
04 qualification and validation overview - DCVMN · May 2015, page 9 Common understanding of the...
16
May 2015, page 1 Qualification and Validation - an Overview - by Dr. Ingrid Walther May 2015, page 2 „Qualification” Action of proving that any equipment works correctly and actually leads to the expected results. The word validation is sometimes widened to incorporate the concept of qualification.“ EU-GMP-Guide, Glossary qualification Action of proving that any premises, systems and items of equipment work correctly and actually lead to the expected results. The meaning of the word “validation” is sometimes extended to incorporate the concept of qualification. WHO Technical Report Series, No. 961, 2011 Qualification is something that has to be done additionally to GEP. It is done for those systems that have a direct impact on product quality. ISPE Baseline Volume 5 Definitions
Transcript of 04 qualification and validation overview - DCVMN · May 2015, page 9 Common understanding of the...

May 2015, page 1
Qualification and Validation - an Overview -
by Dr. Ingrid Walther
May 2015, page 2
„Qualification” Action of proving that any equipment works correctly and actually leads to the expected results. The word validation is sometimes widened to incorporate the concept of qualification.“
EU-GMP-Guide, Glossary
qualification Action of proving that any premises, systems and items of equipment work correctly and actually lead to the expected results. The meaning of the word “validation” is sometimes extended to incorporate the concept of qualification. WHO Technical Report Series, No. 961, 2011
Qualification is something that has to be done additionally to GEP. It is done for those systems that have a direct impact on product quality.
ISPE Baseline Volume 5
Definitions

May 2015, page 3
Definitions
Qualification provides documented evidence that equipment is designed and works as it should:
Qualification
➩ equipment-related
Validation provides documented evidence that processes lead to product of the desired quality and safety:
Validation
➩ process-related
Example: baking oven / car
May 2015, page 4
Regulatory Background
! WHO-Guideline, WHO Technical Report Series, No. 961, 2011 Section 4
! EU Guide to Good Manufacturing Practice Part 1, Chapters 5 + 6 Annex 11: Computerised Systems Annex 15: Qualification and Validation, current Version 2001, new version October 2015
Part 2: GMP for Active Pharmaceutical Ingredients
! US-FDA Regulations – FDA 21 CFR Parts 210 and 211: CGMP-Regulations – Process Validation, General Principles and Practice, January 2011 – Guides for Inspection
! GAMP 5 (Computerised Systems) ! ...further national regulations and laws

