
file
122
Research Collection Doctoral Thesis Zur Identitäts-, Reinheits- und Gehaltsprüfung neuerer Sulfonamid-Substanzen und -Tabletten Author(s): Hafner, Franca Publication Date: 1965 Permanent Link: https://doi.org/10.3929/ethz-a-000095031 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection . For more information please consult the Terms of use . ETH Library
Transcript of file

Research Collection
Doctoral Thesis
Zur Identitäts-, Reinheits- und Gehaltsprüfung neuererSulfonamid-Substanzen und -Tabletten
Author(s): Hafner, Franca
Publication Date: 1965
Permanent Link: https://doi.org/10.3929/ethz-a-000095031
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Prom. Nr. 3598
Zur Identitäts-, Reinheits¬
und Gehaltsprüfung neuerer
Sulfonamid-Substanzen und -Tabletten
Von der
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
zur Erlangung
der Würde eines Doktors der Naturwissenschaften
genehmigte
PROMOTIONSARBEIT
vorgelegt von
FRANCA HAFNER
eidg. dipl. Apotheker
von Zürich
Referent: Herr Prof. Dr. J. Büchi
Korreferent: Herr Prof. Dr. X. Perlia
Steinbauer & Rau, Mündien 2
1965

Meiner lieben Mutter
Dem Andenken meines guten Vaters

Meinen sehr verehrten Lehrern Herrn Prof. Dr. J. Büchi und Herrn Prof.
Dr. X. Perlia möchte ich für die vielen wertvollen Ratschläge und für das
mir entgegengebrachte Wohlwollen bestens danken.
Auch Herrn R. Schwegler danke ich herzlich für seine stete Hilfsbereit¬
schaft.

INHALTSÜBERSICHT
Seite
1. Einleitung und Problemstellung 1
2. Allgemeiner Teil 3
2.1. Bewertung und Wahl von Methoden zur Gehaltsbestimmungvon Arzneistoffen und -Präparaten 3
2.1.1. Genauigkeit 3
2.1.2. Empfindlichkeit 4
2.1.3. Spezifität 4
2.1.4. Schwierigkeitsgrad 4
2.1.5. Aufwand an Zeit, Apparatur und Material 5
2.2. Anwendung der Statistik bei der Gehaltsbestimmung von
Arzneistoffen und -Präparaten 5
2.2.1. Allgemeines 5
2.2.2. Ausführungen und Berechnungen 8
2.2.2.1. Vergleich verschiedener Methoden 8
Bestimmungen mit Streuungszerlegung 8
Sicherung des Bestimmungsbereiches 11
2.2.3. Festlegung der Gehaltslimiten 12
2.2.4. Berechnung der Regressionsgeraden 1?
2.3. Anwendung der Statistik bei der Gehaltsbestimmungvon Tabletten 21
3. Spezieller Teil 25
3.1. Ausarbeitung von Prüfungsvorschriftenfür einige neuere Sulfonamide 2">
3.1.1. Carbutamidum ili
3.1.2. Tolbutamidum 41
3.1.3. Chlorpropamidum 51
3.1.4. Acetazolamidum 60
3.1.5. Probenecidum 68
3.1.6. Sulfamethizolum 76
3.2. Ausarbeitung von Prüfungsvorschriftenfür einige Sulfonamid-Tabletten 85
3.2.1 Allgemeines 85
3.2.2. Aufbau der Prüfungsvorschriften für Tabletten 85
3.2.3. Allgemeine Bemerkungen zu den einzelnen Prüfungsabschnitten 86
3.2.4. Prüfungsvorschriften der einzelnen Sulfonamid-Tabletten .... 91

- II -
Seite
3.2.4.1. Compressi Carbutamidi 500 mg 91
3.2.4.2. Compressi Tolbutamid! 500 mg 95
3.2.4.3. Compressi Acetazolamidi 250 mg 98
3.2.4.4. Compressi Probenecid! 500 mg 101
3.2.4.5. Compressi Sulfamethizoli 500 mg 105
3.2.4.6. Compressi Trisulfonamidici 500 mg 108
4. Zusammenfassung 113
5. Literatur 114

- 1 -
1. Einleitung und Problemstellung
Im Zusammenhang mit der Neubearbeitung der Pharmakopoea Helvetica
Sexta (Ph.Helv.Vl) wurde die Ausarbeitung der Identitäts-, Reinheits- und
Gehaltsbestimmung folgender Sulfonamide übernommen:
Reinsubstanzen
Carbutamidum
Tolbutamidum
Chlorpropamidum
Acetazolamidum
Probenecidum
Sulfamethizolum
Tabletten
Compressi Carbutamidi
Compressi Tolbutamidi
Compressi Acetazolamidi
Compressi Probenecidi
Compressi Sulfamethizoli
Compressi Trisulfonamidici
Dabei stellten wir uns unter anderem die Aufgabe, die Wahl der Methode zur
Gehaltsbestimmung und die Festlegung der Gehaltslimiten am Beispiel von
Carbutamidum durch statistische Bewertung vorzunehmen.

Leer - Vide - Empty

- 3 -
2. Allgemeiner Teil
2.1 Bewertung und Wahl der Methoden zur Gehaltsbestimmung von
Arzneistoffen und Präparaten
Wir haben uns vorerst Rechenschaft zu geben über die Frage, welche Gesichts¬
punkte bei der Wahl einer geeigneten Gehaltsbestimmungsmethode für eine
Pharmakopöe-Monographie berücksichtigt werden müssen.
2.1.1 Genauigkeit
Eine genau arbeitende quantitative Bestimmungsmethode soll Analysenresul¬
tate ergeben, welche dem Sollwert, resp. dem effektiven Wert möglichst nahe
liegen.
Mit der Begründung, dass eine Wirkstoffdifferenz von i 2% keinen Einfluss
auf die therapeutische Wirkung ausübe, wird eine allzu grosse Bestimmungs¬
genauigkeit und eine hohe Gehaltsforderung an die Arzneimittel oft als nicht
erforderlich betrachtet. Ohne genaue Analysenmethode ist unseres Erachtens
eine sinnvolle Gehaltsbestimmung nicht möglich. Aus folgenden Gründen be¬
fürworten wir die Wahl möglichst genauer Gehaltsbestimmungsmethoden für
Pharmakopöezwecke:
a) Die Gehaltsbestimmung stellt auch eine Reinheitsprüfung dar und limi¬
tiert eventuell vorhandene unerwünschte Nebenstoffe und Zersetzungs¬
produkte.
b) Arzneistoffe mit geringer therapeutischer Breite müssen genau dosiert
werden können.
c) Der Wirkstoffgehalt in Arzneipräparaten wird durch die Arzneiformung
zusätzlich beeinträchtigt.
d) Die Tendenz des Arzneimittelhandels meistens die untere Grenze der
Gehaltsforderung einzuhalten.
Um die Genauigkeit einer Bestimmungsmethode eindeutig zu ermitteln, sind
die Resultate statistisch zu bewerten. Eine solche statistische Auswertung be¬
dingt aber infolge der vielen erforderlichen Bestimmungen und den Berech¬
nungen einen grossen Arbeitsaufwand.

_ 4 -
2.1.2 Empfindlichkeit
Unter Empfindlichkeit kann man unseres Erachtens
a) die Erfassung möglichst kleiner Stoffmengen
oder
b) die Erfassung von kleinen Mengenunterschieden
verstehen. Der erste Fall ist für die Pharmakopöebestimmungen nur bei sehr
teuren Arzneistoffen (Oekonomie) von Bedeutung, wobei der durch die Ver¬
dünnung bewirkte Multiplikationsfehler bewusst in Kauf genommen werden
muss. Bei der Gravimetrie und der Titrimetrie ist die Grösse des Umrech¬
nungsfaktors ein Mass der Empfindlichkeit. Die Empfindlichkeit gemäss b
kann durch grössere Substanzeinwaagen erhöht werden. Die photometrischen
Methoden erfassen sehr kleine Substanzmengen (Empfindlichkeit gemäss a
gross) und ist deshalb für Bestimmungen von verdünnten Lösungen geeignet;
sie erreicht aber die Empfindlichkeit gemäss b der Gravimetrie und der
Titrimetrie nicht. Subjektive Bestimmungen beeinträchtigen selbstverständ¬
lich die Empfindlichkeit, z.B. Kolorimetrie, Polarimetrie.
Der empfindlicheren Methode ist nur dann der Vorzug zu geben, wenn sie
auch genau ist, d.h. wenn sie keinen systematischen Fehler aufweist, also
der Durchschnittswert nicht wesentlich vom Sollwert abweicht.
2.1.3 Spezifität
Ein arbeitstechnisch genau festgelegtes Verfahren ist spezifisch, wenn es
bestimmte Elemente oder Molekeln ohne Störung durch die überwiegende
Mehrzahl anderer Elemente oder Molekeln erfasst. Die Selektivität, d.h. die
Spezifität in Gemischen genau bekannter Zusammensetzung, und die Spezifi¬
tät ist besonders bei Arzneipräparaten wichtig, da sie gestatten die Arznei¬
stoffe neben den zur Arzneiformung benützten Hilfsstoffen zu bestimmen.
2.1.4 Schwierigkeitsgrad
Die einfach arbeitenden und durchführbaren Verfahren sind vorzuziehen,
weil sie weniger Fehlerquellen besitzen und zudem Zeit gespart werden kann.
Die physikalischen Methoden sind meistens einfacher als die chemischen Ver¬
fahren und sind vor allem den Methoden mit Ausschüttelungen und jenen mit
Fällungsreaktionen vorzuziehen.

- 5 -
2.1.5 Aufwand an Zeit, Apparatur und Material
Beim heutigen Personalmangel fällt der Zeitaufwand am meisten ins Gewicht
und rechtfertigt die Anschaffung von Apparaturen zur Abkürzung der Bestim¬
mungszeit. Der Materialkosten wegen wird bei sehr teuren Arzneistoffen, wie
Cyanocobalamin und Ergometrin, durch Verwendung einer Methode mit hoher
Empfindlichkeit gemäss a (z.B. Spektrophotometrie) Rechnung getragen.
Obige Überlegungen führen uns dazu, einfachen und spezifischen Methoden
den Vorzug zu geben, auch wenn sie kostspieligere Apparaturen und mehr
Substanz erfordern würden.
2.2 Anwendung der Statistik bei der Gehaltsbestimmung von Arzneistoffen
2.2.1 Allgemeines
Bei der Bearbeitung eines Pharmakopöeartikels stellt sich die Frage, wie
in nützlicher Zeit diejenige Vorschrift zur Gehaltsbestimmung gefunden wer¬
den kann, welche statistisch gesichert die genauesten Werte ergibt und auf
Grund deren Auswertung eine vernünftige Gehaltslimite gefordert werden kann.
Wir überprüfen vorerst bereits in Vorschlag gebrachte oder schon verwendete
Methoden anhand einer Standardsubstanz, weil die Pharmakopöemethoden
meist aus den schon vorhandenen Bestimmungsmethoden ausgewählt werden.
Wir sind uns bewusst, dass mit folgender Anordnung nicht alle systematischen
Fehler aufgedeckt werden. Durch die Anwendung reiner Standardsubstanzen
und chemisch verschiedener Bestimmungsmethoden, welche sicher nicht die
gleichen Fehler bewirken, sowie durch die Sicherung des Bestimmungsberei¬
ches werden die mit dieser Anordnung nicht erfassbaren Fehler ausgeschlos¬
sen.
Wir führen dann kurz die theoretisch sicherste Auswertung aus, welche unse¬
res Erachtens unumgänglich ist, wenn eine einzige oder chemisch gleiche
Methoden durchgeführt wird und wenn keine Standardsubstanz erhältlich ist.
In der Theorie beschränken wir uns auf die für unsere Untersuchungen not¬
wendigen Kenntnisse und Gleichungen der Statistik.
Fehlerquellen bei Gehaltsbestimmungen
Die Fehler setzen sich aus vielen Faktoren zusammen und können in zwei
Gruppen aufgeteilt werden, in systematische Fehler mit einer bestimmbaren
Grösse und zufällige Fehler mit einer undefinierbaren Grösse.

- 6 -
Systematische Fehler sind durch Bestimmungsverfahren, Arbeitsgerät, Ver¬
unreinigungen der Substanz und Arbeitstechnik verursachte Abweichungen,
die praktisch vermeidbar und rechnerisch eliminierbar sind.
Hierher gehören:
Fehler der Methode: Gravimetrie, Löslichkeit der Fällung,
Nachfällung,
Flüchtigkeit.
Titrimetrie, unvollständige, chemische Reaktion,
Nebenreaktionen.
Fehler des Analytikers: z.B. Objektivität,
Ungenauigkeit,
Unkenntnis.
Fehler der Apparatur: z.B. Kalibrierung (innerer Fehler),
Temperatur (äusserer Fehler).
Die systematischen Fehler sind entweder additiv (z.B. Löslichkeit der Fäl¬
lung) oder proportional (z.B. Verunreinigung einer Standardlösung). Um den
proportionalen Fehler aufzuzeigen, muss die Bestimmung immer mit ver¬
schiedenen Substanzeinwaagen durchgeführt werden.
Zufällige Fehler sind beim gewählten Bestimmungsverfahren, Arbeitsgerät
und personell bedingter Arbeitstechnik unabhängig vom Willen des Analytikers.
Sie lassen sich durch eine grosse Anzahl Bestimmungen verkleinern. Bei Nor¬
malverteilung bilden die Häufigkeiten eines Wertes eine Gauss'sche Glocken¬
kurve.
Musterentnahme
Eine wichtige Voraussetzung für eine einwandfreie Arzneimittelprüfung ist
die Verwendung eines Analysenmusters, welches ein repräsentatives Durch¬
schnittsmuster der gesamten zu untersuchenden Stoffmenge darstellt. Für die
Musterentnahme können keine allgemein gültigen Regeln aufgestellt werden,
da diese von der Art und Menge des Untersuchungsmaterials abhängen (18, 19).
Während die Ph.Helv.V lediglich vorschreibt, dass die Untersuchungen der
Arzneimittel an zweckmässig entnommenen Durchschnittsproben vorzuneh¬
men sind, wird die Musterentnahme in der Ph.Helv.VI genauer umschrieben.
Die Vorschriften für chemische Arzneistoffe werden lauten:

- 7 -
"Für Arzneimittel in nichtabgeteilter Form gelten die folgendenVorschriften:
Der Inhalt von einzelnen Packungen, welche bis 5 kg Nettoge¬wicht feste Arzneimittel mit Einzelpartikeln von höchstens 1 cm
Durchmesser enthalten sowie von flüssigen und salbenartigenArzneimitteln ist vor der Probeentnahme gut durchzumischen.
Dieser Mischung wird ein Analysenmuster entnommen.
Enthält eine Packung mehr als 5 kg, so sind aus jedem zu un¬
tersuchenden Behälter an drei gleichmässig über das ganze Vo¬
lumen verteilten Orten angemessene Mengen zu entnehmen, wo¬
zu vorteilhaft ein Musterstecher oder ein Saugrohr verwendet
wird. Die drei, wenn nötig zerkleinerten Proben sind sorgfältigzu vermischen. Diese Mischung stellt das Analysenmuster dar.
Teilweise erstarrte, flüssige (z.B. Olivenöl) oder teilweise ver¬
flüssigte, feste Arzneimittel müssen vor der Probeentnahme ge¬schmolzen werden. Bei Flüssigkeiten, in denen sich Kristalle
ausgeschieden haben, sind diese durch vorsichtiges Erwärmenin Lösung zu bringen.
Liegen vom gleichen Arzneimittel mehrere Packungen vor, so
sind die Proben aus den verschiedenen Packungen gemäss unten¬
stehenden Angaben zu entnehmen und zu einem Gesamtmuster zu
mischen.
Gesamtzahl der Packungen Anzahl Packungen aus
denen Muster zu ent¬
nehmen sind:
2 - 5 mindestens 2
6-10 mindestens 3
11-25 mindestens 4
26 - 50 mindestens 5
51 -100 mindestens 8
über 100 mindestens 10
Lässt schon die Sinnenprüfung bei der Musterentnahme erkennen,dass die einzelnen Packungen von sehr ungleicher Beschaffenheit
sind (z.B. Farbe, Geruch, Aggregatzustand), so müssen noch wei¬
tere Packungen gemustert werden.
Bei hygroskopischen oder rasch verwitternden Arzneimitteln ist
durch rasches Arbeiten und Verwendung dicht schliessender Behäl¬
ter nach Möglichkeit jede Veränderung während der Probeentnahme
zu vermeiden.
In den allgemeinen Bestimmungen, allgemeinen Artikeln und Mono¬
graphien können besondere Vorschriften für die Probeentnahme ge¬
macht werden, die von diesen Bestimmungen abweichen."
Die von uns untersuchten Arzneistoffmuster erhielten wir direkt von den Her¬
stellerfirmen in Mengen von 10-20 g, weshalb wir sie, nach gründlicher
Durchmischung alle als Analysenmuster betrachten konnten.

Verwerfung stark abweichender Resultate
Nach JENKINS, CHRISTIAN und HAGER (18) darf ein Bestimmungswert ver¬
worfen werden, wenn mindestens vier Bestimmungen vorliegen und das ab¬
weichende Resultat eine vierfache Abweichung im bezug auf die durchschnitt¬
liche Abweichung zeigt; nach KOLTHOFF und ELVING (19) darf dies erfol¬
gen, wenn die Differenz zum nächstliegenden Wert das 1,5fache der ganzen
Variationsbreite betragt. Wir arbeiteten nach der Auffassung von LINDER
(21), der einen Wert nur verwirft, wenn ein bewusster konkreter Grund da¬
für vorliegt.
2.2.2. Ausführungen und Berechnungen
2.2.2.1 Vergleich verschiedener Methoden
2.2.2.1.1. Bestimmungen mit deren Streuungszerlegung
Die Substanz "Carbutamidum" durch Umkristallisation gereinigt und durch
Elementaranalyse geprüft, bestimmten wir nach den vier folgenden Bestim¬
mungsmethoden:
Argentometrie (nach Ph.Helv.V "Sulfadimidin")
Bromometrie, , ,, , .„,
nach den Vorschriften
Acidimétrie des Artikels "Carbuta-
TT, c . „.. i-midum" S. 33, 34, 35
Wasserfreie Titration
Mit der chemisch optimalen Substanzmenge sind mindestens fünf Bestim¬
mungen durchzuführen. Wir entschlossen uns deren 10 vorzunehmen.
Argentometrie Bromometrie Acidimétrie Wasserfreie
Titration
100 % = 100 % = 100 % = 100 % =
250 mg 100 mg 500 mg 200 mg
ai bi ci di
98,24 % 100,20 % 99,56 °'o 99,94 %
98,91 100,28 99,75 99,99
98,91 100,22 99,95 99,96
97,31 100,05 99,45 100,25
98,91 99,66 100,40 100,42
98,68 99,85 100,08 100,95
98,04 99,52 100,76 100,08
98,74 99,42 100,18 100,83
98,51 99,74 100,01 100,07
98,74 99,70 99,54 100,13
984,99 = A 998,64 = B 999,68 = C 1002,62 = D

- 9 -
Obiges Zahlenmaterial unterwarfen wir der Streuungsverlegung, um beurtei¬
len zu können, ob wesentliche Unterschiede zwischen den Durchschnitten be¬
stehen.
I = Summe der Quadrate aller einzelnen Bestimmungen
= a? + a|+
...+ a2 + b? + b§ +
...+ b2 + c2 + ci +
...+ c2 + df +
...+ d21 ^
na nb nc nd
II = Summe der Quadrate der Summe der Gruppenbestimmungen dividiert
durch die Anzahl Bestimmungen pro Gruppe (n)
na nb nc nd
III = Quadrat der Summe aller Bestimmungen dividiert durch die Anzahl
aller Bestimmungen (N).
(A + B + C + D)2=
(A + B + C + D)2=
J?na + nb + nc + nd
"
M-n* N
gilt, wenn na=
nb=
nc=
nd .
Ursache Freiheitsgrad Summenquadrat Durchschnitts¬
FG SQ quadrat DQ
zwischen
Gruppen
M - 1 II - inWZT
- °Q(G>
innerhalb
Gruppen
N - M I - II nIm = °Q(R)
M - 1 = Anzahl Gruppen - 1
N - M = Anzahl aller Bestimmungen - Anzahl Gruppen
.
DQ(G)DQ(R)
mit nt = FG zwischen Gruppen und
na = FG innerhalb Gruppen
Fber.
FTab.

- 10 -
Streuungszerlegung (4 Methoden)
Ursache FG SQ DQ Fber.
zwischen
Gruppen
innerhalb
Gruppen
4-1 = 3
40-4 = 36
19,4626
5,9104
6,4875
0,1642
39,515
FTab.: Fo-05; 3; 36= 2'88
^o,oi; 3 ;36= 4,41
Fo.ooj; 3 ;36= 6,81
Fber. >> F0-00*
Schlussfolgerung; Es besteht mindestens ein stark gesicherter Unterschied
zwischen den Durchschnittswerten. Der tiefere Durchschnittswert der argen-
tometrischen Bestimmung lässt vermuten, dass diese Methode den Fehler be¬
wirkt. Dies wurde durch die Streuungszerlegung der restlichen drei Methoden
bewiesen, indem diese keinen signifikanten Unterschied ergaben.
Streuungszerlegung (3 Methoden)
Ursache FG SQ DQ Fber.
zwischen 3-1 = 2 0,8522 0,4261
Gruppen
3,23innerhalb 30-3 = 27 3,5632 0,1320
Gruppen
rTab.: Fo,o5; 2; 27= 3,354
Fo,oi; 2; 27= 5,488
Fo.ooi; 2; 27= 9,020
?ber. < Fo,o5
Schlussfolgerung: Es besteht kein wesentlicher Unterschied zwischen den
Durchschnittswerten der bromometrischen, acidimetrischen und wasser¬
freien Bestimmung. Wir verwarfen die argentometrische Methode, weil
Änderungen in der Bestimmung zu keiner Verbesserung führten.

- 11 -
2.2.2.1.2 Sicherung des Bestimmungsbereiches
Da die Substanz-Einwaage nach Ph.Helv.V und i 10% schwanken darf und auch
der proportionalen Fehler wegen müssen wir prüfen, ob die Methoden mit den
gleichen Reagenzienmengen in diesem Mengenbereich genaue Resultate ergibt.
Deshalb ermittelten wir je fünf weitere Werte mit vorgeschriebener Substanz¬
einwaage (Werte siehe S. 8) zu bzw. abzüglich 20% und unterwarfen die pro
Methode bestimmten Resultate der Streuungszerlegung.
Bromometrie
98,86 %
99,49
98,64
99,17
98,85
49 3,12 = 5x
Acidimétrie
99,70 %
99,61
100,71
100,54
99,75
500,31 = 5x
Wasserfreie
Titration
Einwaage 80 mg Einwaage 480 mg Einwaage 200 mg
100,33 %
100,27
100,70
100,81
100,09
502,20 = 5x
Einwaage 120 mg
98,00 %
98,95
98,41
99,11
98,65
493,12 5x
Einwaage 720 mg
99,31 %
100,31
99,41
99,31
99,03
497,37 = 5x
Einwaage 300 mg
99,44 %
99,94
100,63
100,06
99,88
499,95 = 5x
Streuungszerlegung (drei Methoden einzeln)
Ursache FC SQ DQ Fber.
Rromometrie
zwischen 3-1 = 2 5,8803 2,9402
Gruppen24,195
innerhalb 20-3 = 17 2,0658 0,1215
Gruppen
Acidimétrie
zwischen 1,0644 0,5322
Gruppen2,531
innerhalb 3,5740 0,2102
Gruppen
Wasserfreie
Titration
zwischen 0,2764 0,1382
Gruppen1,031
innerhalb 2.2790 0,1341
Gruppen

- 12 -
FTab.: F°i°3>' 2; 17= 3,592
^0,01; 2; 17= 6,1 12
Fo.001; 2; 17= 10,659
Schlussfolgerung: Aus den obigen Werten ist ersichtlich, dass alle drei
Methoden innerhalb der Gruppen eine kleine Standardabweichung zeigen, dass
aber die bromometrische zwischen den Gruppen signifikant verschieden ist.
Dies ist auch chemisch erklärbar, da der Bromüberschuss von Bedeutung ist.
Die bromometrische Methode ist auch zu verwerfen, wenn nicht eine ganz ge¬
naue Substanzeinwaage gefordert werden kann.
Ob die acidimetrische oder die wasserfreie Titration vorgeschlagen werden
soll, kann statistisch nicht gelöst werden; die Grösse der Standardabwei¬
chung, die Spezifität der Methode, der Arbeits- und Kostenaufwand werden
hier ausschlaggebend sein. Der Einfachheit, der Irrtumsmöglichkeiten und
des Aufwandes an Zeit und Material wegen ist der acidimetrischen Methode
den Vorzug zu geben. Die wasserfreie Bestimmung weist dafür eine kleinere
Standardabweichung auf.
2.2.3 Festlegung der Gehaltslimiten
Durch den zufälligen Fehler einer Gehaltsbestimmung ist der Gehaltslimite
eine Grenze gesetzt, welche wie der Vertrauensbereich des Durchschnittes
durch die Anzahl der Bestimmungen in der Grössenordnung 1/VTT gegen Null
konvergiert. Dienten zur Berechnung von s resp. von dessen absolutem Wert
mehr als Tausend Bestimmungen, so ist der Vertrauensbereich (95%) einer
einzigen Bestimmung 2 Cf.
Prüfung von Handelsmustern
Bei der Synthese oder Gewinnung von Arzneimitteln sind dem Reinheitsgrad
bestimmte Grenzen gesetzt (geringe Stabilität, zu hohe Reinigungskosten etc.).
Um festzustellen, ob die handelsüblichen Qualitäten vom Sollwert (= 100% Ge¬
halt) abweichen, führten wir an vier Carbutamid-Handelsmustern folgende Ge-
haltsbestimmungen und Berechnungen durch.

