
(8!15!13) Free Radical Injury, Acute & Chronic Inflammation OUTLINE
10
SRB 1 Pathology: Free Radical Injury, Acute and Chronic Inflammation August 15 | Dr. DeRisio (and Dr. Sattar) Free Radical Injury Free radical = chemical species w/ unpaired electron in outer orbit that can do damage within the cell Physiologic Generation of Free Radicals Occurs during Oxidative Phosphorylation Cytochrome c oxidase transfers electrons to O 2 as final e - acceptor during ox phos o Drives proton gradient which drives production of ATP o O 2 accepts 4 electrons, after which water is generated If it doesn’t accept all 4, free radicals are generated: If it accepts 1 superoxide (O 2 ∙ - ) If it accepts 2 peroxide (H 2 O 2 ) If it accepts 3 hydroxyl ion (∙OH) radicals If it accepts 4 water (H 2 O) Pathogenic Generation of Free Radicals Ionizing radiation – radiation hits water in tissues causing formation of hydroxyl free radical (∙OH) o Of all free radicals, hydroxyl free radical is the most damaging Inflammation o When neutrophils come in to battle an infection, two mechanisms can happen to kill microbe: (1) Oxygen-dependent rxn starts w/ “oxidative burst”; free radicals are generated O 2 is acted on by NADPH oxidase to become superoxide Superoxide is acted on by superoxide dismutase to hydrogen peroxide Hydrogen peroxide is acted on by myloperoxidase to become bleach (HOCl) (2) Oxygen-independent Metals (ie: copper and iron) o Copper and iron are usually tightly bound in the body Iron is bound as soon as it enters the blood b/c when free, free radicals are generated Fenton reaction allows Fe to generate hydroxyl free radical (most damaging) o If copper or iron builds up, we get diseases Fe buildup = hemochromatosis Tissue damage (cirrhosis of liver); excess iron generates free radicals Cu buildup = Wilson’s disease Tissue damage bc of generation of free radicals Sample question: What’s the underlying mechanism of damage in hemochromatosis or in Wilson’s Disease? Pathologic generation of free radicals Drugs and chemicals o Acetaminophen – goes to liver, gets converted by P450 and free radicals are generated High dose acetaminophen can be taken to commit suicide via liver necrosis, hepatocytes convert it and free radicals are generated o CCl 4 (see below)
description
free radical
Transcript of (8!15!13) Free Radical Injury, Acute & Chronic Inflammation OUTLINE

SRB 1
Pathology: Free Radical Injury, Acute and Chronic Inflammation
August 15 | Dr. DeRisio (and Dr. Sattar)
Free Radical Injury
Free radical = chemical species w/ unpaired electron in outer orbit that can do damage within the cell
Physiologic Generation of Free Radicals Occurs during Oxidative Phosphorylation
Cytochrome c oxidase transfers electrons to O2 as final e- acceptor during ox phos
o Drives proton gradient which drives production of ATP
o O2 accepts 4 electrons, after which water is generated
If it doesn’t accept all 4, free radicals are generated:
If it accepts 1 superoxide (O2∙-)
If it accepts 2 peroxide (H2O2)
If it accepts 3 hydroxyl ion (∙OH) radicals
If it accepts 4 water (H2O)
Pathogenic Generation of Free Radicals
Ionizing radiation – radiation hits water in tissues causing formation of hydroxyl free radical (∙OH)
o Of all free radicals, hydroxyl free radical is the most damaging
Inflammation
o When neutrophils come in to battle an infection, two mechanisms can happen to kill microbe:
(1) Oxygen-dependent rxn starts w/ “oxidative burst”; free radicals are generated
O2 is acted on by NADPH oxidase to become superoxide
Superoxide is acted on by superoxide dismutase to hydrogen peroxide
Hydrogen peroxide is acted on by myloperoxidase to become bleach (HOCl)
(2) Oxygen-independent
Metals (ie: copper and iron)
o Copper and iron are usually tightly bound in the body
Iron is bound as soon as it enters the blood b/c when free, free radicals are generated
Fenton reaction allows Fe to generate hydroxyl free radical (most damaging)
o If copper or iron builds up, we get diseases
Fe buildup = hemochromatosis
Tissue damage (cirrhosis of liver); excess iron generates free radicals
Cu buildup = Wilson’s disease
Tissue damage bc of generation of free radicals
Sample question: What’s the underlying mechanism of damage in hemochromatosis or in
Wilson’s Disease? Pathologic generation of free radicals
Drugs and chemicals
o Acetaminophen – goes to liver, gets converted by P450 and free radicals are generated
High dose acetaminophen can be taken to commit suicide via liver necrosis, hepatocytes convert
it and free radicals are generated
o CCl4 (see below)

