
Activation of -adrenoceptors contributes to intermittent ... · tion and infarction (5, 18, 37, 49,...
13
Activation of 1B -adrenoceptors contributes to intermittent hypobaric hypoxia-improved postischemic myocardial performance via inhibiting MMP-2 activation Ling Gao, 1 Le Chen, 1 Zhi-Zhen Lu, 2 Hong Gao, 1 Lan Wu, 1 Yi-Xiong Chen, 1 Cai-Mei Zhang, 1 Yu-Kun Jiang, 1 Qing Jing, 1 You-Yi Zhang, 2 and Huang-Tian Yang 1 1 Key Laboratory of Stem Cell Biology and Laboratory of Molecular Cardiology, Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences and Shanghai Jiao Tong University School of Medicine Shanghai, China; and 2 Institute of Vascular Medicine, Peking University Third Hospital and Key Laboratory of Molecular Cardiovascular Sciences Ministry of Education, Beijing, China Submitted 7 October 2013; accepted in final form 1 April 2014 Gao L, Chen L, Lu ZZ, Gao H, Wu L, Chen YX, Zhang CM, Jiang YK, Jing Q, Zhang YY, Yang HT. Activation of 1B - adrenoceptors contributes to intermittent hypobaric hypoxia-improved postischemic myocardial performance via inhibiting MMP-2 activa- tion. Am J Physiol Heart Circ Physiol 306: H1569 –H1581, 2014. First published April 4, 2014; doi:10.1152/ajpheart.00772.2013.— Inhibition of matrix metalloproteinases-2 (MMP-2) activation renders cardioprotection from ischemia/reperfusion (I/R) injury; however, the signaling pathways involved have not been fully understood. Inter- mittent hypobaric hypoxia (IHH) has been shown to enhance myo- cardial tolerance to I/R injury via triggering intrinsic adaptive re- sponses. Here we investigated whether IHH protects the heart against I/R injury via the regulation of MMP-2 and how the MMP-2 is regulated. IHH (PO 2 84 mmHg, 4-h/day, 4 wk) improved postisch- emic myocardial contractile performance, lactate dehydrogenase (LDH) release, and infarct size in isolated perfused rat hearts. More- over, IHH reversed I/R-induced MMP-2 activation and release, dis- orders in the levels of MMP-2 regulators, peroxynitrite (ONOO ) and tissue inhibitor of metalloproteinase-4 (TIMP-4), and loss of the MMP-2 targets -actinin and troponin I. This protection was mim- icked, but not augmented, by a MMP inhibitor doxycycline and lost by the 1 -adrenoceptor (AR) antagonist prazosin. Furthermore, IHH increased myocardial 1A -AR and 1B -AR density but not 1D -AR after I/R. Concomitantly, IHH further enhanced the translocation of PKC epsilon (PKCε) and decreased the release of mitochondrial cytochrome c due to I/R via the activation of 1B -AR but not 1A -AR or 1D -AR. IHH-conferred cardioprotection in the postischemic con- tractile function, LDH release, MMP-2 activation, and nitrotyrosine as well as TIMP-4 contents were mimicked but not additive by 1 -AR stimulation with phenylephrine and were abolished by an 1B -AR antagonist chloroethylclonidine and a PKCε inhibitor PKCε V1–2. These findings demonstrate that IHH exerts cardioprotection through attenuating excess ONOO biosynthesis and TIMP-4 loss and se- quential MMP-2 activation via the activation of 1B -AR/PKCε path- way. intermittent hypobaric hypoxia; ischemia-reperfusion injury; matrix metalloenzymes-2; 1 -adrenoceptors; mitochondria MATRIX METALLOPROTEINASES (MMPs) belong to the zinc-depen- dent endopeptidases that are best recognized for their ability to degrade components of the extracellular matrix (29). Among MMPs, MMP-2 has been shown to contribute to myocardial ischemia/reperfusion (I/R) injury via the proteolysis of specific protein targets within the cardiomyocytes, whereas the inhibi- tion of MMP-2 improves the postischemic recovery of myo- cardial contractile function (1, 10, 29). MMP-2 activity is tightly controlled by endogenous inhibitors, such as tissue inhibitors of metalloproteinase (TIMPs), especially TIMP-4, in the heart with or without I/R (29, 33, 44). Meanwhile, MMP-2 is directly activated by peroxynitrite (ONOO ), the reaction product of nitric oxide (NO) and superoxide (O 2 · ), which is overgenerated upon first reperfusion following ischemia (29, 48, 53). Ischemic preconditioning (IPC) or other conditioning forms significantly resist the I/R-induced ONOO overproduc- tion or TIMP-4 loss and thus exert myocardial protection via sequentially diminishing the activation of MMP-2 (8, 15, 31, 45). However, the signaling pathway regulating the ONOO and TIMP-4 during cardioprotection has not yet been fully clarified. 1 -Adrenoceptor ( 1 -AR) activation has been shown to mimic IPC and trigger intrinsic adaptive responses to protect the heart against subsequent I/R-induced myocardial dysfunc- tion and infarction (5, 18, 37, 49, 52). We previously demon- strated that 1 -AR stimulation with phenylephrine alleviates I/R injury via protecting the mitochondrial function through the 1B -AR-activated PKC epsilon (PKCε)-mediated mitochon- dria ATP-sensitive potassium (mito-K ATP ) channel opening in rat cardiomyocytes (18), while it is unknown whether the activation of 1 -ARs regulates MMP-2 in the cardioprotection against I/R injury. Myocardial tolerance to ischemia has also been observed in people living at a high altitude (Peru, 4,000 m) (27) and in experimental animals using a simulated model of chronic intermittent hypobaric hypoxia (IHH) to mimic the physiolog- ical adaptation to altitude in the laboratory (3, 9, 34, 38, 58, 59, 61, 62, 68). This form of cardioprotection is reproducible in improving postischemic recovery of contractile function, re- ducing the incidence and severity of arrhythmias, and limiting the infract size (3, 9, 34, 38, 58, 61, 62, 68), and it has the merits of maintaining the cardioprotection longer than classic IPC and having less adverse effects such as right ventricular hypertrophy compared with the chronic continuous hypoxia (3, 58, 59, 69). Therefore, IHH provides us a unique model to better understand how the intrinsic defense system is triggered and developed. We found that IHH has a better improvement in the I/R-suppressed cell contraction than in the Ca 2 transients (9), an indication of the preservation of myofilament during I/R Address for reprint requests and other correspondence: H.-T. Yang, Labo- ratory of Molecular Cardiology, Institute of Health Sciences, SIBS, CAS & SJTUSM, 320 Yue Yang Rd., Biological Research Bldg. A, IHS Mail Box 115, Shanghai, 200031, China (e-mail: [email protected]). Am J Physiol Heart Circ Physiol 306: H1569–H1581, 2014. First published April 4, 2014; doi:10.1152/ajpheart.00772.2013. 0363-6135/14 Copyright © 2014 the American Physiological Society http://www.ajpheart.org H1569 by 10.220.33.2 on May 30, 2017 http://ajpheart.physiology.org/ Downloaded from
Transcript of Activation of -adrenoceptors contributes to intermittent ... · tion and infarction (5, 18, 37, 49,...

Activation of �1B-adrenoceptors contributes to intermittent hypobarichypoxia-improved postischemic myocardial performance via inhibitingMMP-2 activation
Ling Gao,1 Le Chen,1 Zhi-Zhen Lu,2 Hong Gao,1 Lan Wu,1 Yi-Xiong Chen,1 Cai-Mei Zhang,1
Yu-Kun Jiang,1 Qing Jing,1 You-Yi Zhang,2 and Huang-Tian Yang1
1Key Laboratory of Stem Cell Biology and Laboratory of Molecular Cardiology, Institute of Health Sciences, ShanghaiInstitutes for Biological Sciences, Chinese Academy of Sciences and Shanghai Jiao Tong University School of MedicineShanghai, China; and 2Institute of Vascular Medicine, Peking University Third Hospital and Key Laboratory of MolecularCardiovascular Sciences Ministry of Education, Beijing, China
Submitted 7 October 2013; accepted in final form 1 April 2014
Gao L, Chen L, Lu ZZ, Gao H, Wu L, Chen YX, Zhang CM,Jiang YK, Jing Q, Zhang YY, Yang HT. Activation of �1B-adrenoceptors contributes to intermittent hypobaric hypoxia-improvedpostischemic myocardial performance via inhibiting MMP-2 activa-tion. Am J Physiol Heart Circ Physiol 306: H1569–H1581, 2014.First published April 4, 2014; doi:10.1152/ajpheart.00772.2013.—Inhibition of matrix metalloproteinases-2 (MMP-2) activation renderscardioprotection from ischemia/reperfusion (I/R) injury; however, thesignaling pathways involved have not been fully understood. Inter-mittent hypobaric hypoxia (IHH) has been shown to enhance myo-cardial tolerance to I/R injury via triggering intrinsic adaptive re-sponses. Here we investigated whether IHH protects the heart againstI/R injury via the regulation of MMP-2 and how the MMP-2 isregulated. IHH (PO2 � 84 mmHg, 4-h/day, 4 wk) improved postisch-emic myocardial contractile performance, lactate dehydrogenase(LDH) release, and infarct size in isolated perfused rat hearts. More-over, IHH reversed I/R-induced MMP-2 activation and release, dis-orders in the levels of MMP-2 regulators, peroxynitrite (ONOO�) andtissue inhibitor of metalloproteinase-4 (TIMP-4), and loss of theMMP-2 targets �-actinin and troponin I. This protection was mim-icked, but not augmented, by a MMP inhibitor doxycycline and lostby the �1-adrenoceptor (AR) antagonist prazosin. Furthermore, IHHincreased myocardial �1A-AR and �1B-AR density but not �1D-ARafter I/R. Concomitantly, IHH further enhanced the translocation ofPKC epsilon (PKCε) and decreased the release of mitochondrialcytochrome c due to I/R via the activation of �1B-AR but not �1A-ARor �1D-AR. IHH-conferred cardioprotection in the postischemic con-tractile function, LDH release, MMP-2 activation, and nitrotyrosine aswell as TIMP-4 contents were mimicked but not additive by �1-ARstimulation with phenylephrine and were abolished by an �1B-ARantagonist chloroethylclonidine and a PKCε inhibitor PKCε V1–2.These findings demonstrate that IHH exerts cardioprotection throughattenuating excess ONOO� biosynthesis and TIMP-4 loss and se-quential MMP-2 activation via the activation of �1B-AR/PKCε path-way.
