
A Novel Kinase Inhibitor of FADD Phosphorylation...
12
Therapeutic Discovery A Novel Kinase Inhibitor of FADD Phosphorylation Chemosensitizes through the Inhibition of NF-kB Katrina A. Schinske 1,4 , Shyam Nyati 1,4 , Amjad P. Khan 4,5 , Terence M. Williams 1,4 , Timothy D. Johnson 3 , Brian D. Ross 2,4 , Ricardo P erez Tom as 6 , and Alnawaz Rehemtulla 1,4 Abstract Fas-associated protein with death domain (FADD) is a cytosolic adapter protein essential for mediating death receptor–induced apoptosis. It has also been implicated in a number of nonapoptotic activities including embryogenesis, cell-cycle progression, cell proliferation, and tumorigenesis. Our recent studies have shown that high levels of phosphorylated FADD (p-FADD) in tumor cells correlate with increased activation of the antiapoptotic transcription factor NF-kB and is a biomarker for aggressive disease and poor clinical outcome. These findings suggest that inhibition of FADD phosphorylation is a viable target for cancer therapy. A high-throughput screen using a cell-based assay for monitoring FADD-kinase activity identified NSC 47147 as a small molecule inhibitor of FADD phosphorylation. The compound was evaluated in live cells and mouse tumors for its efficacy as an inhibitor of FADD-kinase activity through the inhibition of casein kinase 1a. NSC 47147 was shown to decrease levels of p-FADD and NF-kB activity such that combination therapy leads to greater induction of apoptosis and enhanced tumor control than either agent alone. The studies described here show the utility of bioluminescent cell–based assays for the identification of active compounds and the validation of drug–target interaction in a living subject. In addition, the presented results provide proof-of-principle studies as to the validity of targeting FADD-kinase activity as a novel cancer therapy strategy. Mol Cancer Ther; 10(10); 1–11. Ó2011 AACR. Introduction Fas-associated protein with death domain (FADD) was first identified as a cytosolic adapter protein essential for mediating death receptor–induced apoptosis (1–3). It links to the cytoplasmic tail of active death receptors, such as Fas, DR4, and DR5, where it binds to procaspases- 8 and -10, leading to the formation of the death-inducing signaling complex (DISC) at the cytosolic side of the cell membrane. DISC formation initiates intracellular proces- sing and activation of procaspases, which, in turn, initi- ates cleavage of the downstream targets, such as caspase- 3, -6, and -7, and subsequently, apoptosis (4–6). Besides its role in regulating death receptor–induced apoptosis, FADD is also implicated in a number of death receptor– induced nonapoptotic activities including embryo- genesis, cell-cycle progression, cell proliferation, and tumorigenesis (6–10). Many of these nonapoptotic activ- ities are determined by the level of phosphorylation of a specific C-terminal serine (Ser194) in a region distinct from the proapoptotic function related to the death domain (11, 12). Recent studies have led to a better understanding of the FADD gene and its location on chromosome 11q13.3, a hot spot for chromosomal amplification in a number of hu- man cancers including esophagus, lung, and head and neck carcinomas (13, 14). Our recent studies provide evidence of overexpression of FADD mRNA and protein in human lung adenocarcinoma and its correlation to NF- kB activation. We have also shown that high levels of phosphorylated FADD (p-FADD), predominantly local- ized to the nucleus in lung tumor cells, is a biomarker for aggressive disease as well as for poor clinical outcome (13). The molecular basis for this correlation stems from the role of p-FADD as a potent mediator of the nonapop- totic transcription factor NF-kB (13, 15), a known regulator of cell fate decisions such as resistance to programmed cell death and lack of proliferation control (16). Phosphorylation of FADD at Ser194 has been shown to be mediated by casein kinase 1a (CK1a; 4) and FADD- interacting serine-threonine kinase/homeodomain-inter- acting protein kinase (FIST/HIPK3; refs. 3, 17), but the exact regulation and role of p-FADD in cancer are not well understood. In this study, we used a bioluminescent cell–based assay to characterize NSC 47147 as a potent Authors' Affiliations: Departments of 1 Radiation Oncology, 2 Radiology, and 3 Biostatistics, 4 Center for Molecular Imaging, and 5 Center for Trans- lational Pathology, University of Michigan, Ann Arbor, Michigan; and 6 Cancer Cell Biology Research Group, Department of Pathology and Experimental Therapeutics, University of Barcelona, Barcelona, Spain Corresponding Author: Alnawaz Rehemtulla, Department of Radiation Oncology, University of Michigan Medical School, Room A528, 109 Zina Pitcher Place, Ann Arbor, MI 48109. Phone: 734-764-4209; Fax: 734-615- 5669; E-mail: [email protected] doi: 10.1158/1535-7163.MCT-11-0362 Ó2011 American Association for Cancer Research. Molecular Cancer Therapeutics www.aacrjournals.org OF1 on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362
Transcript of A Novel Kinase Inhibitor of FADD Phosphorylation...

Therapeutic Discovery
A Novel Kinase Inhibitor of FADD PhosphorylationChemosensitizes through the Inhibition of NF-kB
Katrina A. Schinske1,4, Shyam Nyati1,4, Amjad P. Khan4,5, Terence M. Williams1,4, Timothy D. Johnson3,Brian D. Ross2,4, Ricardo P�erez Tom�as6, and Alnawaz Rehemtulla1,4
AbstractFas-associated protein with death domain (FADD) is a cytosolic adapter protein essential for mediating
death receptor–induced apoptosis. It has also been implicated in a number of nonapoptotic activities
including embryogenesis, cell-cycle progression, cell proliferation, and tumorigenesis. Our recent studies
have shown that high levels of phosphorylated FADD (p-FADD) in tumor cells correlate with increased
activation of the antiapoptotic transcription factor NF-kB and is a biomarker for aggressive disease and poor
clinical outcome. These findings suggest that inhibition of FADD phosphorylation is a viable target for cancer
therapy. A high-throughput screen using a cell-based assay for monitoring FADD-kinase activity identified
NSC 47147 as a small molecule inhibitor of FADD phosphorylation. The compoundwas evaluated in live cells
and mouse tumors for its efficacy as an inhibitor of FADD-kinase activity through the inhibition of casein
kinase 1a. NSC 47147 was shown to decrease levels of p-FADD and NF-kB activity such that combination
therapy leads to greater induction of apoptosis and enhanced tumor control than either agent alone. The
studies described here show the utility of bioluminescent cell–based assays for the identification of active
compounds and the validation of drug–target interaction in a living subject. In addition, the presented results
provide proof-of-principle studies as to the validity of targeting FADD-kinase activity as a novel cancer
therapy strategy. Mol Cancer Ther; 10(10); 1–11. �2011 AACR.
Introduction
Fas-associated protein with death domain (FADD) wasfirst identified as a cytosolic adapter protein essential formediating death receptor–induced apoptosis (1–3). Itlinks to the cytoplasmic tail of active death receptors,such as Fas, DR4, andDR5, where it binds to procaspases-8 and -10, leading to the formation of the death-inducingsignaling complex (DISC) at the cytosolic side of the cellmembrane. DISC formation initiates intracellular proces-sing and activation of procaspases, which, in turn, initi-ates cleavage of the downstream targets, such as caspase-3, -6, and -7, and subsequently, apoptosis (4–6). Besidesits role in regulating death receptor–induced apoptosis,FADD is also implicated in a number of death receptor–induced nonapoptotic activities including embryo-genesis, cell-cycle progression, cell proliferation, and
tumorigenesis (6–10). Many of these nonapoptotic activ-ities are determined by the level of phosphorylation of aspecific C-terminal serine (Ser194) in a region distinctfrom the proapoptotic function related to the deathdomain (11, 12).