SRB 2
Free Radical Damage
Peroxidation of lipids (lipid membrane)
Oxidation of DNA and proteins
o Results in damage to the cell
Particularly important in oncogenesis
o Antioxidants fight off free radicals to prevent cancer and fight off aging
Elimination of Free Radicals
Antioxidants
Vitamin A, C, E
Enzymes
(1) Superoxide dismutase (SOD): removes superoxide
(2) Catalase: removes peroxide
(3) Glutathione peroxidase: removes hydroxyl ions
Metal carrier proteins
Copper and iron can create free radicals (iron via Fenton reaction)
o Thus, we have carrier proteins to hide the metals to free radicals aren’t generated
Transferrin in blood tightly binds Fe
Ferratin in liver tightly binds Fe
Free Radical Injury
CCl4 (carbon tetrachloride)
Found in dry cleaning industry
When in blood, converted to CCl3∙ in the P450 system of the liver
o CCl3∙ is a free radical that causes reversible damage (cell swelling)
When cell swells, rough ER will also swell and ribosomes
will pop off to reduce protein synthesis
Decrease in protein synthesis results in lack of
apolipoproteins, which remove fat from liver
Fat comes in the liver but can’t leave classic presentation = FATTY LIVER
o Image above: red circle indicates hepatocyte, white bubbles are fat cells
Reperfusion Injury
When blood supply to an organ is cut off, tissue dies and cell membrane is damaged
o Ex: coronary artery occlusion heart tissue dies enzymes (troponins) leak into blood
If blood is returned to the organ, oxygen + inflammatory cells return with it
o Inflammatory cells reacting w/dead tissue + presence of oxygen = free radicals are generated
Free radicals further damage cardiac myocytes cardiac enzymes continue to rise after opening
artery/returning blood to injured organ (classic manifestation of reperfusion injury)
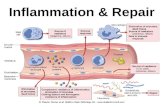
Inflammation
Definition = taking cells FROM within a blood vessel and bring them OUT to the tissue (usually in response to infection)
Acute inflammation: when neutrophils leave blood vessel into tissue space
Chronic inflammation: when lymphocytes leave blood vessel and into tissue space
REMEMBER THIS:
If O2 accepts 1 e- superoxide (O2∙
-)
If O2 accepts 2 e- peroxide (H2O2
)
If O2 accepts 3 e- hydroxyl ion (∙OH)
If O2 accepts 4 e- water (H2O)

SRB 3
BV BV N N
N N
E
E
Acute Inflammation
Characterized by Presence of (1) Edema and (2) Neutrophils in Tissue
Edema (E): fluid from blood vessels (BV) that fills the interstitial space
Neutrophils (N): key inflammatory cells that leave blood vessels (BV)
and get into tissue space (hallmark of acute inflammation)
Arises in Response to (1) Infection or (2) Tissue Necrosis
Goal is to eliminate pathogen or clear necrotic debris
o Neutrophils destroy by eating/consuming the infectious pathogen
Difference between necrosis v. apoptosis: necrosis is followed by acute inflammation
Ex: myocardial infarction causes increase in patients white count (↑ neutrophils)
o 12 hours after
o 24 hours after: neutrophils and acute inflammation
Immediate Response with Limited Specificity
Response is immediate, caveat is that it has limited specificity
o Generalized response to stimulus, not a specific attack to a particular antigen
Innate immunity = includes: epithelium covering body surfaces, mucous secreted by cells, complement system
(series of protein in inactive state in serum that can be activated), mast cells, macrophages (consume pathogens
and present them to further immune response), neutrophils, basophils, eosinophils; broad, nonspecific system to
defend host against microbes
Adaptive immunity = longer but more specific response; includes: lymphocytes (produce Abs and have T-cell
receptors for a specific target)
Mediated by Several Factors
Toll-like Receptors (TLRs)
Present on cells of innate immune system (macrophages and dendritic cells)
o Recognize PAMPS (pathogen associated molecular patterns) – patterns of molecules on pathogens
recognized by TLRs and lets the body know there is an invader
CD14 on macrophages recognizes PAMP called LPS on outer membrane of gram neg bacteria
TLR activation results in upregulation of NF-κB (on switch to turn on acute inflammatory response)
o Leads to production of multiple immune mediators
Present on cells of adaptive immune system
o Mediate chronic inflammation
Arachidonic Acid
Released from the phospholipid cell membrane by phospholipase A2
Acted on by (1) cyclooxygenase or (2) 5-lipooxygenase
o Cyclooxygenase produces prostaglandins (PGs)
PGI2, PGD2, PGE2 mediate vasodilation (in arteriole) and increased vascular permeability (occurs
at post-capillary venule)
PGE2 also mediates fever and pain (fEEEEEEver for PGEEEEEE2)
o 5-lipooxygenase produces leukotrienes (LTs)
LTB4 attracts and activates neutrophils
REMINDER
There are 4 key mediators for
attracting neutrophils: LTB4,
C5a, IL8, bacterial products

