
456 | P a g e International Standard Serial Number (ISSN ...ijupbs.com/Uploads/31....
14
456 | Page International Standard Serial Number (ISSN): 2319-8141 Full Text Available On www.ijupbs.com International Journal of Universal Pharmacy and Bio Sciences 3(2): March-April 2014 INTERNATIONAL JOURNAL OF UNIVERSAL PHARMACY AND BIO SCIENCES IMPACT FACTOR 1.89*** ICV 5.13*** Pharmaceutical Sciences RESEARCH ARTICLE……!!! FORMULATION AND IN-VITRO EVALUATION OF FAST DISSOLVING TABLET OF OLMESARTAN MEDOXOMIL Zinkal K Patel*, Rahul R. Patel, Dr K. R. Patel, Dr M. R. Patel. Department of Pharmaceutics, Shri B. M. Shah College of Pharmaceutical Education and Research Dhansura Road College campus Modasa, Dist:- Arvali. Pin code:- 383315 Gujarat, (India). KEYWORDS: Olmesartan medoxomil; solid-dispersion; FDT; Solubility; dissolution; superdisintegrants. For Correspondence: Zinkal K Patel* Address: Shri B. M. Shah College of pharmaceutical education and research Modasa-383315, Dist: Arvali. Gujarat, India E-Mail id:- [email protected] om ABSTRACT The aim of this investigation was to formulation and in-vitro evaluation of fast dissolving tablet of olmesartan medoxomil by direct compression method. Oral bioavailability of olmesartan medoxomil is very low (26%), due to its poor water solubility. For this purpose, initially solubility of olmesartan medoxomil was improved by using PEG 6000 and PVP K30 as a carriers by solid dispersion techniques. The prepared solid dispersion powder were evaluated for flow properties, solubility and in-vitro drug release, FTIR analysis, DSC. Then optimized batch of solid dispersion(P4) was used in formulation of fast dissolving tablet using different superdisintegrants. Various batches of tablets were evaluated for pre-compression and post-compression parameter of tablet. Optimized batch contain 93.91CPR, 100.0% drug content, 17sec.disintegration time. optimized batch compared with marketed formulation.
Transcript of 456 | P a g e International Standard Serial Number (ISSN ...ijupbs.com/Uploads/31....

456 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
International Journal of Universal Pharmacy and Bio Sciences 3(2): March-April 2014
INTERNATIONAL JOURNAL OF UNIVERSAL
PHARMACY AND BIO SCIENCES IMPACT FACTOR 1.89***
ICV 5.13*** Pharmaceutical Sciences RESEARCH ARTICLE……!!!
FORMULATION AND IN-VITRO EVALUATION OF FAST DISSOLVING
TABLET OF OLMESARTAN MEDOXOMIL
Zinkal K Patel*, Rahul R. Patel, Dr K. R. Patel, Dr M. R. Patel.
Department of Pharmaceutics, Shri B. M. Shah College of Pharmaceutical Education and
Research Dhansura Road College campus Modasa, Dist:- Arvali. Pin code:- 383315 Gujarat,
(India).
KEYWORDS:
Olmesartan medoxomil;
solid-dispersion; FDT;
Solubility; dissolution;
superdisintegrants.
For Correspondence:
Zinkal K Patel*
Address:
Shri B. M. Shah College
of pharmaceutical
education and research
Modasa-383315, Dist:
Arvali. Gujarat, India
E-Mail id:-
om
ABSTRACT
The aim of this investigation was to formulation and in-vitro
evaluation of fast dissolving tablet of olmesartan medoxomil by
direct compression method. Oral bioavailability of olmesartan
medoxomil is very low (26%), due to its poor water solubility. For
this purpose, initially solubility of olmesartan medoxomil was
improved by using PEG 6000 and PVP K30 as a carriers by solid
dispersion techniques. The prepared solid dispersion powder were
evaluated for flow properties, solubility and in-vitro drug release,
FTIR analysis, DSC. Then optimized batch of solid dispersion(P4)
was used in formulation of fast dissolving tablet using different
superdisintegrants. Various batches of tablets were evaluated for
pre-compression and post-compression parameter of tablet.
Optimized batch contain 93.91CPR, 100.0% drug content,
17sec.disintegration time. optimized batch compared with
marketed formulation.

457 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
INTRODUCTION1, 2, 3, 4
Oral delivery is currently the gold standard in the pharmaceutical industry where it is regarded as the safest,
most convenient and most economical method of drug delivery. Oral route of drug administration become
popular route for systemic effects due to ease of ingestion, accurate dosage, self-medication, pain
avoidance. Also solid oral delivery systems do not require sterile conditions Fast dissolving drug delivery
system are Novel Drug Delivery techniques aim for designing dosage forms, convenient to be manufacture
and administer without water, free of side effects, offering immediate release and enhanced bioavailability,
so as to achieve better patient compliance.1 This segment of formulation is especially designed for
pediatric, geriatric, bedridden, psychotic patients who are unable to swallow or refuse to swallow
conventional oral formulation and also for active patients who are busy and traveling and may not have
access to water. United states Food And Drug Administration (FDA) defined fast dissolving tablet (FDT)
as “a solid dosage form containing medicinal substance or active ingredient which disintegrate or dissolve
rapidly within seconds when placed upon the tongue.” Fast dissolving tablets are also known as mouth-
dissolving tablets, rapid dissolving, melt-in mouth tablets, Orodispersible tablets, rapimelts, porous tablets,
quick dissolving, quick melt, quick disintegrating tablets.2
FDT prepared by various techniques like direct
compression, lyophilization, spray drying, sublimation, test masking etc. But commonly used method to
prepare FDT is direct compression. It is easiest way to manufacture tablets. Conventional equipments,
commonly available excipients and a limited number of processing steps are involved in direct
compression.
