
Accumulation of Immune Complexes in Glomerular Disease...
11
Accumulation of Immune Complexes in Glomerular Disease Is Independent of Locally Synthesized C3 Neil S. Sheerin, Katsushige Abe, Paul Risley, and Steven H. Sacks Department of Nephrology and Transplantation, Guy’s Hospital, King’s College London, London, United Kingdom Although complement activation can make immune complex glomerulonephritis worse, the third complement component also can solubilize immune complexes and thus reduce the severity of disease. How C3 that is produced within the kidney contributes to this balance is unknown. This study therefore investigated the relative roles of systemic and local C3 production in a model of glomerular immune complex disease. Injection of sheep anti– glomerular basement membrane antibody into preimmunized mice resulted in accumulation of immune complexes and progressive loss of function over 14 d that was much more marked in C3-deficient (C3 / ) mice. In C3-sufficient mice that received a transplant of a C3 / mouse kidney and in C3 / mice with C3-sufficient mouse kidney transplants, the severity and the pattern of injury went with the complement status of the recipient. That is, mice with deficient circulating C3 developed severe glomerular immune complex disease, whereas those with a high level of circulating C3 had well-preserved glomerular structure and function. It is concluded that circulating C3 is a critical factor in reducing the glomerular accumulation of immune complexes. Local synthesis of C3 did not have a major influence on this aspect of glomerular disease. J Am Soc Nephrol 17: 686 – 696, 2006. doi: 10.1681/ASN.2004070515 I t is widely known that complement plays a role in the pathogenesis of immune complex disease. However, the nature of this role is controversial, because complement activation can mediate inflammatory injury (1–3), whereas de- ficiency, especially of the early components, can predispose toward immune complex disease. In particular, C1q deficiency has been associated with reduced clearance of apoptotic cells (4) and the development of antibody-mediated glomerulone- phritis (5). C3 has an important role in the removal of immune complexes both from the circulation and from the glomerulus (6 – 8), and failure can result in glomerulonephritis. It is likely that the effect of complement in the pathogenesis of glomerular disease may be dependent on the stage of the disease (9). Acute glomerular inflammation after a single injection of anti– glo- merular basement antibody is reduced in the absence of com- plement (1,10), whereas the slower accumulation of glomerular complexes that are formed by autologous antibody may be increased (9). A further complexity is that whereas a large amount of complement, produced in the liver (11), is present in the circu- lation, smaller, although significant, quantities can be synthe- sized locally in the extravascular space. Circulating compo- nents are instantly available, whereas tissue-specific components are produced in a regulated manner after the induction of disease or after exposure of cultured cells to var- ious inflammatory stimuli. In the kidney, glomerular epithelial (12), mesangial (13,14), and endothelial cells (15) have the capacity to synthesize C3, a pivotal component of the com- plement cascade that is able to mediate the clearance of immune complexes as well as induce inflammation. In addi- tion, renal tubule cells can produce C3 (16). After the induc- tion of glomerular disease in rats, tissue expression of C3 mRNA is increased, and the degree of protein leak from the injured kidney correlates positively with the level of C3 gene expression (17). It therefore has been proposed that locally synthesized C3 may have a role in the pathogenesis of renal injury and that this function may differ from that of circu- lating complement. To assess the contribution of systemic and local production of C3, we investigated the development of immune complex glo- merulonephritis after the administration of anti– glomerular basement antibody to mice. We specifically examined the de- velopment of this disease in mice that lack either circulating C3 or renally produced C3, achieved by transplanting kidneys between mice that are C3 deficient and C3 sufficient. Materials and Methods Mice The C3-deficient (C3 / ) mice were originally generated by homol- ogous recombination in the laboratory of Prof. M.C. Carroll (18). The mice have no detectable C3 by ELISA. Although originally generated on a mixed C57Bl/6 129 background, the C3 / mice that were used in these experiments had been backcrossed six generations onto a C57Bl/6 background. Skin that was engrafted from these mice onto C57Bl/6 mice showed no evidence of rejection (data not shown). C3- sufficient C57Bl/6 (C3 / ) mice were bred in our own facility. All animal experiments were performed in accordance with United King- dom Home Office regulations. Received July 1, 2004. Accepted December 7, 2005. Published online ahead of print. Publication date available at www.jasn.org. Address correspondence to: Dr. Neil Sheerin, Department of Nephrology and Transplantation, 5th Floor, Thomas Guy House, Guy’s Hospital, St. Thomas’ Street, London, SE1 9RT. Phone: 44-207-955-4305; Fax: 44-207-955-4303; E- mail: [email protected] Copyright © 2006 by the American Society of Nephrology ISSN: 1046-6673/1703-0686
-
Upload
doannguyet -
Category
Documents
-
view
215 -
download
1
Transcript of Accumulation of Immune Complexes in Glomerular Disease...

Accumulation of Immune Complexes in Glomerular DiseaseIs Independent of Locally Synthesized C3
Neil S. Sheerin, Katsushige Abe, Paul Risley, and Steven H. SacksDepartment of Nephrology and Transplantation, Guy’s Hospital, King’s College London, London, United Kingdom
Although complement activation can make immune complex glomerulonephritis worse, the third complement component alsocan solubilize immune complexes and thus reduce the severity of disease. How C3 that is produced within the kidneycontributes to this balance is unknown. This study therefore investigated the relative roles of systemic and local C3 productionin a model of glomerular immune complex disease. Injection of sheep anti–glomerular basement membrane antibody intopreimmunized mice resulted in accumulation of immune complexes and progressive loss of function over 14 d that was muchmore marked in C3-deficient (C3�/�) mice. In C3-sufficient mice that received a transplant of a C3�/� mouse kidney and inC3�/� mice with C3-sufficient mouse kidney transplants, the severity and the pattern of injury went with the complementstatus of the recipient. That is, mice with deficient circulating C3 developed severe glomerular immune complex disease,whereas those with a high level of circulating C3 had well-preserved glomerular structure and function. It is concluded thatcirculating C3 is a critical factor in reducing the glomerular accumulation of immune complexes. Local synthesis of C3 did nothave a major influence on this aspect of glomerular disease.
