
2012 Southwest Theoretical Chemistry Conference Presentations 2012 Southwest Theoretical Chemistry...
73
2012 Southwest Theoretical Chemistry Conference October 26-28, 2012 Department of Chemistry Texas A&M University College Station, TX
-
Upload
nguyenphuc -
Category
Documents
-
view
217 -
download
2
Transcript of 2012 Southwest Theoretical Chemistry Conference Presentations 2012 Southwest Theoretical Chemistry...

2012 Southwest Theoretical Chemistry Conference
October 26-28, 2012
Department of Chemistry Texas A&M University
College Station, TX

Welcome to SWTCC2012
We are pleased to welcome you to the 2012 Southwest Theoretical Chemistry Conference, hosted by the Department of Chemistry at Texas A&M University. The conference will be held primarily in the Integrated Life Sciences Building (ILSB), with the poster session in the Rudder Exhibit Hall and the conference banquet in the Gates Ballroom of the Memorial Student Center (MSC). A small map is provided below for your convenience. A more complete campus map can be found on the inside back cover.
We are grateful to our corporate sponsors, whose financial support has allowed us to keep registration costs low. We also thank the Department of Chemistry for subsidizing undergraduate registration fees.
Please let us know if we can provide any assistance, and enjoy the conference.
Sincerely,
Steven E. Wheeler

Conference Agenda Friday, October 26
6:00 - 7:00 PM Registration and Welcome Reception (ILSB Lobby) Session 1 (Session Chair: Steven E. Wheeler)
7:00 PM Welcome 7:15 PM Gustavo Scuseria, “Symmetry breaking and restoration”
8:00 PM Danny Yeager, “Aspects of Complex scaling for electron-atom/molecule resonances using MCSCF, MCSTEP and MCTDHF”
8:20 PM Dieter Cremer, “Local Vibrational Modes: A new tool to describe the electronic structure of molecules”
Saturday, October 27 8:00 - 8:30 AM Registration and Continental Breakfast (ILSB Lobby)
Session 2 (Session Chair: Robert Lucchese) 8:30 AM Angela Wilson, “Multireference wavefunction composite strategies” 9:00 AM Bill Poirier, “Exact quantum dynamics calculations using phase space wavelets” 9:20 AM Eric Bittner, “Quantum origins of molecular recognition and olfaction in drosophila”
9:50 Carlos Jimenez-Hoyos, “TDA and RPA approximations to excited states based on a symmetry-projected HF ground state”
10:10 AM Coffee Break Session 3 (Session Chair: TBA)
10:30 AM Kevin Shuford, “Nanoplasmonics: Sensing and energy applications” 11:00 AM Perla Balbuena, “Electron transfer from electrode to electrolyte in Li-ion batteries”
11:20 AM Matthew Wander, “Water hydrogen bonding behavior of alkali-halide solutions at a graphite interface”
11:40 PM D. Balamurugan, “Multiscale simulation of charge-transfer triad molecule in an organic solvent: exploring the conformations, fluctuations and the free energy landscape”
12:00 - 1:30 PM Lunch (ILSB Lobby)
Session 4 (Session Chair: Kameron Jorgensen) 1:30 PM Lan Cheng, “Relativistic theory for chemical shieldings” 1:50 PM Thomas Sommerfeld, “Characterizing the excess electron of Li(NH3)4”
2:10 PM Marie Laury, “Examining the 4d transition metals and the lower p-block with a pseudopotential-based composite method: Atomic and molecular applications of rp-ccCA”
2:30 PM Coffee Break Session 5 (Session Chair: Tom Schmalz)
3:00 PM Feng Wang, “Predicting the melting temperature of ice-Ih with only electronic structure information”
3:30 PM Jhenny Galan, “Conformational preferences of furan- and thiophene-based arylamides: A combined computational and experimental study”
3:50 PM Diego Gomez-Gualdron, “Nanocatalyst structure as a template to control chirality during chemical vapor deposition synthesis of single-walled carbon nanotubes”
4:10 PM Jerry Darsey, “A hybrid Monte-Carlo molecular modeling approach to predicting the 3-D structure of proteins”

Poster Session and Conference Banquet 5:30 – 7:00 PM Poster Session (Rudder Exhibit Hall) 7:00 – 9:00 PM SWTCC2012 Banquet (Gates Ballroom, Memorial Student Center)
Sunday, October 28 8:00 - 8:30 AM Continental Breakfast (ILSB Lobby)
Session 6 (Session Chair: Lisa Pérez) 8:30 AM Jan Martin, “A simple DFT-based diagnostic for nondynamical correlation” 9:00 AM Sebastian Kozuch, “Double hybrid DFT - the slow quest for perfection” 9:20 AM Ben Janesko, “Nonlocal ingredients for exchange-correlation density functionals” 9:50 AM Coffee Break
Session 7 (Session Chair: Dan Singleton) 10:10 AM Steven Bachrach, “Computational studies of organic superbases”
10:40 AM Elfi Kraka, “The decisive role of hydrogen-bonds in chiral discrimination - unraveled by quantum chemical means”
11:00 AM Kevin Riley, “Halogen bonds in protein-ligand complexes”
11:30 AM Charles Edwin Webster, “The many phosphoryl transfers of Phospholipase D superfamily: a quantum mechanical theoretical study”
11:50 AM Closing Remarks 12:00 - 1:00 PM Lunch and Annual Meeting (ILSB Lobby)
All events will be in the integrated life sciences building (ILSB) auditorium unless noted otherwise.

Abstracts for Oral Presentations

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Computational studies of organic superbases
Steven M. Bachrach
Department of Chemistry, Trinity University, San Antonio, TX 78212
Typical strong bases are salts and can have limited solubility in organic solvents. Proton sponge 1 has inspired the development of a series of organic strong bases, typically by placing two or more amino groups in near proximity. Kass has suggested that an extended network of hydrogen bonding can also serve to stabilize ammonium cations, potentially offering a new means for creating strong bases. In this talk, I will discuss a number of proposed organic superbases that take advantage of both types of mechanisms, such as 3 and 4. The basicity of these compounds is evaluated using density functional theory in both the gas- and solution-phases.
1
2 3

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Multiscale simulation of charge-transfer triad molecule in an organic solvent: exploring the conformations, fluctuations and
the free energy landscape D. Balamurugan,a Adelia Aquino,b Hans Lischka,b Francis De Dios,a Lionel Flores Jr.,a
and Margaret Cheunga aDepartment of Physics, University of Houston, TX 77004
[email protected] bDepartment of Chemistry, Texas Tech University, Lubbock, TX 79409
Molecular triad is an artificial analogue of natural photosynthetic system that is composed of fullerene, porphyrin, and carotene functional groups. Triad is considered for applications in solar energy conversion since it produces a strong and stable photo-induced charge separated state. A multiscale simulation that combines quantum chemical calculations and classical molecular dynamics simulations is employed to characterize the influence of structural fluctuations and dynamics of a polar organic solvent, namely tetrahydrofuran (THF) on the structural stability of the charge-separated triad. The quantum chemical calculations were performed on triad using two methods - algebraic diagrammatic construction through second order (ADC(2)) method and time-dependent density functional theory. Molecular dynamics simulations were performed on triad in a box of THF solvent with the replica exchange enhanced sampling technique to explore the free energy landscape. We have analyzed the free energy landscape, structural fluctuations, solvent orientations, and the long range electrostatic interactions between triad and solvent molecules. The results suggest that the polarity and re-organization of the solvent is critical in stabilization of charge-separated state in triad.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Electron transfer from electrode to electrolyte in Li-ion batteries
Perla B. Balbuena
Department of Chemical Engineering, Texas A&M University, College Station, TX 77843
Formation of a solid-electrolyte interphase at the electrode surface of Li-ion batteries takes place due to electrochemical reduction of the solvent and salt present in the electrolyte. A thin film is formed which is composed of organic and inorganic products, and keeps growing until its thickness does not allow quantum tunneling. The most common solvents are ethylene and propylene carbonate, and a typical salt is LiPF6. We used density functional theory to examine the electron transfer reaction and its possible products, as well as the role of certain chemical species (additives) that present in small amounts are able to significantly improve the film properties. We discuss the possible electron transfer mechanisms as a function of the film growth.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Quantum origins of molecular recognition and olfaction in drosophila Eric R. Bittner
Department of Chemistry, University of Houston, Houston, TX 77204
The standard model for molecular recognition of an odorant is that receptor sites discriminate by molecular geometry as evidenced that two chiral molecules may smell very differently. However, recent studies of isotopically labeled olfactants indicate that there may be a molecular vibration-sensing component to olfactory reception, specifically in the spectral region around 2300 cm–1. Here we present a donor-bridge-acceptor model for olfaction which attempts to explain this effect. Our model, based upon accurate quantum chemical calculations of the olfactant (bridge) in its neutral and ionized states, posits that internal modes of the olfactant are excited impulsively during hole transfer from a donor to acceptor site on the receptor, specifically those modes that are resonant with the tunneling gap. By projecting the impulsive force onto the internal modes, we can determine which modes are excited at a given value of the donor-acceptor tunneling gap. Only those modes resonant with the tunneling gap and are impulsively excited will give a significant contribution to the inelastic transfer rate. Using acetophenone as a test case, our model and experiments on D. melanogaster suggest that isotopomers of a given olfactant give rise to different odorant qualities. These results support the notion that inelastic scattering effects play a role in discriminating between isotopomers, but that this is not a general spectroscopic effect.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Relativistic theory for chemical shieldings
Lan Cheng and John F. Stanton
Institute for Theoretical Chemistry, Department of Chemistry and Biochemistry,The University of Texas at Austin, Austin, TX 78712
The combination of the magnetic-balance condition [1] and the gauge-including atomic orbital ansatz [2] has recently emerged as an effective approach to obtain a fast basis-set convergence in fully relativistic calculations of chemical shieldings based on the Dirac-Coulomb (DC) Hamiltonian. [3, 4] On the other hand, due to the high cost of the DC approach, it is worthwhile to explore cost-effective approximate schemes for relativistic chemical-shielding calculations.
In the present work, we report on the formulation and implementation of a spin-free exact two-component theory at the one-electron level (SFX2c-1e) [5] for chemical shieldings. The X2c-1e Hamiltonian and derivative integrals relevant to the chemical-shielding calculations are constructed via a block-diagonalization [5, 6, 7] of the matrix representation of the Dirac equation in terms of magnetically balanced gauge-including atomic orbitals. The separate treatment of spin-free and spin-dependent terms [8] serves to facilitate efficient relativistic coupled-cluster calculations of chemical shieldings. Here we present calculations of the 129Xe chemical shifts in the series of xenon fluorides, XeF2, XeF4, and XeF6 to demonstrate the applicability of the present SFX2c-1e approach.
1. G. A. Aucar, T. Saue, L. Visscher, and H. J. A. Jensen, J. Chem. Phys. (110), 6208 (1999). 2. K. Wolinski, J. F. Hinton, and P. Pulay, J. Am. Chem. Soc. (112), 8251 (1990). 3. L. Cheng, Y. Xiao and W. Liu, J. Chem. Phys. (131), 244113 (2009). 4. S. Komorovsky, M. Repisky, O. L. Malkina, and V. G. Malkin, J. Chem. Phys. (132), 154101 (2010). 5. K. G. Dyall, J. Chem. Phys. 115, 9136 (2001). 6. L. Cheng and J. Gauss, J. Chem. Phys. 135, 084114 (2011). 7. L. Cheng and J. Gauss, J. Chem. Phys. 135, 244104 (2011). 8. K. G. Dyall, J. Chem. Phys. 100, 2118 (1994).

