14.139.121.10614.139.121.106/pdf/2013/may13/5.pdf14.139.121.106
180
DESIGN AND DEVELOPMENT OF PROTEASE INHIBITORS AS POTENTIAL ANTIMALARIAL AGENTS A THESIS SUBMITTED TO THE MAHARAJA SAYAJIRAO UNIVERSITY OF BARODA FOR THE AWARD OF THE DEGREE OF Doctor of Philosophy in Pharmacy BY UTTAM R. MANE UNDER THE GUIDANCE OF Prof. M. R. YADAV Pharmacy Department, Faculty of Technology and Engineering, The M. S. University of Baroda, Vadodara-390 001. NOVEMBER 2012
Transcript of 14.139.121.10614.139.121.106/pdf/2013/may13/5.pdf14.139.121.106

DESIGN AND DEVELOPMENT OF PROTEASE INHIBITORS AS POTENTIAL
ANTIMALARIAL AGENTS
A THESIS SUBMITTED TO
THE MAHARAJA SAYAJIRAO UNIVERSITY OF BARODA
FOR THE AWARD OF THE DEGREE OF
Doctor of Philosophy
in
Pharmacy
BY
UTTAM R. MANE
UNDER THE GUIDANCE OF
Prof. M. R. YADAV
Pharmacy Department,
Faculty of Technology and Engineering,
The M. S. University of Baroda,
Vadodara-390 001.
NOVEMBER 2012

Date:
CERTIFICATE
This is to certify that the thesis entitled “DESIGN AND DEVELOPMENT
OF PROTEASE INHIBITORS AS POTENTIAL ANTIMALARIAL AGENTS”
submitted for the award of Ph. D. Degree in Pharmacy by Mr. UTTAM RAJARAM
MANE incorporates the original research work carried out by him under my
supervision.
Supervisor
(Prof. M. R. Yadav)
HEAD DEAN
Pharmacy Department Faculty of Technology & Engineering,
The M.S. University of Baroda,
Vadodara -390 001

DECLARATION
I hereby declare that the topic entitled “DESIGN AND DEVELOPMENT
OF PROTEASE INHIBITORS AS POTENTIAL ANTIMALARIAL AGENTS”
submitted herewith to The Maharaja Sayajirao University of Baroda, Vadodara, for
the award of the degree of DOCTOR OF PHILOSOPHY IN PHARMACY is the
result of the work carried out by me in Pharmacy Department, Faculty of
Technology and Engineering, The M. S. University of Baroda, Vadodara.
The result of this work has not been previously submitted for any
degree/fellowship.
Date:
Place: Vadodara UTTAM R. MANE

Dedicated to my FAMILY

ACKNOWLEDGEMENT
I am deeply indebted to my supervisor Prof. M. R. Yadav, Head, Department of Pharmacy,
Faculty of Technology & Engineering, The M. S. University of Baroda, Vadodara, for
guiding me to accomplish this dissertation work. It was my privilege and pleasure to work
under his able guidance. A person who strongly believes in the principle of perfection and
enriches the true researchers’ spirit with intuition, logical reasoning and his well versed
knowledge has made him an oasis of ideas. I am indeed grateful to him for providing helpful
suggestions, from time to time. Due to his constant encouragement and inspiration, I am able
to present this thesis work.
I would like to thank Prof. A. N. Mishra, Dean, Faculty of Technology and Engineering, The
M. S. University of Baroda, for providing the facilities for research.
I convey my deepest gratitude to Prof. Rajani Giridhar, Coordinator, Q. I. P. Cell,
Pharmacy Department, for her moral support during the course of the study.
I gratefully acknowledge Dr. C. Dutt, Director and Dr. S. S. Nadkarni, ED, Dr. V.
Chauthaywale, VP, Torrent Research Centre for their enduring support to carry out this
dissertation work.
I sincerely acknowledge Dr. R. C. Gupta, GM, Torrent Research Centre for his never ending
support and fruitful suggestions at the hours of need.
My special thank go to Prof. Li Huang and Prof. Honglin Li, East China University of
Science and Technology, for carrying out biological studies of this research work.
I am also thankful to Dr. R. Shah, GM, Dr. Shailesh, GM, Dr. A. Mandhare, AGM and Dr.
Sanjay, AGM, my Seniors and Colleagues at Torrent Research Centre for their moral
support.
I am thankful to A. Shinde, Prakash and Anurag for their contributions.
I convey my thanks to Prashant, P. Bhaver, Suhas, Amit and Mahesh for giving me
valuable support.
I express my sincere thanks to teaching and non-teaching staff of Pharmacy Department for
their co-operation during the course of the work.
My parents deserve special mention for their support and prayers. I convey my deepest
gratitude to my late mother, who inculcated the fundamentals of learning and in true sense she
is the only source of my inspiration. My father is an ideal person in my life whom I follow
every step of my life. I owe thanks to my ever supporting wife Archana and my cute little
baby Anvi. I also owe so much to my sister, brother and Vahini and in laws, my late uncle
and aunty and other family members, friends and relatives for their encouragement and
support throughout my life. I express my deepest sentiments of affection and regards to them
for whatever I am today.

Finally, I would like to thank everybody who directly or indirectly involved in the successful
completion of this thesis, as well as expressing my apology that I could not mention
personally one by one.
Finally, Thanks to almighty for his blessings on me forever……………
Uttam R. Mane

ABBREVIATIONS
__________________________________________________________
Aq. : Aqueous
DMF : Dimethyl formamide
DMSO : Dimethyl sulphoxide
EtOAc : Ethyl acetate
DIEA : N.N-diisopropyl ethyl amine
TEA : Triethylamine
EMME : Diethyl ethoxymethylenemalonate
DPE : Diphenyl ether
conc. HCl : Concentrated hydrochloric acid
ECF : Ethyl chloroformate
HOBT : 1-Hydroxy benzotriazole hydrate
EDC : 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide
KBr : Potassium bromide
NaN3 : Sodium azide
RT : room temperature
ppt. : precipitate
Anal. : Analytical
TLC : Thin layer chromatography
Rf : Retention factor
ppm : Parts per million
M. P. : Melting point
Mass : Mass spectroscopy
M+
: Molecular ion
FP : Falcipain
FP-2 : Falcipain-2
FP-3 : Falcipain-3
IR : Infrared spectroscopy 1HNMR (PMR) : Proton nuclear magnetic resonance spectroscopy

INDEX
Certificate i
Declaration ii
Dedication iii
Acknowledgement iv
Abbreviations v
1. Introduction 1-57
2. Research envisaged 58-59
3. Results and Discussion 60-99
3.1 Chemical studies 60-96
3.1.1 Synthesis of 4H-pyrido[1,2-a]pyrimidin-4-one intermediates (4a-c & 6a-c) 61-66
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a) 61
Synthesis of 3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a) 63
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4b) 63
Synthesis of 3-isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (6b) 64
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4c) 65
Synthesis of 7-chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6c) 66
3.1.2 Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives (Series I) 66
Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives 66
Synthesis of 8-methyl-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives 74
Synthesis of 7-chloro-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivative 76
3.1.3 Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives (Series II)
79
Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)-piperazine-1-
carboxamide derivatives
79
Synthesis of N-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives
80
Synthesis of N-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives
81
3.1.4 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-ylcarbamate derivatives (Series III)
83
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives 83
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives 85
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives 85
3.1.5 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives (Series IV)
86
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives 87
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives 92
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives 93
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamide 94

intermediates
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamides
derivatives
94
3.2. Biological evaluation 96-99
4. Experimental work 100-143
4.1 Chemical studies 100-143
4.1.1 Synthesis of 4H-pyrido[1,2-a]pyrimidin-4-one intermediates (4a-c & 6a-c) 100
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a) 100
Synthesis of 3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a) 101
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4b) 102
Synthesis of 3-isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (6b) 103
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4c) 104
Synthesis of 7-chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6c) 105
4.1.2 Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives (Series I) 106
Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives 106
Synthesis of 8-methyl-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives 114
Synthesis of 7-chloro-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivative 116
4.1.3 Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1- carboxamide derivatives (Series II) 121
Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)-piperazine-1-
carboxamide derivatives
121
Synthesis of N-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives
123
Synthesis of N-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives
124
4.1.4 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-ylcarbamate derivatives
(Series III)
126
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives 126
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives 128
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives 129
4.1.5 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives (Series IV)
130
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives 130
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives 137
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives 138
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamide
intermediates
140
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamides
derivatives
141
4.2. Biological work 143
5. Summary 144-151
6. References 152-165

1. Introduction

Chapter-1 Introduction
1
Parasite-borne diseases are having devastating impact on human
civilizations. However, none of the parasitic diseases has had as enormous an
impact on humans as that of malaria, which is one of the major causes of
morbidity and mortality of human civilizations in the past, and even today it
remains one of the deadliest diseases on the planet. It displays full explosive
power of vector-borne infections, erupting suddenly and with intensity that can
overwhelm vulnerable communities.1 Even today malaria, particularly the one
caused by Plasmodium falciparum, remains a serious health problem in Africa,
South America and many parts of Asia.2 As per the recent WHO report malaria is
having devastating impact on the lives of more than 3 billion people of the world,
about half of the humanity.3
An estimated 600 million people are at risk of contracting malaria
infection and the disease kills more than 1 million children and a large number of
pregnant women every year. About 90% of these casualties occur in tropical
Africa and majority of them are children under the age of five. Exacerbated by
poverty, malaria exerts a high toll on populations that can least afford a cure.4
Despite a heavy toll of tropical parasitic diseases on human life in the developing
world, present antimalarial therapy still rely on drugs developed decades ago.
There are several reasons for re-emergence and spread of the infection across the
globe as a major public health burden; the most common ones are complex life
cycle of the parasite, resistance developed by parasite against common
antimalarial drugs, inadequate control of the mosquito vector, and despite
tremendous impetus and thrust instituted legitimately, the dream of developing an
effective, safe and economical vaccine is a big illusion till date. The other major
contributors to the re-emergence of the disease and its spread in the new locations
and populations are increased population density, global warming, continuing
poverty and political instability. The business travels to malaria endemic areas
further added the spread of the disease to the developed countries where it has
been thought to be totally eradicated a long time ago. The prophylaxis of malaria
is also complex because of resistance, safety and tolerability issues of the existing
drugs, so there are very limited antimalarial drug options available.5
WHO aims for the global roll back of malaria with intensified efforts in
providing access to safe, affordable and effective antimalarial combination
treatments worldwide along with preventive measures.6 Science still has no magic

Chapter-1 Introduction
2
bullet for malaria and doubt remains whether such a single solution will ever exist.
In many cases, the clinical usefulness of new compounds is limited due to poor
efficacy, toxicity and constant attrition rate of drugs due to development of
resistance. The absence of profit incentives has resulted in the lack of investment
by the pharmaceutical industry in developing new chemotherapies7 because the
disease is more prevalent among the people of the developing countries who
hardly have any purchasing power. Efforts in the direction of vaccine
development and control/eradication of the mosquito vectors have either failed or
met with limited success. Development of new antimalarial drugs only remains an
economically and environmentally viable alternative.
Overall, there is an alarming situation as the parasite has developed
resistance to all of the available drugs except artemisinins, and to prevent
resistance development against artemisinins, these are generally used in
combination with other antimalarials. But some recent reports have challenged the
effectiveness and use of artemisinins alone or in combination due to reports on
their reduced efficacy on Thai-Combodian border, historically a site of emerging
antimalarial drug resistance and also because vaccine development has not yet
yielded any fruitful results.6 Hence there is an urgent need for identification and
characterization of new targets for antimalarial chemotherapy. Fortunately the P.
falciparum genome sequencing has revealed a number of new targets for drug and
vaccine development. But only the development of newer antimalarial drugs
remains an economically and environmentally viable alternative to fight out the
menace of the disease.8
1.1 Replicative life cycle of malaria
Malaria is caused by four species of protozoan parasites of the
Plasmodium genus. These are Plasmodium falciparum, P. vivax, P. ovale and P.
malariae, each of which presents slightly different clinical symptoms.
Geographically P. falciparum is the most widespread and the most pernicious of
the four species causing the major malaria related morbidity and mortality.9
In 1897, Ronald Ross first reported that the parasite Plasmodium could
infect the female anopheles mosquito, parasites are transmitted from one person to
another by this insect vector, thus completing parasite cycle. The malaria parasite
exhibits a complex life cycle (Fig. 1) involving an insect vector (mosquito) and a

Chapter-1 Introduction
3
vertebrate host (human). The major phases of the life cycle are: liver stage, blood
stage, sexual stage and sporogony.9
Figure 1: The life cycle of malaria parasite
Malaria infection is initiated when sporozoites are injected into the blood
of human host along with the saliva of a feeding mosquito. Sporozoites are carried
by the circulatory system to the liver where they invade parenchymal cells of the
hepatocytes (1), a process involving receptor ligand mediated adhesion. The
intracellular parasite undergoes an asexual replication known as exoerythrocytic
schizogony within the hepatocyte (2-4). Exoerythrocytic schizogony culminates in
the production of merozoites, which are released into the bloodstream (5). During
development and multiplication in the liver the host is asymptomatic. In
nonrelapsing malaria (falciparum, malariae) no parasites are left in the liver as
merozoites infection moves entirely in the blood. In relapsing malaria, a
proportion of the liver-stage parasites from P. vivax and P. ovale go through a non
dividing dormant stage known as hypnozoite (not shown in Fig. 1) instead of
immediately going through the asexual replication (i.e., temporarily stay at step
2). These hypnozoites are reservoir of parasites which will reactivate after several
weeks to months (or years) after the primary infection and are responsible for
relapses. Merozoites invade erythrocytes (6) and undergo a trophic period in

Chapter-1 Introduction
4
which the parasite enlarges (7-8). The early trophozoite is often referred to as 'ring
form' because of its morphology. Trophozoite enlargement is accompanied by an
active metabolism including the ingestion of host cytoplasm and the proteolysis of
hemoglobin into constituent amino acids. Multiple rounds of nuclear division
manifest the end of the trophic period without cytokinesis resulting in a schizont
(9). Merozoites bud from the mature schizont, also called a segmenter (10) and the
merozoites are released following rupture of the infected erythrocyte (11).
Invasion of erythrocytes reinitiates another round of the blood-stage replicative
cycle (6-11). The time intervals between the intermittent fever and chills often
associated with malaria is due to the synchronous rupture of infected erythrocytes
and release of merozoites, parasitic waste and the cell debris. The debris is the
cause of the episodes of fever and chills. Trophozoite and schizont infected
erythrocytes are rarely found in the peripheral circulation during P. falciparum
infections. Erythrocytes infected with these stages adhere to endothelial cells and
sequester in the microvasculature of vital organs, especially brain, heart and lungs.
Sequestration in the brain is a contributing factor in cerebral malaria. As an
alternative to the asexual replicative cycle, the parasite can differentiate into
sexual forms known as macro or microgametocytes (12). The gametocytes are
large parasites, which fill up the erythrocyte, but only contain one nucleus.
Ingestion of gametocytes by the mosquito vector induces gametogenesis (i.e., the
production of gametes) and their escape from the host erythrocyte. Factors which
participate in the induction of gametogenesis include a drop in temperature, an
increase in carbon dioxide concentration and mosquito metabolites. Microgametes
formed by a process known as exflagellation (13) are flagellated forms, which will
fertilize the macrogamete (14) leading to a zygote (15). The zygote develops into
a motile ookinete (16), which penetrates the gut epithelial cells and develops into
an oocyst (17). The oocyst undergoes multiple rounds of asexual replication (18)
resulting in the production of sporozoites (19). Rupture of the mature oocyst
releases the sporozoites into the hemocoel (body cavity) of the mosquito (20). The
sporozoites migrate to and invade the salivary glands (not shown in Fig. 1), thus
completing the life cycle. In summary, the malaria parasite undergoes three
distinct asexual replicative stages exoerythrocytic schizogony, blood stage
schizogony and sporogony, resulting in the production of invasive forms
(merozoites and sporozoites). Sexual reproduction occurs with the switch from

Chapter-1 Introduction
5
vertebrate to invertebrate host and leads to the invasive ookinete. All invasive
stages are characterized by the apical organelles typical of apicomplexan species.
The invasive stages differ in regard to the types of cells or tissues they invade and
their motility.
1.2. Currently available antimalarial drugs
A systemic malaria control program began after discovery of the malaria
parasite by Laveran (Nobel Prize for medicine, 1907) in 1880 and the
demonstration by Ross in 1897 that the female anopheles mosquito was the vector
of malaria. The mosquito control combined with therapeutic and
chemoprophylatic approaches in parallel are used in the successful disease
control. The efforts of complete eradication of the disease were limited by the
resistance developed by the parasite against common antimalarial drugs and also
by the mosquito vector.9 Mostly, antimalarial drugs are targeted against the
asexual erythrocytic stage of the parasite. The parasite degrades hemoglobin in its
acidic food vacuole,10
producing free heme, capable of reacting with molecular
oxygen and thus generating reactive oxygen species as toxic by-products. A major
pathway of detoxification of heme moieties is polymerization as malarial
pigment.11
Majority of antimalarial drugs act by disturbing the polymerization
(and/or the detoxification by any other way) of heme, thus killing the parasite with
its own metabolic waste.12
The main classes of active schizonticides are 8-
aminoquinolines, 4-aminoquinolines, arylalcohols including quinoline alcohols
and antifolate compounds which inhibit the synthesis of parasitic pyrimidines. A
newer class of antimalarials is based on the natural endoperoxide artemisinin, its
semisynthetic derivatives and synthetic analogs. Some antibiotics are also used,
generally in association with quinoline-alcohols.13
Very few compounds are active
against gametocytes and also against the intra-hepatic stages of the parasite.
1.2.1 8-Aminoquinolines
This is the only class of gametocytocides available to the therapeutic
purposes. Primaquine has been widely used for the treatment of the hypnozoites
(liver reservoirs) responsible for the relapsing forms of P. vivax and P. ovale.
However, primaquine was recently reconsidered for malaria chemoprophylaxis to
eliminate P. falciparum at the early stage of infection, when parasite develops in

Chapter-1 Introduction
6
the liver, thus preventing the clinical disease. Despite its good oral absorption, this
molecule has a short half life and needs to be administered daily. Serious toxicity
can be a major problem in patients with glucose-6-phosphate dehydrogenase
deficiency. Primaquine is interfering with the mitochondrial function of
Plasmodium. Tafenoquine (WR 238605) is a primaquine analog with a longer
elimination half-life (14 days compared to 4 hours for primaquine). It also has a
higher therapeutic index than primaquine. This molecule may be useful for
chemoprophylaxis of P. falciparum and for prevention of relapses of vivax
malaria.
1.2.2 4-Aminoquinolines
The main antimalarials are the 4-aminoquinolines because they have proven to be
a highly successful class of compounds for the treatment and prophylaxis of
malaria. They are easy to synthesize, cheap and generally well tolerated. These
compounds, as well as the quinoline alcohols, are active against the intra-
erythrocytic stages of the parasite. The 4-aminoquinolines are able to accumulate
in high concentrations within the acid food vacuole of Plasmodium, to kill the
parasite.14
Chloroquine (CQ) was introduced in 1944-1945 and soon became the
mainstay of therapy and prevention, as this drug was cheap, nontoxic and active
against all strains of malaria parasites. In 1994, CQ was the third most widely
consumed drug in the world after aspirin and paracetamol.15
The precise mode of
action of the quinoline antimalarials and the mechanism of parasite resistance are
still not completely understood. On the mode of action of CQ, one can cite 14
many hypotheses like (i) direct-heme binding, (ii) inhibition of an unidentified
heme ferriprotoporphyrin- IX “polymerase”, (iii) inhibition of vacuolar
phospholipase, (iv) inhibition of protein synthesis and (v) interaction with DNA.
However, the main mode of action of CQ seems to be related to the accumulation
of this weak base in the acidic lysosome and binding to ferriprotoporphyrin-IX
polymerase, thereby preventing the detoxification of ferriprotoporphyrin-IX by
polymerization and thus killing the parasite.16
CQ resistance was observed in
Southeast Asia and South America at the end of the 1950s and in Africa in the late
1970s. Resistant parasites accumulate CQ less avidly than do sensitive ones.
Resistance can be reversed in vitro using drugs known to reverse drug resistance
in tumor cells, such as verapamil.17
The observation that CQ resistance appeared

Chapter-1 Introduction
7
rather late, about 10 years after its widespread use, has been considered as an
argument to support the hypothesis that CQ resistance has genetic basis.18
NH
CH3
N
NH2
CH3
O F3C
NH
CH3
N
NH2
N
NH
CH3
N
CH3
CH3
Cl
N
OH
N
CH2
ClCl
CF3 OH
N CH3
CH3
N
NH
Cl
OH
N CH3
CH3
O
CH3
OR
CH3
HCH
3
H
O
O
N
OHNH
CF3
CF3
N
N
NH2
CH3
Cl
NH2
O
OH
O
Cl
NN
NH
S
NH2
O O
S
NH2
NH2
OO
O
O
CH3
CH3
OH CH3
CH3
OH
O
O
CH3
CH3
CF3
CF3
OOOCH
3
CH3
CH3
CH3
OH
Tafenoquine (WR 238605) Primaquine ChloroquineQuinine
HalofantrineAmodiaquine
Quinoline class of antimalarials
R = H DihydroartemisininR = CH3 Artmether
R = Et Arteether
Artemisinin antimalarials
Mefloquine
Quinoline class of antimalarials
Pyrimethamine
Atovaquone
Arylalcohol class of antimalarials
Folate antagonist class of antimalarials
Other antimalarials
Sulfadoxine Dapsone
Yingzhaosu A ArtefleneYingzhaosu C
OCH3
OCH3
OCH3
H3CO
OCH3
H3CO
Figure 2: Examples of some common antimalarial drugs
In spite of its reduced efficacy, CQ is still the most widely used
antimalarial drug in Africa, both for the reason of cost and because of the
widespread prevalence of resistance among people infected with the parasite
towards a large number of other antimalarial drugs. Moreover, tumor necrosis
factor (TNF), a cytokine responsible for some cerebral damages, which is

Chapter-1 Introduction
8
produced by immune system during the malaria crisis, has been proven to have a
synergistic effect with chloroquine, thus enhancing the effect of the drug.19
Amodiaquine is chemically related to CQ, but is more effective than CQ for
clearing parasitemia in cases of uncomplicated malaria, even against some
chloroquine-resistant strains.20
However, drug resistance and potential hepatic
toxicity limit its use. Amodiaquine has been shown to bind to heme and to inhibit
heme polymerization in vitro, with a similar efficiency as CQ.21
Furthermore,
amodiaquine exhibits cross-resistance with CQ suggesting that it exerts its activity
by a similar mechanism.22
1.2.3 Quinolinemethanols
Quinine, the active ingredient of cinchona bark, introduced into Europe
from South America in the 17th
century, had the longest period of effective use
and relieved more human suffering than any other drug for almost 3 centuries. But
now there is an overall decline in its clinical response for P. falciparum in some
areas. As on today quinine is not a first line treatment drug because of its small
therapeutic index, longer period of treatment and the toxicity associated with it.
Nevertheless, it remains an essential antimalarial drug for severe falciparum
malaria and intravenous infusion is, in this case, the preferred route of its
administration. The addition of a single dose of artemisinin enhances the parasite
elimination rate and thus increases the cure rate.23
Quinine interacts weakly with
heme, but has been shown to inhibit heme polymerization in vitro. The
mechanism of resistance to quinine is unknown, but a similar one as that of
mefloquine has been suggested.21
Combination of quinine and clindamycin24
or
quinine and allopurinol25
significantly shorten the duration of treatment with
respect to quinine used alone.
Mefloquine is structurally related to quinine and its long half-life (14-21
days) has probably contributed to the rapid development of resistance towards this
drug. For this reason, mefloquine should be used in combination with other
antimalarial agents. It binds with high affinity to membranes, causes
morphological changes in the food vacuole of Plasmodium and interacts relatively
weakly with free heme. The plasmodial P-glycoprotein 1 (Pgh 1) plays a role in
mefloquine resistance and it may also be the target of this drug.26,27
Lumefantrine
was synthesized by the Academy of Military Medical Sciences in Beijing. It

Chapter-1 Introduction
9
belongs to the quinoline alcohol group that includes quinine and mefloquine. Its
mechanism of action involves the interaction of the drug with heme and it is active
against CQ resistant P. falciparum infection. The antibiotics like tetracycline are
used in combination with quinine for multidrug resistant P. falciparum infection,28
and doxycycline and azithromycin29
are used for the purpose of prophylaxis.
Fluoroquinolone antibiotics showed activity only in the in vitro tests.
1.2.4 Other arylalcohols
Halofantrine is effective against chloroquine-resistant malaria.30
Despite
this, cardiotoxicity has limited its use as a therapeutic agent.31
Mefloquine usage
appears to lead to selection of parasites resistant to halofantrine.32
Furthermore, it
is an expensive drug without availability of parenteral formulations. Pyronaridine,
an acridine derivative, is a synthetic drug widely used in China that may have
utility for multiresistant falciparum malaria.33
Its current Chinese oral formulation
is reported to be effective and well tolerated, but its oral bioavailability is low and
this contributes to an unacceptably high cost of the treatment. It seems likely that
drug resistance would emerge rapidly if pyronaridine was used in monotherapy.
As reported above, resistance to a lot of antimalarial drugs has been observed in
clinical isolates, but resistance to mefloquine, quinine and halofantrine appears to
be inversely correlated with resistance to chloroquine and amodiaquine,
suggesting that the development of a high level of resistance to chloroquine makes
the parasite more sensitive to the arylmethanols.34
1.2.5 Folate antagonists
These compounds inhibit the synthesis of parasitic pyrimidines and thus
of parasitic DNA. There are two groups of antifolates (i) the dihydrofolate
reductase (DHFR) inhibitors like the antimetabolite antimalarial drugs,
pyrimethamine and proguanil and (ii) the dihydropteroate synthase (DHPS)
inhibitors having sulfones and sulphonamides like sulfadoxine and dapsone.
Due to a marked synergistic effect, a drug of the first group is usually
used in combination with a drug of the second one. Unfortunately, resistance is
widespread in Asia and now in Africa.35
Pyrimethamine-sulfadoxine (SP,
Fansidar) is the most widely used combination. It is cheap, practicable (only one
dose is needed because of the slow elimination of the drugs from the body) and

Chapter-1 Introduction
10
currently efficient in many parts of Africa. However, it is poorly active against
highly chloroquine-resistant strains. SP is also particularly prone to rapid
emergence of resistance. The mechanism of resistance for this combination has
been shown to be due to mutations in the genes of DHFR and DHPS.36
Proguanil
has also been combined with CQ in some oral formulations. It should be
mentioned that proguanil is a prodrug, its P-450 metabolite cycloguanil is the
active compound. Chlorproguanil is a chlorinated analog of proguanil that is also
metabolized as an active triazine compound. It is more efficient and has a larger
therapeutic index than proguanil and its combination with dapsone is eliminated
more rapidly than SP, offering the possibility of lowering the selection pressure
for resistance.37
Finally, the development of a combination of proguanil and
atovaquone (Malarone) would provide another useful way for the treatment of
malaria.38
Atovaquone, a hydroxynaphthoquinone derivative, is an analog of
ubiquinone, a parasite mitochondrial electron-carrier which is a cofactor of the
dihydroorotate dehydrogenase enzyme. Atovaquone acts by inhibiting parasite
mitochondrial electron transport. However, the mechanism of synergy of
proguanil with atovaquone is complex. This combination is well tolerated and
more effective than CQ alone, CQ-SP 39
or mefloquine,40
against acute
uncomplicated multidrug resistant P. falciparum. It is also effective in regions
where proguanil alone is ineffective due to resistance. Unfortunately, atovaquone
is expensive and not easily affordable in most of the African countries.
1.2.6 Artemisinin derivatives
To date, resistance has emerged to all classes of antimalarial drugs
except artemisinins41,42
and therefore, most of the African countries have adopted
artemisinin-based combination therapy (ACT) as the first-line treatment for
uncomplicated malaria but some recent reports have challenged credibility and use
of ACTs due to reduced efficacy of certain artemisinin-based combination
therapies.6 Artemisinin derivatives are the fastest acting antimalarial drugs.
43 Four
compounds that have been used are the parent artemisinin extracted from Chinese
herb ‘qinghao’ (Artemisia annua.)44
and three derivatives that are actually more
active than artemisinin itself.43
One of them is a water-soluble hemisuccinate
artesunate and the other two are oil-soluble ethers, artemether and arteether. All of
them are readily metabolized to the biologically active metabolite,

Chapter-1 Introduction
11
dihydroartemisinin. Artemisinin is active in nanomolar concentrations in vitro on
both CQ-sensitive and resistant P. falciparum strains. These drugs are fast acting
and act against gametocytes, the sexual forms of the parasite that infect
mosquitoes. The treatment of several million patients with artemisinin derivatives
for acute malaria failed to detect any significant toxicity,45
even for pregnant
women,46
despite the fact that neurotoxicity was observed in animals with higher
doses than used clinically. Artemisinin and its derivatives appear to be the best
alternative for the treatment of severe malaria47
and arteether has been included in
the WHO List of Essential Drugs for the treatment of severe multidrug resistant P.
falciparum infection. Arteether is having advantage over artemether as it is more
lipophilic and gives less toxic metabolite (ethanol versus methanol). In this
family, The Walter Reed Institute of Research has patented a stable, water-soluble
derivative called artelinic acid that is now being tested in animals. When
compared with other artemisinin derivatives, artelinic acid had the highest plasma
concentration, the highest oral bioavailability, the longest half-life, the lowest
metabolism rate and the lowest toxicity at equivalent doses.48
A key advantage of
these endoperoxide containing antimalarial agents, which have been used for
nearly two decades is the absence of any drug resistance. When several strains of
P. berghei or P. yoelii were exposed to a selection pressure by artemisinin or
synthetic analogs within infected mice, resistance proved very hard to induce. A
low level of resistance has been observed which disappeared as soon as the drug
selection pressure was withdrawn. Furthermore, with a synthetic analog of
artemisinin, BO7, resistance to the drug was lost when drug pressure was
removed; it was not regained once drug pressure was re-applied.49
Remarkably,
the introduction of artemisinin derivatives in routine treatment in some areas of
Southeast Asia has been associated with a significant reduction of incidences of
falciparum malaria. In fact, artemisinin derivatives prevent gametocyte
development and therefore reduce the transmission.
The major drawback of artemisinin derivatives is their short half-life (3-
5 hours). When used in monotherapy, a treatment as long as 5 days is required for
complete elimination of the parasites. They are then preferentially used in
combination with other antimalarial agents such as sulfadoxine-pyrimethamine,50
benflumetol,45
mefloquine51
or chlorproguanil-dapsone to increase cure rates and
to shorten the duration of therapy in order to minimize the emergence of resistant

Chapter-1 Introduction
12
parasites. Combination of artemether or artesunate and mefloquine has been used
in areas of multidrug resistance in Southeast Asia. When associated with
lumefantrine (benflumetol, a slow eliminated oral drug,) artemether is as effective
as the artesunate-mefloquine combination and better tolerated. Artemether clears
most of the infection and the lumefantrine concentration that remains at the end of
the 3 to 5-day treatment course is responsible for eliminating the residual
parasites. This combination is safe in patients with uncomplicated falciparum
malaria and even in children. It clears parasites rapidly and results in fewer
gametocytes carriers. The only limiting factor of artemisinins in comparison to
common antimalarials is their treatment cost and duration of course in
monotherapy, which make them least affordable in case of the low income
countries sharing world’s major malaria episodes and its associated deaths.52
The
relatively high cost and erratic supply of the natural parent compound artemisinin
make it necessary to develop new synthetic and cheap endoperoxide-based
antimalarials.53,54
1.2.7 Other antimalarials
Yingzhaosu A and Yingzhaosu C, two naturally occurring peroxides
with bisabolene skeleton have been isolated from the roots of the plant yingzhao
(Artabotrys uncinatus), both showing significant antimalarial activity in vitro.54-56
Arteflene a synthetic analog of Yingzhaosu A is under clinical developement. The
compound has low human toxicity as it interacts with specific parasite proteins,
fast acting and equipotent to artemisinin and has better chemical stability. Its
single dose can lasts for longer period of time.57-59
1.3 New antimalarial drug development approaches
Due to complex life cycle and different causative strains of the parasite
and high mutability of the genome of Plasmodium falciparum, development of
malaria vaccine has met with failures time and again hence, identification of new
targets have been explored for the development of new drugs acting via different
mechanisms in order to avoid fast development of resistance.60
The P. falciparum
genome sequencing has revealed a number of new targets for drug and vaccine
development.61,62
But discovery of new targets, the developmental time and costs
are also likely to be too high for such an approach. Inhibition of the proteases that

Chapter-1 Introduction
13
specifically degrade hemoglobin in the parasite food vacuole, the plasmepsins,
falcipains and falcilysins, is an interesting area of research.8 Also very promising
are targets associated with pathways specific to the apicoplast, such as the
shikimate pathway and the 2-deoxy-D-xylulose-5-phosphate (DOXP) pathway.63
Cellular and biochemical targets in the mosquito vector are worthy of
consideration as well.64
One group of workers has demonstrated the viability of
using a molecular connectivity QSAR approach to identify new active antimalarial
agents.65
1.3.1 Old targets new compounds
The main challenges associated with antimalarial drug development
program are high costs for developing new targets, development of resistance to
the available drugs by the parasite and general lack of interest among the major
pharma players, as the disease belongs to the poor third world countries. Other
challenges associated with it are the cost, efficacy, target specificity, oral-
bioavailability and safety. A new antimalarial drug must meet all standard drug
development criteria of a new molecule so, an alternative strategy is the
exploitation of the existing targets.66
As the major target population is from Africa
and other developing countries, any new drug development program must address
the challenge of cost and drug resistance along with all standard drug development
criteria. The free heme liberation in the parasite food vacuole is also an “old” but
always an attractive pharmacological target. It could be the most specific target
that can be exploited since it comes from the hemoglobin digestion by the parasite
that occurs only in infected erythrocytes.
Many chemical entities are directed toward this well known target, such
as CQ and artemisinin derivatives. CQ is a cheap and easy-to-prepare molecule
that has proved its credibility as it is highly effective, safe and well-tolerated drug
for treatment and prophylaxis. However, the spread of resistance has resulted in a
huge reduction in the utility of CQ. Many chemical modifications have been
attempted to obtain a molecule as affordable as CQ and equally active even on
resistant strains. These modifications carried out are substitutions in the quinoline
nucleus, variations in the side chain, synthesis of bisquinolines and more recently,
the introduction of a ferrocenyl moiety.67
A little modification could provide a

Chapter-1 Introduction
14
new compound that would be active on resistant strains; but the wait for a safe and
effective chloroquine alternative still continues.
Artemisinin and its derivatives (artemether, arteether and artesunate) are
increasingly used in Asia and Africa where multidrug-resistant P. falciparum is
prevalent. They are rapidly effective and well-tolerated treatments, but the total
synthesis is too complex to be exploited and the yield of extraction from the plant
is too low, despite research efforts toward the enhancement of artemisinin or one
of its precursor’s productions in A. annua. As a result, they remain expensive
treatments that are hardly accessible to people in endemic areas. The cost will also
be limitative for sophisticated artemisinin derivatives.68
Synthetic trioxanes,
simplified analogs of artemisinin retaining the crucial endoperoxide bridge have
been developed, but none of them could enter into clinical trials successfully.69
1.3.2 Proteases-general introduction
Proteases or proteolytic enzymes found in plants, bacteria, virus,
protozoa, fungi and mammals are essential for replication and are involved in a
series of essential metabolic and catabolic processes, such as protein turnover,
Table I: Proteases in disease propagation and their functions70
Protease Function Involved in disease
HIV-1 Protease HIV replication AIDS
Renin Generation of angiotensin I Hypertension
Thrombin Blood coagulation Stroke, vascular clots
Tryptase Phagocytosis Asthma
Cathepsin K Bone resorption Osteoporosis
ACE Generation of angiotensin II Hypertension
Neutral endoprotease Release of ANP Hypertension
Rhinovirus 3C protease Viral replication Common cold
Falcipain Proteolysis Malaria
Plasmepsin I & II Proteolysis Malaria
Cruzain Proteolysis Chagas’ disease

Chapter-1 Introduction
15
digestion, blood coagulation, apoptosis, fertilization, cell differentiation and the
immune response system.71
Proteases have enormous commercial importance in
industrial enzyme applications, mainly in detergent, food and leather industries.72
Proteases selectively catalyze hydrolysis of large polypeptides into smaller
peptides and further into amino acids thus facilitating their absorption by the
cells.73
Their virtual control over protein synthesis in human beings and other
microorganisms enable them to regulate physiological processes.74
They have
been implicated in many health problems (Table-I) involving the circulatory
system, immune system, allergies, sports injuries and infectious diseases. They
catalyze important proteolytic steps in tumor invasion or in infection cycle of a
number of pathogenic microorganisms and viruses. This makes proteases a
valuable target for the development of newer drugs.
1.3.3 Protease inhibitors as drugs
Proteases are involved in a number of diseased states and thus have
gained tremendous attention as therapeutic targets with regard to viral and
parasitic infections, stroke, cancer, Alzheimer’s disease, neuronal cell death and
arthritis. They are designated either as endo or exopeptidases that cleave peptide
bonds within a protein or peptide and remove amino acids from the N or the C-
terminus. Depending on the catalytic residue responsible for peptide hydrolysis,
the proteases have been divided into classes like serine, cysteine, aspartic and
metalloproteases.71,75
NNH
NH
O
O
OH
N
O CH3
N
O
NH2
CH3
CH3 S
N
N NH
NH
NHS
N
CH3
CH3
CH3
O
CH3
CH3
O OH
O
O
NSH
O
CH3
COOH
NNH
O
CH3
COOH
Saquinavir Ritonavir
Captopril Enalapril
EtOOC
Figure 3: Protease inhibitor drugs available in market
Proteases are crucial for propagation of certain diseases and are
emerging as promising therapeutic targets to be used in the treatment of these

Chapter-1 Introduction
16
diseases. These diseases include inflammation, tumor progression, parasitic,
fungal and viral infections (e.g. malaria, schistosomiasis, C. albicans, HIV, herpes
and hepatitis), immunological, respiratory, neurodegenerative and cardiovascular
disorders. Protease inhibitors thus, have considerable potential utility for
therapeutic intervention in a variety of disease states.73,76
There are many
examples of protease inhibitors as highly successful drugs.
The human immunodeficiency virus protease (HIV-1 protease) has
proved to be an attractive target due to its essential role in the replicative cycle of
HIV. Several low molecular weight HIV-1 protease inhibitors (MW < 1000 Da)
are now used as drugs, including saquinavir, ritonavir, indinavir, nelfinavir and
amprenavir. These are among the first successful examples of receptor/structure
based designer drugs. Keeping in mind the prior knowledge of inhibition of other
aspartic proteases (eg. renin) these compounds were designed and developed by
docking these structures on the active site of HIV-1 protease.70
Captopril and
enalapril are the examples of inhibitors of a metalloprotease, angiotensin-
converting enzyme (ACE) used successfully for the control of hypertension.77
1.3.4 Cysteine proteases and related aspects
Cysteine (thiol) proteases exist in three structurally distinct classes,75
which are papain-like (e.g. cathepsins), ICE-like (caspases) and picorna-viral type.
Several members of the cysteine protease family of enzymes have been implicated
as possible causative agents in a variety of diseases. More notable examples
include cathepsin K in degradation of bone matrix, cathepsin-L and S for MHC-II
antigen presentation, the caspases in programmed cell death, rhinovirus 3C
protease for viral processing, falcipain and cruzain in parasitic infections and the
possible role played by the gingipains in periodontal diseases. The biggest
problem in designing inhibitors for cysteine proteases is the similarity in their
substrate affinities and proteolytic mechanisms with the serine proteases.
Although, the spatial configurations of the catalytic triads of serine and cysteine
proteases are quite similar, it appears that the oxyanion hole and the negatively
charged tetrahedral intermediate are the central features of the catalytic
mechanisms of serine proteases, while cysteine proteases stabilize the later more
neutral acyl intermediate with the help of imidazole ring of histidine. This

Chapter-1 Introduction
17
mechanistic difference could be exploited in the development of potent, reversible
and selective transition state analogs as potential drugs.
Like serine proteases, cysteine proteases tend to have relatively shallow
solvent exposed active sites that can accommodate short substrate/inhibitor
segments of protein loops or strands. Most of the inhibitors developed to date tend
to be 2-4 amino acids or their equivalents in chain length, interacting with the
nonprime subsites of the enzymes and terminating with various electrophilic
isosteres. Recently it has been convincingly demonstrated, for a wide range of
proteases including cysteine protease, that they universally bind their
inhibitors/substrates in extended or β-strand conformations.
1.4 Malarial cysteine proteases: new target for chemotherapy
1.4.1 Malarial cysteine proteases’ role in hemoglobin degradation
Erythrocytic stage of the malaria parasite P. falciparum has developed
complex proteolytic machinery for haemoglobin digestion.78
Malaria parasite
ingests and degrades most of the host cell hemoglobin during the morphologically
separate phases inside the erythrocyte (ring, trophozoite and schizont stages).79
Hemoglobin degradation is essential to the survival of the parasite, as interruption
of this process with a variety of protease inhibitors leads to death.80
The amino
acids derived from hemoglobin degradation are incorporated into parasite proteins
or utilized for energy metabolism.81,82
It has also been suggested that removal of
hemoglobin through degradation provides space in the red blood cell for the
growing parasite and prevents premature erythrocyte lysis.83
Massive degradation of hemoglobin generates a large amount of toxic
heme, which causes membrane damage due to its peroxidative properties.84
The
released hematin is detoxified to a cyclic dimer, -hematin. These dimers
crystallize to give hemozoin (insoluble malaria pigment).11
This massive catabolic
feat is carried out in a specialized organelle, the food vacuole and is most
pronounced during the trophozoite stage of development.10
More recent studies
indicate that the crystallization process from -hematin to hemozoin takes place in
or closely associated with, neutral lipid nanospheres in the aqueous content of the
vacuole.85
A major function of the food vacuole is to degrade the host red cell
hemoglobin sequestered through the cytostome machinery and provide amino

Chapter-1 Introduction
18
acids to the parasite. This is brought about by a variety of proteases and the
inhibition of this process is well investigated as a drug target.86,87
These enzymes
act at an optimum pH in the range of 4.5-5.0, i.e. pH of the digestive vacuole.78
A
low digestive vacuolar pH further promotes biomineralization of heme to
hemozoin.88
Endocytosis of host cell cytosol
Oxyhemoglobin
Plasmepsins I, II
(>20AA) Peptides
Falcipains (2, 3)
Small (6-8AA) Peptides
Falcilysins
DPAP
Amino Acids
Ferroproto-
porphyrinHeme
Polymerization
Hemazoin
Food Vacuole
2O-2H2O2
H+O2
H2O+1/2O2
Amino peptidaseSmall Peptides
Globin
Plasmepsin (IV & HAP)
Figure 4: Role of cysteine proteases in hemoglobin digestion89,90
In P. falciparum, proteases involved in hemoglobin degradation have
been proposed to act in a semi-ordered fashion as outlined in Fig. 4. Ten aspartic
proteases named plasmepsins (PMs) were identified in the genome of P.
falciparum and four of them, namely palsmepsin I, II and IV and histo-aspartic
protease (HAP) participate in hemoglobin degradation.89,90
Plasmepsin I and II
have been postulated to initiate hemoglobin degradation, by hydrolyzing the
peptide bond between residues Phe33 and Leu34 of native hemoglobin. 90,91
This
initial cleavage is thought to promote unfolding and release of the heme moiety
followed by further degradation by the plasmepsins,87
the cysteine proteases
(named falcipain-2 and falcipain-3),92-94
a metalloprotease (named falcilysin)95
and the lately discovered dipeptidyl aminopeptidase (DPAP).96
However, the
precise sequence of events particularly whether a plasmepsin or a falcipain
catalyzes the initial degradation step is still under debate.80
Disruption of each plasmepsin gene was achieved and these studies
confirmed that all these four vacuolar enzymes play important roles for parasite

Chapter-1 Introduction
19
development though not an essential one, as PMs knockouts possess the ability to
compensate for the loss of individual plasmepsin function.97
On the other hand, it
is known that inhibition of cysteine proteases turns out to be lethal for parasites.
Cysteine protease inhibitors irreversibly block the rupture of host cell membrane,
which prevent parasites to invade fresh erythrocytes.98
The metalloprotease,
falcilysin is only able to cleave small polypeptides, up to 20 amino acids,
producing even shorter oligopeptides.95
DPAP1 cleaves off dipeptides from
hemoglobin-derived oligopeptides in the food vacuole.96
Finally, small peptides
may be pumped out of the food vacuole into the cytoplasm and an amino
peptidase activity provides amino acids essential for parasite’s survival.99,100
1.4.2 Falcipains’ role in proteolysis
The best characterized P. falciparum cysteine proteases initially known
as trophozoite cysteine protease (TCP) and later termed as falcipains share
sequence identity and other features with papain family cysteine proteases.
Falcipain-1 (FP-1),92
nearly identical copies of two falcipain-2 (FP-2) (falcipain-2
and falcipain-2’, also known as falcipain-2A and falcipain-2B, respectively)93,101
and falcipain-3 (FP-3)94
are all expressed during the erythrocytic stage of the
Plasmodium life cycle.102
Falcipain-2’ is not fully understood and the knock out
studies have revealed that there is no significant change in its absence in parasite’s
function in erythrocytic stage.101
FP-1, located on chromosome 14, shares approximately 38-40%
sequence identity with FP-2 and FP-3. The function of FP-1 remains uncertain
because of its low abundance and inadequate systems for the production of
recombinant enzyme. FP-1 has been proposed to play a role in parasite invasion as
a relatively specific inhibitor of FP-1 blocked parasite invasion of host
erythrocytes independent of hemoglobin degradation.103
However, a subsequent
study showed that FP-1 knockout parasites developed normally in erythrocytes,
suggesting that this protease alone is neither required for parasite invasion nor
during the intracellular development within erythrocytes.92
The gene disruption
studies clearly demonstrated an important role for FP-1 in oocyst production
during parasite development in the mosquito midgut, but not an essential role in
asexual or gametocyte/gamete development. FP-1 could be directly involved in
the actual transition from gamete to oocyst by activating proteins by proteolytic

Chapter-1 Introduction
20
processing or, if secreted, it could degrade the peritrophic matrix or midgut
endothelium facilitating the migration of the ookinete.104
In contrast, the well-characterized function of FP-2 and FP-3 includes
degradation of host hemoglobin required for sustaining the metabolic needs of
rapidly growing parasite.93
They show a high level of amino acid sequence
homology (65%) but a different substrate specificity,105
although both enzymes
favor substrates with Leu at P2.93,94
FP-2 and FP-3 are primarily localized in the
food vacuole, consistent with their roles in hemoglobin digestion. Both falcipains
are synthesized over fairly long interval during the erythrocytic cycle as
membrane-bound pro-forms that are processed to soluble mature forms. However,
FP-2 is synthesized earlier, in the beginning of early trophozoites and is processed
more quickly than FP-3 which showed peak expression in late trophozoites/early
schizonts stage. Cysteine protease inhibitors and the lactone antibiotic brefeldin-
A, blocked the processing of both enzymes, suggesting that FP-2 and FP-3 are
trafficked to the food vacuole via the endoplasmic reticulum/Golgi network and
that they are processed by autohydrolysis (which occurs at neutral pH) before
arrival at the food vacuole.106
Genetic ablation of FP-2 gene, located on chromosome 11 underscored
the critical importance of this protease in hemoglobin degradation,107
which is
actually considered the prime target for discovery of novel antimalarial drugs.
During the late trophozoite and schizont stages, FP-2 is involved in the
degradation of erythrocyte membrane skeletal proteins, including ankyrin and the
band 4.1 protein.108
This activity displays a pH optimum in the range of 7.0-7.5
and is thought to contribute to destabilization of the erythrocyte membrane,
leading to host cell rupture and release of the mature merozoites, hence FP-2
shows stage specific activity i. e. in early trophozoite stage it degrades
hemoglobin in acidic pH and in neutral pH it cleaves the host erythrocytic skeletal
membrane proteins ankirin and protein 4.1, vital for the stability of RBC
membrane. 93,109
The disruption of FP-2 gene revealed that there is transient block in
hemoglobin hydrolysis but the loss of this enzyme alone is not sufficient to cause
parasite lethality, the parasite recovered from the defects later presumably due to
expression of FP-3 in the later stage of life cycle of the parasite, thus suggesting
the participation of additional cysteine proteases for parasite invasion and growth

Chapter-1 Introduction
21
in human erythrocytes. Alternatively, the development of parasite lethality might
require concomitant deletion of multiple cysteine proteases. Unlike other
falcipains, the FP-3 gene could not be disrupted, but its replacement with a tagged
functional copy has been recently achieved.110
These studies revealed that FP-3 is
essential to erythrocytic parasites, suggesting that efforts to develop cysteine
protease inhibitors as antimalarial drugs should probably be focused on FP-2 and
FP-3.
FP-2’ was the last cysteine protease to be identified through analysis of
genetic variability among isolates of P. falciparum. These studies revealed that P.
falciparum contains a nearly identical copy of the FP-2 gene, also located on
chromosome 11, encoding a distinct enzyme. These two genes are actually
paralogs, which share 96% identity at the nucleotide level and 93% identity at the
amino acid level. The products of these paralogs were designated as FP-2 and FP-
2’.111
Their amino acid sequences differ at seven positions within their mature
forms, including three amino acid replacements localized close to residues that are
predicted to interact with substrate and consequently may affect its affinity or
specificity.112
Recombinant FP-2’ protein expressed in bacteria exhibits protease
activity as FP-2 in erythrocytic parasites but importantly, the recombinant FP-2’
cleaves host ankyrin but not protein 4.1. Functional analysis revealed similar
biochemical properties to those of FP-2, including pH optima (pH 5.5-7.0),
reducing requirements and substrate preference. Notwithstanding its predicted
hemoglobinase function, the P. falciparum FP-2’ may contribute and orchestrate
selective proteolytic events during the exit of malaria parasite from human red
blood cells.113
However, gene disruption studies demonstred that FP-2’ knockout
parasites showed no differences in morphology neither in sensitivities to cysteine
or aspartic proteases compared to the wild-type parasites.110
These results indicate
a subordinate role for FP-2’ in parasite development.
All falcipains except FP-1 are involved in the conversion of
proplasmepsins into their active forms. FP-2’ and FP-3 are expressed later in the
erythrocytic cycle of the parasite. This would suggest that FP-2 might serve as the
dominant proplasmepsin maturase, although the contribution of FP-2’ and FP-3
cannot be ruled out.114

Chapter-1 Introduction
22
1.4.3 General structural features of falcipain-2
The mature FP-2 is a single polypeptide chain of 241 amino acids,
synthesized during the trophozoite stage as a membrane-bound proenzyme
comprising 484 amino-acid residues.93
The proenzyme is transported to the food
vacuole through the endoplasmatic reticulum/Golgi system and during this
process the N-terminal 243 residues containing the membrane anchor are
proteolytically removed. An autoproteolytic processing mechanism was suggested
on the basis of inhibitor studies110
and from the observation that the recombinant
proenzyme undergoes spontaneous processing during in vitro refolding.93
The crystal structure of the free FP-2 (inactivated by iodoacetamide, pdb
code 2GHU)115
and in complexation with cystatin (1YVB pdb code)116
(Fig. 5)
has been deposited in the Protein Data Bank. More recently, the crystal structures
of FP-2 in complexation with epoxysuccinate E-64 (3BPF) and FP-3 with
aldehyde leupeptin have also been reported.117
FP-2 generally adopts a classic papain-like fold in which the protease is
divided into L (left) and R (right) domains. The catalytic residues Cys42, His174
and Asn204 (Cys25, His159 and Asn175 following papain convention) are located
in a cleft at the junction between the two domains (Fig. 6). Like papain and the
majority of C1A proteases, FP-2 also has the disulfide bond Cys168-Cys229 to fix
the upper loops defining the S2 and S1’ substrate-binding sites. An additional
Cys99-Cys119 disulfide bond, predicted on the basis of homology modeling
studies,118
appears to play an important role together with the Cys73-Cys114
disulfide in stabilizing the long 4/ 2 loop (~30 residues) that blankets the lateral
surface of the L-domain.
The most extraordinary features of FP-2 are two unique structural
elements: a ‘‘noselike’’ projection (at the N-terminus) connecting the L and R
domains and an ‘‘arm-like’’ structure (near the C-terminus of the mature protein)
extending away from the protease surface.116
FP-2 nose consists of 17 amino acids
and deletional analysis of this sequence119
confirms that FP-2 nose is fundamental
for proper folding; generally it possesses a short significant element of secondary
structure and the amino acid Glu138 forms a buried hydrogen bond with Tyr4 and
a salt bridge with Arg5. This clearly indicates that FP-2 nose, even if shorter than
the standard papain family prodomain, may play a critical role as chaperone for
protease folding. Furthermore, in a different way from the other members of the

Chapter-1 Introduction
23
a
b
Figure 5: a) Crystal structure of FP-2 and cystatin complex. b) Overall structure of FP-2-cystatin is
depicted in stereo in a conversational orientation for papain-like cysteine proteases: with
the L domain (Left), the R domain (Right) and the active site on the top facing
forward.116
papain family, interactions of a residue in the protein core (Glu138) with the FP-2
nose may provide the additional binding energy essential to stabilize initial
interactions for folding.

