A Standard Tool for Density-Functional Calculations · A Standard Tool for Density-Functional...
32
FHI Workshop 2003 1 Jörg Behler DMol 3 A Standard Tool for Density-Functional Calculations Fritz-Haber-Institut der Max-Planck-Gesellschaft Berlin, Germany Paul-Scherrer-Institut, Zürich, Switzerland Bernard Delley
Transcript of A Standard Tool for Density-Functional Calculations · A Standard Tool for Density-Functional...

FHI Workshop 2003 1
Jörg Behler
DMol3
A Standard Tool for Density-Functional Calculations
Fritz-Haber-Institut der Max-Planck-Gesellschaft
Berlin, Germany
Paul-Scherrer-Institut,Zürich, Switzerland
Bernard Delley

FHI Workshop 2003 2
What is DMol3?
Introduction
• all-electron DFT-code• basis functions are localized atomic orbitals (LCAO) • can be applied to:
• free atoms, molecules and clusters• solids and surfaces (slabs)
Topic of this talk
Characteristics of the DMol3 approach to DFT (selected aspects)

FHI Workshop 2003 3
“DMol” = density functional calculations on molecules
History
• first fragments early 1980’s(full potential electrostatics,numerical atomic orbitals)
• total energy functional 1983• electrostatics by partitioning 1986• first release of DMol by Biosym 1988• forces 1991• parallel version 1992• DSolid 1994• geometry optimization 1996• unification of DMol and DSolid
⇒ DMol3 1998• molecular dynamics 2002
Main author:Bernard Delley
Further contributions by:J. Andzelm R.D. King-SmithJ. Baker D. EllisG. Fitzgerald many more...

FHI Workshop 2003 4
• Gaussian type orbitals (GTO’s)• Slater type orbitals (STO’s)• numerical atomic orbitals (AO’s)
Localized Basis Sets
Most DFT codes use numerical integration (i.e. for XC-part)⇒ in DMol3: numerical techniques used wherever possible⇒ AO‘s can be used
• high accuracy (cusp, asymptotic behaviour)• high efficiency (few basis functions ⇒ small matrix size)
Why?
What?
Idea:

FHI Workshop 2003 5
• exact DFT-spherical-atomic orbitals of• neutral atoms• ions• hydrogenic atoms
• radial functions are calculated in the setup (free atom) • implemented as numerically tabulated functions
Properties:• maximum of accuracy for a given basis set size• infinitely separated atoms limit treated exactly• small number of additional functions needed for polarization
• square integrability, cusp singularities at the nuclei
DMol3 Basis Functions

FHI Workshop 2003 6
minimal: Al 1s, 2s, 2p, 3s, 3p ⇒ E = -242.234806 Ha
Basis Set Example: Al
dn: Al2+ 3s, 3p ⇒ E = -242.234845 Ha
dnd: z = 5 3d ⇒ E = -242.235603 Ha
dnp: z = 4 3p, z = 7.5 4f ⇒ E = -242.235649 Ha
9 AOs
13 AOs
18 AOs
28 AOs
Atomic energy
Quality test:lowering of total energy by adding basis functions
(variational principle)

FHI Workshop 2003 7
Basis Set Example: AlRadial Basis Functions
-3.00
-2.50
-2.00
-1.50
-1.00
-0.50
0.00
0.50
1.00
1.50
2.00
0.0 2.0 4.0 6.0 8.0 10.0
r / bohr
Rad
ial B
asis
Fun
ctio
n
Al 1s
Al 2s
Al 2p
Al 3s
Al 3p
Al+ 3s
Al+ 3p
3d z=5
4f z=7.5
3p z=4

FHI Workshop 2003 8
Atomic orbitals
Basis Set ConvergencePlane waves
quality ⇒ basis set extension ⇒ rcut
quality ⇒ Ecut
extension ⇒ infinte
Basis test for the oxygen molecule
O basis functionsO 1sO 2sO 2p O2+ 2sO2+ 2pz = 3 3dz = 5 3d
-150.5600
-150.5595
-150.5590
-150.5585
-150.5580
-150.5575
-150.55706 7 8 9 10 11 12 13 14 15
rcut / bohr
Eto
t / H
a
dndall
alldnd