J Am Soc Nephrol 17: 686–696, 2006. doi: 10.1681/ASN.2004070515
I t is widely known that complement plays a role in thepathogenesis of immune complex disease. However, thenature of this role is controversial, because complement
activation can mediate inflammatory injury (1–3), whereas de-ficiency, especially of the early components, can predisposetoward immune complex disease. In particular, C1q deficiencyhas been associated with reduced clearance of apoptotic cells(4) and the development of antibody-mediated glomerulone-phritis (5). C3 has an important role in the removal of immunecomplexes both from the circulation and from the glomerulus(6–8), and failure can result in glomerulonephritis. It is likelythat the effect of complement in the pathogenesis of glomerulardisease may be dependent on the stage of the disease (9). Acuteglomerular inflammation after a single injection of anti–glo-merular basement antibody is reduced in the absence of com-plement (1,10), whereas the slower accumulation of glomerularcomplexes that are formed by autologous antibody may beincreased (9).
A further complexity is that whereas a large amount ofcomplement, produced in the liver (11), is present in the circu-lation, smaller, although significant, quantities can be synthe-sized locally in the extravascular space. Circulating compo-nents are instantly available, whereas tissue-specificcomponents are produced in a regulated manner after theinduction of disease or after exposure of cultured cells to var-ious inflammatory stimuli. In the kidney, glomerular epithelial
(12), mesangial (13,14), and endothelial cells (15) have thecapacity to synthesize C3, a pivotal component of the com-plement cascade that is able to mediate the clearance ofimmune complexes as well as induce inflammation. In addi-tion, renal tubule cells can produce C3 (16). After the induc-tion of glomerular disease in rats, tissue expression of C3mRNA is increased, and the degree of protein leak from theinjured kidney correlates positively with the level of C3 geneexpression (17). It therefore has been proposed that locallysynthesized C3 may have a role in the pathogenesis of renalinjury and that this function may differ from that of circu-lating complement.
To assess the contribution of systemic and local production ofC3, we investigated the development of immune complex glo-merulonephritis after the administration of anti–glomerularbasement antibody to mice. We specifically examined the de-velopment of this disease in mice that lack either circulating C3or renally produced C3, achieved by transplanting kidneysbetween mice that are C3 deficient and C3 sufficient.
Materials and MethodsMice
The C3-deficient (C3�/�) mice were originally generated by homol-ogous recombination in the laboratory of Prof. M.C. Carroll (18). Themice have no detectable C3 by ELISA. Although originally generatedon a mixed C57Bl/6 � 129 background, the C3�/� mice that were usedin these experiments had been backcrossed six generations onto aC57Bl/6 background. Skin that was engrafted from these mice ontoC57Bl/6 mice showed no evidence of rejection (data not shown). C3-sufficient C57Bl/6 (C3�/�) mice were bred in our own facility. Allanimal experiments were performed in accordance with United King-dom Home Office regulations.
Received July 1, 2004. Accepted December 7, 2005.
Published online ahead of print. Publication date available at www.jasn.org.
Address correspondence to: Dr. Neil Sheerin, Department of Nephrology andTransplantation, 5th Floor, Thomas Guy House, Guy’s Hospital, St. Thomas’Street, London, SE1 9RT. Phone: �44-207-955-4305; Fax: �44-207-955-4303; E-mail: [email protected]
Copyright © 2006 by the American Society of Nephrology ISSN: 1046-6673/1703-0686

Measurement of Anti-Sheep IgG TitersTo determine whether C3�/� and C3�/� mice had similar anti-sheep
IgG antibody titers, we injected 1 mg of sheep IgG in complete Freund’sadjuvant (CFA) into 10 mice from each strain. Blood was taken at days5, 12, and 19. Ninety-six-well plates were coated with 20 �g/ml sheepIgG in PBS overnight at 4°C. Plates were blocked with 1% BSA, andserial dilutions of serum in PBS–1% BSA were added for 1 h at 37°C.After washing, the plates were incubated with 1:1000 dilution of goatanti-mouse IgG horseradish peroxidase (HRP) conjugate (Stratech Sci-entific Ltd, Soham, UK) or rabbit isotype-specific alkaline phosphateconjugate (Southern Biotechnology Associates Inc., Birmingham, AL)for 1 h at 37°C, washed, and developed with O-phenylenediamine orp-Nitrophenyl phosphate.
Induction of Glomerular Injury and the Transplant ModelAnti–glomerular basement membrane (GBM) antibody–mediated
glomerular injury was induced in 6- to 8-wk-old female C3�/� andC3�/� mice (13 mice per group). Five days before the injection ofanti-GBM antiserum, mice were immunized subcutaneously with 1 mgof sheep IgG (Sigma, Dorset, UK) in CFA. Before the injection ofanti-GBM antiserum, mice were housed in metabolic cages to measurebaseline albuminuria, and blood was taken to measure serum albuminand urea concentrations. Mice received an injection of 50 �l of sheepanti-GBM antiserum (Dr. D.J. Salant, Boston, MA) intravenously on 3consecutive days. Urine and blood were collected at days 7 and 14 afterthe first injection of anti-GBM antiserum. Mice were killed at day 14,and their kidneys were harvested for histologic analysis.
Heterotropic kidney transplantation was performed in two donor–recipient combinations: Group 1, C3�/� donor mouse to C3�/� mouserecipient (n � 4); and group 2, C3�/� donor mouse to C3�/� mouserecipient (n � 4). The resulting mice either were deficient in locallyproduced C3 in the transplanted kidney but with intact systemic pro-duction (group 1) or had intact local C3 production but deficientsystemic production (group 2). The donor and recipient aorta and venacava were joined by end-to-side anastomosis. A patch of donor bladderwas attached to the recipient bladder (19). Unilateral recipient nephrec-tomy was performed at the time of transplantation. After a period ofrecovery (10 d), a second native nephrectomy was performed so thatthe only source of renal function was from the transplanted kidney.Preliminary studies demonstrated that the transplanted kidney washistologically normal 14 d after transplantation, with donor and graftsurvival for at least 3 mo with no evidence of rejection. Fourteen daysafter transplantation, anti-GBM antibody–mediated glomerular injurywas induced.