intermittent hypobaric hypoxia; ischemia-reperfusion injury; matrixmetalloenzymes-2; �1-adrenoceptors; mitochondria
MATRIX METALLOPROTEINASES (MMPs) belong to the zinc-depen-dent endopeptidases that are best recognized for their ability todegrade components of the extracellular matrix (29). AmongMMPs, MMP-2 has been shown to contribute to myocardial
ischemia/reperfusion (I/R) injury via the proteolysis of specificprotein targets within the cardiomyocytes, whereas the inhibi-tion of MMP-2 improves the postischemic recovery of myo-cardial contractile function (1, 10, 29). MMP-2 activity istightly controlled by endogenous inhibitors, such as tissueinhibitors of metalloproteinase (TIMPs), especially TIMP-4, inthe heart with or without I/R (29, 33, 44). Meanwhile, MMP-2is directly activated by peroxynitrite (ONOO�), the reactionproduct of nitric oxide (NO) and superoxide (O2
·�), which isovergenerated upon first reperfusion following ischemia (29,48, 53). Ischemic preconditioning (IPC) or other conditioningforms significantly resist the I/R-induced ONOO� overproduc-tion or TIMP-4 loss and thus exert myocardial protection viasequentially diminishing the activation of MMP-2 (8, 15, 31,45). However, the signaling pathway regulating the ONOO�
and TIMP-4 during cardioprotection has not yet been fullyclarified.
�1-Adrenoceptor (�1-AR) activation has been shown tomimic IPC and trigger intrinsic adaptive responses to protectthe heart against subsequent I/R-induced myocardial dysfunc-tion and infarction (5, 18, 37, 49, 52). We previously demon-strated that �1-AR stimulation with phenylephrine alleviatesI/R injury via protecting the mitochondrial function through the�1B-AR-activated PKC epsilon (PKCε)-mediated mitochon-dria ATP-sensitive potassium (mito-KATP) channel opening inrat cardiomyocytes (18), while it is unknown whether theactivation of �1-ARs regulates MMP-2 in the cardioprotectionagainst I/R injury.
Myocardial tolerance to ischemia has also been observed inpeople living at a high altitude (Peru, 4,000 m) (27) and inexperimental animals using a simulated model of chronicintermittent hypobaric hypoxia (IHH) to mimic the physiolog-ical adaptation to altitude in the laboratory (3, 9, 34, 38, 58, 59,61, 62, 68). This form of cardioprotection is reproducible inimproving postischemic recovery of contractile function, re-ducing the incidence and severity of arrhythmias, and limitingthe infract size (3, 9, 34, 38, 58, 61, 62, 68), and it has themerits of maintaining the cardioprotection longer than classicIPC and having less adverse effects such as right ventricularhypertrophy compared with the chronic continuous hypoxia (3,58, 59, 69). Therefore, IHH provides us a unique model tobetter understand how the intrinsic defense system is triggeredand developed. We found that IHH has a better improvement inthe I/R-suppressed cell contraction than in the Ca2� transients(9), an indication of the preservation of myofilament during I/R
Address for reprint requests and other correspondence: H.-T. Yang, Labo-ratory of Molecular Cardiology, Institute of Health Sciences, SIBS, CAS &SJTUSM, 320 Yue Yang Rd., Biological Research Bldg. A, IHS Mail Box115, Shanghai, 200031, China (e-mail: [email protected]).
Am J Physiol Heart Circ Physiol 306: H1569–H1581, 2014.First published April 4, 2014; doi:10.1152/ajpheart.00772.2013.
0363-6135/14 Copyright © 2014 the American Physiological Societyhttp://www.ajpheart.org H1569
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

by IHH. Moreover, cardiac sympathetic stimulation and �1-ARactivation have been shown to play an important role in thecardioprotective effect of IHH (35, 56), while it is unclearwhether IHH exerts cardioprotection against MMP-2 activationvia �1-ARs. Considering a critical role of MMP-2 in theproteolysis of myofilament, such as troponin I (TnI) and�-actinin (2, 4, 47, 54), we hypothesized that the inhibiting ofMMP-2 activation might contribute to the chronic IHH-con-ferred cardioprotection against I/R injury through the activa-tion of �1-AR.
To address those questions, the present study was designed1) to determine whether IHH protects the heart via the regu-lation of MMP-2 activation; 2) to clarify whether �1-ARsregulate MMP-2 and its regulators during cardioprotectionagainst I/R injury; and 3) to identify the signaling pathwayimplicated in the regulation of MMP-2 during cardioprotec-tion.
MATERIALS AND METHODS
Animal care. The animals used in this study were maintained inaccordance with the National Institutes of Health Guide for the Careand Use of Laboratory Animals (NIH Pub. No. 85–23), and allprocedures were approved by the Institutional Review Board of theInstitute of Health Sciences, Shanghai Institutes for Biological Sci-ences, Chinese Academy of Sciences and School of Medicine, Shang-hai Jiao Tong University (Shanghai, China).
IHH adapted rat model. To establish animal models adapted toIHH, male Sprague-Dawley rats were exposed to hypoxia in a hypo-baric chamber (equivalent to an altitude of 5,000 m; barometricpressure � 404 mmHg, PO2 � 84 mmHg) for one 4-h period each dayfor 4 wk as previously described (58). During this period, their bodyweight rose from 100–130 g to 310–360 g. Age-matched normoxicanimals were maintained in the normoxic environment for a corre-sponding period. All animals had free access to water and a standardlaboratory diet and were raised at room temperature (23–26°C) with a12-h light-dark cycle. Body weights of rats were measured weekly.
Heart perfusion. The rats were anesthetized with sodium pentobar-bital (45 mg/kg ip). The hearts were promptly excised and perfusedwith Krebs-Henseleit buffer containing the following (in mM): 118NaCl, 4.7 KCl, 1.2 MgSO4·7H2O, 2.5 CaCl2, 1.2 KH2PO4, 25NaHCO3, 0.026 Na2-EDTA, and 11.1 glucose, gassed with 95%O2-5% CO2 (pH 7.4) at 37°C by using the Langendorff techniqueunder a constant pressure of 80 mmHg as previously described (58,66). Left ventricle (LV) developed pressure (LVDP), LV end-diastolicpressure (LVEDP), maximal rate of pressure development (�dP/dtmax), and decay over time (�dP/dtmax), heart rate and rate-pressureproduct (RPP) were synchronously monitored with PowerLab system(AD Instrument, New South Wales, Australia).
Experimental protocols for isolated rat hearts. After equilibrationperfusion, the hearts from normoxic or IHH rats were subjected to 30min no-flow global ischemia followed by 30 or 120 min of reperfu-sion. The MMP inhibitor doxycycline (100 �M) (10) and the �1-ARantagonist prazosin (1 �M) (25, 55, 56) were perfused for 10 minbefore ischemia, and for the first 10 min of reperfusion, and the �1-ARagonist phenylephrine (10 �M) (5, 18, 40, 42, 52) was perfused for 5min followed a 10-min washout before ischemia. The antagonists5-methylurapidil (5-MU; 1 �M) (18, 21, 22, 65, 67) for �1A-AR andBMY7378 (1 �M) (18, 67) for �1D-AR were perfused for 10 minbefore ischemia and maintained during the reperfusion period, and the�1B-AR antagonist chloroethylclonidine (CEC; 10 �M) (18, 63) wasperfused for 15 min before ischemia. The above reagents were fromSigma (St. Louis, MO). Myristoylated PKCε V1–2 (a PKCε-specificinhibitor; 10 �M; Biomol, Hamburg, Germany) (18, 23, 28, 58) wasperfused for 5 min followed a 5-min washout before ischemia. All
above reagents were dissolved in the deionized distilled water andadded to perfusate at corresponding concentrations. At the end of theexperiments, the hearts were freeze clamped and stored at �80°C.Coronary effluents were collected at 1 min before ischemia and at 3and 30 min of reperfusion and stored at �80°C.
Measurement of LDH. LDH was spectrophotometrically measuredin the coronary effluent at preischemia and at 3 and 30 min ofreperfusion using a LDH assay kit (Jianchen Bioengineering Institute,Nanjing, China) as previously described (58, 66).
Infarct size estimation. The hearts subjected to I/R (30 min/120min) were frozen and cut into 2-mm-thick slices as previously de-scribed (15, 58). Sections were then incubated in 1% triphenyltetra-zolium chloride for staining. The infarct size was measured byplanimetry (Image Pro Plus, Media Cybernetics, MD) and representedas a percentage of the LV area at risk.
Measurement of the activity of MMP-2 in coronary effluent. Theactivity of MMP-2 in coronary effluent was determined by thefluorescent method as previously described (11) using GenMedMMP-2 Fluorimetric Assay Kit (GenMed Scientifics, Shanghai,China) according to the manufacturer’s instruction. Briefly, the col-lected coronary effluent samples were concentrated in Amicon UltraCentrifugal 4 ml-10 K concentrating vessels (4,000 g, 4°C; Millipore).Then, proteins of concentrated samples were measured by the BCAmethod and balanced to an equal protein concentration by addingKrebs-Henseleit buffer. Twenty-microliter balanced samples (5 �gtotal proteins) were added to an 80-�l reaction mixture containingMMP fluorescence resonance energy transfer (FRET) peptide sub-strate [Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2, where Mca is (7-methoxy-coumarin-4-yl) acetyl and Dpa is N-3-(2,4-dinitrophenyl)-L-�,�-diaminopropionyl]. The fluorescence intensity was measured at2-min intervals for 1 h at 37°C using a SPECTRAmax Gemini XPSfluorescence microplate reader (�ex: 328 nm; �em: 393 nm; MolecularDevices). The specific activity of MMP-2/�g protein in the coronaryeffluent was calculated as the OA-Hy-nonsensitive activity subtractedfrom the total activity. One unit of MMP-2 activity was defined as theamount of OA-Hy-sensitive enzyme that hydrolyzes 1 nM of peptidesubstrate per min at 37°C (pH 7.5).
