
9780471729259.mc06a05s26
51
UNIT 6A.5 Detection, Isolation, and Identification of Vibrio cholerae from the Environment Anwar Huq, 1 Bradd J. Haley, 1 Elisa Taviani, 1 Arlene Chen, 1 Nur A. Hasan, 1 and Rita R. Colwell 1,2 1 Maryland Pathogen Research Institute, Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, College Park, Maryland 2 University of Maryland Institute of Advanced Computer Studies, University of Maryland, College Park, Maryland, and Bloomberg School of Public Health, Johns Hopkins University, Baltimore, Maryland ABSTRACT Recent molecular advances in microbiology have greatly improved the detection of bac- terial pathogens in the environment. These improvements and a downward trend in the cost of molecular detection methods have contributed to increased frequency of detection of pathogenic microorganisms where traditional culture-based detection methods have failed. Culture methods also have been greatly improved, and the confluence of the two suites of methods provides a powerful tool for detection, isolation, and characteriza- tion of pathogens. While molecular detection provides data on the presence and type of pathogens, culturing methods allow a researcher to preserve the organism of interest for “-omics” studies, such as genomic, metabolomic, secretomic, and transcriptomic analy- sis, which are rapidly becoming more affordable. This has yielded a clearer understanding of the ecology and epidemiology of microorganisms that cause disease. In this unit, we present commonly accepted methods for isolation, detection, and characterization of V. cholerae, providing more extensive knowledge of the ecology and epidemiology of this organism. This unit has been fully revised and updated from the earlier version with the latest knowledge and additional information not previously included. Curr. Protoc. Microbiol. 26:6A.5.1-6A.5.51. C 2012 by John Wiley & Sons, Inc. Keywords: Vibrio cholera isolation identification detection characterization INTRODUCTION It is well established that Vibrio cholerae, the causative agent of cholera, is autochthonous to the aquatic environment globally and is not confined to cholera endemic areas (Kenyon et al., 1984; Louis et al., 2003; Schuster et al., 2011). Further, V. cholerae cells encoding genes and mobile elements known to cause disease in humans and confer resistance to antibiotics are found in the absence of cholera. When appropriate methods are used, V. cholerae can be detected in these aquatic environments throughout the year. V. cholerae culture–negative water samples do not definitively indicate absence of this organism, as cells can enter into a viable but nonculturable (VBNC) state in which they may not form colonies on traditional bacteriological culture plates (Xu et al., 1982; Roszak and Colwell, 1987). In the VBNC state, cell densities may be extremely low; therefore, if a small amount of water sample is collected, it may not contain a sufficient number of cells for detection. Thus, it is important that appropriate volumes of water be examined, using appropriate methods to determine the presence of V. cholerae in a given sample. This unit describes several widely accepted and highly cited methods used to detect, cultivate, enumerate, and characterize V. cholerae cells in environmental samples, such as water, sediment, plankton, and seafood, e.g., oysters. In developing countries, expo- sure to V. cholerae typically occurs via consumption of natural surface water containing Current Protocols in Microbiology 6A.5.1-6A.5.51, August 2012 Published online August 2012 in Wiley Online Library (wileyonlinelibrary.com). DOI: 10.1002/9780471729259.mc06a05s26 Copyright C 2012 John Wiley & Sons, Inc. Nonenteric Gamma Proteobacteria 6A.5.1 Supplement 26
-
Upload
giuseppegnr -
Category
Documents
-
view
215 -
download
2
Transcript of 9780471729259.mc06a05s26

UNIT 6A.5Detection, Isolation, and Identification ofVibrio cholerae from the Environment
Anwar Huq,1 Bradd J. Haley,1 Elisa Taviani,1 Arlene Chen,1
Nur A. Hasan,1 and Rita R. Colwell1,2
1Maryland Pathogen Research Institute, Department of Cell Biology and MolecularGenetics, University of Maryland, College Park, College Park, Maryland2University of Maryland Institute of Advanced Computer Studies, University of Maryland,College Park, Maryland, and Bloomberg School of Public Health, Johns Hopkins University,Baltimore, Maryland
ABSTRACT
Recent molecular advances in microbiology have greatly improved the detection of bac-terial pathogens in the environment. These improvements and a downward trend in thecost of molecular detection methods have contributed to increased frequency of detectionof pathogenic microorganisms where traditional culture-based detection methods havefailed. Culture methods also have been greatly improved, and the confluence of the twosuites of methods provides a powerful tool for detection, isolation, and characteriza-tion of pathogens. While molecular detection provides data on the presence and type ofpathogens, culturing methods allow a researcher to preserve the organism of interest for“-omics” studies, such as genomic, metabolomic, secretomic, and transcriptomic analy-sis, which are rapidly becoming more affordable. This has yielded a clearer understandingof the ecology and epidemiology of microorganisms that cause disease. In this unit, wepresent commonly accepted methods for isolation, detection, and characterization ofV. cholerae, providing more extensive knowledge of the ecology and epidemiology ofthis organism. This unit has been fully revised and updated from the earlier version withthe latest knowledge and additional information not previously included. Curr. Protoc.Microbiol. 26:6A.5.1-6A.5.51. C© 2012 by John Wiley & Sons, Inc.
Keywords: Vibrio cholera � isolation � identification � detection � characterization
INTRODUCTION
It is well established that Vibrio cholerae, the causative agent of cholera, is autochthonousto the aquatic environment globally and is not confined to cholera endemic areas (Kenyonet al., 1984; Louis et al., 2003; Schuster et al., 2011). Further, V. cholerae cells encodinggenes and mobile elements known to cause disease in humans and confer resistanceto antibiotics are found in the absence of cholera. When appropriate methods are used,V. cholerae can be detected in these aquatic environments throughout the year. V. choleraeculture–negative water samples do not definitively indicate absence of this organism, ascells can enter into a viable but nonculturable (VBNC) state in which they may notform colonies on traditional bacteriological culture plates (Xu et al., 1982; Roszak andColwell, 1987). In the VBNC state, cell densities may be extremely low; therefore, if asmall amount of water sample is collected, it may not contain a sufficient number of cellsfor detection. Thus, it is important that appropriate volumes of water be examined, usingappropriate methods to determine the presence of V. cholerae in a given sample.
This unit describes several widely accepted and highly cited methods used to detect,cultivate, enumerate, and characterize V. cholerae cells in environmental samples, suchas water, sediment, plankton, and seafood, e.g., oysters. In developing countries, expo-sure to V. cholerae typically occurs via consumption of natural surface water containing
Current Protocols in Microbiology 6A.5.1-6A.5.51, August 2012Published online August 2012 in Wiley Online Library (wileyonlinelibrary.com).DOI: 10.1002/9780471729259.mc06a05s26Copyright C© 2012 John Wiley & Sons, Inc.
NonentericGammaProteobacteria
6A.5.1
Supplement 26

Detection of Vibriocholerae from the
Environment
6A.5.2
Supplement 26 Current Protocols in Microbiology
Table 6A.5.1 Overview of Methods Presented in this Unit for Isolation and Detection of V. cholerae
Protocol Included methodology Protocol ID
Isolation of presumptive V. cholerae andconfirmation, using traditional methods
Specimen collection and transportation. Basic Protocol 1
Conventional bacteriological culture method forV. cholerae
Basic Protocol 2
Alternate method for presumptive identification Alternate Protocol 1
Serogroup determination Basic Protocol 3
Molecular methods for detection ofV. cholerae Isolates
Preparation of DNA templates and confirmationof suspected or presumptive V. cholerae by PCR
Support Protocol 1 andBasic Protocol 4
Real-time PCR Basic Protocols 6 and 7
Colony blot hybridization with labeled DNA orRNA hybridization probes
Basic Protocol 9
Molecular methods for direct detection ofV. cholerae in environmental samples
Fluorescence in situ hybridization (FISH) Basic Protocol 10
Direct PCR Basic Protocol 8
Direct detection and/or enumeration ofV. cholerae using immunological methods
Direct fluorescent antibody—direct viable count(DFA-DVC)
Basic Protocol 11
Indirect fluorescent antibody (IFA) method Basic Protocol 12
V. cholerae in sufficient numbers to cause disease, whereas in developed countries, ex-posure to V. cholerae often occurs via consumption of raw or undercooked shellfish ortravel to regions where cholera outbreaks occur frequently. Recent outbreaks of shellfish-related vibrioses and cholera in developed countries demonstrate the need to define areliable method for detection of these organisms in any environmentally derived foodsource (Onifade et al., 2011). Both plankton and sediment play an important role inthe occurrence and survival of V. cholerae in water and oysters. Sediment and plank-ton both can occur suspended within the water column. Sediment is known to harborV. cholerae (due to its polar electrical charge). Plankton are known to be natural reser-voirs of V. cholerae, and zooplankton blooms facilitate increases in V. cholerae density.Furthermore, an infectious dose of V. cholerae (6 × 106 cells; Cash et al., 1974) can bepresent on a single plankton body (Huq et al., 1984; Rawlings et al., 2007). Presence ofV. cholerae in water, sediment, and plankton will also result in the presence of V. choleraein shellfish, as these organisms filter feed all three through their bodies and concentratehigh numbers of V. cholerae. It is, therefore, important to consider the type of sampleand desired outcome (detection, isolation, or enumeration) when choosing the appro-priate bacteriological/molecular methods discussed here. For the readers’ convenience,Table 6A.5.1 and Figure 6A.5.1 provide an overview of the methods provided in thisunit.
STRATEGIC PLANNING
As stated above, the type of sample and the desired outcome are integral in determiningwhich method(s) should be used to achieve satisfactory results. Furthermore, the skillset of the laboratory members and laboratory resources are all essential in determiningwhich tests can be conducted, as all of these facets can vary dramatically from laboratoryto laboratory as well as over time within one laboratory.
Traditional culturing methods, although yielding only a small subset of culturable bacteriain the original sample (<0.1% of total bacteria), do allow researchers to archive individualstrains and run a variety of informative bench-top studies at a later date. These include

NonentericGammaProteobacteria
6A.5.3
Current Protocols in Microbiology Supplement 26
collect samples(Basic Protocol 1)
conventionalbacteriological culturemethod for V. cholerae
(Basic Protocol 2)
serogroupdetermination
(Basic Protocol 3)
preparation of DNA templateand confirmation of suspected
or presumptive V. choleraeby PCR (Support Protocol1, Basic Protocol 4, and
Tables 6A.5.3 and 6A.5.4)
or
preparation of DNA templateand real-time PCR
(Support Protocol 1 and BasicProtocols 6 or 7)
alternative method forpresumptive identification
(Alternate Protocol 1)
colony blot hybridizationwith labeled DNA or RNA
hybridization probes(Basic Protocol 9)
direct PCR(Basic Protocol 8)
direct fluorescentantibody – direct
viable count(Basic Protocol 10)
indirect fluorescentantibody method
(Basic Protocol 12)
fluorescence in situhybridization – FISH(Basic Protocol 10)
Figure 6A.5.1 Flowchart of methods (protocols) used to detect and/or isolate and characterize V. cholerae from theenvironment.
genetic characterization by PCR, reconstruction of metabolic pathways by phenotypicanalyses, gene expression assays, and whole genome sequencing, among many others.However, as stated earlier, reliance on traditional culturing methods only focuses one’sscope on those bacteria that are culturable while inherently ignoring those bacterial cellsthat cannot grow on media but which do remain a significant public health threat, as theyare capable of causing disease (Colwell, 1991).
To evaluate the presence of these organisms regardless of their culturability, a directdetection method should be employed. Two microscopy methods are reliable, notablyfluorescent antibody coupled with direct viable count (FA-DVC), or fluorescence in situhybridization (FISH), a nucleic acid marker–based method of detection. FISH can beutilized to detect genomic DNA sequences specific to V. cholerae regardless of serogroup,while FA-DVC can be used to detect V. cholerae of serogroups O1 and O139, serogroupsassociated with pandemic cholera. The direct fluorescence antibody (DFA) detection ofserogroups O1 and O139 is more reliable than attempting to detect these strains viaculture methods, as targeting one serogroup in the environment is rather difficult todo, considering the vast serodiversity of V. cholerae in the environment. Both methodsallow the researcher to quantify the target if no pre-enrichment step is used. Subsequentbacterial isolation is not possible when using this method unless a parallel sample iscollected and processed using traditional culturing methods.
Direct molecular detection using the polymerase chain reaction (PCR) can also be em-ployed to evaluate the presence of these organisms in environmental samples. Thesemethods rely on detection of species-specific nucleotide sequences, followed by ampli-fication of the targeted nucleic acid and visualization on an agarose gel (conventionalPCR) or in an amplification plot using computer software (real-time PCR). Direct de-tection using these methods can be applied directly to environmental samples, as well asto samples following a pre-enrichment step in alkaline peptone water (APW; see recipein Reagents and Solutions) or estuarine peptone water (EPW; autoclaved APW with 1%
Detection of Vibriocholerae from the
Environment
6A.5.4
Supplement 26 Current Protocols in Microbiology
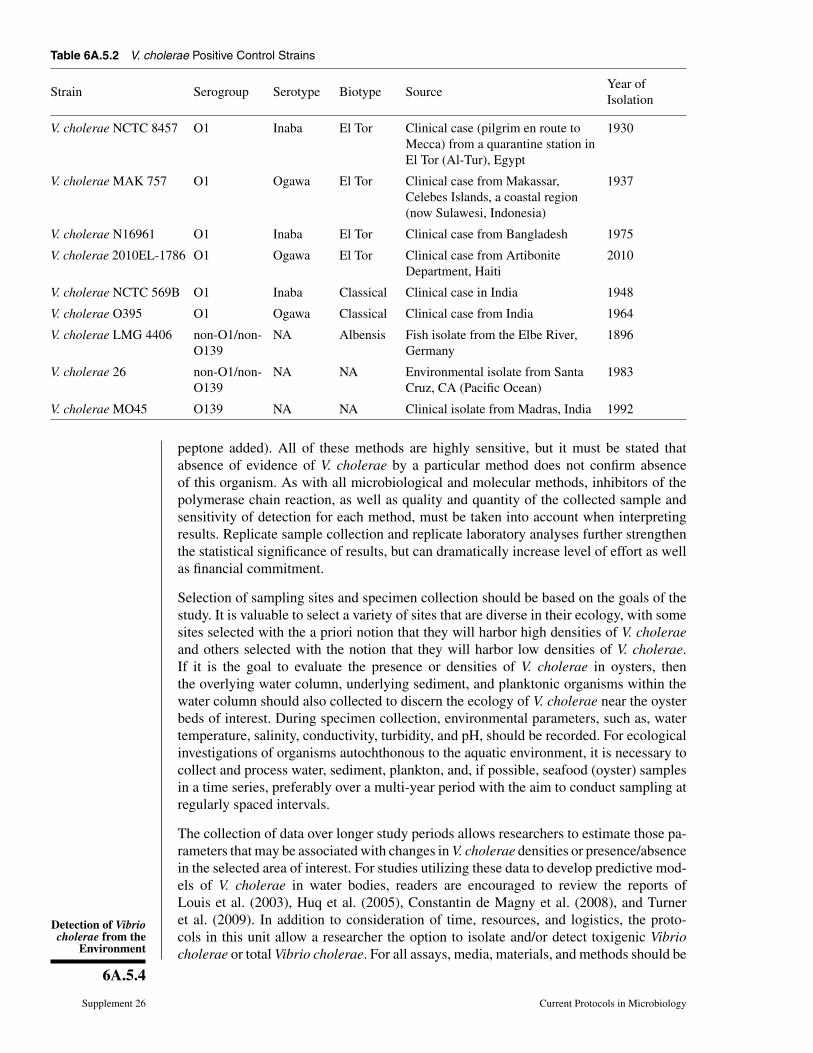
Table 6A.5.2 V. cholerae Positive Control Strains
Strain Serogroup Serotype Biotype SourceYear ofIsolation
V. cholerae NCTC 8457 O1 Inaba El Tor Clinical case (pilgrim en route toMecca) from a quarantine station inEl Tor (Al-Tur), Egypt
1930
V. cholerae MAK 757 O1 Ogawa El Tor Clinical case from Makassar,Celebes Islands, a coastal region(now Sulawesi, Indonesia)
1937
V. cholerae N16961 O1 Inaba El Tor Clinical case from Bangladesh 1975
V. cholerae 2010EL-1786 O1 Ogawa El Tor Clinical case from ArtiboniteDepartment, Haiti
2010
V. cholerae NCTC 569B O1 Inaba Classical Clinical case in India 1948
V. cholerae O395 O1 Ogawa Classical Clinical case from India 1964
V. cholerae LMG 4406 non-O1/non-O139
NA Albensis Fish isolate from the Elbe River,Germany
1896
V. cholerae 26 non-O1/non-O139
NA NA Environmental isolate from SantaCruz, CA (Pacific Ocean)
1983
V. cholerae MO45 O139 NA NA Clinical isolate from Madras, India 1992
peptone added). All of these methods are highly sensitive, but it must be stated thatabsence of evidence of V. cholerae by a particular method does not confirm absenceof this organism. As with all microbiological and molecular methods, inhibitors of thepolymerase chain reaction, as well as quality and quantity of the collected sample andsensitivity of detection for each method, must be taken into account when interpretingresults. Replicate sample collection and replicate laboratory analyses further strengthenthe statistical significance of results, but can dramatically increase level of effort as wellas financial commitment.
Selection of sampling sites and specimen collection should be based on the goals of thestudy. It is valuable to select a variety of sites that are diverse in their ecology, with somesites selected with the a priori notion that they will harbor high densities of V. choleraeand others selected with the notion that they will harbor low densities of V. cholerae.If it is the goal to evaluate the presence or densities of V. cholerae in oysters, thenthe overlying water column, underlying sediment, and planktonic organisms within thewater column should also collected to discern the ecology of V. cholerae near the oysterbeds of interest. During specimen collection, environmental parameters, such as, watertemperature, salinity, conductivity, turbidity, and pH, should be recorded. For ecologicalinvestigations of organisms autochthonous to the aquatic environment, it is necessary tocollect and process water, sediment, plankton, and, if possible, seafood (oyster) samplesin a time series, preferably over a multi-year period with the aim to conduct sampling atregularly spaced intervals.
The collection of data over longer study periods allows researchers to estimate those pa-rameters that may be associated with changes in V. cholerae densities or presence/absencein the selected area of interest. For studies utilizing these data to develop predictive mod-els of V. cholerae in water bodies, readers are encouraged to review the reports ofLouis et al. (2003), Huq et al. (2005), Constantin de Magny et al. (2008), and Turneret al. (2009). In addition to consideration of time, resources, and logistics, the proto-cols in this unit allow a researcher the option to isolate and/or detect toxigenic Vibriocholerae or total Vibrio cholerae. For all assays, media, materials, and methods should be

NonentericGammaProteobacteria
6A.5.5
Current Protocols in Microbiology Supplement 26
validated with a positive control and a negative control. Several commonly usedV. cholerae positive-control strains that are typically available from culture collections,such as the American Type Culture Collection (ATCC), are listed in Table 6A.5.2.
CAUTION: V. cholerae is a Biosafety Level 2 (BSL-2) pathogen. Follow all appropriateguidelines and regulations for the use and handling of pathogenic microorganisms. SeeUNIT 1A.1 and other pertinent resources (APPENDIX 1B) for more information.
ISOLATION AND IDENTIFICATION OF V. CHOLERAE USINGTRADITIONAL METHODS
Although developed several decades ago, traditional culturing methods continue to be im-proved, with the result of more reliable detection of V. cholerae from environmental sam-ples. The process involves either directly plating the study sample (water, plankton, sedi-ment, shellfish) onto TCBS, TTGA, or CHROMagar Vibrio (http://www.chromagar.com),or pre-enrichment of the sample in alkaline peptone water followed by streaking for iso-lation onto one or a combination of these three agars. For environmental samples, itis highly recommended to use a pre-enrichment step to improve detection or isolation.Following an incubation period, presumptive V. cholerae can be isolated from these me-dia and confirmed either by biochemical analyses (Huq et al., 2006) or immediately byPCR. Isolates can then be serogrouped as O1, O139, or non-O1/non-O139 by a slideagglutination assay using antisera for the O1 and O139 antigens or by PCR using primersdeveloped to target O1 and O139 coding regions of the genomic DNA. These V. choleraeisolates can then be archived with replicates in nutrient agar containing 0.5% NaCl over-laid with sterile mineral oil or as a glycerol stock at −70◦C. Stock cultures of V. choleraeshould be periodically re-streaked for isolation to ensure their viability and purity.
BASICPROTOCOL 1
Specimen Collection and Transportation
If the aim is to detect and isolate V. cholerae in water samples, then screening of concen-trated water samples and plankton samples and “plankton-free” water is recommended,since a combination of all types of samples provides a higher probability of V. choleraedetection as well as a better understanding of the role of plankton in V. cholerae dynamicsin the study region (Huq et al., 1990; Binsztein et al., 2004; Huq et al., 2005; Turneret al., 2009; Lizarraga-Partida et al., in preparation). Water collection bottles should becleaned with detergent; however, the detergent must not leave residue and should notbe antibacterial. The bottles should be pre-sterilized in an autoclave for 15 to 20 min at121◦C prior to use. Polypropylene bottles should be used for water samples and glassbottles should be used for plankton samples. A sufficient volume of water and plank-ton should be collected to ensure that appropriate analyses can be performed. Planktonsamples should be collected by passing water through a 200, 64, and/or 20-μm pore sizeplankton net by towing the net manually or behind a boat. Alternatively, a known volumeof water can be manually passed through the net by bucket pouring or pumping, whichcan be useful for quantitative analysis (Huq et al., 2005).
Processing of samples should begin soon after collection (typically within 24 hr of col-lection). If enumeration of V. cholerae is desired, then the sample should be stored in acool box at a temperature of 10◦ to 15◦C until processing begins (not to exceed 8 hr;Clesceri et al., 1998). However, a recent study has demonstrated that transporting samplesat ambient air temperature prior to processing can enhance the culturability of V. choleraein the sample (Alam et al., 2006a,b). Based on type of examination, samples may requiretreatments, such as addition of direct viable count (DVC) reagents, before proceedingwith further examination and testing. It is recommended that basic physiochemical pa-rameters, e.g., temperature, salinity, pH, dissolved oxygen, and conductivity of the waterbe measured on site at the time of collection, as it is known that V. cholerae densitiescan be influenced by such parameters. If the study site is influenced by tides, coasts, and