Assessment of Renal Functional InjuryUrine albumin concentration was measured by ELISA, and the 24-h
urinary albumin excretion was calculated. Ninety-six-well plates werecoated with 5 �g/ml goat anti-mouse albumin (Nordic ImmunologicalLaboratories, Tilburg, The Netherlands) in carbonate buffer (pH 9.6)overnight at 4°C. After blocking with 2% BSA at 37°C for 2 h, samplesor control dilutions of mouse albumin (Sigma) in blocking buffer wereadded to the plates for 2 h at 37°C. A secondary HRP-conjugated goatanti-mouse albumin (Nordic) in blocking buffer was added for 2 h at37°C. Absorbance was measured after incubation with O-phenylenedi-amine. Serum urea and albumin concentrations were measured usingSigma kits according to the manufacturer’s protocols.
Assessment of Histologic InjuryTo assess histologic injury we stained formalin-fixed, wax-embedded
kidney sections (2 �m thick) with periodic acid-Schiff reagent (PAS).
The severity of injury was scored by a blinded observer according tothe following scheme: 0, normal; 1, mild (small areas of glomerularabnormalities); 2, moderate (�50% of the glomerulus affected by ne-crosis or crescent formation); and 3, severe (�50% of the glomerulusaffected by necrosis or crescent formation) (20). Fifty glomeruli wereassessed for each animal.
Immunohistochemical StainingCryostat sections (5 �m) were fixed with acetone at 4°C for 10 min.
Deposition of mouse IgG, mouse IgM, and sheep IgG was detected withHRP-conjugated anti-mouse IgG, HRP-conjugated anti-mouse IgM(both Stratech), and HRP-conjugated anti-sheep IgG (Serotec, Oxford,UK; preabsorbed against sheep and mouse Ig, respectively, and lack ofcross-reactivity tested by ELISA [data not shown]). For C3 staining, theprimary antibody was rabbit anti-human C3d (Dako Ltd, Cam-bridgeshire, UK) and a secondary HRP-conjugated goat anti-rabbit IgG.For C4 staining, the primary antibody was rat anti-mouse C4 and asecondary HRP-conjugated donkey anti-rat (Stratech). HRP-antibodywas detected with diaminobenzidine. Sections were counterstainedwith methyl green.
Fifty glomeruli from each animal were examined, and a semiquan-titative assessment of the intensity of the immunochemical staining wasperformed using a scale from 0 to 3 (0, negative; 1, weak; 2, moderate;3, strong staining) according to the methods described previously (21).
Electron MicroscopyTissue was prefixed with 2.5% glutaraldehyde and postfixed with
osmium tetroxide. Sections were stained with lead citrate and viewedon a transmission electron microscope (Hitachi H7000; Hitachi, Berk-shire, UK). A minimum of three glomeruli were assessed in eachmouse.
Demonstration of Renal C3 SynthesisUsing standard phenol-chloroform methods, RNA was extracted
from the renal cortex of transplanted kidney, and cDNA was synthe-sized. C3 gene expression was detected by reverse transcription–PCRusing the primer sequences 5�-TCACACACCGAAGAAGACTGCC-3�
and 5�-GTGGCTGATGAACTTGCGTTGC-3� (product size 407 bp).�-Actin gene expression (5�-GAGCAAGAGAGGTATCCTGACC-3�
and 5�-GGATGCCACAGGATTCCATACC-3�) was used as an internalcontrol. All amplifications were performed in the linear phase of am-plification (28 cycles: 94°C for 1 min; 60°C for 1 min; 72°C for 2 min).The specific PCR product was not seen in non–reverse-transcribedmRNA or genomic DNA controls with the primers used (data notshown).
In situ hybridization for C3 mRNA was performed using a 30-baseantisense oligonucleotide probe, corresponding to bases 167 to 196 ofmouse C3 cDNA. The probe was labeled using digoxigenin (DIG)oligonucleotide tailing kit according to the manufacturer’s instructions(Boehringer Mannheim, Lewes, UK). Frozen sections (4 �m) were fixedwith 4% paraformaldehyde in PBS. The sections were deproteinized byusing HCl and proteinase K, prehybridized, and then hybridized withDIG-labeled oligonucleotide probe in prehybridization buffer at 37°Covernight. After washing with 2� SSC, the DIG-labeled probe wasvisualized using HRP-conjugated sheep polyclonal anti-DIG antibody(Boehringer Mannheim) and diaminobenzidine (22). Control studieswith a sense probe and competitive binding studies with sense andantisense probe confirmed specificity.
J Am Soc Nephrol 17: 686–696, 2006 C3 and Renal Immune Complex Clearance 687

Statistical AnalysesData were expressed as mean � SD. Differences between different
groups were tested for statistical significance using one-way ANOVAwith Scheffe’s F test. P � 0.05 was taken as statistically significant.
ResultsIn the first phase, anti-GBM antibody–mediated glomerulo-
nephritis was studied in the C3�/� and C3�/� back-crossedmice. The second phase used a transplantation strategy todefine whether locally synthesized C3 plays any role in thedevelopment of injury.
Assessment of Anti-Sheep IgG Titers in C3�/� andC3�/� Mice
C3�/� and C3�/� mice developed similar anti-sheep IgGtiters 12 (Figure 1A) and 19 (Figure 1B) days after immuniza-tion with 1 mg of sheep IgG in CFA. Titers were low in bothgroups at day 5. In addition, the titers of IgG1 and IgG2b weresimilar in the two groups of mice (Figure 1, C and D). Very littleIgG2a or IgG3 was detected in either group of mice (data notshown).
Assessment of Antibody-Mediated Injury in C3�/� andC3�/� Mice
Before the injection of anti-GBM antiserum, C3�/� andC3�/� mice had equivalent low levels of albuminuria. Sevendays after the first injection, C3�/� mice had significantlygreater albuminuria than the C3�/� mice (57.5 � 30.1 versus4.3 � 3.6 mg/24 h, respectively; P � 0.01). This differencepersisted to day 14 (62.3 � 17.4 versus 9.9 � 4.5 mg/24 h; P �
0.01). By day 14 after the induction of disease, the serum ureawas significantly higher in the C3�/� mice than in the C3�/�
mice (31.2 � 8.0 and 9.5 � 2.1 mmol/L, respectively; P � 0.01).These observations confirm greater disruption to glomerularfiltration function in the mice that are deficient in C3.
