
© 2015 Kori Lynn Dunn - IDEALS
160
© 2015 Kori Lynn Dunn
Transcript of © 2015 Kori Lynn Dunn - IDEALS

© 2015 Kori Lynn Dunn

ENGINEERING ZYMOMONAS MOBILIS FOR THE PRODUCTION OF BIOFUELS
AND OTHER VALUE-ADDED PRODUCTS
BY
KORI LYNN DUNN
DISSERTATION
Submitted in partial fulfillment of the requirements
for the degree of Doctor of Philosophy in Chemical Engineering
in the Graduate College of the
University of Illinois at Urbana-Champaign, 2015
Urbana, Illinois
Doctoral Committee:
Associate Professor Christopher V. Rao, Chair
Associate Professor Yong-Su Jin
Associate Professor Hyunjoon Kong
Associate Professor Mary Kraft

ii
Abstract
Zymomonas mobilis is a promising organism for lignocellulosic biofuel
production as it can efficiently produce ethanol from simple sugars using unique
metabolic pathways. Z. mobilis displays what is known as the “uncoupled growth”
phenomenon, meaning cells will rapidly convert sugars to ethanol regardless of their
energy requirements for growth. This makes Z. mobilis attractive not only for ethanol
production, but for alternative product synthesis as well.
One limitation of Z. mobilis for cellulosic ethanol production is that this organism
cannot natively ferment the pentose sugars, like xylose and arabinose, which are present
in lignocellulosic hydrolysates. While it has been engineered to do so, the fermentation
rates of these sugars are still extremely low. In this work, we have investigated sugar
transport as a possible bottleneck in the fermentation of xylose by Z. mobilis. We showed
that transport limits xylose fermentations in this organism, but only when the starting
sugar concentration is high. To discern additional bottlenecks in pentose fermentations by
Z. mobilis, we then used adaptation and high-throughput sequencing to pinpoint genetic
mutations responsible for improved growth phenotypes on these sugars. We found that
the transport of both xylose and arabinose through the native sugar transporter, Glf, limits
pentose fermentations in Z. mobilis, thereby confirming our previous results. We also
found that mutations in the AddB protein increase plasmid stability and can reduce
cellular aggregation in these strains. Consistent with previous research, we found that
reduced xylitol production improves xylose fermentations in Z. mobilis. We also found
that increased transketolase activity and reduced glyceraldehyde-3-phosphate
dehydrogenase activity improve arabinose fermentations in Z. mobilis.

iii
In order for Z. mobilis to prosper as an industrial host for alternative product
synthesis, the genetic techniques utilized in this organism must be improved. Toward this
goal, we have adapted the λ Red recombinase system for use in Z. mobilis. We have
shown that this system increases the frequency of double crossover events for the purpose
of constructing gene knockouts in the strain. We have also constructed an expression
system for the type II CRISPR/CRISPR-associated (Cas) bacterial adaptive immunity
system in Z. mobilis. We have shown that the Cas9 nuclease can be directed by small
RNAs to target the Z. mobilis genome for the purpose of genome editing, and that this
system can also likely be used to facilitate gene knockouts in this organism. Finally,
toward improving heterologous protein production in Z. mobilis, we have constructed a
constitutive promoter library that leads to a range of gene expression levels in the strain.
Collectively, our results provide the framework for the development of an industrial
production process utilizing Z. mobilis as the microbial host.

iv
Acknowledgements
I am forever indebted to the many people who have helped me throughout my
years in graduate school. First and foremost, I am especially grateful for my advisor, Dr.
Christopher Rao. Thank you for being patient with me as I figured out Zymomonas, for
affording me the independence that I prefer, and for always trusting me to solve problems
in my own way. I am leaving your lab today with a wealth of knowledge that I couldn’t
have gotten under the direction of anyone else. It has been a privilege working with you.
Thank you also to Dr. Yong-Su Jin, Dr. Mary Kraft, and Dr. Hyunjoon Kong for being on
my committee and for providing me with valuable advice on my projects, and the Energy
Biosciences Institute for funding my research.
My days in lab were enjoyable thanks to the members of the Rao lab, past and
present. Ahmet Badur, Santosh Koirala, and Shuyan Zhang, I couldn’t have asked for
better people with whom to share all 6 years of my PhD. Thank you for always being on
my side and for sharing my sense of humor. Thank you for appreciating the real me, and
for making me feel like that person was someone special. To the many other members of
the Rao lab, thank you for the frequent fun times and the acceptance. To my dearest
friends outside of lab, Ritika Mohan and Dawn Eriksen, thank you for making the days
less daunting. My years in graduate school will be remembered as some of the best of my
life, thanks to all of you.
To my sister, Briana, thank you for always being my biggest fan. Thank you for
being by my side through it all. We made it this far together, and we will make it even
further together, too. Nothing in my life means more to me than you. I don’t know what I
would do without you.

v
To my counterpart, Justin Bradley, thank you for moving to Illinois for me. Thank
you for giving up your life to join mine. Thank you for making the most of the situation,
for taking the opportunity to better yourself, and of course for never complaining when I
had to work late.
Thank you also to my family for making the drive to Illinois and for
understanding that education is important. Thank you to my grandparents for the
encouragement and for making me feel loved. Finally, thank you to my brothers, Evan
and Nathan, for bringing me so much happiness in my life. I hope you will both read this
document and aim to follow in my footsteps.

vi
Table of Contents
Chapter 1: Introduction ................................................................................................... 1
1.1 Motivation ................................................................................................................. 1
1.2 The physiology of Zymomonas mobilis .................................................................... 2
1.3 Genetically modifying Z. mobilis .............................................................................. 9
1.4 Z. mobilis for ethanol production from lignocellulosic biomass ............................. 12
1.5 Z. mobilis for alternative product synthesis ............................................................ 17
1.6 Conclusions ............................................................................................................. 21
1.7 Figures ..................................................................................................................... 23
Chapter 2: Expression of a xylose-specific transporter improves ethanol production
in metabolically engineered Zymomonas mobilis ......................................................... 27
2.1 Introduction ............................................................................................................. 27
2.2 Materials and methods ............................................................................................ 28
2.3 Results ..................................................................................................................... 33
2.4 Discussion ............................................................................................................... 36
2.5 Tables and figures ................................................................................................... 39
Chapter 3: High-throughput sequencing reveals adaptation-induced mutations in
pentose-fermenting strains of Zymomonas mobilis ...................................................... 44
3.1 Introduction ............................................................................................................. 44
3.2 Materials and methods ............................................................................................ 45
3.3 Results ..................................................................................................................... 56
3.4 Discussion ............................................................................................................... 63
3.5 Tables and figures ................................................................................................... 68
Chapter 4: Genetically modifying Z. mobilis using the λ Red recombinase system
and a CRISPR-associated nuclease ............................................................................... 76
4.1 Introduction ............................................................................................................. 76
4.2 Materials and methods ............................................................................................ 80
4.3 Results ..................................................................................................................... 88
4.4 Discussion ............................................................................................................... 94
4.5 Tables and figures ................................................................................................. 100
Chapter 5: Development of a promoter library for use in Z. mobilis....................... 111

