
2017 Vol. 38 - USTC
21
Transcript of 2017 Vol. 38 - USTC


2017
Vol. 38 No. 10
2017年
第38卷 第10期
In This Issue
Cover: Tang and coworkers in their Article on pages 1668–1679 reported a Fe–N–C catalyst via one–step pyrolysis in the presence of anhydrate iron chloride using cheap and nontoxic riboflavin as both carbon and nitrogen precursors. The picture shows the processes of catalyst preparation and oxygenreduction reaction, as well as the morphology of the catalyst.
封面:唐水花等报道了以廉价无毒的核黄素作为碳氮源, 一步热解制得
Fe–N–C 催化剂并将其应用于燃料电池阴极氧还原反应. 图片给出了催化剂
制备和氧还原反应过程以及催化剂形貌. 见本期第 1668–1679 页.
About the Journal
Chinese Journal of Catalysis is an international journal published monthly by Chinese Chemical Society, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, and Elsevier. The journal publishes original, rigorous, and scholarly contributions in the fields of heterogeneous and homogeneous catalysis in English or in both English and Chinese. The scope of the journal includes: New trends in catalysis for applications in energy production, environmental protection, and production of new materials, petroleum
chemicals, and fine chemicals; Scientific foundation for the preparation and activation of catalysts of commercial interest or their representative models; Spectroscopic methods for structural characterization, especially methods for in situ characterization; New theoretical methods of potential practical interest and impact in the science and applications of catalysis and catalytic reaction; Relationship between homogeneous and heterogeneous catalysis; Theoretical studies on the structure and reactivity of catalysts. The journal also accepts contributions dealing with photo-catalysis, bio-catalysis, and surface science and chemical kinetics issues
related to catalysis.
Types of Contributions
Reviews deal with topics of current interest in the areas covered by this journal. Reviews are surveys, with entire, systematic, and important information, of recentprogress in important topics of catalysis. Rather than an assemblage of detailedinformation or a complete literature survey, a critically selected treatment of thematerial is desired. Unsolved problems and possible developments should also bediscussed. Authors should have published articles in the field. Reviews should havemore than 80 references.
Communications rapidly report studies with significant innovation and major academicvalue. They are limited to four Journal pages. After publication, their full-text papers can also be submitted to this or other journals.
Articles are original full-text reports on innovative, systematic and completed researchon catalysis.
Highlights describe and comment on very important new results in the originalresearch of a third person with a view to highlight their significance. The results shouldbe presented clearly and concisely without the comprehensive details required for anoriginal article.
Perspectives are short reviews of recent developments in an established or developingtopical field. The authors should offer a critical assessment of the trend of the field, rather than a summary of literatures.
Viewpoints describe the results of original research in general in some area, with a viewto highlighting the progress, analyzing the major problems, and commenting thepossible research target and direction in the future.
Impact Factor
2016 SCI Impact Factor: 2.813 2016 SCI 5-Year Impact Factor: 2.163 2015 ISTIC Impact Factor: 1.179
Abstracting and Indexing
Abstract Journals (VINITI) Cambridge Scientific Abstracts (CIG) Catalysts & Catalysed Reactions (RSC) Current Contents/Engineering, Computing
and Technology (Thomson ISI) Chemical Abstract Service/SciFinder
(CAS) Chemistry Citation Index
(Thomson ISI) Japan Information Center of Science and
Technology Journal Citation Reports/Science Edition
(Thomson ISI) Science Citation Index Expanded
(Thomson ISI) SCOPUS (Elsevier) Web of Science (Thomson ISI)

ChineseJournalofCatalysisVol.38,No.10,October2017 催化学报2017年第38卷第10期|www.cjcatal.org
a v a i l a b l e a t www. s c i e n c ed i r e c t . c om
j o u r n a l h omep a g e : www. e l s e v i e r. c om / l o c a t e / c h n j c
ChineseJournalofCatalysis
GraphicalContents
PerspectiveChin.J.Catal.,2017,38:1659–1663 doi:10.1016/S1872‐2067(17)62894‐8
Titanocenedichloride:Anewgreenreagentinorganicchemistry
AntonioRosalesMartínez*,MaríaCastroRodríguez,IgnacioRodríguez‐García,LauraPozoMorales,RomanNicolayRodríguezMaeckerUniversityofSevilla,Spain;UniversityofAlmería,Spain;UniversidaddelasFuerzasArmadas‐ESPE,Ecuador
ThisreviewpresentsCp2TiClasanewgreenreagentwidelyusedinorganicchemistry,withpotentialapplicationsinfinechemistry,polymerchemistryandotherfields.
CommunicationChin.J.Catal.,2017,38:1664–1667 doi:10.1016/S1872‐2067(17)62901‐2DirectconstructionofsulfenylatedpyrazolescatalyzedbyI2atroomtemperature
Shuang‐HongHao,Li‐XiaLi,Dao‐QingDong,Zu‐LiWang*QingdaoAgriculturalUniversity
Aniodine‐catalyzedsulfenylationofpyrazolesatroomtemperatureisdescribed,inwhichavarietyofpyrazoleswerewelltoleratedandthedesiredproductsobtainedingoodtoexcellentyields.
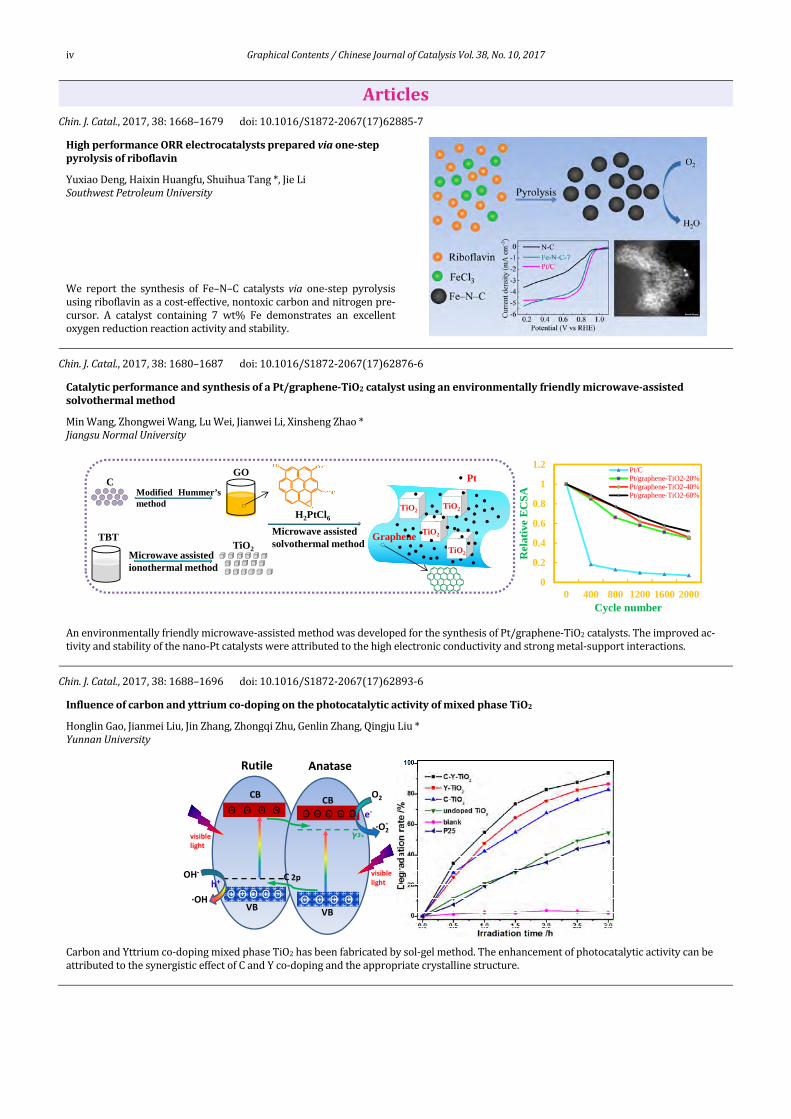
iv GraphicalContents/ChineseJournalofCatalysisVol.38,No.10,2017
ArticlesChin.J.Catal.,2017,38:1668–1679 doi:10.1016/S1872‐2067(17)62885‐7
HighperformanceORRelectrocatalystspreparedviaone‐steppyrolysisofriboflavin
YuxiaoDeng,HaixinHuangfu,ShuihuaTang*,JieLiSouthwestPetroleumUniversity
We report the synthesis of Fe–N–C catalysts via one‐step pyrolysisusingriboflavinasacost‐effective,nontoxiccarbonandnitrogenpre‐cursor. A catalyst containing 7 wt% Fe demonstrates an excellentoxygenreductionreactionactivityandstability.
Chin.J.Catal.,2017,38:1680–1687 doi:10.1016/S1872‐2067(17)62876‐6CatalyticperformanceandsynthesisofaPt/graphene‐TiO2catalystusinganenvironmentallyfriendlymicrowave‐assisted solvothermalmethod
MinWang,ZhongweiWang,LuWei,JianweiLi,XinshengZhao*JiangsuNormalUniversity
GO
Modified Hummer’smethod
Microwave assisted ionothermal method
H2PtCl6
Microwave assisted solvothermal method
Graphene
Pt
TiO2
C
TBT
0
0.2
0.4
0.6
0.8
1
1.2
0 400 800 1200 1600 2000
Rel
ativ
e E
CS
A
Cycle number
Pt/CPt/graphene-TiO2-20%Pt/graphene-TiO2-40%Pt/graphene-TiO2-60%
TiO2TiO2
TiO2
TiO2
Anenvironmentallyfriendlymicrowave‐assistedmethodwasdevelopedforthesynthesisofPt/graphene‐TiO2catalysts.Theimprovedac‐tivityandstabilityofthenano‐Ptcatalystswereattributedtothehighelectronicconductivityandstrongmetal‐supportinteractions.
Chin.J.Catal.,2017,38:1688–1696 doi:10.1016/S1872‐2067(17)62893‐6
Influenceofcarbonandyttriumco‐dopingonthephotocatalyticactivityofmixedphaseTiO2
HonglinGao,JianmeiLiu,JinZhang,ZhongqiZhu,GenlinZhang,QingjuLiu*YunnanUniversity
AnataseRutile
visible light
visible light
O‐O‐O‐O‐O‐
O+ O+ O+ O+ O+
CB
OH‐
∙OH
h+
e‐
O2
∙O2‐
CB
VB VB
C 2p
O‐ O‐O‐O‐
O+ O+ O+ O+
CarbonandYttriumco‐dopingmixedphaseTiO2hasbeenfabricatedbysol‐gelmethod.TheenhancementofphotocatalyticactivitycanbeattributedtothesynergisticeffectofCandYco‐dopingandtheappropriatecrystallinestructure.