SRB 4
LTC4, LTD4, and LTE4 mediate vasoconstriction, bronchospasm, and increased vascular
permeability
Basically, these cause smooth muscle to contract
o Smooth muscle in arteriole contracts vasoconstriction
o Smooth muscle in bronchus bronchospasm
o Pericytes (layer of cells underneath epithelium) have smooth muscle contractile
function and when they contract, they pull apart the epithelial cells allowing for
fluid to leak from post-capillary venule into the interstitial space increased
vascular permeability
Mast Cells
Widely distributed throughout connective tissue in body
Activated by:
o (1) Tissue trauma – mast cells get activated to help initiate immune response
o (2) Complement proteins: C3a and C5a activate mast cells
o (3) Cross-linking of cell-surface IgE by antigen
Mast cells express IgE on their surface, if an antigen comes by and cross-links these IgE, the mast
cell is activated
Once activated, the mast cells undergo an immediate response:
o Release of preformed histamine granules
Mediates (1) vasodilation of arterioles; (2) increased vascular permeability at postcapillary venule
After releasing the histamine granules, the mast cell undergoes a delayed response:
o Production of arachidonic acid metabolites (particularly leukotrienes)
Complement Proteins
Proinflammatory serum proteins
“Complement” inflammation
Circulate as inactive precursors
Activation can occur via three different pathways:
o (1) Classical pathway C1 binds to IgG or IgM that is bound to antigen (“GM makes classic cars”)
o (2) Alternative pathway microbial products directly activate complement
o (3) Mannose-binding lectin pathway MBL binds mannose on microorganisms to activate complement
Result of activation = generation of C3 convertase
o C3 convertase converts C3 C3a & C3b
C3b helps C5 convertase
o C5 convertase converts C5 C5a & C5b
C5b complexes with C6 and C9 to form MAC
o Formation of membrane attack complex (MAC)
Produces a hole in the membrane for lysis of the microbe
Key products of complement pathway:
o C3a & C5a: trigger mast cell degranulation
o C5a: chemotactic for neutrophils
o C3b: opsonin for phagocytosis
When neutrophils get in tissue, they consume and then destroy whatever they eat
Phagocytosis is usually blind, but opsonins can assist and tell neutrophil what to consume

