
Ab-Initio Molecular Dynamics Modeling of Molten...
23
Mark Asta Department of Chemical Engineering & Materials Science Ab-Initio Molecular Dynamics Modeling of Molten Superalloys ES09 Workshop University of California, Davis June 25, 2009
Transcript of Ab-Initio Molecular Dynamics Modeling of Molten...

Mark Asta
Department of Chemical Engineering & Materials Science
Ab-Initio Molecular Dynamics Modeling of Molten Superalloys
ES09 Workshop University of California, Davis
June 25, 2009

Acknowledgements
Stefano Angioletti-Uberti1, Mike Finnis1, Peter Lee1, James Lill2, Dallas Trinkle3, Chris Woodward4
1Department of Materials, Imperial College London, UK 2High Performance Technologies Inc, WPAFB, Dayton, OH
3Department of Materials Science and Engineering, University of Illinois Urbana-Champaign
4Materials and Manufacturing Directorate, Air Force Research Laboratory, Wright-Patterson Air Force Base
Research Support US DOE (Office of Basic Energy Sciences)
AFOSR (Materials Engineering for Affordable New Systems Program)
Supercomputing Resources DoD ASC-MSRC Challenge Grant

1969 1972 1982 1984 1988 1989 1994 2000
R80
R12
5
R80
H
N4
R14
2
N5
N6
MX
4
800
900
1000
1100
Year of Introduction
Tem
pera
ture
Cap
abili
ty (o
C)
Evolution of Temperature Capability
Higher-T Materials Higher Efficiency
Ni-Based Superalloys

1st-Principles Thermo & Kinetic Modeling
• Design of High-Temperature Materials
– Requires Data for Bulk and Interfacial Free Energies, Diffusion Kinetics, Lattice Parameters, … – In Many Cases Data Difficult to Measure (e.g., Metastable Phases, High T)
• Role of 1st-Principles Calculations – Predictive Framework for Computing Structural, Thermodynamic and Kinetic Properties – Applicable to Both Stable and Metastable Phases, Bulk and
Interfaces, High & Low T
CMSX-4 Superalloy A.Dlouhy and G.Eggeler
€
Ni62.936Cr7.5Mo0.38Al12.7Co9.9W2.1Ti1.3
Ta2.2Re0.95Hf0.034