GraphicalContents/ChineseJournalofCatalysisVol.38,No.10,2017 v
Chin.J.Catal.,2017,38:1697–1710 doi:10.1016/S1872‐2067(17)62891‐2
Coke‐tolerantSiW20‐Al/Zr10catalystforglyceroldehydrationtoacrolein
AminTalebian‐Kiakalaieh,NorAishahSaidinaAmin*UniversitiTeknologiMalaysia(UTM),Malaysia
SiW20‐Al/Zr10
Glycerol Acrolein
Glycerol Conversion (%)
Acrolein Selectivity (%)
Time
Catalyst
Acoke‐tolerantcatalyst(SiW20‐Al/Zr10),whichgave87.3%selectivityforacroleinand97%glycerolconversionundertheconditions300°C,0.5wt%catalyst,and10wt%feedconcentration,wassynthesized.
Chin.J.Catal.,2017,38:1711–1718 doi:10.1016/S1872‐2067(17)62907‐3One‐stepsynthesisofgraphiticcarbonnitridenanosheetsforefficientcatalysisofphenolremovalundervisiblelight
WangDing,SuqinLiu*,ZhenHe CentralSouthUniversity
Asimplemethodwasdevelopedtoprepareg‐C3N4nanosheetsbythermalpolymerizationofcyanuricacidandmelamineinair.Theg‐C3N4nanosheetsshowedsuperiorphotocatalyticactivitiesthanbulkg‐C3N4.
g-C3N4 nanosheets
550 ºC
Air
Chin.J.Catal.,2017,38:1719–1725 doi:10.1016/S1872‐2067(17)62884‐5
Novel‐structuredMo‐Cu‐Fe‐Ocompositeforcatalyticairoxidationofdye‐containingwastewaterunderambienttemperatureandpressure
YinXu*,HenanShao,FeiGe,YunLiuXiangtanUniversity
Air
O2
H2O
H2O
O2
Water
CO2+H2O
Cationic dye+•
Cationic dye•OH
Mo-Cu-Fe-O
(e-)
Anovel‐structuredMo‐Cu‐Fe‐Ocompositeshowedsuperiorcatalyticactivityforthedegradationofcationicdyesinwastewaterthroughthegenerationofhydroxylradicals.
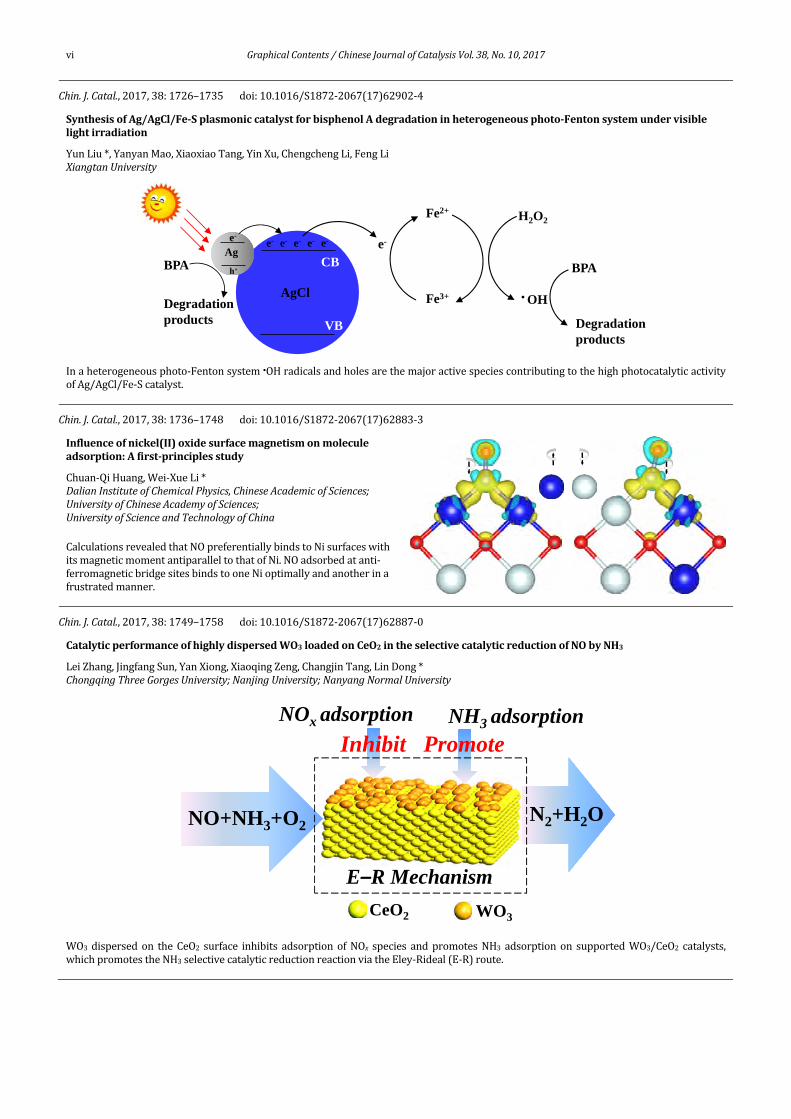
vi GraphicalContents/ChineseJournalofCatalysisVol.38,No.10,2017
Chin.J.Catal.,2017,38:1726–1735 doi:10.1016/S1872‐2067(17)62902‐4
SynthesisofAg/AgCl/Fe‐SplasmoniccatalystforbisphenolAdegradationinheterogeneousphoto‐Fentonsystemundervisiblelightirradiation
YunLiu*,YanyanMao,XiaoxiaoTang,YinXu,ChengchengLi,FengLiXiangtanUniversity
e- e- e- e- e-
CB
VB
AgCl
e-
Ag
h+
e-
Fe2+
Fe3+
H2O2
• OH
BPA
Degradationproducts
Degradationproducts
BPA
Inaheterogeneousphoto‐Fentonsystem•OHradicalsandholesarethemajoractivespeciescontributingtothehighphotocatalyticactivityofAg/AgCl/Fe‐Scatalyst.
Chin.J.Catal.,2017,38:1736–1748 doi:10.1016/S1872‐2067(17)62883‐3Influenceofnickel(II)oxidesurfacemagnetismonmolecule adsorption:Afirst‐principlesstudy
Chuan‐QiHuang,Wei‐XueLi*DalianInstituteofChemicalPhysics,ChineseAcademicofSciences; UniversityofChineseAcademyofSciences; UniversityofScienceandTechnologyofChina
CalculationsrevealedthatNOpreferentiallybindstoNisurfaceswithitsmagneticmomentantiparalleltothatofNi.NOadsorbedatanti‐ferromagneticbridgesitesbindstooneNioptimallyandanotherinafrustratedmanner.
Chin.J.Catal.,2017,38:1749–1758 doi:10.1016/S1872‐2067(17)62887‐0
CatalyticperformanceofhighlydispersedWO3loadedonCeO2intheselectivecatalyticreductionofNObyNH3
LeiZhang,JingfangSun,YanXiong,XiaoqingZeng,ChangjinTang,LinDong*ChongqingThreeGorgesUniversity;NanjingUniversity;NanyangNormalUniversity
NOx adsorption NH3 adsorptionInhibit Promote
N2+H2ONO+NH3+O2
E–R Mechanism
CeO2 WO3
WO3dispersedon theCeO2 surface inhibits adsorptionofNOx species andpromotesNH3 adsorptionon supportedWO3/CeO2 catalysts,whichpromotestheNH3selectivecatalyticreductionreactionviatheEley‐Rideal(E‐R)route.

GraphicalContents/ChineseJournalofCatalysisVol.38,No.10,2017 vii
Chin.J.Catal.,2017,38:1759–1769 doi:10.1016/S1872‐2067(17)62890‐0
Anovelprocessofozonecatalyticoxidationforlowconcentrationformaldehyderemoval
BinZhu,Xiao‐SongLi,PengSun,Jing‐LinLiu,Xiao‐YuanMa,XiaobingZhu*,Ai‐MinZhu*DalianMaritimeUniversity;DalianUniversityofTechnology
Apromisingnovelprocessofcycledstorage‐ozonecatalyticoxidation(OZCO)wassuccessfullyemployedtoremovelowconcentrationfor‐maldehyde(HCHO)fromairovertheoptimalMnOx/Al2O3catalyst(manganeseacetateprecursorand10wt%Mnloading)atroomtemper‐ature.
Chin.J.Catal.,2017,38:1770–1779 doi:10.1016/S1872‐2067(17)62888‐2Visiblelight‐responsivecarbon‐decoratedp‐typesemiconductorCaFe2O4nanorodphotocatalystforefficientremediationof organicpollutants
XinLiu,YuhongZhang,YushuaiJia*,JunzheJiang,YabinWang,XiangshuChen*,TianGui JiangxiNormalUniversity
Carbon‐decoratedp‐typesemiconductorCaFe2O4nanorodsexhibitanenhancedphotocatalyticactivity forthedegradationofMBowingtothe simultaneous improvement of MB adsorption, separation andtransferofphotogeneratedchargesandlightabsorption. 0
1
2
3
4
5
6
7
C/CaFe2O4
Rat
e co
nst
ant
(10-3
min
- 1)
CaFe2O4
CB
VB
e-e- e-
h+h+h+
CaFe2O4
e-
e-
e-
Carbon
O2
.O2-
MB
Products
e-
MB
Products 4.8 times

中国科学院科学出版基金资助出版
月刊 SCI 收录 2017 年 10 月 第 38 卷 第 10 期
目 次
1659
Cp2TiCl: 有机化学中广泛使用的新绿色试剂 Antonio Rosales Martínez , María Castro Rodríguez, Ignacio Rodríguez-García, Laura Pozo Morales, Roman Nicolay Rodríguez Maecker
1664
室温下碘催化直接构建硫化吡唑啉酮 郝双红, 李丽霞, 董道青, 王祖利
1668
一步热解核黄素制备高性能氧还原反应电催化剂 邓玉潇, 皇甫海新, 唐水花, 李杰
1680
微波辅助溶剂热法制备Pt/石墨烯-TiO2及其催化性能 王敏, 王中伟, 韦露, 李建伟, 赵新生
1688
碳和钇共掺杂对混相TiO2光催化性能的影响 高洪林, 刘健梅, 张瑾, 朱忠其, 张艮林, 柳清菊
1697
容碳的SiW20-Al/Zr10催化剂用于甘油脱水制丙烯醛 Amin Talebian-Kiakalaieh, Nor Aishah Saidina Amin
1711
一步法合成g-C3N4纳米片用作苯酚可见光降解高效催化剂 丁望, 刘素琴, 何震
1719
常温常压湿式催化氧化染料废水的Mo-Cu-Fe-O新型复合
催化材料 许银, 邵贺楠, 葛飞, 刘云
1726
Ag/AgCl/铁-海泡石异相可见光Fenton催化降解双酚A 刘云, 毛妍彦, 唐宵宵, 许银, 李程程, 李峰
1736
NiO表面磁性对分子吸附的影响的第一性原理研究 黄传奇, 李微雪
1749
CeO2表面分散态WO3的氨选择性催化还原性能 张雷, 孙敬方, 熊燕, 曾小清, 汤常金, 董林
1759
臭氧催化氧化脱除低浓度甲醛的新方法 朱斌, 李小松, 孙鹏, 刘景林, 马晓媛, 朱晓兵, 朱爱民
1770
可见光响应的碳修饰纳米棒状p型CaFe2O4半导体光催化降解有机污染物 刘鑫, 张玉红, 贾玉帅, 姜君哲, 王亚斌, 陈祥树, 桂田
英文全文电子版(国际版)由Elsevier出版社在ScienceDirect上出版 http://www.sciencedirect.com/science/journal/18722067 http://www.elsevier.com/locate/chnjc www.cjcatal.org 在线投审稿网址 https://mc03.manuscriptcentral.com/cjcatal
视 角
快 讯
论 文

