Advances in behavioral genetics: mouse models of autismsswang/CB-pediatric/...model for autism, the...
23
FEATURE REVIEW Advances in behavioral genetics: mouse models of autism SS Moy 1 and JJ Nadler 2 1 Neurodevelopmental Disorders Research Center, Department of Psychiatry, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA and 2 Neurodevelopmental Disorders Research Center, Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA Autism is a neurodevelopmental syndrome with markedly high heritability. The diagnostic indicators of autism are core behavioral symptoms, rather than definitive neuropathological markers. Etiology is thought to involve complex, multigenic interactions and possible environmental contributions. In this review, we focus on genetic pathways with multiple members represented in autism candidate gene lists. Many of these pathways can also be impinged upon by environmental risk factors associated with the disorder. The mouse model system provides a method to experimentally manipulate candidate genes for autism susceptibility, and to use environmental challenges to drive aberrant gene expression and cell pathology early in development. Mouse models for fragile X syndrome, Rett syndrome and other disorders associated with autistic-like behavior have elucidated neuropathology that might underlie the autism phenotype, including abnormalities in synaptic plasticity. Mouse models have also been used to investigate the effects of alterations in signaling pathways on neuronal migration, neurotransmission and brain anatomy, relevant to findings in autistic populations. Advances have included the evaluation of mouse models with behavioral assays designed to reflect disease symptoms, including impaired social interaction, communication deficits and repetitive behaviors, and the symptom onset during the neonatal period. Research focusing on the effect of gene-by-gene interactions or genetic susceptibility to detrimental environmental challenges may further understanding of the complex etiology for autism. Molecular Psychiatry (2008) 13, 4–26; doi:10.1038/sj.mp.4002082; published online 11 September 2007 Keywords: Angelman syndrome; fragile X; repetitive behavior; Rett syndrome; serotonin signaling; social interaction Introduction Autism is a severe neurodevelopmental disorder that is typically diagnosed by age 3. Twin studies have provided evidence for a markedly strong genetic component for autism, with concordance rates as high as 70–80% between monozygotic twins. 1 Hetero- geneity of the clinical syndrome suggests that the autism domain may encompass several disorders with different genetic profiles. Core symptoms of autism include profound deficits in social interac- tion and communication, restricted interests, stereo- typed responses and other repetitive patterns of behavior. 2,3 Other abnormalities include high preva- lence of mental retardation, with rate estimates of 40–55% or higher, 4,5 and co-morbid epilepsy, observed in approximately 30% of autistic subjects. 6 These symptoms underscore the catastrophic consequences of the genetic inheritance for brain function and behavior. Disease etiology is thought to involve an interaction between genetic susceptibility, mediated by multiple genes, and possible environmental factors, leading to aberrant neurodevelopment. 7–11 A complex combination of genetic predisposition and environmental contribution may underlie the broad range and differential severity of symptoms in autism. In recent years, mouse models have been developed that reflect genetic alterations associated with autism. Some mutant lines are based on monogenic aberra- tions, such as loss of Fmr1, methyl-CpG-binding protein-2 (Mecp2) or ubiquitin protein ligase 3A (Ube3A) function, that underlie syndromes associated with autistic-like behavior. Other mutant lines are relevant to loci for autism susceptibility, identified by association or linkage studies in human populations. Mouse models have also been produced by prenatal or neonatal environmental challenges, including early exposure to valproic acid or inflammatory agents, that have been suggested as autism risk factors by clinical surveys. This review describes recent advances in the behavioral validation of mouse models for autism, and how mutant lines have been used to elucidate the molecular mechanisms underlying the functional effects of genetic and other changes. The review also examines how models for alterations in signaling pathways can indicate novel genetic targets for studies of autism spectrum disorders. Received 21 March 2007; revised 23 July 2007; accepted 31 July 2007; published online 11 September 2007 Correspondence: Dr SS Moy, Neurodevelopmental Disorders Research Center, Department of Psychiatry, CB# 7146, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: [email protected] Molecular Psychiatry (2008) 13, 4–26 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp
Transcript of Advances in behavioral genetics: mouse models of autismsswang/CB-pediatric/...model for autism, the...
-
FEATURE REVIEW
Advances in behavioral genetics: mouse models of autismSS Moy1 and JJ Nadler2
1Neurodevelopmental Disorders Research Center, Department of Psychiatry, University of North Carolina at Chapel Hill,Chapel Hill, NC, USA and 2Neurodevelopmental Disorders Research Center, Department of Genetics, University of NorthCarolina at Chapel Hill, Chapel Hill, NC, USA
Autism is a neurodevelopmental syndrome with markedly high heritability. The diagnosticindicators of autism are core behavioral symptoms, rather than definitive neuropathologicalmarkers. Etiology is thought to involve complex, multigenic interactions and possibleenvironmental contributions. In this review, we focus on genetic pathways with multiplemembers represented in autism candidate gene lists. Many of these pathways can also beimpinged upon by environmental risk factors associated with the disorder. The mouse modelsystem provides a method to experimentally manipulate candidate genes for autismsusceptibility, and to use environmental challenges to drive aberrant gene expression andcell pathology early in development. Mouse models for fragile X syndrome, Rett syndrome andother disorders associated with autistic-like behavior have elucidated neuropathology thatmight underlie the autism phenotype, including abnormalities in synaptic plasticity. Mousemodels have also been used to investigate the effects of alterations in signaling pathways onneuronal migration, neurotransmission and brain anatomy, relevant to findings in autisticpopulations. Advances have included the evaluation of mouse models with behavioral assaysdesigned to reflect disease symptoms, including impaired social interaction, communicationdeficits and repetitive behaviors, and the symptom onset during the neonatal period. Researchfocusing on the effect of gene-by-gene interactions or genetic susceptibility to detrimentalenvironmental challenges may further understanding of the complex etiology for autism.Molecular Psychiatry (2008) 13, 4–26; doi:10.1038/sj.mp.4002082; published online 11 September 2007
Keywords: Angelman syndrome; fragile X; repetitive behavior; Rett syndrome; serotoninsignaling; social interaction
Introduction
Autism is a severe neurodevelopmental disorder thatis typically diagnosed by age 3. Twin studies haveprovided evidence for a markedly strong geneticcomponent for autism, with concordance rates ashigh as 70–80% between monozygotic twins.1 Hetero-geneity of the clinical syndrome suggests that theautism domain may encompass several disorderswith different genetic profiles. Core symptomsof autism include profound deficits in social interac-tion and communication, restricted interests, stereo-typed responses and other repetitive patterns ofbehavior.2,3 Other abnormalities include high preva-lence of mental retardation, with rate estimates of40–55% or higher,4,5 and co-morbid epilepsy, observedin approximately 30% of autistic subjects.6 Thesesymptoms underscore the catastrophic consequencesof the genetic inheritance for brain function andbehavior. Disease etiology is thought to involve aninteraction between genetic susceptibility, mediated
by multiple genes, and possible environmentalfactors, leading to aberrant neurodevelopment.7–11
A complex combination of genetic predispositionand environmental contribution may underlie thebroad range and differential severity of symptomsin autism.
In recent years, mouse models have been developedthat reflect genetic alterations associated with autism.Some mutant lines are based on monogenic aberra-tions, such as loss of Fmr1, methyl-CpG-bindingprotein-2 (Mecp2) or ubiquitin protein ligase 3A(Ube3A) function, that underlie syndromes associatedwith autistic-like behavior. Other mutant lines arerelevant to loci for autism susceptibility, identified byassociation or linkage studies in human populations.Mouse models have also been produced by prenatalor neonatal environmental challenges, including earlyexposure to valproic acid or inflammatory agents, thathave been suggested as autism risk factors by clinicalsurveys. This review describes recent advances in thebehavioral validation of mouse models for autism,and how mutant lines have been used to elucidate themolecular mechanisms underlying the functionaleffects of genetic and other changes. The review alsoexamines how models for alterations in signalingpathways can indicate novel genetic targets forstudies of autism spectrum disorders.
Received 21 March 2007; revised 23 July 2007; accepted 31 July2007; published online 11 September 2007
Correspondence: Dr SS Moy, Neurodevelopmental DisordersResearch Center, Department of Psychiatry, CB# 7146, Universityof North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA.E-mail: [email protected]
Molecular Psychiatry (2008) 13, 4–26& 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00
www.nature.com/mp
http://dx.doi.org/10.1038/sj.mp.4002082mailto:[email protected]://www.nature.com/mp
-
Behavioral phenotyping of mouse models
The primary diagnostic indicators of autism areabnormal behaviors, rather than biochemical, neuroa-natomical or other physiological indices.3 Determin-ing whether a proposed mouse model for autismrecapitulates one or more of the core clinical symp-toms can provide valuable insight as to the functionalimpact of altered genes or environment.12–16 However,the development of mouse behavioral assays fordetecting aberrant social responses, restricted inter-ests, or repetitive behavior reflective of autism hasproved challenging. Our research group has proposeda set of behavioral tests that can be used to assesssocial deficits and repetitive behavior in mice.17–19
The testing screen includes assays for social approachand preference for social novelty, in which miceare offered a choice between different types of socialand non-social stimuli. These choice tasks haveprovided evidence that sociability and social avoid-ance are dependent on genetic background. Forexample, mice from C57BL/6J and FVB/NJ inbredstrains, but not from the A/J, BALB/cByJ or BTBRTþ tf/J inbred strains, demonstrated significant pre-ference for proximity to another mouse rather thanbeing alone.17–19 Overall, inbred strain phenotypesvary across a continuum of social behavior, withextremes of high social preference and overt socialavoidance.17,20–22
The symptom of repetitive behavior encompassesboth ‘lower-order’ motoric stereotypy and self-injury,and ‘higher-order’ responses reflecting general cogni-tive rigidity, such as restricted, obsessive interests andstrong resistance to environmental change.13,23,24 Bothcomponents of the repetitive behavior domain tend toco-occur in children with autism or related disor-ders.23,25–27 A recent study examining the relationshipbetween core symptoms of autism in twins providedevidence that, while highly heritable, the domains ofsocial impairment and repetitive behavior were ge-netically heterogeneous.28,29 These findings raise theintriguing possibility that mouse models reflectingdifferent components of the autism behavioral pheno-type might be used to distinguish the specific geneticpathways that mediate stereotypy and cognitiveinflexibility from those underlying deficiencies insocial interaction or communication. Lewis et al.13
provide a comprehensive overview of animal modelsfor repetitive behavior, including both the lower-orderand higher-order response clusters. In our character-ization of inbred mouse strains, we have used homecage observations to detect persistent, stereotypedmotoric behavior. To model more higher-order defi-ciencies, we have evaluated reversal learning in T-maze or water maze tasks.17 Selective impairment inreversal learning has been reported in autistic chil-dren,30 suggesting that this type of task may serve as anindex for resistance to change a learned pattern ofbehavior, relevant to the disease profile.
