
Chaptershodhganga.inflibnet.ac.in/bitstream/10603/973/9/09_chapter 4.pdfchapter N Isomvs of (CHW (X...
61
Chapter IV: Isomers of (CH)sX (X = N, P, As and SiH) and C4Si2H6
Transcript of Chaptershodhganga.inflibnet.ac.in/bitstream/10603/973/9/09_chapter 4.pdfchapter N Isomvs of (CHW (X...

Chapter IV: Isomers of (CH)sX (X = N, P, As and SiH) and C4Si2H6

Chapter IV: Isomers of (CH)*X (X = N, P, As and SiH) and C4Sil&
4.0: Abstract
Ah mlho HF, MP2 and CCSD(T) and hybnd denslty functional theory (B3LYP)
calculat~ons were performed on the valence Isomers of benzene, group V heterobenzenes
and s~labenzene A benchmark study on the benzene valence lsomers was done at varlous
levels of theory In each of the Group V heterobenzene potentla1 energy surface, ten
mlnlmum energy structures were located The broad relatlve energy orderlng of the
valence lsomers of benzene and the group V heterobenzenes rematn the same Flfteen
valence lsomers were Identified on the sllabenzene potentla1 energy surface, out of whlch
twelve were found to be minima, two were transltlon states and the other was found Lo be
a second order saddle polnt A stark contrast 1s notlced In slla- and d~s~lahen/ene valence
Isomers compared to those of group V heterobenzenes, several mlnlma on the potential
energy surface are observed whlch are wlth~n 30 kcallmol Two new valence lsomcrs
were located, namely Vla, and Vlb,, they were found to 11e only about 20 kcallmol
hlgher than the global mlnlma, sllabenzene The reactlvlty of the sllabenzene Isomers IS
assessed based on the absolute chemical hardness values Benchmark calculat~ons on the
three dlsllabenzenes were performed at vanous levels lncludlng the HF. B3LYP. MP2
CCSD(T), CASSCF and CASPT2 uslng varylng sues of bass sets Based on the results.
B3LYP method w ~ t h 6-3IG' level was chosen to model the other d~s~lahewene lsomcrs
Totally, 78 statlonary polnts were ldentlfied of whlch, 61 were characterlrcd ds mlnlmd
on the potent~al energy surface Several unconvent~onal structures are hlghly colnpctltlvc
In energy compare to the global mlnlma

chapter N Isomvs o f (CHW (X = N. P. As and s i H ) d C+SI& 107
4.1: Introdaction
The valence Isomers of benzene, whlch differ only by carbon-carbon connccanty
of the SIX CH umts, represent the most Important sub-class of benzene wmers among
more than 200 posslble Isomers, owng to the enormous expenmental and theoret~cal
Interest I J Scheme 4 1 depicts the five fundamental valence lsomenc forms, whch arc
benzene (B), benzvalene (V), Dewar benzene (D). pnsmane (P) and blcyclopropenyl (C)
Recently, Johnson and Daoust confirmed that trans-Dewar benzene and Motblus benzene
are also mlnlma on the (CH)6 potentla1 energy surface3 In addlt~on to these Isomers,
Balahan's henzmob~usstnpane, tw~sted pnsmane proposed by Karl and Bauer, and Claus'
benzene, all of wh~ch have the ~dentlcal connectlvlty, are other poss~b~l~t~es (Scheme 4 2).
however, these are chemically unreallstlc '* The syntheses of all the classrcal Isomers
except for trans-Dewar benzene (T) have been achieved, and these formed the subjects of
a number of theoretical and expenmental Investcgatlons
Scheme 4.1
benzene (B). D, benzvalene (V), C,, Dcwar benzene (D), C,,
pnsmane (P), D,, 3,3'-btcyclopropmyl ( C), C,, rrons-Dewar benzene (T). C,,
Scheme 4.2

Chapter IV h m a - s of (CHN (X - N, Pa As and SIH) md &St+& 108
Phosptume, srsabmzene and nlabenzene, multed tn replacing the methlne
p u p by P. As and SIH respectively were also demonstrated as aromatlc compounds "' Most of the valence Isomers of phosphlnme, B1P (Scheme 4 3) were synthesized and
were found to undergo ~nterestlng rearrangement react~ons and possess novel blndlng
capablhtles as hgands to transltron metal templates lo' ' I b Translent generation of the
Dewar pyndlne (DIN) through W lrradlat~on of pyndlne 1s reported recently I' It 1s to
be noted that the substltuted Dewar pyndlnes and azapnsmanes are well known
However, the data on aza-benzvalenes (VIN, V2N or V3N) and 3.3'-aza-b~cyclopropenyl
Isomers or then substltuted Isomers are scarce Although, arsabenzene IS known for qulte
some t~mqthe chem~stry of 11s other valence lsomenc forms IS relatively unknown ' I
Scheme 4.3
B IX V I X V2X V3X DIX
D2X P I X C IX C2X T2X
X=N,PandAs
The analogy between carbon and slhcon appears to be stra~ghtfonvard, desp~te the
substant~al contrasts In the chemlstly of hydrocarbons and their s~hcon subst~tuted
counterparts The s~hcon-organ~c compounds have been favonte huntlng grounds for
the theoretical and computat~onal chemistry for more than three decades The slmplest
sll~con analogue of benzene - sllabenzene (B1) - has been a subject of great Interest for
qulte some tune (Scheme 4 4)2126 Although the parent compound of sllabenzene
remalned an elusive specles, an unambiguous charactenzat~on of substltuted sllabenzene
1s ach~eved employtng the strategy of employng bulky groups attached to the SI

Chapter N: Isomers of (CHN (X = N. P. As md SiH) md &Si& 109
center!334 Nonetheless, various spectroscopic studies on the identification of silabatzene
and its isomerization to other products were known for quite some time.2s26 The wnccpt
of silaaromaticity is one of the most intensely studied topics in the last three decades.""
A large number of theoretical studies wnfess aromaticity on silabenzene, where one of
the methine groups is replaced by s ~ H ! ~ ~ ~ ~ ~ ~ The synthesis and characterization of
substituted silabenzene was unambiguously achieved only recently, while the data on the
other valence isomeric forms is scarce!' However, the infra-red, electronic and
photoelectronic spectroscopic characterization of silabenzene and I-methylsilabenzene in
argon matrices were reported quite a while ago!"he strategy of employing bulky
substituents resulted in a fluny of synthetic accomplishments in the silaaromatics, which
are otherwise elusive in their pristine form.23"' Maier and co-workers reported the
photochemical isomerizations of silabenzene to Dewar silabenzene, in a matrix isolation
study." While the silabenzene was unambiguously characterized, the characterization of
the isomerization product, benzvalene was based only on the NMR data, which was not
definitive, as acknowledged by the authors themselves, and these class of compounds are
too reactive for elaborate experimental investigation using the conventional techniques
such as X-ray diffraction.'! Chandresekhar and Schleyer have done calculations on B1,
Dl and other three isomers of silabenzene." Recent studies indicate that silabenzene is
only slightly less aromatic than benzene.27.30-32 Wakita et al along with their experimental
investigation on the isomerization of silabenzene to silabenzvalene reported B3LYP16-
31G* energetics on the five (Bl, V2, V3, Dl, DZ) valence isomeric forms?"
Among the disilabenzene isomers, hexamethyl substituted 1,4-disilabenzcne (3)
has been synthesized as early as 1987 and several photochemical reactions were
studied." Also, recent studies indicate the existence of silabenzene and 1,bdisilabenzene
as ruthenium complexes.36 Ando et al, have suc~essfully synthesized 40, which yields 25
and 49 on heating, and proposed that the possible common intermediate for both the
conversions to be 42?l Even though 37 and 46 are not valence isomers, we have
considered in this study as their derivatives have been synthesized recently?8e39
Considering the experimental interest in the isomerization reactions among the various
forms of disilabenzene, we ventured into a detailed theoretical study on disilabenzene
isomers. Baldridge and Gordon reported theoretical studies on the three disilabenzenes, 1,

2 and 3 uslng the HF method wth the STO-3G bas~s set "O In 1985. Chandrasckhar and
Schleyer have reported the HF13-21G* energehcs of 3, 10.11 and 25 " Recently, Ando
and w-workers used the HF method wth the 6-3 IG* bass set to evaluate the energet~cs
of 3, 25, 49, 40 and 42 " Yoshzawa and w-workers reported a detaled computat~onal
study on the lsomenzat~on reactlon between 13-d~s~labenzene (3) and 1.4-dls~la Dcwar
benzene (25) uslng the dens~ty funct~onal B3LYP and CASSCF procedures employing a
double-< bas~s set 42 The last part of the chapter concentrates on the vanous d~stlabenzene
Isomers, whlch are classified as monocychc, b~cycltc, tncycl~c and tetracycl~c Isomers
(Scheme 4 5 - 4 8)
Scheme 4.4

chnpter I V Isomers o f ( C H H (X = N. P, As and SIH) and &Sl2H6 111
Scheme 4.5
c': Q /
Sl

Chapter I V Isomers of (CHM (X = N. P, As ond SIH) d C.SI~H( 112

Chapter IV Isomers of (CHM O( - N. P. As and SIH) and C.SIII+ 113
Scheme 4.7
Scheme 4.8
I:'::

Chapter I V I s o w s of (CHhX (X = N. P. AS and SIH) and C.SI& 114
The chemistry of the valence Isomers of &(X = N, P. AS, and SIH) where all the
methtne groups are replaced by ~sovalent groups was extenstvely studled by
computat~onal means 4' However, httle attention 1s pald to the theoret~cal stud~es on the
valence Isomers of group V heterobenzenes, sllabenzene and d~s~labenzene, desplte the
expenmental Interest m these classes of compounds Examlnatlon of the equ~llbnum
geometnes, charactenzatlon of the mlnlma on the potentla1 energy surface, and the
relative energles and thelr comparison w ~ t h the parent benzene counterparts provlde a
basis to assess the vanatlon Induced by the heteroatom Benchmark calculat~ons are done
on benzene to test the sultablllty of the popular theoret~cal models by comparing with the
ava~lable experimental and other hlgh level theoret~cal calculat~ons Ah mir~o and hybnd
density funct~onal theory calculat~ons were performed on the group V heterobenzene,
sllabenzene and d~s~labenzene valence lsomers and other related lsomers of d~s~labenzene
and the results are presented In the same order
4.2: Valence lsomers of Group V Heterobenzenes
Thls sectlon presents the computat~onal results obta~ned for the group V
heterobenzene valence Isomers Deta~led calculat~ons on benzene valence Isomers, and
companson of the geometric parameters and energetics computed by vanous procedures
with the hlgh level calculat~ons and expenmental data are presented first The effect of
replacement of methlne groups of the benzene valence lsomers by lsovalent group V
elements (N, P and As) on the equil~bnum geometnes and the relatlve energles is studled
4.2.1: Computational Details
All the calculat~ons were done uslng the Gausslan 98 sulte of programs44 The
gwmetnes of all the structures were fully opt~m~zed wlthln the symmetry constraints
~nltlally at the HFl6-3lG* level 45 Further refinement of geometnes 1s done at B~LYP*"
and MP2 levels uslng thc 6-31G* bass set The effect of addlng a ser of polarization
functions to the peripheral hydrogens on the geometry of these compounds 1s tested by
dolng optlmlzatlon uslng the 6-31G** basis set at the MP2 level The MP216-31G' and
MP216-31G** geometnes are virtually ~dent~cal lndlcat~ng that 6-31G* 1s adequate In
glvlng proper equlllbnurn geometnes The nature of the statlonary po~nts obtalned was
charactenzed by frequency calculat~ons at HF and B3LYP levels, which des~gnate all the
valence isomers considered In the study as mlnlma The present and prevlous stud~es

