
13C NMR studies of heterocyclic naphtho- and anthraquinones. Deshielding effect on the carbonyl...
4
191 Spectral Assignments and Reference Data Received: 31 May 2007 Revised: 4 October 2007 Accepted: 26 October 2007 Published online in Wiley Interscience: 19 December 2007 (www.interscience.com) DOI 10.1002/mrc.2156 13 C NMR studies of heterocyclic naphtho- and anthraquinones. Deshielding effect on the carbonyl carbon with methyl substitution Cristian Salas, Veronica Armstrong, Claudio L ´ opez and Ricardo A. Tapia ∗ 13 C chemical shift assignment of several methyl substituted heterocyclic naphtho- and anthraquinones, including dihydronaphthofuranquinones, azaanthraquinones, benzopyrroloquinolinediones and benzothiophenoquinolinediones, are described. A deshielding effect due to a methyl group was observed over the neighbouring carbonyl carbon, in every case studied. 13 C assignments were based on 2D experiments. Copyright c 2007 John Wiley & Sons, Ltd. Keywords: 13 C NMR; heterocycles; naphthofuranquinones; azaanthraquinones; benzopyrroloquinones; benzothiophenoquinones; methyl substitution Introduction Heterocyclic 1,4-benzo- and 1,4-naphthoquinones as well as 5,10- anthraquinones containing oxygen and nitrogen atoms have been reported to exhibit a broad range of bioactivities. [1 – 6] Complete 13 C NMR studies have been published for methyl substituted anthraquinones: the deshielding experienced by the carbonyl carbon because of methyl substitution has been reported [7] and explained as reduced contributions of the enolate resonance structure to the overall electronic distribution because of peri interactions hindering co-planarity of the carbonyl group with the aromatic system. [8] Despite these studies, there is no evidence in the literature, according to our knowledge, of this effect being used for structure assignment. Moreover, influence of methyl substitution on chemical shifts ( 13 C) has not been studied, until now, in heterocyclic quinonic compounds. Several 13 C NMR spectra have been described and completely assigned for various oxygen and nitrogen derivatives, [9,10] never- theless, the methyl deshielding effect has received little attention. Although, for cleistopholine (5) the more deshielded carbonyl resonance was attributed to C-10; this could be determined [9] because of an NOE interaction between C-5 and the protons of the C-4 methyl. Amazingly, no similar interactions were evident in the compounds studied by us. Prompted by the interesting biological behaviour of these classes of compounds, we have performed regio- and stereoselec- tive syntheses of heterocyclic azaanthraquinones, and naphthofu- ranquinones derivatives, through Diels – Alder cycloadducts. [11,12] Therefore, we describe in this paper the 13 C NMR assignments for ten methyl substituted heterocyclic quinones. In every case, a downfield shift is observed for one of the carbonyl carbons, when compared to the same heterocycle in the absence of the methyl substitution. These sets of data might serve as reference for future assignments of structurally related substances. Results and Discussion The chemical structures and the numbering system of dihydron- aphthofurandiones (1 – 4) are presented in Fig. 1. 13 C NMR data and assignments for quinones (1 – 4) are listed in Table 1. The effect exerted over the carbonyl shift, by the oxygen atom in the heterocycle is well observed in the unsubstituted analogue (1) and in the symmetrical compound (2) (δ 182.4 and 183.0, respectively for C-4; δ 178.2 and 178.7 for C-9). An influence of the methyl substitution is evidenced by 2.9 ppm deshielding for C-4 carbonyl in the 5-methyl derivative (3) in relation to C-4 in the unsubstituted naphthofuranquinone (1). Moreover, C-9 in the 8-methyl derivative (4) appears 2.4 ppm downfield with respect to C-4 in the same compound (4), in spite of the oxygen influence. The chemical structures and the numbering system of azaan- thraquinones (5 – 13) are presented in Fig. 2. 13 C NMR data and assignments for quinones (6 – 13) are listed in Table 2. For the azaanthraquinones series, the effects observed are quite similar to those previously discussed for dihydronaphthofuran- diones. The influence exerted by the nitrogen atom is evidenced in the unsubstituted compound (6) and the chemical shift of C-5 is downfield related to that of C-10. In the 6-methyl derivative (7), C-5 appears 1.7 ppm downfield with respect to C-5 in (6). Moreover, the chemical shifts of C-5 and C-10 in the 9-methyl derivative (8) are almost the same, in spite of the influence of the nitrogen. The 1-aza-4-methylanthraquinone (cleistopholine, 5) represents a different example, previously characterised. [9] Thus, the carbonyl carbon shift enabled the unambiguous assignment of the methyl position in the three regioisomers (5, 7 and 8). In the case of the more substituted azaanthraquinones (9) and (10), the same chemical shift inversion is observed for C-5 and C-10, as a result of the methyl influence. In the 6-methyl ∗ Correspondence to: Ricardo A. Tapia, Facultad de Química, Pontificia Universi- dad Cat ´ olica de Chile, Casilla 306, Santiago22, Chile. E-mail: [email protected] Facultad de Química, Pontificia Universidad Cat´ olica de Chile, Casilla 306, Santiago22, Chile Magn. Reson. Chem. 2008; 46: 191 – 194 Copyright c 2007 John Wiley & Sons, Ltd.