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Local Vibrational Modes A new tool to describe the electronic structure of molecules
Dieter Cremer and Wenli Zou
CATCO, Department of Chemistry, Southern Methodist University, Dallas, TX, 75275
http://smu.edu/catco
[email protected] The normal vibrations of a molecule depend on its geometry, its bonding framework, and its electronic structure. If exactly determined, their properties should be sufficient to characterize a molecule in detail. However, molecular vibrations are coupled and therefore delocalized. They reflect both kinematic and electronic effects, which are difficult to separate. We will prove that the normal vibrational modes correspond to a unique set of local vibrational modes, the properties of which fully characterize chemical bonding and electronic structure. Also, we will show how local vibrational modes can be derived from measured or calculated vibrational frequencies, how normal modes and vibrational modes are related by an adiabatic connection scheme (see figure) that quantifies mode-mode coupling by coupling frequencies, and how the local mode intensities can be related to effective atomic charges reflecting the electron density distribution in a molecule. Examples are given that demonstrate the usefulness of local mode properties for describing nonclassical bonding, H-bonding, dihydrogen bonding, halogen and agostic bonding.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
A Hybrid Monte-Carlo Molecular Modeling Approach to Predicting the 3-D Structure of Proteins
Jerry Darsey
Department of Chemistry, University of Arkansas, Little Rock, AR, 72204
Protein structure prediction is one of the most important goals pursued by bioinformatics and theoretical chemistry; it is highly important in medicine (for example, in drug design) and biotechnology (for example, in the design of novel enzymes). Identifying a protein's structure is the key to understanding a protein’s biological function and role in health and disease. It could be said that correctly modeling the 3-D structure of a protein is the “Holy Grail” of computational molecular modeling. Therefore, the ultimate goal for modeling is to be able to predict the native structure of a protein based on nothing more than the sequence of amino acids. Unfortunately, the way from the amino acid sequence to the corresponding protein structure is not always straightforward - it is still impossible, using computational chemistry, to calculate the tertiary structure directly from the protein sequence. There are many reasons why understanding the three-dimensional (3-D) structure of proteins is so vital. Some of the most important reasons are: (1) In the pharmaceutical industry, understanding holds the prospects of greatly reducing the cost and expense of developing new therapeutic drugs. (2) Many proteins have more than one structure. One structure produces normal biological function; the alternative structure many-times lead to diseases, such as in Parkinson disease, Alzheimer's, etc. In many modeling studies used to predict 3-D structure, the torsion angles φ, ψ of the amino acids are modeled. The angle ω is usually held fixed because of partial double bond character within the amino acid (See Fig. 1). In our study, taking into account the angle ω was found to be very important in predicting the overall 3-D structure of a protein molecule. We feel that Monte-Carlo modeling is the best procedure for developing structures which can directly account for non-180º angles for ω. It is also the only modeling technique which can probabilistically bias sampling of the entire conformational space of a protein molecule.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Conformational Preferences of Furan- and Thiophene-based Arylamides: A Combined Computational and Experimental Study
Jhenny Galan,a Zhiwei Liu,b Chi Ngong Tang,b Shubhashis Chakrabarty,b Vojislava Pophristic,b and Guillermo Moynab
aDepartment of Marine Sciences, Texas A&M at Galveston, Galveston, TX 77554
[email protected] bDepartment of Chemistry and Biochemistry, University of the Sciences, Philadelphia, PA
19104
Foldamers, synthetic oligomers that adopt defined, stable secondary structures in solution, have been the focus of intense research efforts in the past two decades. We examine the conformational preferences of furan- and thiophene-based arylamides using a combination of computational methods and NMR experiments. Our study quantifies the differences in the conformational rigidity of the compounds. Specifically, we demonstrate the effects of intramolecular H-bonding, geometrical patterns and solvent polarity on arylamide conformations by comparing the furan-based, thiophene-based and ortho-substituted arylamides. We will present detailed quantum mechanical study of the backbone torsions as well as conformational distributions from MD simulations in various solvent environments. To corroborate the computational results,1D-NOESY spectroscopy has been performed on some of the model compounds and has confirmed our findings. This study shows how the interplay of several forces can modulate the conformational flexibility of an arylamide foldamer. The quantitative information on backbone conformational preferences will enhance the rational design of arylamide foldamers. Another vital aspect of this study is the re-parameterization of critical force field parameters based on high-level QM potential energy profiles, as well as validation using experimental data on conformational preferences in solution.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Nanocatalyst structure as a template to control chirality during chemical vapor deposition synthesis of single-walled carbon
nanotubes Diego A. Gomez-Gualdron and Perla Balbuena
Chemical Engineering Department, Texas A&M University, College Station, TX 77843
For two decades single-walled carbon nanotubes (SWCNTs) have captured the imagination of the research community due to their outstanding properties, and their potential to revolutionize a number of fields. These properties are strongly dependent on the nanotube structure, wherein the metallic or semiconductor character (and associated band gap) depends on nanotube helicity or chirality. Since synthesis of chirality-wise heterogeneous nanotube mixtures hinders the exploitation of nanotube properties for a particular target application. Therefore, achieving chiral selectivity has constituted one of the most sought-after goals in nanotube synthesis.
In this work, density functional theory (DFT), and reactive molecular dynamics (RMD) are used to study the viability of using the structure of the nanoparticles supporting growing nanotubes during chemical vapor deposition (CVD) synthesis as a template to guide nanotube growth toward desired chiralities. The structural correlation between early-formed nanotube chiral caps, and metal nanoparticles is discussed. Carbon structures nucleated on controlled nanoparticle facet structures anticipate the preferential formation of nanotube of specific chiralities depending on the nanoparticle structure. Thus, designing the catalyst/support interaction is proposed to alter nanoparticle structure and dynamics leading to preferential formation of different chiralities through a direct template effect. Nonetheless, the effectiveness of such effect has shown a non-monotonic relation with catalyst/support interaction and catalyst size, which denotes the necessity of fine-tune reaction conditions to effectively exploit such direct template effect.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Nonlocal ingredients for exchange-correlation density functionals
Benjamin G. Janesko
Department of Chemistry, Texas Christian University, Fort Worth, TX 76129
Density functional theory (DFT) with approximate exchange-correlation (XC) functionals has become a standard tool in electronic structure theory. DFT calculations on molecules typically use “hybrid” XC functionals built from the square of the nonlocal one-particle density matrix γ(r,r’). These are typically more accurate than simple approximations built directly from the electron density ρ(r), but are often computationally expensive in large systems. We have explored XC functionals constructed from functions depending linearly on the density matrix. These incorporate useful nonlocal information at reduced computational cost. Numerical results from some nonempirical and few-parameter approximations illustrate the utility of this approach.
12
′ , ′ , ′
| ′|

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
TDA and RPA approximations to excited states based on a symmetry-projected HF ground state
Carlos A. Jimenez-Hoyos,a R. Rodriguez-Guzman,a,b and Gustavo E. Scuseriaa,b a Department of Chemistry, Rice University, Houston TX 77005
[email protected] b Department of Physics and Astronomy, Rice University, Houston TX 77005
Our recent work has focused on variational approximations to the ground state by means of a symmetry-projected HF state [1, 2]. In Ref. [2] we have also shown how excited states can be obtained by a sequence of variational calculations. Here, we discuss the approximate description of low-lying excited states as particle-hole vibrations out of the reference symmetry-projected ground state.
1. Jimenez-Hoyos, C. A., et al., J. Chem. Phys. 136 (2012), 164109. 2. Rodriguez-Guzman, R., et al., Phys. Rev. B 85 (2012), 245130.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Double hybrid DFT – The slow quest for perfection
Sebastian Kozucha and Jan Martinb a Weizmann Institute of Science, Rechovot, Israel (Present Address: University of North
Texas, Denton, TX 76203)
[email protected] b Department of Chemistry and Center for Advanced Scientific Computing and Modeling
(CASCaM), University of North Texas, Denton, TX 76203
Double hybrid (DH) DFT has shown to be a successful method for the accurate energy estimation of small and medium sized molecules. DH-DFT in its more typical formulation mixes an exact exchange term ("HF-like") with the DFT exchange functional as simple hybrids, but adds a perturbational correlation term ("MP2-like") to the DFT correlation in the basis of the Kohn-Sham orbitals. Herein we will seek for the best combination of exchange-correlation functional for the double hybrid method, including dispersion correction and spin-component scaled MP2.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
The decisive role of hydrogen-bonds in chiral discrimination - unraveled by quantum chemical means
Elfi Kraka, Marek Freindorf, and Dieter Cremer
CATCO, Department of Chemistry, Southern Methodist University, Dallas, TX 75275
http://smu.edu/catco
[email protected] Chiral discrimination of pairs of homo- and heterochiral molecules is a challenge for both experimentalists and computational chemists. Limited success has been accomplished by analyzing molecular properties such as chirodiastaltic energy, geometry, NMR magnetic shieldings or NMR spin-spin coupling constants. Vibrational spectroscopy is a sensitive tool for detecting H-bonding and therefore it should also distinguish H-bonding in pairs of homo- and heterochiral molecules.
The information contained in the vibrational spectra is difficult to decode, which complicates the discrimination of H-bonding in homo- and heterochiral molecules. However, the normal vibrational modes of a molecule provide all information needed to determine its local vibrational modes. The local vibrational modes, as first described by Konkoli and Cremer, lead to an accurate description of bonding, which can be exploited when distinguishing H-bonding in pairs of homo- and heterochiral molecules or to assess the role of H-bonding in chiral recognition.[1, 2]
1. Freindorf M, Kraka E, Cremer D. A Comprehensive Analysis of Hydrogen Bond Interactions Based on Local Vibrational Modes. Int. J. Quant. Chem., doi = 10.1002/qua.24118 2. Kraka E, Freindorf M, Cremer D. Chiral Discrimination by Vibrational Spectroscopy utilizing Local Modes. Chirality, in press
CH3OC
OCO
H H3C
H
C
O
H
H
H3C
C
OH
H
H3C
CH3OC
OCO
H
H3C
H
CH3
OCO
COH
CH3
H
C
OH
H
H3C
CH
OCO
COH
CH
HC
OH
H
H3CRS RR RS RR
S or R methyl-lactate, conf. 1
S or R methyl-lactate, co

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Examining the 4d transition metals and the lower p-block with a pseudopotential-based composite method: Atomic and
molecular applications of rp-ccCA Marie L. Laury and Angela K. Wilson
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, TX 76203
The correlation consistent composite approach (ccCA) has proven to be an effective first-principles-based composite approach for main group and first-row transition metal species. By combining relativistic pseudopotentials and ccCA, accurate energetic and thermodynamic data for heavier elements, including transition metals and lower p-block (5p and 6p) elements, are obtainable. Relativistic pseudopotential ccCA (rp-ccCA) is formulated and tested on a subset of the G3/05 set that contains 4p elements (Ga-Kr). The time savings and accuracy of the methodology, as compared to ccCA and ccCA-TM, is gauged before extending the method to the 4d transition metals and lower p-block elements. The TM-4d set, composed of 30 experimental enthalpies of formation, is employed for the second row transition metal study, while the LP80 set, composed of atomic ionization potentials and electron affinities and molecular dissociation energies and enthalpies of formation, is utilized for the lower p-block study. The accuracy and utility of rp-ccCA for energetic and thermodynamic studies of elements further down the periodic table will be demonstrated for 4d metals and lower p-block molecules.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
A simple DFT-based diagnostic for nondynamical correlation
Uma R. Fogueri,a Sebastian Kozuch,b Amir Karton,c and Jan M.L. Martina,b aDepartment of Chemistry and Center for Advanced Scientific Computing and Modeling
(CASCaM), University of North Texas, Denton, TX 76201
bDepartment of Organic Chemistry, Weizmann Institute of Science, 76100 Rehovot, Israel
cSchool of Chemistry, University of Sydney, Sydney, NSW 2006, Australia
We propose[1] a simple DFT-based diagnostic for nondynamical correlation effects, namely Aλ=(1–TAE[XλC]/TAE[XC])/λ where TAE stands for the molecular total atomization energy, XC is a pure-DFT exchange-correlation functional, and XλC represents the corresponding hybrid with 100λ % Hartree-Fock-type exchange. The diagnostic is a good predictor for sensitivity of energetics to the level of theory, unlike most wavefunction-based diagnostics. For GGA functionals, Aλ values approaching unity indicate severe nondynamical correlation, while values between 0 and about 0.1 indicate systems where correlation is predominantly dynamical in character (or entirely absent). The diagnostic is only weakly sensitive to the basis set (beyond polarized valence double zeta) and can easily be applied to problems beyond the practical reach of wavefunction ab initio methods required for other diagnostics. We also propose a simple measure for the importance of dynamic correlation.
1. U. R. Fogueri, S. Kozuch, A. Karton, and J. M. L. Martin, Theor. Chem. Acc., in press (2012)