Chapter IV: Isomus of (CUM (X = N. P. As and SiH) and C4SizH6 115
indicate that MP2 gives better equilibrium geometries; hence MPU6-31G* optimized
geometries were taken for further single point calculations at CCSD(T)/6-31G* and
MP216-31 1+G0* levels. A recent study on the diphosphinines and their valence isomers
indicates that a single determinantal approach is adequate to get reliable results for this
class of compounds." The enthalpy correction to the total energy is obtained from the
vibrational frequency data obtained at the B3LYP level. The best estimates for the
relative energies were obtained for all the molecules considered in this study, using eqn.
4.1.
AE = AEccswr, + AE(~~2,6311ffi*.- M P U ~ ~ I G * ) + AH - ... eq. 4.1.
AH is the enthalpy correction factor obtained by frequency calculations at the
B3LYPI6-31G* method. Although, the CCSD(T) method is a reliable theory, the basis
set of 6-31G* quality is not adequate. Performing CCSD(T) calculations with triple-<
quality basis sets uniformly for all the species considered in this study is beyond our
available computational facilities. Therefore our scheme of best estimates, which
accounts for the CCSD(T) basis set deficiency at MP2 level, is designed to circumvent
this problem. This scheme seems to be in excellent agreement with the high level
calculations on benzene isomers and the available experimental results (vlde infro).
4.2.2: Results and Discussion
The discussion on the valence isomers of benzene is done as a reference as well as
to test the suitability of the adopted computational tools in modeling the systems under
~tudy. The equilibrium geometries of all the valence isomers are compared and contrasted
among themselves as well as with the reference pristine isomers. Similarly, the relative
energies of the valence isomers of benzene are taken as reference to assess the relative
energy ordering in its group V counterparts. The discussion on the relative stability
orderings is presented next.
4.2.2.1: Equilibrium Geometries
Figure 4.1 depicts the MP2/6-31G* optimized geometries of the valence isomers
of benzene along with the experimental data, wherever available. The MP2 geometries
are in excellent agreement (maximum deviation of 0.012A in bond length) with the
QCISDl6-311G* geometries.7 The computed geometries are in reasonable agreement
with the available experimental numbers, and will form our reference values to estimate

RDpter IV: Isomers of (CHW (X = N. P, As and SIH) and C4Si2H6 116
the perhubahon m the skeletal bond lengths upon replacing the methine groups w~th the
lsovalent atoms. A went computat~onal study, wh~ch cons~dered the five classical
valence Isomers, c o n f i s the sultablllty of MP2 level In obtaining reliable geometnes ' The less explored trans-Dewar benzene (T) Isomer has a substantially shorter central
slngle bond and fauly longer double bond lengths '
Figure 4.1 The pnnc~pal geornetnc parameters of the valcnce Isomers of benzene obtalned at the MP216-31Gt level Expenmental parameters are glven In parenthesls wherever avalable The bond lengths are In A and the angles are In degree
The pnnc~pal geometnc parameters of the substituted benzenes (BIX),
benzvalenes (VlX, V2X and V3X), Dewar benzenes (DlX and DZX), pnsmanes (PIX),
b~cyclopropenyls (ClX and C2X) and trans-Dewar benzenes (T2X) obta~ned at the HF,
B3LYP and MF'2 levels of theory w~th 6-31G* basls set are glven In F~gures 4 2.4 3.4 4,
4 5, 4 6 and 4 7 respectively The numbering for the lndlv~dual pos~t~onal Isomers has to
be dec~phered h m Scheme 4 1 Wlule Hartree-Fock method consistently underest~mates
bond lengths for well known reasons, B3LYP and MP2 are In farly good agreement w~th

Chaptv W. ISOMS o f (CHW (X = N, P, As md SiH) and C,Si21+ 117
each other in most of the cases. In general B3LYP method overestimates bond lengths
especially for P and As valence isomers, with maximum deviation of 0.033 A in the bond
lengths and 1 .So in bond angles. Employing 6-31G9* basis set at the MP2 level did not
result in any noticeable changes in geometric parametm, with almost identical results to
first three decimal places in bond lengths (maximum deviation 0.005 A) and to the first
decimal for bond angle (maximum deviation 0.2'). The discussion on the structures will
be based on the MP216-31Gt geometries throughout the rest of the study unless
otherwise stated.
Figure 4.2: The principal geometric parameters of pyridine (BIN), phosphinine (BlP) and arsabenzene (BIAS) obtained at the HF (ordinary), B3LYP (underlined) and MP2 (bold) levels using the 6-31G' basis set. All values are given in A.
Pyridine (BIN), phosphinine (BIP) and arsabenzene (BIAS) are found to have
bond lengths corresponding to aromatic compounds, a result which is consistent with
previous studies. The C 4 bond lengths in the three compounds are similar to the C-C
bond length in benzene with a maximum deviation of only 0.005 A in BIAS. In addition
to this, the C-X bond lengths in these compounds are in between the normal C-X single
and double bond lengths witnessing full delocalization in terms of bond length
equali~ation.~~ In general, among the benzvalene isomers, the substitution pattern or the
type of substitution does not seem to perturb the skeleton significantly in a majority of
the cases. Among the benzvalene valence isomers all the C-C bonds in the positional

Chapter I V . Isomers of (CHH (X = N, P, As ond 8H) and C,S2H6 118
~somcrs are very smtlar and are closer to the correspond~ng pnstlne molecules
Obv~ously, the C-X bonds are d~fferent due to the d~fferent atomlc tad11 of the substituted
atom X The bndge-head C-C bond of the b~cyclobutane molety In VZN 1s substant~ally
shorter compared to the parent molecules, while every other Isomer. VZP. VZAs, V3N,
V3P and V3As have bond lengths s ~ m ~ l a r to the unsubstltuted benzvalene
A closer look at the Dewar benzene pos~t~onal isomers ~nd~ca te that the
heteroatom subst~tut~on does not make any s~gn~ficant perturbat~on to the skeleton In
terms of bond lengths In general. the computed C-C, srngle and double bond lengths are
essent~ally ~dent~ca l to the parent molecule lrrespect~ve of the subst~tuent slze and slte
The angle between the two planes (0) 1s slmllar, except In DIP and Dl As, when P and As
are subst~tuted at the bndge head posltlon result~ng in reduct~on of 0 by approx~mately
10' The pnsmane valence Isomers also do not show any slgnlficant devlat~ons In the
bond lengths compared to the parent molecules In CZN, the C-N s~ngle bond length IS
found to be longer than the normal C-N bond length Even In the same compound, the C-
C s~ngle bond IS computed to be much less compared to the C-N s~ngle bond
Interestingly, the bond lengths In CIX and CZX (X = P and As) are found to be
comparable to the correspond~ng standard bond lengths
In all T2X, the bndg~ng bond IS substant~ally shrunk compared to the bndglng
bond In the correspond~ng crs-lsomer (DZX), w ~ t h TZN e x h ~ b ~ t ~ n g the maxlmum effect
Sim~lar to the sltuatlon In parent Isomers, most other bond lengths exhlb~t exactly the
opposlte trend, I e , elongation when compared to the correspond~ng DZX Isomers Thc
C-N s~ngle bond length in TZN 1s substant~ally elongated, and T2P and T2As also show
s ~ m ~ l a r trends albeit to a smaller extent T h ~ s feature IS lnd~catlve of straln 111 the system
and lndlcates the p o s s ~ b ~ l ~ t y of nng openlng through C-X cleavage Therefore, the present
analys~s ~nd~ca tes that the replacement of methlne group by N. P, or As on the skeletons
of the valence Isomers of benzene does not Induce not~ceable skeletal perturbat~ons

Figure 4.3 The pnnctpal geometric parameters of the benzvalene tsomen (VIX, VZX and V3X) obta~ned at the HF (ord~nary), B3LYP (underlined) and MP2 (bold) levels ustng the 6-31G* bas~s set The bond lengths are ~n A and the angles are in degree

Chapter IV: Isomcrs of (&I)& (X : N. P. As rmd SiH) md C,SilH6 120
DIN. C, (0) DIP. C, (0) D l h , C, (0)
Figure 4.4: The principal geometric parameters of the Dewar benzene isomers (D1X and DZX) obtained at the HE (ordinary), B3LYP (underlined) and MP2 (bold) levels using the 6-31G* basis set. The bond lengths are in A and the angles are in degree.
PIN. C, (0) PIP, c, (0) PIAs. C, (0)
Figure 4.5: The principal geometric parameters of the prismane isomers (PIX) obtained at the HF (ordinary), B3LW (underlined) and MP2 @old) levels using the 6-31G* basis set. All values are given in A.

Chapter I V Isomers of (CHM (X = N. P, AS and SIH) a d C,SI~Y 121
F~gure 4.6 The pnnc~pal geornetnc parameters of the b~cyclopropenyl Isomers (CIX and CZX) ohtamed at the HF (ordinary), B3LYP (underlmed) and MP2 @old) levels usrng the 6-31G* basis set All values are glven In A
Figure 4.7 The pnnc~pal geornetnc parameters of the trans-Dewar benzene Isomers (T2X) obta~ned at the HF (ordinary), B3LYP (underlmed) and MP2 @old) levels uslng the 6-3 lG* bas,$ set The bond lengths are ~n A and the angles are ln degree

Chapter I V Isomers of ( C H M (X = N P As and SIH) and C1S~2H6 122
4.2.2.2: Relative Energies
In t h ~ s sechon taklng the valence lsomers of benzene, on wh~ch the expenmental
or highly rel~able theoret~cal data 1s ava~lable, the performance of vanous levels of theory
is assessed In glving the rehable energetlcs for thls class of compounds A comparison
will enable us to assess the strengths and drawbacks of vanous levels of theory and help
us In chooslng a rlght cho~ce of method, whlch can be appl~ed on the valence lsomers of
the compounds under study The relat~ve energles for all the valence lsomers of benzene
ohtamed at different levels of theory are glven In Table 4 1 Previously reported relat~ve
energles from G2 methods and expenmental heats of format~on wherever ava~lable are
also glven In the same table 6 7 Expectedly, the HF method was not good for quantltat~ve
results and the dynamic electron correlation 1s essent~al In obtaming rehable energetlcs
Surpr~smgly, the B3LYPl6-31G* method also cons~derably overestimate the stab~llty of
the benzene wh~ch results In the consistent underestlmatlon of all other Isomers Thls
problem may be traced to the Ilmitat~ons of the B3LYP, as well as most DFT based
methods, In comparing the energles between x-delocal~zed and locallzed structures along
the potent~al energy surface V~rtually same values are obta~ned at MP216-31GL and
MP216-31G** levels for all the lsomers Very s~m~lar relat~ve energles are obtalned at
CCSD(T)I6-31G* level The Inadequate basis set, 6-31G*, used at the CCSD(T) level IS
remed~ed as the bass set correct~on 1s done at the MP2 level (eqn 4 1) Thus, excellent
agreement 1s observed between the best est~mates of the relat~ve energles (eqn 4 1) and
the reported hlgh level calculat~ons Therefore, the relatlve energles obta~ned lor the
valence lsomers of pyndine (BIN), phosphln~ne (B1P) and arsabenzene (BIAS) by t h~s
procedure are llkely to be very slmllar at further h~gher levels of theory
The relatlve energles obta~ned at varlous levels of theory for all the valence
Isomers of pwdme (BIN), phosphn~ne (BIP) and arsabenzene (BIAS) considered are
glven In Tables 4 2, 4 3 and 4 4 respect~vely Both the HF and the B3LYP levels
consistently overestimate the stabil~ty of the delocallzed con~ugated systems While

