
3.MATERIALS AND METHODS - Shodhgangashodhganga.inflibnet.ac.in/bitstream/10603/10663/8/08...
18
98 3.MATERIALS AND METHODS The present study was undertaken in the Department of Microbiology, College of Basic Sciences, CSK Himachal Pradesh Krishi Vishvavidyalaya, Palampur. It is located in the mid-hills and sub-humid agro-climatic zone of Himachal Pradesh, India. This town is located at 32º6’N, 76º18’E and 1300 m msl. The chemicals and media components used were of analytical grade (AR) obtained from Merck limited - India, Sigma-Aldrich Inc. USA, and Hi Media Laboratories, Bombay, India. The materials used and the methods employed are presented below under the following sub-headings: 3.1 Standard microorganisms 3.2 Collection of rhizosphere soil samples 3.3 Isolation of Nitrogen fixing and phosphate solubilizing microorganisms 3.4 Qualitative assay of phosphate solubilizing activity 3.5 Quantitative assay of phosphate solubilizing activity 3.6 Quantitative assay of Nitrogenase activity 3.7 Detection of Indole acetic acid (IAA) production 3.8 Detection of siderophore production 3.9 Detection of ammonia production 3.10 Characterization, identification and maintenance of isolated microbial strains 3.10.1 Morphological and Biochemical characterization 3.10.2 Molecular characterization of efficient strains 3.11 Development of liquid formulations 3.11.1 Liquid carriers for formulations 3.11.2 Effect of stress conditions on liquid formulation 3.12 Statistical analysis of data
Transcript of 3.MATERIALS AND METHODS - Shodhgangashodhganga.inflibnet.ac.in/bitstream/10603/10663/8/08...

98
3.MATERIALS AND METHODS
The present study was undertaken in the Department of Microbiology, College of
Basic Sciences, CSK Himachal Pradesh Krishi Vishvavidyalaya, Palampur. It is located
in the mid-hills and sub-humid agro-climatic zone of Himachal Pradesh, India. This town
is located at 32º6’N, 76º18’E and 1300 m msl.
The chemicals and media components used were of analytical grade (AR)
obtained from Merck limited - India, Sigma-Aldrich Inc. USA, and Hi Media
Laboratories, Bombay, India. The materials used and the methods employed are
presented below under the following sub-headings:
3.1 Standard microorganisms
3.2 Collection of rhizosphere soil samples
3.3 Isolation of Nitrogen fixing and phosphate solubilizing microorganisms
3.4 Qualitative assay of phosphate solubilizing activity
3.5 Quantitative assay of phosphate solubilizing activity
3.6 Quantitative assay of Nitrogenase activity
3.7 Detection of Indole acetic acid (IAA) production
3.8 Detection of siderophore production
3.9 Detection of ammonia production
3.10 Characterization, identification and maintenance of isolated microbial strains
3.10.1 Morphological and Biochemical characterization
3.10.2 Molecular characterization of efficient strains
3.11 Development of liquid formulations
3.11.1 Liquid carriers for formulations
3.11.2 Effect of stress conditions on liquid formulation
3.12 Statistical analysis of data

99
3.1 Standard microorganisms
Standard strains of Pseudomonas striata (PSB), Azotobacter chroococum
(Nitrogen Fixer) and Azospirillum brasilense (Nitrogen Fixer) were procured from the
Institute of Microbial Technology (IMTECH), Chandigarh. Pseudomonas striata MTCC
1259 strain was maintained on nutrient agar, Azotobacter chroococum MTCC 446 was
maintained on Jensen’s medium and Azospirillum brasilense MTCC 125 was maintained
on nitrogen free malate (NFb) medium (Appendix I).
3.2 Collection of rhizosphere soil samples
Soil samples were collected from the rhizosphere of five crop plants (Plate 3.1)
viz., Triticum aestivum (Wheat), Zea mays (Maize) and Solanum tuberosum (Potato) and
two medicinal plants Aloe barbadensis (Aloevera) and Bacopa monnieri (Brahmi) grown
in the Model Organic Farm, CSK HPKV, Palampur. For this purpose, the plants were
uprooted carefully, shoots were cut off and roots along with rhizosphere soils were
brought to the laboratory in polythene bags. The soil samples were processed
immediately or stored at 4-8 ºC for the isolation of microorganisms.
3.3 Isolation of Nitrogen fixing and Phosphate solubilizing microorganisms
3.3.1 Processing of rhizosphere soil samples for isolation of Azotobacter
Isolation of Azotobacter from the above mentioned soil samples were done on
Jensen’s medium (Norris and Chapman 1968) by dilution plate technique. One gram of
each soil sample was transferred to 9 ml sterile dilution blank under aseptic conditions
and serial dilutions were made accordingly. Appropriate dilutions were plated on
Jensen’s medium and incubated at 30 ± 1 ºC for 2-3 days. The isolated strains were
stocked on Jensen’s medium slant for further studies.
