
1 La bioinformatique de l'identification microbienne et de la diversité, à l'ère de la...
42
1 La bioinformatique de l'identification microbienne et de la diversité, à l'ère de la métagénomique et du séquençage massivement parallèle Richard Christen CNRS UMR 6543 & Université de Nice [email protected] http://bioinfo.unice.fr
-
date post
18-Dec-2015 -
Category
Documents
-
view
216 -
download
1
Transcript of 1 La bioinformatique de l'identification microbienne et de la diversité, à l'ère de la...

1
La bioinformatique de l'identification microbienne et de la diversité, à l'ère de la
métagénomique et du séquençage massivement parallèle
Richard ChristenCNRS UMR 6543 & Université de Nice
[email protected]://bioinfo.unice.fr

2
Tasks and problems
• Identification of a new isolate: the 16S “gold standard”.
• Other genes.
• Typing a strain.
• Studying biodiversity: new approaches.

3
0
10,000
20,000
30,000
40,000
50,000
60,000
70,000
80,000
90,000
100,000
0 500 1000 1500 2000 2500
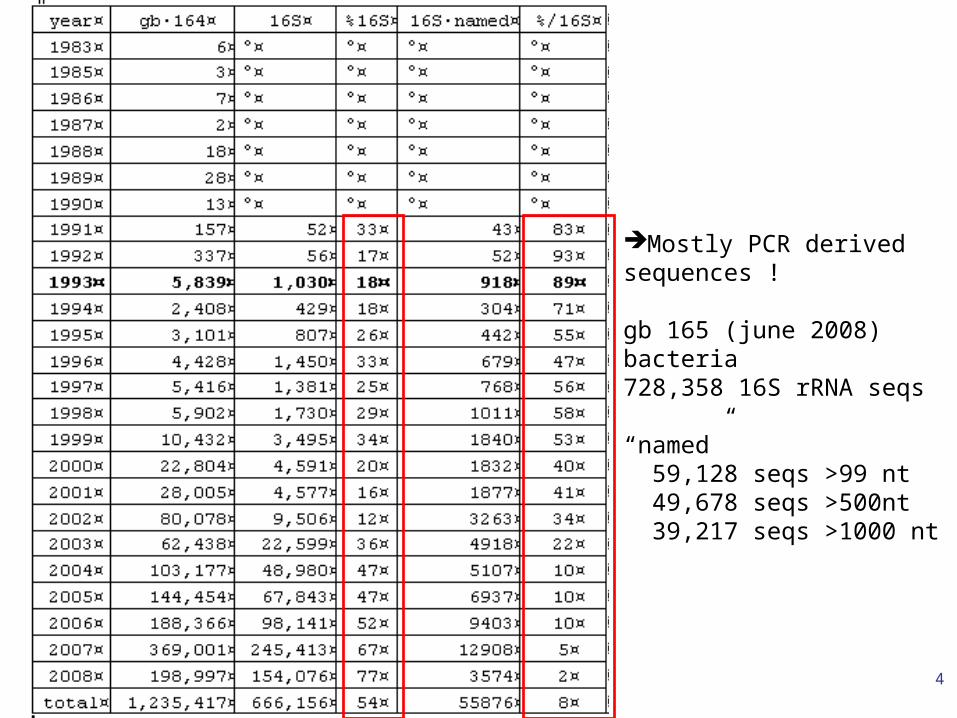
Some long sequences correspond to badly annotated sequences such as Z94013, annotated with keywords "16S ribosomal RNA; 16S rRNA gene" when in fact it is a 23S rRNA sequence...
The 16S “gold standard”
4
Mostly PCR derivedsequences !
gb 165 (june 2008)bacteria728,358 16S rRNA seqs
“named” 59,128 seqs >99 nt 49,678 seqs >500nt 39,217 seqs >1000 nt