Ctmpter I V Isomers of (CH)3( (X = N. P. As and SIH) and CISI~H) 123
adding a set of polanzat~on funct~ons on per~pheral hydrogens, golng to 6-31 I+G**
qual~ty b a s s set marg~nally Improves the energet~cs, espec~ally for the 3,3'-
bicyclopropenyl and trans-Dewar benzene Isomers In contrast, MP2 g~ves a cons~stently
better agreement and the bans set qual~ty over the 6-31G* have only a mlnor
~mprovement In the relat~ve energles Therefore. dynam~c electron correlat~on 1s essent~al
In ob ta~n~ng the accurate energet~cs, and the bas~s sets of double-< quahty augmented
w ~ t h a set of polanzat~on funct~ons are expected to y~eld rel~able energles " However. In
t h ~ s class of compounds the conxent~onal ah ~ n ~ t t o methods perform much better than the
currently popular dens~ty funct~onal theory based methods
The d~scusslon on energetlcs w~ll be based on the best estlmates unless otherw~se
specified The plot of the best estlmates of all the valence lsomers of benzene, pyndlne,
phosph~nine and arsabenzene 1s glven In F~gure 4 8 As reported earher, the five valence
lsomers of benzene span a w ~ d c range of stab~lrt~es up to 150 4 kcal mol ' w ~ t h ~rnns-
Dewar benzene (T) be~ng the least stable (Table 4 1 ) ' ( I 7 T h ~ s d~fference between the
energles of the most stable and the least stable Isomer decreases wh~le go~ng from
benzene farn~ly to the p y r ~ d ~ n e fam~ly, wh~ch 1s 147 0 kcal mol ' T h ~ s further decreases
to a greater extent for the valence lsomers of phosph~n~ne (121 8 kcal mol ' ) and reduces
a httle for the valence isomers of arsabenzene ( 1 13 3 kcal mol ') (Tables 4 2, 4 3 and
4 4) In all the class of compounds cons~dered here benzene Isomer (BIX) IS computed to
be the most stable and the I~OIIJ-Dewar benzene Isomer (T2X) to be the least stable
Isomer The valence lsomers of phosph~n~ne follow the same relatlve energy order~ng as
that of arsabenzene Absolutely no crossover In the relat~ve cncrgy ordcr~ngs IS found In
golng from the valence Isomers of phosphln~ne to arsabenzene (F~gure 4 8) Although the
framework 1s ch~efly respons~ble for the relat~ve stab~ht~es of the subst~tuted valence
Isomers, the energy perturbat~ons by skeletal subst~tut~ons are substantla1 among the
benzvalene lsomers VZN, where the heteroatom IS In the sp2 centcr, 1s the most stable
among the azabenzvalenes whereas the correspond~ng PlAs subst~tuted Isomer,

Chaptv IV : Isomers of (CH)& (X = N. P. As and SiH) and C,SI&+~ 124
VZPNZAs is the least stable. Similarly, in case of Dewar benzene, the stability ordering
of D1X and DZX is reverse when we go h m X = N to P or N to As. In contrast, in the
3.3'-bicyclopropenyl isomers, the reversal in the stability is not seen between C1X and
C2X when going 'om X = N to P or As. However, the energy difference between C l X
and C2X for X = N is 37.0 kcal mol-' which is only 6.0 and 7.6 kcal mol" for X = P and
As respectively. The N substitution always prefer to occupy the sp2 center compared to
the sp3, as is evident from the higher stability of V3N, D2N and CZN compared to their
other positional isomers. In contrast, the P and As substitutions prefer the sp3 centers in
benzvalene and Dewar benzene isomers. However, the bicyclopropenyl isomers, the sp2
center substituted isomers are marginally more stable. The relative energy orderings of
the phosphinine and arsabenzene valence isomers are identical and the energy gap
between various isomers decreases slightly in going from the former to the later.
Figure 4.8: The plot of the best estimates of the relative energies of the valence isomers of benzene and Group V heterobenzenes.


Chapter IV Isomers of (CHW (X = N, P, As ond SOH) and CISIZH~ 127

chapter I V Isomers of ( C U M (X = N, P. As and SIH) and CCSa2H6 128

chapter IV: Isomers of (CHkX (X = N. P. As and SiH) and C,Si,& 129
4.3: Valence Isomers of Silabenzene
A total of eleven (CH)sSiH isomers are considered, the stationary points located,
their nature on the silabenzene potential energy surface, their relative energies and the
reactivities assessed by the chemical hardness values are discussed in this section.
4.3.1: Computational Details
All the structures (Scheme 4.4) were fully optimized within the symmetry
constraints at the B3LYP level of theory with the 6-31G* basis set initially. The
stationary points thus obtained were characterized based on the frequency calculations.
The geometries were further refined with the cc-pVDZ and 6-31 1 +G** basis sets at the
B3LYP level. The geometries were also evaluated at the MP216-31G** level. These were
followed by single point calculations at the MP216-31 I+G** and CCSD(T)/6-31G*
levels on the MP216-31G** optimized geometries. However, 6-31G* basis set is
probably not adequate at the CCSD(T) level and therefore we have considered a bigger
basis set at MP2 level and this scheme (equation 4.2) was quite impressive in giving good
fits with the higher level calculations (Section 4.2.2.2).
AE = A E c c s q ~ ~ + AE(MPM-31 I+G.. - ~ ~ 2 1 6 - 3 1 ~ ' . ) + AH ... eq. 4.2
AH is the enthalpy correction factor obtained by frequency calculations at the
B3LYPl6-31 1+G** method. Harmonic frequencies were computed using 6-31G*, cc-
pVDZ, and 6-31 1+G** basis sets at the B3LYP level. The differences in the computed
harmonic frequencies obtained using various basis sets were very small. Thus the basis
set employed to obtain the harmonic frequencies is adequate. Most of the B3LYP
optimizations were carried out using the Jaguar 4.1 program package initially.52
However, for the sake of uniformity all the reported calculations were done using the
Gaussian 98 suite of programs.M
4.3.2: Results and Discussion
All the valence isomeric forms of silabenzene given in Scheme 4.4 were fully
optimized within the symmetry constraints initially at the B3LYP level with 6-31G* basis
set. All the structures were characterized as minima showing all real frequencies except
for the benzvalene isomer, V1. Attempts to locate V1 lead to one or other of the
stationary points, V l a and V l b and both of them turned out to be first order saddle
points. The normal modes corresponding to the imaginary frequencies are followed and

Chapter I V : Isomers o f (CUM (X = N, P, As ond SiH) and C4Si2H6 130
minimum energy struchuw, Vla, and Vlb., were obtained. The two new structures
Vla, and Vlb, are distinctly distorted when compared to benzvalene and correspond to
bicyclic structures. Energetically, these two isomers lie closer to the benzenoid
compound, B1 compared to the other isomers (vide infra). This prompted us to
investigate the esthetically appealing C5, pyramidal structure, PY, where the Si is bound
to all the five carbons and a stationary point was located. The frequency calculation
characterizes this as a second order saddle point. Following one of the two imaginary
frequencies lead to the first order saddle point, Vla and Vlb, which further, lead to
minima (Vla, and Vlb,). Although, it is necessary to have two electrons less to
stabilize such a nido system, the relatively low energy of PY and its connectivity to the
novel minimum energy structures, Vla. and Vlb,, prompted us to explore this part of
the potential energy hypersurface in detail. The cyclopentadienyl moiety is virtually flat
In all the cases, PY, Vla, Vlb, Vla, and Vlb,, depicted in Scheme 4.9. While SiH is
bound in ~l~-fashion in PY, it is in ~l'-fashion in Vla and Vlb and ?12-fashion in Vla,
and Vlb,. Similarly, a closer examination of the optimized structure of CZ reveals that,
C-Si single bond was broken in the Si containing three membered ring, leading to an
open structure, trans-C2 with a divalent Si (Scheme 4.10). The corresponding cis isomer,
cis-CZ was located and characterized as a minimum on the potential energy surface.
Several attempts to locate a stationary point corresponding to TI failed at both B3LYP
and MP2 levels and the putative structures collapsed to Dl upon optimization. Attempts
were made to locate T I at the HF, B3LYP and MP2 levels, and in each case all the
putative structures converged to the cis-Dewar benzene and thus indicating that T I
structure does not correspond to a stationary point on the potential energy surface of
(CH)$iH. Thus the present study identifies fifteen important stationary points on the
potential energy surface of silabenzene where thirteen are minima, two are transition
states and one structure is a second order saddle point. All the fifteen structures were then
reoptimized and recharacterized by frequency calculations at the B3LYP level using the
cc-pVDZ and 6-311+G** basis sets. The geometry optimization were also carried out at
the MP216-31G8 level. The assignment of the stationary points with higher basis sets was
found to be identical with the 6-31G* basis set. The equilibrium geometries are discussed
first and then the relative energies, vibrational spectra and the reactivity.

dropter IV : Isomers of (CHH (X = N, P. As and SIH) and C.SI~HI 131
Scheme 4.9
H ,
Scheme 4.10

Chapter I V ISO~CTS of (CHM (X = N, P. AS and SIH) and C,S1&I6 132
4.3.2.1: Equilibrium Geometries
Figure 4 9 deplcE the pnnclpal optlm~zed geometnc parameters of all the
structures cons~dered m the study at B3LYPl6-31G8, B3LYPIcc-pVDZ. B3LYP16-
311+G**, and MP216-31G** levels of theory In general, the geometnes obtalned at the
B3LYP and MP2 levels are m good agreement w~th each other However, B3LYP
method consistently overestimates the C-SI slngle bond lengths w~th all the bas~s sets
compared to the MP2 level The geometnc parameters obtalned uang the 6-31G* and the
6-31 1+G** bass sets are essennally ~dent~cal and therefore, 6-31G* quality bas16 set may
be assumed to be qulte adequate for the geometnes The d~scuss~on on the equlllbnum
geometnes from here throughout the rest of the text will be based on those obtalned at the
MP216-31G** level
The C-C bond lengths In sllabenzene, B1 are equal and also the C-SI bond length
IS found to Ile between the C-SI slngle and double bond lengths The bond length
equallzat~on tn skeletally substituted benzenes IS elaborately discussed In Chapter 111 In
Vla and Vlb, the five membered nng formed by the carbon atoms 1s found to be
virtually planar All the C-C bond lengths are closer to the aromatlc bond lengths whereas
the C-SI bonds are substant~ally elongated It occurred to us that the sltuatlon m~ght
correspond to the one where a cyclopentad~enyl anlon 1s coordlnated In a rl'-fash~on w~ th
SIH' cap Indeed, the natural population analysls at the HFl6-31G*llMP2/6-31G'
lnd~cates that there 1s a charge polanzat~on of +O 4546 In Vla and +O 4678 In V l b on
SIH group lndlcatlng that the five membered rlng has substant~al aromat~c stab~llzat~on
However, the benzvalene isomers only VZ and V3 retaln the skeleton and V1 does not
correspond to a statlonary polnt and spontaneously collapses to Vla and V l b T h ~ s 1s In
contrast w~ th the valence Isomers of pyndlne, phosph~nlne and arsabenzene, where all the
three benzvalene Isomers were charactenzed as mmmma (Sect~on 4 2 2) These tn.0
structures are transltlon states, the five membered nng formed by the carbon atoms In the
corresponding mlmmum energy structures, Vla, and Vlb, exhlblts slm~lar properties It
IS lnterest~ng to note that the SIH unlt 1s coordlnated to only two carbon atoms The
bndglng C-C bond of the cyclobutane molety m V2 1s 1 523 A, whlch 1s elongated
compared to 1 453 A In the pnstlne compound at the same level of theory Thls may be
due to the release of stran caused by the presence of Sl In the blc~clobutane moiety In

Chapter I V : Isomers o f (CHW (X = N, P, As and SIH) and C4Si+i6 133
V2. The comparable bond length in V3 with the corresponding C-C bond length in
pristine benzvalene confirms this.
In C1, trans-C2 and cis-C2, the bond lengths in the three membered rings are
shorter when compared to the corresponding standard bond lengths. The bridging C-C
bond in T 2 is substant~ally shrunk compared to the C-C bond in the cis isomer, D2,
whereas all other bonds are elongated. 1401
V l b . C , ( I ) Vl a,. C, (0)
Figure 4.9: The principal geometric parameters in the valence isomers of silabenzene obtained at the B3LYP level using 6-31G* (normal), cc-pVDZ (underlined) and 6- 311+G** (italics) basis sets and MP216-31G** level (bold). All values are given in A. The number of imaginary frequencies is given in parenthesis, the point group is also given.

Choptcr I V : Isoms of ( C H M (X : N, P. As and SIH) and &sa2u6
Figure 4.9 (contd.): The pnnc~pal geometric parameters in the valence ~somers of silabenzene obtained at the B3LYP level using 6-31Gf (normal), cc-pVDZ (underlined) and 6-31 1+G** (itahcs) bas~s sets and MP216-31G** level (bold). All values are given in A. The number of imaginary frequencies is given in parenthesis, the point p u p is also given.

