which is a much more accurate reflection of molecular reality capable of demonstrating motion and solvent effects [3]. Apart from this, theoretical calculations permit the computation of free binding energies and other relevant molecular properties. The term "molecular modelling" has expanded over the last decades from a tool to visualize three dimensional (3D) molecular structures & to simulate, predict & analyze the properties and behaviour of molecules on an atomic level to a data- mining & in-silico drug design platform to organize many compounds & their properties into databases & to perform virtual drug screening via 3D database searches for novel drug compounds [3-5]. Jun M Kalita*, Ashmita Saha, Dipankar Nath, Uddhav Patangia Department of Pharmaceutical Chemistry, Himalayan Pharmacy Institute, Majhitar, Rangpo, E. Sikkim - 737136, INDIA Corresponding Author: www.ujpsr.com 17 nd 2 Issue
Transcript of ADVANCES IN COMPUTER AIDED DRUG DESIGNujpsr.com/sites/default/files/articles/UJPSRMN-05.pdf · F...

REVIEW ARTICLE
Department of Pharmaceutical Chemistry
Kalita et.al / UJPSR / 1 (2), 2015, 17-22
e ISSN: 2454-3764Print ISSN: 2454-3756
ADVANCES IN COMPUTER AIDED DRUG DESIGN
Abstract
Key words
In the early ages of drug design and discovery system, very less information was available with respect to the structure of ligands and the targets. With advancement in the technological and information system now it is possible to simulate the drug-target interactions inside the computer chips. Computer-aided drug design (CADD) is a modern, rationale and diverse discipline where various aspects of basic and applied knowledge are used to design new drugs. There are different strategies, protocols and algorithms included under computer aided drug design. It is the researcher who can choose the perfect Strategy for CADD depending on the extent of structural and other information available regarding the targets (enzyme/receptor) and the ligands. In the present review we reported a brief history of CADD together with its progress in the last few years.
CADD, Structure Based Drug Design, Molecular Modelling, Docking.
ARTICLE INFO:Article history:Received: 01 August 2015Received in revised form:09 September 2015Accepted: 1 November 2015Available online: 10 November 2015
Jun Moni KalitaDepartment of Pharmaceutical ChemistryHimalayan Pharmacy InstituteMajhitar, E-Sikkim-737136, INDIAE-mail: [email protected]: +919508980893
and its commercialization is a tedious and time consuming process and moreover the cost has increased drastically during the past thirty-four years. In 1962, the total cost was $4 million, which became $350 million in 1996. It has been observed that only small amount of drugs which has been synthesized will be examined in clinics and among them few will be marketed. In 1950, it has been estimated that about 7000 compounds are isolated or synthesized and then tested for therapeutic activity for each one that became a pharmaceutical product. The challenge is becoming more difficult when 10,000 compounds had to be evaluated in 1979, and this number could be as high as 20,000 today [1, 2]. So, due to this reason now the entire world's major pharmaceutical and biotechnology
The discovery of a drug, its development
INTRODUCTION
companies started using computational design tools. These tools are used to replace the crude mechanical models by displaying of structure which is a much more accurate reflection of molecular reality capable of demonstrating motion and solvent effects [3]. Apart from this, theoretical calculations permit the computation of free binding energies and other relevant molecular properties. The term "molecular modelling" has expanded over the last decades from a tool to visualize three dimensional (3D) molecular structures & to simulate, predict & analyze the properties and behaviour of molecules on an atomic level to a data-mining & in-silico drug design platform to organize many compounds & their properties into databases & to perform virtual drug screening via 3D database searches for novel drug compounds [3-5].
Jun M Kalita*, Ashmita Saha, Dipankar Nath, Uddhav Patangia
Department of Pharmaceutical Chemistry, Himalayan Pharmacy Institute, Majhitar, Rangpo, E. Sikkim - 737136, INDIA
Corresponding Author:
www.ujpsr.com 17nd2 Issue
In the past few years, the field of computer-aided drug design (CADD) has grown speedily, enhancing the perceptive of multifaceted and difficult biological processes. With the help of computer programs, it is now possible to study and predict experimental results with sound accuracy in
a short duration making it both cost and time effective. In the recent few decades computational methods has been extensively used in the field of drug design and discovery. Few examples of drugs as a result of computer aided drug design are included in table 1 [6].