Hypertension is fast becoming a major public health problem in world. Olmesartan medoxomil (OLM) is
currently used as an alternative therapeutic antihypertensive agent for patient intolerant of angiotensin
converting enzyme inhibitor. It is known that diabetic patient are at increased risk of cardiovascular disease
and reduce vascular resistance and oxidative stress, addition to significant reducing BP in type -2 diabetic
patients.5, 6
OLM is a poorly aqueous soluble drug. Its solubility is reported to be 0.0077mg/ml. Rapid onset of action
is desirable to provide fast relief in treatment of heart failure. Therefore, it is necessary to enhance the
aqueous solubility and dissolution rate of OLM to obtain faster on set of action and improve its overall oral
bioavailability102
. Solubility of OLM is improve by solid dispersion because it cause reduction in particle
size, improve drug porosity, increase wettability and convert crystalline drug in to amorphous state. OLM
is a selective AT1 subtype angiotensin–II receptor antagonist that has been approved by US Food and Drug
Administration (FDA) for treatment of hypertension. OLM dose dependently reduces blood pressure

458 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
through arterial vasodilatation and reduce sodium retention. It is a prodrug that is rapidly de-esterified
during absorption from GIT to produce an active metabolite olmesartan.OLM is BCS class-II drug. i.e. low
solubility and high permeability. Oral bioavailability is very low (26%) due to its poor aqueous solubility.
OLM given orally in single daily doses of 20mg-40mg. Having log p value 5.9 i.e. drug is lipophilic in
nature. Elimination half life of OLM is 13hrs and 99%plasma protein binding. Pka value is 4.37, 8, 9
MATERIALS AND METHOD
Olmesartan Medoxomil was obtained from Sun Pharma Ltd. Baroda. Kyron T-314 was obtained from
Corel Pharma Chem., Ahmadabad. Cross Carmellose Sodium, Crosspovidone, Avicel pH101, Aerosil was
obtained from Orbicular Pharma. Tech. Pvt Ltd. PEG 6000 was obtained from Modern chemical corp.,
Mumbai. PVP K30 was obtained from Oxford lab. Mumbai. Mannitol was obtained from Finar chemicals
ltd, Mumbai.Aspartame was obtained from Lesar Chemicals, Ahmedabad.
METHOD :
Preparation of the Solid Dispersions10, 11
Solid dispersion of olmesartan medoxomil (OLM) were prepared by solvent evaporation and fusion
method using PEG 6000 and PVP K30 as a carrier.
Solvent evaporation method
In this method, 1 : 1 ratio of PEG 6000 : drug (SE1) and PVP K30 : drug (SE2) were carefully transferred
into two beaker seperately and dissolved in methanol. The solution was transferred to a petridish and
solvent was allow to evaporate at room temperature for 1 hr and then dried at 600c for 6 hr in a hot air
oven. The mass obtained in each case was crushed, pulverized and sifted the 80#. All the solid dispersion
was preserved in well closed glass container till use.
By fusion method
In this method, 1 : 1 ratio of drug : PEG 6000 (FM1) and drug : PVP K30 : (FM2) was heated in two
different china dish to a temperature just above its melting point. The melt is solidified on an ice bath
under vigorous stirring. The mass obtained in each case was crushed, pulverized and sifted the 80#. All the
solid dispersion was preserved in well closed glass container till use. Solid dispersion prepared by PEG
6000 by fusion method give good solubility. So, it used in different ration like 1: 1(P1), 1: 2(P2), 1 : 3(P3),
1: 4(P4), 1: 5(P5), 1 : 6(P6).
Evaluation of solid dispersion12, 13
A. Saturation solubility studies
Saturation solubility was performed according to method reported by higuchi and cannors. Excess quantity
of solid dispersion, equivalent to 20 mg of drug was added to 100 ml conical flask containing 20ml 0.1N

459 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
Hcl and mixtures were shaken for 24 hour at 100rpm at room temperature in orbital shaker and filter
through whatman filter paper No.41. The filtrate was suitably diluted and analyzed spectrophotometrically
at 257nm against blank using UV visible spectrophotometer. The studies were carried out in triplicate and
average value (±SD) was noted
B. Dissolution study of solid dispersion
It was performed as described in Indian Pharmacopoeia 2010 using USP apparatus II (paddle). Quantity of
solid dispersion equivalent to 20 mg of drug was kept in a flask of the dissolution apparatus containing
900ml of 0.1N HCl as a dissolution media maintaining the temperature at 37±0.50C and at a speed of 50
rpm. Aliquot of dissolution medium (5ml) was withdrawn at a specific time intervals and the samples were
replaced with fresh dissolution medium. Sample was filter through whatman filter paper No.41. The filtrate
was suitably diluted and analyzed spectrophotometrically at 257nm against blank using UV visible
spectrophotometer.
C. Differential scanning calorimetry
Calorimetric measurements were performed with DSC instrument, over the temperature range from 30 to
300oC at heating and cooling rates of 50
oC/min. The temperature scale was calibrated with high purity
standards. The glass transition was reported as the point of inflection of specific heat increment. Samples of
OLM and solid dispersion powder were analysed in the aluminium pan and their DSC spectra were
recorded.
D. Fourier transform infrared (FTIR)
Drug - carrier interaction play a vital role in the release of drug from formulation. FTIR has been used to
study the physical and chemical interaction between drug and carriers. FTIR spectra of OLM and carrier
were recorded using KBR mixing method on FTIR instrument.
FORMULATION OF FAST DISSOLVING TABLET14
Fast Dissolving Tablet of OLM were prepared by direct compression method according to the formula
given in below table using optimized batch of solid dispersion(P4). Various superdisintigrants like CCS,
Crosspovidone, Kyrone T-314 are use in the range of 2%, 4%, 6% to select the optimum concentration of
superdisintigrants. All the required material were passed through sieve no.60# and mixed homogeneously
for 15 min. Finally magnesium stearate and aerosil were added and mixed for 1 min. This blend was
compressed using 8 mm size flat faced punch on rotary compression machine.
Mannitol act as diluent. Microcrystalline cellulose act as binder. Aspartame was used as a sweetener.
Aerosil was used as glidant and Magnesium stearate was used as lubricant.
