
A new interpretation of total column BrO during Arctic spring · A new interpretation of total...
9
A new interpretation of total column BrO during Arctic spring R. J. Salawitch, 1,2,3 T. Canty, 1 T. Kurosu, 4 K. Chance, 4 Q. Liang, 5 A. da Silva, 6 S. Pawson, 6 J. E. Nielsen, 7 J. M. Rodriguez, 6 P. K. Bhartia, 6 X. Liu, 4,5 L. G. Huey, 8 J. Liao, 8 R. E. Stickel, 8 D. J. Tanner, 8 J. E. Dibb, 9 W. R. Simpson, 10 D. Donohoue, 10 A. Weinheimer, 11 F. Flocke, 11 D. Knapp, 11 D. Montzka, 11 J. A. Neuman, 12,13 J. B. Nowak, 12,13 T. B. Ryerson, 13 S. Oltmans, 13 D. R. Blake, 14 E. L. Atlas, 15 D. E. Kinnison, 11 S. Tilmes, 11 L. L. Pan, 11 F. Hendrick, 16 M. Van Roozendael, 16 K. Kreher, 17 P. V. Johnston, 17 R. S. Gao, 13 B. Johnson, 13 T. P. Bui, 18 G. Chen, 19 R. B. Pierce, 20 J. H. Crawford, 19 and D. J. Jacob 21 Received 30 April 2010; revised 24 August 2010; accepted 27 August 2010; published 3 November 2010. [ 1] Emission of bromine from sea ‐ salt aerosol, frost flowers, ice leads, and snow results in the nearly complete removal of surface ozone during Arctic spring. Regions of enhanced total column BrO observed by satellites have traditionally been associated with these emissions. However, airborne measurements of BrO and O 3 within the convective boundary layer (CBL) during the ARCTAS and ARCPAC field campaigns at times bear little relation to enhanced column BrO. We show that the locations of numerous satellite BrO “hotspots” during Arctic spring are consistent with observations of total column ozone and tropopause height, suggesting a stratospheric origin to these regions of elevated BrO. Tropospheric enhancements of BrO large enough to affect the column abundance are also observed, with important contributions originating from above the CBL. Closure of the budget for total column BrO, albeit with significant uncertainty, is achieved by summing observed tropospheric partial columns with calculated stratospheric partial columns provided that natural, short‐lived biogenic bromocarbons supply between 5 and 10 ppt of bromine to the Arctic lowermost stratosphere. Proper understanding of bromine and its effects on atmospheric composition requires accurate treatment of geographic variations in column BrO originating from both the stratosphere and troposphere. Citation: Salawitch, R. J., et al. (2010), A new interpretation of total column BrO during Arctic spring, Geophys. Res. Lett., 37, L21805, doi:10.1029/2010GL043798. 1. Introduction [2] Regions of elevated BrO at high northerly latitudes during spring associated with the autocatalytic release of bromine from sea‐salt aerosol, frost flowers, ice leads, and snow, commonly called the “bromine explosion”, cause complete removal of surface ozone [e.g., Barrie et al., 1988; Platt and Hönninger, 2003]. Satellite observations of enhanced column BrO during spring, which we term “BrO hotspots” (regions where total column BrO is elevated by 2 to 3 × 10 13 cm −2 relative to the zonal mean), have long been associated with the surface release of bromine and ozone depletion events (ODEs) [e.g., Chance, 1998; Richter et al., 1998; Wagner et al., 2001]. [3] There is also widespread interest in atmospheric BrO due to its role as a catalyst for loss of ozone in the strato- sphere [e.g., Salawitch et al., 2005] and upper troposphere [e.g., von Glasow et al., 2004]. Oxidation by reaction with atomic Br could be the dominant sink for elemental mer- cury, with important consequences for mercury deposition [e.g., Holmes et al., 2006]. Reaction with BrO could be a significant sink for dimethlysulfide in the marine boundary layer, reducing subsequent production of SO 2 and new cloud condensation nuclei [e.g., von Glasow et al., 2004]. [4] Satellite observations provide the best constraint on the global distribution of BrO. Measurements of the vertical column abundance of BrO (BrO VC ) reveal much higher amounts (i.e., factor of 2 or 3 more) than found in standard 1 Department of Atmospheric and Oceanic Science, University of Maryland, College Park, Maryland, USA. 2 Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland, USA. 3 Earth System Science Interdisciplinary Center, University of Maryland, College Park, Maryland, USA. 4 Harvard-Smithsonian Center for Astrophysics, Cambridge, Massachusetts, USA. 5 GEST, University of Maryland Baltimore County, Greenbelt, Maryland, USA. 6 NASA Goddard Space Flight Center, Greenbelt, Maryland, USA. 7 Science Systems and Applications, Inc., Lanham, Maryland, USA. 8 School of Earth and Atmospheric Science, Georgia Institute of Technology, Atlanta, Georgia, USA. 9 Complex Systems Research Center, University of New Hampshire, Durham, New Hampshire, USA. 10 Department of Chemistry and Biochemistry, University of Alaska Fairbanks, Fairbanks, Alaska, USA. 11 National Center for Atmospheric Research, Boulder, Colorado, USA. 12 CIRES, University of Colorado at Boulder, Boulder, Colorado, USA. 13 Earth System Research Laboratory, NOAA, Boulder, Colorado, USA. 14 Department of Chemistry, University of California, Irvine, California, USA. 15 RSMAS, University of Miami, Miami, Florida, USA. 16 Belgian Institute for Space Aeronomy, Brussels, Belgium. 17 NIWA Lauder, Omakau, New Zealand. 18 NASA Ames Research Center, Moffett Field, California, USA. 19 NASA Langley Research Center, Hampton, Virginia, USA. 20 NESDIS, NOAA, Madison, Wisconsin, USA. 21 School of Engineering and Applied Sciences, Harvard University, Cambridge, Massachusetts, USA. Copyright 2010 by the American Geophysical Union. 0094-8276/10/2010GL043798 GEOPHYSICAL RESEARCH LETTERS, VOL. 37, L21805, doi:10.1029/2010GL043798, 2010 L21805 1 of 9
Transcript of A new interpretation of total column BrO during Arctic spring · A new interpretation of total...

A new interpretation of total column BrO during Arctic spring
R. J. Salawitch,1,2,3 T. Canty,1 T. Kurosu,4 K. Chance,4 Q. Liang,5 A. da Silva,6
S. Pawson,6 J. E. Nielsen,7 J. M. Rodriguez,6 P. K. Bhartia,6 X. Liu,4,5 L. G. Huey,8
J. Liao,8 R. E. Stickel,8 D. J. Tanner,8 J. E. Dibb,9 W. R. Simpson,10 D. Donohoue,10
A. Weinheimer,11 F. Flocke,11 D. Knapp,11 D. Montzka,11 J. A. Neuman,12,13
J. B. Nowak,12,13 T. B. Ryerson,13 S. Oltmans,13 D. R. Blake,14 E. L. Atlas,15
D. E. Kinnison,11 S. Tilmes,11 L. L. Pan,11 F. Hendrick,16 M. Van Roozendael,16
K. Kreher,17 P. V. Johnston,17 R. S. Gao,13 B. Johnson,13 T. P. Bui,18 G. Chen,19
R. B. Pierce,20 J. H. Crawford,19 and D. J. Jacob21
Received 30 April 2010; revised 24 August 2010; accepted 27 August 2010; published 3 November 2010.