FHI Workshop 2003 9
BSSE
DMol3: very small BSSE because of nearly perfect basis set for the separated atoms limit=> excellent description of weak bonds (but DFT limitation)
Basis Set Superposition Error
Definition:BSSE is a lowering of the energy when the electrons of each atom spread into the basis functions provided by the other atoms due to an incomplete basis set.
GTO/STO Codes: BSSE can be a serious problem
Plane waves: BSSE does not appear

FHI Workshop 2003 10
• no systematic way to improve basis set quality• careful tests required to construct a basis set
Summary Basis Functions
• very efficient (small) basis ⇒ fast calculations• allows for calculations without periodic boundary conditions or less dense systems (slabs with large vacuum)
• easy physical interpretation of basis functions• (almost) no basis set superposition error• different basis sets for different elements possible
Advantages
Disadvantages

FHI Workshop 2003 11
Numerical Integration3 Step approach
1. Decomposition of the total integral in 3D into a sum of independent integrals for atomic contributions ⇒ partitioning
2. Decomposition of each atomic integral into a radial and an angular part ⇒ spherical polar coordinates
3. Integration of the angular part on a sphere or decomposition into separate integrations for ϑ and φ

FHI Workshop 2003 12
Partition Functions
Partition functions pα are used to rewrite integrals over all space:
( ) ( ) ( ) ( )∫ ∫∑∫∑ == rrrrrrr dpfdfdfα
αα
αα
1. Step: Decomposition into atomic contributions
Enables integration using spherical polar coordinates!
( ) ( )∫ ∑∫=α
αα rrrr dfdf⇒ Sum of atomic integrals

FHI Workshop 2003 13
Partition Functions
Example:
Definition: The partition function for a center α:
( ) ( )( )∑
=
ββ
αα rg
rgrp ∑ =α
α 1p
• choose one peaked function gα for each center α
• calculate the partition function for each center α
Normalization:
αg
αR
2
=
α
αα
ρr
gExample:

FHI Workshop 2003 14
-3 -2 -1 0 1 2 3
-3 -2 -1 0 1 2 3-3 -2 -1 0 1 2 3
Partition FunctionsTotal function:
Partition functions: Decomposed functions:
Ap Af Bf
Peaked functions gAand gB:
A B
-3 -2 -1 0 1 2 3
A B
A B
Bp
fAg Bg
1
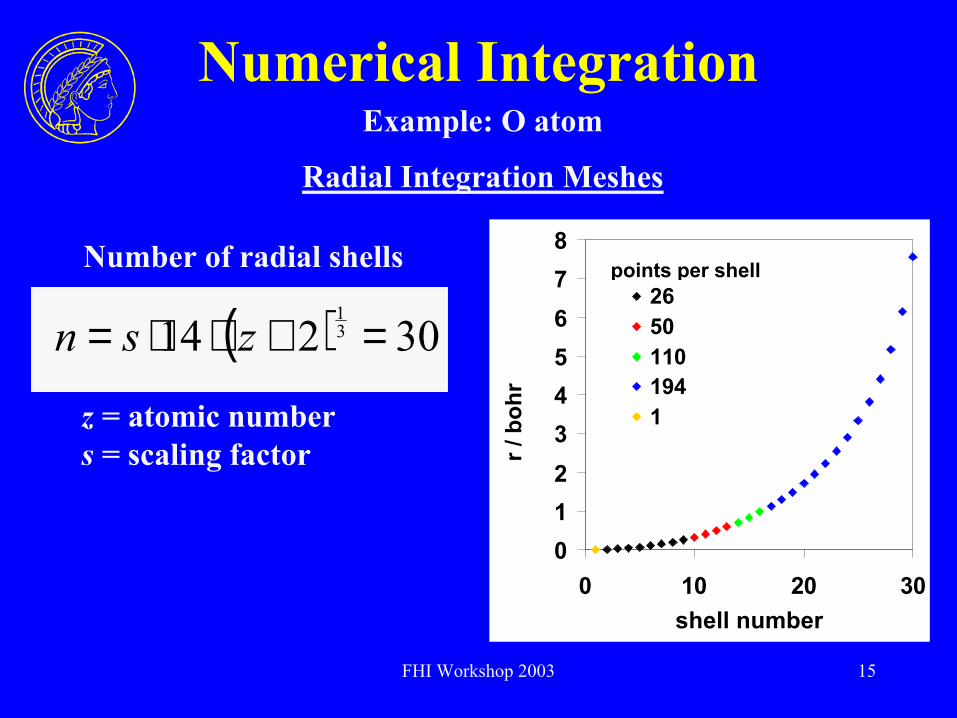
FHI Workshop 2003 15
Numerical IntegrationExample: O atom
points per shell
012345678
0 10 20 30shell number
r / b
ohr
26501101941
( ) 30214 31
=+⋅⋅= zsn
Number of radial shells
Radial Integration Meshes
z = atomic numbers = scaling factor