5
>NF001CTCTCTCTCGCATTCGTCAGTGCTGGAGGCTGTTGACCCCCAACCCTTTCTTAACGAGTGACAGTGGTTTACAACCCGAAGGCCTTCATCCCACACGCGGCGTCGCTCCGTCAAGCTTGCGCTCATTGCGGAAGATCCTCGACTGCAGCCTCCCGTAGGAGTTTGGGCAGTGTCTCAGTCCCAATGTGGCCGGACACCCGCTAAGGCCGGCTACCCGTCAATGCCTTGGTGGGCCATTACCCTCACCAACTAGCTGATAGGACATAGATCCCTCCCCGAGCGGGAGCATCTTCAGAGGCCTCCTTTAGTCACCGAACCAGGCGATCCAGTGACCCCATCCGGTCTTAGCTCCGGTTTCCCGGAGTTATCCCGGTCTCGGGGGCAGGTTATCTATGCATTACTACCCTTCGCACTAACACCCGTATTGCTACGGTGTCCGTTCGTCTTGCATGCCTAATCACGCCGCTGGCGTTCGTTCTGAGCCAGGATCCAAACTCTATCCGG
The 16S “gold standard”
A case study: identification of a “DGGE” band using the “usual” Blast servers
EBI
NCBI
DDBJ

6
NCBI
...

7
DDBJ

8
DDBJ improved

9
DDBJ improved
...
Now
Previous

10
EBI “standard”
Similar to NCBI nr
...

11
EBI improved
Select the database excluding sequences from the “ENV” division

12
EBI improved

13
Blast on cultured strains
Select by minimal lengthSelect two sequences only by species
http://bioinfo.unice.fr/blast/

14
Blast on cultured strains
The taxonomy bar-code :17
15
1

15
Blast on type strains
http://210.218.222.43:8080
This Blast does not take parameters

16
Blast 2 TreeDyn
Download sequences and annotations

17
Clustal - Phylip - TreeDyn
About one hour for an expert !(Not including alignments and calculations of trees)
Ready for publication !
http://www.treedyn.org/

18
Identify 16S rRNA sequences: LOL ?
16S (LSU) RIBOSOMAL RNA 16S LARGE RIBOSOMAL RNA16S LARGE SUBUNIT RIBOOSMAL RNA16S LARGE SUBUNIT RIBOSOMAL RNA
?

19
Tasks and problems
• Identification of a new isolate: the 16S “gold standard”.
• Other genes.
• Typing a strain.
• Studying biodiversity: new approaches.
20
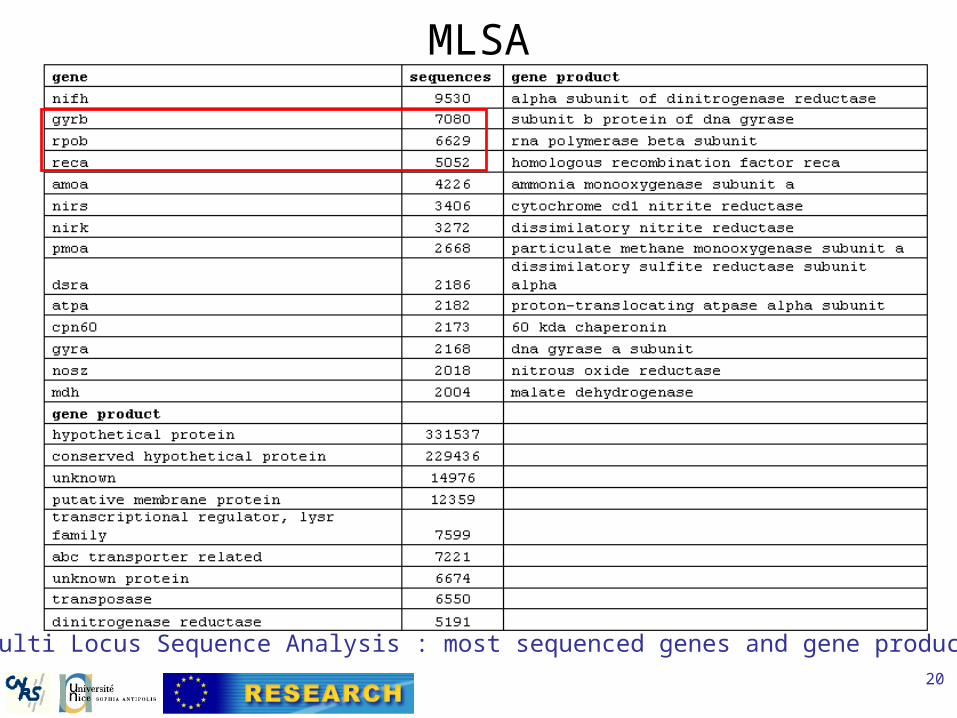
MLSA
Multi Locus Sequence Analysis : most sequenced genes and gene products.