[1] Emission of bromine from sea‐salt aerosol, frostflowers, ice leads, and snow results in the nearly completeremoval of surface ozone during Arctic spring. Regions ofenhanced total column BrO observed by satellites havetraditionally been associated with these emissions.However, airborne measurements of BrO and O3 withinthe convective boundary layer (CBL) during the ARCTASand ARCPAC field campaigns at times bear little relationto enhanced column BrO. We show that the locations ofnumerous satellite BrO “hotspots” during Arctic spring areconsistent with observations of total column ozone andtropopause height, suggesting a stratospheric origin tothese regions of elevated BrO. Tropospheric enhancementsof BrO large enough to affect the column abundance arealso observed, with important contributions originatingfrom above the CBL. Closure of the budget for totalcolumn BrO, albeit with significant uncertainty, isachieved by summing observed tropospheric partialcolumns with calculated stratospheric partial columnsprovided that natural, short‐lived biogenic bromocarbonssupply between 5 and 10 ppt of bromine to the Arcticlowermost stratosphere. Proper understanding of bromineand its effects on atmospheric composition requiresaccurate treatment of geographic variations in column BrOoriginating from both the stratosphere and troposphere.Citation: Salawitch, R. J., et al. (2010), A new interpretation oftotal column BrO during Arctic spring, Geophys. Res. Lett., 37,L21805, doi:10.1029/2010GL043798.
1. Introduction
[2] Regions of elevated BrO at high northerly latitudesduring spring associated with the autocatalytic release ofbromine from sea‐salt aerosol, frost flowers, ice leads, andsnow, commonly called the “bromine explosion”, causecomplete removal of surface ozone [e.g., Barrie et al., 1988;Platt and Hönninger, 2003]. Satellite observations ofenhanced column BrO during spring, which we term “BrOhotspots” (regions where total column BrO is elevated by 2to 3 × 1013 cm−2 relative to the zonal mean), have long beenassociated with the surface release of bromine and ozonedepletion events (ODEs) [e.g., Chance, 1998; Richter et al.,1998; Wagner et al., 2001].[3] There is also widespread interest in atmospheric BrO
due to its role as a catalyst for loss of ozone in the strato-sphere [e.g., Salawitch et al., 2005] and upper troposphere[e.g., von Glasow et al., 2004]. Oxidation by reaction withatomic Br could be the dominant sink for elemental mer-cury, with important consequences for mercury deposition[e.g., Holmes et al., 2006]. Reaction with BrO could be asignificant sink for dimethlysulfide in the marine boundarylayer, reducing subsequent production of SO2 and newcloud condensation nuclei [e.g., von Glasow et al., 2004].[4] Satellite observations provide the best constraint on
the global distribution of BrO. Measurements of the verticalcolumn abundance of BrO (BrOVC) reveal much higheramounts (i.e., factor of 2 or 3 more) than found in standard
1Department of Atmospheric and Oceanic Science, University ofMaryland, College Park, Maryland, USA.
2Department of Chemistry and Biochemistry, University ofMaryland, College Park, Maryland, USA.
3Earth System Science Interdisciplinary Center, University ofMaryland, College Park, Maryland, USA.
4Harvard-Smithsonian Center for Astrophysics, Cambridge,Massachusetts, USA.
5GEST, University of Maryland Baltimore County, Greenbelt,Maryland, USA.
6NASA Goddard Space Flight Center, Greenbelt, Maryland, USA.7Science Systems and Applications, Inc., Lanham, Maryland, USA.8School of Earth and Atmospheric Science, Georgia Institute of
Technology, Atlanta, Georgia, USA.
9Complex Systems Research Center, University of New Hampshire,Durham, New Hampshire, USA.
10Department of Chemistry and Biochemistry, University of AlaskaFairbanks, Fairbanks, Alaska, USA.
11National Center for Atmospheric Research, Boulder, Colorado,USA.
12CIRES, University of Colorado at Boulder, Boulder, Colorado, USA.13Earth System Research Laboratory, NOAA, Boulder, Colorado,
USA.14Department of Chemistry, University of California, Irvine,
California, USA.15RSMAS, University of Miami, Miami, Florida, USA.16Belgian Institute for Space Aeronomy, Brussels, Belgium.17NIWA Lauder, Omakau, New Zealand.18NASA Ames Research Center, Moffett Field, California, USA.19NASA Langley Research Center, Hampton, Virginia, USA.20NESDIS, NOAA, Madison, Wisconsin, USA.21School of Engineering and Applied Sciences, Harvard University,
Cambridge, Massachusetts, USA.
Copyright 2010 by the American Geophysical Union.0094-8276/10/2010GL043798
GEOPHYSICAL RESEARCH LETTERS, VOL. 37, L21805, doi:10.1029/2010GL043798, 2010
L21805 1 of 9

models. Considerable debate has centered on the relativerole of contributions from the stratosphere and troposphereto this difference [e.g., Richter et al., 1998; Salawitch et al.,2005].[5] Traditionally, the tropospheric BrO burden has been
obtained by subtracting a background from the measuredsatellite signal, accounting for different air mass factors(ratio of light path through the atmosphere to a vertical path)of the stratospheric and tropospheric components [e.g.,Wagner et al., 2001]. The background is commonly basedon a longitudinally invariant stratosphere, although potentialerrors of this approach have been noted [e.g., Richter et al.,1998]. Significant effort has been devoted to measuring thenear surface mixing ratio of BrO during Arctic spring [e.g.,Platt and Hönninger, 2003, and references therein]. Priormeasurements in the Arctic from aircraft [McElroy et al.,1999] and ground‐based [Hönninger et al., 2004] instru-ments suggest the presence of significant levels of tropo-spheric BrO above the top of the convective boundary layer(CBL, characterized by constant potential temperature withrespect to altitude), further complicating our ability to relatesatellite observations of BrOVC to surface ODEs. Quantita-tive closure of the budget for BrOVC has heretofore not beenachieved [e.g., Ridley et al., 2007].[6] This paper is focused on quantification of contribu-
tions from the troposphere and the stratosphere to BrOVC
during Arctic spring. Enhancements of BrO large enough tobe recorded as a satellite “hotspot” are associated with thecompression of stratospheric air to high pressure in regionsof a low altitude tropopause. Consequently, the notion thatthe stratospheric contribution to total column BrO can beapproximated by a constant background is flawed. Ourobservations demonstrate that the tropospheric burden ofBrO can also contribute to BrOVC at a magnitude consistentwith satellite “hotspots” and that a significant portion of thetropospheric signal originates from above the top of theCBL, a region not typically sampled by ground based in-struments. We show that closure of the budget for BrOVC
can be achieved by summing observed tropospheric partialcolumn BrO with calculated stratospheric partial columnBrO, albeit with significant uncertainty in each term. Thisrepresents a significant step forward in our understanding ofatmospheric BrO.
2. Observations and Model Description
2.1. OMI BrO
[7] OMI is on the NASA Aura platform in a sun‐synchronous orbit with a 1:38 pm equator crossing time(ascending node). BrOVC is retrieved from reflected sunlightobserved in nadir. The algorithm is based on non‐linear,least‐squares fitting of radiances in the 319 to 347.5 nmwindow [Chance, 1998]. BrOVC is found using wavelengthdependent air mass factors, computed with a multiple scat-tering radiative transfer model. Contributions from the O2
dimer have been neglected, resulting in lower noise andsmaller fitting uncertainties than the operational OMI BrOproduct. Surface albedo is based on a geographically varying,monthly mean climatology derived from OMI observations[Kleipool et al., 2008]. Typically, the fitting residual leads toa ±22% uncertainty (1s) for BrOVC.