460 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
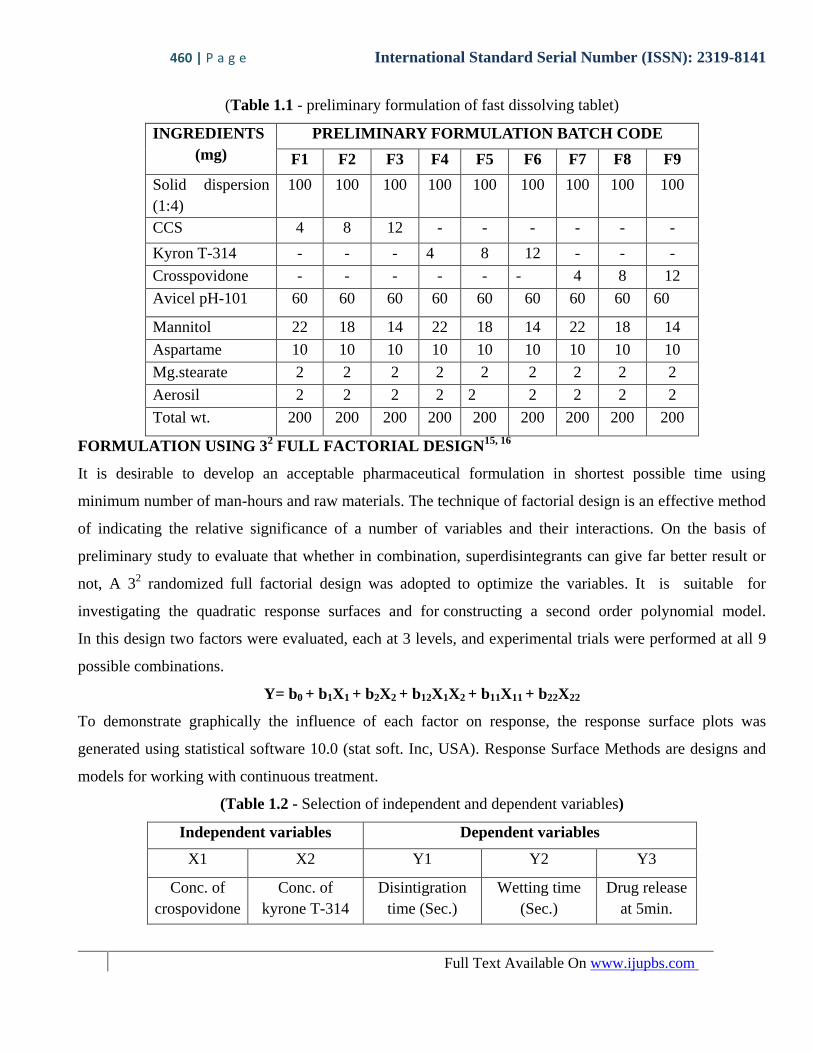
(Table 1.1 - preliminary formulation of fast dissolving tablet)
INGREDIENTS
(mg)
PRELIMINARY FORMULATION BATCH CODE
F1 F2 F3 F4 F5 F6 F7 F8 F9
Solid dispersion
(1:4)
100 100 100 100 100 100 100 100 100
CCS 4 8 12 - - - - - -
Kyron T-314 - - - 4 8 12 - - -
Crosspovidone - - - - - - 4 8 12
Avicel pH-101 60 60 60 60 60 60 60 60 60
Mannitol 22 18 14 22 18 14 22 18 14
Aspartame 10 10 10 10 10 10 10 10 10
Mg.stearate 2 2 2 2 2 2 2 2 2
Aerosil 2 2 2 2 2 2 2 2 2
Total wt. 200 200 200 200 200 200 200 200 200
FORMULATION USING 32 FULL FACTORIAL DESIGN
15, 16
It is desirable to develop an acceptable pharmaceutical formulation in shortest possible time using
minimum number of man-hours and raw materials. The technique of factorial design is an effective method
of indicating the relative significance of a number of variables and their interactions. On the basis of
preliminary study to evaluate that whether in combination, superdisintegrants can give far better result or
not, A 32 randomized full factorial design was adopted to optimize the variables. It is suitable for
investigating the quadratic response surfaces and for constructing a second order polynomial model.
In this design two factors were evaluated, each at 3 levels, and experimental trials were performed at all 9
possible combinations.
Y= b0 + b1X1 + b2X2 + b12X1X2 + b11X11 + b22X22
To demonstrate graphically the influence of each factor on response, the response surface plots was
generated using statistical software 10.0 (stat soft. Inc, USA). Response Surface Methods are designs and
models for working with continuous treatment.
(Table 1.2 - Selection of independent and dependent variables)
Independent variables Dependent variables
X1 X2 Y1 Y2 Y3
Conc. of
crospovidone
Conc. of
kyrone T-314
Disintigration
time (Sec.)
Wetting time
(Sec.)
Drug release
at 5min.