3.3.2 Processing of rhizosphere soil samples for isolation of Azospirillum
Azopirillum was isolated from the above mentioned soil samples in semi-solid
nitrogen free malate (NFb) medium. Serial dilutions of soil samples were made as
mentioned above and one ml of appropriate dilution was inoculated in semi-solid NFb
medium (Dobereiner 1992) and incubated at 30 ± 1 ºC for 2-3 days. Formation of white,
dense pellicle in semi-solid NFb medium and change of color of medium from green to

100
blue indicated the presence of Azospirillum. The culture was purified by streaking white
pellicle on NFb agar plate and stocked on NFb agar slant for further studies.
A B
D C

101
Plate 3.1 Selected crop/medicinal plants which were used to isolate nitrogen fixing and
phosphate solubilizing bacterial (PSB) isolates (A) Triticum aestivum (Wheat), (B) Zea
mays (Maize), (C) Solanum tuberosum (Potato), (D) Aloe barbadensis (Aloevera) and (E)
Bacopa monnieri (Brahmi)
E

102
Processing of soil samples for isolation of Phosphate solubilizing microrganisms
Phosphate solubilizing microorganisms (PSM) were isolated from the above
mentioned soil samples by dilution plate technique using Pikovskaya’s medium
(Pikovskaya 1948) containing tri-calcium phosphate (TCP). Appropriate soil dilutions
were plated on Pikovskaya’s agar medium by spread plate technique and incubated at 30
± 1 ºC for 2-3 days. The colonies forming halo zone of clearance (Pikovskaya’s
medium) around them were counted as P-solubilizers. All the bacterial colonies
exhibiting halo zones were selected, purified and maintained on nutrient agar slants for
further studies.
3.4 Qualitative assay of phosphate solubilizing activity
Pure cultures of phosphate solubilizing bacteria were spot inoculated at the centre
of already prepared plates of Pikovskaya’s agar medium. The plates were incubated at 30
± 1 ºC for 7-10 days. The colonies forming more than 5.0 mm zone of solubilization were
stocked. The zone of phosphate solubilization (mm) formed around colonies was
recorded after every 24 hours for 10 days.
The solubilizing efficiency of the microorganisms was calculated using following
formula:
Z – C
Solubilizing efficiency (% S.E) = x 100
C
Z = Solubilization zone (mm)
C = Colony diameter (mm)
3.5 Quantitative assay of phosphate solubilizing activity
3.5.1 Estimation of solubilized P by bacterial isolates
The quantitative estimation of solubilized P by bacterial isolates was done by the
vanadomolybdophosphoric yellow color method (Subba Rao 1988) in NBRIP (National
Botanical Research Institute’s Phosphate growth medium) broth (Nautiyal 1999; Mehta
and Nautiyal 2001) containing 1000 µg/ml tri-calcium phosphate (TCP).

103
Method
NBRIP broth (Appendix I) in 100 ml aliquots containing 1000 µg P/ml in the
form of TCP was inoculated aseptically with 1 ml of culture broth having OD of 1.5 at
600 nm. The aliquots were incubated with shaking at 28 ºC up to 11 days. Five ml of the
growth medium from each flask was taken out on 3rd
, 5th
, 7th
and 11th
day, filtered
through Whatman No. 1 filter paper, and centrifuged (R-24 REMI) at 10,000 r.p.m for 20
minutes. Phosphorus in the cell free culture supernatant was determined by above
mentioned method. For this, 0.5 ml or 1 ml of the supernatant was taken, 2.5 ml of
Barton’s reagent was added and volume was made up to 50 ml with double distilled
water (ddw). After 10 minutes, the intensity of yellow color was read on
spectrophotometer (UV-VIS Spectrophotometer- SL-159, Elico, India) at 430 nm and the
amount of P-solubilized was extrapolated from the standard curve.
3.5.2 Standard curve
3.5.2.1 Standard stock P solution
Weigh 0.2195 g of potassium dihydrogen orthophosphate AR grade (dried in oven
at 60ºC for 1 hr and cooled in a dessicator) and dissolved in one liter of ddw. For
preparation of working solution, 150 ml of this solution was taken and volume was made
up to 250 ml with ddw.