-
Upload
cristian-salas -
Category
Documents
-
view
216 -
download
1
Transcript of 13C NMR studies of heterocyclic naphtho- and anthraquinones. Deshielding effect on the carbonyl...

19
1
Spectral Assignments and Reference DataReceived: 31 May 2007 Revised: 4 October 2007 Accepted: 26 October 2007 Published online in Wiley Interscience: 19 December 2007
(www.interscience.com) DOI 10.1002/mrc.2156
13C NMR studies of heterocyclic naphtho- andanthraquinones. Deshielding effect on thecarbonyl carbon with methyl substitutionCristian Salas, Veronica Armstrong, Claudio Lopez and Ricardo A. Tapia∗
13C chemical shift assignment of several methyl substituted heterocyclic naphtho- and anthraquinones, includingdihydronaphthofuranquinones, azaanthraquinones, benzopyrroloquinolinediones and benzothiophenoquinolinediones, aredescribed. A deshielding effect due to a methyl group was observed over the neighbouring carbonyl carbon, in every casestudied. 13C assignments were based on 2D experiments. Copyright c© 2007 John Wiley & Sons, Ltd.
Keywords: 13C NMR; heterocycles; naphthofuranquinones; azaanthraquinones; benzopyrroloquinones; benzothiophenoquinones;methyl substitution
Introduction
Heterocyclic 1,4-benzo- and 1,4-naphthoquinones as well as 5,10-anthraquinones containing oxygen and nitrogen atoms have beenreported to exhibit a broad range of bioactivities.[1 – 6]
Complete 13C NMR studies have been published for methylsubstituted anthraquinones: the deshielding experienced bythe carbonyl carbon because of methyl substitution has beenreported[7] and explained as reduced contributions of the enolateresonance structure to the overall electronic distribution becauseof peri interactions hindering co-planarity of the carbonyl groupwith the aromatic system.[8] Despite these studies, there is noevidence in the literature, according to our knowledge, of thiseffect being used for structure assignment.
Moreover, influence of methyl substitution on chemical shifts(13C) has not been studied, until now, in heterocyclic quinoniccompounds.
Several 13C NMR spectra have been described and completelyassigned for various oxygen and nitrogen derivatives,[9,10] never-theless, the methyl deshielding effect has received little attention.Although, for cleistopholine (5) the more deshielded carbonylresonance was attributed to C-10; this could be determined[9]
because of an NOE interaction between C-5 and the protons of theC-4 methyl. Amazingly, no similar interactions were evident in thecompounds studied by us.
Prompted by the interesting biological behaviour of theseclasses of compounds, we have performed regio- and stereoselec-tive syntheses of heterocyclic azaanthraquinones, and naphthofu-ranquinones derivatives, through Diels–Alder cycloadducts.[11,12]
Therefore, we describe in this paper the 13C NMR assignmentsfor ten methyl substituted heterocyclic quinones. In every case, adownfield shift is observed for one of the carbonyl carbons, whencompared to the same heterocycle in the absence of the methylsubstitution. These sets of data might serve as reference for futureassignments of structurally related substances.