Chapter-1 Introduction
24
FP-2 arm is composed of 14 amino acids located at the distal end of the
R domain (residues 183-196). It is a protruding arm-like structure of two extended
strands connected by an abrupt turn and generally it does not establish any
contact with the protease core. Mutagenesis studies suggest that FP-2 arm may
interact with the natural substrate of FP-2, hemoglobin. However, because FP-2
arm is > 25A˚ away from the protease active site, FP-2 arm probably functions in
hemoglobin hydrolysis through distal-site binding rather than direct participation
in substrate cleavage.
Figure 6: Crystal structure of FP-2 and FP-3 bound to small molecule inhibitors; implications
for substrate specificity 117
The 14-residue hemoglobin-binding -hairpin exhibits a high degree of
conformational flexibility when freely exposed to solvent bulk. The large
positional deviations exhibited by the -hairpin, while comparing the structure of
the free FP-2 and that of the FP-2-cystatin complex further underscores an
inherent flexibility and plasticity of this element that may have functional
implications in hemoglobin binding.
Recently, the co-crystallized FP-3-leupeptin structure (3BPM) has been
reported; FP-3 also possesses the two unique insertions discussed above, that
distinguishes plasmodial cysteine proteases from all other structurally
characterized papain family enzymes: the noselike domain located at the N-

Chapter-1 Introduction
25
terminus (AA 1-25) and the flexible 14-residue -hairpin at the C-terminus
outside of the two unique motifs discussed above. The rest of FP-2/3 is
structurally similar to homologous proteases in the C1A family. The active site is
located in a cleft between the structurally distinct domains of the papain-like fold.
The S2 pocket is the major determinant of specificity for most cysteine proteases
and its predominantly hydrophobic nature in FP-2 is confirmed by the fact that
FP-2 has a strong preference for substrates with a hydrophobic residue,
particularly Leu, at the P2 position.93
The crystal structure reveals that although
the S2 subsite is generally hydrophobic, the electronegative side chain of Asp234
is positioned very deep in the S2 pocket. Therefore, the presence of basic side
chain in the P2 position could form a salt-bridge interaction with Asp234, adding
further insights useful to explore the structural determinants of FP-2 specificity.
1.5 Falcipain-2 inhibitors
To date, many chemotypes have been identified as inhibitors of FP-2
such as peptides, peptidomimetics, isoquinolines, thiosemicarbazones, nonpeptidic
chalcones, pyrimidinenitriles and others which are able to inactivate the enzyme in
a reversible or irreversible manner. The majority of malarial cysteine protease
inhibitors are peptidic or peptidomimetic compounds in which the hydrolysable
amide bond is replaced by an electrophilic functionality. In this way, the catalytic
thiol of the enzyme reacts with the inhibitor to form a covalent complex. Until
recently, most potent cysteine protease inhibitors were irreversible inhibitors, in
which the electrophilic “warhead” would alkylate the enzyme, through
nucleophilic displacement or conjugate addition. Examples of this approach
include fluoromethyl ketones, acyloxymethyl ketones, vinyl sulfones, or
epoxysuccinates.
Alternatively, potent reversible inhibition can be achieved through
highly electrophilic warheads like aldehydes, ketoamides and nitriles, which form
reversible, covalent bonds to the thiol active site. As synthetic peptide inhibitor of
the P. falciparum schizont cysteine protease, Pf 68 inhibited erythrocyte invasion
by cultured parasites120
. The most effective peptide, GlcA-Val-Leu-Gly-Lys-
NHC2H5 inhibited the protease and blocked parasite development at high
micromolar concentrations (IC50 = 900 μM)120
. The natural triterpene betulinic
acid and its analogs (betulinic aldehyde, lupeol, betulin, methyl betulinate and

Chapter-1 Introduction
26
betulinic acid amide) caused concentration dependent alterations of erythrocyte
membrane shape towards stomatocytes or echinocytes according to their hydrogen
bonding properties.121
Thus, analogs with a functional group having a capacity of
donating a hydrogen bond (COOH, CH2OH and CONH2) caused formation of
echinocytes, whereas those lacking this ability (CH3, CHO, COOCH3) induced
formation of stomatocytes. Both kinds of erythrocyte alterations were prohibitive
with respect to P. falciparum invasion and growth; all compounds were inhibitory
with IC50 values in the range of 7-28 M and the growth inhibition correlated well
with the extent of membrane curvature changes assessed by transmission electron
microscopy. Although these results do not demonstrate the levels of inhibition
expected to be therapeutically relevant, they suggest that a specific protease
activity is required for erythrocyte invasion by malaria parasites and thus is a
potential target for antimalarial drugs.
The active site of papain and related proteases is large and can be
divided into pockets at each side of the catalytic site, S1-S4 and S1’-S3’, with each
pocket accommodating one amino acid residue of the peptide substrate.122
The
general structure of a cysteine protease inhibitor (Fig. 8) should comprise an
electrophilic moiety for interaction with the cysteine residue of the enzyme and
one or two series of substituents, P1-P4 and/or P1’-P3’, responsible for interactions
with the different binding pockets of the protease.
The irreversible inhibitors are better characterized by means of kinetic
constants: the dissociation constant Ki measures the affinity of inhibitor for the
enzyme, the kinac describes the velocity of formation of covalent enzyme inhibitor
complex and the second-order rate constant of inhibition k2nd that determines their
potency.
1.5.1 Peptide-based falcipain-2 inhibitors
1.5.1.1 Peptidyl fluoromethyl ketones
Fluoromethyl ketones are in general, highly reactive and selective
irreversible inhibitors of cysteine proteases.123
A number of peptidyl fluoromethyl
ketones were tested against FP-2,124
previously named as P. falciparum
trophozoite cysteine proteinase (TCP). In this context CBZ-Phe-Arg-CH2F (1)
(Fig. 7) proved to be the most potent inhibitor of FP-2 at picomolar concentration
(IC50 = 0.36 nM), to block the hemoglobin degradation (IC50 = 0.10 mM) and to

Chapter-1 Introduction
27
inhibit the development of cultured malaria parasites in the nanomolar range (IC50
= 64 nM).125
CBZ-Phe-Arg-CH2F (1) was effective at nanomolar concentrations
on four different strains of P. falciparum (Itg2, FCR3, W2, D6) and proved to be
nontoxic to four human cell lines.
A number of fluoromethyl ketones characterized by the presence of a
morpholineurea (Mu) group at P3 position was tested against FP-2; the most
effective inhibitors were Mu-Phe-HomoPhe-CH2F (2) (IC50 = 3 nM) and Mu-Leu-
HomoPhe-CH2F (3) (IC50 = 0.42 nM). Mu-Phe-HomoPhe-CH2F (2), which proved
to be the most effective inhibitor against P. vinckei cysteine protease (IC50 = 5.1
nM against P. vinckei protease) is biochemically similar in terms of molecular
mass, pH optimum, substrate specificity and inhibitor sensitivity to FP-2. It was
also tested in vivo in a murine malaria model.126
When administered parenterally
N NH
NH
CH2F
O
O
O
O
O NH
NH
CH2F
O
O
NH
O
NH2
NH
N NH
NH
CH2F
O
O
O
O
CH3
CH3
(1)(2)
(3)
Figure 7: Structures of peptidyl fluoromethyl ketones against FP-2
four times a day for 4 days to P. vinckei infected mice, the inhibitor (2) cured 80%
of the treated mice. However, the use of peptidyl fluromethyl ketones in therapy
had an important limitation due to their proteolysis by serum, tissue and gut
proteinases. These in vivo assays clearly evidenced that the serum half-life of 2
was short and that the efficacy of the inhibitor could be improved only by its
frequent administrations
1.5.1.2 Peptidyl vinyl sulfones
Vinyl sulfones (VS) well known covalent inhibitors of FP-2 are able to
inactivate the enzyme by irreversible addition of the thiol group of active site
Cys42 to the electrophilic vinyl sulfone moiety, which behaves as a Michael
acceptor.127
It is proposed that a hydrogen bond from the protonated active site
His174 to one of the sulfone oxygens might polarize the vinyl group, thus further
activating the -carbon proposed that a hydrogen bond from the protonated active
site His174 to one of the sulfone oxygens might polarize the vinyl group, thus

Chapter-1 Introduction
28
further activating the -carbon proposed that a hydrogen bond from the protonated
active site His174 to one of the sulfone oxygens might polarize the vinyl group,
thus further activating the -carbon toward nucleophilic attack (Fig. 9).123
NH
NH
O
O
SR
1'
OO
R3
R1
R2
P1'
P1
P2
P3
R1' = Phenyl, O-R, NH-R R1 = Ala, homoPhe, Phe, Arg, ValR2 = Phe, LeuR3 = Morpholine, N-Me-piperazine
Figure 8: General structure of vinyl sulfones, vinyl sulfonate esters and vinyl sulfonamides
The obtained negative charge at the -carbon is eliminated via protonation by the
histidinium residue.128
RNH
S
R
O
O
R
H-His174
H-Asp204
S
Cys42
S
Cys42
RNH
S
R
O
O
R
H-His174
H-Asp204
His174
S
Cys42
RNH
S
R
O
O
R
+ +
-
Figure 9: Mechanism of FP-2 inhibition by vinyl sulfones
Furthermore vinyl sulfones proved to be unreactive toward serine
proteases, metalloproteases, aspartyl proteases, nonactive-site cysteines and
circulating thiols such as glutathione; moreover, substituted vinyl sulfones proved
to be less reactive toward nucleophiles than the analogous vinyl ketones or esters
and sufficiently inert without the target.127
N NH
NH
O
OO
SO
O
N NH
NH
O
OO
CH3
CH3
SO
O
N NH
NH
O
ON
CH3
CH3
SR
OO
CH3
(4) (5) (6) R = Phenyl
(7) R = 2-Naphthyl
Figure 10: Structures of vinyl sulfones containing morpholineurea
A series of vinyl sulfones as novel peptide-based FP-2 cysteine
proteinase inhibitors were reported in P. vinckei.129
For most of the compounds,
inhibitory effects against FP-2 and P. vinckei proteases were similar; however, in
some cases efficacy against the two related enzymes proved to be different and
this clearly indicated a critical role of the peptide portion of the inhibitors in their

Chapter-1 Introduction
29
specificity toward similar enzymes. In this context, Mu-Phe-HomoPhe-VSPh (4)
(Fig. 10) has been proven to be a mid-nanomolar inhibitor of both proteinases
(IC50 = 0.08 M against FP-2 and IC50 = 0.1 M against P. vinckei protease); on
the contrary, the structurally similar peptide Mu-Leu-HomoPhe-VSPh (5)
containing a Leu residue at P2 site improved effectiveness against FP-2, while
activity against the P. vinckei enzyme decreased (IC50 = 0.003 M against FP-2
and IC50 = 0.2 M against P. vinckei protease).
An improved in vivo effectiveness resulted when the morpholineurea
group was replaced with the N-methylpiperazineurea (N-Pipu) group in
compound (6) due to increased aqueous solubility and bioavailability. The phenyl
vinyl moiety was also replaced with a 2-naphthalene group.130
Both of the
compounds (6 and 7) (Fig. 10) showed antimalarial effects correlating with
potency against FP-2. Both of them strongly inhibited hemoglobin degradation,
development and metabolic activity in cultured P. falciparum parasites in the
nanomolar range. The compounds (6 and 7) displayed better activity than the
parent molecule (5) in the inhibition of FP-2 and this clearly showed positive
effect of the replacement of morpholine nucleus with the N-methylpiperazine.
Also the extension of the aromatic area at the P1’ site (i.e. 7) resulted more
fruitful. When the most active compound (7) was administered orally to mice, it
markedly delayed the progression of murine malaria and cured about 40% of the
treated animals. However, many problems common to peptide-based inhibitors
still remained, like: (i) poor pharmacological profile, (ii) reduced absorption
through cell membranes, (iii) susceptibility to protease degradation and (iv)
limited bioavailability.
The structure-activity relationships of a new series of peptidyl vinyl
sulfones, vinyl sulfonate esters and vinyl sulfonamides (Table-II, III) for
inhibition of the protease FP-2 and of P. falciparum development has been
reported.131
Compounds containing the core sequence Phe-homoPhe showed
modest activity against FP-2 and substitutions at position P3 (Fig. 11) in these
compounds had relatively little impact on activity. Compounds with the core
sequence Phe-O-(phenyl)Ser had similar activities, with IC50 for the inhibition of
FP-2 in the mid-to-high nanomolar range. The Leu-homoPhe compounds revealed
to be very active against FP-2, with IC50 values generally in the high picomolar to
low nanomolar range. Identical compounds, except for the P1’ substituent

Chapter-1 Introduction
30
displayed the general rank order of activity: vinyl sulfonate esters > vinyl
sulfonamides > phenyl vinyl sulfones (Tables-II, III). The vinyl sulfones (8) were
potent inhibitors with IC50 in the low nanomolar range.
NNH
ON
OH
N
N
CH3
N
N
OH
N
O
N
O
OCH
3
N
OH
N
N
R NH
NH
O
O
CH3
CH3
SO
O
R NH
NH
O
O
CH3
CH3
SNH
OO
a b c d
e f g h
(9)(8)R =
Figure 11: P3 (R) substituents of peptidyl vinyl sulfones (8) and vinyl sulfonamides (9)
Sulfonates (10) and sulfonamides (9), despite their greater activity
against FP-2, were generally less active against the parasite. In this case, for
compounds with identical structures and with variable P1’ portion, the
antiparasitic activity order was generally (8) > (9) > (10); an order which was
opposite with respect to the activity against FP-2 (Table-I, II). In the vinyl
sulfonates (10) antiparasitic order was observed with ‘X’ group as OCH3 > F > H,
opposite to that reported for the enzymatic inhibition (Table-III).
It is noteworthy that P3 substituents (Fig. 11) showed a marked impact
on the activity against parasite cultures. Three of the six vinyl sulfonate esters (10)
with the highest antiparasitic activities contained an isonipecotic ester moiety (8f,
9f, 10f), while the other ones contained a tertiary amine (8c, 8h and 9c). It is
interesting to observe that compounds containing the isonipecotic ester moiety
proved to be active both against the enzymes as well as the cultured parasites. On
the contrary, those compounds containing a prolinol substituent at P3 (8b, 9b and
10b) were typically potent against the enzymes but showed relatively poor
antiplasmodial activity. The measured second-order rate constants (kassM-1
sec-1
)

Chapter-1 Introduction
31
Table II: Effects of R substituents (P3 Unit) on the activity of peptidyl vinyl sulfones (8) and vinyl
sulfonamides (9) toward FP-2 development of P. falciparum
R
Compound 8 Compound 9
FP-2
IC50
(nM)
P. falciparum
IC50 (nM)
FP-2
IC50
(nM)
P. falciparum
IC50 (nM)
a 35 200 14 1500
b 27 1800 3.6 220
c 8.7 4.5 2.3 4.4
d 9.2 15 2.5 22
e 3 22 2.2 46
f 6.9 3.9 2.2 1.6
g 9.9 21 - -
h 6.7 1.6 - -
Note : Compounds (8e) and (8c) correspond to compounds (5) and (6), respectively.
R NH
NH
SO
O
O
OOX
CH3
CH3
(10)
Table III: Effects of R substituents (P3 Unit) on the activity of vinyl sulfonate esters (10) toward
FP-2 and development of P. falciparum
R X FP-2 IC50 (nM) P. falciparum IC50 (nM)
b H 0.8 390
b F 0.8 394
b CH3O 0.9 220
e H 0.7 49
e F 0.7 84
e CH3O 0.7 14
f H 0.7 42
f F 0.7 120
f CH3O 0.9 9.7
for the inhibition of FP-2 and FP-3 of the most potent vinyl sulfone (8),
sulfonamide (9) and sulfonate (10) inhibitors were generally similar against both

Chapter-1 Introduction
32
of the recombinant enzymes. As seen with IC50 determinations, the vinyl sulfonate
esters were the most potent inhibitors, with kass values against both the proteases
being >105M
-1 sec
-1 for all of the tested compounds. The second-order rate
constants for the phenyl vinyl sulfones and vinyl sulfonamides were lower but
they were consistently >104 M
-1 sec
-1.
1.5.1.3 Peptidyl aldehyde and -ketoamide derivatives
It was found that peptidyl aldehydes and -ketoamide derivatives were
effective to inhibit the enzymatic activity of FP-2 at very low nanomolar range.
The best two inhibitors were revealed to be the aldehyde (11) and the ketoamide
(12) with an IC50 value of 1 nM. The other representative inhibitor was Z-Leu-
homoPhe-al (13) exhibiting an IC50 value of 2 nM.132
Compounds (11-13) (Table-
IV) proved to be active against multiple strains of P. falciparum with the best
activity shown by compound (11) (Table-IV).132
In agreement with older studies of vinyl sulfone inhibitors,129
compounds containing Leu at the P2 position were about an order of magnitude
more potent than those with Phe at this position. The key difference between the
plasmodial cysteine proteases and many other papain family cysteine proteases,
including the abundantly available host proteases cathepsin B and cathepsin L
suggests the potential for specific inhibition of plasmodial enzymes.133
The lead inhibitors (11-12) (Table-IV) when evaluated against
recombinant FP-2 and FP-3 proved to be similarly active against both of the
enzymes. Cysteine proteases of rodent malaria parasites that appear to be
orthologs of FP-2 and FP-3 were also characterized. A single ortholog of the two
P. falciparum proteases appeared to be present in P. vinckei and in three other
rodent malaria parasites.134
Interestingly, all the rodent parasite cysteine proteases
contain an unusual active site substitution that, in the case of the P. vinckei
protease vinckepain-2, mediates unusual kinetics.135
Taking into consideration that
assays in mouse models are generally a key step of malaria drug discovery, it was
of interest to check the activity of peptidyl aldehydes against P. falciparum and P.
vinckei cysteine proteases. The obtained results revealed that vinckepain-2 was
substantially less inhibited by the best inhibitors by about an order of magnitude;
inhibition was particularly poor with a compound containing Phe at P2 consistent
with literature data.

Chapter-1 Introduction
33
N NH
NH
H
O
O
O
O
CH3
CH3
N NH
NH
NH
O
O
O
O O
NH2
O
CH3
CH3
O NH
NH
H
O
O
O
CH3
CH3
(11)(12) (13)
Table IV: Falcipain-2 inhibition and activity against different strains of P. falciparum of peptidyl
aldehyde and a-ketoamide derivatives. (11-13)
Compound FP-2 IC50
(nM)
IC50 (nM)
W2 ItG D6 Dd2 HB3
11 1 1.1 0.96 1.4 1.9 2.6
12 1 2.9 5.3 2.5 2.4 3.1
13 2 8.2 19 12 19 16
Although activity against vinckepain-2 was weak, the in vivo
antimalarial activity of the lead compound Mu-Leu-homoPhe-al (11) was checked
against P. vinckei malaria and these studies demonstrated for this inhibitor: (i)
very poor pharmacokinetic profile, (ii) problems with its formulation and (iii)
need of subcutaneous infusion pumps for administration. This clearly emphasizes
that differences between P. falciparum and P. vinckei targets may contribute to the
limited in vivo efficacies of some cysteine protease inhibitors, if it is considered
that antimalarial drug discovery is based on validation with rodent models before
advancement to a full scale development.
1.5.1.4 E-64
The N-(L-3-trans carboxyoxiran-2-carbonyl)-L-leucyl)-amido(4-
guanido) butane (E-64, Fig. 12), was the first epoxysuccinyl peptide isolated from
a culture of Asperigillus japonicus,136
and its structure was determinated by
Hanada in 1978.137
E-64 is an irreversible inhibitor of papain-like cysteine
proteases (clan CA, family C1), including papain, bromelain, ficin,136
cathepsin
B,138
cathepsin L,139
calpain II 140,141
and cruzain,142
but it does not inhibit clan CD
proteases such as caspases,143
legumains,144
and gingipains,145
and inhibits
clostripain very slowly.146
It shows a second-order rate constant (k2nd) of FP-2
inhibition of 1.16x103
M-1
sec-1
and IC50 values of 0.015 M and of 0.075 M
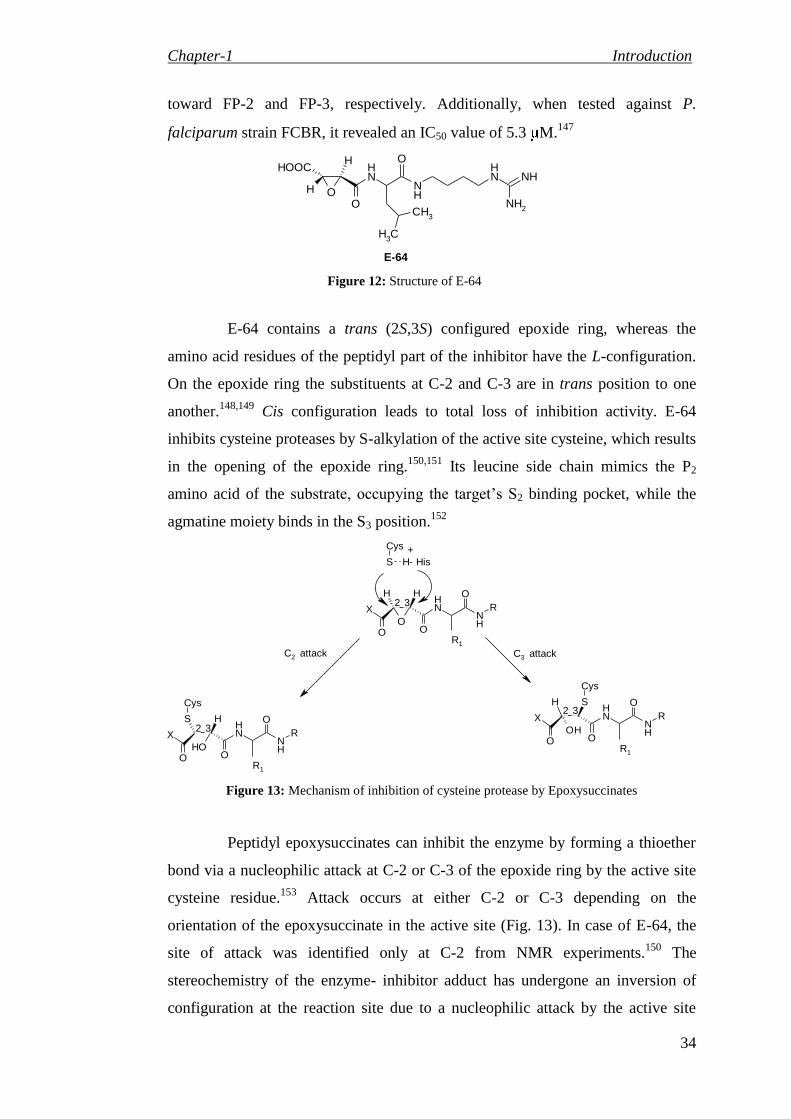
Chapter-1 Introduction
34
toward FP-2 and FP-3, respectively. Additionally, when tested against P.
falciparum strain FCBR, it revealed an IC50 value of 5.3 M.147
O
NH
NH
NH
O
CH3
CH3
O
H
H
HOOCNH
NH2
E-64
Figure 12: Structure of E-64
E-64 contains a trans (2S,3S) configured epoxide ring, whereas the
amino acid residues of the peptidyl part of the inhibitor have the L-configuration.
On the epoxide ring the substituents at C-2 and C-3 are in trans position to one
another.148,149
Cis configuration leads to total loss of inhibition activity. E-64
inhibits cysteine proteases by S-alkylation of the active site cysteine, which results
in the opening of the epoxide ring.150,151
Its leucine side chain mimics the P2
amino acid of the substrate, occupying the target’s S2 binding pocket, while the
agmatine moiety binds in the S3 position.152
O
NH
NH
O
R
O
HH
X
O
S
Cys
H- His
S
Cys
OH
NH
NH
O
R
O
H
X
O
S
Cys
OH
NH
NH
O
R
O
H
X
O
R1
2 3
+
R1
2 3
R1
2 3
C2 attack C3 attack
Figure 13: Mechanism of inhibition of cysteine protease by Epoxysuccinates
Peptidyl epoxysuccinates can inhibit the enzyme by forming a thioether
bond via a nucleophilic attack at C-2 or C-3 of the epoxide ring by the active site
cysteine residue.153
Attack occurs at either C-2 or C-3 depending on the
orientation of the epoxysuccinate in the active site (Fig. 13). In case of E-64, the
site of attack was identified only at C-2 from NMR experiments.150
The
stereochemistry of the enzyme- inhibitor adduct has undergone an inversion of
configuration at the reaction site due to a nucleophilic attack by the active site

Chapter-1 Introduction
35
thiolate in an SN2 reaction. For instance, E-64, which has the 2S,3S configuration
before the nucleophilic attack, will become 2R,3R after the covalent bond between
the cysteine residue and C-2 of the oxirane ring is formed.
Initially, it was postulated that when E-64 inhibited papain, the oxirane
ring would be protonated by His159. This suggestion was rejected by Varughese
and co-workers,154,155
from the crystal structure of papain inhibited by E-64. They
argued that the epoxide was more likely protonated by water (Fig. 14) because of
the distance (5.5A˚) of the resulting hydroxyl group from His159.156
1.5.1.5 Peptidyl aziridines
Aziridines are aza-analogs of epoxides, that are equally susceptible to
ring opening by nucleophiles.123,157
This class of inhibitors has been tested against
several types of proteases, including serine proteases, aspartyl proteases and
metalloproteases, but aziridines are irreversible and selective inhibitors of cysteine
proteases.158
Further, aziridines such as aziridine-2-carboxylates and aziridine-2,3-
dicarboxylates, are hydrolyzed by serine proteases.159,160
There are three types of
aziridinyl peptides (Table-V) that are classified according to their structural
differences. 123
Type I are N-acylated aziridines with the aziridine moiety located
on the C-terminus of the peptides or amino acids. Type II are aza-analogs of
epoxysuccinyl peptides that have the aziridine-2,3-dicarboxylic acid moiety at the
N-terminus of the amino acid. Type III are N-acylated aziridines such as type I,
but the aziridine moiety is located in the middle of the peptide chain. Besides the
common inhibition mechanism, several differences can be found between the
epoxide and the aziridine heterocycles. These include reactivity, the influence of
the pH of medium, selectivity and stereospecificity of inhibition.161
The stereospecificity of the inhibition is one major difference between
aziridines and epoxides. Aziridines have been shown to be more reactive at low
pH,162
with maximum activity at pH 4, while the epoxides are more reactive at pH
6-7 due to protonation of aziridine nitrogen atom, which translates into the
increased reactivity of the inhibitor toward papain. Another difference between
aziridines and epoxides is the hydrogen-bonding abilities.163
Aziridines (Type-II)
are H-bond donors, whereas the epoxides are H-bond acceptors. These differences
suggest that the two classes of inhibitors may have different binding modes and
possibly show variable interactions with cysteine proteases. The aziridine ring can
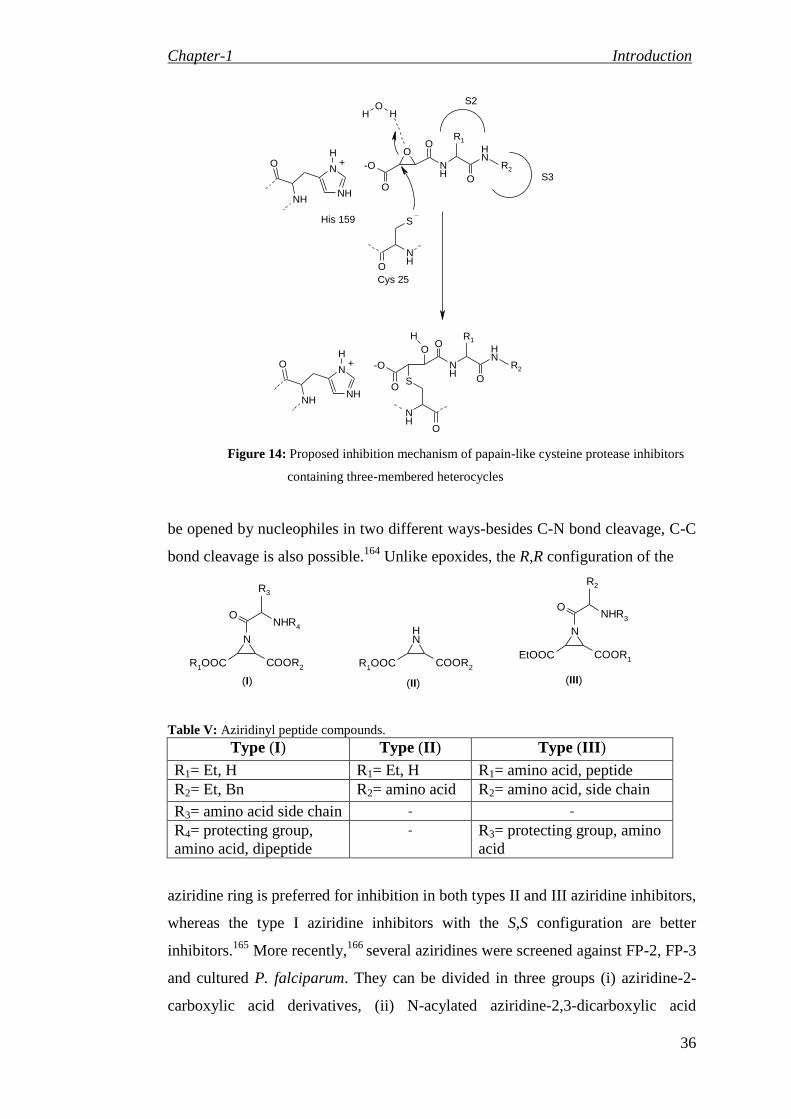
Chapter-1 Introduction
36
N
NHNH
OH
HO
H
O
NH
NH
-O
O
O
O
NH
S
O
NH
NH
NH
-O
O
O
O
O
H
S
O
N
NHNH
OH
+
His 159
Cys 25
R1
R2
S2
S3
R1
R2+
Figure 14: Proposed inhibition mechanism of papain-like cysteine protease inhibitors
containing three-membered heterocycles
be opened by nucleophiles in two different ways-besides C-N bond cleavage, C-C
bond cleavage is also possible.164
Unlike epoxides, the R,R configuration of the
COOR2
N
NHR4
R1OOC
O
COOR2
NH
R1OOC
COOR1
N
O
EtOOC
NHR3
R3
R2
(I) (II) (III)
Table V: Aziridinyl peptide compounds.
Type (I) Type (II) Type (III)
R1= Et, H R1= Et, H R1= amino acid, peptide
R2= Et, Bn R2= amino acid R2= amino acid, side chain
R3= amino acid side chain - - R4= protecting group,
amino acid, dipeptide
- R3= protecting group, amino
acid
aziridine ring is preferred for inhibition in both types II and III aziridine inhibitors,
whereas the type I aziridine inhibitors with the S,S configuration are better
inhibitors.165
More recently,166
several aziridines were screened against FP-2, FP-3
and cultured P. falciparum. They can be divided in three groups (i) aziridine-2-
carboxylic acid derivatives, (ii) N-acylated aziridine-2,3-dicarboxylic acid

Chapter-1 Introduction
37
derivatives and (iii) aziridine-2-carboxylates containing a lysine residue. Within
the series of aziridine-2-carboxylic acid derivatives with free 3-position, inhibitor
(14) (Fig. 15) is the most active one with IC50 value of 2.2 M against FP-2.
Among the N-acylated aziridine-2,3-dicarboxylic acid derivatives, 15 and 16
R
O
N
O
N
OR
O
O
O
O
NO
(14) R = (S)-PheOBn
(16) R = Boc-(S)-Leu-(S)-Pro
(15) R =
Boc-(S)-Leu
OCH3
Figure 15: Aziridine-2-carboxylic and 2,3-dicarboxylic acid derivatives.
offered the most potent inhibitors of FP-2 (IC50 = 0.079 and 5.4 M, respectively)
and FP-3 (IC50 = 0.25 and 39.8 M, respectively).
1.5.1.6 The 1,2,4-trioxolane compounds
In a prodrug approach, 1,2,4-trioxolane carbonyl protected compounds
were reported to act by their cleavage/decomposition by iron chelation. The
peptidic carbonyl protected (aldehyde) morpholineurea (17) (Fig. 17) was reported
OO
ONH
HO
CH3
CH3
NH
R
R1
(17) R = Morphlineurea, R1 = CH2Ph
Figure 16: 1,2,4-Trioxolane compounds
to be weak falcipain inhibitor than parent aldehyde but it showed good
antimalarial activity with IC50 value of 9.3 nM in P. falciparum 3D7 strain.167
1.5.1.7 The γ-lactam or pyrrolidinone isosteres
The γ-lactam or pyrrolidinone isostere has proven to be an effective
conformational constraint for inhibitors of other classes of proteases.168
A series of
conformationally constrained pyrrolidinone inhibitors attached with electrophilic

Chapter-1 Introduction
38
aldehyde169
were reported as inhibitors of falcipain. Compound (18) (Fig. 17)
exhibited excellent binding affinity (50 nM) for falcipain.
NSNH
O
O
O
H
O
(18)
Figure 17: The γ-lactam or pyrrolidinone derivative
1.5.1.8 Peptidic fumaric acids and other peptidic analogs
Fumaric acid derived oligopeptide compounds library was successfully
identified from a high-throughput screening of a solid phase-bound combinatorial
library with the glycine, asparagine or phenylalanine in P1 and phenylglycine and
cyclohexylglycine in P2 site. These fumaric acid derivatives from the library were
found to be the most potent.170
This information was used for synthesis of more
potent vinylogous Michael-type peptidic cysteine protease inhibitors, displaying
different inhibition mechanisms depending on the configuration of the double
bond and the peptide sequence. It has been reported that the E configured
vinylogous Gln-tert-esters are time-dependent inhibitors and on the other hand, Z
or E configured maleic and fumaric acid derivaites containing two phenyl residues
are reversible inhibitors. Some of the compounds show preferential selectivity to
FP-2 over FP-3.171
1.5.1.9 Azapeptide nitriles derivatives
Azadipeptide nitrile cysteine protease inhibitors display structure
dependent antimalarial activity against both chloroquine sensitive and chloroquine
O NH
NN N
O
O
CH3
R2
(19a) R1 = -i.Pr, R2 = -Methyl
(19b) R1 = -CH2Ph, R2 = -Pentyl
(19c) R1 = -CH2Ph, R2 = -(CH2)2Ph
R1
Figure 18: Azapeptide nitrile derivatives
resistant P. falciparum malaria parasites. Inhibition of parasite’s hemoglobin
degrading cysteine proteases was also investigated, revealing the azadipeptide

Chapter-1 Introduction
39
nitriles as potent inhibitors of FP-2 and 3. A correlation between the cysteine
protease inhibiting activity and the antimalarial potential of the compounds was
observed. The FP-2, IC50 for the compound derivatives are more potent than
phenylalanine peptides suggesting that the S2 pocket of the enzyme is deep rather
than shallow.172
1.5.2 Peptidomimetic falcipain-2 inhibitors
1.5.2.1 Peptidomimetics based on a 1,4-benzodiazepine scaffold
Even though several peptide-based FP-2 inhibitors have been identified,
their utility as therapeutic agents is limited due to their susceptibility to protease
degradation and poor absorption through cell membranes; a common strategy to
improve pharmacokinetic and pharmacodynamic parameters is to lock a defined
conformation of the peptide into a rigid scaffold, which could mimic protein’s
secondary structure. In this context the -turn is a structural motif that has been
postulated in many cases for the biologically active form of linear peptides and in
recent years several scaffolds mimicking -turns have been reported.173-176
The benzodiazepine (BZD) nucleus with its similar molecular
dimensions has been proven to be a good -turn mimetic.177,178
Coupled with the
fact that BZDs possess good oral bioavailability as a drug class that are certainly
well tolerated; some novel peptidomimetic FP-2 inhibitors have been designed.
They are based on a 1,4-BZD scaffold 179
as -turn mimetics introduced internally
to a peptide sequence which mimics the dipeptide D-Ser-Gly, and on a C-terminal
aspartyl aldehyde building block, which inhibits the enzyme by forming a
reversible covalent bond with the active site cysteine. The serine hydroxyl group
was functionalized with various aryl isocyanates.
All new peptidomimetics displayed a significant inhibition of both FP-
2A and FP-2B with IC50 values in the range of 8-26 M. In this context compound
(20) (Fig. 19) proved to be the most active inhibitor with an IC50 = 8.2 M for the
FP-2A and of 11.2 M for FP-2B. Trying to define a structure-activity
relationship, in this test series the phenyl derivative proved to be the least potent
inhibitor (IC50 about 20 M in both the enzymes); the introduction of an electron-
donating or an electron-withdrawing group at position 4 of the phenyl ring of the
carbamoyl moiety increased the inhibitory activity in both the cases. The best

Chapter-1 Introduction
40
results were obtained when a methyl (20) or a trifluoromethyl (21) group was
introduced at position 2 of 4-chloro derivative. Also the extension of the arom-
NNH
ON
ONH
OO
R
Cl
O
O
OH
FP-2A FP-2B
(IC50 in mM)
(20) R = CH3 8.2 11.2
(21) R = CF3 8.7 25.9
Figure 19: Inhibition of FP-2A and FP-2B by peptidomimetic 1,4-benzodiazepine
compounds.
atic area seemed to be fruitful. It is interesting to see that all new peptidomimetics
inhibit both FP-2A and FP-2B at a similar level and this is a key step in the
development of a FP-2 inhibitors, as it is well known that the disruption of FP-2A
gene and in this case a decrease of the expression of this paralog seems to be
compensated by the increased expression of FP-2B.86
Furthermore, all compounds
proved to possess good selectivity when tested against other cysteine proteases;
such a panel of active recombinant human caspases (i.e., caspases 1-9) did not
show any inhibitory activity up to 50 M. The peptidomimetics bearing a
protected aspartyl aldehyde warhead led to the thioacylals (22) (Fig. 20) and the
acylals (23) with increased antiplasmodial activity in comparison to the parent
molecule (21).180
NNH
ON
ONH
OO
CF3
Cl
O
O
R
FP-2 FP-2
Ki ( M) IC50 in M
(22) R = S-t-Bu 8.2 28.5
(23) R = OBn 8.9 49.5
Figure 20: Thioacylal and acylal compounds
In order to further investigate the structural requirements of this class of
inhibitors, the peptidomimetics were modified by replacing the P1 aspartyl

Chapter-1 Introduction
41
aldehyde in the lead compound (20) with a vinyl sulfone moiety which was able to
interact as classical Michael acceptor with the active site cysteine leading to an
irreversible inactivation of the target enzyme as described above. In particular,
two different series of vinyl sulfones were synthesized,181
first one containing, at
NNH
S
O O
ON
ONH
OO
CF3
Cl
(24) R1 = H
(25) R1 = CH2CH2Ph
R1
R2
Table VI: Inhibition of FP-2 and antiplasmodial activity of compounds (25)
Compound R1 k2nd
(M-1
min-1
)
kinacM-1
/kinac M
P.
falciparum
IC50 (nM)
25a Methyl 432,000 0.16/0.39 55.4
25b Ethyl 307000 0.1/0.32 9.1
25c Phenyl 175000 0.11/0.60 9.2
25d 4-OCH3-C6H4 634000 0.034/0.053 59.6
the P1 site, a homoPhe residue (e.g. 25, Table-VI), an amino acid known to
strongly increase the potency of FP-2 inhibitors and second one containing in the
same position, a glycine (e.g. 24, Table-VI), useful to evaluate the relevance of the
amino acid side chain in the process of recognition of the ligand by the enzyme.130
It was observed that all of the compounds (24-25) exhibited high second-order
rate constants in the range of 161,000-634,000M-1
min-1
, in particular compounds
(25) (Table-VI) generally showed higher second order rate constants with respect
to analogs (24). Furthermore, it was possible to observe that the introduction, at
the P1’ site, of a methoxy group in position 4 of the phenyl ring, strongly
stabilized the enzyme-inhibitor interaction, probably acting as hydrogen bond
acceptor, as shown in the most active compound (25d) with the highest value of
the second order rate constants of inhibition (K2nd = 634,000M-1
min-1
) and the
lowest dissociation constant value (Kinac = 53 nM). Although there was no clear
correlation between the antiplasmodial activity and the inhibitory potency,
compounds (25) proved to be more active than their glycine analogs (24). It is
interesting to point out that compound (25c), one of the lesser potent inhibitors
against the enzyme, displayed the highest antiplasmodial activity, while the

Chapter-1 Introduction
42
presence of the methoxy group para to the phenyl ring (i.e. 25d), basic for the
enzyme inhibitor interaction, decreased the activity against the parasite, probably
reducing the penetration through cell membranes.
Selectivity assays were also performed against papain family human
cysteine proteases cathepsins B and L by demonstrating good selectivity of all
vinyl sulfones toward FP-2. Other efforts were made to further optimize the
pharmacophore portion by replacing the vinyl sulfone moiety with a vinyl
phosphonate one, generating unsatisfactory results; a decrease of potency and
selectivity was obtained.182
NNH
ON
ONH
OO O
NH
CF3
Cl
NNH
ON
O
OO O
R1
(27)
R2
R3
(26)
OCH3
OCH3
Table VII: Inhibition of falcipain-2 and antiplasmodial activity of compounds (27).
Comp. R1 R2 R3 k2nd (M-
1 min
-1)
kinacM-1
/kinac M
P. falciparum
IC50 (nM)
27a H 4-Cl, 2-CF3-C6H4 Br 24,500 0.038/1.56 6.33
27b H 4-Cl, 2-CF3-C6H4 H 3570000 0.06/0.017 12
27c H 1-Adamantyl H 1160000 0.013/0.014 10.7
27d H 1-Naphthyl H * * 17.6
* Time independent inhibition.
Based on the constrained peptidomimetic vinyl ester (26) (IC50 12
M)183
an irreversible inhibitor of FP-2, the constrained vinyl ester
peptidomimetics were evaluated; compound (27a) (Table-VII) was found to be the
most potent one having an IC50 of 10.7 nM. Any further modification in the parent
compound leads to decrease in FP-2 inhibition.184
1.5.2.2 Peptidomimetics based on a pyridine/pyrimidone ring scaffold
Recently, another nonpeptidic scaffold was incorporated on a peptide
sequence suitable for FP-2 recognition, in a way to lock the amino acid backbone.
The pyridone ring core was selected as a replacement for the peptidic leucine
residue, at the P2 position, within the inhibitor’s framework. Aldehyde, vinyl
sulfone or vinyl ester moieties were the chosen pharmacophores.185
The new

Chapter-1 Introduction
43
synthesized peptidomimetics showed in vitro antimalarial activity against the 3D7
parasite strain in the range of 5-40 M, with the vinyl sulfone (28) (Fig. 21) as the
best inhibitor in the series (IC50 = 5.7 M).
Consistently, with binding features previously reported, compounds
bearing a homoPhe residue at P2 site proved to be more active than those bearing a
phenylalanine in the same position. Some of the synthesized peptidomimetics
were also evaluated on recombinant FP-2 and FP-3. In this evaluation they proved
to be weak inhibitors of FP-2 (IC50 10-20 M) and inactive toward FP-3 (IC50 >
25 M). The best inhibitory profile against FP-2 was shown by compound (29)
(IC50 = 10.9 M), whereas it was not possible to test the most active compound
(28) because of its low solubility.
N R
O
O NH
O
N
N
NH
NH
O CH3
OCOCH3
OO
Ph
(28) R = CH=CHSO2Ph
(29) R = CHO
(30)
Z OCH3
Figure 21: Pyridone and pyrimidone compounds
A new class of pyrimidinyl peptidomimetics (30) was reported as
malarial cysteine protease inhibitors. The core structure of the new agents consists
of a substituted 5-aminopyrimidone ring and a Michael acceptor side chain methyl
2-hydroxymethyl-but-2-enoate. The most effective compound (30) of the series
(IC50 = 9 ng/ml) showed comparable efficacy to that of CQ (IC50 = 6 ng/ml). In
general, this class of compounds exhibited weak to moderate in vitro cytotoxicity
against neuronal and macrophage cells and less toxicity in colon cell line.
Preliminary results indicated that compound (30) is active against P. bergehi
prolonging the life span of the parasite-bearing mice from 6 days for untreated
control to 16-24 days for drug treated animals.186
1.5.3 Nonpeptidic falcipain-2 inhibitors
1.5.3.1 Chalcones
Modeling studies on acylhydrazones have been made on a model
structure of FP-2, designed on the basis of the X-ray structures of papain and
actinidin. This investigation on the most active compound of this class, the oxalic

Chapter-1 Introduction
44
bis[(2-hydroxy-1-naphthylmethylene)hydrazide], clearly revealed that one
naphthyl group filled the hydrophobic S2 site while the other one interacted with
the Trp177 of the S1’ site. On the basis of these results, the length of the backbone
was shortened; the aromatic rings being replaced with heteroaromatic nucleus in
order to improve the water solubility and to enhance interaction with His67 of the
S2 pocket. Finally, to improve the metabolic stability, the hydrazide linker was
replaced with an , -unsaturated ketone backbone leading to calchones.162
Some
chalcones were thought to inhibit malarial cysteine proteases and today their
ability to inhibit the protease of P. falciparum FP-2 is well known. Chalcones are
biosynthetic precursors of flavonoids.
Licochalcone A (Fig. 22), a natural product isolated from Chinese
liquorice roots, was the first compound of this family to exhibit in vivo and in
vitro antimalarial activity.187,188
Chalcones-artemisinins combination therapy have
been reported to have synergistic or additive effect in the treatment of malaria.189
O
OH
CH3
CH3
CH2
OH
OCH3
Figure 22: Licochalcone
First study on synthetic chalcones was performed by Li and co-workers
in 1995.190
The most active chalcone derivative, 1-(2,5-dichlorophenyl)-3(4-
quinolinyl)-2-propen-1-one (31) (Fig. 23) showed an IC50 value of 200 nM against
both CQ-resistant (W2) and CQ-sensitive (D6) strain of P. falciparum. The main
conclusions of this work were: (i) the C2-C3 double bond was revealed to be
essential for the inhibitory properties, because it kept the molecular structure in
extended conformation leading to a better interaction with the active site, (ii)
substitutions on the bridge portion caused a pronounced decrease in the inhibitory
activity, probably due to steric interactions, (iii) introduction of halogens on ring
A (fluorine and chlorine at 2,3 or 2,4-positions e.g. 33) and electron-donating
groups on the B ring increased the antimalarial activity and (iv) the presence of
quinolinyl group in ring A resulted in increased activity. Under acidic conditions,
nitrogen-containing heterocycles can be protonated within the acidic food vacuole
of the parasite facilitating the interaction with His67 of FP-2. Additionally, this

Chapter-1 Introduction
45
chemical entity which is common in established antimalarials may interfere with
heme detoxification affecting the whole process.
O F
FNH
NH
OCl
O
N
Cl
Cl
O
N
(31) (32)
(33)
B A
OCH3H3CO
OCH3
Figure 23: Chalcone compounds.
Later, a broad series of alkoxylated and hydroxylated chalcones have
been synthesized by Liu and co-workers in 2001,191
with the most active
compound 1-(2’,3’,4’-trimethoxyphenyl)-3-(3-quinolinyl)-2-propan-1-one (32)
(Fig. 23) showing an in vitro IC50 value of 2 M against a strain of CQ-resistant
human malaria parasite, P. falciparum (K1). The whole series was not tested
directly against FP-2. Its FP-2 inhibitory activity was presumed. The results
indicated that (i) the substitution pattern on the B ring could influence the
antimalarial activity, (ii) hydrophobicity and size factors, respectively, for the A
and B rings were important parameters for activity, (iii) presence of halogens on A
ring did not necessarily increase the antimalarial activity while the influence of the
latter groups on A ring seemed to be influenced by substitution pattern on B ring
and (iv) an interesting observation was the association of good antimalarial
activity with the presence of a 3-quinolinyl A ring. This was observed with 3-
quinolinyl A ring and several B ring series of compounds except 4’-
hydroxylchalcone. The 4-quinolinyl A ring derivatives were significantly less
active than their 3-quinolinyl counterparts indicating that a steric element is also
involved in the interaction with the target.
Dominguez et al. reported phenylurenyl chalcones to possess good
inhibitory properties against FP-2.192
The best inhibitor of this series, was 1-[4’-N-
(N’-p-chlorophenylurenyl)phenyl]-3-(3,4,5-trimethoxyphenyl)-2-propen-1-one
disp-laying an IC50 value of 1.76 M toward the protease, coupled with a
satisfactory inhibition of cultured malaria parasite development (IC50 = 3 M).