SRB 5
o MAC: lyses microbes by creating a hole in the cell membrane
C5b joins with C6 and C9 to make MAC
Hageman Factor
Inactive proinflammatory protein produced in liver
Activated upon exposure to subendothelial or tissue collagen
Plays an important role in DIC
Activates:
o Coagulation and fibrinolytic systems*
o Complement
o Kinin system – cleaves HMWK to bradykinin increased vascular permeability, vasodilation, & pain
Remember: pain is mediated by BRADYKININ and PGE2
Symphony of Mediators Result in Cardinal Signs of Inflammation
Redness (rubor) and Warmth (calor)
Due to vasodilation, which results in increased blood flow
Occurs via relaxation of arteriolar smooth muscle
Key mediators: histamine*, PGs, and bradykinin
Swelling (tumor)
Due to leakage of fluid from postcapillary venules into interstitial space
Key mediators: histamine*, tissue damage
Pain (dolor)
Key mediators: bradykinin, PGE2
Sensitize sensory nerve endings
Fever
Pyrogens cause macrophages to release IL-1 and TNF
Increase COX activity in perivascular cells of hypothalamus
Increased PGE2 raises temperature set point creating fever
Neutrophil Arrival and Function
Step 1: Margination
Vasodilation slows blood flow in postcapillary venules
Cells marginate from center of flow to the periphery
Step 2: Rolling
Cells that have marginated need to slowdown
o So they hit “selectins” (speed bumps) which are upregulated on endothelial cells
P-selectin is released from Weibel-Palade bodies (mediated by histamine)
Weibel-Palade bodies contain 2 important proteins: van Willebrand factor & P-selectin
E-selectin is induced by TNF and IL-1
Selectins
o Bind sialyl Lewis X on leukocytes
Three phases of acute inflammation:
(1) F: fluid phase; (2) N: neutrophil phase (after
24hr); (3) M: macrophage phase

SRB 6
o Interaction results in rolling of leukocytes along vessel wall
“Leukocytes” is generic WBC, but we’re talking about neutrophils when we discuss rolling
Step 3: Adhesion
Cellular adhesion molecules (CAMs: VCAM & ICAM) upregulate on endothelium by TNF and IL-1
Integrins upregulated on leukocytes by C5a and LTB4
Interaction results in firm adhesion to vessel wall
Clinical Correlation: Leukocyte Adhesion Deficiency
Autosomal recessive defect of integrins (CD18 subunit)
No integrin = no adhesion = never going to be drawn into tissue
o Findings:
Delayed separation of umbilical cord
Increased circulating neutrophils
50% of neutrophils hang out in blood vessels in the lung
o If needed, they can come out into the blood
o Without adhesion, they float around in the blood
Recurrent bacterial infections that lacks pus formation
What is pus? Dead neutrophils sitting in fluid
If no neutrophils can get in the tissue, no pus can be formed so they will have recurrent
bacterial infections that lacks pus
Step 4: Transmigration and Chemotaxis
Leukocytes transmigrate across endothelium of POSTCAPILLARY VENULES
Move toward chemical attractants (chemotaxis)
o Neutrophils are attracted by bacterial products (IL8, C5a, LTB4)
Step 5: Phagocytosis
Consumption of pathogens or necrotic tissue
Enhanced by opsonins (IgG and C3b)
o Molecules neutrophils recognize and say “oh, this is what I am supposed to eat”
Pseudopods from leukocytes extend to form phagosomes
o Internalized and merged with lysosomes to form phagolysosomes
Lysosomes contain highly degradative enzymes to break stuff down
Clinical Correlation: Chediak-Higashi Syndrome
Protein trafficking defect (autosomal recessive)
o Microtubule defect, phagosome cannot travel to the lysosome
Characterized by impaired phagolysosome formation
Clinical features:
o Increased risk of pyogenic infections b/c organisms can’t destroy what the neutrophil has consumed
since the phagosome cannot get to lysosome (train tracks are broken)
o Neutropenia – cells cannot divide properly so you don’t get the total number you should be getting
o Giant granules in leukocytes