ChineseJournalofCatalysis38(2017)1736–1748 催化学报2017年第38卷第10期|www.cjcatal.org
a v a i l a b l e a t www . s c i e n c e d i r e c t . c om
j o u r n a l h omep a g e : www . e l s e v i e r . c om / l o c a t e / c h n j c
Article
Influenceofnickel(II)oxidesurfacemagnetismonmolecule adsorption:Afirst‐principlesstudy
Chuan‐QiHuanga,b,Wei‐XueLia,b,c,*aStateKeyLaboratoryofCatalysis,DalianInstituteofChemicalPhysics,ChineseAcademicofSciences,Dalian116023,Liaoning,ChinabUniversityofChineseAcademyofSciences,Beijing100049,ChinacDepartmentofChemicalPhysics,CollegeofChemistryandMaterialsScience,iChEM,CASCenterforExcellenceinNanoscience,HefeiNationalLaboratoryforPhysicalSciencesattheMicroscale,UniversityofScienceandTechnologyofChina,Hefei230026,Anhui,China
A R T I C L E I N F O
A B S T R A C T
Articlehistory:Received27May2017Accepted26June2017Published5October2017
Theinfluenceofthemagnetismoftransitionmetaloxide,nickel(II)oxide(NiO),onitssurfacereac‐tivityandthedependenceofsurfacereactivityonsurfaceorientationandreactantmagnetismwerestudied by density functional theory plus U calculations.We considered five different antiferro‐magnetically ordered structures and one ferromagnetically ordered structure, NiO(001) andNi(011)surfaces,paramagneticmoleculeNO,andnonparamagneticmoleculeCO.Thecalculationsshowedthatthedependenceofsurfaceenergiesonmagnetismwasmodest,rangingfrom49to54meV/Å2 forNiO(001) and from 162 to 172meV/Å2 forNiO(011). OnNiO(001), bothmoleculespreferredthetopsiteof theNicationexclusively forallNiOmagneticstructuresconsidered,andcalculatedadsorptionenergiesrangedfrom−0.33to−0.37eVforCOandfrom−0.42to−0.46eVforNO.OnNiO(011),bothmoleculespreferredthebridgesiteoftwoNicationsirrespectiveoftheNiOmagnetism.Itwasfoundthatratherthanthelong‐rangemagnetismofbulkNiO,thelocalmagneticorderoftwocoordinatedNicationsbindingtotheadsorbedmoleculehadapronouncedinfluenceonadsorption.ThecalculatedNOadsorptionenergyatthe(↑↓)bridgesitesrangedfrom−0.99to−1.05eV,andbecomestrongerat the (↑↑)bridge siteswithvaluesof−1.21 to−1.30eV.ForCO,althoughthecalculatedadsorptionenergiesatthe(↑↓)bridgesites(−0.73to−0.75eV)wereveryclosetothoseatthe(↑↑)bridgesites(−0.71to−0.72eV),theirelectronhybridizationswereverydifferent.Thepresentworkhighlightstheimportanceofthelocalmagneticorderoftransitionmetaloxidesonmolecularadsorptionatmulti‐foldsites.
©2017,DalianInstituteofChemicalPhysics,ChineseAcademyofSciences.PublishedbyElsevierB.V.Allrightsreserved.
Keywords:MagnetismSurfaceorientationMoleculeadsorptionFirst‐principlestheoryElectronicstructure
1. Introduction
Transition metal oxide (TMO) surfaces currently have awide range of applications in energy and environmental sci‐ence,suchascatalystsforcleanenergyconversion,cleanupofairpollutants,andsensorsforchemicalandbiomedicaldevices[1,2]. Understanding the surface chemistry and struc‐
ture–activity relationship of TMOs is therefore important forthefurtherdevelopmentoftheseapplications[3].Inparticular,3dTMOsarenotablebecauseoftheirinvolvedspinstatesandmagnetism,whichissensitivetosurfaceorientationandparti‐cle size [47]. For instance, the spin statesofCo ions (ratherthannumberof3delectrons)inperovskiteoxidesplayacriti‐cal role in their oxygen evolution activity [8]. In addition to
*Correspondingauthor.Tel:+86‐551‐63600650;E‐mail:[email protected] work was supported by the National Natural Science Foundation of China (91645202), the National Key R&D Program of China(2017YFB602205), theNationalBasicResearchProgramofChina (2013CB834603), and theFrontierScienceKeyProjectofChineseAcademyofSciences(QYZDJ‐SSW‐SLH054).DOI:10.1016/S1872‐2067(17)62883‐3|http://www.sciencedirect.com/science/journal/18722067|Chin.J.Catal.,Vol.38,No.10,October2017

Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748 1737
oxides, thespinstatesofmoleculessuchasO2 influence theircorrespondingdissociationactivityonoxides[9]andevensim‐ple metal surfaces [10]. Magnetism can be induced in metaloxidesthatareparamagneticinbulkbysurfaceadsorptionandreaction [11,12]. It is therefore interesting to study the influ‐enceofthespinstatesandmagnetismofoxidesontheirsurfacechemistry.
Late 3d transition metal monoxides (MnO, FeO, CoO, andNiO)withcubicrocksaltstructure(Fm3m)[13,14]areofpar‐ticular importance in the study of antiferromagnetic (AFM)TMOs,andareoftenconsideredasmodelAFMsystems.BesidestheirrelativelyhighNeeltemperature(TN)andchemicalstabil‐ity,theirlargebandgap(EG)andinsulatingelectronicstructureallow theirmagnetic properties to be accurately described intermsofmagneticmomentslocalizedontransitionmetalcati‐ons [15,16]. The anisotropic interaction betweenneighboringmagnetic moments leads to long‐range magnetic ordering,known as type‐2 antiferromagnetism (AFM2), below TN[1720]andstackingofferromagnetic(FM)(111)planeswithopposite spin directions in adjacent layers, as identified byneutron diffraction [21]. Using magnetic exchange force mi‐croscopy, the magnetic order of surface nickel ions on theNiO(001) surface has been measured directly in real space[2226].DespitethedefiniteAFMorderofmagneticionsintheperfectbulkcrystalbelowTN,theorderingofmagneticionsonthesurfacecanbechangedunderdifferentconditions.Forex‐ample,byengineeringdefects in the latticeofAFMNiO, aFMorderingdislocationcanbe introduced[27].WhenthesizeofAFMNiOdecreasestothenanoscale,theresultingdecreaseofcoordinationand latticedistortioncandrive thephase transi‐tiontoaFM‐likephase[27,28].
The magnetism of metal ions in late 3d TMOs originatesfromtheirpartiallyfilled3dstates,whicharefamousfortheirhighly correlated nature [29]. It remains challenging to accu‐rately describe strongly localized and correlated d electronsusing standard density functional theory (DFT) approachessuchas the localdensity approximation (LDA)or generalizedgradientapproximation(GGA)[30,31].BoththeLDAandGGAoften fail topredictEGof theseTMOs [32].For instance,bothCoOandFeOwerepredictedtobemetallic.AlthoughNiOandMnO were predicted to have EG, the calculated values wereseverelyunderestimated[33].Nevertheless,EGof thesemate‐rialscanbeopenedincalculationsbyaddingaHubbardUtermdescribingthestrongon‐siteCoulombrepulsiontotheLDAorGGA(DFT+U)[34,35].Comparedtohigher‐leveltheoriessuchas the hybrid functional or many‐body perturbation [31,36],DFT+U can substantially improve the calculation accuracywithoutincreasingthecomputationalcost,asdemonstratedfortheadsorptionofCOandNOonNiO(001)[37,38].Simulationofwater dissociative adsorption on NiO(111) was reported re‐cently,andthecalculationwasfoundtobefairlyinsensitivetothechoiceoftheDFT+Ufunctional[39].
Chemicallyequivalentoxidesurfacesmaybecomeinequiv‐alent when considering the different symmetry of variouslong‐range magnetisms. Because surface chemistry is largelydecidedbylocalenvironment(geometryandcomposition),itiscurrentlyunclearhowthedifferentmagnetismsaffectsurface
chemistry.Furthermore, the influenceofdifferent surfacesonmagnetisms is stillunknown.This influencemaybemediatedbyreactantswithdifferentspinand/ormagneticmoments.Toclarify these issues,hereNiOasamodel system is studiedbytheGGA+Umethod.Anumberofmagnetic phases, includingAFM1, AFM2, AFM3, AFM4, AFM5, and FM, are considered.NiO(001)andNiO(110)surfacesareusedtostudythesensitiv‐ity of these magnetic phases to structure. While CO and NOadsorbedonNiO(001)ofAFM2hasbeenstudiedtoverifytheDFT+U scheme treatment for strongly correlated systems[37,38],hereweusetheadsorptionofparamagneticmoleculeNOandnonparamagneticmoleculeCOtoprobetheinfluenceofmagnetismonmoleculeadsorptionbyNiOsurfaces.
2. Computationaldetails
DFTcalculationswereperformedwiththeViennaabinitiosimulation package (VASP) [40] based on the projected aug‐mented wave (PAW) method [41,42]. The spin‐polarizedPerdew Burke Ernzerhof (PBE) [43] exchange‐correlationfunctional and on‐site Coulomb repulsion U term for d elec‐tronsinthespiritofHubbardaccordingtoDudarev’sapproach[35]wereusedtodescribeelectronexchangeandcorrelation.AneffectiveUof5.3eVforNiwasusedthroughoutthepresentwork, because it could reproduce well bulk properties andCO/NOadsorptiononNiO(001)surfaces[37].
Becausepartiallyoccupieddorbitalsarestrongly localizedatNi2+,anetspinmagneticmomentwasassignedtoeachNi2+.For the cubic sublattice of magnetic ions, all magnetic ionshavingparallelspinmeansthere isFMorder(Figure1(a)). In
(b) AFM1p=1, G=(001)
(c) AFM2p=1, G=(111)
(e) AFM4p=2, G=(001)
(d) AFM3p=2, G=(201)
(f) AFM5p=2, G=(101)
(a) FM
Fig.1.SchematicillustrationofNiOferromagnetismFM(a),antiferro‐magnetismAFM1(b),AFM2(c),AFM3(d),AFM4(e)andAFM5(f).Linecross points represent O, blue balls represent Ni spin up, and whiteballsrepresentNispindown.