May 2015, page 5
Regulatory Background
Guidelines (no legal obligation, but should be followed):
! PIC-Document PI 006-3 (Title: „Validation Master Plan, Installation and Operational Qualification, Non-sterile Process Validation, Cleaning Validation“, 25 September 2007)
! ISPE Baseline, Vol. 5, Commissioning and Qualification, 2001 January 2008 – Draft for comments – no new version published yet!
Common understanding in all Regulations and Guidelines:
" Qualification and Validation activities are needed to achieve the target of reliable product quality and safe products!
May 2015, page 6
Regulatory Overview
VMP RA
DQ IQ OQ PQ PV CV Comp. Val.
EU-GMP-Guide x x x x x x x x x
WHO x x x x x x x x
PIC Document PI 006-3
x - x x x x x -
FDA-Guide-lines x - - x x x x x x
EU-GMP-Guide Part 2
Validation Policy
Identi-fication
of critical para-
meters
x x x x x x x
ISPE- Baseline
Commis-sioning
Plan / VMP
Impact Assess-
ment
En-hanced Design Review (EDR)
x x x - - -
GAMP 5 x x x x x x
New: URS is expected in EU-GMP-Guide!
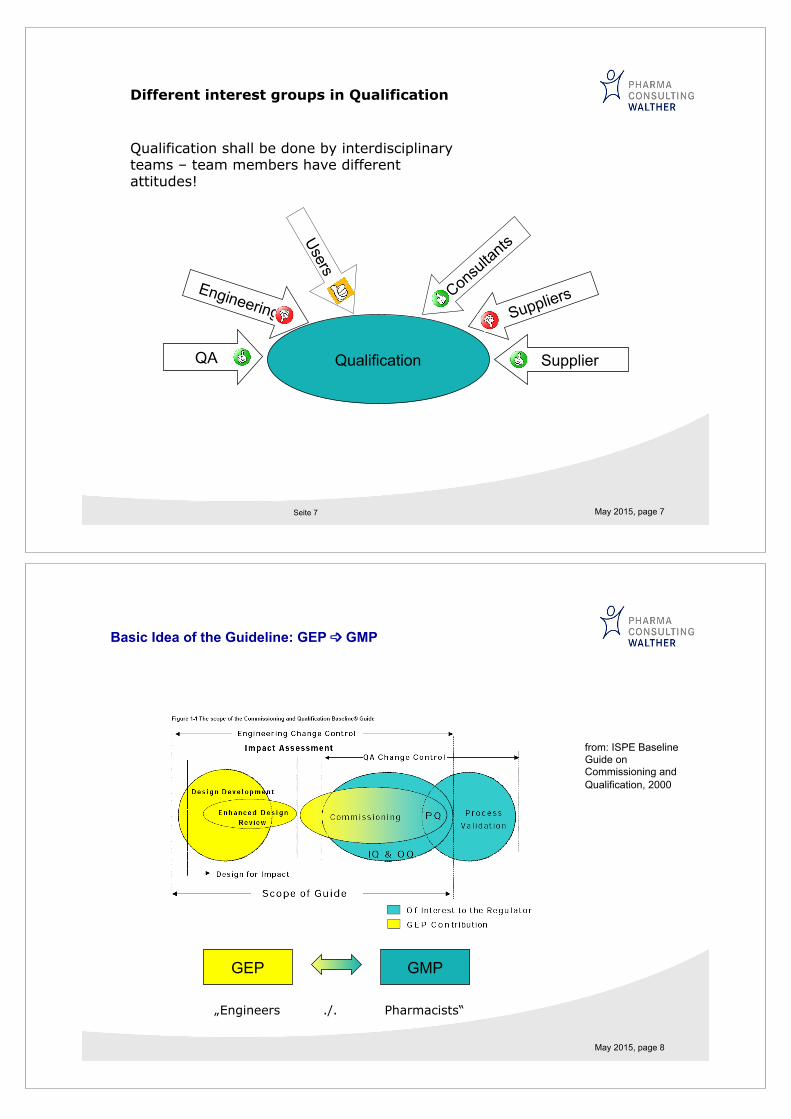
May 2015, page 7 Seite 7
Different interest groups in Qualification
Qualification shall be done by interdisciplinary teams – team members have different attitudes!
Qualification QA
Engineering
Supplier
Suppliers
May 2015, page 8
GEP GMP
„Engineers ./. Pharmacists“
from: ISPE Baseline Guide on Commissioning and Qualification, 2000
Basic Idea of the Guideline: GEP ➩ GMP

May 2015, page 9
Common understanding of the GEP and GMP Regulations
" Qualification and Validation activities are needed to achieve the target of reliable product quality and safe products!
• ISPE Baseline, Vol. 5, Commissioning and Qualification, 2001
• GAMP 5 (Computerized Systems)
• ASTM E2500-7, Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment, 2007
Different Regulations – with different focuses!
GEP
GEP GMP
May 2015, page 10
Qualification and Validation Overview
DQ
IQ
OQ
PQ
VMP
Qualification Computer-Val. Validation
DQ
IQ
OQ
PQ
Method Validation
Process Validation
Cleaning Validation
Impact Assessment
Risk Analyses Risk Analyses Risk Analyses