461 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
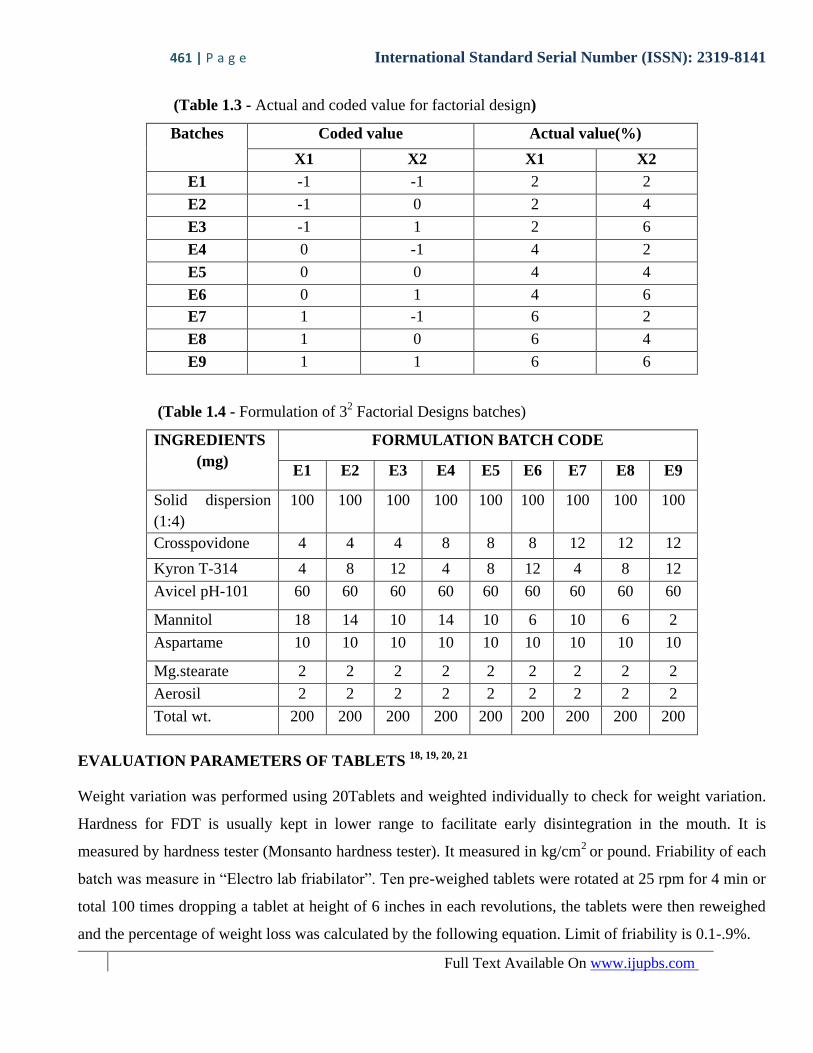
(Table 1.3 - Actual and coded value for factorial design)
Batches Coded value Actual value(%)
X1 X2 X1 X2
E1 -1 -1 2 2
E2 -1 0 2 4
E3 -1 1 2 6
E4 0 -1 4 2
E5 0 0 4 4
E6 0 1 4 6
E7 1 -1 6 2
E8 1 0 6 4
E9 1 1 6 6
(Table 1.4 - Formulation of 32 Factorial Designs batches)
INGREDIENTS
(mg)
FORMULATION BATCH CODE
E1 E2 E3 E4 E5 E6 E7 E8 E9
Solid dispersion
(1:4)
100 100 100 100 100 100 100 100 100
Crosspovidone 4 4 4 8 8 8 12 12 12
Kyron T-314 4 8 12 4 8 12 4 8 12
Avicel pH-101 60 60 60 60 60 60 60 60 60
Mannitol 18 14 10 14 10 6 10 6 2
Aspartame 10 10 10 10 10 10 10 10 10
Mg.stearate 2 2 2 2 2 2 2 2 2
Aerosil 2 2 2 2 2 2 2 2 2
Total wt. 200 200 200 200 200 200 200 200 200
EVALUATION PARAMETERS OF TABLETS 18, 19, 20, 21
Weight variation was performed using 20Tablets and weighted individually to check for weight variation.
Hardness for FDT is usually kept in lower range to facilitate early disintegration in the mouth. It is
measured by hardness tester (Monsanto hardness tester). It measured in kg/cm2
or pound. Friability of each
batch was measure in “Electro lab friabilator”. Ten pre-weighed tablets were rotated at 25 rpm for 4 min or
total 100 times dropping a tablet at height of 6 inches in each revolutions, the tablets were then reweighed
and the percentage of weight loss was calculated by the following equation. Limit of friability is 0.1-.9%.
462 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
Disintegration time was carried out on 6 tablets using the apparatus specified in I.P. Distilled water at 37ºC
± 2ºC was used as a disintegration media and time requied for complete disintegration of tablet with no
palatable mass remaining in the apparatus was measured .
Dissolution test was performed in 0.1N HCl using USP apparatus-II (paddle). Tablet was kept in a flask of
the dissolution apparatus containing 900ml of 0.1N HCl maintaining at 37±0.50C and 50 rpm. Aliquot of
dissolution medium (5ml) was withdrawn at a specific time intervals and the samples were replaced with
fresh dissolution medium. Sample was filter through whatman filter paper No.41. The filtrate was suitably
diluted and analyzed spectrophotometrically at 257nm against blank using UV visible spectrophotometer.
In Drug content test ten tablets were weighed and powdered in a glass mortar. Quantity of powder
equivalent to 20mg was transferred in a 100 ml volumetric flask. Make final volume in volumetric flask up
to 100ml using 0.1N HCl and filter through whatman filter. The filtrate was suitably diluted and analyzed
spectrophotometrically at 257nm against blank using UV visible spectrophotometer.
Wetting time was closely related to the inner structure of the tablets and to the hydrophilicity of the
excipients. To measure wetting time, five circular tissue papers of 10cm diameter are placed in a petridish
with a 10cm diameter. 10ml of water containing eosin, a water soluble dye, is added to petridish. A tablet is
carefully placed on the surface of the tissue paper. The time required for water to reach upper surface of the
tablet is noted as a wetting time.
The stability studies were carried out on the most satisfactory formulations as per ICH guidelines Q1C. The
samples of optimized batch were kept at 40°C ± 5°C and 75% relative humidity for one month in vial
which is sealed with rubber cap. At the end of study the tablet were withdrawn and analyzed for physical
characterization such as Hardness, Drug content, Wetting time, Disintegrations, and Dissolution etc.