Defects in Directional Solidification
Freckle Chain in Superalloy (Pollock and Murphy, 1996)
“Channels” in NH4Cl (Wooster, 1997)
Directionally Solidified Superalloy Ingot
(A. F. Giamei, UTRC)
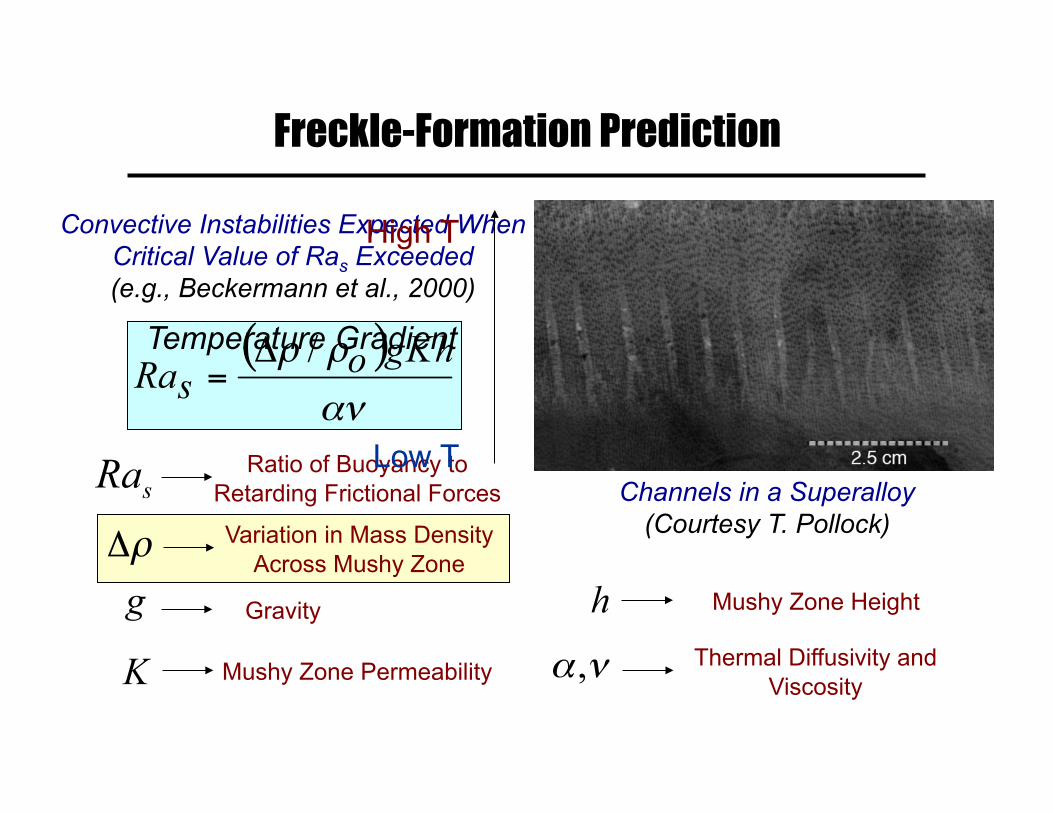
Freckle-Formation Prediction
Convective Instabilities Expected When Critical Value of Ras Exceeded (e.g., Beckermann et al., 2000)
Channels in a Superalloy (Courtesy T. Pollock) Variation in Mass Density
Across Mushy Zone
Mushy Zone Permeability
Mushy Zone Height
Thermal Diffusivity and Viscosity
Ratio of Buoyancy to Retarding Frictional Forces
Gravity
Temperature Gradient
Low T
High T

Mass Density
Accurate Parametrization of Temperature and Composition Dependent Molar Volume and Solute Partitioning Required to
Predict Density Inversions Across Mushy Zone

• Assume Vα Depends Only on xα and T
– Neglects Multicomponent Interactions
• Molar Volumes Fit to Experimentally Measured Binary Alloy Data
– Large Variations of Partial Molar Volumes w/ Temperature & Composition Obtained from Fits
Molar-Volume Parametrization K. Mukai, Z. Li and K. C. Mills, Met. Trans. B, vol. 36B, 255 (2005)