[8] Retrievals of BrOVC from OMI compare extremelywell with estimates from ground‐based instruments locatedin Harestua, Norway (60.2°N, 11°E) and Lauder, NewZealand (45.0°S, 169.7°E). The ground‐based and satellitemeasurements agree within 15%, with no discernable bias.The auto‐correlation of errors in the state vector elements ofBrOVC and O3 column, from a simultaneous retrieval, isnegligible. We therefore conclude regions of enhancedBrOVC are not an artifact caused by the treatment of O3 inthe retrieval algorithm. Further details of the retrieval andthese comparisons are given in the auxiliary material.1
2.2. Aircraft BrO and Related Species
[9] A variety of aircraft observations are used. The DC‐8 andWP‐3D aircraft flown during the NASA ARCTAS (ArcticResearch of the Composition of the Troposphere from Air-craft and Satellites) and NOAA ARCPAC (Aerosol, Radi-ation, and Cloud Processes affecting Arctic Climate)campaigns carried in situ instruments that measured BrO,BrCl, and Br2 using chemical ionization mass spectrometry(CIMS) [Neuman et al., 2010]. The measurements of BrOare accurate to ± 40% + 1 ppt with a precision of 3 ppt for a2 sec integration time. The DC‐8 measurements are reportedwith 30 sec time resolution and the WP‐3D measurementsare reported with 2 sec resolution. The detection limit wastypically ∼2 ppt for the WP‐3D instrument and between 2 to5 ppt for the DC‐8 instrument. Laboratory [Neuman et al.,2010] and field comparisons [Liao et al., 2010] indicatethat BrO is not produced or lost on the inlets of the CIMSinstruments. The presence of Br2 during daylight is thoughtto result from the conversion of HOBr to Br2 on theinstrument inlet [Neuman et al., 2010]. We show time seriesof BrO + BrCl + 2 × Br2, which we term BrOx. Solublebromide was measured in a mist chamber and includesnumerous condensable species. Detailed descriptions of thevarious instruments are given by Neuman et al. [2010].[10] Measurements of organic bromocarbons and CFC‐12
were acquired by Whole Air Sampler (WAS) instrumentsonboard the NASA DC‐8 aircraft during ARCTAS and TC4
(Tropical Composition, Cloud and Climate Coupling) andthe NASA WB‐57 aircraft during TC4. Details of the WASinstruments are given by Schauffler et al. [1999]. The WASdata are discussed primarily in the auxiliary material. In situO3 was measured using chemiluminescence on the WP‐3Dand DC‐8 and using a dual beam UV photometer on theWB‐57.
2.3. Ground Based BrO
[11] Observations of tropospheric BrO over Barrow,Alaska (71.3°N, 156.8°W) are provided by a Max‐DOAS(Multi Axis Differential Optical Absorption Spectroscopy)instrument. The differential slant column density of BrO(BrOdSCD) is found as a function of elevation angle (EA) ofthe acquired spectra [Hönninger et al., 2004]. A radiativetransfer program is used to model the variation of BrOdSCD
with EA, for various assumptions regarding the height dis-tribution of BrO. This technique provides a strong constrainton the distribution of BrO within the lowest several km ofthe troposphere.
1Auxiliary materials are available in the HTML. doi:10.1029/2010GL043798.
SALAWITCH ET AL.: FRONTIER L21805L21805
2 of 9

2.4. Stratospheric Model BrO
[12] Calculation of the vertical column abundance ofstratospheric BrO (BrOSTRAT) is central to this paper. Thecalculation begins with global profiles of CFC‐12 found bya GEOS‐5 assimilation conducted for ARCTAS, with sur-face emission and stratospheric destruction of CFC‐12[Liang et al., 2008]. CFC‐12 is output on a 0.5° × 0.67°(lat/lon) grid for 72 pressure levels, from the surface to0.01 hPa, every 6 hours. Comparison to aircraft observations(see auxiliary material) demonstrates that modeled CFC‐12 isaccurate to within ±4% in the lower stratosphere. Values ofBry (total inorganic bromine) are found from CFC‐12, usingthe method of Wamsley et al. [1998]. The baseline value forBry assumes supply of stratospheric inorganic bromine (Bry)from CH3Br, halons, and CH2Br2.[13] We also conduct simulations of BrO assuming supply
of an additional 5 and 10 ppt of Bry from very short lived(VSL) bromocarbons, termed Bry
VSL. Since our baseline valueof Bry includes a contribution from CH2Br2 by source gasinjection (SGI) into the stratosphere, our definition of BryVSL
differs from that used by World Meteorological Organization(WMO) [2007]. The relation of our definition of Bry
VSL toWMO [2007] and justification for use of 5 and 10 ppt levelsof BryVSL based on the WAS measurements are discussed inthe auxiliary material. Briefly, CBry (total organic bromine)was observed to reach upwards of 30 ppt in the tropicalmarine boundary layer and to exceed 25 ppt in the region ofconvective outflow in the tropical upper troposphere duringTC4. These observations, together with WAS measurementsthat show direct injection of VSL species into the Arcticlowermost stratosphere (LMS), support the plausibility thatBry in the Arctic LMSwas 5 to 10 ppt higher than our baselinevalue of Bry.[14] Once Bry is specified, BrO is found using the BrO/
Bry ratio from a run of WACCM (Whole AtmosphereCommunity Climate Model) [Garcia et al., 2007] conductedfor the START08 (Stratosphere‐Troposphere Analyses ofRegional Transport 2008) campaign (April to June 2008).WACCM output, provided every 3 hrs on a 1.9° × 2.5° (lat/lon)grid for 89 pressure levels ranging from 1000 to 4.5 ×10−6 hPa, is interpolated to the finer GEOS‐5 grid for thetime of OMI overpass. A single run of WACCM that con-sidered supply of Bry from only CH3Br and halons is used;we adjust Bry outside of WACCM and rely on WACCM forthe BrO/Bry ratio (which is insensitive to Bry for the range ofvariations used in this study). We use this procedure becauseBrO/Bry is sensitive to O3 and NO2 [e.g., Theys et al., 2009]and the START08 WACCM run provides an estimate of O3
and NO2 for conditions specific to spring 2008. WACCMcalculations of O3, NOx and NO compare extremely well toobservations of these species obtained during ARCTAS andSTART08. A description of WACCM and demonstration ofits performance, including excellent evaluation of the BrO/Bry ratio, is given by Chipperfield and Kinnison [2010].BrOSTRAT is found by integrating model profiles of BrOfrom the tropopause (WMO definition of thermal tropo-pause) to the top of the model atmosphere, for the time ofOMI overpass.[15] A photochemical steady state model is used to assess
the uncertainty in BrOSTRAT. This model has been con-strained to profiles of O3, NOy, Bry, etc. from WACCM asdescribed by Chipperfield and Kinnison [2010]. The rate
constant of each reaction that affects BrO/Bry is varied usinguncertainties of Sander et al. [2006]. The total uncertainty inBrOSTRAT is found from a root‐sum‐squares combination ofthe individual chemical kinetics terms (including J values)and propagation, through the model, of the impact of a 4%error in the specification of CFC‐12 in the lowermoststratosphere. The reaction of BrO + NO2 forming BrNO3,which is uncertain by a factor of 2 at 220 K, contributesmost to the overall uncertainty [e.g., Hendrick et al., 2008].The 1s uncertainty in BrOSTRAT for the Bry
VSL = 10 pptsimulation is ±30% (globally) and ±45% (region of lowaltitude tropopause), with larger uncertainties near the lowaltitude tropopause due to increased importance of BrO +NO2.