21
MLSA Vibrios
Mostly short PCR sequences !

22
Using a pathogenicity gene as target
Legionella pneumophila: the mip gene.
Analyses of 2006 publications !
URL: http://bioinfo.unice.fr/ohm

23
Using a pathogenicity gene as target
Wrong primer used in publications of year 2006 !

24
Tasks and problems
• Identification of a new isolate: the 16S “gold standard”.
• Other genes.
• Typing a strain.
• Studying biodiversity: new approaches.

25
Use tandem repeat sequences
http://minisatellites.u-psud.fr.
Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA)VAN BELKUM Alex (1) ; FEMS immunology and medical microbiology ISSN 0928-8244 2007, vol. 49, no1, pp. 22-27

26
Tasks and problems• Identification of a new isolate: the 16S
“gold standard”.
• Other genes.
• Typing a strain.
• Studying biodiversity: new approaches.

27
Genome Res. 2006 16: 316-322
The “classic” approach
• Use PCR with “universal” primers.• Clone.• Random sequence ... 200 clones.

28
Biodiversity analyses - classic
PCR – clone - sequence : too tedious for most labs !

29
30 years Roadmap to «Global Sequencing»• 1975 First complete DNA genome : bacteriophage φX174• 1977 Maxam and Gilbert "DNA sequencing by chemical degradation" • 1977 Sanger "DNA sequencing by enzymatic synthesis".• 1982 Genbank starts as a public repository of DNA sequences.
• 1985 PCR• 1986
• First semi-automated DNA sequencing machine.
• BLAST algorithm for sequence retrieval.• Capillary electrophoresis.
• 1991 Venter expressed genes with ESTs • 1992 Venter leaves NIH to set up The Institute for Genomic Research (TIGR).
• BACs (Bacterial Artificial Chromosomes) for cloning.• First chromosome physical maps published: Y & 21• Complete mouse genetic map• Complete human genetic map
• 1993 Wellcome Trust and MRC open Sanger Centre, near Cambridge, UK. – The GenBank database migrates from Los Alamos (DOE) to NCBI (NIH).
• 1995 Haemophilus influenzae • S. cerevisiae • RIKEN : first set of full-length mouse cDNAs.• ABI : the ABI310 sequence analyzer.
• 1997 E. coli • Venter starts “Celera”• Applied Biosystems introduces the 3700 capillary sequencing machine.
• C. elegans
• Human chromosome 22 • 2000 Drosophila melanogaster
• H.s. chromosome 21• Arabidopsis thaliana
• 2001 HGP consortium : Human Genome Sequence• Celera : the Human Genome sequence.
• 2005 420,000 human sequences (Applied VariantSEQr).– Pyrosequencing machine
• 2007 A set of closely related species (12 Drosophilidae) sequenced• Craig Venter publishes his full diploid genome: the first human genome to be sequenced completely.
30
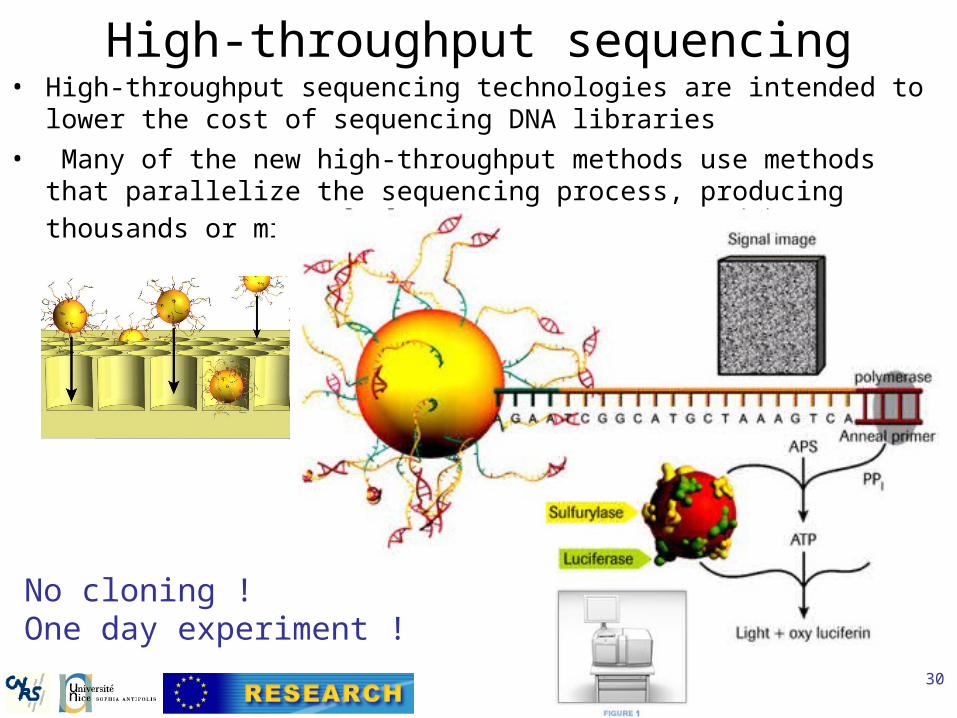
High-throughput sequencing• High-throughput sequencing technologies are intended to lower the cost of
sequencing DNA libraries
• Many of the new high-throughput methods use methods that parallelize the sequencing process, producing thousands or millions of sequences at
once.
No cloning ! One day experiment !