Quantum MD for Molten Ni-Based Alloys
• Simulation Details – (NVT) Dynamics at T=1750 & 1830 K – Time Step: 0.002 or 0.003 ps – ~5-10 ps Simulation Time – 500 Atom System Size – VASP Code on 64 Processors – Ultrasoft Pseudopotentials and PAW
• Volumes – 3 or 4 Volumes: 0.95 Vref – 1.05 Vref – Equations of State and Equilibrium
Volume
• Systems – Elemental Ni and Al – Binary Ni-Al, Ni-W, Ni-Re, Ni-Ta – Ternary Ni-Al-W, Ni-Al-Re, Ni-Al-Ta
Snapshot of Simulation for Ni436Al50W14
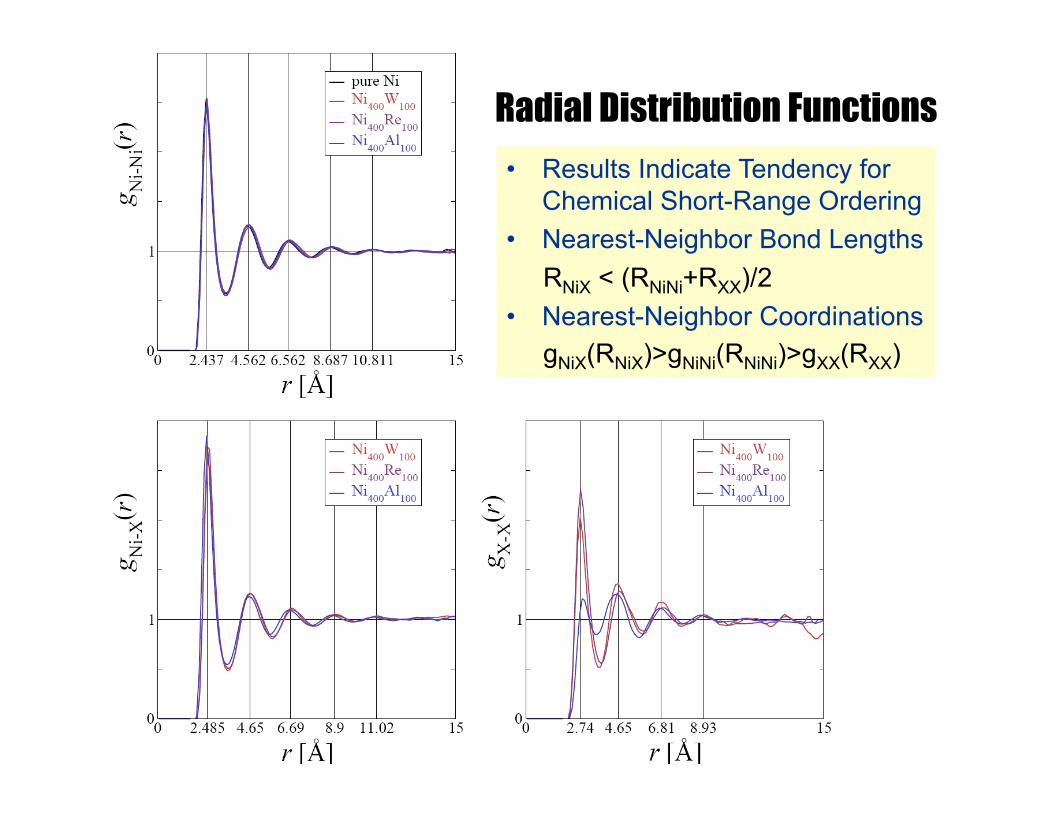
Radial Distribution Functions • Results Indicate Tendency for
Chemical Short-Range Ordering • Nearest-Neighbor Bond Lengths RNiX < (RNiNi+RXX)/2
• Nearest-Neighbor Coordinations gNiX(RNiX)>gNiNi(RNiNi)>gXX(RXX)

Liquid Structure Ni, Ni473W27 and Ni473Re27 at 1830 K
Liquid Structures Display Short Range Order Featuring Predominantly Icosahedral and BCC Local Structures
Bond Angle Distributions Common-Neighbor Analysis
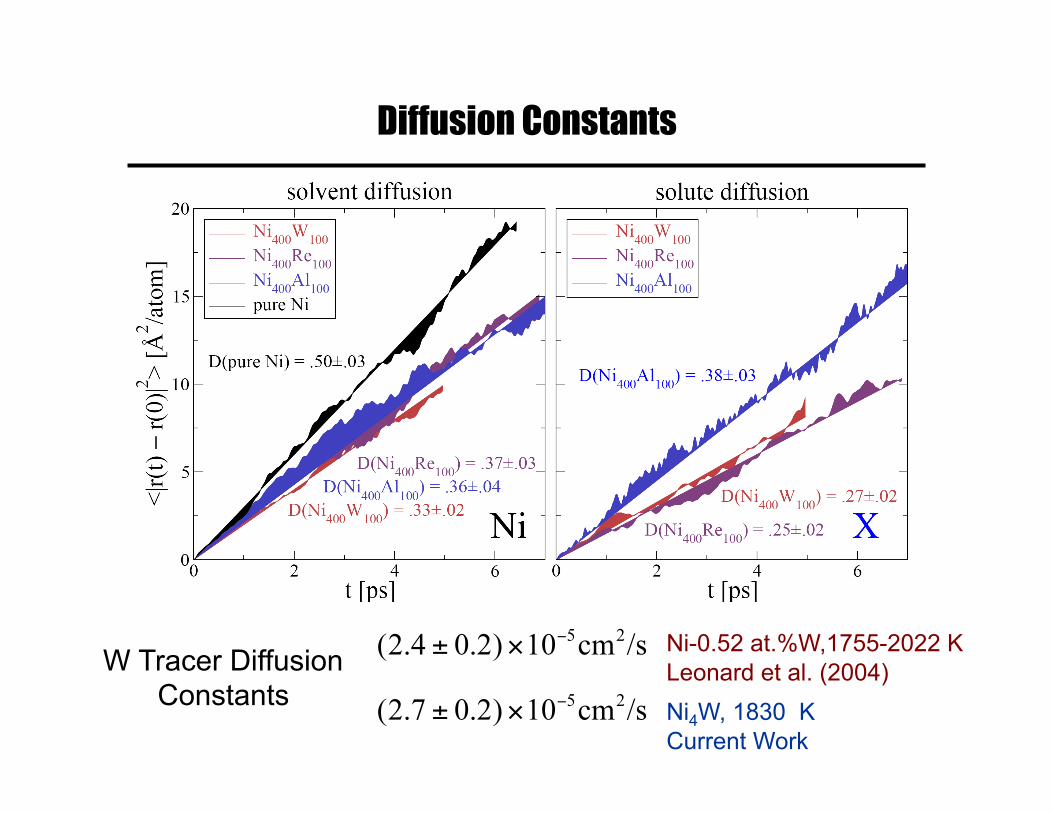
Diffusion Constants
W Tracer Diffusion Constants Ni4W, 1830 K
Current Work
Ni-0.52 at.%W,1755-2022 K Leonard et al. (2004)

