
1.1 Synthesis of Oligonucleotides 1.1.1...
36
Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques 1.1 Synthesis of Oligonucleotides 1.1.1 INTRODUCTION Deoxyribonucleic acid (DNA) is a nucleic acid that encodes entire hereditary information and controls the growth and division of cells of all known living organisms and some viruses. 1,2 It contains the information needed to construct other components of cell such as RNA molecules and proteins. DNA was first isolated in 1869 from the nuclei of white blood cells. As this material was found in the nucleus and was acidic, it was called nucleic acid. Eventually, scientist found that nuclei of all cells contain DNA and it acts as carrier of genetic information. In 1953, James Watson and Francis Crick described the three dimensional structure of DNA - the famed Double helix, for which they were awarded 1962 Nobel price in medicine. 3 This led to many important developments in the field of DNA. One such important milestone which received great deal of interest was chemical synthesis of DNA or oligonucleotide. The intension of artificially synthesizing DNA by H. G. Khorana and coworkers in 1967 raised many doubts regarding practical applications of such artificial DNA. However, the numerous uses developed for synthetic DNA in recent years have dispelled all such doubts and the ability to synthesize DNA fragments rapidly has become an important asset in many laboratories. This chapter gives a brief account of different structural aspects of DNA, chemical strategies used for synthesis of oligonucleotides, and spin labeling techniques. 1
Transcript of 1.1 Synthesis of Oligonucleotides 1.1.1...

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
1.1 Synthesis of Oligonucleotides
1.1.1 INTRODUCTION
Deoxyribonucleic acid (DNA) is a nucleic acid that encodes entire hereditary
information and controls the growth and division of cells of all known living
organisms and some viruses.1,2 It contains the information needed to construct other
components of cell such as RNA molecules and proteins. DNA was first isolated in
1869 from the nuclei of white blood cells. As this material was found in the nucleus
and was acidic, it was called nucleic acid. Eventually, scientist found that nuclei of
all cells contain DNA and it acts as carrier of genetic information. In 1953, James
Watson and Francis Crick described the three dimensional structure of DNA - the
famed Double helix, for which they were awarded 1962 Nobel price in medicine.3
This led to many important developments in the field of DNA. One such important
milestone which received great deal of interest was chemical synthesis of DNA or
oligonucleotide. The intension of artificially synthesizing DNA by H. G. Khorana
and coworkers in 1967 raised many doubts regarding practical applications of such
artificial DNA. However, the numerous uses developed for synthetic DNA in recent
years have dispelled all such doubts and the ability to synthesize DNA fragments
rapidly has become an important asset in many laboratories. This chapter gives a
brief account of different structural aspects of DNA, chemical strategies used for
synthesis of oligonucleotides, and spin labeling techniques.
1

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
1.I.2 Structure of DNA
Primary Structure of DNA
Chemically DNA consists of two long polymeric strands of simple units called
nucleotides. Each nucleotide in turn is made up of nucleoside and phosphate
molecule. Nucleoside further consists of a pentose sugar attached to nitrogen atom
of heterocyclic molecules called as nitrogen bases. The pentose sugar observed in
DNA is 2-deoxy-D-ribose, which discriminates it from another type of nucleic acid,
ribonucleic acid (RNA) containing D-ribose sugar. The nitrogen bases observed in
DNA are either substituted purines (adenine and guanine) or pyrimidines (thymine
and cytosine) (Figure 1.1). In case of RNA thymine is replaced by uracil. The
nitrogen bases are bonded to the anomeric carbon of sugar via β-glycosidic linkage
forming the corresponding nucleoside (Figure 1.2). Nucleotides are the phosphate
ester of nucleosides (Figure 1.3).2
NH
N
N
N
NH2
NH
N
N
NH
NH2
O
NH
N
O
NH2
NH
NH
O
OCH3
NH
NH
O
O
adenine guanine cytosine thymine uracil
1
2
34
5 67
8
9 1
2
345
6
Figure 1.1
2

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
N
N
O
OH
OHN
N
NH2
N
N
O
OH
OHN
NH
O
NH2N
N
O
OH
OH
NH2
O N
NH
O
OH
OH
O
O
CH3
2' - deoxyadenosine 2' - deoxyguanosine 2' - deoxycytosine thymidine
1'
2'3'
4'5'
Figure 1.2
N
N
O
OH
ON
N
NH2
PO
O
O
N
N
O
OH
ON
NH
O
NH2PO
O
O
N
N
O
OH
O
NH2
OPO
O
ON
NH
O
OH
O
O
O
CH3
PO
O
O
2' - deoxyadenosine- 5' - monophosphate (dA)
2' - deoxyguanosine- 5' - monophosphate (dG)
2' - deoxycytosine- 5' - monophosphate (dC)
thymidine- 5' - monophosphate (dT)
--
--
--
--
Figure 1.3
Two nucleotide units attached to each other forms a dinucleotide. An
oligonucleotide consists of three to ten nucleotide subunits and polynucleotide
contains many nucleotide subunits. A single strand of DNA is a polynucleotide
consisting of either of four nucleosides attached to each other via phosphodiester
linkage connecting 5'-hydroxyl group of one nucleotide to 3'-hydroxyl group of
another nucleoside. This forms the primary structure of DNA, uniqueness of which
solely resides in sequence of nitrogen bases. The nitrogen bases in DNA are capable
of existing in tautomeric forms (either amino - imine or keto - enol or both) which
are alternative structures differing in the location of hydrogen atom. It has been
established by UV, NMR and IR studies that all nitrogen bases exist
overwhelmingly (99.99 %) in amino - and keto - tautomeric forms at physiological
pH. The primary structure of oligonucleotide containing four nucleotides with
3

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
nitrogen bases adenine (A), thymine (T), guanine (G), cytosine (C) can be shown as
in (Figure 1.4).
N
N
O
O
NH2
O
N
N
O
O
N
NH
O
NH2
N
NH
O
O
O
O
CH3
N
N
O
O
ON
N
NH2
PO
PO
OO
PO O
O
PO
O
O
OO -
-
-
-
3'
5'
A
T
G
C
Figure 1.4
Watson and Crick Model of DNA
James D. Watson and Francis Crick who, using x-ray diffraction data collected by
Rosalind Franklin, proposed the double helix or spiral staircase structure of the
DNA molecule in 1953. According to Watson and Crick, the DNA molecule consist
of two very long single polynucleotide strands wrapped around each other in the
form of a double helix.3 The width of the double stranded helix is 20 Å. Each turn of
helix is 34 Å and contains 10 nucleotides pairs. The twisting of strands results in
4