Detection of Vibriocholerae from the
Environment
6A.5.6
Supplement 26 Current Protocols in Microbiology
estuaries, for example, the prevailing tidal level at the time of sampling should berecorded. Furthermore, in addition to water temperature, significant correlation of waterdepth, rainfall, conductivity, and copepod density with cholera outbreaks has been re-ported, with lag periods from 0 to 8 weeks from optimum environmental conditions tocholera outbreaks (Huq et al., 2005). These parameters can be measured on site usingportable meters (such as HACH Model CO150 conductivity meter, HACH ChemicalCompany, or dissolved oxygen and pH meter Model 210A, Orion Laboratories). It isimportant to note that in all studies of the ecology of microorganisms in the environment,several rounds of “practice” sampling and processing should be considered to optimizethe volumes of water, plankton, or other biotic and abiotic media to be collected inorder to detect or recover the specific microorganism of interest, as well as to identifymissing equipment and reagents and evaluate the ability to comprehend and adhere tothe standard methods described herein. It is hard to keep the sample collection volumesconstant, as the amounts (volumes) may need to be adjusted depending on the quality ofwater due to presence of seasonal plankton. The authors of this unit strongly encourageinvestigators to use several methods and several types of media throughout the durationof their studies. All methods have some error or detection limits associated with themand, therefore, use of multiple approaches will allow investigators to converge on themost accurate answer. Water, sediment, and oysters isolated from different environments,both on a global and local scale, may have different chemistry associated with them, and,therefore, the different media requirements discussed in this unit may perform more orless effectively over time and space. Diversifying one’s approach to gathering data on theecology of V. cholerae will help overcome these challenges. The authors also encouragethe investigators to use a range of incubation temperatures for APW enrichment andgrowth on selective agar plates. It is well known that V. cholerae can grow within awide range of temperatures, and that this group of organisms is phenotypically heteroge-neous. Therefore, utilizing two incubation temperatures may increase the probability thatV. cholerae is detected and/or isolated. It should be known that this may effectively doublethe amount of work downstream, but the investigators should also be aware that severalmethods of isolation and detection will allow convergence on an accurate analysis.
Materials
Phosphate-buffered saline (PBS; APPENDIX 2A), sterilePre-sterilized 500-ml glass (Qorpak) and 1000-ml polypropylene containers
(Nalgene)Simple plankton nets, 200-μm, 64-μm, and 20- μm, or nets of different mesh sizes
(for size filtration of plankton); Aquatic Research Instruments(http://www.aquaticresearch.com/) or SEA-GEAR (http://www.sea-gear.net/);see Fig. 6A.5.2)
Portable meter(s) that measure temperature, dissolved oxygen, pH, turbidity, andsalinity (HACH Company; Geo Scientific; YSI/Xylem); turbidity and salinitycan be measured ex situ, i.e., in the laboratory, preferably immediately aftersample collection.
Bucket of known volume: .g., 4 or 5 liters (optionally used for pouring waterthrough plankton nets)
Piston corer (Aquatic Research Instruments; http://www.aquaticresearch.com/) or aPeterson grab (Wildlife Supply Company; http://www.wildco.com/)
Wide-bore serological pipetsOyster tongs or dredge≥92-oz. Whirl-Pak Bags (eNasco; http://www.enasco.com/)Autoclaved and pre-weighed 100-ml polypropylene container (optionally used
when collecting sediment from an oyster bed)Scooper/spoon that has been wrapped in aluminum foil and autoclaved (optionally
used when collecting sediment from an oyster bed)

NonentericGammaProteobacteria
6A.5.7
Current Protocols in Microbiology Supplement 26
To collect water sample
1a. Uncap pre-sterilized plastic bottle, submerge bottle to fill. Fill to half of volume ofbottle, re-cap, shake to rinse, and discard. Repeat 3 to 5 times. Rinse downstream ofwater sample collection site if possible.
2a. Remove the sample bottle from the water. Cap, leaving enough air in the bottle foragitation and mixing.
To collect plankton1b. Rinse plankton net and cod-end of the net (see Fig. 6A.5.2) in the body of water to
be sampled (downstream of sample collection site if possible).
2b. Filter 10 to 100 liters water through the plankton net by towing, using a calibrated,net-mounted flow meter, or by pouring known volumes through the net with a smallerbucket (the bucket should be clean, but without detergent, and rinsed several timeswith sample water, downstream of the sample collection site, immediately prior tosample collection).
3b. For each plankton fraction of interest, rinse inside the net of the plankton net,followed by the cod-end, with sterile PBS using a spray or squirt bottle, or withexcess plankton-free water collected after pouring sample water through the net.
This allows any plankton adhering to the net to be flushed down to the cod-end forcollection.
4b. Remove cod-end from the plankton net and decrease volume to 100 ml by continuingto filter.
This can be done by gently swirling the cod end so that water passes through the meshsiding of that cod end.
5b. Measure (with a sterile graduated cylinder) and decant plankton fraction into a sterileglass bottle, making sure to label that bottle with the collected plankton fraction size.
6b. Repeat this process for all plankton fractions of interest.
7b. Finally, collect 500 ml to 1000 ml water that passes through the 20-μm plankton net.
This is “plankton-free” water.
To collect sediment sample1c. Collect sediment with a piston corer or a Peterson grab.
2c. Sample a sediment-water interface with a wide-bore serological pipet and put ina sterile bottle (this sediment-water interface sample can be treated as a separate
cod-end metal ringthree-point
bridle
Figure 6A.5.2 Simple plankton net (Aquatic Research Instruments). The net is composed ofa metal ring and bridle, a heavy-duty nylon net of variable mesh size, and a PVC cod-end orcollecting bucket which can be removed for easy sample collection. A flow meter may be mountedin the mouth of the net to the metal ring to measure volume when the net is towed.

Detection of Vibriocholerae from the
Environment
6A.5.8
Supplement 26 Current Protocols in Microbiology
sediment sample and processed alongside the other media collected, following thesame protocols).
3c. Remove the remaining sediment from the sampling apparatus aseptically and putinto a sterile bottle.
To collect shellfishShellfish collection in many areas is regulated by local or state governments. It is imper-ative to check these regulations with your local Fish and Wildlife department or otherrelevant government bodies prior to sample collection. Collection of shellfish may re-quire purchase of a permit and, in some locations, may be done only on certain days. Theoyster processing protocols described here can be used to evaluate store-bought oysters.However, these oysters may have been depurated and stored for some time, and resultsfrom these analyses should be described with respect to post-harvesting conditions, notthe environment from which these oysters were harvested. Additionally, since oystershells may be sharp, it is advised to wear gloves whenever handling oysters in order toavoid injury.
1d. Collect oysters from boats by using oyster tongs or a dredge or hand collect at lowtide when the oyster beds are easily accessible.
2d. Examine oysters for viability in the field, keeping only those oysters that are tightlyclosed. Rinse sediment off of the oyster shells with surface water, but do this onlyafter water and plankton samples have been collected.
Tapping the mid-shell point of an oyster on a hard surface will allow determination ofwhether oyster meat is present in the oyster shell, because a hollow sound will be heardwhen an empty oyster is tapped. Rinse sediment off of the oyster shells with surface water,but do this only after water and plankton samples have been collected.
3d. Place viable oysters in ventilated ≥92-oz. Whirl-Pak Bags and transport in a coolcontainer, but not immediately next to ice or cool packs. Do not re-immerse oystersin water after collection.
To collect sediment from an oyster bedIf oysters are already being collected during sample collection, it is possible to collectsediment samples at the same time in order to avoid using a piston corer, as directsediment sample collection from a hard oyster bed (also known as an oyster reef) maybe difficult. When determining whether or not oysters are present in the shell by tapping,hollow oyster shells may contain sediment inside; therefore it is imperative to open theoyster shells to check for sediment. If sediment is found, use a scooper that has beenpreviously wrapped in aluminum foil and autoclaved to scrape the sediment off the shell.Place sediment in a sterile, pre-weighed 100-ml polypropylene container.
Transportation and/or processingTransport samples to the laboratory for processing or, preferably, begin processing onsite within 1 hr of collection. For samples that require transport to the laboratory to beprocessed after collection, store and/or transport the samples in a cold box at 10◦ to15◦C. Alternatively, samples can be kept in the dark at ambient air temperature (22◦ to25◦C) after collection for up to 24 hr, as this may enhance recovery of Vibrio species asshown in a recent study (Alam et al., 2006a,b), and can be a useful approach for detectingvibrios in those environments where the organism is predominantly in the viable butnon-culturable (VBNC) state (Alam et al., 2006a,b). This aspect of the protocol mayneed to be optimized for the water source and environmental conditions.

NonentericGammaProteobacteria
6A.5.9
Current Protocols in Microbiology Supplement 26
BASICPROTOCOL 2
Conventional Bacteriological Culture Method
Conventional culture methods for isolating V. cholerae from environmental water samplesrely on an enrichment step(s) in broth and plating on selective media, followed by confir-mation using a series of biochemical tests or PCR and serological tests to determine strainserotype. Alternatively, biochemical tests can be bypassed, and confirmation can be doneusing PCR if adequate supplies of PCR reagents are available. Most water and planktonsamples require some amount of concentration. Running several practice rounds at thedesired study site will help estimate appropriate volumes of sample that should processedin order to achieve desired goals. However, it should be pointed out that environmentalV. cholerae densities are not static, so volumes deemed to be appropriate at one samplingpoint may not yield the same results at another time. We address this inconsistency byurging the investigator to use multiple methods (if resources are available) to isolate,detect, and characterize environmental V. cholerae during the course of the study. Watersamples, both whole water and plankton-free water, should be concentrated by filtrationusing 0.2-μm polycarbonate membrane filters. A good starting volume is 500 ml; 1000ml can be used if water passes easily through the polycarbonate membrane. If the 0.2-μmpolycarbonate membrane filter clogs quickly, multiple filters can be used per sample. Itis preferable to use 0.2-μm pore size filters as opposed to 0.45-μm pore-size filters, asVBNC bacteria are often <0.45-μm in size. Polycarbonate membranes are preferable tonitrocellulose membranes, as the cells sit on top of the polycarbonate filters and are easilyremoved by vortexing, while cells get caught in the fibers of nitrocellulose membranes.It is imperative that the cells be physically agitated off of the membrane so that they maymigrate to the microaerophilic layer of the enrichment broth discussed below.
Overnight enrichment is performed using alkaline peptone water (APW), pH 8.6, andsome investigators recommend two successive enrichments. Surface aliquots from themicroaerophilic pellicle layer are streaked for isolation onto selective bacteriological me-dia. Three commonly used selective media for V. cholerae isolation are thiosulfate citratebile-salts sucrose (TCBS) agar, tellurite taurocholate gelatin agar (TTGA), also known asMonsur medium (Monsur, 1961), and CHROMagar Vibrio (http://www.chromagar.com/).V. cholerae appears as translucent, flat, yellow colonies with elevated centers on TCBS; ascolorless colonies on TTGA, often with a characteristic dark center after 2 days growth,surrounded by a halo, which appears due to the hydrolysis of gelatin; and as turquoisecolonies on CHROMagar Vibrio (Fig. 6A.5.3). If possible, use more than one medium,as that may allow convergence on the best results. APW enriches for many bacteria otherthan V. cholerae; therefore, direct plating of samples onto TCBS, TTGA, and/or CHRO-Magar Vibrio can be done in parallel with APW enrichment, and may yield V. choleraecolonies that can be used for further analyses. This direct plating method is beneficial ininstances when overgrowth of non-target bacteria occurs on solid media after overgrowthin APW.
TCBS is a highly selective medium that may, in rare instances, inhibit growth ofV. cholerae and at the same time allow growth of non-Vibrio organisms, such asAeromonas sp., Staphylococcus sp., and Shewanella sp. TCBS is commercially avail-able and produced by several companies that have global distribution. TTGA mediumis considered to be less inhibitory to V. cholerae cells; however longer incubation timesare needed (48 hr) to observe the dark center that is characteristic of V. cholerae onthis medium. It may be difficult to distinguish V. cholerae from V. parahaemolyticus onthis medium; therefore, modified TTGA medium can be used in which V. choleraecolonies appear as brilliant blue with fluorescence in 24 hr or less, by adding β-galactosidase (4-methylumbelliferyl-β-�-galactosidase) (O’Brien and Colwell, 1985).CHROMagar Vibrio, primarily used for the detection of V. cholerae, V. parahaemolyticus,V. vulnificus, and V. alginolyticus, distinguishes these organisms from each other basedon a proprietary chromogenic reaction that precludes making it in the laboratory from

Detection of Vibriocholerae from the
Environment
6A.5.10
Supplement 26 Current Protocols in Microbiology
A
B
C
Figure 6A.5.3 Growth of V. cholerae O1 on (A) TCBS, (B) TTGA, (C) CHROMagar Vibrio,courtesy of Dr. Munir Alam, International Center for diarrheal Disease Research, Bangladesh.
scratch. TCBS and CHROMagarVibrio need not be autoclaved, and incubation times forthese media are shorter (24 hr) than for TTGA. TTGA medium must be autoclaved priorto use. Once presumptive strains are purified on a nonselective medium, such as gelatinagar, modified nutrient agar, or Luria Bertani (LB) agar with 1% NaCl, they are con-firmed and serogrouped by PCR or by simple slide agglutination, employing polyclonalV. cholerae O1, O139 and monoclonal Inaba and Ogawa antiserum.
Materials
Water, plankton, oyster, or sediment samples (Basic Protocol 1)10× and 1× alkaline peptone water (APW), pH 8.6 (see recipe)Thiosulfate citrate bile-salts sucrose (TCBS) agar plates (see recipe)

NonentericGammaProteobacteria
6A.5.11
Current Protocols in Microbiology Supplement 26
Tellurite taurocholate gelatin agar (TTGA) plates (see recipe)CHROMagar Vibrio (CA; http://www.chromagar.com/)Phosphate-buffered saline (PBS), pH 7.4 (APPENDIX 2A), sterile95% alcohol for flame sterilization of forceps, loops, needle, bacterial cell spreaderGelatin agar (GA; see recipe), modified nutrient agar (see recipe), or Luria-Bertani
agar (LB) plates (see APPENDIX 2C)Oxidase reagent: 1% (w/v) N,N,N′N′-tetramethyl-p-phenylenediamine
dihydrochloride (e.g., Sigma)
Filter membranes, 47-mm diameter, 0.22-μm pore size (Millipore)Filter apparatus with vacuum sourceForceps50-ml centrifuge tubes (e.g., BD Falcon)Inoculating loops, needles, and cell spreaders (see UNIT 4A.1)Tissue homogenizer (hand-held) to homogenize plankton (Kimble Chase Life
Science and Research Products)Enrichment flask (sterile 150-ml Erlenmeyer flasks)Cut-resistant oyster shucking glove (Chef Revival, http://www.chefrevival.com/)Oyster knife (Dexter-Russell; http://www.dexter1818.com/)Autoclavable kitchen blender (Waring, 700G)Filter paper
Sample processingIf not specified, assume APW is 1× APW.
1a. For plankton samples: Filter concentrated plankton sample through a 47-mm, 20-μmpore size polycarbonate filter (this step may take a while depending on the abundanceof plankton in the sampled water). Add the filter(s) with attached bacteria to a 50-mlcentrifuge tube with 25 ml of sterile PBS.
i. Vortex the centrifuge tube in order to remove cells from the filter.ii. Add 1-ml whole plankton to 25 ml of 1× APW.
iii. Add 100 to 1000 μl of whole plankton directly onto TCBS and/or CA and TTGAplates and spread using a bacterial cell spreader.
iv. Homogenize 10-ml of concentrated plankton samples using a glass tissue grinderby moving the pestle up and down in the tube, while rotating, 10 to 20 times. Add1 ml homogenized plankton to an enrichment flask containing 25 ml APW.
v. Add 100 to 1000 μl of homogenized plankton directly onto TCBS and/or CA andTTGA plates and spread using a bacterial cell spreader.
1b. For water samples: Filter 100 to 1000 ml water (depending on bacterial density, seeBasic Protocol 1) through a 47-mm, 0.22-μm pore size polycarbonate filter. Add thefilter(s) with attached bacteria to a 50-ml centrifuge tube with 12 ml of sterile PBS.Vortex vigorously for ∼5 min to detach the bacteria from the filter.
i. Add 100 to 1000 μl of the PBS containing detached bacterial cells directly ontoTCBS and/or CA and TTGA plates and spread until plates appear dry, using abacterial cell spreader.
ii. Add 1 ml PBS containing detached bacterial cells to an enrichment flask containing25 ml APW.
This step may be done in replicate to increase the probability of isolating V. cholerae.However, it should be known that replication at this step dramatically increases themagnitude of analyses downstream.
Large volumes or highly turbid sample water may require more than one polycarbonatefilter, as they will clog.

Detection of Vibriocholerae from the
Environment
6A.5.12
Supplement 26 Current Protocols in Microbiology
The remaining PBS containing detached bacterial cells can be stored at −20◦C for otherdirect molecular detection methods, including metagenomic analysis.
1c. For oyster samples: Rinse and scrub exterior of oysters in sterile deionized water,making sure to remove sediment from the oyster hinge. With extreme caution,wearing a cut-resistant oyster-shucking glove, shuck oysters with a sterile oysterknife.
It may help to also hold the oysters level on a table with a towel between your glove andthe oyster. This may add protection and stability.
i. When shucking, with the curved-side of the knife facing downwards, trace theknife along the upper shell, making sure to sever the adductor muscle which holdsthe oyster shells shut. Shuck enough oysters to produce 250 g of oyster meat aswell as liquid surrounding the oyster meat and add to an autoclavable kitchenblender.
ii. Add an equal volume of sterile PBS and homogenize for 90 sec.iii. Add 20 ml of homogenized oyster sample to an enrichment flask containing 80 ml
of 10× APW.iv. Add 100 to 1000 μl of oyster homogenate to TCBS and/or CA and TTGA plates
and spread using a bacterial cell spreader.
This step may be further adjusted to include both direct plating of 100 to 1000 μl ontoagar as well as plating serial dilutions of homogenized oyster meat in sterile PBS.
If an autoclavable blender is not available, the interior of the blender pitcher can be ster-ilized with 10% bleach or 70% alcohol followed by multiple rinses with sterile deionizedwater or PBS. It is imperative that all bleach or alcohol be removed prior to adding theoyster meat. Alternatively, a Stomacher (http://www.seward.co.uk/) could be used.
To sterilize the oyster knife, dip blade into 95% ethanol and then pass through a flame.If the oyster knife is autoclavable, it can be wrapped in aluminum foil and sterilized at121◦C for 15 min. Oysters can be shucked when the blade is cool.
1d. For sediment samples: Weigh sediment samples and add an equal volume of sterilePBS based on the weight of the sediment and an assumed density of 1.0, and vortexor shake well.
i. Add 100-ml sediment/PBS slurry to an enrichment flask containing 11 ml 10×APW.
ii. Additionally, add 100 to 1000-μl sediment/PBS slurry directly onto TCBS and/orCA and TTGA plates, and spread using a bacterial cell spreader.
This step may be further adjusted to include both direct plating of 100 to 1000 μl ontoagar as well as plating serial dilutions of sediment in sterile PBS.
2. Incubate the APW enrichment flasks statically for 16 hr at 30 to 35◦C (overnight).
In all cases, APW enrichment flasks should not be disturbed or agitated during or afterincubation, as Vibrio species tend to migrate to the liquid-air interface.
A range of incubation temperatures can be used, as V. cholerae is known to grow between12◦ and 42◦C. Lower incubation temperatures for longer periods of time may allow formore V. cholerae cells to grow while also allowing for other competitive bacteria to growas well. Higher incubation temperatures may inhibit growth of some environmentallystressed V. cholerae cells but will also inhibit growth of competitive bacteria species. Itmay be beneficial to consider testing two incubation temperatures and choosing whichyields better results for your sample area of interest. Alternatively, replicate flasks canbe incubated at each temperature to increase the probability of isolating V. cholerae (seeitalicized note immediately after step 1b, above).