Histologic analysis 14 d after the induction of disease dem-onstrated that severity of the injury was dependent on thecomplement status of the mice: The C3�/� mice showed onlyminor glomerular changes (Figure 2, A and C). In contrast, thekidneys from C3�/� mice showed significant glomerular injurywith glomerular hypercellularity and crescent formation. Therewas also capillary wall thickening with deposition of PAS-positive material (Figure 2, B and C).
Immunohistochemical Analysis of Diseased KidneysThe kidneys from C3�/� and C3�/� mice 14 d after disease
induction were examined for the presence of C3, C4, sheep IgG,and mouse IgG and IgM. C3, C4, and IgM can be detected in theglomeruli of normal mice in a mesangial distribution (data notshown). Increased C3 deposition was detected in the glomeruliof C3�/� mice after disease induction. C3 was deposited inboth a pericapillary and mesangial distribution (Figure 3A). Asexpected, there was no C3 deposition in the kidneys fromC3�/� mice (Figure 3B). C4 staining was seen in the glomeruliof both C3�/� and C3�/� mice predominantly in a mesangialdistribution (Figure 3, C and D, respectively). The intensity ofstaining was greater, as assessed on a semiquantitative scale, inthe C3�/� mice (Table 1).
Staining for sheep IgG (Figure 3, E and F), mouse IgG (Figure
Figure 1. Anti-sheep antibody titers in C3�/� (f) and C3�/� (Œ) mice. IgG titers were measured 12 (A) and 19 (B) days after theinjection of 1 mg of sheep IgG in complete Freund’s adjuvant (CFA). IgG1 (C) and IgG2b (D) titers were also assessed in the day19 sample. Serum from nonimmunized mice was also included (�). n � 10 per group.
688 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 686–696, 2006

3, G and H), and mouse IgM (Figure 3, I and J) was moreintense in the C3�/� mice than in the C3�/� mice. Antibodystaining was predominantly in a pericapillary distribution, al-though some mesangial staining was also seen. Using a semi-quantitative scoring system, this reached statistical significancein all three cases (Table 1). Increased deposition of Ig in theglomeruli of C3�/� mice could explain the increased amountsof C4 detected in these mice. We previously reported that theC3�/� mice have immune complexes within their glomerulimore frequently than do control C3�/� mice (9). This was trueagain with this group of mice (data not shown) and is consis-tent with the increased glomerular deposition of Ig and C4 inC3�/� mice.
Assessment of Functional Injury in Mice with a TransplantThe data above suggest that, after intravenous injection of
anti-GBM antibody, mice that are totally deficient in C3 accu-mulate immune-reactive material in their glomeruli and subse-quently develop more severe renal injury. To dissect the rela-tive importance of circulating and locally synthesized C3, weused a renal transplantation model.
Fourteen days after the second native nephrectomy, the micethat received a transplant in both groups 1 and 2 seemedhealthy. Before the induction of anti-GBM antibody–mediatedglomerulonephritis, the serum urea was equivalent in both
groups but approximately two-fold higher than in normal un-manipulated mice (Table 2), as a consequence of the animals’surviving on a single transplanted kidney. The serum albuminwas also higher in the mice that received a transplant before theinduction of disease. Before disease induction, there was min-imal albuminuria in mice from both transplant groups (Figure4). After the induction of disease, albuminuria developed inboth groups but was significantly greater in mice from group 2(C3�/� donor: C3�/� recipient) at both days 7 and 14 (P � 0.01at both time points). Fourteen days after the induction of dis-ease, C3�/� mice that received a kidney from a C3�/� donor(group 2) had severe functional disturbance with a significantrise in serum urea and reduction in serum albumin (P � 0.01;Table 2). In contrast, the renal function of mice in group 1 wasnot altered significantly after the induction of disease. There-fore, the functional disturbance seen after disease induction inthe mice that received a transplant was dependent on thecomplement status of the recipient rather than that of the donorkidney.
Assessment of Histologic Injury in Kidney TransplantsThe glomeruli of the group 1 mice were hypercellular and
exhibited areas of minor sclerosis (Figure 5A). The glomeruli ofmice in group 2 showed marked histologic damage, includingcapillary occlusion with PAS-positive material and areas of
Figure 2. Histology of mouse kidney 14 d after the induction of anti–glomerular basement membrane (anti-GBM) antibody(Ab)-mediated glomerulonephritis. Representative histology of C3�/� mice (A) and C3�/� mice (B) is shown. (C) Semiquanti-tative analysis of the severity of glomerular damage was performed on a minimum of 50 glomeruli from all mice (n � 13) in eachgroup. Magnification, �400 in A and B, periodic acid-Schiff (PAS) staining.
J Am Soc Nephrol 17: 686–696, 2006 C3 and Renal Immune Complex Clearance 689

focal proliferation and crescent formation (Figure 5B). Scoringof the glomerular histologic changes confirmed the greaterinjury in group 2 mice (2.5 � 0.6) compared with group 1 mice(1.4 � 0.2; P � 0.01). Similarly, the tubulointerstitium in C3�/�
mice that received a C3�/� kidney (group 1) was well pre-served in contrast to mice from group 2 that had tubulardilation, epithelial cell flattening expansion of the interstitialcompartment with evidence of a mononuclear cell infiltrate.
Immunohistochemical Analysis of Transplanted KidneysComplement C3 was deposited in the glomeruli of mice from
both transplant groups 14 d after disease induction (Figure 6, Athrough D). Because mice from group 2 lack systemic C3 pro-duction, the C3 that is deposited in the glomeruli must be ofdonor kidney origin, presumably derived from resident renal
Figure 3. Immunohistochemistry of mouse kidney after theinduction of anti-GBM Ab-mediated glomerulonephritis. Stain-ing for C3 (A and B), C4 (C and D), sheep IgG (E and F), mouseIgG (G and H), and mouse IgM (I and J) was performed onC3�/� mice (A, C, E, G, and I) and C3�/� mice (B, D, F, H, andJ). Magnification, �400.