vii
5.1 Introduction ........................................................................................................... 111
5.2 Materials and methods .......................................................................................... 112
5.3 Results ................................................................................................................... 116
5.4 Discussion ............................................................................................................. 118
5.5 Tables and figures ................................................................................................. 120
Chapter 6: Conclusions and recommendations ......................................................... 128
6.1 Contributions ......................................................................................................... 128
6.2 Future directions .................................................................................................... 128
References ...................................................................................................................... 135

1
Chapter 1: Introduction
1.1 Motivation
Microorganisms have historically been used for the production of a variety of
value-added products, including pharmaceuticals, foods, and biofuels (da Silva et al.
2013). The yeast Saccharomyces cerevisiae and the bacterium Escherichia coli are
popular hosts for product synthesis because they are well-characterized and are highly
amenable to genetic manipulation (Leonelli and Ankeny 2013). However, researchers in
recent decades have realized the potential of less common, non-model organisms for
answering key biological questions and, consequently, for producing chemicals and fuels
(Armengaud et al. 2014). The study of these organisms has allowed for the analysis of a
diversity of mechanistic phenomena that would be lost if focus was placed strictly on the
commonly used hosts.
One of the most intriguing non-model organisms is the bacterium Zymomonas
mobilis. Z. mobilis is the only bacterium that uses the Entner-Doudoroff pathway
anaerobically in combination with the pyruvate decarboxylase and alcohol
dehydrogenase enzymes (Swings and De Ley 1977). Combining this with the fact that Z.
mobilis displays the “uncoupled growth” phenomenon, which enables it to metabolize
sugars rapidly regardless of its requirements for growth, makes Z. mobilis an outstanding
ethanol producer (Kalnenieks 2006). This capacity for ethanol production has prompted
interest in Z. mobilis for industrial biofuel production since the early 1980s (Baratti and
Bu'lock 1986). Around that time, oil embargoes were threatening the United States’ fuel
supplies. In response, the development of alternative energy sources became a priority,
and the Department of Energy created the Office of Alcohol Fuels (OAF). The OAF was

2
charged with accelerating the scale-up of ethanol production by funding research projects
aimed at producing the fuel cheaply in an environmentally-conscious manner (Wyman
2001). This research brought Z. mobilis into the spotlight as an organism with the
potential to bring the United States out of its energy crisis, commencing years of
investigation into this organism’s interesting physiology.
1.2 The physiology of Zymomonas mobilis
1.2.1 Central metabolic pathways
Z. mobilis is a Gram-negative, obligately fermentative, motile, rod-shaped
bacterium frequently found in tropical plant saps and spoiled cider (Swings and De Ley
1977). It produces ethanol and carbon dioxide in equimolar, near-theoretical amounts
from the sugars glucose, fructose, and sucrose using the Entner-Doudoroff (ED) pathway,
also known as the 2-keto-3-deoxy-6-phosphogluconate (KDPG) pathway, in combination
with the pyruvate decarboxylase (Pdc) and alcohol dehydrogenase (Adh) enzymes
(Dawes et al. 1966; Gibbs and Demoss 1954). In contrast to other bacteria, Z. mobilis
uses the ED pathway anaerobically to produce only one mole of ATP per mole of
glucose, in place of the Embden-Meyerhof-Parnas (EMP) glycolysis pathway which
produces 2 (Conway 1992; Kalnenieks 2006). Z. mobilis is restricted to the ED pathway
for the metabolism of sugars because its genome lacks sequences for the 6-
phosphofructokinase enzyme of the EMP pathway and the 2-oxoglutarate dehydrogenase
complex and malate dehydrogenase enzymes of the tricarboxylic acid (TCA) cycle (Raps
and Demoss 1962; Seo et al. 2005). Genes for most pentose phosphate pathway (PPP)
enzymes are also not present on the Z. mobilis chromosome (Seo et al. 2005). The TCA
cycle and PPP enzymes that are present, along with phosphoenolpyruvate carboxylase

3
and malic enzyme are used for anaplerotic reactions (Bringermeyer and Sahm 1989;
Sprenger 1996). Similar to glucose and fructose, sucrose metabolism also proceeds
through the ED pathway in Z. mobilis, with the sugar first being degraded into glucose
and fructose monomers by extracellular levansucrases, frequently with the formation of a
levan byproduct (Goldman et al. 2008; Gunasekaran et al. 1990; Kannan et al. 1995a;
Kannan et al. 1995b; Kyono et al. 1995; Senthilkumar et al. 2009; Silbir et al. 2014; Song
et al. 1993; Song et al. 1994). Typically little more than 2% of carbon source utilized by
Z. mobilis is converted into biomass (Belauich and Senez 1965; McGill and Dawes
1971). This low growth yield is a characteristic of Z. mobilis that makes it ideal for
product synthesis.
1.2.2 Uncoupled growth
A distinguishing characteristic of Z. mobilis is that it displays what is known as
the “uncoupled growth” phenomenon, meaning cells will consume sugars rapidly
regardless of their requirements for growth (Belauich and Senez 1965). The catabolic
rates in this organism are up to 5 times faster than those seen in yeast, reaching up to 1.0
µmol glucose mg dry wt.-1
min-1
during anaerobic exponential growth (Kalnenieks 2006).
All ED enzymes are highly expressed, most of them constitutively, and ED proteins
comprise up to half of the cells’ total protein (Algar and Scopes 1985; An et al. 1991).
Although the ATP yield of the ED pathway is the lowest of the fermentative pathways,
the high catabolic rate in Z. mobilis prevents the organism from suffering from a lack of
ATP (Kalnenieks 2006). In fact, due to its low biomass yield, Z. mobilis has a surfeit of
ATP that must somehow be exhausted. The energy consumption reactions for exhausting
this ATP balance well with energy production reactions during log phase growth,