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
formation of deep and shallow spiral grooves. The double helix structure is
analogous to a coiled ladder, outsides of which are formed by the pentose-phosphate
backbones. The purine and pyrimidine bases of each strand are facing inward
towards each other and interacting thus forming the rungs of the ladder and holding
the strands together. Each base on one chain interacts with complementary base on
the second chain via hydrogen bonds. This interaction was found to be specific as
adenine (A) interacts with thymine (T) via two hydrogen bonds and vice versa.
Whereas, guanine (G) interacts with cytosine (C) via three hydrogen bonds and vice
versa (Figure 1.5).
Figure 1.5
5

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
Different Conformers of DNA
The DNA duplex can exist in three major conformations namely A-DNA, B-DNA
and Z-DNA depending upon the sequence and the environment (Figure 1.6).4,5 The
A and B - DNA are both right-handed helices, whereas Z-DNA is a left handed
helix. The A-helix is the predominant form in nonpolar solvents, while B-helix is
the predominant form in aqueous solution. Nearly all the DNA in living organisms
is in B-helix form. Z-DNA is observed in regions where there is high content of G-C
base pairs. The A-helix is shorter (for a given number of base pairs) and about 3%
broader than B-helix, which is shorter and broader than Z-helix. A-DNA has 11 base
pairs per turn with rise (distance between adjacent base pairs) of 2.3 Å. B-DNA has
10 base pairs per turn and rise of 3.4 Å. Z-DNA has 12 base pairs per turn with rise
of 3.8 Å.
Figure 1.6
6

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
In the late 1950s and early 1960s it was shown that double stranded DNA or
RNA containing purines (Pu) in one strand and pyrimidines (Py) in another, forms
triple stranded structure containing either one polypurine strand and two
polypyrimidine strands (Py*Pu-Py) or one polypyrimidine strand and two
polypurine strands (Pu*Py-Pu). Watson-Crick interaction between two strands of
duplex is represented by (-) and the interaction between duplex and third strand
called as Hoogstein H-bonding is represented by (*) (Figure 1.7). The
oligonucleotides that invade dulplex DNA and form triple helix are termed as
Triplex Forming Oligonucleotides (TFOs).6
N
NN
N
NH
HOH
R
N
NN
N
R NH
H
H
O NN
O R
CH3NH
H
C - G * G
G
G
C
N
NN
N
NH
R
H
H
N
NN
N
R NH
H
H
O NN
O R
CH3NH
H
C - G * A
A
G
C
N
N
R
OO
CH3
H
NNN
N
R
NH
H
NN
O
H
O R
CH3
T - A * T
T
A T
N
N
N
N
NH
H
R
NNN
N
R
NH
H
NN
O
H
O R
CH3
T - A * A
A T
T
Figure 1.7
7

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
1.I.3 Chemical Synthesis of Oligonucleotides
Discovery of DNA, understanding of its structure and functions was followed by its
applications in variety of areas such as site-directed mutagenesis, recombinant DNA
technology, cloning, chip based DNA-microarrays, PCR technologies, etc. This led
to the exponential increase in the demand for synthetic oligonucleotides. Most of the
applications mentioned above require smaller quantities (< 1 μg) of synthetic
oligonucleotides. However, much larger quantities of highly pure DNA are required
for structural studies involving NMR-spectroscopy or X-ray crystallography.7
Oligonucleotides can be synthesized via solid phase synthesis or classical
solution phase synthesis. Solid phase synthetic route is useful, practicable, and
reasonably economical for preparation of oligonucleotides up to about 50 mg.
Beyond this amount, classical solution phase synthesis is more appropriate but it is
relatively laborious. Isolation of pure oligonucleotides in both the routes is highly
time consuming.
The key step in DNA synthesis is the specific and sequential formation of
phosphate linkage between 3' - O of one nucleoside and 5' - O of another nucleoside.
Since a deoxyribonucleoside monomer contains two hydroxyl groups (primary
hydroxyl at 5' and secondary hydroxyl at 3' position) one must be chemically
protected, while the other is specifically phosphorylated or phosphitylated and then
coupled to the next deoxyribonucleoside unit. In the process other reactive
functional groups present must be protected. Various strategies developed for the
synthesis of oligonucleotides involve two types of protecting groups.
• Permanent protecting groups, which remain attached to the oligonucleotide
throughout the synthesis and are removed at the end of the synthesis.
8

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
• Temporary protecting groups, which are used to obtain specificity in a single
reaction and are removed immediately.
Permanent protection is required for the heterocyclic bases in the nucleosides
during DNA synthesis. The exocyclic amino group needs to be protected due to its
reactive nature. The most commonly used amino protecting group is benzoyl for
adenine and cytosine and isobutyryl for guanine.8 Thymine usually requires no
protecting group. Recently, it was observed that during extended synthesis side
reactions take place at 6 - O position of guanine. Thus various protecting groups are
proposed for 6 - O position of guanine such as, phosphinothioyl9, 2-nitrophenyl10, β-
cyanoethyl11, nitrophenylethyl11, etc.
Most of the synthesis strategies for oligonucleotides involve selective
phosphorylation or phosphitylation of 3' - OH group thus 5' - OH group needs to be
protected temporarily. The 4,4'-dimethoxytrityl chloride (DMT-Cl) remains the best
choice for this purpose.12 Due to the bulky nature of DMT- group, it can be
introduced selectively at 5'-position of sugar unit and can be removed easily under
very mild acidic conditions as strong acidic conditions results in depurination.
Further efficiency of deprotection step can be monitored easily by
spectrophotometer due to strong visible absorption of the cationic species produced
upon detritylation.
The phosphate group needs to be permanently protected as it may compete
with sugar hydroxyl group during chain extension in oligonucleotide synthesis. Thus
development of permanent phosphate protecting groups selectively removable at the
end of the synthesis has received a lot of importance. Earlier chlorophenyl13 and
methyl14 groups were used as phosphate protecting groups. Chlorophenyl group can
be cleaved selectively by use of mild nucleophile, an oximate anion and the methyl
9

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
group by the thiophenate anion. Recently these protecting groups were replaced by
β-cyanoethyl group15, which can be easily removed by β-elimination with mild
ammonia solution. The other phosphate protecting groups such as (9-
fluorenyl)methyl16, 2,2,2-trichloroethyl17, 2,2,2-trichloro-1,1-dimethylethyl18.19,
2,2,2-tribromoethyl20, 2-(p-nitrophenyl)ethyl21, 2-(2-pyridyl)ethyl22, 2-
(methylsulfonyl)ethyl23, 2-(phenylsulfonyl)ethyl24, o-nitrobenzyl25, o-(t-
butyl)phenyl26, o-or p-chlorophenyl27, 8-quinolyl28 and allyl29 have been found to be
functional in place of β-cyanoethyl group.
Michelson and Todd Synthesis
The first chemical synthesis of dinucleotide phosphate ([d(TpT)] thymidylyl-
(3'→5')-thymidine) and a dinucleotide ([d(pTpT)] 5'-O-phosphorylthymidylyl-
(3'→5') thymidine) with natural 3' to 5' internucleotide linkages was reported by
Michelson and Todd in 1955.30 It involves phosphorylation of 3'-O-acetylthymidine
with 3'-O-phosphorochloridate of 5'-O-acetylthymidine (Scheme 1.1). Phosphate
functionality was protected by benzyl group. However the approach did not received
much attention and no further work was carried out on this approach.
10