Results and Discussion
The chemical structures and the numbering system of dihydron-aphthofurandiones (1–4) are presented in Fig. 1. 13C NMR dataand assignments for quinones (1–4) are listed in Table 1.
The effect exerted over the carbonyl shift, by the oxygen atomin the heterocycle is well observed in the unsubstituted analogue(1) and in the symmetrical compound (2) (δ 182.4 and 183.0,respectively for C-4; δ 178.2 and 178.7 for C-9). An influence ofthe methyl substitution is evidenced by 2.9 ppm deshielding forC-4 carbonyl in the 5-methyl derivative (3) in relation to C-4 inthe unsubstituted naphthofuranquinone (1). Moreover, C-9 in the8-methyl derivative (4) appears 2.4 ppm downfield with respect toC-4 in the same compound (4), in spite of the oxygen influence.
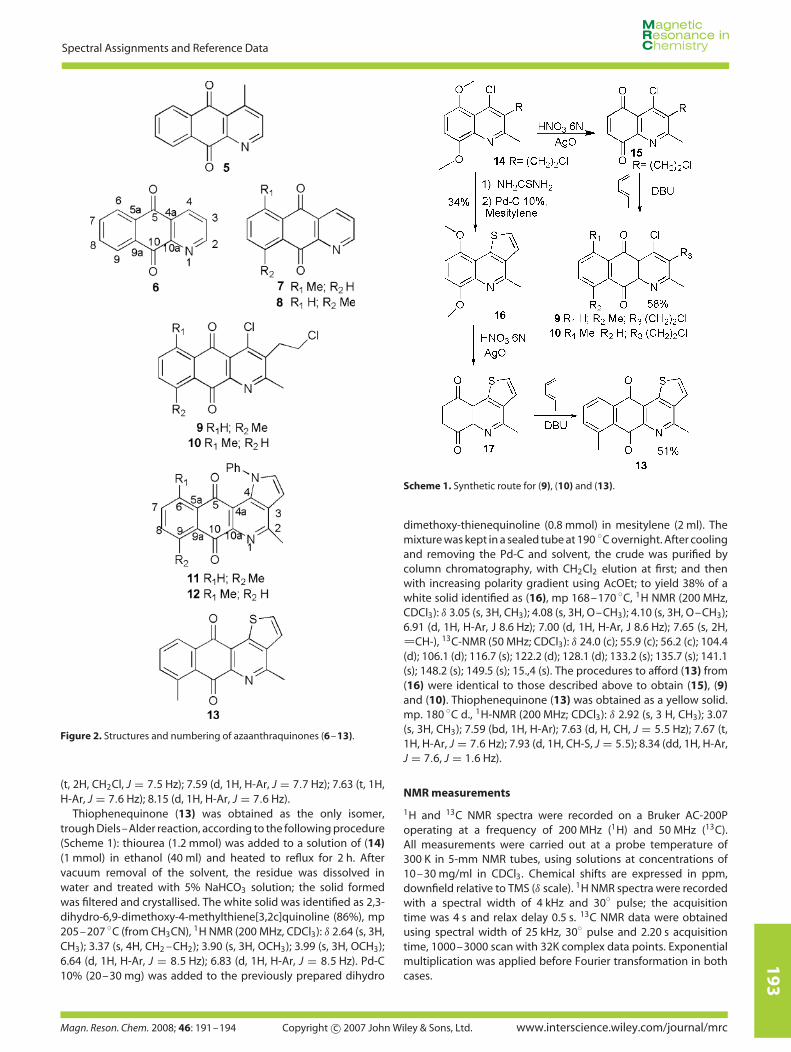
The chemical structures and the numbering system of azaan-thraquinones (5–13) are presented in Fig. 2. 13C NMR data andassignments for quinones (6–13) are listed in Table 2.