Chaptcr I V Isomers of (CHhX (X = N, P, As and SIH) and C,Sl2H6 135
4.3.2.2: Relative Energies
Table 4 5 glves the relatlve energles obta~ned at vanous levels of theory,
~ncludmg the best estlmates calculated uslng equatlon 4 2 The trends obtalned at vanous
levels of theory are essentially ~dentlcal w ~ t h rnlnor quanntatlve d~fferences The
tendency of the dens~ty funct~onal methods to overestimate the stab~llzatlon of the n-
delocal~zed structure compared to the nondelocallzed structures IS reflected In the present
case also " The d~scuss~on on the relat~ve energles will be based on the best estlmates,
throughout the rest of thls sect~on unless othenv~se spec~fied
Table 4.5 The relative energles obtained at vanous levels of theory and the best estlmates of the relatlve energles, of the valence Isomers of s~labenzene All values are given In kcallmol -- --
B3LYPl B3LYPI B3LYPI MP2I MP21 CCSD(T)/ Best Shucture 6-31G* cc-p\'DZ 6-31 ItGI* 6-31G** 6-31 l+G**' 6-3 IG*' ~s t lmatc~ ' B1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 Vla 25 2 240 268 205 202 222 210 1'1 b 30 5 290 315 280 275 289 275 Vla, 23 4 22 2 25 0 20 1 199 212 205 Vlb, 276 265 291 244 242 254 249 V2 480 496 490 419 404 432 404 V3 78 0 78 4 79 3 69 5 68 9 71 1 68 8 Dl 427 449 434 392 385 375 355 D2 72 1 729 729 659 649 648 624 PI 89 1 902 913 831 815 848 813 C1 85 4 879 840 842 829 822 782 trans-C2 85 1 84 6 85 1 890 89 6 83 4 82 1 cis-C2 88 0 874 880 919 924 860 846 T2 1323 131 8 1323 1280 1253 1248 1203 PY - 612 590 619 594 563 617 558 Slngle polnt calculat~ons on MP216-31G** opt~m~zed geomeines I l e best estlmates are calculated uslng equatlon 4 2 The enthalpy correction obtalned at the B3LYPl6-31 1+G8* level, scaled w~th a factor of 0 98
Flgure 4 10 ~llustrates the vanatron of the relatlve energles of the vanous Isomers
cons~dered In the study The best estlmates of the relatlve energles for the comspondlng
alence lsomers of benzene were also glven In the sanle figure for cornpanson The two
translt~on states (Vla and Vlb) and the pyram~dal structure (PY) are excluded, as they do
not Correspond to mlmrna on the potentla1 energy surface However, all s~labenzene

Chaptcr IV komcrs of ( C H N (X = N. P, AS and SIH) end C,SI~H( 136
~somers Ite closer to the reference compound, s~labenzene mndaatlng that the suhst~tut~on
reduces the energy gap among the vanous valence Isomers Thus, the transltlon state
structures, V l a and V l b he ~ u s t 21 0 and 27 5 kcallmol h ~ g h energet~cally above the most
stable s~labenzene, B1 It IS to be noted that the correspond~ng energy d~fference between
benzene and benzvalene 1s as h ~ g h as 71 7 kcaUmol The two correspond~ng non-class~cal
valence lsomenc mlnlmum energy structures, Vla. and Vlb, are less stable than
s~labenzene only by around 20 and 25 kcaVmol respect~vely The non-classmal nature of
Vla, and Vlb, coupled w ~ t h the11 prox~m~ty to the s~labenzene warrants expenmental
attempts towards them V3 IS found to be thermodynam~cally less stable than the other
benlvalene Isomers, the reason be~ng the weak rr-bondlng a b ~ l ~ t y of SI w ~ t h C The
framework seems to broadly declde the relat~ve energ~es to a greater extent compared to
the substltutlon pattern Thus the relatlve energy ordenng IS qu~te s ~ m ~ l a r to those
observed In the valence Isomers of benzene There 1s a stnk~ng d~fferentlat~on In the
stab~l~zat~on of the pos~t~onal Isomer, depending on the nature of the replacement slte,
thus replacement by SI at the saturated scte 1s ovenvhelm~ng preferred compared to the
unsaturated sltes Thus, although the skeletal replacement by SI causes substantla1
stablhzat~on among the valence Isomers of s~labenzene In general, the stabll~zat~on 1s
rnarglnal In the lsomers contalnlng C=SI S~m~larly the weak C=SI" 55 bond In D2 makes
~t energet~cally above Dl C1, wh~ch has two straned three membered nngs, IS expected
to be less stable than trums-CZ and cis-C2, both hav~ng only one three membered nng
But, C1 IS observed to 11e below both the C2 Isomers, whlch may be due to the presence
of d~valent s ~ l ~ c o n In the latter The C=SI present In the trans-Dewar benzene Isomer, TZ
renders lt as a least stable lsomer and IS computed to Ile about 120 kcallmol w ~ t h respect
to s~labenzene Interestingly, PY, whtch 1s a hlgher order saddle po~nt was computed to
11e only 55 S kcallmol h~gher and becomes more stable than many other valence Isomers
o i sllabenzene T h ~ s adds to the amazlng list of contrasts that were witnessed In the
chemlstnes of organlc and sllaorganlc compounds In all cases, there 1s a s~gnlficant
destab~l~zat~on In the Isomers where the C=SI exlsts, a result In aaeement wlth the
class~cal double bond rule

thaptcr I V : ISOMS of (CHW (X : N, P. AS md SiH) 4 C4Si2& 137
Structure
Figure 4.10: The correlation of the best estimates of relative energies of the valence isomers of silabenzene. The relative energies of corresponding benzene valence isomers are given for comparison.
4.3.2.3: Chemical Hardness
Absolute chemical hardness (7) has been used as a measure of kinetic stability or
the reactivity of organic ~ o m ~ o u n d s . ~ ~ ~ ~ ' Within the Koopman's approximation, hardness
( 7 ) is defined as half of the magnitude of the energy difference between the energies of
the HOMO and the LUMO.
7 = (ELIJMO - EHOMO)R . . .eq. 4.3
The frontier orbital energies and the chemical hardness computed at B3LYPl6-
311+G8* level is given in Table 4.6. While the thermodynamic stabilities of the
compounds under study are controlled by the skeleton of their structure, the kinetic
stability seems to be dictated by the bonding type of Si atom. C1, V2, PI and Dl, where
the Si occupies the sp3 centre, are kinetically stable than the other isomers. Whereas B1,
D2, V3, Vla,, T2 and V2b, are more reactive; in all these cases Si is tri-coordinated.
Compounds containing divalent Si, rransC2 and cis-C2 are found to be the least stable.
SO the kinetic stability exactly follows the order: Isomers containing tetracoordinated

Chapter IV: 1~0tners of (CHhX (X = N, P. AS and SiH) and C4SizH6 138
Si>isomers containing hicoordinated Si>isomers containing dicoordinated Si. The
relative stabilities and the hardness values do not have a linear relationship. Thus, quite a
few valence isomers (Vl, Dl, P1 and V1) have much higher hardness values than the
benzvalene isomers, Vla, and Vlb,, which are very stable energetically, according to
the hardness criteria correspond to least stable compounds.
Table 4.6: The frontier orbital energies along with the Mulliken symbols and the absolute chemical hardness (7) of the valence isomers of silabenzene obtained at the B3LYP16- 3 1 I S * * level. All values are given in eV.
Structure €HOMO ELUMO tl
B1 -6.02 (B,) -1.14 (B!) 2.44
Vln. -6.24 (A') -2.16 (A") 2.04
Vlb, -5.70 (A') -2.36 ( A ) 1.67
V2 -6.58 (A") -0.75 (A") 2.91
V3 -5.23 (A") -l.IO(A") 2.07 Dl -6.80 (A') -1.32 (A') 2.74
D2 -5.81(A) -1.35(A) 2.23 PI -5.87 (A") -0.30 (A') 2.78
C1 -6.78 (A') -0.78 ( A ) 3 .OO
trans-C2 -5.88 (A') -2.62 ( A ) 1.63
cis-C2 -5.81 (A') -2.74 (A") 1.54
TZ -5.32 (A) -1.83 (A) 1.75
4.4: Isomers of Disilabenzene
The present study reporls B3LW and CCSD(T) computed results of
Jisilabenzene valence isomers and some related structures (Scheme 4.5-4.8). By no
means we have considered all the possible isomers. See, e.g., benzene has more than 200
isomers. Thus C4SiZH6 will have well over 1000 isomers. But, in this study a systematic
attempt is made to consider most of the stable isomeric forms. The structures considered
are classified as (a) monocyclic, (b) bicyclic, (c) tricyclic and (d) tetracyclic. The
equilibrium geometries of all the stationary points located 011 the potential energy surface
and their relative energies are discussed in the above order. Comparisons were made with
the corresponding benzene, silabenzene and diphosphabenzene valence isomers in some

w t w I V Isomers of (CHhX (X = N. P. As and SIH) and C , S , ~ H ~ 139
4.4.1: Computational Details
All the structures glven In Schemes 4 5-4 8 were fully optlmlzed w~thln the
symmetry constra~nts at the B3LYP level"47 w ~ t h 6-31G+ bas~s set The statlonary polnts
obtalned uslng the default gradlent procedures were charactenzed based on the frequency
calculat~ons The normal modes corresponding to the lmaglnary frequencies of the
transltlon states and hlgher order saddle polnts were followed and the true mlnlma were
located Slngle polnt energy calculat~ons at the B3LYPIcc-pVTZ level on the B3LYP16-
31G* optlmlzed geometnes were done to estlmate the effect of bass set on the relat~ve
energies Couple cluster method was found to y~eld excellent energetics for thls class of
compounds 4951 Therefore, we have performed CCSD(T) slngle polnt calculat~ons w ~ t h
the 6-31G* bass set on the B3LYP opt~mlzed geometr~es The best est~mate, AE glven In
following equatlon 1s expected to y~eld results slmllar to CCSD(T)lcc-pVTZ level, even
though the effect of Increase In the qual~ty of the basls set 1s evaluated only at the B3LYP
level
AE = AEcrsnii + AE(BILYPIC~ PVTZ BXYPM JIG.) + AH eq 4 3
Enthalpy correction values (AH) are Included from the frequency data obtalned at the
B3LYPl6-31G* level All the theoret~cal methodolog~es employed are based on the slngle
detenn~nantal approach Hence, we have performed CASSCF and CASPTZ" calculat~ons
with 6-3 IG* and cc-pVDZ bass sets by lncludlng the x-system In the actlve space for I ,
2 and 3 The coefficients of the major Slater deternunant are found to be more than 0 92
In all the cases at the CASSCF level e~ther wlth 6-31G* or cc-pVDZ bas~s set Thls
~nd~cates that non-dynam~c electron correlat~on 1s not declslve and single determlnantal
approaches adequately descrlbe the electron~c structure and bondlng In thls class of
compounds All denslty functional theory calculat~ons were performed uslng the Jaguar
4 1 program package Gauss~an 98 su~te of program was used to perform the CCSD(T).
CASSCF and CASPT2 calculat~ons " The graphical Interface program, Moplot was used
to examlne the equll~bnum geometnes 59
4.4.2: Results and Discussion
A total of s~xty SIX Isomers were consldered ln~t~ally (Scheme 4 5-4 8) Upon
geometry opt~m~zatlon and charactenzat~on of the nature of the statlonary po~nts of all the
structures consldered. 61 of them were confirmed as the true mlnlma possessing all real