1738 Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748
contrast, the series of common AFM configurations of Ni2+AFM1–AFM5(asshown inFigure1(bf))canbedescribedasoppositely stackedFM layers indirectionGwithsuper latticeperiodicitypinwhichNi2+maintainparallelspinwithinplay‐ers[44].BelowTN=523K,theground‐statemagneticconfigu‐ration of NiO is AFM2 [21].We considered AFM1–AFM5 andFM magnetic order as a starting point to construct surfaceswith different surface magnetic order by truncation of bulkNiO.
FourlayersofanNiO(001)slab( 2 2 2 2)surfacesuper‐cellandfivelayersofanNiO(011)slab(44)surfacesupercellwereused tomodel theadsorptionofmoleculesonNiO(001)andNiO(011) surfaces, respectively. Slabswere separated by12Åof vacuumwith thebottom two layers fixed at thebulktruncated structure, and dipole correction perpendicular toeach slabwas imposed. The layer thickness and vacuum sizewere found to converge for adsorption of CO/NO on AFM2.Surface energy was evaluated with symmetric slabs of theminimumsurfacecell foreachsurfaceuntiltheyconvergedtolessthan1meV/Å2withrespecttoslabthickness.Aplanewavecutoff of 400 eV and k‐point grids of (441) for NiO(001)‐( 2 2 2 2)and(321) forNiO(011)‐(2×4)wereused.Geo‐metricaloptimizationwasperformeduntiltheforceonrelaxedatomswaslessthan0.02eV/Å.
3. Resultsanddiscussion
3.1. NiObulk
Table1liststheoptimizedequilibriumlatticeconstant,en‐ergydifferenceperNiOformulawithrespecttothelowesten‐ergystate(AFM2),localizedmagneticmomentonNi,andbandgapforthevariousmagneticphasesconsidered.Theenergeti‐callymostfavorablelong‐rangemagneticphaseisAFM2,whichisconsistentwiththestructureofNiObelowTNdeterminedbyneutron diffraction. The energy difference between differentmagneticphasescanbeaslargeas102meV/NiO.AFM2hasthesmallestlatticeconstant(a=4.19Å)andmagneticmomentofNi(m=1.71μB),butthelargestEGof3.25eVforthemagneticphases.Thepresentworkagreeswellwiththeresultsofapre‐viousSGGA+Ucalculation(a=4.20Å,m=1.72μB,EG=3.2eV)[37].For theotherAFMphases, the latticeconstantandmag‐neticmoment increaseby0.02Åand0.10μBatmost,respec‐tively,butEGdecreasesby1.16eV.Itisclearthatthelowertheenergyof themagneticphase, thesmaller the latticeconstant
and magnetic moment and the larger the EG. These trendsoriginate from the superexchange interaction of the secondnearestneighbor(NN)Ni2+,asrationalizedinanearliertheoryforrocksalttransitionmetalmonoxides[33,45].
To understand the origin of the AFM behavior of NiO,wefitted the calculated energy in Table 1 to the Ising model ofmagneticmomentinteraction[19].Thefittingresultgives i‐thNNmagneticmomentexchangepairinteractionsJi(i=1,2,3,and4)of0.71,15.58,0.24,and0.07meV,respectively.Thismeansthat thesecondNNinteraction(J2) is thedominant in‐teraction and the negative value indicates the preference forAFM order, similar to the behavior of CoO andMnO [18,19].Because J1, J3, and J4 aremuch smaller than J2, thenumberofsecondNNpairsdeterminestherelativestabilityofthevariousAFMphases.ForAFM2,allthesecondNNpairsareantiparallel,soitisenergeticallythemostfavorablestateofNiO.
Inthiswork,aNiatomwithanupward(orpositive)mag‐netic moment (↑) means that the electron population of thespin‐up component is larger than that of the spin‐down,whereas a Ni atomwith a downward (or negative)magneticmoment (↓) means that the electron population of thespin‐down component is larger than that of the spin‐up. Thespin‐resolved density of states (SDOS) of Ni (↑) in bulkNiO‐AFM2isillustratedinFigure2.Forthe3dxy,yz,xz(t2g)band,boththespin‐upandspin‐downcomponentsaredistributedinthevalenceband.Comparedwith thespin‐upcomponent, theelectrons in the spin‐down component mainly populate theupper/toppartofthevalenceband.Forthe3dx2–y2,z2(eg)band,themarked exchange splitting causes the eg states to bewellseparated(by~9.69eV). Inparticular, thespin‐downcompo‐nents are distributed in the conduction band. Most of thespin‐up components are distributed in the lower part of thevalenceband,andasmallamountaredistributedclosetothetop of the valence band. The gap between the valence bandmaximum(VBM)andconductionbandminimum(CBM)oftheNi3dband is3.23eV,which isnearlysamewithas theEGofbulkNiO (3.25 eV). ForNi(↓), the corresponding componentsaresimplyreversed.Asshownbelow,thesefeaturesareessen‐tialforthehybridizationbetweenNiandCO/NOmolecules.
3.2. NiO(001)
3.2.1. SurfacepropertiesForrocksaltcrystals,the(001)planeischemicallyequiva‐
lentwith(010),(100),(001),(010),and(100):allbelongtothe{001} plane family, and the corresponding primitive cell is(1×1).A long‐rangemagneticphase lowersthecorrespondingsymmetry and decreases the degeneracy of the {001} planefamily.Thenonequivalentplanesdependedonthe long‐rangemagnetic phase (AFM1–AFM5), as classified in Table 2 andplottedinFigure3.EachofAFM1,AFM3,AFM4,andAFM5hadtwononequivalentplanes,whereasAFM2hadonlyonenone‐quivalent plane. This led to nine nonequivalent surfaceswithdifferentprimitivecells,althoughtheyareallchemicallyequiv‐alent.
TakingAFM1‐(100) (Figure3(b)) as an example, themag‐neticmomentsofthefirstNNNipairareexclusivelyantiparal‐
Table1Energy difference per NiO formula with respect to the lowest state(AFM2) (ΔE), equilibrium lattice constant (a), localizedmagneticmo‐mentonNi(M),andenergybandgap(EG)ofvariousmagnetismphases.
ΔE/meV a/Å M/μB EG/eVAFM1 101 4.21 1.81 2.20AFM2 0 4.19 1.71 3.25AFM3 65 4.20 1.77 2.51AFM4 61 4.20 1.77 2.26AFM5 30 4.19 1.74 2.88FM 91 4.21 1.80 2.09

Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748 1739
lel (↑↓). Similar behavior was found for AFM3‐(001) (Figure3(d)). In contrast, for AFM1‐(001) (Figure 3(a)) andAFM4‐(001) (Figure 3(f)), themagneticmoments of the firstNNNipairareexclusivelyparallel(↑↑).Fortheremaining ivesurfaces shown in Figure3, both the (↑↓) and (↑↑) con igura‐tionscoexistinthefirstNNNipairs, formingdifferentsurfacemagnetism patterns. The different localmagnetic pairs alongwith their long‐range magnetic phases provide a good play‐ground to investigate the influence of magnetism on surfaceproperties. Figure 4 plots the SDOS of a surface Ni atom (↑)fromAFM2‐(001).Theoverallfeaturesremainthesameasthatof AFM2 bulk (Figure 2). The main difference is the gap be‐tween theVBMandCBMof theNi 3dband,which decreasesfromthebulkvalueof3.23to2.81eV.
The calculated surface energies are presented in Table 2.
Thesurfaceenergiesrangefrom49to54meV/Å2,andthedif‐ference between different magnetic phases is less than 5meV/Å2. To rationalize this small variation, we note that thesurface energy increases mainly from the cost of NiO bondcleavage when creating a new surface. Considering the ex‐change coupling between magnetic cations, cleavage of theNiObondmightintroduceanadditionalcostoriginatingfromthelossofthecorrespondingexchangeinteraction.BecausetheexchangeinteractionbetweenNi2+islargelydeterminedbyJ2,theestimatedcostofbreakingtheJ2interactionwhencreatinganewsurface is~1.7meV/Å2.Namely, thecontribution fromthe long‐range AFM phases to the surface energy is indeedsmall. Compared to the calculated overall surface energy, thecontributionfrommagnetism(~3%)isnegligible.
3.2.2. AdsorptionofCOandNOmoleculesCOandNOadsorptiononthesenonequivalentsurfacesata
coverage of 0.25 monolayer with respect to surface Ni was
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
z2
Ni 3dx2-y2
SD
OS
(a.u
.)
E-EF/eV
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
xy
Ni 3dyz
Ni 3dxz
SD
OS
(a.u
.)
E-EF/eV
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
SD
OS
(a.u
.)
E-EF/eV
x
y
(b)
(a)
(c)
Fig.2.Spin‐resolvedprojecteddensityofstates(SDOS) forNi inbulkAFM2NiO.(a)SDOSprojectedontoNi3dz2,3dx2–y2; (b)SDOSprojectedontoNi3dxy,3dyz,3dxz; (c)TotalSDOSofNi3d.The referencecoordi‐nates forDOSprojection is shown in insetof (a),wherex‐axis repre‐sentsfor[100]directionandy‐axisfor[010]direction.
Table2NonequivalentsurfacesofrocksaltNiO{001}familytakingintoaccountofmagnetism, though ineachcolumnthe surfacesremainequivalent.Correspondingsurfaceenergyγ(meV/Å2)isgiven,andasreferenceforFM(001),γ=50meV/Å2. AFM1 AFM2 AFM3 AFM4 AFM5γ 49 50 54 54 51 54 52 52 54 (001)(001)
(100)(100)(010)(010)
(001)(001)(010)(010)(100)(100)
(001)(001)
(010)(010)(100)(100)
(001)(001)
(010)(010)(100)(100)
(001)(001)(100)(100)
(010)(010)
Fig.3.TopviewconfigurationofvariousnonequivalentNiO(001)sur‐faces taking into account of magnetism. Surface primitive cells aremarkedbyframeofreddashline.