3.5.2.2 Preparation of the standard curve
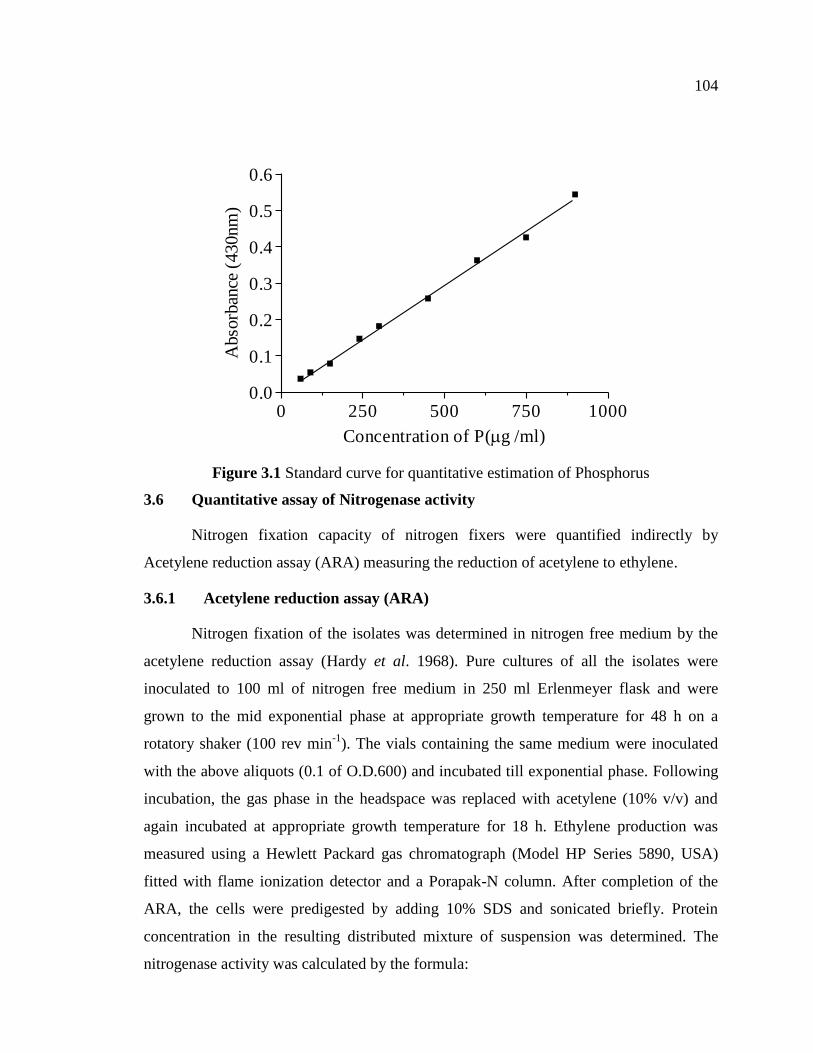
Standard curve was prepared by taking 2, 3, 5, 8, 10, 15, 20, 25 and 30 ml of the
working solution in each 50 ml volumetric flask, and 2.5 ml of Barton’s reagent was
added to each flask and the volume was made up to 50 ml with ddw. After 10 minutes,
the intensity of yellow color developed was read at 430 nm spectrophotometrically.
Standard curve was prepared by plotting absorbance at 430 nm Vs concentration of P
(Figure 3.1).
3.5.3 Determination of pH
The pH of the growth medium was determined at regular intervals by using digital
pH meter (Decibel India Ltd).
104
0 250 500 750 10000.0
0.1
0.2
0.3
0.4
0.5
0.6
Concentration of P(g /ml)
Abso
rban
ce (
430nm
)
Figure 3.1 Standard curve for quantitative estimation of Phosphorus
3.6 Quantitative assay of Nitrogenase activity
Nitrogen fixation capacity of nitrogen fixers were quantified indirectly by
Acetylene reduction assay (ARA) measuring the reduction of acetylene to ethylene.
3.6.1 Acetylene reduction assay (ARA)
Nitrogen fixation of the isolates was determined in nitrogen free medium by the
acetylene reduction assay (Hardy et al. 1968). Pure cultures of all the isolates were
inoculated to 100 ml of nitrogen free medium in 250 ml Erlenmeyer flask and were
grown to the mid exponential phase at appropriate growth temperature for 48 h on a
rotatory shaker (100 rev min-1
). The vials containing the same medium were inoculated
with the above aliquots (0.1 of O.D.600) and incubated till exponential phase. Following
incubation, the gas phase in the headspace was replaced with acetylene (10% v/v) and
again incubated at appropriate growth temperature for 18 h. Ethylene production was
measured using a Hewlett Packard gas chromatograph (Model HP Series 5890, USA)
fitted with flame ionization detector and a Porapak-N column. After completion of the
ARA, the cells were predigested by adding 10% SDS and sonicated briefly. Protein
concentration in the resulting distributed mixture of suspension was determined. The
nitrogenase activity was calculated by the formula:

105
C x PS x V
Nitrogenase activity (nmole C2H4 h-1
mg-1
protein) =
PStd x T x P
C = conc. of ethylene in nmoles
PS = peak height of sample
V= volume of air space in the assay vial
PStd = peak height of standard
T = time of incubation in hrs
P = protein concentration of bacterial cell in mg
3.6.2 Protein Measurement of Bacterial cells
The protein concentration of the bacterial cell was determined by Lowry method
(Lowry et al. 1951). To 0.2 ml of predigested bacterial cell suspension, distilled water
was added to make the volume 1 ml. To this, 5 ml of reagent “C” was added with
immediate vortexing and kept for 10 minutes at room temperature. Finally, 0.5 ml of
reagent “D” was added and incubated in the dark for 30 minutes. The intensity of blue
color developed was read on spectrophotometer (UV-VIS Spectrophotometer-SL-159,
Elico, India) at 660 nm and the amount of protein was extrapolated from the standard
curve. The reagents used in Lowry Method are given in Appendix I.
3.6.3 Standard Curve
3.6.3.1 Standard stock Protein (BSA) solution
Standard stock solution of Bovine Serum Albumin (100 μg/ml) was prepared in
distilled water.