- 13 -
Acidimétrie
Handelsmuster
I II HI IV
99,42 %
99,19
99,58
3xj
99,53 %
99,55
100,62
98,78 %
98,80
99,91
99,71
45 in
99,56 %
99,59
100,65
100,39
298,19 = 299,70 = 3xrj 397,28 = 400,19 = 4xrv
99,40 = % 99,90 =
xj 99,32 = 5 HI 100,05 =
xry
0,148 =
SI 0,582 =
sTT 0,623 =
SIH 0,556 =
Sjy
Die Standardabweichung s der einzelnen Muster bestimmten wir nach folgen¬
der Gleichung:
S (x.. - x.)2LI i
N. - 1l
Wasserfreie Titration
Handelsmuster
I II HI IV
99,47 %
99,45
99,73
99,58
4XI
100,45 %
100,19
100,20
100,25
100,05
100,00
6xn
99,39 %
99,61
99,84
100,00
45III
100,51
99,77
99,77
100,39
%
398,23 = 601,14 = 298,84 = 400,44 = 4xIV
99,58 = XI 100,19 = 5II 99,71 =
XHI 100,11 =
xiv
0,13 =
SI 0,16 =
SII 0,27 =
SIII 0,39 =
siv
Vergleich der Streuungen der untersuchten Carbutamid-Handelsmuster
Bevor wir den Durchschnitt der einzelnen Handelsmuster mit dem Sollwert
vergleichen können, müssen wir prüfen, ob die Standardabweichungen der
Handelsmuster von dem der Standardsubstanz signifikant abweichen.
Die Standardabweichung der Standardsubstanz berechnen wir aus den Werten
der Streuungszerlegung (siehe S. 11) wie folgt:

- 14 -
st= wh [(M-1)DQ(G) + (N-M)DQ(R)]
st, Acidimétrie= °*49 st, wasserfreie Titration
= °'37
Diese Gleichung gilt nur, sofern der F-Test für Gruppen (siehe S. 11) auf
Homogenität schliessen lasst.
Da bei der wasserfreien Titration nur ein Handelsmuster eine grössere
Standardabweichung zeigt als die der Standardsubstanz st=
st . • (nur diese
müssen geprüft werden), eignet sich der F-Test zur Feststellung eines even¬
tuellen Unterschiedes.
s2„
_
Handelsmuster
*ber.~
ö Handelsmuster > st
st
mit FGt = Anzahl Musterbestimmungen - 1
FTab
mit FG2 = Anzahl Standardbestimmungen - 2
s2__,
Handelsmuster IV. „c
Fber.= =
1'65
s
t, wasserfreie Titration
FTab. 0,05; 3; 1 8" 3'1 6
Schlussfolgerung: Die Standardabweichung des Handelsmusters IV ist nur
zufällig von der der Standardsubstanz verschieden. Besser ist es jedoch auch
mit den Werten der Handelsmuster die Streuungs Zerlegung auszuführen, weil
dadurch die verschiedenen Handelsmuster miteinander verglichen werden.

- 15 -
Streuungszerlegung (Handelsmuster)
Ursache FG SQ DQ Fber.
Acidimétrie
zwischen 3 1,4428 0,4809
Gruppen
1,633innerhalb 10 2,9450 0,2945
Gruppen
Wasserfreie
Titration
zwischen 3 1,283 0,427
Gruppen
6,97innerhalb 14 0,8595 0,0614
Gruppen
Fo,o5; 3; io= 3''71 Fo.oi; 3; 14
" 5,563
Die mit der acidimetrischen Methode bestimmten Handelsmusterdurchschnitte
zeigen keinen signifikanten Unterschied, wohl aber die mit der wasserfreien
Titration gefundenen.
Die Standardabweichung der Handelsmuster wird aus den Werten der Streu¬
ungszerlegung, wie oben erwähnt, berechnet.
Handelsmuster, Acidimétrie
^Handelsmuster, Wasserfreie Titration
=2
0,17
Handelsmuster, Acidimétrie
s2t, Acidimétrie
Fo,OS;12-18 = 2,34
= 1,408 = Fiber.
Fber. < Fo,o
Schlussfolgerung: Die Standardabweichungen der Handelsmuster weichen bei
beiden Methoden nur zufällig von jener der Standardsubstanz ab.

- 16 -
Abweichung der Handelsmusterdurchschnitte vom Sollwert
(100 - x) Vi?
atW.
n = Anzahl Handelsmusterbestimmungen
N = Anzahl Standardbestimmungen
FTab mit FGj = n-1 und FG2 = N-2
Acidimétrie
Wasserfreie Titration
^.0,01:18= 2>861
I II III IV
,156 0,3535 2,776 0,2041
,257 1,2576 1,567 0,5946
tTab.O, 05:; 182 ,101
Schlussfolgerung: Obige Resultate zeigen, dass der Durchschnitt des Handels¬
muster I bei beiden Methoden eine wahrscheinliche Abweichung vom Sollwert
ergibt. Der Durchschnitt des Musters III weist nur bei der acidimetrischen
Methode eine wahrscheinliche Abweichung vom Sollwert auf.
Der Vertrauensbereich des Handelsmusterdurchschnittes, welcher angibt
in welchen Bereich der Durchschnitt von n Bestimmungen bei einer ange¬
nommenen Wahrscheinlichkeit p und einer bekannten Standardabweichung s
fällt, zeigt einen signifikanten Unterschied vom Sollwert an, wenn dieser
Sollwert ausserhalb des berechneten Vertrauensbereiches liegt.
Vertrauensbereich =
-, t • sx ±
Vn1
Wo.os.-is= 2'101
Ismus3ter Acidimétrie Wasserfreie Titration
I 99,40 + 0,59% 99,58 ± 0,39%
II 99,90 i 0,59 100,19 i 0,32
III 99,32 ± 0,52 99,71 + 0,39
IV 100,05 ± 0,52 100,11 + 0,39
Wir schlagen vor, dass der Sollwert minus die durchschnittliche Abweichung
+ t • s/3 gefordert werden soll, wenn die Hälfte der Handelsmuster einen si¬
gnifikanten Unterschied aufweist.

- 17 -
Wir schlagen auch vor, dass von der Parmakopöe drei Gehaltsbestimmungen
gefordert werden sollen, weil der Vertrauensbereich von einer bis drei Be¬
stimmungen merklich abnimmt. Auf eine vierte Bestimmung kann verzichtet
werden, weil diese vierte Bestimmung den Vertrauensbereich nur noch wenig
verkleinert. Die untenstehende Aufstellung gibt die Verhältnisse für die Acidi¬
métrie wieder:
Anzahl Bestimmungen Vertrauensbereich
1 x+ 1,03%
2 x+ 0,73
3 x+0,59
4 x+ 0,52
Wenn ein kleiner Vertrauensbereich erwünscht ist, eignet sich die wasser¬
freie Titration besser als die acidimetrische und die Gehaltslimiten könnten
für 3 Bestimmungen auf 99,5 - 100,5 festgelegt werden.
2.2.4 Berechnung der Regressionsgeraden
Im folgenden führen wir auch noch die theoretisch sicherste statistische Aus¬
wertung zur Bestimmung des systematischen prozentualen Fehlers aus, welche
sehr zeitraubend ist und nur auszuführen ist, wenn nur eine einzige oder
chemisch gleiche Methoden durchführbar ist oder wenn keine Reinsubstanz zur
Verfügung steht.
Es wurden 10 acidimetrische Bestimmungen mit verschiedenen Substanzein¬
waagen (Carbutamid-Standardsubstanz) durchgeführt. Vorteilhaft ist es die
Einwaage so zu wählen, dass eine arithmetische Reihe entsteht.
Acidimetrische Bestimmung
eingewogene Menge x bestimmte Mer
305,29 mg 302,48 mg360,05 359,25
423,94 421,63
494,15 491,96
568,69 567,26
593,33 590,09
650,80 655,75
713,95 710,59
781,58 775,98
925,93 927,72

- 18 -
Wenn die Bestimmungen weder systematische noch zufällige Fehler enthielten,
würde die Darstellung der eingewogenen Menge als Abszisse und der Bestim¬
mungen y als Ordinate Punkte ergeben, die genau auf der Geraden liegen wür¬
den, welche den Winkel zwischen den Koordinaten halbiert, mit folgender
Gleichung (Fall a):
y= a + bx
in der das konstante Glied gleich Null und der sogenannte Regressionskoeffizient
b gleich 1 zu setzen wären (siehe Abb. 1).
Als Fall b können wir annehmen, dass alle Bestimmungen einen systemati¬
schen Fehler vom gleichen Betrag a enthalten. Dieser von der Einwaage un¬
abhängige Fehler kann selbstverständlich positiv oder negativ sein. Die Glei¬
chung lautet:
y =
a0 + x
Einen weiteren Fall c erhalten wir, wenn wir annehmen, dass der systema¬
tische Fehler proportional zur Einwaage ist;
y = box
bQ kann kleiner oder grösser als 1 sein. Dann ist der proportionale (relative)
Fehler konstant.
Kommen proportionale und konstante Fehler vor, Fall d, so gilt folgende
Gleichung:
y = ao + box
Bei einer guten Bestimmungsmethode müssten a0 gleich Null oder sehr klein
und b0 gleich 1 oder sehr wenig davon verschieden sein.
Ausser den systematischen Fehlern müssen wir nun die zufälligen betrachten.
Auch hier können wir von der Einwaage unabhängige, Fall e, und proportio¬
nale Fehler annehmen, Fall f.
Wie kann man nun bei den gegebenen Werten beurteilen, ob systematische
Fehler vorhanden sind und welcher der obigen Fälle a - f vorliegt?
Diese Frage ist einfach zu beantworten, wenn die zufälligen Fehler unabhän¬
gig von der Einwaage sind. Wir berechnen die Regressionsgerade, y = a + bx,
d.h. die Gerade, von welcher die Abstände zu allen Punkten ins Quadrat erho¬
ben und summiert ein Minimum ergeben.

- 19 -
y N - -
b = a =
y - bx
Ix2 ^^
Für obige Werte erhalten wir folgende Regressionsgrade:
y = a + bx = -3,021 331 + 1,002 615x
Da der Regressionskoeffizient und das absolute Glied von 1 resp, von 0 wenn
auch wenig abweichen, bleibt zu prüfen, ob diese Abweichungen zufallsbedingt
sind.
Fb = t2 =-*b^l! (Ix^CIx]2) FGt = 1 FG2 = N-2
s2
In unserem Beispiel ist der theoretische Wert (i = 1.
Fber. = °>242 FTab. = 5'32
Wie zu erwarten war, ist die Abweichung b-1 nicht statistisch gesichert. Dies
kann auch anders ausgedrückt werden, indem man für b die sogenannten Ver¬
trauensgrenzen berechnet. Zu diesem Zweck setzt man in der obigen Beziehung
für den Wert F jenen von F005 (oder einen anderen Tabellenwert von F) ein
und löst die Gleichung nach (? auf.
Vertrauensgrenze von b
Kos = b + Vs2 Fo^/Ix2 - (Ix)2'
= bls-Vos/V^-tx)2'
In unserem Beispiel ist für FGi 1 und für FG2 8 zu setzen.
#>,05 = 1,002 615 i 9,594-5,317/339 100,077
= 1,002 615 + 0,012 275
= 0,990 350 1,014 980
Der Wert b = 1 liegt somit innerhalb der Vertrauensgrenzen, was den selben
Schluss erlaubt, wie der oben ausgeführte Test.

- 20 -
Den Ausdruck sïZx2 - (Zx)2 = 0,005 319 nennt man den Standardfehler von b
und schreibt auch 1,002 616 t 0,005 319 als summarischen Ausdruck für b und
seine Genauigkeit.
Will man prüfen, ob a vom theoretischen Wert a wesentlich abweicht, benützt
man folgende Formel:
Fa = t2(a-«)2-N LIxMlx)2] FGt = 1
2 (Ix?) FG2 = N - 2
Für unser Beispiel gilt, da « gleich Null ist:
=(3,021 331)10(339 100,077)
_ 8g6
9,564 3723675
Man kann also annehmen, dass der Fall a vorliegt. Streng genommen ist es
nicht ausgeschlossen, dass der Fall b vorliegen könnte. Um dies zu entschei¬
den, müsste die Anzahl der acidimetrischen Bestimmungen grösser sein.
Zu entscheiden, ob es sich um den Fall e oder f handelt (zufällige Fehler),
ist besonders bei einer kleinen Anzahl Bestimmungen nicht leicht. Da aber
diese Fälle für unsere Problemstellung wenig wichtig sind, unterliessen wir
diese Berechnungen.
Zu Vergleichszwecken berechneten wir auch die Regressionsgeraden und ihre
eventuellen Abweichungen von 0 resp. 1 für die drei übrigen Methoden.
Bromometrische Bestimmung
Regressionsgerade: y = 0,658 527 + 0,985 638x
Standardabweichung: s2 = 0,7354
Vertrauensbereich von b:ß0>os = 0,985 638 ± 0,014 675
00,05= 0,970963 .... 1,000313
Fb = 5,09 3 Fa = 0,621
Wasserfreie Bestimmung
Regressionsgerade: y2 =-0,055 325 + 0,998 001x
Standardabweichung: s = 0,7 354
Vertrauensbereich von b: ß„,05 = 0,998 001 +0,004 354
= 0,993 647.. . 1,000 236
Fb = 1,122

- 21 -
Argentometrische Bestimmung
Regressionsgerade y2= -27,984 800 t 0,934 684x
Standardabweichung s2 = 2485,8513
Vertrauensbereich von b: ß0>05 = 0,934 684 +0,126 573
= 0,808 111... 1,061 257
Fb =1,416 Fa = 0,264
Schlussfolgerung: Obige Werte sagen aus, dass alle drei Regressionsgeraden
nur zufällig nicht durch den Nullpunkt gehen und nur zufällig eine von 1 ab¬
weichende Steigung aufweisen.
Bemerkungen: Diese Schlussfolgerung befriedigt uns nicht, denn es zeigte
sich, dass die Berechnung der Regressionsgeraden für unsere Aufgabestel¬
lung nicht geeignet ist. Eine grosse Standardabweichung bewirkt einen grossen
Vertrauensbereich, welcher leicht ß = 1 einschliesst. Um die Standardabwei¬
chung möglichst zu verkleinern, sind sehr viele Bestimmungen nötig. Bei
sehr kleinen Standardabweichungen aber kann unter Umständen ein signifi¬
kanter Unterschied berechnet werden, auch wenn die Werte dem Sollwert
näher liegen, als die Werte der mehr streuenden Methode.
2.3. Bestimmung des Wirkstoffes in Tabletten
Die vorstehenden Überlegungen mit Bezug auf die Reinsubstanz gelten auch
für die Arzneiformen.
Die Arzneiformung bewirkt eine zusätzliche Abweichung, nämlich die Dosie¬
rungsgenauigkeit des Gewichtes. Berechnung siehe MÜNZEL, BÜCHI und
SCHULTZ (28).Die relative Standardabweichung setzt sich wie folgt zu¬
sammen:
s2
s2 = -^ + s2 + s2N y p
s = relative Standardabweichung des Gewichtes,
s = relative Standardabweichung der Analysenmethode,
s = relative Standardabweichung der zugelassenen
Verunreinigungen und
N = Anzahl gewogener Tabletten.

- 22 -
Abbildung 1
(a) Keine Fehler.
(b) Systematischer Fehler, unabhängig von Einwaage.
(c) Systematischer Fehler, proportional der Einwaage.
(d) Konstante und proportionale systematische Fehler.
(e) Zufallsfehler, unabhängig von Einwaage,
(f ) Zufallsfehler, proportional der Einwaage.
y = x
,*ï = *y = x

- 23 -
Wir führten 10 Bestimmungen durch mit Einwaagen einer Menge Tabletten¬
pulver, welche 500 mg Wirkstoff entsprachen und erhielten bei der acidime-
trischen Bestimmung der Compressi Carbutamidi (Handelsmuster HI) einen
Durchschnittswert von 99,06 und ein sre^ der Analysenmethode von 0,3%.
Diese Werte und der Vergleich mit den Werten der drei anderen Methoden
veranlassten uns, die acidimetrische Bestimmung als Pharmakopöemethode
vorzuschlagen.
Die Streuungszerlegung der obigen 10 Werte mit den Werten der Reinsub¬
stanz (S. 8 ) ergab einen verständlichen signifikanten Unterschied
(Fber = 34,43; F^ab 0 001-1 18= 15,38)und es ist sinnlos diese durch¬
zuführen.
Wir führten auch 10 Bestimmungen mit steigender Menge Tablettenpulver,
entsprechend 300 bis 1000 mg Wirkstoff durch und bestimmten die Regressi¬
onsgerade mit
y= -2,8 mg + 0,994 x.
Die Regressionsgerade geht nur zufällig nicht durch den Nullpunkt und zeigt
nur zufällig eine von 1 abweichende Neigung.
Wie wir schon bei der Reinsubstanz (siehe S. 21 ) feststellten, sagt uns die
Regressionsgerade wenig aus, und wir würden bei weiteren statistischen
Untersuchungen auf ihre Bestimmung verzichten.

-
24-
3. Spezieller Teil
3.1. Ausarbeitung von Prüfungsvorschriften für einige neuere Sulfonamide
3.1.1. Auswahl der zu bearbeitenden Arzneistoffe
Die nachfolgend aufgeführten Sulfonamide wurden bearbeitet, weil sie für die
Series medicaminum der neuen Ausgabe der schweizerischen Pharmakopoe
vorgesehen sind. Entsprechend den Richtlinien für die Aufnahme neuer Arz¬
neistoffe in die Ph.Helv.VI werden sie in der Schweiz therapeutisch gebraucht,
auch haben sie sich bis zu einem gewissen Grade medizinisch bewährt und
werden von mehr als einem Hersteller in den Handel gebracht. Ihrer therapeu¬
tischen Verwendung gemäss, sind diese Sulfonamide wie folgt einzuordnen:
a) Antidiabetica :
1. Carbutamidum
2. Tolbutamidum
3. Chlorpropamidum
b) Diuretica:
4. Acetazolamidum
5. Probenecidum
c) Chemotherapeuticum:
6. Sulfamethizolum
Unsere Bearbeitung dieser chemisch recht einheitlichen Arzneistoffgruppe
stellt eine Fortsetzung der Promotionsarbeit von ENGLER (10) dar. Die von
ihm vorgeschlagenen Prüfungsvorschriften wurden zum grössten Teil in die
Supplemente der Ph.Helv.V aufgenommen.
3.1.2. Massgebende Richtlinien für Bearbeitung der Monographien
Die Ausarbeitung der Prüfungsvorschriften und die Redaktion der Monogra¬
phien hatte weitgehend die Grundsätze der Eidg. Pharmakopöekommission für
die Ph.Helv.VI zu befolgen. Die Prüfungsvorschriften hatten zu umfassen
1. Nomenklatur (lateinische Hauptbezeichnung, höchstzulässige Abkürzungen,
landessprachliche Bezeichnungen).
2. Definition des Arzneistoffes (Strukturformel, Bruttoformel und Molekular¬
gewicht nach der Atomgewichtstabelle 1961).

- 25-
3. Gehaltsforderung (Minimal- und Maximalgehalt unter Berücksichtigung
eines eventuellen Feuchtigkeits- und Kristallwassergehaltes).
4. Prüfungsteil (mit Herstellung der Stammlösung, Sinnenprüfung, Identitäts -
reaktionen, Reinheitsprüfungen und Gehaltsbestimmung).
5. Rezepturvorschriften (Herstellung von Arzneiformen, antimikrobielle Be¬
handlung von Lösungen usf., Beschriftungs- und Abgabevorschriften).
6. Aufbewahrungsvorschriften.
7. Einteilung (in Innocua, Separanda, Venena, Stupefiacientia, Radiopharmaka).
8. Maximaldosen (für Erwachsene, nach Applikationsart).
9. Nützliche Angaben (Gebrauchsdosen für Erwachsene und nach Applikations¬
art, Löslichkeit und Mischbarkeit, Inkompatibilitäten, andere Bezeichnun¬
gen, Offizinelle Präparate und Markenpräparate).
Die PrufungsvorSchriften hatten nach Möglichkeit mit den Analysenmethoden
(Identitäts- und Reinheitsprüfungen, physikalische, chemische und physika¬
lisch-chemische Prüfungsverfahren) sowie den Apparaturen und der Reagen¬
zienliste entsprechend dem Entwurf der Ph.Hel.VI auszukommen. Wir mach¬
ten vor allem vermehrten Gebrauch von spektrophotometrischen Identitäts¬
und Gehaltsprüfungen; damit konnte vor allem auf unspezifische Farbreaktio¬
nen und Reaktionen mit unbekanntem Reaktionsverlauf verzichtet werden.
Selbstverständlich war die Verwendbarkeit neuer Speziaireaktionen für die
Identitäts- und Reinheitsprüfung sowie neuer allgemeiner analytischer Me¬
thoden experimentell zu überprüfen. Sie konnten nur dann in die Prüfungsvor¬
schriften übernommen werden, wenn sie eine Lücke schlössen oder Besseres
leisteten als die bereits in den Allgemeinen Bestimmungen vorgesehenen Vor¬
schriften.
Im allgemeinen handelt es sich um eine kritische Sichtung der Prüfungsvor¬
schriften aus der Literatur, der herstellenden Industrie, der halbamtlichen
und amtlichen Arzneibücher.
Die zur Beschaffung der Prüfungsunterlagen erforderlichen Arzneistoffe be¬
schafften wir uns direkt von der herstellenden pharmazeutisch-chemischen
Industrie (Handelsmuster), die uns die benötigten Analysenmaterialien in
grosszügiger Weise zur Verfügung stellten. Anhand der Handelsmuster und
des Ausfalls der in Aussicht genommenen Prüfungsvorschriften waren die
möglichen Anforderungen abzuwägen. Fast durchwegs entsprachen die

- 26 -
Handelsmuster strengen Reinheits- und Gehaltsforderungen, so dass wir
praktisch keine Konzessionen machen mussten bezüglich der in Aussicht ge¬
nommenen Anforderungen.
3.1.3. Arbeitsgang bei der Ausarbeitung der Monographien
Der theoretischen Bearbeitung, der chemischen Zusammensetzung sowie der
exakten chemischen Nomenklatur und Formulierung zwecks eindeutiger Defi¬
nition der bearbeiteten Sulfonamide wurde die grösste Aufmerksamkeit ge¬
schenkt. Es folgte eine möglichst lückenlose Dokumentation über die Herstel¬
lung zwecks Festlegung der möglichen Verunreinigungen sowie der physikali¬
schen und chemischen Eigenschaften als Grundlage für die verschiedenen Prü¬
fungen.
Nach diesem Studium der chemischen und pharmazeutischen Fachliteratur
werteten wir die folgenden Quellen für Arzneistoff-Prüfungsvorschriften aus:
a) Prüfungsvorschriften der herstellenden Industrie, die uns immer in gross¬
zügiger Weise auf Anfrage hin zur Verfügung gestellt wurden;
b) Fachpublikationen mit Rubriken über neue Arzneistoffe
"Medicamenta Nova" im Arch.Pharm.Chemie,
"DAK-Vorschriften" im Arch.Pharm.Chemie,
"Prüfungsvorschriften" der Subsidia Pharmaceutica SAV.
c) Veröffentlichte Vorschläge der Ph.Kommissionen
"OnderzoekvorSchriften" der niederländischen Pharmakopöekommission
im Pharm.Wbl.,
"Pro Pharmacopoea" der französischen Pharmakopöekommission in den
Ann.pharm.franc.,
"Drug Standards" der American Pharmaceutical Association,
"Monographie-Entwürfe für das DAB 7" Deutsche Pharmakopöekommission
in Dtsch.Apoth.Ztg., Pharm.Ztg. und Pharm.Zhalle.
d) Halbamtliche Arzneibücher
British Pharmaceutical Codex 1963
National Formulary 1960
Ergänzungsband 1959 zum DAB 6;