Pressure-Volume Relations Ni400Al100 at T=1830 K

T=1750 K T=1830 K
Composition Mukai AIMD VAIMD/VEXPT Mukai AIMD VAIMD/VEXPT
Ni500 7.4597 7.57(1) 1.015 7.5724 7.62(1) 1.006
Ni400Al100 7.7628 7.88(1) 1.015 7.8492 7.94(1) 1.012
Ni473W27 7.5534 7.66(1) 1.014 7.6520 7.70(1) 1.006
Ni400W100 6.5192 7.94(1) 7.3294 7.98(1)
Ni473Re27 7.4940 7.65(1) 7.6499 7.69(1)
Ni400Re100 7.5869 7.91(1) 7.8593 7.93(1)
Ni473Ta27 7.3310 7.70(1) 7.5053 7.75(1)
Ni400Ta100 1.1043 8.14(1) 4.1440 8.18(1)
Ni436Al50W14 7.6920 7.77(1) 7.7655 7.80(1)
Ni436Al50Re14 7.6921 7.7510 7.80(1)
Ni436Al50Ta14 7.6911 7.79(1) 7.7553 7.84(1)
All Volumes in Units of cm3/mole
Molar Volumes for Molten Ni-Based Alloys MD Calculations, Mukai Model and Experiment

Molar Volume Predictions Results for Ternary Ni-Al-W Alloys at T=1830 K
Ni Ni 4 Al Ni 436 Al 50 W 14 Ni 473 W 27 Ni
Model
MD Calculations
Ni
Ni 4 Al
Ni 473 W 27
Ni 436 Al 50 W 14
Ni
Ni 4 Al
Ni 473 W 27
Ni 436 Al 50 W 14
Ni
Ni 4 Al
Ni 473 W 27
Ni 436 Al 50 W 14
Similar results obtained for Ni-Al-Re and Ni-Al-Ta
Results suggest high accuracy of published parameterization for compositions bounded by experimental measurements

Ni-W T=1830 K
Ni-Re T=1830 K
Ni-Ta T=1830 K
Model
MD Calcs
Molar Volume Predictions Results for Binary Alloys
MD results demonstrate limitations in accuracy of published parameterizations when extrapolated beyond bounds of
experimental measurements