3.6.3.2 Preparation of the standard curve
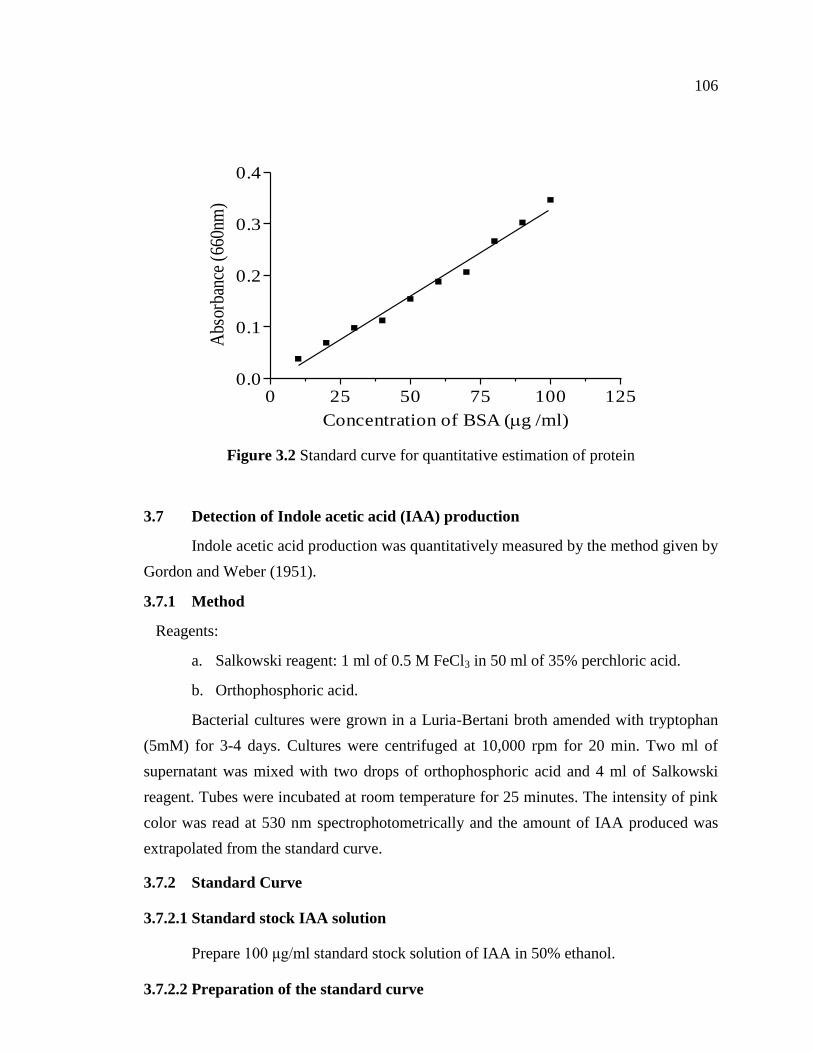
Standard curve was prepared by taking 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and
1.0 ml of standard BSA solution in test tubes. The volume was made to 1 ml with
distilled water, and to this, 5 ml of reagent “C” was added with immediate vortexing and
kept for 10 minutes at room temperature. To this, 0.5 ml of reagent “D” was added and
incubated in the dark for 30 minutes. The intensity of blue color developed was read at
660 nm spectrophotometrically. Standard curve was prepared by plotting absorbance Vs
concentration of Protein (Figure 3.2).
106
0 25 50 75 100 1250.0
0.1
0.2
0.3
0.4
Concentration of BSA (g /ml)
Abs
orba
nce
(660
nm)
Figure 3.2 Standard curve for quantitative estimation of protein
3.7 Detection of Indole acetic acid (IAA) production
Indole acetic acid production was quantitatively measured by the method given by
Gordon and Weber (1951).
3.7.1 Method
Reagents:
a. Salkowski reagent: 1 ml of 0.5 M FeCl3 in 50 ml of 35% perchloric acid.
b. Orthophosphoric acid.
Bacterial cultures were grown in a Luria-Bertani broth amended with tryptophan
(5mM) for 3-4 days. Cultures were centrifuged at 10,000 rpm for 20 min. Two ml of
supernatant was mixed with two drops of orthophosphoric acid and 4 ml of Salkowski
reagent. Tubes were incubated at room temperature for 25 minutes. The intensity of pink
color was read at 530 nm spectrophotometrically and the amount of IAA produced was
extrapolated from the standard curve.
3.7.2 Standard Curve
3.7.2.1 Standard stock IAA solution
Prepare 100 μg/ml standard stock solution of IAA in 50% ethanol.