Western blotting. The total protein fraction was prepared as de-scribed previously (58). Briefly, LV tissues were homogenized in lysisbuffer containing the following (in mM): 20 Tris, 150 NaCl, 2.5EDTA, 50 NaF, 0.1 Na4P2O7, 1 Na3VO4, 1 PMSF, 1 DTT, 1 �g/mlleupeptin, and 1% Triton X-100 (pH 7.4, 4° C) using a Polytron PT3100 homogenizer (Kinematica, Littau-Lucerne, Switzerland). Thehomogenate was kept on ice for 30 min and then centrifuged at 10,000g for 20 min at 4°C, and the supernatant was saved as total proteinfraction. For examining PKCε translocation, the membrane and cyto-sol fractions were separated as previously described (58). LV tissueswere homogenized in lysis buffer containing the following (in mM):20 Tris·HCl (pH 7.4), 250 sucrose, 1 EDTA, 1 EGTA, 1 NaF, 1Na3VO4, 1 PMSF, and 1 DTT with 0.2% (vol/vol) protease inhibitorcocktail and centrifuged at 1,000 g for 10 min at 4°C. The supernatantwas centrifuged at 10,000 g for 30 min at 4°C, and the pellet waswashed once with lysis buffer by centrifugation, resuspended with0.5% Triton X-100 in lysis buffer, sonicated on ice, and then centri-fuged at 20,000 g for 10 min at 4°C. The resultant supernatant wasdefined as the membrane fraction. The supernatant after the membranefraction had been pelleted and was centrifuged at 100,000 g for 1 h at4°C, and the resultant supernatant was defined as the cytosolicfraction. Triton X-100 was added at a final concentration of 0.5%. Foranalysis of cytochrome c release, the mitochondrial and cytoplasmiccompartments were fractionated as previously described (18). Briefly,frozen LV tissues were homogenized in lysis buffer containing thefollowing (in mM): 20 HEPES (pH 7.5), 10 KCl, 1.5 MgCl2, 1 EDTA,1 EGTA, 1 DTT, 0.1 PMSF, 1 leupeptin, and 250 sucrose. Thehomogenate was centrifuged at 750 g at 4°C. The supernatant wasaspirated and centrifuged at 10,000 g at 4°C. The pellet containing themitochondrial fraction was resuspended in lysis buffer. The superna-
H1570 REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

tant was centrifuged at 100,000 g at 4°C to remove any mitochondrialcontamination and saved as cytoplasmic compartments. The proteinconcentrations were determined by BCA method. The specific proteinmarker and contamination control for each fractions were assayed byimmunoblotting.
The standard Western blot was performed as previously described(58). Equal protein fractions were separated by electrophoresis onSDS-PAGE gels. After being transferred to PVDF, proteins wereprobed with primary specific antibodies [anti-MMP-2, 72 kDa, 1:500,Chemicon, CA; anti-TnI, anti-voltage-dependent anion-selectivechannel proteins-1 (VDAC-1) and anti-cytochrome c, 1:1,000, Santa
Cruz, TX; anti-�-actinin and anti-PKCε, 1:1,000, Sigma; anti-Na�/Ca2� exchanger-1 (NCX-1), 1:1,000, Abcam, Cambridge, UK; andGAPDH, 1:10,000; KangChen, Shanghai, China] overnight at 4°C.The immunoreaction was visualized using an enhanced chemilumi-nescent detection kit (Amersham, London, UK), exposed to X-rayfilm, and quantified by densitometry with a video documentationsystem (Gel Doc 2000; Bio-Rad, Hercules, CA).
RT-PCR. Total RNA was extracted from LV tissue with TrizolReagent (Invitrogen, Carlsbad, CA). cDNA was prepared by reversetranscription of 1 �g total RNA using the oligo (dT) primer andReverTra Ace reverse transcriptase (Toyobo, Osaka, Japan). PCR was
DC
A
Normoxia+doxycycline (7)
Normoxia (10) IHH (10)
Normoxia+prazosin (6) IHH+prazosin (6)
IHH+doxycycline (8)
Time (min) 10 30 30 Pre Ischemia Reperfusion Pre Ischemia Reperfusion
Time (min) 10 30 30
†
##
†
-10 0 10 20 30 40 50 60 Time (min)
LVD
P (m
mH
g)
0 20 40 60 80
120 140
Time (min) -10 0 10 20 30 40 50 60
+dP/
dtm
ax (m
mH
g/s)
0 600
1200 1800 2400 4000
##
Time (min) -10 0 10 20 30 40 50 60
-3000 -2400
-1200
-600
0 †
-dP/
dtm
ax (m
mH
g/s)
##
†
B
#
LVED
P (m
mH
g)
Time (min) 0
40 60 80
100 120
-10 0 10 20 30 40 50 60
†
Normoxia+doxycycline (7)
Normoxia (10) IHH (10)
IHH+doxycycline (8) Normoxia+prazosin (6) IHH+prazosin (6)
†
†
†
E
0
120
240
360
Pre R30
Hea
rt ra
te (b
pm)
* *
F
Rat
e-pr
essu
re p
rodu
ct
(mm
Hg
x m
in-1
x 1
03)
0
10
20
30
40
Pre R30
* *
##
†
†
Fig. 1. Effects of intermittent hypobaric hypoxia (IHH) on the left ventricular (LV) functional performance in isolated rat hearts subjected to 30 min of no-flowglobal ischemia followed by 30 min of reperfusion with or without the matrix metalloproteinase (MMPs) inhibitor doxycycline (100 �M) and �1-adrenoceptor(�1-AR) antagonist prazosin (1 �M) treatment. A: LV developed pressure (LVDP). B: LV end-diastolic pressure (LVEDP). C: maximal rate of LV pressuredevelopment (�dP/dtmax). D: maximal rate of LV pressure decline (�dP/dtmax). E: heart rate. F: rate-pressure product. Pre, preischemia; R30, 30 min ofreperfusion. The number of individual rat hearts per group is indicated in parentheses. **P 0.01 vs. corresponding preischemic values; #P 0.05, ##P 0.01vs. corresponding normoxic values; †P 0.05 vs. corresponding I/R control values.
H1571REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

carried out for 30 cycles using Taq DNA Polymerase (Takara, Shiga,Japan) as previously described (6). The following primers were used:MMP-2 sense, 5=-CCCCTATCTACACCTACACCAAGAAC-3=;MMP-2 antisense, 5=-CATTCCAGGAGTCTGCGATGAGC-3=;GAPDH sense, 5=-ACGACCCCTTCATTGACC-3=; and GAPDH anti-sense, 5=-TGCTTCACCACCTTCTTG-3=.
Cardiac nitrotyrosine measurement. As a marker of the cardiacONOO� formation, the nitrotyrosine level of LV tissue extracts at 30min of reperfusion was measured using an enzyme-linked immu-nosorbent assay (Cell Biolabs) as previously described (20, 50).Nitrotyrosine content was normalized to protein content of myocar-dium extracts and expressed as picogram per milligram of protein.
Radioligand binding assay. The preparation of crude LV mem-brane fraction and radioligand binding assay were performed asdescribed previously (67). The �1-AR antagonist 2-b(4-hydroxyphen-yl)-ethylaminomethyl-tetralone (BE2254; Beiersdorf, Hamburg, Ger-many) was radioiodinated with 125I-Na� (Beijing Institute of AtomicEnergy, Beijing, China) using the chloramine T method. Then, themembrane preparation (1 mg/ml) was incubated with 125I-BE2254(2,200 Ci/mM) for 20 min at 37°C in the presence or absence ofcompeting drugs, and the incubation was terminated by 10 mMTris·HCl. The mixture was filtered, washed, dried, and measured forits radioactivity and protein concentration. Nonspecific binding wasdetermined in the presence of 10 �M of phentolamine (Sigma). Todetermine the affinity (KD) and the maximal binding capacity (Bmax)
of 125I-BE2254 to cardiac �1-ARs, saturation curves were determinedby incubating membrane preparations with increasing concentrationsof 125I-BE2254 (35–640 pM, 2.8–52.1 MBq) and the data wereanalyzed by the method of Scatchard. About 50 pM 125I-BE2254 (4MBq) was incubated with 100-�l membrane preparations in thepresence of 16 concentrations of 5-MU and BMY7378 to gain thecompetitive binding curve. The Bmax of �1A- and �1D-subtypes werecalculated from the percentage of high affinity site of the 5-MU andBMY7378 using Prism software, and the rest of the total Bmax wasevaluated as of �1B-AR subtype.
Statistic analysis. Data are presented as means SE. Statisticalsignificance was determined using ANOVA or repeated ANOVA formultiple comparisons or repeated measurements. P 0.05 wasregarded as statistically significant.
RESULTS
IHH improves the postischemic recovery of myocardialperformance and cell survival via inhibiting MMP-2activation. To determine whether IHH protects the heartagainst I/R injury via the regulation of MMPs, we first exam-ined hemodynamic changes during I/R in Langendorff-per-fused rat hearts with or without a MMP inhibitor doxycycline(10). The LV functional performance, characterized by LVDP,
* * #
# †
A
0
100
200
300
400
Pre R3 R30
LDH
rele
ase
(U/L
) * * †
† †
B
0 50
100
150
D
0 10 20 30 40
LVD
P (m
mH
g)
Rat
e-pr
essu
re p
rodu
ct
(mm
Hg
x m
in-1
x 1
03)
IHH (10) Normoxia (10)
Normoxia+doxycycline (6) IHH+doxycycline (6) Normoxia+prazosin (6) IHH+prazosin (6)
* *
- - Dox Pra
Pre R120
C
Hea
rt ra
te (b
pm)
0
120
240
360 IHH Normoxia
- - Dox Pra
Pre R120
- - Dox Pra
Pre R120
In
farc
t siz
e
(%
of a
rea
at ri
sk)
E
0 10 20 30 40
#
†
†
- Dox Pra
IHH Normoxia
IHH Normoxia
IHH Normoxia
* * * *
* * * * ##
##
†† ††
†† ††
R120
Fig. 2. Effects of IHH on cell necrosis inisolated rat hearts subjected to I/R with orwithout doxycycline (Dox) or prazosin (Pra)treatment. A: LDH activity in coronary efflu-ent. The number of preparations from indi-vidual rat hearts per group is indicated inparentheses. B: LV developed pressure.C: heart rate. D: rate-pressure product. E: LVinfarct size; n � 5 hearts from individual ratper group. R3, 30, and 120: 3, 30, and 120min of reperfusion. **P 0.01 vs. corre-sponding preischemic values; #P 0.05,##P 0.01 vs. corresponding normoxic val-ues; †P 0.05, ††P 0.01 vs. correspond-ing I/R control values.