May 2015, page 11 Seite 11
Separate responsibilities and target!
Entirety of functions of an equipment
GMP-relevant Functions
Qualification (only a part of the technical acceptance testing!)
technical documents / Technical Acceptance Testing
May 2015, page 12
Why does the split between GEP and GMP make sense?
• Pharmaceutical responsibility lies with a pharmacist: He / she is obliged to ensure that all quality relevant process parameters and conditions are under control
• Approval of Qualification documents " pharmacist / biotechnologist may not be able to evaluate ALL technical requirements
• Technical equipment is often very complex " Fokusing on GMP-aspects in Qualification allows the view on the essential
• In case of authority inspections: " GMPrequirements must be fulfilled and THIS must be documented (the amount of paper does NOT count!)
• In production routine: " Formal Change Control is only required for GMP-relevant aspects
• Reduce documentation effort by separating GEP from GMP!
GMP-compliant?
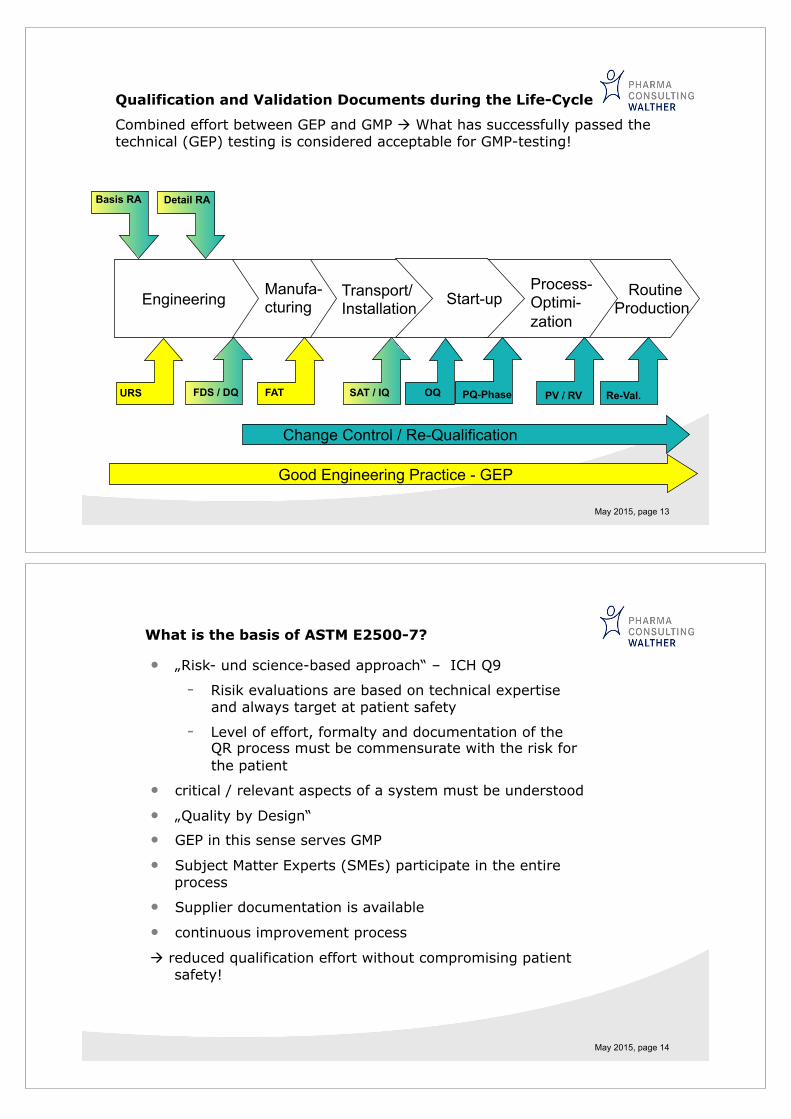
May 2015, page 13
Qualification and Validation Documents during the Life-Cycle
Combined effort between GEP and GMP " What has successfully passed the technical (GEP) testing is considered acceptable for GMP-testing!
Engineering
Routine
Production Transport/ Installation
Process- Optimi-zation
FDS / DQ PV / RV PQ-Phase SAT / IQ FAT OQ
Detail RA
Change Control / Re-Qualification
Manufa- cturing Start-up
URS
Basis RA
Re-Val.
Good Engineering Practice - GEP
May 2015, page 14
What is the basis of ASTM E2500-7?
• „Risk- und science-based approach“ – ICH Q9
- Risik evaluations are based on technical expertise and always target at patient safety
- Level of effort, formalty and documentation of the QR process must be commensurate with the risk for the patient
• critical / relevant aspects of a system must be understood
• „Quality by Design“
• GEP in this sense serves GMP
• Subject Matter Experts (SMEs) participate in the entire process
• Supplier documentation is available
• continuous improvement process
" reduced qualification effort without compromising patient safety!
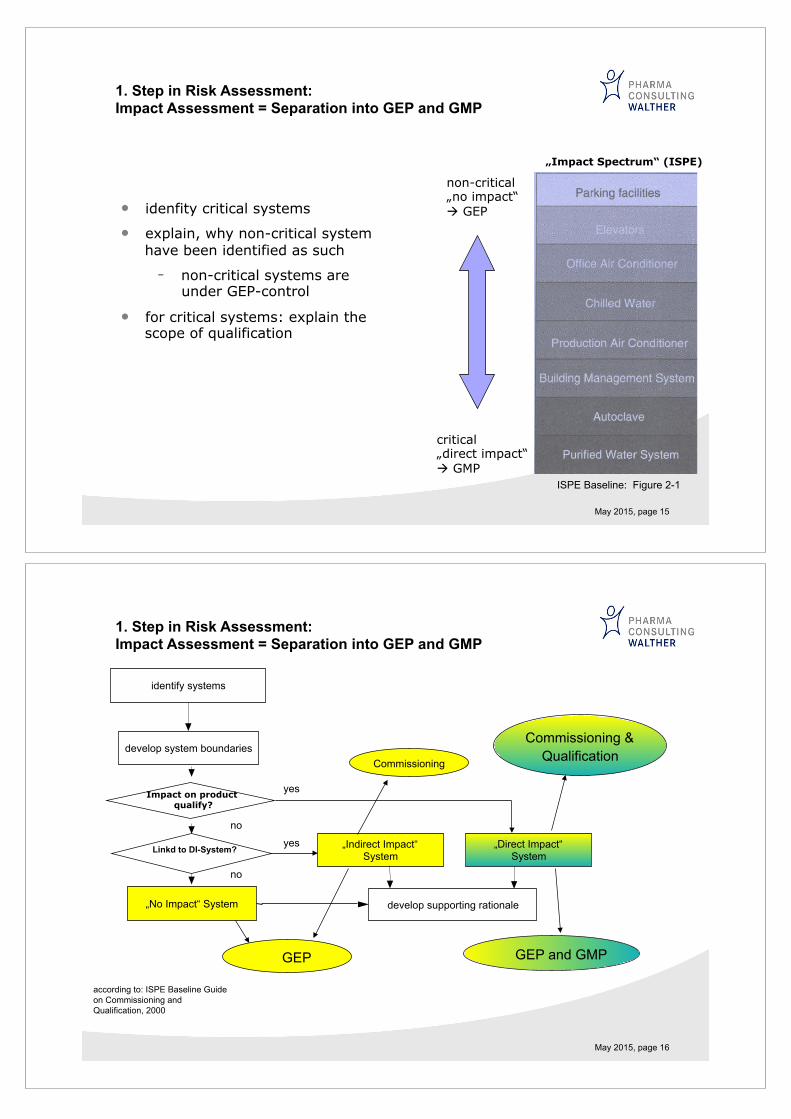
May 2015, page 15
• idenfity critical systems
• explain, why non-critical system have been identified as such
- non-critical systems are under GEP-control
• for critical systems: explain the scope of qualification
1. Step in Risk Assessment: Impact Assessment = Separation into GEP and GMP
„Impact Spectrum“ (ISPE)
ISPE Baseline: Figure 2-1
non-critical „no impact“ " GEP
critical „direct impact“ " GMP
May 2015, page 16
identify systems
„No Impact“ System
„Direct Impact“ System
develop supporting rationale
develop system boundaries
„Indirect Impact“ System
Impact on product qualify?
Linkd to DI-System?
no
no
yes
yes
Commissioning & Qualification Commissioning
GEP GEP and GMP
according to: ISPE Baseline Guide on Commissioning and Qualification, 2000
1. Step in Risk Assessment: Impact Assessment = Separation into GEP and GMP