3. Results
3.1. Comparison of Airborne and Satellite BrO
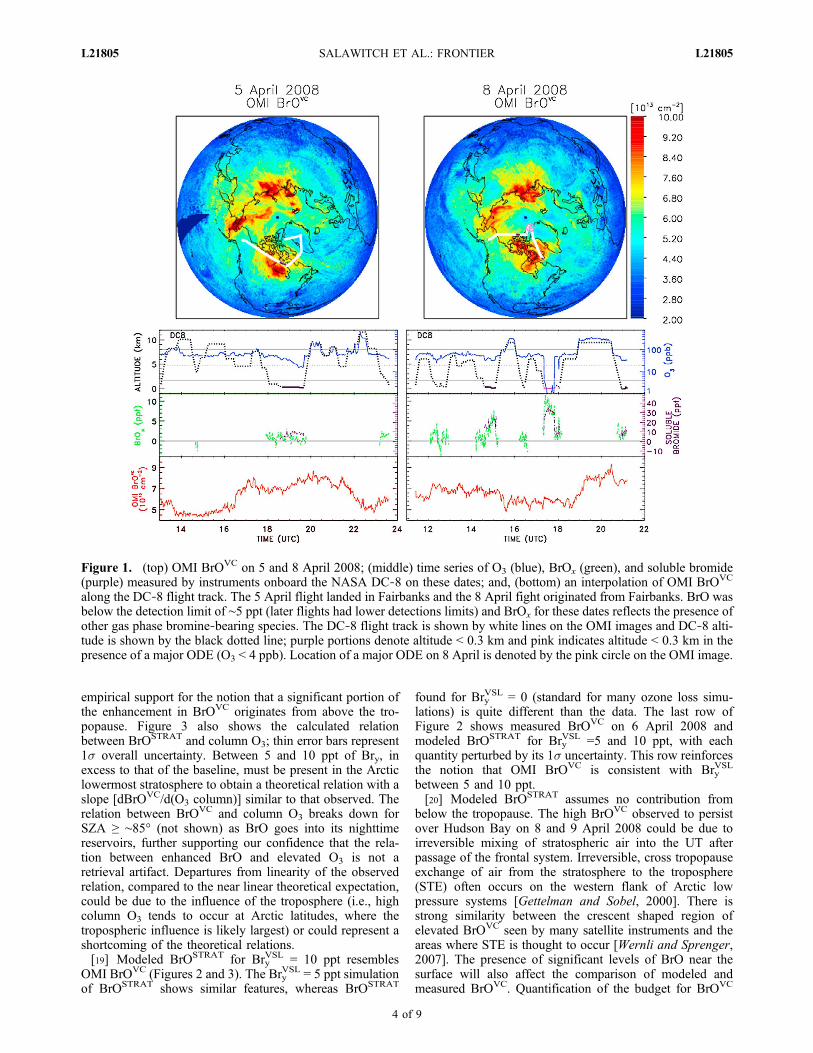
[16] Observations of enhanced BrOx and soluble bromideas well as depleted ozone obtained by the DC‐8 aircraftduring ARCTAS often bear little relation to the location ofOMI BrOVC “hotspots”. Measurements obtained on 5 and8 April 2008 are shown in Figure 1. Several extended lowaltitude legs (aircraft below 0.3 km) were targeted for re-gions of elevated BrO, based on analysis of OMI observa-tions from the prior day. These flight portions are denotedby purple line segments for the flight track and altitudetraces. The instruments recorded little perturbation toambient O3, BrOx, and soluble bromide in the regions whereOMI BrOVC was highly enhanced. Furthermore, a majorODE co‐located with highly enhanced BrOx and solublebromide was observed on 8 April 2008 near 83°N, 65°W,far from the region of elevated BrOVC (pink circle). Inter-polation of OMI BrOVC along the DC‐8 flight track (bottompanel) exhibits no meaningful correlation with any of theDC‐8 measurements. This set of observations, representa-tive of the majority of data obtained during ARCTAS andARCPAC, challenges pre‐conceived notions of the relationbetween ODEs and satellite measurements of enhancedcolumn BrO.
3.2. Importance of the Stratosphere
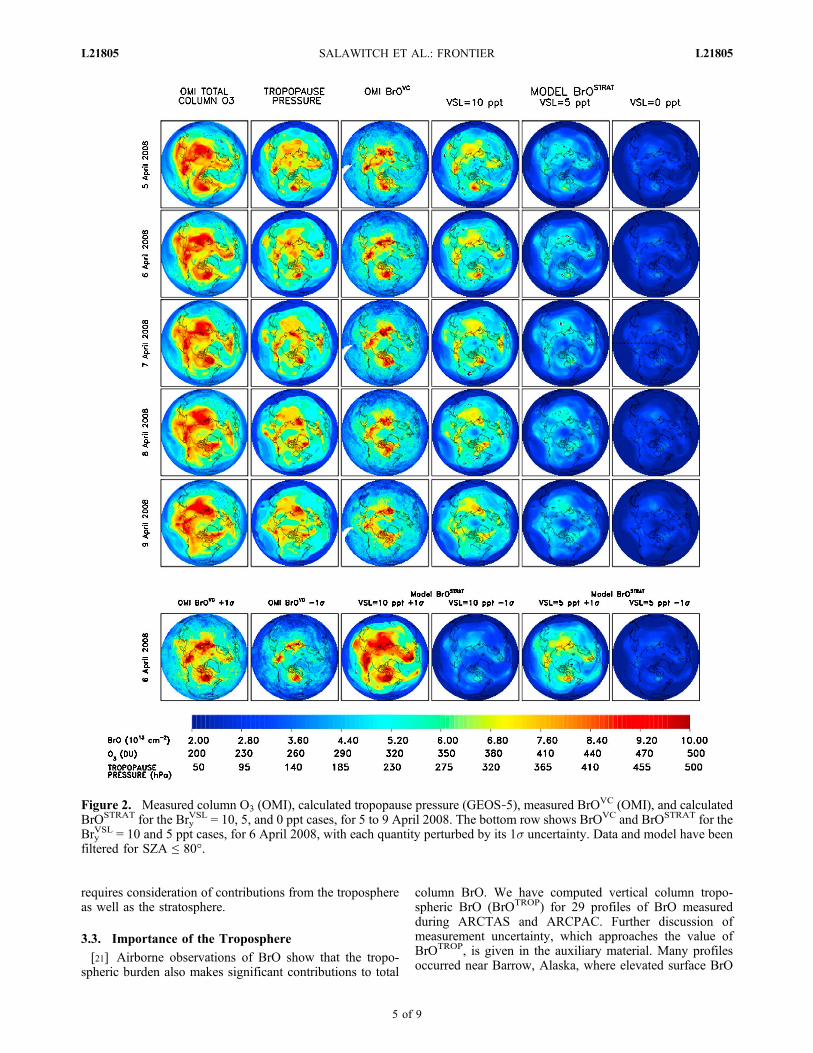
[17] Figure 2 shows total column O3, tropopause pressure,BrOVC, and modeled BrOSTRAT for 5 to 9 April 2008.ColumnO3 is from the standardOMI product, available at http://disc.sci.gsfc.nasa.gov/Aura/data‐holdings/OMI/omto3d_v003.shtml. The region of elevated BrO over Hudson Bay iscoincident with a low tropopause (∼5 km altitude or ∼450 hPapressure) and high total column O3 (∼450 DU). These fea-tures progress in a similar counter‐clockwise fashion, sug-gesting the enhancements in BrOVC originate from abovethe tropopause. Enhanced BrOSTRAT is associated with asynoptic weather pattern characterized by a low altitudetropopause and high column O3. The preponderance of priorobservations of elevated column BrO over Hudson Bayduring spring may be related to a weather pattern known asthe Hudson Bay low that is responsible for the depressedtropopause [Liu and Moore, 2004].[18] An important feature of the satellite data is the
compact, monotonic relation between BrOVC and columnO3 for data acquired with solar zenith angle (SZA) ≤ 80°(Figure 3). The monotonic nature of this relation (i.e.,BrOVC rises as column O3 increases) provides strong
SALAWITCH ET AL.: FRONTIER L21805L21805
3 of 9
empirical support for the notion that a significant portion ofthe enhancement in BrOVC originates from above the tro-popause. Figure 3 also shows the calculated relationbetween BrOSTRAT and column O3; thin error bars represent1s overall uncertainty. Between 5 and 10 ppt of Bry, inexcess to that of the baseline, must be present in the Arcticlowermost stratosphere to obtain a theoretical relation with aslope [dBrOVC/d(O3 column)] similar to that observed. Therelation between BrOVC and column O3 breaks down forSZA ≥ ∼85° (not shown) as BrO goes into its nighttimereservoirs, further supporting our confidence that the rela-tion between enhanced BrO and elevated O3 is not aretrieval artifact. Departures from linearity of the observedrelation, compared to the near linear theoretical expectation,could be due to the influence of the troposphere (i.e., highcolumn O3 tends to occur at Arctic latitudes, where thetropospheric influence is likely largest) or could represent ashortcoming of the theoretical relations.[19] Modeled BrOSTRAT for Bry
VSL = 10 ppt resemblesOMI BrOVC (Figures 2 and 3). The Bry
VSL = 5 ppt simulationof BrOSTRAT shows similar features, whereas BrOSTRAT
found for BryVSL = 0 (standard for many ozone loss simu-
lations) is quite different than the data. The last row ofFigure 2 shows measured BrOVC on 6 April 2008 andmodeled BrOSTRAT for Bry
VSL =5 and 10 ppt, with eachquantity perturbed by its 1s uncertainty. This row reinforcesthe notion that OMI BrOVC is consistent with Bry
VSL
between 5 and 10 ppt.[20] Modeled BrOSTRAT assumes no contribution from
below the tropopause. The high BrOVC observed to persistover Hudson Bay on 8 and 9 April 2008 could be due toirreversible mixing of stratospheric air into the UT afterpassage of the frontal system. Irreversible, cross tropopauseexchange of air from the stratosphere to the troposphere(STE) often occurs on the western flank of Arctic lowpressure systems [Gettelman and Sobel, 2000]. There isstrong similarity between the crescent shaped region ofelevated BrOVC seen by many satellite instruments and theareas where STE is thought to occur [Wernli and Sprenger,2007]. The presence of significant levels of BrO near thesurface will also affect the comparison of modeled andmeasured BrOVC. Quantification of the budget for BrOVC