FHI Workshop 2003 16
Numerical IntegrationExample: O atom
Order 17110 points
Order 23194 points
Order 726 points
Order 1150 points
Angular Integration Meshes
(projections of points on sphere into plane)
• integration is done on Lebedev Spheres• integration scheme with octahedral symmetry

FHI Workshop 2003 17
Numerical IntegrationExample: O atom (cutoff 10 bohr)
Total Grid: Remarks: • user defined grids• higher order angular
schemes are possible • symmetry is used to reduce
number of points • a weight is assigned to each
point • in a molecule or solid a
superposition of atomicmeshes is used
3287 points

FHI Workshop 2003 18
Numerical IntegrationEffect of mesh quality: total energy of the Al atom
-6600.8
-6600.6
-6600.4
-6600.2
-6600.0
-6599.86 8 10 12 14 16 18 20
cutoff radius / bohr
Eto
t / e
V
fine gridcoarse grid

FHI Workshop 2003 19
ElectrostaticsClassical electrostatic contribution in Hamiltonian
[ ] ( ) ( )21
12
21
21 rrrr dd
rJ ∫ ∫=
ρρρ
Hartee-Fock
DFT
( ) ( ) ( ) ( )21
12
2211
1 1
rrrrrr
ddr
PJL L
∫ ∫∑∑= =
= σλνµ
λ σλσµν
χχχχ
Matrix elements
( ) ( ) ( )21
12
211 rrrrr
ddr
J ∫ ∫=ρχχ νµ
µν

FHI Workshop 2003 20
Electrostatics
Density-fitting using an auxiliary basis set { }kω
( ) ( ) ( )∑=≈k
kkc rrr ωρρ ~ ( ) Nd =∫ rrρ~
constraint
The basis functions are of the same type as for the wavefunction expansion.
{ }kω
( ) ( ) ( )21
12
211 rrrrr
ddr
cJk
k ∫ ∫∑=ωχχ νµ
µν
auxiliary density
Matrix elements
Common procedure in DFT using localized basis functions:
O(N3)

FHI Workshop 2003 21
Approach to electrostatics in DMol3: Overview
Electrostatics
Partitioning: decompose the electron density into atomic componentsStep 1
Step 2Projection onto Ylm functions yields multipoles attached to the atoms
Step 3 Solve Poisson’s equation for each multipole(only 1-dimensional problem)
Step 4 Assemble electrostatic potential from all multipoles and atoms
DMol3: No basis set required for density expansion!

FHI Workshop 2003 22
Electrostatics
2. Step: Multipole expansion of each ρα
( )rlmαρ• truncation of expansion at lmax• reduction to 1-dimensional radial density
1. Step: Partitioning of total density ρ into atomic densities ρα
⇒ like numerical integration
( ) ( ) ( ) φϑφϑρφϑπ
ρ ααα ddrYl
r lmlm ,,,12
141
∫+=
( ) ( ) ( )∑ +≈max
,124,,l
lmlmlm Yrlr φϑρπφϑρ ααα

FHI Workshop 2003 23
Electrostatics
Single center Poisson’s equation
( ) ( )φϑπρφϑ αα ,,4,,2 rrV −=∇
2
2
2
22 11
rl
rrr−
∂∂
=∇
( ) ( ) ( )∑=max
,1,,l
lmlmlm YrV
rrV φϑφϑ ααspherical Laplacian
potential expansion
∑= lmαα ρρ
⇒ set of equations for numerical evaluation of all
density decomposition
( )rV lmα
3. Step: Calculation of the potential contributions