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
O
O
TRO
POOBn
Cl
O
AcO
TOH
O
O
AcO
TOPO
RO T
O OBn
O
O
OH
TOPO
R'O T
O O
OPOOBn
OBn
OPOOH
O
2,6-lutidineMeCN
Deprotection
-
+
R = -OAc or
R' = -H or-
i. H2SO4, EtOH, Waterii. Ba(OH)2, H2O
Scheme 1.1 Michelson and Todd synthesis
The Phosphodiester Approach
The phosphodiester approach for oligonucleotide synthesis was introduced by H. G.
Khorana in 1956. It involved the condensation of appropriate nucleoside with
nucleotide derivative in presence of dicyclohexylcarbodiimide (DCC) or
arylsulfonyl chloride as the activating agent.31 The approach was simple and
dominated the field of oligonucleotide synthesis for almost 20 years. However it
suffers from disadvantage offered by the presence of anionic phosphodiester
11

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
function in the backbone of DNA fragment used in the synthesis at which side
reaction occurs, leading to complications in isolation of products. Moreover it
requires large excess of nucleoside reagents and coupling yields are usually low.
Thus the approach is now only of historical and developmental interest.
O
OH
B1RO O
AcO
B2OPOH
O
O
O
O
AcO
B2OPO
RO B1
O O
O
O
OH
B2OPO
OH B1
O O
Coupling agent
Deprotection
-
--
B1 and B2 are A Bz, C Bz, C Ibu or T
Coupling agent is DCC or triisopropylbenzenesulfonyl chloride or mesitylsulfonyl chloride
+
Scheme 1.2 Phosphodiester Approach
The Phosphotriester Approach
The phosphotriester approach involves protection of the anionic site in the
phosphate backbone during oligonucleotide synthesis.32 In this approach, a
phosphotriester is generated by condensation of phosphotriester intermediate of one
12

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
nucleoside with another protected nucleoside or oligonucleotide. The 5'- O and 3'- O
protecting groups can be selectively removed to extend the synthesis in either
direction. After oligonucleotide chain has formed, all the P - O protecting groups are
removed in one step.
O B1RO
OH
PO
ClCl
OR'O B1RO
OP OR'OO
O B2OH
R''O
O
O B1RO
OP OR'OO B2
R''O
O
O B1OH
OP OOO B2
OH
i)
ii) Hydrolysis
Base
Methylsulfonylchloride / base
Deprotection
R' is 2-cyanoethyl, phenyl or o-chlorophenyl
B1 and B2 are A Bz, C Bz, C Ibu or TR is trityl or substituted trityl
-
Scheme 1.3 The Phosphotriester Approach
The phosphitetriester Approach
R. L. Letsinger has introduced the phosphotriester approach for the synthesis of
oligonucleotides in 1975.33 It involves use of highly reactive phosphate reagent for
coupling two deoxyribonucleosides. Suitably protected nucleoside is reacted with
bifunctioal phosphodichloridite to form nucleoside-3'-phosphomonochloridite which
13

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
is then reacted with protected nucleoside in which 5'-hydroxyl group is free to react.
Upon coupling the trivalent phosphorous is converted into more stable pentavalent
phosphorous via mild oxidation method. The procedure is summarized in Scheme
1.4. Later on this approach was used in solid phase synthesis of oligonucleotides
using deoxyribonucleoside-3'-phosphomonochloridite or monotetrazolides and
deoxyribonucleoside derivatised silica gel as the insoluble support. Although
monochloridite and monotetrazolide had the rapid and efficient coupling potential,
they were not ideal reagents for oligonucleotide synthesis. These reagents were
exceptionally sensitive to hydrolysis and air oxidation, difficult to prepare and
isolate, required special handling techniques, had limited life time in solution and
often contains appreciable amount of inert 3'-3'dinucleoside phosphine.34
O B1RO
OH
PClCl
OR'O B1RO
OP
R'O Cl
O B2OH
AcO
O
O B1RO
OP OR'O B2
R''O
O
O B1OH
OP OOO B2
OH
Base
Base
ii) Deprotection
R' is 2-cyanoethyl, phenyl or o-chlorophenyl
B1 and B2 are A Bz, C Bz, C Ibu or TR is trityl or substituted trityl
i) Oxidation, I2/H2O
-
Scheme 1.4 The Phosphitetriester Approach
14

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
The Phosphoramidite Approach
The problems faced by monochloridite (Figure 1.8, X = Cl) and monotetrazolide
(Figure 1.8, X = tetrazole) trivalent phosphorous reagents in phosphitetriester
approach were resolved by Beaucage and Caruthers by introducing a new class of
deoxyribonucleoside phosphites called as deoxyribonucleoside-3'-O(N,N-
dimehylamino) phosphoramidites35 (Figure 1.8, X = NMe2). These reagents had
improved stability and were resistant to hydrolysis by water or oxidation to air.
Further they were much easier to prepare and use.
O BDMTO
OP
MeO X
NN
NN
ON
X
= N Me2
= N Et2
= N iPr2
= Cl
=
=
Figure 1.8
Unlike their phosphomonochloridite and monotetrazolide counterparts
phosphoramidite reagents were less reactive and could not react directly with
another 5'-hydroxyl containing deoxyribonucleoside. They need to be activated by
treatment with weak acid such as tetrazole. Adams et al. observed that increase in
the steric hindrance around the nitrogen in phosphoramidites leads to increase in
their stability.36 McBride and Caruthers found that morpholino and diisopropyl
phosphoramidites are the reagents of choice as they are nonhygroscopic, stable at
room temperatures as dry powders as well as in anhydrous acetonitrile solution for
at least a week, and can be purified easily on a silica gel column.37 With the
15