Recent studies have led to a better understanding of theFADD gene and its location on chromosome 11q13.3, a hotspot for chromosomal amplification in a number of hu-man cancers including esophagus, lung, and head andneck carcinomas (13, 14). Our recent studies provideevidence of overexpression of FADD mRNA and proteinin human lung adenocarcinoma and its correlation to NF-kB activation. We have also shown that high levels ofphosphorylated FADD (p-FADD), predominantly local-ized to the nucleus in lung tumor cells, is a biomarker foraggressive disease as well as for poor clinical outcome(13). The molecular basis for this correlation stems fromthe role of p-FADD as a potent mediator of the nonapop-totic transcription factorNF-kB (13, 15), a knownregulatorof cell fate decisions such as resistance to programmed celldeath and lack of proliferation control (16).
Phosphorylation of FADD at Ser194 has been shown tobe mediated by casein kinase 1a (CK1a; 4) and FADD-interacting serine-threonine kinase/homeodomain-inter-acting protein kinase (FIST/HIPK3; refs. 3, 17), but theexact regulation and role of p-FADD in cancer are notwell understood. In this study, we used a bioluminescentcell–based assay to characterize NSC 47147 as a potent
Authors' Affiliations: Departments of 1Radiation Oncology, 2Radiology,and 3Biostatistics, 4Center for Molecular Imaging, and 5Center for Trans-lational Pathology, University of Michigan, Ann Arbor, Michigan; and6Cancer Cell Biology Research Group, Department of Pathology andExperimental Therapeutics, University of Barcelona, Barcelona, Spain
Corresponding Author: Alnawaz Rehemtulla, Department of RadiationOncology, University of Michigan Medical School, Room A528, 109 ZinaPitcher Place, Ann Arbor, MI 48109. Phone: 734-764-4209; Fax: 734-615-5669; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-11-0362
�2011 American Association for Cancer Research.
MolecularCancer
Therapeutics
www.aacrjournals.org OF1
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

inhibitor of FADD phosphorylation and to evaluate itspotential as a therapeutic agent in cancer treatment.
Materials and Methods
Antibodies and reagentsNSC 47147 was synthesized as previously described
(18). SP600125 was purchased from Calbiochem (EMDChemicals), CKI-7 from Toronto Research Chemicals,and bortezomib from Sigma Aldrich. The NCI DiversitySet was acquired from the NCI/DTP Open ChemicalRepository (http://dtp.cancer.gov). Cisplatin wasobtained from the University of Michigan pharmacy,and D-luciferin was purchased from Promega. Rabbitpolyclonal antibodies to phospho-FADD (Ser194), phos-pho-c-Jun (Ser63), phospho-b-catenin (Ser45), phospho-IkBa (Ser32/36), b-catenin, IkBa, caspase-3, cleavedcaspase-3 (Asp175), glyceraldehyde-3-phosphate dehy-drogenase (GAPDH), and mouse monoclonal antibodyto c-jun were purchased from Cell Signaling Technol-ogy. Goat polyclonal antibody to CK1a was purchasedfrom Santa Cruz Biotechnology and mouse monoclonalantibody to FADD from BD Pharmingen.
Cell cultureA549 (lung epithelial carcinoma), Jurkat (T lympho-
cyte), and SW620 (colorectal adenocarcinoma) cells werepurchased from the American Type Culture Collection(ATCC). Cell cultures were maintained in a humidifiedincubator at 37�C and 5%CO2. A549 and Jurkat cells weregrown in RPMI 1640 and SW620 cells in Leibovitz’s L-15medium. Each cell culture was supplemented with 10%heat-inactivated FBS (Invitrogen) and 100 units/mL pen-icillin. ATCC cell lines were tested routinely for Myco-plasma and purity. All ATCC lines were expandedimmediately upon receipt, and multiple vials of low-passage cells were maintained in liquid N2. No vial ofcells was cultured for more than 1 to 2 months. A549-FKRand SW620-BGCR cells have been previously described(19, 20). A549-FKR findings were validated with freshlyobtained A549 cultures from the ATCC. Cultures weremaintained in a humidified incubator at 37�C and 5%CO2, and all cell culture experiments were done inserum-containing media. For in vitro and in vivo experi-ments, cells were removed from tissue culture disheswith 0.05% trypsin containing EDTA. Cell cultureswere between 70% and 90% confluent at the time ofharvest.
Western blot analysisA549 and Jurkat cells were seeded at the appropriate
density in 6-well plates 24 hours before compoundtreatment. A549 cells were treated, washed twice withice-cold PBS, and lysed with extraction buffer [1% NP-40, 150 mmol/L NaCl, and 25 mmol/L Tris (pH 8.0)supplemented with complete phosphatase and proteaseinhibitor cocktail (Roche Diagnostics)]. Cell lysates wererocked at 4�C for 30 minutes. Particulate material was
removed by centrifugation at 13,000 rpm for 15 minutesat 4�C. The supernatants were collected, and proteincontent was estimated with a detergent-compatibleprotein assay kit from Bio-Rad. Whole-cell lysates con-taining equal amounts of protein (10–20 mg) were sep-arated by 12% Bis-Tris polyacrylamide gels (Invitrogen)and transferred to polyvinylidene difluoride mem-branes. The membranes were probed against specificprimary antibodies followed by horseradish peroxi-dase–conjugated secondary antibodies and visualizedwith the Enhanced Chemiluminescence Plus WesternBlotting System (GE Healthcare).
Bioluminescent FADD-kinase reporter assayThe bioluminescent FADD-kinase reporter (FKR) assay
was executed as previously described (18). Briefly, A549cells-expressingFKRcellswere seeded (1� 105 cells/well)in opaque 96-well plates, 24 hours before assaying. Com-pound stocks were prepared in dimethyl sulfoxide(DMSO) and diluted 1:100 in PBS. Intermediate stocks(10 mL) were added to the assay plates using the BeckmanBiomek NXP Laboratory Automation Workstation (Beck-man Coulter). Unless otherwise noted, cells were incu-bated with test compound at 37�C, 5% CO2 for 1 hour(CKI-7) and 6 hours (SP600125 and NSC 47147) at theindicated concentrations. Live cell luminescence imagingwas read with an EnVision Xcite Multilabel Reader (Per-kinElmer) 10 minutes after the addition of D-luciferin (100mg/mL final concentration) to the assay medium. Thepercentage of change in FKR activity was calculated as(Acontrol/Asample) � 100.
CK1a inhibition assaysCK1a enzymatic activity was evaluated by the Lance
Ultra CK2a1/b Kinase Assay (PerkinElmer) according tothe manufacturer’s instructions. Recombinant CK1a waspurchased from ProQinase. Serial dilutions of NSC 47147(1–100 mmol/L) and CKI-7 (1–300 mmol/L) were incu-bated with 25 nmol/L CK1a enzyme, 50 nmol/L ULight-Topo-IIa (Thr1342) peptide, and 1 mmol/L ATP, and finalconcentrations of inhibitors were in 2% DMSO. Kinasereactions were terminated after 30 minutes by the addi-tion of EDTA. Experiments were carried out in triplicate,and the data were derived from a minimum of 3 inde-pendent experiments. The percentage of inhibition wascalculated as % inhibition ¼ [(rate with no inhibitor) �(rate with inhibitor)/(rate with no inhibitor)] �100.