- 27 -
e) Pharmakopoen und Supplemente
Um eine gewisse Vereinheitlichung der Anforderungen der verschiedenen
Pharmakopoen zu erreichen, versuchten wir uns den Vorschriften der
Brit.Ph. 1963, der USP XIV und der Pharm.Austr.IX anzugleichen.
Unter Auswertung dieser vielseitigen Dokumentationsmöglichkeiten wurde
eine Laboratoriumsvorschrift für das betreffende Sulfonamid aufgestellt
und diese anhand der beschafften Handelsmuster experimentell erprobt.
Die für die experimentelle Bearbeitung verwendeten Substanzen wurden durch
wiederholte Umkristallisation bis zum konstanten Schmelzbereich gereinigt,
und deren Elementaranalyse bestimmt. Diese Muster hatten als Bezugs¬
substanzen Vergleichszwecken zu dienen. Im übrigen hielten wir uns streng
an die im "Entwurf zur Ph.Helv.VI" niedergelegten Allgemeinen Bestim¬
mungen.
Die Redaktion der Monographien erfolgte nach den "Redaktionellen Normen
für die Ph.Helv.VI".
- 28 -
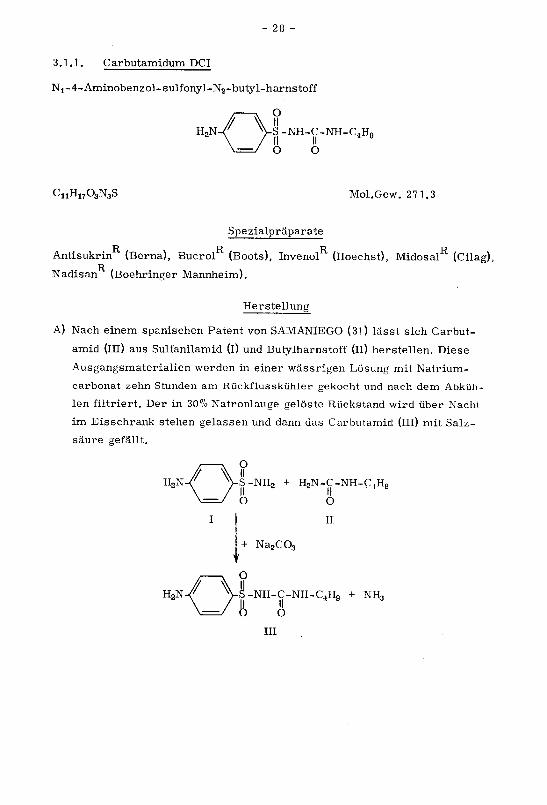
3.1.1. Carbutamidum DCI
Ni-4-Aminobenzol-sulfonyl-N2-butyl-harnstoff
n2N-r Vs-nh-c-nh-c4h9\=y o o
CuH1703N3S Mol.Gew. 271.3
Spe zialpräparate
R R R RAntisukrin (Berna), Bucrol (Boots), Invenol (Hoechst), Midosal (Cilag),
TD
Nadisan (Boehringer Mannheim).
Herstellung
A) Nach einem spanischen Patent von SAMANIEGO (31) lässt sich Carbut-
amid (IE) aus Sulfanilamid (I) und Butylharnstoff (II) herstellen. Diese
Ausgangsmaterialien werden in einer wässrigen Lösung mit Natrium-
carbonat zehn Stunden am Rückflusskühler gekocht und nach dem Abküh¬
len filtriert. Der in 30% Natronlauge gelöste Rückstand wird über Nacht
im Eisschrank stehen gelassen und dann das Carbutamid (III) mit Salz¬
säure gefällt.
OOII
S-NH2 + H2N-C-NH-C_,H9
o o
+ Na2C03
O
H2N-<f \-S -NH-C-NH-C4H9 + NH3
\=/o O
III

- 29 -
B) Butylamin (V) lässt sich ohne Lösungsmittel unter Erwärmen mit Nt-Sulf-
anilylisocyanat oder Nj-Sulfanilylharnstoff resp. Nt-Sulfanilylurethan (IV)
umsetzen (5, 7). Das Reaktionsprodukt wird in Ammoniak gelöst und das
Carbutamid (III) mit Essigsäure gefällt.
H2N S-NH-C-0-C2Hs + H2N-C„H9
o o
IV
/—\°
H2N-T \s -T>. _ -NH-C-NH-C4H9 + C2H5OH
\ / II II
III
Mögliche Verunreinigungen
Ausgangsstoffe und Schwefelsäure, schweflige Säure, Salzsäure, Essigsäure,
Phosphorsäure, Kohlensäure, Alkalien, Ferrichlorid, Ammoniak.
Sinnenprüfung
Muster IV war gelblich gefärbt, alle anderen weiss. Die Gelbfärbung des
Pulvers machte sich auch bei der Prüfung der Azeton-Lösung bemerkbar.
Identitätsprüfung
Carbutamid ist noch in keinem Arzneibuch aufgenommen. Zur Identitätsprü-
fung wurden deshalb die für Sulfonamide gebräuchlichen, sowie einige in der
Industrie verwendeten Reaktionen geprüft (aromatische Aminogruppe, Sulfon-
gruppe und Butylamin-Rest).
Nachweis von Butylamin; Bei Sulfanilamid und Sulfaguanidin entwickelt sich
nach dem Schmelzen Ammoniak, bei Carbutamid dagegen Butylamin. Wie
Sulfanilamid zeigt auch Carbutamid nach dem Schmelzen plötzlich eine vio¬
lette Farbe, welche bei Sulfaguanidin langsam auftritt.

- 30 -
Untersuchung von Sulfanilamid: Zur Differenzierung von Sulfanilamid, welches
keine Fluoreszenz zeigt, führen wir die Fluoreszenzreaktion nach LUCATELLI
(24) durch, die sich für Pharmakopöe-Zwecke eignet.
Schmelzbereich: Wie untenstehende Zusammenstellung zeigt, lassen sich
Carbutamid, Sulfanilamid und Sulfaguanidin gut durch ihren Schmelzbereich
unterscheiden.
Carbutamid 139-142°
Sulfanilamid 162-165°
Sulfaguanidin 185-188°
Im Gegensatz zu ENGLER (10), der bei Sulfanilamid keine Schmelzpunkts¬
depression nach Zusatz von 1-10% Sulfanilsäure feststellen konnte, erhielten
wir bei Carbutamid durch Zumischen von 1% Sulfanilamid eine Depression von
4°, bei 10% eine solche von 14°. Der Schmelzbereich kann also auch als Rein¬
heitskriterium gelten.
Reaktion: HÄUSSLER und HAJDU (16) bestimmten die Dissoziationskonstante K
von Carbutamid zu 5,1 • 10-6 was einem pKs-Wert von 5,3 + 0,1 entspricht. Die
von uns ermittelten pH-Werte der drei Handelsmuster lagen mit 4,8 - 5,2 in
sehr engen Grenzen.
Löslichkeit: Nach HÄUSSLER und HAJDU (16) nimmt die Löslichkeit von pH 6
an stark zu. Des amphoteren Charakters wegen löst sich Carbutamid in Säuren
und Basen, in letzteren aber besser.
Lösungsverhalten: Diese Prüfung auf gefärbte Verunreinigungen wird in
Aceton durchgeführt, weil sich Carbutamid darin am besten löst und eine
möglichst grosse Stoffkonzentration erlaubt.
Quantitative Bestimmungen
Handelsmuster
I
II
III
IV
nungsversuche ergaben folgende Resultate:
Vakuumexsikkator Trockenschrank
Phosphorpentoxid 103-105°
0,08% 4, 39 %
0,07% 3,43%
0,08% 4,86%
0,08% 4,90%

- 31 -
Die hohen Resultate der Trocknung bei 103-105° dürften auf eine gewisse
Flüchtigkeit oder Zersetzlichkeit von Carbutamid zurückzuführen sein. Die
Firma Boehringer Mannheim ermittelt den Wassergehalt auch im Vakuum bei
Zimmertemperatur. Interessanterweise zeigt das fast gleich gebaute Tolbut¬
amid keine solchen Differenzen.
Verbrennungsrückstand: Unwägbar bei allen Handelsmustern.
Gehaltsbestimmung
1. Bromometrische Bestimmung
Die Bromometrie wurde für die meisten Sulfonamide untersucht
[ENGLER (10), WOLFFBRANDT und NIELSEN (43) und CONWAY (8)].
Die verschiedenen Vorschriften unterscheiden sich im Bromatüberschuss
und in der Säurekonzentration.
Um die Verwendbarkeit dieser Bestimmungsart zu prüfen ermittelten wir
eine Bromverbrauchskurve nach WOLFFBRANDT und NIELSEN (43) als
Funktion der Bromierungszeit. Daraus kann die minimale Bromierungs-
zeit abgelesen werden. Wie die unten wiedergegebene Abb. 2 zeigt, bleibt
der Bromverbrauch von Carbutamid von 5 Sekunden bis 20 Minuten kon¬
stant. Stark saures Milieu beschleunigt die Reaktion.
Abbildung 2
Bromverbrauch von Carbutamid
molBr2
A,1
4,0
M
20* 40* 1' 10' 20' Zeit

- 32 -
Da die direkte Titration für nicht routinierte Analytiker unsicher ist, wur¬
den verschiedene Rücktitrationen ausgearbeitet, welche wir kurz zusammen¬
stellen:
a) Kaliumjodid+Natriumthiosulfat nach CONWAY (8),
b) Arsenige Säure, potentiometrisch nach CONWAY (8),
c) Indikatoren: Methylorange, Methylrot, Bordeauxrot.
Die Prüfungsvorschrift Boehringer, Mannheim, verwendet für 100 mg
Carbutamid 30 ml Eisessig R, 10 ml Salzsäure 37% RS und einen Uberschuss
von 2-3 ml 0,1 N Bromat.
BRÄUNINGER und DUDA (6) lösen 100 mg Carbutamid in 10 ml Eisessig R
und 10 ml Salzsäure 37% RS und titrieren direkt oder mit einem Uberschuss
von ca. 1 5 ml 0,1 Bromat.
Um den Einfluss der Säurekonzentration und des Bromatüberschusses zu
prüfen, variierten wir die Reagenzienmenge folgendermassen:
100 mg Carbutamid + 10 ml Salzsäure 37% RS +
a) 10 ml Eisessig R
20,00 ml 0,1 N Bromid-Bromat,
b) 10 ml Eisessig R
25,00 ml 0,1 N Bromid-Bromat,
c) 10 ml Essigsäure 12% RS
25,00 ml 0,1 N Bromid-Bromat.
Nach der Bromatzugabe wurde sofort 0,5 g Kaliumjodid zugefügt und mit
0,1 N Natriumthiosulfat zurücktitriert.
d) Methode der Ph.Helv.V für Sulfaguanidin.
40 mg Carbutamid (genau gewogen) werden in einem Erlenmeyerkolben
von 300 ml Inhalt mit Glasstopfen in 25 ml Wasser und 1 ml Ammoniak
7% RS unter Erwärmen gelöst. Der Lösung werden 20,00 ml 0,1 N Bromid-
Bromat und eine Mischung von 5 ml Salzsäure 37% RS und 20 ml Wasser
zugefügt. Der Kolben wird sofort geschlossen und unter Umschütteln 40
Minuten im Dunkeln stehen gelassen. Nach Zugabe von 1 g Kaliumjodid R
und 100 ml Wasser wird kräftig durchgeschüttelt und das ausgeschiedene
Jod sofort mit 0,1 N Natriumthiosulfat titriert. Gegen Ende der Titration
werden 30 Tropfen Stärke RS zugefügt.

- 33 -
1 ml 0,1 KBrOa entspricht 6,784 mg Carbutamid
Die Resultate sind der folgenden Zusammenstellung zu entnehmen:
d
104,47%
104,47 %
105,15%
Beurteilung der Resultate
Methode a); Aus den Resultaten und nach Vergleich mit anderen Bestim¬
mungsmethoden ist ersichtlich, dass diese, für die Ph. vorgeschlagene,
die genaueste ist.
Methode b): Zu grosser Bromüberschuss ergab zu hohe Werte, was schon
CONWAY (8) feststellte. BRÄUNINGER und DUDA (6) erhielten mit diesem
Überschuss Werte von 99,42%.
Methode c): Gibt schwach tiefere Resultate. In stark saurem Milieu wird
schneller bromiert und die Fällung unvollständig bromierter Produkte ver-
unmöglicht.
Methode d): Ist unbrauchbar. Nach Kommentar Suppl. I+II Ph.Helv.V bedingt
allein der Essigsäurezusatz eine Erniedrigung von 0,5 - 1,0%. Der Substi¬
tuent in p-Stellung bedingt die Fällung unvollständig bromierter Produkte.
2. Acidimetrische Titration
Carbutamid ist mit einem pKg-Wert von 5,3 genügend sauer, um in Äthanol
titriert werden zu können. Die Prüfungsvorschrift von Boehringer, Mann¬
heim, lautet folgendermassen:
"Ca. 600 mg Carbutamid werden in 30 ml Methanol gelöst und mit
20 ml ausgekochtem Wasser versetzt. Dann wird nach Zugabe von
3 Tropfen Phenolphthaleinlösung mit 0,1 Natronlauge bis kurz vor
den zu erwartenden Endpunkt titriert. Nach Zugabe von weiteren
30 ml ausgekochtem Wasser wird bis zum endgültigen Umschlagtitriert. Ein Blindwert wird bestimmt und dieser Wert abgezogen".
Handelsmuster
I II III IV
Mittelwerte 99,46% 99,99% 100,02% 99,32%
Handels-
muster a
I 99,70%
II 100,20%
III 100,46%
IV 99,91%
Methode
b c
100,43% 99,22%
100,29%
100,93% 100,39%
100,49%

- 34 -
3. Wasserfreie Titration
Carbutamid kann theoretisch der Aminogruppe wegen als Base, der Sulfa-
minogruppe wegen als Säure bestimmt werden. FRANCHI (12) erhielt mit
beiden Methoden gute Werte. Zur Bestimmung als Base löst er in Eisessig
und titriert mit Perchlorsäure gegen Kristallviolett.
Nach seiner Vorschrift erhielten wir aber schon mit 1 /10 der berechneten
Perchlorsäuremenge den Farbumschlag. Auch für Probeneeid, Sulfanil-
amid und Sulfadicramid ergab die Titration als Base keine brauchbaren
Werte. Auch FABER (10) erhielt bei der Titration der Sulfonamide als
Basen schlechte Resultate. Wir bestimmten Carbutamid als Säure nach der
Vorschrift von BRÄUNINGER und DUDA (6), welche einen Durchschnitt von
99,86 % erhielten, wie folgt:
"Ca. 200 mg Carbutamid (genau gewogen) werden in 25 ml neutralem
Dimethylformamid gelöst und nach Zugabe von 5 Tropfen Thymolblau-
lösung mit 0,1 N Natriummethylat bis zur Blaufärbung titriert".
1 ml 0,1 N CH3ONa entspricht 27,1 3 mg Carbutamid
Bei der Herstellung und Einstellung der Titrationsflüssigkeit arbeiteten
wir nach der Vorschrift von BECKETT und TINLEY (4). Den Umschlags¬
punkt stellten wir mit Thymolblau als Indikator sowie potentiometrisch fest.
In dieser Weise konnten wir auch die genaue Farbnuance des Thymolblaus
beim Umschlagspunkt ermitteln. Wir verwendeten die in der Ph.Helv.
Suppl.HI für die K.Fischer Titration beschriebene Apparatur.
Resultate:
Handelsmuster
Methode I H III IV V
potentiometrisch 99,65% 100,76% 100,14% 99,92% 100,43%Mittelwerte
mitlndikator 99,40% 100,42% 100,18% 99,93% 100,23%Mittelwerte
Wir stellten fest, dass der Umschlag von Blau nach Blauviolett dem Titra¬
tionspunkt am nächsten steht, gut ersichtlich ist und genaue Werte gibt.

- 35 -
Anwendung:
Bei einfachem, nicht jugendlichem Diabetes mellitus. Der Blutzuckerspiegel
wird nur gesenkt, wenn die Pankreas ß-Zellen noch Insulin bilden können.
Als Nebenerscheinungen können Nausea, Erbrechen, Dermatosen und Diarrhöe
auftreten (37). Wegen der häufigen Allergieerscheinungen ist Tolbutamid dem
Carbutamid vorzuziehen (9). Mit Carbutamid soll auch Besserung und sogar
Heilung der Schizophrenie erreicht worden sein (13).
Kontraindikationen: Leber- und Nierenerkrankungen.
Gravidität, Infektionen und alle Zustände mit erhöhter endokriner Belastung.
Inkompatibilitäten: Eisen(lll)-Salze, Silber- und Quecksilbersalze bewirken
im Gegensatz zu anderen Sulfonamiden keine Fällung.
Tabelle 1
Übersicht verschiedener Prüfungsvorschriften
CarbutamidumBoehringer
1957
Lucatelli
1956
VorschlagPh.Helv.VI
Nachweis der
Aminogruppe= = =
Schmelze = =
Nachweis der
Sulfongruppe
Fluoreszenz = =
Schmelzbereich 138 - 141°
DAB 6
140 - 141°
korr.
139 - 142°
pH von S 4,4 - 5,6
Lösungsverhaltenin Azeton 5 ml
max.FVL "A"
USP XV
max.FVL BG5
Schwefelsäure-
Porbe
max.FVL "A" max.FVL BG<
Arsen nicht nach¬
weisbar
nicht nach¬
weisbar
Schwermetalle Grenzreakt. a I
Chlorid max. 20 mg% Grenzreakt. a I
Sulfat max. 50 mg % Grenzreakt. a II
Wassergehalt max. 0,5 % max. 0,1 7«
Verbrennungs¬rückstand max. 0,1 % max. 0,1 %
Gehalt acidimetrisch
99 - 101 %
bromometrisch
99 - 101%
= bedeutet; den Anforderungen des Vorschlages entsprechend

- 36 -
4. Argentometne
SZABOLTOS (39) sowie STIVIC und MARINOW (38) beschreiben eine argen-
tometnsche Bestimmung fur Tolbutamid, welche mit Carbutamid Fehler von
26-30% ergeben soll. Wir erhielten fur beide Substanzen nach obiger Vor¬
schrift gleiche, ca. 5% zu hohe Werte. Darauf prüften wir die in der Ph.Helv.
übliche Silbermtratmethode. Trotz Änderung des Saurezusatzes und der
Zeit des Stehenlassens, konnte die grosse Streuung nicht ausgemerzt wer¬
den. Die besten Resultate erhielten wir bei einer Losung der Substanz m
3 ml Ammoniak 10% und Wasser und einem Zusatz von 0,35 ml Essigsaure
12%
Resultate: Handelsmuster
II V
Mittelwerte 99,87% 98 50%
5. Kolonmetrische Bestimmung eignet sich zur Bestimmung kleiner Mengen.
Durch Diazotierung und Kuppelung mit Thiocol z.B. können Mengen von
10 ug/ml bei 470 nm bestimmt werden (15).
Beurteilung der Methoden zur quantitativen Bestimmung
Auf Grund der statistischen Auswertung der Einzelresultate der vorstehenden
Bestimmung (siehe Kap.2.2) sowie der übrigen Gesichtspunkte bei der Wahl
von Bestimmungsmethoden (siehe Kap. 2.1) kommen wir zu folgender Beurtei¬
lung.
1. Bromometrische Titration. Die Streuung ist klein und der Arbeitsaufwand
massig; die Einwaage sollte möglichst genau 100 mg betragen.
2. Acidimetrische Titration. Die Titration ist weniger spezifisch, die Ein¬
waage und auch die Streuung sind etwas grosser, obwohl nicht signifikant
verschieden.
3. Wasserfreie Titration. Die Streuung ist klein der Arbeitsaufwand fur ein¬
zelne Bestimmungen aber gross.
4. Argentometrie: Diese Methode ist ungeeignet, da sie verschiedene Quellen
für systematische Fehler, wie Filtration, voluminöse Fallung und Saure-
konzentration, aufweist und viele Gefasse gebraucht werden.
Fur die Vorschrift der Ph.Helv.VI schlagen wir aus obigen Gründen die
bromometrische Titration vor.

- 37 -
Tabelle 2
Zusammenstellung der Untersuchungsresultate
Carbutamidum Handels¬
muster
I
Handels¬
muster
II
Handels¬
muster
III
Handels¬
muster
IV
VorschlagPh.Helv.VI
Sinnenprüfung = - = =
Nachv/eis der
Aminogruppe = = = =
Nachweis der
Sulfongruppe = = = =
Nachweis von
Butylamin = = = =
Fluoreszenz = = = =
Schmelzbereich 137-139° 140.5-142° 140.5-142° 140.5-142° 139-142°
pH von S 5,2 4,8 5,0 4,8 4,8- 5,2
Lösungsverhalten
in Azeton
farblos farblos farblos FVL BG2 max.FVL BGB
Schwefelsäure¬
probe
FVL BG4 farblos farblos FVL BG5 max.FVL BG4
Arsen - - - - -
Schwermetalle all al al all al
Chlorid al al mehr als
al
al al
Sulfat all all all all all
Wassergehalt 0,08% 0,07 % 0,08% 0,08% max. 0,1 %
Verbrennungs¬rückstand = = = = max. 0,1 %
Gehalt
bromometrisch
99,76%99,68%99,65%
100,22%
99,9 3%
100,45%
100,27%99,53%99,92%
100,77%100,47%100,14%
99-101 %
= bedeutet: den Anforderungen des Vorschlages entsprechend.
- bedeutet: abwesend, bzw. unwägbar.

- 38 -
Carbutamidum (PCI)
(Carbutam.)
Carbutamid Carbutamide Carbutamido
H2N-^ \s-NH-C-NH-C4H9O O
CnH1703N3S MoLGew. 271,3
Ni-4-Aminobenzol-sulfonyl-Ng-butyl-harnstoff mit einem Gehalt von minde¬
stens 99,0 (99,0-101,0) % CaH1703N3S.
Prüfung
Stammlösung (S): 1,0 g wird in 50 ml ausgekochtem Wasser 1 Minute lang
gekocht, erkalten gelassen und filtriert. Das Filtrat, das farblos sein muss,
dient für die Prüfungen 7, 11, 12 und 1 3.
1. Sinnenprüfung: Farblose Kristalle oder weisses bis gelblich-weisse« Pul¬
ver von schwach bitterem Geschmack.
2. Nachweis der primären aromatischen Aminogruppe: 10 mg werden in
5 ml Salzsäure 7% RS gelöst und mit 2 ml Wasser verdünnt. Der Lösung wer¬
den 2 Tropfen Natriumnitrit RS zugefügt. Beim Zutropfen einer Lösung von
ß-Naphthol RS entsteht sofort ein orangefarbener Niederschlag, dann eine
tiefrote Färbung.
3. Nachweis der Sulfongruppe: 50 mg werden mit 0, 4 g getrocknetem Na-
triumcarbonat R vermischt und in einem Reagenzglas ohne zu glühen bis zum
Verkohlen der organischen Substanz erhitzt. Nach dem Erkalten wird der Rück¬
stand in 3 ml Wasser aufgenommen, mit 5 Tropfen Bleiazetat RS versetzt und
mit Salzsäure 7% RS angesäuert. Nach dem Abklingen der Kohlendioxid-Ent¬
wicklung wird das Reagenzglas mit einem mit Fuchsin-Formaldehyd RS ge¬
tränkten Filterpapierscheibchen bedeckt und im Wasserbad erhitzt. Das
Filterpapier muss sich blau oder blauviolett färben.

- 39 -
4. Nachweis von Butylamin; Ca. 50 mg werden in einem kleinen Reagenzglas
vorsichtig erwärmt. Bei weiterem Erwärmen tritt nach dem Schmelzen ein
aminartiger Geruch auf und die Schmelze wird plötzlich violett.
5. Unterschied gegenüber Sulfanilamid: Zu 0,1 g werden einige Kristalle
Resorzin R und 1 ml Schwefelsäure 98% RS zugefugt und diese Losung ca.
3 Minuten lang unter Vermeidung von Verkohlung erwärmt. Nach dem Erkal¬
ten wird vorsichtig mit 25 ml Wasser verdünnt und mit Natriumhydroxid 30%
RS alkalisch gemacht. Im UV wird eine gelb-grüne Fluoreszenz sichtbar,
welche beim Ansäuern verschwindet und beim Alkalisieren wieder erscheint.
6. Schmelzbereich: 139-142°.
7. Reaktion: pH von S = 4,4 - 5,6.
8. Losungsverhalten: 0,4 g müssen sich in 2 ml Azeton klar losen. Die Lo¬
sung darf nicht starker gefärbt sein als Farb-Vergleichslosung BG5.
9. Schwefelsäureprobe: Die Losung von 0, 1 g wird während 5 Minuten im
Wasserbad erwärmt. Die Losung darf nicht stärker gefärbt sein als Farb-
Vergleichslosung BG}.
10. Arsen: 0,5 g müssen der Grenzprufung genügen.
1 1. Schwermetalle: 10 ml S müssen den Anforderungen der Grenzreaktion a I
genügen.
12. Chlorid. 10 ml S müssen den Anforderungen der Grenzreaktion a I ge¬
nügen.
13. Sulfal : 10 ml S müssen den Anforderungen der Grenzreaktion a II genügen.
14. Wassergehalt: Höchstens 0,1%, bestimmt mit 0,5 g (Vakuumexsikkator).
15. Verbrennungsruckstand: Höchstens 0, 1%, bestimmt mit 0,5g.
16. Gehalt: Ca. 0,1 g der im Vakuumexsikkator wahrend 4 Stunden getrock¬
neten Substanz (genau gewogen) wird in 10 ml Eisessig R und 10 ml Salzsaure
37% RS gelost. Unter Umschwenken werden 20,00 ml 0.1 N Bromid-Bromat
zugefugt und nach Zugabe von 0,5 g Kaliumjodid R sofort mit 0,1 N Natrium-
thiosulfat bis zur Entfärbung zurucktitnert. Gegen das Ende der Titration
werden 10 Tropfen Stärke RS zugefugt.
1 ml 0,1 N KBr03 entspricht 6,784 mg CnH^O^S

- 40 -
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
S ep ar andum
Gebrauchsdosen: Einzeldosis = 500 mg
Tagesdosis = 500 mg - 1,5 g
Löslichkeit: Sehr schwer löslich in Wasser, löslich in 20 T. Äthanol 94%,
in 4 T. Azeton; schlecht löslich in Äther, Chloroform und Tetrachlorkohlen¬
stoff; löslich in Alkalien und Mineralsäuren.
Offizinelles Präparat: Compressi Carbutamidi 500 mg,
R R R R RMarkennamen: Antisukrin
,Bucrol
,Invenol
,Midosal
,Nadisan
,
Oranil.