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Exact quantum dynamics calculations using phase space wavelets
Bill Poirier and Tom Halverson
Department of Chemistry and Biochemistry, and Department of Physics, Texas Tech University, Lubbock TX 79409
[email protected] In a series of earlier papers [1-5], the authors introduced the first exact quantum dynamics method that defeats exponential scaling of CPU effort with system dimensionality. The method uses a “weylet” basis set (orthogonalized Weyl-Heisenberg wavelets), combined with a phase space truncation scheme first proposed by M. Davis and E. Heller [6]. Here [7], we use a related but simpler wavelet basis consisting of momentum-symmetrized phase space Gaussians. Despite being non-orthogonal, symmetrized Gaussians exhibit collectively locality, allowing for effective phase space truncation, and the defeat of exponential scaling. Application to both isotropic uncoupled harmonic oscillators and coupled anharmonic oscillators are discussed. Results for uncoupled systems up to 15 dimensions are compared with our previous weylet calculations, and found to be essentially just as efficient. For the coupled case, a “universal” and remarkably simple code has been written, which is dimensionally independent, and which also exploits massively parallel algorithms. Using the new codes, coupled and uncoupled calculations up to 27 dimensions and 2300 cores have been achieved. Lastly, symmetrized Gaussian calculations for coupled anharmonic oscillators are analyzed, and compared to first order degenerate perturbation theory.
Support from The Robert A. Welch Foundation (D-1523) and the National Science Foundation (CHE-0840493) are gratefully acknowledged, as is the Texas Tech University High Performance Computing Center, and the Texas Advanced Computing Center Lonestar facility.
1. R. Lombardini and B. Poirier, J. Chem. Phys. 124, 144107 (2006) 2. R. Lombardini and B. Poirier, Phys. Rev. E, 74, 36705 (2006) 3. B. Poirier, J. Theo. Comput. Chem. 2, 65 (2003) 4. B. Poirier and A. Salam, J. Chem. Phys. 121, 1690 (2004) 5. B. Poirier and A. Salam, J. Chem. Phys. 121, 1704 (2004) 6. M. J. Davis and E.J. Heller, J. Chem. Phys. 71, 3383 (1979) 7. T. Halverson and B. Poirier, J. Chem. Phys. (submitted).

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Halogen Bonds in Protein-Ligand Complexes
Kevin E. Riley
Department of Chemistry, Xavier University of Louisiana, New Orleans, LA 70125
Surveys of crystal structures from the Protein Data Bank have shown that halogen bonds may play a very important role in a great number of protein-ligand complexes, including native interactions involving (for example) hormones and interactions of pharmaceutical compounds with protein receptors. Very little is known, however, of the energetic contributions that halogen bonds make to stabilization of protein-ligand complexes. Obtaining such energetic information is somewhat complicated because, in general, force-field based methods and semiempirical QM methods fail to describe the anisotropic halogen charge distribution responsible for halogen bonding. Thus it is generally necessary to use full QM methods, such as DFT-D or MP2, to describe these interactions. In this laboratory we have developed a semiempirical method, PM6-DH2X, that can describe halogen bonding interactions at a qualitative level.
We have performed MP2/aug-cc-pVDZ binding energies on a series of halogen bonding systems derived from crystal structures of protein-ligand complexes. We analyze the difference between HF and MP2 binding energies in order to gain insight into the nature of the halogen bonding interaction (ie dominated by electrostatics or dispersion). It is found that the orientation of a halogen bond has a large effect on the strength and nature of the noncovalent interaction. The strongest halogen bonds have (gas phase) binding energies comparable to those of the hydrogen bond in the water dimer (~5.0 kcal/mol)

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Symmetry breaking and restoration
Gustavo E. Scuseria
Department of Chemistry and Department of Physics and Astronomy, Rice University, Houston, TX 77005
I will present our recent results on the calculation of symmetry-projected wave functions [1,2] for electronic structure theory. For Hartree-Fock and spin projection, this is a 50-year old problem in quantum chemistry, going all the way back to Löwdin and his “extended HF” theory. For Hartree-Fock-Bogoliubov and number projection, our approach offers new perspectives on Antisymmetrized Geminal Power (AGP) wavefunctions that were the focus of much attention in the 1980s. In our work, all molecular symmetries (electron number, spin S2 and Sz , point group, and complex conjugation) are deliberately broken and restored in a self-consistent variation-after-projection approach. The resulting method yields a comprehensive black-box treatment of static correlation with one-electron (mean-field) computational cost. The ensuing wave function is of high quality multireference character competitive with CASSCF. The method can also be applied to calculate excited states and spectral functions [3]. The curse of the thermodynamic limit and the quest for a low-cost treatment of residual correlations will also be addressed. 1. G. E. Scuseria, C. A. Jimenez-Hoyos, T. M. Henderson, J. K. Ellis, and K. Samanta, J. Chem. Phys. 135, 124108 (2011). 2. C. A. Jimenez-Hoyos, T. M. Henderson, and G. E. Scuseria, J. Chem. Phys. 136, 164109 (2012). 3. R. Rodrıguez-Guzman, K. W. Schmid, C. A. Jimenez-Hoyos, and G. E. Scuseria, Phys. Rev. B 85, 245130 (2012).

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Nanoplasmonics: Sensing and energy applications
Kevin L. Shuford
Department of Chemistry and Biochemistry, Baylor University, Waco, TX 76798
Noble metals have loosely bound electrons that collectively oscillate when excited by an electromagnetic field. These plasmon resonances reside in the visible and infrared portions of the electromagnetic spectrum for silver and gold nanoparticles. At the resonance frequencies, the nanoparticle is highly polarizable, and the oscillating charge density results in a strongly enhanced field near the surface. The large field enhancements and the ability to selectively tune the resonance wavelength via particle morphology make plasmonic nanostructures ideal for many sensing and energy applications. This talk will briefly introduce plasmonics and common methods used to calculate the optical properties of nanostructures. We will then examine some of the more subtle aspects of how nanoparticles couple to each other as well as to molecules, and how the nature of the coupling can affect the properties of the system as a whole. Specific examples include nanostructures that utilize surface enhanced Raman spectroscopy for sensing, spontaneous emission of molecules near nanoparticles, and plasmon enhanced photovoltaic devices. All of these examples highlight the importance of understanding the interplay between system components in sensing and energy applications based on plasmonics.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Characterizing the excess electron of Li(NH3)4
Thomas Sommerfeld and Katelyn M. Dreux
Department of Chemistry and Physics, Southeastern Louisiana University, Hammond, LA 70402
Small lithium-ammonium clusters are model systems for the dissociation of metals into solvated cations and electrons. Here the smallest cluster with a complete solvation shell is used to investigate the seemingly conflicting findings that solvated electrons in metal-ammonia systems form, on the one hand, cavities showing essentially Rydberg character, but exhibit, on the other hand, large spin densities on the N atoms, which is more in-line with a localized radical character.
The first question investigated is whether different theoretical characterizations of the “excess electron” lead to different conclusions about it. Fairly small differences are found between orbital-based and spin density-based approaches as well as between SCF and coupled-cluster-based method. Natural orbitals from EA-EOM-CCSD calculations are then used to analyze the excess electron’s distribution in Li(NH3)4 with particular emphasis on the portion of the density closely associated with the N atoms. It is found that only 6% of the excess electron are closely associated with any of the atoms, with about 1% being closely associated with an individual N atom, which clearly demonstrates the Rydberg character. It is also shown that the spin density at the N nuclei is nevertheless substantial, and the apparent conflict is resolved by showing how the magnetic observations used to support a localized-radical picture can be explained in terms of the cavity model.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Water hydrogen bonding behavior of alkali-halide solutions at a graphite interface
Matthew C. F. Wander and Kevin L. Shuford
Department of Chemistry and Biochemistry, Baylor University, Waco TX 76701
In this paper, a variety of alkali halide, aqueous electrolyte solutions in contact with charged, planar-graphite slit-pores simulations using classical molecular dynamics are discussed. Size trends in structure and transport properties are examined by varying the choice of ions among the alkali metal and halide series. As with the uncharged pores, system dynamics are driven by changes in water hydration behavior and specifically by variations in the number of hydrogen bonds per water molecule. Size trends in structure and transport properties are examined by varying the choice of ions. The intermediate atomic weight ions within each group are found to diffuse faster than the larger or smaller ions. System dynamics are driven by changes in water hydration behavior and, specifically, by variations in the number of hydrogen bonds per water molecule. However, the larger ions diffuse more rapidly under high surface charge conditions than the smaller ions. In particular, for the 1 nm slit, ion diffusivity increased by a factor of 4 compared to the uncharged case.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Predicting the melting temperature of ice-Ih with only electronic structure information
Feng Wang
Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville, AR 72701
[email protected] The melting temperature of ice-Ih was calculated with only electronic structure information as input by creating a problem-specific force field with the adaptive force matching (AFM) method. The force field, WAIL, was created by fitting to post-Hartree-Fock quality forces obtained with a Quantum Mechanics and Molecular Mechanics (QM/MM) scheme. WAIL utilizes simple energy expressions similar to TIP4P and predicts a melting temperature of ice-Ih very close to the experimental value. The model also predicts the densities of ice and water, the temperature of maximum density of water, the heat of vaporizations, and the radial distribution functions for both ice and water in good agreement with experiments. Without fitting to any experimental property, WAIL outperforms all existing water potentials for describing ice and water near equilibrium.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
The many phosphoryl transfers of Phospholipase D superfamily: a quantum mechanical theoretical study
Charles Edwin Webster and Nathan J. DeYonker
Department of Chemistry, The University of Memphis, Memphis, TN 38152
Members of the HKD Phospholipase D (PLD) superfamily catalyze the hydrolysis of the P-O bond of phosphatidylcholine to form phosphatidic acid (PA) and choline. PA is the essential precursor for the biosynthesis of many other lipids, plays a role in the modulation of membrane curvature, and acts as a signaling lipid with activity in cell survival and apoptosis. Previously reported experimental in vitro studies with Streptomyces sp. Strain PMF and a model substrate (1,2-dibutyrylphosphatidylcholine) had a short-lived five-coordinate phosphohistidine intermediate and a “dead-end” four-coordinate phosphohistidine species. More recently, in experimental studies with Tyrosyl-DNA phoshodieasterase I a four-coordinate phosphohistidine species has also been isolated. In the current computational study, the mechanism of PLD is studied using a fully quantum mechanical, geometrically constrained enzyme model. Density Functional Theory (DFT) is used to acquire thermodynamic and kinetic data for 839 unique stationary points (including 255 transition states) to map out forty-eight unique mechanisms along the catalytic pathway, including the initial phosphoryl transfer, subsequent hydrolysis, and finally, the formation of the experimentally-observed "dead-end" phosphohistidine product. The persistence of the short-lived five-coordinate phosphorane intermediate is explained.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Multireference Wavefunction Composite Strategies
Wanyi Jiang and Angela K. Wilson
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, TX 76203
The ab initio correlation consistent Composite Approach (ccCA) is a thermochemical model devised to achieve energetics (i.e. enthalpies of formation, ionization potentials, proton affinities) akin to those obtained using electronic correlation methods such as CCSD(T) at the complete basis set limit, but as much greater computational efficiency. While the approach has proven to be of great utility, the prediction of features of potential energy surfaces, bond formation and bonding breaking, and excited states can warrant a multireference wavefunction-based approach. We describe developments towards the description of nondynamical electron correlation, via multireference composite strategies (MR-ccCA), efficient alternatives to such approaches, and diagnostics for the potential need for multireference wave function-based treatment.

Oral Presentations
2012 Southwest Theoretical Chemistry Conference
Aspects of Complex Scaling for Electron-Atom/Molecule Resonances using MCSCF, MCSTEP and MCTDHF
Danny L Yeager
Department of Chemistry,Texas A&M University, College Station, TX 77843
Electron-atom/molecule resonances are temporarily bound states which lie in the continuum part of the Hamiltonian.
If the electronic coordinates of the Hamiltonian are scaled (“dilated”) by a complex parameter, η = αexp(iθ) (α, θ real), then its complex eigenvalues represent the scattering states (resonant and non-resonant) while the eigenvalues corresponding to the bound states and the ionization and the excitation thresholds remain real and unmodified. These make the study of these transient species amenable to the bound state methods.
We developed a quadratically convergent multiconfigurational self-consistent field method (MCSCF), a well-established bound-state technique, combined with a dilated Hamiltonian to investigate resonances. This is made possible by the adoption of a second quantization algebra suitable for a set of “biorthogonal” spin orbitals and a modified step-length constraining algorithm to control the walk on the complex energy hypersurface while searching for the stationary point using a multidimensional Newton-Raphson scheme.
I will outline our latest developments and results with the complex MCSCF (CMCSCF), the complex multiconfigurational spin tensor electron propagator (CMCSTEP) and the complex multiconfigurational time dependent Hartree-Fock (CMCTDHF) methods.