SRB 7
Granules within the leukocytes are produced by golgi apparatus and then move out to the
periphery; however, without a railroad system, they can’t move and bunch together as one big
granule
o Defective primary hemostasis – dependent on platelets
o Albinism
Pigment of the skin occurs by melanocytes which distributes pigment to the keratinocytes
Without being able to hand them off (due to railroad system defect), no pigment goes anywhere
o Peripheral neuropathy
Railroad/protein trafficking defect, so long nerves don’t get what they need distally from their
nucleus
Step 6: Destruction of Phagocytosed Material
Can occur in two ways:
(1) oxygen dependent killing
o Most effective method
o HOCl generated by oxidative burst in phagolysosomes destroys phagocytosed microbes
o Mechanism:
O2 converted to superoxide by NADPH oxidase [aka “oxidative burst”]
Superoxide converted to hydrogen peroxide by superoxide dismutase (SOD)
Hydrogen peroxide is converted to bleach (HOCl) by myeloperoxidase (MPO)
HOCl is key molecule that destroys phagocytosed materials
Clinical Correlation: Chronic Granuloma Disease (CGD)
o Poor O2-dependent killing
o Due to NADPH oxidase defect (X-linked or autosomal recessive)
Patients cannot produce HOCl from the normal pathway
However, they CAN take H2O2 from the microbe and convert that to HOCl
o Catalase destroys H2O2 though, so catalase + bacteria will destroy its H2O2, so
that cannot be used either and HOCl will NEVER get formed
o Leads to infection and granuloma formation w/ catalase positive organisms:
S. Aureus, P. cepacia*, S. marcescens, Nocardia, Aspergillus
o Nitroblue tetrazolium test (NBT test): can we convert oxygen to superoxide?
Used to screen for CGD
Turns blue if NADPH oxidase can convert O2 to O2-
Remains colorless if NADPH oxidase is defective (ie: patients with CGD)
Clinical Correlation: MPO deficiency
o Patients cannot form HOCl b/c of an MPO deficiency (defective conversion of H2O2 to HOCl∙)
o Remain relatively asymptomatic; but are prone to candida infections
o NBT test would be normal in these patients
(2) oxygen-independent killing
o Less effective method
o Occurs via enzymes present in leukocyte secondary granules
ex: lysozyme and major basic protein

SRB 8
P
L
Step 7: Resolution
Neutrophils undergo apoptosis**
o Pus = dead neutrophils within fluid
Disappear within 24 hours after resolution of inflammatory stimulus
Macrophage Predominate After Neutrophils (third phase, see p. 5)
Peak 2-3 days after inflammation begins
Derived from monocytes in blood
o Arrive via margination, rolling, adhesion, and transmigration
Ingest via phagocytosis
Destroy phagocytosed material using enzymes in secondary granules
o Lysozyme: very important
Manage the next step of the acute inflammatory response
o Resolution and healing: IL-10 and TGF-β these both shutdown the inflammatory process
o Continued acute inflammation: IL-8 cytokine produced by macrophages to call in neutrophils*
“Acute” inflammation not defined by time, it’s defined by mediators
IL-8 calls on more neutrophils, this is still considered “acute inflammation” even though it could
still be going on 6 months later
o Abscess: area of fibrosis
Macrophages can form a wall of fibrosis around area of infection so area of inflammation gets
blocked in
o Chronic inflammation
Macrophages ingest antigens of microbe and present it on MHCII which results in activation by
helper T-cells in chronic inflammation
Chronic Inflammation
Characterized by Lymphocyte and Plasma Cells in Tissue
Delayed response, but more specific (adaptive immunity)
Inflammation = getting what’s on the inside of a blood vessel into the surrounding tissue
o Acute inflammation – neutrophils go from BVs tissue
o Chronic inflammation – lymphocytes (L) (can also become plasma cells) go from BVs tissue
Cells are mononuclear, don’t have multi-
lobulated nucleus like a neutrophil
Plasma cells (P): characterized by offset clock-
face nucleus w/glossy clearing next to it
Stimuli for Chronic Inflammation
Persistent infection *most common
Infection with viruses, mycobacteria, parasites, and fungi
Autoimmune disease
Foreign material
Some cancers