H1572 REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

A M
MP-
2 ac
tivity
in c
oron
ary
ef
fluen
t (%
of n
orm
oxic
Pre
)
0
100
200
300
Pre R3 R30
Pre R30 R30 R30 Pre R30 R30 R30
C IHH
Pre R30 Pre R30
Normoxia
MM
P-2
mR
NA
/GA
PDH
(%
of n
orm
oxic
Pre
)
IHH Normoxia
0
40
80
120
R30 Pre
MMP-2
GAPDH
B
MMP-2
Normoxia IHH
GAPDH
- - + - - + Prazosin Doxycycline - - - + - - + -
Pre R30 R30 R30 Pre R30 R30 R30
-Actinin
TnI
D
GAPDH
Normoxia IHH
Prazosin - - + - - - - + Doxycycline - - - + - - + -
31 kDa
36 kDa
100 kDa
72 kDa
36 kDa
- -
IHH (10) Normoxia (10)
Normoxia+doxycycline (6) IHH+doxycycline (6) Normoxia+prazosin (6) IHH+prazosin (6)
Vent
ricul
ar M
MP-
2/G
APD
H
(% o
f nor
mox
ic P
re)
#
IHH Normoxia
0
40
80
120
†
R30 R30 Pre R30
**
Doxycycline - -
Prazosin + -
- - + -
†
Doxycycline
TnI/G
APD
H
(% o
f nor
mox
ic P
re)
0
40
80
120
IHH Normoxia
#
†
- - Prazosin
+ - - - + -
R30 R30 Pre R30
**
-A
ctin
in/G
APD
H
(% o
f nor
mox
ic P
re)
0
40
80
120
IHH Normoxia
#
†
Doxycycline - -
Prazosin + -
- - + -
R30 R30 Pre R30
**
†
†
†
* * † * * #
22 kDa
Fig. 3. Effects of IHH on the release, protein, and mRNA levels and sensitive substrates of MMP-2 during I/R with or without doxycycline and prazosin treatment.A: MMP-2 activity in coronary effluent. The number of preparations from individual rat hearts per group is indicated in parentheses. B: MMP-2 protein contentin LV tissue. C: MMP-2 mRNA content in LV tissue. D: �-actinin and troponin I (TnI) protein contents in LV tissue; n � 5 hearts from individual rat per group.**P 0.01 vs. corresponding preischemic (Pre) values; #P 0.05, ##P 0.01 vs. corresponding normoxic values; †P 0.05 vs. corresponding I/R controlvalues.
H1573REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

LVEDP, dP/dtmax, and RPP, was suppressed by I/R (30min/30 min), although the heart rate remained unchanged (Fig. 1).These suppressions were significantly improved by IHH, whilethe preischemic values and heart rate were comparable be-tween the two groups (Fig. 1). Consistently, IHH significantlyattenuated I/R-induced increases of LDH release in coronaryperfusate (an indicator of cell damage) at 3 and 30 min ofreperfusion and LVDP and RPP suppression and myocardialinfarct size after 2 h of reperfusion without affecting the heartrate (Fig. 2). Doxycycline (100 �M) mimicked the cardiopro-tective effects of IHH on the postischemic myocardial perfor-mance, LDH release, and infarct size, but it had no additiveprotective effects to the IHH and did not affect the preisch-emic values and heart rate during I/R (Figs. 1 and 2).Besides, the weight gain of normoxic and IHH rats wascomparable (IHH: from 120.5 1.4 to 323.5 2.4 g vs.normoxia: from 120.9 1.3 to 325.3 2.6 g, n � 30 each)as observed previously (58).
Because MMP-2 has been shown to play an important rolein early myocardial I/R injury (29), we next analyzed whetherIHH affects the MMP-2 activity and expression during I/R.The MMP-2 activity in coronary effluent was significantlyincreased at 3 min of reperfusion in the normoxic control group(Fig. 3A). Concomitantly, the protein content of 72-kDaMMP-2 in LV tissue from normoxic group was decreased (Fig.3B) because of its activation and release from myocytes duringreperfusion for the purpose of cell self-rescue (10), while themRNA level of MMP-2 remained unchanged after I/R (Fig.3C). In contrast, IHH attenuated I/R-induced MMP-2 activa-tion and release and preserved the myocardial MMP-2 proteincontent during reperfusion without affection the MMP-2mRNA level (Fig. 3, A–C). Moreover, �-actinin and TnI, twosensitive proteolysis substrates of MMP-2 (47, 54), were de-creased after I/R. The lower molecular mass fragment of TnI(�22 kDa) after I/R was also detected (Fig. 3D). Thesechanges were reversed by IHH or doxycycline, but both ofthem had no additive protective effects (Fig. 3, A, B, and D),indicating that the cardioprotection of IHH is mediated throughthe inhibition of MMP-2 activation during I/R.
IHH inhibits MMP-2 activation via reversing I/R-inducedONOO� overgeneration and TIMP-4 loss. ONOO� overgen-eration and TIMP-4 loss have been reported to undertake theresponsibility for the activation of MMP-2 (29, 44, 53). Wethus examined the effects of IHH on the alternations ofONOO� formation and TIMP-4 level due to I/R. I/R induceda marked increase of nitrotyrosine content (Fig. 4A), reflectingthe ONOO� formation (50), and a decrease of TIMP-4 proteinlevel in the LV (Fig. 4B), while these disorders were signifi-cantly attenuated by IHH (Fig. 4). Thus IHH reduced MMP-2activation against I/R injury through the inhibition of overgen-eration of ONOO� and loss of TIMP-4.
�1B-AR mediates the IHH-conferred cardioprotection on thecontractile function, cell survival, and MMP-2 activation.Because the activation of �1-ARs mimics IPC against I/Rinjury by activating endogenous adaptive responses and con-tributes to IHH-afforded cardioprotection (5, 18, 37, 52, 56),we then investigated a potential involvement of �1-ARs in theIHH-regulated MMP-2 activation. The �1-ARs antagonist pra-zosin at 1 �M abolished IHH-conferred improvement of post-ischemic myocardial performance, LDH release, and myocar-dial infarction, but it did not affect I/R-induced disorders (Figs.
1 and 2). Consistently, IHH-attenuated I/R-induced the activa-tion and release of MMP-2, reduction of �-actinin and TnIprotein contents, increase of nitrotyrosine content, and de-crease of TIMP-4 protein content were also abolished byprazosin (Fig. 3 and 4).
To determine if there were changes in the �1-AR subtype,we then performed competition ligand binding experiment asreported previously (67). The total density and affinity of�1-ARs at preischemia were comparable between the LV fromnormoxic and IHH rats, while IHH not only reversed I/R-decreased density of �1-ARs but also enhanced it further at 30min of reperfusion, although it did not alter the affinity of�1-ARs before or after I/R (Fig. 5, A and B). The densities of�1A-AR and �1D-AR at 30 min of reperfusion were lower thanthe values at preischemia, whereas the density of �1B-ARremained unchanged (Fig. 5C). However, IHH significantlyincreased the densities of �1A-AR and �1B-AR at both preisch-emia and reperfusion, but it decreased the density of �1D-ARonly at preischemia (Fig. 5C). Therefore, IHH changed the
†
TIMP-4
GAPDH
Pre R30 Pre R30 Pre R30 Pre R30
Normoxia IHH
TIM
P-4/
GA
PDH
(%
of n
orm
oxic
Pre
)
0
40
80
120
B
Prazosin - - + + - - + +
A
Nitr
otyr
osin
e co
nten
t (p
mol
/mg
prot
ein)
Normoxia IHH
0 1 2 3 4 5
## **
†
- - Prazosin + + R30 R30 Pre Pre
#
Normoxia IHH
- - Prazosin + + R30 R30 Pre Pre
**
22 kDa
36 kDa
*****
****
Fig. 4. Effects of IHH on the nitrotyrosine content and tissue inhibitor ofmetalloproteinases-4 (TIMP-4) protein level after I/R with or without prazosintreatment. A: nitrotyrosine content; n � 10 each in normoxic and IHH groupsand 6 each in normoxia � prazosin and IHH � prazosin groups fromindividual rat hearts in each group; B: protein levels of TIMP-4; n � 5 heartsfrom individual rat per group. **P 0.01 vs. corresponding preischemic (Pre)values; #P 0.05, ##P 0.01 vs. corresponding normoxic values; †P 0.05vs. corresponding I/R control value.