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
commercial availability of these reagents synthesis of oligonucleotides in laboratory
has become accessible even to non-chemists particularly via solid phase method.
O B1DMTO
O
O B2DMTO
OP
RO N
O
O B2DMTO
OP ORO B1
O
O
O B2OH
OP OOO B1
OH
O B1OH
O
CPG
ii) Deprotection
R is 2-cyanoethyl or methyl
B1 and B2 are A Bz, C Bz, C Ibu or T
i) Oxidation, I2 / H2O
-
Trichloroaceticacid / CH2Cl2
CPG CPG
CPG is Controlled pore glass
1H-tetrazole / ACN
Scheme 1.5 The Phosphoramidite Approach
The H-phosphonate Approach
The H-phosphonate approach for oligoncleotide synthesis involves reaction of
suitably protected nucleoside-3'-H-phosphonate with 5'-hydroxyl group of protected
nucleoside. As the H-phosphonate is less reactive, it is first activated by treatment
with acyl chloride. The acyl chlorides used are pivaloyl chloride or 1-admantoyl
chloride. Use of these activating agent may lead to side reactions involving
modification of heterocyclic bases.38 A new activating reagent, dipentafluorophenyl
16

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
carbonate was introduced by Efimov et al. which gives high coupling efficienty with
considerable decrease in the side reactions.39 In H-phosphonate chemistry H-
phosphonate moity acts as protecting group for phosphorus through out the
synthesis. After completion of the synthesis all H-phosphonate moieties are
simultaneously oxidized by iodine solution to the corresponding phosphates.
O B1DMTO
O
O B2DMTO
OPOO
H
O
O B2DMTO
OPO B1
O
O H
O
O B2OH
OP OOO B1
OH
O B1OH
O
CPG
ii) Deprotection
B1 and B2 are A Bz, C Bz, C Ibu or T
i) Oxidation, I2 / H2O
-
Trichloroaceticacid / CH2Cl2
CPG CPG
CPG is Controlled pore glass Pivaloyl chloridebase
- TEA+
Scheme 1.6 The H-phosphonate Approach
17

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
1.I.4 Purification of Synthetic Oligonucleotide
All the protecting groups used in the oligonucleotide synthesis needs to be removed
in correct order. Temporary protecting groups are removed at each cycle during
chain elongation process, whereas permanent protecting groups are removed at the
end of synthesis. Various techniques are used for the purification of synthesized
oligonucleotide sequence such as polyacrylamide gel electrophoresis (PAGE) and
chromatographic techniques. The PAGE technique is useful for isolating the product
directly from the small scale reactions or at the end of chromatography. Various
chromatography techniques are also useful for purifying the oligonucleotides. High
performance liquid chromatography technique proves to be much useful for
oligonucleotide purifications. Rapid and efficient separations can be obtained with
the anion exchanger pellinox AL WAX,40 Permaphase AAX,41 Pellionex SAX,42
reverse phase column μ-Bondapak,43 Partsil ODS-244 and RP C-18 column.45
1.II Spin labeling Technique
1.II.1 Introduction
The concept of labelling a biological system with some external probe was
first introduced by Burr and Koshland. The technique involves the introduction of
the probe at specific site of the system to be studies. By monitoring the alternation
in the spectroscopic properties of the group, inferences can be drawn about the
molecular architecture in the vicinity of the probe, also commonly known as a
“reporter group” and the technique involves referred to as reporter group techniques.
Such external probes can be in the form of a fluorescent dye such as biotin,
radioactive labels such as 2H, 13C, 31P and 19F etc or a paramagnetic species called as
spin label and variety of spectroscopic methods have been used in the past to study
18

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
the dynamics, conformational mobility and other structural properties of
biomolecules.
Requirement of a Reporter Group:
The probe used in such methods should fulfill the following requirements:
1. It should be sensitive to changes in environment of the site of intrest and must
subsequently ‘report’ the changes to the external detector.
2. It should have well characterized spectroscopic or physical properties which are
either unique or distinct from the properties of the system under investigation.
3. The introduction of the probe should not alter the structure and function of the
system under study.
1.II.2 Spin Labeling Technique
McConnell46 first introduced spin labeling in 1965 and since then it has been
rapidly emerged as one of the most relible and extensively used tool for research in
biophysics. Owing to the relatively low content of stable paramagnetic species in
biological systems, a paramagnetic reporter group which is ESR-sensitive
constitutes a physically distinct moiety from the rest of system. A paramagnetic
reporter group is termed as a spin label. The technique which involves the
introduction of spin labels as a paramagnetic probe into a system followed by the
monitoring of the changes in its ESR spectra is called spin labeling. This method has
distinct advantages over other methods due to the fact that extremely low
concentration of spin labels can be used in ESR experiments. Paramagnetic species
which have been used for spin labeling studies are nitric oxides, lanthanide ions and
a few organic radicals for specific experiments. The great versatility, sensitivity,
immense stability and the variety of information available from nitroxide radicals
19

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
has made it synonymous with spin labeling. It is necessary at this point to make a
distinction between the terms spin labels and spin probes. The term spin label is
used to describe a molecule to which a nitroxide is covalently attached. On the
other hand a spin probe is a nitroxide containing molecules which is not attached to
molecules of the system under study. A spin probe can be a paramagnetic metal ion
almost always of transition or lanthanide series. The distinction is hard and fast, as
some times a spin label may be used as a spin probe to study more complex systems.
Nitroxides in spin labeling
Nitroxide or nitroxyl radicals are compounds containing the >N-O group.
This has one unpaired electron. Although the inorganic nitroxide Fremy’s salt47 A
had know since 1845, it was one in 1901 that the first organic nitroxide was isolated
and characterised by Piloty and Schwerin.48 This heterocyclic free radical,
porphyrexide B was formulated at that time as C a derivative of “quadrivalent
nitrogen”.
O NSO3K
SO3K
. NH
N NH
NH
O
NH
N NH
NH
O
A B C Figure 1.9
Electronic Structure of nitroxide
Nitroxides may be considered as derivatives of the stable inorganic radical
nitric oxide. The possible resonating forms of nitric oxide are shown below:
20