Given that symptoms in autism emerge early inchildhood, it is important to develop mouse beha-
vioral phenotyping protocols that can evaluatewhether a specific mouse model recapitulates thetime course of disease onset.31–33 Wagner and collea-gues34,35 investigated abnormalities in neonatal andjuvenile mice relevant to neurodevelopmental dis-orders using sets of behavioral tasks for sensorimotorabilities, learning and memory, and other functionaldomains. The researchers utilized a test for juvenileplay to reveal reduced social interaction in a geneticmodel for autism, the Engrailed 2 null mouse.34 Adultmutant and wild-type mice were assayed with thesame interaction test, as well as a resident-intruderparadigm, to confirm chronic changes in socialbehavior in the mouse model. Other researchers havemeasured ultrasonic vocalizations in mouse pupsseparated from their mothers as a test foraltered emotional behavior early in postnatallife (Table 1).36–46 In most (but not all) of thesestudies, mouse pups with genetic alterations relevantto social behavior had decreased levels of ultrasonicvocalizations. These findings suggest that the vocali-zation assay may be useful to measure attenuatedresponses to social isolation in neonates. Further,since the vocalizations may be viewed as distresscalls to elicit maternal intervention, deficits maymodel the early impairments in communicationcharacteristic of autism.
However, studies in early development are particu-larly challenging to conduct. For example, Hahn andLavooy,47 in a review of methodology for ultrasonicvocalization and maternal pup retrieval studies,enumerate the many subject and experimental vari-ables that need to be carefully considered in thedesign of neurodevelopmental evaluations. Difficul-ties with neonatal behavioral assessments include alimited behavioral repertoire in very young pups,stressful effects of repeated handling or maternalseparation, and alterations in dam behavior causedby repeated disturbance of the home cage. Ognibeneet al.,38 in a study on neonatal behavior in reeler(Relnrl/rl) mice, evaluated behavior in both pups anddams. The researchers reported profound deficits inthe number of ultrasonic vocalizations emitted bymale Relnrl/rl mice, in comparison to Relnþ /þ orRelnrl/þ mice. These phenotypic differences in ultra-sonic vocalizations, and less overt changes in earlyactivity levels, were dependent on length of theperiod of maternal separation before the test. Longperiods of maternal separation (5 h per day acrosspostnatal days 2–6) did not decrease rates of sniffing,licking or nursing pups by the dams, suggesting thatthe deficient vocalization in the male reeler pups wasdue to intrinsic changes in response to isolation, andnot to disrupted maternal behavior. The findings alsodemonstrated the importance of gender as a studyvariable, since changes in ultrasonic vocalization arenot seen in the female Relnrl/rl mice. The enhancedsusceptibility to effects of deficient Reln function inmale pups may reflect sex differences in rates ofautism, which has an overall male/female occurrenceratio of approximately 4:1.4
Mouse models of autismSS Moy and JJ Nadler
5
Molecular Psychiatry
-
Mouse models of genetic clinical disorders withautism symptomatology
The Fmr1-null mouse
In humans, mutations in the FMR1 gene, the under-lying abnormality in fragile X syndrome (FXS), areassociated with mental retardation, facial dysmor-phology, macroorchidism, seizures, and symptoms ofautism.48 The Fmrp (fragile X mental retardationprotein)-deficient mouse, a model for FXS, exhibitsmarked susceptibility to audiogenic seizures49–51 andevidences the enlarged testes, although not the facialdysmorphology, characteristic of the human dis-ease.51–54 Most studies report that the loss of Fmrpin mice does not lead to overt motor impairment,severe learning deficiencies, or a lack of socialapproach. Alterations in the behavioral phenotypeof the Fmr1-null mouse include increased levels ofsocial anxiety,55 reduced social interaction,56 hyper-activity,52,57–60 and deficits in spatial learning on a
radial arm maze57 and reversal learning in the Morriswater maze task52,53,61 (see also Ref. 62).
Deficient FMR1 function in humans can havedevastating consequences for normal behavior. There-fore, the relative mildness of the behavioral pheno-type in Fmr1-null mice is problematic for the validityof the model. Some studies have found unchangedbehavioral responses in FXS-model mice in tests ofanxiety and activity,63,64 fear conditioning,58,62,64,65
spatial learning,54,58 and aggression.57 Dobkin et al.65
have suggested that differences in behavioral pheno-type may be attributed, in part, to the effect ofdifferent genetic backgrounds of the mouse strainsused for the Fmr1-null mice, with fewer effectsobserved in mice on a C57BL/6 background andgreater effects in mice with a mixed backgroundincluding the 129 strain (see also61). A systematicevaluation of the mutation on two inbred back-grounds (C57BL/6J and FVB/NJ), including F1 hybridmice carrying the X-linked Fmr1-null allele, also
Table 1 Ultrasonic vocalizations in genetic mouse models relevant to autism and mental retardation
Human locus Mouse genotype Change in total USVs (postnatal day)in comparison to wild-type mice
DVL140 (Dishevelled1) Dvl1�/� — (PD6–8)
FOXP239 (Forkhead box) Foxp2�/� k (PD6)Foxp2þ /� k (PD6)
Mouse model of Rett syndromeMECP236 (methyl-CpG-bindingprotein-2)
MeCP21lox/Y male (null) m (PD5) — (PD3,4,6,7)MeCP21lox/þ female (heterozygous) m (PD7) — (PD3–6)
OXT46 (oxytocin neuropeptide) Oxt�/� k (PD7–8)a
OXTR41 (oxytocin neuropeptidereceptor)
Oxtr�/� males k (PD7)
RELN38 (Reelin) Relnrl/rl males (reeler) k (PD7)b
Relnrl/rl females (reeler) — (PD7)Relnrl/þ males (heterozygous) — (PD7)Relnrl/þ females (heterozygous) — (PD7)
5-HT1A43 Htr1a�/� k (PD7–8)c
(serotonin receptor subtype HTR1A)
5-HT1B42,44 (serotonin receptor
subtype HTR1B)Htr1b�/� k (PD6–9,12,15)
Mouse model of Down syndromeTs2145 (Trisomy of humanchromosome 21)
Ts65Dn (Trisomy of analogous regionon mouse chromosome 16)
Maturational delay (PD3–13)d
Abbreviation: USVs, ultrasonic vocalizations.—, no change in USV number; k, decrease; m, increase in USV number.aDecreases observed in obligate (single genotype) and non-obligate litters.bDecreases not observed following repeated 5-h maternal separation.cDecreases observed in obligate and non-obligate litters.dDecreased or increased numbers of USVs may have occurred on some postnatal days, comparison with diploid littermates.
Mouse models of autismSS Moy and JJ Nadler
6
Molecular Psychiatry
-
found a generally mild behavioral phenotype, in-creased seizure susceptibility, and macroorchidism.51
The same study provided evidence that the Fmr1-mutant allele is a hypomorph, suggesting the possi-bility of remaining residual function.51 This wouldnot explain behavioral changes in the Fmr1-null micethat are opposite to those associated with FXS andautism. For example, Frankland et al.66 reported thathuman subjects with FXS showed marked deficits inprepulse inhibition of acoustic startle responses.Similar impairments in sensorimotor gating have alsobeen reported in adults with autism67 or Aspergersyndrome.68 Parallel studies conducted by Franklandet al.66 confirmed that the Fmr1-null mice haveenhanced prepulse inhibition of acoustic startleresponses, in line with previous findings.49,63 Inaddition, the Fmr1-null mice also had enhancedlearning in complex operant conditioning tasks.66,69
These divergent findings underscore the premise thatmouse models may not reflect all components of ahuman clinical syndrome; however, recapitulatingendophenotypes can allow exploration of neuro-pathology underlying specific behavioral alterationsin the disease symptomatology.70
The Mecp2-mutant mouseRett syndrome is characterized by normal develop-ment in the first months of life, followed by regressionof social, language, and cognitive function, and theemergence of unusual, stereotyped hand movements,gait and other motoric impairment, and decrements inbrain growth.71 Similar to FXS, Rett is an X-linkeddisorder, but, unlike FXS and autism, Rett syndromeis observed primarily in girls. Most cases areattributed to mutations in a single gene, MECP2.72–74
The Mecp2-null mouse, a genetic model for thedisease, shows overtly normal development for aboutthe first month of life, followed by increasingly severeneurological abnormalities, and death by approxi-mately 10 weeks of life.75,76 The mutant behavioralphenotype includes hypoactivity, body trembling,gait ataxia, and limb clasping. Picker et al.36 utilizeda neonatal screen of tests for sensorimotor develop-ment and ultrasonic vocalizations in the Rett syn-drome-model mice . Male pups carrying the X-linkednull allele and heterozygous female pups werenormal for many somatic and somatosensory mea-sures, but had delays in a few behavioral reflexes, andalso emitted higher numbers of ultrasonic calls onsome postnatal days (see also77). A related allele thatmimics one found in Rett patients, Mecp2308, pro-duces a truncated protein and confers lower pene-trance of the lethality phenotype.78 Mecp2308/y malesdemonstrate overtly normal early development, butby 6 weeks of age, the mice begin to exhibitprogressive tremor, hypoactivity, seizure-like re-sponses, and stereotyped forelimb movements remi-niscent of the repetitive hand wringing observed inchildren with Rett syndrome.78 By around 8 weeks ofage, male mutant mice demonstrate significant signsof motor impairment in a wire suspension task.78,79
Moretti et al.79 conducted a systematic set ofexperiments to investigate social behavior in 10-week-old Mecp2308/y mice. While no differences wereobserved in the resident-intruder challenge (seealso78), the mutant mice demonstrated significantsocial approach deficits in a partition test, in whicha wild-type conspecific was located behind a clear,perforated barrier. Both the Mecp2308/y mice and thecontrols preferred to investigate a novel conspecificversus a more familiar mouse; however, lower levelsof social investigation were evident in the Rettsyndrome model animals for both familiar andunfamiliar conspecifics. These results indicated thatdeficient Mecp2 function did not prevent socialrecognition, but did lead to reduced social approach.In a standard cage setting, Mecp2308/y mice exhibiteddeficits in social investigation of a juvenile malemouse, but not in the investigation of a novel object.79
The mutant mice have also been characterized bydeficits in long-term social memory, observed follow-ing the repeated presentation of a juvenile mouseacross several days.80,81
Other behavioral abnormalities in Mecp2308/y miceinclude deficits in nest building and other home cageactivity, alterations in diurnal motor patterns,79 andimpaired learning and memory.81 An abnormal phe-notype is still observed when the loss of Mecp2 islimited to forebrain areas and to postnatal develop-ment.75,80 In particular, the conditional Mecp2-nullmice still show forelimb and hindlimb clasping,motor impairment and ataxic gait, and decreasedsocial preference.75,80 However, some behavioralalterations emerge at a later time point than observedwith prenatal loss of Mecp2 function,75 or, in the caseof general hypoactivity or reduced context-dependentfear conditioning, are not observed.80 These studiesdemonstrate that embryonic loss of Mecp2 is notnecessary to induce behavioral changes, which isrelevant to the normal early development seen in theclinical disorder.