31
Advantages and Disadvantages
• 454 Sequencing runs at 20 megabases per 4.5-hour run (1 day: from sampling to sequences).
• G-C rich content is not as much of a problem.• Unclonable segments are not skipped. • Detection of mutations in an amplicon pool at a low
sensitivity level.
• Each read of the GS20 is only 100 base pairs long (2005-2006);
• The new FLX system does 200-300 base pairs (2007)
• 454 has said they expect 500 in '08.

32
Biodiversity, examples
• Huber, J. A., D. B. Mark Welch, et al. (2007). "Microbial population structures in the deep marine biosphere." Science 318(5847): 97-100.
• Sogin, M. L., H. G. Morrison, et al. (2006). "Microbial diversity in the deep sea and the underexplored "rare biosphere"." Proc. Natl. Acad. Sci. U S A 103(32): 12115-20.
• Roesch, L. F., R. R. Fulthorpe, et al. (2007). "Pyrosequencing enumerates and contrasts soil microbial diversity." ISME J. 1(4): 283-90.

33
Possible variable domains in the 16S rRNA gene sequences

34
Tag dereplication
1
10
100
1000
10000
100000
1 1970 3939 5908 7877 9846 11815 13784 15753 17722 19691
FS396
FS312

35
Clustering tags into OTU
Total calculation time : 7 minutes
• Usual manner : align (Muscle), compute distances, phylogeny or cluster.• Better : cluster according to words frequencies
• No alignement• Much faster• Much better

36
Assign each tag to a taxon
• GreenGenes. The greengenes web application provides access to a 16S rRNA gene sequence alignments for browsing, blasting, probing, and downloading. URL: http://greengenes.lbl.gov
• RDP. The Ribosomal Database Project (RDP) provides ribosome related data services to the scientific community, including online data analysis, rRNA derived phylogenetic trees, and aligned and annotated rRNA sequences. URL: http://rdp.cme.msu.edu/
• Silva. SILVA provides comprehensive, quality checked and regularly updated databases of aligned small (16S/18S, SSU) and large subunit (23S/28S, LSU) ribosomal RNA (rRNA) sequences for all three domains of life (Bacteria, Archaea and Eukarya). URL: http://www.arb-silva.de/
Assignments done using first hit of blast.