Chapter I V . ISOM of (CUM (X N, P, AS d SIH) and C ~ S I ~ H ~ 140
harmonic fnquencles However, some are found to be transltlon states, some are lugher
order saddle polnts and some do not correspond to a statlonary point on the dlsllabenzene
~otentlal energy surface The normal modes corresponding to the tmaglnary fiequencles
of the saddle polnts were followed In each case and the mlnlmum energy structures were
ohtamed. whlch Increases the total number of statlonary polnts to 78 Among them, 61
were charactenzed as mlnlma, 12 as transltlon states, 4 as second order saddle polnts and
one structure was found to be a thlrd order saddle polnt The equ~l~bnum geometnes of
the opt~mlzed statlonary po~nts and thelr nature on the potentla1 energy surface are
discussed first They-are arranged m the follow~ng order, vlz monocycl~c, blcycl~c.
tncycl~c and tetracycllc Isomers Then, the relatlve energles of the vanous isomers are
d~scussed Vanous factors, controlling the relatlve energy ordenngs, are addressed bnefly
In general as well as In each group of the pos~t~onal lsomers
4.4.2.1: Equilibrium Geometries
4.4.2.1.1 : Monocyclic Isomers
F~gure 4 11 deplcts the important geometnc parameters of the dls~lahenzenes and
other monocycllc Isomers obtnned at the B3LYPl6-31G' level For the dlmlabenzenes
(1-3), the reported bond lengths obtalned at the B3LYPicc-pVTZ level are given m
parentheses The geometnc parameters obtalned uslng the two bass sets are very slmllar
w~th a maxlmum dev~at~on of 0 007 A
We also have cons~dered some monocycl~c structures (4-15) where the rr-
lelocahzat~on m the three dlsllabenzenes 1s d~smpted and concomltantly one or both the
SI atoms become dlvalent Among the monocycltc Isomers w~th one d~valent sllrcon (4-
11 ), the planar forms of those structures where the dlvalent slllcon 1s present adjacent to a
CHI group are found to be trans~tlon states (5, 7.8 and 11) The corresponding mrnlmum
energy structures (Sm, 7m, 8m and I lm) were then rdentrfied Importantly, at the
CCSD(T) level, 8m and l l m he hgher in energy compared to 8 and 11 respectively
Th~s reveals that the planar forms are very l~kely to be mlnlma at the CCSD(T) level
~ndlcatlng that the B3LYP method incorrectly designates the nature of the planar
molecules ~h~ lndlcates that the out-of-plane dlstortron may be traced to the llmltat1on
of B3LYp method and slmllar observabon was made earher All the m~lecules with two
dlvalent slllcon atoms are found to be h~gher order saddle Points (12-15) The mmlmum

Chapter IV : ISOWS of (CHM (X = N, P, As and SiH) and C4SizHs 141
energy structures 12m. 13m and 14m correspond to pyramidal like structures, with one
of the Si atoms forming the apex of the pyramid. Unlike in the previous case, the planar
forms lie much higher in energy compared to the non-planar structure. 16, where one of
the hydrogens is bridged between the two Si atoms, is found to be a transition state. The
Si-Si bond length is too long (2.922 A) and the connection between the two may be
traced mainly through the bridging hydrogen atom. The normal mode of the imaginary
frequency corresponds to the puckering of the bridged hydrogen, which was then
followed and 16m was obtained. Therefore, 16 may be treated as a transition state
interconnecting the two identical forms of 16m. In 16, all the C-C and C-Si bonds are
almost comparable to the m a t i c bond lengths, whereas in the minimum energy
structure (16m) the bonds are localized. Similar to 16, the Si-Si bond length in 17 is
substantially elongated with the bridging hydrogens strongly bound to both the silicon
atoms. 18 is found to be a transition state and following the normal mode corresponding
to the imaginary frequency yielded 48.
4.4.2.1.2: Bicyclic Isomers
The equilibrium geometries of the bicyclic isomers (Scheme 4.6) considered in
the present study are discussed in this section. Figure 4.12 gives the principal geometric
parameters of the bicyclic isomers obtained at the B3LYPi6-31G* level. In our previous
study on the silabenzene valence isomers, we identified two bicyclic structures where a
SiH unit is bound to a cyclopentadienyl species in a r12 fa~hion.~'' These unusual
s*.uctures were found to be the next stable species to silabenzene in the (CH)5SiH
potential energy surface. We have considered similar structures, 19-24 (Scheme 4.6) out
of which, stationary points corresponding to 19, 20.22 and 24 could be located and were
found to be minima. Initial structures of 21 and 23 upon optimization collapsed to 48
indicating that these are not stationary points on the potential energy surface. In 24, the
C-Si bond with the r12-bound SiH moiety seems to be too long and in contrast Si-Si is
quite normal indicating that its connectivity is close to that found in 18. The greater
stabilization in 24 compared to 18 may be traced to the higher delocalization in the five
.nembered ring in the former.
The stationary points corresponding to the six Dewar benzene isomers (25-30)
were obtained and the frequency calculations indicate that all the Dewar benzene isomers

Chnptcr I V I s o m o f (CHhX (X = N, P, As and SIH) and C,SlzH6 142
arc nuluma except 28 In all the cases except ~n 25, the bndgng bond IS longer compared
to the cowpond~ng standard bond lengths, a feature whlch 1s observed In the parent
Dewar benzene itself The SI-SI d~stance m 27 1s computed to be too short However, the
bond order calculated usmg Atoms m Molecules AIM)^ at the B3LYPl6-31G8 level of
theory 1s only 0 04 suggestmg that bond between the two SI atoms does not exlst The
normal mode of the rmaglnary frequency In 28 corresponds to the out-of-plane d~stortion
of the hydrogen atoms connected to the two SI atoms The mlnlmum energy structure
correspondlng to 28 IS obwned following the dlrectlon of the lmagtnary frequency
normal mode 28m 1s found to have a twlsted double bond where the hydrogens
connected to the SI atoms stay out-of-plane Th~s IS s~mllar to the sltuatlon ~n the slhcon
analog of ethylene where the D2h fonn of H2S1=S1H2 IS a transltlon state, the
correspondlng mlnlmum energy structure tnvolves substantial pyramidal~zat~on at the SI
center b' Thus there seem to an Inherent tendency to have puckered geometry for the
SI=SI, but considering the fact that the planar 1,2-dls~labenzene (1) corresponds to
mmima, thls puckering could be prevented when a stronger symmetrlzlng force 1s
operatlve All of our attempts to locate a statlonary compound correspondlng to 32 were
fut~le, upon geometry optlmizatlon the lnltlal structures collapsed to 48 The frequency
calculations designate the five statlonary polnts for the trans-Dewar benzenes as mlnlma
on the potent~al energy surface In all the Isomers except m 33, the bndgmg bond 1s short
compared to that In the correspondlng as-compound Most of the computed peripheral
bond lengths In the trans-Dewar benzene Isomers are longer compared to those in the cry
Isomers, which 1s lnd~catlve of hlgher stratn In the trans lsomers S~milar to 27, despite
the Si-SI Interatomic dlstance of 2 476 A In 33, the bond order calculat~on does nor
support any bond between the SI atoms
Among the b~cyclopropenyl Isomers (40-45), 43 1s computed to be a transltlon
state The normal mode of the lmaglnary frequency corresponds to the out-of-plane
dlstortlon of two hydrogens connected to the SI atoms Opt~mizlng 43 wlth the two
hydrogen atoms out-of-plane, lead to I-43m, where one of the C-SI bonds becomes too
long to glve a monocycl~c compound, w~th only one three membered nng In the process,
the sp3 carbon has become an sp2 c h n and one of the SI atoms becomes dlvalent The
correspondlng cls Isomer, c43m was also opt~mized and was charactenzed as a mlnlma

C h a p t ~ L W W S of (CHM (X = N. P, As and SiH) Md C4SilH6 143
SimilWl~, 42 On optimization leads to a monocyclic compound, r-42 with one divalent
silicon; the cis compound, c-42 was also then identified. ~ 0 t h 44 and 45 upon
optimization collapses to an acyclic compound, n-44 with two divalent silicon atoms; the
corresponding cis-tram isomers (cc-44 and 13-44) were then characterized as minima on
the disilabenzene potential energy surface. This conIrast with the parent bicyclopropenyl
can be explained based on the instability of divalent carbene in contrast to the divalent Si.
The propensity of three membered rings containing a tricoordinated Si for ring opening
may be understood based on the following points: (1) it relieves strain in the three
membered ring, (2) one C-Si n-bond is replaced by C-C n-bond, (3) it leads to divalent
Si. Isomers, 37 and 46, where all the hydrogens are replaced by tertiary butyl groups have
been synthesized recently and photochemical rearrangement reaction has been observed
between these two c o m p o ~ n d s . ' ~ ~ ~ ~ In both cases especially in 46, the Si-C single bond
lengths are slightly shortened compared to the standard bond length. 38 is computed to be
minima while its positional isomer, 39 is a transition state. Following the nonnal mode
corresponding to the imaginary frequency and optimizing th~s collapsed to 22.
4.4.2.1.3: Tricyclic Isomers
The principal geometric parameters of all the tricyclic obtained at the B3LYP/6-
31G* level are given in Figure 4.13. Initially, efforts were made to locate the stationary
points for the seven possible disilabenzvalene isomers (48-54) depicted in Scheme 4.7.
However, exhaustive attempts could not yield a stationary point corresponding to 53. All
the putative structures of 53, upon optimization relaxed to 48. Thus, the computations
Imply that 53 is not a stationary point on the disilabenzene potential energy surface. All
other stationary points were characterized as minima, except 54, which was characterized
as a transition state. Seven disilabenzvalenes were located, out of which six were
characterized as minima. In 48 the Si atom, which is in the bridgehead of the
bicyclobutane moiety, is weakly bound to two carbon atoms and the other silicon.
However, this isomer is the one, which is energetically more stable, compared to the
other benzvalene isomers. The five membered ring formed by one of the Si atoms and the
four cabon atoms is virtually planar and the bond distances are substantjally short
compared to the standard single bond lengths. Thus, 48 may be considered
as an S ~ H + ion bound to the silacyclopentadienyl species in an ?' fashion to the CCSi

Chaptv I V : IsomuS of (CHW (X = N. P. As and SiH) and C,SizH6 1 44
ort ti on. The computed bond lengths in 49, 51 and 52 exhibit normal bond distances
except for the bridging bond of the bicyclobutane moiety in 49, which is slightly
elongated. In 49, the Si-Si distance is computed to be only 2.639 A. In 50, the computed
Si-Si and C-C single bond lengths are shorter compared to the normal bond lengths. The
frequency calculation characterizes 54 as a transition state, and the imaginary frequency
normal mode corresponding to out-of-plane distortion of the hydrogens attached to the
Si=Si, similar to the situation in 28 and 43. Following this normal mode, the minimum
energy structure 54m is reached where the silicon center is pyramidal. In both 54 and
541% the bridging bond of the bicyclobutane moiety is found to be very short compared
to the corresponding C-C bond in other isomers.
Benzvalene isomer, 54, with one hydrogen bridging between the two Si atoms
(55) was computed to be a second order saddle point. Following the normal modes of the
imaginary frequencies the structure 55m was obtained, which has a tetravalent and a
divalent Si. However, similar structure, 56 with two bridging hydrogens was conlputed to
be a minimum energy structure. In 56, the Si-Si bond length is too long compared to the
normal Si-Si single bond distance. Similar to the structures, 19-24, we have considered
57-63, where two SiH units are bound to a cyclobutadien~ moiety in various possible
modes. Among these structures, 61 and 63 are minima; 57,58 and 60 are computed to be
transition states. Many attempts to locate a stationary point corresponding to 59 failed
and all the initial geometries collapsed to 20 upon optimization. 57m and 58m
correspond to distorted forms of 57 and 58 respect~vely where, two C-SI bonds are
significantly elongated. These compounds can be viewed as cyclobutenes, where t u o
hydrogens connected to sp' carbon atoms are substituted by SiH units. D~s ton~ng the
skeleton of 60 following the normal mode of the imaginary frequency and opt1n1171ng
lead to 20. Initial structures of 62 on optimization leads to a molecule where one of the SI
is tetravalent with two hydrogens connected and the other is divalent Si (Flgure 4.13).