SRB 9
WAYS TO ACTIVATE CASPASES
Intrinsic mitochondrial pathway:
cytochrome C leaks out of
mitochondria to activate caspases
Extrinsic receptor pathway:
Fas/Fas ligand
CD8+ cytotoxic T cell:
releases granzyme to activate
caspases
Lymphocytes can be divided into T cells and B cells:
T Lymphocytes
Produced in the bone marrow as progenitor T cells
Further develop in the thymus (where the T cells “go to college”)
o TCR undergoes rearrangement
o Cells can become CD4+ helper T cells or they can “graduate” to become CD8 cytotoxic T cells
Use TCR complex for antigen surveillance
o TCR complex (complex includes CD3) recognizes antigens presented on MHC
T cells can only recognize antigens presented on MHC
CD4+ T cells MHC Class II
CD8+ T cells MHC Class I
Activation of T cells requires: (1) binding of antigen/MHC complex and (2) additional 2nd
signal
CD4+ T cell activation
o Extracellular antigen is phagocytosed, processed, and presented via MHC class II (APCs)
APCs = macrophages, dendritic cells
o 2nd
activation signal = B7 on APC binds CD28 on CD4+ T cells
o What do activated CD4+ helper T cells do?
Secrete cytokines that “help” inflammation
Divided into two subsets: one that helps B cells & one that helps CD8+ cytotoxic T cells
TH1 subset [helps the CD8+ T cells]
o IL-2: T-cell growth factor and CD8+ T cell activator
o IFN-ϒ : macrophage activator
TH2 subset [helps the B cells]
o *IL-4: class switching to IgG and IgE
o *IL-5:
Eosinophil chemotaxis and activation
Maturation of B cells plasma cells
Class switching to IgA
o IL-10: inhibits TH1 subset
CD8+ cytotoxic T cell activation
o Intracellular antigen is processed and presented on MHCI
o 2nd
activation signal = IL-2 from CD4+ TH1 cell provides
o Cytotoxic T cells are activated for killing
o What do activated CD8+ cytotoxic T cells do? KILL THEIR TARGET!
o Secretion of (to make a hole) and perforins granzyme
induces apoptosis of the target cell
[ are the enzymes that mediate apoptosis] CASPASES
o Expression of FasL binds Fas on target cell to activate apoptosis
B Lymphocytes
Immature B cells are produced in bone marrow
Undergo Ig rearrangement to become naïve B cells that express surface IgM and IgD
Activation of B cells:
o (1) antigen binding by surface IgM (which causes B cell to become plasma cell that secretes IgM) OR
o (2) B-cell consumes and presents antigen to CD4+ helper T cells via MHCII

SRB 10
G
EH EH
EH
EH
L
Noncaseating granuloma. Note: EH cells have nuclei and are not
necrotic.
Caseating granuloma. Note: Big clump of dead cells in the middle
surrounded by EH cells.
2nd
activation signal = CD40 receptor on B cells binds CD40L on helper T cell
o What do activated B cells do?
Helper T cell now secretes IL-4 and IL-5
These mediate B-cell isotype switching, hypermutation, and maturation to plasma cells
so that they can secrete IgM, IgD, IgG, IgE, etc
Chronic inflammation is divided into granulomatous and non-granulomatous inflammation:
Granulomatous Inflammation
Characterized by granuloma
Key cell: *epithelioid histiocytes (EH)* (macrophages with
abundant pink cytoplasm)
o Aggregation of these cells = “granuloma”
Surrounded by giant cells (G) and rim of lymphocytes (L)
Divided into noncaseating and caseating subtypes
Noncaseating granulomas lack central necrosis.
These types of granulomas can be caused by:
Reaction to foreign material
o Ex: pt has breast cancer removed followed by breast
implants. They leak and lead to a reaction leading to
enlarged axillary lymph nodes
Sarcoidosis
o Distinguishing feature of this is noncaseating granulomas in different areas of the body (particularly in the
lung)
Beryllium exposure
Crohn’s disease
o Distinguishing feature of this is noncaseating granulomas
o NOTE: distinguishing feature of ULCERATIVE COLITIS is crypt abscesses
Cat scratch disease
o Distinguishing feature of this is noncaseating stellate-shaped granulomas in the neck
Caseating granulomas exhibit central necrosis.
These types of granulomas can be caused by
TB
o AFB stain checks for TB
Fungal infections
o GMS stain checks for fungus
Steps Involved in Granuloma Formation [both caseating AND noncaseating are formed this way!]
Macrophages present antigen via MHCII to CD4+ helper T cells
Macrophages secrete IL-12, inducing CD4+ helper cells to
differentiate into TH1 subtype
TH1 cells secrete IFN-ϒ, which converts macrophages to
epithelioid histiocytes and giant cells














![Skin Inflammation, [Acute, Suppurative, Chronic, Chronic ... · Skin – Inflammation, [Acute, Suppurative, Chronic, Chronic Active, Granulomatous] presence of mononuclear cells (lymphocytes,](https://static.fdocuments.in/doc/165x107/5f0eb0c97e708231d44075f1/skin-inflammation-acute-suppurative-chronic-chronic-skin-a-inflammation.jpg)