Rett syndrome-model mice also have alterations inbehavior and neurophysiology linked to stress re-sponses, such as increased anxiety-like behavior,80,82
higher levels of corticosterone release followingrestraint, enhanced expression of corticotropin-re-leasing hormone82 and increased expression of genesregulated by glucocorticoids.83 These findings havesuggested that dysregulation of the hypothalamic–pituitary–adrenal axis during development plays asignificant role in symptoms of Rett syndrome.82
Mouse models for chromosome 15q11–13 disordersAlterations in the chromosomal region 15q11–13 havebeen associated with autism, and with two otherneurodevelopmental disorders: Angelman syndrome(AS) and Prader–Willi syndrome (PWS).84–86 Thesedisorders are linked to a similar chromosomal region,but differ in phenotype based on genomic imprinting.Genes are inherited in two copies, one paternally andthe other maternally; imprinted genes show expres-sion from only one of these copies rather than both.
Mouse models of autismSS Moy and JJ Nadler
7
Molecular Psychiatry
-
Often, if the copy of the gene that should be expressedis deleted, the second copy cannot compensate,producing an effective loss of function.
Diagnostic indicators for AS include some symp-toms that overlap with the autism clinical phenotype,such as profound language deficits, hand flappingmovements, seizures, and mental retardation, andother characteristics not associated with autism, suchas motor ataxia, microcephaly, and a happy, sociabledisposition. The disorder has been linked to maternaldeficiency of an imprinted region on 15q11–13, andspecifically to loss of UBE3A function. Reducedexpression of UBE3A in cerebral tissue has beenreported for autism and Rett syndrome, as well as AS,albeit in a small number of samples.87 Mice withmaternal deficiency of Ube3a (m�/pþ ) have deficitsin motor coordination and context-dependent fearconditioning,88–90 reduced spatial learning in theMorris water maze task,88,90 and enhanced seizuresusceptibility.88,89 More severe symptoms in AS mayinvolve the contribution of other genes in the 15q11–13 region, including GABRB3, which encodes the b3-subunit of the g-aminobutyric acid (GABA) type Areceptor. Deletion of Gabrb3 in mice leads to highrates of neonatal mortality.91 Surviving offspringevidence many markers for neuropathology, includ-ing enhanced seizure susceptibility, abnormal motorcoordination, hyperactivity and stereotyped circlingbehavior, and impaired learning and memory.91,92
PWS, arising from the paternal deficiency of animprinted region on 15q11–13 different from thatassociated with AS, is characterized by mentalretardation, early-onset obesity and autistic-like re-petitive behavior, including compulsions, rituals, andresistance to environmental change.93 Unfortunately,mouse models for the multigene deletion associatedwith PWS have shown early postnatal lethality.94,95
Other mouse lines have been developed with morespecific targeted disruptions, focusing on the expres-sion of a single gene from the imprinted region,Necdin (Ndn). In one study, the majority of mice withpaternal inheritance of a Ndn null allele died, mostlikely of respiratory depression, within hours ofbirth.96 The rate of mortality was much greater inthe male mice (95%) than in female mice (40%) withthe paternally deleted Ndn allele, and was dependenton background strain of the wild-type dams. Musca-telli et al.97 constructed a similar mouse line withNdn disruption, but with only partial lethality in theearly postnatal period. These mice showed an alteredbehavioral phenotype, with higher levels of sponta-neous ‘skin scraping’ in an open field and enhancedlearning in the Morris water maze task, as well asreduced levels of oxytocin-expressing neurons in thehypothalamus. The investigators noted that thesechanges might reflect characteristics of PWS, includ-ing repetitive ‘skin-picking’ responses, intact or evenadvanced skills in visual–spatial tasks and jigsawpuzzles, and deficiencies in oxytocin-expressingneurons. The alterations in the mouse model mayalso be relevant to autism. Repetitive self-injury23,98
and decreased levels of oxytocin in blood plasma99
have been observed in autistic children. In addition,patients with autism spectrum disorders can showhigh levels of performance for some visual–spatialtasks.100 Overall, the findings suggest that the genesaltered in PWS may be relevant to specific changes inautism.84
Synaptic dysregulation in genetic mouse models forautism
Although the mouse model for FXS does not fullyrecapitulate the behavioral phenotype of the clinicaldisease, there are very interesting symmetries be-tween findings of abnormal dendritic spine morphol-ogy, including alterations in length and density, inbrain of human patients and in Fmr1-null mice.101,102
Further work has shown that Fmrp loss has markedeffects on measures of synaptic plasticity in mutantmice, with significant enhancement of mGluR(group 1 metabotropic glutamate receptor) dependentlong-term depression (LTD) in hippocampus,103–105
and decreased cortical long-term potentiation(LTP).106,107 The molecular mechanisms underlyingthese changes have yet to be elucidated, but arethought to be related to the role of FMRP in mRNAtransport and translation.108–111 In particular, the‘mGluR theory’ proposes that activation of group 1mGlu receptors during long-term depression is asso-ciated with protein synthesis, followed by FMRP-mediated repression of mRNA translation.112,113
Under these circumstances, loss of FMRP would leadto prolonged mGluR signaling, with fundamentalalterations of experience-dependent synaptic devel-opment and function.
Mecp2-null mice have also been found to have age-dependent abnormal synaptic plasticity in hippocam-pus.114 Similarly, impaired synaptic plasticity hasbeen reported for hippocampus, and motor andsensory cortex, in Mecp2308/y mice.81 MECP2 has beenlinked to both DNA methylation and histone deace-tylation,115 two processes which regulate transcrip-tion in brain. Protein levels can also be regulated bydegradation. UBE3A encodes an ubiquitin ligase, E6-AP, which may facilitate the degradation of proteinsrelated to synaptic function through the ubiquitina-tion process. Overt deficiencies in LTP are found inUbe3A (m�/pþ ) mice.88,89,116 Overall, loss of func-tion of FMRP, MECP2, or UBE3A could lead todysregulation of protein synthesis or degradation atthe synapse, and subsequent changes in neurotrans-mission, including changes in cortical excitability.Alteration in the balance of excitation and inhibitionin brain has been proposed as a fundamentalmechanism underlying autism,117–119 and may involveenhanced glutamatergic signaling and/or a decreasein GABA-mediated neurotransmission. As previouslynoted, AS is associated with anomalies in the regionof chromosome 15q11–13, which includes GABRB3and a cluster of other GABA-related genes. Samacoet al.87 have shown that the expression of GABRB3 in
Mouse models of autismSS Moy and JJ Nadler
8
Molecular Psychiatry
-
brain is deficient in subjects with AS, autism or Rettsyndrome, but not in subjects with Down syndrome.Reduced expression of specific GABA(A) receptorsubunits120–122 and aberrant GABAergic neural circui-try123 have been reported in the FXS model mouse. Inaddition, disruption of Necdin, implicated in PWS,leads to reduction in forebrain GABAergic neuronaldevelopment.124 These findings support the view thatcompromised GABAergic function may be linked toautism and related syndromes. However, one caveatto the hypothesis of an excitatory/inhibitory imbal-ance in brain is that the direction of the alteration maynot be an overall increase in cortical excitation. Forexample, one study found decreased spontaneousfiring in pyramidal neurons of Mecp2-null mice. Theauthors attributed this change to a shift towardreduced cortical excitability and an enhanced inhibi-tory drive in the model of Rett syndrome, rather thanto any intrinsic anomaly in the neurons them-selves.125
Caþ 2/calmodulin-dependent protein kinase II(CaMKII) is an important mediator of LTP126–130 andother forms of synaptic plasticity.131 Stimuli thatinduce LTP also persistently activate CaMKII, whichmay be critical for the molecular memory of synapticevents.132 There is evidence that CaMKII is regulatedby Fmrp, since wild-type mice, but not Fmr1-nullmice, demonstrate activity-dependent increases inlevels of hippocampal aCaMKII, the a-subunit ofCaMKII, following mGluR-LTD.103 Induction of aCaM-KII protein synthesis following N-methyl-d-aspartate(NMDA)/glutamate stimulation is also absent insynaptoneurosome preparations from FXS-modelmice, in comparison to wild-type controls.133 Thedysregulation of CaMKII translation could have directeffects on the maintenance of synaptic memory, andmight also alter function in other genes relevant tohuman disorders with autism symptomatology. Onestudy demonstrated that Mecp2 regulation of dendri-tic patterning and other components of neuronalconnectivity is dependent on phosphorylation in-duced by neuronal activation.134 The application of aselective CaMKII inhibitor, but not inhibitors of otherkinases (CaMKK, protein kinase A, protein kinase C,mitogen-activated protein kinase, cyclin-dependentkinase 5 (CDK5), or phosphatidylinositol 3-kinase(PI3K)), blocked the stimulation-dependent phos-phorylation of Mecp2, suggesting that CaMKII mod-ulates the state of Mecp2 activation during synaptictransmission.134 In addition to altered translation,dysregulation of CaMKII could occur through pro-cesses involved in autophosphorylation. Weeber etal.116 have reported that Ube3A (m�/pþ ) mice havehigher levels of aCaMKII phosphorylation at a siteinhibitory for enzymatic activity. Adding a mutationto prevent the inhibitory phosphorylation of CaMKIIallows the rescue of LTP deficits in the AS mousemodel.88 In addition, Ube3A (m�/pþ ) mice withattenuated CaMKII inhibitory phosphorylation alsodemonstrate a startling reversal of the aberrantbehavioral phenotype with normalized rotarod per-
formance, spatial learning in a Morris water mazetask, and context-dependent fear conditioning, aswell as a markedly reduced susceptibility for sei-zures.88 These findings emphasize that dysregulationof proteins important in synaptic plasticity may befundamental to the clinical phenotype of humandisorders, and that reversal of altered synapticfunction may have therapeutic benefits.