3.7.2.2 Preparation of the standard curve
107
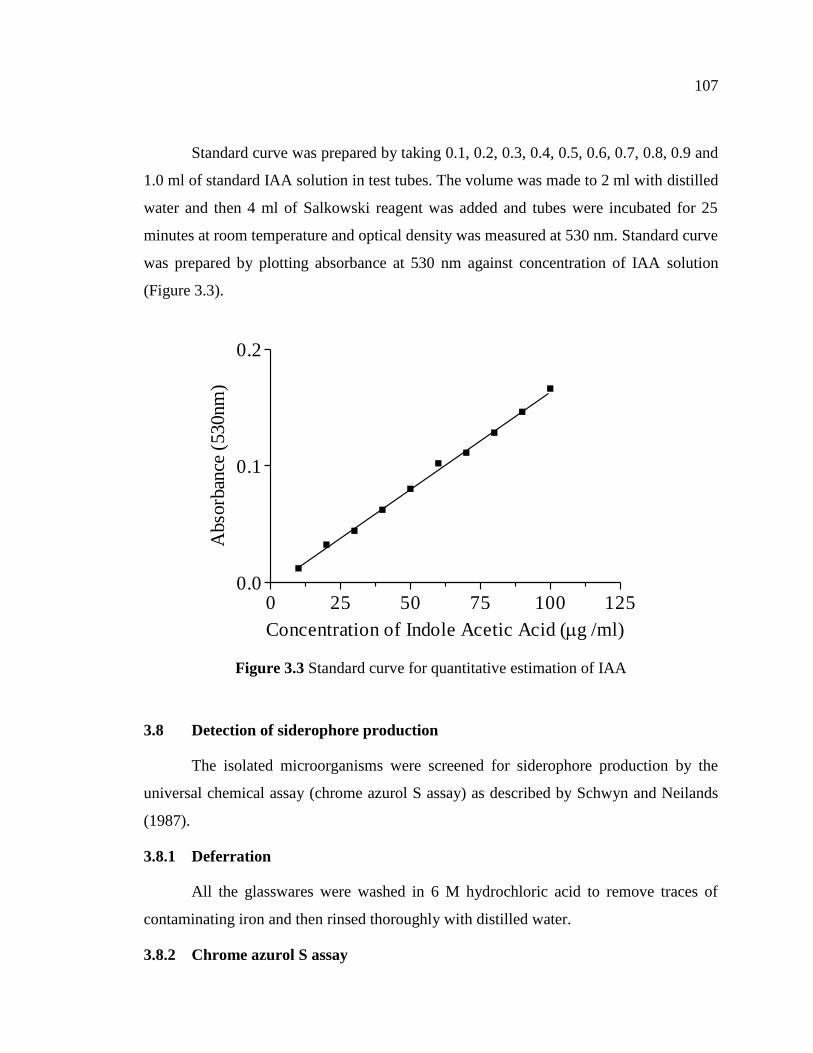
Standard curve was prepared by taking 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and
1.0 ml of standard IAA solution in test tubes. The volume was made to 2 ml with distilled
water and then 4 ml of Salkowski reagent was added and tubes were incubated for 25
minutes at room temperature and optical density was measured at 530 nm. Standard curve
was prepared by plotting absorbance at 530 nm against concentration of IAA solution
(Figure 3.3).
0 25 50 75 100 1250.0
0.1
0.2
Concentration of Indole Acetic Acid (g /ml)
Abso
rban
ce (
530nm
)
Figure 3.3 Standard curve for quantitative estimation of IAA
3.8 Detection of siderophore production
The isolated microorganisms were screened for siderophore production by the
universal chemical assay (chrome azurol S assay) as described by Schwyn and Neilands
(1987).
3.8.1 Deferration
All the glasswares were washed in 6 M hydrochloric acid to remove traces of
contaminating iron and then rinsed thoroughly with distilled water.
3.8.2 Chrome azurol S assay

108
The chrome azurol sulfonate (CAS) assay was used for the detection of
siderophores. This assay uses an iron-dye complex which changes color on loss of iron.
Siderophores, which have more affinity for the iron than the dye, remove the iron,
resulting in a change in color of the dye to orange. The iron-dye complex used to detect
the siderophore thus can be incorporated into King’s B medium
CAS-HDTMA solution
CAS-HDTMA solution was prepared by dissolving 121 mg chrome azurol sulfate
(CAS) in 100 ml of distilled water, and to this, 20 ml of 1 mM FeCl3.6H2O solution (1
mM FeCl3.6H2O solution prepared in 10 mM HCl) was added. It was slowly added to 20
ml hexadecyltrimethylammonium bromide (HDTMA) solution (729 mg HDTMA in 400
ml distilled water) and autoclaved at 121ºC for 15 minutes.
To 900 ml sterilized King’s B medium, 100 ml CAS-HDTMA solution was added
drop wise and mixed gently to avoid foaming. Plates were poured and incubated for 24
hrs at 30ºC for checking any contamination. All the isolates were spot inoculated on these
prepared plates of CAS agar and incubated at optimum growth temperature for 3-4 days.
The isolates producing orange color in the form of halo zone around the colonies were
considered as siderophore producers.
3.9 Detection of ammonia production
Qualitative detection of ammonia production was done by the method given by
Bakker and Schippers, (1987).
3.9.1 Method
Nessler’s reagent: 100 g Mercuric iodide and 70 g potassium iodide were
dissolved in a small quantity of distilled water and this mixture was added slowly, with
stirring, to a 500 ml cooled solution of sodium hyroxide (160 g sodium hyroxide
dissolved in 500 ml distilled water) and finally, this solution was diluted to 1 liter with
distilled water. Reagent was stored in rubber-stoppered borosilicate glassware in darkness
to maintain its stability for up to a year under normal laboratory conditions.
Bacterial isolates were grown in peptone water for 2-3 days at optimum growth
temperature. After incubation, 1ml of Nessler’s reagent was added in each tube. Tubes

109
showing faint yellow color indicated small amount of ammonia, and deep yellow to
brownish color indicated maximum amount of ammonia.