1740 Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748
studied. Various adsorption sites were considered, and theresultsforadsorptionattheenergeticallymostfavorablesitesare given in Table 3. For the nonequivalent surfaces consid‐ered, calculated CO adsorption energies varied from−0.33 to−0.37eV,whileNOadsorptionenergiesweremoreexothermicand ranged from −0.42 to −0.46 eV. The small change of ad‐sorption energies (0.04 eV; less than 10%) reveals the weakinfluenceofthelong‐rangemagneticphasesonbothCOandNOadsorption. The calculated adsorption energies are in goodagreementwiththoseobtainedfromapreviousGGA+Ucalcu‐lationona(001)surfaceofNiOwithAFM2magneticorderandthermodesorptionandinfraredexperiments(−0.30to−0.45eVforCOand−0.52to−0.57eVforNO)[37,38,4648].
Inlinewithitsweakbindingstrength,COadsorptioninduc‐eslittlechangeofthemagnetism(lessthan0.02μB)ofCOandcoordinated Ni atom. As shown in Figure 5(a), CO adsorbednearlyperpendicularlyat thetopofaNiatom.TheoptimizedNi−Cbondlengthvariedfrom2.02to2.07A,whereashorterbond length indicates stronger binding. Meanwhile, the C−Obond length of 1.14Åwas the same as that of gas‐phase CO.Small tilting of the angle between the Ni−C and C−O bondscouldbeenergetically favorable;however,thevariationofthecorrespondingpotentialenergysurfacewas less than10meVfortiltanglesupto30[37].
ThebondingmechanismbetweenCOandacoordinatedNiatomcanbe inferred fromthecorrespondingSDOSplotted inFigure 5(b). The hybridization is mainly between the CO 5σorbitalandNi3dz2bandinthebottomofthevalenceband(do‐nation). In addition, there is weak hybridization between CO2π*andNi3dx2–y2intheconductionband(backdonation).Thefact that both hybridizations appear far from the Fermi levelrationalizes well the weak binding strength of adsorbed CO.
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
z2
Ni 3dx2-y2
SD
OS
(a.u
.)
E-EF/eV
x
y
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
xy
Ni 3dyz
Ni 3dxz
SD
OS
(a.u
.)
E-EF/eV
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
SD
OS
(a.u
.)
E-EF/eV
(b)
(a)
(c)
Fig. 4. Spin‐resolved projected density of states (SDOS) for upwardmagneticmomentNi(↑) of cleanNiOAFM2(001). (a) SDOS projectedontoNi3dz2,3dx2–y2;(b)SDOSprojectedontoNi3dxy,3dyz,3dxz;(c)TotalSDOSofNi3d.ThereferencecoordinatesforDOSprojectionisshownininsetof (a),wherex‐axisrepresents for [100]directionandy‐axis for[010]direction.
Table3Magnetismandstructure informationofCOadsorptiononNiO{001}.Magneticmoment(MNi,MCO)isgiveninunitofμB.AdsorptionenergyofCOEadsisgivenineV.Bondlength(dNi‐C)isgiveninÅ.
MNi(clean)(↑)/μB MNi(↑)/μB MCO/μB dNi‐C/Å Eads/eVAFM1(001) 1.80 1.78 0.01 2.04 0.34AFM1(100) 1.81 1.78 0.02 2.05 0.34AFM2(001) 1.72 1.69 0.01 2.07 0.36AFM3(001) 1.78 1.76 0.02 2.05 0.33AFM3(100) 1.77 1.74 0.01 2.04 0.34AFM4(001) 1.78 1.76 0.01 2.05 0.34AFM4(100) 1.77 1.74 0.02 2.04 0.35AFM5(001) 1.74 1.70 0.01 2.02 0.37AFM5(100) 1.75 1.72 0.02 2.04 0.34FM(001) 1.80 1.77 0.02 2.05 0.35
(c)
Ni OO
C
O+0.01e/A3
‐0.01e/A3
(d)
Ni OO
C
O
CO/AFM2‐(001)(a) (b)
-8 -4 0 4
-3
0
3
Ni 3d CO 2p
SD
OS
(a.u
.)
E-EF/eV
Fig.5.COadsorptiononNiOAFM2(001).(a)Adsorptionconfiguration;(b)SDOSofNi3dandCO2porbitals; (c) Isosurfaceview;(d)Sectioncontour viewof difference of electrondensity for CO adsorption. Forisosurface view, yellow/green color means electron accumulation/depletionrespectively.

Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748 1741
Directvisualizationof thebondingbetweenCOandNicanbebetterseenfromthedifferenceofelectrondensity(Figure5(c)and (d)). Pronounced charge accumulation between CO andcoordinated Ni is observed, which indicates the dominatingroleofthedonationinteractioninCOadsorptiononaNiO(001)surface.
ComparedwiththecaseforCOadsorption,NObindsmorestrongly to Ni with a tilted configuration (Figure 6(a)), inagreementwithpreviousstudiesofNOadsorptiononNiO(100)[38] and transition metal surfaces [49,50]. For instance, onAFM2‐(001),thetiltangleis10.7fortheNi−Nbondand49.5fortheN−Obond.Dependingonthemagneticphase(Table4),the optimized NiN bond length varies in the range of1.98–2.05A, and the con igurationwith a shorterNi−N bondlength is consistent with stronger binding. The N−O bondlength(1.17–1.18Å)isnearlythesameasthatofthegasphase(1.17Å).AfterNOadsorption,themagneticmomentsofNOandcoordinatedNidecreasebyabout0.10and0.20μB,respective‐ly. Interestingly, themagnetic moment of adsorbed NO is al‐ways antiparallelwith coordinatedNi irrespective of the sur‐facesmagneticphase;thiswillbediscussedfurtherlater.
UponNOadsorption,thehybridizationbetweentheNO5σorbitalandNi3dz2bandinthebottomofthevalencebandde‐creasesslightly(Figure6(b)).ComparedtotheCO2π*orbital,whichiscompletelyunoccupied,theNO2*orbitalinthetopofthevalencebandispartiallyoccupied.ForNi(↑),itismainlytheNi 3dxz,yz spin‐down component populated in the top/upperpart of the valence band. To maximize the hybridization be‐tweenNO2*andNi3dxz,yzinthetop/upperpartofthevalenceband,theoccupiedNO2π*mustpopulatethespin‐downcom‐ponent.Inotherwords,thefavorableconfigurationofmagneticmomentbetweenNOandcoordinatedNiisantiparallel,name‐ly, NO(↓) and coordinated Ni(↑), which is also found for NOadsorptiononNiO(011).
Regarding the decrease of the magnetic moment of ad‐sorbedNO,wenotethatthepeakintensityofNO2π*atthetopof the valence band is smaller than those of the three otherpeaks of NO 2π* in the conduction band. This indicates theelectron depletion in the corresponding states (spin‐downcomponent), and accordingly, the NO magnetic moment de‐creases.Similarly,thedecreaseofthemagneticmomentofco‐ordinated Ni can be rationalized by the depletion of Ni 3dz2states(lowerpeakintensity)atthebottomofthevalenceband(spin‐upcomponent).
Compared to CO adsorption, the extra hybridization be‐tweenNO2*andNi3dxz,yzinthevalencebandstrengthensthebindingbetweenNOand the coordinatedNi atom.ThedirectparticipationoftheNO2π*orbitalcanbeclearlyseenfromthedifference of electron density plotted in Figure 6(c) and (d).Moreover, themagnitudeof thedifferenceofelectrondensityinducedbyNOadsorptionisaboutfivetimeslargerthanthatofCO(Figure6(d)versusFigure5(d)).Thelargerextentofelec‐tron redistribution induced by NO than by CO corroborateswellwithitsstrongerbindingstrength.ForbothCOadsorption(Figure6(c)and(d))andNOadsorption(Figure5(c)and(d)),theadsorption‐induced charge redistribution is limited to thesingly coordinated Ni atom, and electrons of the NN Ni arehardlyaffected.Inotherwords,thelong‐rangemagneticphasehaslittleinfluenceonNOandCOadsorptiononNiO(001).
3.3. NiO(011)
3.3.1. Surfaceproperties
Without considering themagnetic phases, the {011} planefamily consists of twelve equivalent planes.When taking intoaccount the magnetism, the symmetry and degeneracy arebroken.AslistedinTable5andplottedschematicallyinFigure7,forAFM1,AMF2,AFM3,andAFM4,therearetwononequiv‐alentsurfaces,andforAFM5,therearethreeinequivalentsur‐faces. This gives a total of elevennonequivalent surfaces. Fornonequivalent AFM1‐(011), AFM2‐(011), AFM3‐(110), andAFM5‐(101) surfaces, the localmagneticmoments of the twoadjacent surface Ni atoms are exclusively antiparallel (↑↓).Conversely, for nonequivalent AFM1‐(110), AFM2‐(011),AFM4‐(110), and AFM5‐(101) surfaces, the local magnetic
(c) (d)
+0.05e/A3
‐0.05e/A3
Ni OO
N
O
-8 -4 0 4
-3
0
3
Ni 3d NO 2p
SD
OS
(a.u
.)
E-EF/eV
NO/AFM2‐(001)(a) (b)
Fig.6.NOadsorptiononAFM2(001).(a)Adsorptionconfiguration;(b)SDOSofNi3dandNO2porbitals;(c)Isosurfaceview;(d)Sectioncon‐tour view of difference of electron density for NO adsorption. Forisosurface view, yellow/green color means electron accumulation/depletionrespectively.
Table4Magnetismandstructure informationofNOadsorptiononNiO {001}surfaces.Magneticmoment(MNi,MNO)isgiveninunitofμB.AdsorptionenergyofNO(Eads)isgivenineV.Bondlength(dNi‐N)isgiveninÅ.
MNi(clean)(↑)/μB MNi(↑)/μB MNO(↓)/μB dNi‐N/Å Eads/eVAFM1(001) 1.80 1.63 0.61 2.00 0.45AFM1(100) 1.81 1.63 0.61 2.00 0.46AFM2(001) 1.72 1.56 0.58 2.05 0.42AFM3(001) 1.78 1.62 0.63 2.01 0.44AFM3(100) 1.77 1.59 0.60 1.98 0.45AFM4(001) 1.78 1.62 0.62 2.00 0.44AFM4(100) 1.77 1.59 0.57 1.98 0.45AFM5(001) 1.74 1.56 0.61 1.98 0.45AFM5(100) 1.75 1.58 0.58 1.99 0.44FM(001) 1.80 1.63 0.56 1.99 0.45