- 41 -
3.1.2. Tolbutamiden PCI
p-Toluolsulfonyl-butyl-harnstoff
CH3-T N\-S-NH-C-NH-C4H9\=/ O O
C12H1803N2S Mol.Gew. 270,4
Spezialpräparate
Artosin (Boehringer Mannheim), Orinase (Upjohn), Rastinon (Hoechst),
Toluvan (Zambeletti).
Prüfungsvorschriften
Brit.Ph.1963, USP XVI, Boehringer Mannheim.
Herstellung
A) LOGEMANN und Mitarbeiter (23) kochen p-Toluolsulfonyl-N-butyl-formyl-
harnstoff (I) mit Natronlauge in Äthanol, wobei Kohlenmonoxid abgespalten
wird. Das entstandene Tolbutamid wird nach Verdünnen mit Wasser durch
Salzsäure gefällt. Die Ausgangssubstanz (I) wird durch Umsetzen von p-
Toluolsulfonylisocyanat mit Butylformamid in wasserfreiem Toluol herge¬
stellt.
S-NH-C-N-C.Ho —»• Tolbutamid + COII II IO O CHO
I IV
B) n-Butylamin (III) wird nach zwei verschiedenen Patentvorschriften mit
1. p-Toluolsulfonyl-urethan (II) (7),
2. p-Toluolsulfonyl-harnstoff (V) (7),
3. p-Toluolsulfonyl-isocyanat (VI) (15)
durch Erwärmen ohne Lösungsmittel umgesetzt, in Ammoniak gelöst und das
Tolbutamid (IV) durch Essigsäure gefällt.

- 42 -
p-Toluolsulfonyl-harnstoff ist von obigen Ausgangs-Substanzen am einfach¬
sten herzustellen.
C) Gemäss Brit.Ph.1963 kondensiert man Butylisocyanat (VIII) mit dem, aller¬
dings schwer herstellbaren p-Toluolsulfonamidnatrium (VII).
B,) •OlII
o
CH, / \»
NH2-C4H9 III
S-NH-C-NH-QHjj + C2H5-OH
=J O O
IV
B2)
O
CH3< \-S-NH-C-NH,o o
V
o
NH2-CjH9 III
CH3-<f \s-NH-C-NH-C4H9 + NH3
=J OII II
o
IV
B3) CH;
O/ V
C) CH3-/ V S-NHNa
NH2-C4H9 \ in
CH3-f >S-NH-C-NH-C4H9
c4h9-n=c=o vm° °
IV
o
VII

- 43 -
Mögliche Verunreinigungen
Ausgangssubstanzen, Säuren, Ammoniak und Natriumhydroxid.
Identitätsprüfung
Nachweis der p-Toluolsulfonamid-Gruppe: Alle Vorschriften verwenden die
Spaltung von Tolbutamid (IV) und den Nachweis der Spaltprodukte, erklären
aber den Reaktionsablauf nicht. Durch Kochen am Rückflusskühler mit Schwe¬
felsäure wird Tolbutamid (IV) gespalten (a). Das in der Kälte auskristallisie¬
rende p-Toluolsulfonylamid (K) wird durch seinen Schmelzbereich bestimmt.
a) CH,/ w
CH,
O
IIS-NH-C-NH-C4HoII IIo o
IV
,
o
/ Vi
IX
+ H2S04
S-NH2 + NH2-C4H9 + C02
III
b) xo2-/ \n=n er
X
N02-/ \n=>
+ NH2-C4H9
III
N-NH-C4H9
Nachweis des Butylamins: Das wasserdampfflüchtige Butylamin (III) wird
durch eine Farbreaktion mit p-Nitrobenzoldiazoniumchlorid (X) nachge¬
wiesen (b).

- 44 -
Bei Verwendung des Ph.Helv.V Puffers pH 8,0 erhielten wir eine schwächere
Färbung, weshalb der in der Prüfungsvorschrift (siehe S. 49) angegebene
Spezialpuffer empfohlen wird. Ist Butylamin in höheren Konzentrationen vor¬
handen, so geht die Kuppelung ohne Pufferzusatz und bei Zimmertemperatur
vor sich. Sulfanilamid zeigt keine Kuppelung mit diazotiertem p-Nitroanilin.
Da beide Spaltprodukte identifiziert werden, verzichten wir auf den Nachweis
der Sulfogruppe. Die gebräuchlichen Nachweisreaktionen sind bei Tolbutamid
nicht charakteristisch (Schmelze und Kupferreaktion) oder undurchführbar
(Diazo-Reaktion).
Reinheitsprüfungen
a) Physikalische und physikalisch-chemische Prüfungen
Lösungsverhalten: Siehe Carbutamidum (S. 31 ).
Schmelzbereich: Eine Beimischung von 1 % Carbutamid gibt eine Depression
von 3°, eine Beimischung von 10% eine solche von 12°. Für p-Toluolsulfon-
amid-Beimischungen bestimmen wir folgende Werte: 1% = 2°; 10% = 12°.
Grössere Mengen organischer Verunreinigungen können also durch den
grösseren und tiefer liegenden Schmelzbereich ermittelt werden.
Gehaltsbestimmung
1 ) Die bromometrische Bestimmung ist nicht möglich, weil die primäre
aromatische Aminogruppe des Carbutamids durch eine Methylgruppe ersetzt
ist.
2) Acidimetrische Methode: Wir arbeiteten nach der bei Carbutamidum (siehe
S. 34) beschriebenen Methode und erhielten folgende Werte:
Handelsmuster
I II III
101,13% 101,42% 100,71%
100,9 % 100,71 % 100,19%100,91 %

- 45 -
3) Wasserfreie Titration: Potentiometrisch bestimmt erhielten wir folgende
Werte:
Handelsmuster
I II in
99,26% 99,92% 99,26%100,69% 100,02% 99,53%100,27% 99,77%
Als Indikator eignet sich Thymolblau, dessen Umschlag von Blau nach Blau¬
violett dem potentiometrischen Wendepunkt sehr nahe liegt (Differenzen
0-0,01 ml). Die Resultate sind in der Tabelle 4 aufgeführt. Sie lassen erkennen,
dass die wasserfreie Titration sich von den untersuchten Verfahren am besten
eignet.
4) Argentometrische Bestimmung: Nach STIVIC und MARINOW (38) erhiel¬
ten wir mit 105,6%, 107,3% und 105,85% zu hohe Werte. Nach der in der
Ph.Helv.V für Sulfonamide üblichen Silbernitrat-Methode lösten wir ca.
1 g Tolbutamid in 3 ml Ammoniak 10% RS und neutralisierten mit Essigsäure
12% RS. Um den Einfluss der Reaktion (pH) zu ermitteln (siehe auch Carbut-
amidum S. 36), wurden verschiedene Mengen Säure zugefügt.
Essigsäure 12% RS Tolbutamid
ml %
0,30 98,85
0,35 99,75
0,40 98,70
0,45 98,47
0,50 98,05
0,60 97,05
1,00 92,70
Dieses Bestimmungsverfahren hat den Nachteil stark pH abhängig zu sein.
5) Die Stickstoff-Bestimmung mit der Kjeldahlapparatur der Ph.Helv.VI gab
wohl nahe beieinanderliegende, aber zu tiefe Werte: 97,90; 97,74; 97,99%
Tolbutamid.

- 46 -
6) Spektrophotometrische Bestimmung: Tolbutamid zeigt in Chloroform ein
Absorptionsmaximum bei 264 nm ( £ = 6922) und ein Absorptionsminimum bei
253 nm (£ = 517 3). In Methanol zeigt Tolbutamid Maxima bei 205 und 228 nm,
wovon letzteres zur quantitativen Bestimmung verwendet wird (E"
~ 500)
und kleinere Maxima bei 257, 262, 267 und 274 nm (36). Die einfachere und
genauere Bestimmung durch wasserfreie Titration Hess auf die Verwendung
der Spektrophotometrie verzichten.
Stabilität: Natriumsalzlösungen von Tolbutamid (pH 9,3) spalten sich bei 80°
leicht in p-Toluolsulfonamid, Dibutylharnstoff und Kohlendioxid. Bei normal
gelagerten Ampullen beträgt die Spaltung ca. 1% (17).
Anwendung: Wie Carbutamid, aber schwächer wirksam. Tolbutamid zeigt
weniger Nebenerscheinungen. Da die primäre aromatische Aminogruppe fehlt,
besitzt Tolbutamid keine Sulfonamidwirkung und schädigt die Darmflora nicht.
Nach einem Bericht des deutschen Bundesgesundheitsamtes (9) besitzt Tol¬
butamid abortive und teratogene Wirkung, welche berücksichtigt werden muss.
Nach neueren Ergebnissen der Diabetes-Forschung ist bei Schwangeren über¬
haupt auf die Verabreichung synthetischer Antidiabetica zu verzichten.

99,0-101,0%
Titration
Wasserfreie
99-101%
Methanol
in
acidimetrisch
min.97,0%
Methanol
in
acidimetrisch
99,0-101,0%
Kjeldahl
Gehalt
max.0,1%
max.0,1%
-
max.0,1%
Verbrennungsrückstand
max.0,2%
max.0,2%
max.0,5%
%1
max.
Wassergehalt
ai
Grenzreaktion
nachw.
nicht
--
Sulfat
all
Grenzreaktion
nachw.
nicht
-
-
Chlorid
all
Grenzreaktion
nachw.
nicht
20/Mill.
nachw.
nicht
Schwermetalle
nachw.
nicht
--
-
Arsen
farblos
(USP)
"A"
FVL
max.
-
-
Schwefelsäureprobe
farblos
klar,
__
_
Azeton
in
verhalten
Lösungs
5,0-5,6
--
-
Svon
pH
127-130°
128-129°
126-132°
128°
Schmelzbereich
++
++
Butylamin
von
Nachweis
137-139°
136-137°
135-138°
ca.136°
luolsulfonamid
p-To-
von
Nachweis
Ph.Helv.VI
Vorschlag
Mannheim
Boehringer
XVI
USP
Bri1.Ph.19G3
Tolbutamidum
Prüfungsvorschriften
verschiedener
Übersicht
3Tabelle
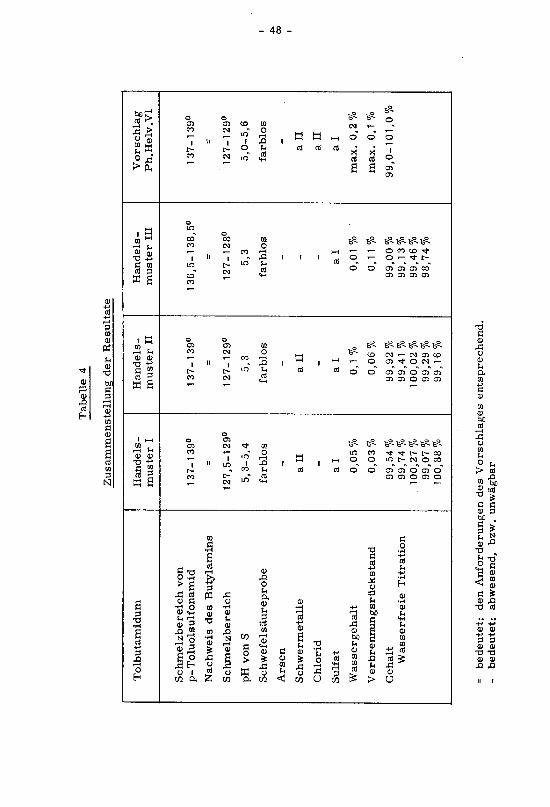
unwägbar
bzw.
abwesend,
bedeutet:
-
entsprechend.
Vorschlages
des
Anforderungen
den
bedeutet:
=
99,0-101,0%
98,74%%
99,46
99,13%
99,00%
99,16%
99,29%
100,02%%
99,41
99,92%
100,88%
99,07%%
100,27
99,74%
99,54%
Titration
Wasserfreie
Gehalt
%0,1
max.
0,11%
0,06%
0,03%
Verbrennungsrückstand
0,2%
max.
%0,01
%0,1
0,05%
Wassergehalt
ai
ai
Ia
Ia
Sulfat
aH
--
-Chlorid
all
-all
aH
Schwermetalle
--
--
Arsen
farblos
farblos
farblos
farblos
Schwefelsäureprobe
5,0-5,6
5,3
5,3
5,3-5,4
Svon
pH
127-129°
127-128°
127-129°
127,5-129°
Schmelzbereich
==
==
Butylamins
des
Nachweis
137-139°
136,5-138,5°
137-139°
137-139°
onamid
Toluolsulf
p-
von
Schmelzbereich
Ph.Helv.VI
Vorschlag
in
muster
Handels¬
II
muster
Handels¬
Imuster
Handels¬
Tolbutamidum
Resultate
der
Zusammenstellung
4Tabelle

- 49 -
Tolbutamidum (PCI)
(Tolbutam.)
Tolbutamid Tolbutamide Tolbutamide
/ So
CHg-f y S-NH-Ç-NH-C4Hg^=/ O O
C^HjgOaNaS Mol.Gew. 270,4
N1-p-Toluolsulfonyl-N2-butyl-harnstoff mit einem Gehalt von mindestens 99,0
(99,0-101,0) %C12H1803N2S.
Prüfung
Stammlösung (S): 1,5 g werden in 75 ml Wasser während einer Minute gekocht,
erkalten gelassen und filtriert. Das Filtrat dient als S für die Prüfungen 5 und
9-11.
1. Sinnenprüfung: Farblose Kristalle oder weisses Pulver; geruchlos und
geschmacklos.
2. Nachweis der p-Toluolsulfonamid-Gruppe: 0,2 g werden mit 16 ml Schwe¬
felsäure 50 X 30 Minuten lang am Rückflusskühler gekocht. Die Hälfte der er¬
haltenen Lösung wird erkalten gelassen. Der Niederschlag wird einmal aus
heissem Wasser umkristallisiert. Schmelzbereich: 137-139°.
3. Nachweis des Butylamin: Die andere Hälfte der unter 2 erhaltenen Lösung
wird mit Natriumhydroxid 30% RS vorsichtig stark alkalisch gemacht und
während 30 Minuten mit Wasserdampf destilliert. Das Destillat wird in 20 ml
0,1 N Salzsäure aufgefangen. Zu 1 ml dieses Destillates werden 0,1 g Natrium¬
azetat R und 10 ml eines Puffers, der 4,08 % primäres Natriumphosphat, 1,6 %
Borax und 1,27 % Natriumhydroxid R enthält, zugefügt und die Mischung 10 Mi¬
nuten in Eis gekühlt. Dann wird 1 ml diazotiertes p-Nitranilin (0,5 g p-Nitrani¬
lin werden in 100 ml heisser 1 N Salzsäure gelöst und auf 5° abgekühlt, dann
werden 2,7 ml Natriumnitrit RS zugefügt; das Reagens ist nach 30 Minuten
gebrauchsfertig) zugesetzt und während 20 Minuten stehen gelassen. Nach
tropfenweiser Zugabe von 2 ml Natriumhydroxid 7 % RS färbt sich die Lösung
orange-rot.

- 50 -
4. Schmelzbereich: 127-130°
5. Reaktion: pH von S = 5,0-5,6.
6. Lösungsverhalten: 0,5 g müssen sich in 2 ml Azeton klar und farblos lösen.
7. Schwefelsäureprobe; 0,2 g müssen sich in 2 ml Schwefelsäure 95% RS klar
und farblos lösen.
8. Arsen: 0,5 g müssen der Grenzprüfung genügen.
9. Schwermetalle: 10 ml S müssen den Anforderungen der Grenzreaktion
a II genügen.
10. Chlorid: 10 ml S müssen den Anforderungen der Grenzreaktion a II
genügen.
11. Sulfat: 10 ml S müssen den Anforderungen der Grenzreaktion a I genügen.
12. Wassergehalt: Höchstens 0,2% bestimmt mit 0,5 g (Trockenschrank).
13. Verbrennungsrückstand: Höchstens 0,1 %, bestimmt mit 0,5 g.
14. Gehalt; Bestimmt mit 0,2 g getrockneter Substanz durch wasserfreie
Titration, Methode c (Allgemeine Bestimmungen, Ph.Helv.VI).
1 ml 0,1 N LiOCH3 entspricht 27,04 mg C12Hlg03N2S.
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
Se par andum
Gebrauchsdosen: Einzeldosis = 0,500 g
Tagesdosis = 0,500 g - 1 g
Löslichkeit: Unlöslich in Wasser, 1 T. löslich in 10 T. Äthanol 94%, in 3 T.
Azeton; leicht löslich in Alkalien, wenig löslich in Mineralsäuren.
Offizinelles Präparat: Compressi tolbutamidi 500 mg.
RR R
Markennamen: Artosin,Orinase
,Rastinon
.

- 51 -
3.1.3 Chlorpropamidum
Nj-U-ChlorbenzolsulfonyO-lN^-propyl-harnstoff
Cl 7 xx -S -NH-C-ÎSrH-C3H7
o o
CjoHuGjNaSCl Mol.Gew. 276,8
Spezialpräparate
R RCaianil (De Angeli), Diabetabs (Wolfs) und Diabinese (Pfizer).
Prüfungsvorschriften
Brit.Ph.1963.
De Angeli, Mailand; Pfizer, Folkestone, England.
Herstellung
Chlorbenzolsulfonsâureamid (I) wird mit Äthylkohlensäurechlorid (II) zum
entsprechenden Sulfonylcarbamat (III) umgesetzt und dieses gibt unter Er¬
hitzen mit n-Propylamin im Vakuum Chlorpropamid (IV) (25):
O
Cl-T XVS-NH2 + Cl-C-OC2Hs
\=y o • o
o
Cl-f > S-NH-C-0-C2H5
\=/ o o
II
Cl-f NVS-NH-C-NH-C3H7\ ß Jlv=/ o ö
IV

- 52 -
Mögliche Verunreinigungen
Ausgangssubstanzen, Ammoniak und Säuren.
Identitätsprüfung
Nachweis der Chlorbenzolsulfonamid-Gruppe:
Die Brit.Ph.1963 lässt durch Kochen mit Schwefelsäure Chlorbenzolsulfon-
amid abspalten und dessen Schmelzpunkt bestimmen (143°). Wir führten diese
Reaktion sinngemäss wie bei Tolbutamidum (siehe S. 43) durch und ermittel¬
ten als Schmelzbereich des Chlorbenzolsulfonamids 145-147° (Forderung
144-147°).
Nachweis von Butylamin: Das bei Spaltung von Chlorpropamid entstehende
Butylamin lässt sich in der unter Tolbutamidum (siehe S. 43) angegebenen
Weise durch eine Farbreaktion nachweisen. Da diese Reaktion einen grossen
Arbeitsaufwand erfordert, konnte das Amin aus der alkalisch gemachten Lo¬
sung mit Lackmus nachgewiesen werden.
Nachweis von Chlor; Nach dem Mineralisieren der organischen Substanz
weist die Brit.Ph.1963 Chlor nach. Wir verzichten auf diese Reaktion, weil
der Nachweis der Spaltsprodukte des Chlorpropamids genügt.
Absorptionsspektrum: Spektrophotometrisch lässt sich Chlorpropamid ein¬
deutig und einfach nachweisen durch Ermittlung der Absorptionsmaxima einer
0,001proz. äthanolischen Losung bei 258, 266, 269 und 277 nm und einer
0,0004proz. äthanolischen Losung bei 233 nm. Das Verhältnis der molaren
Extinktionen bei 233 und 265 nm beträgt ca. 23.

- 53 -
Abbildung 3
Absorptionsspektrum von Chlorpropamid in 0,01 N HCl
2N nm
Schmelzbereich: Isomere Verbindungen lassen sich durch den Schmelzbereich
unterscheiden (25):
Isomeres
N^-2-Chlorbenzolsulfonyl-N2-propylharnstoff
Nj-4-Chlorbenzolsulfonyl-
N2-isopropylharnstoff
Nj-4-Chlorbenzolsulfonyl-N2-propylharnstoff
(Chlorpropamid)
Schmelzbereich
176 - 176
144 - 146°
126 - 129°

- 54 -
GehaltsbeStimmung
Der Gehalt liesse sich auch spektrophotometrisch bei 233 nm mit einer
0,0008proz. Lösung bestimmen, welche wie jene für die Identitätsprüfung
herzustellen wäre. E1 = 596 (siehe Tab. 6). Der grösseren Genauigkeit
und Durchführungsmöglichkeit im Apothekenlaboratorium wegen, schlagen
wir die Gehaltsbestimmung durch wasserfreie Titration vor. Resultate
siehe Tab. 6.
Aufbewahrung
Chlorpropamid ist am Licht und an der Luft stabil.
Anwendung
Blutzuckersenkung bei Diabetes mellitus.

- 55 -
rschlag 4-147" +>P 6-129" 0-5,0 an ail
|
ail x.0,5%
x.0,1%
serfreie tration -101,0%o •s- CM ., cd co CO .H o
> * S ar* C33
II
TT« 8,5" end end end
|
euo x
^ freie ion 1,0%Angi + >
P 7-12 bwes bwesco
eu
rtX)
ax.1, .Fisc ax.O, sser itrat 0-10tu
0CM cd cd cd aw a llfH •
AngeliI
+ >127" /Mill. /Mill. 0,5
%
.105" 0,1
% irfreie ition 39,0%P H H x3 x" œ h GCM
o o cd co cd CO .rt .*eu
0•* o
•*S* a «Hg
C So O
o•H-r> ^
Isorpti ,97,0%izer -130 reak .0,5ttH
LU 126
|
Grenz maxp
en
CD ion #(Jî o -r> r-i O
oo00
m
rit.Ph 143 + + Ê 126-1 4,0-5
•enzre: Kjeldi 9,0-10ffl
OCD
cm CO 0
midum •eichdes lsulfonamidei
es
Butylamin phenylhydraz äspektrum eich ot.)
chwermetaller>I—1tri
[gsrückstandhlorpropa
ri o9 n•° c
ȣ
^3achweisd
,4-Dinitro hlor .bsorption! chmelzberH
vonS
(p
hlorid ulfat Wassergeh; erbrennur ehaltO CßU 3 (M o <q CO a co O en > Ü

%99,0-101,0
%100,59
%100,03
%100,08
%100,40
%100,19
%98,94
%99,09
%99,94
%99,66
%100,03
%97,97
%100,74
%99,09
%100,05
%101,08
%101,50
100,45%
Titration
Wasserfreie
spektrophotometrisch
Gehalt:
%max.0,5
%0,22
%0,1
%0,12
0,2%
K.Fischer
103-105°
Wassergehalt
%max.0,1
%0,02
%0
%0
Verbrennungsrückstand
all
all
II
aall
Sml
2Sulfat
all
all
aH
all
Sml
2Chlorid
aH
all
all
all
Sml
2Schwermetalle
farblos
+klar
mlfarblos
0,2g/1
O,2g/2mlfarblos
farblos
ml
0,2g/2
Azeton
in
Lösungsverhalten
4,0-5,0
4,4
3,95
4,6
4,4
4,2
4,75
4,4
4,1
4,4
kolorimetrisch
filtriert
nicht
pot.
(pot
.)S
von
pH
126-129°
127,5-129°
128-129°
sublimiert)
50%
ca.
118
(bei
125,5-126,5°
Schmelzbereich
nm
277
nm
277
nm
277
nm
277
nm
269
nm
266
nm
269
nm
265,5
nm
269
nm
265,5
nm
269
nm
265,5
Maxima
nm
258
nm
258
nm
258
nm
258
nm
257
nm
256,5
nm
257
nm
257
Minimum
Absorptionsspektrum
•=
==
Butylamins
des
Nachweis
144-147°
146-147°
146-147°
145-147°
ulfonamids
benzols
Chlor¬
des
Schmelzbereich
Ph.Helv.VI
Vorschlag
in
muster
Handels¬
II
muster
Handels¬
Imuster
Handels¬
Chlorpropamidum
Resultate
der
Zusammenstellung
6Tabelle

- 57 -
Chlorpropamidum
(Chlorpropam.)
Chlorpropamid Chlorpropamide Chloropropamide
O
Cl-T VS-NH-C-NH-CgB,\=/ o o
C10H13O3N2SCl Mol.Gew. 276,8
Nj-4-Chlorbenzolsulfonyl-N2-propyl-harnstoff mit einem Gehalt von minde¬
stens 99,0 (99,0-101,0) % C10H13O3N2SCl.
Prüfung
Stammlosung (S): 0,1 g wird mit 10 ml Wasser geschüttelt und filtriert. Das
Filtrat, das farblos sein muss, dient als S fur die Prüfungen 6-9.
1. Sinnenprufung. Weisses kristallines, geruch- und geschmackloses Pulver.
2. Nachweis der Chlorbenzolsulfonamid-Gruppe. 0,2 g werden mit 16 ml
Schwefelsaure 50 °o RS 30 Minuten lang am Ruckflusskuhler gekocht. Die
Hälfte der erhaltenen Losung wird erkalten gelassen. Der entstandene Nieder¬
schlag wird einmal lus heissem Wasser umkristallisiert. Schmelzbereich.
144-147°.
3. Nachweis des Butylamin. Die andere Hälfte, der unter 2 erhaltenen Losung
wird mit Natriumhydroxid 30 % vorsichtig alkalisch gemacht und wahrend
30 Minuten mit Wasserdampf destilliert. Das Destillat wird in 20 ml 0,1 N
Salzsaure aufgefangen. Zu 1 ml dieses Destillates werden 0,1 g Natriumazetat R
und 10 ml eines Puffers, der 4,08 % primäres Natriumphosphat, 1,6 % Borax
und 1,27 % Natriumhydroxid R enthalt, zugefugt, und die Mischung 10 Minuten
in Eis gekühlt. Dann wird 1 ml diazotiertes p-Nitranilm (0,5 g p-Nitranilm
werden in 100 ml heisser 1 N Salzsaure gelost. Ist die Losung auf 5° abge¬
kühlt werden 2,7 ml Natriumnitrit 10ffo RS zugefugt, das Reagens ist nach
30 Minuten gebrauchsfertig) zugesetzt und wahrend 20 Minuten stehen gelas¬
sen. Nach tropfenweiser Zugabe von 2 ml Natriumhydroxid 7 °o RS färbt sich
die Losung orangerot.