Abstracts for Poster Presentations

Poster Presentations Poster 1
2012 Southwest Theoretical Chemistry Conference
Origin of Sequence Selectivity in the Anti-ssDNA Autoantibody ED-10: Role of Hydrogen-Bonding, XH/π and π-Stacking
Interactions
Yi An, Rajesh K. Raju, and Steven E. Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77842
[email protected] We present a detailed computational analysis of the binding of four dinucleotides to a highly-sequence selective single-stranded DNA (ss-DNA) binding antibody (ED-10) and selected point-mutants. Anti-DNA antibodies are central to the pathogenesis of systemic lupus erythematosus (SLE), and a detailed understanding of the mode of binding of DNA and other ligands will be necessary to elucidate the role of anti-DNA Abs in the kidney inflammation associated with SLE. Classical molecular mechanics based molecular dynamics (MD) simulations and density functional theory (DFT) computations were applied to investigate the origin of selectivity for the 5’ nucleotide. In particular, the strength of interactions between each nucleotide and surrounding residues were computed using MM/GBSA as well as DFT applied to a cluster model of the binding site. The results agree qualitatively with experimental binding free energies and indicate that stacking, CH/π, NH/π, and hydrogen bonding interactions all contribute to the sequence selectivity of ED-10. Finally, MD simulations and DFT computations were also performed to quantify how the 5’ nucleotide binding enthalpy changes as TrpH50 of ED-10 is mutated to AlaH50, PheH50 and TyrH50, and hence shed light on the role of TrpH50 in sequence selectivity. DFT results show that these mutations induce changes in hydrogen bonding interactions with other binding site residues.
Figure 1. Nucleotide binding sites in ED-10. The dinucleotide is colored in yellow; the aromatic amino acids are colored in green; and all the other residues are colored in pink.

Poster Presentations Poster 2
2012 Southwest Theoretical Chemistry Conference
Racing carbon atoms. Atomic motion reaction coordinates and structural effects on newtonian kinetic isotope effects
Ivonne L. Andujar-De Sanctis and Daniel A. Singleton
Department of Chemistry, Texas A&M University, College Station, TX 77842
Our group has recently described a new form of kinetic isotope effect that has its origin in classical mechanics rather than in quantum mechanical phenomena. These "Newtonian kinetic isotope effects" arise from the greater acceleration of light masses in response to a constant force, leading to a selective formation of products from greater motion of the lighter of two competing isotopes. We describe here a second system, in the thermal dimerization of methacrolein, where this novel form of kinetic isotope effects has an impact on the product selectivity. Each 13C-containing isotopomer of methacrolein can lead to the formation of two isotopomeric cycloadducts, and their ratio constitutes an intramolecular KIE that reflects the selectivity among trajectory outcomes on the “bifurcating energy surface” of the reaction. The intramolecular KIEs show a preference for 12C at the newly formed sigma bonds, consistent with a greater acceleration of light masses in response to a constant force as atoms cross the transition state. Experimental results on the selectivity between the two isotopomeric products cannot be explained based on the transition state theory because the pair of products arises from a single transition state. Nonetheless, trajectories are in agreement with experimental observations and support the influence of classical physics and dynamic factors in the selectivity of this reaction. Atomic motion reaction coordinate diagrams, represented below, provide insight into both the direction of the isotope effects and their magnitude.

Poster Presentations Poster 3
2012 Southwest Theoretical Chemistry Conference
Understanding the physical nature of substituent effects in XH/π interactions
Jacob W. G. Bloom, Rajesh K. Raju, and Steven E. Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77843
XH/π interactions (e.g.: CH/π, OH/π, etc.) are found throughout chemical and biochemical contexts. Many prior studies exist, but are limited in the selection of substituents or selection of XH group. Herein, we address this lack of studies on a wide array of substituents and XH groups by studying model BH/π, CH/π, NH/π, OH/π, and FH/π interactions (e.g.: BH3···C6H5Y, CH4···C6H5Y, etc.) with 22 monosubstituted benzenes. Estimated CCSD(T)/aug-cc-pVTZ interaction energies as well as symmetry-adapted perturbation theory (SAPT) results were used to elucidate the underlying physics behind these interactions. It is shown that the interaction becomes stronger with increasing polarization of the XH bond. In fact, the nonpolar BH/π and CH/π are driven by dispersion, while electrostatic effects become increasingly important for the increasingly polar NH/π, OH/π, and FH/π. Additionally, for aryl substituent effects, Hammett constants (σm) correlate well with the electrostatic portion of the interactions and both molar refractivities (MR) and interaction distances are needed to capture the dispersion component.[1]
1. J. W. G. Bloom, R. K. Raju, and S. E. Wheeler, J. Chem. Theory Comput. 8, 3167 (2012).

Poster Presentations Poster 4
2012 Southwest Theoretical Chemistry Conference
Quantum dynamical kinetic study of H atom transfer via Lengevin dynamics and inverse classical control
Paul Brown,a Kevin Shuford,a and Michael Messinab aDepartment of Chemistry and Biochemistry, Baylor University, Waco, TX 76798
b Department of Chemistry and Biochemistry, University of North Carolina at Wilmington, Wilmington NC 28403
In this study we further advance recent research concerning enzymatic H atom transfer coupled to innumerable degrees of freedom comprising the surrounding enzyme active-site, and apply nonlinear optimal control theory to assess the influence of various enzymatic modulations. The low dimensional model studied herein presents a Quantum Generalized Langevin equation (QGLE) and explores the necessity of vibrational relaxation. We show that a novel quantum mechanical friction operator can be applied to the general quantum theory of H atom transfer, which is formulated for a simple vibrational Gaussian wavepacket (GWP). This generalization is applied to single and two-state non-adiabatic harmonic and anharmonic quantum dynamics. As we show the application of the friction operator reproduces results observed earlier in the research of quantum dissipation for H atom transfer. Furthermore, we explore the question of how enzymatic H atom transfer takes place under the influence of donor-acceptor modulation via Inverse Classical Control (ICC). We also incorporate the QGLE into the dynamics of H atom transfer for various a priori tracks to observe enhanced transfer of H atom reactant probability density.

Poster Presentations Poster 5
2012 Southwest Theoretical Chemistry Conference
Computed molecular frame photoelectron angular distributions of valence ionization of ground-state nitromethane
Ralph Carey and Robert R. Lucchese
Department of Chemistry, Texas A&M University, College Station TX 77843
The electron and photon absorption spectrum and fragmentation pathways of the smallest nitroalkane, nitromethane (CH3NO2), have been investigated extensively through the years [Kilic et al., J. Phys. Chem. A 101, 817 (1997)], yet until recently there have been few known experimental or computational photoelectron angular distributions available for this energetic molecule.
Electron angular distributions resulting from hν = 400 nm = 3.1 eV multiphoton absorption of CH3NO2 leading to the production of ionic fragments CH3
+, NO2+, and e3
– show that photoelectron density is strongly directed towards the methyl fragment [Vredenborg et al., Chem. Phys. Chem. 12 1459 (2011)]. From the total energy E = 15.5 eV stemming from n = 5 absorbed photons, the internal energy of the ionic fragments, and the Ek = 0.56 kinetic energy of the photoelectron, Vredenborg et al. propose that the distributions result from excitations from the (9a′)–1 shell, which has an ionization energy near EIP = 14.7 eV,
We have computed photoionization cross sections in the (X1 A′) electronic ground state of Cs symmetry nitromethane from the HOMO through HOMO-4 valence shells (6a′′)2(10a′)2(5a′′)2(9a′)2(4a′′)2 from the respective ionization thresholds to a photoelectron kinetic energy of Ek = 20 eV. These calculations were performed with EPOLYSCAT within the frozen-core Hartree-Fock approximation [Gianturco et al., J. Chem. Phys. 100, 6464, Natalense and Lucchese, J. Chem. Phys. 111, 5344 (1999)]. We have also generated photoelectron angular distributions in the recoil (RFPAD) and the molecular frame (MFPAD) for Ek = 0.5 eV photoelectrons with a recoil axis consisting of the CN bond for each valence excitation. The computed RFPADs and MFPADs make use of linearly polarized light. We find that the parallel transition HOMO-1 (10a′)–1 Ek = 0.5 eV angular distribution matches those of Vredenborg et al. but from a higher-energy valence orbital. These results illustrate the breakdown of Koopmans approximation for valence ionization due to orbital relaxation on ionization, as is evident in the discrepancy between the computed SCF MO energies and the He(I) PES assignments of experiment [Chin et al, J. Electron Spectrosc. Relat. Phenom. 60, 101 (1992)].

Poster Presentations Poster 6
2012 Southwest Theoretical Chemistry Conference
Equilibrium geometry determination by ccCA energy gradients
Matthew J. Carlson, Wanyi Jiang, and Angela K. Wilson
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, TX 76203
The correlation consistent Composite Approach (ccCA) has been shown to predict energetic data such as enthalpies of formation, ionization energies, and electron affinities within 1 kcal/mol, on average, from experiment for main group species, and at a reduced computational cost as compared to CCSD(T) with a large basis set. We have investigated the use of ccCA energy gradients to optimize equilibrium geometries of a set of closed-shell molecules. Within ccCA, several different schemes have been considered for extrapolation of properties (energies, and now gradients) to the complete basis set (CBS) limit. Of these schemes, the Peterson extrapolation has been determined to provide equilibrium geometries that most closely match experimental data. In order to reduce computation time, the scalar relativistic correction was not included in the gradient extrapolation, with an insignificant effect on the quality of results. The structures determined using the ccCA gradient-based optimization are in close agreement with experimental data, with a mean absolute deviation (MAD) of 0.10 picometers for bond lengths and 0.2 degrees for bond angles for the set of molecules considered.

Poster Presentations Poster 7
2012 Southwest Theoretical Chemistry Conference
Dynamically complex enzyme-catalyzed [6+4] and [4+2] cycloadditions in the biosynthesis of spinosyn A
Zhuo Chen and Daniel Singleton
Department of Chemistry, Texas A&M University, College Station, TX 77842
A recent discovery of a key transannular Diels-Alder reaction in the biosynthesis of spinosyn A has promoted extensive studies towards its mechanism. The traditional definition of a Diels-Alder reaction does not take into account of a number of complications in the mechanism that have come into light in recent years, especially with respect to the dynamics effects on the rate and selectivity. We present here the importance of such complications in the transannular Diels-Alder reaction catalyzed by SpnF, including the role of a bispericyclic transition state, a bifurcating energy surface, a dynamically stepwise cycloaddition, an entropically-delineated intermediate, transition state recrossing and a competing [6+4] cycloaddition in the mechanism. This reaction is not its caricature from classical mechanistic analysis and it is not well described by either concerted or stepwise labels. Instead, the mechanism is richer and can only be understood with consideration of dynamics.

Poster Presentations Poster 8
2012 Southwest Theoretical Chemistry Conference
Polypeptides in alpha-helix conformation perform as diodes
Dahiyana Cristancho and Jorge Seminario
Department of Chemical Engineering, Texas A&M University, College Station, TX 77843
Molecules that resemble a semiconductor diode depletion zone are those with an intrinsic electric dipole, which were suggested as potential electronic devices. However, so far, no single molecule has met such a goal because any electron donor-acceptor linker strongly diminishes any possibility of diode behavior. We find an intrinsic diode behavior in polypeptides such as poly(L-alanine) and polyglycine in alpha-helix conformation, explained in terms of molecular orbital theory using ab initio methods. The application of an antiparallel electric field with respect to the molecular dipole yields a gradual increase in current through the junction because the valence and conduction orbitals approach each other reducing their gap as the bias increases. However, a parallel field makes the gap energy increase, avoiding the pass of the electrons.