Chapter-1 Introduction
46
Quantum chemical QSAR study on the chalcone derivatives confirmed
that their activity against W2 and D6 P. falciparum strains depended upon steric
and hydrophobic factors. Generally, a width limiting chemical substituent on A
ring is optimal for activity on W2 and D6 strains. Molecular weight, which is
related to molecular volume, appears to influence only the activity of D6 strain.
This study also indicates that chalcones are capable of taking resonance structures
along unsaturated chain, thus they can be good Michael acceptors. This would
result in irreversible inhibition. The oxygen of the carbonyl is subject to the
protonation in the internal acidic environment of the digestive vacuole of
plasmodium, intensifying the positive partial charges on carbons C-1 and C-3 and
favoring the nucleophilic addition at these carbons. However, these studies were
performed in the absence of crystal structure of the parasitic cysteine protease,
giving a rough evaluation for enzyme-inhibitor interaction mechanism.193
O
O
N
NN
OH
R
S CH3
CH3
O
O
N
O
O
Cl
R1 = 4-methoxy, 2,4-dimethoxy, 2,3,4-trimethoxy
R1
ThiolactonesIsatins
R =
Figure 24: -Amino alcohol thiolactone-chalcone and isatin-chalcone derivatives
Hans et al. reported two types of chalcone, thiolactone and isatin
scaffold- containing compounds (Fig. 24). A 36-member -aminoalcohol triazole
library showed that the thiolactone-chalcones, with IC50 ranging from 0.68 to 6.08
M, were more active against W2 strain of P. falciparum than the isatin-chalcones
with IC50 values of 14.9 M or less. Interestingly, isatin-chalcones diaplayed FP-2
inhibitory activity whereas the thiolactone-chalcones lacked enzyme inhibitory
activity. No correlation was observed between the antiplasmodial activity and the
FP-2 enzyme inhibition suggesting that it may not be the primary mode of action
of the chalcones.194

Chapter-1 Introduction
47
1.5.3.2 The -pyranochalcones and pyrazoline derivatives
Wanare G et al. reported the -pyranochalcones and pyrazoline
derivatives as falcipain inhibitors. Docking studies (GLIDE) on (E)-3-(3-(2,3,4-
O O
O
O
O
O
OH
O O
N N
O
O
CH3
CH3
R R
O O
N NCH
3
O
O
O
CH3
CH3
(34) (35)
(36)(37)
Figure 25: Chalcone compounds
Table VIII: -Pyranochalcones and pyrazoline falcipain inhibitors
Compound R
Antimalarial activity P. falciparum (mg/ml)
3D7
IC50
3D7
IC80
RKL9
IC50
Resistance index
(IC50 D7/IC50 RKL9)
34a 3-OCH3- 11 24 4.9 2.2
34b 3,4-di OCH3- 26.7 100 - -
34c 2,3,4-tri OCH3- 3.1 6.2 1.1 2.8
35a 2-Cl 20 25 13.5 1.5
35b 2,5-di OCH3- >100 - - -
36 - 10 10 9 1.1
37 - 10 44 7.6 1.3
trimethoxyphenyl)-acryloyl)-2H-chromen-2-one with active site residue Cys42 of
falcipain enzyme revealed the requirement of hydrogen bond acceptor in ring B.
The compound (34c) with trimethoxy substituent (Table-VIII) was found to be the
most potent compound.195
1.5.3.3 Isoquinoline derivatives
Isoquinolines derivatives (Fig. 26) were designed as FP-2 inhibitors
according to homology modeling studies and subsequent validation by docking. It
was found that isoquinoline framework was not essential for the activity against
FP-2.196
The planarity of the isoquinolines was unimportant as both the
dihydroisoquinoline and isoquinoline cores were found to have IC50 values in the

Chapter-1 Introduction
48
same range. Moreover, all of the compounds bearing a p-methoxyphenyl
substituent at position 1 of the isoquinoline core were almost equipotent to the
N
R
N
O
O
O
(38) R= OMe, IC50 3 M
(39) R= OBu, IC50 3 M
(40) IC50 3 M
Figure 26: Isoquinoline derivatives
earlier reported ligands.197
On the other hand, compounds with p-benzyloxyphenyl
group (38-40, Fig. 26) docked well into the active pocket of the homology-
modeled FP-2 and were found to be more potent than the corresponding hydroxyl
analogs suggesting that S2 pocket of the enzyme prefered to accommodate the
hydrophobic groups.196
1.5.3.4 Thiosemicarbazones
1.5.3.4.1 2-Acetylpyridine-4-phenyl-3-thiosemicarbazone
The first thiosemicarbazone synthesized as potential antimalarial agent
was 2-acetylpyridine-4-phenyl-3-thiosemicarbazone (41, Fig. 27). Klayman and
co-workers198
prepared a series of thiosemicarbazone derivatives from the parent
compound (41) and tested them against P. berghei in mice. The main conclusions
of this work were (i) the 2-pyridylethylidene group was important for antimalarial
NN
NH
NH
S
CH3
(41)
Figure 27: 2-Acetylpyridine-4-phenyl-3-thiosemicarbazone
activity and (ii) at N-4 position the presence of unsubstituted phenyl, benzyl,
phenethyl or cycloalkyl groups such as adamantyl and cyclohexyl could also
contribute to antimalarial activity.
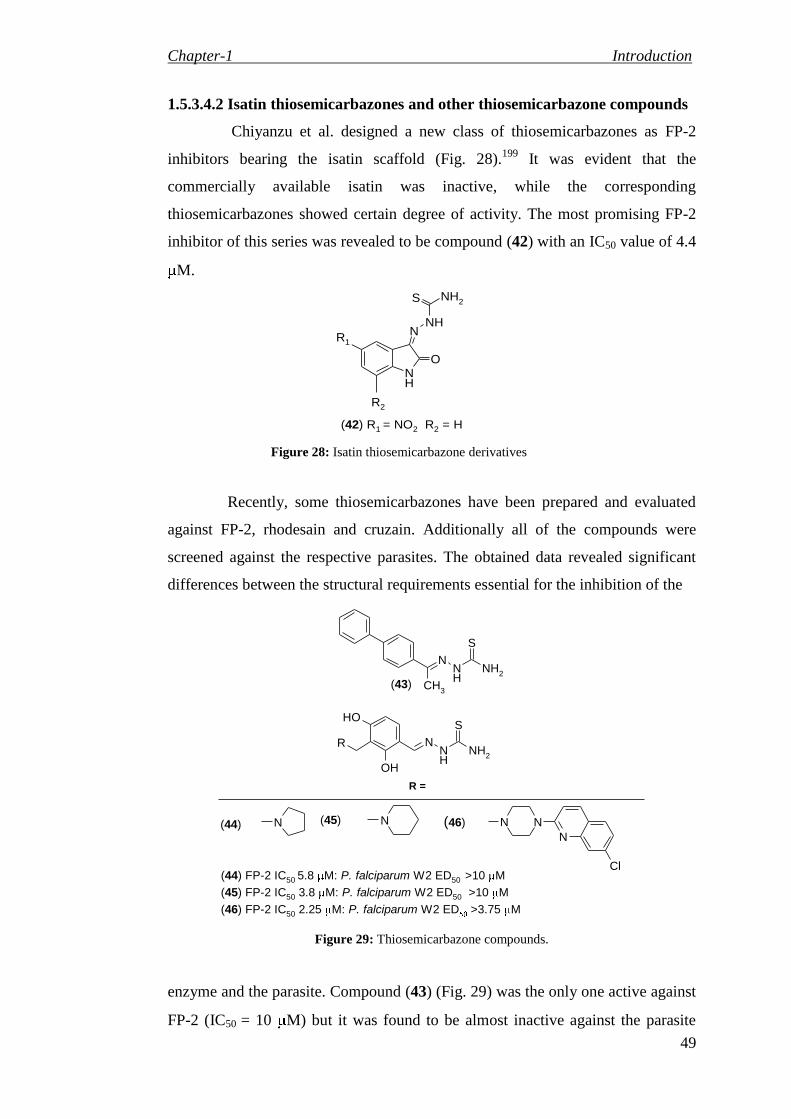
Chapter-1 Introduction
49
1.5.3.4.2 Isatin thiosemicarbazones and other thiosemicarbazone compounds
Chiyanzu et al. designed a new class of thiosemicarbazones as FP-2
inhibitors bearing the isatin scaffold (Fig. 28).199
It was evident that the
commercially available isatin was inactive, while the corresponding
thiosemicarbazones showed certain degree of activity. The most promising FP-2
inhibitor of this series was revealed to be compound (42) with an IC50 value of 4.4
M.
NH
N
O
NH
NH2S
R1
R2
(42) R1 = NO2 R2 = H
Figure 28: Isatin thiosemicarbazone derivatives
Recently, some thiosemicarbazones have been prepared and evaluated
against FP-2, rhodesain and cruzain. Additionally all of the compounds were
screened against the respective parasites. The obtained data revealed significant
differences between the structural requirements essential for the inhibition of the
NNH
NH2
CH3
S
NNH
NH2
OHS
R
OH
N N N N
N
Cl
(43)
(44) (45) (46)
(44) FP-2 IC50 5.8 M: P. falciparum W2 ED50 >10 M
(45) FP-2 IC50 3.8 M: P. falciparum W2 ED50 >10 M
(46) FP-2 IC50 2.25 M: P. falciparum W2 ED >3.75 M
R =
Figure 29: Thiosemicarbazone compounds.
enzyme and the parasite. Compound (43) (Fig. 29) was the only one active against
FP-2 (IC50 = 10 M) but it was found to be almost inactive against the parasite

Chapter-1 Introduction
50
(ED50 20 M). On the contrary, all other compounds (44-46) (Fig. 29) showed
good activity against FP-2. As the corresponding aldehyde precursors were
practically inactive against FP-2, it was reasonable to assume that the (thio)
semicarbazide moiety played an important role in the inhibition of FP-2; but
compounds without mannich bases i. e. phenolic aldehydes and
(thio)semicarbazides independently were found inactive against both FP-2 and the
parasite. This clearly suggested that the activity of the discussed compounds was
not due to the thiosemicarbazide side chain alone, but it was due to the combined
effects of both, the mannich base of aldehyde and thiosemicarbazide
components.200
More recently, new mannich bases of phenolic benzaldehyde and
4-aminoquinoline-(thio)semicarbazide were synthesized. These small compounds
showed good activity against CQ-resistant P. falciparum W2 strain but were very
weak FP-2 inhibitors hence they primarily act some by some mechanism other
than FP-2 inhibition.201
1.5.3.4.3 Gold thiosemicarbazones
The gold (I) thiosemicarbazone derivatives were evaluated for their FP-2
inhibiton. There was no correlation of antiplasmodial activity and FP-2 inhibition
of the parent ligands hence these compounds were assumed to exhibit the
antiplasmodial activity by inhibition of multiple targets.202
1.5.3.5 Acridinediones
Acridinediones like compound (47) (Fig. 30), have been shown to have
antimalarial activity, but their mechanism of action remains unknown. From a
N
OO
Cl
OHCF
3
NH
S
OO
Cl
O
(47) (48)H3CO
OCH3
Figure 30: Acridione and isostere analog
series of chalcones, previously identified as inhibitors of falcipain, a
conformationally constrained series was synthesized. This conformationally
constrained series utilized a sulfur isostere of acridindione to afford the

Chapter-1 Introduction
51
phenothiazine (48) that blocked parasite metabolism and development at low
micromolar concentrations.203
1.5.3.6 2-Cyanopyrimidines
Coteron et al. have reported a detailed study of optimization of the FP-2
and FP-3 activity on 5-chloro/bromo-2-cyanopyrimidine, (Fig. 31) an active
scaffold. Based on initial studies on 4-chloro/bromo-2-cyanopyrimidines, it has
been identified as an active scaffold at micromolar concentrations which was
N
N
NNH
X
N
O
N
Nalkyl
O
NN
CH3
S
N
NCH
3
O
X = Br, Cl
P2
P3
P2 =
P3 =
Figure 31: The 2-cyanopyrimidine derivatives
further subjected to optimization studies by varying the groups at X, P2 and P3
positions (Fig. 33) to obtain potent FP-2/FP-3 inhibitors. The FP-2/FP-3 inhibition
concentrations were found to be in low nanomolar to picomolar range (IC50 0.2
nM & 1nM in Pf W2 strain).204
1.5.3.7 Cryptolepine derivatives
Cryptolepine derivatives (Fig. 32) containing basic side chains at the C-
11 position containing propyl, butyl and cycloalkyl diamine groups significantly
increased activity against chloroquine-resistant P. falciparum strains with less
cytotoxicity, compared to chloroquine. Cryptolepine derivatives containing linear
alkyl (eg. ethyl, propyl) or cyclic groups with terminal secondary or tertiary
amines were found to be potent compounds with IC50 values of 20-90 nM. Any
branching led to decrease in FP-2 inhibitory activity.205

Chapter-1 Introduction
52
N
NH
CH3
R2
R3
R1+
R1, R2 = H/Cl
R3 = NH-Linker-NR4R5
R4, R5 = H, Alkyl, Cycloalkyl
. Cl
Figure 32: Cryptolepine derivatives
1.5.3.8 Indolin-2-one derivatives
The 3-methyleneindolin-2-one derivatives (Fig. 33) showed moderate
antimalarial activity (IC50 = 10 M). The recognition moiety interaction of the
Leu-iso-amyl sequence with the S2 pocket plays important role in activity.
Furthermore, the antiplasmodial activity could be significantly improved when a
NH
O
NH
CH3
CH3
N
O
R1
R2
R3
R1 = H, methyl
R2 = H, Cl
R3 = H, isobutyl, phenyl ethyl/methyl
Figure 33: 3-Methyleneindolin-2-one derivatives
4-aminoquinoline moiety was coupled to the 3-methylene moiety of the indolin-2-
one scaffold (IC50 = 140 nM).206
1.5.3.9 Allicin derivatives
Allicin (S-allyl-2-propenylthiosulfinate) and its derivatives were studied
for FP-2 inhibition (Fig. 34). The mode of action is through the attack of active-
site Cys residue on the primary carbon atom in vicinity to the thiosulfinate sulfur.
Compounds 49 (allicin, Ki = 1.04 M, IC50 = 5.21 M), 55 (Ki = 1.8 M, IC50 =
10.9 M) and 51 (Ki = 4.5 M, IC50 = 30.3 M) showed good FP-2 inhibition.207
RS
SR
O
SS
O
CH3
CH3
CH3
(49-55) (56)
Figure 34: Allicin derivatives

Chapter-1 Introduction
53
Table IX: Allicin derivatives and their antimalarial activity
Compound R FP-2 Ki ( M) P. falciparum IC50 ( M)
49 allyl 1.04 5.21
50 n-propyl 26.4 78.3
51 benzyl 4.5 30.3
52 tert-butyl >100 72.5
53 cyclohexyl >100 34.4
54 iso-pentyl 3.04 54.7
55 n-hexyl 1.8 10.9
56 - >100 52.9
1.5.4 Molecular modeling studies
1.5.4.1 Virtual screening of ChemBridge database
Virtual screening of ChemBridge database (consisting of approximately
2,41,000 compounds) was performed in an attempt to identify nonpeptide
inhibitors of parasitic cysteine proteases as novel drugs.208
The compounds were
screened against homology models of falcipain-2 and falcipain-3 in three
consecutive stages of docking. A total of 24 diverse inhibitors were identified, out
NH
N
NNH
COOH
N
Cl
N
OH
OHOH
OH
NH
O
NNH
O
CN
FO
(57)
HOOC (58)
(59)
Figure 35: FP-2 and FP-3 dual inhibitors identified by virtual screening of
ChemBridge database
of which 12 compounds appeared to be dual inhibitors of FP-2 and FP-3 (Fig. 35).
Some of them showed preferential selectivity for FP-2 over FP-3. 203
1.5.4.2 Docking studies on compounds of SPECS database
In another virtual screening study, the SPECS database (2,87,000) compounds
were screened and reduced to about 80,000 using the drug likeness filter and
subsequently docked by two methods Glide and GAs Dock, with different scoring

Chapter-1 Introduction
54
functions to identify leads for novel FP-2 inhibitors. The top 1000 compounds
were selected using Glide with XP docking used for further accurate docking. The
NN
N
OH
SN
CH3
O
Cl
ON
NN
ON
NH
CH3
ClNH
N
CH3
Cl
N
CH2
NS
O
SNH
O
S
ONH
2(60)
(61)
(62)
(60) 53.1 2.4
(61) 71.4 5.8
(62) 56.5 6.4
Inhibition rate(%) IC50 M
Figure 36: Potent compounds identified by docking studies on of SPECS database
[Note: Inhibition rate (%) calculated with respect to percent inhibition of control (E64)]
top 200 compounds were subjected to visual inspection of the docking geometry
by (1) complementarity between the ligand and the hydrophobic S2 pocket of the
protein and (2) formation of hydrogen bonds between the ligand and residues near
the catalytic cysteine (Cys42), resulting into identification of 53 compounds. In
GAs Dock method top druglike 1000 compounds were selected by its energy
score. The compounds were further evaluated and ranked using the CSCORE
module of the Sybyl package and 154 compounds were then selected with the
consensus score of 5. By visual analysis of the 154 docked poses by elimination
criteria, 56 compounds were selected. Total 81 compounds were then screened for
in vitro evaluation of FP-2 inhi bition. The compounds (60-62) (Fig. 36) were
found to be the most potent ones.209
1.5.4.3 2-Amido-3-(1H-indol-3-yl)-N-substitued propanamides
2-Amido-3-(1H-indol-3-yl)-N-substitued-propanamides were designed
and synthesized based on structure-based virtual screening in conjunction with

Chapter-1 Introduction
55
surface plasmon resonance (SPR) based binding assays. The compounds showed
moderate FP-2 inhibition activity, with IC50 values ranging from 10.0-39.4 μM.210
NH
NHNH
O
R1
R2
Figure 37: 2-Amido-3-(1H-indol-3-yl)-N-substitued propanamides
1.5.4.4 2-(3,4-Dihydro-4-oxothieno[2,3-d]pyrimidin-2-ylthio)acetamides
A series of novel small molecule FP-2 inhibitors have been designed and
synthesized on the basis of structure-based virtual screening in conjunction with
an enzyme inhibition assay. All of the 2-(3,4-dihydro-4-oxothieno[2,3-
d]pyrimidin-2-ylthio)acetamide derivatives showed high inhibitory activity
against FP-2 with IC50 values ranging from 1.46-11.38 μM.211
N
N
S
O
SNH
O
R4
R1
R2
R3
Figure 38: 2-(3,4-Dihydro-4-oxothieno[2,3-d]pyrimidin-2-ylthio)acetamides
1.5.5 Combination or multi-target/hybrid structural approaches
Combination therapies are generally used to combat malaria that can
minimize the chances of developing dug resistance. Certain combinations of
falcipain inhibiting scaffolds and other antimalarial drugs or multi-target/hybrid
therapy approaches have been studied in recent papers.
Aspartic and cysteine protease inhibitors act synergistically to degrade
hemoglobin in vitro212
therefore inhibitors of these enzymes would possess
synergistic effects in inhibiting the growth of cultured parasites. So combination
therapy of inhibitors of malarial cysteine and aspartic proteases may provide the
most effective chemotherapeutic regimen and could possibly limit the
development of parasite resistance to protease inhibitors in the best possible way.
Hans et al. reported two types of chalcones, thiolactone and isatin
scaffolds containing hybrid compounds (Fig. 24) as antimalarial agents. 194
Chalcone-azole derivatives and artemisinins combination therapy have been
reported to have synergistic or additive effect in the treatment of malaria. The

Chapter-1 Introduction
56
synergistic combinations decrease hemozoin formation in parasitized erythrocytes.
These combinations do not affect new permeation pathways induced in the host
cells. The combinations showed very potent FP-2 inhibition with IC50 values
ranging from 0.29 to 10.63 M. The binding affinities and interaction modes were
studied by molecular docking studies using AutoDock 3.05 for the potent
compounds.189
O O
O
O
O N
O
N SO O
R3
R2
R1
R1 = (CH2)2Ph, CH2Ph
R2 = CH2CHMe2, CH2Ph, H
R3 = Ph, Me
(63)
H3CCH3
CH3
H3C
Figure 39: Artemisinin-dipeptidyl vinylsulfone hybrids
Peptidomimetic-thioacylals and acylals bearing protected aspartyl
protease aldehyde warhead were studied for inhibiton of protozoal cysteine
proteases falcipain-2 and rhodesain. Compounds showed potent anti-tripanosomal
activity.180
Artemisinin-dipeptidyl vinylsulfone hybrid molecules (63) were acting
in the parasites food vacuole via endoperoxide activation and falcipain inhibition.
Compounds showed FP-2 inhibition in micromolar range but almost all the
compounds displayed IC50 values ranging from 2-6 nM in P. falciparum W2
strain.213
In another combination strategy through a single prodrug approach,
1,2,4-trioxolane carbonyl protected compounds were reported to act by their
cleavage/decomposition by iron chelation. Peptidic aldehydes and esters were
used to get hybrid compounds. The aldehyde endoperoxide hybrids showed
activity in nanomolar concentration in 3D7 strain of P. falciparum but were
observed to be not good falcipain inhibitors when compared with the parent
peptidic aldehydes.167

Chapter-1 Introduction
57
SNH
NH
O
F
SN
N
CH3
O O
IC50 FP-2 = 7 M, DHFR = 6.3 M
Figure 40: FP-2 and DHFR duel inhibitor
In a multitarget approach a series of small molecules were designed and
synthesized. The compounds showed dual inhibition of falcipain-2 and
dihydrofolate reductase as antimalarial agent and their potency increased 6 to 8
times in comparison to the individual inhibitors. The IC50 values for FP-2 ranged
from 2.7–13.2 μM and for DHFR IC50 values ranged from 1.8–19.8 μM. The most
potent compound (Fig. 40) exhibited moderate in vivo antimalarial activity in a
dose dependent fashion. The molecular docking studies have been performed to
identify the key structural information to attain dual inhibitory activity214
The existing armamentarium of antimalarial drugs is not sufficient to
combat malaria, primarily because of resistance developed by the parasite. P.
falciparum genome sequencing has revealed a number of drug targets. Although
the enzymes of lethal malaria parasite species still has not drawn much attention
of medicinal chemists, biologist and clinicians’ worldwide to the extent it is
required for the development of new drug. Ultimately, a better understanding of
the biochemical and biological roles played by malarial proteases will foster the
development of protease inhibitors that would specifically inhibit malaria parasite
enzymes. There exist a great potential in the exploration of cysteine proteases
falcipain inhibitor and thus might prove to be the most suitable drug target for the
chemotherapy of malaria. This thesis work has been directed towards design and
synthesis of FP-2 inhibitors. The details about design strategy, synthetic
methodologies and biological screening results are described in the Chapter-2
Research Envisaged, Chapter-3 Result and discussion and Chapter-4 Experimental
work sections.

Introduction 2. Research Envisaged

Chapter-2 Research envisaged
58
As discussed earlier, majority of the antimalarial drugs were discovered
during the Second World War when the soldiers of the western countries while
fighting in the topical countries started contracting malaria in mosquito infested areas.
Over a long period of their usage against malaria caused development of resistance in
the parasite against these drugs. The problem got further compounded due to non-
enrichment of antimalarial drug inventory as the cash rich western countries were not
interested in finding new therapeutics for a disease which was considered to be the
disease of the poor third world tropical countries. Unfortunately for these poor
malaria infested countries an effective antimalarial vaccine also could not be
developed due to fast and constant mutations in the parasite genome. All these factors
led to a woefully thin antimalarial drug development pipeline with little chemical
diversity. There exist a great potential in the exploration of cysteine proteases
falcipain inhibitor and thus might prove to be the most suitable drug target for the
chemotherapy of malaria.
Genome mapping of the malaria parasite offered a number of attractive
drug targets including plasmepsins and falcipains, the enzymes involved in parasite
metabolism and in providing nutrients to the parasite to meet energy requirements.
Falcipains 2 and 3 especially were more attractive targets as these enzymes degrade
haemoglobin to small sized peptides in the acidic environment of the vacuole and
degrade the membrane proteins of the host cells and help the parasite rupture the host
cells in the alkaline environment. So, inhibition of FP-2/3 enzymes could effectively
contain the growth of the parasite and control their progression in the host system.
A large number of peptidic and non-peptidic motifs like chalcones, (thio)
semicarbozones, hydrazides, heterocycles, oxiranes and succinates have been reported
in the literature to exhibit FP-2/3 inhibitory activity. Unfortunately none of such
compounds have
N
N
X
O
AX = Urea, carbamate, amide
A = Alkyl/aryl/heteroaryl
R = H, CH3, Cl, 4-C6H4OCH3
R
(M)
crossed the clinical phase of evaluation as antimalarial agents. There are some reports
of some heterocyclic215
and polycyclic216
pyridopyrimidines exhibiting antimalarial
activity. Taking the basic information from literature about the enzyme inhibitors into
consideration, it was planned to synthesize pyrido[1,2-a]pyrimidin-4-one class of

Chapter-2 Research envisaged
59
compounds (M). It is worth noting that the circled pharmacophore may act as Michael
acceptor for reversible/irreversible binding to the thiol nucleophile present in the FP-
2/3 enzymes. While the groups, X and R were selected in such a way that compounds
could exhibit better binding profile in the P2 and P3 binding pockets of the falcipain
enzyme that could translate in to enhanced potency of the compounds.
It was planned to synthesize the given four (Series-I to IV) of
compounds and to evaluate them against FP-2 enzyme in the in vitro tests. FP-3
enzyme having a high degree of homology to FP-2 was presumed to react with all
such compounds which would prove to be powerful inhibitors of FP-2.
N
N
N
O
O
NRR N N
N
N
N
O
O
N
N
O
N
O
AR
N
N
N
O
O
OR
I II
AA
R1
IV
R1
III
A
Where: R = H, 8-CH3, 7-Cl, 4-C6H4OCH3
R1 = H, Alkyl
A = Alkyl/heteroaryl or alkyl

3. Results and Discussion
Envisaged

Chapter-3 Results and discussion
60
The research work carried out towards achieving the proposed plan has
been discussed under the following two main headings:
3.1 Chemical studies
3.2 Biological evaluation
3.1 Chemical studies
3.1.1 Synthesis of 4H-pyrido[1,2-a]pyrimidin-4-one intermediates (4a-c & 6a-c)
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a)
Synthesis of 3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a)
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid
(4b)
Synthesis of 3-isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (6b)
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid
(4c)
Synthesis of 7-chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6c)
3.1.2 Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives
(Series I)
Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives
Synthesis of 8-methyl-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
derivatives
Synthesis of 7-chloro-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
derivative
3.1.3 Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives (Series II)
Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)-
piperazine-1-carboxamide derivatives
Synthesis of N-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives
Synthesis of N-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-
2-yl)piperazine-1-carboxamide derivatives
3.1.4 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives
(Series III)
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate derivatives

Chapter-3 Results and discussion
61
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate
derivatives
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate
derivatives
3.1.5 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives
(Series IV)
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide derivatives
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide
derivatives
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide
derivatives
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-
carbox-amide intermediates
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-
carbox-amides derivatives
Compounds belonging to these individual categories have been
synthesized by following six major schemes as described below:
3.1.1 Synthesis of 4H-pyrido[1,2-a]pyrimidin-4-one intermediates
(4a-c & 6a-c)
The acids (4a-c) and the isocyanates (6a-c) are the key intermediates in
the preparation of the targeted compounds. Their synthesis was accomplished by
following the key steps i) Gould and Jacob reaction ii) cyclization, iii) hydrolysis
and iv) Curtius rearrangement reaction. Synthesis of theses intermediates is
accomplished by following the reaction sequence as outlined in the Scheme-1.
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a)
2-Aminopyridine (1a) was reacted with diethyl ethoxymethylene-
malonate affording the diethyl 2-pyridylaminomethylenemalonate (2a).
Compound (2a) offered characteristic peaks at 3274 (N-H str.), 1686 (C=O, str.)
and 1248 cm-1
(C-O, str.) in its IR spectrum. The compound (2a) displayed

Chapter-3 Results and discussion
62
characteristic signals at 1.22-1.29 (m, 6H, CH3(8/10)), 4.12-4.18 (q, 2H, CH2(7/9)),
4.20-4.25 (q, 2H, CH2(7/9)), 7.14-7.17
N
NH2
N
N
O
O
CH3
O
N
N
O
OH
O
N
N
N
O
ON
N
O O
N
N+
N
R
R R
R
R
N
NH
O
O
O
CH3
O
CH3
R
(1a-c)(2a-c)
(3a-c) (4a-c)
(5a-c)(6a-c)
a. R = H, b. R = 8-CH3, c. R = 7-Cl
i. ECF, TEA, DMF, 0OC
ii. NaN3, H2O,
EMME, Reflux
DPE,Reflux
HCl, Reflux
Toluene, Reflux
a. R = H, b. R = 4-CH3, c. R = 5-Cl
Scheme 1
(m, 1H, Ar-H(2)), 7.39-7.41 (d, 1H, Ar-H(4)), 7.79-7.83 (m, 1H, Ar-H(3)), 8.36-
8.37 (d, 1Ar-H(1)), 9.05-9.08 (d, 1H, Ar-H(6)) and 10.76-10.80 (d, 1H, NH(5)) in its
NMR spectrum. The (M++1) peak was observed at (m/z) 265 in its mass spectrum.
N
1
4
2
3 6NH
5
O
7
O
O
9
CH3
10
O
CH3
8
N
1
4
2
3 5N
O
O
O6
CH3
7N
1
4
2
3 5N
O
OH
O
(2a) (3a) (4a)
The diester (2a) was refluxed in diphenyl ether to get the cyclized
product ethyl 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (3a). Its IR
spectrum displayed characteristic peaks at 1730 (C=O str.) and 1103 cm-1
(C-O
str.). The cyclized product showed characteristic signals at 1.29-1.32 (3H,
CH3(7)) and 4.25-4.30 (m, 2H, CH2(6)), 7.56-7.59 (m, 1H, Ar-H(2)), 7.84-7.87 (d,

Chapter-3 Results and discussion
63
1H, Ar-H(4)), 8.19-8.23 (m, 1H, Ar-H(3)), 8.87 (s, 1H, Ar-H(5)) and 9.16-9.18 (d,
1H, Ar-H(1)) in its NMR spectrum. The (M++1) peak was obtained at (m/z) 219 in
its mass spectrum.
4-Oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a) was prepared
by acid catalyzed ester hydrolysis of the compound (3a) using concentrated
hydrochloric acid. Characteristic peaks were displayed at 3091 (O-H str.) and
1757 cm-1
(C=O str.) in its IR spectrum. The 1H-NMR displayed characteristic
signals at 7.67-7.71 (m, 1H, Ar-H(2)), 7.97-8.00 (d, 1H, Ar-H(4)), 8.31-8.35 (m,
1H, Ar-H(3)), 8.96 (s, 1H, Ar-H(5)) and 9.22-9.23 (d, 1H, Ar-H(1)). The (M++1)
peak was observed in its mass spectrum at (m/z) 191.
Synthesis of 3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a)
The carbonyl azide (5a) was prepared by reacting the acid (4a) with
ethyl chloroformate to get the mixed anhydride which was treated in situ with
aqueous sodium azide offering 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-
carbonylazide (5a). Characteristic absorption bands were observed at 2140 for the
azide and 1724 cm-1
(C=O, str.) in its IR spectrum. The 1H-NMR showed peaks
at 7.61-7.64 (m, 1H, Ar-H(2)), 7.87-7.91 (d, 1H, Ar-H(4)), 8.24-8.35 (m, 1H, Ar-
H(3)), 8.94 (s, 1H, Ar-H(5)) and 9.10-9.28 (d, 1H, Ar-H(1)). The mass spectrum of
the compound (5a) showed (M+-1) peak at (m/z) 214.
N
1
4
2
3 5N
O
N
O
N+
N
N
1
4
2
3 5N
O
N=C=O
(5a) (6a)
The azide (5a) was refluxed in toluene to get the Curtius rearrangement
product 3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a). Characteristic peaks
were observed at 2140 (N=C=O str.) and 1687 cm-1
(C=O str.) in its IR spectrum.
The mass spectrum displayed (M++1) peak at (m/z) 188.
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic
acid (4b)
Similarly, diethyl (4-methyl-2-pyridylamino)methylenemalonate (2b)
was prepared by reacting compound (1b) with diethyl ethoxymethylenemalonate

Chapter-3 Results and discussion
64
as described above for compound (2a). It gave strong characteristic absorption
bands at 1682 (C=O str.) and 1218 cm-1
(C-O str.) in its IR spectrum. The mass
spectrum displayed (M++1) peak at (m/z) 279.
N
1
4
2
6NH
5
O
7
O
O
9
CH3
10
O
CH3
8
CH3
3
N
1
4
2
5N
O
O
O6
CH3
7
CH3
3
N
1
4
2
5N
O
OH
O
CH3
3
(2b) (3b) (4b)
Ethyl 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (3b)
was prepared from compound (2b) as described above for compound (3a). Strong
characteristic peaks were observed at 1689, (C=O str.) and 1143 cm-1
(C-O str.) in
its IR spectrum. Its 1H-NMR displayed signals of the ethyl ester groups at 1.30
(t, 3H, CH3(7)), 2.51 (s, 3H, CH3(3)), 4.23-4.29 (m, 2H, CH2(6)), 7.42-7.44 (m, 1H,
Ar-H(2)), 7.67 (s, 1H, Ar-H(4)), 8.82 (s, 1H, Ar-H(5)), 9.04-9.05 (d, 1H, Ar-H(1)).
The mass spectrum showed (M++1) peak at (m/z) 233.
8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4b) was
obtained on acidic hydrolysis of the ester (3b). It gave characteristic absorption
bands at 2645 (O-H str.) and 1764 cm-1
(C=O str.) in its IR spectrum. 1H-NMR
displayed characteristic signals for methyl group at 2.62 (s, 3H, CH3(3)) and acid
at 10.90 (b, 1H, COOH). The aromatic protons were observed at 7.62-7.64 (m,
1H, Ar-H(2)), 7.89 (s, 1H, Ar-H(4)), 8.88-8.90 (d, 1H, Ar-H(5)) and 9.14-9.16 (d,
1H, Ar-H(1)). The compound gave (M+-1) peak at (m/z) 203 in its mass spectrum.
Synthesis of 3-isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one
(6b)
The synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-
carbonylazide (5b) was accomplished by treating (4b) with ethyl chloroformate
and sodium azide as outlined in (Scheme 1). Characteristic IR peaks were
observed at 2142 (azide str.) and 1712 cm-1
(C=O str.) in its IR spectrum. Its 1H-
NMR signals appeared at 2.56 (s, 3H, CH3(3)), 7.49-7.51 (d, 1H, Ar-H(2)), 7.75
(s, 1H, Ar-H(4)), 8.84 (s, 1H, Ar-H(5)) and 9.09-9.13 (d, 1H, Ar-H(1)). The mass
spectrum showed (M++1) signal at (m/z) 230.

Chapter-3 Results and discussion
65
N
1
4
2
5N
O
N
O
N+
NCH
3
3
N
1
4
2
5N
O
CH3
3
N=C=O
(5b) (6b)
The acid azide (5b) was refluxed in toluene as described above for (6a)
yielding 3-isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (6b). Strong
absorption bands were observed at 2223 (N=C=O str.) and 1670 cm-1
(C=O str.) in
its IR spectrum. The mass showed (M++1) peak at (m/z) 201.
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic
acid (4c)
Diethyl (5-chloro-2-pyridylamino)methylenemalonate (2c) was
synthesized by reacting compound (1c) with diethyl ethoxymethylenemalonate as
described above for compound (2a). Its IR spectrum showed strong absorption
bands at 1676 (C=O
N
1
4
2
3 6NH
5
O
7
O
O
9
CH3
10
O
CH3
8
ClN
1
4
2
3 5N
O
O
O6
ClCH
37
N
1
4
2
3 5N
O
OH
O
Cl
(2c) (3c) (4c)
str.) and 1219 cm-1
(C-O str.). The characteristic NMR signals were displayed at
1.11-1.29 (m, 6H, CH3(8/10)), 4.13-4.26 (m, 4H, CH2(7/9)), 7.47-7.49 (d, 1H, Ar-
H(3)), 7.91-7.94 (m, 1H, Ar-H(4)), 8.41 (s, 1H, Ar-H(1)), 8.94-8.97 (d, 1H, Ar-H(6))
and 10.79-10.82 (d, 1H, NH(5)). Molecular ion peak (M++1) was observed at (m/z)
299 in its mass spectrum.
Ethyl 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (3c)
was prepared from compound (2c) as described above for compound (3a). Strong
peaks were observed in its IR spectrum at 1681 (C=O str.) and 1222 cm-1
(C-O str.).
The mass spectrum displayed a prominent (M++1) peak at (m/z) 253.