A bioluminescent reporter cell line expressing a con-struct designed to monitor changes in CK1a activityrecently described by our laboratory (SW620 BGCR)was used to provide additional evidence that NSC47147 inhibits FADD phosphorylation through inhibitionof CK1a (20). In brief, SW620-BGCR cells were plated in12-well plates and treated with 250 mmol/L CKI-7 and 3mmol/L NSC 47147 for 3 hours. Bioluminescence wasacquired on an IVIS 200 imaging platform (Caliper LifeScience) after adding 100 mg/mL D-luciferin (Xenogen).Region of interest values were calculated for each
Schinske et al.
Mol Cancer Ther; 10(10) October 2011 Molecular Cancer TherapeuticsOF2
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

exposure and analyzed. Bioluminescence measurementswere carried out in triplicate.
Cell viability assayA549 cells were monitored for viability based on ATP
levels 24 hours post–NSC 47147 treatment. NSC 47147/bortezomib and cisplatin sensitization experimentswere preincubated with cisplatin for 24 hours, followedby the addition of various doses of NSC 47147 orbortezomib for 24 hours. The percentage of viabilitywas measured by the CellTiter-Glo Luminescent CellViability Assay (Promega) and measured with the En-Vision Xcite Multilabel Reader (PerkinElmer). The per-centage of viability was calculated as (Asample/Acontrol)� 100.
Phospho-IkB functional assayPhospho-IkBlevelsweremonitoredinwhole-cell lysates
using AlphaScreen SureFire p-IkB (p-Ser32/Ser36) AssayKit (PerkinElmer), a homogenous bead-based assaydesigned to measure the phosphorylation of endogenousp-IkB (Ser32/Ser36) in cell lysates, according to the man-ufacturer’s protocol. TNFa-mediated IkBa phosphoryla-tion was measured in cells treated with 10 ng/mL TNFa(Invitrogen) for 0, 5, 15, and 30 minutes following 6-hourpretreatment with 6 mmol/L NSC 47147.
Flow cytometryA549 cells were pretreated with cisplatin (10 mmol/L)
or control vehicle for 18 hours, followed by treatmentwith 6 mmol/L NSC 47147 for 6 hours after which cellswere trypsinized, counted, and pelleted at 1 � 106 cellsper sample. The pellet was resuspended in PBS, fixed bydropwise addition of an equivalent volume of ice-cold100% ethanol. The samples were placed on ice for 20minutes and stored at 4�C until day of analysis. On theday of analysis, the cells were pelleted, ethanol decanted,and pellet resuspended in propidium iodide/RNase(50/100 mg/mL) PBS solution and analyzed at the FlowCytometry Core Facility at the University of MichiganCancer Center.
In vivo studiesTumor xenografts expressing FKR were established by
implanting 2� 106 stably transfected A549-FKR cells ontoboth flanks of nu/nu CD-1 male nude mice (Charles RiverLaboratories). When tumors reached a volume of approx-imately 100 to 150 mm3, treatment was initiated. Allmouse experiments were approved by the UniversityCommittee on the Use and Care of Animals of theUniversity of Michigan.
In vivo bioluminescence imaging and tumor growthstudiesFor bioluminescence imaging, mice bearing A549-FKR
xenograft were given a single intraperitoneal injection of0.5 mg/kg NSC 47147 or vehicle control (DMSO). Fol-lowing treatment, the mice were anesthetized with 2%
isofluorane/air mixture and given a single intraperito-neal injection of 150 mg/kg D-luciferin in PBS. Biolumi-nescent images were acquired beginning 5 minutes afterluciferin injection and at designated times posttreatment.Relative luminescence was calculated as the ratio ofbioluminescence at each time point over bioluminescencepretreatment.
For the in vivo tumor growth studies, tumor-bearingmice were randomized into 4 groups as follows: vehicle(DMSO), cisplatin (2 mg/kg), NSC 47147 (3 mg/kg),and a combination of cisplatin and NSC 47147(2 and 3 mg/kg, respectively). Cisplatin was dissolvedin water and NSC 47147 in 20% DMSO/water. Bothagents were administered via intraperitoneal injection.NSC 47147 was given daily for 8 days either alone orcoadministered in combination with cisplatin. Cisplatinwas given on days 1 and 7 in all cisplatin-containingregimens. Tumor volume was calculated according tothe equation for a prolate spheroid as follows: tumorvolume¼ (p/6)� (L�W2), where L andW represent thelonger and shorter dimensions of the tumor,respectively. Data are expressed as the ratio of tumorvolume at various times posttreatment compared withtumor volume on day 1. Mice were monitored daily,and tumor size and animal weight were measured every2 or 3 days until day 13.
ImmunohistochemistryA549-FKR tumor–bearing mice were treated each
day for 4 days with vehicle, cisplatin, NSC 47147, orcisplatin with NSC 47147. On the final day, mice weresacrificed 2 hours after injection and tumors wereexcised and fixed in 10% neutral-buffered formalinfor 48 hours (n ¼ 6 tumors for each of the 4 groups).Tumor slices (5 mm) were prepared according tostandard procedures and stained with hematoxylin–eosin. Level of apoptosis was detected with the Apop-Tag Peroxidase In Situ Apoptosis Detection Kit(Chemicon, Inc.), which stains for apoptosis by labelingand detecting DNA strand breaks by the terminaldeoxynucleotidyl transferase–mediated dUTP nickend labeling (TUNEL) method. Representative imageswere taken on an Olympus Bx-51 microscope with �20magnification.
Statistical analysisAll data were expressed as the mean � SEM from at
least 3 independent experiments. GraphPad Prism v5(GraphPad Software) nonlinear regression analyses andsigmoidal dose–response (variable slope) was used togenerate the IC50 values. For tumor growth analysis,ANOVAwas done on the proportional change of volumefrom baseline at each time point. If there was a significantdifference, all pairwise comparisons were run adjustingfor the multiple comparisons using Tukey’s HSD test.Differences between groups were considered significantat P � 0.05.
A Novel Inhibitor of FADD Phosphorylation
www.aacrjournals.org Mol Cancer Ther; 10(10) October 2011 OF3
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

Results
Identification of a FADD-kinase inhibitor, NSC47147
Using the A549-FKR assay to screen anNCI compounddiversity set, we identified NSC 47147, a tripyrrole alka-loid compound that inhibits FADD phosphorylation(Fig. 1). The compound increases FKR activity in a con-centration-dependent manner with an IC50 value of 2.0mmol/L (Fig. 2A). Comparatively, the c-jun-NH2-kinaseinhibitor (SP600125), known to downregulate FIST/HIPK3 activity, and the CK1a inhibitor (CKI-7) also showa concentration-dependent increase in FKR biolumines-cence with IC50 values of 22 and 143 mmol/L, respectively(Fig. 2A). These results are supported by a concomitantdecrease in p-FADD expression following treatment withSP600125 and CKI-7 (Fig. 2B). Inhibition of CK1a andJNK activity was validated based on decrease in p-b-cate-nin and p-c-jun levels, respectively. Changes in expres-sion levels were not due to differences in protein loadingas shown by expression of GAPDH. It is important to notethat in response to inhibition of FADD phosphorylation,corresponding levels of total FADD also show a decrease.We have previously shown that inhibition of FADDphosphorylation results in its ubiquitin-dependent deg-radation. Using MG132, a proteasome inhibitor, we con-firmed that a decrease in total FADD levels in response toFADD dephosphorylation (i.e., in the presence of aFADD-kinase inhibitor) can be reversed by inhibitingproteasomal degradation (19).