RESULT AND DISCUSSION
Phase Solubility Study of Preliminary Batches of Solid Dispersion
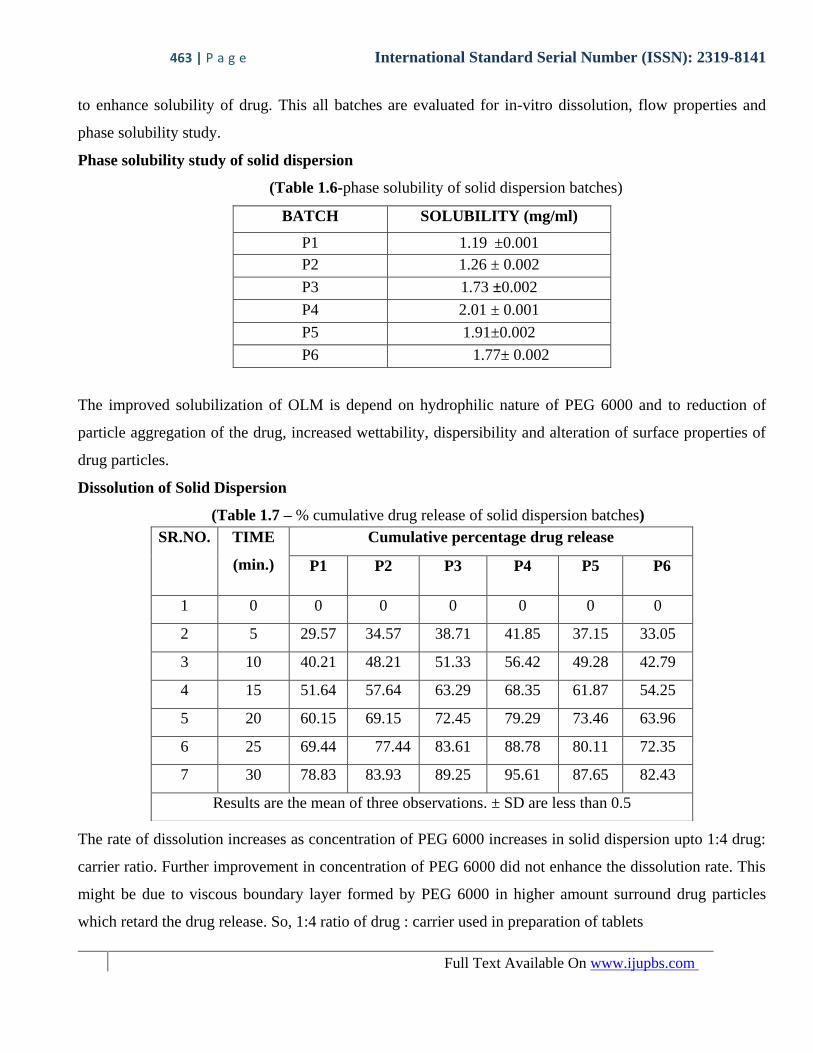
(Table 1.5 - phase solubility of preliminary batches of solid dispersion)
BATCH SOLUBILITY (mg/ml)
Pure drug 0.526 ±0.002
SE1 0.94 ±0.002
SE2 0.86 ±0.002
FM1 1.19 ±0.001
FM2 0.91 ±0.002
From above evaluation it was concluded that solid dispersion by fusion method with PEG 6000 give good
solubility. So, OLM : PEG 6000 in different ratio like 1:1, 1:2, 1:3, 1:4, 1:5, 1:6 were used in further study
463 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
to enhance solubility of drug. This all batches are evaluated for in-vitro dissolution, flow properties and
phase solubility study.
Phase solubility study of solid dispersion
(Table 1.6-phase solubility of solid dispersion batches)
BATCH SOLUBILITY (mg/ml)
P1 1.19 ±0.001
P2 1.26 ± 0.002
P3 1.73 ±0.002
P4 2.01 ± 0.001
P5 1.91±0.002
P6 1.77± 0.002
The improved solubilization of OLM is depend on hydrophilic nature of PEG 6000 and to reduction of
particle aggregation of the drug, increased wettability, dispersibility and alteration of surface properties of
drug particles.
Dissolution of Solid Dispersion
(Table 1.7 – % cumulative drug release of solid dispersion batches)
The rate of dissolution increases as concentration of PEG 6000 increases in solid dispersion upto 1:4 drug:
carrier ratio. Further improvement in concentration of PEG 6000 did not enhance the dissolution rate. This
might be due to viscous boundary layer formed by PEG 6000 in higher amount surround drug particles
which retard the drug release. So, 1:4 ratio of drug : carrier used in preparation of tablets
SR.NO. TIME
(min.)
Cumulative percentage drug release
P1 P2 P3 P4 P5 P6
1 0 0 0 0 0 0 0
2 5 29.57 34.57 38.71 41.85 37.15 33.05
3 10 40.21 48.21 51.33 56.42 49.28 42.79
4 15 51.64 57.64 63.29 68.35 61.87 54.25
5 20 60.15 69.15 72.45 79.29 73.46 63.96
6 25 69.44 77.44 83.61 88.78 80.11 72.35
7 30 78.83 83.93 89.25 95.61 87.65 82.43
Results are the mean of three observations. ± SD are less than 0.5
464 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
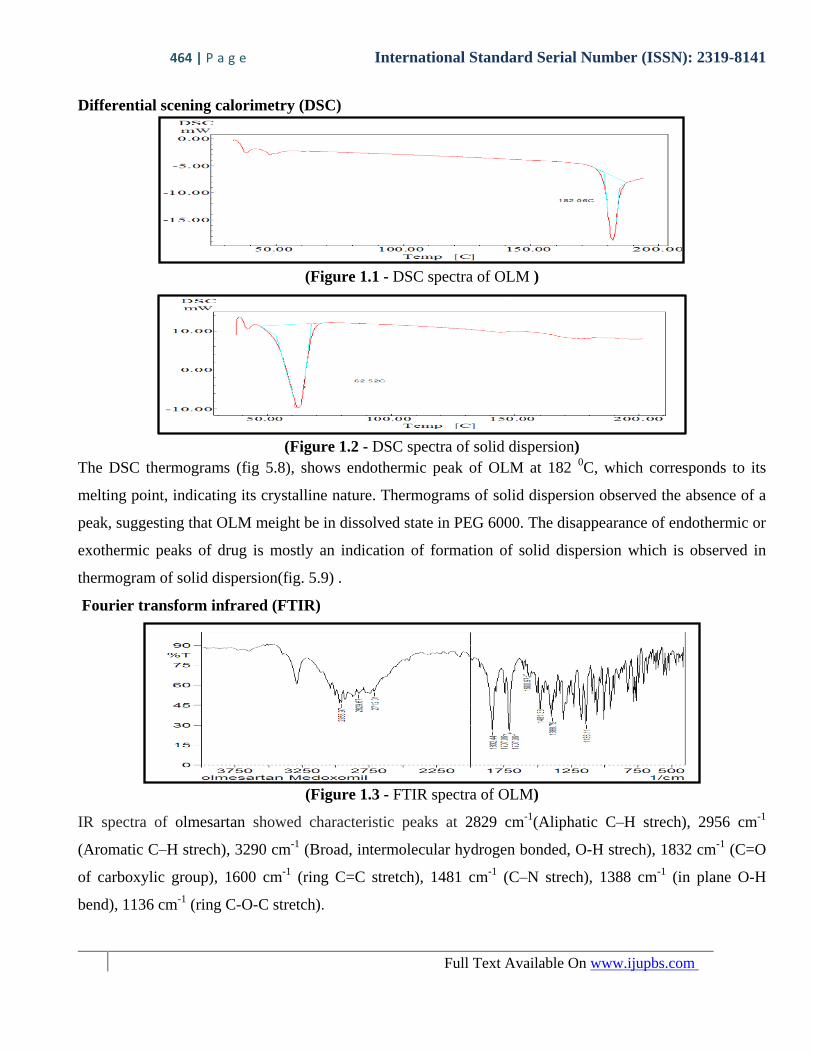
Differential scening calorimetry (DSC)
(Figure 1.1 - DSC spectra of OLM )
(Figure 1.2 - DSC spectra of solid dispersion)
The DSC thermograms (fig 5.8), shows endothermic peak of OLM at 182 0C, which corresponds to its
melting point, indicating its crystalline nature. Thermograms of solid dispersion observed the absence of a
peak, suggesting that OLM meight be in dissolved state in PEG 6000. The disappearance of endothermic or
exothermic peaks of drug is mostly an indication of formation of solid dispersion which is observed in
thermogram of solid dispersion(fig. 5.9) .