NonentericGammaProteobacteria
6A.5.13
Current Protocols in Microbiology Supplement 26
3. Incubate TCBS and CA plates at 30 to 35◦C for 24 ± 2 hr and TTGA plates at 30◦to 35◦C for 48 hr.
The same approach as stated under step 2 for incubation temperatures of APW inoculatescan be applied, and should be considered for incubation of agar plates.
4. Subculture smooth, flat, sucrose-fermenting, yellow colonies from TCBS and/orturquoise blue colonies from CA, and translucent, dark-centered colonies with halozones from TTGA (see Fig. 6A.5.3) onto LB, GA, and/or modified nutrient agarplates.
Selective plating post-APW enrichment5. After enrichment in APW, collect surface growth (which may be present as a whitish
film) from the enrichment flask with an inoculating loop and streak onto TCBSand/or CA and TTGA plates.
6. Incubate the plates 16 to 24 hr at 30◦ to 35◦C for TCBS and CA or 48 hr at 30◦ to35◦C for TTGA.
7. Subculture smooth, flat, sucrose-fermenting, yellow colonies from TCBS and/orturquoise blue colonies from CA, and translucent, dark-centered colonies with halozones from TTGA (see Fig. 6A.5.3) onto LB, GA, and/or modified nutrient agarplates, respectively.
To subculture presumptive V. cholerae colonies from these selective media, touch thetop-center of the colony using a sterile inoculating loop. Take care not to touch thesurface of the agar or any other surrounding bacteria (even if they too are presumptiveV. cholerae). If plates are heavily overgrown with bacteria, try to touch the top-center ofa presumptive V. cholerae colony and re-streak onto selective media to isolate a singlecolony.
If TCBS is used, morphology should be registered only on recent and well isolated colonies.
Perform oxidase test1% (w/v) tetramethyl-p-phenylenediamine dihydrochloride solution (oxidase reagent)should be prepared the same day the test is being performed.
8. Moisten filter paper with 1% tetramethyl-p-phenylenediamine solution. Using aplatinum loop, pick colonies and spread them onto the moistened filter paper. Lookfor a purple or violet color appearing in 10 sec, which indicates a positive read.
ALTERNATEPROTOCOL 1
Bacteriological Culture Method for Situations of Limited Resources
This alternative method can be used to presumptively estimate the presence of culturableV. cholerae in surface waters in regions where even general microbiology resourcesare limited. Bacteriological methods are based on the work of Choopun et al. (2002).Yellow colonies isolated from TCBS that give negative reactions (no color change)in esculin hydrolysis and arginine dihydrolase assays can be presumptively identifiedas V. cholerae.
Sterilization procedures
Glassware can be used for assays and can be sterilized by wrapping in aluminum foilor newspaper and then heated in an oven at 180◦C for 2 hr. Sterilization can also beaccomplished by microwaving materials for 5 min. Investigators must make sure thatno non-microwavable materials are used (e.g., aluminum foil). Mineral oil should besterilized by heating in an oven at 170◦C for 1 to 2 hr. If it is not possible to procureautoclavable sample-collection bottles, then store-bought drinking water bottles may beused. It is important to rinse these bottles with surface water from the collection site threeto five times prior to collecting the sample for analysis.

Detection of Vibriocholerae from the
Environment
6A.5.14
Supplement 26 Current Protocols in Microbiology
Materials
Water sample (Basic Protocol 1)10× and 1× alkaline peptone water (APW), pH 8.6 (see recipe)Thiosulfate citrate bile-salts sucrose (TCBS) agar plates (see recipe)Tellurite taurocholate gelatin agar (TTGA) plates (see recipe)CHROMagar Vibrio (CA; http://www.chromagar.com/)Brain-heart infusion agar stabs (see recipe) containing 0.1% (w/v) esculin and
0.05% (w/v) ferric chlorideLuria-Bertani broth containing 1% (w/v) L-arginine (pH 6.8) and phenol red
powder added as an indicatorSterile mineral oilInoculating loops and needles, sterile (see UNIT 4A.1)
1. Add 100-ml of water to be tested to a sterile bottle containing 900-ml 10× APW andshake or swirl well.
2. Incubate at 30◦ to 35◦C overnight.
3. With a sterile loop or toothpick, dab the upper pellicle layer of the incubated sampleand streak for isolation onto a selective medium for V. cholerae isolation (TTGA,TCBS, or CA). Incubate plates at 30◦ to 35◦C overnight (or 48 hr if TTGA agar isused).
If incubators are not available, then the 10× APW inoculation can be left to sit at roomtemperature for 24 hr and agar plates can be inverted (agar-side up) and left to sit in thedark until growth occurs (up to 2 days).
4. For esculin hydrolysis assay, gently touch the top and center of a yellow non-swarming colony with a sterile loop or toothpick and inoculate into brain-heartinfusion agar stabs containing 0.1% esculin and 0.05% ferric chloride.
A non-swarming colony is one that is symmetrical and <3 mm in diameter.
5. For arginine dihydrolase assay, simultaneously inoculate Luria-Bertani broth con-taining 1% (w/v) L-arginine (pH 6.8) and phenol red powder (Difco) added as anindicator. After inoculation, cover with ∼0.5 cm sterile mineral oil.
6. Incubate esculin hydrolysis assays at 30◦ to 35◦C for 72 hr, or at room temperaturefor approximately 96 hr, and arginine dihydrolase assays for 24 hr at 35◦C or 48 to72 hr at room temperature.
Blackening of the esculin hydrolysis medium after incubation indicates that that therewas a positive reaction, while appearance of a red color after incubation of the argininedihydrolase assay is considered a positive reaction. For both assays, a negative control(uninoculated assays) should be incubated at the same temperature for the same time,as a comparator. Used materials, including inoculated media, can be sterilized by mi-crowaving for 5 min. Used materials can also be sterilized in a dry oven at 180◦C for2 to 3 hr.
BASICPROTOCOL 3
Serogroup Determination
More than 200 serogroups of V. cholerae have been described to date based on anti-genic properties of cell surface polysaccharides, of which serogroups O1 and O139 havebeen implicated in epidemics of cholera while non-O1/non-O139 serogroups are knownto cause sporadic outbreaks. However, serogroup O37 was responsible for a localizedoutbreak of cholera in Czechoslovakia and Sudan. The remaining serogroups, collec-tively and commonly termed as non-O1/non-O139, predominate among the strains ofV. cholerae isolated from the aquatic environment (Sack et al., 2003). Although reported

NonentericGammaProteobacteria
6A.5.15
Current Protocols in Microbiology Supplement 26
mostly in O1 and O139 serogroup clinical isolates, the cholera toxin gene can be found innon-O1/non-O139 strains from the aquatic environment globally, as well as other Vibriospecies (Chakraborty et al., 2000; Malayil et al., 2010). However, because of the epidemicpotential, the method to determine O1 and O139 serogroup is mentioned below. Strainsother than O1 or O139 serogroup need not be serogrouped unless there is a special need.In such cases, those strains should be sent to a Reference Center for serotyping, sinceantisera for serogroups other than O1 and O139 are not commercially available.
Materials
Bacterial growth (6- to 16-hr subculture on nonselective medium; see BasicProtocol 2)
Phosphate-buffered saline (PBS; APPENDIX 2A)Antiserum for serogroup O1 and O139 V. cholerae
Glass slidesWax pencils
1. Draw two squares approximately 6 cm × 6 cm on a microscope slide with a waxpencil. Add one drop of PBS in each square .
2. Add a loopful of fresh growth (6- to 16-hr subculture on non-selective media, e.g.,LB, GA, or modified nutrient agar) into each drop and resuspend.
3. Add an equal-sized drop of group O1 polyvalent antiserum (undiluted) to one of thedrops.
4. Mix the antiserum–culture suspension by tilting the slide back and forth. Determineif the reaction clumps (i.e., agglutinates) within 0.5 to 1 min, indicating a positiveresult.
Agglutination can be better visualized by looking through microscope slide with a brightlamp in the near background.
Autoagglutination, clumping in the saline solution without antiserum, is indicative of a“rough” morphotype and cannot be typed by antisera.
5. Test non-O1 serogroup colonies with O139 antisera, following steps 1 to 4, above,replacing the O1 antiserum with O139 antiserum.
MOLECULAR METHODS FOR DETECTION AND IDENTIFICATION OFV. CHOLERAE ISOLATES
Conventional PCR and real-time PCR have recently been used to characterize V. choleraestrains, along with confirming a strain as V. cholerae or confirming the presence ofV. cholerae in environmental or clinical samples. Further, the increase in genomesequences now available has allowed development of PCR-based typing schemesfor analysis of variants of strains, genes, and mobile genetic elements, includ-ing pathogenicity islands. The body of knowledge derived from the V. choleraepangenome continues to provide researchers with targets useful in strain identification andcharacterization.
Identification and characterization of suspected or presumptive V. cholerae isolatesby PCRThe polymerase chain reaction (Kramer and Coen, 2001) is a useful alternative to labor-intensive biochemical tests, which are occasionally difficult to interpret, often requiringreplication as well as several days of incubation and media preparation. Ideally, at leastan oxidase test (Basic Protocol 2) should be done on presumptive colonies to reduce

Detection of Vibriocholerae from the
Environment
6A.5.16
Supplement 26 Current Protocols in Microbiology
Table 6A.5.3 PCR Targets and Primers for V. cholerae
Target Primer Sequence (5′-3′) Amplicon Reference
ITS pVC-F2 TTAAGCSTTTTCRCTGAGAATG 295-310 Chun et al. (1999)
PVCM-R1 AGTCACTTAACCATACAACCCG
ctxA PCTA-94F CGGGCAGATTCTAGACCTCCTG 563 Fields et al. (1992)
PCTA-614R CGATGATCTTGGAGCATTCCCAC
toxR pToxR-101F CCTTCGATCCCCTAAGCAATAC 778 Rivera et al. (2001)
pToxR-837R AGGGTTAGCAACCGATGCGTAAG
tcpA pTcpA-72F CACGATAAGAAAACCGGTCAAGAG 452 Keasler and Hall (1993)
pTcpAET-477R CGAAAGCACCTTCTTTCACGTTG 621
pTcpACL-647R TTACCAAATGCAACGCCGAATG
zot PZot-225F TCGCTTAACGATGGCGCGTTTT 946 Rivera et al. (2001)
PZot-1129R AACCCCGTTTCACTTCTACCCA
ompU pOmpU-80F ACGCTGACGGAATCAACCAAAG 868 Rivera et al. (2001)
pOmpU-906R GCGGAAGTTTGGCTTGAAGTAG
ctxA VCT1 ACAGAGTGAGTACTTTGACC 308 Hoshino et al. (1998)
VCT2 ATACCATCCATATATTTGGGAG
O1-rfb O1F2-1 GTTTCACTGAACAGATGGG 192 Hoshino et al. (1998)
O1R2-2 GGTCATCTGTAAGTACAAC
O139-rfb O139F2 AGCCTCTTTATTACGGGTGG 449 Hoshino et al. (1998)
O139R2 GTCAAACCCGATCGTAAAGG
ompW ompW-F CACCAAGAAGGTGACTTTATTGTG 588 Nandi et al. (2000)
ompW-R GAACTTATAACCACCCGCG
ctxA CtxA-F CTCAGACGGGATTTGTTAGGCACG 302 Shirai et al. (1991);Nandi et al. (2000)
CtxA-R TCTATCTCTGTAGCCCCTATTACG
16S rDNAa 16S-F CAGCMGCCGCGGTAATWC 888 Amann et al. (1995)
16S-R ACGGGCGGTGTGTRCaCurrently, there are no published 23S rDNA PCR primers specific for V. cholerae.
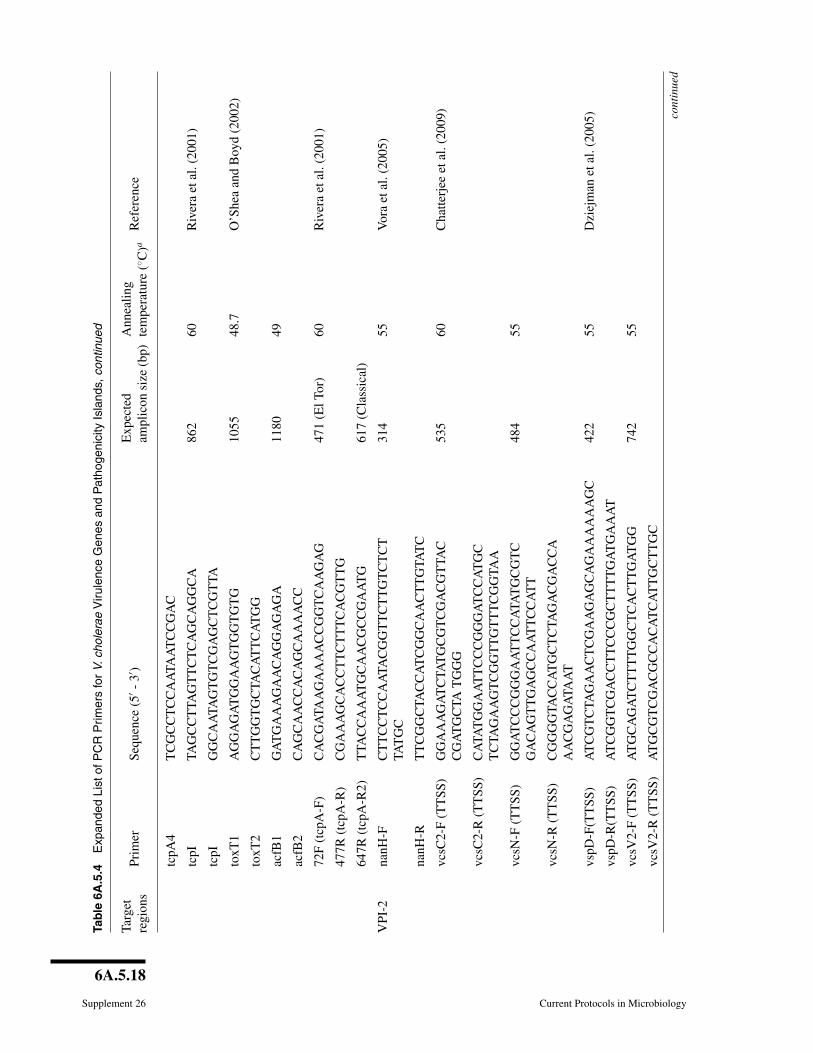
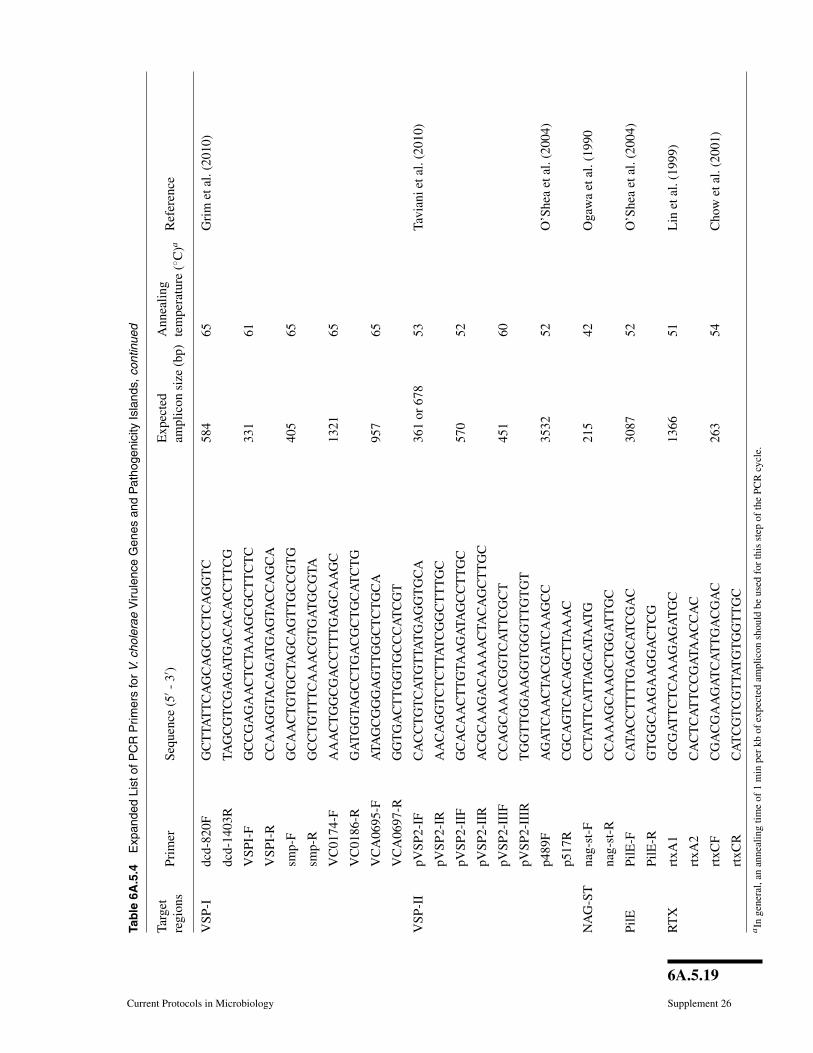
the number of presumptive V. cholerae isolates that are not V. cholerae. In this method,crude template is prepared by boiling to lyse the cells. Genomic regions within thistemplate are amplified using PCR primers specific to V. cholerae and targeting theinternal transcribed spacer (ITS) region between 16S and 23S rDNA or the outer mem-brane protein subunit W (ompW). Confirmed V. cholerae strains can be screened for thegenes associated with virulence and their variants (Table 6A.5.3). A more comprehen-sive list of relevant targets, with PCR conditions and expected amplicons, is provided inTable 6A.5.4 (http://www.currentprotocols.com/protocol/mc06A05). PCR products areanalyzed by gel electrophoresis and visualized under UV light with ethidium bromide.Positive and negative controls should be run in parallel and should include eubacterial16S rDNA PCR reaction for each sample to test template quality (see Table 6A.5.3for PCR primers, expected amplicon size and reference). Further, phenotypic and ge-netic screens on V. cholerae isolates can be done following the methods described inUNIT 6A.3.