To delineate possible FADD-kinase targets of NSC47147, we examined whether the compound inhibitedphosphorylation of b-catenin, a downstream target ofCK1a. Western blot analysis of A549 and Jurkat cellsfollowing 6-hour treatment with NSC 47147 shows adecrease in phosphorylated b-catenin (p-b-catenin) pro-tein (Fig. 2C). These data show a trend similar to theinhibition of p-b-catenin expression following treatmentwith CKI-7 (Fig 2B).
Three-hour treatment of 250 mmol/L CKI-7 and 3mmol/L NSC 47147 of our recently described CK1a
reporter cell line (SW620-BGCR) resulted in 10-fold in-duction in bioluminescence activity, indicative of CKIainhibition (Fig 3A). In this assay, CKI-7 resulted in a 7.5-fold increase in activity. To examine further the effect ofNSC 47147 on CK1a inhibition, we directly evaluatedCK1a enzymatic activity in response to the compound(Fig. 3B). It is worth mentioning that maximal inhibitionby NSC 47147 was not observed because of its limitedaqueous solubility. However, the resulting IC50 value forNSC 47147, 6 mmol/L, supports the ability of NSC 47147to inhibit CK1a. Taken together, these results substantiateour finding that NSC 47147 inhibits phosphorylation ofFADD through inhibition of FADD-kinase CK1a.
Effect of NSC 47147 on cell viabilityTo examine the correlation of NSC 47147 activation of
the FKR and corresponding decrease in p-FADD on cellviability, A549 lung carcinoma cells were treated withincreasing concentrations of NSC 47147 and ATP levelsweremeasured at 24 hours posttreatment. The increase inFKR activity directly correlates with a decrease in thepercentage of cell viability. Results show similar IC50
values (2 mmol/L) for both inhibition of p-FADD, asindicated by increased reporter activity, and correspond-ing decrease in cell viability (Fig. 4).
NSC 47147 inhibits NF-kB activation in A549 lungcarcinoma cells
Our previous studies have shown a close correlationbetween levels of p-FADD and NF-kB activation (13). Onthe basis of this evidence, we sought to examine the effectof NSC 47147 on the inhibition of both endogenous andTNFa-induced levels of phosphorylated IkBa (p-IkBa),critical to the transcriptional activity of NF-kB (16). Treat-ment of A549 cells with 1, 3, and 6 mmol/L NSC 47147resulted in 31%, 63%, and 76% decrease in p-IkBa activ-ity, respectively, along with a corresponding decrease inprotein expression of both p-IkBa and total IkBa (Fig. 5Aand B). A549 cells stimulated with TNFa, followingpretreatment with NSC 47147, show that the inhibitoralso blocks TNFa-induced IkBa phosphorylation(Fig. 5C). Western blot analysis shows that NSC 47147inhibits TNFa-induced expression of p-IkBa at 5 minutespoststimulation (Fig. 5D) as well as the continued phos-phorylation and turnover of newly synthesized IkBa 30minutes after TNFa stimulation (Fig. 5D; ref. 21).
NSC 47147 as a chemosensitizing agentWe hypothesized that p-FADD–mediated NF-kB inhi-
bition by NSC 47147 would sensitize tumor cells tochemotherapeutic agents and enhance tumor cell death.To test this hypothesis, A549 cells were treated with NSC47147 or bortezomib, a small-molecule NF-kB inhibitor,in combination with cisplatin, a chemotherapeutic agentknown to induce apoptosis. The cells were pretreatedwith cisplatin (10 mmol/L) for 18 hours followed by 24-hour treatment with NSC 47147 or bortezomib followingwhich, ATP levels were measured. As shown in Fig. 6A,
MeO
NH
HN
CH3
C5H11
N
Figure 1. Compound structure for NSC 47147.
Schinske et al.
Mol Cancer Ther; 10(10) October 2011 Molecular Cancer TherapeuticsOF4
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362
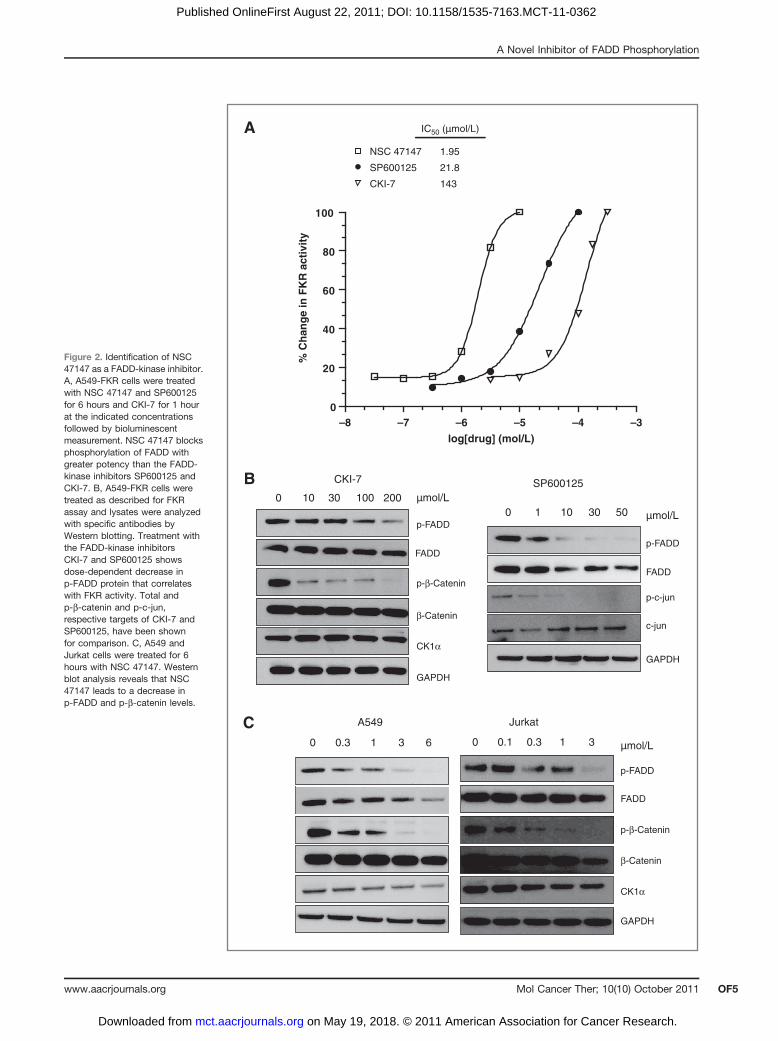
Figure 2. Identification of NSC47147 as a FADD-kinase inhibitor.A, A549-FKR cells were treatedwith NSC 47147 and SP600125for 6 hours and CKI-7 for 1 hourat the indicated concentrationsfollowed by bioluminescentmeasurement. NSC 47147 blocksphosphorylation of FADD withgreater potency than the FADD-kinase inhibitors SP600125 andCKI-7. B, A549-FKR cells weretreated as described for FKRassay and lysates were analyzedwith specific antibodies byWestern blotting. Treatment withthe FADD-kinase inhibitorsCKI-7 and SP600125 showsdose-dependent decrease inp-FADD protein that correlateswith FKR activity. Total andp-b-catenin and p-c-jun,respective targets of CKI-7 andSP600125, have been shownfor comparison. C, A549 andJurkat cells were treated for 6hours with NSC 47147. Westernblot analysis reveals that NSC47147 leads to a decrease inp-FADD and p-b-catenin levels.