Figure 1. (top) OMI BrOVC on 5 and 8 April 2008; (middle) time series of O3 (blue), BrOx (green), and soluble bromide(purple) measured by instruments onboard the NASA DC‐8 on these dates; and, (bottom) an interpolation of OMI BrOVC
along the DC‐8 flight track. The 5 April flight landed in Fairbanks and the 8 April fight originated from Fairbanks. BrO wasbelow the detection limit of ∼5 ppt (later flights had lower detections limits) and BrOx for these dates reflects the presence ofother gas phase bromine‐bearing species. The DC‐8 flight track is shown by white lines on the OMI images and DC‐8 alti-tude is shown by the black dotted line; purple portions denote altitude < 0.3 km and pink indicates altitude < 0.3 km in thepresence of a major ODE (O3 < 4 ppb). Location of a major ODE on 8 April is denoted by the pink circle on the OMI image.
SALAWITCH ET AL.: FRONTIER L21805L21805
4 of 9
requires consideration of contributions from the troposphereas well as the stratosphere.
3.3. Importance of the Troposphere
[21] Airborne observations of BrO show that the tropo-spheric burden also makes significant contributions to total
column BrO. We have computed vertical column tropo-spheric BrO (BrOTROP) for 29 profiles of BrO measuredduring ARCTAS and ARCPAC. Further discussion ofmeasurement uncertainty, which approaches the value ofBrOTROP, is given in the auxiliary material. Many profilesoccurred near Barrow, Alaska, where elevated surface BrO
Figure 2. Measured column O3 (OMI), calculated tropopause pressure (GEOS‐5), measured BrOVC (OMI), and calculatedBrOSTRAT for the Bry
VSL = 10, 5, and 0 ppt cases, for 5 to 9 April 2008. The bottom row shows BrOVC and BrOSTRAT for theBry
VSL = 10 and 5 ppt cases, for 6 April 2008, with each quantity perturbed by its 1s uncertainty. Data and model have beenfiltered for SZA ≤ 80°.
SALAWITCH ET AL.: FRONTIER L21805L21805
5 of 9

and ODEs are often observed. Most profiles were obtained inclear sky. Profiles acquired on 16 April 2008 by the DC‐8 areshown in Figure 4. The flight track is superimposed on OMIBrOVC (Figure 4a) and modeled BrOSTRAT (Figure 4b).Elevated OMI BrOVC is remarkably well aligned with theunderlying sea, suggesting an association with surfacerelease of bromine. The largest value of BrOTROP observedthis day, 3.9 × 1013 cm−2, is co‐located with a local maxi-mum in OMI BrOVC.[22] The most significant contribution to BrOTROP on 16
April 2008 was from altitudes well above the CBL. For thehighlighted profile, the CBL had a ceiling of ∼0.2 km(Figure 4e). The WP‐3D aircraft sampled extensively aboveand below this altitude on 16 April and other days, at timesdescending to 60 m above the surface [Neuman et al., 2010].The importance of BrO above the CBL to BrOTROP iscommon to all profiles acquired during ARCTAS andARCPAC (auxiliary material). ARCTAS and ARCPACaircraft measurements reveal lower abundances of BrO inthe CBL than reported in past ground‐based studies [e.g.,Platt and Hönninger, 2003]. The aircraft flights, by design,often sampled O3 depleted air within the CBL. Inorganicbromine shifts from BrO into other inorganic species asambient O3 falls below ∼4 ppb, which was often the case forCBL air sampled during ARCTAS and ARCPAC [Neumanet al., 2010], perhaps accounting for the tendency for ourmeasurements of BrO to be lower than prior observations inthe CBL.[23] Figure 5 shows BrOdSCD vs EA for Max‐DOAS data
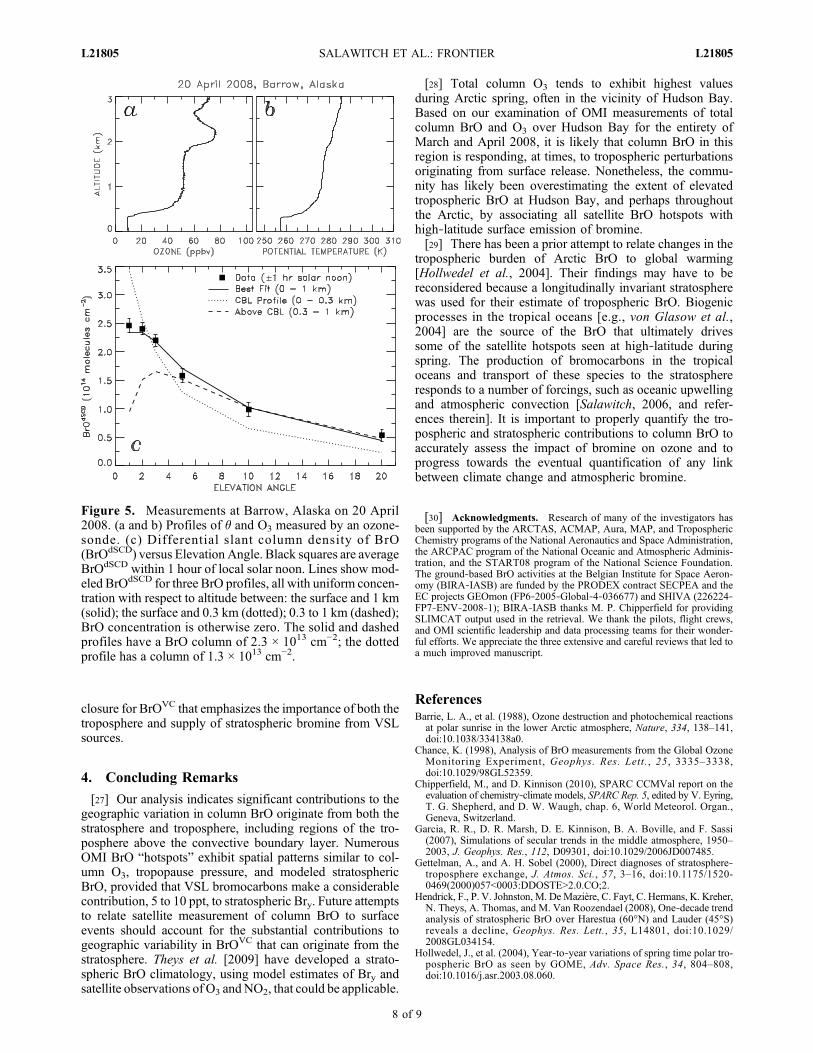
acquired on 20 April 2008. Best agreement between mod-
eled and measured BrOdSCD is found assuming a constantconcentration of BrO within the lowest 1 km of the atmo-sphere, with a column of 2.3 × 1013 cm−2. An ozonesondeshowed a classic CBL extending to 0.3 km, with O3 presentuniformly at ∼10 ppb. A model placing all of the BrO withinthe CBL overestimates observed BrOdSCD at low EA andunderestimates BrOdSCD at high EA, suggesting BrO hadvented above the CBL. A significant amount of BrO wasalso present within the CBL, as demonstrated by theinability to properly simulate BrOdSCD by placing all of theBrO between 0.3 and 1 km. Max‐DOAS observationsconducted during clear sky conditions throughout Marchand April 2008 (not shown) reveal contributions to BrOVC