Chapter-3 Results and discussion
66
7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4c) was
prepared from compound (3c) as described above for compound (4a).
Characteristic IR bands were observed at 3063 (O-H str.), 1756 cm-1
(C=O str.) in
its IR spectrum. The mass spectrum showed (M++1) peak at (m/z) 225.6.
Synthesis of 7-chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one
(6c)
Reaction of compound (4c) with ethyl chloroformate and sodium azide
was done as described above for compound (5a) to get 7-chloro-4-oxo-4H-
pyrido[1,2-a]pyrimidine-3-carbonylazide (5c). Its IR spectrum showed absorption
bands at 2158 (azide str.) and 1732 cm-1
(C=O str.). A prominent (M++1) peak
was observed at (m/z) 250.6 in its mass spectrum.
N
1
4
2
3 5N
O
N
O
N+
N
Cl
N
1
4
2
3 5N
O
Cl N=C=O
(5c) (6c)
7-Chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6c) was
synthesized as described above for compound (6a). It displayed characteristics
peaks at 2243 (N=C=O str.) and 1668 cm-1
(C=O str.) in its IR spectrum. The
mass spectrum showed (M++1) peak at 222.6.
3.1.2 Synthesis of substituted (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)urea derivatives (Series I)
The ureas Series-I (I-1 to I-34) were prepared by coupling of suitable
isocyanate (6a-c) with amine derivatives of type R1NHA in toluene under reflux
conditions following the procedure as depicted in Scheme 2 or Scheme 3.
Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives
The synthesis of 1-(4-chlorobenzyl)-3-(4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)urea (I-1) has been carried out by refluxing the isocyanate
derivative (6a) (Scheme 1) and 4-chlorobenzylamine in toluene as depicted in
Scheme 2 to get the compound (I-1). Its IR spectrum showed characteristic peaks

Chapter-3 Results and discussion
67
at 3318 (N-H str.) and 1693 cm-1
(C=O str.). The 1H-NMR displayed
characteristic signals at 4.39-4.41 (d, 2H, CH2(8)), 7.26-7.28 (m, 1H, Ar-H(2)),
N
N
NH
O
O
NR R N NN
N
NH
O
O
NH N A,
N
N
O
N
CF3
CF3
NH
CH3
O
N N
O
CF3
FF
NO
CH3
CH3
N
N
ClN
ONN
O
CH3
CH3
O
OCH
3
N
N
O
O
O
CH3
NH2
O
O
OSS
O
OCH
3
CH3
CH3
6 (a-c)
Toluene, reflux
A A
R1NHA, X=
R1
Toluene, reflux
(I-1 to I-34)
N=C=O
OCH3
A =
R1 = H, i.Pr, Cycloproyl, Benzyl
OCH3
OCH3
a. R = H, b. R = 8-CH3, c. R = 7-Cl
(II-1 to II-11)
Scheme 2
7.30-7.35 (m, 2H, Ar-H(9,10)), 7.41-7.47 (m, 2H, Ar-H(11,12)), 7.56 (t, 1H, NH(7)),
7.61-7.64 (d, 1H, Ar-H(4)), 7.68-7.71 (m, 1H, Ar-H(3)), 8.55 (s, 1H, NH(6)), 8.85-
8.87 (d, 1H, Ar-H(1)) and 9.16 (s, 1H, Ar-H(5)). The mass spectrum displayed
molecular ion peak (M+) at 328.9 and (M
++2) at (m/z) 330.8 characteristic of the
chloro compound.
1-(4-Methoxybenzyl)-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-
2) was prepared by reacting the isocyanate (6a) with 2-methoxyethylamine as

Chapter-3 Results and discussion
68
described above for compound (I-1). Characteristic signals appeared at 3361 (N-H
str.), 1639 (C=O str.) and 1180 cm-1
(C-O str.) in its IR spectrum. 1H-NMR
spectrum showed peaks at 3.73 (s, 3H, OCH3(13)), 4.24–4.25 (d, 2H, CH2(8)),
6.89–6.91 (d, 2H, Ar-H(9,10)), 7.22–7.27 (m, 3H, Ar-H(11,12) & Ar-H(2)), 7.40–7.43
(t, 1H, NH(7)), 7.61–7.63 (d, 1H, Ar-H(4)), 7.68–7.72 (m, 1H, Ar-H(3)), 8.39 (s, 1H,
NH(6)), 8.84–8.86 (d, 1H, Ar-H(1)) and 9.18 (s, 1H, Ar-H(5)). A molecular ion peak
(M++1) was observed at (m/z) 325 in its mass spectrum.
N
1
4
2
3 5N
O
NH
6NH
7
O8 9
11
12
10 Cl
N
1
4
2
3 5N
O
NH
6NH
7
O8 9
11
12
10 OCH3
(I-1) (I-2)
The synthesis of 1-(2-methoxyethyl)-3-(4-oxo-4H-pyrido[1,2-a]pyri-
midin-3-yl)urea (I-3) was accomplished by reacting the isocyanate derivative (6a)
with 2-methoxyethylamine as described above for compound (I-1). It gave
characteristic IR absorption bands at 3362 (N-H str.), 1639 (C=O str.) and 1099
cm-1
(C-O str.). Its 1H-NMR showed characteristic signals at 3.25-3.28 (m, 5H,
OCH3(10) & CH2(9)), 3.70-3.39 (t, 2H, CH2(8)), 7.16-7.17 (m, 1H, NH(7)), 7.23-7.27,
(m, 1H, Ar-H(2)), 7.60-7.63 (d, H, Ar-H(4)), 7.67-7.72 (m, 1H, Ar-H(3)), 8.41 (s,
1H, NH(6)), 8.45-8.86 (d, 1H, Ar-H(1)) and 9.16 (s, 1H, Ar-H(5)). The mass
spectrum displayed (M++1) peak at (m/z) 263.
N
1
4
2
3 5N
O
NH
6NH
7
O8
9
OCH3
10
N
1
4
2
3 5N
O
NH
6NH
7
O8
9
NH
10
O
CH3
11
(I-3) (I-4)
The isocyanate (6a) was reacted with 2-acetamidoethylamine as
described above for compound (I-1) affording N-{2-[3-(4-Oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)ureido]ethyl}acetamide (I-4). It showed characteristic peaks at
3317 (N-H str.), 1633 cm-1
(C=O str.) in its IR spectrum. The characteristic peaks
were observed at 1.81 (s, 3H, CH3(11)), 3.11-3.16 (m, 4H, CH2(8,9)), 7.09-7.11 (m,
1H, NH(7)), 7.23-7.27 (m, 1H, Ar-H(2)), 7.61-7.63 (d, 1H, Ar-H(4)), 7.68-7.72 (m,
1H, Ar-H(3)), 7.94 (b, 1H, NH(10)), 8.35 (s, 1H, NH(6)), 8.84-8.86 (d, 1H, Ar-H(1))

Chapter-3 Results and discussion
69
and 9.17 (s, 1H, Ar-H(5)) in its NMR spectrum. The compound (I-4) offered
(M++1) peak at (m/z) 290 in its mass spectrum.
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(2-trifluoromethylbenzyl)-
urea (I-5) was prepared by reacting the isocyanate (6a) with 2-trifluoromethyl-
benzylamine as described above for compound (I-1). The compound showed
strong peaks at 3315 (N-H str.) and 1667 cm-1
(C=O str.) in its IR spectrum. The
characteristic signals were displayed at 4.51-4.52 (d, 2H, CH2(8)), 7.25-7.28 (m,
1H, Ar-H(2)), 7.47-7.51 (m, 1H, NH(7)), 7.56-7.64 (m, 3H, Ar-H(4,11,12)), 7.68-7.74
(m, 3H, Ar-H(3,9,10)), 8.59 (s, 1H, NH(6)), 8.86-8.88 (d, 1H, Ar-H(1)) and 9.17 (s,
1H, Ar-H(5)) in its NMR spectrum. The compound gave (M
++1) peak at (m/z) 363
in its mass spectrum.
N
1
4
2
3 5N
O
NH
6NH
7
O8
10
12
11
9
CF3
N
1
4
2
3 5N
O
NH
6NH
7
O8 9
11
12
10 CF3
(I-5) (I-6)
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(4-trifluoromethylbenzyl)-
urea (I-6) displayed characteristic peaks at 3328 (N-H str.), 3048 (Ar-H str.) and
1670 cm-1
(C=O str.) in its IR spectrum. The NMR spectrum displayed
characteristic signals at 4.42-4.44 (d, 2H, CH2(8)), 7.25-7.28 (m, 1H, Ar-H(2)),
7.51-7.54 (d, 2H, Ar-H(9,10)), 7.59-7.64 (m, 2H, Ar-H(3,4)), 7.69-7.73 (m, 3H, Ar-
H(11,12) & NH(7)), 8.50 (s, 1H, NH(6)), 8.86-8.87 (d, 1H, Ar-H(1)) and 9.16 (s, 1H,
Ar-H(5)). The (M+-1) peak was observed at (m/z) 361 in its mass spectrum.
The compound 1-(2,4-difluorobenzyl)-3-(4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)urea (I-7) showed characteristic IR signals at 3328 (N-H str.)
and 1676 cm-1
(C=O str.). The 1H-NMR displayed characteristic peaks at 4.35-
N
1
4
2
3 5N
O
NH
6NH
7
O8
10
11
9F
F
10 11
12S9
N
1
4
2
3 5N
O
NH
6NH
7
O8
(I-7) (I-8)
4.37 (d, 2H, CH2(8)), 7.14-7.20 (m, 2H, Ar-H(9,11)), 7.24-7.28 (m, 2H, Ar-H(2,10)),
7.52-7.55 (t, 1H, NH(7)), 7.62-7.64 (d, 1H, Ar-H(4)), 7.69-7.74 (m, 1H , Ar-H(3)),

Chapter-3 Results and discussion
70
8.50 (s, 1H, NH(6)), 8.85-8.87 (d, 1H, Ar-H(1)) and 9.14 (s, 1H, , Ar-H(5)). The
molecular ion (M++1) peak was observed at (m/z) 331 in its mass spectrum.
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(thiophen-2-yl)ethyl]-
urea (I-8) showed characteristic IR signals at 3213 of urea (N-H str.), 3033 (Ar-H
str.) and 1656 cm-1
(C=O str.). The compound gave the signals at 2.97-3.01 (t,
2H, CH2(9)), 3.37-3.4 (t, 2H, CH2(8)), 6.92-6.93 (b, 1H, Ar-H(10)), 6.97-6.99 (m,
1H, Ar-H(11)), 7.15-7.18 (t, 1H, NH(7)), 7.23-7.27 (m, 1H, Ar-H(2)), 7.35-7.37 (d,
1H, Ar-H(12)), 7.61-7.63 (d, 1H, Ar-H(4)), 7.68-7.72 (m, 1H, Ar-H(3)), 8.41 (s, 1H,
NH(6)), 8.84-8.85 (d, 1H, Ar-H(1)) and 9.19 (s, 1H, Ar-H(5)) in its NMR spectrum.
The mass spectrum showed (M++1) peak at (m/z) 315.
The compound 1-[3-(morpholin-4-yl)propyl]-3-(4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)urea (I-9) displayed characteristic peaks at 3297 (N-H str.) and
3072 (Ar-H str.), 1663 (C=O str.) and 1118 cm-1
(C-O str.) in its IR spectrum. The
signals were displayed at 1.55–1.62 (m, 2H, CH2(9)), 2.28–2.32 (m, 2H, CH2(10)),
2.34 (b, 4H, CH2(11/12)), 3.10–3.15 (m, 2H, CH2(8)), 3.56–3.58 (t, 4H, CH2(13/14)),
7.01–7.04 (m, 1H, NH(7)), 7.23–7.27 (m, 1H, Ar-H(2)), 7.60–7.63 (d, 1H, Ar-H(4)),
7.67–7.71 (t, 1H, Ar-H(3)), 8.29 (s, 1H, NH(6)), 8.84–8.86 (d, 1H, Ar-H(1)) and 9.17
(s, 1H, Ar-H(5)) in its NMR spectrum. The compound gave (M++1) peak at (m/z)
332 in its mass spectrum.
N
1
4
2
3 5N
O
NH
6NH
7
O8
9
10
N
11 13
O
1412
N
1
4
2
3 5N
O
NH
6NH
7
O8
9
10
1211
1413
15
17
16
19
18
20
(I-9) (I-10)
1-(3,3-Diphenyl)propyl-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
(I-10) displayed strong IR signals at 3324 (N-H str.) and 1664 cm-1
(C=O Str.).
The signals were observed at 2.18-2.23 (m, 2H, CH2(9)), 2.97-3.05 (m, 2H,
CH2(8)), 4.01-4.05 (t, 1H CH(10)), 7.11-7.34 (m, 12H, Ar-H(2,11-20) & NH(7)), 7.60-
7.63 (d, 1H, Ar-H(4)), 7.67-7.74 (m, 1H, Ar-H(3)), 8.32 (s, 1H, NH(6)), 8.85-8.92
(d, 1H, Ar-H(1)) and 9.15 (s, 1H, Ar-H(5)) in its 1H-NMR spectrum. The (M
++1)
peak was displayed at (m/z) 399 in the mass spectrum.

Chapter-3 Results and discussion
71
The IR spectrum of the compound 1-(4-oxo-4H-pyrido[1,2-a]pyrimidin-
3-yl)-3-[3-(n.pentyloxy)pyridin-2-yl]urea (I-11) displayed characteristic bands at
3350 (N-H str.), 1665 (C=O str.) and 1220 cm-1
(C-O Str.). The 1H-NMR signals
were displayed at 0.90-0.94 (t, 3H, CH3(15)), 1.32-1.47 (m, 4H, CH2(13,14)), 1.78-
1.85 (m, 2H, CH2(12)), 4.07-4.11 (t, 2H, CH2(11)), 7.05-7.08 (m, 1H, Ar-H(9)), 7.30-
7.34 (m, 1H, Ar-H(2)), 7.44-7.46 (d, 1H, Ar-H(8)), 7.67-7.69 (d, 1H, Ar-H(4)), 7.76-
7.81 (m, 1H, Ar-H(3)), 7.89-7.91 (dd, 1H, Ar-H(10)), 8.35 (s, 1H, NH(6)), 8.92-8.94
(d, 1H, Ar-H(1)), 9.29 (s, 1H, Ar-H(5)) and 12.15 (s, 1H, NH(7)). The compound
gave (M++1) peak at (m/z) 368 in its mass spectrum.
Characteristic peaks were displayed in the IR spectrum of 1-[3-
(benzyloxy)pyridin-2-yl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-12) at
3436 (N-H str.), 1675 (C=O str.) and 1229 cm-1
(Ar-O str.). 1H-NMR signals
appeared at 5.29 (s, 2H, CH2(11)), 7.07 (m, 1H, Ar-H(9)), 7.33-7.42 (m, 3H, Ar-
H(14-16)), 7.51-7.57 (m, 3H, Ar-H(2,12,13)), 7.70-7.76 (m, 1H, Ar-H(4)), 7.77-7.79 (m,
1H, Ar-H(3)), 7.91-7.92 (d, 1H, Ar-H(8)), 8.46 (s, 1H, NH(6)), 8.90-8.94 (m, 1H, Ar-
H(10)), 9.22-9.28 (d, 1H, Ar-H(1)), 9.52 (s, 1H, Ar-H(5)) and 12.06 (s, 1H, NH(7)).
The compound offered (M++1) peak at (m/z) 388 in its mass spectrum.
N
1
4
2
3 5N
O
NH
6NH
7
O N10
9
8
O
11
12
13
14
CH3
15
N
1
4
2
3 5N
O
NH
6NH
7
O N
8
10
9
O
11
12 14
16
1513
(I-11) (I-12)
N
1
4
2
3 5N
O
NH
6NH
7
O
N
8
9
10
N
O
14
13
12
11
(I-13)
1-[6-(Morpholin-4-yl)pyridin-2-yl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimi-
din-3-yl)urea (I-13) displayed characteristic Ibands at 3118 (N-H str.), 1688
(C=O str.) and 1229 cm-1
(C-O str.). Its NMR spectrum showed characteristic
signals at 3.66-3.68 (b, 4H, Ar-H(11,12)), 3.95-3.97 (b, 4H, Ar-H(13,14)), 6.17-6.20
(d, 1H, Ar-H(10)), 6.29-6.31 (d, 1H, Ar-H(8)) and 11.48 (s, 1H, NH(7)). The mass
spectrum showed (M++1) peak at (m/z) 367.
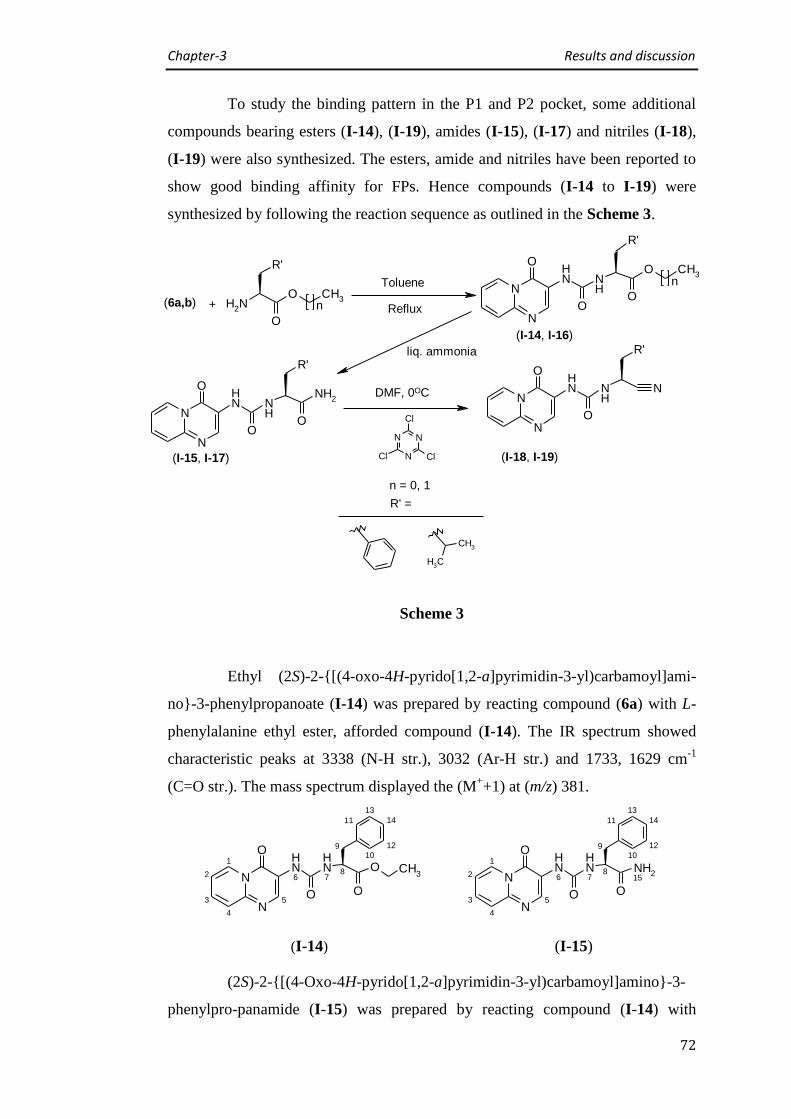
Chapter-3 Results and discussion
72
To study the binding pattern in the P1 and P2 pocket, some additional
compounds bearing esters (I-14), (I-19), amides (I-15), (I-17) and nitriles (I-18),
(I-19) were also synthesized. The esters, amide and nitriles have been reported to
show good binding affinity for FPs. Hence compounds (I-14 to I-19) were
synthesized by following the reaction sequence as outlined in the Scheme 3.
NH2
R'
O
O CH3
n
NH
R'
O
O
N
N
NH
O
O
CH3
n
NH
R'
O
NH2
N
N
NH
O
O
CH3
CH3
NH
R'
N
N
NH
O
O
N
N
N
N
Cl
Cl
Cl
liq. ammonia
(6a,b) +
Toluene Reflux
(I-14, I-16)
R' =
n = 0, 1
DMF, 0OC
(I-15, I-17) (I-18, I-19)
Scheme 3
Ethyl (2S)-2-{[(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]ami-
no}-3-phenylpropanoate (I-14) was prepared by reacting compound (6a) with L-
phenylalanine ethyl ester, afforded compound (I-14). The IR spectrum showed
characteristic peaks at 3338 (N-H str.), 3032 (Ar-H str.) and 1733, 1629 cm-1
(C=O str.). The mass spectrum displayed the (M++1) at (m/z) 381.
N
1
4
2
3 5N
O
NH
6NH
7
O
O8
9
O
10
12
14
13
11
CH3
N
1
4
2
3 5N
O
NH
6NH
7
O
NH2
158
9
O
10
12
14
13
11
(I-14) (I-15)
(2S)-2-{[(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]amino}-3-
phenylpro-panamide (I-15) was prepared by reacting compound (I-14) with

Chapter-3 Results and discussion
73
ammonia solution yielding the compound (I-15). Its IR spectrum showed peaks at
3329 (N-H str,) and 1634 cm-1
(C=O str.). The mass spectrum displayed (M++1)
signal at (m/z) 352.
Methyl (2S)-4-methyl-2-{[(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carba-
moyl]amino}n.pentanoate (I-16) was obtained by reacting the compound (6a)
with L-leucine methyl ester. Its IR spectrum showed characteristic peaks at 3325
(N-H str.) and 1745, 1657 cm-1
(C=O str.). The peak (M++1) was observed at
(m/z) 333 in its mass spectrum.
N
1
4
2
3 5N
O
NH
6NH
7
O
8
9 10
O
CH3
12
CH3
11
OCH313
N
1
4
2
3 5N
O
NH
6NH
7
O
NH2
138
9 10
O
CH3
12
CH3
11
(I-16) (I-17)
(2S)-4-Methyl-2-{[(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]-
amino}n.pentanamide (I-17) was prepared by treating the compound (I-16) with
ammonia solution affording the compound (I-17). Its IR spectrum displayed
characteristic peaks at 3361 (N-H str.) and 1672, 1628 (C=O str.). The compound
offered (M++1) peak at (m/z) 318 in its mass spectrum.
Dehydration of the amide compound (I-16) was done using cyanuric
chloride in dimethylformamamide to get the nitrile product 1-[(1S)-1-(cyano-2-
phenyl)ethyl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-18). Characteristic
peaks at 3.09-3.19 (m, 2H, CH2(9)), 4.92-4.98 (m, 1H, CH(8)), 7.26-7.34 (m, 2H,
Ar-H(2,14)), 7.36-7.37 (m, 4H, Ar-H(10-13)), 7.63-7.67 (m, 2H, Ar-H(4) & NH(7)),
7.72-7.77 (t, 1H, Ar-H(3)), 8.58 (s, 1H, NH(6)), 8.85-8.87 (d, 1H, Ar-H(1)) and 9.10
(s, 1H, Ar-H(5)) were observed in its PMR. The mass spectrum showed (M++1)
peak at (m/z) 334.
N
1
4
2
3 5N
O
NH
6NH
7
O
8
910
12
14
13
11
N
N
1
4
2
3 5N
O
NH
6NH
7
O
8
9 10 CH3
12
CH3
11
N
(I-18) (I-19)

Chapter-3 Results and discussion
74
1-[(1S) 1-(Cyano-3-methyl)butyl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-
3-yl)urea (I-19) was obtained by the dehydration of the compound (I-17) using
cyanuric chloride affording the compound (I-19). The 1H-NMR gave peaks at
0.91-0.96 (m, 6H, CH3(11,12)), 1.64-1.80 (m, 3H, CH2(9) & CH(10)), 4.65-4.71 (m,
1H, CH(8)), 7.27-7.31 (m, 1H, NH(2)), 7.65-7.67 (d, 2H, Ar-H(4) & NH(7)), 7.73-
7.77 (m, 1H, Ar-H(3)), 8.49 (s, 1H, NH(6)), 8.86-8.88 (d, 1H, Ar-H(1)) and 9.12 (s,
1H, Ar-H(5)). The mass spectrum gave (M++1) peak at (m/z) 300.
9
11
13
12
10
N
1
4
2
3 5N
O
NH
6N7
O8
14
CH3
16
CH3
15
(I-20)
1-Benzyl-1-isopropyl-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-
20) showed strong signals at 3373 (N-H str.) and 1641 cm-1
(C=O str.) in its IR
spectrum. Characteristic NMR peaks appeared at 1.14-1.16 (d, 6H, CH3(15,16)),
4.51-4.52 (m, 1H, CH(14)), 4.55 (s, 2H, CH2(8)), 7.25-7.29 (m, 2H, Ar-H(2,13)), 7.36-
7.37 (d, 4H, Ar-H(9-12)), 7.50 (s, 1H, NH(6)), 7.63-7.65 (d, 1H, Ar-H(4)), 7.73-7.77
(m, 1H, Ar-H(3)), 8.78-8.80 (d, 1H, Ar-H(1)) and 8.96 (s, 1H, Ar-H(5)). The
compound offered (M++1) peak at (m/z) 337 in its mass spectrum.
Synthesis of 8-methyl-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
derivatives
1-(2-Methoxyethyl)-3-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)urea (I-21) showed characteristic peaks at 3305 (N-H str.), 1633 (C=O str.) and
1228 cm-1
(C-O str.) in its IR spectrum. Characteristic signals appeared at 2.39
(s, 3H, CH3(3)), 3.22-3.36 (m, 7H, CH2(8,9) & OCH3(10)), 7.09-7.7.11 (m, 2H, Ar-
H(2) & , NH(7)), 7.41(s, 1H, Ar-H(4)), 8.30 (s, 1H, NH(6)), 8.73-8.75 (d, 1H, Ar-H(1))
and 9.06 (s, 1H, Ar-H(5)) in its 1HNMR. The compound displayed (M
+-1) peak at
(m/z) 277 in its mass spectrum.
N-{2-[3-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)ureido]ethyl}-
acetamide (I-22) gave strong IR signals at 3356 (N-H str.) and 1633 cm-1
(C=O
str) in its IR spectrum. The characteristic signals were displayed at 1.81 (s, 3H,
CH3(11)), 2.41 (s, 3H, CH3(3)), 3.1-3.15 (m, 4H, CH2(8,9)), 7.03-7.06 (t, 1H, NH(7)),

Chapter-3 Results and discussion
75
7.11-7.13 (dd, 1H, Ar-H(2)), 7.43 (s, 1H, Ar-H(4)), 7.93-7.94 (t, 1H, NH(10)), 8.26
(s, 1H, NH(6)), 8.76-8.78 (d, 1H, Ar-H(1)) and 9.09 (s, 1H, Ar-H(5)) in its NMR.
The mass showed the (M++1) peak at (m/z) 304.
NH
7
9
8N
1
4
2
5N
O
NH
6
OCH
3
3
OCH3
10
N
1
4
2
5N
O
NH
6NH
7
O8
9
NH
10
O
CH3
11
CH3
3
(I-21) (I-22)
The characteristic IR signals of 1-(8-methyl-4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)-3-[2-(thiophen-2-yl)ethyl]urea (I-23) appeared at 3278 (N-H
str) and 1634 cm-1
(C=O str.). The characteristic peaks at 2.41 (s, 3H, CH3(3)),
2.95-2.98 (m, 4H, CH2(8,9)), 6.92 (s, 1H, Ar-H(10)), 6.98 (b, 1H, Ar-H(11)), 7.11-
7.12 (m, 2H, Ar-H(2) & NH(7)), 7.35-7.37 (d, 1H, Ar-H(12)), 7.43 (s, 1H, Ar-H(4)),
8.32 (s, 1H, NH(6)), 8.75-8.77 (d, 1H, Ar-H(1)) and 9.11 (s, 1H, Ar-H(5)) in its
NMR spectrum. The mass spectrum displayed the (M++1) peak at (m/z) 329.
10 11
12S9
N
1
4
2
5N
O
NH
6NH
7
O8
CH3
3
N
10 12
O
1311
N
1
4
2
5N
O
NH
6NH
7
O8
9
CH3
3
(I-23) (I-24)
The IR spectrum of 1-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
3-[2-(morpholin-4-yl)ethyl]urea (I-24) showed characteristic peaks at 3334 (N-H
str.), 1632, (C=O str.) and 1145 cm-1
(C-O str.). The 1H-NMR displayed peaks at
2.37-2.41 (m, 9H, CH3(3) & CH2(9,10,11)), 3.21-3.24 (m, 2H, CH2(8)), 3.58-3.59 (b,
4H, CH2(12,13)), 6.98-6.99 (m, 1H NH(7)), 7.11-7.12 (d, 1H Ar-H(2)), 7.42 (s, 1H
Ar-H(4)), 8.37 (s, 1H NH(6)), 8.76-8.78 (d, 1H Ar-H(1)) and 9.08-9.09 (d, 1H Ar-
H(5)). The mass spectrum displayed (M++1) peak at (m/z) 332.
1-Benzyl-1-isopropyl-3-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)urea (I-25) gave characteristic bands at 3353 (N-H str.) and 1642 cm-1
(C=O
str.) in its IR spectrum. The characteristic
1HNMR peaks were observed at 1.13-
1.15 (d, 6H, CH3(15,16)), 2.41 (s, 3H, CH3(3)), 4.49-4,51 (m, 1H, CH(14)), 4.54 (s,
2H, CH2(8)), 7.13-7.15 (d, 1H, Ar-H(2)), 7.25- 7.26 (m, 1H, Ar-H(13)), 7.35-7.36 (d,
4H, Ar-H(9-12)), 7.45 (b, 2H, NH(6) & Ar-H(4)), 8.70-8.72 (d, 1H, Ar-H(1)) and 8.87

Chapter-3 Results and discussion
76
(s, 1H, Ar-H(5)). The compound displayed (M++1) peak at (m/z) 351 in its mass
spectrum.
9
11
13
12
10
N
1
4
2
5N
O
NH
6N7
O8
14
CH3
16
CH3
15
CH3
3
(I-25)
Synthesis of 7-chloro-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
derivatives
IR spectrum of 1-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-
(morpholin-4-yl)ethyl] urea (I-26) gave characteristic peaks at 3285 (N-H str.),
1729, 1646 (C=O str.) and 1284 cm-1
(C-O str.). The signals appeared at 2.39 (b,
6H, CH2(9,10,11)), 3.23-3.24 (m, 2H, CH2(8)), 3.60 (b, 4H, CH2(12,13)), 7.08 (b, 1H,
NH(7)), 7.63-7.65 (d, 1H, Ar-H(4)), 7.70-7.73 (dd, 1H, Ar-H(3)), 8.57 (s, 1H, NH(6)),
8.84 (s, 1H, Ar-H(1)) and 9.17 (s, 1H, Ar-H(5)) in its 1H-NMR. The compound
showed (M+) peak at 352.1 and (M
++2) at (m/z) 354.1 in its mass spectrum.
N
10 12
O
1311
N
1
4
2
3 5N
O
NH
6NH
7
O8
9Cl
10 11
12S9
N
1
4
2
3 5N
O
NH
6NH
7
O8
Cl
(I-26) (I-27)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(thiophen-2-
yl)ethyl]urea (I-27) in its IR spectrum showed characteristic signals at 3387 (N-H
str.) and 1651 cm-1
(C=O str.). 1H-NMR displayed the peaks at 2.97-3.00 (t, 2H,
CH2(9)), 3.37-3.40 (t, 2H, CH2(8)), 6.92-6.93 (d, 1H, Ar-H(10)), 6.97-6.99 (m, 1H,
Ar-H(11)), 7.18-7.20 (m, 1H, NH(7)), 7.35-7.37 (m, 1H, Ar-H(12)), 7.63-7.66 (d, 1H,
Ar-H(4)), 7.71-7.74 (dd, 1H, Ar-H(3)), 8.51 (s, 1H, Ar-H(6)), 8.83-8.84 (d, 1H, Ar-
H(1)) and 9.19 (s, 1H, Ar-H(5)). The mass spectrum showed the molecular ion peak
(M+) at 349 and (M
++2) at (m/z) 350.9.
The IR spectrum of 1-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
3-(thiophen-2-yl)methylurea (I-28) gave characteristics IR signals at 1702, 3272
(N-H str.) and 1652 cm-1
(C=O str.). The compound showed peaks at 4.49-4.50

Chapter-3 Results and discussion
77
(d, 2H, CH2(8)), 6.97-6.98 (m, 1H, Ar-H(10)), 7.00 (b, 1H, Ar-H(9)), 7.40-7.41 (d,
1H, Ar-H(11)), 7.56 (b, 1H, NH(7)), 7.64-7.66 (d, 1H, Ar-H(4)), 7.72-7.74 (d, 1H,
Ar-H(3)), 8.51 (s, 1H, NH(6)), 8.89 (s, 1H, Ar-H(1)) and 9.18 (s, 1H, Ar-H(5)) in its
NMR spectrum. The compound showed (M+) peak at (m/z) 335.34 and (M
++2) at
(m/z) 337.38 in its mass spectrum.
S11
109
N
1
4
2
3 5N
O
NH
6NH
7
O8
Cl
N
1
4
2
3 5N
O
NH
6NH
7
O8
9Cl
OCH3
10
(I-28) (I-29)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(2-methoxyeth-
yl)urea (I-29) displayed characteristic peaks 3370 (N-H str.), 1649 (C=O str.) and
1229 cm-1
(C-O str.) in its IR spectrum.
NMR spectrum displayed characteristic
peaks at 3.24-3.39 (m, 7H, CH2(8,9) & CH3(10)), 7.19-7.21 (t, 1H, NH(7)), 7.63-
7.65 (d, 1H, Ar-H(4)), 7.70-7.73 (dd, 1H, Ar-H(3)), 8.51 (s, 1H, NH(6)), 8.83-8.84
(b, 1H, Ar-H(1)) and 9.16 (s, 1H, Ar-H(5)). The mass spectrum gave (M+) peak at
(m/z) 297 and (M++2) at (m/z) 299.
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(4-trifluorome-
thyl)benzylurea (I-30) gave characteristic peaks at 3397 (N-H str.) and 1692, 1656
cm-1
(C=O str.) in its IR spectrum. Characteristic signals appeared at 4.42-4.44
(d, 2H, CH2(8)), 7.57-7.66 (m, 6H, NH(7) & Ar-H(4,9-12)), 7.72- 7.75 (dd, 1H, Ar-
H(3)), 8.58 (s, 1H, NH(6)), 8.84-8.85 (d, 1H, Ar-H(1)) and 9.16 (s, 1H, Ar-H(5)) in its
NMR spectrum. The compound offered (M+) at (m/z) 397.34 and (M
++1) at (m/z)
399.38.
CF3
9
11
12
10
2
ClN
1
4
3 5N
O
NH
6NH
7
O8
N
1
4
2
3 5N
O
NH
6NH
7
O8 9
12
11
10
ClCF
3
(I-30) (I-31)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(3-trifluorometh-
yl)benzylurea (I-31) displayed characteristic peaks at 3312 (N-H str.) and 1677,
1646 cm-1
(C=O str.) in its IR spectrum. 1
HNMR spectrum signals appeared at
4.42-4.43 (d, 2H, CH2(8)), 7.51-7.53 (d, 2H, Ar-H(4,10)), 7.61-7.63 (m, 2H, NH(7)
& Ar-H(11)), 7.70-7.75 (m, 3H, Ar-H(3,9,12)), 8.60 (s, 1H, NH(6)), 8.84-8.85 (d, 1H,
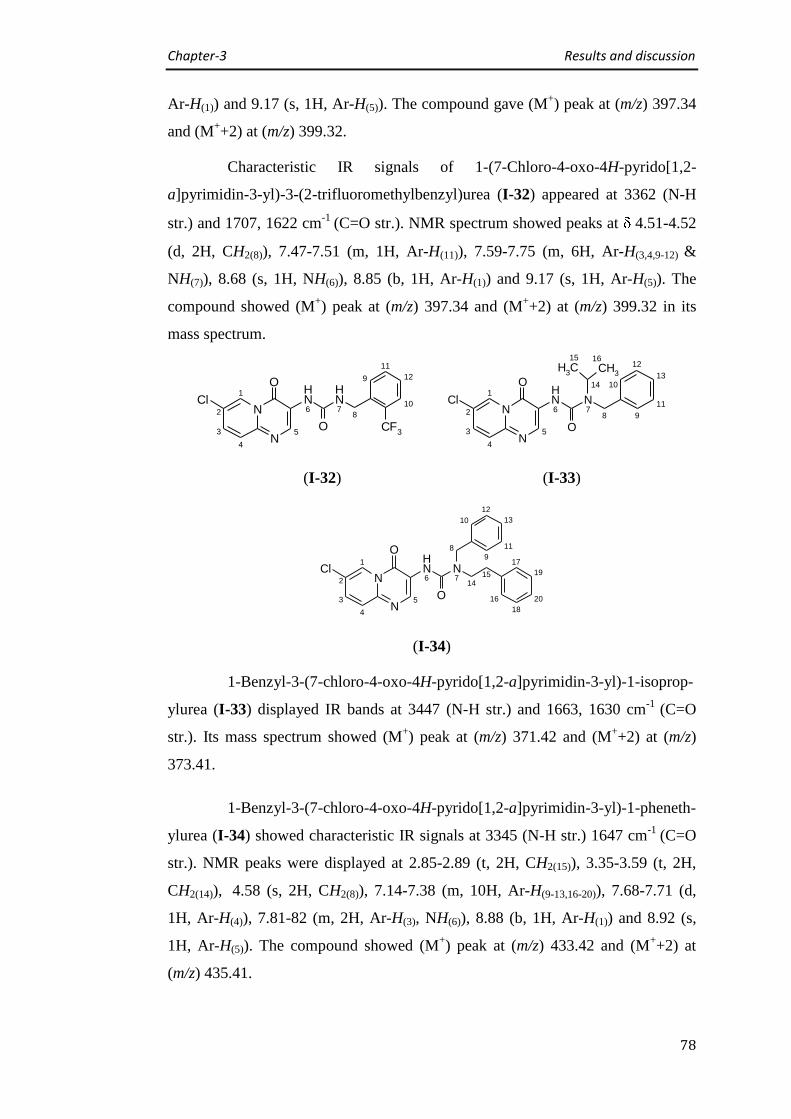
Chapter-3 Results and discussion
78
Ar-H(1)) and 9.17 (s, 1H, Ar-H(5)). The compound gave (M+) peak at (m/z) 397.34
and (M++2) at (m/z) 399.32.
Characteristic IR signals of 1-(7-Chloro-4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)-3-(2-trifluoromethylbenzyl)urea (I-32) appeared at 3362 (N-H
str.) and 1707, 1622 cm-1
(C=O str.). NMR spectrum showed peaks at 4.51-4.52
(d, 2H, CH2(8)), 7.47-7.51 (m, 1H, Ar-H(11)), 7.59-7.75 (m, 6H, Ar-H(3,4,9-12) &
NH(7)), 8.68 (s, 1H, NH(6)), 8.85 (b, 1H, Ar-H(1)) and 9.17 (s, 1H, Ar-H(5)). The
compound showed (M+) peak at (m/z) 397.34 and (M
++2) at (m/z) 399.32 in its
mass spectrum.
N
1
4
2
3 5N
O
NH
6NH
7
O8
10
12
11
9
CF3
Cl
N
1
4
2
3 5N
O
NH
6N7
O8
14
CH3
16
CH3
15
Cl
9
11
13
12
10
(I-32) (I-33)
N
1
4
2
3 5N
O
NH
6N7
O
Cl14
15
8
16
18
20
19
179
11
13
12
10
(I-34)
1-Benzyl-3-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-1-isoprop-
ylurea (I-33) displayed IR bands at 3447 (N-H str.) and 1663, 1630 cm-1
(C=O
str.). Its mass spectrum showed (M+) peak at (m/z) 371.42 and (M
++2) at (m/z)
373.41.
1-Benzyl-3-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-1-pheneth-
ylurea (I-34) showed characteristic IR signals at 3345 (N-H str.) 1647 cm-1
(C=O
str.). NMR peaks were displayed at 2.85-2.89 (t, 2H, CH2(15)), 3.35-3.59 (t, 2H,
CH2(14)),
4.58 (s, 2H, CH2(8)), 7.14-7.38 (m, 10H, Ar-H(9-13,16-20)), 7.68-7.71 (d,
1H, Ar-H(4)), 7.81-82 (m, 2H, Ar-H(3), NH(6)), 8.88 (b, 1H, Ar-H(1)) and 8.92 (s,
1H, Ar-H(5)). The compound showed (M+) peak at (m/z) 433.42 and (M
++2) at
(m/z) 435.41.

Chapter-3 Results and discussion
79
3.1.3 Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives
(Series II)
Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives (piperazine urea analogs) of the Series-II
(II-1 to II-11) were synthesized by coupling of isocyanate (6a-c) with suitably
substituted piperazine derivatives under reflux conditions in toluene as depicted in
Scheme 2.
Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine-1-carboxamide derivatives
The synthesis of compound N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
4-(pyridin-2-yl)piperazine-1-carboxamide (II-1) was done by reacting compound
(6a) with 4-(2-pyridyl)piperazine as described for compound (I-1). Characteristic
IR peaks were observed at 3393 (N-H str.) and 1659 cm-1
(C=O) str.). The NMR
spectrum showed characteristic peaks at 3.57-3.58 (b, 8H, CH2(7-10)), 6.65-6.69
(m, 1H, Ar-H(14)), 6.86-6.88 (d, 1H, Ar-H(11)), 7.31-7.35 (m, 1H, Ar-H(2)), 7.54-
7.58 (m, H, Ar-H(13)), 7.67-7.69 (d, 1H, Ar-H(4)), 7.80-7.84 (m, 1H, Ar-H(3)), 8.08
(s, 1H, NH(6)), 8.13-8.14 (d, 1H, Ar-H(12)), 8.77 (s, 1H, Ar-H(5)) and 8.90-8.93 (d,
1H, Ar-H(1)). The compound displayed (M++1) peak at (m/z) 351 in its mass
spectrum.
N
7 9
N
108
N12
14
1311
N
1
4
2
3 5N
O
NH
6
O
N
7 9
N
108
11 13
1412
N
1
4
2
3 5N
O
NH
6
O
OCH3
15
(II-1) (II-2)
The IR spectrum of 4-(4-methoxyphenyl)-N-(4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-yl)piperazine-1-carboxamide (II-2) showed characteristic bands at
3396 (N-H str.), 1649 (C=O str.) and 1229 cm-1
(C-O str.). The characteristic
signals appeared at 2.99 (b, 4H CH2(9,10)), 3.61(b, 4H CH2(7,8)), 3.81 (s, 3H,
OCH3(15)), 6.87-7.01 (m, 4H, Ar-H(11-14)), 7.32-7.35 (m, 1H, Ar-H(2)), 7.67-7.69 (d,
1H, Ar-H(4)), 7.80-7.84 (t, 1H, Ar-H(3)), 8.06 (s, 1H, NH(6)), 8.77 (s, 1H, Ar-H(5))
and 8.91-8.93 (d, 1H, Ar-H(1)) in its NMR spectrum. The (M++1) peak was
observed at (m/z) 380 in its mass spectrum.

Chapter-3 Results and discussion
80
N-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-[3-(N-cyclopropylcarbo-
xamido)pyridine-2-yl]piperazinecarboxamide (II-3) offered the peaks at 3296 (N-
H str.) and 1654 cm-1
(C=O str.). The 1H-NMR gave characteristic signals at
0.55-0.58 (m, 2H, CH2(16/17)), 0.69-0.72 (m, 2H, CH2(16/17)), 2.82-2.83 (m, 1H,
CH(15)), 3.31-3.36 (t, 4H, CH2(9,10)), 3.57-3.59 (t, 4H, CH2(7,8)), 6.89-6.92 (m, 1H,
Ar-H(12)), 7.32-7.36 (m, 1H, Ar-H(2)), 7.65-7.67 (m, 2H, Ar-H(4,11)), 7.81-7.85 (m,
1H, Ar-H(3)), 8.08 (s, 1H, NH(6)), 8.23-8.25 (dd, 1H, Ar-H(13)), 8.51-8.53 (d, 1H,
NH(14)), 8.76 (s, 1H, Ar-H(5)) and 8.91-8.93 (d, 1H, Ar-H(1)). The (M++1) peak
was observed at (m/z) 434 in its mass spectrum.
N
1
4
2
3 5N
O
NH
6N
O
N
10
9
8
7
N13
12
11
NH14
O
15
16 17
N
1
4
2
3 5N
O
NH
6N
O
11
13
12
15
14
16
18
17 20
19 21
N
9
10
7
8
(II-3) (II-4)
4-Benzhydryl-1-[N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)]carboxami-
dopiperazine (II-4) displayed characteristic signals at 3403 (N-H str.) and 1657
cm-1
(C=O Str.). The 1H-NMR displayed characteristic peaks at 2.33-2.36 (b,
4H, CH2(9,10)), 3.48-3.49 (b, 4H, CH2(7,8)), 4.35 (s, 1H, CH(11)), 7.19-7.22 (m, 2H,
Ar-H(16,21)), 7.30-7.34 (m, 5H, Ar-H(2,14,15,19,20)), 7.45-7.47 (d, 4H, Ar-
H(14,15,19,20)), 7.66-7.68 (d, 1H, Ar-H(4)), 7.79-7.83 (m, 1H, Ar-H(3)), 7.93 (s, 1H,
NH(6)), 8.74 (s, 1H, Ar-H(5)) and 8.88-8.90 (d, 1H, Ar-H(1)). The compound gave
(M++1) peak at (m/z) 440 in its mass spectrum.
Synthesis of N-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives
N-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)-
piperazine-1-carboxamide (II-5) offered characteristic bands at 3395 (N-H str.)
and 1654 cm-1
(C=O str.) in its IR spectrum. Characteristic peaks appeared at
2.45 (3H, CH3(3)), 3.57 (b, 8H, CH2(7-10)), 6.65-6.68 (t, 1H, Ar-H(14)), 6.86-6.88 (d,
1H, Ar-H(11)), 7.20-7.22 (d, 1H, Ar-H(2)), 7.50 (s, 1H, Ar-H(4)), 7.54-7.58 (m, 1H,
Ar-H(13)), 8.05 (s, 1H, NH(6)), 8.13-8.14 (d, 1H, Ar-H(12)), 8.66 (s, 1H, Ar-H(5)) and

Chapter-3 Results and discussion
81
8.82-8.84 (d, 1H, Ar-H(1)) in its NMR spectrum. The mass spectrum displayed the
(M++1) peak at (m/z) 365.
N
7 9
N
108
N12
14
1311
N
1
4
2
3 5N
O
NH
6
OCH
3
N
7 9
N
108
11 13
1412
N
1
4
2
3 5N
O
NH
6
OCH
3
OCH3
15
(II-5) (II-6)
N
1
4
2
3 5N
O
NH
6N
O
11
13
12
15
14
16
18
17 20
19 21
N
9
10
7
8
CH3
(II-7)
N-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-[(4-methoxy-
phenyl)]piperazine-1-carboxamide (II-6) gave characteristic IR signals at 3393
(N-H str.), 1648 cm-1
(C=O str.) and 1241 (C-O str.). Its PMR spectra displayed
characteristic peaks at 2.44 (s, 3H, CH3(3)), 2.97-2.99 (b, 4H, CH2(9,10)), 3.58-
3.61 (b, 4H, CH2(7,8)), 3.80 (s, 3H, OCH3(15)), 6.87-7.00 (m, 4H, Ar-H(11-14)), 7.19-
7.22 (dd, 1H, Ar-H(2)), 7.50 (s, 1H, Ar-H(4)), 8.00 (s, 1H, NH(6)), 8.66 (s, 1H, Ar-
H(5)), 8.82-8.84 (d, 1H, Ar-H(1)) in its NMR spectrum. The mass spectrum
displayed (M++1) peak at (m/z) 394.
N-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-benzhydrylpip-
erazine-1-carboxamide (II-7) displayed characteristic IR bands at 3390 (N-H str.),
1644 cm-1
(C=O str.). The compound displayed characteristic signals at 2.32-
2.34 (b, 4H, CH2(9,10)), 2.43 (s, 3H, CH3(3) ), 3.46-3.48 (b, 4H, CH2(7,8)), 4.34 (s,
1H, CH(11)), 7.18-7.22 (m, 3H, Ar-H(2 & diphenyl)), 7.29-7.33 (m, 4H, Ar-H(diphenyl)),
7.44-7.48 (m, 5H, Ar-H(4 & diphenyl), 7.87 (s, 1H, NH(6)), 8.63 (s, 1H, Ar-H(5)) and
8.79-8.81 (d, 1H, Ar-H(1)) in its NMR spectrum. The (M++1) peak was observed at
(m/z) 454 in its mass spectrum.
Synthesis of N-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)piperazine]-1-carboxamide (II-8) was prepared by refluxing compound (6c)

Chapter-3 Results and discussion
82
with 4-(pyridin-2-yl)piperazine as depicted in Scheme 1. Characteristic peaks
were observed in its IR spectrum at 3397 (N-H str.) and 1729, 1643 cm-1
(C=O
str.). Characteristic signals were observed at 3.57-3.58 (m, 8H, CH2(7-10)), 6.65-
6.68 (m, 1H, Ar-H(14)), 6.86-6.88 (d, 1H, Ar-H(11)), 7.54-7.58 (m, 1H, Ar-H(13)),
7.69-7.71 (d, 1H, Ar-H(4)), 7.83-7.86 (dd, 1H, Ar-H(3)), 8.13-8.14 (d, 1H, Ar-
H(12)), 8.16 (s, 1H, NH(6)), 8.82 (s, 1H, Ar-H(5)) and 8.89-8.90 (d, 1H, Ar-H(1)) in
its NMR spectrum. The compound gave (M+) peak at (m/z) 384.8 and (M
++2) at
(m/z) 386.5.
N
7 9
N
108
N12
14
1311
N
1
4
2
3 5N
O
NH
6
O
Cl
N
7 9
N
108
11 13
1412
N
1
4
2
3 5N
O
NH
6
O
ClOCH3
15
(II-8) (II-9)
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(4-methoxyphe-
nyl) piperazine]-1-carboxamide (II-9) was synthesized by reacting (6c) with 4-(4-
methoxyphenyl)piperazine. The compound offered characteristic IR peaks at 3393
(N-H str.), 1665 (C=O str.) and 1236 cm-1
(C-O str.). Characteristic signals were
observed at 2.97-2.99 (b, 4H, CH2(9,10)), 3.60-3.61 (b, 4H, CH2(7,8)), 3.80 (s, 3H,
OCH3(15)), 6.87-7.00 (m, 4H, Ar-H(11,14)), 7.69-7.71 (d, 1H, Ar-H(4)), 7.83-7.86 (m,
1H, Ar-H(3)), 8.13 (s, 1H, NH(6)), 8.89 (s, 1H, Ar-H(5)) and 8.90 (s, 1H, Ar-H(1)) in
its NMR spectrum. The mass spectrum displayed (M+) peak at 414.4 and (M
++1)
at (m/z) 416.2 in its mass spectrum.
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-(4-tert-butyloxy-
carbonyl)piperazine]-1-carboxamide (II-10) gave strong absorption bands at 3393
(N-H str.), 1691, 1650 cm-1
(C=O str.). NMR spectrum displayed characteristic
peaks at 1.42 (s, 9H, CH3(11)), 3.39 (b, 4H, CH2(9,10)), 3.44-3.45 (b, 4H, CH2(7,8)),
7.68-7.71 (d, 1H, Ar-H(4)), 7.83-7.85 (d, 1H, Ar-H(3)), 8.13 (s, 1H, NH(6)), 8.78 (s,
1H, Ar-H(5)) and 8.88-8.89 (d, 1H, Ar-H(1)). The compound gave (M+) peak at
(m/z) 408.41 and (M++2) at (m/z) 410.39 in its mass spectrum.
N
7 9
N
108
N
1
4
2
3 5N
O
NH
6
O
O 11 CH3
O CH3
CH3
Cl
O13
1211
N
7 9
N
108
N
1
4
2
3 5N
O
NH
6
O O
Cl
(II-10) (II-11)

Chapter-3 Results and discussion
83
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(furan-2-carbo-
nyl)-piperazine]-1-carboxamide (II-11) displayed peaks at 3446 (N-H str.) and
1626 cm-1
(C=O str.) in its IR spectrum. 1HNMR peaks were displayed at 3.57-
3.58 (b, 4H, CH2(9,10)), 3.74 (b, 4H, CH2(7,8)), 6.65 (b, 1H, Ar-H(11)), 7.04-7.05 (m,
1H, Ar-H(12)), 7.69-7.71 (d, 1H, Ar-H(4)), 7.83-7.86 (m, 1H, Ar-H(3)), 7.87 (s, 1H,
Ar-H(13)), 8.18 (s, 1H, NH(6)), 8.80 (s, 1H, Ar-H(5)) and 8.89 (b, 1H, Ar-H(1)). The
compound gave molecular ion (M+) peak at (m/z) 402.37 in its mass spectrum.
3.1.4 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-
carbamate derivatives (Series III)
Synthesis of the carbamate derivatives of Series-III (III-1 to III-9) have
been accomplished by reaction of the isocyanate (6a-c) and suitable alcohol
derivative (A-HO) in toluene under reflux conditions following the procedure as
outlined in Scheme 4.
N
N
O
R N
N
NH
O
O
OR
N F
O
O
N
6 (a-c)
A HO-A
Toluene, reflux
N=C=O
(III-1) - (III-9)
a. R = H, b. R = 8-CH3, c. R = 7-Cl
A =
Scheme 4
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate
derivatives
The isocyanate derivative (6a) and benzyl alcohol were refluxed in
toluene offering the compound benzyl (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)carbamate (III-1). The compound (III-1) displayed characteristic peaks at 3239
(N-H str.), 1718, 1650 cm-1
(C=O str.) and 1116 cm-1
(C-O str.) in its IR spectrum.
The 1H-NMR showed characteristic peaks at 5.17 (s, 2H CH2(7)), 7.32-7.44 (m,
6H, Ar-H(2,8-12)), 7.69-7.71 (d, 1H Ar-H(4)), 7.84-7.89
(m, 1H Ar-H(3)), 8.70 (b,
1H, Ar-H(5)), 8.92-8.94 (d, 1H Ar-H(1)) and 9.05 (b, 1H, NH(6)). The mass
spectrum showed (M++1) peak at (m/z) 296.