0 1 10
SP600125
30 50
p-c-jun
c-jun
p-FADD
FADD
GAPDH
µmol/L
0 10
CKI-7
p-FADD
FADD
30 100 200
CK1α
β-Catenin
p-β-Catenin
GAPDH
µmol/L
A549
0 0.3 1 3 6 µmol/L0 0.1 0.3 1 3
p-FADD
FADD
GAPDH
CK1α
β-Catenin
p-β-Catenin
Jurkat
A
–8 –7 –6 –5 –4 –30
20
40
60
80
100
% C
han
ge
in F
KR
act
ivit
y
log[drug] (mol/L)
IC50 (µmol/L)
CKI-7 143
SP600125 21.8
NSC 47147 1.95
B
C
A Novel Inhibitor of FADD Phosphorylation
www.aacrjournals.org Mol Cancer Ther; 10(10) October 2011 OF5
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

NSC 47147 treatment in combination with cisplatin leadsto an increase in cell death (*, P � 0.05) as compared withNSC 47147 treatment alone. As shown in Fig. 6B, treat-ment of A549 cells with bortezomib and cisplatin alsoresulted in amarked (*, P� 0.05) decrease in cell viability.These data suggest that the enhanced sensitivity to cis-platin in response to NSC 47147 or bortezomib treatmentis due to inhibition of NF-kB.
To further confirm the cooperative effects of cisplatinand NSC 47147, A549 cells pretreated with cisplatin
(10 mmol/L) for 18 hours followed by 6-hour treatmentwith NSC 47147 (6 mmol/L) were subjected to cell-cycleanalysis. Figure 6C shows 12.3% of cells in sub-G1 phasein response to NSC 47147/cisplatin combination treat-ment as compared with vehicle control (1.8%), NSC 47147(1.6%), and cisplatin (5.2%) alone. Western blot analysison similarly treated cells resulted in an increase in cas-pase-3 cleavage with cisplatin, no effect with NSC 47147alone, and a marked increase in cleaved caspase-3 levels(17- and 19-kDa bands) with combined NSC 47147 andcisplatin treatment (Fig. 6D), thus supporting the capacityof NSC 47147 to augment cisplatin-induced apoptosis.
Effect of NSC 47147 in tumor xenograft modelsHaving established the efficacy of NSC 47147 toward
suppression of FADD phosphorylation and subsequentNF-kB inhibition in cells, we next sought to investigatethe effects of the compound in a mouse tumor model. Toestablish tumors, we implanted A549-FKR cells into theflanks of nude mice. When tumors reached a volumebetween 100 and 150 mm3, we monitored biolumines-cence over time inmice treatedwith NSC 47147 or vehiclecontrol. Bioluminescence imaging shows an increase inFKR activity following treatment with NSC 47147; max-imum reporter activity was reached within 6 hours fol-lowed by sustained, although lower, activity up to 36hours (Fig. 7A). Representative images of NSC 47147–treated mice are shown in Fig. 7B.
The chemosensitizing effects of NSC 47147 on tumorprogression were evaluated in A549-FKR mouse xeno-grafts treated with NSC 47147 (3 mg/kg) once daily for8 days with or without cisplatin (2 mg/kg) intraperito-neal injections on days 1 and 7. Significant reduction(P � 0.05) in tumor volume was observed at day 13 forthe NSC 47147/cisplatin combination therapy as com-pared with control or either chemical agent alone(Fig. 7C). Notably, mice treated with the combinationtherapy had no increase in tumor volume through the
Rel
ativ
e lu
min
esce
nce
(fold
indu
ctio
n)
12
10
8
6
4
2
0
Control NSC 47147 CKI-7 –7 –6 –5 –4 –30
20
40
60
80
100
log[drug] (mol/L)
IC50 (µmol/L)
CKI-7 22.3
NSC 47147 6.36
Perc
ent i
nhib
ition
A B
Figure 3. NSC 47147 inhibitsCK1a. A, SW620 CK1a reportercells were treated with 3 mmol/LNSC 47147 and 250 mmol/L CKI-7for 3 hours. Data show 10-foldincrease in bioluminescenceindicative of CK1a inhibition inresponse to NSC 47147treatment. CKI-7, a CK1ainhibitor, is shown as a positivecontrol. B, NSC 47147 wasevaluated by a TR-FRETbiochemical assay for its directeffect on CK1a enzymatic activity.The in vitro assay reveals directinhibition of CK1a activity by NSC47147.
FKR assay
Cell viability
IC50 (µmol/L)
–8 –7 –6 –5 –40
20
40
60
80
100
0
20
40
60
80
100
log[NSC 47147] (mol/L)
% V
iab
ility
% C
han
ge
in F
KR
act
ivit
y
1.95
2.04
Figure 4. Inhibition of FADD phosphorylation correlates with decreasedcell viability. Concentration-dependent inhibition by NSC 47147 in A549-FKR cell–based assay is shown plotted against the percentage of cellviability. Cells were treated with NSC 47147 for 6 hours in the FKR assayand 24 hours to assess viability. ATP levels were quantified followingtreatment with Promega CellTiter-Glo. Identical IC50 values for eachanalysis suggest a correlation between inhibition of FADD phosphorylationand decreased cell viability.
Schinske et al.
Mol Cancer Ther; 10(10) October 2011 Molecular Cancer TherapeuticsOF6
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

duration of the study, whereasmice treatedwith cisplatinor NSC 47147 alone resumed tumor growth at day 13 andday 7, respectively.On the basis of these results, we evaluated the degree of
apoptosis in the tumor xenografts following treatmentwith NSC 47147 alone or in combination with cisplatin.Immunohistologic staining for apoptosis shows areas ofcell death within tumors of cisplatin-treated mice, a smallamount of apoptosis in tumors of NSC 47147–treatedmice, and a pronounced increase in apoptosis throughoutthe tumors of mice receiving the combination of cisplatinand NSC 47147 (Fig. 7D).
Discussion
Although FADD was originally identified as a keymediator of apoptosis, it has recently been identified asa modulator of a number of death receptor–dependentand -independent nonapoptotic activities includingembryogenesis, cell-cycle progression, cell proliferation,and tumorigenesis (6, 8, 9). Previous research from ourlaboratory has revealed p-FADD as a biomarker for poor
clinical outcome in human lung adenocarcinoma. Ourstudies have shown a strong correlation between highlevels of p-FADD and elevated NF-kB activity, the anti-apoptotic actions of which lead to the formation of ag-gressive phenotypes, resistance to chemotherapeuticagents, and poor clinical outcome (13, 15, 22). In thisstudy, we provide evidence that abrogation of NF-kBsignaling through small-molecule inhibition of FADDphosphorylation is a novel and viable approach for can-cer therapy.