H1574 REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from
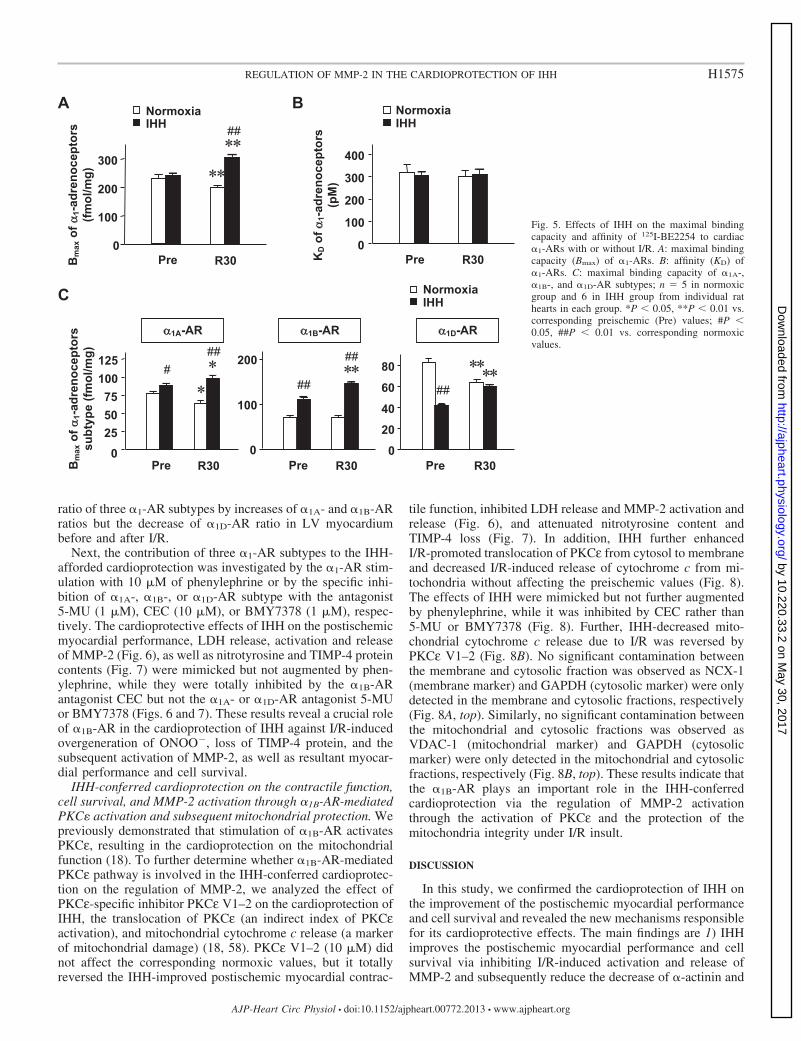
ratio of three �1-AR subtypes by increases of �1A- and �1B-ARratios but the decrease of �1D-AR ratio in LV myocardiumbefore and after I/R.
Next, the contribution of three �1-AR subtypes to the IHH-afforded cardioprotection was investigated by the �1-AR stim-ulation with 10 �M of phenylephrine or by the specific inhi-bition of �1A-, �1B-, or �1D-AR subtype with the antagonist5-MU (1 �M), CEC (10 �M), or BMY7378 (1 �M), respec-tively. The cardioprotective effects of IHH on the postischemicmyocardial performance, LDH release, activation and releaseof MMP-2 (Fig. 6), as well as nitrotyrosine and TIMP-4 proteincontents (Fig. 7) were mimicked but not augmented by phen-ylephrine, while they were totally inhibited by the �1B-ARantagonist CEC but not the �1A- or �1D-AR antagonist 5-MUor BMY7378 (Figs. 6 and 7). These results reveal a crucial roleof �1B-AR in the cardioprotection of IHH against I/R-inducedovergeneration of ONOO�, loss of TIMP-4 protein, and thesubsequent activation of MMP-2, as well as resultant myocar-dial performance and cell survival.
IHH-conferred cardioprotection on the contractile function,cell survival, and MMP-2 activation through �1B-AR-mediatedPKC� activation and subsequent mitochondrial protection. Wepreviously demonstrated that stimulation of �1B-AR activatesPKCε, resulting in the cardioprotection on the mitochondrialfunction (18). To further determine whether �1B-AR-mediatedPKCε pathway is involved in the IHH-conferred cardioprotec-tion on the regulation of MMP-2, we analyzed the effect ofPKCε-specific inhibitor PKCε V1–2 on the cardioprotection ofIHH, the translocation of PKCε (an indirect index of PKCεactivation), and mitochondrial cytochrome c release (a markerof mitochondrial damage) (18, 58). PKCε V1–2 (10 �M) didnot affect the corresponding normoxic values, but it totallyreversed the IHH-improved postischemic myocardial contrac-
tile function, inhibited LDH release and MMP-2 activation andrelease (Fig. 6), and attenuated nitrotyrosine content andTIMP-4 loss (Fig. 7). In addition, IHH further enhancedI/R-promoted translocation of PKCε from cytosol to membraneand decreased I/R-induced release of cytochrome c from mi-tochondria without affecting the preischemic values (Fig. 8).The effects of IHH were mimicked but not further augmentedby phenylephrine, while it was inhibited by CEC rather than5-MU or BMY7378 (Fig. 8). Further, IHH-decreased mito-chondrial cytochrome c release due to I/R was reversed byPKCε V1–2 (Fig. 8B). No significant contamination betweenthe membrane and cytosolic fraction was observed as NCX-1(membrane marker) and GAPDH (cytosolic marker) were onlydetected in the membrane and cytosolic fractions, respectively(Fig. 8A, top). Similarly, no significant contamination betweenthe mitochondrial and cytosolic fractions was observed asVDAC-1 (mitochondrial marker) and GAPDH (cytosolicmarker) were only detected in the mitochondrial and cytosolicfractions, respectively (Fig. 8B, top). These results indicate thatthe �1B-AR plays an important role in the IHH-conferredcardioprotection via the regulation of MMP-2 activationthrough the activation of PKCε and the protection of themitochondria integrity under I/R insult.
DISCUSSION
In this study, we confirmed the cardioprotection of IHH onthe improvement of the postischemic myocardial performanceand cell survival and revealed the new mechanisms responsiblefor its cardioprotective effects. The main findings are 1) IHHimproves the postischemic myocardial performance and cellsurvival via inhibiting I/R-induced activation and release ofMMP-2 and subsequently reduce the decrease of �-actinin and
KD o
f 1-
adre
noce
ptor
s (p
M)
Normoxia IHH
B
100
200
300
400
Pre R30 0
Bm
ax o
f 1-
adre
noce
ptor
s (fm
ol/m
g)
Normoxia IHH ##
A
Pre 0
100
200
300
R30
* *
C
Bm
ax o
f 1-
adre
noce
ptor
s
subt
ype
(fmol
/mg)
Normoxia IHH
Pre R30 Pre R30
##
0
25 50 75
100 125
###
Pre R30 0
20
40
60
80
##
0
100
200
##
1A-AR 1B-AR 1D-AR
* * * * * * * *
* *
Fig. 5. Effects of IHH on the maximal bindingcapacity and affinity of 125I-BE2254 to cardiac�1-ARs with or without I/R. A: maximal bindingcapacity (Bmax) of �1-ARs. B: affinity (KD) of�1-ARs. C: maximal binding capacity of �1A-,�1B-, and �1D-AR subtypes; n � 5 in normoxicgroup and 6 in IHH group from individual rathearts in each group. *P 0.05, **P 0.01 vs.corresponding preischemic (Pre) values; #P 0.05, ##P 0.01 vs. corresponding normoxicvalues.
H1575REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

TnI; 2) the inhibitory effect of IHH on the MMP-2 activationis dependent on the reversal of I/R-induced ONOO� overgen-eration and TIMP-4 loss; and 3) IHH inhibits MMP-2 activa-tion and release through the attenuation of I/R-inducedONOO� overgeneration and TIMP-4 loss via �1B-AR andsubsequently activated PKCε pathway. A schematic represen-tation for the IHH-stimulated protective pathway in the regu-lation of MMP-2 activation during I/R is shown in Fig. 9.These findings extend previous knowledge and provide a newinsight into the importance of �1B-AR/PKCε pathway on the
regulation of MMP-2 activation in the cardioprotection againstI/R injury.
Inhibiting MMP-2 activation plays a crucial role in thecardioprotection afforded by IHH. Cumulated evidence showsthat MMP-2 is involved in the acute myocardial I/R injury (1,10, 29, 54). MMP-2 widely locates in the sarcomere, cytosol,mitochondrion, nucleus, and other organelles of cardiomyocyte(12, 26, 29). Upon first reperfusion, MMP-2 is largely activatedand cleaves a series of proteins, such as TnI, myosin lightchain-1 (MLC-1), and titin, resulting in severe contractile
* *
D
LDH
rele
ase
(U/L
)
0
100200
300
400
#
* * # # †
†
†
500
E
MM
P-2
activ
ity in
cor
onar
y ef
fluen
t (%
of n
orm
oxic
Pre
)
0
100
200
300
#
* * # # †
400 †
†
0
50
100
150LV
DP
(mm
Hg)
A Normoxia IHH
##
* * # #
††
†
†
- - 5-MU CEC BMY V1-2 Phe
Pre R30
0
120
240
360
Hea
rt ra
te (b
pm)
B
0
10
30
40
Rat
e-pr
essu
re p
rodu
ct
(mm
Hg
x m
in-1
x 1
03)
C
- - 5-MU CEC BMY V1-2 Phe
Pre R30
- - 5-MU CEC BMY V1-2 Phe
Pre R3
- - 5-MU CEC BMY V1-2 Phe
Pre R3
- - 5-MU CEC BMY V1-2 Phe
Pre R30
Normoxia IHH
Normoxia IHH Normoxia IHH
Normoxia IHH
* *
*
20
* * ## * *
# # ††
†
†
Fig. 6. Effects of IHH on I/R-induced alternations of LV functional performance, LDH release and MMP-2 activation with or without the �1A-AR antagonist5-methylurapidil (5-MU; 1 �M), �1B-AR antagonist chloroethylclonidine (CEC; 10 �M), �1D-AR antagonist BMY7378 (BMY; 1 �M), PKCε inhibitor PKCεV1–2 (V1–2, 10 �M), or �1-AR agonist phenylephrine (Phe; 10 �M) treatment. A: LVDP. B: heart rate. C: rate-pressure product. D: LDH activity in coronaryeffluent. E: MMP-2 activity in coronary effluent; n � 10 in normoxic group; 7 in IHH group; 4 each in normoxia � 5-MU, IHH � 5-MU, normoxia � BMY7378,IHH � BMY7378, normoxia � V1–2 group, and IHH � V1–2 groups; 5 each in normoxia � CEC, normoxia � Phe, and IHH � Phe groups, and 6 in IHH �CEC group from individual rat hearts in each group. *P 0.05, **P 0.01 vs. corresponding preischemic (Pre) values; #P 0.05, ##P 0.01 vs. correspondingnormoxic values; †P 0.05, ††P 0.01 vs. corresponding I/R control values.