Table 1. Semiquantitative assessment of the intensity ofstaining for complement and Ig in the C3�/� and C3�/�
mice 14 d after disease inductiona
C3�/� Mice C3�/� Mice
C3 � �C4 0.6 � 0.2 1.0 � 0.4b
Sheep IgG 1.9 � 0.4 2.6 � 0.5b
Mouse IgG 0.8 � 0.4 1.3 � 0.4b
Mouse IgM 1.2 � 0.3 1.7 � 0.4b
aData are mean � SD.bP � 0.01 versus C3�/� mice.
Table 2. Serum urea and albumin in mice that receiveda transplanta
Group 1 Group 2
Serum albumin (g/dl)day �1 4.7 � 0.2 4.6 � 0.3day 14 4.3 � 2.5 1.6 � 0.4b
Serum urea (mmol/L)day �1 17.3 � 2.8 18.6 � 6.4day 14 11.8 � 7.2 129 � 10.7c
aData are mean � SD, n � 4 for both groups.bP � 0.05 versus group 1 at day 14.cP � 0.01 versus group 1 at day 14.
Figure 4. 24-hour urinary albumin excretion (mg/24 h) in themice that received a transplant. Albuminuria in group 1 mice(C3�/� donor: C3�/� recipient; dotted line) was significantlyless than group 2 mice (C3�/� donor: C3�/� recipient; solidline); n � 4 per group.
690 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 686–696, 2006

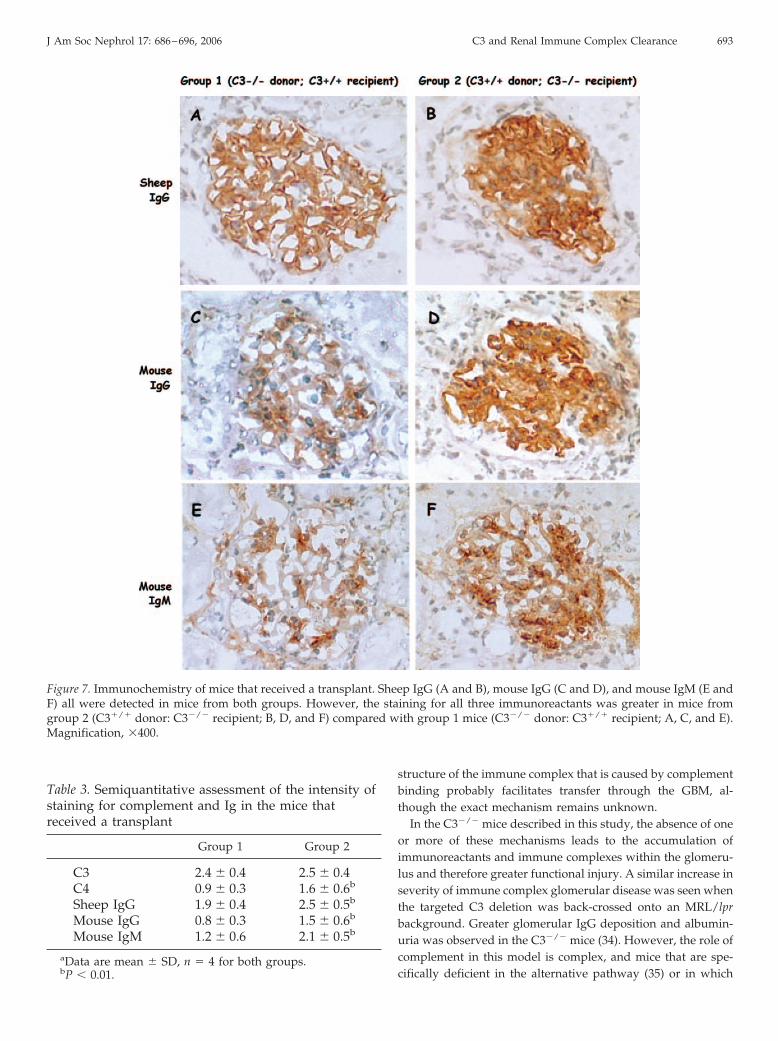
cells. Staining for C3 was present in a capillary wall and mes-angial distribution. C4 was also seen in the glomeruli of micefrom both groups, as in the mice that did not received a trans-plant, in a mesangial distribution, suggesting activation of theclassical pathway of complement (Figure 6, E and F). Sheep IgGand mouse IgG and IgM were deposited in the glomeruli ofmice from both groups in both the capillary walls and themesangium (Figure 7). Semiquantitative assessment of the in-tensity of staining showed greater intensity of C4, sheep IgG,and mouse IgG and IgM in group 2 mice (Table 3). Therefore,as with the functional changes, the pattern of deposition ofimmunoreactive proteins followed the complement status ofthe recipient. We cannot exclude an effect of nonspecific proteintrapping in the glomeruli of the more proteinuric mice in group2, in particular the mesangial IgM.
Electron Microscopy on Kidneys from Mice with aTransplant
The kidneys from mice in group 1 (Figure 8A) and group 2(Figure 8B) were examined by electron microscopy. In theglomeruli of mice from group 2, there were electron-denseimmune complexes in all mice examined (three of three). Inaddition, there was epithelial cell foot-process effacement andendothelial cell swelling. Evidence of immune complex depo-sition was detected in only one mouse (one of four) from group1, and the structural changes were less severe than those seen ingroup 2.
C3 Production within Transplanted KidneysPCR was used to analyze reverse-transcribed mRNA from
the renal cortex of mice that received a transplant. C3 mRNAwas detected in the cortex of both groups (Figure 9A), suggest-ing that C3 gene expression was occurring in both infiltratingcells (group 1) and native renal cells (group 2). To localizecortical C3 gene expression further, we performed in situ hy-bridization. In kidney transplants that were derived from aC3�/� donor, C3 message was detected only in infiltrating cells
around damaged tubules (Figure 9B). When the kidney wasderived from a C3�/� donor, glomerular and tubular C3 geneexpression was evident (Figure 9C). The result with sense probeis shown as a control (Figure 9D). Therefore, during the devel-opment of renal injury, C3 that is synthesized within the kidneycould be derived from both native and infiltrating renal cells.
DiscussionThis work supports a protective role for the complement
system in the later, autologous phase of anti-GBM antibody–mediated glomerulonephritis. The protective effect is mediatedby circulating complement rather than complement that is pro-duced from resident glomerular cells.