For the azaanthraquinones series, the effects observed are quitesimilar to those previously discussed for dihydronaphthofuran-diones. The influence exerted by the nitrogen atom is evidencedin the unsubstituted compound (6) and the chemical shift of C-5 isdownfield related to that of C-10. In the 6-methyl derivative (7), C-5appears 1.7 ppm downfield with respect to C-5 in (6). Moreover,the chemical shifts of C-5 and C-10 in the 9-methyl derivative (8)are almost the same, in spite of the influence of the nitrogen.The 1-aza-4-methylanthraquinone (cleistopholine, 5) represents adifferent example, previously characterised.[9] Thus, the carbonylcarbon shift enabled the unambiguous assignment of the methylposition in the three regioisomers (5, 7 and 8).
In the case of the more substituted azaanthraquinones (9)and (10), the same chemical shift inversion is observed for C-5and C-10, as a result of the methyl influence. In the 6-methyl
∗ Correspondence to: Ricardo A. Tapia, Facultad de Química, Pontificia Universi-dad Catolica de Chile, Casilla 306, Santiago22, Chile. E-mail: [email protected]
Facultad de Química, Pontificia Universidad Catolica de Chile, Casilla 306,Santiago22, Chile
Magn. Reson. Chem. 2008; 46: 191–194 Copyright c© 2007 John Wiley & Sons, Ltd.

19
2
Cristian Salas et al
Figure 1. Structures and numbering of dihydronaphthofurandiones (1–4).
Table 1. 13C NMR chemical shifts (δ, ppm) of dihydronaphthofuran-diones (1–4)a
C 1 2 3 4
2 91.8 91.8 91.6 91.7
3 40.0 40.1 40.5 39.9
3a 123.4 122.9 124.8 121.5
4 182.4 183.0 185.3 180.14a 131.5 131.6 133.2 128.9
5 125.9 127.2 140.8 124.8
6 134.1 143.9 138.9 133.2
7 132.8 142.3 131.9 137.0
8 126.2 127.4 125.4 141.7
8a 133.0 129.5 130.2 134.8
9 178.2 178.7 178.5 182.59a 158.7 158.9 157.4 159.7
10 28.3 28.4 28.4 29.7
11 28.3 28.4 28.4 29.7
a Signal assignments based on 2D experiments.
derivative (10), C-5 is 2.2 ppm downfield with respect to C-10 inthe same compound, which is consistent with the effect exertedby the nitrogen atom, in spite of possible influence from otherheteroatoms present. The chemical shifts of C-5 and C-10 (δ 182.2and 182.6, respectively) in the 9-methyl derivative (9) are almostthe same, reproducing the same pattern observed between (7)and ( 8).
The methyl-deshielding effect is observed even in more complexheterocyclic quinones, such as pyrrole and thiophene derivatives(11–13). The chemical shift of C-5 in the 6-methyl derivativeof benzopyrroloquinolinedione (12) is 2.2 ppm downfield withrespect to that of C-10 in the same compound, which isconsistent with the effect exerted by the nitrogen of thequinolinedione system. The chemical shift of C-10 in the 9-methyl derivative (11) is 1.0 ppm downfield with respect to thatof C-5 in the same compound; the more deshielded carbonylresonance corresponding to the opposite of what is expectedfrom the quinolinedione nitrogen influence. The same behaviouris exhibited by the carbonyl carbons of (13), where C-10 appears3.3 ppm downfield with respect to C-5 in the same compound.
From preliminary DFT calculations performed with the GAUS-SIAN 03 package programmes, at the B3LYP level of theory with the6–31G basis set, geometries have been optimised, confirming thatthe minimum energy conformations of some of these compoundsare flat. Results of NBO calculations and electrostatic potentialsuggest that the presence of methyl displaces the electronic den-sity towards the carbonyl carbon in the opposite position, thusdeshielding the carbonyl in ortho position.