Poster Presentations Poster 9
2012 Southwest Theoretical Chemistry Conference
A QTAIM investigation of the anomeric effect
Jordan Dodson and Preston MacDougall
Department of Chemistry, Middle Tennessee State University, Murfreesboro, TN 37132
The anomeric effect can be defined as the preference of an electronegative substituent at the anomeric carbon to favor the axial rather than the equatorial position. Extending previous work on the generalized anomeric effect, which also includes acyclic systems, we show that the alignment of critical points (CPs) in the Laplacian of the electronic charge density, , is closely linked to the stability of compounds exhibiting the anomeric effect. Whereas past investigations of this type have focused on the magnitude of these CPs, we find that their alignment and separation is a very consistent indicator of all anomeric interactions. Also, using the theory of atoms in molecules, we show in several species exhibiting the anomeric effect that the atomic energy of the anomeric carbon is strongly correlated to the molecular energy. This atomic-molecular energy trend is unique to the anomeric carbon.
Figure 1. An instance of CP alignment as seen down the exocyclic C-O bond in 2-methoxytetrahydrofuran. The spheres nearest to the atom labeled O6 are (3,+3) CPs around oxygen, and the smaller spheres just beyond these are (3,-1) CPs around carbon, which is hidden by oxygen. The envelope around the atoms is a zero-value isosurface of .
Financial support is acknowledged from the Office of Science, U.S. Department of Energy.

Poster Presentations Poster 10
2012 Southwest Theoretical Chemistry Conference
Developing density functional approximations for noncovalent interactions
Jessie Girgisa and Benjamin Janesko,b aDepartment of Physics, University of Dallas, Irving, TX 75062
[email protected] bDepartment of Chemistry, Texas Christian University, Fort Worth, TX 76129
Density functional theory (DFT) is a method used to approximate molecular electronic structure. Weak dispersion (van der Waals) interactions are important in many chemical processes, and not well approximated by standard DFT approximations. These chemical processes can be treated by adding an empirical dispersion correction to standard DFT methods. We tested an empirical dispersion correction for some standard DFT methods, and for new 'Rung 3.5' DFT approximations. We used benchmark calculations on noble gas dimers to fit an adjustable parameter in the dispersion correction, as well as parameters in the 'Rung 3.5' functionals. We then tested the parameterized methods for standard sets of accurately known gas-phase small molecule interaction energies (hydrogen bonds, charge-transfer, and so on). We conclude that dispersion corrected 'Rung 3.5' functionals are as accurate as standard dispersion-corrected DFT for weak interactions.

Poster Presentations Poster 11
2012 Southwest Theoretical Chemistry Conference
Singlet and triplet energy differences for bpyNi(II) complexes
Hector E. Gonzalez and Thomas R. Cundari
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, TX 76203
A study of catalytic hydroarylation using bpyPt(II) complexes was expanded to include Ni(II)-based catalytic systems. Singlet and triplet states were modeled to determine the ground state of the Ni stationary points. As there were a couple of points on the free energy surface that showed < 5 kcal/mol difference for the two spin states, higher correlation methods, e.g., CCSD(T), were used to corroborate the B3LYP numbers.
NiN
N
NiN
N
NiN
N
H
NiN
N
NiN
N
+
+
+
+
+
NiN
N
+
NiN
N
+

Poster Presentations Poster 12
2012 Southwest Theoretical Chemistry Conference
Empirical correction of DFT for non-dynamical correlation
Chris Jeffrey, Wanyi Jiang, and Angela K. Wilson
Department of Chemistry and Center for Advanced Scientific sComputing and Modeling (CASCaM), University of North Texas Denton, TX 76203
Density functional theory (DFT) has become popular due to its low computational cost, relative to wavefunction-based methods, as well as its predictive abilities for a wide range of chemical systems and properties. Despite the successes in accounting for dynamical correlation, DFT is unable to reliably describe systems with a significant amount of non-dynamical correlation. Such systems typically require multi-reference wavefunction-based treatment, which can become quite computationally costly.

Poster Presentations Poster 13
2012 Southwest Theoretical Chemistry Conference
Amine scrubbers: Substituent effects on CO2 interactions
Kameron R. Jorgensen,a Thomas R. Cundari,b and Angela K. Wilsonb
aDepartment of Biology and Chemistry, Texas A&M International University, Laredo, TX 78041
[email protected] bDepartment of Chemistry and Center for Advanced Scientific Computing and Modeling
(CASCaM), University of North Texas, Denton, Texas 76203
Organic amine compounds (e.g., monoethanolamine and diethanolamine) are being used for the capture of CO2 from flue gas; however, much energy is required for the regeneration of these amine compounds via the removal of CO2. To aid in the enhancement of amine carbon capture agents the nature of the CO2 interactions with amine compounds were investigated on a molecular level. Effects of the addition of methyl, silyl, and trifluoromethyl substituent groups on the amine compound were considered. Interaction energies of CO2 with amine complexes were calculated via two methods: a) an ab initio composite method, the correlation consistent Composite Approach (ccCA); and b) density functional theory, B3LYP/aug-cc-pVTZ and B97D/aug-cc-pVTZ. The calculations suggested two different binding modes, hydrogen-bonding and acid-base interactions, which arise from the modification of the amine substituents, echoing previous work by our group on modeling of protein•CO2 interactions. Recommendations have been noted for the development of improved amine scrubber complexes.
CO2•NH2CH3 CO2•NH2SiH3 CO2•NH2CF3

Poster Presentations Poster 14
2012 Southwest Theoretical Chemistry Conference
Valence shell photoionization of SF6 and high harmonic generation
J. Jose and R. R. Lucchese
Department of Chemistry, Texas A&M University, College Station, TX 77842
When an atom/molecule is exposed to highly intense laser fields, the target can emit coherent radiation at photon energies which are multiples of incident laser energy. This process is known as High-order harmonic generation (HHG). There has been experimental and theoretical investigation of HHG for atoms. However, HHG has been few such studies for molecules.
In the current work, we investigate HHG for SF6 theoretically. We employ quantitative rescattering theory (QRS) which makes use of the magnitude phase of the dipole transition matrix elements for photoionization. For calculating dipole transition matrix elements we employ the iterative Schwinger variational method. The main features of the QRS calculation result for HHG signal are (1) a minimum centered around 20th harmonic and (2) another shallow minimum centered on 50th harmonic. Current results are compared with available experimental result and qualitative agreement between both is found. The calculation is repeated for different polarization of incident laser and different intensities. The analysis helps us to find the HHG characteristics of SF6.

Poster Presentations Poster 15
2012 Southwest Theoretical Chemistry Conference
Theoretical study of the reactivity of Ir(II) sulfides, Ir2(μ-S)2(PPh3)4 and Ir2(μ-S)2(PH3)4
Stella Kritikou and Michael B. Hall
Department of Chemistry, Texas A&M University, College Station, TX 77843
Soluble metal sulfido complexes react with H2, forming SH ligands. These reactions have previously been investigated by Rauchfuss et al.[1] In 2001 Rauchfuss reported an unusual Iridium (II) sulfide species, Ir2(μ-S)2(PPh3)4, that undergoes both homolytic and heterolytic addition of two hydrogen equivalents. We started investigating the activation of this Ir(II) complex, and of the corresponding model complex Ir2(μ-S)2(PH3)4, in order to determine the details of this pathway. Density functional theory (DFT) calculations, implementing the ωB97XD functional with Pople type basis sets and effective core potentials were used to predict the potential energy surface as well as possible alternative products of these reactions.
1. R. C. Linck, R. J. Pafford, T. B. Rauchfuss, J. Am. Chem. Soc. 123, 8856-8857 (2001).

Poster Presentations Poster 16
2012 Southwest Theoretical Chemistry Conference
Comparison of atomic and electronic environment of the rhodopsin retinal binding before and after isomerization
Carlos Kubli-Garfias,a Blanca M. Cabrera-Vivas,b Juan Carlos Ramírez,b
Flor P. Pineda,b and Ricardo Vázquez-Ramíreza
aInstituto de Investigaciones Biomédicas, UNAM, México D.F.
[email protected] bFacultad de Ciencias Químicas, BUAP, Puebla MEXICO
Retinal the natural ligand of rhodopsin changes its conformation from cis to trans by photon energy. The isomerization reaction has been studied extensively. However, a comparison of the cis pre-isomerization state and the trans state may help to understand the post-isomerization visual cycle of the retinal-opsin complex. Atomic and molecular properties of both retinal conformations were analyzed starting with the orbital contribution of each atom to the frontier orbitals. DFT calculations using the time-dependent (TD) method, were applied to analyze the electronic transitions. The N-atom from Lys305 was conserved as part of the chromophore. Other properties such as: HOMO, LUMO, ionization potential, electronic affinity, hardness, softness, polarizability, atomic charges, electrostatic potential, dipole moment, solvation energy, area and volume were calculated. The squid rhodopsin crystals: PDB ID 2Z73 and 3AYM were retrieved and the free energy of retinal bound to 14 residues enveloping the ligand in a hydrophobic environment at 4 Å of distance was calculated. The cavity occupied by the trans ligand is larger than that occupied by the cis isomer, and its free energy is lower by 42.11 kJ/mol. Figure 1 shows important atomic contributions to the transition molecular orbitals.
Figure 1. Orbital atomic populations of cis and trans-retinal. Carbon atomic orbitals participate alternatively in both conformations of retinal.
Based on those data it may be concluded that the clear differences between cis and all-trans conformers of retinal may help to gain some insight in the bleaching process and retinal metabolism in the visual cycle, as well as, the transduction process of vision.

Poster Presentations Poster 17
2012 Southwest Theoretical Chemistry Conference
What controls the regioselectivity in rhodium catalyzed hydroformylation of unsaturated substrates? DFT computations
offer unprecedented mechanistic insight Manoj Kumar,a,b Bibhas Sarkar,b Raghunath V. Chaudhari,b and Timothy A. Jackson,a,b
aDepartment of Chemistry, University of Kansas, Lawrence, KS 66045
[email protected] bCenter for Environmentally Beneficial Catalysis, University of Kansas, Lawrence, KS
66045
Rhodium (Rh) catalyzed hydroformylation of alkenes, first developed by Wilkinson and later implemented by Union Carbide (now Dow Chemicals), is one of the most important applications of homogenous catalysis as far as the industrial production of aldehydes and alcohols is concerned. Despite being discovered about 70 years ago, this catalytic process continues to remain the focal point of experimental and theoretical investigations. This is mainly attributed to the fact that the origin of regioselectivity is not well understood i.e., a comprehensive understanding of the factors controlling the linear/branched ratio of aldehyde remains elusive. Herein we have performed DFT calculations to reinvestigate the issue while covering a broad range of substrates and different ligand structures of Rh catalyst. The calculations suggest that the linear aldehyde is predominantly produced in the case of simple linear alkenes (e.g., propylene) while the branched is the dominant product in the case of heteroatom or aromatic substituted substrates. The analysis of the transition state structures explains this differential regioselective behavior: the transition state leading to the linear aldehyde in the case of simple alkenes is better stabilized due to the dispersion containing aromatic-aromatic (ligand-ligand) and aromatic-CH (ligand-substrate) interactions that are either absent or significantly weakened in the corresponding transition state of the branched aldehyde. However, the transition state leading to the branched isomer is better stabilized in the case of heteroatom or aromatic substituted substrates due to the enhanced ligand-substrate interactions. These computations suggest that, for the series explored herein, it is the steric crowding-induced ligand-ligand and ligand-substrate interactions, but not intraligand interactions that determine the regioselectivity in Rh catalyzed hydroformylation.