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
N O+ -
: :... ..
N O: :. .. +-
N O:.. .
.. N O. . .: ,
1 2 3 4
Figure 1.10 Resonance hybrid of nitroxide
Another mode of representation is by a structure with five bonding electrons where
there is a sigma bond and a three electron bond between nitrogen and oxygen. By
analogy, nitroxides can also be represented as a resonance hybrid of the forms a and
b by c with a three electron bond.
-N O
R
RN O
R
R
+
N OR
R.
. . .
a b c Figure 1.11 Resonating forms of nitroxides
The contributions of the above resonating forms differ in the ground state depending
on the conjugation and polarity of the medium. In the present thesis, for the purpose
of convenience in representation and familiarity with representations from literature,
nitroxides will be represented as 5a, regardless of their actual electronic structure.
Stability of Nitroxides
Johnson and coworkers49 have suggested that nitroxides are rendered stable
by the presence of tertiary carbon atom flanking the >N-O groups. Such nitroxides,
unlike most other free radicals, are extremely stable towards purification, handling
and storage. The instability of nitroxides owing to the presence of hydrogen on α-
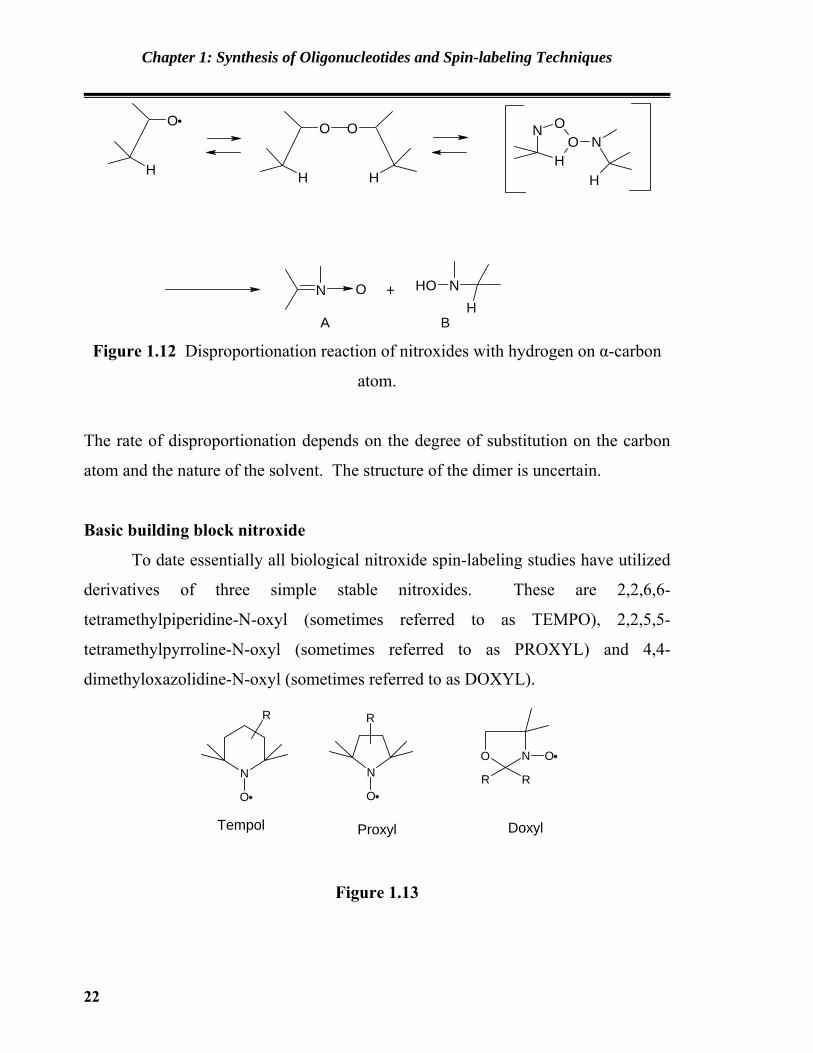
carbon atom leads to a disproportionation reaction50 producing a nitrone A and
hydroxylamine B via the formation of a dimer.
21
Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
H
O
H
O
H
O N
HO
ON
H
N O OH NH
+
A B Figure 1.12 Disproportionation reaction of nitroxides with hydrogen on α-carbon
atom.
The rate of disproportionation depends on the degree of substitution on the carbon
atom and the nature of the solvent. The structure of the dimer is uncertain.
Basic building block nitroxide
To date essentially all biological nitroxide spin-labeling studies have utilized
derivatives of three simple stable nitroxides. These are 2,2,6,6-
tetramethylpiperidine-N-oxyl (sometimes referred to as TEMPO), 2,2,5,5-
tetramethylpyrroline-N-oxyl (sometimes referred to as PROXYL) and 4,4-
dimethyloxazolidine-N-oxyl (sometimes referred to as DOXYL).
N
O
R
N
O
R
NO
R R
O
Tempol Proxyl Doxyl
Figure 1.13
22

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
Typically these structures bear functional group at the postion 4 and/ or 3
capable of undergoing chemical reaction with some functional group of the
biomolecules to be spin labeled. Some of the examples51 of nitroxides are
NX
O
XN O
NO
O
NO
I NO
CH2Br
NO
ONO
OO N
O
O
NO
NHCOCH2X
NO
COOH
NO
COOHNH2
X = OSO2CH3, I etc
NO
OPX
NN
X = O,S
N O
N
O
OP
O
CH2ClO
N
O
NHCOCH2Br
NO
O
O
NO
Br
NO
CHO
Figure 1.14
23

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
Characterization of Nitroxides:
Nitroxides are deeply coloured solids or liquids. However, bis-
[trifuloromethyl]-nitroxyl is a gas under normal condition.
1. Infrared Spectra: Generally all nitroxides shows characteristic IR band in the
range of 1310 to 1370 cm-1. This is attributed to the stretching of N-O..
However it has been proposed that the infrared stretching frequency of several
piperidine notroxides to be in the range of 1373 ± 7 cm-1.
2. UV-Visible Spectra: Nitroxides are orange-yellow in colour owing to an
absorption band in the region 410-450 nm (ε~ 20). A much more intense
absorption band is observed at about 230 (ε~ 3000). The visible band exhibits a
hypsochromic shift with highly polarity of the solvent and hence, is ascribed to
n→π* transition, whereas the absorption in the UV region is due to π→π*
transition. Other than the yellow and orange colours observed in some
nitroxides, α-nitrosonitroxides are blue in colour and has absorption bands at
238, 263, 360 nm. Resonance between the nitroxides group and the aromatiaac
ring, expectedly leads to bathochromic shifts up to the extent of 335 to 340 nm
and 490 to 510 nm.
3. Circular Dichroism: The circular dichroism spectra of several optically active
nitroxides are reported. The octant rule as used for ketones is also applicable in
the case.
4. NMR Spectra: The presence of unpaired electron in the nitroxide group
leads to the broadening of signals in the NMR spectra. Interaction of the
unpaired electron with nuclear spins (both intermolecular as well as
intramolecular), shortens the lifetime of spins in excited state and hence, leads to
line broadening. To overcome this difficultly NMR spectra are run at high
concentration viz. of the order of 3M, so that the electron spin relaxation time is
24

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
considerably shortened compared to the inverse of proton hyperfine coupling
constant. Alternatively though less conveniently, one may employ paramagnetic
solvent such as di-tert-butly nitroxide or 2-doxyl propane or corresponding
completely duterated analogues as spin-relaxer solvents for NMR studies of
nitroxide free radicals.
NO O N
O
Figure 1.15
5. Mass spectra: The mass spectra of several types of nitroxides have been
reported. The five membered pyrrolidine nitroxide substituted with –CH2OH,
OH or –NH2 exhibits (M+ 1) peak along with peaks corresponding to the loss of
methyl, isobutylene and nitric oxide. Six membered nitroxide shows a (M+1)
molecular ion peak along with a peak corresponding to the loss of methyl group.
Nitroxides of the doxyl type fragments producing a protonated ketone which
constitutes the base peak in the mass spectra.
25