Chapter-3 Results and discussion
84
12
8
10
11
9
N
1
4
2
3 5N
O
NH
6O
O7
11
N9
10
8
N
1
4
2
3 5N
O
NH
6O
O7
(III-1) (III-2)
The compound pyridin-2-ylmethyl (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)carbamate (II-2) displayed characteristic bands at 3245 (N-H str.), 1713, 1666
(C=O str.) and 1234 cm-1
(C-O str.) in its IR spectrum. The 1H-NMR showed
characteristic signals at 5.22 (s, 2H, CH2(7)), 7.32-7.37 (m, 2H, Ar-H(2,11)), 7.51-
7.53 (d, 1H, Ar-H(4)), 7.69-7.72 (d, 1H, Ar-H(8)) , 7.83-7.89 (m, 2H, Ar-H(3,10)),
8.55-8.56 (d, 1H, Ar-H(9)), 8.72 (s, 1H, Ar-H(5)), 8.94-8.95 (d, 1H, Ar-H(1)) and
9.21 (b, 1H, NH(6)) in its NMR spectrum. The (M++1) peak was observed in the
mass spectrum at (m/z) 297.
3-Fluorobenzyl (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamate (III-
3) was obtained by reacting isocyanate derivative (6a) with 3-fluorobenzyl alcohol
as described above for compound (III-1). Characteristic absorption bands were
observed at 3416 (N-H str.), 1716, 1659 (C=O str.) and 1234 cm-1
(C-O str.) in its
IR spectrum. The NMR signals appeared at 5.19 (s, 2H, CH2(7)), 7.14-7.19 (m,
1H, Ar-H(10)), 7.27-7.37 (m, 3H, Ar-H(2,9,11)), 7.41-7.47 (m, 1H, Ar-H(8)), 7.69-
7.71 (d, 1H, Ar-H(4)), 7.85-7.89 (m, 1H, Ar-H(3)), 8.72 (b, 1H, Ar-H(5)), 8.93-8.95
(d, 1H, Ar-H(1)) and 9.16 (b, 1H, NH(6)). The mass spectrum gave (M++1) signal
at (m/z) 314.
11
8
10
9
N
1
4
2
3 5N
O
NH
6O
O7 F
8
10
9
N
1
4
2
3 5N
O
NH
6O
O7
O
11O
(III-3) (III-4)
The IR spectrum of 3,4-methylenedioxybenzyl(4-oxo-4H-pyrido[1,2-
a]pyrimdin-3-yl)carbamate (III-4) showed characteristic peaks at 3242 (N-H str.),
1717, 1666 (C=O str.) and 1235 cm-1
(C-O str.). The compound (III-4) displayed
characteristic signals at 5.06 (s, 2H, CH2(7)), 6.02 (s, 2H, CH2(11)), 6.92 (s, 2H,
Ar-H(8,10)), 7.02 (s, 1H, Ar-H(9)), 7.33-7.37 (m, 1H, Ar-H(2)), 7.69-7.71 (d, 1H, Ar-

Chapter-3 Results and discussion
85
H(4)), 7.84-7.89 (m, 1H, Ar-H(3)), 8.70 (b, 1H, Ar-H(5)), 8.92-8.94 (d, 1H, Ar-H(1))
and 9.01 (b, 1H, NH(6)) in its NMR spectrum. The mass spectrum showed (M++1)
peak at (m/z) 340.
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbam-
ate derivatives
Benzyl (8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamate (III-
5) gave characteristic signals at 3242 (N-H str.), 1717, 1635 (C=O str.) and 1224
cm-1
(C-O str). The
compound (III-5) offered characteristic peaks at 2.45 (s, 3H,
CH3(3)), 5.15 (s, 2H, CH2(7)), 7.21-7.23 (d, 1H, Ar-H(2)), 7.31-7.44 (m, 5H, Ar-H(8-
11)), 7.52 (s, 1H, Ar-H(4)), 8.60 (b, 1H, Ar-H(5)), 8.83-8.85 (d, 1H, Ar-H(1)) and
8.97 (b, 1H, NH(6)) in its NMR spectrum. The (M++1) peak was observed at (m/z)
310 in its mass spectrum.
12
8
10
11
9
N
1
4
2
5N
O
NH
6O
O7
CH3
3
11
N9
10
8
N
1
4
2
5N
O
NH
6O
O7
CH3
3
(III-5) (III-6)
Pyridin-2-ylmethyl (8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)carbamate (III-6) displayed characteristic peaks at 3247 (N-H str.), 1721, 1667
(C=O str.) and 1244 cm-1
(C-O str) in its IR spectrum. The 1H-NMR displayed
characteristic peaks at 2.45 (s, 3H, CH3(3)), 5.20 (s, 2H, CH2(7)), 7.22-7.24 (d,
1H, Ar-H(2)), 7.33-7.36 (m, 1H, Ar-H(11)), 7.49-7.51 (m, 1H, Ar-H(8)), 7.53 (b, 1H,
Ar-H(4)), 7.83-7.87 (m, 1H, Ar-H(10)), 8.55-8.56 (d, 1H, Ar-H(9)), 8.62 (b, 1H, Ar-
H(5)), 8.85-8.87 (d, 1H, Ar-H(1)) and 9.14 (b, 1H, NH(6)). The mass spectrum
showed (M++1) peak at (m/z) 311.
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carba-
mate derivatives
2-Pyridinylmethyl (7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)carbamate (III-7) gave characteristic peaks at 3451 (N-H str.), 1724, 1652
(C=O str.) and 1236 cm-1
(C-O str) in its IR spectrum. Characteristic signals were
appeared at 5.23 (s, 2H, CH2(7)), 7.33-7.36 (t, 1H, Ar-H(11)), 7.51-7.53 (d, 1H,

Chapter-3 Results and discussion
86
Ar-H(8)), 7.71-7.73 (d, 1H, Ar-H(4)), 7.83-7.87 (t, 1H, Ar-H(10)), 7.88-7.91 (dd, 1H,
Ar-H(3)), 8.55-8.56 (d, 1H, Ar-H(9)), 8.76 (s, 1H, Ar-H(5)), 8.92-8.93 (d, 1H, Ar-
H(1)) and 9.32 (s, 1H, NH(6)) in its NMR spectrum. The compound showed (M+)
peak at (m/z) 331.37 and (M++1) at (m/z) 333.35 in its mass spectrum.
3-Pyridinylmethyl (7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
carbamate (III-8) displayed characteristic IR absorption bands at 3431 (N-H str.),
1722, 1664 (C=O str.) and 1232 cm-1
(C-O str) in its IR spectrum. The PMR gave
peaks at 5.22 (s, 2H, CH2(7)), 7.41-7.45 (m, 1H, Ar-H(10)), 7.70-7.73 (d, 1H, Ar-
H(4)), 7.86-7.91 (m, 2H, Ar-H(3,9)), 8.54-8.55 (d, 1H, Ar-H(11)), 8.66 (b, 1H, Ar-
H(8)), 8.74 (b, 1H, Ar-H(5)), 8.91- 8.92 (d, 1H, Ar-H(1)) and 9.16 (s, 1H, NH(6)).
The signals, (M+) at (m/z) 331.37 and (M
++2) at (m/z) 333.35 displayed in its mass
spectrum.
11
N9
10
8
N
1
4
2
3 5N
O
NH
6O
O7
Cl
11
8
N
10
9
N
1
4
2
3 5N
O
NH
6O
O7
Cl
(III-7) (III-8)
11
8
10
9
N
1
4
2
3 5N
O
NH
6O
O7 F
Cl
(III-9)
3-Fluorobenzyl (7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carba-
mate (III-9) displayed characteristic bands at 3276 (N-H str.), 1726, 1660 (C=O
str.) and 1229 cm-1
(C-O str) in its IR spectrum. Its NMR signals were observed
at 5.15 (s, 2H, CH2(7)), 7.20-7.24 (m, 2H, Ar-H(9,10)), 7.48-7.51 (m, 2H, Ar-
H(8,11)), 7.70-7.72 (d, 1H, Ar-H(4)), 7.88-7.90 (d, 1H, Ar-H(3)), 8.74 (s, 1H, Ar-
H(5)), 8.91 (s, 1H, Ar-H(1)), 9.18 (s, 1H, NH(6)). The compound showed (M+) peak
at (m/z) 348 and (m/z) 350 (M++2) in its mass spectrum.
3.1.5 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxa-
mide derivatives (Series IV)

Chapter-3 Results and discussion
87
Synthesis of the amide derivatives of Series IV (IV-1 to IV-26) have
been accomplished by reacting the acid (4a-c) with suitable amine (R’-NHA)
using EDC as a coupling agent as outlined in Scheme 5 and Scheme 6.
N
N
O
OH
O
N
N
O
N
O
ARR
CF3
CF3
N
O
CF3
FF
N
ON
NO
CH3
N O
O
CH3
O
CH3
N
O
O
CH3
N
O
CH3
F
FOCH
3
N
O
4 (a-c) (IV-1) - (IV-20)
EDC, HOBT, DIEA, 0OC/ RT
R'-NHA
R'
a. R = H, b. R = 8-CH3, c. R = 7-Cl
OCH3
A =
OCH3
R' = H, i.Pr, Cyclopropyl, Benzyl
Scheme 5
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide
derivatives
N-(4-Methoxybenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxam-
ide (IV-1) was prepared by the carbodimide coupling reaction. The acid derivative
(4a) was coupled with 4-methoxybenzylamine using 1-(3-dimethylaminopropyl)-
3-ethylcarbodiimide affording the compound (IV-1). Its IR spectrum showed
characteristic peaks at 3336 (N-H str.) and 1679 cm-1
(C=O str.). The NMR
spectrum of the compound (IV-1) displayed characteristic peaks at 3.80 (3H,
OCH3(12)), 4.62-4.64 (m, 2H, CH2(7)), 6.86-6.90 (d, 2H, Ar-H(8,9)) 7.31-7.33 (d,
2H, Ar-H(10,11)) 7.34-7.38 (m, 1H, Ar-H(2)) 7.84-7.86 (d, 1H, Ar-H(4)) 7.92-7.97
(m, 1H, Ar-H(3)) 9.19-9.21 (d, 1H, Ar-H(1)), 9.30 (b, 1H, NH(6)) and 9.38 (s,
1H,Ar-H(5)). The mass spectrum displayed (M++1) peak at (m/z) 310.

Chapter-3 Results and discussion
88
N-(3-Fluorobenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxamide
(IV-2) in its IR spectrum displayed peaks at 3327 (N-H str.), 1662 cm-1
(C=O
str.). The 1H-NMR showed characteristic peaks at 4.59-4.61 (d, 2H, CH2(7)),
7.06-7.11 (m, 1H, Ar-H(10)), 7.15-7.21 (m, 2H, Ar-H(9,11)), 7.36-7.41 (m, 1H, Ar-
H(8)), 7.59-7.63 (m, 1H, Ar-H(2)), 7.90-7.92 (d, 1H, Ar-H(4)), 8.19-8.23 (m, 1H,
Ar-H(3)), 9.05 (s, H, Ar-H(5)), 9.19-9.21 (d, 1H, Ar-H(1)) and 9.43-9.46 (t, 1H,
NH(6)). The mass spectrum gave (M++1) peak at (m/z) 298.
8
10
11
97
N
1
4
2
3 5N
O
NH
6
O
OCH3
12
118
10
97
F
N
1
4
2
3 5N
O
NH
6
O
(IV-1) (IV-2)
108
11
9
7
CF3
N
1
4
2
3 5N
O
NH
6
O
118
10
97CF
3N
1
4
2
3 5N
O
NH
6
O
(IV-3) (IV-4)
4-Oxo-N-[2-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-3) displayed characteristic peaks at 3299 (N-H str.), 1695 cm-1
(C=O str.) in its IR spectrum. 1H-NMR showed characteristic signals at 4.76-
4.78 (d, 2H, CH2(7)), 7.48-7.52 (m, 1H Ar-H(2)), 7.59-7.69 (m, 3H, Ar-H(8,10,11)),
7.75-7.77 (d, 1H, Ar-H(9)), 7.91-7.93 (d, 1H, Ar-H(4)), 8.20-8.24 (m, 1H, Ar-H(3)),
9.05 (s, 1H, Ar-H(5)), 9.21-9.23 (d, 1H, Ar-H(1)) and 9.48-9.51 (t, 1H, NH(6)). The
mass spectrum displayed the (M++1) peak at (m/z) 348.
The compound 4-oxo-N-[3-(trifluoromethyl)benzyl]-4H-pyrido[1,2-
a]pyrimidin-3-carboxamide (IV-4) displayed characteristic peaks at 3328 (N-H
str.), 1691, 1645 and 1490 cm-1
(C=O str.). The NMR spectrum of the compound
(IV-4) displayed characteristic peaks at 4.66-4.67 (d, 2H, CH2(7)), 7.56-7.63 (m,
3H, Ar-H(2,8,10)), 7.66-7.68 (d, 1H, Ar-H(11)), 7.72 (s, 1H, Ar-H(9)), 7.90-7.92 (d,
1H, Ar-H(4)), 8.18-8.23 (m, 1H, Ar-H(3)), 9.05 (s, 1H, Ar-H(5)), 9.20-9.21 (d, 1H,
Ar-H(1)) and 9.50-9.53 (t, 1H, NH(6)). The mass spectrum showed (M++1) peak at
(m/z) 348.
4-Oxo-N-[4-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-5) displayed characteristic IR signals at 3336 (N-H str.), 1663

Chapter-3 Results and discussion
89
(C=O str.) and 1123 cm-1
(C-O str.). The compound showed characteristic peaks at
4.75-4.76 (d, 2H, CH2(7)), 7.38-7.41 (m, 1H, Ar-H(2)), 7.49-7.51 (d, 2H, Ar-
H(8,9)), 7.58-7.60 (d, 2H, Ar-H(10,11)), 7.86-7.88 (d, 1H, Ar-H(4)), 7.96-8.00 (m, 1H,
Ar-H(3)), 9.21-9.23 (d, 1H, Ar-H(1)), 9.37-9.38 (s, 1H, Ar-H(5)) and 9.47 (b, 1H,
NH(6)) in its NMR spectrum. Its mass spectrum displayed the (M++1) peak at (m/z)
348.
8
10
11
97
CF3
N
1
4
2
3 5N
O
NH
6
O
8
10
9
7
F
F
N
1
4
2
3 5N
O
NH
6
O
(IV-5) (IV-6)
The compound N-(2,4-Difluorobenzyl)-4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-carboxamide (IV-6) displayed characteristic IR peaks at 3328 (N-H
str.) and 1664 cm-1
(C=O str.) in its IR spectrum. The NMR signals appeared at
4.60-4.62 (d, 2H, CH2(7)), 7.14-7.21 (m, 2H, Ar-H(8,10)), 7.24-7.29 (m, 1H, Ar-
H(9)), 7.59-7.63 (m, 1H, Ar-H(2)), 7.89-7.92 (d, 1H, Ar-H(4)), 8.19-8.23 (m, 1H,
Ar-H(3)), 9.03 (s, 1H, Ar-H(5)), 9.20-9.23 (d, 1H, Ar-H(1)) and 9.42-9.45 (t, 1H,
NH(6)). The mass spectrum showed (M++1) peak at (m/z) 316.
N
10
9
12
11
ON
1
4
2
3 5N
O
NH
6
O7
8
N
1
4
2
3 5N
O
NH
6
O
7
8
9
N
O10
11
12
13
CH3
14
(IV-7) (IV-8)
N-[2-(Morpholin-4-yl)ethyl]-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-7) displayed characteristic IR peaks at 3423 (N-H str.), 1690
(C=O str.) and 1115 cm-1
(C-O str.). Characteristic 1H-NMR signals were
observed at 2.43 (b, 4H, CH2(9,10)), 2.50-2.51 (m, 2H, CH2(8)), 3.45-3.50 (m, 2H,
CH2(7)), 3.59-3.61 (t, 4H, CH2(11,12)), 7.57-7.61 (m, 1H, Ar-H(2)), 7.84-7.90 (d, 1H,
Ar-H(4)), 8.17-8.21 (m, 1H, Ar-H(3)), 9.02 (s, 1H, Ar-H(5)), 9.15-9.16 (t, 1H, NH(6))
and 9.20-9.22 (d, 1H, Ar-H(1)). The mass spectrum showed (M++1) peak at (m/z)
303.
4-Oxo-N-[3-(n.pentyloxy)pyridin-2-yl]-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-8) displayed characteristic peaks at 3297 (N-H str.) 1693 (C=O
str.) and 1218 cm-1
(C-O str.) in its IR spectrum. The 1H-NMR spectrum

Chapter-3 Results and discussion
90
displayed characteristic signals at 0.89-0.93 (t, 3H, CH3(14)), 1.36-1.43 (m, 2H,
CH2(13)), 1.49-1.57 (m, 2H, CH2(12)), 1.78-1.84 (m, 2H, CH2(11)), 4.09-4.13 (t, 2H,
CH2(10)), 7.95-7.97 (d, 2H, Ar-H(4,9)), 8.24-8.28 (m, 1H, Ar-H(3)), 9.13 (s, 1H, Ar-
H(5)), 9.21-9.24 (d, 1H, Ar-H(1)) and 11.51 (s, 1H, NH(6)). The (M++1) peak
appeared in mass spectrum at (m/z) 353.
N-[3-(Benzyloxy)pyridin-2-yl]-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-9) IR spectrum showed characteristic peaks at 3480 (N-H str.),
1699 (C=O str.) and 1285 cm-1
(Ar-O str.). The characteristic signals were
appeared at 5.28 (s, 2H, CH2(10)), 7.00-7.04 (m, 1H, Ar-H(8)), 7.58-7.60 (d, 2H,
Ar-H(11,12)), 8.16-8.18 (m, 1H, Ar-H(7)), 9.27-9.31 (m, 1H, Ar-H(1)), 9.49 (s, 1H,
Ar-H(5)), 11.76 (s, 1H, NH(6)) in its NMR spectrum. (M++1) peak was observed in
the mass spectrum at (m/z) 373 in its mass spectrum.
11
13
15
14
12
N
1
4
2
3 5N
O
NH
6
O
7
8
9
N
O10
7
8
N
9
10
12
11
O
O
13
CH3
14
O
15
CH3
16
NH
6
N
1
4
2
3 5N
O O
(IV-9) (IV-10)
The IR spectrum of N-[(2,5-diethoxy)-4-(morpholin-4-yl)phenyl]-4-oxo-
4H-pyrido[1,2-a]pyrimidine-3-carboxamide (IV-10) displayed characteristic
peaks at 3263 (N-H str.), 1685 (C=O str.) and 1199 cm-1
(C=O str.). Characteristic
signals were observed at 1.44-1.47 (t, 3H, CH3(14/16)) and 1.54-1.58 (3H,
CH3(14/16)), 3.09-3.10 (t, 4H, CH2(9,10)), 3.89-3.91 (t, 4H, CH2(11/12)), 6.60 (s, 1H,
Ar-H(8)), 7.37-7.41 (m, 1H, Ar-H(2)), 7.85-7.87 (d, 1H, Ar-H(4)), 7.95-7.99 (m, 1H,
Ar-H(3)), 8.40 (s, 1H, Ar-H(7)), 9.33-9.34 (d, 1H, Ar-H(1)), 9.42 (s, 1H, Ar-H(5)) and
11.49 (s, 1H, NH(6)) in its NMR spectrum. The mass showed (M++1) peak at (m/z)
439.
The IR spectrum of N-(3,5-Dimethyl-1,2-oxazol-4-yl)-4-oxo-4H-
pyrido[1,2-a]pyrimidin-3-carboxamide (IV-11) displayed characteristic peaks at
3253 (N-H str.) and 1694 cm-1
(C=O str.). The characteristic peaks were displayed
at 2.15 (s, 3H, CH3(8)), 2.33 (s, 3H, CH3(7)), 7.65-7.69 (m, 1H, Ar-H(2)), 7.95-
7.97 (d, 1H, Ar-H(4)), 8.24-8.29 (m, 1H, Ar-H(3)), 9.09 (s, 1H, Ar-H(5)), 9.26-9.27

Chapter-3 Results and discussion
91
(d, 1H, Ar-H(1)) and 10.20 (s, 1H, NH(6)) in its NMR spectrum. The (M++1) peak
at (m/z) 285 was present in its mass spectrum.
CH3
8
N
O
CH3
7
NH
6
N
1
4
2
3 5N
O O
N
1
4
2
3 5N
O
NH
6
O
7
9
8
11
10
CH3
12
OCH3
(IV-11) (IV-12)
N-[1-(4-Methoxyphenyl)ethyl]-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-12) gave peaks at 3315 (N-H str.), 1667 (C=O str.), 1240 (C-O
str.) and 1025 cm-1
in its IR spectrum. Characteristic peaks were displayed 1.61-
1.66 (m, 3H, CH3(12)), 3.79 (s, 3H, OCH3(13)) and 5.28-5.35 (m, 1H, CH(7)), 6.87-
6.91(d, 2H, Ar-H(8,9)), 7.34-7.38 (m, 3H, Ar-H(2,10,11)), 7.83-7.86 (d, 1H, Ar-H(4)),
7.92-7.97 (m, 1H, Ar-H(3)), 9.20-9.22 (d, 1H, Ar-H(1)) and 9.36 (s, 1H, Ar-H(5),
NH(6)) in its NMR spectrum. The mass spectrum showed (M++1) peak at (m/z)
324.
The IR spectrum of N-(1-benzylpiperidin-4-yl)-4-oxo-4H-pyrido[1,2-
a]pyrimidin-3-carboxamide (IV-13) showed characteristic peaks at 3288 (N-H
str.) and 1695 cm-1
(C=O str.). Characteristic signals appeared at 1.52-1.58 (m,
2H, CH2(8,9)), 1.89-1.92 (b, 2H, CH2(8,9)), 2.18-2.22 (t, 2H, CH2(10,11)), 2.70 (b, 2H,
CH2(10,11)), 3.50 (s, 2H, CH2(12)), 3.88 (b, 1H, CH(7)) and 9.20-9.22 (d, 1H, NH(6))
in its NMR. The (M++1) signal was displayed at (m/z) 363 in mass spectrum.
N-(3,4-Methylenedioxybenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-
carboxamide (IV-14) IR spectrum displayed 3260 (N-H str.), 1666 (C=O str.) and
1224 cm-1
(C-O str.). Characteristic peaks were observed at 4.46-4.48 (d, 2H,
CH2(7)), 5.98 (s, 2H, CH2(11)), 6.82-6.88 (m, 2H, Ar-H(8,10)), 6.93 (s, 1H, Ar-H(9)),
7.58-7.62 (m, 1H, Ar-H(2)), 7.89-7.91 (d, 1H, Ar-H(4)), 8.17-8.22 (m, 1H, Ar-H(3)),
9.05 (s, 1H, Ar-H(5)), 9.17-9.19 (d, 1H, Ar-H(1)) and 9.27-9.33 (t, 1H, NH(6)). in its
NMR spectrum. Its mass spectrum showed (M++1) peak at (m/z) 324.
N
1
4
2
3 5N
O
NH
6
O
7
8 10
N
119
12
13
15 17
16
14N
1
4
2
3 5N
O
NH
6
O7 9
8
10O
11
O
(IV-13) (IV-14)

Chapter-3 Results and discussion
92
N
1
4
2
3 5N
O
N
O6
7
10
8
911
12 13
F
(IV-15)
N-Cyclopropyl-N-(2-fluorobenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-15) displayed characteristic peaks at 1669, 1636 cm-1
(C=O, str.)
in its IR spectrum. The 1H-NMR at 0.58-0.61 (d, 4H, CH2(12,13)), 3.06 (b, 1H,
CH(11)) and 4.93 (s, 2H, CH2(6)). The mass spectrum showed (M++1) peak at (m/z)
338.
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxa-
mide derivatives
N-(4-Fluorobenzyl)-8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-16) was prepared by treating (4b) with 4-fluorobenzylamine as
described above for compound (IV-1) yielding (IV-16). IR spectrum of the
compound displayed characteristic peaks at 3287 (N-H str.) and 1677 cm-1
(C=O
str.). The 1H-NMR displayed characteristic peaks at 2.58 (s, 3H, CH3(3)), 4.65-
4.66 (d, 2H, CH2(7)), 7.00-7.04 (t, 2H, Ar-H(8,9)), 7.18-7.21 (m, 1H, Ar-H(2)), 7.34-
7.37 (m, 2H, Ar-H(10,11)), 7.63 (s, 1H, Ar-H(4)), 9.08-9.09 (d, 1H, Ar-H(1)), 9.33 (s,
1H, Ar-H(5)) and 9.35 (b, 1H, NH(6)). The mass spectrum showed the (M++1) peak
at (m/z) 312.
N
1
4
2
5N
O
NH
6
O7 9
8
11
10FCH
3
3
N
1
4
2
5N
O
NH
6
O7 9
8
10
11
CH3
3
CF3
(IV-16) (IV-17)
The IR spectrum of N-(4-trifluorobenzyl)-8-methyl-4-oxo-4H-
pyrido[1,2-a]pyrimidin-3-carboxamide (IV-17) displayed characteristic peaks at
3414 (N-H str.), 1695 (C=O str.) cm-1
. Its 1H-NMR showed characteristics peaks
at 2.55 (s, 3H, CH(3)), 4.65-4.67 (d, 2H, CH(7)), 7.46-7.49 (dd, 1H, Ar-H(2)),
7.55-7.57 (d, 2H, Ar-H(8,9)), 7.69-7.73 (m, 3H, Ar-H(4,10,11)), 9.00 (s, 1H, Ar-H(5)),
9.08-9.10 (d, 1H, Ar-H(1)) and 9.45-9.48 (t, 1H, NH(6)) The compound gave
(M++1) peak at (m/z) 362 in its mass spectrum.

Chapter-3 Results and discussion
93
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxami-
de derivatives
7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4c) was
reacted with 3,4-methylenedioxy benzylamine) as described above for compound
(IV-1) yielding the compound (IV-18) Characteristic IR peaks were observed at
3300 (N-H str.) and 1672 cm-1
(C=O str.). Characteristic NMR signals were
present at 4.46-4.48 (d, 2H, CH2(7)), 5.98 (s, 2H, CH2(11)), 6.81-6.87 (m, 2H, Ar-
H(8,10)), 6.92 (s, 1H, Ar-H(9)), 7.90-7.93 (d, 1H, Ar-H(4)), 8.24-8.26 (dd, 1H, Ar-
H(3)), 9.04 (s, 1H, Ar-H(5)), 9.15-9.16 (d, 1H, Ar-H(1)) and 9.23-9.26 (t, 1H, NH(6)).
The compound gave molecular ion peak (M+) at (m/z) 358.3 and (M
++2) peak at
(m/z) 360.4.
N
1
4
2
3 5N
O
NH
6
O7 9
8
10O
11
OCl
N
1
4
2
3 5N
O
NH
6
O7 9
8
11
10CF
3
Cl
(IV-18) (IV-19)
7-Chloro-4-oxo-N-[4-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimid-
ine-3-carboxamide (IV-19) gave characteristic IR signals at 3315 (N-H str.) and
1674 cm-1
. The compound (IV-19) gave characteristic peaks at 4.67-4.68 (d,
2H, CH2(7)), 755-7.57 (d, 2H, CH2(8,9)), 7.70-7.72 (d, 2H, Ar-H(10,11)), 7.92-7.95 (d,
1H, Ar-H(4)), 8.26 -8.29 (dd, 1H, Ar-H(3)), 9.04 (s, 1H, Ar-H(5)), 9.17-9.18 (d, 1H,
Ar-H(1)) and 9.43-9.46 (t, 1H, NH(6)) in its NMR spectrum. The mass spectrum
displayed (M+) at (m/z) 382.2 and (M
++2) (m/z) 384.2.
N
1
4
2
3 5N
O
N
O6
7
10
8
911
12 13
F
Cl
(IV-20)
The characteristic signals were displayed in IR spectrum of 7-chloro-N-
cyclopropyl-N-(2-fluorobenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxamide
(IV-20) at 3102 (N-H str.) and 1630 cm-1
(C=O str.). The characteristic signals
were displayed at 0.51-0.55 (d, 4H, CH2(12,13)), 2.87 (b, 1H, CH(11)), 4.75 (b, 2H,
CH2(6)), 7.21-7.24 (m, 2H, Ar-H(7,8)), 7.33-7.36 (m, 1H, Ar-H(9)), 7.63 (b, 1H, Ar-
H(4)), 7.84-7.87 (d, 1H, Ar-H(3)), 8.14-8.17 (dd, 1H, Ar-H(1)), 8.47 (s, 1H, Ar-H(5))

Chapter-3 Results and discussion
94
and 9.14 (s, 1H, NH(6)) in its NMR spectrum. The mass spectrum displayed
(M++1) peak at (m/z) 372.4.
7-(4-Methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic
acid intermediate
The 7-chloro ester (3c) was reacted with p-methoxyphenylboronic acid
through Suzuki coupling offering the ester (7) which on acid hydrolysis yielded
the acid (8). The acid derivative (8) was coupled with suitable amines to get the
targeted compounds (IV23 - IV24) as depicted in Scheme 6. Ethyl 7-(4-
methoxyphenyl)-4-
ON
1
4
3 5N
O O
CH3
H3CO
N
1
4
2
3 5N
O
OH
OH3CO
(7) (8)
oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (7) displayed characteristic IR
signals at 1744 (C=O str.) and 1113 cm-1
(C-O str.) in its IR spectrum. The
compound gave (M++1) peak at (m/z) 325 in its mass spectrum.
The ester (7) on acidic hydrolysis furnished the free acid (8) 7-(4-
methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (8). Its IR
spectrum displayed characteristic bands at 3087 (COO-H str.) and 1772 (C=O str.)
cm-1
. The (M
++1) peak at (m/z) 297 was observed in its mass spectrum.
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-
3-carboxamide derivatives
In order to study the hydrophobic binding interactions by increasing the
steric bulk of the group attached to the B-ring (i.e. pyrido) of the pyridopyrimidine
ring, some additional compounds (IV23 - IV24) were synthesized. Synthesis of 7-
(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamide derivatives
of (Series IV) has been was accomplished by using the intermediates of Scheme 1
and by following the reaction sequence outlined in Scheme 6.

Chapter-3 Results and discussion
95
N
N
O
O
O
ClCH
3 N
N
O
O
O
CH3
N
N
O
OH
O
N
N
O
NH
O
A
B(OH)2
CF3
CF3
F O
(3c) (7)
DME, P(Ph3)4, K2CO3,
Reflux
HCl, Reflux
(8) (IV-21) - (IV-24)
EDC, HOBT, DIEA,
A-NH2, 0OC/ RT
H3COH3CO
H3CO
A =
H3CO
Scheme 6
N-(3-Fluorobenzyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyri-
midine-3-carboxamide (IV-21) was synthesized by reacting compound (8) with 3-
fluorobenzylamine as described for compound (IV-1). Its IR spectrum displayed
characteristic peaks at 3319 (N-H str.), 1688 (C=O str.) and 1264 cm-1
(C-O str.).
The compound gave characteristic signals at 3.89 (s, 3H, OCH3(16)), 4.60-4.62
(d, 2H, CH2(7)), 7.07-7.16 (m, 3H, Ar-H(9-11)), 7.20-7.22 (m, 2H, Ar-H(14,15)), 7.37-
7.42 (m, 1H, Ar-H(8)), 7.79-7.81 (d, 2H, Ar-H(12,13)), 7.95-7.98 (d, 1H, Ar-H(4)),
8.54-8.57 (dd, 1H, Ar-H(3)), 9.04 (s, 1H, Ar-H(5)), 9.27-9.28 (d, 1H, Ar-H(1)) and
9.45-9.48 (t, 1H, NH(6)) in its NMR spectrum. A strong peak (M+-1) peak was
observed at (m/z) 403 in its mass spectrum.
13
15
14
12N
1
4
3 5N
O
NH
6
O7 9
8
10
11
F
13
15
14
12N
1
4
3 5N
O
NH
6
O7 9
8
10
11
CF3
H3CO16
H3CO16
(IV-21) (IV-22)
Characteristic bands were observed for the compound N-(3-
trifluoromethylbenzyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-
3-carboxamides (IV-22) in its IR spectrum displayed characteristic bands at 3414
(N-H str.), 1692 (C=O str.) and 1163 (C-O str.) cm-1
. The PMR spectrum gave

Chapter-3 Results and discussion
96
characteristic peaks at 3.83 (s, 3H, OCH3(16)), 4.67-4.68 (d, 2H, CH2(7)), 7.11-
7.14 (d, 2H, Ar-H(14,15)), 7.57-7.64 (m, 2H, Ar-H(9,10)), 7.78-7.81 (d, 2H, Ar-
H(12,13)), 7.67-7.69 (m, 1H, Ar-H(11)), 7.72 (b, 1H, Ar-H(8)), 7.95-7.97 (d, 1H, Ar-
H(4)), 8.53-8.56 (dd, 1H, Ar-H(3)), 9.04 (s, 1H, Ar-H(5)), 9.26 (s, 1H, Ar-H(1)) and
9.51-9.54 (t, 1H, NH(6)). Its mass spectrum gave (M++1) peak at (m/z) 454.
13
15
14
12N
1
4
3 5N
O
NH
6
O7 9
8
11
10CF
3
H3CO16
13
15
14
12
O
8
9 10
11N
1
4
3 5N
O
NH
6
O7
H3CO16
(IV-23) (IV-24)
N-(4-Trifluoromethylbenzyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido-
[1,2-a]pyrimidine-3-carboxamide (IV-23) gave strong peaks at 3425 (N-H str.),
1689 (C=O str.) and 1263 cm-1
(C-O str.) in its IR spectrum. Characteristic peaks
were observed in its NMR at 3.84 (s, 3H, OCH3(16)), 4.68-4.69 (d, 2H, CH2(7)),
7.13-7.15 (d, 2H, Ar-H(14,15)), 7.57-7.59 (d, 2H, Ar-H(8,9)), 7.71-7.73 (d, 2H, Ar-
H(10,11)), 7.80-7.82 (d, 2H, Ar-H(12,13)), 7.96- 7.99 (d, 1H, Ar-H(4)), 8.55-8.58 (dd,
1H, Ar-H(3)), 9.04 (s, 1H, Ar-H(5)), 9.28-9.29 (d, 1H, Ar-H(1)) and 9.52-9.55 (t, 1H,
NH(6)). Its mass spectrum gave (M++1) signal at (m/z) 454.
N-(Tetrahydrofuran-2-ylmethyl)-7-(4-methoxyphenyl)-4-oxo-4H-
pyrido [1,2-a]pyrimidine-3-carboxamide (IV24) displayed IR bands at 3453 (N-H
str.), 1689 (C=O str.) and 1180 cm-1
(C-O str.) in its IR spectrum.
Characteristic
peaks were appeared at 3.84 (s, 3H, OCH3(16)), 3.96-3.99 (m, 1H, CH(8)), 7.12-
7.15 (d, 2H, Ar-H(14,15)) and 7.79-7.84 (d, 2H, Ar-H(12,13)), 7.91-7.97 (d, 1H, Ar-
H(4)), 8.54-8.56 (dd, 1H, Ar-H(3)), 9.03 (s, 1H, Ar-H(5)), 9.17-9.19 (t, 1H, NH(6))
and 9.29-9.30 (d, 1H, Ar-H(1)) in its NMR spectrum. The compound gave (M++1)
signal at (m/z) 380 in its mass spectrum.
3.2 Biological evaluation
The aim of the current study was to develop new antimalarial agents,
ideally directed against the targets which were not previously exploited in the
antimalarial chemotherapy. Malarial cysteine protease falcipain-2 is such a target.
The synthesized compounds were tested for falcipain inhibition-2 (in vitro
inhibition assay) following the reported procedure.93

Chapter-3 Results and discussion
97
Compounds were evaluated for their inhibitory activity against
recombinant FP-2 using Cbz-Phe-Arg-AMC as a fluorogenic substrate.
Preliminary screening was performed at 10 M concentration. An equivalent
concentration of DMSO was used as negative control and the irreversible standard
inhibitor of clan CA family C1 cysteine proteases (papain family), namely E-64
was used as positive control. Assays were performed to determine the percentage
inhibition of the enzyme at a concentration of 10 M. FP-2 activity is assessed by
cleavage of the fluorogenic substrate Cbz-Phe-Arg-AMC releasing the fluorescent
AMC group. Hence decrease in fluorescence intensity in a sample represents
inhibition of enzyme activity. The IC50 values were determined for those
compounds only which showed more than 40 % inhibition at 10 M
concentration.
3.2.1 Assay of falcipain-2 inhibition
The diluted soluble Plasmodium falciparum FP-2 (30 nM) was incubated
for 10 min at room temperature in 100 mM sodium acetate buffer of pH 5.5, 10
mM dithiothreitol (DTT), with a fixed/different concentration of the compounds
to be tested or the standard (E64). Compound solutions were prepared from a
stock in DMSO. After 10 min incubation, the substrate Z-Phe-Arg-AMC was
added to a final concentration of 25 µM. The fluorescence intensity was
monitored (excitation 355 nm; emission 460 nm) for 10 min at room temperature
with a SynergyTM
4 Multi-Mode Microplate Reader (BioTek). The inhibition rate
(%) is calculated using the given equation:
% Inhibition = [1 - (F test / F stand.)] × 100
Where (F test is the fluorescence intensity of the test compound, F
standard is the fluorescence intensity of the standard compound (E64). All values
are the means of three independent determinations and the deviations are <10 %
of the mean value. Fluorescence was monitored for 15 min at room temperature
in a Labsystems Fluoroskan Ascent spectrofluorometer. IC50 values were
determined from plots of percents of activity over the compound concentration
using Graphpad Prism Software. . The IC50 values were determined for those
compounds only which showed 40 % or more of enzyme inhibition at 10 μM. The
results of the study are summarized in Table 1.

Chapter-3 Results and discussion
98
Table 1: In vitro falcipain-2 inhibition activity data of the test compounds
Comp. No.
Falcipain-2
Inhibition
Ratio (%)
IC50
(μM) Comp. No.
Falcipain-2
Inhibition
Ratio (%)
IC50
(μM)
I-1 38.83 NP II-5 37 NP
I-2 18.35 NP II-6 46.14 18.87
I-3 31.11 NP II-7 23.2 NP
I-4 46.37 16.08 II-8 41.34 12.16
I-8 44.43 14.27 II-9 36.01 NP
I-9 25.12 NP III-1 11.52 NP
I-21 62.51 6.36 III-2 17.53 NP
I- 22 15.28 NP III-5 30.71 NP
I-23 45.09 21.16 III-6 41.91 18.32
I-24 27.21 NP IV-14 7.4 NP
I-26 67.97 6.93 IV-16 5.56 NP
I-27 41.81 23.35 IV-18 23.65 NP
II-1 46.13 14.84 E-64 92 (2 M) 0.02
II-2 42.79 NP DMSO 0
NP- not performed
The enzyme inhibition data given in Table 1 shows that the pyrido[1,2-
a]pyrimidin-4-ones show FP-2 inhibition in micromolar range. Based on the result
of evaluated compounds a broad structure activity relationship could be deduced
for this series of the compounds. Urea moiety has the potential to provide potent
FP-2 inhibitors as the carbamate and carboxamide derivatives are much less active
in comparison to the substituted ureas.
Ethylmorpholine (I-26, IC50 6.93 M) and methoxyethyl (I-21, IC50 6.36
M) urea derivatives proved to be the most potent compounds. 2-Thiophenethyl
(I-8, IC50 14.27 M; IV-23, IC50 21.16 M; I-27, IC50 23.35 M), acetamidoethyl
(I-4, IC50 16.08 M) and 4-methoxyphenylpiperazine (II-5, IC50 18.87 M) were
the other groups which gave active compounds when attached to the other end of
the urea moiety. 2-Pyridylpiperazine is another moiety which offered quite active
compounds (II-8, IC50 12.16 M; II-1, IC50 14.84 M).
In general 8-methyl and 7-chloro substituted derivatives offered more
potent compounds indicating that increasing the steric bulk of the group attached
to the B-ring (i.e. pyrido) is likely to provide more potent FP-2 inhibitors. An

Chapter-3 Results and discussion
99
electron rich environment at the end of the carbon chain attached to the urea
nitrogen also seems to be conducive for better activity as indicated by the
compounds having morpholine, 2-thiophenenyl, 2-pyridyl, 4-methoxyphenyl,
acetamidoethyl and 2-methoxyethyl groupings. But bulky groupings (II-7,
benzhydrylpiperazine and I-9, 4-morpholinopropyl) may not offer potent
derivatives.
The above given SAR points have been framed on the basis of available
biological data for a limited number of compounds. For the remaining compounds
the biological data is yet to be obtained. A detailed structure activity relationship
would be framed once the biological data of all the synthesized compounds is
available at hand.
With the availability of biological activity for compounds the current
work has proved the potential of pyrido[1,2-a]pyrimidin-4-one derivatives as
potent FP-2 inhibitors. Based on the activity data of the synthesized compounds it
could be claimed that pyrido[1,2-a]pyrimidin-4-one can serve as a lead series for
future investigations. By appropriate structural optimizations the potency of this
series of compounds can be improved substantially against FP-2 enzyme which in
turn can serve to be potential antimalarial agents. Further optimization of the lead
structures and biological screening is in progress in the laboratory.