Clwpter I V . Isomers of (CHM (X = N, P. As and SOH) and C I S I ~ H ~ 145
4, C,; 17.5 (0) 5, C,; 27.2 (1) 5m, CI; 25.1 (0)
6 , C,; 30.0 (0) 7, C,; 21.0 (1) 7m, C1; 20.9 (0)
8, C,; 16.3 (1) 8m, C1; 16.1 (0) 9, C,; 17.5 (0)
I'tgure 4.1 1 (contd.) The prlnclpal geometric parameter? of Lhc mooocycl~c Isomers ohtnlned at the B3LYPl6-31G* level Those ohtalned at the BiLYPIcc-pVTZ are glven 111 parentheses only for 1, 2 and 3 All values are glven In A The polnl groups, best emnate of the relat~ve energles and number of tmaglnary tiequencies are glven

10, C2v; 13.8 (0) 11, C,; 34.4 (1) l l m , C1; 34.7 (0)
2 j40
14, C,; 34.4 (2) 14m, C1; 23.8 (0)
Figure 4.11 (contd.) The pnnc~pal geometnc parameters ol the m o n o ~ y ~ l l ~ Isomer\ obtalned at the B3LYPl6-3IG* level Those obtalned at the B3LYP/cc-pV7Z arc glvcn In parentheses only for 1, 2 and 3 All values are glven In A The po~nt groups, best estlmate of the relat~ve energles and number of imaginary frequcnc~es are glven

Chapter I V . Isomers of (CHM (X = N. P, As and SIH) and C I S I ~ H ~ 147
16, C,; 47.0 (1) 16m, CI; 16.6 (0) 17, C2,; 32.8 (0)
18, C,; 71.7 (1)
Figure 4.11 The pnnc~pal geometric parameters of the rnonocycl~c Isomers obtatned at hc B3LYPi6-31G* level Those ohtamed at the B3LYP:cc-pVTL are glvcn In ~drenthcscs only for 1, 2 and 3 All values are given In A The polnt group, best estlmate IS the relatlve energles and number of Irnagmary Srequencles are glven

Chapter I V : Isomers of (CHhX (X = N, P. As ond SIH) and C4SlzHb 148
19, C,; 30.8 (0) 20, C,; 35.3 (0) 22, C1; 22.2 (0)
27, C1; 21.9 (0) 28, C,; 49.3 ( 1 ) 28m, Cl. 47 4 (0)
29, C2; 64.8 (0) 30, C,; 61.1 (0) 31, Czh, 88.5 (0)
Figure 4.12 (contd.) The pnnc~pal geometnc parameters (11 the ~ I L Y L I I C isomers
obtmned at the B3LYPl6-31G* level All values are glven In A The point groups, bcst estlmate of the relatlve energles and number of Imaginary frequencies are glven

Chapter I V : Isomers of ( C H M (X 1 N, P. As and SIH) and C4S~&t6 149
36, c,; 106.1 (0) 37, C2h; 26.0 (0) 38, C,; 66.0 (0)
40, czh; 48.6 (0) 41, cI; 75.0 (0) C-42, c,; 56.8 (0)
t-42, C,; 54.4 (0) 43, C,; 98.5 ( I ) c-43,, C,; 85.9 (0)
Figure 4.12 (contd.) The pnnc~pal geometnc parameters of the b ~ c y c l ~ c Isomers ohtamed at the B3LYPi6-31G* level All values are glven In A The polnt groups, best estlmate of the relat~ve energ~es and number of lmaglnary frequenc~es are glven
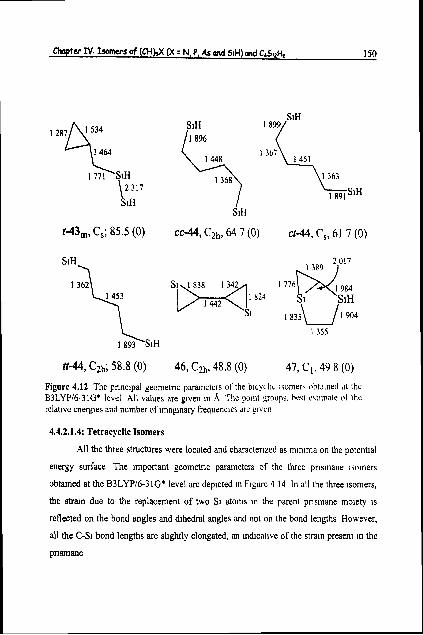
Chapter I V . Isomers of (CH)3( (X = N. P, As and SiH) and C ~ S I Z H ~ 150
SiH 13x9
1 362 slms: 824 776&i8i
Figure 4.12 The pnnc~pal geometric parameters of the ~ I L Y L ~ I L IwIIior\ obtillned at thc B3LYPl6-31G' level All values are glven In A Thc polnt groups, ho~t c\tlmate ol tho relatlve energles and number of lmaglnary frequencies 'Ire g1vc11
4.4.2.1.4: Tetracyclic Isomers
All the three structures were located and character~zed as mlnlma on the potentla1
energy surface The Important geometric parameters of the three prlsmane Isomers
obta~ned at the B3LYPl6-31G* level are deplcted In Flgurc 4 14 In all the three Isomers,
the straln due to the replacement of two SI atoms In the parent prlsmane molety IS
reflected on the bond angles and d~hedral angles and not on the bond lengths However,
all the C-Si bond lengths are sl~ghtly elongated, an lndtcatlve of the straln present In the

Chapter IV: Isomers of (CHM (X = N. P. As and SIH) md G S I Z H ~ 151
51, '2,; 38.3 (0) 52, C,; 48.4 (0) 54, Czv; 60.2 (1)
Figure 4.13 (cootd.) The principal geometric parameters of the tricyclic Isomers obta~ned at the B3Ll-P/6-31G* level All values are glven In A The point groups, best estlmate of the relati\ e energles and number of maginary frequenc~es are glven

Chaptv IV . Isomcrs of (CHM (X = N. P, As and SIH) and C4S2H6 152
61, CzV; 109.3 (0) 62, C,; 72.8 (0) 63, CZ; 72.6 (0)
Figure 4.13 The pnnclpal geometnc parameters of the t n c y c l ~ ~ Iwtners ohtalned at the B3LYPl6-31G* level All values are glven In ,A Thc polnt group\, best cstlmate ot'thc relatlve energles and number of ~maglnary frequcncle~ arc given
64, C,; 56.3 (0) 65, C2v; 67.0 (0) 66, C2, 56.0 (0)
Figure 4.14 The principal geometr~c parameters of the t e t rncy~ l l~ Isomers obtained at the B3LYPi6-31G* level All values are glvcn In A The polnt groups. bust estlmate of the relatlve energles and number of lmaglnary frequen~~es are glverl

C b p t ~ IV I S O ~ W S of (CHbX (X : N, P. As and SIH) and C,!j~~b 153
4.4.2.2: Relative Energies
The relatlve energ~es of the three dlsllabenzenes have been studled extensively by
theoretical calculat~ons of vanous levels of soph~st~catlon, the results of whlch are glven
In Table 4 7 The complex~t~es In choosing the rehable computat~onal procedures for
s~labenzenes was thoroughly lnvestlgated by Baldndge et al and thelr systematic and
elaborate calculat~ons on a serles of sllabenzenes inferred that B3LYP level w ~ t h cc-
pVTZ qual~ty bas~s set IS the rel~able approach for getting the correct energetlcs
compared to the conventional HF or MP2 methodolog~es~' B3LYPicc-pVTZ method,
wh~ch was shown to y~elds results comparable to that of the CCSD(T)/cc-pVTZ level IS
chosen as the method of cholce In thls study to model the other non-planar Isomers
cons~dered In the study Consldenng the lack of mult~determmantal nature of the
wavefunct~ons, the CASSCF and CASPT2 approaches do not seem to be appropnate for
t h ~ s class of compounds Table 4 7 reflects that the qualitat~ve agreement of MP2 and
B3LYP IS much better w ~ t h h~gher level calculat~ons compared to that of CASPT2
The B3LYPl6-31G*, B3LYPIcc-pVTZ, CCSD(T)I6-31G* relatlve energ~es and
thc best estlnlates of the relatlve energies obtalned uslng equatlon I are given In Table
4 8 The B3LYP level computed relattve energles uslng the 6-31G* and the cc-pVTZ
hasls sets are m close agreement wlth each other with a maximum deviation of 3 5
kcallmol The qual~tat~ve trend of the relatlve s tab~l~t~es obtalned at all the levels of
theory employed IS essent~ally the same The dlscuss~on on the energetlcs wlll be based
on the best estimates of the relatlve stabllitles method unless othenvlse stated

Chapter IV: Isomers of (CHW (X = N, P, As and SiH) a d C,S,H, 154
Table 4.7: The relative energies of 2 and 3 with respect to 1 obtained at various levels of theory. All values are given in kcal/mol.
Method 2 3
HFl6-3 IG* -2.6 13.4
Thls work. Taken from Ref. 27.
" From Chapter 111.. Slngle point calculations on the MP216-31G** optimized geometries. Single point calculations on the B3LWl6-3 IG* optimized geometnes. Single point calculat~ons on the B3LWIcc-pVTZ optimized geometries.

Chaptv IV: Isomers of ( C H H (X : N. P. As and S i n ) ond C4Si2H, 155
Table 4.8: The relative energies (kcaVmol) of the isomers of disi labeme obtained at the B3LYP and CCSD(T) levels, the best estimates of relative energies. The number of ~maginaty frequencies (NIMG) obtained at the B3LYPl6-31Gg level is also given.
B3LYPl CCSD(T)I Best SbUcmre NIMG !-%g =-pVTZ1 6-31C*' bsflmatcb
1 0 0.0 0.0 0.0 0.0 2 3 4 5 Sm 6 7 7m 8 8m 9 10 11 l l m 12 1 2m 13 13m 14 14m 1s 15m 16 16m 17 111 19 20 22 24 25 26 27 28 28m 29 30 31

66 0 63 2 62 3 58 9 56 0 Slngle polnt calculations on B3LYPi6-31G* opt~m~zed geometnes

Chapter IV Isomers o f ( C H M (X = N, P, As and S I H ) and C4S~,H6 157
4.4.2.2.1: Monocyclic Isomers
Among the three dlsllabenzenes (Figure 4 11). only the denvatlve of 1.4-
d~sllabenzene, 3 1s expenmentally known and rearrangement reactions have b m studred
Interestlngly, thls 1s the least stable Isomer among the three dlsllabenzenes 1.3-
dlsllabenzcne (2) 1s found to be more stable than 12-dtsrlabenzcne (1) by about 3
kcalimol Thls 1s In contrast to the sltuatlon In d~phosphabenzenes, where 1.2-
dlphosph~ntne was found to be the global mlnlma 48 The relatlve stablhty ordenng of the
skeletally subst~tuted benzenes has been one of the most lntngulng aspects and several
mutually Independent factors were found to be respons~ble (See Chapter 111)
Among the monocycl~c isomers, where the rr-delocal~zat~on 1s disrupted (4-IS),
10 1s found to be the most stable one and IS found to he only about 5 kcal/mol above tts
aromatlc counterpart. 3 Whereas, 4 1s less stable than 11s dlsllabenzene counterpart, 1 by
about 17 kcaVmol T h ~ s lndlcates that ~t 1s more expensive to dlsmpt the rr-delocal~zat~on
In 1 than In 3 Among those structures, where one of the slhcon 1s dlvalent and the other
St 1s tncoordlnated (5-9, l l ) , 7m, 8m and 9 are more stable than the rest The orlgtn may
be traced to the preference of SI atom to occupy 1,3 posltlon for electrostat~c reasons
Among the planar forms of the compounds conta~nlng two dlvalent slllcon atoms (12-IS),
14 1s the most stable one where the two SI atoms occupy 1,3 posltlons These structures
were computed to be hlgher order saddle points Among the non-planar Isomers, 13m 1s
the most stable, where one of the SI atoms 1s bound to the other SI and three carbon
atoms 16m 1s less stable than 1 by 16 kcalimol even though they possess equivalent
skcleton Thls may be due to the fact that the brldgtng bnngs In locallzat~on In the
benzene skeleton, whlch 1s reflected In the geometries Thls effect 1s furthered In the
d~br~dged structure, 17 whlch l ~ e s around 32 kcaYmol htgher than 1
4.4.2.2.2: Bicyclic Isomers
In addltlon to the class~cal valence lsomenc analogues, Dewar benzene and
blcyclopropenyl, several other Isomers were cons~dered In tbls category S ~ m ~ l a r to the
case In a monosllabenzene Isomer, where SIH 1s coordinated to cyclopentadlenyl molety
11: a tll fashlon, 22 and 24 he only about 25 kcaVmol hlgher than the global mlnlma The
charge analysts lnd~cates that the five membered nng formed by one SI and the four
carbon atoms IS negatively charged and the SIH possesses Posltlve charge The high