Patients with circulating antibody directed against the GBMdevelop a severe, rapidly progressive glomerulonephritis. An-tibody is deposited along the basement membrane, activatingthe complement system, components of which can be demon-strated on the GBM by immunohistochemistry. This led to thehypothesis that complement activation was important in thegeneration of injury, either by causing direct cell damage or bypromoting an inflammatory cell infiltrate. Early studies in an-imal models using complement depletion (23,24) supportedthis view; however, this was not a universal finding, particu-larly in the mouse (25,26). More recent studies using C3�/�
mice support a role for complement activation. After the injec-tion of anti-GBM antibody, there is rapid complement activa-tion and neutrophil influx. At this stage of the disease, comple-ment activation seems to have a harmful effect as deficiency(1,27) or inhibition (28) reduces injury. However, as the diseaseprogresses and the animal develops autologous antibodyagainst the heterologous serum, the absence of C3 seems tohave a detrimental effect, both on renal function and on histo-logic injury (9).
The results presented here suggest that the reason for thegreater functional injury in the C3�/� mice is the accumulationof immune complexes in the glomerular capillary wall. This
Figure 5. Histology of the transplanted kidneys. Representative glomerular histology from group 1 mice (C3�/� donor: C3�/�
recipient; A) and group 2 mice (C3�/� donor: C3�/� recipient; B) is shown. The glomeruli from group 2 mice showed severehistologic damage including capillary occlusion with PAS-positive material and areas of focal proliferation and crescent formation.Magnification, �100, PAS staining.
J Am Soc Nephrol 17: 686–696, 2006 C3 and Renal Immune Complex Clearance 691

could be explained by either reduced clearance of circulatingimmune complexes or reduced clearance of complexes fromwithin the glomerulus. C3 is readily incorporated into immunecomplexes because of the high density of Ig Fc regions, therebydisrupting immune complex structure and increasing solubility(29). In addition, in some mammals (not rodents), C3 withinimmune complexes acts as the ligand for complement receptor1 on erythrocytes, binding to which facilitates immune complextransport to phagocytic cells of the reticuloendothelial system(30). However, the model of anti-GBM antibody–mediated glo-merulonephritis that is used in this study relies on affinity ofthe heterologous serum for the GBM. Therefore, the immune
complex disease seen in this model is probably strongly depen-dent on in situ immune complex formation. This does notexclude the possibility that reduced clearance of circulatingcomplexes plays a role in the pathogenesis of glomerular dis-ease in the C3�/� mice.
Complement also has a function in the solubilization of im-mune complexes from within the glomerulus (31) and thetransfer of immune complexes across the GBM. Studies inanimals have shown that, in contrast to the normal passage ofantigen and antibody through the GBM, in animals that aredepleted of complement with cobra venom factor, antigen re-mains in a subendothelial position (32,33). The disruption to the
Figure 6. Immunochemistry of transplanted kidneys. C3 staining was demonstrated in mice from both group 1 (A and C) andgroup 2 (B and D). Glomerular C4 staining was also demonstrated in both groups (E and F) but with greater intensity in group2 mice (C3�/� donor: C3�/� recipient; F). Magnification, �160 in A and B, �400 in C through F.
692 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 686–696, 2006
structure of the immune complex that is caused by complementbinding probably facilitates transfer through the GBM, al-though the exact mechanism remains unknown.
In the C3�/� mice described in this study, the absence of oneor more of these mechanisms leads to the accumulation ofimmunoreactants and immune complexes within the glomeru-lus and therefore greater functional injury. A similar increase inseverity of immune complex glomerular disease was seen whenthe targeted C3 deletion was back-crossed onto an MRL/lprbackground. Greater glomerular IgG deposition and albumin-uria was observed in the C3�/� mice (34). However, the role ofcomplement in this model is complex, and mice that are spe-cifically deficient in the alternative pathway (35) or in which
Table 3. Semiquantitative assessment of the intensity ofstaining for complement and Ig in the mice thatreceived a transplant
Group 1 Group 2
C3 2.4 � 0.4 2.5 � 0.4C4 0.9 � 0.3 1.6 � 0.6b
Sheep IgG 1.9 � 0.4 2.5 � 0.5b
Mouse IgG 0.8 � 0.3 1.5 � 0.6b
Mouse IgM 1.2 � 0.6 2.1 � 0.5b
aData are mean � SD, n � 4 for both groups.bP � 0.01.
Figure 7. Immunochemistry of mice that received a transplant. Sheep IgG (A and B), mouse IgG (C and D), and mouse IgM (E andF) all were detected in mice from both groups. However, the staining for all three immunoreactants was greater in mice fromgroup 2 (C3�/� donor: C3�/� recipient; B, D, and F) compared with group 1 mice (C3�/� donor: C3�/� recipient; A, C, and E).Magnification, �400.
J Am Soc Nephrol 17: 686–696, 2006 C3 and Renal Immune Complex Clearance 693

complement activation is inhibited (36) have reduced diseaseseverity. Overall, this suggests a predominant role for the clas-sical pathway of complement activation in protecting fromglomerular immune complex disease.
We next addressed the issue of the source of C3 that reduces thebuild-up of immune complexes in the glomerulus. Native cells
within the glomerulus, including mesangial (13,37), epithelial (38),and endothelial cells (15), as well as tubular epithelial cells (16,39)have the capacity to synthesize C3. Although it is well docu-mented that glomerular expression of C3 mRNA is increased inglomerulonephritis, the data presented here provide no evidencefor a functional role for intraglomerular complement synthesis.
Figure 8. Electron microscopy of transplanted kidneys. Representative glomerular changes in group 1 (C3�/� donor: C3�/�
recipient; A) and group 2 (C3�/� donor: C3�/� recipient; B) mice is shown. Group 2 mice demonstrated accumulation ofelectron-dense material in the subendothelial space (arrows). Bar � 500 nm. Magnification, �20,000.
Figure 9. C3 mRNA analysis in transplanted kidneys. Reverse transcription–PCR was used to determine the presence of C3 genetranscription in mice from both group 1 and group 2. In situ hybridization was used to identify the site of C3 gene expression inmice from group 1 (B) and group 2 (C). Staining with sense probe of group 2 tissue is shown as control (D). G, glomerulus; T,tubule. Magnification, �300 in B, �400 in C, �250 in D.