4. Experimental

Chapter-4 Experimental work
100
All the reagents and solvents required for synthesis were purified by
general laboratory techniques before use. Compounds were purified by passing
them through silica gel H (100-200 mesh) purifying column using mixture of ethyl
acetate and hexane or methanol as eluent. Melting points were determined using a
Labindia make melting point apparatus (heating block type) and are uncorrected.
Purity of the compounds and completion of reactions were monitored by thin layer
chromatography (TLC) on silica gel plates (60 F254; Merck), visualizing with
ultraviolet light or iodine vapors. The yields reported here are un-optimized. The
IR spectra were recorded using KBr disc method on a Bruker FT-IR, model alpha.
The 1H-NMR spectra (on a Bruker 400 MHz spectrometer) were recorded in
DMSO-d6 (chemical shifts in δ ppm). The assignments of exchangeable protons
were confirmed by the D2O exchange studies wherever required. Mass spectral
data was obtained on a Waters Micromass ESCi spectrometer.
4.1. Chemical studies
4.1.1 Synthesis of 4H-pyrido[1,2-a]pyrimidin-4-one intermediates
(4a-c & 6a-c)
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a)
Diethyl 2-pyridylaminomethylenemalonate (2a)
A mixture of 2-aminopyridine (1a) (10 g, 10.6 mmole) and diethyl
ethoxymethylenemalonate (24.47 g, 10.6 mmole) was refluxed for 2 h. The
reaction mixture was cooled to RT to get (2a) as a light brown solid (14 g, 50%),
m. p. 68-69 °C (Lit. 217,218
67.5-68 °C).
Anal.:
TLC : Rf 0.85 (EtOAc)
IR : 3274, 1686, 1649, 1601, 1558, 1414, 1248 and 1147 cm-1
1H-NMR : 1.22-1.29 (m, 6H), 4.12-4.18 (q, 2H), 4.20-4.25 (q,
2H), 7.14-
7.17 (m, 1H), 7.39-7.41 (d, 1H), 7.79-7.83 (m, 1H), 8.36-8.37
(d, 1H), 9.05-9.08 (d, 1H), 10.79-10.82 (d, 1H)
MS : (m/z) 265 (M++1)

Chapter-4 Experimental work
101
Ethyl 4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (3a)
The pyridine derivative (2a) (13 g, 5.9 mmole) was refluxed in diphenyl
ether (100 ml) using air condenser for 8 h in a two-neck round-bottom flask (250
ml). The reaction mixture was cooled to RT and hexane (500 ml) was added in to
it to get a solid precipitate. The precipitated solid was filtered, washed with
hexane and dried to obtain the compound (3a) (10 g, 93.1%), m. p. 112-13 °C
(Lit.217
110-11 °C).
Anal.:
TLC : Rf 0.48 (EtOAc)
IR : 1730, 1482, 1288, 1227 and 1103 cm-1
1H-NMR : 1.29-1.32 (t, 3H), 4.25-4.30 (m, 2H), 7.56-7.59 (m, 1H), 7.84-
7.87 (d, 1H), 8.19-8.23 (m, 1H), 8.87 (s, 1H), 9.16-9.18 (d,
1H)
MS : (m/z) 219 (M++1)
4-Oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4a)
A solution of the ester (3a) (10 g, 5.2 mmole) in conc. HCl (15 ml) was
refluxed for 6 h. The reaction mixture was cooled to RT to get a solid precipitate.
The solid precipitate was filtered, washed with ether and dried to get compound
(4a) as a dark brown solid (8 g, 85.5%), m. p. 268 °C (decomp.) [Lit.217
265 °C
(decomp.)].
Anal.:
TLC : Rf 0.1 (EtOAc)
IR : 3221, 3091, 1757, 1677, 1636, 1519, 1483, 1275, 1247 cm-1
1H-NMR : 7.67-7.71 (m, 1H), 7.97-8.00 (d, 1H), 8.31-8.35 (m, 1H), 8.96
(s, 1H), 9.22-9.23 (d, 1H)
MS : (m/z) 191 (M++1)
Synthesis of 3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a)
4-Oxo-4H-pyrido[1,2-a]pyrimidine-3-carbonylazide (5a)
In a two-neck round-bottom flask (250 ml), compound (4a) (8 g, 4.2
mmole) and triethylamine (8.5 g, 8.4 mmole) were dissolved in DMF (25 ml). The
reaction mixture was cooled to 0 oC. Ethyl chloroformate (5.03 g, 4.6 mmole) was
added to the reaction mixture and stirred for 15 min followed by addition of

Chapter-4 Experimental work
102
aqueous solution of sodium azide (10.95 g, 16.8 mmole) resulting into a yellow
solid precipitate. The solid precipitate was filtered, washed with cold water,
hexane and then air dried affording the compound (5a) as brown solid (6.8 g,
75.1%) m. p. 130-31 °C.
Anal.:
TLC : Rf 0.28 (EtOAc)
IR : 2310, 1725, 1472, 1185 and 1012 cm-1
1H-NMR : 7.61-7.64 (m, 1H), 7.87-7.91 (d, 1H), 8.24-8.35
(m, 1H), 8.94
(s, 1H), 9.10-9.28 (d, 1H)
MS : (m/z) 214 (M+-1)
3-Isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6a)
The acid azide (5a) (4 g, 1.8 mmole) in toluene was refluxed for 2 h.
Work up of the reaction mixture was done by removing toluene under vacuum to
get the compound (6a) (3.2 g, 40.6%), m. p. 180-81 °C.
Anal.:
TLC : Rf 0.5 (EtOAc)
IR : 2140, 1687, 1650, 1575, 1550, 1490 and 1190 cm-1
MS : (m/z) 188 (M++1)
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic
acid (4b)
Diethyl (4-methyl-2-pyridylamino)methylenemalonate (2b)
4-Methyl-2-aminopyridine (10 g, 9.25 mmole) was reacted with diethyl
ethoxymethylenemalonate (21.29 g, 9.25 mmole) as described above for
compound (2a), offering compound (2b) (15 g, 58.2%), m. p. 73-74 °C (Lit.217
72-
73 °C).
Anal.:
TLC : Rf 0.9 (EtOAc)
IR : 1731, 1682, 1648, 1605, 1548, 1374 and 1218 cm-1
MS : (m/z) 279 (M++1)

Chapter-4 Experimental work
103
Ethyl 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (3b)
In analogy to compound (3a), 2b (10 g, 4.31 mmole) was refluxed in
diphenyl ether to obtain compound (3b) (10 g, 88.4%), m. p. 174-75 °C (Lit.217
171-72 °C).
Anal.:
TLC : Rf 0.55 (EtOAc)
IR : 1689, 1639, 1556, 1501, 1284 and 1143 cm-1
1H-NMR : 1.30 (t, 3H), 2.51 (s, 3H), 4.23-4.29 (m, 2H), 7.42-7.44 (m,
1H), 7.67 (s, 1H), 8.82 (s, 1H), 9.04-9.05 (d, 1H)
MS : (m/z) 233 (M++1)
8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4b)
A solution of the ester (3b) (10 g, 4.3 mmole) in conc. HCl (15 ml) was
refluxed for 2 h. The reaction mixture was cooled to RT to get a solid precipitate.
The solid so obtained was filtered, washed with ether (100 ml) and dried to offer
8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxylic acid (4b) (8.5 g, 96.7%),
m. p. 225 °C [Lit.219
223 °C].
Anal.:
TLC : Rf 0.11 (EtOAc)
IR : 2645, 1764, 1696, 1620, 1522, 1268, 1204 and 1152 cm-1
1H-NMR : 2.62 (s, 3H), 7.62-7.64 (m, 1H), 7.89 (s, 1H), 8.88-8.90 (d,
1H), 9.14-9.16 (d, 1H), 10.9 (b, 1H)
MS : (m/z) 203 (M+-1)
Synthesis of 3-isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one
(6b)
8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbonylazide (5b)
Compound (5b) was prepared by reacting the acid (4b) (8 g, 3.9 mmole)
with ethyl chloroformate (4.68 g, 4.3 mmole) and sodium azide (10.19 g, 15.7
mmole) as described above for compound (5a) resulting in to compound (5b) (7 g,
77.9%), m. p. 148-50 °C.
Anal.:
TLC : Rf 0.35 (EtOAc)
IR : 2142, 1712, 1640, 1569, 1485, 1285 and 1172 cm-1

Chapter-4 Experimental work
104
1H-NMR : 2.56 (s, 3H), 7.49-7.51 (d, 1H), 7.75 (s, 1H), 8.84 (s, 1H),
9.09-9.13 (d, 1H)
MS : (m/z) 230 (M++1)
3-Isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (6b)
The azide (5b) (4 g) was refluxed in toluene for 2 h. The reaction
mixture was cooled to RT and the solvent was removed under vacuo to get 3-
isocyanato-8-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (6b) (3.34 g, 95.1%), m. p.
234-35 °C.
Anal.:
TLC : Rf 0.61 (EtOAc)
IR : 2353, 2316, 2223, 1670, 1639 and 1482 cm-1
MS : (m/z) 201 (M++1)
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic
acid (4c)
Diethyl (5-chloro-2-pyridylamino)methylenemalonate (2c)
Compound (2c) was prepared by reacting compound (1c) (10 g, 7.8
mmole) with diethyl ethoxymethylenemalonate (17.96 g, 7.8 mmole) as described
above for compound (2a), affording the compound (2c) (15 g, 42.9%), m. p. 133-
34 °C (Lit.220
131-32 °C).
Anal.:
TLC : Rf 0.91 (EtOAc)
IR : 3270, 1676, 1644, 1588, 1552, 1395, 1219 and 1007 cm-1
1H-NMR : 1.11-1.29 (m, 6H), 4.13-4.26 (m, 4H), 7.47-7.49 (d, 1H),
7.91-7.94 (m, 1H), 8.41 (s, 1H), 8.94-8.97 (d, 1H), 10.79-
10.82 (d, 1H)
MS : (m/z) 299 (M++1)
Ethyl 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate (3c)
With a procedure similar to that adopted for compound (3a) compound
(3c) was prepared from compound (2c) (15 g, 5.03 mmole) providing the titled
compound (3c) (10 g, 78.8%), m. p. 146-47 °C (Lit.220
147-48 °C).
Anal.:

Chapter-4 Experimental work
105
TLC : Rf 0.54 (EtOAc)
IR : 1681, 1645, 1591, 1552, 1466, 1366, 1222 and 1091 cm-1
MS : (m/z) 253 (M++1)
7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4c)
In analogy to compound (4a), compound (4c) was obtained by
employing the same procedure using 3c (10 g, 3.9 mmole) to get 7-chloro-4-oxo-
4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (4c) (8 g, 89.9%), m. p. 256-57 °C.
Anal.:
TLC : Rf 0.12 (EtOAc)
IR : 3063, 1756, 1661, 1612, 1510, 1418, 1263 and 1163 cm-1
MS : (m/z) 225.6 (M++1)
Synthesis of 7-chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one
(6c)
7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbonylazide (5c)
Compound (5c) was prepared by reacting compound (4c) (8 g, 6.25
mmole) with ethyl chloroformate (6.78 g, 6.87 mmole) and sodium azide (16.25 g,
25 mmole) as described above for compound (5a) producing the compound (5c)
(6.5 g, 74.2%), m. p. 159-60 °C.
Anal.:
TLC : Rf 0.25 (EtOAc)
IR : 2158, 1732, 1563, 1472, 1296, 1201 and 1023 cm-1
MS : (m/z) 250.6 (M++1)
7-Chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-one (6c)
5c (4 g, 1.8 mmole) was refluxed in toluene for 2 h. The reaction mixture
was concentrated to obtain 7-chloro-3-isocyanato-4H-pyrido[1,2-a]pyrimidin-4-
one (6c) (3.34 g, 98.5%), m. p. 312-13 °C.
Anal.:
TLC : Rf 0.65 (EtOAc)
IR : 2317, 2243, 1668, 1625, 1547, 1525, 1475 and 1182 cm-1
MS : (m/z) 222.6 (M++1)

Chapter-4 Experimental work
106
4.1.2 Synthesis of substituted (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
yl)urea derivatives (Series I)
Synthesis of (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea derivatives
1-(4-Chlorobenzyl)-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-1)
The isocyanate (6a) (0.5 g, 0.27 mmole) and 4-chlorobenzylamine
(0.227 g, 0.16 mmole) in toluene (25 ml) were refluxed together for 6 h. The
reaction mixture was cooled to RT to get a solid precipitate. The solid precipitate
was filtered and washed with hexane. The product was purified by column
chromatography using hexane (20%) in ethyl acetate as eluent to get the titled
compound (I-1) (0.2 g, 37.9%), m. p. 250-51 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)
IR : 3318, 3051, 1693, 1637, 1536, 1479, 1220 and 1136 cm-1
1H-NMR : 4.39-4.41 (d, 2H), 7.26-7.35 (m, 3H), 7.41-7.47 (m, 2H), 7.56
(t, 1H), 7.61-7.64 (d, 1H), 7.68-7.71 (m, 1H), 8.55 (s, 1H),
8.85-8.87 (d, 1H), 9.16 (s, 1H)
MS : (m/z) 328.9 (M+), 330.8 (M
++2)
1-(4-Methoxybenzyl)-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-2)
Compound (I-2) was prepared by treating the isocyanate (6a) (0.3 g,
0.16 mmole) with 4-methoxybenzylamine (0.172 g, 0.16 mmole) as described
above for compound (I-1). The crude product was purified by column
chromatography using methanol (1%) in ethyl acetate as eluent offering the
compound (I-2) (0.15 g, 51.9%), m. p. 229-30 °C.
Anal.:
TLC : Rf 0.68 (1% MeOH in EtOAc)
IR : 3361, 3277, 3038, 1639, 1242, 1220, 1180 and 1122 cm-1
1H-NMR : 3.73 (s, 3H), 4.24-4.25 (d, 2H), 6.89-6.91 (d, 2H), 7.22-7.27
(m, 3H), 7.40-7.43 (t, 1H), 7.61-7.63 (d, 1H), 7.68-7.72 (m,
1H), 8.39 (s, 1H), 8.84-8.86 (d, 1H), 9.18 (s, 1H)
MS : (m/z) 325 (M++1)

Chapter-4 Experimental work
107
1-(2-Methoxyethyl)-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-3)
Compound (I-3) was prepared by reacting the isocyanate (6a) (0.3 g,
0.16 mmole) with 2-methoxyethylamine (0.119 g, 0.16 mmole) as described
above for compound (I-1). The crude product after chromatographic purification
using methanol (0.9%) in ethyl acetate as eluent provided compound (I-3) (0.5 g,
61.1%), m. p. 196-97 °C.
Anal.:
TLC : Rf 0.65 (1% MeOH in EtOAc)
IR : 3362, 1696, 1639, 1548, 1463, 1226, 1192 and 1099 cm-1
1H-NMR : 3.25-3.28 (m, 5H), 3.70-3.39 (t, 2H), 7.16-7.17 (m, 1H), 7.23-
7.27 (m, 1H), 7.60-7.63 (d, 1H), 7.67-7.72 (m, 1H), 8.41 (s,
1H), 8.45-8.86 (d, 1H), 9.16(s, 1H)
MS : (m/z) 263 (M++1)
N-{2-[3-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)ureido]ethyl}acetamide (I-4)
The isocyanate (6a) (0.3 g, 0.16 mmole) was reacted with 2-
acetamidoethylamine (0.62 g, 0.16 mmole) as described above for compound (I-
1). Chromatographic purification of the crude product using methanol (0.9%) in
ethyl acetate as eluent furnished compound (I-4) (0.5 g, 61.1%), m. p. 247-49 °C.
Anal.:
TLC : Rf 0.45 (5% MeOH in EtOAc)
IR : 3317, 3251, 1726, 1633, 1536, 1467, 1219 and 1120 cm-1
1H-NMR : 1.81 (s, 3H), 3.11-3.16 (m, 4H), 7.09-7.11 (m, 1H), 7.23-7.27
(m, 1H), 7.61-7.63 (d, 1H), 7.68-7.72 (m, 1H), 7.94 (b, 1H),
8.35 (s, 1H), 8.84-8.86 (d, 1H), 9.17 (s, 1H)
MS : (m/z) 290 (M++1)
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(2-trifluoromethylbenzyl)urea
(I-5)
Compound (I-5) was prepared by reacting the isocyanate (6a) (0.3 g,
0.16 mmole) with 2-trifluoromethylbenzylamine (0.281 g, 0.16 mmole) as
described above for compound (I-1). Work-up of the reaction mixture followed by
chromatographic purification of the crude product using hexane (10%) in ethyl

Chapter-4 Experimental work
108
acetate as eluent earned the titled compound (I-5) (0.22 g, 37.8%), m. p. 243-45
°C.
Anal.:
TLC : Rf 0.52 (EtOAc)
IR : 3315, 3047, 1667, 1638, 1548, 1478, 1328 and 1224 cm-1
1H-NMR : 4.51-4.52 (d, 2H), 7.25-7.28 (t, 1H), 7.47-7.51 (m, 1H), 7.56-
7.64 (m, 3H), 7.68-7.74 (m, 3H), 8.59 (s, 1H), 8.86-8.88 (d,
1H), 9.17 (s, 1H)
MS : (m/z) 361 (M+-1)
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(4-trifluoromethylbenzyl)urea
(I-6)
The isocyanate (6a) (0.3 g, 0.16 mmole) was treated with 4-
trifluoromethylbenzylamine (0.281 g, 0.16 mmole) as described above for
compound (I-1). The crude product after chromatographic purification using
hexane (10%) in ethyl acetate as eluent offered the titled compound (I-6) (0.21 g,
34.4%), m. p. 256-57 °C.
Anal.:
TLC : Rf 0.48 (EtOAc)
IR : 3328, 3048, 1670, 1636, 1478, 1332, 1222 and 1107 cm-1
1H-NMR : 4.42-4.44 (d, 2H), 7.25-7.28 (m, 1H), 7.51-7.54 (d, 2H), 7.59-
7.64 (m, 2H), 7.69-7.73 (m, 3H), 8.50 (s, 1H), 8.86-8.87 (d,
1H), 9.16 (s, 1H)
MS : (m/z) 363 (M++1)
1-(2,4-Difluorobenzyl)-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-7)
The coupling reaction of the isocynate (6a) (0.3 g, 0.16 mmole) was
done with 2,4-difluorobenzylamine (0.229 g, 0.16 mmole) as described above for
compound (I-1). Work-up of the reaction mixture followed by chromatographic
purification of the crude product using hexane (5%) in ethyl acetate as eluent
yielded compound (I-7) (0.1 g, 18.8%), m. p. 170-71 °C.
Anal.:
TLC : Rf 0.48 (EtOAc)
IR : 3328, 1676, 1558, 1483, 1236 and 1127 cm-1

Chapter-4 Experimental work
109
1H-NMR : 4.35-4.37 (d, 2H), 7.14-7.20 (m, 2H), 7.24-7.28 (m, 2H),
7.52-7.55 (t, 1H), 7.62-7.64 (d, 1H), 7.69-7.74 (m, 1H), 8.50
(s, 1H), 8.85-8.87 (d, 1H), 9.14 (s, 1H)
MS : (m/z) 331 (M++1)
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(thiophen-2-yl)ethyl]urea (I-8)
A mixture of isocyanate (6a) (0.3 g, 0.16 mmole) and 2-(2-
thiophenyl)ethylamine (0.308 g, 0.16 mmole) were reacted together as described
above for compound (I-1). The crude product was purified by column
chromatography using methanol (2%) in ethyl acetate as eluent to obtain the
compound (I-8) (0.2 g, 39.6%), m. p. 216-17 °C.
Anal.:
TLC : Rf 0.5 (1% MeOH in EtOAc)
IR : 3213, 3033, 1656, 1525, 1469, 1218, 1182 and 1127 cm-1
1H-NMR : 2.97-3.01 (t, 2H), 3.37-3.4 (t, 2H), 6.92-6.93 (b, 1H), 6.97-
6.99 (m, 1H), 7.15-7.18 (t, 1H), 7.23-7.27 (m, 1H), 7.35-7.37
(d, 1H), 7.61-7.63 (d, 1H), 7.68-7.72 (m, 1H), 8.41 (s, 1H),
8.84-8.85 (d, 1H), 9.19 (s, 1H)
MS : (m/z) 315 (M++1)
1-[3-(Morpholin-4-yl)propyl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
(I-9)
6a (0.5 g, 0.27 mmole) and 3-(4-morpholino)propylamine (0.227 g,
0.16 mmole) were reacted together as described above for compound (I-1). The
crude product after chromatographic purification using hexane (10%) in ethyl
acetate as eluent offered the titled compound (I-9) (0.18 g, 40.6%), m. p. 189-90
°C.
Anal.:
TLC : Rf 0.67 (EtOAc)
IR : 3297, 3072, 1663, 1549, 1479, 1228 and 1118 cm-1
1H-NMR : 1.55-1.62 (m, 2H), 2.28-2.32 (m, 2H), 2.34 (b, 4H), 3.10-3.15
(m, 2H), 3.56-3.58 (t, 4H), 7.01-7.04 (m, 1H), 7.23-7.27 (m,
1H), 7.60-7.63 (d, 1H), 7.67-7.71 (m, 1H), 8.29 (s, 1H), 8.84-
8.86 (d, 1H), 9.17 (s, 1H)

Chapter-4 Experimental work
110
MS : (m/z) 332 (M++1)
1-(3, 3-Diphenyl)propyl-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-10)
The isocyanate (6a) (0.25 g, 0.13 mmole) was reacted with 3,3-
diphenylpropylamine (0.282 g, 0.13 mmole) as described above for compound (I-
1). The chromatographic purification of the crude product using hexane (10%) in
ethyl acetate as eluent afforded the urea derivative (I-10) (0.21 g, 39.4%), m. p.
259-60 °C.
Anal.:
TLC : Rf 0.62 (EtOAc)
IR : 3324, 3133, 3024, 1664, 1631, 1559, 1476 and 1241cm-1
1H-NMR : 2.18-2.23 (m, 2H), 2.97-3.05 (m, 2H), 4.01-4.05 (t, 1H), 7.11-
7.34 (m, 12H), 7.60-7.63 (d, 1H), 7.67-7.74 (m, 1H), 8.32 (s,
1H), 8.85-8.92 (dd, 1H), 9.15(s, 1H)
MS : (m/z) 399 (M++1)
1-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[3-(n.pentyloxy)pyridin-2-yl]-
urea (I-11)
Compound (I-11) was prepared by reacting the isocynate (6a) (0.3 g,
0.16 mmole) with 3-n.pentyloxy-2-aminopyridine (0.289 g, 0.16 mmole) as
described above for compound (I-1). The crude product after chromatographic
purification using methanol (1%) in ethyl acetate as eluent offered the desired
product (I-11) (0.2 g, 33.96%), m. p. 178-80 °C.
Anal.:
TLC : Rf 0.47 (EtOAc)
IR : 3387, 3350, 3067, 1665, 1550, 1478 and 1220 cm-1
1H-NMR : 0.90-0.94 (t, 3H), 1.32-1.47 (m, 4H), 1.78-1.85 (m, 2H), 4.07-
4.11 (t, 2H), 7.05-7.08 (m, 1H), 7.30-7.34 (m, 1H), 7.44-7.46
(d, 1H), 7.67-7.69 (d, 1H), 7.76-7.81 (m, 1H), 7.89-7.91 (dd,
1H), 8.35 (s, 1H), 8.92-8.94 (d, 1H), 9.29 (s, 1H), 12.15 (s,
1H)
MS : (m/z) 368 (M++1)

Chapter-4 Experimental work
111
1-[3-(Benzyloxy)pyridin-2-yl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
(I-12)
Compound (6a) (0.3 g, 0.16 mmole) was treated with 2-amino-3-
benzyloxypyridine (0.319 g, 0.16 mmole) as described above for compound (I-1).
The crude product was purified by column chromatography using methanol (1%)
in ethyl acetate as eluent offering the titled compound (I-12) (0.19 g, 30.6%), m.
p. 249-50 °C.
Anal.:
TLC : Rf 0.46 (EtOAc)
IR : 3133, 1675, 1558, 1482, 1192 and 1118 cm-1
1H-NMR : 5.29 (s, 2H), 7.07 (m, 1H), 7.33-7.42 (m, 3H), 7.51-7.57 (m,
3H), 7.70-7.76 (m, 1H), 7.77-7.79 (m, 1H), 7.91-7.92 (d, 1H),
8.46 (s, 1H), 8.90-8.94 (m, 1H), 9.22-9.28 (d, 1H), 9.52 (s,
1H), 12.06 (s, 1H)
MS : (m/z) 388 (M++1)
1-[6-(Morpholin-4-yl)pyridin-2-yl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
urea (I-13)
Compound (I-13) was prepared by treating the isocyanate (6a) (0.3 g,
0.16 mmole) with 2-amino-6-morpholinopyridine (0.287 g, 0.16 mmole) as
described above for compound (I-1). The chromatographic purification of the
crude product using methanol (1%) in ethyl acetate as eluent offered compound
(I-13) (0.25 g, 42.5%), m. p. 275-76 °C.
Anal.:
TLC : Rf 0.45 (EtOAc)
IR : 3118, 1688, 1558, 1484, 1440, 1229 and 1113 cm-1
1H-NMR : 3.66-3.68 (b, 4H), 3.95-3.97 (b, 4H), 6.17-6.20 (d, 1H), 6.29-
6.31 (d, 1H), 7.11-7.15 (m, 1H), 7.48-7.52 (m, 2H), 7.58-7.62
(m, 1H), 7.67-7.70 (d, 1H), 8.97-8.99 (d, 1H), 9.53 (s, 1H),
11.48 (s, 1H)
MS : (m/z) 367 (M++1)

Chapter-4 Experimental work
112
Ethyl (2S)-2-{[(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]amino}-3-
phenylpropanoate (I-14)
The isocyanate (6a) (1.94 g, 6.1 mmole) and L-phenylalanine ethyl ester
(2 g, 6.1 mmole) in toluene (25 ml) were refluxed together for 6 h. The reaction
mixture was cooled to RT to yield a solid precipitate. The solid precipitate was
filtered, washed with hexane and dried to get the titled compound (I-14) (2 g,
62.5%), m. p. 158-59 °C.
Anal.:
TLC : Rf 0.75 (EtOAc)
IR : 3338, 3032, 1733, 1688, 1629, 1132 and 1185 cm-1
MS : 381(M++1)
Methyl (2S)-4-methyl-2-{[(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]-
amino}n.pentanoate (I-15)
Compound (I-15) was obtained by reacting compound (6a) (2.85 g, 1.37
mmole) with L-leucine methyl ester (2 g, 1.37 mmole) as described above for
compound (I-14). The crude product was recrystallized from methanol to yield
compound (I-15) (2.1 g, 45.8%), m. p. 129-30 °C.
Anal.:
TLC : Rf 0.78 (EtOAc)
IR : 3325, 3276, 1745, 1657, 1634, 1550, 1528 and 1191 cm-1
MS : (m/z) 333 (M++1)
(2S)-2-{[(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]amino}-3-phenyl-
propanamide (I-16)
A mixture of compound (I-14) (2 g, 0.52 mmole) and ammonia solution
was stirred for 12 h. The solid precipitate so obtained was filtered, washed with
water and recrystallized from methanol to furnish compound (I-16) (1.8 g,
97.4%), m. p. 239-40 °C.
Anal.:
TLC : Rf 0.35 (EtOAc)
IR : 3329, 1634, 1519, 1477, 1244 and 1191 cm-1
MS : (m/z) 352 (M++1)

Chapter-4 Experimental work
113
(2S)-4-Methyl-2-{[(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamoyl]amino}-
n.pentanamide (I-17)
Compound (I-15) (2 g, 0.52 mmole) was stirred in ammonia solution for
12 h to get the solid precipitate. The precipitated recrystallized from methanol to
obtain compound (I-17) (1.6 g 83.7%), m. p. 243-44 °C.
Anal.:
TLC : Rf 0.4 (EtOAc)
IR : 3361, 3320, 1672, 1628, 1528, 1469, 1437 and 1234 cm-1
MS : (m/z) 318 (M++1)
1-[(1S)-1-(Cyano-2-phenyl)ethyl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
urea (I-18)
The amide (I-16) (1.2 g, 0.34 mmole) was dissolved in
dimethylformamide (2.5 ml) and cooled to 0°C. Cyanuric chloride (1.26 g, 0.68
mmole) was added in small portions and stirred for 2 h. The reaction mixture was
quenched in ice-cold water (20 ml) to get solid precipitate. The solid precipitate
was filtered and dried. The crude product so obtained was purified by column
chromatography to earn the compound (I-18) (0.34 g, 29.8%), m. p. 223-25 °C.
Anal.:
TLC : Rf 0.45 (EtOAc)
IR : 3303, 2210, 1686, 1631, 1530, 1433, 1227 and 1133 cm-1
1H-NMR : 3.09-3.19 (m, 2H), 4.92-4.98 (m, 1H), 7.26-7.34 (m, 2H),
7.36-7.37 (m, 4H), 7.63-7.67 (m, 2H), 7.72-7.77 (m, 1H), 8.58
(s, 1H), 8.85-8.87 (d, 1H), 9.10 (s, 1H)
MS : (m/z) 334 (M++1)
1-[(1S) 1-(Cyano-3-methyl)butyl]-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-
urea (I-19)
Compound (I-19) was prepared by treating (I-17) (2.0 g, 0.63 mmole)
with cyanuric chloride (2.33 g, 1.26 mmole) as described above for compound (I-
18). The chromatographic purification of the crude product using hexane (25%) in
ethyl acetate as eluent offered compound (I-19) (0.35 g, 18%), m. p. 237-39 °C.
Anal.:
TLC : Rf 0.55 (EtOAc)

Chapter-4 Experimental work
114
IR : 3342, 2210, 1642, 1557, 1510, 1465, 1434 and 1235 cm-1
1H-NMR : 0.91-0.96 (d, 6H), 1.64-1.80 (m, 3H), 4.65-4.71(m, 1H), 7.27-
7.31 (m, 1H), 7.65-7.67 (d, 2H), 7.73-7.77 (m, 1H), 8.49 (s,
1H), 8.86-8.88 (d, 1H), 9.12 (s, 1H)
MS : (m/z) 300 (M++1)
1-Benzyl-1-isopropyl-3-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea (I-20)
Isocyanate (6a) (0.25 g, 0.13 mmole) and N-isopropylbenzylamine
(0.199 g, 0.13 mmole) were reacted as described above for compound (I-1). The
crude product by chromatographic purification using hexane (10%) in ethyl
acetate offered the titled compound (I-20) (0.15 g, 30.6%), m. p. 145-46 °C.
Anal.:
TCL : Rf 0.61 (EtOAc)
IR : 3373, 1641, 1527, 1483, 1410, 1232, 1176, and 940 cm-1
1HNMR : 1.14-1.16 (d, 6H), 4.51-4.52 (m, 1H), 4.55 (s, 2H), 7.25-7.29
(m, 2H), 7.36-7.37 (d, 4H), 7.50 (s, 1H), 7.63-7.65 (d, 1H),
7.73-7.77 (m, 1H), 8.78-8.80 (d, 1H), 8.96 (s, 1H)
MS : (m/z) 337 (M+-1)
Synthesis of 8-methyl-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
derivatives
1-(2-Methoxyethyl)-3-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
(I-21)
Compound (I-21) was prepared by reacting compound (6b) (0.3 g, 0.149
mmole) with 2-methoxyethyl amine (0.111 g, 0.149 mmole) as described above
for compound (I-1). Work up of the reaction mixture followed by
chromatographic purification using hexane (5%) in ethyl acetate as eluent yielded
compound (I-21) (0.16 g, 38.84%), m. p. 233-34 °C.
Anal.:
TLC : Rf 0.75 (1% MeOH in EtOAc)
IR : 3388, 3305, 2890, 2360, 1633, 1534, 1452, 1228, 1179,
1088, 1006, 935, 809 and 630 cm-1
1HNMR : 2.39 (s, 3H), 3.22-3.25 (t, 4H), 3.35 (s, 3H), 7.09-7.11 (m,
2H), 7.41 (s, 1H), 8.30 (s, 1H), 8.73-8.75 (d, 1H), 9.06 (s, 1H)

Chapter-4 Experimental work
115
MS : (m/z) 277 (M+-1)
N-{2-[3-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)ureido]ethyl}aceta-
mide (I-22)
Compound (I-22) was prepared by reacting compound (6b) (0.3 g, 0.149
mmole) with 2-acetamidoethylamine (0.151 g, 0.149 mmole) as described above
for compound (I-1). Chromatographic purification of the crude product was done
using hexane (5%) in ethyl acetate as eluent to yield the desired porduct (I-22)
(0.2 g, 44.2%), m. p. 269-70 °C.
Anal.:
TLC : Rf 0.7 (1% MeOH in EtOAc)
IR : 3356, 3289, 1633, 1524, 1451, 1239 and 1004 cm-1
1H-NMR : 1.81 (s, 3H), 2.41 (s, 3H), 3.1-3.15 (m, 4H), 7.03-7.06 (m,
1H) 7.11-7.13 (dd, 1H), 7.43 (s, 1H), 7.93-7.94 (m, 1H), 8.26
(s, 1H), 8.76-8.78 (d, 1H), 9.09 (s, 1H)
MS : (m/z) 304 (M++1)
1-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(thiophen-2-yl)ethyl]
urea (I-23)
Compound (6b) (0.3 g, 0.149 mmole) was treated with 2-(2-
thiophenyl)ethylamine (0.188 g, 0.149 mmole) as described above for compound
(I-1). Work-up of the reaction mixture followed by chromatographic purification
of the crude product using methanol (0.2%) in ethyl acetate as eluent produced 1-
(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(thiophen-2-yl)-ethyl]urea
(I-23) (0.2 g, 40.8%), m. p. 249-50 °C
Anal.:
TLC : Rf 0.78 (1% MeOH in EtOAc)
IR : 3278, 1634, 1518 and 1449 cm-1
1H-NMR : 2.41 (s, 3H), 2.95-2.98 (m, 4H), 6.92 (s, 1H), 6.98 (d, 1H),
7.11-7.12 (m, 2H), 7.35-7.37 (d, 1H), 7.43 (s, 1H), 8.32 (s,
1H), 8.75-8.77 (d, 1H), 9.11 (s, 1H)
MS : (m/z) 329 (M++1)

Chapter-4 Experimental work
116
1-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(morpholin-4-yl)eth-
yl]urea (I-24)
The isocyanate derivative (6b) (0.3 g, 0.149 mmole) was reacted with 2-
(4-morpholinyl)ethylamine (0.163 g, 0.149 mmole) as described above for
compound (I-1). Chromatographic purification of the crude product using
methanol (0.5%) in ethyl acetate as eluent offered compound (I-24) (0.21 g,
42.5%), m. p. 225-26 °C.
Anal.:
TLC : Rf 0.64 (1% MeOH in EtOAc)
IR : 3334, 1632, 1569, 1460, 1235, 1145, 1104 and 1002 cm-1
1H-NMR : 2.37-2.41 (m, 9H), 3.21-3.24 (m, 2H), 3.58-3.59 (b, 4H),
6.98-6.99 (m, 1H), 7.11-7.12 (d, 1H), 7.42 (s, 1H), 8.37 (s,
1H), 8.76-8.78 (d, 1H), 9.08-9.09 (d, 1H)
MS : (m/z) 332 (M++1)
1-Benzyl-1-isopropyl-3-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
(I-25)
Isocyanate derivative (6b) (0.25 g, 0.12 mmole) in toluene (25 ml) was
refluxed with N-isopropylbenzylamine (0.185 g, 0.12 mmole) for 4 h to get a
solid. The solid so obtained was filtered and washed with hexane.
Chromatographic purification of the crude product using hexane (5%) in ethyl
acetate provided the titled compound (I-25) (0.16 g, 36.7%), m. p. 161-62 °C.
Anal.:
TLC : Rf 0.62 (EtOAc)
IR : 3353, 3031, 2971, 1642, 1546, 1486, 1403 and 1242 cm-1
1HNMR : 1.13-1.15 (d, 6H), 2.41 (s, 3H), 4.49-4,51 (m, 1H), 4.54 (s,
2H), 7.13-7.15 (d, 1H), 7.25- 7.26 (m, 1H), 7.35-7.36 (d, 4H),
7.45 (b, 2H), 8.70-8.72 (d, 1H), 8.87 (s, 1H)
MS : (m/z) 349 (M+), 350.9 (M
++2)
Synthesis of 7-chloro-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)urea
derivatives

Chapter-4 Experimental work
117
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(morpholin-4-
yl)ethyl]urea (I-26)
Compound (I-26) was prepared by reacting compound (6c) (0.3 g, 0.135
mmole) with 2-(4-morpholinyl)ethylamine (0.176 g, 0.135 mmole) as described
above for compound (I-1). Chromatographic purification of the crude product
using methanol (2%) in ethyl acetate as eluent furnished the titled compound (I-
26) (0.22 g, 46.2%), m. p. 226-27 °C.
Anal.:
TLC : Rf 0.5 (2% MeOH in EtOAc)
IR : 3285, 1729, 1646, 1552, 1464, 1284, 1247 and 1117 cm-1
1H-NMR : 2.39 (b, 6H), 3.23-3.24 (m, 2H), 3.60 (b, 4H), 7.08 (b, 1H),
7.63-7.65 (d, 1H), 7.70-7.73 (dd, 1H), 8.57 (s, 1H), 8.84 (s,
1H), 9.17 (s, 1H)
MS : (m/z) 352.1 (M+), 354.1 (M
++2)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-[2-(thiophen-2-yl)eth-
yl]urea (I-27)
Compound (I-27) was obtained by treating compound (6c) (0.3 g, 0.135
mmole) with 2-(2-thiophenyl)ethylamine (0.17 g, 0.135 mmole) as described
above for compound (I-1). Work-up of the reaction mixture followed by
chromatographic purification of the crude product using hexane (10%) in ethyl
acetate as eluent offered compound (I-27) (0.24 g, 50.8%), m. p. 216-17 °C.
Anal.:
TLC : Rf 0.75 (1% MeOH in EtOAc)
IR : 3387, 1651, 1544, 1510, 1470, 1224 and 1189 cm-1
1H-NMR : 2.97-3.00 (t, 2H), 3.37-3.40 (t, 2H), 6.92-6.93 (d, 1H), 6.97-
6.99 (m, 1H), 7.18-7.20 (m, 1H), 7.35-7.37 (m, 1H), 7.63-7.66
(d, 1H), 7.71-7.74 (dd, 1H), 8.51 (s, 1H), 8.83-8.84 (d, 1H),
9.19 (s, 1H)
MS : (m/z) 349 (M+), 350.9 (M
++2)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(thiophen-2-yl)methyl-
urea (I-28)
Isocyanate derivative (6c) (0.25 g, 0.11 mmole) in toluene was refluxed

Chapter-4 Experimental work
118
with (2-thiophenyl)methylamine (0.127 g, 0.11 mmole) to get a solid.
The solid so obtained was filtered and washed with hexane. The crude product
was purified by column chromatography using 1% methanol in ethyl acetate to
obtain the compound (I-28) (0.15 g, 39.76%), m. p. 285-87 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)
IR : 3272, 3024, 1652, 1562, 1472, 1337, 1240, and 1155 cm-1
1HNMR : 4.49-4.50 (d, 2H), 6.97-6.98 (m, 1H), 7.00 (s, 1H), 7.40-7.41
(d, 1H), 7.56 (b, 1H), 7.64-7.66 (d, 1H), 7.72-7.74 (d, 1H),
8.51 (s, 1H), 8.89 (s, 1H), 9.18 (s, 1H)
MS : (m/z) 335.34(M++1), 337.38 (M
++2)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(2-methoxyethyl)urea
(I-29)
A mixture of isocyanate derivative (6c) (0.25 g, 0.11 mmole), 2-
methoxyethylamine (0.162 g, 0.11 mmole) and toluene was refluxed for 4 h to get
solid precipitate. The solid so obtained was filtered and washed with hexane.
Chromatographic purification of the product using hexane (15%) in ethyl acetate
accomplished the desired compound (I-29) (0.22 g, 65.74%), m. p. 254-56 °C.
Anal.:
TLC : Rf 0.65 (5% MeOH in EtOAc)
IR : 3370, 3116, 2972, 1702, 1649, 1557, 1528, 1434, 1354, 1229,
1199, 1106, 817, 757 and 593 cm-1
1HNMR : 3.24-3.39 (m, 7H,), 7.19-7.21 (m, 1H), 7.63-7.65 (d, 1H),
7.70-7.73 (dd, 1H), 8.51 (s, 1H), 8.83-8.84 (b, 1H), 9.16 (s,
1H)
MS : (m/z) 297.39 (M+), 299.38 (M
++2)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(4-trifluoromethyl)ben-
zylurea (I-30)
A mixture of isocyanate derivative (6c) (0.25 g, 0.11 mmole) and 4-
trifluoromethylbenzylamine (0.197 g, 0.11 mmole) in toluene was refluxed for 4
h. Solid precipitate so obtained was filtered and washed with hexane.

Chapter-4 Experimental work
119
Chromatographic purification of the crude product using 1% methanol in ethyl
acetate yielded the titled compound (I-30) (0.23 g, (51.4%), m. p. 287-89 °C.
Anal.:
TLC : Rf 0.61 (1% MeOH in EtOAc)
IR : 3397, 3187, 1692, 1656, 1632, 1364 and 1226 cm-1
1HNMR : 4.42-4.44 (d, 2H), 7.57-7.66 (m, 6H), 7.72-7.75 (dd, 1H), 8.58
(s, 1H), 8.84-8.85 (d, 1H), 9.16 (s, 1H)
MS : (m/z) 397.34(M+), 399.38 (M
++2)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(3-trifluoromethyl)ben-
zylurea (I-31)
A mixture of isocyanate derivative (6c) (0.25 g, 0.11 mmole) and 3-
trifluoromethylbenzylamine (0.197 g, 0.11 mmole) and toluene (50 ml) was
refluxed for 4 h. Solid precipitate so obtained was filtered and washed with
hexane. The crude product was purified by column chromatography using hexane
(5%) in ethyl acetate to furnish the titled compound (I-31) (0.22 g, 49.1%), m. p.
260-62 °C.
Anal.:
TLC : Rf 0.6 (1% MeOH in EtOAc)
IR : 3312, 3282, 3085, 1677, 1646, 1564, 1488, 1433, 1331, 1243,
1160, 1107, 1016, 821, 765 and 644 cm-1
1HNMR : 4.42-4.43 (d, 2H), 7.51-7.53 (d, 2H), 7.61-7.63 (m, 2H), 7.70-
7.75 (m, 3H), 8.60 (s, 1H), 8.84-8.85 (d, 1H), 9.17 (s, 1H)
MS : (m/z) 397.34(M+), 399.32 (M
++2)
1-(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-3-(2-trifluoromethyl)ben-
zylurea (I-32)
Isocyanate derivative (6c) (0.25 g, 0.11 mmole) in toluene was refluxed
with 2-trifluoromethylbenzylamine (0.197 g, 0.11 mmole) to get solid precipitate.
The solid so obtained was filtered and washed with hexane. Chromatographic
purification of the crude product using hexane (5%) in ethyl acetate furnished the
titled compound (I-32) (0.23 g, 51.4%), m. p. 276-78 °C.
Anal.:

Chapter-4 Experimental work
120
TLC : Rf 0.62 (1% MeOH in EtOAc)
IR : 3362, 3126, 1707, 1622, 1558, 1461, 1311 and 1225 cm-1
1HNMR : 4.51-4.52 (d, 2H), 7.47-7.51 (m, 1H), 7.59-7.75 (m, 6H), 8.68
(s, 1H), 8.85 (b, 1H), 9.17 (s, 1H)
MS : (m/z) 397.34 (M+), 399.32, (M
++2)
1-Benzyl-3-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-1-isopropylurea
(I-33)
Isocyanate derivative (6c) (0.25 g, 0.11 mmole) in toluene (25 ml) was
refluxed with N-isopropylbenzylamine (0.168 g, 0.11 mmole) for 5 h to get solid
precipitate. Chromatographic purification of the crude product using hexane
(10%) in ethyl acetate offered the desired compound (I-33) (0.2 g, 47.8%), m. p.
158-60 °C.
Anal.:
TLC : Rf 0.64 (EtOAc)
IR : 3447, 1663, 1630, 1521, 1487, 1234, 1114 and 1070 cm-1
MS : (m/z) 371.42 (M+), 373.41(M
++2)
1-Benzyl-3-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-1-phenethylurea
(I-34)
Reaction of isocyanate derivative (6c) (0.25 g, 0.11 mmole) and N-
benzyl phenylethylamine (0.238 g, 0.11 mmole) was done as described above for
compound (I-1). The crude product was purified by column chromatography
using 2% methanol in ethyl acetate to obtain the titled compound (I-34) (0.18 g,
36.8%), m. p. 195-97 °C.
Anal.:
TLC : Rf 0.55 (1% MeOH in EtOAc)
IR : 3345, 3028, 1647, 1550, 1493, 1435, 1366 and 1233 cm-1
1HNMR : 2.85-2.89 (t, 2H), 3.35-3.59 (t, 2H),
4.58 (s, 2H), 7.14-7.38
(m, 11H), 7.68-7.71 (d, 1H), 7.81-7.82 (b, 2H), 8.88 (b, 1H),
8.92 (s, 1H)
MS : (m/z) 433.42 (M+), 435.41 (M
++2)

Chapter-4 Experimental work
121
3.1.3 Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives (Series II)
Synthesis of N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-
yl)- piperazine-1-carboxamide derivatives
N-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)piperazine-1-
carboxamide (II-1)
Compound (II-1) was prepared by reacting compound (6a) (0.3 g, 0.16
mmole) with 4-(2-pyridyl)piperazine (0.262 g, 0.16 mmole) as described above
for compound (I-1). The crude product was purified by column chromatography
using hexane (10%) in ethyl acetate as eluent to afford the compound (II-1) (0.19
g, 33.8%), m. p. 194-95 °C.
Anal.:
TLC : Rf 0.51 (1% MeOH in EtOAc)
IR : 3393, 3107, 1659, 1596, 1537, 1482 and 1238 cm-1
1H-NMR : 3.57-3.58 (b, 8H), 6.65-6.69 (m, 1H), 6.86-6.88 (d, 1H), 7.31-
7.35 (m, 1H), 7.54-7.58 (m, 1H), 7.67-7.69 (d, 1H), 7.80-7.84
(m, 1H), 8.08 (s, 1H), 8.13-8.14 (d, 1H), 8.77 (s, 1H) 8.90-
8.93 (d, 1H)
MS : (m/z) 351 (M++1)
4-(4-Methoxyphenyl)-N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)piperazine-1-
carboxamide (II-2)
The urea derivative (II-2) was obtained by refluxing the isocyanate (6a)
(0.3 g, 0.16 mmole) with 1-(4-methoxyphenyl piperazine (0.308 g, 0.16 mmole) in
toluene for 6 h. The reaction mixture was cooled to RT and the solid precipitate
was filtered. The chromatographic purification of the crude product using hexane
(10%) in ethyl acetate as eluent yielded the titled compound (II-2) (0.19 g 31.2%),
m. p. 143-45 °C.
Anal.:
TLC : Rf 0.52 (1% MeOH in EtOAc)
IR : 3396, 3031, 2926, 1649, 1479, 1437, 1229 and 1021 cm-1
1H-NMR : 2.99 (b, 4H), 3.61(b, 4H), 3.81 (s, 3H), 6.87-7.01 (m, 4H),

Chapter-4 Experimental work
122
7.32-7.35 (m, 1H), 7.67-7.69 (d, 1H), 7.80-7.84 (m, 1H), 8.06
(s, 1H), 8.77 (s, 1H), 8.91-8.93 (d, 1H)
MS : (m/z) 380 (M++1)
N-(4-Oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-[3-(N-cyclopropylcarboxamido)
pyridine-2-yl]piperazinecarboxamide (II-3)
Compound (II-3) was prepared by reacting the isocyanate (6a) (0.3 g,
0.16 mmole) with N-cyclopropyl-2-(piperazine-1-yl)pyridine-3-carboxamide
(0.319 g, 0.16 mmole) as described above for compound (I-1). The crude
compound was purified by column chromatography using methanol (3%) in ethyl
acetate as eluent to get the desired compound (II-3) (0.22 g, 31.6%), m. p. 140-42
°C.
Anal.:
TLC : Rf 0.4 (EtOAc)
IR : 3296, 3035, 1654, 1530, 1484, 1434, 1234 and 1134 cm-1
1H-NMR : 0.55-0.58 (m, 2H), 0.69-0.72 (m, 2H), 2.82-2.83 (m, 1H),
3.31-3.36 (t, 4H), 3.57-3.59 (t, 4H), 6.89-6.92 (m, 1H), 7.32-
7.36 (m, 1H), 7.65-7.67 (m, 2H), 7.81-7.85 (m, 1H), 8.08 (s,
1H), 8.23-8.25 (dd, 1H), 8.51-8.53 (d, 1H), 8.76 (s, 1H), 8.91-
8.93 (d, 1H)
MS : (m/z) 434 (M++1)
4-Benzhydryl-1-[N-(4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)]carboxamido-
piperazine (II-4)
The isocyanate (6a) (0.3 g, 0.16 mmole) was reacted with
benzhydrylpiperazine (0.404 g, 0.16 mmole) as described above for compound (I-
1). The crude product was purified by column chromatography using hexane (5%)
in ethyl acetate as eluent offering the titled compound (II-4) (0.2 g, 42.5%), m. p.
253-55 °C.
Anal.:
TLC : Rf 0.45 (EtOAc)
IR : 3403, 3032, 1657, 1527, 1484, 1294, 1232 and 1110 cm-1
1H-NMR : 2.33-2.36 (t, 4H), 3.48-3.49 (t, 4H), 4.35 (s, 1H), 7.19-7.22

Chapter-4 Experimental work
123
(m, 2H), 7.30-7.34 (m, 5H), 7.45-7.47 (d, 4H), 7.66-7.68 (d,
1H), 7.79-7.83 (m, 1H), 7.93 (s, 1H), 8.74 (s, 1H), 8.88-8.90
(d, 1H)
MS : (m/z) 440 (M++1)
Synthesis of N-(8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives
N-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)piperazi-
ne-1-carboxamide (II-5)
Compound (6b) (0.3 g, 0.149 mmole) was reacted with 4-(2-
pyridyl)piperazine (0.243 g, 0.149 mmole) as described above for compound (I-
1). The chromatographic purification of the crude product was done using
methanol (2%) in ethyl acetate as eluent to get the product (II-5) (0.18 g, 33.1%),
m. p. 160-61°C.
Anal.:
TLC : Rf 0.75 (1% MeOH in EtOAc)
IR : 3395, 3045, 1654, 1596, 1531, 1484, 1245, and 1192 cm-1
1H-NMR : 2.45 (s, 3H), 3.57 (b, 8H), 6.65-6.68 (m, 1H), 6.86-6.88 (d,
1H), 7.20-7.22 (d, 1H), 7.50 (s, 1H), 7.54-7.58 (m, 1H), 8.05
(s, 1H), 8.13-8.14 (d, 1H), 8.66 (s, 1H), 8.82-8.84 (d, 1H)
MS : (m/z) 365 (M++1)
N-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-[(4-methoxyphenyl)]-
piperazine-1-carboxamide (II-6)
Compound (II-6) was prepared by reacting compound (6b) (0.3 g, 0.149
mmole) with 4-(4-methoxyphenyl)piperazine (0.287 g, 0.149 mmole) as described
above for compound (I-1). Chromatographic purification of the crude product
using methanol (2%) in ethyl acetate as eluent provided the titled compound (II-6)
(0.195 g, 33.2%), m. p. 134-35 °C.
Anal.:
TLC : Rf 0.78 (1% MeOH in EtOAc)
IR : 3393, 1648, 1530, 1483, 1241, 1188 and 1153 cm-1
1H-NMR : 2.44 (s, 3H), 2.97-2.99 (b, 4H), 3.58-3.61 (b, 4H), 3.80 (s,
3H), 6.87-7.00 (m, 4H), 7.19-7.22 (dd, 1H), 7.50 (s, 1H), 8.00

Chapter-4 Experimental work
124
(s, 1H), 8.66 (s, 1H), 8.82-8.84 (d, 1H)
MS : (m/z) 394 (M++1)
N-(8-Methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-benzhydrylpiperazine-
1-carboxamide (II-7)
The isocyanate derivative (6b) (0.3 g, 0.149 mmole) and benzhydryl
piperazine (0.376 g, 0.147 mmole) were reacted together as described above for
compound (I-1). The crude product was purified by chromatographic purification
using methanol (0.5%) in ethyl acetate as eluent to obtain the compound (II-7)
(0.2 g, 29.5%), m. p. 233-35 °C.
Anal.:
TLC : Rf 0.54 (EtOAc)
IR : 3000, 1644, 1529, 1481, 1237 and 1186 cm-1
1H-NMR : 2.32-2.34 (b, 4H), 2.43 (s, 3H), 3.46-3.48 (b, 4H), 4.34 (s,1H),
7.18-7.22 (m, 3H), 7.29-7.33 (m, 4H), 7.44-7.48 (m, 5H),
7.87 (s, 1H), 8.63 (s, 1H), 8.79-8.81 (d, 1H)
MS : (m/z) 454 (M++1)
Synthesis of N-(7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-
(pyridin-2-yl)piperazine-1-carboxamide derivatives
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(pyridin-2-yl)pipera-
zine]-1-carboxamide (II-8)
Compound (II-8) was prepared by reacting compound (6c) (0.3 g, 0.135
mmole) with 4-(2-pyridyl)piperazine (0.221 g, 0.135 mmole) as described above
for compound (I-1). The chromatographic purification of the crude product was
done using methanol (2%) in ethyl acetate as eluent to earn the compound (II-8)
(0.21 g, 40.3%), m. p. 189-90 °C.
Anal.:
TLC : Rf 0.45 (10% MeOH in EtOAc)
IR : 3397, 3088, 1729, 1643, 1531, 1482, 1243 and 1124 cm-1
1H-NMR : 3.57-3.58 (m, 8H), 6.65-6.68 (m, 1H), 6.86-6.88 (d, 1H),
7.54-7.58 (m, 1H), 7.69-7.71 (d, 1H), 7.83-7.86 (dd, 1H),
8.13-8.14 (d, 1H), 8.16 (s, 1H), 8.82 (s, 1H), 8.89-8.90 (d,
1H)