Autism candidate genes and synaptic function
Given the complex, multigenic etiology proposed forautism, it is possible that small or moderate perturba-tions in sets of genes related to synapse formation andplasticity might, through an accumulation of detri-mental effects, result in global brain deficiencies (forexample, Ref. 135). In some idiopathic cases, thedisease phenotype might be driven by mild epigeneticabnormalities involving imprinted regions of 15q11–13, in combination with one or more loci conferringsusceptibility to core symptoms.84,136 Candidate genesderived from association or familial linkage studiesinclude multiple genes relevant to synaptic genesisand function, including GABRB3, GLRB (glycinereceptor, b), several genes encoding glutamatereceptors, and NLGN3 and NLGN4 (neuroligin 3and 4).11,137,138 The neuroligin family, in particular,has been found to have an important role in excitatoryand inhibitory synaptic contacts.139,140 Neuroligins,located in the postsynaptic region, function as trans-synaptic cell adhesion molecules, connecting withpresynaptic b-neurexin or, in some cases, a-neurexinpartners. Five neuroligin genes have been identifiedin humans, and three (Nlgn1, 2 and 3) in rodents.Aberrations of chromosomal regions containingNLGN1 and NLGN2, and a point mutation in NLGN3,have been linked to autism or Asperger syndrome(reviewed in Lise and El-Husseini140).141 A recentpopulation study found a hemizygous microdeletionwithin coding regions of NRXN1 (neurexin 1) in twosisters diagnosed with autism spectrum disorder.142
Thus, both neuroligin genes and neurexin 1, encodingthe binding partner, have been implicated in autism.Targeted disruption lines have been generated forNlgn1, 2 and 3.143 The single and double-null miceproved to be viable, but triple deletion resulted inperinatal death. Examination of Nlgn mutant linesindicated changes in some measures of synapticfunction, but no overall reduction of synapse num-bers, suggesting that Nlgn1, 2 and 3 were critical fornormal synaptic maturation, but not synaptogen-esis.143 Functional characterization of the single ordouble Nlgn null lines will be of great interest to thebehavioral genetics field. Similar to the studies withNlgn, triple-null mutations of a-neurexin (Nrxn) 1, 2and 3 resulted in mortality for newborn pups,while the majority of Nrxn double-null mice died inthe first week of life.144 Single Nrxn1, 2 or 3 nullmice were viable, but had respiratory impairment.Further work with a neurexin-binding partner, neur-exophilin 3 (Nxph3), showed that Nxph3-null mice
Mouse models of autismSS Moy and JJ Nadler
9
Molecular Psychiatry
-
had significant behavioral changes, including en-hanced startle responses, deficits in prepulse inhibi-tion and impaired motor coordination on a rotarodtask.145 The Nxph3 null animals did not have deficitsin either acquisition or reversal in the Morris watermaze task, and had significantly higher swim speedsthan wild-type mice. In the case of genetic mousemodels with milder phenotypes, it is possible thatcombining the targeted disruption of, for example,Fmr1 with the disruption of Nxph3, might reveal aphenotype with more widespread alterations insynaptic function, and possible recapitulation of coresymptoms in autism.
Genetic studies in human populations have sug-gested that the RELN gene may be associated withautism susceptibility,146–149 although not all findingshave been positive.150–152 Fatemi et al.153,154 haveshown that levels of RELN mRNA and Reelin proteinare significantly deficient in the brain of autisticsubjects. RELN plays multiple roles in brain, includ-ing cell guidance during embryonic development,and mediation of neurotransmission and synapticplasticity in adulthood.9,155 In adult mice, Reelinprotein is synthesized and secreted from GABAergicinterneurons in cortex and hippocampus, with extra-cellular localization to dendrites and dendriticspines.156 Loss of Reln function in mice leads to overtmotor impairment, increased anxiety, learning defi-cits and abnormal neuroanatomy,157–160 as well asaberrant striatal LTP.159 As described previously,reeler mouse pups have markedly lower rates ofultrasonic vocalization, dependent on gender andhistory of maternal separation.38 Examination ofhippocampal neurons in reeler embryos has shownreduced glutamatergic synapse formation, whichcould be reversed by the administration of reelin tothe cell culture.161 Mice with null or mutant alleles forthe receptors mediating reelin signaling, the very low-density lipoprotein (VLDL) receptor and apolipopro-tein E receptor 2 (apoER2), have deficits in learning,deficient hippocampal LTP and altered neuronalmigration during brain development.162,163
Relnrl/þ mice, which retain approximately 50% ofnormal Reln expression, do not show the reeling gaitcharacteristic of the Relnrl/rl animals. There arevariable reports of an abnormal behavioral phenotypein Relnrl/þ mice, including findings of selectiveimpairments in reversal learning,164 increased anxi-ety-like behavior, decreases in prepulse inhibition ofacoustic startle responses,165 deficits in odor discri-mination166 and impaired contextual fear condition-ing.167 However, other researchers have found normalreversal learning, contextual fear conditioning andworking memory,168 and unchanged sensorimotorgating, social responses and other indexes of cogni-tive function157,169 in heterozygous mice.
The RELN gene has also been implicated in otherneuropsychiatric syndromes, including schizophre-nia and obsessive-compulsive disorder. Studies inpost-mortem brain from schizophrenia subjects haveshown reductions in both Reelin protein and glutamic
acid decarboxylase 67 (GAD67), an enzyme with a keyrole in the synthesis of GABA. Marrone et al.159 haveprovided evidence that deficient GABAergic neuro-transmission in reeler mutant mice might underlie theaberrant induction of LTP observed in reeler striatalsynapses. Relnrl/þ mice have reduced GAD67-positiveneurons, decreased density of dendritic spines170 andseveral abnormalities in synaptic function, includingimpaired long-term depression and LTP.167 Thus, evenpartial loss of Reelin can lead to significant alterationsin synaptic plasticity. In addition, Relnrl/þ mice havedecreases in the numbers of oxytocin receptors inseveral brain areas,171 which may reflect reducedoxytocin levels in blood samples from autisticchildren.99,172
Mouse models for altered serotonergicneurotransmission
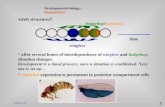
Many lines of evidence implicate serotonin signalingin the etiology of autism. One of the most consistentphysiological findings among patients with autism ishyperserotonemia (for example, Ref. 173). Numerousgenes involved in 5-HT (serotonin) signaling havebeen identified in genome scans of autistic popula-tions, including the serotonin transporter (SERT orSLC6A4), monoamine oxidase A (Maoa), which isinvolved in catabolism of 5-HT, and two serotoninreceptors: 5-HT2A (HTR2A) and 5-HT7 (HTR7).
11,137,138
An example of a 5-HT signaling pathway is shown inFigure 1. At least 15 genes have been cloned inmammalian brain that encode 5-HT receptors, withmost of the receptors classified as metabotropicG-protein-coupled receptors, signaling through thesecond messengers adenylate cyclase and cAMP. Twoexceptions are the 5-HT2 (HTR2) family, G-protein-coupled receptors that signal through phospholipaseC, and the 5-HT3 (HTR3) family, whichare ionotropic (ligand-gated channel) receptors.174,175
5-HT signaling is involved with multiple neurodeve-lopmental processes, including neurogenesis,migration, differentiation, axon branching, dendrito-genesis, synaptogenesis, plasticity, and cell survival.Alterations of 5-HT signaling reveal that the neuro-developmental functions of this pathway havepotential roles in etiology of autism-relevant behaviorand pathology.
One model for disruption of this pathway is thedepletion of serotonergic neurons in the mouse byinjection of the specific neurotoxin 5,7-dihydroxy-tryptamine into the bilateral medial forebrain bundleat birth. This lesion results in decreased densityof 5-HT containing fibers in cortical regions and thehippocampus, persisting through 2 months of age.Lesioned mice also exhibit widening of specificcortical regions, perhaps similar to increased corticalvolume in autistic children.176 As adults, micewith neonatal loss of serotonergic neurons demon-strate impaired social learning, increased repetitivedigging and grooming behaviors, and increased socialaggression.177
Mouse models of autismSS Moy and JJ Nadler
10
Molecular Psychiatry
-
The consequences of other alterations in 5-HT sig-naling can be seen in the phenotypes of mouse linesbearing targeted disruptions in the pathway.42–44,178,179
Many of the genes involved in 5-HT signaling havebeen mutated in the mouse, including receptors,metabolic and catabolic enzymes, SERT, and others.Most of these cause behavioral changes related toanxiety, depression, and aggression. Some also causechanges in spatial learning and memory, response toreward, hyperphagia, and seizure incidence. Oneinteresting mutant line has a deletion of 5-HT1A(Htr1a). Htr1a-null mice demonstrate increased anxi-ety-like behavior, which can be reversed by chronictreatment with the tricyclic antidepressants imipra-
mine and desipramine, but not the 5-HT reuptakeinhibitor fluoxetine.180 5-HT1A is mainly expressed inthe hippocampus and raphe nucleus during embryo-nic development.181 Depletion of 5-HT during theearly postnatal period leads to the reduction ofnumber and length of dendritic spines in thehippocampus in an 5-HT1A-dependent fashion.
182
This receptor is also responsible for hippocampalneurogenesis and dendritic maturation in the adult.Conditional expression of 5-HT1A in hippocampaland cortical regions is sufficient to normalize anxiety-like behavior in mutant mice.178 Interestingly, theconditional loss of forebrain 5-HT1A function does notinduce the anxiety-like phenotype when the deletion
PTEN PIP2
PI3K
BDNF
TrkB
GRB2CRK
Myoinositol
VPA
RasRafMEK
VPA
nucleus
cell membrane
AKTSGK
SGK
TSC1
TSC2RHEB
AKT FOXO1
FOXO3A
Transcription
TORProtein
Synthesis
5-HT
5-HTR
cAMP
AC
PKA/CAMK
TRP53
FOXO4
PP2A
PDK1
PIP3
ERK
CREB
ERK
Figure 1 Interaction between several pathways implicated in the etiology of autism. 5-HT (serotonin) signaling (shown ingreen) is mediated by several different seven-pass transmembrane receptors (HTR). Most 5-HT receptors are coupled to Gproteins (GPCRs) and signal through second messengers AC (adenylate cyclase) and cAMP (cyclic AMP).174,175 Receptortyrosine kinase signaling, typified by BDNF (brain-derived neurotrophic factor), is shown in yellow. BDNF or other ligandbinds a homo- or heterodimer of the receptor (for example, TrkB) that, in turn, phosphorylates proteins downstream, such asGRB2 (growth factor receptor-bound protein). This initiates a cascade of phosphorylation through Ras/Raf/Mek and ERK(extracellular signal-regulated kinase), which finally translocates to the nucleus and effects transcription. Signaling viaphosphatidylinositol 3-kinase (PI3K) of PTEN (phosphatase and tensin homolog on chromosome ten)215 is shown in blue.PI3K is activated by a multitude of mechanisms, including stimulation by growth factors through receptor tyrosine kinases,such as TrkB. PI3K phosphorylates PIP2 (phosphatidylinositol 4,5-biphosphate) to produce PIP3 (phosphatidylinositoltriphosphate). The presence of PIP3 recruits PDK1 (phosphatidylinositol-dependent kinase 1) to the membrane, which thenphosphorylates AKT (also known as protein kinase B). AKT signals through the tuberous sclerosis complex, TSC1 and TSC2(tuberin and hamartin, respectively), RHEB (Ras-homolog enriched in brain) and TOR (target of rapamycin). TOR increasesprotein synthesis by relieving inhibition of eukaryotic initiation factor 4 E and activation of ribosomal S6 kinase. AKT alsotranslocates to the nucleus and inhibits the FOXO (forkhead transcription factors), which transcribe mediators of apoptosisand cell-cycle arrest. Furthermore, AKT phosphorylates other targets, such as CREB (cAMP-responsive element bindingprotein), which are downstream of TrkB, and GSK-3b, a member of the WNT signaling pathway (not shown in Figure). PTENinhibits the PI3K pathway by converting PIP3 to PIP2. Signaling pathways can be affected by exposure to valproic acid (VPA).Pathways have been greatly simplified for the purposes of this review; all interactions are not illustrated. Protein kinaseA/CAMK, protein kinase A/calcium/calmodulin-dependent protein kinase; CRK, v-crk sarcoma virus CT10 oncogenehomolog; Ras, Harvey rat sarcoma virus oncogene; Raf, v-raf-leukemia viral oncogene; MEK, MAPK/ERK kinase; SGK, serum/glucocorticoid regulated kinase; PP2A, protein phosphatase 2A; TRP53, transformation-related protein 53. MAPK, mitogen-activated protein kinase.