Comprehensive studies recently conducted in A549and Jurkat cell lines have provided insight into themechanistic basis for FADD and its involvement inNF-kB activation and tumorigenesis (13, 23, 24). Inaddition, it was in Jurkat cells that CK1a was identifiedas the kinase that phosphorylates FADD (4). Researchfrom our laboratory has shown these lines to express highlevels of p-FADD protein, thereby providing the impetustoward selecting these models for the identification andevaluation of a potential FADD-kinase inhibitor. UsingA549 lung cancer cells expressing FKR (19), we con-ducted a high-throughput screen with an NCI diversity
NSC 47147
TNF-α (min) 0 5 15 30 0 5 15 30
Lum
ine
sce
nce
(R
LU
)
(-)
(+)
TNF-α (min)
- - - - + + + +
0 1 3 6
NSC 47147 (µmol/L)
0
20,000
40,000
60,000
80,000
NSC 47147
NSC 47147
IκB
p-IκB
GAPDH
NSC 47147 (µmol/L) 0 1 3 6
IκB
p-IκB
GAPDH
0 5 15 30
8,000
6,000
4,000
2,000
0
Lum
ines
cenc
e (R
LU)
A
C
B
D
Figure 5. NSC 47147 attenuation of NF-kB activity. The phosphorylation status of IkBa was evaluated by AlphaScreen p-IkB assay (A) and byWestern blot analysis (B) in A549 cells following 6-hour treatment with 0, 1, 3, and 6 mmol/L NSC 47147. Results showed a decrease in luminescenceindicative of p-IkB inhibition. Western blotting confirmed decreasing levels of p-IkB proteins and total IkBa proteins in response to NSC 47147.A549 cells were stimulated with 10 ng/ml TNFa at the indicated timepoints following pretreatment with 6 mmol/L NSC 47147. Cellular lysates were subjectedto analysis by p-IkB AlphaScreen (C) and Western blotting (D).
A Novel Inhibitor of FADD Phosphorylation
www.aacrjournals.org Mol Cancer Ther; 10(10) October 2011 OF7
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

compound collection and identified NSC 47147 as apotent inhibitor of FADD phosphorylation. NSC 47147is a prodigiosin, a family of natural red pigments syn-thesized by a variety of microorganisms. Prodigiosinshave been shown to possess antineoplastic properties,showing an ability to initiate cell-cycle arrest and apo-ptosis (25, 26).
Treatment of A549-FKR cells with NSC 47147 showed aconcentration-dependent increase in reporter activity,with 1 to 2 logs greater potency than the previously
described FADD-kinase inhibitors SP600125 and CKI-7(IC50 ¼ 2, 22, and 150 mmol/L, respectively). The increasein reporter bioluminescence following treatment withinhibitors of FADD-kinases, FIST/HIPK3, and CK1asuggests that the mechanism of action of NSC 47147could be, in part, due to its inhibition of either of thesekinases. Western blot analysis of A549 and Jurkat cellstreated with NSC 47147 shows a dose-dependent deple-tion of p-FADD with a concomitant decrease in p-b-cate-nin, also a CK1a substrate (27). No decrease in c-jun
–9 –8 –7 –6 –5 –4
20
40
60
80
100
NSC 47147
NSC 47147 + cisplatin
log[NSC 47147] (mol/L)
% V
iab
ility
Control NSC 47147
Cisplatin NSC 47147 + cisplatin
A
% G1 = 51.7% S = 36.9% G2 = 11.4% sub-G1 = 1.8
% G1 = 43.0% S = 37.8% G2 = 19.1% sub-G1 = 1.6
% G1 = 5.1% S = 67.9% G2 = 27.0% sub-G1 = 5.2
% G1 = 8.0% S = 68.4% G2 = 23.6% sub-G1 = 12.3
Cleaved caspase-3
p-FADD
GAPDH
Caspase-3
FADD
- + - +- - + +
Cisplatin
NSC 47147
B
0
100 80
50
0
0
80
0
0 200 400 600 800 1,000
0 200 400 600 800 1,000 0 200 400 600 800 1,000
0 200 400 600 800 1,000
40
60
80
100
% V
iab
ility
–10 –9 –8 –7 –6
log[Bortezomib] (mol/L)
Bortezomib
Bortezomib + cisplatin
* **
*
*
**
*
C D
Figure 6. NSC 47147 chemosensitizes A549 lung cancer cells to cisplatin-induced apoptosis. A and B, A549 cells were preincubated with 10 mmol/Lcisplatin for 24 hours followed by increasing concentrations of NSC 47147 or bortezomib for 24 hours. ATP levels following treatment were quantifiedwith Promega CellTiter-Glo. Data show that inhibition of NF-kB by NSC 47147 or bortezomib in combination with cisplatin leads to greater cell deaththan NSC 47147 or bortezomib alone (*, P � 0.05). C, A549 lung cancer cells treated with NSC 47147 in the absence or presence of cisplatin weresubjected to cell-cycle analysis. Data show more cells in sub-G1 with NSC 47147/cisplatin combination therapy (12.3%) than with control (1.8%),NSC 47147 (1.6%), and cisplatin alone (5.2%). D, A549 cells treated with NSC 47147, cisplatin, or both agents were evaluated for inhibition of p-FADDlevels and induction of apoptosis by assessing the levels of cleaved caspase-3.
Schinske et al.
Mol Cancer Ther; 10(10) October 2011 Molecular Cancer TherapeuticsOF8
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

activation was apparent, indicating that the compoundobtrudes CK1a activity but does not influence FIST/HIPK3 (data not shown). The dose-dependent decreasein p-FADD and p-b-catenin proteins in both cell linessuggests that NSC 47147 may affect p-FADD levels byeither direct inhibition of CK1a or by impinging on an
upstream signaling event. The results presented herereveal inhibitory activity in a CK1a cell–based assay aswell as direct inhibition in a CK1a biochemical assay,thereby providing mechanistic evidence that NSC 47147is a CK1a inhibitor. CK1a regulates multiple oncogenicpathways, in addition to its proposed involvement in
Control
NSC 47147
Cisplatin
Cisplatin + NSC 47147
B
0.0
0.5
1.0
1.5
2.0
1 3 5 7 10 13
Re
lativ
e tu
mor
vol
ume
Time (d)
Control
NSC 47147
Cisplatin
NSC 47147 + cisplatin
A
Treatment
0.0
1.0
2.0
3.0
4.0
0 0.5 3 6 18 36
Rel
ativ
e lu
min
esce
nce
Time (h)
C D
6,000
5,000
4,000
3,000
2,000
1,000
0 h
3 h
6 h
18 h
NSC 47147
Control
Figure 7. NSC 47147 inhibits FADD phosphorylation in an A549-FKR xenograft model and sensitizes tumors to an apoptotic stimulus. A, athymic nudemice bearing A549-FKR–expressing xenografts were treated with vehicle control (DMSO) or NSC 47147 (0.5 mg/kg) by intraperitoneal injection.Mice were imaged for bioluminescence at the indicated times. Relative luminescence was calculated as the ratio of bioluminescence at each timepoint to the basal bioluminescence before treatment. Data points represent mean � SEM relative luminescence. B, representative bioluminescenceimages of tumor-bearing mice pretreatment (basal) and 3, 6, and 18 hours posttreatment with NSC 47147 (0.5 mg/kg). C, tumor-bearing mice weretreated with 3 mg/kg NSC 47147 once daily for 8 days and/or 2 mg/kg cisplatin on days 1 and 7 by intraperitoneal injection, and tumor volume wascalculated by day 13. Data are plotted as mean � SEM relative tumor volume. D, immunohistochemical staining of A549-FKR xenografts afteronce daily treatment for 4 days with 3 mg/kg NSC 47147 and/or 2 mg/kg cisplatin. Tumors were harvested and fixed on day 4; sections werestained for apoptosis as described in Materials and Methods. Figure shows duplicate images of representative fields.