58 -
4. Absorptionsspektrum: 0,1 g wird in einem Messkolben von 100 ml Inhalt
in Äthanol 94 % RS gelöst und mit Äthanol 94 % RS bis zur Marke ergänzt.
5,0 ml dieser Lösung werden in einem Messkolben von 50 ml Inhalt mit 0,01 N
Salzsäure bis zur Marke ergänzt. Die Lösung wird gegen eine Mischung von
5,0 ml Äthanol 94% RS und 45,0 ml 0,01 N Salzsäure als Blindprobe mit einem
geeigneten Spektrophotometer gemessen. Sie muss bei 258; 266, 269 und 277 nm
Maxima zeigen. 4,0 ml dieser Lösung werden in einem Messkolben von 100 ml
Inhalt mit 0,01 N Salzsäure verdünnt und ihre Extinktion bei 233 nm gegen£ 233
0,01 N Salzsäure als Blindprobe gemessen. Das Verhältnis -z muss ca. 23
betragen.
5. Schmelzbereich: 126-129°
6. Reaktion: pH von S = 4,0-5,0
7. Lösungsverhalten: 0,2 g werden in 2,0 ml Azeton R gelöst.
Diese Lösung muss klar und farblos sein.
8. Schwermetalle: 2 ml S müssen den Anforderungen der Grenzreaktion a II
genügen.
9. Chlorid: 2 ml S müssen den Anforderungen der Grenzreaktion a II genügen.
10. Sulfat: 2 ml S müssen den Anforderungen der Grenzreaktion a II genügen.
11. Wassergehalt: Höchstens 0,5%, bestimmt mit 0,25 g (Trockenschrank).
12. Verbrennungsrückstand: Höchstens 0,1 %, bestimmt mit 0,5 g.
13. Gehalt: Bestimmt mit ca. 0,2 g durch wasserfreie Titration, Methode C
(Allgemeine Bestimmungen Ph.Helv.Vl).
1 ml 0,1 N LiOCH3 entspricht 27,68 mg C10H13O3N2SCl
AufbeWährung
In gut verschlossenem Behälter.
Maximaldosen Einzeldosis 400 mg
(Vorschlag) Tagesdosis 1,2 g
S epar andum

- 59 -
Gebrauchsdosen; Tagesdosis 100 - 500 mg
(Vorschlag)
Löslichkeit: 1 T. löst sich in 5 T. Azeton, 14 T. Äthanol 94%, 6 T. Benzol,
9 T Chloroform, 120 T. Äther, 250 T. Wasser; löslich in Alkalien und unlös¬
lich in Säuren.
R R R
Markennamen: Catanil,Diabetabs
,Diabinese
.

- 60 -
3.1.4. Acetazolamidum DCI
2-Acetylamino-5-sulfamidyl-1,3,4-thiadiazol
N N0
CHg-C-NH—U JJ— S-NH,
oö
o
C^C*,!^ Mol.Gew. 222,3
Spezialpräparate
RR RDiamox (Lederle), Natrionex (Glutan-Chemie) Oedemin (Astra).
Prüfungsvorschritten
Brit.Ph.1963, USP XVI.Pro Pharmacopoea franc. (29).
Herstellung
Die meisten Autoren (1, 3, 30, 44) gehen vom 2-Acetylamino-5-mercapto-
1,3,4-thiadiazol (I) aus, welches nach GUHA (14) aus Diacetyl-dithioharn-
stoff (II) in Gegenwart von Phosgen oder aus Acetyl-Thiosemicarbazid (III)
mit Schwefelkohlenstoff und Kaliumhydroxid erhalten werden kann. Das
Mercaptan oder dessen Disulfid (2) werden mit Brom oder Chlor in saurer
Lösung zum Sulfonylhalogenid (IV) oxydiert, und dieses mit flüssigem
Ammoniak zu Acetazolamid (V) umgesetzt.

- 61 -
CH3-C-NH-C-NH-NH-C-NH-C-CH3
O S o
CH3-C-NH-C-NH-NH,Il IIO S
II
COCl2
Toluol
140°
N N
H2S + CH3-C-NH-
O
-SH_,
> CH3-C-NH-C12 II
-LM- 4 HCl
CH,-C-NH-IIO
Mögliche Verunreinigungen
Ausgangsstoffe, Ammoniak, Salzsäure, Schwefelkohlenstoff, Kalium- und
Natriumbromide.

- 62 -
Identitätsprüfungen
Nachweis von Acetazolamid: Wie schon von ENGLER (10) beschrieben, ist
die Kupferreaktion pH-abhängig. Unter pH 10,5 tritt praktisch keine Fällung
und Färbung auf, oberhalb pH 12,5 aber Blaufärbung durch Kupferhydroxid.
Nachweis der primären aromatischen Aminogruppe: Durch Salzsäure 7% RS
wird die Acetylgruppe nicht abgespalten und die Diazoreaktion verläuft nega¬
tiv. Wir verwenden deshalb Salzsäure 37% RS.
Absorptionsspektrum: Wir ziehen die Bestimmung der Absorptionsmaxima
im UV und deren Grössenverhältnis vor, weil sie mehr aussagen, als unab¬
geklärte Farbreaktionen.
In Äthanol, Methanol und 0,1 N Salzsäure, sowie in Ph.Helv.V Pufferlösungen
von pH 8 und 10 zeigt Acetazolamid nur ein Maximum bei 265 nm, weil die
nicht enolisierte Form vorliegt. Mit Lösungen in 0,1 N Natriumhydroxid er¬
hielten wir 2 Maxima bei 241 und 292 nm; die Werte aber zeigten grosse
Schwankungen, welche wir auf die Spaltung des Säureamids zurückführten.
Beim Vergleich der Werte in 0,1 N Natriumhydroxid, bestimmt nach 0,4,8
und 24 Stunden sahen wir, dass die £-Werte zunahmen. Dies ist verständ¬
lich, da das einsame Elektronenpaar des Stickstoffs bei der desazetylierten
Verbindung nur noch auf den Ring wirkt. Mit Lösungen in Ph.Helv.V Puffer¬
lösungen pH 11,4 erhielten wir die beiden Maxima der enolisierten Form,
deren £-Werte auch nach 24 Stunden konstant waren (292 nm: £,.= 12 000;
241 nm: £ = 3 700). Nach YOUNG, WOOD, EICHLER, VAUGHAN und
ANDERSON (44) ist Acetazolamid zwischen den pH 11 und 1 3 durch Anionen-
bildung der enolisierten Carbonylgruppe stabil.

- 63 -
Abbildung 4
Absorptionsspektren von Acetazolamid
Eltf3
I I I I 1 I I I 1
210 230 2S0 270 290 nm
in Äthanol in HCl 0,1 N
in Puffer pH 11,4 in NaOH 0,1 N
Gehaltsbestimmung
1. Wasserfreie Titration: Acetazolamid reagiert als zweibasische Saure,
zeigt aber kleine Potentialsprunge. Beide potentiometrischen Wendepunkte
ergaben einen Wert von 99,85^0 Acetazolamid. Die potentiometrische Titra¬
tion ist langwierig und nur in geschlossenen Apparaturen durchfuhrbar. Azo-
violett schlägt beim ersten Potentialsprung um, o-Nitroanilin, Alizarin¬
gelb G und Alizaringelb R beim zweiten. Der Umschlag der Alizarinfarbstoffe
scheint uns weniger scharf, weshalb wir o-Nitroanilin vorschlagen. Als Lo¬
sungsmittel verwendeten wir Dimethylformamid.

- 64 -
Indikator Handelsmuster Mittelwert
Azoviolett: rotviolett 98,63%; 100,21%; 98,83% 99,22
rosa 102,09%; 100,91%; 102,04% 101,34
Alizaringelb R 98,63%; 98,15%; 98,44%; 98,92% 98,54
Alizaringelb G 99,23%; 98,81%; 99,63%; 98,69%
98,54%; 99,19%; 99,34%; 99,66%
97,93% 98,99
o-Nitroanilin: 100,51%: 100,93%; 100,51% 100,62
KRACMAR und VACEK (20) verwenden Pyridin als Lösungsmittel.
2. Spektrophotometrie : Die quantitative Bestimmung ergab folgende Werte,
bestimmt bei 265 nm in 0,1 N Salzsäurelösung (E, = 461, £ = 10246): 103,14%;° 1cm
96,46%; 99,92%.
Löslichkeit: Die Angaben der USP XVI, der Brit.Ph.1963 und der Pro Phar-
macopoea (29) weichen stark voneinander ab. Unsere Versuche deckten sich
mit jenen der Brit. Ph. 1963.
Anwendung
Diureticum, durch Beeinflussung der Carboanhydrase in den Nierentubuli.

Vorschlag.
im
als
anders
aber
durchgeführt,
Prüfung
bedeutet:
+
entsprechend.
Vorschlages
des
Anforderungen
den
bedeutet;
=
99,0-101.0%
Titration
Wasserfreie
%100,62
Titration
Wasserfreie
min.98,5%
Titration
Wasserfreie
9,0%
9min.UV
98,0-102,0%
IR
Gehalt
%1
max.O,
0%
1%
max.0,
max.0,1%
%max.0,1
Verbrennungsrückstand
5%
max.O,
0,2%
5%
max.O,
max.0,5%
KF
%max.0,5
Wassergehalt
all
all
Grenzprüfung
max.400/Mill.
Sulfat
Ia
Ia
140/Mill.
max.
Chlorid
all
Ha
Grenzprobe
20/Mill.
max.
Schwermetalle
B5
FVL
B5
FVL
Schwefelsäureprobe
4,4-5,6
5,0
4,0-5,0
Svon
pH
251-252°
258°
ca.
Schmelzbereich
Standard
wie
IR-Spektrum
11,4
pH
nm
291
und
242
Maximum
=
HCl
in
nm
265
Maximum
Absorptionsspektrum
=+
+Schwefel
von
Nachweis
==
Aminogruppe
aromatischen
primären
der
Nachweis
==
blaugrün
=+
Acetazolamid
von
Nachweis
Ph.Helv.VI
Vorschlag
Handelsmuster
(29)
Pharm.Franc.
Pro
3Brit.Pharm.196
XVI
USP
Acetazolamidum
Prüfungsresultate
und
Prüfungsvorschriften
der
Zusammenstellung
7Tabelle

- 66 -
Acetazolamidum (DCI)
(Acetazolamid.)
Acetazolamid Ace'tazolamide Acetazolamide
N N0
IIO O
-NH2
C4H603N4S2 Mol.Gew. 222,3
2-Acetylamino-5-sulfamidyl-1,3,4-thiadiazol mit einem Gehalt von mindestens
99,0 (99,0-101,0) %, bezogen auf die getrocknete Substanz.
Prüfung
Stammlosung: (S): 0,5 g werden mit 25 ml Wasser 1 Minute gekocht und nach
dem Erkalten filtriert. Das Filtrat, das farblos sein muss, dient als S fur
die Prüfungen 4 und 6-9.
1. Sinnenprufung: Weisses, höchstens schwach gelbliches, kristallines Pul¬
ver, geruchlos und von schwach bitterem Geschmack.
2. Nachweis von Acetazolamid: 50 mg werden in 5 ml 0,1 N Natriumhydroxid
gelöst und mit 3 Tropfen Kupfersulfat RS verset/t. Es entsteht eine bestandige
türkisblaue Fallung.
3. Nachweis der primären aromatischen Aminogruppe: 10 mg werden in 1 ml
Salzsäure 37% RS gelost. Durch 2 Tropfen Natriumnitrit RS färbt sich die
Losung gelb und nach Zusatz von 2 ml (3-Naphthol RS rot.
4. Nachweis von Schwefel: 50 mg werden mit 0,4 g getrocknetem Natrium-
carbonat R vermischt und in einem Reagenzglas ohne zu glühen bis zur Ver¬
kohlung erhitzt, wobei der Geruch nach Schwefelwasserstoff auftritt. Nach
dem Erkalten wird der Ruckstand in 3 ml Wasser aufgenommen, mit 5 Tropfen
Bleiazetat RS versetzt und mit Salzsäure 37% RS angesäuert. Nach Abklingen
der Kohlendioxid-Entwicklung wird das Reagenzglas mit einem mit Fuchsin-

- 67 -
Formaldehyd RS getränkten Filtrierpapier bedeckt und in ein Wasserbad ge¬
stellt. Das Filtrierpapier färbt sich blau oder blauviolett.
5. Absorptionsspektrum: 0,2 g werden in 250 ml Puffer pH 11,4 RS gelöst.
1,00 ml dieser Lösung wird mit 100 ml Puffer pH 11,4 RS versetzt und deren
Absorption bei 292 und 241 nm bestimmt. Das Verhältnis der Extinktionen muss
ca. 3,55 betragen.
6. Reaktion: pH von S = 4,4-5,6
7. Schwefelsàureprobe: 0,1 g darf nicht stärker gefärbt sein als Farbver-
gleichslösung Bs.
8. Schwermetalle : 2 ml S müssen den Anforderungen der Grenzreaktion a II
genügen.
9. Chlorid: 2 ml S müssen den Anforderungen der Grenzreaktion a I genügen.
10. Sulfat: 2 ml S müssen den Anforderungen der Grenzreaktion a II genügen.
11. Wassergehalt: Höchstens 0,5%, bestimmt mit 0,5 g (Trockenschrank).
12. Verbrennungsrückstand: Höchstens 0,1 fo bestimmt mit 0,5 g.
13. Gehalt: Bestimmt mit 0,1 g getrockneter Substanz durch wasserfreie
Titration, Methode C (Allgemeine Bestimmungen Ph.Helv.Vl) unter Ver¬
wendung von 3 Tropfen o-Nitroanilin RS bis zum Umschlag von Grüngelb nach
Orangegelb titriert.
1 ml 0,1 N LiOCH, entspricht 1 1,1 2 mg C4H603N4S2
Aufbewahrung: In gut verschlossenem Behälter, unter Lichtschutz
SEPARANDUM

- 68 -
3.1.5 Probenecidum DCI
N-(Dipropyl-p-sulfamyl)-benzoesäure
f-\? /3H7HO-C-
O \ /Ö
^H,11 \ / « \3 \=/ O C
Ci3H1904NS Mol.Gew. 285,4
Spezialpräparate
R TD
Benemid (Merck, Sharpe und Dohme), Perdurine (Pharma Union Belgique),R R
Probecid (Astra Schweden), Probenzen (Vi-Be Italia).
Prüfungsvorschriften
Brit.Ph.1963, USPXVI.
Herstellung
A) Durch Kondensation von p-Carboxybenzol-sulfonylchlorid mit Dipropyl-
amin (35).
B) Durch Kondensation von p-Cyanbenzol-sulfonylchlorid (I) mit Di-n-propyl-
amin (II). Die Cyangruppe des erhaltenen p-Cyanbenzol-sulfonyl-di-n-propyl-
amins (III) wird mit Natriumhydroxid zu Probenecid (IV) verseift.

- 69 -
OCnH-
NC-T VS-C1 + HNfO
C^
3n7
3H7
II
o
ON/C3H7
s-n;
OC3H7
III
NaOH + 2 H20
HO-C-C VS-N^
o\=/ O^
+ NH3 + NaOH
IV
C) DAL MONTE CASONI (27) beschreibt die Synthese ausgehend von Toluol (V)
über p-Toluolsulfonsäurechlorid (VI) und dessen Amid (VII). Die durch Oxy¬
dation erhaltene p-Sulfamido-benzoesäure (VIII) kondensiert er mit Propyl-
amin in alkalischer Lösung zu Probeneeid (IV).
CH,
V
CISOoOH
CH3
o=s=oI
Cl
VI
COOH
I
o=s=o
C^-N-C,!!,
NH,
C3H7NH2
CH,
o=s=o1
NH,
VII
COOH
o=s=o!
NH2
Oxydât.
IV VIII

- 70 -
Mögliche Verunreinigungen
Ausgangsstoffe, Mineralsäuren und Ammoniak, Alkalihydroxide.
Identitätsprüfungen
Nachweis des Benzoesäurerestes; Probeneeid lässt sich mit Kalciumcarbonat
in wässriger Lösung unter Kochen zu Benzoesäure aufspalten. Diese wird im
Filtrat als Eisen(in)-benzoat nachgewiesen.
Nachweis des Dipropylaminrestes: Die Reaktion auf tertiäre Amine fällt nega¬
tiv aus, weil der Stickstoff amidartig an die Sulfogruppe gebunden ist. Die
hydrolytische Abspaltung des Amins ist für eine Nachweisreaktion zu lang¬
wierig. Es müsste mit Raney-Nickel gearbeitet werden, oder einige Stunden
in Säure oder Base gekocht werden. Da Dipropylamin einen Sdp. von 111° be¬
sitzt, wurde Probeneeid mit Ätznatron vermischt und in einem Glühröhrchen
erhitzt. Die Dämpfe bläuten rotes Lackmuspapier.
Obige chemische Nachweise genügen aber nicht zur Identifizierung. Coron-
amid [p-(Benzylsulfonamido)-benzoesäure] z.B. unterscheidet sich von
Probeneeid nur durch die Absorptionskurve im UV und die Diazotierung nach
hydrolytischer Spaltung.
Absorptionsspektrum: Die Absorptionskurve im UV einer äthanolischen Lö¬
sung zeigt 2 Maxima im Gegensatz zur Lösung in Chloroform und Natrium¬
hydroxid. Die Maxima der äthanolischen Lösung liegen bei 247 nm (£ = 9964)
und 225 nm (£ = 9484) und sollten als Identitätsnachweis genügen (siehe Abb. 5).

E1tf>
- 71 -
Abbildung 5
Absorptionsspektren von Probenecid
11
10
9
-
^- \ /'/ / v
/\ \
---'" / \ \ \
t ~ /
/
\ \ \
\ \ \
7 -
/ \ \ \
\ \ \
\ \ \
« ~ \ \ \
'> \ \
5 -
\ \ \
\ \ \
4 - \ \
3
\2
1
1 1 1 1 1 1 1
210 230 250 270 290 im
in Äthanol
in 0,1 N Natriumhydroxid
in Chloroform
Gehaltsbestimmung
1) Acidimétrie: Die acidimetrische Bestimmung ist einfach auszuführen und
ergibt, wie aus der Tabelle 8 zu ersehen ist, ebenso genaue Werte, wie die
wasserfreie Titration und verdient deshalb den Vorzug.
2) Wasserfreie Titration: 0,3 g Probenecid werden wie Carbutamidum
(siehe S. 35), aber mit dem unten angeführten Indikator durchgeführt.
1 ml 0,1 N LiOCH3 entspricht 28,54 mg CuH1904NS

- 72 -
Wie untenstehende Resultate zeigen sind alle vier Indikatoren brauchbar.
Alizaringelb R, auch Alizaringelb G genannt, eignet sich aber mit seinem
Farbumschlag von Gelborange nach Violett besser als Alizaringelb GG,
welches von Gelb nach Orange umschlägt.
Handelsmuster III
(umkristallisiert)
Indikator
100,07%;100,47%;
100,33%;100,39%.
Mittelwert
Alizaringelb G oder R 100,32%
Alizaringelb GG 99,52%;100,54%.
99,34%; 99,80%
Azoviolett 100,18%;100,45%.
100,31%; 100,31%
o-Nitroanilin 99,42%; 100,46%. 99,94%
3) Spektrophotometrie: Diese Methode weist bekanntlich einen grösseren
Fehlerbereich auf und ist, da spezifisch, ev. bei der Bestimmung des Wirk¬
stoffes in Tabletten zu gebrauchen (siehe Identitätsprüfung S. 70 ).
Anwendung: Diureticum. Setzt die Resorption von Uraten und die Ausscheidung
von Penicillin, p-Aminosalizylsäure und Phenolphthalein herab.

entsprechend.
Vorschlags
des
Anforderungen
den
bedeutet:
=
99,0-101,0%
99,89%
100,
37%)
99,23%
min.98,0%
min.98,0%
acidimetrisch
Gehalt
unwägbar
unwägbar
unwägbar
unwägbar
1%
max.O,
max.0,1%
ruckstand
Verbrennungs-
5%
max.O,
00,3%
0,3%
5%
max.O,
5%
max.O,
Wassergehalt
all
all
all
II
aSulfat
Ia
all
ai
II
aChlorid
all
II
aall
II
amax.20/Mill.
max.10/Mill.
metalle
Schwer
G4
FVL
farblos
Gs
FVL
Gs
FVL
probe
äure-
Schwefels
4,6-5,4
195-198°
5,2
197-198°
4,8
195-196°
4,8
194-196°
198-200°
198-200°
Svon
pH
Schmelzbereich
nm
225
247,
Maxima
==
=Standard
wie
1cm
36)
3ca.
=(E^°
nm
225
248,
Maxima
spektrum
Absorptions¬
==
==
Schwefels
des
Nachweis
==
==
Dipropylamin
von
Nachweis
__
==
Benzoesäure
von
Nachweis
Ph.Helv.VI
Vorschlag-
III
Handelsmuster
II
Handelsmuster
I
Handelsmuster
XVI
USP
Brit.Ph.1963
Probenecidum
Untersuchungsresultate
und
Prufungsvorschriften
der
Zusammenstellung
8Tabelle

- 74 -
Probenecidum
(Probenec.)
Probenecid Probénécide Probenecido
0°/C3H7
S-N
Ci3H1904NS Mol.Gew. 285,4
N-(Dipropyl-4-sulfamyl)-benzoesäure mit einem Gehalt von mindestens 99,0
(99,0-101,0) % C^HtgC^NS, bezogen auf die getrocknete Substanz.
Prüfung
Stammlösung (S): 1,0 g wird in 50 ml Wasser 1 Minute lang gekocht, erkalten
gelassen und filtriert. Das Filtrat, das farblos sein muss, dient als S für die
Prüfungen 7 und 9-11.
1. Sinnenprüfung : Weisses, geruchloses und schwach bitter schmeckendes
Pulver.
2. Nachweis des Benzoesäureesters: 50 mg werden mit 0,2 g Kalciumcarbonat
R gekocht und nach Abklingen der Kohlendioxidentwicklung filtriert. Das Filtrat
gibt mit 5 Tropfen Eisen(lIl)-chlorid RS einen fleischfarbenen Niederschlag.
3. Nachweis des Dipropylaminrestes: 50 mg werden mit 0,2 g Natriumhydro¬
xid R vermischt und in einem Glühröhrchen erhitzt. Die Dämpfe bläuen rotes,
befeuchtetes Lackmuspapier.
4. Nachweis von Schwefel: 50 mg werden mit 0,4 g wasserfreiem Natrium-
carbonat ohne zu glühen bis zur Verkohlung erhitzt. Nach dem Erkalten wird
der Rückstand in 3 ml Wasser aufgenommen, mit 5 Tropfen Bleiazetat RS ver¬
setzt und mit Salzsäure 7% RS angesäuert. Nach dem Abklingen der Kohlen¬
dioxidentwicklung wird das Reagenzglas mit einem mit Fuchsin-Formaldehyd RS
befeuchteten Filtrierpapier bedeckt und in ein Wasserbad gestellt. Das Filtrier¬
papier färbt sich blau oder blauviolett.