Fourier transform infrared (FTIR)
(Figure 1.3 - FTIR spectra of OLM)
IR spectra of olmesartan showed characteristic peaks at 2829 cm-1
(Aliphatic C–H strech), 2956 cm-1
(Aromatic C–H strech), 3290 cm-1
(Broad, intermolecular hydrogen bonded, O-H strech), 1832 cm-1
(C=O
of carboxylic group), 1600 cm-1
(ring C=C stretch), 1481 cm-1
(C–N strech), 1388 cm-1
(in plane O-H
bend), 1136 cm-1
(ring C-O-C stretch).
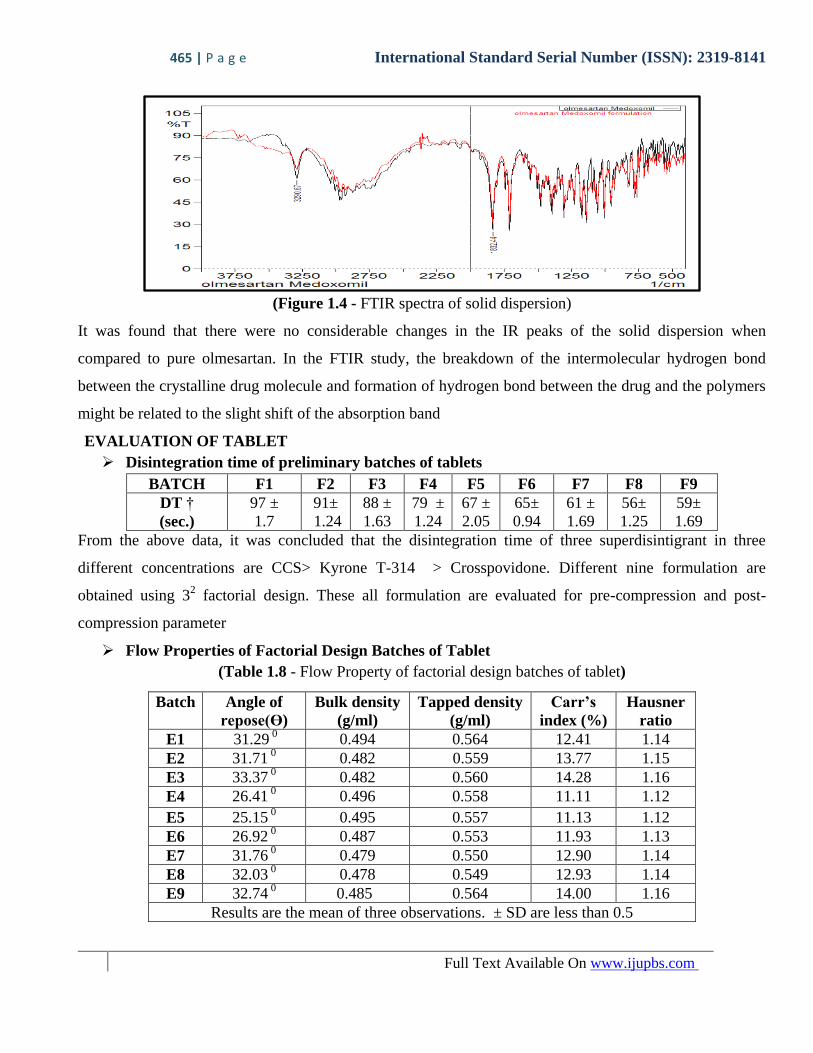
465 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
(Figure 1.4 - FTIR spectra of solid dispersion)
It was found that there were no considerable changes in the IR peaks of the solid dispersion when
compared to pure olmesartan. In the FTIR study, the breakdown of the intermolecular hydrogen bond
between the crystalline drug molecule and formation of hydrogen bond between the drug and the polymers
might be related to the slight shift of the absorption band
EVALUATION OF TABLET
Disintegration time of preliminary batches of tablets
BATCH F1 F2 F3 F4 F5 F6 F7 F8 F9
DT †
(sec.)