Mouse models of autismSS Moy and JJ Nadler
11
Molecular Psychiatry
-
occurs in adulthood.178 Furthermore, adult mice withloss of 5-HT1A function do not have enhancedanxiety-like behavior if 5-HT1A expression occurredearlier in life, between postnatal day 5 and 80.178 Thisis an elegant example of how normal adult behaviormay be dependent on signaling events early indevelopment.
Mice with disruption of Maoa, the enzyme thatmetabolizes 5-HT during postnatal development,have a ninefold increase in brain 5-HT, as well asaberrant aggressive behavior.183 Increased 5-HT causesdisorganization in the somatosensory and visualcortices due to disruption in the clustering andsegregation of thalamocortical fibers.184,185 Similarly,mice with a targeted disruption of Sert also have anexcess of 5-HT in the extracellular space, leading todisruption of somatosensory cortex formation, reduc-tion of apoptosis in the telencephalon186,187 andvariable alterations in cortical layer thickness andneuronal cell density, dependent on backgroundstrain.188 Sert-null mice also show an altered beha-vioral phenotype, including hypolocomotion, markedreductions in exploration, and reduced social inter-action.189,190 As with the altered parameters of braingrowth,188 observation of behavioral changes in Sert-null mice may be dependent on background strain.191
Serotonergic signaling and brain-derived neurotrophicfactorIt is possible that perturbations of serotonergicsignaling, in combination with one or more othergenetic anomalies relevant to the etiology of autism,lead to the neuropathological symptoms of thedisease. Brain-derived neurotrophic factor (BDNF)has been identified as a candidate gene for autismsusceptibility.11,138 BDNF plays a critical role duringneurodevelopment, with effects on dendritic growthand spine maturation, synaptogenesis, and neuronalplasticity.192–194 Work with mouse lines characterizedby deficient Bdnf has shown that these trophic effectsare important for normal serotonergic neurotransmis-sion.195–200 In addition, several lines of evidence haveprovided support for a role of Mecp2 in the regulationof activity-dependent transcription of Bdnf,134,201,202
suggesting that alterations in 5-HT signaling couldarise from deficient Mecp2 function, through dysre-gulation of Bdnf. Bdnf-null mice die soon after birth,but mutants have been developed with conditionaldisruption of Bdnf, limited to either the prenatal orpostnatal period.196 The conditional null mice de-monstrate significant behavioral changes, includingmarked hyperactivity and enhanced aggression, aswell as selective deficits in serotonergic neurotrans-mission.196,203 One study has reported that thebehavioral effects of Bdnf disruption in forebrain,whether in late embryogenesis or during the postnatalperiod, are dependent on gender, with only maleconditional null mice showing increases in activity,and only female mutants exhibiting depression-likeresponses, measured as greater immobility in a forcedswim test.204
Monteggia et al.205 used an inducible Bdnf-nullmutation to compare forebrain-specific neurotrophinloss during development and in adulthood. Theembryonic disruption of Bdnf led to hyperactivityand more extensive impairment in fear conditioning,in comparison to disruption in adulthood. PrenatalBdnf loss also caused a significant diminution of 5-HT1A receptor function.
198 In contrast to the signifi-cant effects on behavior and serotonin signaling,neither embryonic nor adult knockout of Bdnf altereddendritic arborization206 or changed expression ofGAD67, a marker for GABAergic function,207 incortical areas. Interestingly, heterozygous mice withone null Bdnf allele have decreased function of theserotonin transporter in hippocampus.195,197 Murphyet al.,208–210 in a series of informative studies on geneinteractions, showed that deficits in Sert nulls wereexacerbated when mice were bred with Bdnf hetero-zygous animals. Male offspring had much greatersusceptibility for the more severe phenotype thanfemale mice.210 Together, these findings of geneticsynergy suggest that relatively subtle changes inMECP2 and BDNF function, when combined withalterations in one or more genes in the 5-HT signalingpathway, could accumulate, with detrimental con-sequences for normal neurodevelopment.
The conditional Pten-null mouse
Human genetic studies have found polymorphisms inthe phosphatase and tensin homolog on chromosometen (PTEN) locus associated with macrocephaly andautistic behaviors.211 These genetic anomalies areusually in association with tumor syndromes suchas Cowden’s and tuberous sclerosis. Mutations inTSC1 and TSC2, which are downstream in the PTENsignaling pathway (Figure 1), cause tuberous sclero-sis, a syndrome associated with greatly increasedincidence of autism.212 A recently reported geneticmouse model for autism is based on disruption ofPten in post-mitotic neurons of the cerebral cortexand dentate gyrus.213 Germline mutation of Ptenresults in embryonic lethality at embryonic day 9.5due to defective chorioallantoic development; em-bryos also have expanded and poorly patternedcephalic and caudal regions.214 However, early mor-tality is not observed with neuron-specific ablation ofPten using Nse-cre.213 These animals exhibit lowsocial approach, increased activity in a novel envir-onment and impaired sensorimotor gating. Brainsfrom conditional Pten-null mice show progressivemacrocephaly, which may reflect the larger headcircumference reported in autistic children.176 On aneuronal level, there is dendritic hypertrophy, ectopicdendrites and increased spine density.213
PTEN acts as a phosphatase on phosphatidylinosi-tol triphosphate to antagonize signaling through thePI3K pathway.215 This pathway (in blue; Figure 1) hasan important role in protein synthesis, as well as inthe regulation of cell size and proliferation. There aremultiple lines of evidence implicating the PI3K
Mouse models of autismSS Moy and JJ Nadler
12
Molecular Psychiatry
-
pathway in brain development and function. Micecarrying homozygous null mutations of the Akt3locus show a 20–25% reduction in brain size as aresult of fewer, smaller cells.216 The brains of theseanimals also have smaller ventricles and thinnerwhite matter tracts connecting the corpus callo-sum.217 In mouse models of tuberous sclerosis, theconditional loss of Tsc1 leads to abnormal dendriticspine morphology and density,218 enhanced corticalexcitability,219 enlarged neurons in the cortex andhippocampus, and seizures.220 The altered excitationstate of cortex in the Tsc1 conditional null mouse isnot associated with tuber formation or changes indistribution of GAD67, used as a marker for GABAer-gic function.219 The PI3K pathway is antagonizedby PTEN, but can be stimulated by glutamate,221
serotonin222 and dopamine.223 BDNF can alsostimulate the pathway through TrkB, and inducesactivation of protein synthesis in neuronal den-drites.224 The pathway is also activated by sodiumvalproate,225 an environmental risk factor for autism(see Figure 1).226–228
Given this body of evidence, genes in the PI3Kpathway are good candidates for further studies inmouse models of autism. This pathway is intimatelyintertwined with many other pathways utilizing lociindicated in autism susceptibility. The interactionsmay be relevant to a model of autism as a disorder ofsmall perturbations of many loci that interact witheach other to ultimately produce a behavioral pheno-type.
Mouse models of environmental contributions toautism etiology
Early onset for neuropathology in autismWhile the clinical syndrome is typically diagnosed bythe age of 3 years,3 retrospective studies of autisticchildren have demonstrated that abnormalities insocial interaction229 and blood biochemistry230,231 canbe detected in the first days or months of life. Theearly emergence of symptoms suggests that theunderlying disturbance in brain development occursduring embryogenesis.232 In line with this premise,clinical surveys have linked complications duringpregnancy, including viral infections and maternalstress, to a higher incidence of autism.233,234 Otherresearchers have noted that autistic children havehigher rates of physical malformations and facialdysmorphology, indicative of prenatal pathology.235–237
These physical manifestations are predictive of great-er symptom severity and neuroanatomical abnormal-ities.235,238,239 Exposure to teratogens during gestationhas also been shown to be a risk factor for autism,with higher disease incidence associated with mater-nal use of valproic acid,226–228,240 thalidomide241 andmisoprostol.242
The detrimental neurodevelopmental effects ofmaternal infection have been attributed, in part, tothe induction of inflammatory cytokines.243 Mousemodels for prenatal exposure to maternal infection or
inflammation have shown that the challenged off-spring demonstrate an altered behavioral phenotype,including deficiencies in social interaction, explora-tion and sensorimotor gating.244–246 Following mater-nal challenge with influenza virus, offspring alsodemonstrate altered gene expression in brain, includ-ing genes related to transcription and neurotransmis-sion.247 Meyer et al.248 have shown that prenatalexposure to the viral mimic PolyI:C (polyriboinosi-nic–polyribocytidilic acid) can result in reversallearning deficits, dependent on prenatal day ofadministration. In addition, the researchers reportedthat PolyI:C led to increased cytokine levels anddecreased numbers of Reelin-positive hippocampalcells. Treatment of mouse dams with lipopolysacchar-ide (LPS), which also elevates levels of proinflamma-tory cytokines, leads to increased expression ofNecdin (Ndn) in the offspring, with upregulationpersisting up to 12 h following LPS exposure.249 Aspreviously noted, NDN is located on the chromoso-mal region 15q11–13 associated with PWS. Thesefindings indicate that prenatal exposure to inflamma-tory agents in mice may provide a model for aberrantgene expression relevant to early abnormal develop-ment in autism.