Chapter I V Isomcrs of (CHM (X = N. P. As and SIH) a d C.SlrHa 158
stab~l~ty of this class of compounds may be attributed to the ammatlc stab~hzatlon of the
five membered nng
Among the Dewar benzene Isomers (25-30), 25 IS the most stable Isomer and l ~ e s
only about 21 kcaVmol hgher than the global mlnlma In ttus Isomer, both the s~llcon
atoms are tetracoord~nated It 1s to be noted that the parent Dewar benzene 1s less stable
than henzene by about 75 kcallmol 27 and 26, where one of the SI 1s coord~nated and the
other 1s tncoord~nated, Ile energet~cally h~gher compared to 25 The least stable lsomers
are the ones where both the SI atoms are tncoord~nated However, 27 IS energet~cally
competltlve to 25 In splte of the fact that one of the SI atoms IS tncoord~nated In case of
d~phosphahenzenes, all the Dewar benzene Isomers Ile very close to each other (44 8-52 4
kcaWmol), whereas Dewar d~sllabenzenes span a w~de range of stabll~t~es from 18 3 to
64 8 kcaVmol As observed In case of the valence lsomers of benzene, sllabenzene and
Group V heterobenzenes, trans-Dewar benzenes are the ones those are least stable among
the valence Isomers However, one of the trans-Dewar benzene Isomer, 33 l~e s below few
b~cyclopropenyl lsomers (41, 43, c-43m and t-43m) Among the trans-Dewar benzene
lsomers (31-36). 33 1s the least stable lsomer and 1s found to be less stable than the global
rnlnlma by only 76 kcaVmol whereas the parent trans-Dewar benzene hes as high as 150
kcalimol above benzene
As observed In the Dewar benzene Isomers, the envlronrnent In wh~ch the SI atom
1s present seems to control the relatlve s t ab~ l~ t~es of the b~cyclopropenyl Isomers One
mlght expect that n-45, wh~ch does not have even a s~ngle three membered nng would be
more stable than the other lsomers hav~ng one or two stmned three membered nngs But,
tt-45 IS found to be less stable than 40, wh~ch has two three membered nngs The relat~ve
stabll~ty ordenng seems to be controlled by the pos~tlon of the Sl atoms T h ~ s lndlcates
that the subst~tutlon pattern plays an important role In dec~d~ng the relat~ve stab~l~ty of the
Isomers m t h ~ s class of compounds In addltlon to the stra~n ~n the skeleton Th~s 1s tn
contrast to the d~phosphtn~ne Isomers, since the most stable b~cyclopropenyl lsomer 1s the
one where both the phosphorous atoms occupy sp2 centers and the least stable Isomer 1s
the one where the P atoms are present In sp3 centers 37 and 46, whose denvat~ves were
synthes~zed recently, are unstable than the global mlnlma only by 29 and 52 kcaVmol

Chopter IV Isomers of (CHM ( X - N, P, As ond SIH) Md C4S01H6 159
respect~vely Both of them have two str;uned four and three membered nngs respectively,
but are energet~cally competltrve w ~ t h the other Isomers
4.4.2.2.3: Tricyclic Isomers
In t h ~ s sect~on, the relat~ve stab~htres of benzvalene lsomers and some other
tncychc non-classical structures are discussed As observed In the Dewar benzene and
b~cyclopropenyl lsomers, molecules hav~ng two tetracoord~nated SI atoms are stable
compared to those contalnlng tncoordinated Si atoms T h ~ s can be attnbuted to the weak
rr-bondmg a h ~ l ~ t y of SI either w ~ t h C or w ~ t h another SI 48, where one of the SI atom 1s
bound to the five membered nng formed by the other s~hcon atom and the four carbon
atoms in a ~l' fashion, IS computed to the most stable Isomer among the benzvalene
isomers 55m, where one of the SI 1s tetravalent and the other 1s dlvalent, IS more stable
than 54m, where both the Si atoms are tncoordlnated, otherw~se shanng an equivalent
skeleton, by 9 2 kcalimol T h ~ s ~nd~cates that SI atoms prefer to be more stable when In
tetravalent and d~valent form compared to be In two tncoordlnated form The drbndged
structure, 56 lies 8 kcalimol blgher In energy than the correspond~ng non-bridged isomer,
54m Among the two SIH unit capped cyclobutadlenyl systems (57-63), the trans
compounds, 57 and 58 are more stable than the correspond~ng CIS compounds 60 and 61
The Isomer, where the two SIH unlts are coord~nated from elther s~des to the oppos~te
comers ofbutadlenyl specles, 63 is found to be most stable among these lsomers
4.4.2.2.4: Tetracyclic Isomers
64 and 66 I I C close to each other w ~ t h 65 lying about 10 kcal mol ' lower
cornpared to the other two Thus, the relat~ve energy ordenng of the pnsmane lsomers 1s
as follows 66 z 64 < 65
4.5: Conclusions
Thls chapter presents a comprehens~ve theoret~cal study on the valence lsomers of
group V heterobenzenes, silabenzene and d~sllabenzene, In add~t~on to some C4SlzHb
lsomers Ten valence Isomers were located on each of the potentla1 energy surface of
(CH)5X (X = N, P and As) and found to be mlnlma The planar benzene analog 1s the
lowest energy Isomer ln all cases and the relative energy ordenng of the vanous classes
of posit~onal isomers resembles that of benzene ~n most cases The relatlve energles
obtnned uslng the MP2 method are consistently in better agreement w ~ t h coupled cluster

Chapter I V Isomers of (GI)& (X = N. P. As md SIH) and C,SIZH~ 160
compared than those obtained at HF or B3LYP levels The hybnd dens~ty funchonal
B3LYP method sl~ghtly overestunates the stablllty of the planar stmctuns wth
delocal~zed rt-systems The geometries obtaned at the MP2 and the B3LYP levels are
very slmllar In most cases and are In good agreement wlth the avalable expenmental
results
Out of the fifteen lsomers Identified on the (CH)sSiH potentla1 energy surface,
twelve of them were charactenzed as minlma on the potentla1 energy surface, two of
them were transltlon states and one 1s a second order saddle polnt The spread of the
relatlve energies of vanous sllabenzene lsomers is substant~ally smaller compared to the
corresponding benzene valence lsomers Interestingly. wh~le benzene hes above the next
stable valence lsomer by about 70 kcal/mol, Vla, is only about 20 kcallmol higher In
energy than the most stable silabenzene (Bl) However, the relat~ve energy ordenng in
silabenzene valence lsomers 1s very slmllar to that In benzene valence lsomers In general
bamng some exceptions The hardness (q) values taken as a measure of reactlvlty
indicates that compounds havlng tetracoordmated SI atoms are reactlve compared to
those havlng tn- and dl-coordmated Si The apparent d~spar~ty In energet~c and hardness
cntena In determlning the stab~l~ty and the hlgh reactlvlty of thls class of compounds are
~nd~cative of lntncacles involved in making pred~ct~ons
S~xty SIX dls~labenzene lsomers were cons~dered, which ~ncludes (CH)I(SIH)?
valence Isomers and many other related Isomers, seventy eight statlonary polnts were
located Of these, slxty one were charactenzed as mlnlmum energy structures, 12 as
transltlon states, 4 as second order saddle polnts and 1 as a thlrd order saddle point In all
the minlmum energy structures of the b~cyclopropenyl Isomers where tncoordlnated Si
atom 1s connected to an sp3 carbon, the three membered nng opens up to y~eld
monocycl~c or acycllc isomers In Dewar benzene, benzvalene and b~cyclopropenyl
isomers, those compounds w~th Sl=Si bond, with the hydrogens connected to them are In-
plane, are computed to be transltlon states The environment in wh~cb the Si atoms are
present and the strain In the skeleton are some guldlng factors In explaining the observed
stab~ht~es In most of the cases, the lsomer where both the slhcon atoms are
tetracoordlnated IS found to be the most stable among the pos~tlonal lsomers So, the
slhcon atom has an Inherent preference to be tetracoordlnated compared to tncoordlnated,

Chapter I V Isomers of (CHW ( X = N, P, As and SIH) and C,SI~H~ 161
whlch IS attnbuted to the weak n-bonding ahrllty of SI w~th C or w~th another SI
Companson of the relat~ve energles of the valence Isomers of d~s~lahenzene and
d~phosphabenzene reveals that the posltlonal lsomers In d~s~labenzene span a wlde range
of relat~ve stah~l~tles compared to the d~phosphabenzene lsomers Thls study h~ghllghts
the nchness of C4S12H6 Isomers and importantly ~nd~cates how close are these lsomers to
each other energetically Thls leads to a sltuatlon where fac~le rearrangement among
vanous lsomerlc forms owlng to the h~gh reactlvlty of this class of compounds
The energy gaps between the varlous valence lsomers ~nvolv~ng P. As and SI
atoms are smaller compared to the benzene valence Isomers. ~ndlcat~ng smoother
rearrangements Also, the smaller magnitudes of the harmon~c trequencles correspond~ng
to skeletal reorganlzatlons. comparcd to the henzene, ~ndlcate that skeleral
rearrangements among the valence lsomers of s~lahenzene are more fac~le compared to
the parent benzene The success w~tnessed In the synthesis of phosphln~nc valence
lsomers should tr~gger synthet~c attempts towards thelr u a - and arsa- analogs and these
valence lsomers have the potentla1 to d~splay lnlrlgulng Isorner~tatton reacttons We lee1
that the present study and the recent advances In the synthes~s of phosphln~ne valence
lsomers and some aza analogs and bulky group protected s~laaromat~cs should enthuse
the expenmental~sts to explore the r~ch potentla1 of the rearrangement reactions of t h ~ s
class of compounds
4.6: References
(1) (a) V~ehe, H G Angew ('hem, lnt Ed 1965 4, 746 ( h ) lrrnelcn F t. !, Anges
Chem, Int Ed 1965, 4, 738 (c) Tarnelen, E E v Act (lrmr Re\ 1972 5 , 186 (d)
Scott, L T , Jones, M Clreal Rev 1972, 72, 181
(2) Nagendrappa, G Resonmrte, May 2001,74
(3) Johnson, R P , Daoust, K J J J Am Chem Suc 1996,118 7381
(4) Balaban, A T Rev Rouni Chrm 1966,11, 1097
(5) Karl, R R lr , Bauer, S H J Mol Stmcl 1975, 25, 1
(6) LI, Z , Rogers, D W , McLafferty, F J , Mandz~uk, M , Podoaenrn, A V J Ph~1.s C'he~r~
A 1999,103,426
(7) Cheung, Y - S , Wong, C - K , LI, W - K J Mol Sfmcf (Theorhem) 1998, 454,17