www.ujpsr.com 18
Table 1: Examples of drugs found with the help of CADD
Year
1989
1997
1998
1999
2007
Drug Name
Zanamivir
Nelfinavir
Raltitrexed
Amprenavir
Raltegravir
Used as
Anti HIV
Anti HIV
Anti Cancer
Anti HIV
Anti HIV
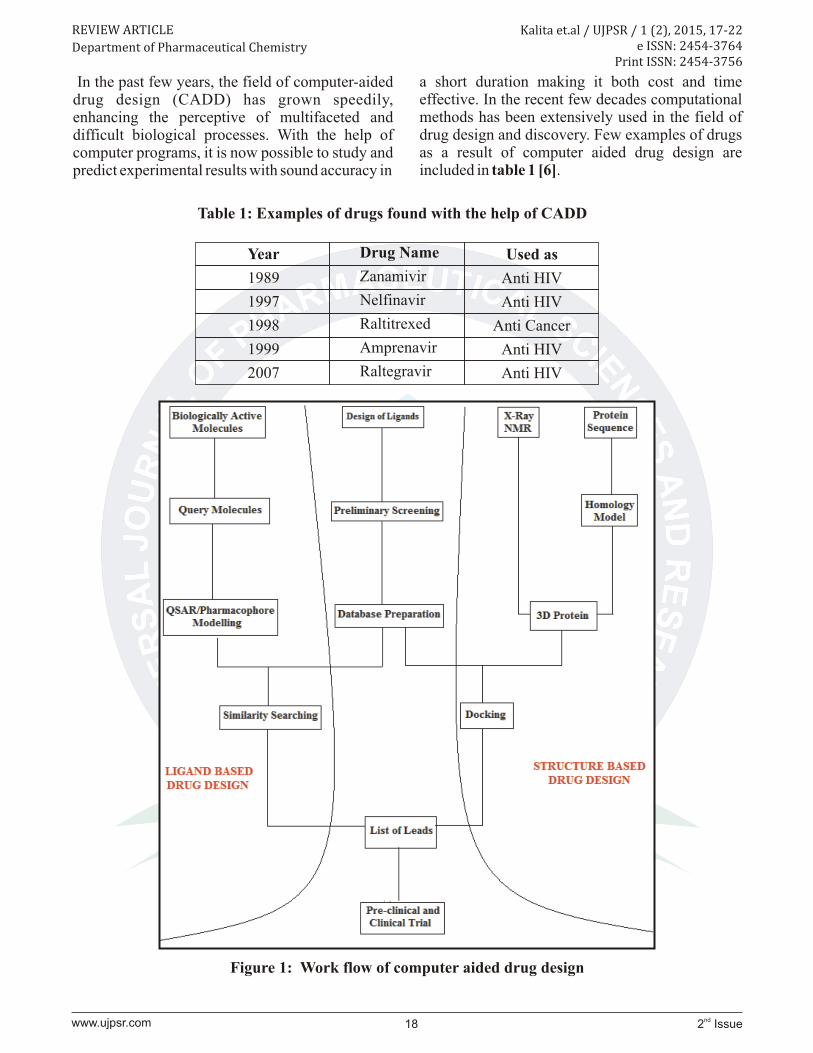
Figure 1: Work flow of computer aided drug design
REVIEW ARTICLE
Department of Pharmaceutical Chemistry
Kalita et.al / UJPSR / 1 (2), 2015,
17-22e ISSN: 2454-3764
Print ISSN: 2454-3756
nd2 Issue

Though the total energy has no physical meaning by itself, but differences in total energy between two different conformations of the same molecule can be compared[9-11].
Quantum mechanics
It determines the complete structural information by considering the molecule up to atomic and electronic level. As its calculation are much complex in nature, quantum mechanical methods are restricted to simple system & the approximation known as molecular orbital theory is mostly used for practical purposes [11].
Virtual Screening
Virtual screening (VS) is a computational technique used in drug design and discovery to search library of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme. These virtual screening techniques can be broadly grouped in two group viz. ligand-based virtual screening and structure based or target-based virtual screening. Methods like pharmacophore modelling and QSAR comes under ligand based virtual screening where else method like docking comes under structure based or target based drug design.
Pharmacophore modelling
A pharmacophore is an abstract description of molecular properties which are necessary for molecular recognition of a ligand by a biological macromolecule . The IUPAC def ines a pharmacophore to be "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response". A typical pharmacophore features include hydrophobic centroids, aromatic rings, hydrogen bond acceptors or donors, cations, and anions.