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
N
O
R
N
O
R
NO O N+
O OO
HCH2O
H
NO
++
Figure 1.16 Mass spectral fragmentation pattern of nitroxide
6. Electron Spin Resonance Spectra: The electron spin resonance (ESR) spectra,
also known as electron paramagnetic resonance (EPR) spectra, of nitroxides
have been studied extensively. It constitutes the subject of numerous books and
review articles and continuous to be a subject of immense interest till today. A
detailed account of the ESR spectra features will be presented in the later
section. The ESR spectrum of nitroxide, which is employed to detect the present
of nitroxides radicals, consists of a three line spectrum in solution. (Figure 1.17)
Figure 1.17 Typical nitroxide ESR spectra
The three lines appears owing to the coupling of the unpaired electron spin in the
free radicals with the nuclear spin (J = 1) of 14N. the (2 J + 1) component which
26

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
results in a three line spectrum of nitroxide with 14N, collapses into a doublet on
replacement of 14N by 15N having J=1/2. Presence of atoms having non-zero J values
in the vicinity of the >N-O group results in finer splitting of the three line spectrum.
Some complex spectra cannot be unambiguously interpreted, though it provides
information regarding the structure of the radical. At this point it is worthwhile to
mention the salient features of the ESR spectra of biracials or polyradicals. As
expected, nonconjugatead biradicals do not give a nitroxide triplet. On the contrary,
a five line spectrum is obtained indicating the interaction of both unpaired electron
with both nitrogen atoms. This phenomenon is termed as ‘exchange interaction’ and
is observed in di- or polynitroxides wherein the >N-O group lies in close proximity
with each other. The structure of the intervening framework between the >N-O
groups in polynitroxdes or binitroxides determine the shape of the ESR spectra. This
phenomenon was explored to determine the stereochemistry of organic molecules.
This involved the reaction of cyclopropane-1,2-cis- and the corresponding trans-
dicarboxylic acid dichlorides. The resulting compound, a dinitroxide diester, in
each case exhibited different ESR spectra. The cis compounds had a five line ESR
spectra where as the trans isomer exhibit a triplet. This reflected the presence of the
>N-O group in close proximity in the former isomer, compared to the trans
compounds where the greater distance inhibits the desired exchange phenomenon.
Applications of Spin labeling technique
The spin labeling field blossoms even more as we head into the next
millenium. It has been applied to systems in food chemistry, nucleic acid and
nucleotide biochemistry, to cell application, in vivo applications with small animals
and most recently as a tool for specifically incorporating spin labels at unique amino
acid residues position in mutated proteins.
27

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
Spin Labelling In Oligonucleotides
Magnetic resonance spectroscopy is well adapted to the study of molecular
structure and dynamics. It’s application to biological molecules fetches information
about the chemical structural and biochemical aspects of the biomolecules. The
electron spin resonance calls for the presence of an unpaired electron on the
biomolecule to make it suitable for the above investigations. The method is
recognized as an extremely specific tool, since it is most unlikely to have unpaired
electron in the biological systems. The range of applications can be extended by
attaching synthetic free radicals (spin labels) in the precise chemical and structural
manner. The most important feature is the anisotropy of the spectrum with respect
to the magnetic field orientation, which can yield detailed structural information.
The line splitting in spectra, fetches information about the quantitative aspects of the
chemical bond. The detection of DNA sequence by using DNA biochip is a, rapidly
developing technology. In its present form, DNA sequences are attached to a chip,
which is then exposed to the solution containing DNA. If the complementary
sequence is present in the solution, it anneals to the chip, the chip is then treated
with a fluorescent probe which detects the double stranded DNA. The technique is
very sensitive mainly due to the method of detection. However, there are some
negative aspects of the method. For example, hybridization may be incomplete, or
may be difficult to optimize. The hybridization condition or the detection of
hybridization may be non selective. It is highly desirable to device a method which
shall provide an insight into the interaction of oligonucleotide on the chip. The spin
labeled DNAs can be used to differentiate the DNAs which are annealed to the solid
bound nucleotide versus the oligonucleotide which are in the solution. This is
because the spin labels are uniquely sensitive to their environment. Spin labelling
also helps in understanding the optimal set of condition (temperature, time and
buffer) under which the annealing process should be conducted.30 In the discussion
that follows we have emphasized on the reports which proved the applicability of
28

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
the DNA spin labelling as applied to the determination of structure, conformation
and detection of hybrid formation. An enormous growth is witnessed by the area of
development of novel Nitroxides, which can be easily used for the labeling of bin
molecules, and at equally high peace the novel method for labeling and detection are
also developed.
Gannett et. al. showed the application of spin probe labeled oligonucleotides
(Figure 1.18) for detecting the oligonucleotide binding and its extent by using EPR.
The authors further demonstrated the utility of same probes for optimizing the
conditions required for annealing of DNA. The method can also serve as a selective
method for detection of oligonucleotide hybridization under the conditions that
model DNA biochip.52
N
NH
N
O
O
O
O
O ODNA
N
NH
N
O
O
O
O O
O
DNA
..
Figure 1.18
In order to achieve an EPR sensitive probe for DNA, Giordanol et. al. linkded 3-
carboxy-Proxy1 free radical to O6of dG through a five-atoms-tether. The modified
base was incorporated into a 30-mer ODN, and then annealed to its complementary
DNA strand ODN in order to verify the possibility to monitor changes in DNA by
EPR spectroscopy.53 Hydrodinamic parameters show only a slight destabilization
with respect to the equivalent unlabeled hybrid. EPR could monitor the hybrid
29

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
formation showing a progressive enlargement of the upfield signal in passing from
the labeled ss- to the ds-30-mer. Several non-isotopic DNA probing techniques
have been developed by means of the insertion of a modified base carrying a group
selected to be easily detectable by spectroscopic methods. Since the probe bound to
O6 of dG protrudes into the central space of the major groove, it is expected to cause
only a minor perturbation of the duplex DNA structure. (Figure 1.19)
N
N
N
ODMTrO
O
N
N
PN O CN
NHR
O NH
O
Figure 1.19
Cekan et. al. reported a rigid spin-labeled nucleoside C*, an analog of deoxycytidine
that base-pairs with deoxyguanosine, was incorporated into DNA oligomers by
chemical synthesis.54 Thermal denaturation experiments and circular dichroism
(CD) measurements showed that C*¸ has a negligible effect on DNA duplex
stability and conformation. Nucleoside C*¸ was incorporated into several positions
within single-stranded DNA oligomers that can adopt two hairpin conformations of
similar energy, each of which contains a fourbase loop. The relative mobility of
nucleotides in the alternating C/G hairpin loops, 5’-d(GCGC) and 5’-d(CGCG), was
determined by electron paramagnetic resonance (EPR) spectroscopy. The most
mobile nucleotide in the loop is the second one from the 5’-end, followed by the
third, first and fourth nucleotides, consistent with previous NMR studies of DNA
hairpin loops of different sequences. The EPR hairpin data were also corroborated
30