- 75 -
5. Absorptionsspektrum: Eine ca. 0,001proz. äthanolische Lösung zeigt
Maxima bei 247 und 225 ran mit dem molaren Extinktionsverhältnis 1,05.
6. Schmelzbereich: 195-198°.
7. Reaktion: pH von S = 4,6-5,4.
8. Schwefelsäureprobe : 0,1 g darf auch nach dem Erwärmen im Wasserbad
keine stärkere Färbung bewirken, als Farbvergleichslösung G4.
9. Schwermetalle: 3 ml S müssen den Anforderungen der Grenzreaktion a II
entsprechen.
10. Chlorid: 3 ml S müssen den Anforderungen der Grenzreaktion a I ent¬
sprechen.
11. Sulfat: 3 ml S müssen den Anforderungen der Grenzreaktion a II ent¬
sprechen.
12. Wassergehalt: Höchstens 0,5%, bestimmt mit 1 g (Trockenschrank).
13. Verbrennungsrückstand: Höchstens 0,1%, bestimmt mit 0,5g.
14. Gehalt: 0,5 g getrocknete Substanz (genau gewogen) werden in 50 ml
Äthanol 94% RS gelöst und unter Verwendung von 3 Tropfen Phenolphthalein
RS mit 0,1 N Natriumhydroxid bis zur Rosafärbung titriert.
1 ml 0,1 N NaOH entspricht 28,54 mg CuH1904NS
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
Gebrauchsdosen: Einzeldosis = 500 mg
Tagesdosis = 500 mg - 2,0 g
Löslichkeit: Unlöslich in Wasser, löslich in 25 T. Äthanol 94%, 12 T Azeton,
in verdünnten Alkalien; unlöslich in Mineralsäuren.
R R R RMarkennamen: Benemid
,Perdurine
,Probeeid
,Probenzen
.

- 76 -
3.1.6 Sulfamethizolum PCI
5-Sulfanilamino-2-methyl-1,3,4-thiadiazol
oN N
/T\ » II II\=/& ^S^
C9H10O2N4S2 Mol.Gew. 270,3
Spezialpräparate
RR RLucosil (Lundbeck), Rufol (Debat France), Thiosulfil (Ayerst), Uroluco-
sil (Lundbeck).
Prüfungsvorschriften
NNR (1957), Ph.Nord. (Dan.) 1964, Analysmetoder XVI (1946), NF XI 1960,
Debat (Paris).
Herstellung
a) Man kondensiert p-Acetylamino-benzol-sulfonylchlorid (I) mit Acetaldehyd-
thiosemicarbazon (II) in Pyridin gelöst (24). Das Zwischenprodukt (III) oxy¬
diert man mit Kalziumferricyanid und erhält dadurch im alkalischen Milieu
den Ringschluss. Durch Azetylabspaltung mit Essigsäure wird Sulfamethizol
gewonnen.
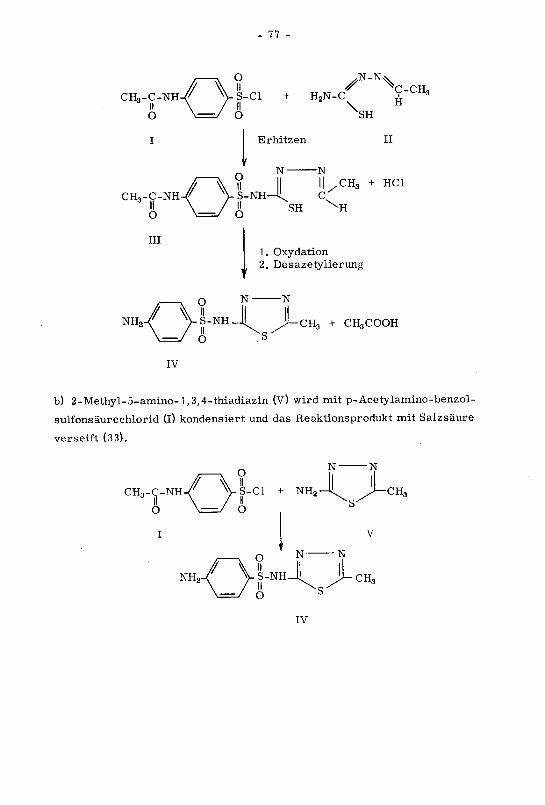
- 77 -
CH3-C-NH
O
/ Vs-Cl + H2N-C^ VÇ-CH3•*"%
o
\H
SH
CH. -C-NH-f >S-NH—H.
Erhitzen
N N
II
,CH, + HCl
III
SH ^H
1. Oxydation2. Desazetylierung
NH2-T VS-NH.
N N
-CH3 + CH3COOH
IV
b) 2-Methyl-5-amino-1,3,4-thiadiazin (V) wird mit p-Acetylamino-benzol-
sulfonsäurechlorid (I) kondensiert und das Reaktionsprodukt mit Salzsäure
verseift (33).
N N
CH3-C-NH^A-Lci + NH.-^ JLCH,
NH;/ V|.NH-
N N
•CK,
IV

- 78 -
Mögliche Verunreinigungen
Ausgangsstoffe, Essigsäure, Salzsäure, Alkalien.
Identitätsprüfungen
Nachweis der Sulfogruppe: Nur die Analysmetoder XVI (1946) fordern auch
diesen Nachweis durch Schwärzung von Bleiacetatpapier. Die im Suppl.III der
Ph.Helv.V bei den Sulfonamiden geforderte Reaktion ist auch bei Sulfamethizol
durchführbar, doch verzichten wir auf diese Reaktion zu Gunsten von spezifi¬
scheren.
Nachweis des Thiadiazolringes: Mit Eisen(lü)-sulfat oder Kupfersulfat, wie
sie die Ph.Nord.1964 (Dan.), NF 1960 und Analysmetoder XVI (1946) verwen¬
den, soll der heterocyclische Ring nachgewiesen werden. Nach ENGLER (10)
sind diese Farbreaküonen stark pH-abhängig und unspezifisch. Bei gleichen
Versuchsbedingungen erhielten wir mit allen Handelsmustern von Sulfa¬
methizol gelb-grüne Niederschläge mit Kupfersulfat RS, welche beim Stehen¬
lassen die Farbe nicht ändern; mit Eisensulfat RS dagegen erhielten wir keinen
deutlichen Unterschied gegenüber dem schon an sich gefärbten Reagenzgemisch.
Wir schlagen deshalb die Kupfersulfat-Reaklion vor.
ReinheitsPrüfungen
Freie Säure: Im Suppl.III der Ph.Helv.V wird bei Sulfamerazinum und Sulfa-
diazinum die freie Säure gegen Methylorange titriert, mit der Begründung
nur die sauren Verunreinigungen und nicht auch die Acidrlät des Sulfonamids
zu erfassen wie bei der Titration gegen Phenolphthalein. Da aber im ersten
Fall nur maximal 0,05 ml 0,1 N Natriumhydroxid verbraucht werden dürfen,
scheint uns die Einhaltung der obigen Forderung auf Kosten der Genauigkeit
zu gehen.
Wir schlagen vor, nur die Reaktion (pH) zu bestimmen.
Lösungsverhalten: Sulfamethizol ist als Natriumsalz löslich; die gelblichen
Pulver zeigten in Natriumhydroxid zum Teil starke Verfärbungen, welche
durch Aufbewahrung am Licht entstehen.
Arsen: Die Notwendigkeit dieser Prüfung wird heute oft in Frage gestellt.
Der Kommentar des 3.Nachtrags zum DAB 6 schliesst bei Sulfanilamid ein
Arsenvorkommen aus, doch scheint uns bei Verwendung technischer Säuren
die Verunreinigung durch Arsen möglich zu sein.

- 79 -
Gehaltsbestimmung
1. Bromometne. Die Firma Debat bestimmt den Gehalt von Sulfamethizol
bromometrisch nach folgender Vorschrift
"Ca 0 6g getrockneter Substanz werden in einem 100 ml Mess¬
kolben in 20 ml Salzsaure 5% RS gelost und mit Wasser bis zur
Marke ergänzt. 10,0 ml werden in einen Erlenmeyerkolben mit
Glasstopfen gegeben und mit Natriumhydroxid 7% RS bis zur Blau¬
färbung von Lackmuspapier alkalisch gemacht Nach Zugabe von
20,0 ml 0,1 N Bromid-Bromat, 30 ml Wasser und 10 ml Salz¬
saure 37% RS wird im Dunkeln 2 Stunden stehen gelassen. Dann
werden 10 ml Kaliumjodid 10% RS zugefugt, mit Wasser auf ca
350 ml verdünnt und das freiwerdende Jod mit 0,1 N Natrium-
thiosulfat titriert"
1 ml 0,1 N KBrGj entspricht 45,04 mg CgHI0O2N4S2
Nach dieser Vorschrift erhielten wir zu tiefe Resultate mit starken
Schwankungen. Diese können auch durch Änderung der Bromierungszeit
(bis 6 Stunden) nicht genügend verbessert werden
2. Acidimetrische Bestimmung. Ph.Nord.1964 (Dan ) bestimmt direkt alka¬
limetrisch gegen Thymolblau wie folgt
"Ca. 125 mg werden durch Erwarmen in 10 ml Äthanol 94% ge¬lost nach Abkühlen und Zufügen von 10 ml Wasser wird mit
0,1 N Natriumhydroxid unter Zusatz von 5 Tropfen ThymolblauRS titriert"
Fur das Handelsmuster III erhielten wir einen Durchschnitt von 100 1%
Der Farbumschlag ist aber nicht sehr deutlich wodurch die beobachteten
Schwankungen bedingt sein können
Der Analysmetoder XVI 1946 bestimmt indirekt alkalimetrisch gegen Phenol¬
phthalein Diese Vorschrift die wir auch fur unsere Pharmakopoe vor¬
schlagen (siehe S 84) ergab uns folgende Werte
Handelsmuster
I II III
Mittelwerte 99 45% 99 75% 100 17%
Der Umschlagspunkt ist sehr deutlich und auch die Herstellung der Losung
rascher als die alkoholische Durch feiner kalibrierte Büretten können
noch genauere Resultate erhalten werden
IV
100 17%

- 80 -
3. Wasserfreie Titration: Siehe Carbutamidum S. 35.
1 ml 0,1 N LiOCH3 entspricht 27,03 mg Sulfamethizol
Für das Handelsmuster III erhielten wir einen Durchschnitt von 100,2%.
4. Argentometrie : Zu Vergleichszwecken haben wir auch die in der Ph.Helv.V
übliche Silbernitratmethode (siehe Carbutamidum S.36) durchgeführt. Va¬
riierende Mengen Salpetersäure 12 % RS von 4,0-4,8 ml ergaben zu stark
streuende Werte von 94,04-102,69%.
Anwendung
Als Chemotherapeuticum; der guten Löslichkeit wegen speziell bei Infektionen
der Harnwege verwendet.

- 81 -
Tabelle 9
Übersicht verschiedener Prüfungsvorschriften
Sulfamethizolum Ph.Nord.
1964
AnalysmetoderXVI 1946
NF XI
1960
VorschlagPh.Helv.VI
Nachweis der primä¬ren aromatischen
Aminogruppe
Schmelzbereich des
Acetylderivâtes 232-235° 241-245°
Nachweis des
Thiadiazolringes = =-
Schmelzbereich 208-212° 204-206° 207-211° 209-212°
pH von S 4,2 4,0-4,8
Lösungsverhalten 5 dg in
3 ml NaOH
5 dg in 3 ml
N NaOH
max. FVL G„
Acidität
5 dg
Methylorange-Methylrot +
max. 0,5 ml
0,1 N NaOH
Arsen - -
Schwermetalle - - 20 T/Mil. all
Chlorid - schw.Opales. 140 T/Mil. ai
Sulfat - 400 T/Mil. a II
Wassergehalt max. 1 % max. 0,5% max. 0,5%
Verbrennungs¬rückstand max. 0,1 % max. 0,2% max. 0,2%
Gehalt direkt acidi-
metrisch ge¬
gen Thymol-blau
99 - 101 %
indirekt acidi-
metrisch gegen
Phenolphthalein98,4-100,6%
Diazo-
tierungmind. 99 %
indirekt
acidime-
trisch ge¬
gen Phenol¬
phthalein99,0-100,5%
= bedeutet: den Anforderungen des Vorschlags entsprechend.
- bedeutet: abwesend, bzw. unwägbar.
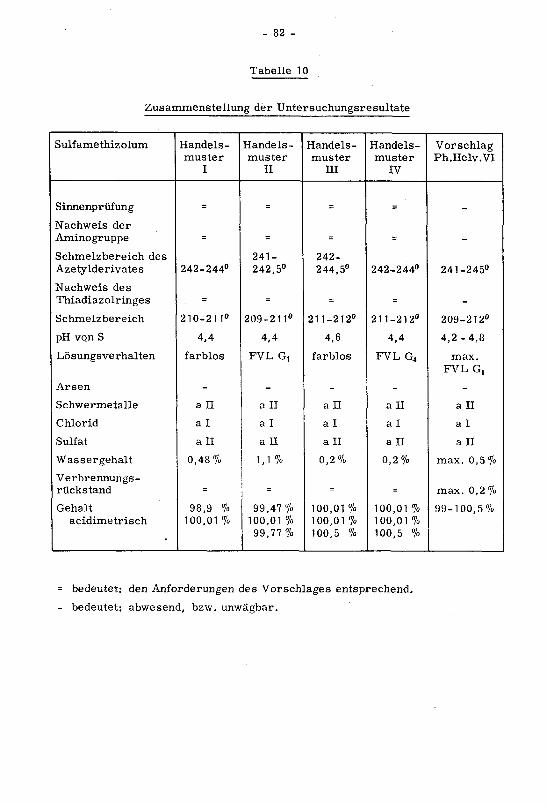
- 82 -
Tabelle 10
Zusammenstellung der Untersuchungsresultate
Sulfamethizolum Handels¬
muster
I
Handels¬
muster
II
Handels¬
muster
HI
Handels¬
muster
IV
VorschlagPh.Helv.VI
Sinnenprüfung - -
= —
_
Nachweis der
Aminogruppe = = = =_
Schmelzbereich des
Azetylderivates 242-244°
241-
242,5°
242-
244,5° 242-244° 241-245°
Nachweis des
Thiadiazolringes = = = = _
Schmelzbereich 210-211° 209-211° 211-212° 211-212° 209-212°
pH von S 4,4 4,4 4,6 4,4 4,2-4,8
Lösungsverhalten farblos FVL Gt farblos FVL G4 max.
FVL G4
Arsen - - - - -
Schwermetalle all all all all an
Chlorid ai ai ai ai a I
Sulfat all all all all all
Wassergehalt 0,48% 1,1 % 0,2% 0,2% max. 0,5%
Verbrennungs¬rückstand =
i= = max. 0,2%
Gehalt
acidimetrisch
98,9 %
100,01 %99,47%100,01 %
99,77%
100,01 %
100,01 %
100,5 %
100,01 %
100,01 %
100,5 %
99-100,5%
= bedeutet: den Anforderungen des Vorschlages entsprechend.
- bedeutet: abwesend, bzw. unwägbar.

- 83 -
Sulfamethizolum (PCI)
(Sulfamethizol.)
Sulfamethizol Sulfamethizol Sulfametizolo
C9Hl0O2ISr^S2 Mol.Gew. 270,3
5-Sulfanilamino-2-methyl-1,3,4-thiadiazol mit einem Gehalt von mindestens
99,0 (99,0-100,5) % C9H10O2N4S2.
Prüfung
Stammlösung (S); 1,0 g wird in 50 ml Wasser während 1 Minute gekocht, er¬
kalten gelassen und filtriert. Das Filtrat dient als S für die Prüfungen 6 und
9-11.
1. Sjnnenprüfung: Farblose Kristalle oder weisses bis gelblich-weisses,
geruchloses Pulver von schwach bitterem Geschmack.
2. Nachweis der primären aromatischen Aminogruppe: 10 mg werden in
2 Tropfen Salzsäure 7 % RS gelöst und mit 1 ml Wasser verdünnt. Diese Lö¬
sung färbt sich auf Zusatz von 2 Tropfen Natriumnitrit RS gelb. Beim Zu-
tropfen von 2 ml p-Naphthol RS entsteht sofort ein orangefarbener Nieder¬
schlag, dann eine tiefrote Färbung.
3. Nachweis des Acetylderivates: 0,2 g werden mit 4 ml Eisessig R gekocht
bis die Substanz gelöst ist, dann mit 5 Tropfen Essigsäureanhydrid R einige
Minuten gekocht und abgekühlt. Durch Reiben an der Glaswand des Reagenzgla¬
ses bildet sich ein weisser Niederschlag, welcher abfiltriert mit Wasser ge¬
waschen und aus Äthanol 94% umkristallisiert wird. Schmelzbereich: 241-245°.
4. Nachweis des Thiadiazolringes: Ca. 50 mg werden in 5 ml Wasser aufge¬
schwemmt und tropfenweise Natriumhydroxid 7% RS zugefügt bis gerade voll¬
ständige Lösung eintritt. Beim Zugeben von 2 Tropfen Kupfersulfat RS ent¬
steht sofort ein gelbgrüner Niederschlag.

- 84 -
5. Schmelzbereich: 209-212°.
6. Reaktion: pH von S = 4,2-4,8.
7. Lösungsverhalten: 0,5 g müssen sich in 3 ml 1 N Natriumhydroxid klar
lösen. Diese Lösung darf nicht stärker gefärbt sein als Farbvergleichslösung
G4.
8. Arsen: In 0,5 g darf kein Arsen nachweisbar sein.
9. Schwermetalle: 10 ml S müssen den Anforderungen der Grenzreaktion a II
genügen.
10. Chlorid: 10 ml S müssen den Anforderungen der Grenzreaktion a I genügen.
11. Sulfat: 10 ml S müssen den Anforderungen der Grenzreaktion a II genügen.
12. Wassergehalt: Höchstens 0,5%, bestimmt mit 0,5 g (Trockenschrank).
13. Verbrennungsrückstand: Höchstens 0,2% bestimmt mit 0,5 g.
14. Gehalt: Ca. 0,25 g getrocknete Substanz (genau gewogen) werden in
15,00 ml 0,1 N Natriumhydroxid gelöst. Nach Zugabe von 2 Tropfen Phenol¬
phthalein RS wird mit 0,1 N Salzsäure bis zur Entfärbung titriert.
1 ml 0,1 N NaOH entspricht 27,03 mg C9Hl0O2N4S2
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
Antimikrobielle Behandlung von Lösungen
Trockenschrank 140°, nach Vortrocknung während 2 Stunden bei 103-105°.
Separandum
Gebrauchsdosen: Einzeldosis = 500 mg
Tagesdosis = 2,0 - 3,0 g
Löslichkeit: 1 T. löst sich in ca. 2000 T. kaltem, in 60 T. kochendem Wasser,
in 30 T. Äthanol 94%, 10 T. Azeton; schwer löslich in Äther und Chloroform;
löslich in Alkalien und Mineralsäuren.
Inkompatibilitäten: Schwermetallsalze (Fällungen).
Offizinelle Präparate: Compressi Sulfamethizoli 500 mg, Iniectabile Sulfa-
methizoli 40%.R RR R
Markennamen; Lucosil,Urolucosil
,Rufol
,Thiosulfil
.

- 85 -
3.2. Ausarbeitung von Prüfungsvorschriften fur einige Sulfonamid-
Tabletten
3.2.1. Allgemeines
Bisher galt nach Ph.Helv.VI die Richtlinie, dass die Zusammenstellung der
offizineilen Tabletten mit Bezug auf Art und Menge Hilfsstoffe vorgeschrieben
waren und entsprechend auf eine einlassliche Prüfung der Tabletten verzichtet
werden konnte. Bereits fur die Supplemente II und III und besonders fur die
Ph.Helv.VI wurde von diesem Grundsatz abgegangen, indem auf die Herstel¬
lungsvorschriften mit Angabe der Art und Menge der Hilfsstoffe verzichtet
wird, um dem Hersteller eine gewisse Bewegungsfreiheit bei der Tabletten¬
bereitung zu lassen. Damit erhält er die Möglichkeit, die physikalisch, che¬
misch und pharmakologisch geeigneten und zulässigen Hilfsstoffe selbst zu
wählen. Um aber die von der Pharmakopoe vorgeschriebene Gehaltsbestimmung
abzusichern, war es notwendig, die folgende Vorschrift in die Allgemeinen Be¬
stimmungen aufzunehmen:
"Zur Herstellung der offizinellen Arzneipräparate dürfen keine
Hilfsstoffe und Hilfsstoffmengen verwendet werden, welche die
vorgeschriebene Gehaltsbestimmung stören".
Diese Vorschrift hat zur Folge, dass die Ausarbeitung der Prufungsvorschrif¬
ten, vor allem der Gehaltsbestimmung, nicht Rucksicht zu nehmen braucht
auf die grosse Zahl der verwendbaren Hilfsstoffe.
3.2.2. Aufbau dor Prufungsvorschriften fur Tabletten
Diese Prufungsvorschriften beschranken sich nach den Richtlinien zur Aus¬
arbeitung der Ph.Helv.VI auf folgende Punkte:
1. Sinnenprufun» Farbe, Geruch und Geschmack.
2. Nachweis der Arzneistoffe
a) Durch Abtrennung der reinen Substanz und Bestimmung ihres
Schmelzbereiches,
b) durch Nachweis mit den in der betreffenden Monographie vorgeschrie¬benen Identitätsreaktionen.
3. Gehalt an deklariertem Arzneistoff.

- 86 -
Entsprechend den Vorschriften der Allgemeinen Monographie "Compressi"
müssen samtliche von uns bearbeiteten Präparate auch geprüft werden auf
1. Dosierungsgenauigkeit des Gewichtes,
2. Zerfallbarkeit.
3.2.3. Allgemeine Bemerkungen zu den einzelnen Prufungsabschnitten
1. Nachweis der Arzneistoffe
Um den Arzneistoff möglichst rein isolieren zu können, bemuhten wir
uns vorerst die wichtigsten und am meisten gebrauchten, bei der Extrak¬
tion loslichen Hilfsstoffe abzutrennen
Die allgemein gebräuchlichen Tablettenhilfsstoffe zeigen folgendes
Losungsverhalten.
Unlöslich in Wasser Agar, kolloides Aluminiumhydroxid, Kalziumstearat,
Fette, kolloide Kieselsaure Magnesiumstearat (4%), Magnesiumtrisilikat,
Paraffinole, Starke (10-25%) (Amylopektm), Stearinsaure (4%), Talk,
Tragant (60-70% unlöslich), Trikalziumphosphat.
Löslich in Wasser. Alginate, Dextrin (5%), Glukose, Glycerin, Laktose
(10-25%), Mannitol, Methylcellulose, Pektin, Polyathylenglykole,
Saccharose (10%), Tragant (0,5%) (30-40% loslich)
In heissem Wasser. Agar, Gelatine, Gummi arabicum, (4%), Starke (10-25%)
(Amylose).
Löslich in organischen Losungsmitteln Fette (Äther, Chloroform, Petrol-
ather), Paraffinole (Äther, Chloroform, Petrolather, Fette Öle), Stearin¬
saure (Azeton, Benzol, Chloroform, Schwefelkohlenstoff, Tetrachlorkoh¬
lenstoff).
Bei den in Chloroform schwer löslichen Arzneistoffen verreiben wir die
Tabletten und extrahieren mit diesem Losungsmittel um Glyzerin, Paraffin
Stearinsaure und andere chloroformlosliche Hilfsstoffe zu beseitigen Bei
den Tolbutamidtabletten wurde auf diese Vorbehandlung verzichtet, weil
Tolbutamid zu gut chloroformloslich 1st
Die Zahlen in Klammern geben den in Tabletten verwendeten höchsten
Prozentgehalt an.