4
regardless of the media used (Lazdunsk.A and Belaich 1972). Lazdunski and coworkers
provided evidence for a membrane-associated ATPase that eliminates the excess ATP
(Lazdunsk.A and Belaich 1972). Reyes and Scopes later purified what they believed to be
the same ATPase, and found that it was of the proton-pumping F0F1-type and that it likely
accounted for 20% of the total ATP turnover in the cells (Reyes and Scopes 1991). The
rest of the ATP turnover is likely due to the action of acid and alkaline phosphatases and
a periplasmic 5’-nucleotidase (Reyes and Scopes 1991). The bacterium Streptococcus
bovis also displays the uncoupled growth phenomenon (Forrest 1967). In this bacterium,
ATP spilling by the ATPase is coupled to a cycling of protons across the membrane
(Cook and Russell 1994). Presumably Z. mobilis has a similar conformation, because its
ATPase does not actually dissipate energy but instead converts it into proton-motive
force (PMF) (Kalnenieks 2006). While a large pH gradient has been observed across the
Z. mobilis membrane (Barrow et al. 1984; Kalnenieks et al. 1987; Osman et al. 1987), no
accurate determination of the transmembrane electric potential has been made to verify
this hypothesis (Kalnenieks et al. 1987; Ruhrmann and Kramer 1992). In addition to the
cycling of protons across the membrane, Kalnenieks and coworkers proposed that the
PMF could be dissipated through the export of bicarbonate anions from the cell
(Kalnenieks 2006). This hypothesis is supported by the presence of the gene for carbonic
anhydrase, which converts carbon dioxide into bicarbonate anions, on the Z. mobilis
chromosome (Seo et al. 2005).
1.2.3 Respiration
Although obligately fermentative, Z. mobilis does possess a constitutive
respiratory chain (Belauich and Senez 1965). The respiration rate is higher in Z. mobilis

5
than in several well-studied organisms, including E. coli and S. cerevisiae, but the
physiological role of respiration in this organism is still uncertain (Kalnenieks 2006).
Experiments using inhibitors like cyanide, chlorpromazine, and myxothiazol to quantify
and differentiate terminal oxidases support a respiratory chain with a branched structure
(Kalnenieks et al. 1998; Kita et al. 1984a; Kita et al. 1984b; Poole 1994). The primary
electron donors are NADH and NADPH (Bringer et al. 1984). Glucose and D-lactate are
also able to donate electrons to the respiratory chain, but to a much lesser extent—the
activity of their dehydrogenases is only about 5% or 10-20% of that of the NADH
dehydrogenases, respectively (Strohdeicher et al. 1989; Strohdeicher et al. 1990;
Strohdeicher et al. 1988). NADH donates electrons to either a NADH:ubiquinone
oxidoreductase complex or a type II NADH dehydrogenase (ndh) (Hayashi et al. 2012;
Seo et al. 2005). The NADH:ubiquinone oxidoreducatse is closely homologous to the
genes of an electron transport complex responsible for nitrogen fixation in the bacterium
Rhodobacter capsulatus (Saez et al. 2001; Schmehl et al. 1993). Evidence of nitrogen
fixation in Z. mobilis was provided recently, after years of failed attempts (Kremer et al.
2015; Seo et al. 2005). The ability to oxidize NADPH in the respiratory chain of bacteria
is rare, but Z. mobilis appears to do so using its type II NADH dehydrogenase
(Kalnenieks et al. 2008). Glucose and D-lactate donate electrons to membrane-bound
glucose dehydrogenase and D-lactate dehydrogenase, respectively (Kalnenieks et al.
1998; Strohdeicher et al. 1989; Strohdeicher et al. 1988). All respiratory dehydrogenases
then pass their electrons onto the vitamin-like substance ubiquinone, or coenzyme Q10.
From Q10 the electrons are passed to terminal oxidases that are able to reduce oxygen to
water. Researchers are confident that a cytochrome bd terminal oxidase is present in Z.

6
mobilis (Kalnenieks et al. 1998; Seo et al. 2005), but the identity of the oxidase that
terminates the other branch of the Z. mobilis respiratory chain is still unclear.
1.2.4 Oxidative phosphorylation
The presence of the confirmed components of the respiratory chain combined
with the confirmed presence of the F0F1-type ATPase mentioned above would indicate
that Z. mobilis is able to perform oxidative phosphorylation for energy production, such
that its aerobic growth yield would be higher than its anaerobic yield (Kalnenieks 2006).
However, this is not the case. Growth yields are typically the same or lower when Z.
mobilis is grown aerobically versus when it is grown anaerobically (Belauich and Senez
1965; Toh and Doelle 1997b). While this implies a non-functional respiratory pathway,
Dawes and Large did show that ATP could be produced by starved aerated Z. mobilis
from ethanol, presumably through oxidative phosphorylation (Dawes and Large 1970).
Kalnenieks and coworkers went on to provide support for this hypothesis by showing that
a proton gradient was present in these cells and that the production of ATP was coupled
to NADH oxidation. They also showed that the Z. mobilis ATPase could directly produce
ATP by providing it with an artificial proton gradient (Kalnenieks et al. 1993). In 1997,
Zikmanis and coworkers showed that they could in fact achieve a higher growth yield
during exponential growth in the presence of oxygen than they could without oxygen
present, but the effect was only seen under precise culture conditions. In addition to the
higher specific growth rate, a lower specific glucose consumption rate and a lower
specific ethanol production rate were seen, providing evidence for the importance of
aerobic ATP production under these conditions (Zikmanis et al. 1997). Toh and Doelle
were also able to achieve higher growth yields in the presence of oxygen, although they