Valproic acid-exposed rodent model for autismRodier et al.250,251 have reported that, in a rat modelfor teratogen exposure, the administration of valproicacid in early development induces morphologicalbrainstem pathology similar to changes sometimesobserved in autism. Alterations in the distribution ofserotonergic neurons in brain, suggestive of abnormalneuronal differentiation and migration, have alsobeen observed in the animal model.252 Further workin rats has shown that the prenatal challenge withvalproic acid induces behavioral changes, includingdelayed maturation, decreased social exploration,deficits in sensorimotor gating, and repetitive, stereo-typed responses in an open field.253 In mice, exposureto valproic acid while in utero leads to behavioralretardation and regression during neonatal andjuvenile development.35
One hypothesis for the mechanism of teratogenicaction for valproic acid is through effects on theexpression of Hox (homeobox) genes.232,254 Thesegenes encode transcription factors that are importantin regulating early development. Mice with disrup-tions of Hoxa1 have profound alterations in hindbrainorganization,255–258 which may reflect brainstem ab-normalities observed in autism.259,260 Evidence fromfamily studies in human populations has suggestedthat HOXA1 is associated with genetic susceptibilityfor autism,261,262 although results have been incon-sistent.263,264 A HOXA1-related disorder, Bosley–Salih–Alorainy syndrome, has been identified in a smallpatient sample, with symptoms that include delayedmaturation and autism.265 Gene expression profiles innormal and Hoxa1�/� embryonic stem cells haveshown that Hoxa1 regulates expression of Bdnf andother genes important for development.266 Stodgell
Mouse models of autismSS Moy and JJ Nadler
13
Molecular Psychiatry
-
et al.254 have shown that, in rats, prenatal exposure tovalproic acid leads to marked increases in embryonicHoxa1 expression, possibly through the inhibition ofhistone deacetylases. In line with this premise, arecent report linked the teratogenic effects of valproicacid to histone deacetylase inhibition.267
By this same mechanism, valproic acid may alsoproduce alterations in the WNT (Wingless-Int) signal-ing pathway,268 which plays multiple roles in cellmigration, proliferation, and survival, as well as indendritic morphogenesis and synapse formation.WNT2 has been identified as a candidate gene forautism susceptibility.11,269 Studies in rat and mouseTSC2 mutants have provided evidence that altera-tions in WNT signaling may be implicated in diseasepathology of tuberous sclerosis,270,271 although rele-vance to symptoms of autism has not been addressed.The targeted disruption of another member of theWNT signaling pathway, Dishevelled-1 (Dvl1), leadsto altered home cage behavior and social interactiondeficits,40,272 reduced dendritic branching273 andchanges in the formation of synapses.274 However,Dvl1-null mutants also demonstrate normal ultrasonicvocalization, spatial learning and hippocampal sy-naptic plasticity.40,272 The mutant mice have variablybeen characterized with impaired272 or normal40
prepulse inhibition.
Genes responsive to environmental factorsAs noted previously, autism risk factors include environ-mental challenges, such as maternal use of pharma-ceutical agents with neurotoxic effects,226–228,241,242
prenatal exposure to viral infections or maternalstress233,234 and, in addition, exposure to high levelsof environmental pollutants, including heavy me-tals.275,276 A recent review by Herbert et al.7 identi-fied 135 genes that have been shown to mediateresponses to environmental challenge, and that arelocated within autism linkage regions. The geneswere derived from several databases, including theNational Institute of Environmental Health Science(NIEHS) Environmental Genome Project, the Com-parative Toxicogenomics Database and the Programfor Genomic Applications SeattleSNPs Database (fo-cused on genes mediating inflammatory responses).
Several paraoxonase genes (PON1, PON2 andPON3) were included as genes located within autismlinkage regions, with a role in responses to environ-mental stimuli (human inflammatory responses).7 Asignificant association has been found betweenvariants of PON1 and autism in a population fromNorth America, but not in an Italian population.277
The authors suggest that this difference in linkage isbased on different levels of exposure to organopho-sphates in the environment, in combination withgenetic susceptibility mediated by variants of RELN(see also9). A recent study determined that rates ofautism spectrum disorders were higher in areas withgreater hazardous air pollutant concentrations, whichincluded the heavy metal mercury.276 Overall, thesefindings suggest that further work in animal models
for prenatal neurotoxin exposure, such as organopho-sphate pesticides or mercury, might provide informa-tion on the interaction between genetic predispositionand environmental challenge in autism.
Another interesting gene identified by Herbertet al.7 is Sonic hedgehog homolog (SHH), derivedfrom the database established by the NIEHS Environ-mental Genome Project. SHH is located at 7q36,within a chromosomal region linked to susceptibilityfor autism.278 Among multiple other roles, SHH isimportant for normal embryonic patterning, includingthe development of midbrain and hindbrain struc-tures.279,280 One study has suggested that the admin-istration of SHH can partially attenuate theneurotoxic effects of valproic acid on early serotoner-gic neuronal development.252 SHH may produceeffects through the PI3K pathway,281 which is alsoimportant for normal brain development.213 An ele-ment of this pathway, AKT2, is an environmentallyresponsive gene located in an autism linkage region.7
Disruption of SHH signaling has been implicated insyndromes of deficient cholesterol biosynthesis, suchas Smith–Lemli–Optiz syndrome (SLOS).282–284 Inter-estingly, SLOS is a neurodevelopmental disordercharacterized by high rates of autism.285,286 Thedisease is caused by mutations in DHCR7, leading toa disruption in cholesterol synthesis and an accumu-lation of precursor sterols.287 In mice, the loss ofDhcr7 function results in severe respiratory impair-ment, failure to feed, and death soon after birth.288–291
Prenatal Dhcr7-null mice evidence marked increasesin measures of serotonin immunoreactivity and amorphological expansion of the serotonergic sys-tem.292 A more viable mouse model for SLOS hasbeen created by generating compound heterozygousanimals, carrying a single null Dhcr7 allele and ahypomorphic Dhcr7T93M allele that reflects a humanmissense mutation.293 The combined Dhcr7 alleles inthis novel SLOS model appear to be embryonicallylethal for approximately 25% of the compoundheterozygotes. Mutant mice show increased ventri-cular size, syndactyly reminiscent of SLOS, and signsof intrinsic biochemical correction of the sterol deficitacross postnatal development. One study has re-ported that cholesterol levels are low in a subset ofautistic children,294 suggesting that genes related tocholesterol biosynthesis may contribute to suscept-ibility for the disease.
ENGRAILED 2, FOXP2, MET, HGFApproximately 10% of the environmentally respon-sive genes in autism linkage regions, listed by Herbertet al.,7 are located on chromosome 7q, including SHHand the paraoxonase genes. This chromosome alsocontains RELN, HOXA1, WNT2 and other candidategenes for autism susceptibility, including EN2 (EN-GRAILED 2) and Forkhead box P2 (FOXP2).9,11,138
Similar to the HOX genes, EN2 encodes a transcrip-tion factor important for neurodevelopment, with acritical role in the formation of specific serotonergicand noradrenergic mid- and hindbrain nuclei,295 and
Mouse models of autismSS Moy and JJ Nadler
14
Molecular Psychiatry
-
in the survival of specific dopaminergic subpopula-tions.296–298 En2 deletion animals have behavioral andneuroanatomical abnormalities that reflect alterationsin autism.15,299 For example, both juvenile and adultEn2-null mice show deficits in social interaction.34
The mutant mice are also characterized by hyper-activity, reduced spatial learning34 and impairedmotor coordination.34,300 Changes in brain morphol-ogy include a smaller cerebellum with aberrantfoliation and reduced numbers of Purkinje andgranule neurons.301,302 The cerebellar neuropathologyemerges during embryonic development of the mu-tant mice.303 In a recent report and overview,Kuemerle et al.299 note that the En2-null mice alsoevidence an anterior shift in the position of amygdalarnuclei, which may reflect a similar shift observed in arat model for prenatal exposure to valproic acid.Disturbances in cerebellar development have beenassociated with deletion of another gene located onchromosome 7q, FOXP2. Foxp2 null mice haveaberrant neuronal organization within Purkinje andgranule cell layers and reduced dendritic arborizationin cerebellum.39 The mutant pups show profounddeficits in ultrasonic vocalizations.39 Interestingly,severe language deficiencies have been associatedwith mutations of FOXP2 in a human popula-tion.304,305
Two other genes of interest, from the c-MET proto-oncogene (MET) and hepatocyte growth factor (HGF),are also found on chromosome 7q. A recent reportdemonstrated a significant association for an allelicvariant of MET in autism families.306 MET encodes theHGF receptor tyrosine kinase Met, which is an initialelement of the HGF signaling cascade. The HGFsignaling pathway regulates cortical neuron migrationduring forebrain development. Segarra et al.307 haveprovided evidence that the effects of HGF signalingon embryonic brain development are mediatedthrough both the PI3K and Ras pathways. Micedeficient in uPAR (the gene encoding urokinaseplasminogen activator receptor), another member ofthe HGF pathway, have marked decreases in neocor-tical GABAergic interneurons, and also demonstrateincreased anxiety-like behavior and enhanced seizuresusceptibility.308 HGF is considered an environmen-tally responsive gene,7 suggesting that HGF signalingmay be particularly sensitive to disruption by neuro-toxin exposure early in development. Overall, mem-bers of the HGF pathway are promising targets forfurther studies relevant to autism.
Epigenetic regulation and autism
Gene expression can be differentially regulated with-out alteration of the DNA code through epigeneticmechanisms. In the case of autism, epigeneticdifferences may underlie enhanced susceptibility forthe disease, through possible mechanisms such asaltered MECP2 regulation of GABAA receptor subunitgenes through DNA methylation,309 or aberrant his-tone acetylation following exposure to a viral agent or
neurotoxin, such as valproic acid.268 Differentialepigenetic modifications may explain why indivi-duals with similar, or even identical, genotypes maybe discordant for the autism phenotype. One studyhas found disparate gene expression in lymphoblas-toid cell lines from monozygotic twins characterizedby different degrees of autistic symptoms.310 A path-way analysis revealed that a majority of the geneswith the most significant alterations in expressionwere important for mediating inflammatory re-sponses. In addition, expression of SERT in thediscordant twins was consistently reduced in thetwin with the most severe autistic symptoms. Whilethe findings from blood-derived cell lines may notreflect molecular events in brain, the results suggestthat epigenetic modifications, reflected in alteredprofiles of gene expression, play a role in autism.