H1576 REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

dysfunction and cell death (1, 29, 47, 54), while the MMP-2inhibition decreases the intracellular protein degradation, im-proves the recovery of myocardial contractile dysfunction, andreduces infarct size in I/R hearts (10, 13, 17). Therefore,inhibiting the activation of MMP-2 appears to be a commondownstream mediator in various forms of cardioprotection-triggered intrinsic adaptive responses, such as IPC, postisch-emia conditioning, noninvasive limb IPC, and pharmacologicalconditioning (rosuvastatin and high-density lipoproteins) (6,13, 15, 31, 32). This view is supported by our findings here byusing of the chronic IHH adaptive model.
Since the first report for the cardiac beneficial effects ofaltitude anoxia in 1958 (30), more and more evidence hasshown that intermittent hypoxia, characterized by repeatedepisodes of hypoxia/reoxygenation, has profound effects on thesusceptibility of the myocardium to I/R injury (3, 34, 58, 61,62, 68). Moreover, IHH improves myocardial perfusion inpatients with severe coronary heart diseases (14) and has atherapeutic effect on the permanent coronary artery ligation-induced animal myocardial infarction by attenuating infarctsize and myocardial fibrosis and improving cardiac perfor-
mance (61). Therefore, IHH may offer beneficial effects topatients with acute myocardial infarction if the mechanisticinsights underlying the cardioprotective effects of IHH areachieved (14, 39). We previously showed that the activation ofprotein kinase A during the end of ischemia and early reper-fusion, phosphoinositide 3-kinase/protein kinase B (PI3K/Akt), calcium/calmodulin-dependent protein kinase II, reactiveoxygen species during the reperfusion, the preservation ofCa2� handing proteins abundance/activity at sarcolamma andsarcoplasmic reticulum, and the mitochondrial structure andfunction contribute to the IHH-conferred cardioprotection (9,58, 60, 62). These findings together with the observation of theregulation of MMP-2 activation by IHH here suggest that theprotective mechanisms in IHH are similar as the classic IPC(64), but the chronic IHH has more significant regulations inthe levels of protein expressions (57).
IHH inhibits MMP-2 activation during I/R through regulat-ing ONOO� generation and TIMP-4 loss. MMP-2 is tightlyregulated at several levels including transcriptional, posttran-scriptional, and posttranslational levels (29), although the spe-cific regulation under various physiological and pathophysio-
†
Pre R30
A N
itrot
yros
ine
cont
ent
(pm
ol/m
g pr
otei
n
Normoxia IHH
0
2
6
4 ## * *
## ##
††
†
- - 5-MU CEC BMY V1-2 Phe
0
40
80
120
TIM
P-4/
GA
PDH
(%
of n
orm
oxic
Pre
)
# # #
TIMP-4
GAPDH
- - 5-MU CEC BMY V1-2 Phe
Pre R30
Normoxia IHH
†
†
* *
B
5-MU CEC V1-2 5-MU BMY V1-2
Normoxia IHH R30
CEC
R30
BMY
22 kDa
Phe
Pre Pre
- - - - Phe
36 kDa
† *
Fig. 7. Effects of IHH on I/R-induced disorders ofnitrotyrosine content and TIMP-4 protein levelwith or without 5-MU, CEC, BMY7378, PKCεV1–2, or phenylephrine treatment. A: nitrotyrosinecontent; n � 10 in normoxic group; 7 in IHHgroup; 4 each in normoxia � 5-MU, IHH � 5-MU,normoxia � BMY7378, IHH � BMY7378, nor-moxia � V1–2 group, and IHH � V1–2 groups; 5each in normoxia � CEC, normoxia � Phe, andIHH � Phe groups; and 6 in IHH � CEC group fromindividual rat hearts in each group. B: TIMP-4protein level; n � 4 hearts from individual rat pergroup. *P 0.05, **P 0.01 vs. correspondingpreischemic (Pre) values; #P 0.05, ##P 0.01vs. corresponding normoxic values; †P 0.05,††P 0.01 vs. corresponding I/R control values.
H1577REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

logical conditions have not yet been fully clarified. Chronichypoxia (48 h) was reported to inhibit MMP-2 activation, butit does not affect the MMP-2 protein expression in humancardiac myofibroblasts (41). Similarly, chronic IHH alone doesnot alter LV MMP-2 protein level (46). This is supported byour data here showing that the chronic IHH does not alter thepreischemic MMP-2 mRNA and protein level but reduces thepostischemic MMP-2 activation in LV.
MMP-2 is synthesized as a proenzyme and can be activatedby proteolytic cleavage of its inhibitory pro-peptide domain(29). A large body of experimental evidence supports that theearly reperfusion leads to an increase in oxidative stress whenoxygen is reintroduced to ischemic tissue. Therefore, simulta-
neous production of NO and O2·� will react to generate
ONOO�, which activates the endogenic MMP-2 by oxidizingthe sulphydryl bond between a cysteine residue of the prodo-main and the Zn2� catalytic center and produces myocardialdysfunction (29, 53). Endogenous inhibitors, such as TIMPs,tightly control MMP-2 activity. Four TIMPs have been iden-tified to date, of which TIMP-4 is expressed predominantly inthe heart compared with others (29, 44). I/R injury-inducedrapid loss of myocardial TIMP-4 also results in an increase ofnet myocardial MMP-2 activity (33, 44). Moreover, the over-generation of ONOO� inhibits TIMP-4 activity, which turns toindirectly activate MMP-2 (16). Interestingly, we found herethat the IHH can reverse I/R-induced activation of MMP-2
Cyto-c
5-MU CEC V1-2 5-MU BMY V1-2
Normoxia IHH R30
CEC
R30
BMY
Mitochondrian Cytosol
Phe
Pre Pre
- - - - Phe
(14 kDa)
C
yto-
c (
Cyt
osol
/m
itoch
ondr
ian)
0.0
0.4
0.8
1.2
- - 5-MU CEC BMY V1-2 Phe
Pre R30
* *
## ## # ††
††
†† Normoxia IHH
PKCCytosol Membrane
5-MU CEC Phe 5-MU BMY Phe
Normoxia IHH R30
CEC
R30
BMY
Pre Pre
- - - -
(82 kDa)
PK
C (
Mem
bran
e/cy
toso
l)
- - 5-MU CEC BMY Phe
Pre R30
Normoxia IHH
*
#### ##
††
0.00.4 0.81.2 1.6
†† * *
Membrane Cytosol A GAPDH
NCX-1
36 kDa
110 kDa
Mito Cytosol B GAPDH
VDAC-1
36 kDa
31 kDa
Fig. 8. Translocation of PKCε from the cytosol tothe membrane fraction and release of mitochondrialcytochrome c (Cyto-c) from the mitochondrian tothe cytoplasmic fraction during preischemia (Pre)and I/R with or without 5-MU, CEC, BMY7378,PKCε V1–2, or phenylephrine treatment. A: repre-sentative immunoblots of the membrane and cyto-solic fractions and purity validated with the cyto-solic marker GAPDH and the membrane markerNCX-1 (top), and representative immunoblots(middle) and analysis (bottom) of PKCε transloca-tion. B: representative immunoblots of the cytosolicand mitochondria fractions and purity validatedwith the cytosolic marker GAPDH and the mito-chondrial marker VDAC-1 (top), and representativeimmunoblot (middle) and analysis (bottom) of mi-tochondrial cytochrome c release; n � 4 heartsfrom individual rat per group. *P 0.05, **P 0.01 vs. corresponding preischemic values; #P 0.05, ##P 0.01 vs. corresponding normoxic val-ues; ††P 0.01 vs. corresponding I/R controlvalues.
H1578 REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

through the posttranslational regulation by modulating theamount of two critical MMP-2 regulators, i.e., reducing theONOO� formation and TIMP-4 loss, and thus subsequentlydecreases I/R-induced MMP-2 activation. Whether othermechanisms are involved in the IHH-regulated MMP-2 acti-vation, such as MMP-2 phosphorylation and endogenous pro-teases (MT-MMPs) (29), needs to be further investigated.
The �1B-AR/PKC� pathway plays an important role in theregulation of MMP-2 activation. The PI3K/Akt and JAK2-ERK pathways have been shown to be involved in the modu-lation of MMP-2 activation under cardioprotection against I/Rinjury (8, 45). Here we showed another signaling pathwaymediated by �1B-AR/PKCε being involved in the regulation ofMMP-2 activation in the cardioprotection against I/R injury. Ithas been well documented that �1-AR activation induces thecardioprotection against I/R injury in animal experiments (5,18, 37, 52), while the role of �1-AR subtypes (�1A-AR,�1B-AR, and �1D-AR) in the cardioprotection remains contro-versial (18, 19, 25, 43). IHH has been shown to increase theactivity of �1-ARs and thus improve the contraction of papil-lary muscle during simulated ischemia (56). Our data herefurther reveal that IHH exerts cardioprotection via the in-creases of postischemic �1B-AR density and activity and thesubsequent activation of PKCε but not other �1-AR subtypes inrat hearts. This is consistent with the reports from other and ourlaboratories showing that the activation of �1B-AR/PKC path-way plays an important role in the IPC and �1-AR stimulation-conferred cardioprotection (18, 25). Consistent with our resultsof the increase in �1-AR density by the chronic IHH, an 8-wktreatment of testosterone has been shown to confer cardiopro-tection against I/R injury by upregulating the cardiac �1-ARand enhancing the effects of �1-AR stimulation (51). However,we could not find reports related to the increase of �1B-ARdensity in classic IPC. Therefore, the activation of �1B-AR/PKCε pathway appears to be a common mechanism in cardio-
protection, while the increase of �1B-AR density seems to bemainly involved in the chronic conditioning. This mechanismmay help the cell to deal with the stress more effectively andneeds to be further investigated for the correlation between theextent and duration of IHH with the magnitude of increases inthe �1B-AR density.