7
were not completely convinced that oxidative phosphorylation was occurring (Toh and
Doelle 1997a). Regardless, it is clear that if oxidative phosphorylation is functional in Z.
mobilis, the efficiency of the process is extremely low. The original consensus among
researchers was that this is due to the accumulation of acetaldehyde in the culture
medium, which is more pronounced in aerobic cultures due to cofactor competition
(Tanaka et al. 1990). Acetaldehyde was believed to inhibit Z. mobilis growth when
present in extremely low amounts (Wecker and Zall 1987). However, it was later
determined that under conditions of environmental shock acetaldehyde can actually be
beneficial (Stanley et al. 1997), and that reports of its inhibitory effects were inconsistent
(Kalnenieks et al. 2000; Zikmanis et al. 1999). It is also known that the inhibition of the
oxidative phosphorylation process itself by acetaldehyde is negligible (Kalnenieks et al.
1993). Therefore, the cause of inefficient respiration in Z. mobilis is still unclear.
1.2.5 Nutrition and environmental effects
While the tolerance of Z. mobilis to the ethanol precursor acetaldehyde may be
low, its tolerance to ethanol is high. Strains have been reported to tolerate more than 100
g/L, higher than the tolerance of the yeast S. cerevisiae (Sprenger 1996; Swings and De
Ley 1977). At ethanol concentrations above this amount, leakage of essential cofactors
and coenzymes through the plasma membrane occurs (Osman and Ingram 1985). Z.
mobilis is also able to tolerate high sugar concentrations of up to 400 g/L (Swings and De
Ley 1977). When growing on sucrose, sorbitol is produced by the Z. mobilis glucose-
fructose oxidoreductase (GFOR) which helps to overcome the osmotic stress of growth in
high sugar concentrations. If sucrose is not the carbon source, the addition of sorbitol to
the culture medium improves growth at high sugar concentrations and also at high

8
ethanol concentrations (Loos et al. 1994; Sootsuwan et al. 2013). Similarly, high salt
concentrations also lead to the formation of sorbitol during growth on sucrose to help
overcome osmotic stress (Bekers et al. 2000). However, Z. mobilis is not able to grow in
media containing more than 20 g/L sodium chloride (Swings and De Ley 1977). The
optimum temperature for the growth of Z. mobilis is 30°C, with most strains also able to
tolerate temperatures up to 36°C fairly well (Fieschko and Humphrey 1983; Swings and
De Ley 1977). Temperatures above this range cause the lipid-to-protein ratio in the
membrane to decline, leading to the leakage of soluble proteins into the surroundings
(Benschoter and Ingram 1986). Growth occurs in a large range of pH values, from about
3.5 to 7.5 (Swings and De Ley 1977). Baumler and coworkers found that the expression
of a proton-buffering peptide in Z. mobilis could improve its survival rate in pH 3 to pH
3.5, but its growth rate at these pH levels was unaffected (Baumler et al. 2006). The need
for pantothenate, a precursor of coenzyme A, in Z. mobilis minimal medium has been
confirmed (Belauich and Senez 1965; Swings and De Ley 1977). In rich medium, yeast
extract is not a suitable carbon source and a fermentable sugar must be added (Swings
and De Ley 1977). All amino acids, ammonium salts, and peptides can act as nitrogen
sources for Z. mobilis (Belauich and Senez 1965). Most assimilated sulfur comes from
sulfate and methionine (Rogers, Lee et al. 1982). The addition of inorganic phosphate and
certain metallic ions can also improve the growth of Z. mobilis (Belauich and Senez
1965; Dawes and Large 1970).
1.2.6 Strain classification
The classification of Z. mobilis into two subspecies, Z. mobilis subsp. mobilis and
Z. mobilis subsp. pomaceae, was proposed by De Ley and Swings in 1976 (Deley and

9
Swings 1976). Nearly all of the strains analyzed by the authors fell into the subspecies
mobilis, while the strains that caused cider sickness belonged to subspecies pomaceae.
The type strain for subspecies mobilis was designated Zymomonas mobilis (Lindner)
Kluyver and van Niel, or ATCC 10988 (Swings and De Ley 1977). While strain ATCC
10988 produced ethanol efficiently, it was shown in 1981 that strain ZM4 (CP4)
produced ethanol from glucose more quickly than ATCC 10988 and also had a higher
tolerance to this ethanol (Skotnicki et al. 1981). Therefore, strain ZM4 was chosen for
further evaluation and engineering and has remained the Z. mobilis strain of choice for
genetic modification and ethanol production since then.
1.3 Genetically modifying Z. mobilis
1.3.1 Early progress
After the ethanol-producing potential of Z. mobilis was brought to light by the
laboratory of P.L. Rogers (Rogers et al. 1979), attempts to modify the strain for industrial
use intensified. In 1980, Skotnicki and coworkers showed that plasmids of the IncP and
IncFII incompatibility groups could be transferred into the strain by conjugation from E.
coli and Pseudomonas aeruginosa (Skotnicki et al. 1980). Realizing that the substrate
range of Z. mobilis would need to be expanded in order for it to prosper in the ethanol
industry, Carey and coworkers used a conjugable vector containing the tetracycline
resistance gene to express a lactose operon in the strain (Carey et al. 1983). In 1984, it
was shown that Z. mobilis could be chemically transformed with a hybrid vector created
by fusing an E. coli vector and a Z. mobilis native vector (Browne et al. 1984). Additional
Z. mobilis native vectors have since been characterized and used in shuttle vectors
(Afendra et al. 1999; Arvanitis et al. 2000; Dally et al. 1982; Drainas et al. 1983; Misawa

10
and Nakamura 1989; Reynen et al. 1990; Scordaki and Drainas 1987; Scordaki and
Drainas 1990; Strzelecki et al. 1987). Conway and coworkers were the first to construct a
vector designed specifically for protein expression in Z. mobilis (Conway et al. 1987a).
The vector contained a broad-host-range origin of replication, a chloramphenicol
acyltransferase gene, and a sequenced Z. mobilis promoter directly upstream of a
restriction site that could be used for gene cloning (Conway et al. 1987a). The team also
sequenced the promoter of pyruvate decarboxylase, one of the most abundant proteins in
Z. mobilis (Conway et al. 1987c). This strong promoter has been used frequently for
heterologous protein expression in the strain since then. Additionally, Conway and
coworkers characterized promoter structure in general in Z. mobilis using lacZ as a
reporter (Conway et al. 1987b). They found that Z. mobilis promoter sequences share
several features with E. coli promoter sequences and contain similar consensus sequences
in the -10 region and partial sequence homology in the -35 region. The sequence AGGA
was also identified as a possible ribosome binding site in Z. mobilis, appearing 8 to 12
base pairs upstream of the start codon (Conway et al. 1987b). In 1997 Pappas and
coworkers were the first to show that transposable elements present on suicide vectors
could be used to mutagenize the Z. mobilis chromosome (Pappas et al. 1997). The authors
successfully used the mini Mu transposon to construct stable Z. mobilis auxotrophs,
thereby demonstrating the feasibility of the method for chromosomal integrations.
1.3.2 Later difficulties with genetic modification
While initial attempts to genetically modify Z. mobilis showed promise, several
difficulties were later realized. Although gene expression from the lactose operon of
Carey and coworkers was confirmed in Z. mobilis, the strain was unable to grow on