6A.5.17
Current Protocols in Microbiology Supplement 26
Tab
le6A
.5.4
Exp
ande
dLi
stof
PC
RP
rimer
sfo
rV.
chol
erae
Viru
lenc
eG
enes
and
Pat
hoge
nici
tyIs
land
s
Targ
etre
gion
sPr
imer
Sequ
ence
(5′ -
3′ )E
xpec
ted
ampl
icon
size
(bp)
Ann
ealin
gte
mpe
ratu
re(◦ C
)aR
efer
ence
CT
Xrs
tA1
AC
TC
GA
TAC
AA
AC
GC
TT
CT
C10
0953
.7W
aldo
ret
al.(
1997
)
rstA
2A
GA
AT
CT
GG
AA
GG
TT
GA
GT
G
orfU
CG
TC
AC
AC
CA
GT
TAC
TT
TT
CG
1072
54.5
Tru
cksi
set
al.(
1993
)
orfU
AG
AA
TG
TAC
GC
CA
TC
GC
zot1
GG
CT
TAA
AC
CT
TG
AA
CG
C10
3654
.7Fa
sano
etal
.(19
91)
zot2
AA
CC
CC
GT
TT
CA
CT
TC
TAC
ctxA
1A
GT
CA
GG
TG
GT
CT
TAT
GC
C10
3751
.2O
’She
aet
al.(
2004
)
ctxB
2T
TG
CC
ATA
CTA
AT
TG
CG
G
tlc3
GG
GA
AT
GT
TG
AG
TT
CT
CA
GT
G15
4855
.5R
ubin
etal
.(19
98)
tlc4
GT
TG
CG
AA
GT
GG
AT
TT
TG
TG
intl4
:3C
CT
TC
AT
TG
GA
TC
AC
TC
G59
751
.9O
’She
aet
al.(
2004
)
intl4
:4G
AC
GG
AA
AA
AG
ATA
GT
GC
C
rstR
rstR
-Cl
CT
TC
TC
AT
CA
GC
AA
AG
CC
TC
CA
TC
400
50Fa
ruqu
eet
al.(
2003
);B
hatta
char
yaet
al.(
2006
);N
usri
net
al.(
2004
)
rstR
2-E
lG
CA
CC
AT
GA
TT
TAA
GA
TG
CT
C47
4
rstR
3-C
alC
TG
TAA
AT
CT
CT
TC
AA
TC
CTA
GG
501
rstR
4-E
nvG
TTA
AC
GC
TT
CA
AG
CC
TG
313
rstA
3-R
TC
GA
GT
TG
TAA
TT
CA
TC
AA
GA
GT
G
RS1
rstC
1A
AC
AG
CTA
CG
GG
CT
TAT
TC
238
52W
aldo
ret
al.(
1997
)
rstC
2T
GA
GT
TG
CG
GA
TT
TAG
GC
Hly
AH
ly(E
lTor
)-F
GG
CA
AA
CA
GC
GA
AA
CA
AA
TAC
C48
160
Riv
era
etal
.(20
01)
Hly
(Cla
)-F
GA
GC
CG
GC
AT
TC
AT
CT
GA
AT
738/
727
Hly
(El-
Cla
)-R
CT
CA
GC
GG
GC
TAA
TAC
GG
TT
TA
VPI
-1tc
pH1
AG
CC
GC
CTA
GA
TAG
TC
TG
TG
2176
51.7
Boy
dan
dW
aldo
r(2
002)
cont
inue
d
6A.5.18
Supplement 26 Current Protocols in Microbiology
Tab
le6A
.5.4
Exp
ande
dLi
stof
PC
RP
rimer
sfo
rV.
chol
erae
Viru
lenc
eG
enes
and
Pat
hoge
nici
tyIs
land
s,co
ntin
ued
Targ
etre
gion
sPr
imer
Sequ
ence
(5′ -
3′ )E
xpec
ted
ampl
icon
size
(bp)
Ann
ealin
gte
mpe
ratu
re(◦ C
)aR
efer
ence
tcpA
4T
CG
CC
TC
CA
ATA
AT
CC
GA
C
tcpI
TAG
CC
TTA
GT
TC
TC
AG
CA
GG
CA
862
60R
iver
aet
al.(
2001
)
tcpI
GG
CA
ATA
GT
GT
CG
AG
CT
CG
TTA
toxT
1A
GG
AG
AT
GG
AA
GT
GG
TG
TG
1055
48.7
O’S
hea
and
Boy
d(2
002)
toxT
2C
TT
GG
TG
CTA
CA
TT
CA
TG
G
acfB
1G
AT
GA
AA
GA
AC
AG
GA
GA
GA
1180
49
acfB
2C
AG
CA
AC
CA
CA
GC
AA
AA
CC
72F
(tcp
A-F
)C
AC
GA
TAA
GA
AA
AC
CG
GT
CA
AG
AG
471
(ElT
or)
60R
iver
aet
al.(
2001
)
477R
(tcp
A-R
)C
GA
AA
GC
AC
CT
TC
TT
TC
AC
GT
TG
647R
(tcp
A-R
2)T
TAC
CA
AA
TG
CA
AC
GC
CG
AA
TG
617
(Cla
ssic
al)
VPI
-2na
nH-F
CT
TC
CT
CC
AA
TAC
GG
TT
CT
TG
TC
TC
TTA
TG
C31
455
Vor
aet
al.(
2005
)
nanH
-RT
TC
GG
CTA
CC
AT
CG
GC
AA
CT
TG
TAT
C
vcsC
2-F
(TT
SS)
GG
AA
AG
AT
CTA
TG
CG
TC
GA
CG
TTA
CC
GA
TG
CTA
TG
GG
535
60C
hatte
rjee
etal
.(20
09)
vcsC
2-R
(TT
SS)
CA
TAT
GG
AA
TT
CC
CG
GG
AT
CC
AT
GC
TC
TAG
AA
GT
CG
GT
TG
TT
TC
GG
TAA
vcsN
-F(T
TSS
)G
GA
TC
CC
GG
GA
AT
TC
CA
TAT
GC
GT
CG
AC
AG
TT
GA
GC
CA
AT
TC
CA
TT
484
55
vcsN
-R(T
TSS
)C
GG
GG
TAC
CA
TG
CT
CTA
GA
CG
AC
CA
AA
CG
AG
ATA
AT
vspD
-F(T
TSS
)A
TC
GT
CTA
GA
AC
TC
GA
AG
AG
CA
GA
AA
AA
AG
C42
255
Dzi
ejm
anet
al.(
2005
)
vspD
-R(T
TSS
)A
TC
GG
TC
GA
CC
TT
CC
CG
CT
TT
TG
AT
GA
AA
T
vcsV
2-F
(TT
SS)
AT
GC
AG
AT
CT
TT
TG
GC
TC
AC
TT
GA
TG
G74
255
vcsV
2-R
(TT
SS)
AT
GC
GT
CG
AC
GC
CA
CA
TC
AT
TG
CT
TG
C
cont
inue
d
6A.5.19
Current Protocols in Microbiology Supplement 26
Tab
le6A
.5.4
Exp
ande
dLi
stof
PC
RP
rimer
sfo
rV.
chol
erae
Viru
lenc
eG
enes
and
Pat
hoge
nici
tyIs
land
s,co
ntin
ued
Targ
etre
gion
sPr
imer
Sequ
ence
(5′ -
3′ )E
xpec
ted
ampl
icon
size
(bp)
Ann
ealin
gte
mpe
ratu
re(◦ C
)aR
efer
ence
VSP
-Idc
d-82
0FG
CT
TAT
TC
AG
CA
GC
CC
TC
AG
GT
C58
465
Gri
met
al.(
2010
)
dcd-
1403
RTA
GC
GT
CG
AG
AT
GA
CA
CA
CC
TT
CG
VSP
I-F
GC
CG
AG
AA
CT
CTA
AA
GC
GC
TT
CT
C33
161
VSP
I-R
CC
AA
GG
TAC
AG
AT
GA
GTA
CC
AG
CA
smp-
FG
CA
AC
TG
TG
CTA
GC
AG
TT
GC
CG
TG
405
65
smp-
RG
CC
TG
TT
TC
AA
AC
GT
GA
TG
CG
TA
VC
0174
-FA
AA
CT
GG
CG
AC
CT
TT
GA
GC
AA
GC
1321
65
VC
0186
-RG
AT
GG
TAG
CC
TG
AC
GC
TG
CA
TC
TG
VC
A06
95-F
ATA
GC
GG
GA
GT
TG
GC
TC
TG
CA
957
65
VC
A06
97-R
GG
TG
AC
TT
GG
TG
CC
CA
TC
GT
VSP
-II
pVSP
2-IF
CA
CC
TG
TC
AT
GT
TAT
GA
GG
TG
CA
361
or67
853
Tavi
anie
tal.
(201
0)
pVSP
2-IR
AA
CA
GG
TC
TC
TTA
TC
GG
CT
TT
GC
pVSP
2-II
FG
CA
CA
AC
TT
GTA
AG
ATA
GC
CT
TG
C57
052
pVSP
2-II
RA
CG
CA
AG
AC
AA
AA
CTA
CA
GC
TT
GC
pVSP
2-II
IFC
CA
GC
AA
AC
GG
TC
AT
TC
GC
T45
160
pVSP
2-II
IRT
GG
TT
GG
AA
GG
TG
GG
TT
GT
GT
p489
FA
GA
TC
AA
CTA
CG
AT
CA
AG
CC
3532
52O
’She
aet
al.(
2004
)
p517
RC
GC
AG
TC
AC
AG
CT
TAA
AC
NA
G-S
Tna
g-st
-FC
CTA
TT
CA
TTA
GC
ATA
AT
G21
542
Oga
wa
etal
.(19
90
nag-
st-R
CC
AA
AG
CA
AG
CT
GG
AT
TG
C
PilE
PilE
-FC
ATA
CC
TT
TT
GA
GC
AT
CG
AC
3087
52O
’She
aet
al.(
2004
)
PilE
-RG
TG
GC
AA
GA
AG
GA
CT
CG
RT
Xrt
xA1
GC
GA
TT
CT
CA
AA
GA
GA
TG
C13
6651
Lin
etal
.(19
99)
rtxA
2C
AC
TC
AT
TC
CG
ATA
AC
CA
C
rtxC
FC
GA
CG
AA
GA
TC
AT
TG
AC
GA
C26
354
Cho
wet
al.(
2001
)
rtxC
RC
AT
CG
TC
GT
TAT
GT
GG
TT
GC
aIn
gene
ral,
anan
neal
ing
time
of1
min
per
kbof
expe
cted
ampl
icon
shou
ldbe
used
for
this
step
ofth
ePC
Rcy
cle.

Detection of Vibriocholerae from the
Environment
6A.5.20
Supplement 26 Current Protocols in Microbiology
SUPPORTPROTOCOL 1
Preparation of Crude DNA Template by Boiling
A crude DNA template, suitable for analysis in Basic Protocol 4, Alternate Protocol 2,and Basic Protocol 5, can be prepared by boiling a small overnight culture or a loopfulon culture from a fresh agar plate.
Materials
1-ml overnight culture or loopful of pure culture from agar plate (Basic Protocol 2)2-ml microcentrifuge tubes, sterileBoiling water bath
1a. From broth: Centrifuge a 1-ml culture (overnight growth at 35◦C) and resuspend in1-ml sterile water.
1b. From agar plates: Resuspend a loopful of pure culture (50 to 100 colonies) ofsuspected or presumptive V. cholerae into 300-μl of sterile water by vigorouslyvortexing.
Plates should be a fresh overnight subculturing demonstrating only one morphology.
2. In a sterile 2-ml microcentrifuge tube, dilute the suspension 1:1000 in sterile water.
Alternatively, a single isolated colony can be resuspended into 20 μl of sterile water.
3. Place the microcentrifuge tube containing the resuspended culture into a boilingwater bath for 10 min.
4. Cool tube to room temperature by allowing tube to sit on bench (approximately30 min).
Treatment of crude DNA template with 10 mg/ml bovine serum albumin (BSA) (4 μlper 100 μl supernatant) may help to limit PCR inhibition and yield more accurateresults. It is also beneficial to make a 1:10 dilution and a 1:1000 dilution (super-natant:sterile deionized water) and use this for PCR; dilution may also help to remove PCRinhibitors.
BASICPROTOCOL 4
Vibrio cholerae–Specific PCR ITS
The following PCR methods (Chun et al., 1999) can be used to confirm isolatesas V. cholerae by targeting the internal transcribed spacer (ITS) region between16S and 23S rDNA or the outer membrane protein subunit W (ompW) specific toV. cholerae.
Materials
Crude DNA template (Support Protocol 1) or extracted genomic DNA (BasicProtocol 8)
10× PCR amplification buffer (APPENDIX 2A)25 mM dNTPs (APPENDIX 2A)20 μM PCR primers (Table 6A.5.3)Taq DNA polymeraseMolecular weight ladder (e.g., Hyperladder IV, Bioline)TAE buffer (APPENDIX 2A)1 μg/ml ethidium bromide staining solution (APPENDIX 2A)
Thermal cycler (BioRad)Microcentrifuge tubesBoiling water bathPCR tubes

NonentericGammaProteobacteria
6A.5.21
Current Protocols in Microbiology Supplement 26
Horizontal agarose gel apparatus, gel tray and comb (see Voytas, 2000)Power supply for gel apparatusUV transilluminator (UVP)
Additional reagents and equipment for agarose gel electrophoresis (Voytas, 2000)
1. Set up V. cholerae-specific ITS (Internal Transcribed Spacer region) PCR reactionmixture in a total reaction volume of 25 μl, containing the following.
5 μl DNA template1× PCR amplification buffer200 μM dNTPs800 nM primers (pVC-F2, pVCM-R1; Table 6A.5.3)0.625 U Taq DNA polymerase.
2. Amplify the V. cholerae-specific ITS target with the following cycling conditions:
Initial denaturation: 1 min 94◦C (initial denaturation)30 cycles: 1 min 94◦C (denaturation)
1 min 60◦C (annealing)1 min 72◦C (extension)
1 cycle: 10 min 72◦C (final extension).
“All-in-one” PCR master mix can be purchased as an alternative to buying the neces-sary reagents separately. This can facilitate screening large numbers of strains. PCRprimers and nuclease-free water will need to be purchased separately. Several prod-ucts include, but are not limited to, GoTaq Green Master Mix (Promega) and Mul-tiplex PCR 5× Master Mix (New England BioLabs) for detecting multiple targetsby PCR.
Using GoTaq Green Master Mix (Promega), prepare the PCR reaction as follows for atotal reaction volume of 25 μl; 5 μl template DNA (step 4, 12.5 μl 2× GoTaq MasterMix (containing GoTaq DNA Polymerase, 400 μM dATP, 400 μM dGTP, 400 μM dCTP,400 μM dTTP, 2 mM MgCl2), 1μl of each primer (20 μM pVC-F2 and pVCM-R1), and5.5 μl nuclease free water.
Perform agarose gel electrophoresis and visualize results3. Run PCR product out on a 1.5% agarose gel in 1× TAE for 1 to 2 hr at 5 V/cm
(Voytas, 2000), along with a molecular weight ladder.
4. Stain the gel in 1 μg/ml ethidium bromide staining solution for 15 min.
Alternative DNA-staining dyes that may be less mutagenic have recently been developedand can be explored as an alternative to ethidium bromide. Dyes include GelRed and Gel-Green (Biotium) and SYBR Safe DNA Gel Stain (Invitrogen). Gels stained with GelGreencan be visualized with a laser-based gel scanner or a Dark Reader that uses visible bluelight for excitation. Gels stained with SYBR Safe DNA Gel Stain can be visualized underblue light as well.
5. Destain the gel in distilled water for 15 min.
6. Visualize the products by viewing the gel using a handheld UV lamp, transillumina-tor, or gel documentation system.
The V. cholerae 16S-23S rDNA intergenic spacer region amplicon is 300-bp in size(Fig. 6A.5.4, lane 2).
7. Screen ITS-PCR confirmed V. cholerae isolates for the virulence-associated factorslisted in Table 6A.5.3 and/or Table 6A.5.4. See Figure 6A.5.4 for image of typicalgel.

Detection of Vibriocholerae from the
Environment
6A.5.22
Supplement 26 Current Protocols in Microbiology
11 2 3 4 5 6 7 8 9 10101 2 3 4 5 6 7 8 9 10
Figure 6A.5.4 Results of PCR assays used to detect and characterize V. cholerae. Lane 1,Hyperladder IV (Bioline); lane 2, V. cholerae-specific ITS; lane 3, ctxA (pCTA); lane 4, tcpA ofV. cholerae O1 Classical, lane 5, tcpA of V. cholerae O1 El Tor; lane 6, tcpA of V. cholerae O139;lane 7, toxR; lane 8, zot; lane 9, ompU; lane 10, O1-O139/ctxA multiplex of V. cholerae O1 andO139.
ALTERNATEPROTOCOL 2
Multiplex PCR Assay for Detection of ompW (V. cholerae-Specific) and ctxA(Toxigenicity)
This protocol (Nandi et al., 2000) can be used in place of Basic Protocol 4, since it detectsthe V. cholerae-specific ompW sequence. It also allows for the detection of the ctxA geneof the cholera toxin.
Materials
Crude DNA template (Support Protocol 1) or extracted genomic DNA (BasicProtocol 8)
10× PCR amplification buffer (APPENDIX 2A)25 mM dNTPs (APPENDIX 2A)OmpW-F and R primersctxA F and R primersTaq DNA polymerase1.5% agarose gel (Voytas, 2000)Molecular weight ladder (e.g., Hyperladder IV, Bioline)TAE buffer (APPENDIX 2A)1 μg/ml ethidium bromide staining solution
Thermal cyclerPCR tubesUV transilluminator
Additional reagents and equipment for agarose gel electrophoresis (Voytas, 2000)
1. Set up ompW-ctxA multiplex PCR in a total reaction volume of 25 μl, containing thefollowing:
10 to 20 ng of crude DNA template or extracted genomic DNA1× PCR amplification buffer

NonentericGammaProteobacteria
6A.5.23
Current Protocols in Microbiology Supplement 26
250-μM dNTPs1.2-pmol/μl of ompW (ompW-F, R) primers0.25-pmol/μl ctxA (ctxA-F, -R) primers0.625 U Taq DNA polymerase.
2. Amplify the targets with the following cycling conditions:
1 cycle: 5 min 94◦C (initial denaturation)30 cycles: 30 sec 94◦C (denaturation)
30 sec 64◦C (annealing)30 sec 72◦C (extension)
1 cycle: 7 min 72◦C (final extension).
3. Run PCR product out on a 1.5% agarose gel in 1× TAE for 1 to 2 hr at 5-V/cm(Voytas, 2000) ), along with a molecular weight ladder.
4. Stain the gel 15 min in 1 μg/ml ethidium bromide staining solution.
5. Destain the gel 15 min in distilled water.
6. Visualize the products by viewing the gel under UV light.
The ompW and ctxA amplicons are 588 and 302-bp in length, respectively.
Screen samples giving a positive result for isolation of V. cholerae using the traditionalculture method as described in Basic Protocol 2, if desired.
BASICPROTOCOL 5
Multiplex PCR Assay for Detection of O1 and O139 Serogroup V. choleraeand ctxA
The multiplex PCR assay (Hoshino et al., 1998) is performed to confirm O1 and O139somatic antigens and for the simultaneous detection of the α-subunit of the cholera toxingene sequence, ctxA in confirmed V. cholerae isolates.
Materials
Crude DNA template (Support Protocol 1) or extracted genomic DNA (BasicProtocol 8)
10× PCR amplification buffer (APPENDIX 2A)20 μM PCR primers (Table 6A.5.3)25 mM dNTPs (APPENDIX 2A)Taq DNA polymeraseMolecular weight ladder (e.g., Hyperladder IV, Bioline)1 μg/ml ethidium bromide solution
Thermal cycler (BioRad)Microcentrifuge tubesBoiling water bathPCR tubesHorizontal gel apparatus, gel tray and comb (see Voytas, 2000)Power supply for gel apparatusUV transilluminator (UVP)
Additional reagents and equipment for agarose gel electrophoresis (Voytas, 2000)

Detection of Vibriocholerae from the
Environment
6A.5.24
Supplement 26 Current Protocols in Microbiology
1. Set up O1/O139-rfb/ctxA multiplex PCR in a total reaction volume of 30 μl, con-taining the following:
10 to 20 ng of crude DNA template or extracted genomic DNA:1× PCR amplification buffer210 μM dNTPs0.5 μM O1-rfb (O1F2-1, O1R2-2) primers0.27 μM O139-rfb (O139F2, O139R2) primers0.17-μM ctxA (VCT1, VCT2) primers0.75 U Taq polymerase.
2. Amplify the targets with the following cycling conditions:
1 cycle: 5 min 94◦C (initial denaturation)35 cycles: 1 min 94◦C (denaturation)
1 min 55◦C (annealing)1 min 72◦C (extension)
1 cycle: 7 min 72◦C (final extension).
3. Run PCR product out on a 2.0% agarose gel in 1× TAE for 1 to 2 hr at 5-V/cm(Voytas, 2000), along with a molecular weight ladder.
4. Stain the gel 15 min in 1 μg/ml ethidium bromide staining solution.
5. Destain the gel 15 min in distilled water.
6. Visualize the products by viewing the gel under UV light.
The O1-rfb, O139-rfb, and ctxA amplicons are 192, 449, and 308-bp in length, respectively(see Fig. 6A.5.4).
REAL-TIME PCR FOR DETECTION OF V. CHOLERAE
DNA-based methods such as PCR have been increasingly employed. These are de-signed for rapid, sensitive analysis of a range of clinical and environmental samples.End-point PCR is not quantitative, and the presence of the PCR product(s) must beverified using a procedure such as southern hybridization and gel electrophoresis (Lyon,2001).
It is important to note that, as in the case of conventional PCR, real-time PCR is notable to discriminate between culturable cells versus VBNC and dead cells, or nakedDNA (that may amplify if the nucleic acid is not completely degraded), which can affectestimation in a quantitative assay. Furthermore, the presence of substances that inhibitPCR, often present in environmental samples, can also result in a false negative (Lyon,2001). Few studies have been designed and validated for a quantitative real-time PCRmethod for detection and quantification of V. cholerae. Oligonucleotide primers andprobes for real-time PCR assays are listed in Table 6A.5.5.
NOTE: We highly recommend conducting all real-time PCR tests in a UV hood,as well as sterilizing all equipment (making sure to not expose reagents, primers,or probes to ultraviolet radiation) used for real-time PCR. This reduces risk ofcontamination.

NonentericGammaProteobacteria
6A.5.25
Current Protocols in Microbiology Supplement 26
Table 6A.5.5 Real-Time PCR Primers and Probes
Primer/probe/beacon name
Sequence Reference
hylA-probe FAM-TCAACCGATGCGATTGCCCAAGA-TAMRAa Lyon (2001)
hylA-F-primer TGCGTTAAACACGAAGCGAT
hylA-R-primer AAGTCTTACATTGTGCTTGGGTCA
MBrtxA CGCGATCACCAGAGCGCCAAGAAGTGACTCGTAGATCGCGb Gubala and Proll (2006)
MBepsM CGCGATGCCACCGACATCGTAACGCTCCGATCGCGc
MBompW CCGAAGAAACAACGGCAACCTACAAAGCTTCGGd
MBtcpA CGCGACGCTGAAACCTTACCAAGGCTGACCAAGTCGCGe
rtxA V1F AGCAAGAGCATTGTTGTTCCTACC
rtxA V1R ACTTCCCTGTACCGCACTTAGAC
epsM V3F TGGTTGATCGCTTGGCGCATC
epsM V3R ATGGCAGCCTTTGAGTGAG
mshA V6F ACACCTGGAACAGTTATTGATGGC
mshA V6R TCACTCGAAGTATCTAGCGTTTGC
tcpA V7F TGCAATGACACAAACTTATCGTAG
tcpA V7R CCCATAGCTGTACCAGTGAAAGaThe TaqMan V. cholerae-specific probe is an oligonucleotide with a 5′ reporter dye (FAM-6-carbooxyfluorescein) and a 3′ quencher dye(TAMRA-6-carboxy-N′ N′N′N′-tetramethylrhodamine).bFAM (6-carboxyfluorescein) fluorophore and Dabcyl quencher.cTexas Red fluorophore and BHQ2 quencher.dCy5 fluorophore and BHQ3 quencher.eCy3 fluorophore and BHQ2 quencher.
BASICPROTOCOL 6
TaqMan Assay for Detection of V. cholerae
The TaqMan assay (Lyon, 2001) is a real-time PCR method based on 5′ exonucleaseactivity of Thermus aquaticus DNA polymerase used to hydrolyze an internal probelabeled with a fluorescent reporter dye (FAM, Cy3, Cy5, TET) and a quencher dye. DuringPCR amplification, hydrolyis of the probe separates the reporter dye from the quencherdye, which results in increasing fluorescence proportional to the amount of template DNApresent in the reaction, thereby giving a quantitative estimation. Lyon (2001) developeda method for a quantitative, sensitive, and rapid detection of V. cholerae in pure cultures,seawater, and raw oysters, with primers and probes directed toward the non-classicalspecific hemolysin (hylA) gene of V. cholerae O1, O139, and non-O1/O139 (Table6A.5.5). Another quantitative TaqMan assay was developed for detection of the ctxAgene in seawater and oysters with the sensitivity of <10 CFU per reaction (Blackstoneet al., 2007).
Materials
100 ng/μl extracted DNA sample (Basic Protocol 8)TaqMan PCR Reagent Kit (Applied Biosystems) containing:
10× TaqMan buffer A25 mM MgCl2dATP, dCTP, dGTP, and dUTPsAmpliTaq Gold DNA polymerase1 U/μl AmpErase uracil N-glycosylase (UNG)
hylA forward primer (Table 6A.5.5)
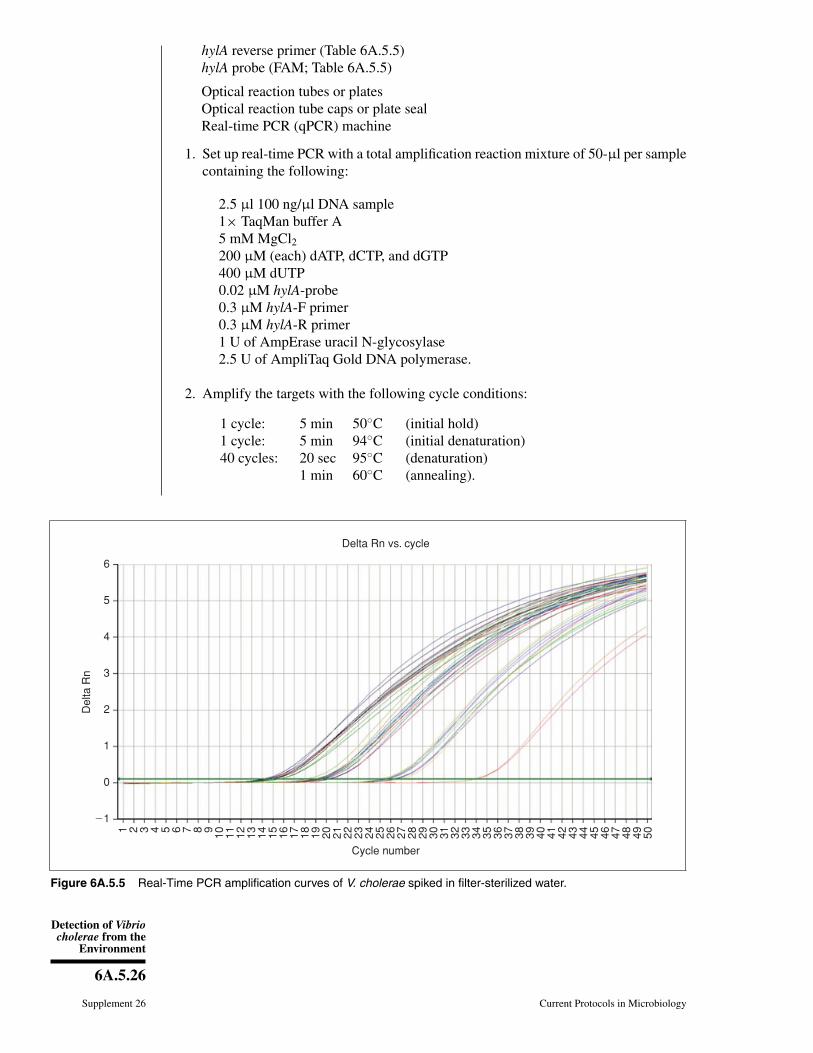
Detection of Vibriocholerae from the
Environment
6A.5.26
Supplement 26 Current Protocols in Microbiology
hylA reverse primer (Table 6A.5.5)hylA probe (FAM; Table 6A.5.5)
Optical reaction tubes or platesOptical reaction tube caps or plate sealReal-time PCR (qPCR) machine
1. Set up real-time PCR with a total amplification reaction mixture of 50-μl per samplecontaining the following:
2.5 μl 100 ng/μl DNA sample1× TaqMan buffer A5 mM MgCl2200 μM (each) dATP, dCTP, and dGTP400 μM dUTP0.02 μM hylA-probe0.3 μM hylA-F primer0.3 μM hylA-R primer1 U of AmpErase uracil N-glycosylase2.5 U of AmpliTaq Gold DNA polymerase.
2. Amplify the targets with the following cycle conditions:
1 cycle: 5 min 50◦C (initial hold)1 cycle: 5 min 94◦C (initial denaturation)40 cycles: 20 sec 95◦C (denaturation)
1 min 60◦C (annealing).
�1
Del
ta R
n
Cycle number
Delta Rn vs. cycle
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50
0
1
2
3
4
5
6
Figure 6A.5.5 Real-Time PCR amplification curves of V. cholerae spiked in filter-sterilized water.