Poster Presentations Poster 18
2012 Southwest Theoretical Chemistry Conference
Ab initio analysis of the interactions of hydrolyzed GaN clusters with atomic-layer-deposited reactants
Paola A. León-Plata,a Mary R. Coan,a and Jorge M. Seminario,a,b,c aDepartment of Chemical Engineering, Texas A&M University, College Station, TX 77842
[email protected] bMaterials Science and Engineering Graduate Program
cDepartment of Electrical and Computer Engineering, Texas A&M University, College Station, TX 77842
It is been calculated and analyzed the interactions of two atomic layer deposition (ALD) reactants, trimethylaluminium (TMA) and tertrakis(ethylmethylamino) hafnium (TEMAH) with hydrolyzed Ga-face of GaN clusters, which could be used as testbeds for the actual Ga-face on GaN crystals of importance in electronics. However, our additional goal is the analysis of the nanoclusters for several other applications in nanotechnology. Our results show that trimethylaluminium spontaneously interacts with hydrolyzed GaN; however it does not follow the atomic layer deposition reaction path unless there is an increase in potential energy by increasing the lengths of various bonds in the structure. Tetrakis (ethylmethylamino) hafnium does not interact with hydrolyzed GaN unless the potential energy of the structure is increased at which point the reaction follows the atomic layer deposition path. The formation of a Ga—N(CH3)(CH2CH3) bond during the atomic layer deposition of HfO2 using tetrakis (ethylmethylamino) hafnium as the reactant without breaking the Hf—N bond could be the key part of the mechanism behind the formation of an interface layer at the HfO2/GaN interface.
Figure 1. Schematic of the Atomic Layer Deposition of trimethylaluminium (TMA) and tertrakis(ethylmethylamino) hafnium (TEMAH) on GaN surface

Poster Presentations Poster 19
2012 Southwest Theoretical Chemistry Conference
Computational insight into the selective amide or imine formation from alcohol and amine catalyzed by the pincer ruthenium(II)-
complexes Haixia Li,a Zhi-Xiang Wang,b and Michael B. Halla
Department of Chemistry, Texas A&M University, College Station, TX 77843
College of Chemistry and Chemical Engineering, Graduate University of the Chinese Academy of Sciences, Beijing 100049, People’s Republic of China
Amides and imines are important organic compounds that are used both in laboratory synthesis and industrial production. Thus, the development of efficient, environmentally benign, and atom-economical synthesis of them is well warranted. Along these lines of thinking, Milstein and coworkers have made significant progress in synthesizing them directly from primary alcohols and amines by using the pincer RuII-complexes. Amides were produced by using the RuII-PNN [PNN = 2-(di-tert-butylphosphanylmethyl)-6-(diethylaminomethyl)pyridine] as a catalyst. Interestingly, in contrast to the amide production, the reaction of primary alcohol and amine, catalyzed by the analogous pincer RuII-PNP [PNP = 2,6-bis(di-tert-butylphosphanylmethyl)pyridine] resulted in imine formation. We have used density functional theory computations to investigate the mechanism, and explicated the essential difference between the RuII-PNP-catalyzed imine synthesis and the RuII-PNN-catalyzed amide formation. The catalytic cycle includes four stages: (stage I) alcohol dehydrogenation to aldehyde, (stage II) coupling of aldehyde with amine to form hemiaminal, (stage III) hemiaminal dehydrogenation to afford amide or dehydration to give imine, and (stage IV) catalyst regeneration by means of H2 elimination of the trans ruthenium dihydride complex produced in stage I. The selectivity of products is determined by stage III: the RuII-PNN-catalyzed amide formation prefers hemiaminal dehydrogenation whereas the RuII-PNP-catalyzed imine synthesis prefers hemiaminal dehydration.

Poster Presentations Poster 20
2012 Southwest Theoretical Chemistry Conference
Transition metal catalyst mediated C-O bond cleavage of the β-O-4 linkages of lignin
Cong Liu and Angela K. Wilson
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, TX 76203
Lignin degradation is one of the most significant challenges that must be addressed to achieve the full potential of lignocellulosic bio-fuels as fossil fuel replacements. To aid in addressing this challenge, the C-O bond activation of the β-O-4 linkage of lignin using transition metal catalysts was investigated. Late 3d and 4d transition metal ion (Fe, Co, Ni, Cu, Ru, Rh, Pd, and Ag) mediated activation of dimethyl ether was considered in order to investigate the intrinsic catalytic properties of metals on organic C-O bond cleavage. A set of density functional (DFT) methods (BLYP, B3LYP, M06, M06-L, B97-1, B97-D, TPSS, and PBE) with aug-cc-pVTZ basis sets were utilized, as was CCSD(T)/CBS, which provided a means of calibrating the DFT calculations. Reaction barriers and energies were determined for possible catalytic pathways. Based on demonstrated thermodynamic favorability, group VIII metals, Fe, Ru, and Os, with “pincer” type ligands were chosen for the catalysis of the C-O bond activation of β-O-4 linkage model compound (as shown in the picture). B3LYP/ CEP-31-G(d) was used for geometry optimizations and energy calculations.

Poster Presentations Poster 21
2012 Southwest Theoretical Chemistry Conference
The complex scaled multiconfigurational spin tensor electron propagator method (MCSTEP) for studying resonant states
Liyuan Liang and Danny L. Yeager
Department of Chemistry, Texas A&M University, College Station, TX 77843
We have recently developed and coded the complex scaled multiconfigurational spin tensor electron propagator (CMCSTEP) method, which is constructed from all the five blocks of the M matrix and uses a CMCSCF state as the initial state, to study resonant states. In real space MCSTEP can produce the electron affinities (EAs) or ionization potentials (IPs) with high accuracy. With the CMCSTEP method, shape and Feshbach resonances of an N-electron system can be obtained with the trajectory method. The CMCSTEP method is applied to study the 2P Be- shape resonances arising from the scattering of low-energy electrons off a neutral Be target. Resonant parameters are obtained, and the results are compared with that obtained by other methods.

Poster Presentations Poster 22
2012 Southwest Theoretical Chemistry Conference
Theoretical study of vibrationally mode-specific photoionization cross sections of acrolein
Jesús A. López-Domínguez and Robert R. Lucchese a Department of Chemistry, Texas A&M University, College Station, TX 77842
In the present work, mode specific photoionization cross sections were calculated for acrolein in the low energy regime. From the total cross section for the 14a’ orbital, a resonant peak is evident near threshold (≈ 1 eV) (see Figure 1). This resonance was further investigated and its geometry dependence was explored for six different normal modes with the two scattering symmetries of the 14a’ orbital, not showing significant changes within geometry variations where Δq = ±3, suggesting its dependence may be ‘occluded’ by the collective displacement of the local modes. Further investigation of geometry dependence was then done by computing local-mode dependent cross sections. The computation of the S-matrix poles for the ionized orbital provided a reliable energy value to calculate the resonant wave function, providing a better understanding of how changes in the acrolein structure modify the resonance appearance during the photoionization of its 14a’ orbital.
Figure 1. Total photoionization cross section from the 14a' orbital of acrolein

Poster Presentations Poster 23
2012 Southwest Theoretical Chemistry Conference
Understanding the inherent stereoselectivity of bipyridine N-oxide catalyzed alkylation reactions
Tongxiang Lu, Mark A. Porterfield, and Steven E. Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77843
Stereoselective allylations and propargylations are key chemical transformations, providing access to optically active homoallylic and homopropargylic alcohols. There has been much work over the last decade to develop metal-free, N-oxide based catalysts for these reactions. While many such catalysts have been developed for allylations, there has only been one highly stereoselective N-oxide propargylation catalyst. The differences between N-oxide catalyzed allylation and propargylation reactions were examined using model bipyridine N-oxides. Although it is often assumed that the reactions proceed via a hexacoordinate silicon intermediate with a trans arrangement of chlorines and a trans relationship between the N-oxide and the nucleophilic alkyl group, we show that there are numerous thermodynamically accessible ligand configurations.[1] Moreover, for allylations, most of these ligands configurations lead to strong stereoselectivity even in the absence of other chiral elements. On the other hand, for propargylations, very few of these ligand configurations lead to high degrees of enantioselectivity. A simple electrostatic model is used to explain the enhanced stereoselectivity of N-oxide catalyzed allylations relative to propargylations. This explains why the development of highly stereoselective N-oxide catalysts for propargylation reactions has proven considerably more difficult than for allylation reactions. Finally, we present guidelines for the development of N-oxide catalysts that should be effective for both allylations and propargylations based on the inherent stereoselectivities of selected chiral ligand arrangements around the hexacoordinate silicon.[2]
1. T. Lu, R. Zhu, Y. An, and S. E. Wheeler, J. Am. Chem. Soc. 134, 3095 (2012). 1. T. Lu, M. A. Porterfield, and S. E. Wheeler, Org. Lett. Article ASAP (2012). DOI: 10.1021/ol302493d

Poster Presentations Poster 24
2012 Southwest Theoretical Chemistry Conference
Heterogeneous nano-catalysts with controllable reactivity
Julibeth M. Martinez and Perla B. Balbuena
Department of Chemical Engineering and Materials Science and Engineering Program. Texas A&M University. College Station, TX 77843
We investigate a new type of heterogeneous nano-catalysts, in which adsorption energies of adsorbates and energy barriers for dissociation can be manipulated to some extent. The proposed catalysts are formed by two metal surfaces separated by distances between 5 and 10 Å. This proximity has been found to result in the presence of electrons in the gap between the surfaces; which modifies the geometric and electronic structure of molecules placed in the gap. The electronic density in the gap between the surfaces, along with the interaction between the surfaces and the adsorbate, help reducing the activation barriers for dissociation of diatomic molecules, compared to those on a single surface.
The influence of the local structure of surfaces forming the nano-catalyst has also been studied. Different gap geometries were found to affect the adsorption strength of molecular oxygen. The proposed catalyst allows certain degree of control of both the extent to which the activation energy decreases and the adsorption energies of adsorbates; therefore, it may be possible to tailor the reactivity of this catalyst toward specific reactions.

Poster Presentations Poster 25
2012 Southwest Theoretical Chemistry Conference
Non-covalent interactions inside carbon nanotube: CNT-benzene/substituted benzene and CNT-cyclohexane complexes
Elango Munusamy, Jacob W. G. Bloom, and Steven E Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77842
We present a theoretical study of endo- and exohedral complexes of a carbon nanotube (CNT) with benzene, substituted benzene and cyclohexane. The calculations were performed at the dispersion corrected density functional theory (DFT) level utilizing B97-D with a TZV(2d,2p) basis set and MO6-2X/6-31G* level of theory. We focus on the endohedral complexes and the nature of the interactions and compare them with the corresponding exohedral complexes. Three different scenarios have been examined for the endohedral complex, (1) aliphatic CH-π interaction using cyclohexane, (2) aromatic CH-π interaction using benzene, and (3) Halogen-π interaction using halogenated benzene. It is observed that the exohedral complex is mediated by a π-π interaction, whereas the endohedral complex is mediated by both π-π and CH-π interactions. For CNT of diameters lower than 9.5Å, encapsulation of benzene and cyclohexane results in the deformation of the CNT. Symmetry adapted perturbation theory (SAPT) provides useful insights into the nature of these non-covalent interactions. Atoms-in-molecules (AIM) theory is employed to understand the nature of interaction via bond critical points. Substituent groups such as -CN, -NO2, -F, -CH3, -OH and –NH2 are studied to understand the role of substitution.
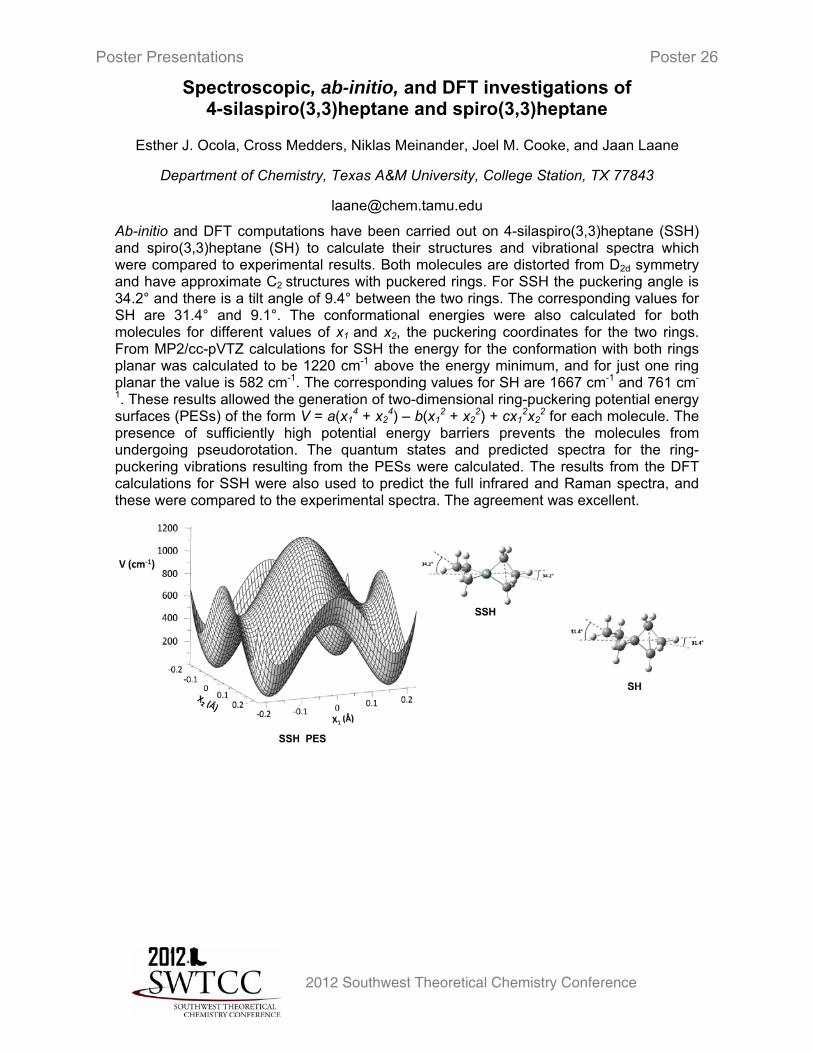
Poster Presentations Poster 26
2012 Southwest Theoretical Chemistry Conference
Spectroscopic, ab-initio, and DFT investigations of 4-silaspiro(3,3)heptane and spiro(3,3)heptane
Esther J. Ocola, Cross Medders, Niklas Meinander, Joel M. Cooke, and Jaan Laane
Department of Chemistry, Texas A&M University, College Station, TX 77843
Ab-initio and DFT computations have been carried out on 4-silaspiro(3,3)heptane (SSH) and spiro(3,3)heptane (SH) to calculate their structures and vibrational spectra which were compared to experimental results. Both molecules are distorted from D2d symmetry and have approximate C2 structures with puckered rings. For SSH the puckering angle is 34.2° and there is a tilt angle of 9.4° between the two rings. The corresponding values for SH are 31.4° and 9.1°. The conformational energies were also calculated for both molecules for different values of x1 and x2, the puckering coordinates for the two rings. From MP2/cc-pVTZ calculations for SSH the energy for the conformation with both rings planar was calculated to be 1220 cm-1 above the energy minimum, and for just one ring planar the value is 582 cm-1. The corresponding values for SH are 1667 cm-1 and 761 cm-
1. These results allowed the generation of two-dimensional ring-puckering potential energy surfaces (PESs) of the form V = a(x1
4 + x24) – b(x1
2 + x22) + cx1
2x22 for each molecule. The
presence of sufficiently high potential energy barriers prevents the molecules from undergoing pseudorotation. The quantum states and predicted spectra for the ring-puckering vibrations resulting from the PESs were calculated. The results from the DFT calculations for SSH were also used to predict the full infrared and Raman spectra, and these were compared to the experimental spectra. The agreement was excellent.