11
lactose as its sole carbon source (Carey et al. 1983). Later attempts to expand the
substrate range of Z. mobilis were also met with difficulty (Byun et al. 1986; Feldmann et
al. 1992; Liu et al. 1988; Yanase et al. 1988). While conjugation was a reliable method
for introducing plasmid DNA into Z. mobilis, chemical transformation and
electroporation were not (Buchholz and Eveleigh 1986; Skotnicki et al. 1982; Sprenger
1993). If plasmid DNA was successfully transformed into the cells, it was often unstable
and readily lost, especially if it did not contain an origin of replication from a Z. mobilis
native vector (Afendra and Drainas 1987b; Bresticgoachet et al. 1990; Dong et al. 2011;
Pappas et al. 1997; Scordaki and Drainas 1987; Strzelecki et al. 1990). Low plasmid
stability in the strain is thought to be due to the presence of both type I and type IV
restriction-modification systems, which are able to degrade foreign DNA (Kerr et al.
2011; Typas and Galani 1992). Protocols for chromosomal integrations and gene
knockouts have not improved since the suicide vector technique of Pappas and coworkers
(Pappas et al. 1997), and targeted methods that utilize suicide vectors require hundreds or
thousands of base pairs of homology to the target region but still yield only a few
transformants (Agrawal et al. 2012b; Delgado et al. 2002; Kerr et al. 2011; Senthilkumar
et al. 2004). Further, an accepted set of promoters and ribosome binding sites (RBSs) for
the precise control of gene expression in Z. mobilis has not been developed. Studies to
date have primarily relied on the use of strong and constitutive promoters for the
uncontrolled expression of heterologous genes, further complicating gene expression
optimization in the strain (Deanda et al. 1996b; Jeon et al. 2005b; Zhang et al. 1995).
Collectively, the resistance of Z. mobilis to genetic modification has led to the emergence
of only a few recombinant strains (Kerr et al. 2011). To date, Z. mobilis has not been

12
accepted as an industrial organism, regardless of its unique physiology and rapid ethanol
production. The acceptance of Z. mobilis for the industrial production of ethanol and
other value-added products will require the improvement and standardization of genetic
techniques.
1.4 Z. mobilis for ethanol production from lignocellulosic biomass
1.4.1 The cellulosic ethanol production process
Traditionally, fuel ethanol was produced by the yeast S. cerevisiae from the six-
carbon sugars found in food crops such as corn and sugarcane (Wheals et al. 1999). More
recently, focus has shifted to producing biofuels from lignocellulosic feedstocks, such as
agricultural and municipal wastes (Schubert 2006). Cellulosic feedstocks are cheap,
abundant, and do not compete with the nation’s food supply (Lynd et al. 1991). They are
made up of three primary components: cellulose, hemicellulose, and lignin (Demirbas
2009). After mechanical degradation and an acid pretreatment, cellulose and
hemicellulose can be broken down enzymatically into a mixture primarily composed of
the six-carbon sugar glucose and the five-carbon sugars xylose and arabinose, which can
then be fermented into ethanol or other biofuels by microorganisms (Carroll and
Somerville 2009; Lawford and Rousseau 2002). The sugar mixture may also contain a
variety of growth inhibitors, including acetic acid, formic acid, furfural, 5-
hydroxymethylfurfural and levulinic acid (Shuai et al. 2010). In order for the cellulosic
ethanol production process to be economical, it is important that the microorganism used
have several important characteristics. It must be able to ferment both five and six-carbon
sugars rapidly to ethanol, in the process tolerating high concentrations of both the sugars
and the ethanol, and it must also have a high tolerance to the inhibitors in the hydrolysate

13
(Bothast et al. 1999; Dien et al. 2003). Further, the organism should be safe, resistant to
contamination, and amenable to genetic manipulation (Nevoigt 2008).
1.4.2 Advantages and disadvantages of Z. mobilis for cellulosic ethanol
production
Due to its naturally high ethanol productivity, Z. mobilis is an obvious contender
in the search for an industrial cellulosic ethanologen. This productivity is up to 5 times
higher than that observed in S. cerevisiae, the traditional host for ethanol production
(Rogers et al. 1979; Zhang et al. 1995). Ethanol yields of up to 97% of theoretical are
possible with Z. mobilis, primarily due to its unique homoethanologenic metabolism and
the low biomass yields obtained with this strain (Belauich and Senez 1965; McGill and
Dawes 1971). Further, Z. mobilis has an extremely high tolerance to both ethanol and to
sugars present in the hydrolysate (Rogers et al. 1979). As an organism frequently used for
beverage making, Z. mobilis is generally regarded as safe for industrial use (Swings and
De Ley 1977). It is resistant to contamination by other microorganisms, and there are no
known viral invaders (Sahm et al. 2006). Its ability to grow at an acidic pH further
renders it robust to contamination (Belauich and Senez 1965; McGill and Dawes 1971).
Finally, Z. mobilis is aerotolerant but does not require oxygen for growth, simplifying
process design requirements and reducing production costs (Swings and De Ley 1977).
In spite of these attractive advantages, several characteristics of Z. mobilis have
prevented it from gaining acceptance for cellulosic ethanol production. Primarily, Z.
mobilis has a very narrow substrate range and is unable to natively digest the five carbon
sugars xylose and arabinose that are present in the lignocellulosic hydrolysate (Swings
and De Ley 1977). Additionally, Z. mobilis has a low tolerance to some of the inhibitors