A Novel Inhibitor of FADD Phosphorylation
www.aacrjournals.org Mol Cancer Ther; 10(10) October 2011 OF9
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

p-FADD–mediated NF-kB regulation, namely, the b-cate-nin/Wnt signaling axis (27, 28), making it a rational targetfor drug therapy.
A decrease in p-FADD levels, as detected by an in-crease in FKR activity, directly correlated with a reduc-tion in cell viability, suggesting that the inhibition ofFADD phosphorylation and increase in cytotoxicity area consequence of the same biological event. It has beenproposed that the molecular basis for the correlationbetween p-FADD levels and cell death stems from itsrole as a potent activator of NF-kB, an antiapoptotictranscription factor (13). Previous research has shownthat overexpression of FADD stimulates NF-kB promoteractivity (15, 29) and that FADD-deficient cells are moresusceptible to viral infection due to defects in NF-kBactivation (30). Furthermore, FADD plays an integral rolein the recently described TRADDosome complex, a cen-tral mediator of NF-kB signaling (31). Although it waspreviously unappreciated, our published work hasshown that increased levels of p-FADD result in elevatedlevels of NF-kB activation (13). The data presented in thisarticle provide further evidence that the phosphorylatedform of FADD rather than FADD is a key component ofthe NF-kB activating complex.
The role of NF-kB in tumorigenesis has been wellestablished and is based on its action as a regulator ofcell fate decisions. It is often dysregulated in cancer cellsleading to uncontrolled cell proliferation and resistanceto therapeutic intervention (16). These data show thatA549 lung carcinoma cells treated with NSC 47147 yield adecrease in NF-kB activation, as measured by the phos-phorylation status of IkBa. Phosphorylation of IkBa byIKK requires recruitment of the TRADDosome complexwherein IKK plays an essential role (31). These resultsconfirm previous reports that TNFa-induced NF-kB ac-tivation is dependent on levels of FADD expression (15,32, 33) and provide further evidence of NSC 47147 as aninhibitor of FADD phosphorylation.
Tumor cells with constitutively active NF-kB areknown to be resistant to chemotherapeutic agents, aresistance that can be alleviated by inhibition of NF-kBsignaling (16). We hypothesized that pretreatment withNSC 47147 would chemosensitize A549 lung cancer cellstoward an apoptotic stimulus leading to enhanced celldeath. Results from our cell-cycle analysis show a higherpercentage of cells in the sub-G1 population with aparallel increase in caspase-3 activity, thereby providingevidence that NSC 47147 sensitizes cells to cisplatinleading to decreased cell viability. We believe the mech-anistic basis for the proposed synergy is that NSC 47147,by inhibiting FADD phosphorylation, abates TRADDo-some-mediated NF-kB activation. In the absence of NF-kB signaling, tumor cells become sensitive to apoptoticstimuli (i.e., in response to cisplatin treatment). In thisregard, our data show that bortezomib-induced NF-kBinhibition in combination with cisplatin also results in adecrease in cell viability. The ability of NSC 47147 toexhibit a similar outcomewhen used in combination with
cisplatin provides additional mechanistic evidence thatthe cytotoxic effects of NSC 47147 are at least in part dueto its inhibition of NF-kB.
Studies from our laboratory, aswell as others, have alsoshown a role of p-FADD in G2–M progression (24, 34).Therefore, we expected NSC 47147 to arrest cells at theG2–M phase of the cell cycle consistent with its proposedmechanism as an inhibitor of p-FADD levels. Asexpected, the fraction of cells in G2–M phase followingNSC 47147 treatment was 19.1% comparedwith 11.4% forcontrol cells, thereby providing additional support thatNSC 47147 inhibits FADD phosphorylation leading tocell-cycle arrest and cell death.
Having shown the in vitro effect of NSC 47147 aloneand in combination with cisplatin, we next examinedthe compounds efficacy as a chemotherapeutic agent ina mouse tumor model. Mouse tumor xenograftsexpressing the FKR showed an increase in biolumines-cence following treatment with NSC 47147, showingthe ability of the compound to inhibit activation ofFADD in an animal model. The combined NSC 47147and cisplatin therapy resulted in a decrease in tumorvolume greater than the effect of either compoundalone. Consistent with the reduction in tumor volume,TUNEL staining revealed enhanced apoptotic activityin mouse tumors treated with combination drug ther-apy compared with either agent alone. Treatment ofmice with 3 mg/kg NSC 47147 did not result in overttoxicity in our treatment groups as well as by otherresearch groups (35, 36). This, combined with thecapacity of NSC 47147 to control tumor burden whenused in combination with a known chemotherapyagent, provides validation for the investigation ofmore efficacious and biologically available inhibitorsof FADD phosphorylation. The identification of com-pounds that, when coadministered with chemothera-peutic drugs, increase their efficacy is of significantclinical importance.
In summary, our previous research has identified p-FADD as a prognostic biomarker for poor clinical out-come. The mechanistic basis for this biomarker is theability of p-FADD to promote the antiapoptotic actions ofNF-kB. The results presented here identify a small mol-ecule inhibitor of FADD phosphorylation that inducescell death through abrogation of NF-kB activity. We haveprovided proof-of-principle studies showing that inhibi-tion of FADD phosphorylation through CK1a may be aviable target for anticancer therapy as a single agent,but more interestingly, in combination with clinicallyapproved chemotherapeutic agents.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank the members of the Center for Molecular Imagingfor their technical help and constructive criticisms.
Schinske et al.
Mol Cancer Ther; 10(10) October 2011 Molecular Cancer TherapeuticsOF10
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

Grant Support
This work was supported by the U.S. NIH research grants R01CA129623(A. Rehemtulla), R21CA131859 (A. Rehemtulla), U24CA083099 (B.D. Ross),P50CA093990 (B.D. Ross), and by an RSNA Resident Seed grant (T.M. Williams).Grant support for synthesis of NSC 47147 was provided by Mi Ministerio deSanidad, Spain, and the European Union (FIS PI061226).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received May 12, 2011; revised July 28, 2011; accepted August 8, 2011;published OnlineFirst August 22, 2011.
References1. Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death
domain-containing protein, interacts with the death domain of Fas andinitiates apoptosis. Cell 1995;81:505–12.
2. Zhang J, Winoto A. Amouse Fas-associated protein with homology tothe human Mort1/FADD protein is essential for Fas-induced apopto-sis. Mol Cell Biol 1996;16:2756–63.
3. Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement ofMACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1-and TNF receptor-induced cell death. Cell 1996;85:803–15.
4. Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, MurmannAE, et al. Phosphorylation of FADD at serine 194 by CKIalpha reg-ulates its nonapoptotic activities. Mol Cell 2005;19:321–32.