694 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 686–696, 2006

Logically, in this context, the local synthesis of C3 should protectagainst immune complex accumulation, a clearly defined physio-logic role of the complement system. In our model of immunecomplex–mediated disease, however, we were unable to demon-strate this effect of locally synthesized C3, despite evidence of C3production in the glomerulus during disease development.
It is possible that because the glomerular capillaries are ex-posed to high concentrations of circulating complement com-ponents, locally synthesized C3 has only minor importance indisease that is induced by anti-GBM antibodies. Moreover, inother models of glomerulonephritis, the glomerular expressionof C3 is time dependent, and maximal expression is achievedafter 14 d (17). Therefore, local synthesis of C3 may have agreater contribution to make in more protracted models ofrenal injury than in the model described here. In models of veryacute injury, such as the heterologous phase of this model, localsynthesis of C3 is even less likely to contribute to diseaseexpression.
It should also be noted that the main site of complement geneexpression in the kidney is the renal tubule (40), and expressionat this site is upregulated during renal injury. It is possible thatlocally synthesized complement that is secreted into the ex-travascular, interstitial compartment has a greater impact onthe development of injury than glomerular-produced comple-ment. For example, there is increasing evidence that comple-ment activation may contribute to the damage to the tubuloin-terstitial compartment in proteinuria (41–43). In addition, renaltubular epithelial cell production of C3 is increased by exposureto serum proteins. However, as yet, there is no direct evidencefor involvement of locally synthesized complement proteins inthis phase of renal disease.
Thus, although local expression of C3 clearly is upregulatedby the induction of immune complex glomerulonephritis, thisstudy does not show that local synthesis of C3 plays a contrib-uting role in the pathogenesis of glomerular dysfunction.Rather, these data suggest that circulating complement plays avital role in prevention of glomerular disease, consistent withthe removal of circulating or planted immune complexes me-diated by C3. It seems likely that locally produced complementmay have greater impact on diseases of the tubulointerstitium,the main intrarenal site of complement synthesis in such dis-orders (44).
References1. Sheerin NS, Springall T, Carroll MC, Hartley B, Sacks SH:
Protection against anti-glomerular basement membrane(GBM)-mediated nephritis in C3- and C4-deficient mice.Clin Exp Immunol 110: 403–409, 1997
2. Couser WG, Schulze M, Pruchno CJ: Role of C5B-9 inexperimental membranous nephropathy. Nephrol DialTransplant 7: 25–31, 1992
3. Topham PS, Haydar SA, Kuphal R, Lightfoot JD, Salant DJ:Complement-mediated injury reversibly disrupts glomer-ular epithelial cell actin microfilaments and focal adhe-sions. Kidney Int 55: 1763–1775, 1999
4. Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, CookHT, Petry F, Loos M, Pandolfi PP, Walport MJ: Homozy-
gous C1q deficiency causes glomerulonephritis associatedwith multiple apoptotic bodies. Nat Genet 19: 56–59, 1998
5. Mitchell DA, Taylor PR, Cook HT, Moss J, Bygrave AE,Walport MJ, Botto M: Cutting edge: C1q protects againstthe development of glomerulonephritis independently ofC3 activation. J Immunol 162: 5676–5679, 1999
6. Schifferli JA: The classical pathway of complement pre-vents the formation of insoluble antigen-antibody com-plexes—Biological implications. Immunol Lett 14: 225–228,1987
7. Schifferli JA, Ng YC, Peters DK: The role of complementand its receptor in the elimination of immune-complexes.N Engl J Med 315: 488–495, 1986
8. Fujigaki Y, Batsford S, Yamashita F, Yonemura K, HishidaA, Kawachi H, Shimizu F, Vogt A: Sequence of events inthe glomerular capillary wall at the onset of proteinuria inpassive Heymann nephritis. Virchows Arch Int J Pathol 438:136–145, 2001
9. Sheerin NS, Springall T, Abe K, Sacks SH: Protection andinjury: The differing roles of complement in the develop-ment of glomerular injury. Eur J Immunol 31: 1255–1260,2001
10. Quigg RJ, Lim A, Haas M, Alexander JJ, He C, Carroll MC:Immune complex glomerulonephritis in C4- and C3-defi-cient mice. Kidney Int 53: 320–330, 1998
11. Alper CA, Johnson AM, Birtch AG, Moore FD: Human C3:Evidence for the liver as the primary site of synthesis.Science 163: 263–288, 1969
12. Sacks SH, Zhou W, Pani A, Campbell RD, Martin J: Com-plement C3 gene expression and regulation in human glo-merular epithelial cells. Immunology 79: 348–354, 1993
13. Sacks S, Zhou W, Campbell RD, Martin J: C3 and C4 geneexpression and interferon-gamma-mediated regulation inhuman glomerular mesangial cells. Clin Exp Immunol 93:411–417, 1993
14. Timmerman JJ, Beersma MF, Gijlswijk-Janssen DJ, van EsLA, van der Woude FJ, Daha MR: Differential effects ofcytomegalovirus infection on complement synthesis by hu-man mesangial cells. Clin Exp Immunol 109: 518–525, 1997
15. Sheerin NS, Zhou W, Adler S, Sacks SH: TNF-alpha regu-lation of C3 gene expression and protein biosynthesis in ratglomerular endothelial cells. Kidney Int 51: 703–710, 1997
16. Brooimans RA, Stegmann AP, van Dorp WT, van der ArkAA, van der Woude FJ, van Es LA, Daha MR: Interleukin2 mediates stimulation of complement C3 biosynthesis inhuman proximal tubular epithelial cells. J Clin Invest 88:379–384, 1991