14
present in this hydrolysate, and is especially sensitive to acetic acid (Jeon et al. 2002;
Kim et al. 2000c; Ranatunga et al. 1997). Finally, as discussed above, researchers have
faced several difficulties genetically modifying Z. mobilis (Kerr et al. 2011). In order for
Z. mobilis to be competitive in the cellulosic ethanol industry, these difficulties must be
overcome.
1.4.3 Engineering Z. mobilis for improved lignocellulosic ethanol production
Early attempts to engineer Z. mobilis specifically for lignocellulosic ethanol
production focused on expanding the organism’s substrate range to include xylose and
arabinose (Rogers et al. 2007a). In 1987, Liu and coworkers expressed xylose isomerase,
xylose permease, and xylulokinase from Xanthomonas XA1-1 in Z. mobilis. Although
enzyme expression was confirmed, the strain was unable to grow on xylose as its sole
carbon source (Liu et al. 1988). Similarly, Feldmann and coworkers expressed xylose
isomerase and xylulokinase from E. coli in Z. mobilis, and used this strain to isolate a
mutant that produced less xylitol phosphate, a potent growth inhibitor produced when
xylose is consumed (Feldmann et al. 1992). They then expressed the E. coli transketolase
gene in the mutant strain, and again, although enzyme activities were confirmed, the
strain was unable to grow on xylose as its sole carbon source. The authors determined
that this was likely due to the absence of detectable transaldolase activity in Z. mobilis,
but since no transaldolase gene had yet been cloned the authors did not attempt to express
this protein in the recombinant strain (Feldmann et al. 1992). It wasn’t until 1995, when
researchers at the National Renewable Energy Laboratory were able to clone and express
the putative E. coli transaldolase gene together with the E. coli xylose isomerase,
xylulokinase, and transketolase genes in Z. mobilis that the strain was able to grow on the

15
sugar as its sole carbon source (Zhang et al. 1995). Shortly thereafter, the same laboratory
was able to engineer Z. mobilis to consume arabinose as its sole carbon source by
expressing the arabinose isomerase, ribulokinase, ribulose-5-phosphate-4-epimerase,
transaldolase, and transketolase genes from E. coli in the strain (Deanda et al. 1996b).
Unfortunately, however, the rates of xylose and arabinose consumption in these
strains were far slower than the rate of glucose consumption, even though the ATP yields
are the same for the two sugars on a molar basis (Agrawal et al. 2011b; De Graaf et al.
1999a; Deanda et al. 1996b; Joachimsthal and Rogers 2000b; Lawford and Rousseau
2000a; Zhang et al. 1995). Consequently, later efforts focused on investigating the
bottlenecks present in xylose and arabinose metabolism in Z. mobilis. The production of
xylitol and xylitol phosphate, which was uncovered by Feldmann and coworkers in 1992,
was determined to play an important role in limiting xylose fermentations by the strain
(Akinterinwa and Cirino 2009; Feldmann et al. 1992; Kim et al. 2000a). At least two
pathways contribute to xylitol formation in Z. mobilis—one involving the glucose-
fructose oxidoreductase of central metabolism, and the other involving an NADPH-
dependent xylose reductase (Feldmann et al. 1992; Zhang et al. 2009). Agrawal and Chen
later found that a strain of Z. mobilis adapted to growth on xylose as its sole carbon
source had a mutation in the NADPH-dependent xylose reductase that greatly decreased
its activity (Agrawal et al. 2011b). They subsequently deleted this reductase from the
chromosome of their xylose-fermenting strain, and found that xylitol production was
decreased with a concomitant increase in the rate of xylose metabolism (Agrawal and
Chen 2011). Aside from xylitol formation, Kim and coworkers found that Z. mobilis
grown on xylose was in a less-energized state than Z. mobilis grown on glucose, possibly

16
due to the weak activities of the heterologously-expressed xylose isomerase and
xylulokinase enzymes (Kim et al. 2000b). Although the studies are unrelated, the adapted
strain of Agrawal and coworkers did have a mutation in the promoter region of xylose
isomerase that increased its activity and presumably expression by roughly fivefold,
revealing the importance of the proper expression level of this enzyme. However, in
another separate study, increased expression of xylulokinase was found to not increase
the rate of xylose metabolism (Jeon et al. 2005a). If anything, it decreased it, possibly due
to the associated increase in xylitol production, although no increase in intracellular
xylitol-5-phosphate was observed in the strain. Less work has been done to determine
bottlenecks present in arabinose metabolism in Z. mobilis. Deanda and coworkers were
not able to discern why their strain did not perform as well on arabinose as it did on
glucose, but the authors did propose that the inefficiency of arabinose transport by the Z.
mobilis native transporter, the glucose facilitated diffusion protein or Glf, may be limiting
arabinose fermentations in the strain (Deanda et al. 1996b).
Work has also been done to increase the tolerance of Z. mobilis to the inhibitors
that are present in lignocellulosic hydrolysates. Ranatunga and coworkers determined that
acetic acid had by far the largest detrimental effect on xylose-fermenting Z. mobilis, with
cells performing at only 9% of maximal in the presence of acetic acid concentrations
typically present in cellulosic hydrolysates (Ranatunga et al. 1997). Far behind were
caproic acid and furfural, which caused cells to perform at only 57 and 58% of maximal,
respectively. Kim and coworkers used nuclear magnetic resonance studies to determine
that acetic acid inhibits Z. mobilis by acidifying the cytoplasm and decreasing the
amounts of nucleotide triphosphates and sugar phosphates present in the cell (Kim et al.

17
2000c). Yang and coworkers later isolated a strain of Z. mobilis with increased acetic acid
tolerance using chemical mutagenesis and by gradually increasing the concentration of
sodium acetate present in the culture medium. They discovered that their strain had
increased expression of the sodium-proton antiporter gene nhaA that likely improved the
sodium tolerance of their strain (Yang et al. 2010a). Chen and colleagues also isolated a
strain of Z. mobilis with improved acetic acid tolerance using adaptation, and separately
isolated strains with improved tolerance to other inhibitors present in lignocellulosic
hydrolysates, also using adaptation (Chen et al. 2009). Skerker and coworkers took a
different approach and constructed a library of Z. mobilis mutants using transposons.
They then analyzed the library for inhibitor tolerance using chemogenomic profiling and
found 44 genes important for hydrolysate tolerance in Z. mobilis. The overexpression of
one of these genes, ZMO1875, improved the specific ethanol productivity of the strain
2.4-fold in the presence of miscanthus hydrolysate (Skerker et al. 2013).
While some bottlenecks in xylose and arabinose fermentations by Z. mobilis have
been uncovered and its tolerance to important inhibitors has been increased, ethanol
production by the strain from lignocellulosic pentose sugars still lags behind ethanol
production from hexose sugars. In order for the industrial ethanol production process to
be economical, pentose sugar fermentations by Z. mobilis must be improved further.
1.5 Z. mobilis for alternative product synthesis
1.5.1 Advantages and disadvantages of Z. mobilis for alternative product
synthesis
Z. mobilis has historically received considerable attention for its high ethanol
productivity from glucose. However, researchers often overlook the fact that Z. mobilis