May 2015, page 17
Technische Dokum ente
Quali fiz ierungsdokum ente
Computer-Validierungs-Rahmenplan
Designqualif izi erungs -P läne
DQ-Bericht
Konzeptstudie
Basic Engineering inkl.Hygienekonzept +
(ev. Druckstufenkonzept )
Lastenhef t
R+I-Schem ata
Leistungsverzeichnisse /Technische Anfragespezif ikation
Bestellung
Good Engineering Practi ces / Change Control
Validierungsmasterplan
Risikoanalysen
Prüfung
gegen di ese
Dokumente
Det.Eng.
ZEIT
Zusammenhang zwischen dertechnischen und der
Qualifizierungsdokumentation
Abarbei tungDQ-Pl äne
Detai l-Layouts
Prozeßablaufpläne
Funkti onsdiagram me
ProtokolleAngebotsverhandlungen
(ggf. Änderungen derAnforderungen)
Quali fiz ierungs-Rahmenplan
(3)
(3)
(3)
(3)
(3)
Einfluß auf techn.Dokumente
P flichtenheft (FS)
Beginn GM P-Change-Control
As-built -Dokumentat ion
RahmenplanRV
Risikoanalyse
P läneProzeß-
Validierung
Risikoanalyse
P läne analyt .Methoden-Validierung
P läneReinigungs--Validierung
Analyt .Methoden-validierung
Prozeß-
validierung
Reinigungs-validierung
Abschluß-bericht
Abschluß-bericht
Abschluß-bericht
Approval ( FDA / EU)
Abschluß-bericht
RahmenplanPV
Abschluß-bericht
ZEIT
Quali fiz ierungs-Abschlußbericht
IQ-P läne
OQ-P läne
Abarbei tungQuali fiz ierung
SDS / HDS
SATS / HATS
FAT
IBN / SAT
Abnahme
Prüfungnach AbschlußIQ-
Protokolle
Prüfung nach AbschlußOQ-
Protokolle
Konst rukt ion / T echn.Dokumentat ion
Pharmaunternehmen
(1) eventuell teilw. Abarbeitung IQ/OQ (Vermeidung von Doppeltestung(2) Überprüfung bei Lieferanten-Audit(3) kein zeitli cher Ablauf , wi cht ige technische Dokumente (Beispiele)
QA-Genehmigungnotwendi g
(2)
Lieferant
Installat ion derAnlage
S tabili täts-chargen
Modul test
Integrationstest
(1)
(1)
Abarbei tung
Quali tätsplan
Complex structure of Qualification and Validation work and flow of information in projects! " Organization required
May 2015, page 18
The Validation Master Plan
Validation work needs to be organised!
Implement a „Validation Project Manual“
➩ the Validation Master Plan
VMP
WHO-Requirement 4.2 The key elements of a qualification and validation programme of a company should be clearly defined and documented in a validation master plan.