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
by fluorescence spectroscopy using oligomers containing reduced C¸ (C¸ f), which
is fluorescent. Furthermore, EPR spectra of duplex DNAs that contained C¸ at the
end of the helix showed features that indicated dipolar coupling between two spins.
These data are consistent with end-to-end duplex stacking in solution, which was
only observed when G was paired to C*, but not when C¸ was paired with A, C or T.
(Figure 1.20)
N
N
N
O
N
O
O
H
N N
N
N
NH H
H
ON
N
NHO
N
O
O
ODMTrO
O
P
N
O
NC
2 C*G
Figure 1.20
In site-directed spin labelling (SDSL), Grant et. al. described a nitroxide moiety
containing a stable, unpaired electron is covalently attached to a specific site within
a macromolecule, and structural and dynamic information at the labeling site is
obtained via electron paramagnetic resonance (EPR) spectroscopy. Successful
SDSL requires efficient site-specific incorporation of nitroxides. Work reported here
presents a new method for facile nitroxide labeling at the 50 terminus of nucleic
acids of arbitrary sizes. T4-polynucleotide kinase was used to enzymatically
substitute a phosphorothioate group at the 50 terminus of a nucleic acid, and the
resulting phosphorothioate was then reacted with an iodomethyl derivative of a
nitroxide. The method was successfully demonstrated on both chemically
synthesized and naturally occurring nucleic acids. The attached nitroxides reported
31

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
duplex formation as well as tertiary folding of nucleic acids, indicating that they
serve as a valid probe in nucleic acid studies.55 (Figure 1.21)
N
OOP-O
O
S
H (or OH)O
B
O
Figure 1.21
Obeid et. al. described the synthesis of two modified 2’-deoxyuridine triphophates
bearing a spin label linked to the base by a rigid linker to ensure a tight coupling of
spin label dynamics. The incorporation of both spin-labeled nucleotides could be
shown in primer extensions reactions in presences of DNA polymerases from
eukaryotic, prokaryotic and archaic origin are competent in the employment of spin
labeled nucleotides as surrogate of natural building blocks in enzymatic DNA
synthesis. This finding opens new opportunities for further advanced applications
e.g. in vivo spin labelling or the generations of complex multi spin systems which
might be useful for DNA-based nanobiotechnology.56 (Figure A.1.22)
N
N
NH
OO
OH
O4P
OO
.
1
NO
N
NH
OO
OH
O4P
O
2
.
Figure A.1.22
32

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
References
1. Avery, O. T.; Macleod, C. M.; McCarty, M. J. Exp. Med. 1981, 79, 137.
2. Alberts, Bruce; Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts,
and Peter Walters (2002). Molecular Biology of the Cell; Fourth Edition New
York and London: Garland Science.
3. Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737.
4. Leslie, A. G.; Arnott, S.; Chandrasekaran, R.; Ratliff, R. L. J. Mol. Biol. 1980,
143(1), 49.
5. Drew, H. R.; Wing, R. M.; Takano, T.; Broka, C.; Tanaka, S.; Itakura, K.;
Dickerson, R. E. Proc. Natl. Acad. Sci. 1981, 78, 2179.
6. Rich, A. Gene 1993, 135, 99.
7. (a) Gait, M. J. Oligonucleotide synthesis a practical approach, 1984, IRL press;
(b) Goforth, S. Scientist 2002, 16(12), 43; (c) Virta, P.; Katajisto, J.; Niittymaki
T; Lonnberg, H. Tetrahedron 2003, 59(28), 5137; (d) Reese, C. B. Org. Biomol.
Chem. 2005, 3, 3851.
8. Agrawal, K. L.; Yamazaki, A.; Cashion, P. J.; Khorana, H. G. Angew. Chem. Int.
Edn. Engl. 1972, 11, 451.
9. Daskalov, H. P.; Sekine, M.; Hata, T. Bull. Chem. Soc. Jpn. 1981, 54, 3076.
10. Jones, S. E.; Reese, C. B.; Sibanda, S.; Ubasawa, A. Tetrahedron Lett. 1981, 22,
4755.
11. (a) Gaffney, B. L.; Jones, R. A. Tetrahedron Lett. 1982, 23, 2257. (b)
Trichtinger, T.; Charubala, R.; Pfleiderer, W. Tetrahedron Lett. 1983, 24, 211.
12. Smith, M.; Rammler, D. H.; Goldberg, I. H.; Khorana, H. G. J. Am. Chem. Soc.
1963, 84, 430.
13. (a) Reese, C. B. Colloques Internationaux du CNRS 1970, 182, 319 ; (b) Silber,
G.; Flockerzi, D.; Varma, R. S.; Charubala, R.; Uhlmann, E.; Pfleiderer, W.
Helv. Chim. Acta 1981, 64, 1704 ; (c) Reese, C. B.; Zard, L. Nucleic Acids Res.
1981, 9, 4611.
33