3.10 Characterization, identification and maintenance of isolated microbial
strains
3.10.1 Morphological and Biochemical characterization
The efficient phosphate solubilizing and nitrogen fixing bacteria were identified
on the basis of morphological, physiological and biochemical characteristics according to
the standard methods described in Bergey’s manual of Systematic Bacteriology (Kreig
and Holf 1984) and Laboratory Manual of Basic Microbiology (Kanwar et al. 1997).
All the isolated and identified cultures were stocked in appropriate agar slants
(nutrient agar, Jensen’s medium and NFb medium or soft agar (0.5% agar) in tight screw
capped sterile vials and incubated for 24-48 h at optimum growth temperature. Vials
having microbial cultures were then preserved at 4 0C.
3.10.2 Molecular characterization of efficient strains
Molecular characterization of most efficient bacterial isolates was done by
sequencing of their 16S rRNA gene.
3.10.2.1 Extraction of genomic DNA
Total genomic DNA of each isolate was extracted using commercial DNA
isolation kit (Real Biogenomics) by following the instruction manual of the manufacturer.
The amount of DNA was quantified by recording the absorbance at 260 nm wavelength
using UV/VIS spectrophotometer (Bio Rad, SmartSpec 3000). DNA was stored at -20oC
for further use.
3.10.2.2 PCR amplification of 16S rRNA gene
The 16S rRNA gene of the target bacterial isolates was amplified by using
universal eubacterial primers as reported by Heddi et al. (1998). The base sequences of
the primers used are presented in Table 3.1. These were custom synthesized (Sigma, Pvt.
Ltd.).

110
Table 3.1: Base sequences of 16S rRNA gene primers
Name of the Primer Sequence (5’ to 3’)
16S rRNA gene F
R
AGA GTT TGA TCA TGG CTC AG
TAC CTT GTT ACG ACT TCA CC
The PCR amplification was carried out in 0.2ml PCR tubes with 25 μl reaction
volume consisting of following components:
Reaction Mixture Quantity(µl)
Buffer 10 X 2.5
MgCl2 (25 mM) 1.5
dNTPs mix (10 mM each) 2.0
Taq DNA polymerase (5U/µl),
(Life Technologies India, Pvt. Ltd)
0.2
Primer forward 20 pmol 1.0
Primer reverse 20 pmol 1.0
Water (SDW) 14.8
DNA (10ng/ µl) 2.0
Total Volume 25.0
Reaction mixture was vortexed and centrifuged in a microfuge (Bangalore Genei,
India). Amplifications were performed using thermal cycler (GeneAmp PCR system
9700, Applied Biosystems, USA) with following temperature transitions:
Steps Temperature (oC) Time

111
1. Initial denaturation 94 5.00 min
2. Denaturation 94 45 sec
3. Annealing 53 45 sec
4. Elongation 72 30 sec
The thermal cycler was programmed for 30 cycles with one cycle of initial
denaturation and steps 2-4 were repeated 30 times and a final extension at 72oC for 30 sec
using fastest ramp time between the temperature transitions.
3.10.2.3 Agarose gel electrophoresis of PCR products
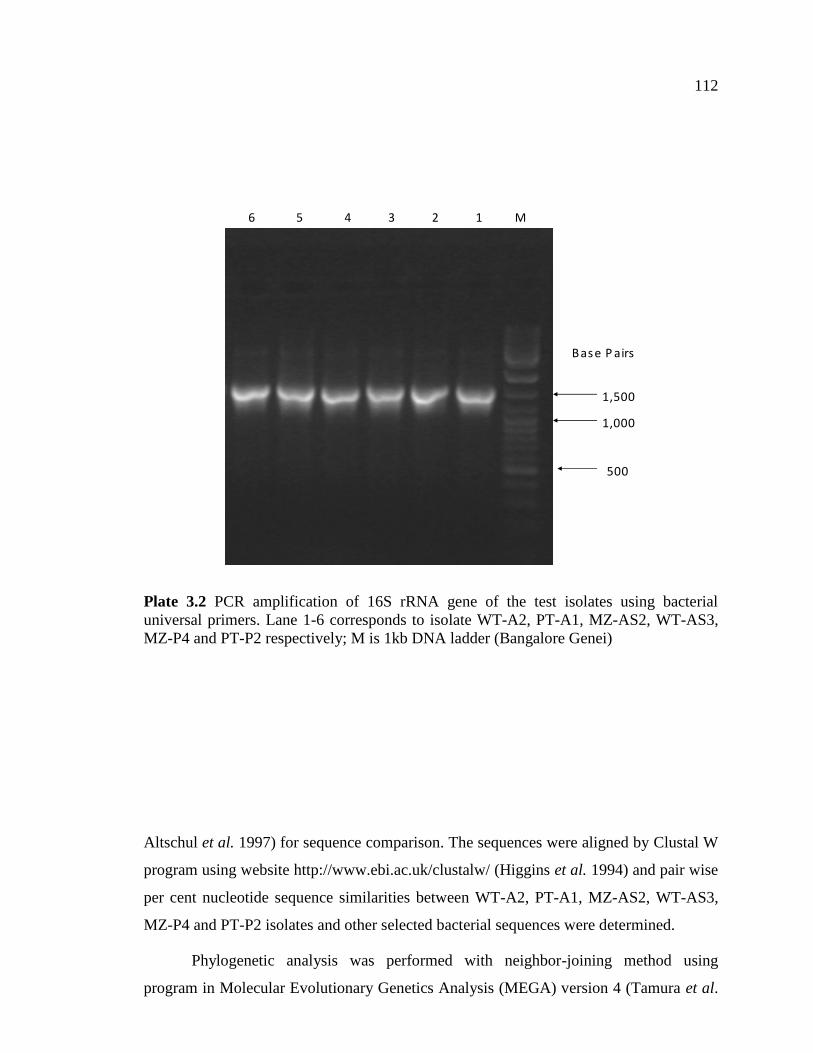
The amplified PCR products (Plate 3.2) were resolved by electrophoresis using 1
per cent agarose gel in 1X Tris acetate EDTA buffer (2 M Tris base, 57.10 ml acetic acid
and 0.5 M EDTA, pH 8.0, 50 X). Agarose gel was mixed with ethidium bromide (0.5 μg/
ml) before pouring. 1 kb DNA ladder (Bangalore Genei) was used as a marker. The gel
was run at 120V for 45 minutes using Bangalore Genei Power Pac system. The ethidium
bromide stained gel was viewed and image captured using gel documentation system
(Alphalmager 2200, Alpha Infotech Corporation, USA).