- 87 -
Zur Extraktion der Sulfonamide benutzen wir Salzsaure 7% RS, wenn
4-Amino-benzol-Denvate die Bildung wasserlöslicher Hydrochloride ge¬
statteten. Diese Methode hat den Vorteil, dass die aus Stearaten frei ge¬
setzte Stearinsaure nicht mitextrahiert wird. Sulfonamide (Probenecid
und Tolbutamid), welche keine Hydrochloride bilden, wurden mit Ammo¬
niak in Ammoniumsalze übergeführt und gelangten so in die wassrige Phase.
Aus den wassrigen Auszügen fällten wir die Sulfonamide durch Neutralisa¬
tion mit Ammoniak, Ammoniumazetat resp. mit Salzsaure 7% RS. Durch
halbstündiges Stehenlassen an einem kühlen Ort forderten wir die Kristal¬
lisation, wuschen mit wenig kaltem Wasser aus und trockneten die erhalte¬
nen Kristalle je nach Sulfonamid im Trockenschrank oder im Vakuumex-
sikkator (Phosphorpentoxid).
Im Vergleich zu den Vorschriften der USP XVI welche die Tabletten
direkt mit Chloroform und der Brit.Ph.1963, welche mit Azeton extra¬
hieren, erhalten wir wie folgender Vergleich der Schmelzbereiche der
isolierten Sulfonamide zeigt, durch Fallen reinere Substanzen.
.Schmelzbereich des
°- isolierten Tolbutamids
mit Chloroform 126-127°
aus Azeton 126-127°
durch Fallen 127-128,5°
Die aus Tabletten zu 500 mg Arzneistoff isolierte Menge Reinsubstanz
betrug bei
Acetazolamid 190 mg
Carbutamid 211 mg
Probenecid 154 mg
Tolbutamid 342 mg
und reichte somit gut aus zur Durchfuhrung der m den Monographien vor¬
geschriebenen Identitatsreaktionen.
2. Gehaltsbestimmung
Wir hielten uns an die Richtlinie, fur die Gehaltsbestimmung der Tabletten
möglichst die Bestimmungsmethode zu verwenden, welche in der Mono¬
graphie des betreffenden Sulfonamids vorgeschrieben ist. Dank der allge¬
meinen Ph.-Vorschrift, dass bei den Tabletten verwendete Hilfsstoffe die

offizineile Gehaltsbestimmungsmethode nicht stören und abfälschen dürfen,
konnten wir auf die mühevolle, kaum zu bewältigende Aufgabe verzichten,
den Einfluss sämtlicher Hilfsstoffe auf die Gehaltsbestimmung zu überprü¬
fen. Immerhin sei im folgenden auf die Möglichkeit einer Beeinflussung der
wichtigsten Bestimmungsverfahren für Sulfonamide durch die hauptsächlich¬
sten Hilfsstoffe hingewiesen.
Extraktion des Wirkstoffes mit Azeton und gravimetrische Bestimmung:
Folgende Gründe veranlassten uns diese Bestimmung nicht weiter zu
prüfen. Die isolierte Reinsubstanz ist gelblich und ihr Schmelzbereich
liegt ca. 2° tiefer als jener der Reinsubstanz. Azeton extrahiert zum Teil
auch Hilfsstoffe.
Sulfadimidin-Tabletten ergaben:
gravimetrischTitration
des Rückstandes
99,0% 95,3%100,2% 96,4%100,9% 97,6%
Bromometrie: MIRIMANOFF (26) zeigte den Einfluss von Glukose, Laktose,
Magnesiumstearat und Natriumbenzoat. Er eliminierte die Laktose bei
Compressi Sulfaguanidini mittels Pyridin. Nach CONWAY (8) werden aber
die Hilfsstoffe in einem Essigsäuremilieu von 50-70% nicht oxydiert. Auch
wir konnten feststellen, dass 300-500 mg Laktose kein Brom verbrauchen.
So kann die unangenehme und umständliche Ausschüttelung umgangen wer¬
den.
Acidimetrische Methode: Diese ist unseres Erachtens für Tablettenbe¬
stimmung geeignet, weil praktisch keine sauer reagierenden Stoffe als
Hilfsstoffe verwendet werden. MIRIMANOFF (26) gibt zwar Stearinsäure
(max. 4%) und Benzoesäure (1-2%) an, doch wurden sie in keiner Firmen-
Deklaration für von uns untersuchte Sulfonamid-Tabletten angeführt.
10 mg Stearinsäure (ca. 1,5%) verbrauchen 0,1-0,15 ml 0,1 N Natrium¬
hydroxid, bei sehr starkem Schütteln 0,2 ml, was bei den Sulfonamid-
Tabletten einen Fehler von nur 0,5-1% ausmachen würde.

- 89 -
Wasserfreie Titration:
Von Basen: Laktose, Stärke und Stearinsäure stören nicht, wohl aber
Polyäthylenglykol, welches ev. mit Chloroform abgetrennt werden kann.
Von Säuren: Nach FABER (11) bewirken Zucker und Gelatine keine, Talk
und Starke nur sehr kleine Fehler. 5 mg Magnesiumstearat sollen 0,05 ml
0,1 N Lithiummethylat verbrauchen. VESPE und FRITZ (41) befürworten
diese Bestimmung fur Tabletten, weil im allgemeinen keine Säuren als
Hilfsstoffe verwendet werden. Auch fur Magnesiumstearat fanden sie nur
einen zu vernachlässigenden Fehler. 0,5 g MagnesiumtrisiJikat und Kaolin
verbrauchen 0,2 ml 0,1 N Lithiummethylat. Alizaringelb eignet sich nicht
gut als Indikator, weil es mit Kalzium und Stearaten inkompatibel ist.
Argentometrie: Stearinsaure und deren Salze stören, können aber leicht
als Bariumsalze eliminiert werden, wie dies bei den Compressi Sulfa-
dimidmi Ph.Helv.V vorgeschrieben ist.
Spektrophotometrische Methode. Diese Bestimmung ist sehr spezifisch,
hat aber den Nachteil des relativ grossen Fehlers (1-3%). Die üblichen
Tablettenhilfsstoffe stören nicht, da sie keine Absorption im UV be¬
sitzen. Da die Aufschwemmung filtriert wird, ist auch die Beeinflussung
durch Lichtstreuung ausgeschlossen.
Die verschiedenen Pharmakopoen schreiben die m Tabelle 1 1 aufgeführ¬
ten Bestimmungsverfahren und Gehaltslimiten vor.

- 90 -
Tabelle 11
Übersicht der Pharmakopöevorschriften
Compressi USP XVI Brit.Ph.
1963
Ph.Nord.
(Dan.) 1964VorschlagPh.Helv.VI
Carbutamidi
500 mg
- - - acidimetr.
95,0-105,0%
Tolbutamidi
500 mg
spektrophot.
95,0-105,0%Kjeldahl92,5-107,5%
- acidimetr.
95,0-105,0%
Acetazolamidi
250 mg
polarograph.92,5-107,5%
spektrophot.92,5-107,5%
- spektrophot.92,0-108,0%
Probenecidi
500 mg
spektrophot.95,0-105,0%
spektrophot.95,0-105,0%
- wasserfrei
95,0-105,0%
Sulfamethizoli
500 mg
- diazometr.
95,0-105,0%acidimetr.
90,0-1 10,0%acidimetr.
95,0-105,0%
Trisulfonamidi
500 mg
diazometr.
95,0-105,0%
diazometr.
95,0-105,0%gravimetr.
103,0-
106,0%
argentometr.96,0-105,0%
Am nützlichsten erschien uns bei der Ausarbeitung der Methode zur Ge¬
haltsbestimmung Muster des betreffenden Sulfonamids mit den mutmasslich
störenden Hilfsstoffen herzustellen und dieses Gemisch analytisch nach der
in der Monographie des Arzneistoffes vorgeschriebenen Methode zu über¬
prüfen. Ergibt diese Untersuchung gute Resultale, so kann auf die Über¬
prüfung weiterer Methoden verzichtet werden. Anderseits wurde angestrebt,
die Gehaltsbestimmung ohne Abtrennung der Hilfsstoffe aus dem Tabletten¬
pulver durchzuführen, weil die Extraktion und Fällung der Sulfonamide die
Methode kompliziert und zudem neue Fehlermöglichkeiten mit sich bringt.
3. Gehaltsforderung
Als Untersuchungsmuster werden 20 Tabletten gesamthaft genau gewogen,
fein pulverisiert und gut durchgemischt. Von dieser Durchschnittsprobe
wird die in den einzelnen Artikeln jeweils vorgeschriebene Menge genau
gewogen und zur Gehaltsbestimmung verwendet. Auf diese Weise wird so¬
mit der Durchschnittswert von 20 Tabletten ermittelt. Aus diesem Grunde
lässt sich eine Gehaltsforderung mit 95,0-105,0% des deklarierten Arz¬
neistoffes rechtfertigen.

- 91 -
3.2.4. Prüfungsvorschriften der einzelnen Sulfonamid-Tabletten
3.2.4.1 Compressi Carbutamidi 500 mg
A) Hilfs stoffdeklaration der Handelsmuster
Hilfsstoffe Handelsmuster
I II III IV
Agar +
Gelatine + +
Glycerin +
Magnesiumstearat + + + +
Natriumlaurylsulfat +
Paraffinöle +
Stärke + + + +
Talk + + + +
B) Untersuchte Bestimmungsmethoden
Wir untersuchten die folgenden, schon bei der Reinsubstanz geprüften
Bestimmungsmethoden: Bromometrie, Acidimétrie, Wasserfreie Titra¬
tion, Argentometrie und zwar ohne Isolierung des Wirkstoffes nach den
bei Carbutamidum (siehe S. 33-36) aufgeführten Vorschriften. Bei der
argentometrischen Bestimmung wurden die Stéarate mit Bariumsulfat
nach der bei "Compressi Sulfadimidini" Ph.Helv.V Suppl. III gegebenen
Vorschrift eliminiert.
C) Vorbereitung des Bestimmungsmaterials
20 Tabletten wurden zur Ermittlung der Dosierungsgenauigkeit des Ge¬
wichtes einzeln und gesamthaft gewogen, fein gepulvert und gut durch¬
mischt.
D) Resultate
Dosierungsgenauigkeit des Gewichts
Handelsmuster
I II III IV
srel 0,30 0,30 0,50 0,55

- 92 -
Die nach MÜNZEL, BÜCHI und SCHULTZ (28) berechneten srel genügen der
von SANDELL (32) für Tabletten aufgestellten Forderung von srel 4,5. Auch
die Vorschrift der Ph.Helv.VI, dass keine von 30 Tabletten mehr als 10%
vom Sollgewicht abweichen dürfe, war erfüllt.
Gehaltsbestimmung
Handelsmuster III
bromometrisch acidimetrisch wasserfreie argentometrisch
Mittel¬
werte
Titration
98,40% 98,66% 99,72% 94,30%98,05 99,23 99,48 97,98
98,26 99,55 99,00 95,80
98,20 99,17 99,70 95,54
98,20 99,13 99,42 96,24
99,30 100,40 96,50
98,90
98,88
98,86
98,89
98,22% 99,06% 99,60% 96,06%
Acidimetrische Methode nach Vorschlag für "Compressi Carbutamidi 500 mg".
Handelsmuster
Mittelwerte 99,0%
II
98,78%
Die bromometrische Methode ergab nach der Vorschrift für "Sulfaguanidinum"
Ph.Helv.V (siehe S. 33) zu hohe Werte von 105%.
Die argentometrische Methode ergab auch bei Verwendung von Reinsübstanz
und Zusatz verschiedener Hilfsstoffe zu tiefe Werte.
Die spektrophotometrische Bestimmung nach Extraktion von Carbutamid be¬
schrieben VITALI und PANCRAZIO (42). Gemessen wird in Äthanol bei
269 ran (E, = 705 + 20) mit einem Fehler von+ 3%.
1cm
Die kolorimetrische Bestimmung lehnen obige Autoren als unbefriedigend ab.

- 93 -
E) Beurteilung und Wahl der Methode
Die beiden acidimetrischen Methoden eignen sich am besten. Auch die
bromometrische Bestimmung gibt gute Resultate, doch braucht es längere
Zeit, den Wirkstoff aus der Tablettenmasse herauszulösen.
Wir schlagen die acidimetrische Methode ohne Eliminierung der Hilfs¬
stoffe als Pharmakopöemethode vor. Der Zusatz von sauerreagierenden
Hilfsstoffen wird durch die Wahl dieser Bestimmungsmethode unzulässig,
da er das Resultat beeinflusst.
Die statistische Auswertung und Beurteilung der acidimetrischen Be¬
stimmungsmethode für Tabletten ist auf S. 23 aufgeführt.

- 94 -
Compressi Carbutamidi 500 mg
Carbutamidtabletten 500 mg Comprimes de carbutamide
à 500 mg
Compresse di carbutamide a 500 mg
Tabletten mit einem Gehalt von 0,5 (0,475-0,525) g Carbutamid (CUH17Q3N3S).
Prüfung
1. Herstellung des Tablettenpulvers: 20 Tabletten werden gesamthaft genau ge¬
wogen, fein gepulvert und gut durchmischt.
2. Sinnenprüfung: Weisse oder höchstens schwach gelbliche geruchlose, fast
geschmacklose Tabletten.
3. Nachweis von Carbutamid:
a) Eine ca. 0,5 g Carbutamid entsprechende Menge Tablettenpulver wird mit
5 ml Chloroform R verrieben und filtriert. Der Rückstand wird mit wei¬
teren 5 ml Chloroform R gewaschen und das Filtrat verworfen. Dann wird
der Rückstand mit 1 0 ml Salzsäure 7% RS verrührt und die durch Filtra¬
tion gewonnene Lösung mit 3 g Ammonazetat R in 3 ml Wasser versetzt
und eine halbe Stunde an einem kühlen Ort stehen gelassen. Der entstan¬
dene Niederschlag wird abfiltriert, mit wenig kaltem Wasser gewaschen
und über Phosphorpentoxid im Vakuumexsikkator getrocknet.
Schmelzbereich: 136-140°.
b) Der Niederschlag muss die in der Monographie "Carbutamidum" vorge¬
schriebenen Identitätsreaktionen zeigen.
4. Gehalt: Eine ca. 0,5 g Carbutamid entsprechende Menge Tablettenpulver
(genau gewogen) wird 2 Minuten lang mit 30 ml Methanol R umgeschwenkt
und dann mit 20 ml ausgekochtem Wasser versetzt. Nach Zusatz von 3 Tropfen
Phenolphthalein RS wird mit 0,1 N Natriumhydroxid bis zur eben auftretenden
Rotviolettfärbung titriert. Nach Zugabe von weiteren 30 ml ausgekochtem Was¬
ser wird bis zur bleibenden Rotviolettfärbung titriert.
1 ml 0,1 N NaOH entspricht 27,1 3 mg CuH1703N3S
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
S eparandum
Gebrauchsdosen: Tagesdosis = 1-3 Tabletten

- 95 -
3.2.4.2 Compressi Tolbutamidi 500 mg
A) Hilfsstoffdeklaration der Handelsmuster
HilfsstoffeHandelsmuster
I II III
Agar +
Aerosil 0,3%
Magnesiumstearat + 0,3% 1%
Natriumlaurylsulfat +
Stärke + 23,3% 25%
Talk + 1,5% 3,5%
Untersuchte Bestimmur.igsmethoden
Wir untersuchten die acidimetrische Methode (Vorschlag Compressi
Tolbutamidi 500 mg, siehe S. 97 ) die wasserfreie Titration (siehe
Carbutamidum S. 35) und die spektrophotometrische Bestimmung nach
dem First USP XVI Supplement 1962 ohne und mit Ausschüttelung.
C) Vorbereitung des Bestimmungsmaterials
20 Tabletten wurden zur Ermittlung der Dosierungsgenauigkeit des Ge¬
wichtes einzeln und gesamthaft gewogen, fein gepulvert und gut durch¬
mischt.
D) Resultate
Dosierungsgenauigkeit des Gewichts
Handelsmuster
I II III
srel 1,3 0,58 0,72
Die nach MÜNZEL, BÜCHI und SCHULZ (28) berechneten srel genügen
der von SANDELL (32) für Tabletten aufgestellten Forderung von srel 4,5.
Auch die Forderung der Ph.Helv.VI, dass keine von 30 Tabletten mehr als
10% vom Sollgewicht abweichen dürfen, war erfüllt.

- 96 -
acidimetrisch
99,02%99,34
99,75
98,82
98,81
Mittelwerte 99,15%
99,60%99,20
101,23
100,42
101,53
100,55
100,73
102,00
100,59
Gehaltsbestimmung
Handelsmuster I
wasserfreie
Titration
99,66%
spektrophoto-metrisch
95,45%96,05
Mittelwerte 100,65%
95,75%
Handelsmuster II
100,46% 98,70%101,82 103,50
101,82 103,90
101,84 103,32
101,80 102,17
101,00
101,20
101,42% 102,32%
E) Beurteilung und Wahl der Methode
Alle Methoden eignen sich, doch ist die acidimetrische weitaus die ein¬
fachste und zeigt kleine Fehler. Bei der spektrophotometrischen Bestim¬
mung erhielten wir ohne und mit Ausschütteln die gleichen Werte mit
dem Maximum bei 264 nm.
Wir schlagen die acidimetrische Methode ohne Eliminierung der Hilfs¬
stoffe als Pharmakopöemethode vor. Der Zusatz von sauerreagierenden
Hilfsstoffen bei der Tablettenherstellung ist demzufolge unzulässig, da
er das Resultat beeinflusst.

- 97 -
Compressi Tolbutamidi 500 mg
Tolbutamidtabletten 500 mg Comprimas de tolbutamide
à 500 mg
Compresse di tolbutamide a 500 mg
Tabletten mit einem Gehalt von 0,5 (0,475-0,525) g (Tolbutamid (C12H1803N2S).
Prüfung
1. Herstellung des Tablettenpulvers: 20 Tabletten werden gesamthaft genau
gewogen, fein gepulvert und gut durchmischt.
2. Sinnenprüfung: Weisse geruchlose und geschmacklose Tabletten.
3. Nachweis von Tolbutamid:
a) Eine ca. 0,5 g Tolbutamid entsprechende Menge Tablettenpulver wird
2 Minuten lang mit 10 ml Ammoniak 3 % RS umgeschwenkt und dann filtriert.
Das Filtrat wird mit 10 ml Essigsäure 12 % RS versetzt und wahrend einer
halben Stunde an einem kühlen Orte stehen gelassen. Der entstandene Nie¬
derschlag wird abfiltriert, mit wenig kaltem Wasser gewaschen und bei
103-105° getrocknet: Schmelzbereich: 126-130°
b) Der Niederschlag muss die in der Monographie "Tolbutamidum" vorge¬
schriebenen Identitätsreaktionen zeigen.
4. Gehalt: Eine ca. 500 mg Tolbutamid entsprechende Menge Tablettenpulver
(genau gewogen) wird 2 Minuten lang mit 30 ml Methanol R umgeschwenkt und
mit 20 ml ausgekochtem Wasser versetzt. Nach Zusatz von 3 Tropfen Phenol¬
phthalein RS wird bis zur eben eintretenden Rotviolettfärbung titriert. Nach
Zugabe von weiteren 30 ml ausgekochtem Wasser wird bis zur bleibenden
Rotviolettfärbung titriert.
1 ml 0,1 N NaOH entspricht 27,04 mg C12H1803N2S
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
Separandum
Gebrauchsdosen: Tagesdosis =1-3 Tabletten

- 98 -
3.2.4.3. Compressi Acetazolamidi 250 mg
A) Handelsmuster
Es war leider nur ein Handelsmuster ohne Deklaration erhältlich.
B) Untersuchte Bestimmungsmethoden
Wir überprüften die wasserfreie Titration wiederum mit verschiedenen
Indikatoren (siehe S. 64) und die spektrophotometrische Bestimmung nach
der Brit.Ph.1963.
C) Vorbereitung des Bestimmungsmaterials
Die vorhandenen 10 Tabletten wurden gesamthaft genau gewogen, fein ge¬
pulvert und gut durchmischt.
D) Gehaltsbestimmung
Wasserfreie Titration
Azoviolett
100,73%100,25
100,88
98,59
Mittelwerte 100,11%
Handelsmuster
o-Nitroanilin
102,10%104,10
101,92
103,63
102,94%
Alizarin
99,57%101,14
100,41
101,20
103,11
101,09%
Spektrophotometrisch
103,26%, 100,44%, 104,99% Mittelwert 102,90%
E) Beurteilung und Wahl der Methoden
Beide untersuchten Methoden eignen sich gut. Wir schlagen die spektro¬
photometrische Bestimmung vor, weil sie spezifischer ist, doch ist sie
der grossen Verdünnung wegen bekanntlich weniger empfindlich. Der Zu¬
satz von Stoffen mit Absorption im UV bei 265 nm in salzsaurer Lösung ist
durch die Wahl dieser Methode unzulässig.

- 99 -
Compressi Acetazolamidi 250 mg
Acetazolamidtabletten 250 mg Comprimés de acetazolamide
à 250 mg
Compressi di acetazolamide a 250 mg
Tabletten mit einem Gehalt von 0,25 (0,23-0,27) g Acetazolamid (C4HeOiSaN4).
.Prüfung
1. Herstellung des Tablettenpulvers: 20 Tabletten werden gesamthaft genau
gewogen, fein gepulvert und gut durchmischt.
2. Sinnenprüfung: Weisse, geruchlose, fast geschmacklose Tabletten.
3. Nachweis von Acetazolamid:
a) Eine ca. 0,5 g Acetazolamid entsprechende Menge Tablettenpulver wird
mit 5 ml Chloroform R verrieben, filtriert, der Rückstand nochmals mit
5 ml Chloroform R gewaschen und das Filtrat verworfen. Dann wird der
Rückstand 2 Minuten lang mit 10 ml Salzsäure 7 % RS umgeschwenkt, die
durch Filtration gewonnene Lösung mit 3 g Ammoniumazetat R in 3 ml
Wasser versetzt und eine halbe Stunde an einem kühlen Orte stehen ge¬
lassen. Der entstandene Niederschlag wird abfiltriert, mit wenig kaltem
Wasser gewaschen und bei 103-105° getrocknet. Schmelzbereich: 253-256°
(Zersetzung).
b) Der Niederschlag muss die in der Monographie "Acetazolamidum" vorge¬
schriebenen Identitätsreaktionen zeigen.
4. Gehalt: Eine ca. 0,25 g Acetazolamid entsprechende Menge Tablettenpul¬
ver (genau gewogen) wird in einen Messkolben von 1000 ml Inhalt in 400 ml
siedendes Wasser gegeben. Die Mischung wird 15 Minuten auf dem Wasser¬
bade erwärmt und nach dem Abkühlen bis zur Marke ergänzt und filtriert.
Die ersten 10 ml des Filtrates werden verworfen. 10,00 ml des Filtrates
werden in einen Messkolben von 250 ml Inhalt gegeben, welcher 25 ml 1 N
Salzsäure enthält und bis zur Marke ergänzt. Die Absorption wird bei 265 nm
1 °*
gemessen ( £ = 10246, E'
= 460,99).

- wo -
Aufbewahrung
In gut verschlossenem Behälter, unter Liehtschutz.
Separandum
Gebrauchsdosen: Tagesdosis = 1-2 Tabletten

- 101 -
3.2.4.4. Compressi Probenecidi 500 mg
A) Hilfsstoffdeklaration des Handelsmuster I
Kalziumstearat 1 %
Gelatine 3 %
Magnesiumcarbonat 9 %
Stärke 18 %
B) Untersuchte Bestimmungsmethoden
Da Probenecid Reinsubstanz sich sehr gut acidimetrisch bestimmen lässt,
versuchten wir den Wirkstoff mit Äthanol 94% zu extrahieren und im Fil¬
trat nach der Methode der Monographie (siehe S. 75) zu bestimmen. Eine
auftretende Opaleszenz verhinderte aber eine genaue Titration. Auch die
in der Monographie "Compressi Carbutamidi" (siehe S.94) vorgeschlagene
Methode ergab zu grosse Schwankungen. Dann prüften wir die wasserfreie
Titration potentiometrisch und unter Verwendung verschiedener Indikatoren.
Spektrophotometrisch bestimmten wir nach den Vorschriften der USP XVI
und der Brit.Ph. 196 3 nach Extraktion mit Äthanol; erstere vergleicht mit
1%einem Standard, die Brit.Ph. fordert bei 248 nm E,
'
= 332. Das Supple-1cm
ment der US XVI extrahiert mit Chloroform, schüttelt mit Natriumcarbo-
nat 1 "o RS aus und extrahiert Probeneeid aus dieser Lösung nach Ansäuern
wiederum mit Chloroform. Die Absorption wird bei 256 nm gemessen und
mit einem Standard verglichen.
C) Vorbereitung des Bestimmungsmaterials
20 Tabletten wurden zur Ermittlung der Dosierungsgenauigkeit des Ge¬
wichts einzeln und gesamthaft genau gewogen, fein gepulvert und gut durch¬
mischt.
D) Resultate
Dosierungsgenauigkeit des Gewichts
Handelsmuster
I II
srel 0,43 1,8
Die nach MÜNZEL, BÜCHI und SCHULZ (28) berechneten srel genügen
der von SANDELL (32) für Tabletten aufgestellten Forderung von srei 4,5.

- 102 -
Auch die Forderung der Ph.Helv.VI, dass keine von 30 Tabletten mehr als
10% vom Sollgewicht abweichen dürfe, war erfüllt.
Gehaltsbestimmung
Wasserfreie Titration
gegen o-Nitroanilin
Handelsmuster
I 99,41%, 103,05%, 103,86%, Mittelwert 102,11 %II 106,83%, 102,80%, 104,81% Mittelwert 104,81 %
gegen Alizaringelb R
Handelsmuster
I gibt FällungII 103,00%, 101,75%, 103,63%, 102,67% Mittelwert 103,68%
potentiometrisch
Handelsmuster II: 100,49 %
Spektrophotometrische Bestimmung nach USP XVI und Brit.Ph. 1963
Handelsmuster
I 98,90%, 95,53%, 97,41% Mittelwert 97,28 %
II 91,59%, 93,31%, 96,56% Mittelwert 9 3,82 %
nach First USP XVI Supplement 1962
Handelsmuster
I 83,96%, 84,42%, 88,60%, 93,17%, 86,39% Mittelwert 87,31 %
II 97,61%, 96,45% Mittelwert 95,06 %
Wurde einmal mehr mit Chloroform ausgeschüttelt, erhielten wir einen
um 1,18% höheren Gehalt.
Wir führten auch zwei Bestimmungen durch ohne Ausschüttelung, d.h.
durch Lösen des Wirkstoffes in Chloroform und Filtration und erhielten
Werte von 93,17 % und 94,41 %.
Da wir bei allen Bestimmungen in Chloroform das Maximum bei 254 nm
fanden, bestimmten wir obige Werte bei dieser Wellenlänge. Mit Rein¬
substanz fanden wir £= 9664, bzw. E,°
= 343.1cm

- 103 -
E) Beurteilung und Wahl der Methode
Beide Methoden streuen stark und befriedigen nicht ganz. Als Pharma-
kopöemethode schlagen wir die wasserfreie Titration vor, wodurch der
Zusatz von sauerreagierenden Stoffen unzulässig wird, da er das Resultat
beeinflusst.
Bei der wasserfreien Titration würde sich Alizaringelb R sehr gut eignen,
da sein Umschlag dem Wendepunkt am nächsten liegt, doch ist dieser Indi¬
kator mit Stearinsäure, Magnesiumstearat und Benzoesäure inkompatibel,
d.h. beim Titrieren wird der Farbstoff mit dem Niederschlag mitgerissen.