A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site. In addition pharmacophore models can be used to identify and screen newly designed molecules for their capability to bind with the same target protein. These pharmacophoric points may be located on the ligand itself or may be projected points presumed to be located in the receptor [12].
Molecular Modelling
Molecular modelling comprises all theoretical and computational methods used to model or mimic the behaviour of small molecules like protein, DNA, ligand etc. These molecular modelling techniques are used in the fields of computational chemistry, drug design, computational biology and materials science for studying different molecular systems. The simplest calculations can be done manually by hand, but computers are required to perform the modelling and complex calculations of any bigger sized systems. The common feature of molecular modelling techniques is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit (the Molecular mechanics approach), or explicitly modelling electrons of each atom (the quantum chemistry approach) [7, 8].
Molecular mechanics
Molecular mechanics considers a molecule to be a compilation of masses interacting with each other through harmonic forces. Thus the atoms in molecules are treated as ball of different sizes and joined together by springs of variable strengths and equilibrium distances (bonds).This simplification enables molecular mechanics as a fast computational model that can be applied to molecules of any size. The total energy of molecule is calculated by adding the different contributions that compute the deviations from equilibrium of bond lengths, angles and torsions plus non-bonded interactions as show in equation I.
E = E + E + E + E + E (I)tot str bend tors vdw ele
Where E refers to total energy of the molecule, E tot str
refers to the bond stretching energy term, E is the bend
angle bending energy term, E is the torsional tors
energy term, E is the van der waals energy term vdw
and E is the electrostatic energy term.elec
The equilibrium values of these bond lengths and bond angles are the corresponding force constants used in the potential energy function defined in the force fields and define a set known as force field parameters. Each deviation from these equilibrium values will result in increasing total energy of the molecule. So, the total energy is a measure of intra molecular strain relative to a hypothetical molecule with an ideal geometry of equilibrium.
www.ujpsr.com 19
REVIEW ARTICLE
Department of Pharmaceutical Chemistry
Kalita et.al / UJPSR / 1 (2), 2015,
17-22e ISSN: 2454-3764
Print ISSN: 2454-3756
nd2 Issue

QSAR
Quantitative Structure Activity Relationship
(QSAR) modelling is the construction of
extrapolative models of pharmacological activities
as a function of structural and molecular
information of a compound library. The idea of
QSAR has normally been used for drug discovery
and development and has gained wide applicability
for correlating molecular information with not only
biological activities but also with other
physicochemical properties, which has therefore
also been termed as Quantitative Structure Property
Relationship (QSPR). Molecular parameters that
are used to contribute for electronic properties,
hydrophobicity, steric effects, and topology can be
determined empirically by experimentation or
theoretically via computational chemistry. A given
collection of data sets is then subjected to data pre-
processing and data modelling through the use of
statistical and/ or machine learning techniques.
Quantitative structure-activity relation- ship
(QSAR) and quantitative structure- property
relationship (QSPR) made it feasible to forecast the
activities/properties of a given compound as a
function of its molecular substituent. By this new
and untested compounds having similar molecular
features as compounds used in the construction of
QSAR/QSPR models are likewise supposed to also
possess similar activities/properties. A number of
successful QSAR/QSPR models have been
published in the last few years which include a wide
span of biological and physicochemical properties.
QSAR/QSPR has gained a great popularity in the
field of drug design and discovery [13].
Docking
Molecular Docking is the study of ability of a ligand
to fit with a receptor or protein. It is a problem like
solving a three dimensional puzzle. It is a method
which predicts the favoured orientation of a
molecule to other when bound to each other to form
a stable complex. Taking the knowledge of the
preferred orientation, strength of association or
binding affinity between two molecules is predicted.
It is often used to guess the binding strength of small
molecule like drug candidates to their protein targets
in order to in turn predict the affinity and activity of
the small molecule. Hence docking plays a very
important role in the era of rational design of drugs.
Molecular docking is a problem of "lock-and-key"
type, where one finds the right relative orientation of
the "key" which opens the "lock". Here, the protein
is thought of as the "lock" and the ligand is thought
of as a "key". Molecular docking may be explained
as an optimization problem, which describes the
"best-fit" orientation of a ligand that binds to a
particular protein. The point up of molecular
docking is to computationally simulate the
molecular recognition process. The main aim of
molecular docking technique is to reach an
optimized conformation for both the protein and
ligand and relative orientation between protein and
ligand with an energy minimized system [14-15].