of 0 to 3 × 1013 cm−2, with much of the day‐to‐day vari-ability related to the direction of prevailing surface winds(higher BrOVC observed when air parcels originate from thenearby sea) (D. Donohoue et al., manuscript in preparation,2010).[24] The Max‐DOAS and aircraft observations of tropo-
spheric BrO are consistent in that they both show significantcontributions to BrOTROP from above the CBL. The prev-alence of elevated BrO above the CBL may be due to vig-orous convection over ice leads driven by warm exposedwater, with BrO then dispersed horizontally by prevailingwinds. This would be consistent with distribution of BrOthroughout the polar boundary layer, a region in which sur-face emissions can be vertically mixed even if the atmosphereappears to be stable with respect to local convection [e.g.,Simpson et al., 2007]. Attempts to relate satellite BrOVC toODEs and surface BrO, common in the literature, are com-plicated by the finding that BrOTROP appears to be dominatedby contributions from above the CBL.
3.4. Budget of Column BrO
[25] We examine here the budget for BrOVC using thestratospheric model and tropospheric aircraft profiles.Figures 4f and 4g show regression plots of BrOMODEL
versus OMI BrOVC, where BrOMODEL is set equal toBrOSTRAT (Figure 4f) or BrOSTRAT+BrOTROP (Figure 4g).Data are shown for all locations where BrOTROP can beestimated from the two aircraft: 29 profiles encompassingobservations acquired on 8 days. Neglecting the tropo-spheric contribution to column BrO, all modeled BrO col-umns show slopes below the 1:1 line, although the modelfor the largest VSL bromine contribution lies close to theline (Figure 4f).[26] Accounting for contributions from both the tropo-
sphere and stratosphere, budget closure is achieved if BryVSL
lies between 5 and 10 ppt (Figure 4g). Closure of the budgetis supported quantitatively by the ratio BrOMODEL/OMIBrOVC encompassing unity, within the standard deviation ofthe mean, for these two simulations. Strictly speaking,budget closure is achieved for values of Bry
VSL ranging from∼1.4 to 13.2 ppt (auxiliary material). Considerable uncer-tainty exists, leading to a wide range of Bry
VSL that could beconsistent with OMI BrOVC, because BrOVC is uncertain at±22%, BrOSTRAT is uncertain at ±45% due to chemicalkinetics, and BrOTROP is uncertain at an amount approach-ing the measured abundance (all 1s). The effects of clouds,which potentially shield a portion of the tropospheric columnfrom view of OMI, have not been considered. Nonetheless,Figure 4g demonstrates a plausible means to achieve budget
Figure 3. BrOVC versus total column O3 from OMI fordata acquired for 5 to 9 April 2008, the five days shownin Figure 2 (black points). The error bars on BrOVC repre-sent the standard deviation, about the mean, of the values inthe respective total column O3 bins (250 to 525 DU, every25 DU). The green, blue, and red points represent calculatedBrOSTRAT for Bry
VSL of 0, 5, and 10 ppt, respectively. Thethick error bar for BrOSTRAT represents the uncertainty inmodel BrO due to errors in GEOS‐5 CFC‐12; the thin errorbar represents the total 1s uncertainty in BrOSTRAT, whichis dominated by the factor of 2 uncertainty in kBrO+NO2+M.The BrOSTRAT points have been displaced slightly, withrespect to the mean O3 of each bin, for clarity of error bars.
SALAWITCH ET AL.: FRONTIER L21805L21805
6 of 9
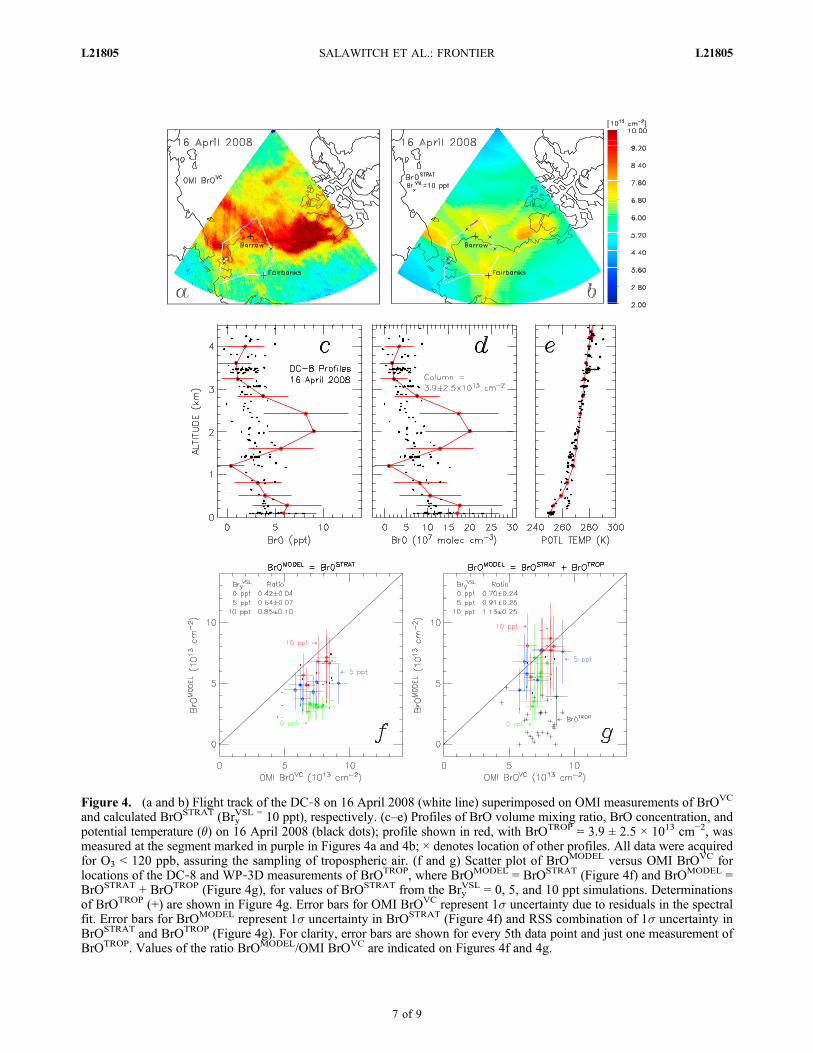
Figure 4. (a and b) Flight track of the DC‐8 on 16 April 2008 (white line) superimposed on OMI measurements of BrOVC
and calculated BrOSTRAT (BryVSL = 10 ppt), respectively. (c–e) Profiles of BrO volume mixing ratio, BrO concentration, and
potential temperature (�) on 16 April 2008 (black dots); profile shown in red, with BrOTROP = 3.9 ± 2.5 × 1013 cm−2, wasmeasured at the segment marked in purple in Figures 4a and 4b; × denotes location of other profiles. All data were acquiredfor O3 < 120 ppb, assuring the sampling of tropospheric air. (f and g) Scatter plot of BrOMODEL versus OMI BrOVC forlocations of the DC‐8 and WP‐3D measurements of BrOTROP, where BrOMODEL = BrOSTRAT (Figure 4f) and BrOMODEL =BrOSTRAT + BrOTROP (Figure 4g), for values of BrOSTRAT from the Bry
VSL = 0, 5, and 10 ppt simulations. Determinationsof BrOTROP (+) are shown in Figure 4g. Error bars for OMI BrOVC represent 1s uncertainty due to residuals in the spectralfit. Error bars for BrOMODEL represent 1s uncertainty in BrOSTRAT (Figure 4f) and RSS combination of 1s uncertainty inBrOSTRAT and BrOTROP (Figure 4g). For clarity, error bars are shown for every 5th data point and just one measurement ofBrOTROP. Values of the ratio BrOMODEL/OMI BrOVC are indicated on Figures 4f and 4g.