NonentericGammaProteobacteria
6A.5.27
Current Protocols in Microbiology Supplement 26
3. Interpret results.
Results are interpreted using software available with real-time PCR machines by monitor-ing increase in fluorescence throughout the amplification cycles and reporting Ct value.The Ct value is the number of amplification cycles required to detect a fluorescent signalabove a given threshold. Ct levels are inversely proportional to the amount of targetnucleic acid in the sample. In general, Ct values <29 are considered strong positive re-actions and are indicative of abundant target nucleic acid in the sample, while Ct valuesof 30 to 35 are positive reactions indicative of moderate amounts of the target, and Ct
values of 38 to 40 are considered weak reactions with little or no target nucleic acid inthe sample (Fig. 6A.5.5).
BASICPROTOCOL 7
SYBR Green Assay for Detection of V. cholerae
An alternative real-time PCR method (Gubala, 2006) involves the use of a dsDNA-binding dye such as SYBR Green I. In this technique the amplification products aredistinguishable by analysis of their melting temperature. This assay was used in twostudies to develop a multiplex real-time PCR of four and six target genes for V. choleraeand other vibrios detected in sea, estuarine, and river water samples, and in seafood(Table 6A.5.5; Gubala, 2006; Gubala and Proll, 2006).
Materials
Extracted DNA sample (Basic Protocol 8; for optimal results, a serial dilution ofvarious concentrations of DNA samples is suggested)
LightCycler FastStart DNA Master SYBR Green I (Roche Diagnostics) including:LightCycler FastStart EnzymeLightCycler FastStart Reaction Mix SYBR GreenMgCl2 Stock Solution, 25 mMPCR-grade Taq DNA polymerase
rtxA forward and reverse primer (Table 6A.5.5)epsM forward and reverse primer (Table 6A.5.5)mshA forward and reverse primer (Table 6A.5.5)tcpA forward and reverse primer (Table 6A.5.5)
Optical reaction tubes or platesOptical reaction tube caps or plate sealSmart Cycler (Cepheid)
1a. For a single target: Prepare 25-μl amplification reaction mixtures containing:
1 μl of template DNA0.2 μM of each primer2.5 μl of LightCycler FastStart DNA Master SYBR Green I3.75 mM MgCl2 (final concentration).
1b. For multiplex PCR: Prepare 25-μl amplification reaction mixtures containing:
1 μl template DNA.0.08 μM each rtxA primer0.15 μM each epsM primer0.40 μM each mshA primer0.20 μM each tcpA primer3 μl of LightCycler FastStart DNA Master SYBR Green I4.0 mM MgCl2 (final concentration).

Detection of Vibriocholerae from the
Environment
6A.5.28
Supplement 26 Current Protocols in Microbiology
2a. For a single target: Amplify the target with the following cycling conditions:
1 cycle: 150 sec 95◦C (initial denaturation)45 cycles: 15 sec 95◦C (denaturation)
30 sec 60◦C (annealing)30 sec 72◦C (extension).
2b. For multiplex assay: Amplify the targets with the following cycling conditions:
1 cycle: 150 sec 95◦C (initial denaturation)35 cycles: 15 sec 95◦C (denaturation)
30 sec 60◦C (annealing)30 sec 72◦C (extension).
3. Interpret results.
Results are interpreted using designated computer software. As with the TaqMan assay,results are expressed in Ct values, the number of amplification cycles required to detect afluorescent signal above a given threshold. In a multiplex SYBR Green real-time PCR, theamplification products are distinguished based on their melting temperature (Tm). TheTm values of each of the products are calculated at the completion of PCR amplification,monitoring the fluorescence in increasing temperature between 60◦C and 95◦C, at a givenrate. The Tm peaks are calculated based on the initial fluorescence curve.
BASICPROTOCOL 8
DIRECT PCR FOR ENVIRONMENTAL SAMPLES
Three types of targets are used to detect V. cholerae in environmental samples by PCR:species-specific genes (16S rDNA, ITS, ompW); serogroup-specific genes (O1 and O139wbe); and toxin and pathogenic factor genes (ctx, tcpA, etc.). Briefly, water, plankton,oyster, and/or sediment samples are collected and concentrated. DNA is extracted fromthe samples, using a modification of the method of Murray and Thompson (1980), andPCR is performed on the extracted DNA using a multiplex (ompW and ctxA) primerarray. The PCR protocol described in Basic Protocol 5 is used, and the DNA template isderived from the DNA extraction method described below. Positive and negative controlsshould be run in parallel, and should include a eubacterial 16S rDNA PCR reaction oneach sample to test template quality. See Table 6A.5.3 for PCR primers and references.
Materials
Water, plankton, sediment, or oyster samplesTE buffer (APPENDIX 2A)10% (w/v) SDS20 mg/ml (2% w/v) proteinase K solution5 M NaClCTAB/NaCl solution (see recipe)25:24:1 phenol/chloroform/isoamyl alcohol (APPENDIX 2A)24:1 chloroform/isoamyl alcoholIsopropanol70% ethanol
Thermal cycler (BioRad)Microcentrifuge tubes65◦C water bath or heat blockBoiling water bathVacuum desiccator or lyophilizer
Additional reagents and equipment for sample collection (Basic Protocol 1),bacteriological culture method for V. cholerae (Basic Protocol 2), and PCRassays (Basic Protocol 4 or 5 or Alternate Protocol 2)

NonentericGammaProteobacteria
6A.5.29
Current Protocols in Microbiology Supplement 26
Collection and concentration of environmental sample
For water and plankton samples
1a. Collect sample as described in the conventional bacterial culture method above(Basic Protocol 1).
2a. Perform enrichment in APW using the conventional bacterial culture method (BasicProtocol 1).
For sediment samples
1b. Add sediment to 100-ml of distilled water until the final volume reaches 200 ml. Mixwell and allow the sediment to settle. Remove particulate matter by centrifuging a10-ml aliquot of the slurry for 8 min at 1000 × g, room temperature.
2b. For sediment samples, enrich 1 ml of the sediment slurry in an APW enrichmentflask, as for water and plankton samples (Basic Protocol 1).
For oyster samples
1c. Collect sample as described in the conventional bacterial culture method above(Basic Protocol 1)
2c. Perform enrichment in APW using the conventional bacterial culture method (BasicProtocol 1).
Processing these samples in 10-fold dilutions and in triplicate or quintuplicate (i.e.,triplicates or quintuplicates of 100, 10, and 1 ml of water), followed by direct PCRon these dilutions, will allow a quantitative estimation of the Most Probable Number(MPN) PCR target in each sample. This can be done for all collected sample types, anddilutions for oysters and sediment would be 10-fold weight increments (i.e., 10, 1, 0.1 gof homogenized oysters). For more information on the Most Probable Number assay seeStandard Methods for the Examination of Water and Wastewater (Clesceri et al., 1998).
It is possible to perform a quick DNA extraction at this point, and proceed directly to PCR(step 14), skipping DNA purification:
1. Remove a 1-ml aliquot from the upper surface (i.e., top 1 to 2 mm) of the APWenrichment.
2. Boil for 10 min.
3. Put on ice, then centrifuge to pellet cell material.
4. Remove ∼700 μl supernatant and add this to a sterile 1.5-ml microcentrifuge tube.
This supernatant contains community DNA.
5. Add 28 μl of 10 mg/ml bovine serum albumin (BSA) (4 μl per 100-μl supernatant).
It is also beneficial to make a 1:10 dilution (supernatant:sterile DI water) and use thisfor PCR as well, as dilution may help to reduce the concentration of PCR inhibitors thatcan be present in community DNA samples.
DNA extraction using phenol, chloroform, and isoamyl alcohol3. Microcentrifuge a 1-ml aliquot from the upper surface (i.e., top 1 tob 2 mm) of the
APW enrichment for 5 min at 12,000 × g, room temperature.
4. Resuspend the cell pellet in 567 μl TE buffer.
5. Add 30 μl of 10% SDS, followed by 3 μl of 20 mg/ml proteinase K solution.
6. Incubate the suspension 1 hr at 35◦C.
7. Add 100 μl of 5 M NaCl, followed by 80 μl of CTAB/NaCl solution.
8. Incubate mixture 10 min at 65◦C.

Detection of Vibriocholerae from the
Environment
6A.5.30
Supplement 26 Current Protocols in Microbiology
9. Add 800 μl phenol/chloroform/isoamyl alcohol (25:24:1), vortex, and microcen-trifuge 5 min at 12,000 × g, room temperature.
10. Transfer aqueous phase (supernatant) to a new microcentrifuge tube. Add 800 μlof chloroform/isoamyl alcohol (24:1), vortex, and microcentrifuge 5 min at 12,000× g, room temperature.
11. Transfer aqueous phase (supernatant) to a new microcentrifuge tube. PrecipitateDNA with an equal volume of isopropanol.
12. Pellet DNA by centrifuging 5 min at 12,000 × g, 25◦C. Wash DNA with 1 ml of70% ethanol and microcentrifuge 5 min at 12,000 × g, 25◦C.
13. Dry pellet in vacuum desiccator or lyophilizer and resuspend in 100 μl TE buffer.
Perform direct PCR14. Perform the PCR protocols described in Basic Protocol 4 or Alternate Protocol 2 or
Basic Protocol 5 on the extracted DNA template.
Additionally, PCR assays described in Table 6A.5.4 can be explored to evaluate thepresence of other V. cholerae virulence genes not targeted in these three protocols.
BASICPROTOCOL 9
COLONY BLOT HYBRIDIZATION WITH LABELED RNA OR DNA PROBES
The colony lift procedure is used to immobilize DNA from bacterial colonies onto ni-trocellulose or nylon filters to allow quick screening of a large number of colonies forgenetic elements of interest by hybridization. The colony blot hybridization procedureis culture based, so it is dependent upon the presence of V. cholerae in the sample as vi-able, culturable cells. Its detection by hybridization precludes the necessity of numerousbiochemical tests. Its advantage over PCR is that isolation is performed simultaneouslywith blot preparation, and enumeration can be performed more easily. Briefly, LB ormodified nutrient agar spread plates are prepared from water samples and incubatedovernight. Other plating media can be used, but the medium should be relatively richand nonselective to allow for vigorous growth and cells with a high RNA content. Nitro-cellulose (or nylon) membranes are overlaid, lifted, and treated to bind RNA (Rehnstamet al., 1989) (or DNA) to the membrane. Plates to be lifted should contain 50 to 150well-defined colonies, 2.0- to 3.0-mm in size. Membranes should be handled with sterileforceps only, and can be sterilized in an autoclave between two pieces of filter paperfor 15 min prior to use. For RNA blots, care should be taken to minimize RNase con-tamination. Blots are then hybridized with labeled probe specific for V. cholerae (and V.mimicus), 5′-ACTTTGTGAGATTCGCTCCACCTCG-3′ (Heidelberg, 1997; Heidelberget al., 2002), or toxigenic V. cholerae (ctxA). V. mimicus is a closely related species toV. cholerae, which was previously described as biochemically atypical V. cholerae (non-sucrose-fermenting, indicated by green colonies instead of yellow colonies on TCBS). V.mimicus produces a variety of toxins, including cholera toxin (potential reservoir), andhas caused sporadic diarrhea (Ramamurthy et al., 1994). Fluorescently labeled probes arepreferred (e.g., see Boyle and Perry-O’Keefe, 1992); however, the DIG system (Roche)offers a good alternative when a variable mode imager (such as Typhoon, GE Health-care) or an alternative machine, such as Dark Reader (Clare Chemical Research), fordetection of the fluorochrome, is not available. The V. cholerae-specific RNA colonyblot/hybridization protocol is presented first, and then a ctxA-DNA colony blot/DIG(Roche) hybridization protocol is presented.
Materials
Bacteria growing in APW enrichment flasks (Basic Protocol 2) or water sample forenumeration

NonentericGammaProteobacteria
6A.5.31
Current Protocols in Microbiology Supplement 26
LB or modified nutrient agar plates (see APPENDIX 2C)10% (w/v) SDS3× SSC (APPENDIX 2A)Pre-washing solution (see recipe)DEPC-treated water (APPENDIX 2A)Hybridization solution base (see recipe)Washing solution (see recipe)Fluorescein-labeled Vchomim1276 probe, reconstituted at 25 ng/μl (see recipe)
85-mm sterile nitrocellulose membranes, 0.22-μm (GE Osmonics)65◦C incubatorFilter paper (Whatman)Pyrex dish slightly larger than membrane70◦C ovenTyphoon scanner (GE Healthcare) or Dark Reader (Clare Chemical)
NOTE: All solutions should be DNase and RNase-free. For RNA colony blot hybridiza-tion, use DEPC-treated water (see APPENDIX 2A) to make hybridization solutions.
Spread-plate preparation1. For detection, prepare spread plates from APW enrichment flasks using APW as
diluent and plating three serial-fold dilutions onto LB or modified nutrient agar plates.
2. For enumeration (and detection), spread plate appropriate dilutions of water sampleonto LB or modified nutrient agar plates on-site without APW enrichment.
Alternatively, 100 to 500 ml of water may be filtered through 0.22-μm nylon membranesand overlaid onto an agar plate. If this method is preferred, incubate membrane and plateovernight at 30◦C and then proceed to step 7.
3. Incubate plates overnight at 30◦C.
Colony lift4. Mark membranes using a lead pencil with a Blot ID (e.g., medium, sample, dilution)
that matches plate to be lifted and orientation marks (asymmetrical).
5. Overlay nitrocellulose membrane, starting from the center, to ensure there are no airbubbles.
6. Allow at least 15 min for transfer.
7. Replica plate the membrane onto a fresh modified nutrient agar plate, transferringorientation markings.
8. Preheat SDS, SSC, and Pyrex dishes (for holding filter paper) to 65◦C.
9. Place membrane, colony-side-up, on the filter paper (cut to just slightly larger thanthe membrane) pre-wetted with 10% SDS, enclosed in a Pyrex dish, for 5 min at65◦C. Cover Pyrex dish containing wetted filter paper and membrane with Saranwrap or equivalent plastic film to prevent the filter paper from drying out.
It is important to use filter paper pre-wetted, but not saturated, with 10% SDS or 3× SSC(see next step) to prevent colonies from over-swelling and losing their circular shape.Pour or pipet the liquid onto the filter paper, let soak briefly, remove air bubbles, and thenpour off excess. Ensure that there is no pooled liquid on the filter paper prior to placingmembrane.
10. Incubate the membrane 15 min at 65◦C on fresh filter paper wetted with 3× SSC.Cover Pyrex dish containing wetted filter paper and membrane with Saran wrap orequivalent plastic film to prevent the filter paper from drying out.

Detection of Vibriocholerae from the
Environment
6A.5.32
Supplement 26 Current Protocols in Microbiology
11. Let membranes air dry 10 min on filter paper.
12. Bake membranes 15 min at 70◦C.
RNA-colony blot hybridization13. Wash membranes three times in adequate pre-washing solution for 15 min at room
temperature.
14. Wash membranes 1.5 hr at 60◦C in pre-washing solution.
15. After pre-washing steps, rinse membranes in DEPC-treated water.
Hybridization solution base will form a precipitation upon maintaining at room temper-ature. If this happens, heat to 40◦ to 50◦C to resuspend.
16. Pre-hybridize the membranes 30 min at 60◦C in hybridization solution base at a ratioof 10-ml solution per 100-cm2 of blot membrane.
85 mm-membranes have a surface area of ∼60-cm2.
17. Pour off solution and add hybridization solution base plus fluorescein-labeledVchomim1276 probe (32 μl of 25 ng/μl probe per 10 ml solution) at a ratio of10 ml solution per 100-cm2 blot.
18. Hybridize overnight (ideally 16 to 20 hr) at 60◦C.
19. After hybridization, wash membranes 30 min at 60◦C in washing solution.
20. View/image the membrane using a Typhoon Scanner or Dark Reader.
21. From replica plates, subculture colonies that were positive by blot hybridization forfurther analysis, if desired.
ALTERNATEPROTOCOL 3
COLONY BLOT HYBRIDIZATION USING DIG-LABELED ctx DNAPROBE
The previous colony blot hybridization protocol (Basic Protocol 9) is used to detectV. cholerae and closely related V. mimicus. This protocol, on the other hand, targets onlytoxigenic strains of V. cholerae. The presence of ctxA is confirmed by hybridization usinga ctxA-specific DNA-probe. There may be some cross-reactivity of the probe with theheat-labile toxin (LT) of E. coli (Dallas and Falkow, 1980). Colony blots are preparedfollowing the method of Pal et al. (1992). The hybridization is done according to theDIG protocol (Roche). The protocol is given here; readers are encouraged to consultthe manual accompanying the High Prime kit. The DIG DNA probe-labeling protocol isgiven in Support Protocol 2. The ctxA probe can be produced by PCR, using the pCTAprimer set (see Table 6A.5.3) or by EcoRI digestion of the plasmid, pKTN901, whichcontains a 540-bp XbaI-ClaI fragment of ctxA (Kaper et al., 1988).
Materials
Bacteria growing in APW enrichment flasks (Basic Protocol 2)Luria-Bertani (LB) agar plates (APPENDIX 4A)Cell lysis buffer (see recipe)Blot neutralization solution (see recipe)1× SSC buffer (APPENDIX 2A)Proteinase K solution (see recipe)DIG High Prime DNA Labeling and Detection Starter Kit II (Roche) kit includes:
DIG Easy Hyb granules10× blocking solutionCSPD

NonentericGammaProteobacteria
6A.5.33
Current Protocols in Microbiology Supplement 26
Anti-digoxigenin-AP conjugateDIG High Prime
Labeled ctxA probe (Support Protocol 2)Stringency wash solution I (see recipe)Stringency wash solution II (see recipe)Maleic acid buffer (see recipe)Antibody solution: 1:10,000 anti-digoxigenin-AP conjugate in 1× blocking
solution (from kit)Washing buffer (see recipe)Detection buffer (see recipe)
Incubators, 37◦C and 42◦CSterile nylon (or nitrocellulose) membranes, 85mm, 0.22-μm (GE Osmonics)Whatman filter paper, #3UV cross-linker or transilluminatorShaking water bathHybridization pouchX-ray film (Kodak or Fuji)Film developer
Blot preparation1. Prepare spread plates as above, using LB agar plates (see Basic Protocol 9, steps 1
to 3).
2. Incubate the plates overnight at 37◦C
3. Mark membranes using a lead pencil, including Blot ID (e.g., medium, sample,dilution) that matches plate to be lifted and orientation marks (asymmetrical).
4. Overlay membrane, starting from the center to ensure there are no air bubbles.
5. Transfer at least 15 min.
6. Transfer the blot onto a new LB plate, keeping colony side up, and incubate 3 hr at37◦C. Wrap the master plate with Parafilm and keep at 10◦ to 15◦C.
Master plates can be kept like this for up to 2 weeks.
7. Place membranes, colony side up, onto #3 Whatman filter paper pre-soaked with celllysis buffer. Incubate 10 min at room temperature.
8. Remove the membrane from cell lysis buffer and place onto #3 Whatman filter paperpre-soaked with blot neutralization solution
9. Repeat step 8.
10. Remove membrane from blot neutralization solution, place onto #3 Whatman filterpaper, and air dry (∼30 min).
11. Immobilize colonies onto the membrane using a UV cross-linker or transilluminator.
Damp membranes should be cross-linked at an output intensity of 120-mJ/cm2, whichcorresponds to the optimal or auto-crosslink setting on commercially calibrated ma-chines. Transilluminators or hand-held UV lamps can be used, if calibrated; however, thefollowing times should be sufficient: 1 min for 254-nm lamps or 3 min for 302-nm lamps.
12. Rinse the blot two times in 1× SSC buffer and air dry (∼30 min).
13. Treat membranes 30 min at 42◦C with 100 ml proteinase K solution using gentleshaking.