37BMC Microbiology 2007, 7:108
Assign each tag to a taxon

38
BMC Microbiology 2007, 7:108
Assign each tag to a taxon
Simulated read resolution for varying read-lengths

39
Numbers of 16S rRNA sequences per species
Most species are known from a single sequence ! Tags taxonomic specificities are over-evaluated. Most species have not been sequenced at all.

40
Main taxa that were not amplified
Primers need to be better designed !
41
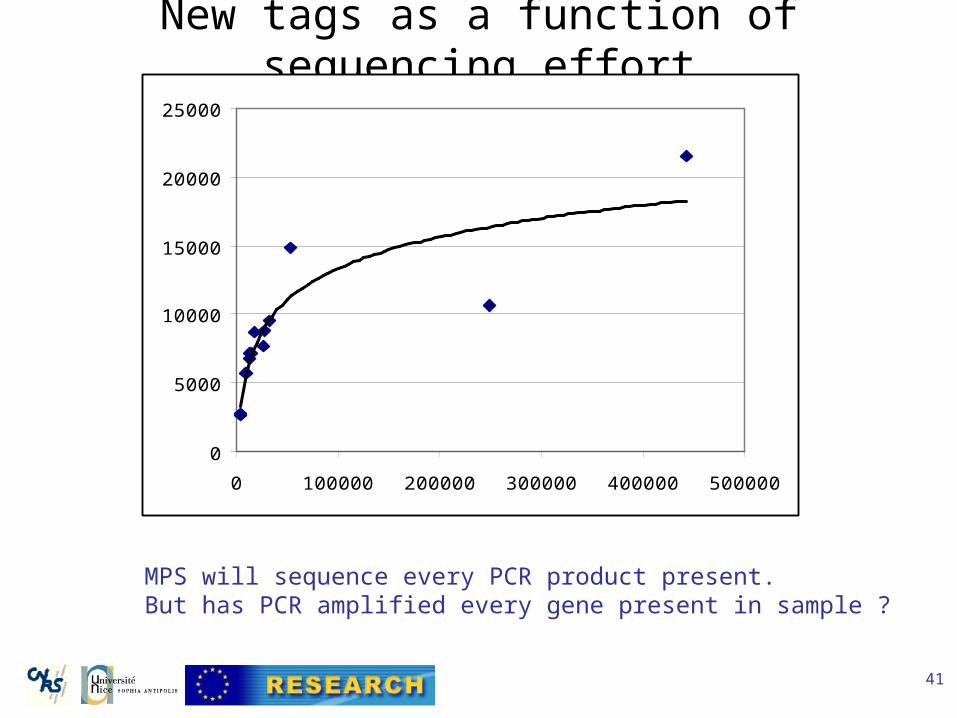
New tags as a function of sequencing effort
0
5000
10000
15000
20000
25000
0 100000 200000 300000 400000 500000
MPS will sequence every PCR product present.But has PCR amplified every gene present in sample ?

42
Conclusions• Identification using 16S rRNA gene sequences is now easy.• MLSA: there is a lack of complete sequences to evaluate published
primers.• MPS on 16S:
– Lack of complete sequences to evaluate primers.– A single sequence available for a majority of species.– Most sequences have a poorly annotated taxonomy.
• 112,509 (16.8 %) only of the 670,401 bacterial 16S rRNA gene sequences of length >100 nt presently deposited have a taxonomic description down to the genus level, while 383,570 sequences (57 %) have "environmental samples" as sole description.
•
– MPS technologies have not been validated against samples of known compositions.
– MPS machines are not calibrated before, during or after a run.– MPS experiments to estimate diversity are not reproduced (duplicated) !– Primers have to be improved– Degenerated primers should NOT be mixed (competition).





![Lake-O-Ukers Ukulele Strum October 25, 2019punchdrunkband.com/songpdfs/LakeOOct25-2019.pdf · [G] La la la la la [B7] laaaa la la [Em] la la la la la la [G7] laaaaaa . La la la la](https://static.fdocuments.in/doc/165x107/5fd12b9fd69a5f331475ceba/lake-o-ukers-ukulele-strum-october-25-g-la-la-la-la-la-b7-laaaa-la-la-em.jpg)