Tremolizzo et al.311 investigated whether the Relnrl/þ
mouse model has enhanced susceptibility for epige-netic effects by using a chronic L-methionine dosingregimen to induce hypermethylation in brain. Theresearchers found that both Relnþ /þ and Relnrl/þ
mice had markedly reduced levels of reelin andGAD67 mRNA levels, as well as alterations inprepulse inhibition of acoustic startle responses,following the protracted exposure to L-methionine.Chronic treatment with valproic acid had an oppositeeffect, leading to upregulation of reelin and GAD67,possibly through inhibition of histone deacetylation.The administration of valproic acid with L-methio-nine blocked the decreased gene expression observedwith L-methionine alone. Since the L-methioninetreatment did not have enhanced effects in the youngadult Relnrl/þmice, these results did not provideevidence for altered sensitivity to epigenetic mechan-isms following haplosufficiency of Reln. However, itis possible that differential susceptibility in themutant mice would have been found with exposureto L-methionine or valproic acid during earlydevelopment. Further work has shown that, innormal mice, chronic L-methionine treatment canlead to reductions in social interaction and impairedsocial recognition.312 Dong et al.313,314 have providedevidence that reduced expression of reelin andGAD67 in the L-methionine-exposed mouse may bemediated by increased binding of Mecp2 to reelin andGAD67 promotor regions, and that valproic acidinterrupts this enhanced association with Mecp2.315
The findings raise the question of whetherthe detrimental prenatal effects of valproic acidexposure may be linked to disruption of normalMECP2 action, with subsequent alterations in RELNand GAD67 expression and dysregulation of GABAer-gic function in cortical regions. Tueting et al.316
provide an excellent overview of how an interactionbetween genes, environment and epigenetic factorsmight underlie aberrant neurodevelopment in thereeler mouse, relevant to autism and other clinicaldisorders.
Tsankova, Nestler, and colleagues,317 in their timelyreview of epigenetic factors in neuropsychiatric
Mouse models of autismSS Moy and JJ Nadler
15
Molecular Psychiatry
-
disorders, outline the importance of chromatin remo-deling, through processes of DNA methylation orhistone modification, as a mechanism for persistentalteration of gene activity. Chromatin remodeling isassociated with several of the genetic disorders withautistic symptomatology, including FXS, Rett syn-drome, AS and PWS.317 Interestingly, genes forchromatin regulation have been shown to regulateexpression of multiple genetic loci, and therefore, canenhance or suppress mutant phenotypes. Lehner etal.318 identified these types of ‘hub’ genes byconstructing a genetic interaction map for the func-tional network underlying development in Caenor-habditis elegans, using systematic RNA interference.Most genes identified were involved in only a smallnumber of interactions. However, some genes hadmultiple interactions, involving diverse signal trans-duction pathways. These genes, classified as ‘hub’genes, all functioned as chromatin regulators. Reduc-tion of activity in the hub genes led to higherpenetrance of specific aberrant phenotypes associatedwith mutations in genes from multiple differentpathways, including the Wnt pathway. In humandisease, it is possible that hub genes serve to bufferthe consequences of specific mutations across diver-gent pathways, which are revealed in cases ofdeficient hub gene function.
Mammalian orthologs of the six most highlyconnected hub genes reported by Lehner et al.318
have been identified. Several of these are expressed inthe brain of the developing and adult mouse. Spen,Mta1 and Hmgb2, in particular, show restrictedpatterns of expression in different regions of thebrain.319 Targeted disruption of hub genes Trrap, Rereor Hmgb1 in the mouse leads to pre- or perinatallethality.320–322 Hsp90, which encodes a 90-kDamember of the heat-shock protein family, is alsothought to have a role in the suppression or bufferingof mutations,323,324 possibly through chromatin mod-ification of genes in the Wnt signaling pathway.325
HSP genes are important for responses to inflamma-tion and toxic environmental stressors. In particular,both hsp90 and hsp70 have critical roles in theregulation of glucocorticoid receptor function.326 In arecent report, HSP90B1 (HSP90 b-member 1) andHSPBP1 (hsp70-interacting protein) had significantlydysregulated expression in lymphoblastoid cell linesfrom male subjects with both autism and FXS.327
Similarly, the expression of HSPA8 (heat shock 70-kDa protein 8) was significantly different between atleast one set of monozygotic twins discordant forseverity of autism symptoms.310 Nuber et al.83 haveshown that the mouse model of Rett syndrome hasupregulation of Hsp105 (related to the Hsp70 family)and Fkbp5, which encodes an immunophilin compo-nent of the hsp90-glucocorticoid receptor complex.Unfortunately, deficiency of Hsp90beta in mice leadsto embryonic mortality due to placental defects,328
although several mouse models with mutations ingenes related to heat shock proteins have beencreated.329 One issue with altering function in genes
with molecular pleiotropy is that the resultingphenotype is often severe or lethal, and may notreveal processes important in human neuropsychia-tric disorders. The creation of mouse lines withconditional or hypomorphic alleles of hub genesmight allow the study of viable models for alterationsin multiple signaling pathways during development,including changes relevant to malfunction of epi-genetic mechanisms.
Discussion
Developing mouse models for disorders with complexmultigenic etiologies has proved challenging, espe-cially for idiopathic autism, a clinical syndrome withno definitive physiological biomarker. Advances havebeen made in the ability to produce viable single-genemouse models, to model imprinting abnormalitiesand in the use of behavioral tests reflecting coredisease symptoms. However, despite its high herit-ability, only approximately 10% of autism cases canbe traced to a known genetic aberration (for example,Ref. 330). Similarly, very few medical histories ofautistic patients include exposure to a knownenvironmental insult. Given that the etiology ofidiopathic autism remains to be elucidated, theobserved association between disease symptoms andspecific genetic disorders or clinical risk factors (suchas maternal use of antiseizure medication) is the basisfor most mouse models of the spectrum disorders.
Validation of autism mouse models can encompassmultiple points of possible congruence, includingreflection of core disease symptoms, developmentalonset, male/female ratio for behavioral or otherabnormalities, neuropathology, genetic contributionand epigenetic factors. The Fmr1-null mouse providesan example of a model with several of these attribu-tions, such as an aberrant behavioral phenotype,association with the X-chromosome, alterations insynaptic function and neuronal morphology, andgenetic aberrations reflective of a human disordercharacterized by autistic symptoms. In fact, recentadvances have included the development of novelmouse models that recapitulate the large repeatexpansions in FMR1331 or mosaic expression ofFMRP332 observed in FXS. The inconsistent findingsof behavioral alterations in the fragile X-model micemay be due, in part, to an enhanced susceptibility foreffects of Fmrp loss in some inbred mouse strains,perhaps relevant to the heterogeneity of symptomsand differential genetic susceptibility observed inautism. However, many other factors may play a rolein discordant behavioral profiles. A human popula-tion study found a significant association betweenincreasing paternal age and incidence of autism in theoffspring,333 suggesting that age of breeding stockcould influence whether progeny demonstrate anaberrant phenotype. Other variables for considerationinclude differences in laboratory procedures, testingand housing conditions, breeding milieu, and diver-sity of environmental stimuli. For example, Restivo
Mouse models of autismSS Moy and JJ Nadler
16
Molecular Psychiatry
-
and colleagues59 found that exposure to an enrichedenvironment can not only reverse some behavioraldeficits in the Fmr1-null mouse, but can also fullyrescue aberrant dendritic morphology in the FXSmodel. On the other hand, alterations in function ofthe hypothalamic–pituitary–adrenal axis, as observedin Fmr1-null mice334 (see also Ref. 335) and in themouse model of Rett syndrome,82,83 suggest that somemutant lines may have abnormal sensitivity tostressful environmental conditions, which could bereflected in measures of anxiety-like responses,activity, social approach and interaction, and othertypes of behavior.
Behavioral phenotyping can provide validation thata genetic mouse model reflects core symptoms of thehuman disease. However, tests that require explora-tion, such as the three-chambered choice tasks used tomeasure social preference, may not be useful fortesting mutant lines with very low activity levels andreduced exploration. General hypoactivity in Sert-null mice has presented difficulties for the interpreta-tion of behavioral changes related to anxiety anddepression-like responses.189,336 In addition, behavior-al and neurological profiles for Sert-null mice candiffer dependent on background strain.188,191 TheC57BL/6 mouse strain has been used as the back-ground for many mutant lines, but studies in thefragile X-model mouse have suggested that this strainhas less susceptibility to the effects of Fmrp defi-ciency.51,61,65 C57BL/6J mice are also characterized byhigh-frequency hearing loss337 and markedly lowacquisition of a T-maze learning task,17 indicatingpossible issues with using this strain as a backgroundfor mutant lines. Some findings suggest that the 129strain confers greater susceptibility to gene disrup-tion,61,65 but, in the case of Ube3A-null mice, alsoleads to higher rates of seizures and mortality.89 The129S1/SvImJ strain demonstrates low social approachin our three-chambered choice task,17 which couldconfound the detection of social deficits in mutantmice with this background.
Besides possible interactions between targetedgenetic alterations and the alleles from the back-ground strains, the observed phenotype for a parti-cular mouse model may also be affected by significantepigenetic factors, including differences in maternalbehavior across inbred mouse strains.338,339 Earlyenvironmental experiences, such as licking andgrooming by the dam or experimenter handling, canhave persistent effects on behavior and gene expres-sion in offspring. Weaver et al.340 have proposed thatthe effects of maternal responses may be mediated bythe changes in serotonergic signaling, leading todifferential levels of glucocorticoid receptor expres-sion and altered regulation of the hypothalamic–pituitary–adrenal axis. Interestingly, rats which re-ceived high levels of licking, grooming and otherforms of maternal attention have increased hippo-campal expression of reelin, BDNF, and markers forsynaptogenesis and survival, as well as increasedNMDA receptor binding,341,342 and demonstrate im-
proved performance in the Morris water maze task.342
There is evidence that persistent effects of earlyexperience on behavior and gene expression can bereversed by DNA methylation or histone deacetylaseinhibition.341,343 Overall, these studies suggest thatexpression of specific genes can be altered by theexperimental presentation of environmental chal-lenges, through epigenetic modifications that may berelevant to autism. At the same time, the findingsindicate the importance of using littermate wild-typemice as the comparison group for mutant mice, tocontrol for the effects of environmental conditions,including maternal behavior, during development.Variability can be reduced further by only testing sex-matched littermate pairs.