Detection of Vibriocholerae from the
Environment
6A.5.34
Supplement 26 Current Protocols in Microbiology
14. Rinse filters three times in 1× SSC in a shaking water bath at room temperature for10 min each. Let air dry (∼30 min).
Prehybridization and hybridization15. Reconstitute DIG Easy Hyb granules according to the manufacturer’s instructions to
prepare DIG Easy Hyb buffer. Preheat DIG-Easy Hyb buffer to 42◦C.
16. Prehybridize blots in preheated DIG-Easy Hyb buffer for 30 min at 42◦C with gentleagitation.
17. Denature the DIG-labeled probe by boiling for 5 min and rapidly cooling on ice.
18. Pour off DIG-Easy Hyb buffer from the blots.
19. Add fresh DIG-Easy Hyb buffer plus denatured, labeled ctxA probe (25 ng/ml solu-tion) and incubate overnight at 42◦C.
Stringency washes20. Pour off probe-containing hybridization solution and store up to 2 months at −20◦C.
Probe-containing hybridization solution can be reused several times.
21. Wash membrane two times in Stringency Wash Solution I for 5 min at room temper-ature under constant agitation.
22. Wash two times in Stringency Wash Solution II for 15 min at 65◦C under constantagitation.
Detection23. Rinse membrane briefly (5 min) in washing buffer.
24. Prepare 1× blocking solution by diluting 10× blocking solution from the kit inmaleic acid buffer. Incubate the membrane 30 min at 25◦C in 1× blocking solution.
25. Incubate the membrane 30 min at 25◦C in antibody solution (1:10,000 dilution ofanti-digoxigenin-AP conjugate) for 30 min.
26. Wash the membrane two times, each time for 15 min, in washing buffer, for 15 mineach.
27. Equilibrate the membrane in detection buffer for 5 min at 25◦C.
28. Place membrane in hybridization pouch. Add CSPD to the membrane, cover, andincubate at room temperature for 5 min.
29. Remove excess liquid from pouch, seal, and incubate at 37◦C for 10 min.
30. Expose the membrane to X-ray film for 20 min at room temperature and develop.
If film is under- or over-exposed, repeat step 30 and vary time of exposure accordingly.
31. From master plates (step 6), subculture colonies that were positive by blot hybridiza-tion for further analysis.
SUPPORTPROTOCOL 2
DIGOXIGENIN LABELING OF ctxA PROBE USING DIG-HIGH PRIME
PCR-or digestion-generated probes can be labeled by DIG-High Prime (Roche), ran-domly incorporating digoxigenin-11-dUTP. A 563-bp ctxA fragment can be produced byamplifying extracted DNA from a toxigenic laboratory reference strain. The PCR productshould be purified by using a PCR clean-up kit or by gel electrophoresis and extraction.Alternately, a PCR product may be labeled during the PCR amplification using the PCRDIG Probe Synthesis Kit (Roche). A 554-bp ctxA probe is also available on a plasmid,

NonentericGammaProteobacteria
6A.5.35
Current Protocols in Microbiology Supplement 26
pKTN901(Kaper et al., 1988), which has been widely used (Pal et al., 1992; Islam et al.,2005). The XbaI-ClaI fragment can be removed from the plasmid by digestion withEcoRI and gel purified. Labeled probes can then be used in the DIG-based hybridizationabove.
Materials
DNA extracted from toxigenic (ctxA+) V. cholerae reference strain (ATCC)Forward and reverse pCTA primers for amplifying ctxA (Table 6A.5.5)Plasmid pKTN901 (available from Dr. James Kaper, [email protected]) or
other source of ctxA probeEcoRI restriction endonuclease (or other restriction enzyme depending on source
of ctxA probe) and restriction buffer DIG-High Prime (Roche)0.2 M EDTA, pH 8.0
Boiling water bath65◦C incubator
Additional reagents and equipment for PCR (Kramer and Coen, 2001), agarose gelelectrophoresis (Voytas, 2000), and isolation and purification of fragments fromagarose gels (Moore et al., 2002)
1. For PCR-generated ctxA target fragment, amplify extracted DNA of a toxigenic(ctxA+) V. cholerae reference strain using the pCTA primer set and the master mixand cycling conditions described in Basic Protocol 4.
2. For digestion-generated ctxA target fragment, digest plasmid, pKTN901, with EcoRIfor 2 hr at 37◦C.
3. Clean-up PCR reaction product or digestion product by agarose gel electrophoresis(Voytas, 2000) followed by gel extraction (Moore et al., 2002).
4. Bring 1-μg of purified ctxA fragment up to a total volume of 16-μl in sterile distilledwater.
5. Denature by placing in a boiling water bath for 10 min, followed by rapidly chillingin an ice bath (5 min).
6. Add 4-μl of DIG-High Prime to the DNA solution, mix, and centrifuge briefly tocollect liquid.
7. Incubate overnight at 37◦C.
8. Stop reaction by adding 2 μl of 0.2 M EDTA, pH 8.0, and/or by heating to 65◦C for10 min.
IMMUNOLOGICAL METHODS FOR DIRECT DETECTION OFV. CHOLERAE IN ENVIRONMENTAL SAMPLES
Conventional culture methods are ineffective when bacterial cells have entered into theviable but non-culturable (VBNC) state. Thus, direct detection becomes extremely im-portant. Despite the ubiquitous nature of V. cholerae, isolation and detection by traditionalmethods are difficult since these methods rely on culturing the organism. The discoveryof monoclonal antibody in the 1980s and, subsequently, the development of a monoclonalantibody against V. cholerae O1, triggered development of direct detection methods forthis bacterial species (Xu et al., 1984; Hasan et al., 1992). Using immunological meth-ods, the mystery concerning the inability to culture V. cholerae in environmental sampleswas reported during an inter-epidemic period in Bangladesh (Roszak and Colwell, 1987;Huq et al., 1990). These difficulties arise from several possible factors: low density,inter-specific competition, cell state, and health (VBNC, starved). Fluorescent In-Situ

Detection of Vibriocholerae from the
Environment
6A.5.36
Supplement 26 Current Protocols in Microbiology
Hybridization (FISH) allows direct detection of taxa-specific nucleic acid followed byvisualization using microscopy, and the polymerase chain reaction offers a molecular-based alternative to the traditional culture and immunological methods (discussed later).
BASICPROTOCOL 10
Fluorescent In Situ Hybridization (FISH) Detection of V. cholerae
Direct quantification methods without the need to enrich and culture can yield highlyaccurate and quick estimates of taxon-specific densities in water bodies. Fluorescence insitu hybridization (FISH) accomplishes this with a fluorescently labeled oligonucleotideprobe that is visualized under epifluorescence or confocal laser scanning microscopy.This method is reliable for accurate estimations of V. cholerae cells in media, as it allowsone to quantify both culturable and non-culturable (VBNC) V. cholerae cells.
Materials
Water for testingPhosphate-buffered saline (PBS; APPENDIX 2A)4% formaldehyde (see recipe)PBS-ethanol solution (1:1 PBS:absolute ethanol)0.01% (w/v) poly-L-lysine (PLL)Ethanol solutions (50%, 80%, and 96%)Hybridization solution (see recipe)FITC-labeled probe: Vchomim1276, 6-FAM (fluorescein phosphoramidite) labeled
probe (5′-[5FITC] ACTTTGTGAGATTCGCTCCACCTCG-3′) at a workingconcentration of 50 ng/μl in sterile water (final concentration, 5 ng/μl)
Washing buffer solution (see recipe)Citifluor AF1 antifade agent (Electron Microscopy Sciences)
Multiwell slidesIncubator or water bath set to 45◦CHumid chamber: 50-ml centrifuge tube containing one to two strips of
hybridization solution–saturated filter paperWhatman filter paper
Sample preparation1. Filter water (∼500 to 1000 ml) through a 0.2 μm polycarbonate membrane, and
resuspend cells attached to membrane in 5 ml PBS.
2. Centrifuge 1 ml of the PBS suspension 10 min at 13,000 × g, room temperature.Discard supernatant and resuspend in 250 μl of PBS.
3. Fix cells by adding 750 μl of fresh 4% paraformaldehyde solution and incubate atroom temperature (22◦ 25◦C) for 1 hr.
4. Next, pellet the cell solution by centrifugation for 5 min at 13,000 × g, roomtemperature. Discard the supernatant and then wash the cell twice with PBS usingthe same centrifugation conditions.
5. Concentrate the cells by centrifugation and resuspend in 100 μl PBS-ethanol solution.
6. Store cells at –20◦C.
Hybridization7. Immerse multiwell slides in 0.01% PLL for 10 min and air dry.
8. Spot 5-μl aliquots of fixed cells onto slides and air dry.
9. Wash slides for 3 min each in successively higher concentration ethanol solutions(50%, 80%, and 96%), and then air dry. Prewarm humid chamber, slides, and filterpaper soaked with hybridization solution at 45◦C for 10 min.

NonentericGammaProteobacteria
6A.5.37
Current Protocols in Microbiology Supplement 26
Figure 6A.5.6 Fluorescent in situ hybridization (FISH) image of V. cholerae cells under epifluo-rescence microscopy.
10. To each well, add 5 μl aliquot of hybridization solution containing 5 ng/μl of labeledprobe.
11. Perform hybridization at 45◦C for 24 hr in the humid chamber using hybridizationsolution–soaked filter paper.
12. Pre-warm the washing buffer solution at 45◦C for 10 min before washing the slidesand incubate at 45◦C for 10 min.
13. Wash slides in sterile deionized water and air dry at room temperature.
14. Add antifade agent to each well and apply a coverslip.
15. Visualize fluorescence under epifluorescence microscopy (Fig. 6A.5.6.)
BASICPROTOCOL 11
Direct Fluorescent Antibody–Direct Viable Count (DFA-DVC) Method
The direct fluorescent antibody staining method for rapid detection of V. choleraeserogroup O1 and O139 is a very useful, direct, and culture-independent method. Coupledwith the direct viable count method of Kogure et al. (1979), it can distinguish culturable,viable cells from viable but non-culturable cells (VBNC) of V. cholerae (Chowdhuryet al., 1994). It is a two-step method where samples are incubated with yeast extract inthe presence of nalidixic acid, after which actively viable, substrate-responsive cells be-come enlarged and elongated (Kogure et al., 1979). Next, an aliquot from this suspensionis air dried on a glass slide and stained with fluorescently labeled monoclonal antibody,raised against ‘A’ factor of V. cholerae O1 lipopolysaccharide, which reacts with bothserotypes, Ogawa and Inaba (Colwell et al., 1990; Hasan et al., 1994). Antibodies againstV. cholerae O139 are also available (Hasan et al., 1995). When observed under an epi-fluorescent microscope, elongated cells of V. cholerae O1 or O139 (based on the type ofantibody used) exhibit a bright-green fluorescing periphery (the outer cell wall) with adark interior (Fig. 6A.5.7). V. cholerae O1 and O139 DVC-DFA positive samples can beconfirmed by PCR (Binsztein et al., 2004). DFA-DVC is a rapid method by which onecan determine the presence of V. cholerae within 8 hr (when the DVC incubation is 6 hr);however, overnight incubation with yeast extract and nalidixic acid is preferred. Kitsfor V. cholerae O1 (CholeraDFA) and O139 (BengalDFA) DFA tests are commerciallyavailable (New Horizon Diagnostics).
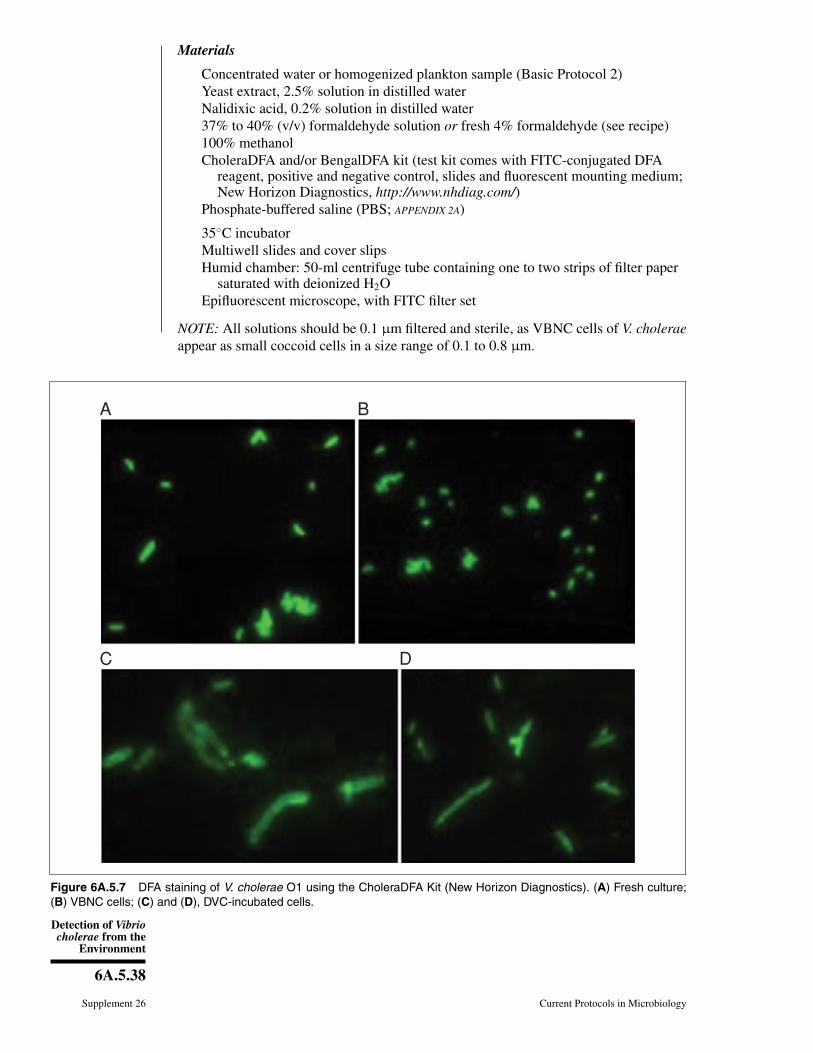
Detection of Vibriocholerae from the
Environment
6A.5.38
Supplement 26 Current Protocols in Microbiology
Materials
Concentrated water or homogenized plankton sample (Basic Protocol 2)Yeast extract, 2.5% solution in distilled waterNalidixic acid, 0.2% solution in distilled water37% to 40% (v/v) formaldehyde solution or fresh 4% formaldehyde (see recipe)100% methanolCholeraDFA and/or BengalDFA kit (test kit comes with FITC-conjugated DFA
reagent, positive and negative control, slides and fluorescent mounting medium;New Horizon Diagnostics, http://www.nhdiag.com/)
Phosphate-buffered saline (PBS; APPENDIX 2A)
35◦C incubatorMultiwell slides and cover slipsHumid chamber: 50-ml centrifuge tube containing one to two strips of filter paper
saturated with deionized H2OEpifluorescent microscope, with FITC filter set
NOTE: All solutions should be 0.1 μm filtered and sterile, as VBNC cells of V. choleraeappear as small coccoid cells in a size range of 0.1 to 0.8 μm.
A B
C D
Figure 6A.5.7 DFA staining of V. cholerae O1 using the CholeraDFA Kit (New Horizon Diagnostics). (A) Fresh culture;(B) VBNC cells; (C) and (D), DVC-incubated cells.

NonentericGammaProteobacteria
6A.5.39
Current Protocols in Microbiology Supplement 26
Perform DVC incubation1. To 1 ml concentrated water or homogenized plankton sample, add 10 μl yeast extract
solution and 10 μl nalidixic acid solution.
2. Freeze (−20◦C) a parallel sample (1 ml) for PCR confirmation.
3. Incubate the mixture at 25◦C for a minimum of 6 hr to overnight.
4. Fix the sample by adding formaldehyde to a final concentration of 3% (v/v) andincubating 30 min at room temperature in the dark.
Fixed samples can be stored at 4◦C in the dark for up to 6 months.
Perform DFA protocol5. Place 5 to 10 μl of the fixed sample onto a glass slide and air dry (∼15 to 20 min).
6. Fix by adding 5 μl methanol and air dry (1 to 5 min).
7. Add 10 μl of reconstituted FITC-conjugated specific DFA reagent (Hasan et al.,1994), as supplied by the Cholera DFA or BengalDFA kit.
8. Incubate 30 min at 37◦C in humid chamber. Protect slide from light.
9. Rinse slide with ∼50 ml PBS (each). Air dry slides in the dark (∼15 to 20 min).
10. Mount slide with one drop of kit-provided fluorescent mounting medium and addcoverslip.
11. Observe under an epifluorescent microscope (see Fig. 6A.5.7).
BASICPROTOCOL 12
Indirect Fluorescent Antibody (IFA) Method
This immunofluorescent method for detection of V. cholerae serogroup O1 in aquaticenvironmental samples was first introduced by Xu et al. (1984). Antiserum specific for O1somatic antigen produced in rabbits was used with fluorescein-isothiocyanate (FITC)-conjugated anti-rabbit globulin goat serum, with rhodamine isothiocyanate (RITC)–conjugated bovine serum albumin as the background stain. This method was found tobe very useful for detecting organisms in samples that gave negative results by culture(Brayton et al., 1986; Huq et al., 1990). It was later optimized and packaged as thedirect fluorescent antibody staining kit for V. cholerae O1, Cholera DFA, by Hasan et al.(1994). The IFA protocol remains useful for laboratories where commercial DFA kits forV. cholerae are not readily available.
Materials
Concentrated water or homogenized plankton sample (Basic Protocol 2)95% ethanolPhosphate-buffered saline (PBS; APPENDIX 2A)FA Rhodamine counterstain (Becton Dickinson)Polyvalent V. cholerae O1 antiserum (BD Difco)FITC-conjugated anti-rabbit globulin goat serum (Sigma)Mounting medium, such as FA (Difco) or Citifluor AF1/AF3 (Electron Microscopy
Sciences)
Multiwell Teflon-coated slidesIncubators, set to 55◦C and 35◦CHumid chamber: 50-ml centrifuge tube containing one to two strips of filter paper
saturated with deionized H2OGlass coverslipsEpifluorescent microscope with FITC bandwidth filter

Detection of Vibriocholerae from the
Environment
6A.5.40
Supplement 26 Current Protocols in Microbiology
Prepare and fix samples1. Add an appropriate amount (dependent on concentration of sample and well size) of
each sample to be tested to a Teflon-coated multiwell slide. Air dry (15 to 20 min)at room temperature.
2. Fix the sample by adding 95% ethanol to each well containing sample. Air dry (5 to10 min) at room temperature.
3. Heat slide 10 min in a 55◦C incubator.
Slides may be stored up to 1 month at −70◦C at this point.
Staining procedure4. Rinse the slide(s) with ∼50 ml PBS and air dry (15 to 20 min).
5. While slide is drying, prepare humid chamber for use. Equilibrate chamber in 35◦Cincubator (∼15 min).
6. To each dry sample well, add 1 to 2 drops of a 1:20 dilution of FA RhodamineCounterstain. Incubate in humid chamber 30 min at 35◦C.
Minimize exposure to light from this step forward.
7. Rinse slide in PBS by gently flooding (∼50 ml), then soak in the same solution10 min at room temperature. Remove and rinse again briefly in PBS.
8. Allow slide to air dry (15 to 20 min).
9. Add 5 to 10 μl of V. cholerae O1-specific antiserum. Incubate in humid chamber30 min at 35◦C.
10. Repeat steps 7 and 8. Use fresh PBS for washing.
11. Add 1 to 2 drops undiluted FITC-conjugated anti-rabbit globulin goat serum andincubate in humid chamber 30 min at 35◦C.
12. Repeat washing steps 7 and 8 using fresh PBS.
13. Mount each slide with a glass coverslip and a low fluorescence, anti-quenchingmounting medium, such as Citifluor AF1.
14. Examine samples immediately using a epifluorescent microscope with a FITC band-pass filter.
REAGENTS AND SOLUTIONSUse deionized, distilled water in all recipes and protocol steps. For common stock solutions, seeAPPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Alkaline peptone water, 1×10 g peptone10 g sodium chlorideAdjust volume to 1 liter with waterAdjust pH to 8.6 with NaOHAutoclave to sterilizeStore up to 6 months at 2◦ to 8◦C
Alkaline peptone water, 10×100 g peptone100 g sodium chloride
continued