Poster Presentations Poster 27
2012 Southwest Theoretical Chemistry Conference
Determining rate of exciton dissociation without Marcus theory: Setting up the calculation
Robert Paredez,a Eric Bittner,a and Sergei Tretiakb aDepartment of Chemistry, University of Houston, Houston, TX 77204
[email protected] bLos Alamos National Laboratory, Los Alamos, NM 87545
Marcus theory is a viable method of calculating electron transfer rates in many cases. However, Marcus theory only takes into account initial and final configurations. An important problem in the study of charge transport in conjugated organic materials is calculating rates of exciton dissociation in between the initial and final configurations, but before that problem can be addressed, the conditions under which there can be a charge transfer state must be determined. Specifically, which solvent should be used, and what is the minimum applied field necessary to have charge transfer states be the lowest in energy?

Poster Presentations Poster 28
2012 Southwest Theoretical Chemistry Conference
Applications of ScalIT to small molecular systems
Corey Petty and Drew Brandon
Department of Chemistry, Texas Tech University, Lubbock, TX 79409
Exact quantum dynamical bound state rovibrational spectroscopy calculations are performed for various molecular systems, using efficient time-independent methods (ScalIT), and massively parallel computers. Some of the molecular systems considered include the HO2 radical, and rare gas clusters. For HO2, all vibrational states are calculated, as well as bound rovibrational states up to the highest J value that are supported (J = 110). In addition, all bound rovibrational states of Ne3 are computed, as well as selected rovibrational states for Ne4 and other rare gas clusters. Many of the results are obtained with greater accuracy than previous studies, and to higher J values. Parallel scalability of ScalIT, both strong and weak, is demonstrated in benchmark tests up to 1200 cores. A new J-shifting approximation scheme, for estimating rovibrational levels based on exact vibrational levels, is also introduced. This new approach appears to be very effective, even for floppy molecules such as HO2.

Poster Presentations Poster 29
2012 Southwest Theoretical Chemistry Conference
Understanding the stereoselectivity of PINDOX catalyzed allylation reactions
Mark A. Porterfield, Tongxiang Lu, and Steven E. Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77843
The stereoselectivity of the allylation and propargylation of benzaldehyde catalyzed by the bipyridine N-oxide PINDOX was studied using density functional theory (DFT) at the B97-D/TZV(2d,2p) level. Experimentally, PINDOX catalyzes the allylation of benzaldehyde to yield the (S) homoallylic alcohol in 90% ee [Org. Lett. 2002, 4, 1047-1049]. This selectivity was originally rationalized based on a transition state (TS) model based on traditional stereoelectronic arguments that suggest a trans arrangement of chlorines and the N-oxide is trans to the nucleophilic allyl group, as is common for N-oxide catalyzed allylations. However, DFT computations paint a starkly different picture, leading to a revised TS model in which the chlorines are cis to each other and the N-oxide is cis to the allyl group. Moreover, detailed studies of a model bipyridine N-oxide catalyst reveal numerous energetically favorable ligand configurations, the vast majority of which lead to strong stereoselectivity even in the absence of other chiral elements. For the PINDOX-catalyzed allylation reactions, the selectivity is attributed to differences in bond lengths of the key transition state bonds. In addition to the allylation reaction, the propargylation of benzaldehyde with PINDOX and the less reactive allenyltrichlorosilane was explored as well. It is predicted that PINDOX will not lead to strong stereoselectivity in propargylation reactions.
O
ArSiCl3
Ar
OH+
N NO
(+)PINDOX
HH

Poster Presentations Poster 30
2012 Southwest Theoretical Chemistry Conference
Agostic interactions in transition metal d8 complexes: Critical role of ancillary ligands on the strength of the bond
Bimal Pudasaini and Benjamin G. Janesko
Department of Chemistry, Texas Christian University, Fort Worth, TX 76129
[email protected] Agostic interaction is an important interaction between a transition metal and a “C-H” bond during various organometallic catalysis and transformations. Described as 3-centers and 2 electrons bond, these interactions are problematic in Pd (or Ni) catalyzed cross coupling reactions involving alkyl reactants. This is because an alkyl ligand in Pd(II) (or Ni(II)) complex can undergo a relatively facile β-elimination, producing side product(s).
Using Density functional theory (DFT) electronic structure calculations, we have exclusively studied the Pd catalyzed Suzuki coupling with possible β-hydride elimination at different stages of the catalytic cycle. Results indicate that the selectivity of the desired product is strongly influenced by how the ligands and other nucleophiles interact with coordinatively unsaturated Pd(II) intermediate. Interestingly, the strength of agostic interaction, which leads to eventual β-elimination, also depends on the nature of ancillary ligands in Pd(II). A stronger agostic interaction would lead to a more facile β-elimination.
Given the weak nature of agostic “C-H” bond and its strong dependence on ancillary ligands, we first performed a systematic benchmark of DFT on model Ni(II) complexes. We also varied the donor-acceptor properties of ligands to exploit the influence of ligands on the agostic bond(s). Our results provide a better understanding of controlling β-elimination via proposed “design rules” on ligand choices. These results could help improve on an already potent synthetic method.

Poster Presentations Poster 31
2012 Southwest Theoretical Chemistry Conference
Origin of enantioselectivity of a helical dual hydrogen-bonding catalyst
Ulises Rangel, Diana Sepulveda-Camarena, Tongxiang Lu, and Steven E. Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77843
We present a preliminary computational study of the enantioselective electrophilic aromatic substitution of 4,7-dihydroindoles and trans-nitroalkenes catalyzed by a novel dual hydrogen-bonding helical catalyst recently developed by Takenaka and co-workers.[1] The mechanistic pathways for this reaction were explored at the PM6 and B97-D/TZV(2d,2p) levels of theory. It is shown that the helical structure of the catalyst induces a stepwise mechanism through non-covalent interactions resulting in kinetic discrimination and high enantioselectivity. Ongoing efforts aim to determine the physical origin of the enantioselectivity through detailed analyses of the competing transition states.
NH
+
R
N+ O
-O1)cat, CH2Cl2
2) p-benzoquinone NH
R
NO2
1. Takenaka, Chen, Captain, Sarangthem, and Chandrakumar, J. Am. Chem. Soc. 2010, 132, 4536.

Poster Presentations Poster 32
2012 Southwest Theoretical Chemistry Conference
C-H activation of cycloalkanes with Cp’Rh(CO) – A lifetime enigma
Amanda L. Renz and Michael B. Hall
Department of Chemistry, Texas A&M University, College Station, TX 77843
The C-H activation of four cycloalkanes (C5H10, C6H12, C7H14, and C8H16) with the Cp’Rh(CO) fragment (Cp’ = η5-C5H5 or η5-C5Me5) have been theoretically investigated with the BMK functional. The reaction mechanism was believed to proceed from the separated species to a σ-complex intermediate then through the C-H activation transition state and to conclude with the oxidative addition product. The calculated free energies for each step in the mechanism were used to compute reaction rates and lifetimes for each cycloalkane reaction and were then compared with the experimental lifetimes. Although the reaction was initially assumed to be straightforward, further scrutiny concluded this was not necessarily true. Since every C-H bond in the cycloalkanes are not electronically equivalent, there are one, two, seven, and ten different σ-complexes for the cyclopentane, cyclohexane, cycloheptane, and cyclooctane, respectively. Each of these σ-complexes have corresponding transition states and oxidative addition products therefore the overall reaction could proceed through simultaneous reactions. However, the σ-complexes can also transition between each other, so instead of simultaneous reactions the intermediates could “hop” to the σ-complex with the lowest activation energy and proceed to product over the lowest. The BMK results favored the “hopping” mechanism, but to be certain this was not an isolated result from a specific functional four other functionals, B3LYP, TPSS, M06-l, and ωB97X-D were tested with the cyclohexane reaction. When the five functionals were compared no clear conclusion could be made, so CCSD single point calculations were done using the TPSS geometries. The CCSD electronic energies supported the results from the BMK calculations which concluded that the transition states between σ-complexes are lower than the activation barriers so the hopping mechanism should occur.

Poster Presentations Poster 33
2012 Southwest Theoretical Chemistry Conference
The Badger-Bauer Rule Revisited: Correlation of Blue Frequency Shifts in the OC Hydrogen Acceptor with Morphed Hydrogen Bond Dissociation Energies in OC-HX (X=F, Cl, Br, I, CN, CCH)
K. Wayne Scott, L.A. Rivera-Rivera, B.A. McElmurry, R.R. Lucchese and J.W. Bevan
Department of Chemistry, Texas A&M University, College Station, TX 77843
Morphing approaches have been applied for investigation of blue frequency shifts in CO, the hydrogen acceptor, on complexation in the hydrogen bonded series OC-HX (X=F, Cl, Br, I, CN, CCH). Linear correlations of morphed hydrogen bonded dissociation energies D0 with experimentally determined Δν0 for such shifts demonstrate consistency with the original tenets of the Badger-Bauer rule. In addition, determination of dissociation energies for other related but different complexes include D0 for H2O-CO, H2S-CO and OC-HOCH3 which are interpolated and found to be 355(13), 171(11) and 377(14) cm-1 respectively from available experimentally determined proton acceptor shifts, Δν0. Results will also be discussed in relation to investigations in which CO has been used as a probe of heme protein active sites.
Figure 1. Correlation (Badger-Bauer rule) for blue shift of CO stretch and dissociation energy D0 obtained from morphing. Interpolation gives D0 H2O-CO=358(13) cm-1.