Chapter-4 Experimental work
125
MS : (m/z) 385 (M++1)
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(4-methoxyphenyl)-
piperazine]-1-carboxamide (II-9)
Compound (6c) (0.3 g, 0.135 mmole) was reacted with 4-(4-
methoxyphenyl)piperazine (0.26 g, 0.135 mmole) as described above for
compound (I-1). Chromatographic purification of the crude product was done
using methanol (2%) in ethyl acetate as eluent yielding the titled compound (II-9)
(0.25 g, 44.72%), m. p. 163-64 °C.
Anal.:
TLC : Rf 0.75 (10% MeOH in EtOAc)
IR : 3393, 1656, 1626, 1525, 1483, 1347, 1236 and 1065 cm-1
1H-NMR : 2.97-2.99 (b, 4H), 3.60-3.61(b, 4H), 3.80 (s, 3H), 6.87-7.00
(m, 4H), 7.69-7.71 (d, 1H), 7.83-7.86 (m, 1H), 8.13 (s, 1H),
8.89 (s, 1H), 8.90 (s, 1H)
MS : (m/z) 414 (M++1)
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-(4-tert-butyloxycarbony-
l)-piperazine]-1-carboxamide (II-10)
Isocyanate derivative (6c) (0.25 g, 11 mmole) in toluene (25 ml) was
refluxed with boc-piperazine (0.209 g, 0.11 mmole) to get a solid precipitate. The
solid so obtained was filtered and washed with hexane. Chromatographic
purification of the crude product using hexane (10%) in ethyl acetate provided the
desired compound (II-10) (0.2 g, 43.4%), m. p. 229-30 °C.
Anal.:
TLC : Rf 0.6 (1% MeOH in EtOAc)
IR : 3393, 1691, 1650, 1629, 1490, 1422, 1233 and 1173 cm-1
1HNMR : 1.42 (s, 9H), 3.39 (b, 4H), 3.44-3.45 (b, 4H), 7.68-7.71 (d,
1H), 7.83-7.85 (d, 1H), 8.13 (s, 1H), 8.78 (s, 1H), 8.88-8.89
(d, 1H)
MS : (m/z) 408.41, 410.39 (M+-1)

Chapter-4 Experimental work
126
N-[(7-Chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)-4-(furan-2-carbonyl)-
piperazine]-1-carboxamide (II-11)
Isocyanate derivative (6c) (0.25 g, 0.11 mmole) was reacted with 1-
(furan-2-carbonyl)piperazine (0.203 g, 0.11 mmole) as described above for
compound (I-1). Chromatographic purification of the crude product using 1%
methanol in ethyl acetate offered the desired compound (II-11) (0.19 g, 41.9%),
m. p. 227-28 °C.
Anal.:
TLC : Rf 0.58 (1% MeOH in EtOAc)
IR : 3446, 3086, 1680, 1626, 1427, 1290, 1242 and 1199 cm-1
1HNMR : 3.57-3.58 (b, 4H), 3.74 (b, 4H), 6.65 (s,
1H), 7.04-7.05 (d,
1H), 7.69-7.71 (d, 1H), 7.83-7.86 (m, 1H), 7.87 (s, 1H), 8.18
(s, 1H), 8.80 (s, 1H), 8.89 (b, 1H)
MS : (m/z) 402 (M+-1)
4.1.4 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-
carbamate derivatives (Series III)
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbamate
derivatives
Benzyl (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamate (III-1)
Compound (6a) (0.3 g, 16 mmole) and benzyl alcohol (0.173 g, 16
mmole) in toluene (25ml) were refluxed for 6 h to get a solid precipitate. The solid
precipitate was filtered and washed with hexane and the crude product so obtained
was purified by column chromatography using hexane (10%) in ethyl acetate as
eluent to offer the titled compound (III-1) (0.5 g, 61.1%), m. p. 158-60 °C (Lit.221
153-55 °C).
Anal.:
TLC : Rf 0.6 (1% MeOH in EtOAc)
IR : 3239, 1718, 1650, 1461, 1274, 1228, 1180 and 1116 cm-1
1H-NMR : 5.17 (s, 2H), 7.32-7.44 (m, 6H), 7.69-7.71 (d, 1H), 7.84-7.89
(m, 1H), 8.70 (b, 1H), 8.92-8.94 (d, 1H), 9.05 (b, 1H)
MS : (m/z) 296 (M++1)

Chapter-4 Experimental work
127
Pyridin-2-ylmethyl (4-oxo-4H-[1,2-a]pyrimidin-3-yl)carbamate (III-2)
Compound (III-2) was prepared by reacting (6a) (0.5 g, 0.26 mmole)
with pyridine-2-methanol (0.175 g, 0.16 mmole) as described above for
compound (III-1). The crude product was subjected to chromatographic
purification using hexane (10%) in ethyl acetate as eluent affording the titled
compound (III-2) (0.24 g, 50.5%), m. p. 178-80 °C.
Anal.:
TLC : Rf 0.6 (1% MeOH in EtOAc)
IR : 3245, 3070, 1713, 1666, 1549, 1477, 1234 and 1125 cm-1
1H-NMR : 5.22 (s, 2H), 7.33-7.38 (m, 2H), 7.51-7.53 (d, 1H), 7.69-7.72
(d, 1H), 7.83-7.89 (m, 2H), 8.55-8.57 (d, 1H), 8.72 (s, 1H),
8.94-8.95 (d, 1H), 9.21 (b, 1H)
MS : (m/z) 297 (M++1)
3-Fluorobenzyl (4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamate (III-3)
A mixture of isocyanate derivative (6a) (0.25 g, 0.13 mmole) and 3-
fluorobenzyl alcohol (0.169 g, 0.13 mmole) were reacted as described above for
compound (III-1). The crude product was purified by column chromatography
using hexane (20%) in ethyl acetate to attain the titled compound (III-3) (0.11 g,
26.2%), m. p. 158-60 °C.
Anal.:
TLC : Rf 0.65 (EtOAc)
IR : 3416, 3264, 1716, 1659, 1480, 1441, 1403, 1234, 1200, 1184,
1134, 796 and 773 cm-1
1HNMR : 5.19 (s, 2H), 7.14-7.19 (m, 1H), 7.27-7.37 (m, 3H), 7.41-7.47
(m, 1H), 7.69-7.71 (d, 1H), 7.85-7.89 (m, 1H), 8.72 (s, 1H),
8.93-8.95 (d, 1H), 9.16 (b, 1H)
MS : (m/z) 314, (M+-1)
3,4-Methylenedioxybenzyl(4-oxo-4H-pyrido[1,2-a]pyrimdin-3-yl)carbamate
(III-4)
6a (0.5 g, 0.26 mmole) was reacted with 3,4-methylenedioxybenzyl
alcohol (0.244 g, 0.106 mmole) as described above for compound (III-1). The
chromatographic purification of the crude product using hexane (10%) in ethyl

Chapter-4 Experimental work
128
acetate as eluent offered the titled compound (III-4) (0.18 g, 33%), m. p. 175-76
°C.
Anal.:
TLC : Rf 0.5 (EtOAc)
IR : 3242, 3031, 1717, 1666, 1485, 1443, 1235 and 1137 cm-1
1H-NMR : 5.06 (s, 2H), 6.02 (s, 2H), 6.92 (s, 2H), 7.02 (s, 1H), 7.33-7.37
(m, 1H), 7.69-7.71 (d, 1H), 7.84-7.89 (m, 1H), 8.70 (b, 1H),
8.92-8.94 (d, 1H), 9.01 (b, 1H)
MS : (m/z) 340 (M++1)
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carbam-
ate derivatives
Benzyl (8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamate (III-5)
Compound (III-5) was prepared by treating the isocyanate derivative
(6b) (0.5 g, 0.26 mmole) with benzyl alcohol (0.161 g, 0.149 mmole) as described
above for compound (III-1). Chromatographic purification of the crude product
using hexane (10%) in ethyl acetate as eluent yielded compound (III-5) (0.18 g,
39%), m. p. 168-70 °C (Lit.221
177-79 °C).
Anal.:
TLC : Rf 0.7 (1% MeOH in EtOAc)
IR : 3242, 1717, 1635, 1546, 1465, 1224 and 1056 cm-1
1H-NMR : 2.45 (s, 3H), 5.15 (s, 2H), 7.21-7.23 (d, 1H), 7.31-7.44 (m,
5H), 7.52 (s, 1H), 8.60 (b, 1H), 8.83-8.85 (d, 1H), 8.97 (b, 1H)
MS : (m/z) 310 (M++1)
Pyridin-2-ylmethyl (8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)
carbamate (III-6)
Compound (6b) (0.5 g, 0.26 mmole) was reacted with pyridine-2-
methanol (0.163 g, 0.149 mmole) as described above for compound (III-1).
Chromatographic purification of the crude product using hexane (10%) in ethyl
acetate as eluent earned the titled compound (III-6) (0.19 g, 41%), m. p. 180-82
°C.
Anal.:
TLC : Rf 0.75 (1% MeOH in EtOAc)

Chapter-4 Experimental work
129
IR : 3429, 3247, 1721, 1667, 1644, 1480, 1244 and 1117 cm-1
1H-NMR : 2.45 (s, 3H), 5.20 (s, 2H), 7.22-7.24 (d, 1H), 7.33-7.36 (m,
1H), 7.49-7.51 (d, 1H), 7.53 (s, 1H), 7.83-7.87 (m, 1H), 8.55-
8.56 (d, 1H), 8.62 (b, 1H), 8.85-8.87 (d, 1H), 9.14 (b, 1H)
MS : (m/z) 311 (M++1)
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carba-
mate derivatives
2-Pyridinylmethyl (7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carba-
mate (III-7)
6c (0.25 g, 0.11 mmole) was reacted with pyridine-2-methanol (0.176 g,
0.135 mmole) as described above for compound (III-1). The crude product was
purified by column chromatography using 2% methanol in ethyl acetate to
accomplish the titled compound (III-7) (0.05 g, 13.4%), m. p. 196-97 °C.
Anal.:
TLC : Rf 0.62 (2% MeOH in EtOAc)
IR : 3451, 3245, 1724, 1652, 1477, 1354, 1236 and 694 cm-1
1HNMR : 5.23 (s, 2H), 7.33-7.36 (d, 1H), 7.51-7.53 (d, 1H), 7.71-7.73
(d,
1H), 7.83-7.87 (t, 1H), 7.88-7.91 (dd, 1H), 8.55-8.56 (d,
1H), 8.76 (s, 1H), 8.92-8.93 (d, 1H), 9.32 (s, 1H)
MS : (m/z) 331.37, 333.35 (M+-1)
3-Pyridinylmethyl (7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carba-
mate (III-8)
6c (0.25 g, 0.11 mmole) and pyridine-3-methanol (0.123 g, 0.11 mmole)
were reacted as described above for compound (III-1) to get solid precipitate. The
solid so obtained was filtered and washed with hexane. Chromatographic
purification of the crude product using hexane (5%) in ethyl acetate provided the
intended compound (III-8) (0.15 g, 40.2%), m. p. 293-95 °C.
Anal.:
TLC : Rf 0.65 (ETOAc)
IR : 3415, 3251, 1722, 1664, 1630, 1402 and 1232 cm-1
1HNMR : 5.22 (s, 2H), 7.41-7.45 (m, 1H), 7.70-7.73 (d, 1H), 7.86-7.91
(m, 2H), 8.54-8.55 (d, 1H), 8.66 (b, 1H), 8.74 (b, 1H), 8.91-

Chapter-4 Experimental work
130
8.92 (d, 1H) 9.16 (s, 1H)
MS : (m/z) 331.37, 333.35, (M+-1)
3-Fluorobenzyl (7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl)carbamate
(III-9)
Compound (III-9) was obtained by reaction of compound (6c) (0.25 g,
0.11 mmole) with 3-fluorobenzyl alcohol (0.142 g, 0.11 mmole) to get solid
precipitate. The solid so obtained was filtered and washed with hexane and
purified by column chromatography using hexane (10%) in ethyl acetate to give
the titled compound (III-9) (0.06 g, 15.3%), m. p. 240-42 °C.
Anal.:
TLC : Rf 0.62 (1% MeOH in EtOAc)
IR : 3431, 3276, 1726, 1660, 1546, 1229 and 1127 cm-1
1HNMR : 5.15 (s, 2H), 7.20-7.24 (m, 2H), 7.48-7.51 (m, 2H), 7.70-7.72
(d, 1H), 7.88-7.90 (d, 1H), 8.74 (s, 1H), 8.91 (s, 1H), 9.18 (s,
1H)
MS : (m/z) 350 (M+-1), 348 (M
+-3)
4.1.5 Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-
carboxamide derivatives (Series IV)
Synthesis of 4-oxo-4H-pyrido[1,2-a]pyrimidin-3-yl-carboxamide
derivatives
N-(4-Methoxybenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamide
(IV-1)
In a two neck round bottom flask (100 ml) equipped with calcium
chloride guard tube, the acid (4a) (0.5 g, 0.26 mmole) was dissolved in
dichloromethane (50 ml) and the solution was cooled to 0 °C. 1-
Hydroxybenzotriazole hydrate (0.355 g, 0.26 mmole), 1-(3-
dimethylaminopropyl)-3-ethylcarbodiimide (0.603 g, 0.31 mmole) and N,N-
diisopropylethylamine (0.679 g, 0.52 mmole) were added to the above solution.
After 20 min 4-methoxybenzylamine (0.397 g, 0.26 mmole) was added and the
reaction mixture was stirred for 1 h at 0 °C and for another 8 h at room

Chapter-4 Experimental work
131
temperature. The reaction mass was quenched by adding cold water (20 ml) and
the medium was neutralized using dilute hydrochloric acid, extracted using ethyl
acetate (3x100 ml), dried over anhydrous sodium sulfate and concentrated to get a
dark brown product. The crude product was purified by column chromatography
using methanol (2%) in ethyl acetate to offer the compound (IV-1) (0.24 g,
28.2%), m. p. 192-94 °C.
Anal.:
TLC : Rf 0.51 (EtOAc)
IR : 3336, 1679, 1489, 1334, 1297, 1243 and 1030 cm-1
1H-NMR : 3.80 (s, 3H), 4.62-4.64 (d, 2H), 6.86-6.90 (m, 2H), 7.31-7.33
(d, 2H), 7.34-7.38 (m, 1H), 7.84-7.86 (d, 1H), 7.92-7.97 (m,
1H), 9.19-9.21 (d, 1H), 9.30 (b, 1H), 9.38 (s, 1H)
MS : (m/z) 310 (M++1)
N-(3-Fluorobenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxamide (IV-2)
Compound (IV-1) was prepared by reacting compound (4a) (0.5 g, 0.26
mmole) with 3-fluorobenzylamine (0.329 g, 0.26 mmole) as described above for
compound (IV-1). The crude product was purified by column chromatography
using hexane (5%) in ethyl acetate as eluent offering the compound (IV-2) (0.32
g, 40.9%), m. p. 168-70 °C.
Anal.:
TLC : Rf 0.64 (EtOAc)
IR : 3327, 3122, 1662, 1623, 1569, 1533, 1500 and 1240 cm-1
1H-NMR : 4.59-4.61 (d, 2H), 7.06-7.11 (m, 1H), 7.15-7.21 (m, 2H),
7.36-7.41 (m, 1H), 7.59-7.63 (m, 1H), 7.90-7.92 (d, 1H), 8.19-
8.23 (m, 1H), 9.05 (s, 1H), 9.19-9.21 (d, 1H), 9.43-9.46 (t,
1H)
MS : (m/z) 298 (M++1)
4-Oxo-N-[2-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimidin-3-carbox-
amide (IV-3)
Compound (IV-3) was prepared by treating compound (4a) (0.5 g, 0.26
mmole) with 2-trifluoromethylbenzylamine (0.461 g, 0.26 mmole) as described
above for compound (IV-1). Chromatographic purification of the crude product

Chapter-4 Experimental work
132
using hexane (5%) in ethyl acetate as eluent yielded compound (IV-3) (0.32 g,
35%), m. p. 158-60 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)
IR : 3299, 3079, 1695, 1630, 1476, 1315, 1167 and 1094 cm-1
1H-NMR : 4.76-4.78 (d, 2H), 7.48-7.52 (m, 1H), 7.59-7.69 (m, 3H),
7.75-7.77 (d, 1H), 7.91-7.93 (d, 1H), 8.20-8.24 (m, 1H), 9.05
(s, 1H), 9.21-9.23 (d, 1H), 9.48-9.51 (t, 1H)
MS : (m/z) 348 (M++1)
4-Oxo-N-[3-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimidin-3-carbox-
amide (IV-4)
Compound (4a) (0.5 g, 0.26 mmole) was reacted with 3-
trifluoromethylbenzylamine (0.461 g, 0.26 mmole) as described above for
compound (IV-1). The chromatographic purification of the crude product using
hexane (5%) in ethyl acetate as eluent furnished the compound (IV-4) (0.33 g,
36.1%), m. p. 139-40 °C.
Anal.:
TLC : Rf 0.63 (EtOAc)
IR : 3328, 3113, 1691, 1645, 1532, 1490, 1415 and 1335 cm-1
1H-NMR : 4.66-4.67 (d, 2H), 7.56-7.63 (m, 3H), 7.66-7.68 (d, 1H), 7.72
(s, 1H), 7.90-7.92 (d, 1H), 8.18-8.23 (m, 1H), 9.05 (s, 1H),
9.20-9.21 (d, 1H), 9.50-9.53 (t, 1H)
MS : (m/z) 348 (M++1)
4-Oxo-N-[4-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimidin-3-carbox-
amide (IV-5)
Compound (IV-5) was prepared by reacting (4a) (0.5 g, 0.26 mmole)
with 4-trifluoromethylbenzylamine (0.461 g, 0.26 mmole) as described above for
compound (IV-1). The chromatographic purification of the crude product using
hexane (5%) in ethyl acetate as eluent yielded compound (IV-5) (0.25 g, 26.2%),
m. p. 167-69 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)

Chapter-4 Experimental work
133
IR : 3336, 3115, 1663, 1600, 1570, 1500, 1325 and 1123 cm-1
1H-NMR : 4.75-4.76 (d, 2H), 7.38-7.41 (m, 1H), 7.49-7.51 (d, 2H), 7.58-
7.60 (d, 2H), 7.86-7.88 (d, 1H), 7.96-8.00 (m, 1H), 9.21-9.23
(d, 1H), 9.37-9.38 (d, 1H), 9.47 (b, 1H)
MS : (m/z) 348 (M++1)
N-(2,4-Difluorobenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxamide
(IV-6)
Compound (IV-6) was prepared by reacting (4a) (0.5 g, 0.26 mmole)
with 2,4-difluoromethylbenzylamine (0.376 g, 0.26 mmole) as described above
for compound (IV-1). The chromatographic purification of the crude product
using hexane (20%) in ethyl acetate as eluent afforded the desired product (IV-6)
(0.21 g, 25.3%), m. p. 172-75 °C.
Anal.:
TLC : Rf 0.62 (EtOAc)
IR : 3328, 3125, 1664, 1500, 1430, 1184 and 1072 cm-1
1H-NMR : 4.60-4.62 (d, 2H), 7.14-7.21 (m, 2H), 7.24-7.29 (m, 1H),
7.59-7.63 (m, 1H), 7.89-7.92 (d, 1H), 8.19-8.23 (m, 1H), 9.03
(s, 1H), 9.20-9.23 (d, 1H), 9.42-9.45 (t, 1H)
MS : (m/z) 316 (M++1)
N-[2-(Morpholin-4-yl)ethyl]-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxamide
(IV-7)
Reaction of compound (4a) (0.5 g, 0.26 mmole) with 2-(4-
morpholinyl)ethylamine (0.342 g, 0.26 mmole) was effected as described above
for compound (IV-1). The chromatographic purification of the crude product
using methanol (0.2%) in ethyl acetate as eluent offered the titled compound (IV-
7) (0.18 g, 22.6%), m. p. 193-95 °C.
Anal.:
TLC : Rf 0.3 (1% MeOH in EtOAc)
IR : 3123, 1690, 1631, 1483, 1400, 1251 and 1115 cm-1
1H-NMR : 2.43 (b, 4H), 2.50-2.51 (m, 2H), 3.45-3.50 (m, 2H), 3.59-3.61
(t, 4H), 7.57-7.61 (m, 1H), 7.84-7.90 (d, 1H), 8.17-8.21 (m,
1H), 9.02 (s, 1H), 9.15-9.16 (t, 1H), 9.20-9.22 (d, 1H)

Chapter-4 Experimental work
134
MS : (m/z) 303 (M++1)
4-Oxo-N-[3-(n.pentyloxy)pyridin-2-yl]-4H-pyrido[1,2-a]pyrimidin-3-
carboxami-de (IV-8)
Compound (IV-8) was prepared by reacting (4a) (0.5 g, 0.26 mmole)
with 2-amino-3-n.pentyloxypyridine (0.474 g, 0.26 mmole) as described above for
compound (IV-1). The chromatographic purification of the crude product using
methanol (2.5%) in ethyl acetate as eluent produced the titled product (IV-8) (0.3
g, 32.3%), m. p. 159-60 °C.
Anal.:
TLC : Rf 0.3 (EtOAc)
IR : 3292, 3237, 1693, 1595, 1470, 1333 and 1218 cm-1
1H-NMR : 0.89-0.93 (t, 3H), 1.36-1.43 (m, 2H), 1.49-1.57 (m, 2H), 1.78-
1.84 (m, 2H), 4.09-4.13 (t, 2H), 6.55 (s, 1H), 7.13-7.16 (m,
1H), 7.46-7.48 (d, 1H), 7.66-7.70 (m, 1H), 7.95-7.97 (d, 1H),
8.24-8.28 (m, 1H), 9.13 (s, 1 H), 9.21-9.24 (d, 1H), 11.51 (s,
1H)
MS : (m/z) 353 (M++1)
N-[3-(Benzyloxy)pyridin-2-yl]-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxam-
ide (IV-9)
Compound (4a) (0.5 g, 0.26 mmole) and 2-amino-3-benzyloxypyridine
(0.526 g, 0.26 mmole) were reacted together as described above for compound
(IV-1). The chromatographic purification of the crude product using methanol
(1%) in ethyl acetate as eluent offered compound (IV-9) (0.23 g, 23.4%), m. p.
193-95 °C.
Anal.:
TLC : Rf 0.55 (EtOAc)
IR : 3480, 3106, 1699, 1598, 1482, 1285 and 1012 cm-1
1H-NMR : 5.28 (s, 2H), 7.00-7.04 (dd, 1H), 7.23-7.26 (dd, 1H), 7.34-
7.38 (m, 1H), 7.41-7.44 (m, 2H), 7.53-7.55 (m, 1H), 7.58-
7.60 (d, 2H), 7.88-7.91 (d, 1H), 7.98-8.02 (m, 1H), 8.10-8.14
(m, 1H), 8.16-8.18 (dd, 1H), 9.27-9.31 (m, 1H), 9.49 (s, 1H),
11.76 (s, 1H)

Chapter-4 Experimental work
135
MS : (m/z) 373 (M++1)
N-[(2,5-Diethoxy)-4-(morpholin-4-yl)phenyl]-4-oxo-4H-pyrido[1,2-a]pyrimid-
ine-3-carboxamide (IV-10)
Compound (IV-10) was prepared by treating compound (4a) (0.5 g,
0.26 mmole) with 2,5-diethoxy-4-morpholin-4-yl-phenylamine (0.7 g, 0.26
mmole) as described above for compound (IV-1). The chromatographic
purification of the crude product using methanol (1%) in ethyl acetate as eluent
provided compound (IV-10) (0.35 g, 30.3%), m. p. 166-67 °C.
Anal.:
TLC : Rf 0.7 (EtOAc)
IR : 3263, 1685, 1487, 1260, 1199, 1113, 1043 cm-1
1H-NMR : 1.44-1.47 (t, 3H), 1.54-1.58 (t, 3H), 3.09-3.10 (t, 4H), 3.89-
3.91 (t, 4H), 4.12-4.17 (m, 4H), 6.60 (s, 1H), 7.37-7.41 (m,
1H), 7.85-7.87 (d, 1H), 7.95-7.99 (m, 1H), 8.40 (s, 1H), 9.33-
9.34 (d, 1H), 9.42 (s, 1H), 11.49 (s, 1H)
MS : (m/z) 439 (M++1)
N-(3,5-Dimethyl-1,2-oxazol-4-yl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carbox-
amide (IV-11)
Compound (IV-11) was prepared by reacting (4a) (0.5 g, 0.26 mmole)
with 3,5-dimethyl-1,2-oxazol-4yl-amine (0.295 g, 0.26 mmole) as described
above for compound (IV-1). The chromatographic purification of the crude
product using hexane (10%) in ethyl acetate as eluent offered compound (IV-11)
(0.34 g, 45.4%), m. p. 243-45 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)
IR : 3253, 3209, 1694, 1486, 1332, 1235 and 1067 cm-1
1H-NMR : 2.15 (s, 3H), 2.33 (s, 3H), 7.65-7.69 (m, 1H), 7.95-7.97 (d,
1H),
8.24-8.29 (m, 1H), 9.09 (s, 1H), 9.26-9.27 (d, 1H), 10.20
(s, 1H)
MS : (m/z) 285 (M++1)

Chapter-4 Experimental work
136
N-[1-(4-Methoxyphenyl)ethyl]-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxa-
mide (IV-12)
Compound (4a) (0.5 g, 0.26 mmole) was reacted with -methyl-4-
methoxybenzylamine (0.397 g, 0.26 mmole) as described above for compound
(IV-1) Chromatographic purification of the crude product using methanol (0.2%)
in ethyl acetate as eluent afforded compound (IV-12) (0.24 g, 28.2%), m. p. 173-
75 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)
IR : 3315, 3128, 1667, 1630, 1506, 1240 and 1025 cm-1
1H-NMR : 1.61-1.66 (d, 3H), 3.79 (s, 3H), 5.28-5.35 (m, 1H), 6.87-6.91
(d, 2H), 7.34-7.38 (m, 3H), 7.83-7.86 (d, 1H), 7.92-7.97 (m,
1H), 9.20-9.22 (d, 1H), 9.33-9.36 (m, 2H)
MS : (m/z) 324 (M++1)
N-(1-Benzylpiperidin-4-yl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carboxamide
(IV-13)
Compound (IV-13) was obtained by reaction of compound (4a) (0.5 g,
0.26 mmole) with 4-amino-1-benzylpiperidine (0.5 g, 0.26 mmole) as described
above for compound (IV-1). The chromatographic purification of the crude
product using hexane (10%) in ethyl acetate as eluent earned the titled compound
(IV-13) (0.25 g, 5%), m. p. 180-83 °C.
Anal.:
TLC : Rf 0.56 (EtOAc)
IR : 3288, 1695, 1544, 1479, 1335 and 1061 cm-1
1H-NMR : 1.52-1.58 (m, 2H), 1.89-1.92 (d, 2H), 2.18-2.22 (t, 2H), 2.70
(b, 2H), 3.50 (s, 2H), 3.88 (b, 1H), 7.26 (b, 1H), 7.33 (b, 4H),
7.60-7.64 (t, 1H), 7.90-7.92 (d, 1H), 8.19-8.23 (t, 1H), 9.04
(b, 2H), 9.20-9.22 (d, 1H)
MS : (m/z) 363 (M++1)

Chapter-4 Experimental work
137
N-(3,4-Methylenedioxybenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbox-
amide (IV-14)
The reaction of compound (4a) (0.5 g, 0.26 mmole) with 3,4-
methylenedioxybenzyl amine (0.397 g, 0.26 mmole) was performed as described
above for compound (IV-1). The chromatographic purification of the crude
product using hexane (20%) in ethyl acetate as eluent produced compound (IV-
14) (0.3 g, 35.2%), m. p. 193-95 °C.
Anal.:
TLC : Rf 0.6 (EtOAc)66
IR : 3260, 3071, 1665, 1627, 1490, 1434, 1246 and 1224 cm-1
1H-NMR : 4.46-4.48 (d, 2H), 5.98 (s, 2H), 6.82-6.88 (m, 2H), 6.93 (s,
1H), 7.58-7.62 (m, 1H), 7.89-7.91 (d, 1H), 8.17-8.22 (m, 1H),
9.05 (s, 1H), 9.17-9.19 (d, 1H), 9.27-9.33 (t, 1H)
MS : (m/z) 324 (M++1)
N-Cyclopropyl-N-(2-fluorobenzyl)-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-
carboxamide (IV-15)
The reaction of (4a) (0.5 g, 0.26 mmole) with N-cyclopropyl-2-fluoro
benzylamine (0.434 g, 0.26 mmole) was performed as described above for
compound (IV-1). The chromatographic purification of the crude product using
hexane (5%) in ethyl acetate as eluent yielded the desired compound (IV-15)
(0.24 g, 27%), m. p. 137-40 °C.
Anal.:
TLC : Rf 0.55 (EtOAc)
IR : 3110, 1669, 1492, 1404, 1090 and 1031cm-1
1H-NMR : 0.58-0.61 (d, 4H), 3.06 (b, 1H), 4.93 (s, 2H), 7.11-7.13 (m,
1H), 7.23-7.25 (m, 1H), 7.32-7.34 (m, 2H), 7.67 (b, 1H), 7.80-
7.82 (d, 1H), 7.90-7.94 (m, 1H), 8.60 (s, 1H), 9.21-9.23 (d,
1H)
MS : (m/z) 338 (M++1)
Synthesis of 8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbox-
amide derivatives

Chapter-4 Experimental work
138
N-(4-Fluorobenzyl)-8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carbox-
amide (IV-16)
Compound (IV-16) was prepared by coupling reaction of (4b) (0.5 g,
0.26 mmole) with 4-fluorobenzylamine (0.306 g, 0.24 mmole) as described above
for compound (IV-1). Chromatographic purification of the crude product using
hexane (10%) in ethyl acetate as eluent furnished the titled product (IV-16) (0.15
g, 51.9%), m. p. 185-87 °C.
Anal.:
TLC : Rf 0.68 (1% MeOH in EtOAc)
IR : 3287, 3014, 1677, 1633, 1481, 1279, 1222 and 1146 cm-1
1H-NMR : 2.58 (s, 3H), 4.65-4.66 (d, 2H), 7.00-7.04 (m, 2H), 7.18-7.21
(m, 1H), 7.34-7.37 (m, 2H), 7.63 (s, 1H), 9.08-9.09 (d, 1H),
9.33 (s, 1H), 9.35 (b, 1H)
MS : (m/z) 312 (M++1)
N-(4-Trifluorobenzyl)-8-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-3-carbox-
amide (IV-17)
Compound (IV-17) was prepared by reacting (4b) (0.4 g, 0.19 mmole)
with 4-trifluoromethylbenzylamine (0.343 g, 0.19 mmole) as described above for
compound (IV-1). Chromatographic purification of the crude product using
methanol (5%) in methylene chloride as eluent yielded the titled product (IV-17)
(0.15 g, 21.2%), m. p. 175-77 °C.
Anal.:
TLC : Rf 0.4 (EtOAc)
IR : 3414, 3294, 1695, 1634, 1545, 1330, 1117, 1015 and 793 cm-1
1H-NMR : 2.55 (s, 3H), 4.65-4.67 (d, 2H), 7.46-7.49 (dd, 1H), 7.55-7.57
(d, 2H), 7.69-7.73 (m, 3H), 9.00 (s, 1H), 9.08-9.10 (d, 1H),
9.45-9.48 (t, 1H)
MS : (m/z) 362 (M++1)
Synthesis of 7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbox-
amide derivatives

Chapter-4 Experimental work
139
N-(3,4-Methylenedioxybenzyl)-7-chloro-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-
carboxamide (IV-18)
Compound (4c) (0.5 g, 0.22 mmole) was coupled with 3,4-
methylenedioxybenzylamine (0.336 g, 0.22 mmole) as described above for
compound (IV-1). Chromatographic purification of the crude product using
hexane (10%) in ethyl acetate as eluent yielded the desired product (IV-18) (0.4 g,
50.2%), m. p. 200-01 °C.
Anal.:
TLC : Rf 0.4 (EtOAc)
IR : 3500, 3300, 1672, 1490, 1434, 1248 and 1066 cm-1
1H-NMR : 4.46-4.48 (d, 2H), 5.98 (s, 2H), 6.81-6.87 (m, 2H), 6.92 (s,
1H), 7.90-7.93 (d, 1H), 8.24-8.26 (dd, 1H), 9.04 (s, 1H), 9.15-
9.16 (d, 1H), 9.23-9.26 (t, 1H)
MS : (m/z) 358.3 (M++1), 360.4 (M
++2)
7-Chloro-4-oxo-N-[4-(trifluoromethyl)benzyl]-4H-pyrido[1,2-a]pyrimidine-3-
carboxamide (IV-19)
Compound (IV-19) was prepared by reacting the acid (4c) (0.5 g, 0.22
mmole) with 4-trifluoromethylbenzylamine (0.39 g, 0.22 mmole) as described
above for compound (IV-1). Chromatographic purification of the crude product
using hexane (10%) in ethyl acetate as eluent offered compound (IV-19) (0.2 g,
23.5%), m. p. 212-14 °C.
Anal.:
TLC : Rf 0.51 (EtOAc)
IR : 3315, 3091, 1674, 1620, 1532, 1498, 1323 and 1134 cm-1
1H-NMR : 4.67-4.68 (d, 2H), 7.55-7.57 (d, 2H), 7.70-7.72 (d, 2H), 7.92-
7.95 (d, 1H), 8.26 -8.29 (dd, 1H), 9.04 (s, 1H), 9.17-9.18 (d,
1H), 9.43-9.46 (t, 1H)
MS : (m/z) 382.2 (M++1), 384.2 (M
++2)
7-Chloro-[N-cyclopropyl-N-(2-fluorobenzyl)]-4-oxo-4H-pyrido[1,2-a]pyrimi-
dine-3-carboxamide (IV-20)
Compound (IV-20) was prepared by reacting the acid (4c) (0.5 g, 0.22
mmole) with N-cyclopropyl-2-fluorobenzylamine (0.367 g, 0.22 mmole) as

Chapter-4 Experimental work
140
described above for compound (IV-1). The chromatographic purification of the
crude product using hexane (10%) in ethyl acetate as eluent provided compound
(IV-20) (0.28 g, 33.8%), m. p. 209-10 °C.
Anal.:
TLC : Rf 0.48 (EtOAc)
IR : 3068, 3019, 1688, 1630, 1481, 1344, 1223 and 1030 cm-1
1H-NMR : 0.51-0.55 (d, 4H), 2.87 (b, 1H), 4.75 (b, 2H), 7.21-7.24 (m,
2H), 7.33-7.36 (m, 1H), 7.63 (b, 1H), 7.84-7.87 (d, 1H), 8.14-
8.17 (dd, 1H), 8.47 (s, 1H), 9.14 (s, 1H)
MS : (m/z) 372.4 (M++1), 374.3 (M
++2)
7-(4-Methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic
acid intermediates
Ethyl 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylate
(7)
Ethyl 7-chloro-4-oxo-4H-pyrido[1,2a]pyrimidine-3-carboxylate (4c) (10
gm, 39.6 mmole) was dissolved in dimethoxyethane (300 ml) and 4-
methoxyphenylboronic acid (6 g, 39.6 mmole), potassium carbonate (8.194 gm,
59.4 mmole) and tetrakis(triphenylphosphene)palladium(0) (0.046 gm, 0.039
mmole) were added to the above reaction mixture under N2 atmosphere. The
reaction mixture was refluxed for 36 h at 90 °C. The reaction mass was filtered
and the filtrate was washed with water and extracted using methylene chloride (3
x100 ml), dried over anhydrous sodium sulfate and concentrated to get a dark
brown product. Chromatographic purification of the crude product using ethyl
acetate (15%) in hexane as eluent offered the titled compound (7) (3.5 g, 27.3%),
m. p. 166-67 °C.
Anal.:
TLC : Rf 0.7 (50% EtOAc in hexane)
IR : 3087, 1744, 1609, 1569, 1487, 1296, 1188, 1113, 1045,
826 and 795 cm-1
MS : (m/z) 325, (M+-1)
7-(4-Methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (8)

Chapter-4 Experimental work
141
A solution of 7 (5 g, 4.3 mmole) in conc. HCl (15 ml) was refluxed for 2
h. The reaction mixture was cooled to RT to get a solid precipitate. The solid so
obtained was filtered, washed with ether (100 ml) and dried to obtain 7-(4-
methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic acid (8) (3.1 g,
99.8%), m. p. 211-12 °C.
Anal.:
TLC : Rf 0.17 (EtOAc)
IR : 3317, 3087, 2474, 1791, 1772, 1606, 1289 and 1187 cm-1
MS : (m/z) 297, (M+-1)
N-(3-Fluorobenzyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-
3-carboxamide (IV-21)
Compound (8) (0.25 gm, 0.08 mmole) and 3-fluorobenzylamine (0.105
g, 0.08 mmole) were reacted as described above for compound (IV-1).
Chromatographic purification of the crude product using hexane (15%) in ethyl
acetate as eluent afforded the titled compound (IV-21) (0.16 g, 47.0%), m. p. 171-
73 °C.
Anal.:
TLC : Rf 0.55 (1% MeOH in EtOAc)
IR : 3445, 3319, 3072, 1688, 1611, 1533, 1482, 1336, 1290, 1264,
1184, 1143, 1060, 941, 832, 793, 691, 649 and 556 cm-1
1HNMR : 3.83 (s, 3H), 4.60-4.62 (d, 2H), 7.07-7.16
(m, 3H), 7.20-7.22
(d, 2H), 7.37-7.42 (d, 1H), 7.79-7.81(d, 2H), 7.95-7.98 (d,
1H), 8.54-8.57 (dd, 1H), 9.04 (s, 1H), 9.27-9.28 (d, 1H), 9.45-
9.48 (t, 1H)
MS : (m/z) 404, (M+-1)
Synthesis of 7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-
3-carboxamide derivatives
N-(3-Trifluoromethylbenzyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-
a]pyrimidine-3-carboxamide (IV-22)
Compound (8) (0.3 gm, 0.10 mmole) was reacted with 3-
trifluoromethylbenzylamine (0.177 g, 0.10 mmole) as described above for
compound (IV-1). Chromatographic purification of the crude product using

Chapter-4 Experimental work
142
hexane (5%) in ethyl acetate as eluent offered the desired compound (IV-22) (0.14
g, 30.49%), m. p. 197-98 °C.
Anal.:
TLC : Rf 0.54 (EtOAc)
IR : 3414, 3290, 3110, 1692, 1563, 1478, 1333, 1262, 1163, 1112,
1066, 828 and 704 cm-1
1HNMR : 3.84 (s, 3H), 4.68-4.69 (d, 2H), 7.13-7.15 (d, 2H), 7.57-7.59
(d, 2H), 7.71-7.73 (d, 2H), 7.80-7.82 (d, 2H), 7.96- 7.99 (d,
1H), 8.55-8.58 (dd, 1H), 9.04 (s, 1H), 9.28-9.29 (d, 1H), 9.52-
9.55 (t, 1H)
MS : (m/z) 454, (M+-1)
N-(4-Trifluoromethylbenzyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-
a]pyrimidine-3-carboxamide (IV-23)
Compound (IV-23) was obtained by reacting (8) (0.3 gm, 0.10 mmole)
with 4-trifluoromethylbenzylamine (0.177 g, 0.10 mmole) as described above for
compound (IV-1). Chromatographic purification of the crude product using
hexane (5%) in ethyl acetate as eluent yielded the titled compound (IV-23) (0.16
g, 45.9%), m. p. 209-10 °C.
Anal.:
TLC : Rf 0.58 (EtOAc)
IR : 3425, 3293, 1689, 1623, 1333, 1263, 792, 583 and 558 cm-1
1HNMR : 3.83 (s, 3H), 4.67-4.68 (d, 2H), 7.11-7.14 (d,
2H), 7.57-7.64
(m, 2H), 7.67-7.69 (m, 1H), 7.72 (b, 1H), 7.78-7.81 (d, 2H),
7.95-7.97 (d, 1H), 8.53-8.56 (dd, 1H), 9.04 (s, 1H), 9.26 (s,
1H), 9.51-9.54 (t, 1H)
MS : (m/z) 454, (M+-1)
N-(Tetrahydrofuran-2-ylmethyl)-7-(4-methoxyphenyl)-4-oxo-4H-pyrido[1,2-
a]pyrimidine-3-carboxamide (IV-24)
Compound (IV-24) (0.25 gm, 0.08 mmole) was reacted with 2-
tetrahydrofuranyl methylamine (0.085 g, 0.08 mmole) as described above for
compound (IV-1). Chromatographic purification of the crude product using

Chapter-4 Experimental work
143
hexane (20%) in ethyl acetate as eluent provided compound (IV-24) (0.1 g,
31.2%), m. p. 176-77 °C.
Anal.:
TLC : Rf 0.61(1% MeOH in EtOAc)
IR : 3453, 3306, 1689, 1611, 1545, 1337, 1264 and 1180 cm-1
1HNMR : 1.54-1.61 (m, 1H), 1.80-1.89 (m, 2H), 1.90-1.99 (m, 1H),
3.38-3.39
(d, 1H), 3.52-3.57 (m, 1H), 3.66-3.69 (m, 1H),
3.80-3.81 (d, 1H), 3.84 (s, 3H), 3.96-3.99 (m, 1H), 7.12-7.15
(d, 2H), 7.79-7.84 (d, 2H), 7.91-7.97 (d, 1H), 8.54-8.56 (dd,
1H), 9.03 (s, 1H), 9.17-9.19 (t, 1H), 9.29-9.30 (d, 1H)
MS : (m/z) 380, (M+-1)
4.2 Biological work
4.2.1 Falcipain-2 enzyme inhibition assay
The diluted soluble Plasmodium falciparum FP-2 (30 nM) was incubated
for 10 min at room temperature in 100 mM sodium acetate, pH 5.5, 10 mM DTT,
with a fixed/different concentration of the compounds to be tested or the standard
(E64). Compound solutions were prepared from stock in DMSO. After 10 min
incubation, the substrate Z-Phe-Arg-AMC was added to a final concentration of
25 µM. The fluorescence intensity was monitored (excitation 355 nm; emission
460 nm) for 10 min at room temperature with a SynergyTM
4 Multi-Mode
Microplate Reader (BioTek). The inhibition rate (%) is calculated using the given
equation:
% Inhibition = [1 - (F test / F stand.)] × 100
where, F test is the fluorescence intensity of the test compound, F control is the
fluorescence intensity of the standard (E64). All values are the means of three
independent determinations and the deviations are <10 % of the mean value. The
IC50 values were determined for those compounds only which showed 40 % or
more of enzyme inhibition at 10 μM.

5. Summary

Summary
144
The existing armamentarium of antimalarial drugs is not sufficient to combat
malaria, primarily because of resistance developed by the parasite. The problem got
further compounded due to non-enrichment of antimalarial drug inventory.
Unfortunately an effective antimalarial vaccine also could not be developed due to
fast and constant mutations in the parasite genome. All these factors led to a woefully
thin antimalarial drug development pipeline with little chemical diversity.
Genome mapping of the malaria parasite offered a number of attractive drug
targets including plasmepsins and falcipains, the enzymes involved in parasite
metabolism and in providing nutrients to the parasite to meet its energy requirements.
There exist a great potential in the exploration of cysteine proteases falcipain
inhibitors. Falcipains 2 and 3 especially were more attractive targets as these enzymes
degrade haemoglobin to small sized peptides in the acidic environment of the vacuole
and degrade the membrane proteins of the host cells and help the parasite rupture the
host cells in the alkaline environment. So, inhibition of FP-2/3 enzymes could
effectively arrest the growth of the parasite and control their progression in the host
system.
A large number of peptidic and non-peptidic motifs like chalcones, (thio)
semicarbozones, hydrazides, heterocycles, oxiranes and succinates have been reported
in the literature to exhibit FP-2/3 inhibitory activity. Unfortunately none of such
compounds have crossed the clinical phase of evaluation as antimalarial agents. There
are some reports of some heterocyclic and polycyclic compounds exhibiting FP-2/3
inhibiting activities. Taking the basic information from literature about the enzyme
inhibitors into consideration, it was planned to synthesize pyrido[1,2-a]pyrimidin-4-
one class of compounds (M).
N
N
X
O
AX = Urea, carbamate, amide
A = Alkyl/aryl/heteroaryl
R = H, CH3, Cl, 4-C6H4OCH3
R
(M)
It is worth noting that the circled pharmacophore may act as Michael
acceptor for reversible/irreversible binding to the thiol nucleophile present in the FP-
2/3 enzymes in the P1 binding pocket. While the groups, X and R were selected in
such a way that compounds could exhibit better binding profile in the P2 and P3

Summary
145
binding pockets of the falcipain enzyme that could translate in to enhanced potency of
the compounds.
It was planned to synthesize the given four (Series-I to IV) of compounds
and to evaluate them against FP-2 enzyme in the in vitro tests. FP-3 enzyme having a
high degree of homology to FP-2 was presumed to react with all such compounds
which would prove to be powerful inhibitors of FP-2.
N
N
N
O
O
NRR N N
N
N
N
O
O
N
N
O
N
O
AR
N
N
N
O
O
OR
I II
AA
R1
IV
R1
III
A
Where: R = H, 8-CH3, 7-Cl, 4-C6H4OCH3
R1 = H, Alkyl
A = Alkyl/heteroaryl or alkyl
All of the derivatives of the given four (Series-I to IV) were synthesized
by following either of the Schemes 1-6 and the compounds were characterized on
the basis of their spectral data. Synthetic methods for preparing these new
chemical entities, spectral (IR, PMR and Mass spectrometry) data and biological
activities of these compounds have been discussed in detail in this thesis work.
The acids (4a-c) and the isocyanates (6a-c) are the key intermediates in
the preparation of the targeted compounds. Their synthesis was accomplished by
following the key steps i) Gould and Jacob reaction ii) cyclization, iii) hydrolysis
and iv) Curtius rearrangement reaction. Synthesis of theses intermediates is
accomplished by following the reaction sequence as outlined in the Scheme 1.
2-Aminopyridines (1a-c) by Gould and Jacob coupling reaction with
diethyl ethoxymethylenemalonate yielded (2a-c), which on cyclization by
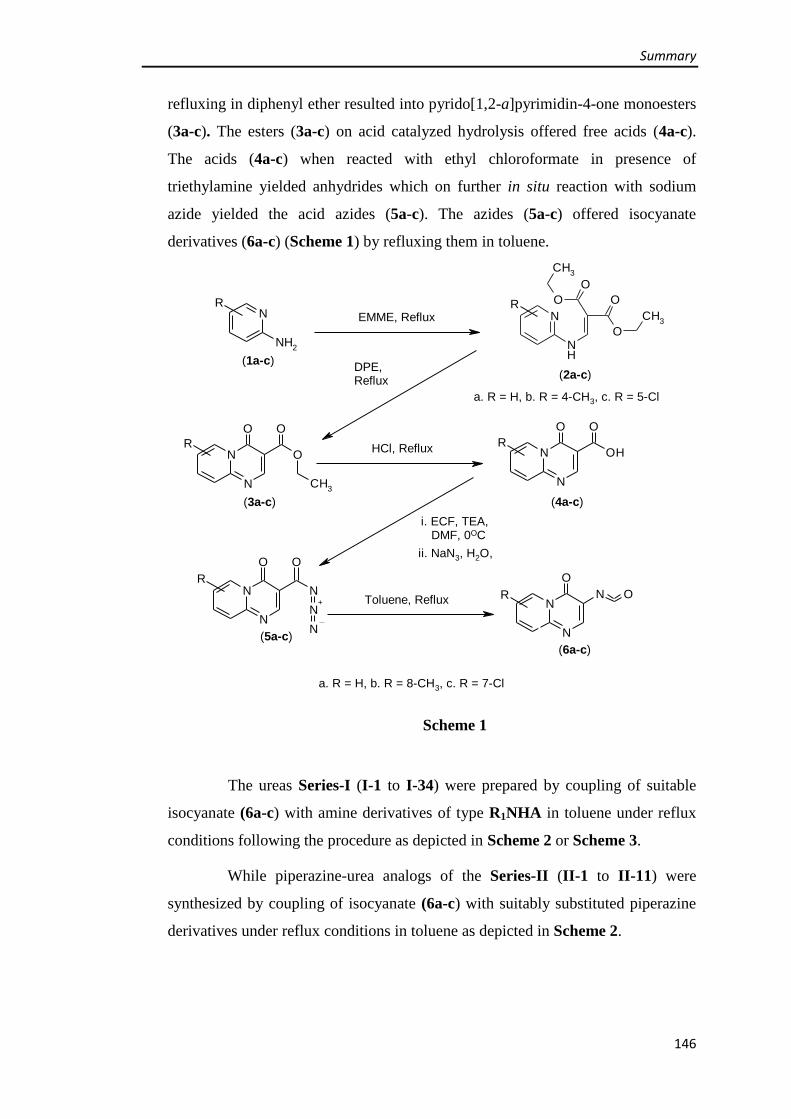
Summary
146
refluxing in diphenyl ether resulted into pyrido[1,2-a]pyrimidin-4-one monoesters
(3a-c). The esters (3a-c) on acid catalyzed hydrolysis offered free acids (4a-c).
The acids (4a-c) when reacted with ethyl chloroformate in presence of
triethylamine yielded anhydrides which on further in situ reaction with sodium
azide yielded the acid azides (5a-c). The azides (5a-c) offered isocyanate
derivatives (6a-c) (Scheme 1) by refluxing them in toluene.
N
NH2
N
N
O
O
CH3
O
N
N
O
OH
O
N
N
N
O
ON
N
O O
N
N+
N
R
R R
R
R
N
NH
O
O
O
CH3
O
CH3
R
(1a-c)(2a-c)
(3a-c) (4a-c)
(5a-c)(6a-c)
a. R = H, b. R = 8-CH3, c. R = 7-Cl
i. ECF, TEA, DMF, 0OC
ii. NaN3, H2O,
EMME, Reflux
DPE,Reflux
HCl, Reflux
Toluene, Reflux
a. R = H, b. R = 4-CH3, c. R = 5-Cl
Scheme 1
The ureas Series-I (I-1 to I-34) were prepared by coupling of suitable
isocyanate (6a-c) with amine derivatives of type R1NHA in toluene under reflux
conditions following the procedure as depicted in Scheme 2 or Scheme 3.
While piperazine-urea analogs of the Series-II (II-1 to II-11) were
synthesized by coupling of isocyanate (6a-c) with suitably substituted piperazine
derivatives under reflux conditions in toluene as depicted in Scheme 2.
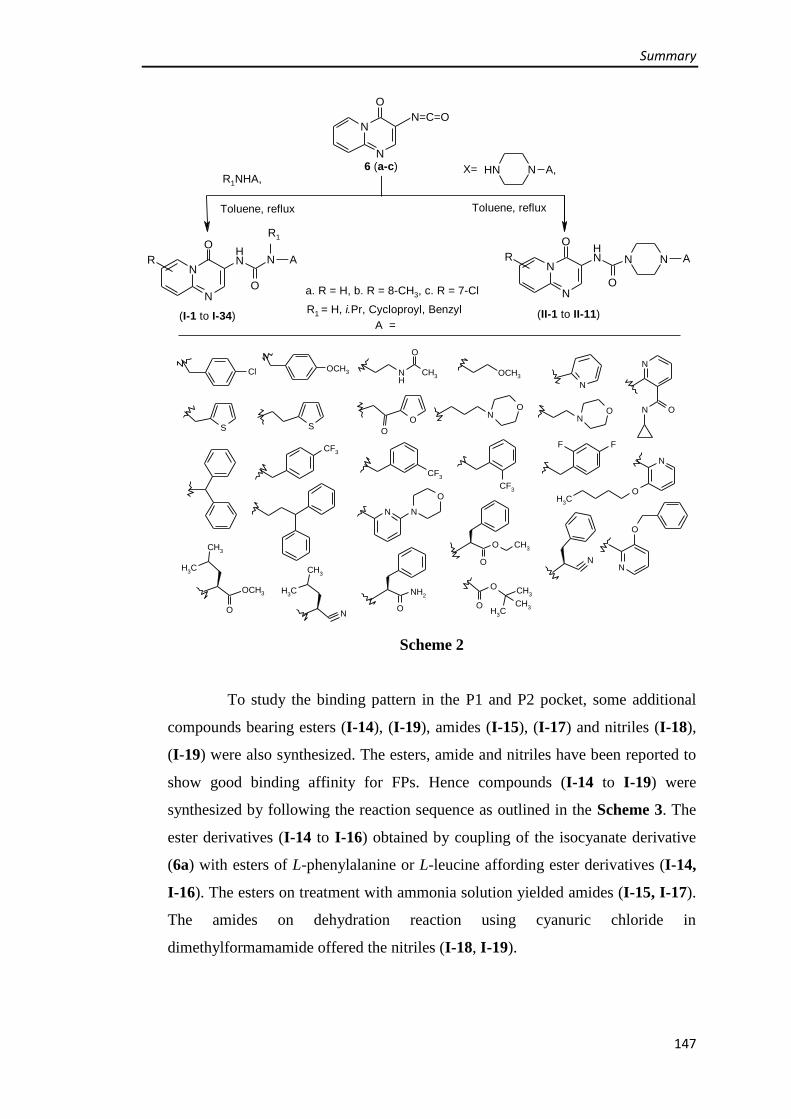
Summary
147
N
N
NH
O
O
NR R N NN
N
NH
O
O
NH N A,
N
N
O
N
CF3
CF3
NH
CH3
O
N N
O
CF3
FF
NO
CH3
CH3
N
N
ClN
ONN
O
CH3
CH3
O
OCH
3
N
N
O
O
O
CH3
NH2
O
O
OSS
O
OCH
3
CH3
CH3
6 (a-c)
Toluene, reflux
A A
R1NHA, X=
R1
Toluene, reflux
(I-1 to I-34)
N=C=O
OCH3
A =
R1 = H, i.Pr, Cycloproyl, Benzyl
OCH3
OCH3
a. R = H, b. R = 8-CH3, c. R = 7-Cl
(II-1 to II-11)
Scheme 2
To study the binding pattern in the P1 and P2 pocket, some additional
compounds bearing esters (I-14), (I-19), amides (I-15), (I-17) and nitriles (I-18),
(I-19) were also synthesized. The esters, amide and nitriles have been reported to
show good binding affinity for FPs. Hence compounds (I-14 to I-19) were
synthesized by following the reaction sequence as outlined in the Scheme 3. The
ester derivatives (I-14 to I-16) obtained by coupling of the isocyanate derivative
(6a) with esters of L-phenylalanine or L-leucine affording ester derivatives (I-14,
I-16). The esters on treatment with ammonia solution yielded amides (I-15, I-17).
The amides on dehydration reaction using cyanuric chloride in
dimethylformamamide offered the nitriles (I-18, I-19).