1742 Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748
momentsoftwoadjacentsurfaceNiatomsareexclusivelypar‐allel(↑↑).ForthenonequivalentAFM3‐(011),AFM4‐(011),andAFM5‐(011)surfaces,both(↑↓)and(↑↑)pairs(notedasX1and
X2,respectively)coexist. Thecalculatedenergiesof{011}surfacesaresummarizedin
Table5.Forthenonequivalentsurfaces,thecalculatedsurfaceenergies vary from 162 to 172 meV/Å2. These energies areaboutthreetimeslargerthanthoseofNiO{001},becausemoreNi−Obonds(perunitarea)arebrokentocreatethenewsur‐facefromthebulkmaterial.Incontrast,thesurfaceenergiesofthenonequivalent surfacesbelonging to the sameAFMphasedifferonlyby1meV/Å2.Thismeans that thevariationof thesurfaceenergiesby10meV/Å2mainlyoriginatesfromthedif‐ferentlong‐rangemagneticphases.Althoughtheabsolutevari‐ationof surfaceenergies is about twice thatofNiO{001} sur‐faces, thecontributiontotheoverallsurfaceenergiesremainssmall(~6%).Thisshowsagaintheweakeffectofthemagneticphasesonsurfaceenergies.
Figure 8 plots the SDOS of a surface Ni atom (↑) ofAFM2‐(011).The3dx2–y2and3dyzstatesarepopulatedmainlyinthebottomofthevalenceband(spinup)andthelowerpartof
Fig. 7. Top view configuration of nonequivalent NiO(110) surfacestakingintoaccountofmagnetism.Niatomsattopandsecondlayerarepresentedwithlargerandsmallerball,respectively.
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
z2
Ni 3dx2-y2
SD
OS
(a.u
.)
E-EF/eV
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
xy
Ni 3dyz
Ni 3dxz
SD
OS
(a.u
.)
E-EF/eV
-8 -4 0 4
-1.6
0.0
1.6 Ni 3d
SD
OS
(a.u
.)
E-EF/eV
x
y
(b)
(a)
(c)
Fig.8.Spin‐resolvedprojecteddensityofstates(SDOS)forNiofcleanAFM2NiO(011). (a)Total SDOSofNi 3d; (b) SDOSprojected ontoNi3dz2, 3dx2–y2; (c) SDOS projected onto Ni 3dxy, 3dyz, 3dxz. The referencecoordinates forSDOSprojection is shown in inset of (a),wherex‐axisrepresentsfor[100]directionandy‐axisfor[011]direction.
Table5Nonequivalent surface of rock salt NiO{011} plane family taking intoaccountofmagnetism.Thesurfaceineachcolumnremainsequivalentfrom each other. Corresponding surface energy γ (meV/Å2) is given,andasreferenceforFMNiO(011),γ=165meV/Å2.
AFM1 AFM2 AFM3 AFM4 AFM5γ 162 163 172 172 166 165 167 167 169 170 170
(011)(011)(011)(011)(101)(101)(101)(101)
(110)(110)(110)(110)
(011)(011)(101)(101)(110)(110)
(011)(011)(101)(101)(110)(110)
(011)(011)(011)(011)(101)(101)(101)(101)
(110)(110)(110)(110)
(011)(011)(011)(011)(101)(101)(101)(101)
(110)(110)(110)(110)
(011)(011)(011)(011)(110)(110)(110)(110)
(101)(101)
(101)(101)

Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748 1743
theconductionband(spindown),while3dz2,3dxz,and3dxyarepopulated in thevalenceband.Theupperpartof thevalenceband originates largely from Ni 3dxy,xz (spin down). AnotherprominentfeatureisthegapbetweentheVBMandCBMoftheNi 3d band of 1.40 eV, which is much smaller than that ofAFM2‐(001)(2.81eV)andAFM2bulk(3.23eV).Thesmaller3dbandgapofthesurfaceNimightfacilitateitshybridizationwithadsorbedmolecules,whichwillbediscussedbelow.
3.3.2. COadsorptionCOadsorptiononthesenonequivalentsurfaceswasstudied,
andtheresultsatthemostfavorablesitesarelistedinTable6.COpreferstoadsorbexclusivelyviatheendoftheCatomatthebridgesiteoftwoadjacentNiatoms,labeledasAandB(Figure9(a) and (c)). Irrespective of the nonequivalent surfaces anddifferent bridge sites (↑↑ and ↑↓) considered, CO has similaradsorption energies at the (↑↓)bridge sites (Figure9(e)); thecalculated adsorption energies vary from −0.73 to −0.75 eV.Conversely, for COadsorption at the (↑↑)bridge sites (Figure9(b)),thecalculatedadsorptionenergiesaresmaller(−0.71to−0.72 eV). Thus, the local magnetic moment and its relativedirectionof twocoordinatedNiatomsareresponsible for thevariationofthecalculatedadsorptionenergiesratherthanthelong‐rangemagnetic phases.The calculatedbinding strengthsof CO onNiO {011} are about twice that of CO on NiO{001},whichpossiblyarises fromthegreaternumberofNi−Cbondsinvolvedandthe lowercoordinationnumberofsurfaceNiat‐oms.
Important geometrical parameters and the bond lengthsbetweenCandtwocoordinatedNiatomsaregiveninTable6.TheoptimizedCNibondlengthsareintherangeof2.13–2.23Å, and longer than those of CO adsorbed on NiO(001)(2.02–2.07Å).Irrespectiveofthemagneticphasesconsidered,the C−O bond lengths stayed at a constant value of 1.15 A,
whichis0.01Ålongerthanthatofgas‐phaseCO.COadsorptionhas little influence on the surface magnetism: the magneticmoment for CO and coordinatedNi atoms decreased by onlyabout0.02and0.05μB,respectively.
SDOS for CO adsorption at the (↑↑)‐bridge sites of AFM2NiO(011)areplottedinFigure10(a).Themagneticmomentsof
Fig.9. Adsorption of CO/NO onNiO AFM2(011) andAFM2(011). (a)AFM2(011)surfacewithbridgesitesofNi(A,↑)andNi(B,↑);(b)COand(c)NOadsorbedconfigurationonAFM2(011); (d)AFM2(011)surfacewithbridgesitesofNi(A,↑)andNi(B,↓); (e)COand(f)NOadsorbedconfigurationonAFM2(011).
Table6MagnetismandstructureinformationofCOadsorptionatthe(↑↑)‐bridgeand(↑↓)‐bridgesitesofNiO{011}surfaces.Ni(A)alwayslabelstheNiwithtotalmagneticmomentspinup.Magneticmoment(MNi,MCO)isgiveninunitofμB.Bondlength(dNi‐C)isgiveninÅ.AdsorptionenergyofCO(Eads)isgivenineV.
MNi(clean)/μB MNi(A)/μB MNi(B)/μB MCO/μB dNi(A)‐C/Å dNi(B)‐C/Å Eads/eV(↑↓)‐bridge (↑) (↓) AFM1(011) 1.77 1.72 1.72 0.01 2.13 2.15 0.73AFM2(011) 1.70 1.66 1.66 0.00 2.18 2.18 0.74AFM3(011)X1 1.75 1.73 1.73 0.04 2.19 2.16 0.75AFM3(110) 1.74 1.68 1.68 0.00 2.13 2.13 0.74AFM4(011)X1 1.75 1.72 1.72 0.05 2.16 2.19 0.72AFM5(011)X1 1.72 1.67 1.66 0.00 2.14 2.12 0.74AFM5(101) 1.72 1.67 1.66 0.01 2.14 2.12 0.74(↑↑)‐bridge (↑) (↑) AFM1(110) 1.78 1.74 1.74 0.02 2.17 2.17 0.71AFM2(011) 1.70 1.68 1.68 0.03 2.17 2.19 0.71AFM3(011)X2 1.75 1.72 1.72 0.05 2.18 2.19 0.71AFM4(011)X2 1.75 1.71 1.73 0.05 2.15 2.23 0.71AFM4(110) 1.74 1.69 1.69 0.00 2.15 2.14 0.72AFM5(011)X2 1.72 1.69 1.69 0.03 2.16 2.19 0.71AFM5(101) 1.73 1.70 1.70 0.06 2.18 2.17 0.71FM(011) 1.78 1.75 1.75 0.06 2.17 2.18 0.71

1744 Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748
both Ni(A) and Ni(B) point upwards, which are denoted asNi(A,↑)andNi(B,↑),respectively.Figure10(a)revealsthatthespin‐up components of Ni(A, ↑) and Ni(B, ↑) are nearly com‐pletely occupied. The hybridization occurs mainly via Ni(A,↑)/Ni(B, ↑) back‐donation towards CO. In contrast, thespin‐down components of Ni(A, ↑) and Ni(B, ↑) are partiallyoccupied, and hybridization occurs via CO donation to Ni(A,↑)/Ni(B,↑).Backdonationinthespin‐upcomponentbutdona‐tion in the spin down can be easily visualized in thespin‐resolved difference of electron density results plotted inFigure10(b)and(c).Forthespin‐upcase,thereispronouncedchargedepletionfrombothNi(A,↑)andNi(B,↑)andaccumula‐tioninCO2π*(backdonation).Forthespin‐downcomponent,thereischargedepletioncausedbyCO5σdonationandaccu‐mulationmainlybetweenCOandNi(A,↑)/Ni(B,↑).
SDOS for CO adsorption at the (↑↓) bridge sites ofAFM2(011) are plotted in Figure 10(d), where the magneticmoments of Ni(A) and Ni(B) respectively point upwards (↑)anddownwards (↓),andaredenotedasNi(A,↑)andNi(B,↓),respectively.Forthespin‐upcomponent,Ni(A,↑)isnearlyfullyoccupied and Ni(B, ↓) is partially occupied. Accordingly, thehybridizationtakesplaceviaNi(A,↑)backdonationtowardCO,butCOdonation towardNi(B, ↓).This canbeobserved in thespin‐resolveddifferenceofelectrondensityofthesystem(Fig‐ure10(e)and(f)).ThereismodestchargedepletionatNi(A,↑)for back donation and pronounced charge accumulation be‐tweenCOandNi(B,↓)forCOdonation.Therelativelargerex‐tentofchargeaccumulationbetweenCOandNi(B,↓)thanthatof charge depletion atNi(A, ↑) indicates that donation ratherthan back donation plays a dominant role in the overall hy‐bridizationbetweenCOandNi.Forthespin‐downcomponent
(Figure10(d)),Ni(B,↓)isnearlyfullyoccupiedandNi(A,↓) ispartially occupied. Accordingly, the hybridization takes placeviaNi(B,↓)backdonationtowardCOandCOdonationtowardNi(A, ↑). The distinct hybridization and charge redistributionforCOatthe(↑↓)bridgesitesfromthatofthe(↑↑)bridgesitesisincontrasttotheirsimilaradsorptionenergies(whichdifferbylessthan0.03eV).
3.3.3. NOadsorptionNOadsorptionalsooccurspreferentiallyatthebridgesiteof
twoadjacentNiatomsvia theendof theNatom(Figure9(c)and (f)). For allnonequivalent surfaces considered, the calcu‐latedadsorptionenergiesforNOatthe(↑↓)bridgesitesareinthe range of −0.99 to −1.05 eV and in the range of −1.21 to−1.30eVatthe(↑↑)bridgesites.DifferentfromCO,thebindingstrengthsatthe(↑↑)bridgesitesareabout0.3eVstrongerthanthose at the (↑↓) bridge sites. Moreover, the NO bindingstrengthonNiO(011)isaboutthreetimesstrongerthanthatofNiO(001).For adsorptionon (↑↑)or (↑↓)bridge sites, thedif‐ference of adsorption energywith variation of bulkmagneticorder and surface index is less than 0.10 eV. In contrast, thevariationofNOadsorptionenergyonallstudiedNiO(001)sur‐facesdidnotexceed0.04eV.Thisindicatesthatthelargervari‐ationofbindingstrengthatall(↑↑)bridgesites(or(↑↓)bridgesites)ofNiO(011)thanthatofNiO(001)maybesimplycausedbythehighersurfaceactivityofNiO(011).However,thediffer‐ence of binding strength between adsorption at (↑↑) bridgesites and that at (↑↓) bridge sites of NiO(011) cannot be at‐tributedtothisreason.Again,theimportanceofthelocalmag‐neticorderratherthanthelong‐rangemagneticphaseisfoundhere.
-8 -4 0 4
-5
0
5 Ni(A) 3d Ni(B) 3d CO 2p
SD
OS
/(a.
u.)
E-EF/eV
CO/AFM2‐(011)(a)
DOWNUP
(b)+0.01e/A3
-0.01e/A3 DOWNUP
Ni(B) Ni(A)
C
O
↑ ↑
(c)
-8 -4 0 4
-5
0
5 Ni(A) 3d Ni(B) 3d CO 2p
SD
OS
/(a.
u.)
E-EF/eV
CO/AFM2‐(0 )(d)
DOWNUP
(e)
DOWNUP
Ni(B) Ni(A)
C
O
↓ ↑
(f)+0.01e/A3
-0.01e/A3
Fig.10.COadsorptiononAFM2(011)andAFM2(011).(a−c)COadsorptiononAFM2(011);(d−f)COadsorptiononAFM2(011);(a,d)SDOSfor3dorbitalsofNi(A)andNi(B)labeledinFigure9and2porbitalsofCO;(b,e)Isosurfaceviewand(c,f)sectioncontourviewofspin‐resolveddifferenceofelectrondensityforCOadsorptionwithspin‐upcomponentlabeledUPandspin‐downcomponentlabeledDOWN,respectively.

Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748 1745
Important geometrical parameters for NO adsorption onNiO(011) are given in Table 7. Irrespective of the differentmagnetic phases considered, theN−O bond lengths stay con‐stantat1.19Å,0.02Ålongerthanthatofgas‐phaseNO.ForNOadsorptionatthe(↑↑)bridgesites,thetwoN−Nibondlengthsarenearlysame(1.96–1.97Å).ThecorrespondingNOmagneticmomentpreferstoalignantiparallelwiththetwocoordinatedNi(↑), namely, as NO(↓), as also found for NO adsorption onNiO(001). The magnetic moments of NO and coordinated Niatomsdecreasedbyatmost0.25and0.13μB,respectively.ForNO adsorption at the (↑↓) bridge sites, the two N−Ni bondlengthswerenotthesame.Theshorteronevariedfrom1.93to1.97Å.ThemagneticmomentsofNOandNicorrespondingtothe shorter N−Ni bond pointed downwards (↓) and upwards(↑), decreasingbyatmost0.43and0.20μB, respectively.ThelongerN−Nibondlengthvariedfrom2.03to2.09Å,andtherewaslittlechangeofthemagneticmomentofcoordinatedNi(↓),which is understandable because of its relative longer N−Nibondlength.
Calculated SDOS for NO at the (↑↑) bridge sites of AFM2NiO(011) is plotted in Figure 11(a),where themagneticmo‐mentsofbothNi(A)andNi(B)pointupwardsandaredenotedasNi(A, ↑) andNi(B, ↑), respectively.As noted above, theNOmagnetic moment prefers to point downward (↓); namely,therearemoreelectronsoccupied in theNOspin‐downcom‐ponent than in the spin‐up component.More electrons in theNO spin‐down componentwould facilitate the correspondingdonationprocess.Thiswasverifiedinthespin‐resolveddiffer‐enceofelectrondensityresultsplottedinFigure11(b)and(c),wherechargeaccumulatedbetweenNOandcoordinatedNi(A,↑)/Ni(B, ↑). For the spin‐up component, less electronoccupa‐tionof theNOorbitalwould facilitate thecorrespondingbackdonation. This can be found in Figure 11(b) and (c), wherechargedepleted from coordinatedNi(A, ↑)/Ni(B, ↑) andaccu‐
mulatedintheNOmolecule.CalculatedSDOSforNOadsorbedatthe(↑↓)bridgesitesof
AFM2(011) is plotted in Figure 11(d), where the magneticmoments of Ni(A) and Ni(B) point upwards and downwardsandaredenotedasNi(A,↑)andNi(B,↓),respectively.TheN−ObondlengthwithNi(A,↑)isshorterthanthatwithNi(B,↓),andthe NOmagneticmoment points downward accordingly. Thehybridization between NO(↓) and Ni(A, ↑) with antiparallelmagnetic configuration is essentially the same as that forNOadsorptiononAFM2(011)becausetheelectronicandmagneticconfigurations are the same. This can be seen further in thecorresponding difference of electron density plots (Figure 11(e)and(f)).Conversely, forNO(↓)andNi(B,↓)withsameori‐entation of magnetic moments, the electronic hybridizationbetweenNOandNi(B)islimited.Indeed,thedifferenceofelec‐trondensity(Figure11(e)and(f))betweenNOandNi(B)be‐comes negligible, particularly in the spin‐down component.This rationalizeswell theweaker binding strength of NO ad‐sorption at the (↑↓) bridge sites than that of NO at the (↑↑)bridgesites.
4. Conclusions
Byconstructing surfacemodelsofNiOwithdifferentmag‐netic phases, and using the paramagnetic molecule NO andnonparamagneticmoleculeCOtoprobethereactivityon(001)and(011)surfaces,weinvestigatedtheinfluenceofmagnetismonthesurfacechemistryofNiO.WhenthefavorableadsorptionsitesinvolveonlyasingleNication,suchasNO/COadsorptionatthetopsiteofaNicationonthe(001)surface,thelong‐rangemagneticphasesmakelittlecontributiontotheoveralladsorp‐tionbehavior.Theadsorptionstrengthisprimarilydeterminedbychemicalenvironment(suchassurfaceorientation,coordi‐nation number, and adsorbed species). When the favorable
Table7MagnetismandstructureinformationofNOadsorptionatthe(↑↑)‐bridgeand(↑↓)bridgesitesofNiO{011}surfaces.Ni(A)alwayslabelstheNiwithtotalmagneticmomentspinup.Magneticmoment(MNi,MNO)isgiveninunitofμB.Bondlength(dNi‐N)isgiveninÅ.AdsorptionenergyofNO(Eads)isgivenineV.
MNi(clean)/μB MNi(A)/μB MNi(B)/μB MNO/μB dNi(A)‐N/Å dNi(B)‐N/Å Eads/eV(↑↓)‐bridge (↑) (↓) (↓) AFM1(011) 1.77 1.58 1.74 0.47 1.93 2.03 1.05AFM2(011) 1.70 1.56 1.71 0.39 1.96 2.07 1.01AFM3(011)X1 1.75 1.54 1.79 0.64 1.95 2.08 1.01AFM3(110) 1.74 1.53 1.78 0.74 1.96 2.09 1.05AFM4(011)X1 1.75 1.57 1.78 0.65 1.97 2.05 1.03AFM5(011)X1 1.72 1.53 1.75 0.63 1.96 2.07 1.02AFM5(101) 1.72 1.54 1.73 0.30 1.94 2.05 0.99(↑↑)‐bridge (↑) (↑) (↓) AFM1(110) 1.78 1.65 1.65 0.68 1.97 1.97 1.30AFM2(011) 1.70 1.57 1.57 0.61 1.97 1.97 1.21AFM3(011)X2 1.75 1.61 1.63 0.49 1.96 1.97 1.26AFM4(011)X2 1.75 1.61 1.74 0.47 1.96 1.97 1.26AFM4(110) 1.74 1.61 1.61 0.72 1.96 1.96 1.30AFM5(011)X2 1.72 1.58 1.59 0.53 1.96 1.97 1.24AFM5(101) 1.73 1.61 1.61 0.53 1.97 1.97 1.23FM(011) 1.78 1.64 1.64 0.59 1.96 1.96 1.29

1746 Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748
adsorption site involves two adjacent Ni cations, such asNO/CO adsorption at the bridge site of two Ni cations inNiO(011),thelocalmagneticorderofNiionscoordinatedtotheadsorbed molecule rather than the long‐range magnetismmakes a considerable contribution to the overall adsorptionbehavior. For nonparamagnetic molecule CO, the orbital hy‐bridization for adsorption at the (↑↑) bridge sites is very dif‐ferentfromthatatthe(↑↓)bridgesites,despitetheirincidentalsimilar adsorption energies. For paramagnetic molecule NO,thehybridizationatthe(↑↑)bridgesitesisdifferentfromthatatthe(↑↓)bridgesites,andthebindingstrengthoftheformeris about 0.3 eV stronger than that of the latter. The presentworkhighlights the importanceof the localmagneticorderofTMO surfaces on molecule adsorption, particularly for para‐magneticmolecules,atmulti‐foldsites.
Acknowledgments
Weare grateful for the fruitful discussionwithProf.HongJiang.
References
[1] U.Diebold, S. C. Li,M.Schmid,Annu.Rev.Phys.Chem.,2010, 61,129148.
[2] J.F.Weaver,Chem.Rev.,2013,113,41644215.[3] G. A. Somorjai, Y. M. Li, Proc. Natl. Acad. Sci. USA, 2011, 108,
917924.[4] S. Shaikhutdinov,H. J. Freund,Annu.Rev.Phys.Chem.,2012, 63,
619633.
GraphicalAbstract
Chin.J.Catal.,2017,38:1736–1748 doi:10.1016/S1872‐2067(17)62883‐3
Influenceofnickel(II)oxidesurfacemagnetismonmoleculeadsorption:Afirst‐principlesstudy
Chuan‐QiHuang,Wei‐XueLi*DalianInstituteofChemicalPhysics,ChineseAcademicofSciences; UniversityofChineseAcademyofSciences; UniversityofScienceandTechnologyofChina
Calculations revealed thatNO preferentially binds toNi surfaceswithitsmagneticmomentantiparalleltothatofNi.NOadsorbedatantiferromagnetic bridge sites binds to oneNi optimally and an‐otherinafrustratedmanner.
-8 -4 0 4
-5
0
5 Ni(A) 3d Ni(B) 3d NO 2p
SD
OS
/(a.
u.)
E-EF/eV
NO/AFM2‐(011)(a)
DOWNUP
(b)+0.05e/A3
-0.05e/A3 DOWNUP
Ni(B) Ni(A)
N
O
↑ ↑
↓
(c)
-8 -4 0 4
-5
0
5 Ni(A) 3d Ni(B) 3d NO 2p
SD
OS
/(a.
u.)
E-EF/eV
NO/AFM2‐(0 )(d)
DOWNUP
(e)
DOWNUP
Ni(B) Ni(A)
N
O
↓
↓ ↑
(f)+0.05e/A3
-0.05e/A3
Fig.11.NOadsorptiononNiOAFM2(011)andAFM2(011).(a−c)NOadsorptiononAFM2(011);(d−f)NOadsorptiononAFM2(011);(a,d)SDOSfor3dorbitalsofNi(A)andNi(B)labeledinFigure9and2porbitalsofNO;(b,e)Isosurfaceviewand(c,f)sectioncontourviewofspin‐resolvedchargeflowdiagramforNOadsorptionwithspin‐upcomponentlabeledUPandspin‐downcomponentlabeledDOWN,respectively.

Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748 1747
[5] H.Kuhlenbeck, S. Shaikhutdinov,H. J. Freund,Chem.Rev.,2013,113,39864034.
[6] D.P.Woodruff,Chem.Rev.,2013,113,38633886.[7] S.M.Zhou,X.B.Miao,X.Zhao,C.Ma,Y.H.Qiu,Z.P.Hu,J.Y.Zhao,L.
Shi,J.Zeng,Nat.Commun.,2016,7,11510.[8] J. Suntivich, K. J. May, H. A. Gasteiger, J.B. Goodenough, Y.
Shao‐Horn,Science,2011,334,13831385.[9] S. Chretien, H. Metiu, J. Chem. Phys., 2008, 129, 074705/1
074705/16.[10] J. Behler, B. Delley, S. Lorenz, K. Reuter, M. Scheffler, Phys.Rev.
Lett.,2005,94,036104/1036104/4.[11] E.Torun,C.M.Fang,G.A.deWijs,R.A.deGroot,J.Phys.Chem.C,
2013,117,63536357.[12] R.V.Mom,J.Cheng,M.T.M.Koper,M.Sprik,J.Phys.Chem.C,2014,
118,40954102.[13] N.C.Tombs,H.P.Rooksby,Nature,1950,165,442443.[14] J.S.Smart,S.Greenwald,Phys.Rev.,1951,82,113114.[15] M.Finazzi,L.Duò,F.Ciccacci,Surf.Sci.Rep.,2009,64,139167.[16] M.Finazzi,L.Duò,F.Ciccacci,in:MagneticPropertiesofAntiferro‐
magneticOxideMaterials,Wiley‐VCH,2010,123.[17] P.W.Anderson,Phys.Rev.,1950,79,350356.[18] H.X.Deng,J.B.Li,S.S.Li,J.B.Xia,A.Walsh,S.H.Wei,Appl.Phys.
Lett.,2010,96,162508/1162508/3.[19] A. Schrön, C. Rödl, F. Bechstedt, Phys. Rev. B, 2010, 82,
165109/1165109/12.[20] T.Archer,C.D.Pemmaraju, S. Sanvito,C. Franchini, J.He,A. Fil‐
ippetti,P.Delugas,D.Puggioni,V.Fiorentini,R.Tiwari,P.Majum‐dar,Phys.Rev.B,2011,84,115114/1115114/14.
[21] C. G. Shull, W. A. Strauser, E. O. Wollan, Phys. Rev., 1951, 83,333345.
[22] U. Kaiser, A. Schwarz, R. Wiesendanger, Nature, 2007, 446,522525.
[23] M. Granovskij, A. Schrön, F. Bechstedt, Phys. Rev. B, 2013, 88,184416/1184416/5.
[24] F.Pielmeier,F. J.Giessibl,Phys.Rev.Lett.,2013,110,266101/1266101/5.
[25] M. Granovskij, A. Schrön, F. Bechstedt, New J. Phys., 2014, 16,23020/123020/18.
[26] A. Schrön, F. Bechstedt, Phys. Rev. B, 2015, 92, 165112/1165112/11.
[27] I.Sugiyama,N.Shibata,Z.C.Wang,S.Kobayashi,T.Yamamoto,Y.Ikuhara,Nat.Nanotechnol.,2013,8,266270.
[28] H.Bi,S.D.Li,Y.C.Zhang,Y.W.Du, J.Magn.Magn.Mater.,2004,277,363367.
[29] V.N.Antonov,L.V.Bekenov,A.N.Yaresko,Adv.Cond.MatterPhys.,2011,2011,1107.
[30] P.Thunstrom,I.DiMarco,O.Eriksson,Phys.Rev.Lett.,2012,109,186401/1186401/5.
[31] L.H.Ye,R.Asahi,L.M.Peng,A. J.Freeman, J.Chem.Phys.,2012,137,154110/1154110/7.
[32] H.Jiang,R.I.Gomez‐Abal,P.Rinke,M.Scheffler,Phys.Rev.B,2010,82,045108/1045108/16.
[33] K.Terakura,T.Oguchi,A.R.Williams,J.Kübler,Phys.Rev.B,1984,30,47344747.
[34] V. I. Anisimov, J. Zaanen,O.K. Andersen,Phys.Rev.B,1991, 44,943954.
[35] S. L.Dudarev,G.A.Botton, S.Y. Savrasov,C. J.Humphreys,A.P.Sutton,Phys.Rev.B,1998,57,15051509.
[36] F.Manghi,V.Boni,J.Electron.Spectrosc.Relat.Phenom.,2015,200,181192.
[37] A. Rohrbach, J. Hafner, G. Kresse, Phys. Rev. B, 2004, 69,075413/1075413/13.
[38] A. Rohrbach, J. Hafner, Phys. Rev. B, 2005, 71, 045405/1045405/7.
[39] W. Zhao, M. Bajdich, S. Carey, A. Vojvodic, J. K. Nörskov, C. T.Campbell,ACSCatal.,2016,6,73777384.
[40] G.Kresse,J.Furthmuller,Comp.Mater.Sci.,1996,6,1550.[41] P.E.Blöchl,Phys.Rev.B,1994,50,1795317979.[42] G.Kresse,D.Joubert,Phys.Rev.B,1999,59,17581775.[43] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett., 1996, 77,
38653868.[44] S.H.Wei,A.Zunger,Phys.Rev.B,1993,48,61116115.[45] T. Oguchi, K. Terakura, A. R. Williams, Phys. Rev. B, 1983, 28,
64436452.[46] M. Schönnenbeck,D.Cappus, J.Klinkmann,H. J. Freund, L.G.M.
Petterson,P.S.Bagus,Surf.Sci.,1996,347,337345.[47] A.Zecchina,D.Scarano,S.Bordiga,G.Ricchiardi,G.Spoto,F.Geo‐
baldo,Catal.Today,1996,27,403435.[48] R.Wichtendahl,M.Rodriguez‐Rodrigo, U.Hartel,H.Kuhlenbeck,
H.J.Freund,Phys.StatusSolidiA,1999,173,93100.[49] Z.H.Zeng,J.L.F.DaSilva,H.Q.Deng,W.X.Li,Phys.Rev.B,2009,
79,205413/1205413/13.[50] Z.H.Zeng,J.L.DaSilva,W.X.Li,Phys.Chem.Chem.Phys.,2010,12,
24592470.
NiO表面磁性对分子吸附的影响的第一性原理研究
黄传奇a,b, 李微雪a,b,c,* a中国科学院大连化学物理研究所催化基础国家重点实验室, 辽宁大连116023
b中国科学院大学, 北京100049 c中国科学技术大学化学与材料科学学院化学物理系, 合肥微尺度物质科学国家重点实验室(筹), 安徽合肥230026
摘要: 过渡金属氧化物广泛应用在当今能源与环境相关的催化领域, 理解其表面化学性质以及结构-反应活性之间的关系
对于先进催化材料的进一步发展以至理性设计至关重要. 3d后过渡系金属(Mn, Fe, Co, Ni)的氧化物以其中金属离子独特
的自旋状态和由此产生的铁磁/反铁磁性为典型特征. 研究过渡金属氧化物的自旋状态以及磁性对表面化学的影响将使我
们更加完整了解这些材料的表面化学. 以NiO为代表的后过渡系金属岩盐结构一元氧化物具有反铁磁性, 被经常作为反铁
磁研究的模型体系. 尽管在低温(低于其Neel温度)下NiO体相的完整晶体具有确定的反铁磁序, 但是一系列最新研究表明,
在条件变化时NiO表面的Ni离子可以产生不同的磁序. 以此为背景, 本工作以NiO为模型体系, 采用DFT+U的第一性原理
方法研究了NiO表面磁序对表面的小分子吸附活性的影响, 包括表面吸附活性对各磁性相的表面取向以及吸附物种磁性的
依赖关系. 我们考察了NiO的5种反铁磁相和一种铁磁相, 两个晶面NiO(001)和NiO(011), 顺磁性分子NO和非顺磁性分子

1748 Chuan‐QiHuangetal./ChineseJournalofCatalysis38(2017)1736–1748
CO. 我们发现表面能受磁性的影响较轻微 , NiO(001)面上从49到54 meV/Å2, NiO(011)面上从162到172 meV/Å2. 在
NiO(001)面上, CO与NO都倾向于在Ni离子的顶位吸附. 对于不同的体相磁序与表面取向, CO吸附能的变化范围为0.33 ~
0.37 eV, NO吸附能的变化范围为0.42 ~ 0.46 eV. 在NiO(011)表面, 两种分子都倾向于吸附在由两个Ni离子构成的桥位.
我们发现相对于NiO不同磁性相的体相长程磁序, 吸附位点处构成桥位的两个Ni离子的局部磁矩相对取向对于分子的吸附
具有更加显著的影响. 计算得到NO在局部磁矩相对取向反平行(↑↓)吸附位点处的吸附能为0.99 ~ 1.05 eV, 在局部磁矩
相对取向平行(↑↑)吸附位点处吸附会增强, 吸附能为1.21 ~ 1.30 eV. 对于CO, 尽管计算的吸附能在(↑↓)吸附位点(0.73 ~
0.75 eV)与在(↑↑)吸附位点(0.71 ~ 0.72 eV)非常接近, 两种吸附位点处的CO吸附时分子轨道杂化方式以及吸附后CO的
局域电子态密度却具有明显不同的特征. 本工作突出揭示了分子在过渡金属氧化物表面的多重吸附位点上吸附时吸附位
点的局域磁矩相对取向对吸附性能的影响.
关键词: 磁性; 表面取向; 分子吸附; 第一性原理; 电子结构
收稿日期: 2017-05-27. 接受日期: 2017-06-26. 出版日期: 2017-10-05.
*通讯联系人. 电话: (0551)63600650; 电子信箱: [email protected]
基金来源: 国家自然科学基金(91645202); 国家重点研发计划(2017YFB602205); 国家重点基础研究发展计划(2013CB834603);
中国科学院前沿科学重点研究项目(QYZDJ-SSW-SLH054).
本文的英文电子版由Elsevier出版社在ScienceDirect上出版(http://www.sciencedirect.com/science/journal/18722067).


