Poster Presentations Poster 34
2012 Southwest Theoretical Chemistry Conference
Asymmetric allylations and propargylations of aldehydes using N,N’-dioxides: Performance of DFT methods and origin of
enantioselectivity
Diana Sepulveda-Camarena, Tongxiang Lu, and Steven E. Wheeler
Department of Chemistry, Texas A&M University, College Station, TX 77843
Computational chemistry, and DFT in particular, have proved invaluable in the analysis of stereoselective, organocatalyzed reactions. However, accurately predicting the stereoselectivity of a given catalyzed reaction can still be challenging. This stems in part from the need to describe the many non-covalent interactions that are operative in competing transition states, including dispersion-dominated interactions that have long been problematic for DFT. We first describe a benchmark study of popular DFT methods for the allylation and propargylation of benzaldehyde catalyzed by the bipyridine N,N′-dioxide (S)-1. The results show that some functionals predict the correct stereoselectivity despite qualitatively incorrect energy ordering of low-lying transition states. We then use B97-D/TZV(2d,2p) to explore the origin of stereoselectivity in these reactions and to explain the reduced selectivity in propargylation reactions compared to allylations. The results reveal a transition state model that is drastically different from that published previously for these reactions, and highlights the benefits of applying reliable DFT methods to problems in asymmetric organocatalysis.

Poster Presentations Poster 35
2012 Southwest Theoretical Chemistry Conference
Boron, the carbon mimic
C. J. Tymczak and Alireza Akbarzadeh
Department of Physics, Texas Southern University, Houston, TX 77004
[email protected] We report on the properties and allotropes of Boron in structures relevant to carbon chemistry. The chemistry of boron is complex, where boron has a propensity to catenate, forming complex structures. This chemistry is known and extensively studied, but what has not been extensively studied is the structures that boron forms when in an environment of excess electrons.
We have initiated investigations of complex boron structures that form when in an electron rich environment. These structures are homomorphic to carbon structures such as benzene rings, hexagonal planes (graphene), bucky-balls and carbon nanotubes. The structures specifically form when: i) boron is in a carbon structure; ii) an electron donor species is present such as an alkali metal. This fact can be used to engineer the electronic structure of boron and boron-carbon nanostructures. Several examples are explored.

Poster Presentations Poster 36
2012 Southwest Theoretical Chemistry Conference
Transition metal catalyzed oxidative cleavage of Cβ-O bond in β–O–4 linkage of lignin
Jiaqi Wang and Angela K. Wilson
Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, TX 76203
Lignin is a potential alternative energy resource. However, lignin is an underused biomass species because of its highly branched structure. To aid in better understanding this species, the oxidative cleavage of the Cβ-O bond in an archetypal arylglycerol β-aryl ether (β–O–4 Linkage) model compound of lignin with Ni, Pt, Rh, Co and Fe species has been investigated. Reaction energies and activation energies have been studied to determine if these reactions are thermodynamically and kinetically feasible. Computational methods that have been applied include several density functionals (B3LYP, B2PLYP, M06, M06L, B97D,PBE) and CCSD(T).

Poster Presentations Poster 37
2012 Southwest Theoretical Chemistry Conference
Theoretical investigation of the OH-initiated oxidation of toluene
Fei Xu and Renyi Zhang
Department of Atmospheric Sciences and Department of Chemistry, Texas A&M University, College Station, TX 77843
Toluene is the most abundant aromatic hydrocarbon in the atmosphere and is emitted primarily from anthropogenic sources. Photochemical oxidation of toluene plays an important role in the formation of tropospheric ozone and secondary organic aerosol (SOA), which profoundly impacts air quality, human health, and climate, but its fundamental chemical mechanism remains largely uncertain. The oxidation of toluene is mainly initiated by the hydroxyl radical OH: the initial OH-toluene reaction results in minor H-abstraction (about 10%) and major OH addition (about 90%) [Molina et al., 1998]. The H-abstraction pathway leads to the formation of benzaldehyde, whose oxidative pathway is well established. The OH addition pathway results in the formation of methylhydroxycyclohexadienyl radicals (the OH-toluene adducts). Under atmospheric conditions, the OH-toluene adducts react with O2 either by O2 addition to form primary peroxy radicals, H-abstraction and subsequent O-bridge formation to aromatic oxide/oxepin, or H- abstraction to yield phenolic compounds [Suh et al., 2002; 2003; 2006]. In this study, the reaction mechanism of toluene with OH was investigated by using hybrid density functional theory (DFT) and the Rice–Ramsperger–Kassel–Marcus (RRKM)/Master equation (ME) method to assess the preferred pathways between the competing toluene-oxide/methyloxepin, peroxy radicals, and phenolic channels, and compare yield of the products from each channel.

Poster Presentations Poster 38
2012 Southwest Theoretical Chemistry Conference
Computational investigation of oxidation of nitric oxide by nonheme iron (IV)-oxo compounds
Zhenggang Xu and Michael B. Hall
Department of Chemistry, Texas A&M University, College Station, TX 77843
Oxidation of Nitric oxide molecule by metal-oxygen species is the key process of NO metabolism in biological systems. Mononuclear nonheme oxo-iron species are a group of important intermediates involved in oxygenation reactions. In our computational study, we looked into the detailed mechanism of the oxidation reaction of NO molecule by a nonheme oxo-iron compound, 2-OAc ([FeIVO(TMC)(OAc)]+; TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetra- azacyclo-tetradecane), which was investigated by Owen et al experimentally [1]. We obtained several noticeable results: 1) 2-OAc has a triplet ground spin state instead of quintet state. 2) Two-state reactivity can be found in this reaction. Although the total spin of reactants in ground state is either doublet or quartet, the reaction goes along the sextet surface, which only has an energy barrier of ~10 kcal/mol. 3) The decay of sextet intermediate ([FeIII(ONO)(TMC)(OAc)]+ affords NO2
- and [FeIII(TMC)(OAc)]2+ if an implicit solvent model (SMD) is used. 4) The decay barrier of intermediates calculated with SMD model in methanol is apparently lower that that in nitromethane, which suggests that the dependence of intermediate decay on the concentration of methanol found in experiments may majorly come from solvent effects. Further work is undergoing to clarify the intrinsic differences of these two solvents and how implicit SMD model catch up these differences in computations.
1. Owen, T. M.; Rohde, J.-U., Inorg. Chem. 2011, 50, 5283-5289.

Poster Presentations Poster 39
2012 Southwest Theoretical Chemistry Conference
Thermodynamics of the carbon dioxide-epoxide copolymerization, and kinetics of the metal-free degradation
Andrew D. Yeung and Donald J. Darensbourg
Department of Chemistry, Texas A&M University, College Station, TX 77843
“Chemically accurate” enthalpies for the reactions of carbon dioxide and epoxides to give polycarbonates and cyclic carbonates were obtained by CBS-4M calculations. Polymer formation is more exothermic than cyclic carbonate formation by 5-8 kcal/mol for ethylene, propylene, chloropropylene, styrene, and cyclohexene polycarbonates.
The metal-free carbonate and alkoxide back-biting reactions yield cyclic carbonates, and CBS-QB3/(+) calculations were used to determine the activation barriers. Under polymerization conditions, the former reaction leads to undesired cyclic carbonate formation. The latter affects the robustness of the product. The SN2 carbonate back-biting reaction has a relatively low activation barrier (ΔG‡ = 18-25 kcal/mol), but it is negligible for poly(cyclohexene carbonate) as a conformational change (ΔG = 4.7 kcal/mol) is required before the activation barrier (ΔG‡ = 21.1 kcal/mol) to cyclization can be traversed.
The alkoxide back-biting proceeds via a tetrahedral alkoxide intermediate, formation of which is barrierless. Further reaction to the cyclic carbonate has a free energy barrier that is 10 kcal/mol less than the carbonate back-biting reaction, making such polycarbonates susceptible to degradation by base.
* We thank Dr. Lisa Pérez for advice, and Texas A&M LMS and Supercomputing Facility for resources.
7.5
0.0
9.58.811.8
‐2.0
0.0
2.0
4.0
6.0
8.0
10.0
12.0
14.0
16.0
Free
ene
rgy (kcal/mol)
Open chain
Intermediate
Cyclic carbonate + alkoxide

Poster Presentations Poster 40
2012 Southwest Theoretical Chemistry Conference
Condensation of exciton polaritons in organic microcavities
Svitlana Zaster,a Eric R. Bittner,a and Carlos Silvab
aDepartment of Chemistry and Physics, University of Houston, Houston, TX 77204
bDepartment of Physics and Regroupement que ́be ́cois sur les mate ́riaux de pointe, Université de Montréal Montreal (Quebec), Canada
Following the observation of polariton condensation in inorganic microcavities we consider a possibility of similar effects in organic semiconductor microcavity, characterized by larger oscillator strength between cavity excitons and the photon field (as compared to their inorganic counterparts). Our approach involves the theoretical considerations of preparation and dynamics of BEC states within a micro-meter scale resonant microcavity following the modeling of macroscopic behavior of exciton-polaritons to predict the conditions under which they could form a macroscopic coherent quantum fluid. Our results establish the criteria for the conditions under which BEC could be achieved in the anthracene microcavity.

Poster Presentations Poster 41
2012 Southwest Theoretical Chemistry Conference
Carboxylation of terminal alkynes assisted by pNHC as Lewis base and Lewis acid
Jia Zhou and Michael B. Hall
Department of Chemistry, Texas A&M University, College Station, TX 77843
CO2 fixation and transformations at mild conditions have drawn a great attention in the past few decades. It is highly desired to develop an efficient catalyst for CO2 utilization. One recent paper (PNAS, 2010, 20184) demonstrates in the experiment that poly-N-heterocyclic carbine (NHC)-Cu system is able to convert CO2 to carboxylic acid in an efficient and economical way.
Here, we use density functional method, along with PCM model to study the mechanism of this catalytic reaction. In contract with the proposed mechanism in the paper, our theoretical calculation shows that pNHC behaves as both Lewis basis and Lewis acid in the reaction.

Visit Microway at SC12 Salt Lake City – Booth 2500
GS-35F-0431N
GSA ScheduleContract Number:GS-35F-0431N
Microway Puts YOU on the Cutting EdgeDesign your next custom configuration with techs who speak HPC. Rely on our integration expertise for complete and thorough testing of your workstations, turnkey clusters and servers. Whether you require Linux or Windows, CUDA® or OpenCL, Tesla K20 or Intel Xeon Phi we’ve been resolving the complicated issues – so you don’t have to – since 1982.
Get the Best Performance, Call Us First!
508-746-7341 or microway.comSign up for technical newsletters at microway.com/newsletterRead our blog at microway.com/hpc-tech-tips
Microway’s Cutting Edge Platforms for Theoretical Chemistry Ready for Intel® Xeon® Phi™ Coprocessors and
NVIDIA® Tesla® K20 GPUs
ANSYS CFD (Fluent)ANSYS MechanicalAutodesk MoldflowAbaqusSIMULIA®
Simulation
MATLABACUSIM AcuSolveTech-X GPULibSeismic City RTMVoxxelGeo
3ds MaxAdobe CS6SolidWorksBunkspeed Shot
Design
AMBERGROMACSNAMDVMDTeraChem
BioTech
Increase performance with CUDA C/C++, PGI CUDA FORTRAN, or these GPU-enabled applications:
WhisperStation™ – Quiet HPC at Your Desk1 to 4 NVIDIA Tesla K20 or Quadro GPUsNVIDIA Quadro® GPUs for visualization + NVIDIA Tesla GPUs for lightning-fast computationUp to 32 x86 Cores and 512GB MemoryHigh clock speed Intel® CPUs; SSDs and/or RAID for fast I/O; up to 8 drives; PCI-E Generation 3.080 PLUS™ certified power supplies, ultra-quiet fans, sound proofing materials and high-efficiency components
All trademarks and logos are property of their respective owners.
Scalable, Fully Integrated ClustersDesigned with Dense 1U/2U Servers and PCI-E Generation 3.08-core Xeon E5-2600 Series CPUs, DDR3-1600 MemoryNVIDIA Tesla K10, K20 or M2090 GPUsReady for Intel® Xeon Phi™ CoprocessorsMellanox ConnectX-3 FDR InfiniBand
Benchmark Remotely on 8 Xeon E5-2600s and 4 Tesla K20s.
Schrödinger on Steroids!Hψ = Eψ GPU


