17. Sasaki O, Zhou W, Miyazaki M, Abe K, Koji T, Verroust P,Tsukasaki S, Ozono Y, Harada T, Nakane PK, Kohno S,Sacks SH: Intraglomerular C3 synthesis in rats with pas-sive Heymann nephritis. Am J Pathol 151: 1249–1256, 1997
18. Wessels MR, Butko P, Ma M, Warren HB, Lage AL, CarrollMC: Studies of group B streptococcal infection in micedeficient in complement component C3 or C4 demonstratean essential role for complement in both innate and ac-quired immunity. Proc Natl Acad Sci U S A 92: 11490–11494, 1995
19. Kalina SL, Mottram PL: A microsurgical technique forrenal transplantation in mice. Aust N Z J Surg 63: 213–216,1993
20. Reynolds J, Tam FWK, Chandraker A, Smith J, Karkar AM,Cross J, Peach R, Sayegh MH, Pusey CD: CD28–B7 block-
J Am Soc Nephrol 17: 686–696, 2006 C3 and Renal Immune Complex Clearance 695

ade prevents the development of experimental autoim-mune glomerulonephritis. J Clin Invest 105: 643–651, 2000
21. Gomez-Guerrero C, Duque N, Casado MT, Pastor C,Blanco J, Mampaso F, Vivanco F, Egido J: Administrationof IgG Fc fragments prevents glomerular injury in experi-mental immune complex nephritis. J Immunol 164: 2092,2000
22. Abe K, Miyazaki M, Koji T, Furusu A, Ozono Y, Harada T,Sakai H, Nakane PK, Kohno S: Expression of decay accel-erating factor mRNA and complement C3 mRNA in hu-man diseased kidney. Kidney Int 54: 120–130, 1998
23. Cochrane CJ, Unanue ER, Dixon FJ: A role of polymorpho-nuclear cells and complement in nephrotoxic nephritis. JExp Med 122: 99–116, 1965
24. Hammer DK, Dixon FJ: Experimental glomerulonephritis.II. Immunologic events in the pathogenesis of nephrotoxicnephritis in the rat. J Exp Med 17: 1019–1034, 2002
25. Huang XR, Holdsworth SR, Tipping PG: Th2 responsesinduce humorally mediated injury in experimental anti-glomerular basement membrane glomerulonephritis. J AmSoc Nephrol 8: 1101–1108, 1997
26. Feith GW, Assmann KJM, Bogman MJJT, VangompelAPM, Schalkwijk J, Koene RAP: Albuminuria in the lateheterologous phase of anti-GBM nephritis in beige mice iscomplement independent but leukocyte dependent. KidneyInt 43: 968, 1993
27. Hebert MJ, Takano T, Papayianni A, Rennke HG, Minto A,Salant DJ, Carroll MC, Brady HR: Acute nephrotoxic se-rum nephritis in complement knockout mice: Relativeroles of the classical and alternate pathways in neutrophilrecruitment and proteinuria. Nephrol Dial Transplant 13:2799–2803, 1998
28. Quigg RJ, Kozono Y, Berthiaume D, Lim A, Salant DJ,Weinfeld A, Griffin P, Kremmer E, Holers VM: Blockade ofantibody-induced glomerulonephritis with Crry-Ig, a sol-uble murine complement inhibitor. J Immunol 160: 4553–4560, 1998
29. Schifferli JA, Woo P, Peters DK: Complement-mediatedinhibition of immune precipitation. 1. Role of the classicaland alternative pathways. Clin Exp Immunol 47: 555–562,1982
30. Birmingham DJ: Erythrocyte complement receptors. CritRev Immunol 15: 133–154, 1995
31. Schifferli JA, Peters DK: Complement, the immune-com-plex lattice, and the patho-physiology of complement-de-ficiency syndromes. Lancet 2: 957–959, 1983
32. Fujigaki Y, Nagase M, Honda N: Intraglomerular base-ment-membrane translocation of immune-complex (IC) in
the development of passive in situ IC nephritis of rats. Am JPathol 142: 831–843, 1993
33. Sawtell NM, Hartman AL, Weiss MA, Pesce AJ, MichaelJG: C-3 dependent, C5 independent immune-complex glo-merulopathy in the mouse. Lab Invest 58: 287–293, 1988
34. Sekine H, Reilly CM, Molano ID, Garnier G, Circolo A,Ruiz P, Holers VM, Boackle SA, Gilkeson GS: Complementcomponent C3 is not required for full expression of im-mune complex glomerulonephritis in MRL/lpr mice. J Im-munol 166: 6444–6451, 2001
35. Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS:Effects of complement factor D deficiency on the renaldisease of MRL/lpr mice. Kidney Int 65: 129–138, 2004
36. Bao LH, Haas M, Boackle SA, Kraus DM, CunninghamPN, Park P, Alexander JJ, Anderson RK, Culhane K,Holers VM, Quigg RJ: Transgenic expression of a solublecomplement inhibitor protects against renal disease andpromotes survival in MRL/lpr mice. J Immunol 168:3601–3607, 2002
37. Montinaro V, Serra L, Perissutti S, Ranieri E, Tedesco F,Schena FP: Biosynthesis of C3 by human mesangial cells.Modulation by proinflammatory cytokines. Kidney Int 47:829–836, 1995
38. Zhou W, Campbell RD, Martin J, Sacks SH: Interferon-gamma regulation of C4 gene expression in cultured hu-man glomerular epithelial cells. Eur J Immunol 23: 2477–2481, 1993
39. Tang S, Zhou WD, Sheerin NS, Vaughan RW, Sacks SH:Contribution of renal secreted complement C3 to the cir-culating pool in humans. J Immunol 162: 4336–4341, 1999
40. Andrews PA, Finn JE, Lloyd CM, Zhou W, Mathieson PW,Sacks SH: Expression and tissue localization of donor-specific complement C3 synthesized in human renal allo-grafts. Eur J Immunol 25: 1087–1093, 1995
41. Hori Y, Yamada K, Hanafusa N, Okuda T, Okada N,Miyata T, Couser WG, Kurokawa K, Fujita T, Nangaku M:Crry, a complement regulatory protein, modulates renalinterstitial disease induced by proteinuria. Kidney Int 56:2096–2106, 1999
42. Nangaku M, Pippin J, Couser WG: C6 mediates chronicprogression of tubulointerstitial damage in rats with rem-nant kidneys. J Am Soc Nephrol 13: 928–936, 2002
43. Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y,Matsuo S: Complement activation products in the urinefrom proteinuric patients. J Am Soc Nephrol 11: 700–707,2000
44. Pratt JR, Basheer SA, Sacks SH: Local synthesis of comple-ment component C3 regulates acute renal transplant rejec-tion. Nat Med 8: 582–587, 2002
696 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 686–696, 2006


