Experimental
Compounds
The syntheses of dihydronaphthofurandiones (1–4) were per-formed through Diels–Alder reactions as described previously.[11]
The azaanthraquinones (6–8) were obtained by reaction ofquinoline-5,8-diones with the appropriate diene following aknown procedure[13]; spectral characteristics for all three com-pounds are in full accordance with those previously described[13];nevertheless, 13C NMR data has not been reported before. Adetailed experimental procedure for the preparation of benzopy-rroloquinolinediones (11) and (12) has been presented recently.[12]
Azaanthraquinones (9) and (10) were prepared through thefollowing procedure (Scheme 1): AgO (180 mg) and HNO3 (6 N,530 µl) were added to a solution of 4-chloro-3-(2-chloroethyl)-5,8-dimethoxy-2-methylquinoline (14)[12] (0.3 mmol in 15 ml CH3CN).After 5 min stirring at room temperature, water (four times theinitial volume) was poured into the reaction mixture. Extractionwith CHCl3 and drying with sulphate were carried out on the crudesolution to which piperylene (penta-1,3-diene, 20 µl) was added.The mixture was kept overnight in a sealed tube at 90 ◦C. Aftercooling, 1,8-diazabicycloundec-7-ene (5 drops) was added and themixture was stirred at room temperature for 4 hours. The solventwas removed and the regioisomers were separated. A yellowcrystalline solid was identified as (9) (36% yield), mp147–149 ◦C,1H NMR (200 MHz, CDCl3): δ 2.84 (s, 3H, CH3); 2.09 (s, 3H, CH3); 3.49(t, 2H, ArCH2, J = 7.5 Hz); 3.80 (t, 2H, CH2Cl, J = 7.5 Hz); 7.59 (d,1H, H-Ar, J = 7.7 Hz); 7.67 (t, 1H, H-Ar, J = 7.7 Hz); 8.15 (d, 1H,H-Ar, J = 7.7 Hz). Isomer (10) was isolated in 22% yield as a yellowcrystalline solid, mp 145–147 ◦C, 1H NMR (200 MHz, CDCl3): δ 2.77(s, 3H, CH3); 2.88 (s, 3H, CH3); 3.47 (t, 2H, ArCH2, J = 7.5 Hz); 3.79
Table 2. 13C NMR chemical shifts (δ, ppm) of azaanthraquinones(6–13)a
C 6 7 8 9 10 11 12 13
2 155.1 154.7 155.1 144.3 144.1 158.7 158.2 159.9
3 127.9 127.9 127.5 136.2 136.6 129.8 130.0 139.4
4 135.5 135.0 135.2 164.4 163.9 117.5 120.6 144.4
4a 130.6 131.6 127.9 124.8 127.6 135.3 134.5 125.7
5 182.6 184.3 183.2 182.2 184.0 183.6 185.4 182.65a 132.7 130.5 133.3 135.5 132.3 135.8 135.4 135.1
6 127.3 142.3 126.0 126.1 141.2 133.1 139.9 127.2
7 134.6 138.6 139.7 138.5 138.5 125.4 137.6 134.0
8 134.8 133.6 138.7 137.9 133.0 137.9 132.9 138.7
9 127.9 126.8 142.9 142.1 125.9 142.2 126.3 142.5
9a 133.5 134.9 127.9 130.2 134.0 130.9 132.8 130.5
10 181.5 182.1 183.3 182.6 181.8 184.6 183.2 185.9
a Signal assignments based on 2D experiments.
www.interscience.wiley.com/journal/mrc Copyright c© 2007 John Wiley & Sons, Ltd. Magn. Reson. Chem. 2008; 46: 191–194
19
3
Spectral Assignments and Reference Data
Figure 2. Structures and numbering of azaanthraquinones (6–13).
(t, 2H, CH2Cl, J = 7.5 Hz); 7.59 (d, 1H, H-Ar, J = 7.7 Hz); 7.63 (t, 1H,H-Ar, J = 7.6 Hz); 8.15 (d, 1H, H-Ar, J = 7.6 Hz).