97 ±
1.7
91±
1.24
88 ±
1.63
79 ±
1.24
67 ±
2.05
65±
0.94
61 ±
1.69
56±
1.25
59±
1.69
From the above data, it was concluded that the disintegration time of three superdisintigrant in three
different concentrations are CCS> Kyrone T-314 > Crosspovidone. Different nine formulation are
obtained using 32 factorial design. These all formulation are evaluated for pre-compression and post-
compression parameter
Flow Properties of Factorial Design Batches of Tablet
(Table 1.8 - Flow Property of factorial design batches of tablet)
Batch Angle of
repose(Ө)
Bulk density
(g/ml)
Tapped density
(g/ml)
Carr’s
index (%)
Hausner
ratio
E1 31.29
0 0.494 0.564 12.41 1.14
E2 31.71 0 0.482 0.559 13.77 1.15
E3 33.37 0 0.482 0.560 14.28 1.16
E4 26.41 0 0.496 0.558 11.11 1.12
E5 25.15 0 0.495 0.557 11.13 1.12
E6 26.92 0 0.487 0.553 11.93 1.13
E7 31.76 0 0.479 0.550 12.90 1.14
E8 32.03 0 0.478 0.549 12.93 1.14
E9 32.74 0 0.485 0.564 14.00 1.16
Results are the mean of three observations. ± SD are less than 0.5
466 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
(Table 1.9 - post compression parameter of factorial design batches)
Batch E1 E2 E3 E4 E5 E6 E7 E8 E9
Hardness*
(kg/cm2)
3.1 ±
0.23
3.0 ±
0.4
3.2 ±
0.23
2.8±
0.23
3.0±
0.40
3.0 ±
0.40
2.8 ±
0.23
3.3 ±
0.23
3.0±
0.40
Thickness*
(mm)
3.9
±
0.04
4.06
±
0.04
4.0
±
0.08
4.0
±
0.00
4.03
±
0.04
4.03
±
0.09
4.03
±
0.12
4.06
±
0.04
3.9
±
0.09
Friability*
(%)
0.66
±
0.04
0.60
±
0.05
0.65
±
0.05
0.47
±
0.02
0.41
±
0.02
0.47
±
0.04
0.76
±
0.03
0.82
±
0.04
0.88
±
0.04
Wt.
variation#
(%)
199
±3.6
200
±5.3
199
±5.3
200
±5.2
200
±3.7
199
±5.1
201
±4.4
201
±4.9
199
±5.7
Drug*
content(%)
97
±2.1
99
±1.2
96
±3.0
98
±1.6
100
±0.9
100
±4.0
99
±3.6
98
±2.0
96
±3.7
DT†
(sec.)
27 ±
1.7
29 ±
1.24
31 ±
1.63
28 ±
1.24
22 ±
2.05
26 ±
0.94
33 ±
1.69
36 ±
1.25
36 ±
1.69
Wett. Time
(sec.)*
22±
1.24
24±
3.74
25±
0.74
21 ±
1.7
17±
1.69
20 ±
0.47
28 ±
1.24
30 ±
0.81
31±
1.25
WAR
(%)*
71.6
± 2.5
63.7
± 1.7
54.6
± 2.9
75.3
± 1.3
83.6
± 2.4
79.7
± 1.6
44.3
± 2.0
39.4
± 1.2
35.6
± 1.2
* All values are mean ± SD, (n =3), † All values are mean ± SD, (n =6), # All
values are mean ± SD, (n =20).
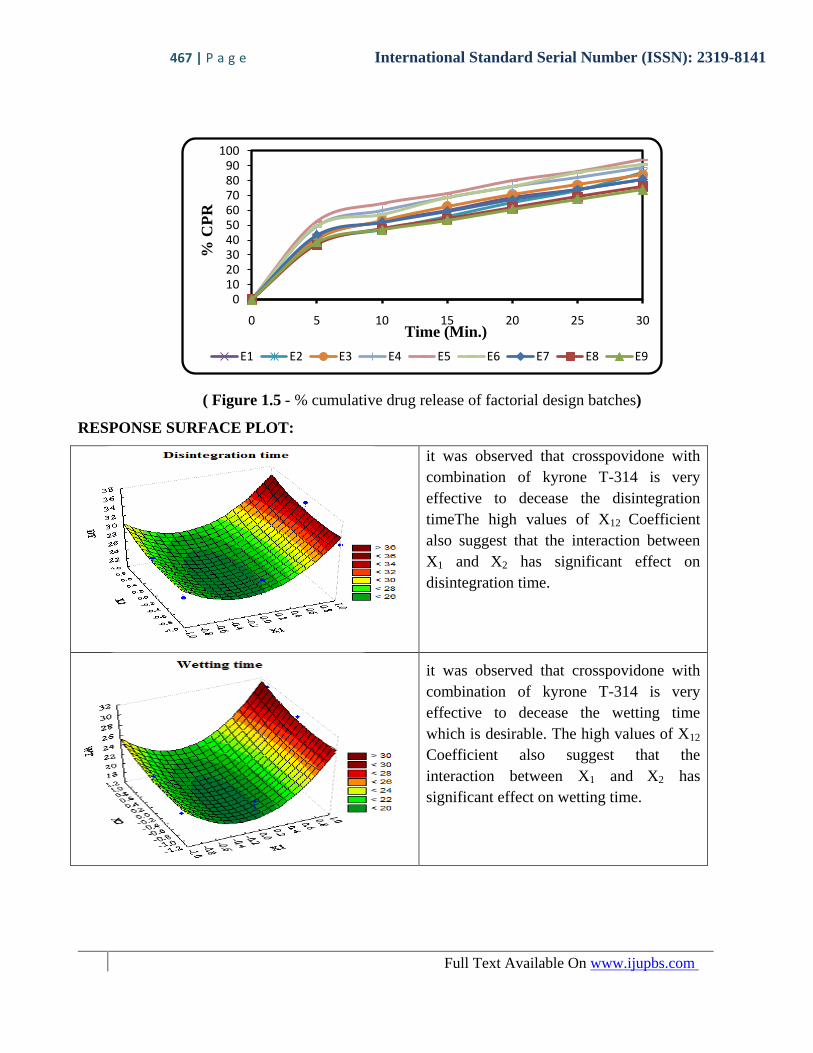
In-Vitro Dissolution of Factorial Design Batches of Tablet
(Table 1.10 - % cumulative drug release of factorial design batches)
TIME
(Min.)