NonentericGammaProteobacteria
6A.5.41
Current Protocols in Microbiology Supplement 26
Adjust volume to 1 liter with waterAdjust pH to 8.6 with NaOHAutoclave to sterilizeStore up to 6 months at 2◦ to 8◦C
Blot neutralization solution
0.5 M Tris·Cl, pH 7.2 (APPENDIX 2A)1.5 M sodium chlorideStore up to 6 months at 2◦ to 8◦C
Brain-heart infusion agar (BHI)
7.7 g calf brain (infusion from 200 g; BD Difco)9.8 g beef heart (infusion from 250 g; BD Difco)10 g proteose peptone (BD Difco)2 g dextrose5 g sodium chloride2.5 g disodium phosphate15 g agar (BD Difco)For use in Alternate Protocol 1 supplement with:1 g esculin (0.1% final; BD Difco)500 mg ferric chloride (anhydrous, 0.05% final)Adjust volume to 1 liter with waterAutoclave to sterilizeAllow to cool to 50◦CPrepare agar stabs by pouring into 4 to 5-ml sterile vials
Cell lysis buffer
0.5 N sodium hydroxide1.5 M sodium chloridePrepare fresh
CTAB/NaCl solution
Add 4 g NaCl to 80 ml water and dissolve. Slowly add 10 g of cetyltrimethylam-monium bromide (CTAB) while heating and stirring at 65◦C. Adjust volume to100-ml with water. Store up to 6 months at room temperature.
Detection buffer
0.1 M Tris·Cl, pH 9.5 (APPENDIX 2A)0.1 M sodium chlorideStore up to 1 month at room temperature
Fluorescein-labeled Vchomim1276 probe
5′-ACTTTGTGAGATTCGCTCCACCTCG-3′, with 5′ fluorescein label
Resuspend probe to a working concentration of 25 ng/μl. Add probe to hybridizationsolution base (see recipe) at a ratio of 32 μl per 10 ml of solution. Store indefinitelyat 4◦C.
Formaldehyde solution, 4%
8 g paraformaldehyde powder200 ml PBS (APPENDIX 2A)Prepare fresh
continued

Detection of Vibriocholerae from the
Environment
6A.5.42
Supplement 26 Current Protocols in Microbiology
In a ventilated hood, heat PBS to 60◦C and add paraformaldehyde powder. Stiruntil dissolved. If some powder remains, slowly raise the pH with 1 N NaOH (finalpH, 6.9). Filter sterilize.
Gelatin agar (GA)
4 g neopeptone1 g yeast extract5 g sodium chloride15 g gelatin15 g agarAdjust volume to 1 liter with waterAutoclave to sterilizeStore up to 1 month at 2◦ to 8◦C
Hybridization solution
0.9 M NaCl20 mM Tris·Cl, pH 7.2 (APPENDIX 2A)0.01% (w/v) SDS35% (v/v) formamideStore up to 1 month at room temperature
Hybridization solution base
Prepare the following in DEPC-treated water (APPENDIX 2A)0. 9 M NaCl50 mM sodium phosphate buffer, pH 8.0 (APPENDIX 2A)5 mM EDTA0.5% (w/v) SDSStore up to 1 month at room temperature
Precipitation will occur upon standing at room temperature. Heat to 45◦ to 50◦C to resus-pend.
Maleic acid buffer
0.1 M maleic acid0.15 M sodium chlorideAdjust pH to 7.5 with solid NaOHPrepare fresh
Modified nutrient agar
3 g beef extract5 g peptone10 g sodium chloride15 g agarAdjust volume to 1 liter with waterAutoclave to sterilizeStore up to 1 month at 2◦ to 8◦C
Pre-washing solution
Freshly prepare 3× SSC (APPENDIX 2A) containing 0.1% (w/v) SDS using RNase-freeingredients and DEPC-treated water (APPENDIX 2A).
Proteinase K solution (40-μg/ml)
Dissolve 4 mg proteinase K in 100 ml 1× SSC (APPENDIX 2A). Store up to 2 monthsat −20◦C

NonentericGammaProteobacteria
6A.5.43
Current Protocols in Microbiology Supplement 26
Stringency wash solution I
2× SSC (APPENDIX 2A)0.1% (w/v) SDSPrepare fresh
Stringency wash solution II
0.5× SSC (APPENDIX 2A)0.1% (w/v) SDSPrepare fresh
Tellurite taurocholate gelatin agar (TTGA)
10 g tryptone10 g sodium chloride5 g sodium taurocholate1 g sodium carbonate30 g gelatin15 g agarAdjust volume to 1 liter with waterBoil to completely dissolve ingredients
Final pH should be 8.5. If it is not, adjust with HCl.
Autoclave and add potassium tellurite to 1% (w/v) finalStore up to 1 month at 2◦ to 8◦C
Thiosulfate citrate bile salts sucrose (TCBS) agar
5 g yeast extract10 g peptone10 g sodium thiosulfate10 g sodium citrate8 g ox bile20 g sucrose10 g sodium chloride1 g ferric citrate0.04 g bromothymol blue0.04 g thymol blue14 g agarAdjust volume to 1 liter with waterBoil to completely dissolve
IMPORTANT NOTE: Do not autoclave.
This medium is also available commercially as a dehydrated powder from Oxoid (Remel)
Washing buffer
0.1 M maleic acid0.15 M sodium chloride0.3% (v/v) Tween 20Adjust pH to 7.5 with solid NaOHPrepare fresh
Washing buffer solution
0.9 M NaCl20 mM Tris·Cl, pH 8 (APPENDIX 2A)0.01% (w/v) SDSStore up to 1 month at 4◦C

Detection of Vibriocholerae from the
Environment
6A.5.44
Supplement 26 Current Protocols in Microbiology
Washing solution
Freshly prepare 1× SSC (APPENDIX 2A) containing 0.1% (w/v) SDS using RNase-freeingredients and DEPC-treated water (APPENDIX 2A).
COMMENTARY
Background InformationUnderstanding the natural ecology of an in-
fectious agent outside of the human body isessential to understanding the epidemiologyof the associated disease and especially nec-essary in any attempt to prevent illness causedby exposure to that pathogen. It is well es-tablished that V. cholerae is autochthonousto the aquatic environment globally and evenin regions where cholera is absent (Haleyet al., 2012). However, these environments arehighly heterogeneous and change over time. Itis also well established that seasonal fluctua-tion in environmental parameters is associatedwith changes in environmental pathogen den-sities and disease prevalence. It is, therefore,necessary to understand the influence of theenvironment on presence and number of theseorganisms. This requires a holistic approachtoward investigation of the microbial ecologyof an infectious disease. Temporal and spatialstudies of V. cholerae in water, planktonic or-ganisms, sediment, and shellfish, coupled withrecording physical and chemical parametersof the study environment (regardless of pres-ence or absence of V. cholerae), allow inves-tigators to estimate changes in the presenceor density of V. cholerae in a particular re-gion. This information can be used to helppredict the public health safety of water bod-ies or seafood, thereby reducing risk of illnessand loss of revenue from inaccurate predictionof the presence of pathogens in water bod-ies used for recreation or shellfish harvesting.Although cholera is a public health threat fordeveloping nations more than for developednations, methods for V. cholerae discussed inthis chapter can be applied to other waterbornepathogens with addition of methods specific tothe microorganism of interest.
As commented above, multiple approachesshould be taken to study the ecology ofpathogens in the environment. The couplingof methods can overcome limitations inherentin any one particular method and allow con-vergence on the precise answer(s). However,increase in the cost and time commitmentneeds to be considered. An investigator shouldfirst determine the data or results needed, ap-propriateness of available methods, and feasi-bility of carrying out the procedures to achieve
the goal. If the microorganism of interest isnot detected, it is not appropriate to concludethat the pathogen is absent from the study, butrather that it was not detected by the methodsused. In this scenario, use of multiple methodsreduces “not detected” results. Furthermore,parallel replication of assays will enhance ac-curacy and statistical power of the results.
Critical Parameters andTroubleshooting
Successful detection and isolation ofV. cholerae is dependent on both its presencein the environment as well as the method usedfor detection. V. cholerae undergoes the VBNCstate at prolonged low temperatures, whichwill influence whether culture of the bac-terium will succeed when water temperaturesare low. Lack of growth on media is indica-tive of V. cholerae in the VBNC state ratherthan an absence of V. cholerae in the waterbody of interest. In such instances, inclusionof direct detection methods (DFA, IFA, FISH,direct PCR) will provide more precise resultswith respect to the presence of V. cholerae.If such methods are used in conjunction withculturing methods, then the methods shouldbe used throughout the entirety of the study,instead of alternating methods based on theprevailing environmental conditions.
The culturing methods presented in this unitare the most commonly used culture-basedprotocols for isolation of V. cholerae (Morriset al., 1979; Rennels et al., 1980). Other meth-ods have been described and may be suitablefor certain study environments. Alkaline bilepeptone water (Spira et al., 1981), Monsur’stellurite taurocholate broth (Monsur, 1961),and sodium-gelatin phosphate broth (Rennelset al., 1980) have been described as enrich-ment media that are effective for culturingV. cholerae. A second enrichment step may beused, but will add an additional 6 to 8 hr to analready lengthy protocol. There is also the con-cern that repeated passaging of cells will allowfor population and genotypic alterations to oc-cur by favoring cells that grow more readilyin enrichment broths, allowing for mutationsto accumulate, i.e., natural mutations occur-ring during cell division. The latter is a con-cern in isolating cells whose genomes will be

NonentericGammaProteobacteria
6A.5.45
Current Protocols in Microbiology Supplement 26
sequenced, as accumulation of SNPs and ge-nomic rearrangements and possibly horizontaltransfer of mobile elements or loss of genomicislands in the enrichment broth will yield inac-curate genomic data about the cells in the en-vironment. Such data may erroneously lead toassumptions on virulence factors, phylogeny,or origin of the strains. Essentially, culturingsteps should be minimized to obtain accurateresults.
As V. cholerae can grow at a wide rangeof temperatures, several incubation tempera-tures in APW and TCBS, TTGA, or CA canbe used for V. cholerae isolation. Incubatingat the lower range of recommended tempera-tures (near 30◦C) may enhance the growth ofenvironmentally stressed cells exposed to lowtemperatures, but this may require longer incu-bation times on solid media. Incubating mediaat higher temperatures (≥37◦C) may inhibitthe growth of some non-Vibrio species but mayalso inhibit the growth of stressed V. choleraecells. If possible, incubate media in parallel atmultiple temperatures. Furthermore, stepwiseincrease in incubation temperature, 25◦C (1to 2 hr) → 30◦C (1 to 2 hr) → 35◦C can beused to sensitize the stressed cells to adapt andgrow.
We strongly advise that a subculturing steponto nonselective media be utilized after iso-lation from TCBS, TTGA, or CA. This stepserves two purposes. First, it is critical thatpure colonies be isolated before proceeding toany genomic or phenotypic characterizationsteps. Secondly, growth on TCBS is not suit-able for the oxidase test, serotyping, or directPCR.
It is known that PCR amplification of targetDNA in environmental samples can be inhib-ited by dissolved organics, such as humic acid.For that reason, we suggest performing a DNAextraction first. This typically adds only 3 to4 hr to the protocol and results in high-yieldDNA with a decrease in inhibition during PCRamplification. For all direct PCR examinations(PCR on extracted or boiled APW), include theeubacterial PCR reaction to confirm templatequality on samples. Further, we strongly sug-gest that direct PCR be done on 1:10, 1:100dilutions (DNA template:TE buffer or DNAtemplate:nuclease free water) in parallel withundiluted samples.
FISH allows visual quantification of allV. cholerae cells, regardless of their cultur-ability, in a sample. One problem with thismethod is observation of autofluorescing con-stituents found in environmental water sam-ples. All “positive” samples should be doc-
umented by digital or film photography, andconfirmation by conventional PCR and/or real-time PCR of a parallel sample (frozen, but notfixed) is advisable.
Table 6A.5.6 outlines some of the morecommon problems that may be experienced inperforming the basic and alternate protocolsfrom this unit. This is not an exhaustive list;others may be encountered. Please consult thetroubleshooting sections from the referencedunits of Current Protocols in Molecular Biol-ogy (see Literature Cited) for further advice.
Anticipated ResultsV. cholerae can be easily isolated from estu-
arine environments during the warm summermonths, even in non-epidemic areas. Chancesfor successful isolation decrease during thecolder winter months, even from samples thatare positive by PCR or DFA. Figure 6A.5.3shows the typical growth of V. cholerae onTCBS, TTGA, and CA media. Growth onTCBS by V. cholerae appears as small (1 to3-mm diameter), flat, yellow colonies (Fig.6A.5.3). V. cholerae appear as medium to large(2 to 5-mm diameter), flat, translucent colonieson TTGA (Fig. 6A.5.3). There will be a zoneof clearing (or halo) surrounding V. choleraecolonies due to hydrolysis of gelatin. A darkcenter usually develops after 24 hr. Growth onCA by V. cholerae appears as medium to large(2- to 5-mm diameter) turquoise colonies.However, V. cholerae is a highly heteroge-neous species and can exhibit various colonymorphologies including a rugose form (White,1940). Several different V. cholerae isolatesshould be used as positive controls to be com-parators for cultured cells. TCBS, TTGA, andCA are all selective, but still allow the growthof other Vibrio species and related bacteria.For example, V. parahaemolyticus will appearas blue-green (occasionally yellow) colonies,V. vulnificus will appear as greenish yellowcolonies, and V. alginolyticus as larger, some-times swarming yellow colonies on TCBS.Therefore, colonies growing on these mediaare not necessarily all V. cholerae; hence, theyare labeled as presumptive until they can beconfirmed by PCR. If several of these solidmedia are available to the investigator, then itmay be beneficial to “dot” or streak for isola-tion presumptive V. cholerae colonies isolatedfrom one medium onto another that is selec-tive for V. cholerae as well (TCBS → CAand TTGA, for instance). Visualizing the colorand morphology of an isolate on one selectivemedium that was recovered from a differentselective medium may help the investigator

Detection of Vibriocholerae from the
Environment
6A.5.46
Supplement 26 Current Protocols in Microbiology
Table 6A.5.6 Common Problems that May be Experienced in Detection, Isolation, and Identification of V. choleraefrom the Environment
Problem Possible solution(s)
Basic Protocol 1
Little or no plankton in cod-endcollecting bucket
Check pore size of plankton net and ensure that it is 64 mm
Zooplankton should be sampled near dawn or dusk (1-2 hr after sunrise orbefore sunset) when they are nearer to the surface.
Filter more water through plankton net
Basic Protocol 2
No V. cholerae growth Adjust dilution volumes and/or incubation temperature
Incubate petri dishes at several temperatures
V. cholerae overgrowth Adjust dilutions or use a smaller volume loop to spread onto agar
Basic Protocol 3
Autoagglutination or clumping in salinewithout antisera
“Rough” morphotypes cannot be serogrouped with antisera. Use O1/O139rfb PCR primers with Basic Protocol 4 to test for toxigenic serogroups.
Support Protocol 1
Low DNA concentration Increase volume of boiled cells
Basic Protocol 4
No PCR product with positive control Ensure all components are added to reaction at the proper concentration
Use fresh dNTPs
Prepare fresh crude template of positive control as it will degrade over time
Dilute crude template 1:5000 or more and repeat the control reaction
Quantify crude template by gel electrophoresis (∼10 ml) to ensuresufficient template concentration
PCR product with negative control Most likely caused by carry-over contamination in one of the reactioncomponents. Make new components.
Basic Protocols 6 and 7
Weak amplification with positive control Ensure all components are added to reaction at the proper concentration
Use fresh dNTPs
Prepare fresh crude template of positive control as it will degrade over time
Dilute crude template 1:5000 or more and repeat the control reaction
Quantify crude template by gel electrophoresis (∼10 ml) to ensuresufficient template concentration
Strong amplification with negativecontrol
Most likely caused by carry-over contamination in one of the reactioncomponents. Make new components.
Basic Protocol 9
Positive control is negative Ensure solutions used are RNase-free
Increase incubation time of lysis step (10% SDS), especially if colonies arelarger than 3 mm
Ensure that the correct microscope filter is used for fluorochrome selected.Other fluorochromes may be used.
continued

NonentericGammaProteobacteria
6A.5.47
Current Protocols in Microbiology Supplement 26
Table 6A.5.6 Common Problems that May be Experienced in Detection, Isolation, and Identification of V. choleraefrom the Environment, continued
Problem Possible solution(s)
Positive control gives weak signal Check scanning settings on detection instrument
Increase probe concentration and/or hybridization time
Alternate Protocol 3
Positive control is negative or weak Ensure that solutions are used in correct order
Overexposure to UV source will degrade DNA template. Consider usingpositively charged nylon membranes, which do not need cross-linking.
Extend hybridization time
Extend development time
Positive control is overdeveloped orbackground is high
Check hybridization temperature
Do not allow membrane to dry
Decrease development time
Positive control is negative or weak Check efficiency of probe labeling reaction
Increase probe concentration (≤ 25 ng/ml)
Basic Protocol 10
Weak fluorescence Increase probe concentration
Check hybridization temperature
Increase hybridization time
Nonspecific staining Perform more stringent wash step
Basic Protocol 11
No PCR product with positive control Ensure all components are added to reaction at the proper concentration
Use fresh dNTPs
Prepare fresh crude template of positive control as it will degrade over time
Dilute crude template 1:5000 or more and repeat the control reaction
Quantify crude template by gel electrophoresis (∼10 ml) to ensuresufficient template concentration
PCR product with negative control Most likely caused by carry-over contamination in one of the reactioncomponents. Make new components.
Positive control is negative Ensure that the proper filter set is used on the fluorescent microscope
Bengal DFA (V. cholerae O139) kit positive control is sometimes poor.Prepare positive control from laboratory reference strain.
Basic Protocol 12
Positive control signal is weak Increase amount of V. cholerae O1 antiserum and FITC conjugate

Detection of Vibriocholerae from the
Environment
6A.5.48
Supplement 26 Current Protocols in Microbiology
determine whether or not to proceed with PCRconfirmation of that isolate.
Table 6A.5.4 lists a comprehensive, yet notexhaustive set of PCR primers used to charac-terize V. cholerae isolates. This table also liststhe reaction temperatures and times as well asthe expected amplicon size. Positive controlstrains known to encode the target region andyielding an appropriate size amplicon shouldbe used for all PCR reactions.
Time ConsiderationsChoosing the appropriate methods dis-
cussed in this unit will determine how rapidlyresults can be achieved. Clinical diagnosisrequires rapid determination of the etiologi-cal agent, and rapid non-culturing techniquesare currently being developed. In the case ofcholera, symptoms are often distinguishablefrom other infections, and stool typically re-quires no enrichment in APW and readilyyields V. cholerae colonies on TCBS agar. En-vironmental monitoring often requires moretime, depending on the method. However, PCRscreening can be performed in 6 to 24 hr, de-pending on the template, and DFA can be donein 4 hr or 24 hr if coupled with DVC. Use ofmultiple methods and genotypic characteriza-tion will significantly increase the amount oftime needed to complete this work. In-depthgenotypic characterization can be done in in-tervals after several rounds of sample collec-tion and V. cholerae isolation. It is importantto confirm any presumptive strain as beingV. cholerae before time and resources are ded-icated to genotypic characterization. As withany proposed work, a daily schedule with esti-mated timeframes may be helpful in efficientlyaccomplishing the investigators’ goals.
AcknowledgementsThis work was partially supported by Na-
tional Science Foundation Grant 0813066,NIH Grant 2RO1A1039129-11A2, NationalInstitutes of Health-Fogarty International Cen-ter Grant 1RC1TW008587-01, U.S. ArmyMedical Research and Material Com-mand Grant W81XWHO9201, NOAA GrantS0660009, and DHS Contract HSHQDC-10-C-00177.
Literature CitedAlam, M., Sadique, A., Nur-, A-Hasan, Bhuiyan,
N.A., Nair, G.B., Siddique, A.K., Sack, D.A.,Ahsan, S., Huq, A., Sack, R.B., and Colwell,R.R. 2006a. Effect of transport at ambienttemperature on detection and isolation of Vib-rio cholerae from environmental samples. Appl.Environ. Microbiol. 72:2185-2190.
Alam, M., Hasan, N.A., Sadique, A., Bhuiyan,N.A., Ahmed, K.U., Nusrin, S., Nair, G.B.,Siddique, A.K., Sack, R.B., Sack, D.A., Huq,A., and Colwell, R.R. 2006b. Seasonal choleracaused by Vibrio cholerae serogroups O1 andO139 in the coastal aquatic environment ofBangladesh. Appl. Environ. Microbiol. 72:4096-4104.
Amann, R.I., Ludwig, W., and Schleifer, K.H. 1995.Phylogenetic identification and in situ detectionof individual microbial cells without cultivation.Microbiol. Mol. Biol. 59:143-169.
Bhattacharya, T., Chatterjee, S., Maiti, D., Bhadra,R.K., Takeda, Y., Nair, G.B., and Nandy, R.K.2006. Molecular analysis of the rstR and orfUgenes of the CTX prophages integrated in thesmall chromosomes of environmental Vibriocholerae non-O1, non-O139 strains. Environ.Microbiol. 8:526-634.
Binsztein, N., Costagliola, M.C., Pichel, M.,Jurquiza, V., Ramırez, F.C., Akselman, R.,Vacchino, M., Huq, A., and Colwell, R. 2004.Viable but nonculturable Vibrio cholerae O1 inthe aquatic environment of Argentina. Appl. En-viron. Microbiol. 70:7481-7486.
Blackstone, G.M., Nordstrom, J.L., Bowen, M.D.,Meyer, R.F., Imbro, P., and DePaola, A. 2007.Use of a real time PCR assay for detection of thectxA gene of Vibrio cholerae in an environmen-tal survey of Mobile Bay. J. Microbiol. Methods68:254-259.
Boyd, E.F. and Waldor, M.K. 2002. Evolutionaryand functional analyses of variants of the toxin-coregulated pilus protein TcpA from toxigenicVibrio cholerae non-O1/non-O139 serogroupisolates. Microbiology 148:1655-1666.
Boyle, A. and Perry-O’Keefe, H. 1992. Label-ing and colorimetric detection of nonisotopicprobes. Curr. Protoc. Mol. Biol. 20:3.18.1-3.18.9.
Brayton, P.R., Roszak, D.B., Palmer, L.M., Huq,S.A., Grimes, D.J., and Colwell, R.R. 1986. Flu-orescent antibody enumeration of V. cholerae inthe marine environment. IFREMER 3:507-514.
Cash, R., Music, S.I., Libonati, J.P., Snyder, M.J.,Wenzel, R.P., and Hornick, R.B. 1974. Responseof man to infection with Vibrio cholerae I. Clin-ical, serologic, and bacteriologic responses to aknown inoculum. J. Infect. Dis. 129:45-52.
Chakraborty, S., Mukhopadhyay, A.K., Bhadra,R.K., Ghosh, A.N., Mitra, R., Shimada, T., Ya-masaki, S., Faruque, S.M., Takeda, Y., Colwell,R.R., and Nair, G.B. 2000. Virulence genes inenvironmental strains of Vibrio cholerae. Appl.Environ. Microbiol. 66:4022-4028.
Chatterjee, S., Ghosh, K., Raychoudhuri,A., Chowdhury, G., Bhattacharya, M.K.,Mukhopadhyay, A.K., Ramamurthy, T., Bhat-tacharya, S.K., Klose, K.E., and Nandy, R.K.2009. Incidence, virulence factors, and clonal-ity among clinical strains of non-O1, non-O139Vibrio cholerae isolates from hospitalizeddiarrheal patients in Kolkata, India. J. Clin.Microbiol. 47:1087-1095.