18
can also be used to produce other valuable metabolites under the correct culture
conditions (Johns et al. 1991). When grown on sucrose, Z. mobilis produces considerably
less ethanol and considerably more byproducts than when grown on glucose, suggesting
that glucose availability and concentration affect whether cells channel their carbon
source into the ED pathway to produce ethanol or elsewhere to produce byproducts
(Johns et al. 1991). Z. mobilis is a promising biocatalyst for alternative product synthesis
because of its low biomass yield and high sugar uptake rate, which allows carbon to
quickly become fuels or chemicals instead of biomass (Belauich and Senez 1965; McGill
and Dawes 1971). It also produces a wide variety of growth-linked byproducts that hold
commercial value, allowing for these products to all be isolated in a single process (Johns
et al. 1991). Further, it is a rich source of many enzymes currently used in diagnostic
analysis and research (Rogers et al. 2007a). As with ethanol production, Z. mobilis is also
a desirable host for alternative product synthesis because it is generally regarded as safe,
it is resistant to contamination, and it has relatively simple growth requirements
(Belauich and Senez 1965; McGill and Dawes 1971; Swings and De Ley 1977).
Regardless of the potential of Z. mobilis for the production of value-added
products, the organism will need to be further engineered to make any of the processes
economical. Cellular metabolism should be shifted away from ethanol production and
toward the production of the desired product—this will require the construction of
knockout and overexpression mutants and the precise control of heterologous gene
expression in Z. mobilis (Rogers et al. 2007a). Therefore, genetic techniques for
constructing these recombinant strains clearly need to be improved in order for the
potential of Z. mobilis for the production of value-added products to be realized.

19
1.5.2 Engineering Z. mobilis for alternative product synthesis
Z. mobilis is natively able to produce the byproducts fructose, sorbitol, gluconate,
fructooligosaccharides, and levan at concentrations exceeding 1 g/L (Johns et al. 1991).
The strain also produces a variety of byproducts in small amounts, including mannitol,
acetaldehyde, glycerol, dihydroxyacetone, and lactic, gluconic, succinic, and acetic acids
(Jobses et al. 1985; Schmidt and Schugerl 1987; Viikari and Gisler 1986; Wecker and
Zall 1987). Z. mobilis has received some attention for the commercial production of
fructose from sucrose with the concomitant production of ethanol, both of which are
value-added products (Doelle and Greenfield 1985). In order for this to be possible at
industrially desirable levels, however, a fructokinase-negative mutant must be used so
that the cells cannot consume the fructose that is being produced by sucrose splitting, and
instead only convert the glucose into ethanol (Pankova et al. 1988). This process is
desirable over the process currently used to produce the food sweetener high-fructose
corn syrup because fructose and ethanol are relatively easy to separate from one another
(Johns et al. 1991). Sorbitol, levan, and fructooligosaccharides are also value-added
products that can be produced by Z. mobilis during growth on sucrose under very specific
culture conditions. These products are commonly used in the food and pharmaceutical
industries. A high rate of sucrose hydrolysis favors sorbitol production, while a low rate
favors levan production (Doelle and Greenfield 1985; Viikari and Gisler 1986).
Fructooligosaccharide production occurs as an alternative to levan production when salt
concentrations are high (Doelle and Doelle 1990; Doelle et al. 1990). Sorbitol production
in Z. mobilis has been improved by overexpressing the glucose-fructose oxidoreductase
enzyme, responsible for producing sorbitol in a single step from glucose and fructose

20
(Liu et al. 2010). Levan production in Z. mobilis has been improved by lowering the rate
of sucrose hydrolysis using a strain with the sacC gene inactivated (Senthilkumar et al.
2004). Z. mobilis can also be used for the isolation of a variety of industrially-relevant
enzymes, including fructokinase, glucose-6-phosphate dehydrogenase, pyruvate kinase,
pyruvate decarboxylase and glucose-fructose oxidoreductase (Johns et al. 1991). A
mutant form of pyruvate decarboxylase was engineered and used in the production
process of an important pharmaceutical (Goetz et al. 2001; Iwan et al. 2001; Pohl 1997;
Rogers et al. 1997), but little else has been done toward improving enzyme yields for
commercial use in Z. mobilis.
While Z. mobilis is promising for industrial fructose production, the process is
tainted by the concomitant production of large amounts of sorbitol. This is detrimental
because the amount of sorbitol allowed in food products is often limited, and fructose and
sorbitol are difficult to separate. While growth condition optimization can be used to
decrease the amount of sorbitol that is produced, careful genetic engineering of the
fructokinase-negative mutants can possibly decrease sorbitol production further, making
the process more industrially feasible. Similarly, it is possible that overexpressing the Z.
mobilis sucrases could improve sorbitol production in the strain and improve its industrial
outlook. In general, all routes of alternative product synthesis in Z. mobilis would benefit
from additional engineering of the strains to decrease ethanol production and increase
production of the desired products. In working toward this goal, the current consensus in
the field is that genetic engineering techniques need to be improved and modernized in
order for Z. mobilis to be considered as a viable platform for future biorefineries (He et
al. 2014).

21
1.6 Conclusions
Z. mobilis clearly holds potential for the commercial production of the biofuel
ethanol in the present and for the commercial production of other value-added products in
the future. In order for these processes to be economical, however, more work needs to be
done. The digestion of lignocellulosic pentose sugars by the strain is still slow and lags
far behind the digestion of hexose sugars; the full reason for this is unknown. Ideally, all
pentose and hexose sugars would be consumed simultaneously. Further, although Z.
mobilis is a promising platform for the isolation of other biofuels and chemicals, little
work can be done to improve these processes with the outdated and inefficient genetic
techniques available to researchers today. Ideally, modern genetic engineering techniques
would be applied for use in Z. mobilis.
In this work, we have attempted to overcome these issues in Z. mobilis. In chapter
2, we describe how we used sugar transport engineering to improve the consumption rate
of the pentose sugar xylose in engineered Z. mobilis. In chapter 3, we explain our use of
adaptive evolution to isolate strains of Z. mobilis capable of more rapid growth on the
pentose sugars xylose and arabinose, and explain how we used high-throughput
sequencing to determine the chromosomal mutations responsible for the altered
phenotypes in order to elucidate additional bottlenecks in pentose sugar metabolism by
the strain. Toward genetic technique improvement, in chapter 4 we describe how we have
applied the lambda Red system combined with the yeast flippase enzyme for constructing
clean chromosomal deletions in Z. mobilis. We also provide proof-of-concept results for
chromosomal editing using the CRISPR/Cas9 system in the strain. In chapter 5, we

22
describe the construction of a promoter library for the precise control of heterologous
gene expression in Z. mobilis. Finally, in chapter 6, we conclude and discuss future work.