One promising direction for research utilizingmouse models of autism has been in the explorationof gene-by-gene interactions. For example, deletion ofboth Engrailed-1 and Engrailed-2 leads to a strikingreduction in serotonergic neurons of the dorsal raphenucleus, and an overt loss of the noradrenergicneurons of the locus coeruleus.295 These abnormal-ities are not evident in the single-null mice, suggest-ing that redundancy of function between genes couldconceal critical roles in neurodevelopment (see alsoSgado et al.296 and Alberi et al.297). Similarly, micenull for both Fmr1 and Fxr2 (which encodes anotherfragile X-related protein) show greater behavioralalterations for some, but not all, measures of function,in comparison to the single-null mice.64 Given thepossible contribution of epigenetic factors to autism,researchers have examined interactions involvinggenes from imprinted regions of chromosome 15q.There has been some evidence that Mecp2 regulatesUbe3A and Gabrb3 expression in the mouse model forRett syndrome,87,344 but this finding has been incon-sistent.345 There are also reports that Mecp2 plays arole in the regulation of Bdnf levels.134,201,202 Reduc-tion in the levels of Bdnf exacerbates the phenotypeof both the Rett syndrome-model mouse346 and theSert deletion line.208–210 In the Rett syndrome-modelmouse, Bdnf overexpression with an inducible Bdnftransgene leads to partial correction of abnormalitieslinked to loss of Mecp2 function.346 As discussedpreviously, suppressing inhibitory CaMKII phosphor-ylation through genetic mutation in the mouse modelof AS can fully or partially normalize components ofthe aberrant phenotype.88 In addition, a recent studyhas shown that the genetic inhibition of a differentkinase, p21-activated kinase, in the forebrain of Fmr1-null mice can fully or partially restore alterations inbehavior and dendritic spine morphology.60 Theseresults demonstrate that gene interaction approachescan be used for the rescue, as well as the exacerbation,of specific endophenotypes relevant to neurodevelop-mental disorders. However, transgenic overexpres-sion of Mecp2347 or Fmr158 in mice has been linked todetrimental effects, providing evidence that altera-tions of tightly regulated gene expression may haveundesired consequences. In line with these findings,Guy et al.348 found that the abrupt restoration of
Mouse models of autismSS Moy and JJ Nadler
17
Molecular Psychiatry
-
Mecp2 function had toxic effects in a mouse model ofRett syndrome, but a more gradual reactivation of thegene led to partial rescue of the aberrant phenotype.
Recent studies on expression profiles in humanpopulations have demonstrated the value of usingpathway analysis to identify sets of genes that operatetogether as networks to generate a disease pheno-type.310,327 Similar approaches can be used withmouse model systems to explore alterations in gene-by-gene interactions underlying abnormal behavioralphenotypes. For example, a targeted disruption mayhave discordant effects on a selected endophenotype,such as impaired social interaction or repetitivebehavior, dependent on genetic background of themouse strain. We can then map modifiers of themutant phenotype by examining established markersof polymorphism between strains. This approach canidentify genes that segregate with the observedbehavioral change and may modulate the effects ofthe targeted mutation on neurodevelopment and brainfunction. A particular domain of behavior, such associal function or reversal learning, can also beinvestigated across multiple inbred mouse strains, todetermine how genetic diversity contributes to thebehavioral phenotype.17 By combining a strain dis-tribution for a particular endophenotype with micro-array expression profiles in brain, genes regulatingcharacteristic behavioral responses can be identi-fied.349 For more detailed mapping, recombinantinbred lines of mice provide a powerful tool. Theselines are the products of a two-generation crossbetween parents of different strains, bred together toproduce stable lines that carry scrambled, homozy-gous segments of each parental genome. Sets ofrecombinant inbred lines are publicly available withassociated mapping data, such as the BXD lines,leaving the investigator to phenotype for the behaviorof interest.350 Congenic strains, also derived from twoinbred backgrounds, provide a different resource toanalyze genetic contribution to behavior. The con-genic lines carry a small, homozygous portion of onechromosome from one strain on the background ofanother. This allows the investigator to examine thecontribution of a very defined genomic region to aspecific endophenotype.351,352
In conclusion, the development of mouse modelsfor autism has advanced through the integration ofmolecular genetic approaches, clinical reports ondisease symptomatology and neuropathology, andfindings from linkage and association studies inhuman populations.15 Clinical reports can also sug-gest interesting targets for further investigation usingmouse model systems. One example is CYFIP1, a genelocated on chromosomal region 15q11–13. One studyhas found that FXS patients which exhibit thePrader–Willi phenotype have significantly reducedexpression of CYFIP1, and markedly high rates ofautistic-like symptoms.353 Altered expression of CY-FIP1 has also been observed in lymphoblastoid cellsfrom males with autism due to chromosomal rearran-gement (a maternally inherited duplication) of 15q11–
13.327 Other genetic targets for future work in theproduction of relevant mutant lines can be drawnfrom signaling pathways that include candidate genesfor autism susceptibility, such as PTEN211 and MET,306
and mediators of neuronal connectivity, such asneurexin.142 Overall, given the many promisingcandidate genes for autism, and armed with tools forgenetic analysis and behavioral assessment, we canbegin to discover the complex network of geneswhich contribute to an autistic phenotype.
Acknowledgments
This work was supported by NIH STAART Grant U54MH66418 and NICHD Grant P30 HD03110.
References
1 Folstein SE, Rosen-Sheidley B. Genetics of autism: complexaetiology for a heterogeneous disorder. Nat Rev Genet 2001; 2:943–955.
2 Lord C, Risi S, Lambrecht L, Cook Jr EH, Leventhal BL, DiLavorePC et al. The autism diagnostic observation schedule-generic: astandard measure of social and communication deficits asso-ciated with the spectrum of autism. J Autism Dev Disord 2000; 30:205–223.
3 American Psychiatric Association. Diagnostic and StatisticalManual of Mental Disorders (DSM-IV-TR). American PsychiatricAssociation: Washington, DC, 2000.
4 Fombonne E. Epidemiological trends in rates of autism. MolPsychiatry 2002; 7(Suppl 2): S4–S6.
5 Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, LevySE et al. The epidemiology of autism spectrum disorders. AnnuRev Public Health 2007; 28: 235–258.
6 Tuchman R, Rapin I. Epilepsy in autism. Lancet Neurol 2002; 1:352–358.
7 Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, Kahler SG et al.Autism and environmental genomics. Neurotoxicology 2006; 27:671–684.
8 Newschaffer CJ, Fallin D, Lee NL. Heritable and nonheritable riskfactors for autism spectrum disorders. Epidemiol Rev 2002; 24:137–153.
9 Persico AM, Bourgeron T. Searching for ways out of the autismmaze: genetic, epigenetic and environmental clues. TrendsNeurosci 2006; 29: 349–358.
10 Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer Jet al. A genomic screen of autism: evidence for a multilocusetiology. Am J Hum Genet 1999; 65: 493–507.
11 Wassink TH, Brzustowicz LM, Bartlett CW, Szatmari P. Thesearch for autism disease genes. Ment Retard Dev Disabil Res Rev2004; 10: 272–283.
12 Insel TR. Mouse models for autism: report from a meeting. MammGenome 2001; 12: 755–757.
13 Lewis MH, Tanimura Y, Lee LW, Bodfish JW. Animal models ofrestricted repetitive behavior in autism. Behav Brain Res 2007;176: 66–74.
14 Moy SS, Nadler JJ, Magnuson TR, Crawley JN. Mouse models ofautism spectrum disorders: the challenge for behavioral genetics.Am J Med Genet C Semin Med Genet 2006; 142: 40–51.
15 Murcia CL, Gulden F, Herrup K. A question of balance: a proposalfor new mouse models of autism. Int J Dev Neurosci 2005; 23:265–275.
16 Rodier PM, Ingram JL, Tisdale B, Croog VJ. Linking etiologies inhumans and animal models: studies of autism. Reprod Toxicol1997; 11: 417–422.
17 Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RPet al. Mouse behavioral tasks relevant to autism: phenotypes of10 inbred strains. Behav Brain Res 2007; 176: 4–20.
18 Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TRet al. Sociability and preference for social novelty in five inbred
Mouse models of autismSS Moy and JJ Nadler
18
Molecular Psychiatry
-
strains: an approach to assess autistic-like behavior in mice.Genes Brain Behav 2004; 3: 287–302.
19 Nadler JJ, Moy SS, Dold G, Trang D, Simmons N, Perez A et al.Automated apparatus for rapid quantitation of autism-like socialdeficits in mice. Genes Brain Behav 2004; 3: 303–314.
20 Brodkin ES, Hagemann A, Nemetski SM, Silver LM. Socialapproach-avoidance behavior of inbred mouse strains towardsDBA/2 mice. Brain Res 2004; 1002: 151–157.
21 Sankoorikal GM, Kaercher KA, Boon CJ, Lee JK, Brodkin ES. Amouse model system for genetic analysis of sociability: C57BL/6Jversus BALB/cJ inbred mouse strains. Biol Psychiatry 2006; 59: 415–423.
22 Bolivar VJ, Walters SR, Phoenix JL. Assessing autism-likebehavior in mice: variations in social interactions among inbredstrains. Behav Brain Res 2007; 176: 21–26.
23 Bodfish JW, Symons FJ, Parker DE, Lewis MH. Varieties ofrepetitive behavior in autism: comparisons to mental retardation.J Autism Dev Disord 2000; 30: 237–243.
24 Turner M. Annotation: repetitive behaviour in autism: a reviewof psychological research. J Child Psychol Psychiatry 1999; 40:839–849.
25 Carcani-Rathwell I, Rabe-Hasketh S, Santosh PJ. Repetitive andstereotyped behaviours in pervasive developmental disorders.J Child Psychol Psychiatry 2006; 47: 573–581.
26 Mooney EL, Gray KM, Tonge BJ. Early features of autism:repetitive behaviours in young children. Eur Child AdolescPsychiatry 2006; 15: 12–18.
27 South M, Ozonoff S, McMahon WM. Repetitive behavior profilesin Asperger syndrome and high-functioning autism. J Autism DevDisord 2005; 35: 145–158.
28 Ronald A, Happe F, Bolton P, Butcher LM, Price TS, WheelwrightS et al. Genetic heterogeneity between the three components ofthe autism spectrum: a twin study. J Am Acad Child AdolescPsychiatry 2006; 45: 691–699.
29 Ronald A, Happe F, Plomin R. The genetic relationship betweenindividual differences in social and nonsocial behaviourscharacteristic of autism. Dev Sci 2005; 8: 444–458.
30 Coldren JT, Halloran C. Spatial reversal as a measure of executivefunctioning in children with autism. J Genet Psychol 2003; 164:29–41.
31 Branchi I, Ricceri L. Transgenic and knock-out mouse pups: thegrowing need for behavioral analysis. Genes Brain Behav 2002; 1:135–141.
32 Branchi I, Bichler Z, Berger-Sweeney J, Ricceri L. Animal modelsof mental retardation: from gene to cognitive function. NeurosciBiobehav Rev 2003; 27: 141–153.
33 Ricceri L, Moles A, Crawley J. Behavioral phenotyping of mousemodels of neurodevelopmental disorders: relevant social beha-vior patterns across the life span. Behav Brain Res 2007; 176: 40–52.
34 Cheh MA, Millonig JH, Roselli LM, Ming X, Jacobsen E, Kamdar Set al. En2 knockout mice display neurobehavioral and neuro-chemical alterations relevant to autism spectrum disorder. BrainRes 2006; 1