Partial Molar Volumes Results for Infinite Dilution
MD Calcs
Model
MD Calcs
Model
T=1830 K
T=1750 K
MD results suggest relatively weak temperature dependencies for partial molar volumes
Ni-W Ni-Re Ni-Ta
Ni-W Ni-Re Ni-Ta
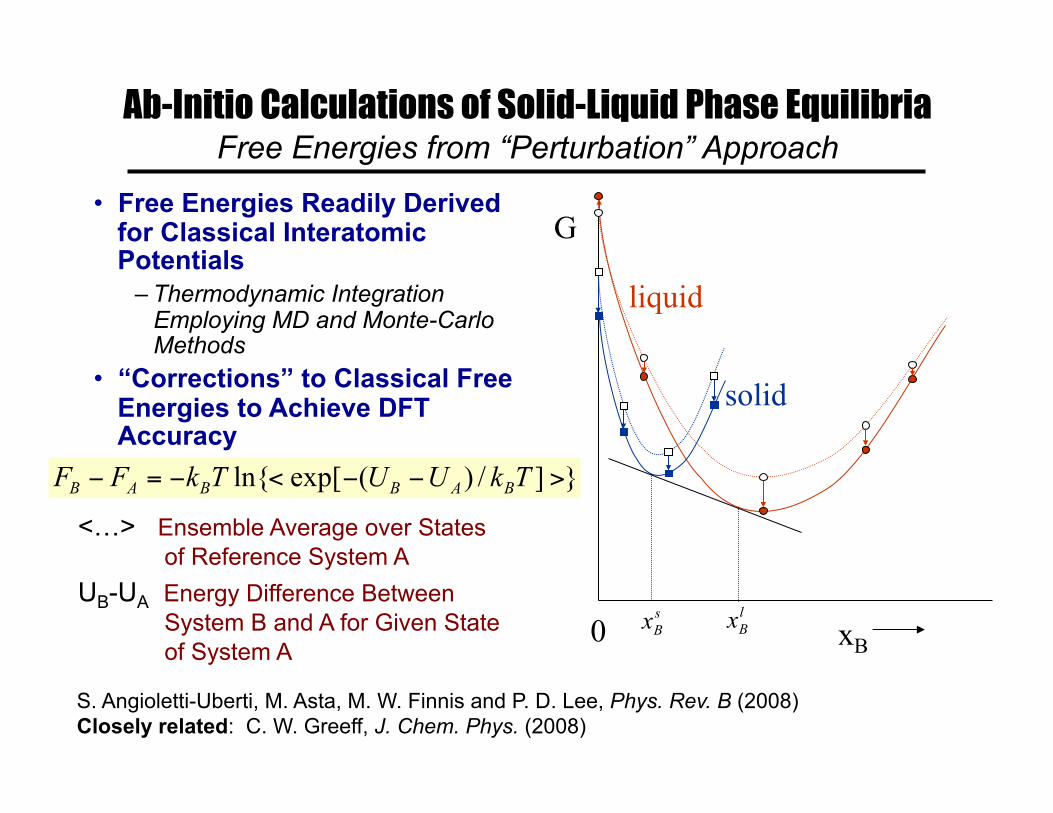
Ab-Initio Calculations of Solid-Liquid Phase Equilibria Free Energies from “Perturbation” Approach
• Free Energies Readily Derived for Classical Interatomic Potentials
– Thermodynamic Integration Employing MD and Monte-Carlo Methods
• “Corrections” to Classical Free Energies to Achieve DFT Accuracy
xB 0
G
liquid
solid
<…> Ensemble Average over States of Reference System A UB-UA Energy Difference Between System B and A for Given State of System A
S. Angioletti-Uberti, M. Asta, M. W. Finnis and P. D. Lee, Phys. Rev. B (2008) Closely related: C. W. Greeff, J. Chem. Phys. (2008)