3.10.2.4 Sequencing and Data Analysis
PCR products of 16S rRNA gene of six efficient bacterial isolates obtained
through amplification with specific primer (section 3.10.2.3) were freeze dried in a
lyophilizer (CHRIST ALPHA I-2LD) and sent for custom sequencing using same
upstream and downstream primers used for the amplification of 16S rRNA gene
(Ocimum Biosolutions, Pvt. Ltd.).
3.10.2.5 Nucleotide sequence analysis
The 16S rDNA sequences of different bacterial isolates were BLAST (Basic
local alignment search tool) searched against the sequences of 16S rRNA of bacterial
isolates available in the Genbank Nucleotide Database (http://www.ncbi.nih.gov/blast;
112
Base P airs
500
1,000
1,500
6 5 4 3 2 1 M
Plate 3.2 PCR amplification of 16S rRNA gene of the test isolates using bacterial
universal primers. Lane 1-6 corresponds to isolate WT-A2, PT-A1, MZ-AS2, WT-AS3,
MZ-P4 and PT-P2 respectively; M is 1kb DNA ladder (Bangalore Genei)
Altschul et al. 1997) for sequence comparison. The sequences were aligned by Clustal W
program using website http://www.ebi.ac.uk/clustalw/ (Higgins et al. 1994) and pair wise
per cent nucleotide sequence similarities between WT-A2, PT-A1, MZ-AS2, WT-AS3,
MZ-P4 and PT-P2 isolates and other selected bacterial sequences were determined.
Phylogenetic analysis was performed with neighbor-joining method using
program in Molecular Evolutionary Genetics Analysis (MEGA) version 4 (Tamura et al.

113
2007). Confidence limits on grouping were done by the bootstrapping technique (1,000
repeats).
3.11 Development of liquid formulations
3.11.1 Liquid carriers for formulations
The following types of materials were used as liquid carriers for formulation
development with efficient isolates.
i) Biogas slurry
ii) Vermiwash
iii) Compost wash or Compost Tea
iv) Matka Khaad
v) Minimal Growth Medium (Peptone water)
Biogas slurry was obtained from Department of Agricultural Engineering, College
of Agricullture, CSK HPKV, Palampur. Vermiwash, Compost Tea and Matka Khaad
were obtained from “Model Organic Farm”, CSK HPKV, Palampur.
Biogas slurry was centrifuged at 10,000 rpm for 15 minutes to remove the
suspended particles and the supernatant was used for formulation development.
Vermiwash, Compost Tea and Matka Khaad were filter through muslin cloth to remove
any suspended particles, and used for formulation development.
The above liquid carriers were tried in combination with different chemicals to
study the survivability of each efficient isolates. To develop liquid formulations the
amendments viz., trehalose (10mM), polyvinylpyrollidone (PVP) (2%), glycerol (10mM)
and polyethyleneglycol (PEG) (1 %) were added separately to different liquid carriers.
These liquid carriers were then sterilized at 121°C for 15 minutes in an autoclave.
3.11.1.1 Mass production of efficient strains
For mass production of inoculant, 500 ml of respective growth medium was
inoculated with 5ml of efficient microbial cultures and incubated for 48-72 h at their
optimum growth temperatures under shaking (120 rev/ min.) conditions. Following
incubation, the cells were harvested by centrifugation (10000 rpm) for 10 min at 4 °C,
washed twice with 100 mM potassium phosphate buffer solution and resuspended in 100
114
mM potassium phosphate buffer, so as to achieve the final concentration of 109 cfu/ml.
This served as mother culture for inoculating various liquid carriers.