PKCε is a most abundant novel PKC isozyme found in adultrat cardiac myocytes and plays an essential role in the devel-opment of cardioprotection (18, 24, 58). Its activation, accom-panying with the translocation from cytosolic to particulatecompartments, has been showed to arouse the intrinsic cardio-protective mechanisms, such as opening mitochondrial ATP-sensitive potassium (mitoKATP) channels and maintaining mi-tochondrial integrity and function against I/R injury (18, 24,58). The protection on mitochondria attenuates oxidative stressand sequential O2
·� and ONOO� overgeneration (7, 48).Moreover, mitochondria protection-mediated decreases of ox-idative stress and intracellular Ca2� overloading alter theactivities of proteases and reduce intracellular protein lysis andrelease (18, 24, 36). Because IHH inhibits the opening ofmitochondrial permeability transition pores and opens mi-toKATP channels to enhance the mitochondria tolerance toreperfusion injury (3, 38, 59, 68), the mitochondrial protectionmediated by the IHH-stimulated �1B-AR/PKCε pathway maysubsequently attenuate O2
·� and sequential ONOO� overgen-eration and inhibit protease activation and sequential TIMP-4loss to reduce MMP-2 activation against I/R injury.
One limitation of the present study is that all of the data werecollected from the use of inhibitors or activator. To excludepotential nonspecific and side effects of those reagents, geneticmanipulation of �1-AR subtypes by overexpression or specificknockdown in the hearts from normoxic and IHH-adapted ratsneeds to be carried out to further confirm those findings.
In conclusion, our results demonstrate that IHH improvesthe postischemic recovery of myocardial dysfunction and celldeath via the suppressing of I/R-induced overgeneration ofONOO� and loss of TIMP-4 through the activation of the�1B-AR/PKCε pathway and the subsequent inhibition ofMMP-2 activation.
ACKNOWLEDGMENTS
We thank Jing-Long Liu, Yan-Jun Zhen, Ji-Liang Tang, Shan-Shan Gu,Xu-Xia Li, and Jing-Xi Wang for the assistance in animal exposure tohypobaric chamber.
GRANTS
This work was supported by the Major State Basic Research DevelopmentProgram of China (2012CB518203), National Science and Technology MajorProject of China (2012ZX09501001), and National Natural Science Founda-tion of China (81170119).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.G., Y.Z., and H.-T.Y. conception and design ofresearch; L.G., L.C., Z.-Z.L., H.G., L.W., Y.-X.C., C.-M.Z., and Y.-K.J.performed experiments; L.G. analyzed data; L.G. interpreted results of exper-iments; L.G. prepared figures; L.G. drafted manuscript; L.G., L.C., Q.J., andH.-T.Y. edited and revised manuscript; L.G., L.C., Z.-Z.L., H.G., L.W.,Y.-X.C., C.-M.Z., Y.-K.J., Q.J., Y.Z., and H.-T.Y. approved final version ofmanuscript.
Ischemia/reperfusion injury
Intermittent hypobaric hypoxia adaptation
Cardiac contractile dysfunction and cell
death
MMP-2 activation
1B-Adrenoceptoractivation
PKCactivation
ONOO-
over-generation
Mitochondrial function
conservation
TIMP-4decrease
Fig. 9. Schematic representation of proposed mechanisms underlying thecardioprotection of IHH against I/R injury through the regulation of MMP-2activation. Full line: induction/activation; dotted line: inhibition.
H1579REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

REFERENCES
1. Ali MA, Cho WJ, Hudson B, Kassiri Z, Granzier H, Schulz R. Titin isa target of matrix metalloproteinase-2: implications in myocardial isch-emia/reperfusion injury. Circulation 122: 2039–2047, 2010.
2. Ali MA, Stepanko A, Fan X, Holt A, Schulz R. Calpain inhibitorsexhibit matrix metalloproteinase-2 inhibitory activity. Biochem BiophysRes Commun 423: 1–5, 2012.
3. Asemu G, Papousek F, Ostadal B, Kolar F. Adaptation to high altitudehypoxia protects the rat heart against ischemia-induced arrhythmias.Involvement of mitochondrial K(ATP) channel. J Mol Cell Cardiol 31:1821–1831, 1999.
4. Aydemir-Koksoy A, Bilginoglu A, Sariahmetoglu M, Schulz R, TuranB. Antioxidant treatment protects diabetic rats from cardiac dysfunctionby preserving contractile protein targets of oxidative stress. J NutrBiochem 21: 827–833, 2010.
5. Banerjee A, Locke-Winter C, Rogers KB, Mitchell MB, Brew EC,Cairns CB, Bensard DD, Harken AH. Preconditioning against myocar-dial dysfunction after ischemia and reperfusion by an alpha 1-adrenergicmechanism. Circ Res 73: 656–670, 1993.
6. Bellosta S, Gomaraschi M, Canavesi M, Rossoni G, Monetti M,Franceschini G, Calabresi L. Inhibition of MMP-2 activation and releaseas a novel mechanism for HDL-induced cardioprotection. FEBS Lett 580:5974–5978, 2006.
7. Camara AK, Bienengraeber M, Stowe DF. Mitochondrial approaches toprotect against cardiac ischemia and reperfusion injury. Front Physiol 2:13, 2011.
8. Chan CY, Chen YS, Lee HH, Huang HS, Lai YL, Chen CF, Ma MC.Erythropoietin protects postischemic hearts by preventing extracellularmatrix degradation: role of Jak2-ERK pathway. Life Sci 81: 717–723,2007.
9. Chen L, Lu XY, Li J, Fu JD, Zhou ZN, Yang HT. Intermittent hypoxiaprotects cardiomyocytes against ischemia-reperfusion injury-induced al-terations in Ca2� homeostasis and contraction via the sarcoplasmic retic-ulum and Na�/Ca2� exchange mechanisms. Am J Physiol Cell Physiol290: C1221–C1229, 2006.
10. Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, SchulzR. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injuryin the heart. Circulation 101: 1833–1839, 2000.
11. Chow AK, Cena J, El-Yazbi AF, Crawford BD, Holt A, Cho WJ,Daniel EE, Schulz R. Caveolin-1 inhibits matrix metalloproteinase-2activity in the heart. J Mol Cell Cardiol 42: 896–901, 2007.
12. Coker ML, Doscher MA, Thomas CV, Galis ZS, Spinale FG. Matrixmetalloproteinase synthesis and expression in isolated LV myocyte prep-arations. Am J Physiol Heart Circ Physiol 277: H777–H787, 1999.
13. D=Annunzio V, Donato M, Erni L, Miksztowicz V, Buchholz B,Carrion CL, Schreier L, Wikinski R, Gelpi RJ, Berg G, Basso N.Rosuvastatin given during reperfusion decreases infarct size and inhibitsmatrix metalloproteinase-2 activity in normocholesterolemic and hyper-cholesterolemic rabbits. J Cardiovasc Pharmacol 53: 137–144, 2009.
14. Del PV, Garcia-Godos F, Woolcott OO, Marticorena JM, RodriguezV, Gutierrez I, Fernandez-Davila L, Contreras A, Valdivia L, RoblesJ, Marticorena EA. Improvement of myocardial perfusion in coronarypatients after intermittent hypobaric hypoxia. J Nucl Cardiol 13: 69–74,2006.
15. Donato M, D’Annunzio V, Buchholz B, Miksztowicz V, Carrion CL,Valdez LB, Zaobornyj T, Schreier L, Wikinski R, Boveris A, Berg G,Gelpi RJ. Role of matrix metalloproteinase-2 in the cardioprotectiveeffect of ischaemic postconditioning. Exp Physiol 95: 274–281, 2010.
16. Donnini S, Monti M, Roncone R, Morbidelli L, Rocchigiani M,Oliviero S, Casella L, Giachetti A, Schulz R, Ziche M. Peroxynitriteinactivates human-tissue inhibitor of metalloproteinase-4. FEBS Lett 582:1135–1140, 2008.
17. Fert-Bober J, Leon H, Sawicka J, Basran RS, Devon RM, Schulz R,Sawicki G. Inhibiting matrix metalloproteinase-2 reduces protein releaseinto coronary effluent from isolated rat hearts during ischemia-reperfusion.Basic Res Cardiol 103: 431–443, 2008.
18. Gao H, Chen L, Yang HT. Activation of alpha1B-adrenoceptors allevi-ates ischemia/reperfusion injury by limitation of mitochondrial Ca2�
overload in cardiomyocytes. Cardiovasc Res 75: 584–595, 2007.19. Gao XM, Wang BH, Woodcock E, Du XJ. Expression of active
alpha(1B)-adrenergic receptors in the heart does not alleviate ischemicreperfusion injury. J Mol Cell Cardiol 32: 1679–1686, 2000.
20. Giricz Z, Lalu MM, Csonka C, Bencsik P, Schulz R, Ferdinandy P.Hyperlipidemia attenuates the infarct size-limiting effect of ischemicpreconditioning: role of matrix metalloproteinase-2 inhibition. J Pharma-col Exp Ther 316: 154–161, 2006.
21. Gross G, Hanft G, Rugevics C. 5-Methyl-urapidil discriminates betweensubtypes of the alpha 1-adrenoceptor. Eur J Pharmacol 151: 333–335,1988.
22. Hanft G, Gross G. Subclassification of alpha 1-adrenoceptor recognitionsites by urapidil derivatives and other selective antagonists. Br J Phar-macol 97: 691–700, 1989.
23. Hassouna A, Matata BM, Galinanes M. PKC-epsilon is upstream andPKC-alpha is downstream of mitoKATP channels in the signal transductionpathway of ischemic preconditioning of human myocardium. Am J PhysiolCell Physiol 287: C1418–C1425, 2004.
24. Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, proteinkinases, mitochondria. Circulation 118: 1915–1919, 2008.
25. Hu K, Nattel S. Mechanisms of ischemic preconditioning in rat hearts.Involvement of alpha 1B-adrenoceptors, pertussis toxin-sensitive G pro-teins, and protein kinase C. Circulation 92: 2259–2265, 1995.
26. Hughes BG, Fan X, Cho WJ, Schulz R. MMP-2 is localized to themitochondria-associated membrane of the heart. Am J Physiol Heart CircPhysiol 306: H764–H770, 2014.
27. Hurtado A. Some clinical aspects of life at high altitudes. Ann Intern Med53: 247–258, 1960.
28. Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinaseC translocation inhibitor as an isozyme-selective antagonist of cardiacfunction. J Biol Chem 271: 24962–24966, 1996.
29. Kandasamy AD, Chow AK, Ali MA, Schulz R. Matrix metalloprotei-nase-2 and myocardial oxidative stress injury: beyond the matrix. Cardio-vasc Res 85: 413–423, 2010.