CONCLUSION
Finally it can be concluded that, the primary goal in
drug design is to predict whether a given molecule
will bind to a target and if so how strongly. Computer
Aided Drug Design, which is the center of attraction
in the modern era of drug design and discovery, can
be also said as rational method of drug design and
discovery.
www.ujpsr.com 20
REVIEW ARTICLE
Department of Pharmaceutical Chemistry
Kalita et.al / UJPSR / 1 (2), 2015,
17-22e ISSN: 2454-3764
Print ISSN: 2454-3756
nd2 Issue

1. Thakur M and Chandan N. Molecular
Modelling Coupled with Computer Aided
Drug Design. Int. Journal of Research in
Pharm and Biomedical Sciences 2012; 3
(1): 23-29.
2. F Ooms. Molecular Modeling and
Computer Aided Drug Design. Examples of
their Applications in Medicinal Chemistry.
Current Medicinal Chemistry 2000; 7: 141-
158.
3. Pranita P., Madhavi M., Rishikesh V.,
Rajesh J., Sandip S. Computer-Aided Drug
Design: An Innovative Tool for Modeling.
Open Journal of Medicinal Chemistry
2012; 2: 139-148
4. Kitchen D. B., Decornez H., Furr J. R.,
Bajorath J. Docking and scoring in virtual
screening for drug discovery: methods and
applications. Nature reviews. Drug
discovery 2004; 3 (11): 935–949.
5. Roy K., Sengupta C and Dey A U.
Theoritical Aspects of Rational Drug
Design – an overview. Journal of Science
and Industrial Research 2001; 60: 699-716.
6. Wei B. Q., Weaver L. H., Ferrari A. M.,
Matthews B. W., Shoichet B. K. Testing a
flexible-receptor docking algorithm in a
model binding site. J. Mol. Biol. 2004; 337
(5): 1161–82.
7. Wilfred F., Gunsteren V., Bakowies D.,
Baron R., Chandrasekhar I., Christen M.,
Daura X., Gee P., Geerke D. P., Glttli A.,
Hnenberger P. H., Kastenholz M. A.,
Oostenbrink C., Schenk M., Trzesniak B.
D., Nico F. A., and Haibo B. Biomolecular
Modeling: Goals, Problems, Perspectives.
Chem. Int. Ed.2006; 45: 4064–4092.
8. Cavasotto C. N. and Pathak S. S. Homology
modeling in drug discovery: current trends
and applications. Drug Discovery Today
2009; 14:
9. Box V.G. The Molecular Mechanics of
Quantized Valence Bonds. J Mol Model
1997; 3 (3): 124–41.
10. Mackerell A. D. Empirical force fields for
biological macromolecules: overview and
issues. J Comput Chem 2004. 25 (13):
1584–604.
11. Parsons J., Holmes J. B., Rojas J. M., Tsai
J., Strauss C. E., Practical conversion from
torsion space to cartesian space for in silico
protein synthesis. J Comput Chem 2005; 26:
1063-1068.
12. Wermuth C. G., Ganellin C. R., Lindberg P.,
Mitscher L. A. Glossary of terms used in
m e d i c i n a l c h e m i s t r y ( I U PA C
Recommendations 1998). Pure and Applied
Chemistry 1998; 70 (5): 1129–1143.
REFERENCES
www.ujpsr.com 21
REVIEW ARTICLE
Department of Pharmaceutical Chemistry
Kalita et.al / UJPSR / 1 (2), 2015,
17-22e ISSN: 2454-3764
Print ISSN: 2454-3756
nd2 Issue

www.ujpsr.com 22
13. Nantasenamat C., Isarankura-Na-Ayudhya
C., Prachayasittikul V. Advances in
computational methods to predict the
biological activity of compounds. Expert
Opin. Drug Discov 2010; 5: 633–54.
14. DvorskyV. R. and Sturdik E. Receptor-
Ligand Interaction and Molecular
Modelling; Gen Physiol Biophysics 1999;
18: 231-248.
15. Lengauer T., Rarey M. Computational
methods for biomolecular docking. Curr.
Opin. Struct. Biol 1996; 6 (3): 402–406.
REVIEW ARTICLE
Department of Pharmaceutical Chemistry
Kalita et.al / UJPSR / 1 (2), 2015,
17-22e ISSN: 2454-3764
Print ISSN: 2454-3756
nd2 Issue


