Cumulative Percentage Drug Release
E1 E2 E3 E4 E5 E6 E7 E8 E9
0 0 0 0 0 0 0 0 0 0
5 41.08 37.51 40.25 48.98 52.51 49.35 43.33 36.75 38.57
10 52.61 46.86 53.44 60.03 64.77 57.19 52.13 47.48 46.85
15 60.23 55.92 62.73 68.47 71.34 68.53 59.24 54.23 53.46
20 68.53 65.44 70.69 76.11 79.82 76.27 66.97 61.94 60.87
25 74.35 73.17 77.24 82.25 86.14 85.29 73.86 69.17 67.42
30 81.03 85.25 84.18 89.09 93.91 91.05 80.74 76.02 73.93
Results are the mean of three observations. ± SD are less than 0.5
467 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
( Figure 1.5 - % cumulative drug release of factorial design batches)
RESPONSE SURFACE PLOT:
it was observed that crosspovidone with
combination of kyrone T-314 is very
effective to decease the disintegration
timeThe high values of X12 Coefficient
also suggest that the interaction between
X1 and X2 has significant effect on
disintegration time.
it was observed that crosspovidone with
combination of kyrone T-314 is very
effective to decease the wetting time
which is desirable. The high values of X12
Coefficient also suggest that the
interaction between X1 and X2 has
significant effect on wetting time.
0102030405060708090
100
0 5 10 15 20 25 30
E1 E2 E3 E4 E5 E6 E7 E8 E9
Time (Min.)
% C
PR

468 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
it was observed that crosspovidone with
combination of kyrone T-314 is very
effective to increase drug release at 5 min
which is desirable. The high values of X12
Coefficient also suggest that the
interaction between X1 and X2 has
significant effect on drug release at 5 min.
CONCLUSION
From the above work it was concluded that the release of drug from the E5 formulation was quick when
compared to other formulation. It shows that the combined effect of crosspovidone and kyrone T-314
gives synergistic effect. Undoubtedly the availability of various technologies and the manifold advantages
of FDT will surely enhance the patient compliance, low dosing, rapid onset of action, increased
bioavailability, low side effect, good stability and its popularity in near future
ACKNOWLEDGEMENT
We are thankful to Sun Pharma Ltd. Baroda, Corel Pharma Chem., Ahmadabad, Orbicular Pharma. Tech.
Pvt Ltd. , Modern chemical corp., Mumbai. , Oxford lab. Mumbai. , Lesar Chemicals, Ahmedabad., Finar
chemicals ltd, Mumbai for the gift samples of the drug and excipients, Department of Pharmaceutics,
Nootan Pharmacy College, Visnagar for providing facilities to carry out DSC study. We are also thankful
to Shri B. M. Shah College of Pharmaceutical Education and Research, modasa. for providing the
opportunity to carry out our research work successfully. I heartly thanks to Patel, Dr k. r. patel, Dr m. r.
patel for providing us Kind support all the time throughout the work.
REFERENCES:
1. Gupta A, Mishra AK, Bansal P, Singh R, Recent trends of fast dissolving tablets –an overview of
formulation technology. Int. J. Pharm. Bio. 2010, 1(1), 1-10.
2. Goel H, Rai P, Rana V, Tiwari AK, Orally disintegrating system: innovation in formulation and
technology. Recent Patent on Drug Delivary And Formulation. 2008, 2(3), 258-274.
3. Goel H, Vora N, Rana V, A novel approach to optimize and formulate fast disintegrating tablets for
nausea and vomitting. AAPS Pharma. Sci. Tech. 2008, 9(3), 774-781.
4. Badgujar BP, Mundada AS, The technologies used for developing orally disintegrating tablets:a
review. Acta Pharmaceutica. 2011, 61(2), 117-13.

469 | P a g e International Standard Serial Number (ISSN): 2319-8141
Full Text Available On www.ijupbs.com
5. http://en.wikipedia.org/wiki/file:o.phg,hypertention
6. Coyne KS, Davis D, Frech F and Hill MN, Health related Quality of Life in Patients treated for
Hypertension. Clinical Therapeutics. 2002, 24(1), 142-69.
7. http://en.wikipedia.org/wiki/file:o.phg,olmesartan medoxomil
8. WWW.drugs.com/monograph/olmesartan medoxomil.html
9. WWW.medguideindia.com/show_generic-php
10. Kumar B, Solid dispersion tech.: a tool for enhancing bioavailability of poorly soluble drug. J.
Chem. Pharm. Sci. 2011, 4(4), 170-178.
11. Reddy K, Development of Domperidone Mouth Dissolving Tablets Using Solid Dispersion
Technique Int. J. Res. Pharm. Bio. Sci. 2013, 4(3), 702-708.
12. Ahmed D, Formulation and evaluation of oral fast dissolving prochloperazine maleate tablet. Iraqi
J. Pharma. Sci. 2012, 21(1), 46-55.
13. Sawarikar P, Formulation and evaluation of fast dissolving/disintegrating tablet of isoxsuprine Hcl.
J. Curr. Pharm. Res. 2010, 3(1), 41-46
14. Dobetti L, Fast melting tablets: development and technology. Pharm. Tech. Drug Del. 2001, 44-50.
15. Patel V, Statistical Evaluation of Influence of Viscosity and Content of Polymer on Dipyridamole
Release From Floating Matrix Tablets: A Technical Note. AAPS Pharm. Sci. Tech. 2007, 8(3), E1-
E5.
16. Schwartz J and Connor R, Optimization techniques in pharmaceutical form-ulation and processing.
3rd Edn, Marcel Dekker, New York, 1997, 727-52.
17. Dr. Y Cheng, The response surface Methodology, 2nd Edition, Nuran Bradley, 2007, 4-6.
18. Velmurugan S, Orally disintegrating tablets: an overview. Int. J. Chem. Pharm. Sci. 2010, 1(2), 1-
12.
19. Patel B, Devlopment and invitro evaluation of FDT of glipizide. Int. J. Pharm. Sci. 2009, 1(1), 145-
150.
20. Garg R, Design optimization of FDT of telmisartan, effect of coprocessed superdisintegrants. Int. J.
Pharm. Bio. 2013, 3(3), 22-26.
21. ICH guidline Q1A-Q1F, www.ich.org/cache/compo/363-272-1.html


