May 2015, page 19
The Validation Master Plan Annex 15 EU-GMP-Guide
i. Qualification and Validation policy; ii. The organisational structure including roles and responsibilities for
qualification and validation activities; iii. Summary of the facilities, equipment, systems, processes on site and the
qualification and validation status;
– equipment to be qualified – processes to be validated ➩ summarised and compiled in a matrix format
iv. Change control and deviation management for qualification and validation; v. Guidance on developing acceptance criteria;
" will be presented in the presentation on Risk Analyses vi. References to existing documents; vii. The qualification and validation strategy, including requalification, where
applicable.
The VMP or equivalent document should define the qualification validation system and include or reference information on at least the following:
May 2015, page 20
The Validation Master Plan
Valididation Master Plan(Validation Policy)
Plant 1 / Line 1 Plant 2 / Line 2 Plant 3 / Line 3 ...
QualificationPlan
ValidationPlan
QualificationPlan
ValidationPlan
QualificationPlan
ValidationPlan
Possible VMP / QP / VP Structure
Everything described in the VMP / QP / VP must be done!

May 2015, page 21
The Validation Master Plan
i. Qualification and Validation policy (firm‘s policy, general description), e.g.:
• Targets • Validity (for which Project - how long?)
• Basic Guidelines (WHO, EU- and US-FDA-cGMP-Guidelines)
• Information about general GMP-interpretation
• GEP issues
May 2015, page 22
The Validation Master Plan
ii. organisational structure of validation activities
• Organisational structure / organisational charts
• Tasks in the project: approve, check or prepare the documents
• Names of responsible personnel
• CVs / background • Matrix of responsibility (Task, function: responsible, carry out the
work, support the work)
• Description of the relationship between commissioning and qualification
• project schedule / reference to the project schedule • capacities / staffing
• training needs
May 2015, page 23
The Validation Master Plan
iii. Summary of the facilities, equipment, systems, processes on site and the qualification and validation status
• Building
• Rooms
• Production equipment
• Media supply
• Processes
• Methods
Detailed description provides good overview ➩ High effort for changes during the project!
Impact Assessment process has to be described!
May 2015, page 24
identify systems
„No Impact“ System
„Direct Impact“ System
develop supporting rationale
develop system boundaries
„Indirect Impact“ System
Impact on product qualify?
Linkd to DI-System?
no
no
yes
yes
Commissioning & Qualification Commissioning
GEP GEP and GMP
according to: ISPE Baseline Guide on Commissioning and Qualification, 2000
1. Step in Risk Assessment: Impact Assessment = Separation into GEP and GMP