- 104 -
Compressi Probenecidi 500 mg
Probenecidtabletten 500 mg Comprimés de probénécide à 500 mg
Compresse di probenecido a 500 mg
Tabletten mit einem Gehalt von 0,5 (0,475-0,525) g Probenecid (CuH1904NS).
Prüfung
1. Herstellung des Tablettenpulvers: 20 Tabletten werden gesamthaft genau
gewogen, fein gepulfert und gut durchmischt.
2. Sinnenprüfung: Weisse, geruchlose, fast geschmacklose Tabletten.
3. Nachweis des Probeneeids:
a) Eine ca. 0,5 g Probenecid entsprechende Menge Tablettenpulver wird mit
5 ml Chloroform R verrieben, filtriert, der Rückstand nochmals mit 5 ml
Chloroform R gewaschen und das Filtrat verworfen. Dann wird der Rück¬
stand 2 Minuten lang mit 10 ml Ammoniak 3% RS verrieben, die durch Fil¬
tration gewonnene Lösung mit 10 ml Essigsäure 12% RS versetzt und eine
halbe Stunde an einem kühlen Ort stehen gelassen. Der entstandene Nieder¬
schlag wird abfiltriert, mit wenig kaltem Wasser gewaschen und bei 10 3-105°
getrocknet. Schmelzbereich: 193-198°.
b) Der Niederschlag muss die in der Monographie "Probenecidum" beschrie¬
benen Identitätsreaktionen zeigen.
4. Gehalt: Eine ca. 0,3 g Probenecid entsprechende Menge Tablettenpulver
(genau gewogen) wird nach 5 Minuten langem Rühren nach den Allgemeinen
Bestimmungen der Ph.Helv.VI für wasserfreie Titration C, unter Verwendung
von 5 Tropfen o-Nitroanilin RS bis zum Farbumschlag von Grüngelb nach
Orangegelb titriert.
1 ml 0,1 N LiOCH3 entspricht 28,54 mg C13H1904NS
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz
Separandum
Gebrauchsdosen: Tagesdosis = 1-4 Tabletten

- 105 -
3.2.4.5. Compressi Sulfamethizoli 500 mg
A) Hilfs stoffdeklaration der Handelsmuster
Hilfsstoffe
Agar
Gelatine
Magnesiumstearat
Milchzucker
Stärke
Talk
Handelsmuster
I
2%
1%
8%
II
Handelsmuster I enthielt 100 mg Wirkstoff
B) Untersuchte Bestimmungsmethoden
Wir untersuchten die acidimetrische, wasserfreie und argentonaetrische
Bestimmung nach den Vorschriften der Reinsubstanz (siehe S. 79 ,35 ,36)
und die gravimetrische Methode nach der Extraktion von Sulfamethizol mit
Azeton.
C) Vorbereitung des Bestimmungsmaterials
20 Tabletten wurden gesamthaft gewogen, fein gepulvert und gut durch¬
mischt.
D) Gehaltsbestimmung
Gravimetrische Bestimmung
250 g Sulfa¬ 500 mg Sulfa¬
Handelsmuster methizol methizol
I II + Hilfsstoffe + Hilfsstoffe
93,83% 92,61 %
93,87 92,00
Acidimetrische Bestimmung
99,02% 96,54% 97,25% 100,30%99,42 97,31 99,79 100,10
96,99 97,54 98,31
96,58 95,98 98,17
99,31 100,20
96,58 98,35
Mittelwerte 98,03% 96,36% 98,67% 100,15%

- 106 -
Handelsmuster
I II
250 g Sulfa-
methizol
+ Hilfsstoffe
500 mg Sulfa-
methizol
+ Hilfsstoffe
Wasserfreie Bestimmung
Mittelwert
95,30%95,56
95,76
95,90
95,63%
Argentometrische Bestimmung
Mittelwert
Mittelwert
98,16%100,32
96,84
97,64
97,95%
98,24%98,80
99,60
100,42
99.26%
mit 1,5 ml Essigsäure 12 % RS
mit 2,2 ml Essigsäure 1 2 % RS
E) Beurteilung und Wahl der Methode
Wir schlagen die acidimetrische Methode als Pharmakopöebestimmung
vor, weil sie sehr einfach und rasch ist sowie der Bestimmungsmethode
der Reinsubstanz entspricht. Durch eine höhere Einwaage oder Verwendung
von 0,05 N Natriumhydroxid könnte der Fehler verkleinert werden. Je 4
Bestimmungen mit Hilfsstoffen ohne Wirkstoff [Vorschrift Debat und
Ph.Nord.1964 (Dan.)] sowie 100 mg Magnesiumstearat verbrauchten kein
Natriumhydroxid.
Der Zusatz von sauerreagierenden Stoffen ist durch die Wahl der acidime-
trischen Methode unzulässig, da er das Resultat beeinflusst. Auch die ar¬
gentometrische Methode eignet sich, doch ist sie umständlich.

- 107 -
Compressi Sulfamethizoli 500 mg
Sulfamethizoltabletten 500 mg Comprimés de sulfaméthizole à 500 mg
Compresse di sulfametizolo 500 mg
Tabletten mit einem Gehalt von 0,5 (0,475-0,525) g Sulfamethizol (C9H10O2N4S2).
Prüfung
1. Herstellung des Tablettenpulvers: 20 Tabletten werden gesamthaft genau
gewogen, fein gepulvert und gut durchmischt.
2. Sinnenprüfung: Weisse, geruchlose, fast geschmacklose Tabletten.
3. Nachweis von Sulfamelhizol:
a) Eine ca. 0,5 g Sulfamethizol entsprechende Menge Tablettenpulver wird mit
5 ml Chloroform R verrieben und filtriert. Der Rückstand wird nochmals
mit 5 ml Chloroform gewaschen und das Filtrat verworfen. Dann wird der
Rückstand in 10 ml Salzsäure 7 *o RS aufgenommen, und die durch Filtra-
lion gewonnene Lösung mit 3 g kristallisiertem Ammonazetat R in 3 ml
Wasser versetzt und während einer halben Stunde an einem kühlen Ort
siehen gelassen. Der entstandene Niederschlag wird abfiltriert, sorgfältig
mil kaltem Wasser gewaschen und während einer Stunde bei 103-105° ge¬
trocknet. Schmelzbereich: 209-212°.
b) Der Niederschlag muss die bei Sulfamethizolum vorgeschriebenen Identi-
lälsreaktionen zeigen.
4. Gehall ; Eine ca. 250 mg Sulfamethizol enthaltende Menge Tablettenpulver
wird in 15.00 ml 0.1 N Natriumhydroxid 5 Minuten lang umgeschwenkt und mit
0,1 M Salzsäure unter Verwendung eines Tropfens Phenolphthalein bis zur
Entfärbung titriert.
1 ml 0,1 NaOH entspricht 27,03 mg CgHi0O2N,S2
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
Separandum
Gebrauchsdosen: Tagesdosis = 4-6 Tabletten

- 108 -
0,12 0,18 0,26
0,17 0,27 0,44
0,26 0,40 0,49
3.2.4.6. Compressi Trisulfonamidici 500 mg
A) Handelsmuster
Es war leider nur ein Handelsmuster mit der Deklaration je 166 mg Sulfa-
diazin, Sulfamerazin und Sulfathiazol und ohne Hilfs Stoffdeklaration erhält¬
lich.
B) Nachweis von Sulfadiazin, Sulfamerazin und Sulfathiazol
Wir versuchten die Auftrennung und den Nachweis der drei deklarierten
Sulfonamide mit Hilfe der Dünnschichtchromatographie.
Es wurden verschiedene Laufmittel geprüft; einzig mit Ammoniak 3% RS
und n-Butanol R (20Vol. %\ 80Vol. %) konnte auch Sulfadiazin und Sulfa¬
merazin getrennt werden. Die Rf-Werte sind stark von der Klimatisie¬
rungsdauer abhängig:
0 Std. 16 Std. 21 Std.
Sulfadiazin
Sulfamerazin
Sulfathiazol
Zum Vergleich führen wir auch die Rf-Werte einiger anderer Sulfon¬
amide an:
Sulfamethizol
Carbutamid
Sulfadimidin
Sulfisomidin
Sulfaguanidin
Sulfanilamid
Die Trennung ist bei einer Klimatisierungsdauer von ca. 16 Stunden am
besten und genügt zum qualitativen Nachweis. Die Sichtbarmachung erfolgt
durch Diazotierung.
C) Gehaltsbestimmung
Argentometrie
Da alle drei Sulfonamide als Reinsubstanzen in der Ph.Helv.V argento-
metrisch bestimmt werden, lag es nahe, diese Methode für die Ermittlung
des Gesamtgehaltes der Tabletten an Sulfonamid zu verwenden. Die neu¬
trale Reaktion des Gemisches ist der Löslichkeit des Silbersalzes wegen
0,25 0,32 0,49
0,25 0,31 0,38
0,22 0,35 0,51
0,29 0,36 0,32
0,42 0,49 0,47
0,55 0,61 0,59

- 109 -
von Wichtigkeit. Bestimmungen nach Zusatz von 0,5 ml Essigsäure 12 % RS
ergaben einen Durchschnitt von 524,5 mg/Tablette, mit 0,3 ml einen sol¬
chen von 496,03 mg/Tablette. Mit dem untersuchten Handelsmuster erhiel¬
ten wir folgende Resultate: 495,76 mg495,76
494,76
497,84
Dünnschichtchromatographie und Spektrophotometrie
Wir versuchten die drei Sulfonamide dünnschicht-chromatographisch zu
trennen und nach Auskratzen kolorimetrisch zu bestimmen. Wir arbeiteten
nach folgender Vorschrift:
"Mit einer Aglaspritze (kleinste Einteilung 0,0002 ml) wurden 0,015 mg
des Sulfonamidgemisches 6mal, rechts und links dieser Startpunkte
gleiche Mengen Testsubstanzen in 0,1proz. Azetonlosungen, aufgetragen.
Das Laufenlassen der Platten erfolgte wie bei der Identitätsreaktion be¬
schrieben, das Entwickeln aber nach Zudecken obiger 6 Laufstrecken.
Auf der Hohe der gewanderten, sichtbar gemachten Testsubstanzen wird
die nicht besprühte Fläche ausgekratzt, mit 10 ml 0,1 N Salzsäure 30 Mi¬
nuten geschüttelt und zentrifugiert. 5 ml der Sulfadiazin resp. Sulfamera-
zinlosung werden in einem Messkolben von 10 ml Inhalt mit 1 ml Natrium¬
nitritlosung 1 %o 3 Minuten geschüttelt, dann mit 1 ml Natriumsulfamat-
losung 5 °oo 2 Minuten und nach Zugabe von 1 ml N-(1 -Naphtyl)-äthylen-
diamin-hydrochloridlosung 1 "oo unter zeitweiligem Schuttein 1 5 Minuten
stehen gelassen. Nach Auffüllen mit 0,1 N Salzsäure bis zur Marke wird
die Absorption fur Sulfadiazin bei 540 nm, fur Sulfamerazin bei 545 nm
gegen den Blindwert gemessen nach der Bestimmung der USP XVI.
1 cm
Sulfadiazin bei 545 nm 2137
Sulfamerazin bei 540 nm 2065
Sulfathiazol kann kolorimetrisch so nicht bestimmt werden, weil eine
Fällung entsteht. Die Lösung von Sulfathiazol in 0,1 N Salzsäure zeigt
bei 280 nm ein Maximum mit E'
= 515,6.

- 110 -
Die Schwierigkeit besteht darin, dass nur ca. 0,005 mg Sulfonamid pro
Flecken aufgetragen werden kann, damit eine saubere Trennung möglich
ist. Um aber eine messbare Absorption zu erhalten, müssen mindestens
6 solcher Flecken gesamthaft bestimmt werden. Auf der gleichen Platte
können also keine Parallelbestimmungen mit Testsubstanzen durchgeführt
werden.
Wie untenstehende Werte zeigen, ist der Fehler bei der quantitativen
Dünnschicht-Chromatographie zu gross:
Sulfadiazin 6 x 0,005 mg 58,61 %
3 x 0,010 83,512 x 0,015 85,09
1 x 0,020 90,251 x 0,020 82,821 x 0,035 86,902 x 0,010 72,53
Aus Tablettenmasse (entsprechend 500 mg Wirkstoff) extrahiert mit
1 x 50 ml Azeton:
Sulfadiazin Sulfamerazin Aufgetragen je
62,94% 50,36% 6 x 0,0066 mg53,03 % 42,86 % 6 x 0,0066 mg
extrahiert mit 2 x 50 ml Azeton:
73,00% 71,67% 6 x 0,005 mg45,86% 43,61% 6 x 0,005 mg
extrahiert mit 3 x 50 ml Azeton:
28,64 % 49,60 % 6 x 0,00S5 mg
30,91% 60,16% 6 x 0,0055 mg.

- 111 -
Compressi Trisulfonamidi 500 mg
Trisulfonamidtabletten 500 mg Comprime's de trisulfonamide à 500 mg
Compresse trisulfonamidici a 500 mg
Tabletten gleiche Mengen Sulfadiazin, Sulfamerazin und Sulfathiazol enthal¬
tend, mit einem Gehalt von 0,5 (0,475-0,525) g des Sulfonamidgemisches.
Prüfung
1. Herstellung des Tablettenpulvers: 20 Tabletten werden gesamthaft genau
gewogen, fein gepulvert und gut durchmischt.
2. Sinnenprüfung: Weisse, höchstens schwach gelbliche, geruchlose und fast
geschmacklose Tabletten.
3. Nachweis von Sulfadiazin, Sulfamerazin und Sulfathiazol: Eine ca. 0,5 g
Wirkstoff entsprechende Tablettenmenge wird 2mal mit 50 ml Azeton R ex¬
trahiert. 0,005 ml dieser Lösung werden auf eine Dünnschicht-Kieselgel¬
platte neben je 0,005 ml von 0,1 % Sulfadiazin-, Sulfamerazin, resp. Sulfa-
thiazollösungen in Azeton aufgetragen. Als Laufmittel dient ein Gemisch von
Ammoniak 3% RS und n-Butanol R (20 Vol.% : 80 Vol.%). Die Wanne ist ca.
1 5 Stunden zu klimatisieren. Die Laufstrecke hat 1 5 cm zu betragen. Nach
dem Trocknen wird die Platte mit einer salzsauren Natriumnitrit R-Lösung
besprüht, wodurch die am weitesten gewanderten Sulfathiazolflecken gelb
gefärbt werden. Durch Besprühen mit ß-Naphthol RS färben sich alle Flecken
rot. Der Tablettenauszug muss drei ungefähr gleich grosse Flecken ergeben,
welche den Rf-Wert der entsprechenden Bezugssubstanzen zeigen.
4. Gehalt an Trisulfonamid: Eine ca. 1 g Sulfonamidgemisch entsprechende
Menge Tablettenpulver (genau gewogen) wird in einem Messkolben von 100 ml
Inhalt eingewogen. Nach Zugabe von 8 ml Ammoniak 3 °o RS und 40 ml Wasser
und einer heissen Lösung von 10 g Bariumnitrat R in 45 ml Wasser wird gut ge¬
schüttelt. Nach dem Erkalten wird mit Wasser bis zur Marke ergänzt. Nach
kräftigem Schuttein werden 25,0 ml unter sorgfältigem Umschwenken in einen
zweiten Messkolben von 100 ml Inhalt gegossen, der 15,00 ml 0,1 N Silber¬
nitrat und 25 ml Wasser enthält. Nach 15 Minuten langem Stehen im Dunkeln
wird Essigsäure 12 °o RS (ca. 0,3 ml) zugefugt bis blaues Lackmuspapier eben

-112-
gerötet wird. Hierauf wird mit Wasser bis zur Marke ergänzt und nach ein¬
stündigem Stehenlassen im Dunkeln durch ein trockenes Filter filtriert, wo¬
bei die ersten 10 ml verworfen werden. 50,00 ml des Filtrates werden mit
5 ml Salpetersäure 12 % RS und 1 ml Eisenammonalaun RS versetzt. Mit
0,1 N Ammoniumrhodanid wird hierauf bis zum Farbumschlag in Rötlichgelb
titriert (Mikrobürette).
1 ml 0,1 N AgN03 entspricht 25,66 mg gleicher Teile C10H10O2N4S, CuH1202N4S
und C9H902N3S2 (mittleres Mol.Gew.).
Gehalt an Sulfonamidgemisch
^ u, « /& ,» 0,01026'qpro Tablette
=(— - b) — — gz p
a = zugefügte Anzahl ml 0,1 N Silbernitrat
b = bei der Rücktitration verbrauchte Anzahl ml 0,1 N Ammoniumrhodanid
p= eingewogene Menge Tablettenpulver in g
q= Gewicht der 20 Tabletten in g
Aufbewahrung
In gut verschlossenem Behälter, unter Lichtschutz.
Separandum
Gebrauchs dosen: 6-8 Tabletten als Stosstherapie, dann
2 Tabletten alle vier Stunden.

- 113 -
4. Zusammenfassung
1. Im allgemeinen Teil wurde die Frage der Bewertung und der Wahl von
Methoden zur Gehaltsbestimmung bei Arzneistoffen und Arzneipräparaten
behandelt, um die Grundlagen für die Beurteilung der von uns untersuch¬
ten Methoden zu schaffen.
2. Wir stellten uns die Aufgabe die Statistik in den Dienst der quantitativen
Arzneimittelprüfung zu stellen, um mit einem geeigneten Arbeitsplan
sichere Voraussetzungen für eine objektive Beurteilung der Untersuchungs¬
resultate zu gewinnen.
Am Beispiel der Carbutamid-Bestimmungsverfahren wendeten wir die
statistische Auswertung an und kamen zum Resultat, dass die acidime-
trischen Methoden der argentometrischen und der bromometrischen vor¬
zuziehen sind.
3. Im speziellen Teil arbeiteten wir die Prüfungsvorschriften (Identität,
Reinheit und Gehalt) der folgenden Sulfonamide und Sulfonamidtabletten
aus:
Reinsubstanzen Tabletten
Carbutamidum Compressi Carbutamidi
Tolbutamidum Compressi Tolbutamidi
Probenecidum Compressi Probenecid!
Acetazolamidum Compressi Acetazolamidi
Sulfamethizolum Compressi Sulfamethizoli
Chlorpropamidum Compressi Trisulfonamidici
Diese Untersuchungen bilden einen Beitrag zur Ausarbeitung unserer neuen
Landespharmakopöe.

- 114 -
5. Literatur
1) American Cyanamid Co., U.S.P.2 554816 (1951).
2) American Cyanamid Co., E.P. 710 614 (1952).
3) American Cyanamid Co., Schweiz.P. 308 809 10 (1952)
4) BECKETT A.H. und TINLEY E.H., Titrations in non-aqueous solvents,BDH Ltd., Poole, England, 3.Ed.
5) BOEHRINGER u. Söhne, Mannheim, D.A.S. 1 011 413 Kl. 12 o (1957).
6) BRÄUNINGER H. und DUDA H., Pharm.Zhalle 97, 305 (1958).
7) CARSTEN E. und VEB Chem.Fabrik v.Heyden, D.P. (DDR) 13 793 Kl. 12 o
(1955).
8) CONWAY S., J.amer.pharm.Ass. (sci.Ed.) 34, 236 (1945).
9) Dtsch.Bundesgesundheitsamt, Bundesgesundheitsblatt 1962, 208;ref. Dtsch.Apoth.Ztg. 1_02, 1008 (1962).
10) ENGLER W.H., Über die Prüfung und Gehaltsbestimmung einigerSulfonamide, Diss. ETH, Zürich 1949.
11) FABER J.S., J.Pharm.Pharmacol. 6, 187(1954).
12) FRANCHI G., Chim.Ind. (Milano) 39, 773 (1957).
13) FROST I., Brit.med.J., (1958)1, 381; ref.Chem.Zbl. 1J29, 9286 (1958).
14) GUHAC, J.amer.chem.Soc. 44, 1502(1922).
15) HÄUSSLER A., Dtsch.Apoth.Ztg. 96, 879(1956).
16) HÄUSSLER A. und HAJDU P., Arch.Pharm. 29J, 531 (1958).
17) HÄUSSLER A. und HAJDU P., Arch.Pharm. 295, 471 (1962).
18) JENKINS G.L., CHRISTIAN J.E. und HAGER G.P., Quantitative
pharmaceutical chemistry, McGraw-Hill Book Comp., New York, 5.Ed.,
1957, S. 7.
19) KOLTHOFF J.M. und ELVING P.J., Treatise of analytical chemistry,Interscience Inc., New York, 1959, Vol.1, part.I, S. 67.
20) KRACMAR J. und VACEK J., Ceskosl.Farm. 9, 497(1960), ref.Chem.Zbl.
133. 567 (1962).
21) LINDER A., Persönliche Mitteilung .
22) LOGEMANN W., ARTINI D., TOSOLINI G. und PICCININI F.,
Chem.Ber. 91, 951 (1958).
23) LUCATELLI I., Farmaco (Ed.prat.) 1J_, 452 (1956).
24) LUNDBECKu. Co., U.S.P. 2 447 702 (1948).
25) MARSHALL F.J. und SIGAL M.V., J.org.Chem. 23, 927 (1958).
26) MIRIMANOFF A., Persönliche Mitteilung aus den Akten der Eidg.Pharmakopöekommission.
27) DAL MONTE CASONI F., Boll.chim.farm. 95, 297 (1956).

- 115 -
28) MÜNZEL K., BÜCHI J. und SCHULTZ O.-E., Galenisches Praktikum,
Wissenschaftliche Verlangsgesellschaft mbH, Stuttgart, 1959.
29) Pro Pharmacopoea, Ann.pharm.franç. 20, 402(1962).
30) ROBLIN R.O. und CLAPP J.W., J.amer.chem.Soc. 72, 4890(1950).
31) SAMANIEGO J.M.A., Span.P. 229 696 (1956).
32) SANDELL E., Farm.Rev. 44, 273 (1945).
33) SCHERING A.G., D.B.P. 957 841 Kl. 12p (1957).
34) SHARP u. DOHME, Schweiz.P. 288 891 (1951).
35) SHARP u. DOHME, U.S.P. 2 608 507 (1952).
36) SPRINGER H. und KAISER F., Arzneim.-Forsch. 6, 760 (1956).
37) SPÜHLER O., Schweiz.Apoth.Ztg. 96, 717 (1958).
38) STIVIC J. und MARINOW V., Acta jugosl.pharm. 8., 93 (1958).
39) SZABOLTCS E., Acta pharm.hung. 25, 17 (1955).
40) VARGA E. und VASTAGH G., Acta.pharm.hung. 28., 44 (1958);ref.Chem.Zbl. 1_30, 9666 (1959).
41) VESPE V. und FRITZ J., J.amer.pharm.Ass. (sci.Ed.) 4J, 197 (1952).
42) VITALI M. und PANCRAZIO G., Farmaco (Ed.prat.) 11., 512 (1956).
43) WOLFBRANDT C.C. und NIELSEN I.S., Dansk T.Farm. 1_4, 113(1940).
44) YOUNG R.W., WOOD K.H., EICHLER J.A., VAUGHAN J.R. jun.
und ANDERSON G.W., J.amer.chem.Soc. 78, 4649 (1956).

Lebenslauf
Am 7. Januar 19 34 wurde ich als Tochter des Franz Hafner und der Chiara,
geb. Bosia in Zürich, das auch mein Bürgerort ist, geboren. Alle Schulen
besuchte ich in Zürich und legte im Herbst 1953 das Maturitätsexamen
(Typ B) an der Töchterschule ab. Sogleich begann ich das Pharmaziestu¬
dium an der Eidgenössischen Technischen Hochschule in Zürich und bestand
die naturwissenschaftliche Prüfung im Herbst 1955. Das Praktikum absol¬
vierte ich bei Herrn W. Baur (Paradiesvogelapotheke), welches ich mit dem
Assistentenexamen im Frühjahr 1957 beendigte. Das Assistentenjahr ver¬
brachte ich bei den Herren M. Baur (Zürich), G. Bordoni (Lugano) und
W. Suter (St. Moritz). Im Frühling 1958 begann ich das Fachstudium an der
ETH und schloss zwei Jahre später mit dem Staatsexamen ab. Im Herbst
1960 begann ich unter der Leitung von Herrn Prof. Dr. Dr. med. h.c.
J. Büchi die vorliegende Promotionsarbeit und war gleichzeitig als Assi¬
stent der Eidg. Pharmakopöekommission tätig. Leider wurde die Beendi¬
gung der Arbeit durch längere Krankheit verzögert.


