5. Elrod HA, Sun SY. Modulation of death receptors by cancer thera-peutic agents. Cancer Biol Ther 2008;7:163–73.
6. Tourneur L, Chiocchia G. FADD: a regulator of life and death. TrendsImmunol 2010;31:260–9.
7. Kabra NH, Kang C, Hsing LC, Zhang J, Winoto A. T cell-specificFADD-deficient mice: FADD is required for early T cell development.Proc Natl Acad Sci U S A 2001;98:6307–12.
8. Imtiyaz HZ, Zhou X, Zhang H, Chen D, Hu T, Zhang J. The deathdomain of FADD is essential for embryogenesis, lymphocyte devel-opment, and proliferation. J Biol Chem 2009;284:9917–26.
9. Zhang J, Kabra NH, Cado D, KangC,Winoto A. FADD-deficient T cellsexhibit a disaccord in regulation of the cell cycle machinery. J BiolChem 2001;276:29815–8.
10. Hua ZC, Sohn SJ, Kang C, Cado D, Winoto A. A function of Fas-associateddeathdomainprotein in cell cycleprogression localized to asingle amino acid at its C-terminal region. Immunity 2003;18:513–21.
11. Tourneur L, Buzyn A, Chiocchia G. FADD adaptor in cancer. MedImmunol 2005;4:1.
12. Werner MH, Wu C, Walsh CM. Emerging roles for the death adaptorFADD in death receptor avidity and cell cycle regulation. Cell Cycle2006;5:2332–8.
13. Chen G, Bhojani MS, Heaford AC, Chang DC, Laxman B, Thomas DG,et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle,and is associated with poor outcome in lung adenocarcinomas. ProcNatl Acad Sci U S A 2005;102:12507–12.
14. Gibcus JH, Menkema L, Mastik MF, Hermsen MA, de Bock GH, vanVelthuysen ML, et al. Amplicon mapping and expression profilingidentify the Fas-associated death domain gene as a new driver in the11q13.3 amplicon in laryngeal/pharyngeal cancer. Clin Cancer Res2007;13:6257–66.
15. Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD,Casper, and caspase-8. J Biol Chem 2000;275:10838–44.
16. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: frominnocent bystander to major culprit. Nat Rev Cancer 2002;2:301–10.
17. Rochat-Steiner V, Becker K, Micheau O, Schneider P, Burns K,Tschopp J. FIST/HIPK3: a Fas/FADD-interacting serine/threoninekinase that induces FADD phosphorylation and inhibits fas-medi-ated Jun NH(2)-terminal kinase activation. J Exp Med 2000;192:1165–74.
18. D'Alessio R, Bargiotti A, Carlini O, Colotta F, Ferrari M, Gnocchi P,et al. Synthesis and immunosuppressive activity of novel prodigiosinderivatives. J Med Chem 2000 Jun 29;43(13):2557–65.
19. Khan AP, Schinske KA, Nyati S, Bhojani MS, Ross BD, Rehemtulla A.High-throughput molecular imaging for the identification of FADDkinase inhibitors. J Biomol Screen 2010;15:1063–70.
20. Nyati S, Ranga R, Ross BD, Rehemtulla A, Bhojani MS. Molecularimaging of glycogen synthase kinase-3beta and casein kinase-1alphakinases. Anal Biochem 2010;405:246–54.
21. Nasuhara Y, Adcock IM, Catley M, Barnes PJ, Newton R. DifferentialIkappaB kinase activation and IkappaBalpha degradation by interleu-kin-1beta and tumor necrosis factor-alpha in human U937 monocyticcells. Evidence for additional regulatory steps in kappaB-dependenttranscription. J Biol Chem 1999;274:19965–72.
22. Monks NR, Biswas DK, Pardee AB. Blocking anti-apoptosis as astrategy for cancer chemotherapy: NF-kappaB as a target. J CellBiochem 2004;92:646–50.
23. Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G,et al. Inhibition of death receptor-mediated gene induction by acycloheximide-sensitive factor occurs at the level of or upstream ofFas-associated death domain protein (FADD). J Biol Chem 2000;275:24357–66.
24. Bhojani MS, Chen G, Ross BD, Beer DG, Rehemtulla A. Nuclearlocalized phosphorylated FADD induces cell proliferation and is as-sociated with aggressive lung cancer. Cell Cycle 2005;4:1478–81.
25. Perez-Tomas R, Vinas M. New insights on the antitumoral propertiesof prodiginines. Curr Med Chem 2010;17:2222–31.
26. Montaner B, Perez-Tomas R. The prodigiosins: a new family ofanticancer drugs. Curr Cancer Drug Targets 2003;3:57–65.
27. Knippschild U, Wolff S, Giamas G, Brockschmidt C, Wittau M, WurlPU, et al. The role of the casein kinase 1 (CK1) family in differentsignaling pathways linked to cancer development. Onkologie 2005;28:508–14.
28. Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. Thecasein kinase 1 family: participation in multiple cellular processes ineukaryotes. Cell Signal 2005;17:675–89.
29. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1,an adaptor triggering RIG-I- and Mda5-mediated type I interferoninduction. Nat Immunol 2005;6:981–8.
30. Balachandran S, Thomas E, Barber GN. A FADD-dependent innateimmune mechanism in mammalian cells. Nature 2004;432:401–5.
31. Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M,Curran J, et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity 2008;28:651–61.
32. Ermolaeva MA, Michallet MC, Papadopoulou N, Utermohlen O, Kra-nidioti K, Kollias G, et al. Function of TRADD in tumor necrosis factorreceptor 1 signaling and in TRIF-dependent inflammatory responses.Nat Immunol 2008;9:1037–46.
33. Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal trans-duction pathways. Cell 1996;84:299–308.
34. Scaffidi C, Volkland J, Blomberg I, Hoffmann I, Krammer PH, PeterME. Phosphorylation of FADD/MORT1 at serine 194 and associationwith a 70-kDa cell cycle-regulated protein kinase. J Immunol 2000;164:1236–42.
35. Yamamoto C, Takemoto H, Kuno K, Yamamoto D, Tsubura A, KamataK, et al. Cycloprodigiosin hydrochloride, a new H(þ)/Cl(�) symporter,induces apoptosis in human and rat hepatocellular cancer cell lines invitro and inhibits the growth of hepatocellular carcinoma xenografts innude mice. Hepatology 1999;30:894–902.
36. Zhang J, Shen Y, Liu J, Wei D. Antimetastatic effect of prodigiosinthrough inhibition of tumor invasion. Biochem Pharmacol 2005;69:407–14.
A Novel Inhibitor of FADD Phosphorylation
www.aacrjournals.org Mol Cancer Ther; 10(10) October 2011 OF11
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362

Published OnlineFirst August 22, 2011.Mol Cancer Ther Katrina A. Schinske, Shyam Nyati, Amjad P. Khan, et al.
BκChemosensitizes through the Inhibition of NF-A Novel Kinase Inhibitor of FADD Phosphorylation
Updated version
10.1158/1535-7163.MCT-11-0362doi:
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://mct.aacrjournals.org/content/early/2011/10/01/1535-7163.MCT-11-0362To request permission to re-use all or part of this article, use this link
on May 19, 2018. © 2011 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst August 22, 2011; DOI: 10.1158/1535-7163.MCT-11-0362


