FHI Workshop 2003 24
Electrostatics
For periodic boundary conditions an Ewald summation is included.
( )φϑα ,,rV ⇒ ( )rVatomic mesh ⇒ full mesh
4. Step: Construction of the total potential fromatomic contributions
O(N2)Calculation of the electrostatic potential:

FHI Workshop 2003 25
Real space:
Ewald Summation for Monopoles
Reciprocal space:
Point charges
Background charge
Negative Gaussians
Positive Gaussians
0
q
background charge + positive Gaussians
point charges + negative Gaussians
r

FHI Workshop 2003 26
1. Generalization for lattices with multipoles
Extensions to Ewald Summation
2. Extension to monopoles and multipoles of finite extent
DMol3 contains an extension to lattices of point multipoles located at the atomic sites:• applied to the ραlm• computationally as demanding as for point charge lattices
Assumption: multipoles are located inside a radius rcut
Ewald terms with r < rcut have to be modified in the real space part,because explicit calculation of the radial details of the extended charge distribution is required.

FHI Workshop 2003 27
Scaling (localized basis sets)
Electrostatics
O(N4)Hartree Fock
DFT in general O(N3)
DMol3 molecular case O(N2)DMol3 solid case O(N3/2)
Electrostatics in DMol3 scale almost linearly with system size for large systems
→ O(N2)Real systems:

FHI Workshop 2003 28
The Harris Functional
• approximate DFT calculations for very large systems• non-selfconsistent energy calculation (1 iteration only)• approximated density = superposition of fragment densities (i.e. atomic
densities)
J. Harris, Phys. Rev B 31 (1985) 1770.B. Delley et al., Phys. Rev B 27 (1983) 2132.
Original idea of the Harris functional:
Harris energy functional
[ ] [ ] [ ] [ ] nn
N
iXCXCHiiHarris ErdEEfE +−+−= ∑ ∫
=1
3ρρµρρερ
Kohn Sham energy functional
[ ] [ ] [ ] [ ] [ ] nnextXCHKS EEEETE ++++= ρρρρρ

FHI Workshop 2003 29
Total Energy in DMol3
• the Harris functional is stationary at the same density as the Kohn-Sham functional and the two are equal in value at this point
• the curvature of EHarris about the stationary point is smaller than the curvature of EKS
• the density in the Harris functional does not have to be V-representable
Reduction of numerical noise
∑−= refatomtotbind EEE
DMol3 uses the Harris functional (scf densities)
Realization: subtraction of atomic densities from total densities in integrands

FHI Workshop 2003 30
• forces• geometry optimization• ab initio molecular dynamics and simulated annealing• COSMO (COnductor-like Screening MOdel ) • transition state search• vibrational frequencies• Pulay (DIIS) charge density mixing• pseudopotentials (optional)
More features ...

FHI Workshop 2003 31
Conclusion• Fast
• very small basis sets, matrix diagonalization O(N3)• no costs for large vacuum or atom / molecules • efficient calculation of electrostatics• O(N) calculation of Hamilton and overlap matrix
• Universal• atom, molecule and cluster calculations• solids and slabs with periodic boundary conditions
• Accurate results• comparable to LAPW ⇒ next talk
• Easy to use• atoms are given in Cartesian coordinates

FHI Workshop 2003 32
• B. Delley, J. Chem. Phys. 92 (1990) 508• B. Delley in “Modern Density Functional Theory: A Tool for Chemistry”,
Theoretical and Computational Chemistry Vol. 2, Ed. by J. M. Seminario and P. Politzer, Elsevier 1995
• B. Delley, J. Chem. Phys. 113 (2000) 7756
• B. Delley, Comp. Mat. Sci. 17 (2000) 122 • J. Baker, J. Andzelm, A. Scheiner, B. Delley, J. Chem. Phys. 101 (1994) 8894• B. Delley, J. Chem. Phys. 94 (1991) 7245• B. Delley, J. Comp. Chem. 17 (1996) 1152• B. Delley, Int. J. Quant. Chem. 69 (1998) 423 • B. Delley, J. Phys. Chem. 100 (1996) 6107• B. Delley, M. Wrinn, H. P. Lüthi, J. Chem. Phys. 100 (1994) 5785
References
Further Details:
Introduction: