
30 Appendices
18
APPENDIX I. OF ORGANIC Appendlces SU BSTITUTIVE NOM ENCLATU RE COMPOUNDS The substitutive name of an organic compound is based on its principal group and principal chain. T\re principal group is assigned according to the following priorities: o il o tl o il -C-X (acid halide) > o ll o il -C-OH (carborylic acid) > -C-O-C- (anhydride) > -C-OR (ester) o il o ll -C-NR2 (amide) > -C-$ (nitrile) > -C-H (aldehyde) > o ll -C- (ketone) > -OH (alcohol, phenol) > -SH (thiol) > -NRz (amine) TI.rc principal chain is identifiedby applying the following criteria in order until a decision can be made. 1. Maximum number of substituents corresponding to the principal group 2. Maximum number of double and triple bonds considered together 3. Maximum length 4. Maximum number of substituents cited as prefixes A-1
description
Loudon Organic Chemistry Appendices
Transcript of 30 Appendices

APPENDIX I.OF ORGANIC
AppendlcesSU BSTITUTIVE NOM ENCLATU RECOMPOUNDS
The substitutive name of an organic compound is based on its principal group and principalchain.
T\re principal group is assigned according to the following priorities:
oil
otl
oil
-C-X (acid halide) >
oll
oil
-C-OH (carborylic acid) > -C-O-C- (anhydride) > -C-OR (ester)
oil
oll
-C-NR2 (amide) > -C-$ (nitrile) > -C-H (aldehyde) >
oll
-C- (ketone) > -OH (alcohol, phenol) > -SH (thiol) > -NRz (amine)
TI.rc principal chain is identifiedby applying the following criteria in order until a decisioncan be made.
1. Maximum number of substituents corresponding to the principal group2. Maximum number of double and triple bonds considered together3. Maximum length4. Maximum number of substituents cited as prefixes
A-1

A,-2 APPE N Dtc ES
1 97 9 Recornrtendatio ns :
1 99 3 Recornntendations:
A principal chain is numbered by applying the following criteria in order until there is noambiguity. Where multiple numbers are possible, comparisons are made at the first point ofdifference.
l. Lowest number for the principal group cited as a suffix-that is, the group on which thename is based
2. Lowest numbers for multiple bonds, with double bonds having priority over triple bondsin case of ambiguity
3. Lowest numbers for other substituents, taking into account the "first point of difference"rule (p. 62, rule 8)
4. Lowest number for the substituent named as a prefix that is cited first in the name
The name ts constructed starting with the hydrocarbon corresponding to the principalchain.
1. Cite the principal group by its suffix and number; its number is the last one cited in thename.
2. lf there is no principal group, name the compound as a substituted hydrocarbon.3. Cite the names and numbers of the other substituents in alphabetical order at the begin,
ning of the name.
These lists cover most of the cases cited in the text. (See Study Problems 8.1-8.3,pp.328-331, for illustrations.) For a more complete discussion of nomenclature, see Nomen-clature of Organic Chemistry, 1979 Edition, by the International Union of Pure and AppliedChemistry, published by Pergamon Press.
In 1993, the IUPAC released A Guide to IUPAC Nomenclature of Organic CompoundsRecommendations 1993, by R. Panico, W. H. Powell, and Jean-Claude Richer (senior editor),Blackwell Science. This publication advocated one major change that affects the nomencla-ture of relatively simple compounds. This change involves the way that principal groups arecited. The 1993 Recommendations cite the principal group or multiple bond position with anumber preceding the suffix itself, whereas the 1979 Recommendarlons (followed in this text)cite the principal group or multiple bond position with a number preceding the hydrocarbonname. These differences are best illustrated by example.
H:C-CHCH2CHTCH-3
I -pentenepent- l -ene
HOCH2CH2CH2CH2CHT
1-pentanolpentan- l -ol
HOCH2CH2CH2CH:CH:
4-penten- l -olpent-4-en- l -ol
The 1993 Recommendationshave not yet been generally adopted. Thus, names that adhereto either set of recommendations are acceptable.
APPENDIX II. INFRARED ABSORPTIONS OF ORGANIC COMPOUNDSThis table presents a summary of the important infrared absorptions discussed in this text. Formore detailed tables, the reader may wish to consult more specialized texts, such as InfraredAbsorption spectroscopy, by Koji Nakanishi and Philippa H. Solomon, San Francisco:Holden-Day, 1977; or Organic Structure Analysis, by Philip Crews, Jaime Rodriguez, andMarcel Jaspars, 1998, Oxford University Press, Chapter 8.

APPENDIX II. INFRARED ABSORPTIONS OF ORGANIC COMPOUNDS A.3
Typeofabsorption FrEquency,Gm-r(lntensity)* Comment
Alkanes
C-H stretch 2850-3000 (m) occurs in all compounds with aliphatic C-H bonds
Alkenes
C:C stretch
-CH:CHz t 640 (m)
16ss (m)C: CHr/others 1660-1025 (w) not observed if alkene is symmetrical
:C-H stretch 3000-31 00 (m)
:C-H bend
-CH:CHz 910-990 (s)
8e0 (s)C:CH,/
960-980 (s)\/C:C/\
H
HH\/r-fL-L/\ 675-730 (s\ position is highly variable
800-8a0 (s)
H\/C:C/\Alcohols and Phenols
O-H stretch 3200-3400 (s)
C-O stretch 1050-1 250 (s) also present in other compounds with C-O bonds:ethers, esters, etc.
AlkynesC:C stretch 2100-2200 (m) not present or weak in many internal alkynes
-C-H stretch 3300 (s) present in 1-alkynes only
Aromatic Compounds
C:C stretch 1 500, t 600 (s) two absorptionsrh
C-H bend 650-750 (s)
overtone 1660-2000 (w)
*(s) - strong; (m) - medium; (w) - weak.
(Table continues)

A-4 APPENDTcES
Type of absorption Frequency, cm-l (lntensity)* Comment
Aldehydes
C:O stretchordinary 1720-1725 (s)a,p-unsaturated 1680-1690 (s)benzaldehydes 1700 (s)
C-H stretch zz20 (m)
Kctones
C:O stretchordinary 1710-171 5 (s) increases with decreasing ring size (Table 2t .3,
p.996)d,&unsaturatd l67G-168O (s)aryl ketones 1680-1690 (s)
Carborylic Acids
C:O stretchordinary t 710 (s)benzoic acids 1680-1690 (s)
O-H stretch 2400-3000 (s) very broad
Esterc and Lactonet
C:O stretch 1735-1745 (s) increases with decreasing ring size (Table 21.3,p.996)
Acid Chlorides
C:O stretch 1800 (s) a second weaker band sometimes observed atr 700-1 750
C:O stretch 1760,1820 (s)
Amldesand Lactams
C:O stretch 165G1655 (s) increases with decreasing ring size (Table 21.3,p.996)
N-H bend 1640 (s)
Anhydrides
two bands; increases with decreasing ring sizein cyclic anhydrides
N-H stretch 3200-3400 (m) doublet absorption observed for someprimary amides
Nitriles
C-N stretch 2200-2250 (m)
Amines
N-H stretch 3200-337s (m) several absorptions sometimes observed,especially for primary amines
*(s) : strong; (m) : medium; (w) : weak.

APPENDIX III. PROTON NMR CHEMICAL SHIFTS IN ORGANIC COMPOUNDS A-5
APPENDIX III. PROTON NMR CHEMICAL SHIFTSIN ORGANIC COMPOUNDS
This appendix is subdivided into a table of chemical shifts for protons that are pa rt of functtonalgroups and a table of chemical shifts for protons that are adjacenfto functional groups.
A. Protons within Functional Groups
Group Chemical shift, ppm Group Chemical shift, ppm
tl-C-C-Htl 4.7 -1 .5
0ll
-c-H 9-1 1
H\/r-a\_-\-/\otl
-C-N-HI
7.s-9.54.6-5.7
-O-H varies with solvent andwith acidity of O-H
-c-c-H 1.7-2.s-c-NH-
I
0.s-1.5
/Y*V 6.5-8.5
B. Protons Adjacent to Functional GroupsIn this table, a range of chemical shifts is given for protons in the general environment
I
H-C-G
in which G is a group listed in column 1, and the two other bonds are to carbon or hydrogen.The remaining columns give the approximate chemical shifts for methyl protons (H3C-G),
methylene protons (-CH2-G), and methine protons (-CH-G), respectively. The shiftsin the following table are typical; some variation with structure of a few tenths of a ppm can beexpected. The chemical shifts of methine protons are usually further downfield than those ofmethylene protons, which are further downfield than methyl protons. Each additional carbonsubstitution increases the chemical shift by 0.3-1.0 ppm.
GNH- 2.s-3.s
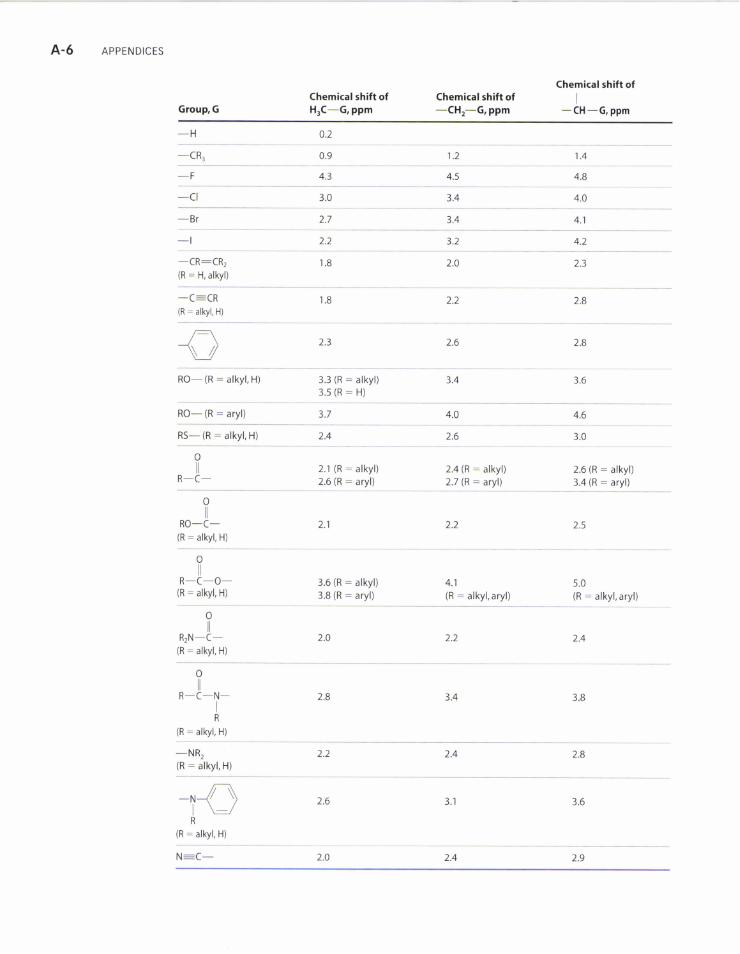
A-6 APPENDTcES
Group, GChemical shift ofH3C-G, ppm
Chemical shift of-CH'-G, ppm
Chemical shift ofI
I
-CH-G,ppm
-H 0.2
-CR, 0.9 1.41.2
-F 4.3 4.5 4.8
-cl 3.0 3.4 4.0
-Br 2.7 4.13.4
-l 2"2 3.2 4.2
-CR:CRz(R : H, alkyl)
1.8 2.4 2.3
-C_CR(R - alkyl, H)
2.2 2.8
RO- (R : alkyl, H) 3.3 (R - alkyl)3.s (R - H)
RO- (R : aryl) 3.7 4.0 4.6
RS- (R : alkyl, H) 2.4 2.6 3.0
otlR-C- 2.1 (R - alkyl)
2.6 (R - aryl)2.4 (R - alkyl)2.7 (R - aryl)
2.6 (R - alkyl)3.4 (R - aryl)
otlRO-C-
(R - alkyl, H)
ollR-C-O-
(R - alkyl, H)3.6 (R - alkyl)3.8 (R : aryl)
4.1(R : alkyl, aryl)
5.0(R : alkyl, aryl)
ollR2N-C-
(R - alkyl, H)
otlR-C-N-
I
R
(R - alVl, H)
2.8
-NR,(R : alkyl, H)
-NnI\JR
(R - alkyl, H)
3.1
N:C-

APPENDIX IV. 13C NMR CHEMICAL SHIFTS IN ORGANIC COMPOUNDS A-7
APPENDIx Iv. 13C NMR CHEMICAL SHIFTSIN ORGANIG GOMPOUNDS
This section is divided into a table of chemical shifts for carbons within functional groups anda table of chemical shifts for alkyl carbons adjacent to functional groups. A typical range ofshifts is siven for each case.
A. Chemical Shifts of Carbons within Functional Groups
Group Chemical shift range' PPm
-CH,-CHr-
I
-CH-
B-23
20-30
21 -33
17 -29I
-c-I
1 05-1 50*\tC:C/\
a-r\-- \- 66-93*
G- 1 25-1 50"
oll
tc -.- 200-220
otl
.CrO.R R: H, alkyl r 70-1 80
R : H, alkyl 165-175
oil
,c--NaRI
R
-C-N 110-120
*Alkyl substitution typically increases chemical shift.
B. Ghemical Shifts of Garbons ediacent to runctional GroupsIn most cases, alkyl substitution on the carbon increases chemical shift. Methyl carbons willhave shifts in the low end ofthe range; tertiary and quaternary carbons will have shifts in theupper end of the range.

A-8 APPENDTcES
Group G Chemical shift of carbon in G - C -I
R,C:CR- 14-40
HC-C- 1 8-28
29-45
F- B3-9 1
cl-Br- 32-6s
5-42
HO- 62-70
RO- R - alkyl,H 70-79
ollR-C- R-alkyl,H 43-50
oilRO-C- R-alkyl,H 33-44
RrN- R-alkyl,H 41-51 (R:H)53-60 (R : alkyl)
N-C- 16-28
APPENDIX V. SUMMARY OF SYNTHETIC METHODS
t-
The following methods are listed in order of their occurence in the text; the section referencefollows each reaction in parentheses. Thus, a review at any point in the text is possible by con-sidering the methods listed for earlier sections.
Don't forget that in many cases, a method can be applied to compounds containing morethan one functional group. Thus, catalytic hydrogenation can be used to convert phenols intoalcohols, but it is listed under "Synthesis of Alkanes and Aromatic Hydrocarbons" becausethe actual transformation is the formation of -CHr-CHr- groups from -CH:CH-groups; the presence of the --{H group is incidental.
Reaction summaries for each chapter are found in the Study Guide.
A. Synthesis of Alkanes and Aromatic Hydrocarbons1. Catalytic hydrogenation of alkenes (4.9A)2. Protonolysis of Grignard or related reagents (8.88)3. Cyclopropane formation by the addition of carbenoids to alkenes (Simmons-Smith re-
action;9.8B)4. Catalytic hydrogenation of alkynes (14.6A)5. Friedel{rafts alkylation of aromatic compounds (16.48)6. Catalytic hydrogenation of aromatic compounds (16.6)7. Stille reaction of aryl triflates and aryl- or alkylstannanes to form substituted aromatic
hydrocarbons (18.10B)

APPENDIX V. SUMMARY OF SYNTHETIC METHODS A.9
8. Wolff-Kishner or Clemmensen reductions of aldehydes or ketones (19.12)9. Reaction of aryldiazonium salts with hypophosphorous acid (23.10A)
B. synthesis of Alkenesl. B-Elimination reactions of alkyl halides or sulfonates (9.5, 10.3A, 17 .38)2. Acid-catalyzed dehydration of alcohols ( I 0' I )3. Catalytic hydrogenation of alkynes (gives cis-alkenes when used with internal alkynes;
14.6A)4. Reduction of alkynes with alkali metals in liquid ammonia (gives trans-alkenes when
used with internal alkenes; 14.68)5. Diels-Alder reactions of dienes and alkenes to give cyclic alkene s (15 .3 , 2'7 .3)
6. Heck reaction of aryl halides and alkenes to give aryl-substituted alkenes (18.6A)7. Suzuki coupling of aryl or vinylic halides with aryl or vinylic boronic acids (18.68)8. Alkene metathesis (18.6C)9. Wittig reaction of aldehydes and ketones ( 19. l3)
10. Aldol condensation reactions ofaldehydes orketones to give a,B-unsaturated aldehydesor ketones (22.4)
11. Hofmann elimination of quaternary ammonium hydroxides (23.8)
c. Synthesis of Alkynes1. Alkylation of acetylenic anions with alkyl halides or sulfonates (14.78)2. B-Elimination reactions of alkyl dihalides or vinylic halides (18.2)
D. Synthesis of Alkyl, Aryl, and Vinylic Halides1. Addition of hydrogen halides to alkenes (4.7, 15.44)2. Addition of halogens to alkenes to give vicinal dihalides (5.2)3. Peroxide-promoted addition of HBr to alkenes (5.6)4. Addition of halogens or HBr to alkynes (14.4)5. Synthesis of dihalocyclopropanes by the addition of dihalomethylene to alkenes (9.8A)6. Reaction of alcohols with HBr, thionyl chloride, or phosphorus tribromide (10.2, 10.3D,
17.1)7. Reaction of sulfonate esters or other alkyl halides with halide ions (10.3,{, 17.4)8. Halogenation of aromatic compounds (16.4A)9. Allylic and benzylic bromination of alkenes or aromatic hydrocarbons (17.2)
10. a-Halogenation of aldehydes, ketones, or carboxylic acids (22.34,C)1 l. Synthesis of aryl halides by the reaction of cuprous chloride, cuprous bromide, or potas-
sium iodide with aryldiazonium salts (Sandmeyer and related reactions; 23.10A)
E. Synthesis of Grignard Reagents and RelatedOrganometallic ComPounds
1. Reaction of alkyl or aryl halides with metals (8.8A)2. Preparation of lithium dialkylcuprates by the reaction of alkyllithium reagents with
cuprous halides (1 1.4C)3. Preparation of acetylenic Grignard reagents by the metal-hydrogen exchange (L4.7 A)4. Preparation of alkyl- and arylstannanes by the reaction of Grignard reagents with
trialkylstannyl chlorides ( I 8.98)

A- 1 0 APPENDTcES
F. Synthesis of Alcohols and Phenols(Syntheses apply only to alcohols unless noted otherwise.)
l. Acid-catalyzed hydration ofalkenes (used industrially, but generally not a good labora-tory method;4.9B)
2. Synthesis ofhalohydrins from alkenes (5.2B)3. Oxymercuration-reduction of alkenes (5.4A)4. Hydroboration-oxidation of alkenes (5.4B)5. Ring-opening reactions of epoxides (1 l.4A,B)6. Reaction of ethylene oxide with Grignard reagents (1 l.4C)7. Reaction of epoxides with lithium organocuprates (l l.4C)8. Reduction of aldehydes orketones (19.8,22.9,24.8D)9. Reactionof aldehydesorketoneswithGrignardorrelatedreagents (19.9,22.10A)
10. Reduction of carboxylic acids to primary alcohols (20. 10)1 l. Reduction of esters to primary alcohols (2I.9A)12. Reaction of esters with Grignard or related reagents (21.10A)13. Aldol addition reactions of aldehydes or some ketones to give B-hydroxy aldehydes or
ketones (22.4)14. Reaction of diazonium salts with water to give phenols (23. 10A)15. Synthesisof phenolsbytheClaisenreanangementof allylicarylethers (27.4B)
c. Synthesis of GlycolsI. Acid-catalyzed hydrolysis of epoxides (I L4B)2. Reaction of alkenes with osmium tetroxide or alkaline potassium permanganate (11.5)
H. Synthesis of Etherg Acetals, and Sulfidesl. Alkylation of alkoxides, phenoxides, or thiolates with alkyl halides or alkyl sulfonates
(Williamson synthesis; I l.l A, 18.78, 24.7)2. Alkoxymercuration-reduction of alkenes ( 1 I . 18)3. Acid-catalyzed dehydration of alcohols ( I 1. I C)4. Acid-catalyzed addition of alcohols to alkenes ( I I . I C)5. Reaction of epoxides with alkoxides and alcohols (11.4A,B)6. Reaction of 2- and 4-nitroaryl halides with alkoxides (18.4)7 . Acetal formation by the acid-catalyzed reaction of alcohols with aldehydes or ketones
(r9.104,24.6)8. Conjugate addition of thiols to d,B-unsaturated carbonyl compounds (22.8A)
l. Synthesis of EpoxidesI . Oxidation of alkenes with peroxycarboxylic acids ( 1 l .2A)2. Cyclization of halohydrins ( I I .2B)3. Asymmetric epoxidation of allylic alcohols (11.10)
J. Synthesis of Disulfides1. Oxidation of rhiols (10.9,26.8)

APPENDIX V. SUMMARY OF SYNTHETIC METHODS 4.11
K. Synthesis of Aldehydes1. Ozonolysis of alkenes (of limited utility because carbon-carbon bonds are broken; 5.5)2. Oxidation of primary alcohols (10.6A)3. Oxidative cleavage of glycols (of limited utility because carbon--carbon bonds are bro-
ken; 11.5B, 24.8C)4. Hydroboration--oxidation of alkynes ( 14.58)5. Oxidation of allylic and benzylic alcohols with MnO, (Sec. 17.54)6. Reduction of acid chlorides (21.9D).7. Aldol addition reactions of aldehydes to give B-hydroxy aldehydes (22.4)8. Aldol condensation reactions ofaldehydes to give a,B-unsaturated aldehydes (22.4)9. Synthesis of aldoses from other aldoses by the Kiliani-Fischer synthesis (24.9) and the
Ruff degradation (24. 10)
L. Synthesis of Ketonesl. Ozonolysis of alkenes (of limited utility because carbon-carbon bonds are broken; 5.5)2. Oxidation of secondary alcohols (10.6A)3. Oxidative cleavage of glycols (of limited utility because carbon--carbon bonds are bro-
ken; 11.5B)4. Mercuric-ion catalyzed hydration of alkynes (14.5A)5. Friedel-Crafts acylation of aromatic compounds (16.4F)6. Oxidation of phenols to quinones (18.8)7. Reaction of acid chlorides with lithium dialkylcuprates (21.10B)8. Aldol condensation reactions of ketones to give a,B-unsaturated ketones (22.4)9. Claisen and Dieckmann condensation reactions of esters to give B-keto esters (22.5A,
B)10. Crossed Claisen condensation reactions of esters to give B-diketones (22.5C)I l. Acetoacetic ester synthesis (22.7C)12. Conjugate addition reactions of a,B-unsaturated ketones (22.8), including the addition
of lithium dialkylcuprate reagents (22.lOB)
M. Synthesis of Sulfoxides and Sulfonesl. Oxidation of sulfides (11.8)
N. Synthesis of carboxylic and Sulfonic Acids(Syntheses apply only to carboxylic acids unless noted otherwise.)
l. Ozonolysis of alkenes (of limited utility because carbon--carbon bonds are broken; 5.5)2. Oxidation of primary alcohols (10.6B)3. Oxidation of thiols to sulfonic acids (10.9)4. Sulfonation of aromatic compounds to give arylsulfonic acids (16.4D, 20.6)5. Side-chain oxidation of alkylbenzenes (17.5)6. Oxidation of aldehydes (19.14)7. Reaction of Grignard or related reagents with carbon dioxide (20.6)8. Hydrolysis of carboxylic acid derivatives, especially nitriles (21.7 , 2l .ll, 26.7)9. Haloform reaction of methyl ketones (of limited utility because carbon--carbon bonds
are broken; 22.38)

A-12 APPENDTcES
10. Malonic ester synthesis (22.64,26.48)11. Strecker synthesis of a-amino acids (26.4C)
O. Synthesis of Esters1. Reaction of alcohols and phenols with sulfonyl chlorides (for sulfonate esters; 10.3,4',
18.10B)2. Acid-catalyzed esterification of carboxylic acids with primary or secondary alcohols
(20.8A, 24;t,26.5)3. Alkylation of carboxylic acids with diazomethane (20.88)4. Alkylation of carboxylate salts with alkyl halides (20.8B)5. Reaction of acid chlorides, anhydrides, or esters with alcohols and phenols (21.8,24.7)6. Claisen and Dieckmann condensation reactions of esters to give B-keto esters (22.54.,8)7. Alkylation of ester enolate ions; includes malonic ester synthesis, acetoacetic ester syn-
thesis, and direct alkylation (22.7)8. Conjugate addition reactions of a,B-unsaturated esters (22.8,22.108)
P. Synthesis of Anhydrides1. Reaction of carboxylic acids with dehydrating agents (20.98)2. Reaction of acid chlorides with carboxylate salts (21.8A)
Q. synthesis of Acid chlorides1. Reaction of carboxylic or sulfonic acids with thionyl chloride, phosphorus pentachlo-
ride, or related reagents (20.9A')2. Synthesis of sulfonyl halides by chlorosulfonation of aromatic compounds (20.9A)
R. Synthesis of Amides1. Reaction of acid chlorides, anhydrides, or esters with amines (21.8,23.7C,26.5)2. Condensation of amines and carboxylic acids with dicyclohexylcarbodiimide (26.6)
S. Synthesis of Nitrilesl. Formation of cyanohydrins from aldehydes and some ketones (19.7A,8,24.9)2. Reaction of alkyl halides or alkyl sulfonates with cyanide ion (21. I i )3. Conjugate addition of cyanide ion to a,B-unsaturated carbonyl compounds (22.8A)4. Reaction of cuprous cyanide with aryldiazonium salts (23.10A)
T. Synthesis of Amines1. Reductionof amides (21.98)2. Reduction of nitriles to primary amines (21.9C)3. Direct alkylation of ammonia or amines (of limited utility because of the possibility of
over-alkylation; 23.7 A, 26.4 A)4. Reductive amination of aldehydes and ketones (23.78)5. Aromatic substitution reactions of aniline derivatives (23.9)6. Gabriel synthesis of primary amines (23.ll{)7. Reduction of nitro compounds (23.11B)

APPENDIX V. SUMMARY OF SYNTHETIC METHODS A-13
8. Pd-catalyzed amination of aryl halides and triflates (23.11C)9. Curtius and Hofmann reuurangements (23.1lD)
U. Synthesis of Nitro Compounds1. Nitration of aromatic compounds (16.4C, 18.9)
APPENDIX VI. REACTIONS USED TO FORMCARBON-CARBON BONDS
Reactions that form carbon-carbon bonds have central importance in organic chemistry, be-cause these reactions can be used to form carbon chains or rings. These reactions are listed inthe order in which they are discussed in the text. The section reference follows each reactionin parentheses.
l. Cyclopropane formation by addition ofcarbenes or carbenoids to alkenes (9.8)2. Reaction of Grignard reagents with ethylene oxide (1 1.4C)3. Reaction of epoxides with lithium organocuprates (11.4C)4. Reaction of acetylenic anions with alkyl halides or sulfonates (14.78)5. Diels-Alder reactions (15.3,21 .3)6. Friedel-Crafts alkylation (16.4E) and acylation reactions (16.4F)7. The Heck reaction of alkenes with aryl halides (18.5F)8. Suzuki coupling of aryl or vinylic halides with aryl or vinylic boronic acids ( I 8.68)9. Alkene metathesis (18.6C)
10. The Stille reaction of organostannanes with aryl triflates (18.9B)I 1 . Cyanohydrin formation (19.7 , 24.9, 26.4C)12. Reaction of Grignard and related reagents with aldehydes and ketones (19.9)13. Wittig alkene synthesis (19.13)14. Reaction of Grignard and related reagents with aldehydes and ketones (20.6)15. Reaction of Grignard and related reagents with esters (21 .10A)16. Reaction of lithium dialkylcuprates with acid chlorides (21.10B)17. Reaction of cyanide ion with alkyl halides or sulfonates (21.11)18. Aldol addition and condensation reactions (22.4)19. Claisen and related condensation reactions (22.5)20. Malonic ester synthesis (22.7 4,26.48)21. Alkylation of ester enolate ions with alkyl halides or sulfonates (22.78)22. Acetoacetic ester synthesis (22.7C)23. Conjugate-addition reactions of cyanide ion (22.8A) or enolate ions (22.8C) to a,B-un-
saturated carbonyl compounds24. Conjugate addition of lithium dialkylcuprate reagents to c,B-unsaturated carbonyl com-
pounds (22.108)25. Reaction of aryldiazonium salts with cuprous cyanide (23.1 0A)26. Formation of rings by electrocyclic reactions (27 .2)27. Claisen realrangement (27 .48)

A-14 APPENDTcES
APPENDIX VII. TYPICAT AGIDITIES AND BASIGITIESOF ORGANIC FUNCTIOIIIAI GROUPS
A. Acldlties of Groups That lonlze to Glve Anionlc Gonlugate Bases
Functional group Structure*Structure ofconjugate base Typical pK"
sulfonic acid
ollR-5-O-Hllo
otlR-S-O-ilo
< 1 (strong acid)
carboxylic acid
0llR-C-O-H
0tlR-C-O- 3-5
phenol*
xkJro-H' *00-t 9-1 1
thiol R-S-H R-5- 9-'l 1
sulfonamide
OHillR-S-N-Rllo
oil_R- S-N-Rtl
o
amide
OHillR-C-N-Rolt_R_C-N-R 15-17
alcohol R-O-H 15-19
aldehyde, ketone
OHlllR-C-CRz
oll-R-C-CR2 17-20
ester
HOttlR2C-C-OR
otl
R2C-C-OR
alkyne R-C-C-H R-C:C-
nitrile
H
I
R2C-C-N R'C-C-N 25
amine R2N-H RrN- 32
R\r -r-lE-ls/\RR
R\/C:C/\R
alkene
benzylic alkylgroup
H
I
Ar-CR2 Ar-C R, 42
alkane R3C-H RrC- 55-60
*ln the structures, R : alkyl or H.The acidic hydrogen is shown in red.tX - a general ring substituent group.

APPENDIX VII. TYPICAL ACIDITIES AND BASICITIES OF ORGANIC FUNCTIONAL GROUPS A-'I 5
B. Basicities of Groups That Protonate to Give CationicGonjugate AcidsOne should be careful to distinguish between the behavior of a particular functional group asan acid and the same functional group as a base. For example, when an alcohol acid acts as anacid, it loses the RO-H proton to form an alkoxide (see the table in part A of this section.)When it acts as a base,it gains a proton to form ROHr. These are very different processes withdifferent pK" values. When we discuss the acidity of an alcohol, the relevant pK^ is that for thealcohol itself (see the table in part A). This same pK" describes the basicity of the alkoxide,RO-, which is the conjugate base of the alcohol. When we discuss the basicity of the alcoholitself, the relevant pK" is the value for the acidity of RdHr, given in the following table.
Functional group Structur€*,*
alkylamine
Structure of Typical conjugate-conjugate acid*,* ".ia p4
R,N+
R3N- H 9-1 1
pyridine 1l*xu O*+-H sxX-l
aromatic amine (,,- ,FNRtx1-l
amide
otl
R-C-NR2
*o/Htl
R-C-NR2
alcohol, ether R-O-RH
IR-O-R+ -2 to -3
ester, carboxylicacid
otlR-C-OR
*o/HllR-C-OR
phenol, aromatic ethert *kJao* -6 to -7
thiol, su lfide R-S- R
H
IR-S-R+ -6 to -7
otl
aldehyde, ketone R-C-R
*o/HilR-C-R -7
*ln the structures, R : alkyl or H. In the conjugate acid, the acidic hydrogen is shown in red.+X = a general ring substituent group.TA phenol or aromatic ether can be protonated on a ring carbon ifthe resulting carbocation can be strongly stabilizedby the substituent groups.
(Table continues)
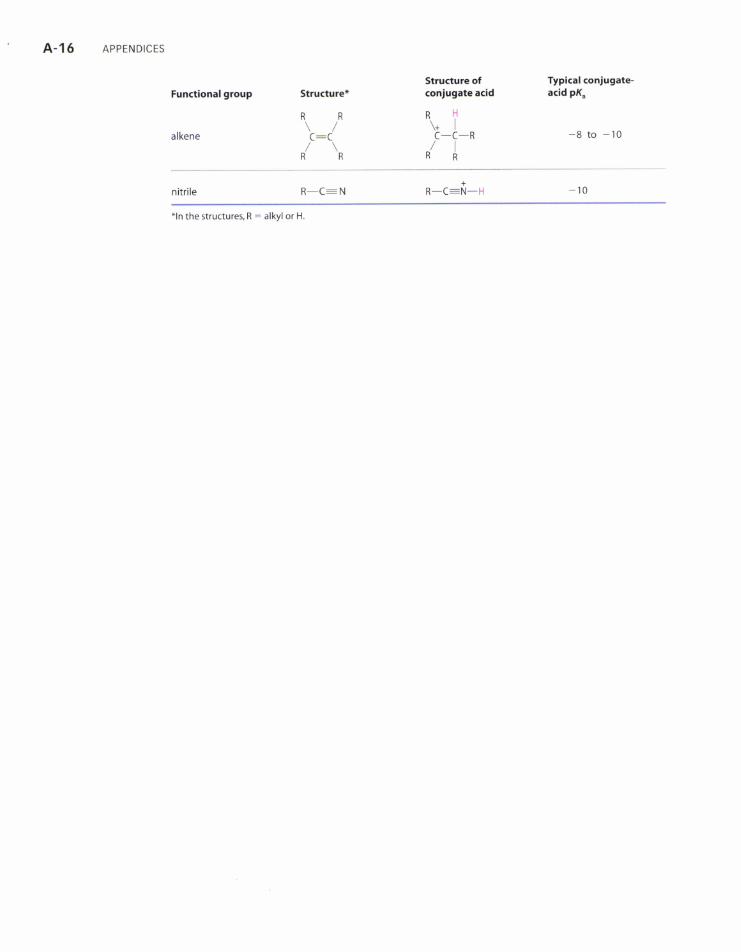
' A-16 APPENDTcES
Structure of Typlcal conjugate'Functionalgroup StructutE* conlugateacid acidpK.
RRRIatkene \:./ \-l-* -8 to -ro/\/li'RRR
nitrile R-C: N R-C-fi-H - 10
*ln the structures, R : alkyl or H.

Credlts
Figure 2.10 Photo copyright @ by Stan Honda/Getty Images.
Figure 2.11 The source of the historical ice-core CO, data is D. M. Etheridge, Division ofAtmospheric Research, Australian Commonwealth Scientific and Industrial Research Organi-zation (CSIRO). The source of the data in the inset is C. D. Keeling of the Scripps Institute ofOceanography, and the National Oceanic and Atmospheric Administration (NOAA).
Figure 2.12 Photo copyright @ by Jenna Wagner/iStockphoto.com
Figure 2.14 The photo of methane digesters is courtesy of Mark Stoermann, Fair OaksFarms. The methanogen in the inset is copyright @ by T. J. Beveridge/Getty Images.
Figure 6.18 Reprinted with permission from G. B. Kauffman and R. D. Myers, Journal ofChemical Education (L975), 52,777 . Copyright @ by the American Chemical Society.
Figure 9.7 Photo courtesy of Tira Bunyaviroch, M.D., and R. Edward Coleman, M.D.Reprinted with permission from the Journal of Nuclear Medicine (20O6), 47, 251. Copyright@ by the Society of Nuclear Medicine.
Figure 12.17 Mass spectra courtesy of Dr. Karl V. Wood and the Purdue University MassSpectrometry Center.
Figure 12.18 Adapted from F. W. Mclafferty, Interpretation of Mass Spectra. Copyright @1977 by the Benjamin-Cummings Publishing Company. Used with permission.
Figure 13.23 DEPT-NMR spectrum of camphor, courtesy of John Kozlowski, Purdue Uni-versity.
Figure 13.19 Reprinted with permission from F. A. Bovey, Chemical and EngineeringNews, August 30, 1965. Copyright @ by the American Chemical Society
Figure L3.26 Courtesy of Dr. Paul J. Keller, Barrow Neurological Institute.
Figure 14.7 Photo copyright @ by Marc Loudon.
Figure 26.5 Mass spectrum courtesy of Prof. Richard Gibbs and Animesh Aditya of PurdueUniversity, Dr. Karl V. Wood, and the Purdue University Mass Spectrometry Center.
Figures 25.5,26.13,26.15, and 26.16 are based on coordinates obtained from the ProteinData Bank, operated by the Research Collaboratory for Structural Biology (RCSB), supportedby the National Science Foundation (NSF), the National Institute of General Medical Sciences(NIGMS), the Office of Science, Department of Energy (DOE), the National Library of Med-icine (NLM), the National Cancer Institute (NCI), the National Center for Research Resources(NCRR), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Na-tional Institute of Neurological Disorders and Sffoke (NINDS), and the National Institute ofDiabetes and Digestive and Kidney Diseases (NIDDK). We gratefully acknowledge the assis-tance of Prof. Markus Lill, Purdue University, in obtaining the images.
c-1

C-2 cREDrrs
Figure 26.16 Coordinates courtesy of Abbott Laboratories.
IR Spectra Adapted fromthe Aldich@ Library of FT-IR Specta, Charles J. Pouchert, Ed-itor. Copyright@ 1997 by the Aldrich Chemical Company, and used with permission. Thesespectra are found in Figures 12.4, 12.7 , 12.10, l2.Il, 12.12, 12.t3, P12.26, P12.27, P12.32,P 12.33, P 13.24, r4.4, r4.5, P 14.3 5, t6.r, 19.3, 20.2, 21 .2, 21.3, and 23.1.
Mass Spectra Mass spectra not separately acknowledged in the foregoing credits are fromthe EPA-NIH Mass Spectral Data Base, published by the National Bureau of Standards,United States Department of Commerce, and used with permission. These spectra are foundin Figures I2.l 4-12. 16, P 12.37, P 13.V1, P 1 6.55a, 19.7, ard P 19.64.
LIV Spectra UV spectra were obtained by the author.
IttMR Spectra We gratefully acknowledge the cooperation of the Purdue Magnetic Reso-nance Laboratory and the able assistance of Dr. Tony Thompson in obtaining the spectra.


