3.11.1.2 Mixing of mother culture with the carrier material
The above prepared bacterial suspension was mixed thoroughly with 100 ml of
liquid carrier at the rate of 1 per cent (%). The inoculated flasks were kept at room
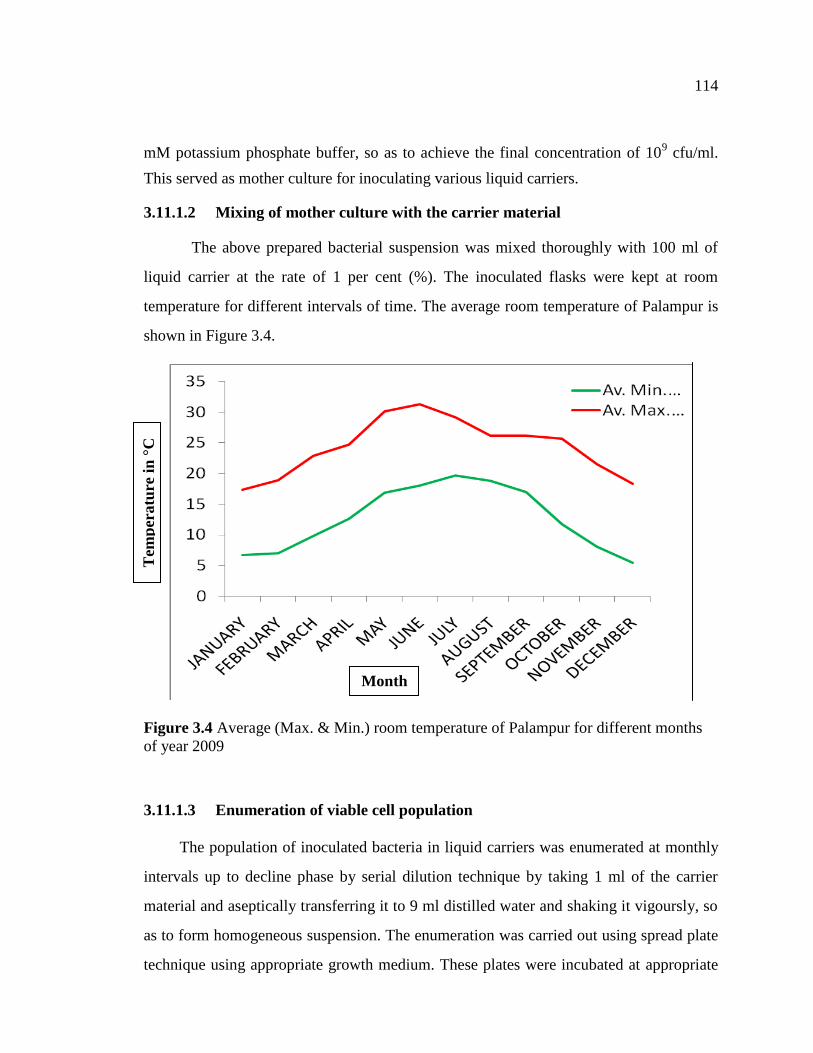
temperature for different intervals of time. The average room temperature of Palampur is
shown in Figure 3.4.
Figure 3.4 Average (Max. & Min.) room temperature of Palampur for different months
of year 2009
3.11.1.3 Enumeration of viable cell population
The population of inoculated bacteria in liquid carriers was enumerated at monthly
intervals up to decline phase by serial dilution technique by taking 1 ml of the carrier
material and aseptically transferring it to 9 ml distilled water and shaking it vigoursly, so
as to form homogeneous suspension. The enumeration was carried out using spread plate
technique using appropriate growth medium. These plates were incubated at appropriate
Month
s
Tempe
rature
in °C
Tem
per
atu
re i
n °
C

115
growth temperature for 24-48 h. Plates containing 30-300 colonies were recorded and the
counts were expressed as log cfu per ml of carrier material.
3.11.2 Effect of stress conditions on liquid formulation
The best formulation was subjected to various stress conditions viz., temperature
(15, 25, 40 and 50 °C), pH (4.5, 5.5, 6.5 and 7.5) and desiccation. The best formulation
was amended with different chemicals and inoculated as described in 3.11.1 and 3.11.1.2.
The viable populations of bacterial cultures were enumerated as described in 3.11.1.3 at
regular intervals of time.
3.11.2.1 Effect of temperature stress on Survivability
The inoculated formulation was incubated at different stress temperatures and
was enumerated for viable cell population at regular intervals of time.
3.11.2.2 Effect of pH stress on Survivability
The best formulation prepared in 3.11.2 was subjected to varying pH, and
incubated at their optimum growth temperature. The population of viable cells was
enumerated at regular intervals of time.
3.11.2.3 Desiccation tolerance of efficient strains
One ml of six fold diluted formulation was taken in sterile 1.5 ml eppendorf
tubes. The tubes were kept open in a sterile box and incubated at 37 °C in an incubator
till these were dried up. The dried cells further incubated and viable cells were
enumerated at regular intervals of time by resuspended in 100 μl of sterile distilled water
with vigorous agitation.
3.12 Statistical analysis of data
Results of the measurements were subjected to analysis of variance (ANOVA) by
Least Significant Difference (LSD) using Windowstat package, Version 8.0 (Windowstat
2008).


