Chapter I V Isomers of (CUM (X = N. P. As and SIH) and C,SI~H, 162
(8) (a) Schulman, J M , Dlsch, R L J Am Chem Soc 1985,107,5059, (b) Newton, M D ,
Schulman, J M , Manus, M M J Am Chem Sac 1974,915, 17
(9) Comprehensrve Heferocycl~c Chemistry 11, Katntzky, A R , Rccs, c w . Smven, E F v , Eds , Elscv~er Science Ltd Oxford, 1996
(10) Markl. G In Muhrple Bonds and Low Coordrnat~on In Phosphoncr C h e m ~ s t ~ ~ . Reg~tz. M ,
Scherer, 0 J , Eds , Th~eme Stungart, Germany, 1990, p 220
(I I) (a) she, A J 111 Acc Chem Res 1978, 11, 153, @) Fr~son, G , Sevm, A . A v a ~ a n , N . Mathey, F . Floch, P L J Org Chem 1999, 64, 5524, (c) Doerksen, R J , Thakkar, A J
J Phys Chem A 1999,103,214l. (d) Brown, E C . Borden, W T Organometallrcs 2000,
19.2208
(12) Baldr~dge, K K , Gordon. M S J Am Chem Soc 1988,110,4204
(13) Dewar, M J S ,Holder, A J Ilereroqcles 1989, 28, 1135
(14) Slmon, J , Bergswaber, U , Regltz, M Chem Commun 1998,867
(15) Blaner, K , Rosch, W . Vogelbacher, U -J , Fmk, J , Reg~tz, M Angew Chem Inr Ed
Engl 1987,26,85
(16) Fmk. J . Rosch. W . Vogelbacher, U -J Regltz, M Angew Chem Inr Ed Engl 1986. 25,
280
(17) Kudoh, S , Takayanagl, M , Nakata, M J Phorochem Phorobrol A 1999, 123, 25
(18) (a) Hees, U , Vogelbacher, U -J , Mlchels, G , Regltz, M Tetruhedron 1989,45,3115, (b)
Chambers, R D , Mtddleton, R , Corbally, R P J Chem Soc, Chem Commun 1975, 731
(19) (a) Grev, R S , Schaefer, H F , III J Chem Phys 1992,97, 7990, (b) Apelolg, Y . Mullcr,
T J Am Chem Soc 1995,117,5363. (c) Zhao, M , G~marc, B M Inorg Chem 19%,35,
5378 (d) Jernm~s, E D , Srlnlvas, G N , Lesz,czynsk~, J , Kapp, J , Korkln, A 4 . Schleyer,
P r R J Am Chem Soc 1995,117, 11361, (e) Jemm~s, E D , Subraman~an, G , Radom,
L J Am Chrm Soc 1992,114,1481
(20) (a) Nagase, S , Teramae, H , Kudo, T J Chem Phys 1987, 86, 4513, (b) Sax, A .
Janoschek, R Angew Chem Inr Ed Engl 1986, 25,651, (c) Clabo, D A , Jr , Schaefer,
H F , 111 J Chenl Phys 1986,84, 1664, (d) Nagase, S , Kudo, T . Aokl, M J Chenl Soc
Chem Commun 198S,ll2l
(21) (a) Raabe, G , M~chl, J Chern Rev 1985,85,419, (b) Tok~toh, N Pure Appl Chem 1999,
71.495
(22) (a) Pnyakumar, U D , Sasby, G N Organomefoll~cs 2002,21, 1493, @) Pr~yakumar, U
D , Sasby, G N OrganometaN~cs, 2002,21,4823, (c) Dhev~, D M , Pnyakumar, U D , Sastry, G N J Mol Sfruct (Theochem) 2002,618,173

Chapter I V . Isomers of (CUM (X = N, P, As cnd SIH) and C.SIIH~ 163
(23) (a) Wahb , K, Tobtoh, N , Okazakl, R , Taka@, N , Nagase, S J Am Chem Soc 2000,
122, 5648, (b) W a k a , K , Tohtoh N , Okazakl, R , Nagasc, S Angew Chem , h t Ed
Engl 2000,39,634
(24) (a) Waklta, K . Tokltoh, N , 0-1, R , Nagase. S , Schleyer, P v R . Jlao, H J Am
Chem Soc 1999, 121, 11336, (b) Tok~toh, N . Waklta, K , Okazakl, R . Nagase, S . Schleyer, P v R , Jlao, H J Am Chem Soc 1997,119,6951
(25) (a) Markl, G , Schlosser, W Angew Chem Int Ed Engl 1988. 27.963. (b) Barton. T J . Bums, G T J Am Chem Soc 1978,100.5246, (c) Barton, T J , Vuper. M J Am Chem
SOC 1981,105,6788
(26) (a) Soloukl, B , Rosmus, P , Bock, H . Maler, G Angew Chem Inr Ed Engl 1980, I 9
51, (b) Maler, G . Mlhm, G , Relsenauer, H P Angew Chem In! Ed Engl 1980, 19, 52
(c) Maler, G , Mlhm G , Baumgartner, R 0 W . Rclsenauer. H P Chem BPT 1984. 117.
2337, (d) Maler, G , Schottler, K , Relsenauer, H P Tetrahedron Lett 1985,26, 4079
(27) Baldndge, K K , Uzan, O , Martm, J M L Orgnnumetalhcs 2000, 19, 1477
(28) Cyanskl. M K , Krygowskl, T M , Kamtzky, A R , Schleyer. P v R J Org Chenl
2002,67,1333
(29) (a) West, R , Buffy, J J , Haaf, M , Muller, 1 . Gehrhus, 9 . Lappert. M t . Apelotg. Y J
Am Chem Soc 1998, 120, 1639, (b) Veszpremt, T , Nyulasz~, L , Karpatl 'I J Phvc
Chem 1996,100,6262, (c) Boehme, C , Frenkmg, G J Am Chem Soc 1996, 118,2039
(30) Baldrldge, K K , Gordon, M S J Am Ckern Soc 1988,110,4204
(31) Dewar, M J S ,Holder, A J Heterocycles 1989.28, 1135
(32) (a) B~rd, C W Tetrahedron 1990, 46, 5697, (b) Herndon, W C Terrahrdron Lell 1979
3283
(33) Maler, G , Mlhm G , Baumgartner, R 0 W , Re~senaucr, H P Chent Ber 1984, 117
2337
(34) Chandrasekhar, 1 , Schleyer, P v R , Baumgartner. R 0 W . Reet~, M T J Org Chml
1983,48,3453
(35) Welsh, K M , Rlch, J D , West, R J Orgar~umet Chem 1987,325. 105
(36) Dysard, J M , T~lley, T D ,Woo, T K Organomerall~c.~ 2001,20, 1195
(37) (a) Ando, W , Shlba, T , Hldaka, T , Monhashl, K , Klkuchl, O J Am Chenl SOL 1997
119, 3629, @) Kabe, Y , Ohkubo, K , Ishlkawa, H , Ando, W J Am Chen~ Soc ZOO0
122,3775
(38) Ostendorf, D , Kumaler, L , Saak, W , Marsmam, H , Weldenbmch, M Eur J Inorg
Chem 1999,2301

Chapter I V Isomers of (CUM (X = N. P. As and SIH) ond C + S I ~ Y 164
(39) Ostendorf, D . Saak, W.. Wcldrnbruch, M , Marsmann. H Organomefall~cr, 2002,21,636
(40) Barldndge. K K , Gordon. M S J Orgunomel Chem 1984,271.369
(41) Chandrasckhar. J . Schleyer, P v R J Organomel Chem 1985,289.51
(42) Kang, S -Y . Yosh~zawa, K , Yamabe, T , Naka. A , Ishlkawa. M J Organomel Chem
2000.611.280
(43) (a) Warren. D S . G~marc. B M , Zhao, M Inorg Chem 1994.33. 710. (b) Glmarc. B M . Zhao. M lnarg Chern . 1996.35,3289. (c) Engelke. R J Phys Chem , 1989,93,5722, (d)
Ndgase, S . Ito. K Chem Pltys Lell 1986. 126, 43, (e) Nguyen, M T . Hegany, A J
Cherrz Soc Cllnit Commurr 1986,383. (0 Saxe, P Schaefer 111. H F J Am Chem SOL
1983,105. 1760
(44) Galrcsran 98. Rev~s~on A l l 2. Fr~sch, M J . Trucks, G W . Schlegel. H B , Scuseria, G
E . Robb, M A Cheeseman. J R , Zakrzewskl, V G ,Montgomery. J r , J A . Stramam.
R E . Burant. J C , Dappnch. S , M~llam. J M , Danlels. A D . Kud~n, K N , Stram. M
C . Farkas. 0 . tom as^, J , Barone, V . Coss~. M . Camm~, R . Mennuccl. B , Pomelll. C ,
Adamo. C , Cl~fford. S , Ochtersk~, J . Petersson. G A , Ayala. P Y . CUI. Q . Morokuma,
K , Rega, N , Salvador, P , Dannenberg, J I . Mal~ck, D K , Rabuck, A D . Raghavachar~.
K . Foresman, J B , C~oslowsk~, J , Ort~z, J V , Raboul. A (3, Stefanov. B B , LIU. G ,
L~ashenko, A , Plskorz, P , Komarom~, I . Gompens, R , Martm, R L , Fox, D J , Ke~th.
T . Al-Laham. M A , Peng. C Y , Nanayakkara, A , Challacombe, M . GIII, P M W .
John5on. B . Chen. W , Wong, M W , Andres, J L , Gonzalez. C , Head-Gordon, M .
Replogle. E S . Pople, J A Gauss~an, lnc P~ttsburgh PA, 2001
(45) IIehre. W J , Radom, L . Schleyer, P v R , Pople. 1 A A6 lnrr~o Molecular Orbrlal
T/?L,oI)', Wlley NewYork, 1986
(46) Becke. A D J Chern Phys 1993, 98.5648
(47) (a) Lee, C , Yang W . Pam, R G Phys REI' B 1988, 37, 785, (b) Mlehllch, B . Savln, A ,
Stoll. H , Preuss H Chem Phys Let1 1989, 157.200
(48) Colombet. L . Volahon, F , M a ~ h e P . H~berty. P C J Am Chem Soc 1999,121,4215
(49) (a) Pr~yakumar. U D , Sasny, G N J Am Chem SOL 2000, 122. 11173. (b) Pr~yakumar.
U D , Sasny. G N J Org Chrm 2002, 67, 271, (c) Pnyakumar, U D . Sasny. G N
Proc Indran Acad SCI (Chernlcal SCI) 2002, m press
(50) Cho~ , C H , Kenesz, M , Karpfen, A P Chem Phys Let1 1997,276,266
(51) Hoffmann, M , Schleyer, P v R , Reg~tz, M Eur J Org Chem 1999,3291
(52) Jaguar 4 1, Schrodmger, Inc , Portland, Oregon, 2000
(53) Kutzeln~gg, W Angew Chem , Int Ed Engl 1984,23,272

C h a p t ~ IV. ISOMS of (CHM (X = N, P. As and SIH) and C ~ S I ~ H ~ 165
(54) Schm~dt, M W , Tmong, P N .Gordon, M S J Am Chem Soc 1987,109,5217
(55) Schleyer, P v R , Kost, D J Am Chem Soc 1988,110,2105
(56) (a) Pearson, R G J Org Chem 1989, 54, 1423 @) Zhou, Z , Pan, R G Tetrahedron
Left 1988, 29, 4843 (c) Zhou, Z , Pam, R G J Am Chem Soc 1989, 111, 7371 (d)
Minsky, A , Meyer, A Y , Rabmov~tz, M Tetrahedron. 1985,41.785 (e) Pearson. R G J
Am Chem Soc 1988,110,2092
(57) Pearson, R G Inorg Chem 1998,27,734
(58) (a) Hegarty, D , Robb, M A Mol Phys 1979,38, 1795, (b) Eade, R H E , Robb, M A
Chem Phys Left 1981.83,362. (c) McDouall, J J , Peasley, K , Robb. M A Chem Phjs
Let1 1988, 148. 183
(59) Bally, T , Albrecht, B , Matzmger, S , Sastry, G M Mop101 3 2. Un~vers~ty of Fribourg
1997
(60) (a) Bader, R F W Atoms m Molecules A Quantum Theory, Oxford Univ Press, Oxford
1990, (b) C~oslowsk~, J , M~xon, S R J Am Chem Soc 1991,113,4142
(61) Hrovat. D A , Sun, H , Borden, W T J Mol Strucr (77teocheni) 1988.163.51
















![Level 8 Book Study Answer Key - goodandbeautiful.com€¦ · Secession [sih - SESH - un]: the act of seceding [SIH - seed - ing] (formal-ly withdrawing from an alliance, political](https://static.fdocuments.in/doc/165x107/603885bc39dafc120b7e952c/level-8-book-study-answer-key-secession-sih-sesh-un-the-act-of-seceding.jpg)