Convergence Properties Description of Test System
• Two classical Embedded Atom potentials for Ni-Cu – Reference: “smf7” potential due to Foiles (1985) – Target: “u3” potential due to Foiles, Baskes & Daw (1984)
• Melting temperature and solidus/liquidus boundaries – Known to be approximately 100 K higher for smf7 than u3 from
previous thermodynamic integration calculations
• Compositions and trajectories – Solid and Liquid Pure Ni: Reference trajectory from NVT MD – Solid and Liquid NiCu Equiatomic Alloy: Reference
trajectory from NVT Monte-Carlo
• Calculated quantities – Free Energy Differences – Differences in Pure-Ni Melting Temperatures
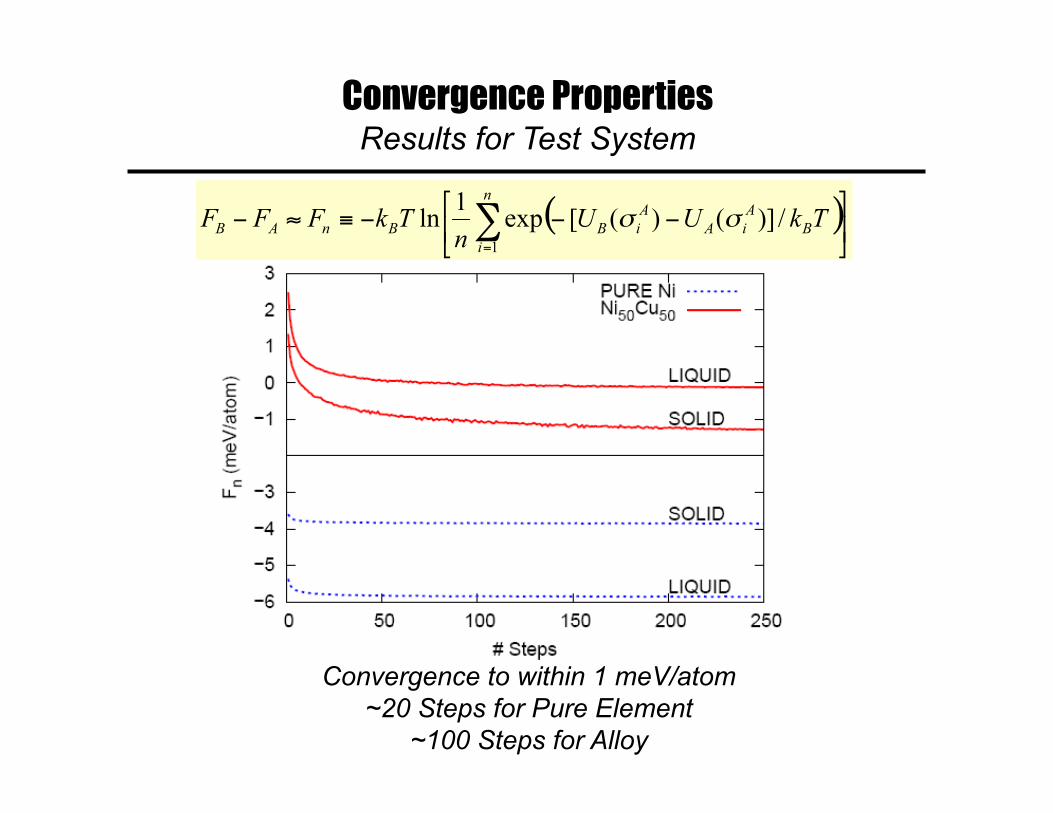
Convergence Properties Results for Test System
Convergence to within 1 meV/atom ~20 Steps for Pure Element
~100 Steps for Alloy

Convergence Properties Some Theoretical Considerations
Lu and Kofke (2001)
€
exp(−βΔFtrue ) − exp(−βΔFcalc,B )exp(−βΔFtrue )
= PA (W )dW−∞
W0
∫
Good
Bad

Summary
• Quantum molecular dynamics simulations of molten superalloys – Good accuracy demonstrated for molar volumes, diffusivities (and
mixing enthalpies) – Results provide data to refine molar-volume parametrizations for
higher length-scale models in materials design
• Towards first-principles calculations of alloy solid-liquid phase equilibria and thermodynamic properties – Free-energy perturbation approach as framework for coupling classical
and first-principles simulations for calculation of free energies with DFT accuracy
– Promising convergence demonstrated for classical model system – Approach requires reasonably accurate classical-potential models


