SALAWITCH ET AL.: FRONTIER L21805L21805
7 of 9
closure for BrOVC that emphasizes the importance of both thetroposphere and supply of stratospheric bromine from VSLsources.
4. Concluding Remarks
[27] Our analysis indicates significant contributions to thegeographic variation in column BrO originate from both thestratosphere and troposphere, including regions of the tro-posphere above the convective boundary layer. NumerousOMI BrO “hotspots” exhibit spatial patterns similar to col-umn O3, tropopause pressure, and modeled stratosphericBrO, provided that VSL bromocarbons make a considerablecontribution, 5 to 10 ppt, to stratospheric Bry. Future attemptsto relate satellite measurement of column BrO to surfaceevents should account for the substantial contributions togeographic variability in BrOVC that can originate from thestratosphere. Theys et al. [2009] have developed a strato-spheric BrO climatology, using model estimates of Bry andsatellite observations of O3 andNO2, that could be applicable.
[28] Total column O3 tends to exhibit highest valuesduring Arctic spring, often in the vicinity of Hudson Bay.Based on our examination of OMI measurements of totalcolumn BrO and O3 over Hudson Bay for the entirety ofMarch and April 2008, it is likely that column BrO in thisregion is responding, at times, to tropospheric perturbationsoriginating from surface release. Nonetheless, the commu-nity has likely been overestimating the extent of elevatedtropospheric BrO at Hudson Bay, and perhaps throughoutthe Arctic, by associating all satellite BrO hotspots withhigh‐latitude surface emission of bromine.[29] There has been a prior attempt to relate changes in the
tropospheric burden of Arctic BrO to global warming[Hollwedel et al., 2004]. Their findings may have to bereconsidered because a longitudinally invariant stratospherewas used for their estimate of tropospheric BrO. Biogenicprocesses in the tropical oceans [e.g., von Glasow et al.,2004] are the source of the BrO that ultimately drivessome of the satellite hotspots seen at high‐latitude duringspring. The production of bromocarbons in the tropicaloceans and transport of these species to the stratosphereresponds to a number of forcings, such as oceanic upwellingand atmospheric convection [Salawitch, 2006, and refer-ences therein]. It is important to properly quantify the tro-pospheric and stratospheric contributions to column BrO toaccurately assess the impact of bromine on ozone and toprogress towards the eventual quantification of any linkbetween climate change and atmospheric bromine.
[30] Acknowledgments. Research of many of the investigators hasbeen supported by the ARCTAS, ACMAP, Aura, MAP, and TroposphericChemistry programs of the National Aeronautics and Space Administration,the ARCPAC program of the National Oceanic and Atmospheric Adminis-tration, and the START08 program of the National Science Foundation.The ground‐based BrO activities at the Belgian Institute for Space Aeron-omy (BIRA‐IASB) are funded by the PRODEX contract SECPEA and theEC projects GEOmon (FP6‐2005‐Global‐4‐036677) and SHIVA (226224‐FP7‐ENV‐2008‐1); BIRA‐IASB thanks M. P. Chipperfield for providingSLIMCAT output used in the retrieval. We thank the pilots, flight crews,and OMI scientific leadership and data processing teams for their wonder-ful efforts. We appreciate the three extensive and careful reviews that led toa much improved manuscript.
ReferencesBarrie, L. A., et al. (1988), Ozone destruction and photochemical reactions
at polar sunrise in the lower Arctic atmosphere, Nature, 334, 138–141,doi:10.1038/334138a0.
Chance, K. (1998), Analysis of BrO measurements from the Global OzoneMonitoring Experiment, Geophys. Res. Lett. , 25, 3335–3338,doi:10.1029/98GL52359.
Chipperfield, M., and D. Kinnison (2010), SPARC CCMVal report on theevaluation of chemistry‐climate models, SPARC Rep. 5, edited by V. Eyring,T. G. Shepherd, and D. W. Waugh, chap. 6, World Meteorol. Organ.,Geneva, Switzerland.
Garcia, R. R., D. R. Marsh, D. E. Kinnison, B. A. Boville, and F. Sassi(2007), Simulations of secular trends in the middle atmosphere, 1950–2003, J. Geophys. Res., 112, D09301, doi:10.1029/2006JD007485.
Gettelman, A., and A. H. Sobel (2000), Direct diagnoses of stratosphere‐troposphere exchange, J. Atmos. Sci., 57, 3–16, doi:10.1175/1520-0469(2000)057<0003:DDOSTE>2.0.CO;2.
Hendrick, F., P. V. Johnston, M. De Mazière, C. Fayt, C. Hermans, K. Kreher,N. Theys, A. Thomas, and M. Van Roozendael (2008), One‐decade trendanalysis of stratospheric BrO over Harestua (60°N) and Lauder (45°S)reveals a decline, Geophys. Res. Lett., 35, L14801, doi:10.1029/2008GL034154.
Hollwedel, J., et al. (2004), Year‐to‐year variations of spring time polar tro-pospheric BrO as seen by GOME, Adv. Space Res., 34, 804–808,doi:10.1016/j.asr.2003.08.060.
Figure 5. Measurements at Barrow, Alaska on 20 April2008. (a and b) Profiles of � and O3 measured by an ozone-sonde. (c) Differential slant column density of BrO(BrOdSCD) versus Elevation Angle. Black squares are averageBrOdSCD within 1 hour of local solar noon. Lines show mod-eled BrOdSCD for three BrO profiles, all with uniform concen-tration with respect to altitude between: the surface and 1 km(solid); the surface and 0.3 km (dotted); 0.3 to 1 km (dashed);BrO concentration is otherwise zero. The solid and dashedprofiles have a BrO column of 2.3 × 1013 cm−2; the dottedprofile has a column of 1.3 × 1013 cm−2.
SALAWITCH ET AL.: FRONTIER L21805L21805
8 of 9

Holmes, C. D., D. J. Jacob, and X. Yang (2006), Global lifetime of elemen-tal mercury against oxidation by atomic bromine in the free troposphere,Geophys. Res. Lett., 33, L20808, doi:10.1029/2006GL027176.
Hönninger, G., et al. (2004), Ground‐based measurements of halogen oxi-des at Hudson Bay by active longpath DOAS and passive MAX‐DOAS,Geophys. Res. Lett., 31, L04111, doi:10.1029/2003GL018982.