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
14. Ogilivie, K. K.; Theriault, N. T.; Seifert, J.; Pon, R. T.; Nemer, M. J. Canad. J.
Chem. 1980, 58, 2686.
15. Sinha, N. D.; Biemat, J.; Koster, H. Tetrahedron Lett. 1983, 24, 5843.
16. Katagiri, N.; Bahl, C. P.; Itakura, K.; Michniewicz, J.; Narang, S. A. J. Chem.
Soc., Chem. Commun. 1973, 803.
17. Hering, G.; Stocklein-Schneiderwind, R.; Ugi, I.; Pathak, T.; Balgobin, N.;
Chattopadhyaya, J. Nucleosides Nucleotides 1985, 4, 169.
18. Eckstein, F.; Scheit, K. H. Angew. Chem. 1967, 79, 317.
19. Letsinger, R. L.; Groody, E. P.; Tanaka, T. J. Am. Chem. Soc. 1982, 104, 6805.
20. Van Boom, J. H.; Crea, R.; Luyten, W. C.; Vink, A. B. Tetrahedron Lett. 1975,
16, 2779.
21. Uhlmann, E.; Pfleiderer, W. Tetrahedron Lett. 1980, 21, 1181.
22. Freist, W.; Helbig, R.; Cramer, F. Chem. Ber. 1970, 103, 1032.
23. Claesen, C.; Tesser, G. I.; Dreef, C. E.; Marugg, J. E.; van de Marcel, G. A.; van
Boom, J. H. Tetrahedron Lett. 1984, 25, 1307.
24. Balgobin, N.; Chattopadhyaya, J. Acta Chem. Scand. 1985, B39, 883.
25. Rubinstein, M.; Amit, B.; Patchornik, A. Tetrahedron 1975, 1445.
26. Hes, J.; Mertes, M. P. J. Org. Chem. 1974, 39, 3767.
27. Reese, C. B.; Titmas, R. C.; Yau, L. Tetrahedron Lett. 1984, 25, 2727.
28. Takaku, H.; Shimada, Y.; Hata, T. Chem. Lett. 1975, 873.
29. Hayakawa, Y. Bull. Chem. Soc. Jpn. 2001, 74(9), 1547.
30. Michelson, A. M.; Todd, A. R. J. Chem. Soc. 1955, 2632.
31. Khorana, H. G.; Tener, G. M. Moffat, J. G. Chem. Ind. 1956, 1523.
32. (a) Letsinger, R. L.; Cruthers, M. H.; Miller, P. S.; Ogilvie, K. K. J. Am. Chem.
Soc.1967, 89, 7146; (b) Eckstein, F.; Rizk, T. Angew. Chem., Intl. Ed. Engl.
1967, 6, 695; (c) Michealson, A. M.; Tood, A. R. J. Chem. Soc. 1955, 2632. (d)
Letsinger, R. L.; Ogilvie, K. K.; Miller, P. S. J. Am. Chem. Soc. 1969, 91, 3360;
(e) Letsinger, R. L.; Ogilvie, K. K. J. Am. Chem. Soc. 1969, 91, 3350.
34

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
33. (a) Letsinger, R. L.; Finnan, J. L.; Heavner, G. A.; Lunsford, W. B. J. Am. Chem.
Soc. 1975, 97, 3278; (b) Letsinger, R. L.; Lunsford, W. B. J. Am. Chem. Soc.
1976, 98, 3655; (c) Finnan, J. L.; Varshney, A.; Letsinger, R. L. Nucleic Acids
Sym. Ser. 1980, 7, 133.
34. Letsinger, R. L.; Groody, E. P.; Tanaka, T. J. Am. Chem. Soc. 1982, 104, 6805.
35. Beaucage, S. L.; Caruthers, M. H. Tetrahedron Lett. 1981, 22, 1859.
36. Adams, S. P.; Kavka, K. S.; Wykes, E. J.; Holder, S. B.; Galluppi, G. R. J. Am.
Chem. Soc. 1983, 105, 661.
37. McBride, L. J. and Caruthers, M. H. Tetrahedron Lett. 1983, 24, 245.
38. (a) Garegg, P. J.; Regberg, T.; Stawanski, J.; Stromberg, R. Chem. Scripta 1985,
25,280; (b) Garegg, P. J.; Regberg, T.; Stawanski, J.; Stromberg, R. Chem.
Scripta 1986, 26, 59; (c) Garegg, P. J.; Lidh, I.; Regberg, T.; Stawanski, J.;
tromberg, R. Chem. Scripta 1986, 27, 4051; (d) Froehler, B. C.; Ng, P. G.;
Matteucci, M. D. Nucleic Acids Res. 1986, 14, 5399; (e) Froehler, B. C.; Ng, P.
G.; Matteucci, M. D. Nucleic Acids Res.1986, 27, 469; (f) Andrus, A.; Efcavitch,
J. W.; Mcbride, L. T.; Giusti, B. Tetrahedron Lett. 1988, 29, 861.
39. Efimov, V. A.; Kalinkina, A. L.; Chakhmakhcheva, O. G. Nucleic Acids Res.
1986, 14, 5399.
40. Miller, P. S.; Cheng, D. M.; Dreon, N.; Jagaraman, K.; Kan, L.; Leutzinger, E.
F.; Pulford, S. M.; Tso, P. O. P Biochemistry 1980, 19, 4688.
41. Van Boom, J. H.; Rooj de, J. Chromatography 1977, 131, 169.
42. Efimov, V. A.; Reverdatto, S. V.; Chakhmakhcheva, O. G. Nucleic Acids
Res.1982, 10, 6675.
43. (a) Matteucci, M. D.; Caruthers, M. H. J. Am. Chem. Soc. 1981, 103, 3185; (b)
Fritz, H. J.; Belagaje, R.; Brown, E. L.; Fritz, R. H.; Jones, R. A.; Lees, R.
F.;Khorana, H. G. Biochemistry 1978, 17, 1257; (c) McFarland, G. D.; Borer, P.
N. Nucleic Acids Res. 1979, 7, 1067.
44. Hillen, W., Klein, R. D.; Wells, R. D. Biochemistry 1981, 20, 3748.
35

Chapter 1: Synthesis of Oligonucleotides and Spin-labeling Techniques
45. Tanaka, T.; Letsinger, R. L. Nucleic Acids Res. 1982, 10, 3240.
46. (a) McConnell, H. M.; Ohnishi, S. J. Am. Chem. Soc. 1965, 87, 2293; (b) Stone,
T.I.; Buckman, T.; Nordio, P. L.; McConnell, H. M. Proc. Nat. Acad. Sci. USA.
1965, 54,1010.
47. Fremy, E. Ann. Chim et Phys., 1845, 15, 408.
48. Piloty, O.; Schwerin, B. Chem. Ber. 1901, 34, 1870.
49. Johnson, D. H.; Rogers, M. A. T.; Trappe, G. J. Chem. Soc. 1965, 1093.
50. Keana, J. F. W. Chem. Rev., 1978, 78, 37.
51. Banerjee, S.; Trivedi, G. K. J. Sci. Ind.Res., 1995, 54, 623; and references cited
therein.
52. Gannett, P. N.; Powell, J. H.; Johnson, E. M.; Darian, E.; Dalal, N. S.; Norton,
M. L.; Budil, D. E. Tetrahedron Lett. 2002, 43, 1931.
53. Giordanol, C.; Pedone, F.; Fattibene, P.; Cellai, L. Nucleosides, Nucleotides &
Nucleic Aacids 2000, 19, 1301.
54. Cekan, P.; Smith, A. L.; Barhate, N.; Robinson, B. H.; Th. Sigurdsson, S.
Nucleic Acids Res. 2008, 36, 5946.
55. Grant, G. P.; Qin, P. Z. Nucleic Acids Res. 2007, 35, 77.
56. Obeid, S.; Yulikov, M.; Jeschke, G.; Marx, A. Nucleic Acids Symposium Series
2008, 52, 373.
36


