Thiophenequinone (13) was obtained as the only isomer,trough Diels–Alder reaction, according to the following procedure(Scheme 1): thiourea (1.2 mmol) was added to a solution of (14)(1 mmol) in ethanol (40 ml) and heated to reflux for 2 h. Aftervacuum removal of the solvent, the residue was dissolved inwater and treated with 5% NaHCO3 solution; the solid formedwas filtered and crystallised. The white solid was identified as 2,3-dihydro-6,9-dimethoxy-4-methylthiene[3,2c]quinoline (86%), mp205–207 ◦C (from CH3CN), 1H NMR (200 MHz, CDCl3): δ 2.64 (s, 3H,CH3); 3.37 (s, 4H, CH2 –CH2); 3.90 (s, 3H, OCH3); 3.99 (s, 3H, OCH3);6.64 (d, 1H, H-Ar, J = 8.5 Hz); 6.83 (d, 1H, H-Ar, J = 8.5 Hz). Pd-C10% (20–30 mg) was added to the previously prepared dihydro
Scheme 1. Synthetic route for (9), (10) and (13).
dimethoxy-thienequinoline (0.8 mmol) in mesitylene (2 ml). Themixture was kept in a sealed tube at 190 ◦C overnight. After coolingand removing the Pd-C and solvent, the crude was purified bycolumn chromatography, with CH2Cl2 elution at first; and thenwith increasing polarity gradient using AcOEt; to yield 38% of awhite solid identified as (16), mp 168–170 ◦C, 1H NMR (200 MHz,CDCl3): δ 3.05 (s, 3H, CH3); 4.08 (s, 3H, O–CH3); 4.10 (s, 3H, O–CH3);6.91 (d, 1H, H-Ar, J 8.6 Hz); 7.00 (d, 1H, H-Ar, J 8.6 Hz); 7.65 (s, 2H,
CH-), 13C-NMR (50 MHz; CDCl3): δ 24.0 (c); 55.9 (c); 56.2 (c); 104.4(d); 106.1 (d); 116.7 (s); 122.2 (d); 128.1 (d); 133.2 (s); 135.7 (s); 141.1(s); 148.2 (s); 149.5 (s); 15.,4 (s). The procedures to afford (13) from(16) were identical to those described above to obtain (15), (9)and (10). Thiophenequinone (13) was obtained as a yellow solid.mp. 180 ◦C d., 1H-NMR (200 MHz; CDCl3): δ 2.92 (s, 3 H, CH3); 3.07(s, 3H, CH3); 7.59 (bd, 1H, H-Ar); 7.63 (d, H, CH, J = 5.5 Hz); 7.67 (t,1H, H-Ar, J = 7.6 Hz); 7.93 (d, 1H, CH-S, J = 5.5); 8.34 (dd, 1H, H-Ar,J = 7.6, J = 1.6 Hz).
NMR measurements
1H and 13C NMR spectra were recorded on a Bruker AC-200Poperating at a frequency of 200 MHz (1H) and 50 MHz (13C).All measurements were carried out at a probe temperature of300 K in 5-mm NMR tubes, using solutions at concentrations of10–30 mg/ml in CDCl3. Chemical shifts are expressed in ppm,downfield relative to TMS (δ scale). 1H NMR spectra were recordedwith a spectral width of 4 kHz and 30◦ pulse; the acquisitiontime was 4 s and relax delay 0.5 s. 13C NMR data were obtainedusing spectral width of 25 kHz, 30◦ pulse and 2.20 s acquisitiontime, 1000–3000 scan with 32K complex data points. Exponentialmultiplication was applied before Fourier transformation in bothcases.
Magn. Reson. Chem. 2008; 46: 191–194 Copyright c© 2007 John Wiley & Sons, Ltd. www.interscience.wiley.com/journal/mrc

19
4
Cristian Salas et al
Carbon multiplicity was established by a DEPT pulse sequenceand signals were assigned based on 2D experiments. All two-dimension spectra were acquired with a Bruker AVANCE 400Spectrometer equipped with a Bruker inverse 5-mm Z gradientprobe. The HMBC spectra were obtained using the inv4gplrndqfpulse sequence in the Bruker software. The spectra widths were5340 Hz (F2) and 30000 Hz (F1) and the delay D2 and D6 were 3.45and 65 ms, respectively. Data were processed using sine windowin F2 and F1.
Acknowledgements
Authors are grateful to FONDECYT (Research Grant 1020874) forfinancial support.