NonentericGammaProteobacteria
6A.5.49
Current Protocols in Microbiology Supplement 26
Choopun, N., Louis, V., Huq, A., and Colwell, R.R.2002. Simple procedure for rapid identificationof Vibrio cholerae from the aquatic environ-ment. Appl. Environ. Microbiol. 68:995-998.
Chow, K.H., Ng, T.K., Yuan, K.Y., and Yam, W.C.2001. Detection of RTX toxin gene in Vibriocholerae by PCR. J. Clin. Microbiol. 39:2594-2597.
Chowdhury, M.A.R., Hasan, J.A.K., Huq, A., Xu,B., and Montilla, R. 1994. DVC-DFA: A sim-plified technique for detection of viable Vib-rio cholerae O1 and O139. Can. J. Microbiol.42:87-93.
Chun, J., Huq, A., and Colwell, R.R. 1999. Analy-sis of 16S-23S rRNA intergenic spacer regionsof Vibrio cholerae and Vibrio mimicus. Appl.Environ. Microbiol. 65:2202-2208.
Clesceri, L., Greenberg, A., and Eaton, A. (eds.).1998. Standard methods for the examination ofwater and wastewater. American Public HealthAssociation, Washington D.C.
Colwell, R. 1991. Viable but non-culturable bacte-ria in the aquatic environment. Culture 12:1-4.
Colwell, R., Tamplin, M. and Brayton, P.R. 1990.Environmental aspects of Vibrio cholerae intransmission of cholera. In Advances in Re-search on Cholera and Related Diarrhoeas (R. B.Sack and Y. Zinnaka, eds.) pp. 327-343. K.T.K.Scientific Publishers, Tokyo.
Constantin de Magny, G., Murtugudde, R., Sapi-ano, M.R., Nizam, A., Brown, C.W., Busalacchi,A.J., Yunus, M., Nair, G.B., Gil, A.I., Lanata,C.F., Calkins, J., Manna, B., Rajendran, K.,Bhattacharya, M.K., Huq, A., Sack, R.B., andColwell, R.R. 2008. Environmental signaturesassociated with cholera epidemics. Proc. Natl.Acad. Sci. U.S.A. 105:17676-17681.
Dallas, W. and Falkow, S. 1980. Amino acid se-quence homology between cholera toxin and Es-cherichia coli heat-labile toxin. Nature 288:499-501.
Dziejman, M., Serrutom, D., Tam, V.C., Sturte-vant, D., Diraphat, P., Faruque, S.M., Rahman,M.H., Heidelberg, J.F., Decker, J., Li, L., Mont-gomery, K.T., Grills, G., Kucherlapati, R., andMekalanos, J.J. 2005. Genomic characterizationof non-O1, non-O139 Vibrio cholerae revealsgenes for a type III secretion system. Proc. Natl.Acad. Sci. U.S.A. 102:3465-3470.
Faruque, S.M., Chowdhury, N., Kamruzzaman, M.,Ahmad, Q.S., Faruque, A.S.G., Abdus Salam,M., Ramamurthy, T., Nair, G.B., Weintraub, A.,and Sack, D. 2003. Reemergence of epidemicVibrio cholerae O139, Bangladesh. Emerg. In-fect. Dis. 9:1116-1122.
Fasano, A., Baudry, B., and Pumplin, D.W. 1991.Vibrio cholerae produces a second enterotoxinwhich affects intestinal tight junctions. Proc.Natl. Acad. Sci. U.S.A. 88:5242-5246.
Fields, P.I., Popovic, T., Wachsmuth, K., andOlsvik, Ø. 1992. Use of polymerase chain re-action for detection of toxigenic Vibrio choleraeO1 strains from the Latin American cholera epi-demic. J. Clin. Microbiol. 30:2118-2121.
Grim, C.J., Choi, J., Chun, J., Jeon, Y.S., Taviani, E.,Hasan, N.A., Haley, B., Huq, A., and Colwell,R.R. 2010. Occurrence of the Vibrio choleraeseventh pandemic VSPI island and a new vari-ant. OMICS 14:1-7.
Gubala, A.J. 2006. Multiplex real-time PCR detec-tion of Vibrio cholerae. J. Microbiol. Methods65:278-293.
Gubala, A. and Proll, D. 2006. Molecular-beaconmultiplex real-time PCR assay for detectionof Vibrio cholerae. Appl. Environ. Micbrobiol.72:6424-6428.
Haley, B.J., Chen, A., Grim, C.J., Clark, P., Diaz,C.M., Taviani, E., Hasan, N.A., Sancomb, E.,Elnemr, W.M., Islam, M.A., Huq, A., Col-well, R.R., and Benediktsdottir, E. 2012. Vibriocholerae in an historically cholera-free country.Environ. Microbiol. Rep. doi:10.1111/j.1758-2229.2012.00332.x.
Hasan, J., Loomis, L. Huq, A., Bernstein, D., Tam-plin, M.L., and Wier, M. 1992. Development ofa fast, simple, and sensitive immunoassay to de-tect Vibrio cholerae O1 from clinical samplesusing SMARTTM Kit. 92nd Genl. Mtg Am. Soc.Microbiol., New Orleans, Louisiana.
Hasan, J.A.K., Bernstein, D., Huq, A., Loomis, L.,Tamplin, M.L., and Colwell, R.R. 1994. CholeraDFA: An improved direct fluorescent mono-clonal antibody staining kit for rapid detectionand enumeration of V. cholerae O1. FEMS Mi-crobiol. Lett. 120:143-148.
Hasan, J., Huq, A., Nair, G.B., Garg, S., Mukhopad-hyay, A.K., Loomis, L., Bernstein, D., and Col-well, R.R. 1995. Development and testing ofmonoclonal antibody-based rapid immunodiag-nostic test kits for direct detection of Vibriocholerae O139 synonym Bengal. J. Clin. Mi-crobiol. 33:2935-2939.
Heidelberg, J. 1997. Seasonal abundance of bacteri-oplankton populations of bacteria, gamma pro-teobacteria, Vibrio/Photobacterium, Vibrio vul-nificus, Vibrio cholerae/Vibrio mimicus, and Vib-rio cincinnatiensis associated with zooplank-ton in the Choptank River, Maryland. Ph.D.thesis. University of Maryland, College Park,Maryland.
Heidelberg, J., Heidelberg, K., and Colwell, R.R.2002. Seasonality of Chesapeake Bay bacte-rioplankton species. Appl. Environ. Microbiol.68:5488-5497.
Hoshino, K., Yamasaki, S., Mukhopadhyay, A.K.,Chakraborty, S., Basu, A., Bhattacharya, S.K.,Nair, G.B., Shimada, T., and Takeda, Y. 1998.Development and evaluation of a multitest PCRassay for rapid detection of toxigenic Vibriocholerae O1 and O139. FEMS Immunol. Med.Microbiol. 20:201-207.
Huq, A., Small, E.B., West, P.A., and Colwell, R.R.1984. The role of planktonic copepods in thesurvival and multiplication of Vibrio choleraein the aquatic environment. In Vibrios in theEnvironment (R.R. Colwell, ed.) pp. 521-534.John Wiley & Sons, New York
Huq, A., Colwell, R.R., Rahman, R., Ali, A.,Chowdhury, M.A., Parveen, S., Sack, D.A., and

Detection of Vibriocholerae from the
Environment
6A.5.50
Supplement 26 Current Protocols in Microbiology
Russek-Cohen, E. 1990. Detection of Vibriocholerae O1 in the aquatic environment by fluo-rescent monoclonal antibody and culture meth-ods. Appl. Environ. Microbiol. 56:2370-2373.
Huq, A., Sack, R.B., Nizam, A., Longini, I.M.,Nair, G.B., Ali, A., Morris, J.G. Jr., Khan, M.N.,Siddique, A.K., Yunus, M., Albert, M.J., Sack,D.A., and Colwell, R.R. 2005. Critical factorsinfluencing the occurrence of Vibrio cholerae inthe environment of Bangladesh. Appl. Environ.Microbiol. 718:4645-4654.
Huq, A., Grim, C., Colwell, R.R., and Nair, G.B.2006. Detection, isolation, and identification ofVibrio cholerae from the environment. Curr.Protoc. Microbiol. 2:6A.5.1-6A.5.38.
Islam, M., Rahman, M.Z., Khan, S.I., Mahmud,Z.H., Ramamurthy, T., Nair, G.B., Sack, R.B.,and Sack, D.A. 2005. Organization of the CTXprophage in environmental isolates of Vibriomimicus. Microbiol. Immunol. 49:779-784.
Kaper, J., Morris, J., and Nishibuchi, M. 1988. DNAprobes for pathogenic Vibrio species. In DNAProbes for Infectious Diseases (F. Tenover, ed.)pp. 65-77. CRC Press, Boca Raton, Fla.
Keasler, S.P. and Hall, R.H. 1993. Detecting andbiotyping Vibrio cholerae O1 with multiplexpolymerase chain reaction. Lancet 341:1661.
Kenyon, J.E., Piexoto, D.R., Austin, B., andGillies, D.C. 1984. Seasonal variations of Vib-rio cholerae (non-O1) isolated from Califor-nia coastal waters. Appl. Environ. Microbiol.47:1243-1245.
Kogure, K., Simidu, U., and Taga, N. 1979. A tenta-tive direct microscopic method for counting liv-ing marine bacteria. Can. J. Microbiol. 25:415-420.
Kramer, M.F. and Coen, D.M. 2001. Enzymaticamplification of DNA by PCR: Standard proce-dures and optimization. Curr. Protoc. Mol. Biol.56:15.1.1-15.1.14.
Lin, W., Fullner, K.J., Clayton, R., Sexton, J.A.,Rogers, M.B., Calia, K.E., Calderwood, S.B.,Fraser, C., and Mekalanos, J.J. 1999. Identi-fication of a Vibrio cholerae RTX toxin genecluster that is tightly linked to the cholera toxinprophage. Proc. Natl. Acad. Sci. U.S.A. 96:1071-1076.
Lizarraga-Partida, M.L., Wong-Chang, I. et al. Vib-rio cholerae O1 and non-O1 in two coastal la-goons of Veracruz. Manuscript in preparation.
Louis, V., Russek-Cohen, E., Choopun, N., Rivera,I.N., Gangle, B., Jiang, S.C., Rubin, A., Patz,J.A., Huq, A., and Colwell, R.R. 2003. Pre-dictability of Vibrio cholerae in ChesapeakeBay. Appl. Environ. Microbiol. 69:2773-2785.
Lyon, W.J. 2001. TaqMan PCR for detection of Vib-rio cholerae O1, O139, non-O1, and non-O139in pure cultures, raw oysters, and synthetic sea-water. Appl. Environ. Microbiol. 67:4685-4693.
Malayil, L., Turner, J.W.,et al. 2010. Novel reser-voirs of the CTX gene. In Vibrios in the Environ-ment. Conference Proceedings, Biloxi, Miss.
Monsur, K. 1961. A highly selective gelatin-taurocholate-tellurite medium for the isolationof Vibrio cholerae. Trans. R. Soc. Trop. Med.Hyg. 55:440-442.
Moore, D., Dowhan, D., Chory, J., and Ribaudo,R.K. 2002. Isolation and purification of largeDNA restriction fragments from agarose gels.Curr. Protoc. Mol. Biol. 59:2.6.1-2.6.12.
Morris, G., Merson, M.H., Huq, I., Kibrya, A.K.,and Black, R. 1979. Comparison of four plat-ing media for isolating Vibrio cholerae. J. Clin.Microbiol. 9:79-83.
Murray, M. and Thompson, W. 1980. Rapid isola-tion of high molecular weight plant DNA. Nu-cleic Acids Res. 8:4321-4325.
Nandi, B., Nandy, R.K., Mukhopadhyay, S., Nair,G.B., Shimada, T., and Ghose, A.C. 2000. Rapidmethod for species-specific identification of Vib-rio cholerae using primers targeted to the geneof outer membrane protein OmpW. J. Clin. Mi-crobiol. 38:4145-4151.
Nusrin, S., Khan, G.Y., Bhuiyan, N.A., Ansaruz-zaman, M., Hossain, M.A., Safa, A., Khan,R., Faruque, S.M., Sack, D.A., Hamabata, T.,Takeda, Y., and Nair, G.B. 2004. Diverse CTXphages among toxigenic Vibrio cholerae O1 andO139 strains isolated between 1994 and 2002 inan area where cholera is endemic in Bangladesh.J. Clin. Microbiol. 42:5854-5856.
O’Brien, M. and Colwell, R.R. 1985. Modifiedtaurochalate-tellurite-gelatin agar for improveddifferentiation of Vibrio species. J. Clin. Micro-biol. 22:1011-1013.
Ogawa, A., Kato, J., Watanabe, H., Nair, B.G., andTakeda, T. 1990. Cloning and nucleotide se-quence of a heat-stable enterotoxin gene fromVibrio cholerae non-O1 isolated from a patientwith traveler’s diarrhea. Infect. Immun. 58:3325-3329.
Onifade, T.J., Hutchinson, R., Van Zile, K.,Bodager, D., Baker, R., and Blackmore, C.2011. Toxin producing Vibrio cholerae O75 out-break, United States, March to April 2011. Euro.Surveill. 16:19870.
O’Shea, Y.A. and Boyd, E.F. 2002. Mobilizationof the Vibrio pathogenicity island between Vib-rio cholerae isolates mediated by CP-T1 gen-eralized transduction. FEMS Microbiol. Lett.214:153-157.
O’Shea, Y., Reen, F.J., Quirke, A.M., and Boyd,E.F. 2004. Evolutionary genetic analysis of theemergence of epidemic Vibrio cholerae isolateson the basis of comparative nucleotide sequenceanalysis and multilocus virulence gene profiles.J Clin. Microbiol. 42:4657-4671.
Pal, A., Ramamurthy, T., Bhadra, R.K., Takeda, T.,Shimada, T., Takeda, Y., Nair, G.B., Pal, S.C.,and Chakrabarti, S. 1992. Reassessment of theprevalence of heat-stable enterotoxin (NAG-ST)among environmental Vibrio cholerae non-O1strains isolated from Calcutta, India, by using aNAG-ST DNA probe. Appl. Environ. Microbiol.58:2485-2489.

NonentericGammaProteobacteria
6A.5.51
Current Protocols in Microbiology Supplement 26
Ramamurthy, T., Albert, M.J., Huq, A., Colwell,R.R, Takeda, Y., Takeda, T., Shimada, T., Man-dal, B.K., and Nair, G.B. 1994. Vibrio mimicuswith multiple toxin types isolated from humanand environmental sources. J Med Microbiol.40:194-196.
Rawlings, T.K., Ruiz, G.M., and Colwell, R.R.2007. Association of Vibrio cholerae O1 El Torand O139 Bengal with the copepods Acartiatonsa and Eurytemora affinis. Appl. Environ. Mi-crobiol. 73:7926-7933.
Rehnstam, A.-S., Norqvist, A., Wolf-Watz, H.,and Hagstrom, A. 1989. Identification of Vib-rio anguillarum in fish by using partial 16SrRNA sequences and a specific 16S rRNAoligonucleotide probe. Appl. Environ. Micro-biol. 55:1907-1910.
Rennels, M., Levine, M.M., Daya, V., Angle, P.,and Young, C. 1980. Selective vs. nonselectivemedia and direct plating vs. enrichment tech-nique in isolation of Vibrio cholerae: Recom-mendations for clinical laboratories. J. Infect.Dis. 142:328-331.
Rivera, I., Chun, J., Huq, A., Sack, R., and Colwell,R.R. 2001. Genotypes associated with virulencein environmental isolates of Vibrio cholerae. J.Appl. Environ. Microbiol. 67:2421-2429.
Roszak, D. and Colwell, R. 1987. Survival strate-gies of bacteria in the natural environment. Mi-crobiol. Rev. 51:365-379.
Rubin, E.J., Lin, W., Mekalanos, J.J., and Waldor,M.K. 1998. Replication and integration of a Vib-rio cholerae cryptic plasmid linked to the CTXprophage. Mol. Microbiol. 28:1247-1254.
Sack, R., Siddique, A.K., Longini, I.M. Jr., Nizam,A., Yunus, M., Islam, M.S., Morris, J.G. Jr., Ali,A., Huq, A., Nair, G.B., Qadri, F., Faruque, S.M.,Sack, D.A., and Colwell, R.R. 2003. A 4-yearstudy of the epidemiology of Vibrio cholerae infour rural areas of Bangladesh. J. Infect. Dis.187:96-101.
Schuster, B.M., Tyzik, A.L., Donner, R.A., Striplin,M.J., Almagro-Moreno, S., Jones, S.H., Cooper,V.S., and Whistler, C.A. 2011. Ecology and ge-netic structure of a northern temperate Vibriocholerae population related to toxigenic isolates.Appl. Environ. Microbiol. 77:7568-7575.
Shirai, U.N., Nishibuchi, M., and Ramamurthy, T.1991. Polymerase chain reaction for detectionof the cholera toxin operon of Vibrio cholerae.J. Clin. Microbiol. 29:2517-2521.
Spira, W., Huq, A., Ahmed, Q.S., and Saeed, Y.A.1981. Uptake of Vibrio cholerae biotype eltorfrom contaminated water by water hyacinth (Ei-chornia crassipes). Appl. Environ. Microbiol.42:550-553.
Taviani, E., Grim, C.J., Choi, J., Chun, J. Ha-ley, B., Hasan, N.A., Huq, A., and Colwell,R.R. 2010. Discovery of novel Vibrio choleraeVSP II genomic islands using comparative ge-nomic analysis. FEMS Microbiol. Lett. 308:130-137.
Trucksis, M., Galen, J.E., Michalski, J., Fasano, A.,and Kaper, J.B. 1993. Accessory cholera toxin(Ace), the third toxin of a Vibrio cholerae vir-ulence cassette. Proc. Natl. Acad. Sci. U.S.A.90:5267-5271.
Turner, J.W., Good, B., Cole, D., and Lipp, E.K.2009. Plankton composition and environmentalfactors contribute to Vibrio seasonality. ISME J.3:1082-1092.
Vora, G.J., Meador, C.E., Bird, M.M., Bopp,C.A., Andreadis, J.D., and Stenger, D.A. 2005.Microarray-based detection of genetic hetero-geneity, antimicrobial resistance, and the viablebut nonculturable state in human pathogenicVibrio spp. Proc. Natl. Acad. Sci. U.S.A.102:19109-19114.
Voytas, D. 2000. Agarose gel electrophoresis. Curr.Protoc. Mol. Biol. 51:2.5A.1-2.5A.9.
Waldor, M.K., Rubin, E.J., Pearson, G.D., Kimsey,H., and Mekalanos, J.J. 1997. Regulation, repli-cation, and integration functions of the Vibriocholerae CTXphi are encoded by region RS2.Mol. Microbiol. 24:917-926.
White, P. 1940. The characteristic hapten andantigen of rugose races of cholera and ElTor vibrios. J. Pathol. Bacteriol. 50:160-164.
Xu, H.S., Roberts, N.C., Singleton, F.L., Attwell,R.W., Grimes, D.J., and Colwell, R.R. 1982.Survival and viability of nonculturable Es-cherichia coli and Vibrio cholerae in the estu-arine and marine environment. Microbiol. Ecol.8:313-323.
Xu, H.S., Roberts, N.C., Adams, L.B., West, P.A.,Siebeling, R.J., Huq, A., Huq, M.I., Rahmand,R., and Colwell, R.R. 1984. An indirect fluores-cent antibody staining procedure for detection ofVibrio cholerae serovar O1 cells in aquatic envi-ronmental samples. J. Microb. Methods 2:221-231.


