Kleipool, Q. L., M. R. Dobber, J. F. de Haan, and P. F. Levelt (2008), Earthsurface reflectance climatology from 3 years of OMI data, J. Geophys.Res., 113, D18308, doi:10.1029/2008JD010290.
Liang, Q., R. S. Stolarski, A. R. Douglass, P. A. Newman, and J. E. Nielsen(2008), Evaluation of emissions and transport of CFCs using surface ob-servations and their seasonal cycles and the GEOS CCM simulation withemissions‐based forcing, J. Geophys. Res., 113, D14302, doi:10.1029/2007JD009617.
Liao, J., et al. (2010), A comparison of Arctic BrO measurements by chem-ical ionization mass spectrometery (CIMS) and long path‐differentialoptical absorption spectroscopy (LP‐DOAS), J. Geophys. Res.,doi:10.1029/2010JD014788, in press.
Liu, A. Q., and G. W. K. Moore (2004), Lake‐effect snowstorms oversouthern Ontario and their synoptic‐scale environment, Mon. WeatherRev., 132, 2595–2609, doi:10.1175/MWR2796.1.
McElroy, C. T., C. A. McLinden, and J. C. McConnell (1999), Evidencefor bromine monoxide in the free troposphere during the Arctic polarsunrise, Nature, 397, 338–341, doi:10.1038/16904.
Neuman, J. A., et al. (2010), Bromine measurements in O3 depleted air overthe Arctic Ocean, Atmos. Chem. Phys., 10, 6503–6514, doi:10.5194/acp-10-6503-2010.
Platt, U., and G. Hönninger (2003), The role of halogen species in the tro-posphere, Chemosphere, 52, 325–338, doi:10.1016/S0045-6535(03)00216-9.
Richter, A., F. Wittrock, M. Eisinger, and J. P. Burrows (1998), GOMEobservations of tropospheric BrO in Northern Hemispheric spring andsummer 1997, Geophys. Res. Lett., 25, 2683–2686, doi:10.1029/98GL52016.
Ridley, B. A., et al. (2007), An ozone depletion event in the sub‐Arctic sur-face layer over Hudson Bay, Canada, J. Atmos. Chem., 57, 255–280,doi:10.1007/s10874-007-9072-z.
Salawitch, R. J. (2006), Biogenic bromine, Nature, 439, 275–277,doi:10.1038/439275a.
Salawitch, R. J., D. K. Weisenstein, L. J. Kovalenko, C. E. Sioris, P. O.Wennberg, K. Chance, M. K. W. Ko, and C. A. McLinden (2005), Sen-sitivity of ozone to bromine in the lower stratosphere, Geophys. Res.Lett., 32, L05811, doi:10.1029/2004GL021504.
Sander, S. P., et al. (2006), Chemical kinetics and photochemical data foruse in atmospheric studies, JPL Publ., 06‐02.
Schauffler, S. M., E. L. Atlas, D. R. Blake, F. Flocke, R. A. Lueb, J. M.Lee‐Taylor, V. Stroud, and W. Travnicek (1999), Distributions of bro-minated organic compounds in the troposphere and lower stratosphere,J. Geophys. Res., 104, 21,513–21,535, doi:10.1029/1999JD900197.
Simpson, W. R., et al. (2007), Halogens and their role in polar boundary‐layer ozone depletion, Atmos. Chem. Phys., 7, 4375–4418, doi:10.5194/acp-7-4375-2007.
Theys, N., et al. (2009), A global stratospheric BrO climatology based on theBASCOE chemical transport model, Atmos. Chem. Phys., 9, 831–848,doi:10.5194/acp-9-831-2009.
von Glasow, R., et al. (2004), Impact of reactive bromine chemistry in thetroposphere, Atmos. Chem. Phys., 4, 2481–2497, doi:10.5194/acp-4-2481-2004.
Wagner, T., C. Leue, M. Wenig, K. Pfeilsticker, and U. Platt (2001), Spa-tial and temporal distribution of enhanced boundary layer BrO concen-trations measured by GOME aboard ERS‐2, J. Geophys. Res., 106,24,225–24,235, doi:10.1029/2000JD000201.
Wamsley, P. R., et al. (1998), Distribution of halon‐1211 in the upper tro-posphere and lower stratosphere and the 1994 bromine budget, J. Geo-phys. Res., 103, 1513–1526, doi:10.1029/97JD02466.
Wernli, H., and M. Sprenger (2007), Identification and ERA‐15 climatologyof potential vorticity streamers and cutoffs near the extratropical tropo-pause, J. Atmos. Sci., 64, 1569–1586, doi:10.1175/JAS3912.1.
World Meteorological Organization (WMO) (2007), Global ozone researchand monitoring project, in Scientific Assessment of Ozone Depletion:2006, Rep. 50, World Meteorol. Organ., Geneva, Switzerland.
E. L. Atlas, RSMAS, University of Miami, 4600 Rickenbacker Cwy.,Miami, FL 33149, USA.P. K. Bhartia, A. da Silva, S. Pawson, and J. M. Rodriguez, NASA
Goddard Space Flight Center, Code 916, Greenbelt, MD 20771, USA.D. R. Blake, Department of Chemistry, University of California, 570
Rowland Hall, Irvine, CA 92697, USA.T. P. Bui, NASA Ames Research Center, Mail Stop 245‐5, Moffett Field,
CA 94035, USA.T. Canty and R. J. Salawitch, Department of Atmospheric and Oceanic
Science, University of Maryland, 2403 Computer and Space SciencesBldg., College Park, MD 20742, USA. ([email protected])K. Chance, T. Kurosu, and X. Liu, Harvard‐Smithsonian Center for
Astrophysics, 60 Garden St., Cambridge, MA 02138, USA.G. Chen and J. H. Crawford, NASA Langley Research Center, Mail Stop
483, Hampton, VA 23681, USA.J. E. Dibb, Complex Systems Research Center, University of New
Hampshire, Morse Hall, 39 College Rd., Durham, NH 03824, USA.D. Donohoue and W. R. Simpson, Department of Chemistry and
Biochemistry, University of Alaska Fairbanks, Fairbanks, AK 99775, USA.R. S. Gao, B. Johnson, J. A. Neuman, J. B. Nowak, S. Oltmans, and T. B.
Ryerson, Earth System Research Laboratory, NOAA, 325 Broadway,Boulder, CO 80305, USA.F. Hendrick and M. Van Roozendael, Belgian Institute for Space
Aeronomy, Ringlaan‐3‐Avenue Circulaire, B‐1180 Brussels, Belgium.L. G. Huey, J. Liao, R. E. Stickel, and D. J. Tanner, School of Earth and
Atmospheric Science, Georgia Institute of Technology, 311 Ferst Dr.,Atlanta, GA 30332, USA.D. J. Jacob, School of Engineering and Applied Sciences, Harvard
University, Pierce Hall, 29 Oxford St., Cambridge, MA 02138, USA.P. V. Johnston and K. Kreher, NIWA, Lauder, Private Bag 50061,
Omakau, New Zealand.D. E. Kinnison, F. Flocke, D. Knapp, D. Montzka, L. L. Pan, S. Tilmes,
and A. Weinheimer, National Center for Atmospheric Research, PO Box3000, Boulder, CO 80305, USA.Q. Liang, GEST, University of Maryland Baltimore County, Code 613.3,
Greenbelt, MD 20771, USA.J. E. Nielsen, Science Systems and Applications, Inc., 10210 Greenbelt
Rd., Ste. 600, Lanham, MD 20706, USA.R. B. Pierce, NESDIS, NOAA, 1225 West Dayton St., Madison, WI
53706, USA.
SALAWITCH ET AL.: FRONTIER L21805L21805
9 of 9


