Summary
148
NH2
R'
O
O CH3
n
NH
R'
O
O
N
N
NH
O
O
CH3
n
NH
R'
O
NH2
N
N
NH
O
O
CH3
CH3
NH
R'
N
N
NH
O
O
N
N
N
N
Cl
Cl
Cl
liq. ammonia
(6a,b) +
Toluene Reflux
(I-14, I-16)
R' =
n = 0, 1
DMF, 0OC
(I-15, I-17) (I-18, I-19)
Scheme 3
Synthesis of the carbamate derivatives of Series-III (III-1 to III-9) have
been accomplished by reaction of the isocyanate (6a-c) and suitable alcohol
derivative (A-HO) in toluene under reflux conditions following the procedure as
outlined in Scheme 4.
N
N
O
R N
N
NH
O
O
OR
N F
O
O
N
6 (a-c)
A HO-A
Toluene, reflux
N=C=O
(III-1) - (III-9)
a. R = H, b. R = 8-CH3, c. R = 7-Cl
A =
Scheme 4
Synthesis of the amide derivatives of Series IV (IV-1 to IV-26) have
been accomplished by reacting the acid (4a-c) with suitable amine (R’-NHA)
using EDC as a coupling agent as outlined in Scheme 5.

Summary
149
N
N
O
OH
O
N
N
O
N
O
ARR
CF3
CF3
N
O
CF3
FF
N
ON
NO
CH3
N O
O
CH3
O
CH3
N
O
O
CH3
N
O
CH3
F
FOCH
3
N
O
4 (a-c) (IV-1) - (IV-20)
EDC, HOBT, DIEA, 0OC/ RT
R'-NHA
R'
a. R = H, b. R = 8-CH3, c. R = 7-Cl
OCH3
A =
OCH3
R' = H, i.Pr, Cyclopropyl, Benzyl
Scheme 5
In order to study the hydrophobic binding interactions by increasing the
steric bulk of the group attached to the B-ring (i.e. pyrido) of the pyridopyrimidine
ring, some additional compounds (IV23 - IV24) were synthesized. The 7-chloro
ester (3c) was reacted with p-methoxyphenylboronic acid through Suzuki
coupling offering the ester (7) which on acid hydrolysis yielded the acid (8). The
acid derivative (8) was coupled with suitable amines to get the targeted
compounds (IV23 - IV24) as depicted in Scheme 6.

Summary
150
N
N
O
O
O
ClCH
3 N
N
O
O
O
CH3
N
N
O
OH
O
N
N
O
NH
O
A
B(OH)2
CF3
CF3
F O
(3c) (7)
DME, P(Ph3)4, K2CO3,
Reflux
HCl, Reflux
(8) (IV-21) - (IV-24)
EDC, HOBT, DIEA,
A-NH2, 0OC/ RT
H3COH3CO
H3CO
A =
H3CO
Scheme 6
The synthesised compounds were submitted for in vitro FP-2 inhibition
evaluation. Compounds were evaluated for their inhibitory activity against
recombinant FP-2 using Cbz-Phe-Arg-AMC as a fluorogenic substrate.
Preliminary screening was performed at 10 M concentration. An equivalent
concentration of DMSO was used as negative control and the irreversible standard
inhibitor of clan CA family C1 cysteine proteases (papain family), namely E-64
was used as positive control. Assays were performed to determine the percentage
inhibition of the enzyme at a concentration of 10 M. FP-2 activity is assessed by
cleavage of the fluorogenic substrate Cbz-Phe-Arg-AMC releasing the fluorescent
AMC group. Hence decrease in fluorescence intensity in a sample represents
inhibition of enzyme activity. The IC50 values were determined for those
compounds only which showed more than 40 % inhibition at 10 M
concentration. The FP-2 enzyme inhibition data of the synthesised compounds
shows that the pyrido[1,2-a]pyrimidin-4-ones show FP-2 inhibition in micromolar
range. Based on the result of evaluated compounds a broad structure activity
relationship could be deduced for this series of the compounds. Urea moiety has
the potential to provide potent FP-2 inhibitors as the carbamate and carboxamide
derivatives are much less active in comparison to the substituted ureas.
Ethylmorpholine (I-26, IC50 6.93 M) and methoxyethyl (I-21, IC50 6.36
M) urea derivatives proved to be the most potent compounds. 2-Thiophenethyl
(I-8, IC50 14.27 M; IV-23, IC50 21.16 M; I-27, IC50 23.35 M), acetamidoethyl

Summary
151
(I-4, IC50 16.08 M) and 4-methoxyphenylpiperazine (II-5, IC50 18.87 M) were
the other groups which gave active compounds when attached to the other end of
the urea moiety. 2-Pyridylpiperazine is another moiety which offered quite active
compounds (II-8, IC50 12.16 M; II-1, IC50 14.84 M).
In general 8-methyl and 7-chloro substituted derivatives offered more
potent compounds indicating that increasing the steric bulk of the group attached
to the B-ring (i.e. pyrido) is likely to provide more potent FP-2 inhibitors. An
electron rich environment at the end of the carbon chain attached to the urea
nitrogen also seems to be conducive for better activity as indicated by the
compounds having morpholine, 2-thiophenenyl, 2-pyridyl, 4-methoxyphenyl,
acetamidoethyl and 2-methoxyethyl groupings. But bulky groupings (II-7,
benzhydrylpiperazine and I-9, 4-morpholinopropyl) may not offer potent
derivatives.
With the availability of biological activity for compounds the current
work has proved the potential of pyrido[1,2-a]pyrimidin-4-one derivatives as
potent FP-2 inhibitors. Based on the activity data of the synthesized compounds it
could be claimed that pyrido[1,2-a]pyrimidin-4-one can serve as a lead series for
future investigations. By appropriate structural optimizations the potency of this
series of compounds can be improved substantially against FP-2 enzyme which in
turn can serve to be potential antimalarial agents. Further optimization of the lead
structures and biological screening is in progress in the laboratory.

6. References

References
152
1. Okafor, E. E., Amzat, J., J. Hum. Ecol., 21, 155-162 (2007).
2. Ettari, R., Bova, F., Zappala, M., Grasso, S., Micale, N., Med. Res. Rev., 30,
136-167 (2010).
3. Wood, C. S., Current Anthropology, 16, 93 (1975).
4. Snow, R. W., Guerra, C. A., Noor, A. M., Myint, H. Y., Hay, S. I., Nature,
434, 214-217 (2005).
5. World Health Organization (2005a). International travel and health, Chapter 7,
132-151 (2005).
6. WHO report (2011, 2012). (http://www.rollbackmalaria.org,
http://www.who.int/malaria- /publications/atoz/arupdate042012.pdf).
7. Caffrey, C. R., Steverding, D., Exp. Opin. Drug Discov., 3, 173-186 (2008).
8. Rosenthal, P. J., New Targ. for Chemother. Emerg. Infect. Diseases, 4, 49-57
(1998).
9. Burgers medicinal chemistry and drug discovery, Vol 5, chemotherapeutic
agents 6th
edn., John Willey & Sons, Inc., pp 920-982 (2003).
10. Francis, S. E., Sullivan, D. J., Goldberg, D. E., Ann. Rev. Microbiol., 51, 97-
123 (1997).
11. Pagola, S., Stephens, P. W., Bohle, D. S., Kosar, A. D., Madsen, S. K., Nature,
404, 307-310 (2000).
12. Egan, T. J., Marques, H. M., Coord. Chem. Rev., 190-192, 493-517 (1999).
13. Pukrittayakamee, S., Chantra, A., Vanijanonta, S., Clemens, R.,
Looareesuwan, S. White, N. J., Antimicrob. Agents Chemother., 44, 2395-
2398 (2000).
14. O’Neill, P. M., Bray, P. G., Hawley, S. R., Ward, S. A., Park, B. K.,
Pharmacol. Ther., 77, 29-58 (1998).
15. Foster, S., Trans. R. Soc. Trop. Med. Hyg., 88, 55-56 (1994).
16. Bray, P. G., Janneh, O., Raynes, K. J., Mungthin, M., Ginsburg, H., Ward, S.
A., J. Cell Biol., 145, 363-376 (1999).

References
153
17. Bray, P. G., Ward, S. A., Pharmacol. Ther., 77, 1-28 (1998).
18. Ridley, R. G., Curr. Biol. 8, R346-349 (1998).
19. Kwiatkowski, D., Bate, C., Trans. R. Soc. Trop. Med. Hyg., 89, 215-216
(1995).
20. Ringwald, P., Bickii, J. Basco, L. K., Trans. R. Soc. Trop. Med. Hyg., 90, 254-
258 (1996).
21. Foley, M., Tilley¸ L., Pharmacol. Ther., 79, 55-87 (1998).
22. Bray, P. G., Hawley, S. R., Ward, S. A., Mol. Pharmacol., 50, 1551-1558
(1996).
23. Vries, P. J., Bich, N. N., Van Thien, H., Hung, L. N., Anh, T. K., Kager, P. A.,
Heisterkamp, S. H., Antimicrob. Agents Chemother., 44, 1302-1308 (2000).
24. Parola, P., Ranque, S., Badiaga, S., Niang, M., Blin, O., Charbit, J. J.,
Delmont, J., Brouqui, P., Antimicrob. Agents. Chemother., 45, 932-935
(2001).
25. Sarma, P. S. A., Mandal, A. K., Khamis, H. J., Am. J. Trop. Med. Hyg., 58,
454-457 (1998).
26. Basco, L. K., Le Bras, J., Rhoades, Z., Wilson, C. M., Mol. Biochem.
Parasitol., 74, 157-166 (1995).
27. Reed, M. B., Saliba, K. J., Caruana, S. R., Kirk, K., Cowman, A. F., Nature,
403, 906- 909 (2000).
28. Plowe, C. V., Wellems, T. E., Adv. Exp. Med. Biol., 390, 197-209 (1995).
29. Steketee, R. W., Nahlen, B. L., Parise, M. E., Menendez, C., Am. J. Trop.
Med. Hyg., 64, 28-35 (2001).
30. Kuile, F. O., Dolan, G., Nosten, F., Edstein, M. D., Luxemburger, C., Phaipun,
L., Chongsuphajaisiddhi, T., Webster, H. K., White, N. J., Lancet, 341, 1044-
1049 (1993).
31. Nosten, F., Kuile, F. O., Luxemburger, C., Woodrow, C., Kyle, D. E.,
Chongsuphajaisiddhi, T., White, N. J., Lancet, 341, 1054-1056 (1993).

References
154
32. Wongsrichanalai, C., Webster, H. K., Wimonwattrawatee, T., Sookto, P.,
Chuanak, N., Thimasarn, K., Wernsdorfer, W. H., Am. J. Trop. Med.Hyg., 47,
112-116 (1992).
33. Ringwald, P., Eboumbou, E. C., Bickii, J., Basco, L. K., Antimicrob. Agents
Chemother., 43, 1525-1527 (1999).
34. Ward, S. A., Bray, P. G., Mungthin, M., Hawley, S. R., Ann. Trop.
Med.Parasitol., 89, 121-124 (1995).
35. Cowman, A. F., Int. J. Parasitol., 31, 871-878 (2001).
36. Nzila, A. M., Mberu, E. K., Sulo, J., Dayo, H., Winstanley, P. A., Sibley, C.
H., Watkins, W. M., Antimicrob. Agents Chemother., 44, 991-996 (2000).
37. Amukoye, E., Winstanley, P. A., Watkins, W. M., Snow, R. W., Hatcher, J.,
Mosobo, M., Ngumbao, E., Lowe, B., Ton, M., Minyiri, G., Marsh, K.,
Antimicrob. Agents Chemother., 41, 2261-2264 (1997).
38. Canfield, C. J., Pudney, M., Gutteridge, W. E., Exp. Parasitol., 80, 373-381
(1995).
39. Bustos, D. G., Canfield, C. J., Canete-Miguel, E., Hutchinson, D. B. A., J.
Infect. Dis., 179, 1587-1590 (1999).
40. Looareesuwan, S., Wilairatana, P., Chalermarut, K., Rattanapong, Y.,
Canfield, C. J., Hutchinson, D. B. A., Am. J. Trop. Med. Hyg., 60, 526-532
(1999).
41. White, N. J., J Clin Invest., 113, 1084-1092 (2004).
42. Cravo, P., Culleton, R., Afonso, A., Ferreira, I. D., Do Rosario, V. E., Anti-
Infect. Agents. Med. Chem., 5, 63-73 (2006).
43. Meshnick, S. R., Taylor, T. E., Kamchonwongpaisan, S., Microbiol.Rev., 60,
301-315 (1996).
44. Jain, D. C., Mathur, A. K., Gupta, M. M., Singh, A. K., Verma, R. K., Gupta,
A. P., Kumar, S. Phytochemistry, 43, 993-1001 (1996).
45. Van Vugt, M., Angus, B. J., Price, R. N., Mann, C., Simpson, J. A., Poletto,
C., Saw, E. H., Looareesuwan, S., White, N. J. Nosten, F., Am. J. Trop.Med.
Hyg., 62, 65-69 (2000).

References
155
46. McGready, R., Nosten, F., Ann. Trop. Med. Parasitol., 93, S19-23 (1999).
47. Dhingra, V., Rao, V. K., Lakshmi-Narasu, M., Life Sci., 66, 279-300 (2000).
48. Li, Q. G., Peggins, J. O., Lin, A. J., Masonic, K. J., Trotman, K. M., Brewer,
T. G., Trans. R. Soc. Trop. Med. Hyg., 92, 332-340 (1998).
49. Peters, W., Robinson, B. L., Ann. Trop. Med. Parasitol., 93, 325-339 (1999).
50. Van-Vugt, M., Brockman, A., Gemperli, B., Luxemburger, C., Gathmann, I.,
Royce, C., Thra S., Looareesuwan, S., White, N. J., Nosten, F., Antimicrob.
Agents Chemother., 42, 135-139 (1998).
51. Price, R. N., Nosten, F., Luxemburger, C., van Vugt, M., Phaipun, L.,
Chongsuphajaisiddhi, T., White, N. J., Trans. R. Soc. Trop. Med. Hyg., 91,
574-577 (1997).
52. Arrow, K. J., Fin. and Dev., 41, 20-21 (2004).
53. White, N. J., Van Vugt, M., Ezzet, F., Clin. Pharmacokinet., 37, 105-125
(1999).
54. Bindschedler, M., Lefevre, G., Ezzet, F., Schaeffer, N., Meyer, I., Thomsen,
M. S., Eur. J. Clin. Pharmacol., 56, 375-381 (2000).
55. Martiney, J. A., Cerami, A., Slater, A. F. G., Mol. Med., 2, 236-246 (1996).
56. Tokuyasu, T., Masuyama, A., Nojima, M., Kim, H. S., Wataya, Y.,
Tetrahedron Lett., 41, 3145-3148 (2000).
57. Williams, T. N., Maitland, K. S. B., Ganczakowski, M., Peto, T. E. A.,
Mewbold, C. I., Bowdens, D. K., Weathrall, D. J., Clegg, J. B., Nature, 383,
522-525 (1996).
58. Girometta, R., Jauch, C., Ponelle, A., Guenzi, R. C., Wiegand, C., Trop. Med.
Parasitol., 45, 272-277 (1994).
59. Weidekamm, E., Dumont, E., Jaquet C., Trop. Med. Parasitol., 45, 278-283
(1994).
60. Anders, R. F., Saul, A., Parasitol. Today, 16, 444-447 (2000).
61. Young, J. A., Winzeler, E. A., Pharmacogenomics, 6, 17-26 (2005).
62. Garcia, Linares, G., E., Rodriguez, J. B., Curr Med Chem, 14, 289-314 (2007).

References
156
63. Ralph, S. A., Ombrain, M. C. D., Mc-Fadden, G. I., Drug Resist. Updates, 4,
145-151 (2001).
64. Shahabuddin, M., Cociancich, S., Zieler, H., Parasitol. Today, 14, 493-497
(1998).
65. Gozalbes, R., Gavez, J., Moreno, A., Garcia-Domenech, R., J. Pharm.
Pharmacol., 51, 111-117 (1999).
66. Calas, M., Ancelin, M. L., Cordina, G., Portefaix, P., Piquet, G., Vidal-
Sailhan, V. and Vial, H., J. Med. Chem., 43, 505-516 (2000).
67. Biot, C., Glorian, G., Maciejewski, L. A., Brocard, J. S., J. Med. Chem., 40,
3715-3718 (1997).
68. Delhaes, L., Biot, C., Berry, L., Maciejewski, L. A., Camus, D., Brocard, J. S.,
Dive, D., Bioorg. Med. Chem., 8, 2739-2745 (2000).
69. Newton, P., White, N., Ann. Rev. Med., 50, 179-192 (1999).
70. Kempf, D. J., Sham H. L., Curr. Pharm. Des., 2, 225-246 (1996).
71. King, C. H., Muchiri, E. M., Ouma, J. H., Emerg. Infect. Dis., 6, 585-94
(2000).
72. Godfrey, T., Industrial Enzymology, 2nd
edition, Godfrey T., Ed. Macmillan
Press, London, pp 1-8 (1996).
73. Ripka, A. S. Rich, D. H., Curr. Opin. Chem. Biol., 2, 453-458 (1998).
74. Seife, C., Science, 277, 1602-1603 (1997).
75. Ripka, A. S. Rich, D. H., Curr. Opin. Chem. Biol., 2, 441-452 (1998).
76. Craik, M. S. Debouck, C., Perspective in Drug Discovery and Design, 2, 1-
125 (1995).
77. Gante, J., Angew.Chem. Int. Ed. Engl., 33, 1699-1720 (1994).
78. Goldberg, D. E., Curr Top Microbiol Immunol, 295, 275-291 (2005).
79. Goldberg, D. E., Semin. Cell. Biol., 4, 355-361 (1993).
80. Liu, J., Istvan, E. S., Gluzman, I. Y., Gross, J., Golberg, D. E., Proc. Natl.
Acad. Sci. USA., 103, 8840-8845 (2006).
81. Sherman, I. W., Tanigoshi, L., Int. J. Biochem., 1, 635-637 (1970).
82. Sherman I. W., Bull WHO, 55, 265-276 (1977).

References
157
83. Lew, V. L., Tiffert, T., Ginsburgm, H., Blood, 101, 4189-4194 (2003).
84. Atamna, H., Ginsburg, H., Mol. Biochem. Parasitol., 61, 231-241 (1993).
85. Pisciotta, J. M., Coppens, I., Tripathi, A. K., Scholl, P. F., Shuman, J., Bajad,
S., Shulaev, V., Sullivan, D. J., Biochem . J., 402, 197-204 (2007).
86. Sijwalai, P. S., Rosenthal, P. J., Proc. Natl. Acad. Sci. USA., 101, 4384-4389
(2004).
87. Banerjee, R., Liu, J., Beatty, W., Pelosof, L., Klemba, M., Goldberg, D. E.
Proc. Natl. Acad. Sci. USA., 99, 990-995 (2002).
88. Pandey, A. V., Singh, N., Tekwani, B. L., Puri, S. K., Chauhan, V. S., J.
Pharm. Biomed. Anal. 20, 203-207 (1999).
89. Gluzman, I. Y., Francis. S. E., Oksman, A., Smith, C. E., Duffin, K. L.,
Goldberg, D. E., J. Clin. Invest., 93, 1602-1608 (1994).
90. Mueller, R., Huerzeler, M., Boss, C., Molecules, 8, 556-564 (2003).
91. Goldberg, D. E., Slater, A. F., Beavis, R., Chait, B., Cerami, A., Henderson,
G. B., J. Exp. Med., 173, 961-969 (1991).
92. Sijwali, P. S., Kato, K., Seydel, K.B., Gut, J., Lehman, J., Klemba, M.,
Goldberg, D. E., Miller, L. H., Rosenthal, P. J., Proc. Natl. Acad. Sci.
USA.,101, 8721-8726 (2004).
93. Shenai, B. R., Sijwali, P. S., Singh, A., Rosenthal, P. J., J. Biol. Chem. 275,
29000-29010 (2000).
94. Sijwali, P. S., Shenai, B. R., Gut, J., Singh, A., Rosenthal, P. J., Biochem. J.,
360, 481-489 (2001).
95. Eggleson, K. K., Duffin, K. L., Goldberg, D. E., J. Biol. Chem., 274, 32411-
32417(1999).
96. Klemba, M., Gluzman, I., Goldberg, D. E., J. Biol. Chem., 279, 43000-43007
(2004).
97. Omara-Opyene, A. L., Moura, P. A., Sulsona, C. R., Bonilla, J. A., Yowell, C.
A., Fujioka, H., Fidock, D. A., Dame, J. B., J. Biol. Chem., 279, 54088-54096
(2004).
98. Glushakova, S., Mazar, J., Hohmann-Marriott, M. F., Hama, E., Zimmerberg,
J., Cell Microbiol., 11, 95-105 (2009).
99. Gavigan, C. S., Dalton, J. P., Bell, A., Mol. Biochem. Parasitol., 117, 37-48
(2001).

References
158
100. Dalal, S., Klemba, M., J. Biol. Chem., 282, 35978-35987 (2007).
101. Singh, N., Sijwali, P. S., Pandey, K. C., Rosenthal, P. J., Exp. Parasitol., 112,
187-192 (2006).
102. Rosenthal, P. J., Int . J. Parasitol., 34, 1489-1499 (2004).
103. Greenbaum, D. C., Baruch, A., Grainger, M., Bozdech, Z., Medzihradszky, K.
F., Engel, J., De-Risi, J., Holder, A. A., Bogyo, M., Science, 298, 2002-2006
(2002).
104. Eksi, S., Czesny, B., Greenbaum, D. C., Bogyo, M., Williamson, K. C., Mol.
Microbiol., 53, 243-250 (2004).
105. Ramjee, M. K., Flinn, N. S., Pemberton, T. P., Quibell, M., Wang, Y., Watts,
J. P., Biochem. J., 399, 47-57 (2006).
106. Dahl, E. L., Rosenthal, P. J., Mol. Biochem.Parasitol., 139, 205-212 (2005).
107. Krungkrai, J., Prapunwattana, P., Krungkrai, S. R., Parasite, 33, 19-26 (2000).
108. Hanspal, M., Dua, M., Takakuwa, Y., Chishti, A. H., Mizuno, A., Blood, 100,
1048-1054 (2002).
109. Dua, M., Raphael, P., Sijwali, P. S., Rosenthal, P. J., Hanspal, M., Mol
Biochem. Parasitol., 116, 95-99 (2001).
110. Sijwali, P. S., Koo, J., Singh, N., Rosenthal, P. J., Mol. Biochem. Parasitol.,
150, 96-106 (2006).
111. Nielsen, K. M., Kasper, J., Choi, M., Bedford, T., Kristiansen, K., Wirth, D.
F., Volkman, S. K., Lozovsky. E. R., Hartl, D. L., Mol. Biol. Evol., 20, 726-
734 (2003).
112. Goh, L. L., Sim, T. S., Biochem. Biophys. Res. Commun., 335, 762-770
(2005).
113. Jeong, J. J., Kumar, A., Hanada, T., Seo, P. S., Li, X., Hanspal, M., Chishti, A.
H.,Blood Cells Mol. Dis., 3, 429-435 (2006).
114. Drew, M. E., Banerjee, R., Uffman, E. W., Gilbertson, S., Rosenthal, P. J.,
Goldberg,D. E., J. Biol. Chem., 283, 12870-12876 (2008).
115. Hogg, T., Nagarajan, K., Herzberg, S., Chen, L., Shen, X., Jiang, H., Wecke,
M., Blohmke, C., Hilgenfeld, R., Schmidt, C. L., J. Biol. Chem., 281, 25425-
25437 (2006).
116. Wang, S. X., Pandey, K. C., Somoza, J. R., Sijwali, P. S., Kortemme, T.,
Brinen, L. S., Fletterick, R. J., Rosenthal, P. J., McKerrow, J. H., Proc. Natl.

References
159
Acad. Sci.USA.,103, 11503-11508 (2006).
117. Kerr, I. D., Lee, J. H., Pandey, K. C., Harrison, A., Sajid, M., Rosenthal, P. J.,
Brinen, L. S., J. Med. Chem., 52, 852-857 (2009).
118. Sabnis, Y., Rosenthal, P. J., Desai, P., Avery, M. A., J. Biomol. Struct. Dyn.,
19, 765-774 (2002).
119. Sijwali, P. S., Shenai, B. R., Rosenthal, P. J., J. Biol. Chem., 277, 14910-
14915 (2002).
120. Mayer, R., Picard, I., Lawton, P., Grellier, P., Barrault, C., Monsigny, M., J.
Med. Chem., 34, 3029-3035 (1991).
121. Hanne, L., Henrik, F., Majid, S., Mehrnoush, T., Mahboubeh, D., Tehrani, K.,
Henry, H., Dan, S. Jerzy, W., Bioorg. Med. Chem., 12, 119-127 (2004).
122. Schechter, I., Berger, A., Biochem. Biophys. Res. Commun., 27, 157-162
(1967).
123. Powers, J. C., Asgian, J. L., Ekiei, O. D., James, K. E., Chem. Rev., 102, 4639-
4750 (2002).
124. Rosenthal, P. J., McKerrow, J. H., Aikawa, M., Nagasawa, H., Leech, J. H., J.
Clin. Invest., 82, 1560-1566 (1988).
125. Rosenthal, P. J., Wollish, W. S., Palmer, J. T., Rasnick, D., J. Clin. Invest., 88,
1467-1472 (1991).
126. Rosenthal, P. J., Lee, G. K., Smith, R. E., J. Clin. Invest., 91, 1052-1056
(1993).
127. Palmer, J. T., Rasnick, D., Klaus, J. K., Bromme, D., J. Med. Chem., 38, 3193-
3196 (1995).
128. Vicik, R., Busemann, M., Baumann, K., Schirmeister, T., Curr Top Med
Chem, 6, 331-353 (2006).
129. Rosenthal, P. J., Olson, J. E., Lee, G. K., Palmer, J. T., Klaus, J. L., Rasnick,
D., Antimicrob. Agents Chemother., 40, 1600-1603 (1996).
130. Olson, J. E., Lee, G. K., Semenov, A., Rosenthal, P. J., Bioorg. Med. Chem., 7,
633-638 (1999).
131. Shenai, B. R., Lee, B. J., Alvarez-Hernandez, A., Chong. P. Y., Emal, C. D.,
Neitz, R. J., Roush, W. R., Rosenthal. P. J., Antimicrob. Agents Chemother.,
47, 154-160 (2003).
132. Lee, B. J., Singh, A., Chiang, P., Kemp, S. J., Goldman, E. A., Weinhouse, M.

References
160
I., Vlasuk, P., Rosenthal, P. J., Antimicrob. Agents Chemother., 47, 3810-3814
(2003).
133. Barrett, A. J., Rawlings, N. D., Woessner, J. F., Handbook of proteolytic
enzymes. San Diego, CA, Academic Press, pp 631-634 (1998).
134. Rosenthal, P. J., Sijwali, P. S., Singh, A., Shenai, B. R., Curr Pharm Des, 8,
1659-1672 (2002).
135. Singh, A., Shenai, B. R., Choe, Y., Gut, J., Sijwali, P. S., Craik, C. S.,
Rosenthal, P. J., Biochem. J., 368, 273-281 (2002).
136. Hanada, K., Tamai, M., Yamagishi, S., Ohmura, S., Sawada, J., Tanaka, I.,
Agric. Biol.Chem., 42, 523-528 (1978).
137. Hanada, K., Tamai, M., Yamagishi, S., Ohmura, S., Sawada, J., Tanaka, I.,
Agric. Biol.Chem., 42, 529-536 (1978).
138. Inaba, T., Hirayama, Y., Fujinaga, N., Agric. Biol. Chem., 43, 655-656 (1979).
139. Towatari, T., Tanaka, K., Yoshikawa, D., Katunuma, N., J. Biochem., 84, 659-
672 (1978).
140. Sugita, H., Ishiura, S., Suzuki, K., Imahori, K., J. Biochem., 87, 339-341
(1980).
141. Suzuki, K., J. Biochem., 93, 1305-1312 (1983).
142. Roush, W. R., Hernandez, A. A., McKerrow, J. H., Selzer, P. M., Hansell, E.,
Engel, J. C., Tetrahedron, 56, 9747-9762 (2000).
143. Buttle, D. J., Saklatvala, J., Tamai, M., Barrett, A. J., Biochem. J., 281, 175-
177 (1992).
144. Kembhavi, A. A., Buttle, D. J., Knight, C. G., Barrett, A. J., Arch. Biochem.
Biophys., 303, 208-213 (1993).
145. Chen, Z., Potempa, J., Polanowski, A., Wikstrom, M., Travis, J., J. Biol.
Chem., 267, 18896-18901 (1992).
146. Nikawa, T., Towatari, T., Katunuma, N., Eur. J. Biochem., 204, 381-393
(1992).
147. Schulz, F., Gelhaus, C., Degel, B., Vicik, R., Heppner, S., Breuning, A.,
Leippe, M., Gut, J., Rosenthal, P. J., Schirmeister, T., Chem. Med. Chem., 2,
1214-1224 (2007).
148. Roush, W. R., Gonzalez, F. V., McKerrow, J. H., Hansell, E., Bioorg. Med.
Chem. Lett., 8, 2809-2812 (1998).

References
161
149. James, H. K., Asgian, J. L., Li, Z. Z., Ekici, O. D., Rubin, J. R., Mikolajczyk,
J., Salvesen, G. S., Powers, J. C., J. Med. Chem., 47, 1553-1574 (2004).
150. Yabe, Y., Guillaume, D., Rich, D. H., J. Am. Chem. Soc., 110, 4043-4044
(1998).
151. Matsumoto, K., Yamamoto, D., Ohishi, H., Tomoo, K., Ishida, T., Inoue, M.,
Sadatome, T., Kitamura, K., Mizuno, H., FEBS Lett., 245, 177-180 (1989).
152. Greenbaum, D., Medzihradszky, K., Burlingame, A., Bogyo, M., Chem. Biol.,
7, 569-581 (2000).
153. Singh, A., Shenai, B. R., Choe, Y., Gut, J., Sijwali, P. S., Craik, C. S.,
Rosenthal, P. J., Biochem. J., 368, 273-281 (2002).
154. Varughese, K. I., Ahmed, F. R., Carey, P. R., Hasnain, S., Huber, C. P., Storer,
A. C., Biochemistry, 28, 1330-1332 (1989).
155. Varughese, K. I., Su, Y., Cromwell, D., Hasnain, S., Xuong, N. H.,
Biochemistry 31, 5172-5176 (1992).
156. Mladenovic, M., Schirmeister, T., Thiel, S., Thiel, W., Engels, B., Chem. Med.
Chem., 2, 120-128 (2007).
157. Schirmeister, T., Kaeppler, U., Mini Rev. Med. Chem., 3, 361-373 (2003).
158. Martichonok, V., Plouffe, C., Storer, A. C., Menard, R., Jones, J. B., J. Med.
Chem., 38, 3078-3085 (1995).
159. Bucciarelli, M., Forni, A., Moretti, I., Prati, F., Torre, G., Tetrahedron
Asymm., 4, 903-906 (1993).
160. Bucciarelli, M., Forni, A., Moretti, I., Prati, F., Torre, G., J. Chem. Soc.
Perkin.Trans., 1, 3041-3045 (1993).
161. Vicik, R., Helten, H., Schirmeister, T., Engels, B., Chem. Med. Chem., 1,
1021-1028 (2006).
162. Dermer, O. C., Ham, G. E., Ethyleneimine and other aziridines. New York,
Academic Press, 106, pp 65-68, (1969).
163. Schirmeister, T., Biopolymers. 51, 87-97 (1999).
164. Schirmeister, T., J. Med. Chem., 42, 560-572 (1999).
165. Moroder, L., Musiol, H. J., Scharf, R., FEBS Lett., 299, 51-53 (1992).

References
162
166. Schulz, F., Gelhaus, C., Degel, B., Vicik, R., Heppner, S., Breuning, A.,
Leippe, M., Gut, J., Rosenthal, P. J., Schirmeister, T., ChemMedChem, 2,
1214-1224 (2007).
167. Gibbons, P., Verissimo, E., Araujo, N. C., Barton, V., Nixon, G. L., Amewu,
R. K., Chadwick, J., Stocks, P. A., Biagini, G. A., Srivastava, A., Rosenthal,
P. J., Gut, J., Guedes, R. C., Moreira, R., Sharma, R., Berry, N., Lurdes, M.,
Cristiano, S., Shone, A. E., Ward, S. A., ONeil,P. M., J. Med. Chem. 53, 8202-
8206 (2010).
168. Roger, M. F., Debra, S. P., Daniel, F. V., J. Org. Chem., 47, 104-109 (1982).
169. Karl, A. S., William, R., McKerrow, J. H., Paul, M., Elizabeth, H., Rosenthal,
P. J., Bioorg. Med. Chem., 6, 2477-2494 (1998).
170. Machon, U., Buchold, C., Stempka, M., Schirmeister, T., Gelhaus, C.,
Leippe, M., Gut, J., Rosenthal, P. J., Kisker, C., Leyh, M., Schmuck, C., J.
Med. Chem., 52, 5662-5672 (2009).
171. Breuning, A., Degel, B., Schulz, F., Buchold, C., Stempka, M., Machon, U.,
Heppner, S., Gelhaus, C., Leippe, M., Leyh, M., Kisker, C., Rath, J., Stich, A.,
Gut, J., Rosenthal, P. J., Schmuck, C., Schirmeister, T., J. Med. Chem., 53,
1951-1963 (2010).
172. Loser, R., Gut, J., Rosenthal, P. J., Frizler, M., Gutschow, M., Andrews, K. T.,
Bioorg. Med. Chem. Lett. 20, 252-255 (2010).
173. Souers, A. J., Ellman, J. A., Tetrahedron, 57, 7431-7448 (2001).
174. MacDonald, M., Aube, J., Curr. Org. Chem., 5, 417-438 (2001).
175. Eguchi, M., Kahn, M., Mini Rev. Med. Chem., 2, 447-462 (2002).
176. Burgess, K., Acc. Chem. Res., 34, 826-835 (2001).
177. Dziadulewicz, E. K., Brown, M. C., Dunstan, A. R., Lee, W., Said, N. B.,
Garratt, P. J., Bioorg. Med. Chem. Lett., 9, 463-468 (1999).
178. Lauffer, D. J., Mullican, M. D., Bioorg. Med. Chem. Lett., 12, 1225-1227
(2002).
179. Micale, N., Kozikowski, A. P., Ettari, R., Grasso, S., Zappala, M., Jeong, J. J.,
Kumar, A., Hanspal, M., Chishti, A. H., J. Med. Chem., 49, 3064-3067 (2006).
180. Ettari, R., Zappala, M., Micale, N., Schirmeister, T., Gelhaus, C., Leippe, M.,
Evers, A., Grasso, S., Eur. J. Med. Chem., 45, 3228-3233 (2010).
181. Ettari, R., Nizi, E., Di Francesco, M. E., Dude, M. A., Pradel, G., Vicik, R.,

References
163
Schirmeister, T., Micale, N., Grasso, S., Zappala, M., J. Med. Chem., 51, 988-
996 (2008).
182. Ettari, R., Nizi, E., Di Francesco, M. E., Micale, N., Grasso, S., Zappala, M.,
Vicik, R., Schirmeister, T., Chem. Med. Chem., 3, 1030-1033 (2008).
183. Ettari, R., Micale, N., Schirmeister, T., Gelhaus, C., Leippe, M., Nizi, E.,
Francesco, M. E., Grasso, S., Zappala, M., J. Med. Chem., 52, 2157-2160
(2009).
184. Bova, F., Ettari, R., Micale, N., Carnovale, C., Schirmeister, T., Gelhaus, C.,
Leippe,M., Grasso, S., Zappala, M., Bioorg. Med. Chem., 18, 4928-4938
(2010).
185. Verissimo, E., Berry, N., Gibbons, P., Cristiano, M. L. S., Rosenthal, P. J.,
Gut, J., Ward, S. A., O’Neill, P. M., Bioorg. Med. Chem. Lett., 18, 4210-4214
(2008).
186. Shuren, Z., Hudson, T. H., Kyle, D. E. Lin, A. J., J. Med. Chem., 45, 3491-
3496 (2002).
187. Chen, M., Theander, T. G., Christensen, B. S., Hviid, L., Zhai, L., Kharazmi
A., Antimicrob. Agents Chemother., 38, 1470-1475 (1994).
188. Chen, M., Christensen, S. B., Zhai, L., Rasmussen, M. H., Theander, T. G.,
Frokjaer, S., Steffansen, S., Davidsen, J., Kharazmi, A., J. Infect. Dis., 176,
1327-1333 (1997).
189. Bhattacharya, A., Mishra, L. C., Sharma, M., Awasthi, S. K., Bhasin, V. K.,
Eur. J. Med. Chem., 44, 3388-3393 (2009).
190. Li, R., Kenyon, G. L., Cohen, F. E., Chen, X., Gong, B., Dominguez, J. N.,
Davidson, E., Kurzban, G., Miller, R. E, Nuzum, E. O., Rosenthal, P. J.,
McKerrow, J. H. J. Med. Chem., 38, 5031-5037 (1995).
191. Liu, M., Wilairat, P., Go, M. L., J. Med. Chem., 44, 4443-4452 (2001).
192. Dominguez, J. N., Leon, C., Rodrigues, J., Dominguez, N. G., Gut, J.,
Rosenthal, P. J., J. Med. Chem., 48, 3654-3658 (2005).
193. Motta, L. F., Gaudio, A. C., Takahata, Y., Int. Electron. J. Mol. Des., 5, 555-
569 (2006).
194. Hans, R. H., Gut, J., Rosenthal, P. J., Chibale, K., Bioorg. Med. Chem. Lett.,
20, 2234-2237 (2010).
195. Wanare, G., Aher, R., Kawathekar, N., Ranjan, R., Kaushik, N., Sahal, D.,

References
164
Bioorg. Med.Chem. Lett., 20, 4675-4678 (2010).
196. Batra, S., Sabnis, Y. A., Rosenthal, P. J., Avery, M. A., Bioorg. Med. Chem.,
11, 2293-2299 (2003).
197. Sabnis, Y., Rosenthal, P. J., Desai, P., Avery, M. A., J. Biomol. Struct. Dyn.,
19, 765-774 (2002).
198. Klayman, D. L., Bartosevich, J. F., Griffin, T. S., Mason, C. J., Scovill, J. P.,
J. Med. Chem., 22, 855-862 (1979).
199. Chiyanzu, I., Hansell, E., Gut, J., Rosenthal, P. J, McKerrowb, J. H. Chibale,
K., Bioorg. Med. Chem. Lett., 13, 3527-3530 (2003).
200. Greenbaum, D. C., Mackey, Z., Hansell, E., Doyle, P., Gut, J., Caffrey, C. R.,
Lehrman, J., Rosenthal, P. J., McKerrow, J. H., Chibale, K., J. Med. Chem.,
47, 3212-3219 (2004).
201. Chipeleme, A., Gut, J., Rosenthal, P. J., Chibale, K., Bioorg. Med. Chem. 15,
273-282 (2007).
202. Khanye, S. D., Smith, G. S., Lategan, C., Smith, P. J., Gut, J., Rosenthal, P. J.,
Chibale, K., J. Inorg. Biochem. 104, 1079-1083 (2010).
203. Dominguez, J. N., Lopez, S., Charris, J., Iarruso, L., Lobo, G. Semenov, A., J.
Med. Chem., 40, 2726-2732 (1997).
204. Coteron, J. M., J. Med. Chem., 53, 6129-6152 (2010).
205. Lavrado, J., J. Med. Chem., 54, 734-750 (2011).
206. Kumar, S. P., Gut, J., Guedes, R. C., Rosenthal, P. J., Santos, M. M., Moreira,
R., Eur. J. Med. Chem. XXX, 1-7 (2011).
207. Waag, T., Gelhaus, C., Rath, J., Stich, A., Leippe, M., Schirmeister, T.,
Bioorg. Med. Chem. Letts., 20, 5541-5543 (2010).
208. Desai, P. V., Panty, A., Sabnis, Y., Tekwani, B., Gut, J., Rosenthal, P. J.,
Srivastava, A., Mitchell, A., J. Med. Chem., 47, 6609-6615 (2004).
209. Li, H., Huang, J., Chen, L., Liu, X., Chen, T., Zhu, J., Lu, W., Shen, W., Li, J.,
Hilgenfeld, R., Jiang, H., J. Med. Chem., 52, 4936-4940 (2009).
210. Zhu, J.; Chen, T.; Chen, L.; Lu, W.; Che, P.; Huang, J.; Li, H.; Li, J.; Jiang, H.
Molecules, 14, 494-508 (2009).
211. Zhu, J.; Chen, T.; Liu, J.; Ma, R.; Lu, W.; Huang, J.; Li, H.; Li, J.; Jiang, H.
Molecules, 14, 785-797 (2009).

References
165
212. Semenov, A., Olson, J. E., Rosenthal, P. J., Antimicrob. Agents Chemother.,
42, 2254 2258 (1998).
213. Capela, R., Oliveira, R., Goncalves, L. M., Domingos, A., Gut, J., Rosenthal,
P. J., Lopes F, Moreira, R., Bioorg. Med. Chem. Letts. 19, 3229-3232 (2009).
215. Huang, H., Lu, W., Li, X., Cong, X., Ma, H., Liu, X., Zhang, Y., Che, P., Ma,
R., Li, H., Shen, X., Jiang, H., Huang, J., Zhu, J., Biorg. Med. Chem Lett. 15,
958-962 (2012).
216. Lesher, G.Y.; Schodack, N. Y. US Patent, US3907798 (1975).
217. Abass, M.; Ismail, M. M.; Abdel-Monem, W. R.; Mayas, A. S. J. Serb.
Chem. Soc. 75, 11–17 (2010).
218. Lappin, G. R. J . Am. Chem. Soc., 70, 3348 (1948).
219. Gannon, W. F.; Steck, E. A. J. Org. Chem. 27, 4137-4140 (1962).
220. Nishigaki, S.; Ichiba, M.; Shinomura, K.; Yoneda, F. J. Het. Chem., 8, 759-
762 (1971).
221. Shur, M.; Israelstam, S. S. J. Org. Chem. 33, 3015-3020 (1968).
222. Simunek, P. Heterocycles, 75, 2477-2491 (2008).


