References
[1] Berdy J, Aszalos A, Bostian M, Mc Nitt KL. Handbook of AntibioticCompounds. CRC Press: Boca Raton, Florida, USA, 1980.
[2] (a) Ribeiro-Rodrigues R, dos Santos WG, Oliveira AB, Snieckus V,Zani CL, Romanha AJ. Bioorg. Med. Chem. Lett. 1995; 5: 1509;(b) Morello A, Pavani M, Garbarino JA, Chamy MC, Frey C, Mancilla J,Guerrero A, Repetto Y, Ferreira J. Comp. Biochem. Physiol. C 1995;112: 119.
[3] Moldeen SVK, Houghton PJ, Rock P, Croft SL, Aboagye-Nyame F.Planta Med. 1999; 65: 536.
[4] Gafner S, Wolfender JC, Nianga M, Stoecki-Evans H, Hostettmann K.Phytochemistry 1996; 5: 1315.
[5] Tapia RA, Prieto Y, Pautet F, Walchshofer N, Fillion H, Fenet B,Sarciron ME. Bioorg. Med. Chem. 2003; 11: 3407.
[6] (a) Tomasz M, Palom Y. Pharmacol. Ther. 1997; 76: 73; (b) Lien JC,Huang LJ, Wang JP, Teng CM, Lee KH, Kuo SC. Bioorg. Med. Chem.1997; 5: 2111; (c) Ryu CK, Song EH, Shim JY, You HJ, Choi KU, Choi IH,Lee EY, Chae MJ. Bioorg. Med. Chem. Lett. 2003; 13: 17.
[7] Bowden BF, Cameron DW, Crossley MJ, Feutrill GI, Griffiths PG,Kelly DP. Aust. J. Chem. 1979; 32: 769.
[8] Stothers JB. Carbon -13 N.M.R. Spectroscopy. Academic Press: NewYork, 1972; 284.
[9] (a) Waterman PG, Muhammad I. Phytochemistry 1985; 24: 523;(b) Tadic D, Cassels BK, Leboeuf M, Cave A. Phytochemistry 1987;26: 537; (c) Seldel V, Baflleul F, Waterman PG. J. Nat. Prod. 2000; 63:6; (d) Chaves MH, Santos LdeA, Lago JHG, Roque NF. J. Nat. Prod.2001; 64: 240.
[10] (a) Rebstock AS, Mongin F, Trecourt F, Queguiner G. Org. Biomol.Chem. 2004; 2: 291; (b) Liebeskind LS, Zhang J. J. Org. Chem. 1991;56: 6379.
[11] Tapia RA, Salas C, Morello A, Maya JD, Toro-Labbe A. Bioorg. Med.Chem. 2004; 12: 2451.
[12] Tapia RA, Lopez C, Morello A, Maya JD, Valderrama JA. Synthesis2005; 6: 903.
[13] Ohgaki E, Motoyoshiya J, Narita S, Kakurai T, Hayashi S, Hirakawa K.J. Chem. Soc., Perkin Trans. 1 1990; 3109.
www.interscience.wiley.com/journal/mrc Copyright c© 2007 John Wiley & Sons, Ltd. Magn. Reson. Chem. 2008; 46: 191–194






![2. Materials and Methods - Oswaldo Cruz Foundation · Echinacea purpurea showed activity against Leishmania enriettii [] and the presence of these ... activity for two anthraquinones](https://static.fdocuments.in/doc/165x107/5c4620cd93f3c34c50618a11/2-materials-and-methods-oswaldo-cruz-foundation-echinacea-purpurea-showed.jpg)



![Synthesis of chemiluminescent biotinyl naphtho[1 ,8-de][ 1 ...nopr.niscair.res.in/bitstream/123456789/30748/1/IJCB 45B(11) 2512-2522.pdfIndian Journal of Chemistry Vol. 45B, November](https://static.fdocuments.in/doc/165x107/5e9ef478b9e87e5f7d7e4aac/synthesis-of-chemiluminescent-biotinyl-naphtho1-8-de-1-nopr-45b11-2512-2522pdf.jpg)