30. Kopecky M, Daum S. Adaptation of the myocardium to altitude anoxia.Cesk Fysiol 7: 218–219, 1958.
31. Lalu MM, Csonka C, Giricz Z, Csont T, Schulz R, Ferdinandy P.Preconditioning decreases ischemia/reperfusion-induced release and acti-vation of matrix metalloproteinase-2. Biochem Biophys Res Commun 296:937–941, 2002.
32. Li SJ, Wu YN, Kang Y, Yin YQ, Gao WZ, Liu YX, Lou JS.Noninvasive limb ischemic preconditioning protects against myocardialI/R injury in rats. J Surg Res 164: 162–168, 2010.
33. Mayers I, Hurst T, Puttagunta L, Radomski A, Mycyk T, Sawicki G,Johnson D, Radomski MW. Cardiac surgery increases the activity ofmatrix metalloproteinases and nitric oxide synthase in human hearts. JThorac Cardiovasc Surg 122: 746–752, 2001.
34. Meerson FZ, Gomzakov OA, Shimkovich MV. Adaptation to highaltitude hypoxia as a factor preventing development of myocardial isch-emic necrosis. Am J Cardiol 31: 30–34, 1973.
35. Meerson FZ, Kopylov I, Baldenkov GN. Increase of alpha 1-adrenore-activity of the rat heart in adaptation to periodic hypoxia. Biull Eksp BiolMed 111: 570–572, 1991.
36. Muller AL, Hryshko LV, Dhalla NS. Extracellular and intracellularproteases in cardiac dysfunction due to ischemia-reperfusion injury. Int JCardiol 164: 39–47, 2013.
37. Naderi R, Imani A, Faghihi M. Phenylephrine produces late pharmaco-logical preconditioning in the isolated rat heart. Eur J Pharmacol 627:203–208, 2010.
38. Neckar J, Szarszoi O, Koten L, Papousek F, Ost’Adal B, Grover GJ,Kolar F. Effects of mitochondrial K(ATP) modulators on cardioprotectioninduced by chronic high altitude hypoxia in rats. Cardiovasc Res 55:567–575, 2002.
39. Przyklenk K, Whittaker P. Cardioprotection via adaptation to hypoxia:expanding the timeline and targets? Basic Res Cardiol 106: 325–328,2011.
40. Rehring TF, Friese RS, Cleveland JC, Meng X, Robertson FG,Harken AH, Bannerjee A. Alpha-adrenergic preservation of myocardialpH during ischemia is PKC isoform dependent. J Surg Res 63: 324–327,1996.
41. Riches K, Morley ME, Turner NA, O’Regan DJ, Ball SG, Peers C,Porter KE. Chronic hypoxia inhibits MMP-2 activation and cellularinvasion in human cardiac myofibroblasts. J Mol Cell Cardiol 47: 391–399, 2009.
42. Rongen GA, Smits P, Thien T. Role of norepinephrine in ischemicpreconditioning in rabbit myocardium. Circulation 89: 2460–2461, 1994.
43. Rorabaugh BR, Ross SA, Gaivin RJ, Papay RS, McCune DF, SimpsonPC, Perez DM. alpha1A- but not alpha1B-adrenergic receptors precon-
H1580 REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from

dition the ischemic heart by a staurosporine-sensitive, chelerythrine-insensitive mechanism. Cardiovasc Res 65: 436–445, 2005.
44. Schulze CJ, Wang W, Suarez-Pinzon WL, Sawicka J, Sawicki G,Schulz R. Imbalance between tissue inhibitor of metalloproteinase-4 andmatrix metalloproteinases during acute myocardial ischemia-reperfusioninjury. Circulation 107: 2487–2492, 2003.
45. Spanikova A, Ivanova M, Matejikova J, Ravingerova T, Barancik M.Influence of ischemia/reperfusion and modulation of PI3K/Akt kinasepathway on matrix metalloproteinase-2 in rat hearts. Gen Physiol Biophys29: 31–40, 2010.
46. Strniskova M, Ravingerova T, Neckar J, Kolar F, Pastorekova S,Barancik M. Changes in the expression and/or activation of regulatoryproteins in rat hearts adapted to chronic hypoxia. Gen Physiol Biophys 25:25–41, 2006.
47. Sung MM, Schulz CG, Wang W, Sawicki G, Bautista-Lopez NL,Schulz R. Matrix metalloproteinase-2 degrades the cytoskeletal proteinalpha-actinin in peroxynitrite mediated myocardial injury. J Mol CellCardiol 43: 429–436, 2007.
48. Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, patho-physiology and development of therapeutics. Nat Rev Drug Discov 6:662–680, 2007.
49. Taliyan R, Singh M, Sharma PL, Yadav HN, Sidhu KS. Possibleinvolvement of alpha1-adrenergic receptor and K(ATP) channels in car-dioprotective effect of remote aortic preconditioning in isolated rat heart.J Cardiovasc Dis Res 1: 145–151, 2010.
50. Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, KochW, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection aftermyocardial ischemia/reperfusion involves the reduction of oxidative/ni-trative stress. Circulation 115: 1408–1416, 2007.
51. Tsang S, Wu S, Liu J, Wong TM. Testosterone protects rat hearts againstischaemic insults by enhancing the effects of alpha(1)-adrenoceptor stim-ulation. Br J Pharmacol 153: 693–709, 2008.
52. Tsuchida A, Liu Y, Liu GS, Cohen MV, Downey JM. Alpha 1-adren-ergic agonists precondition rabbit ischemic myocardium independent ofadenosine by direct activation of protein kinase C. Circ Res 75: 576–585,1994.
53. Wang W, Sawicki G, Schulz R. Peroxynitrite-induced myocardial injuryis mediated through matrix metalloproteinase-2. Cardiovasc Res 53:165–174, 2002.
54. Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G,Schulz R. Intracellular action of matrix metalloproteinase-2 accounts foracute myocardial ischemia and reperfusion injury. Circulation 106: 1543–1549, 2002.
55. Wang Y, Ashraf M. Activation of alpha1-adrenergic receptor duringCa2� preconditioning elicits strong protection against Ca2� overloadinjury via protein kinase C signaling pathway. J Mol Cell Cardiol 30:2423–2435, 1998.
56. Wang YP, Cui F, Zhang LP, Yang CY, Guan Y, Zhou ZN, Zhang Y.Effect of chronic intermittent hypobaric hypoxia on alpha(1)-adrenergic
receptor of myocardium participates in the cardioprotection. Sheng Li XueBao 61: 21–26, 2009.
57. Wang ZH, Cai XL, Wu L, Yu Z, Liu JL, Zhou ZN, Liu J, Yang HT.Mitochondrial energy metabolism plays a critical role in the cardioprotec-tion afforded by intermittent hypobaric hypoxia. Exp Physiol 97: 1105–1118, 2012.
58. Wang ZH, Chen YX, Zhang CM, Wu L, Yu Z, Cai XL, Guan Y, ZhouZN, Yang HT. Intermittent hypobaric hypoxia improves postischemicrecovery of myocardial contractile function via redox signaling duringearly reperfusion. Am J Physiol Heart Circ Physiol 301: H1695–H1705,2011.
59. Xie Y, Zhu WZ, Zhu Y, Chen L, Zhou ZN, Yang HT. Intermittent highaltitude hypoxia protects the heart against lethal Ca2� overload injury. LifeSci 76: 559–572, 2004.
60. Xie Y, Zhu Y, Zhu WZ, Chen L, Zhou ZN, Yuan WJ, Yang HT. Roleof dual-site phospholamban phosphorylation in intermittent hypoxia-in-duced cardioprotection against ischemia-reperfusion injury. Am J PhysiolHeart Circ Physiol 288: H2594–H2602, 2005.
61. Xu WQ, Yu Z, Xie Y, Huang GQ, Shu XH, Zhu Y, Zhou ZN, YangHT. Therapeutic effect of intermittent hypobaric hypoxia on myocardialinfarction in rats. Basic Res Cardiol 106: 329–342, 2011.
62. Yang H, Zhang Y, Wang ZH, Zhou ZN. Protective effects of chronicintermittent hypoxia against myocardial ischemia reperfusion injury. In:Intermittent Hypoxia and Human Diseases, edited by Lei X, Serebrovs-kaya TV. New York: Springer, 2012.
63. Yang HT, Endoh M. Dissociation of the positive inotropic effect ofmethoxamine from the hydrolysis of phosphoinositide in rabbit ventricularmyocardium: a comparison with the effects of phenylephrine and thesubtype of the alpha-1 adrenoceptor involved. J Pharmacol Exp Ther 269:732–742, 1994.
64. Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection byearly ischemic preconditioning. Cardiovasc Drugs Ther 24: 225–234,2010.
65. Yu GS, Han C. Role of alpha 1A- and alpha 1B-adrenoceptors inphenylephrine-induced positive inotropic response in isolated rat leftatrium. J Cardiovasc Pharmacol 24: 745–752, 1994.
66. Zhang CM, Gao L, Zheng YJ, Yang HT. Berbamine protects the heartfrom ischemia/reperfusion injury by maintaining cytosolic Ca2� homeo-stasis and preventing calpain activation. Circ J 76: 1993–2002, 2012.
67. Zhang YY, Xu KM, Han C. Alpha(1)-adrenoceptor subtypes mediatinginotropic responses in rat heart. J Pharmacol Exp Ther 291: 829–836,1999.
68. Zhu WZ, Xie Y, Chen L, Yang HT, Zhou ZN. Intermittent high altitudehypoxia inhibits opening of mitochondrial permeability transition poresagainst reperfusion injury. J Mol Cell Cardiol 40: 96–106, 2006.
69. Zhuang J, Zhou Z. Protective effects of intermittent hypoxic adaptationon myocardium and its mechanisms. Biol Signals Recept 8: 316–322,1999.
H1581REGULATION OF MMP-2 IN THE CARDIOPROTECTION OF IHH
AJP-Heart Circ Physiol • doi:10.1152/ajpheart.00772.2013 • www.ajpheart.org
by 10.220.33.2 on May 30, 2017
http://ajpheart.physiology.org/D
ownloaded from


