May 2015, page 25
The Validation Master Plan iii. summary of facilities, systems, equipment and processed to be validated
Equipment System Explanation
City water Only raw material for pharmaceutical water; quality defined according tonational regulations
Chilled water Only for indirect cooling; no direct contact with product or productcontacts parts; direct measurement of product temperature wherenecessary
Iced water Only for indirect cooling; no direct contact with product or productcontacts parts; direct measurement of product temperature wherenecessary
Plant steam Only for indirect heating; no direct contact with product or productcontacts parts; direct measurement of product temperature wherenecessary
Humidification for HVAC system Requirements of steam to be specified during RA ‚HVAC system‘
Waste water system Not in contact with production equipment; risk for contamination ofequipment will be covered during qualification of production equipmentand rooms
Electrical power supply / UPS - system No direct influence on product quality; reaction on failure of power supplywill be checked during qualification of equipment
Building Management System; IT – systems andtelecommunication
GMP relevant data will not be handled by these systems, only for systemcontrols; GMP relevant data will be covered by separate systems
HVAC system for packaging area To be decided during Risk analysis ‚HVAC‘ system
Example for �supporting rationale� for indirect and no impact systems
May 2015, page 26
The Validation Master Plan
iii. List of systems and processes, e.g.:
Description of Validation Activities during the Phases: • Process transfer(from existing to new plant) • Process Optimisation • Cleaning Validation • Process Validation
Similar to Qualification:
• Operational Organisation (Organisational charts, tasks...) • Processes to be validated (Matrix) • Document format

May 2015, page 27
Qualification Matrix – Result of the Impact Assessment
Qualification Steps System / Equipment
RA DQ IQ IOQ OQ PQ
Formulation system incl. CIP/SIP
Formulation Tank 1
Formulation Tank 2
Diluent tank
Al-Gel diluent tank
x x x x
Weighing scales
x
x - x - - Stopper treatment unit and transfer station x x x - x x Steam autoclave x x x - x x Clean steam generation and distribution x x x - x x LF-units x x x - x x HVAC-System x - x x Environmental Monitoring System x - x - Rooms
x x
- x - x Compressed Air Generation and Distribution x x x - x x Purified Water Generation and Distribution x x - x x WFI-Generation and Distribution
x
x x - x x
The Validation Master Plan
Example to show possible structure!
May 2015, page 28
The Validation Master Plan
v. Change control and deviation management for qualification and validation;
• Reference to existing procedure (SOP?) • Definition of new and project specific process
– Define project stage for start of CC – Who has to inform about changes? – Who has to approve the changes?
– Deviation management may be different from routine process because deviations during qualification and validation do not impact on patient safety!
➩ Simple process for quick decisions!

May 2015, page 29
The Validation Master Plan
vi. Reference to existing documents
• SOPs • List of abbreviations • Definitions • Literature, Codes and Standards, Guidelines • Attachments (Layouts, P&IDs)
vi. The qualification and validation strategy, including requalification, where applicable.
• frequency of requalification • when to revalidate
May 2015, page 30
The Validation Master Plan
Further possible and common subjects:
• general acceptance criteria
• how to choose and involve suppliers
• training of personnel
• how to carry out Risk Analyses
• documentation format - document structure
- numbering system
- example document
- way from protocol " report

May 2015, page 31
The Validation Master Plan
The VMP
• overview
• official GMP-document
• is required due to regulations
• available to all project team members
• read and recognised by all project team members
• importance for equipment suppliers
The VMP is a summary document: ➩ brief, concise and clear!
May 2015, page 32
Disclaimer: • This presentation has been prepared upon best knowledge and it presents the view and
experience of Pharma Consulting Walther.
• The presentation reflects current guidelines, knowledge and thinking. Changes of regulatory guidelines or interpretation thereof or new regulatory expectations may change the applicability of the contents of this presentation.
• Parts of the presentation may not be understood or misinterpreted without the verbal explanation given during the seminar.
• Although the statements made in the presentation in general have been presented to and discussed with inspectors, a different approach may be required and adequate depending on the individual situation.
• The information contained herein may be changed without prior notice.
Contact: Pharma Consulting Walther Dr. Ingrid Walther Rosenweg 36 61381 Friedrichsdorf – Germany +49-(0)6172-958281 +49-(0)172 654 8834 www.consulting-walther.com [email protected]


