23
1.7 Figures
Figure 1.1 Image of Z. mobilis cells taken using differential interference contrast (DIC)
microscopy (bar = 10 µm).

24
Figure 1.2 Pathways for glucose, xylose, and arabinose metabolism in engineered Z.
mobilis. Xylose and arabinose enter central metabolism through the pentose phosphate
pathway (left), while glucose enters the Entner-Doudoroff pathway directly (right).
Heterologous enzymes are shown in bold.

25
Figure 1.3 The lignocellulosic ethanol production process. Cellulosic feedstocks, such as
switchgrass and miscanthus, are first broken down into smaller particles. Dilute acid or
steam is then used to break these particles into polysaccharides that become primarily the
sugars glucose, xylose, and arabinose during the hydrolysis step. These sugars can then
be fermented by engineered Z. mobilis into ethanol that is purified using distillation.

26
Figure 1.4 Pathways for byproduct formation in Z. mobilis.

1Portions of this chapter were reprinted from “Expression of a xylose-specific transporter improves ethanol
production by metabolically engineered Zymomonas mobilis” in the journal Applied Microbiology and
Biotechnology, August 2014, Volume 98, Issue 15, pp 6897-6905, by Dunn and Rao with kind permission
from Springer Science and Business Media.
27
Chapter 2: Expression of a xylose-specific transporter
improves ethanol production in metabolically engineered
Zymomonas mobilis1
2.1 Introduction
Z. mobilis produces ethanol from glucose at rates and yields that surpass those of
other microorganisms (Rogers et al. 2007b; Yang et al. 2010b). It has also been
engineered to consume the cellulosic pentose sugar xylose (Zhang et al. 1995), but it does
so very inefficiently (Agrawal et al. 2011a; De Graaf et al. 1999b; Joachimsthal and
Rogers 2000a). The fermentation rates are almost an order of magnitude slower than they
are for glucose even though the molar yields of ATP are the same for the two sugars
(Lawford and Rousseau 2000b). These strains are also unable to simultaneously ferment
glucose and xylose. Rather, in a manner reminiscent of catabolite repression, they
sequentially ferment these two sugars (Gao et al. 2002; Lawford and Rousseau 1999).
This selective utilization is not thought to be due to any known intrinsic regulatory
system but rather to inefficiencies in the metabolism of xylose.
As discussed in chapter 1, a number of studies have investigated bottlenecks in
xylose metabolism by Z. mobilis. One aspect of xylose metabolism that has not been
investigated in detail is transport. Xylose enters the cell through the promiscuous glucose
facilitated diffusion protein, Glf (Dimarco and Romano 1985; Parker et al. 1995). While
the affinity of Glf for xylose is lower than for glucose (KM=40 mM versus KM=4.1 mM),
the Vmax is twice as great (Weisser et al. 1996a). However, these measurements were
made in a strain of E. coli where the endogenous xylose transporters were still present,

28
possibly leading to a biased estimate of the Vmax. In addtition, glucose may block the
uptake of xylose by the Glf transporter through a competitive inhibitory mechanism.
Such a mechanism would explain why glucose is preferentially utilized by xylose-
fermenting strains of Z. mobilis (Deanda et al. 1996a). Therefore, one potential solution
to improve xylose fermentation, both as a single carbon source and in mixtures with
glucose, would be to introduce pentose-specific transporters into the cell.
In this chapter, we tested how the expression of a xylose-specific transporter in a
xylose-fermenting strain of Z. mobilis affects its ability to utilize xylose, both as a single
carbon source and in mixtures with glucose. Our results show that the introduction of a
xylose-specific transporter increases the rate of xylose utilization at high sugar
concentrations. The increase is most pronounced when xylose is the sole carbon source.
We also found that over-expression of the native Glf transporter improved the efficiency
of xylose utilization at high concentrations when it was the sole carbon source. These
results demonstrate that transport is a bottleneck for xylose utilization by Z. mobilis and
that the expression of xylose-specific transporters can help relieve this bottleneck.
2.2 Materials and methods
2.2.1 Bacterial strains, media, and growth conditions
All experiments were performed in Zymomonas mobilis subsp. mobilis ZM4
(ATCC 31821). RM liquid and solid medium (10 g/L yeast extract and 2 g/L potassium
phosphate, dibasic) supplemented with glucose or xylose at the designated concentrations
were used for all Z. mobilis experiments. All Z. mobilis cultures were grown at 30°C.
Antibiotics were used at the following concentrations: spectinomycin, 100 µg/mL; and
chloramphenicol, 100 µg/mL.

29
2.2.2 Plasmid construction
All plasmids used in this study are listed in Table 2.1. All primers used in this
study are listed in Table 2.2. To construct a plasmid containing the xylose metabolism
and pentose phosphate pathway genes, the xylose isomerase (xylA), xylulokinase (xylB),
transaldolase (talB), and transketolase (tktA) genes were isolated from E. coli MG1655
genomic DNA by PCR synthesis using primers KD002F, KD002R, KD004F, KD004R,
KD005F, and KD005R, respectively. To express the xylose isomerase and xylulokinase
genes (xylAB) in Z. mobilis, they were fused from the xylA start codon to the xylB
terminator sequence to the strong and constitutive 310 bp Z. mobilis glyceraldehyde-3-
phosphate dehydrogenase (gap) promoter, amplified from Z. mobilis ZM4 genomic DNA
using primers KD001F and KD001R, by overlap extension PCR (Horton et al. 1989).
Specifically, overlap extension PCR reactions were performed in a 50 µL volume
containing 500 ng of the larger gene fragment and an equimolar amount of the second.
No single-stranded DNA primers were added to the splicing reaction. Double-stranded
DNA fragments were annealed and extended using Phusion high-fidelity DNA
polymerase (New England Biolabs) during 25 PCR cycles with an annealing temperature
of 50°C. The resulting 3264 bp Pgap-xylAB synthetic operon was then inserted into
plasmid pJS71 to form plasmid pKLD1. To express the transaldolase and transketolase
genes in Z. mobilis, the transaldolase gene from its start to stop codon was first fused to
the strong and constitutive 308 bp Z. mobilis enolase (eno) promoter, amplified from Z.
mobilis ZM4 genomic DNA using primers KD003F and KD003R, using overlap
extension PCR. The constructed 1264 bp Peno-talB fragment was then inserted into
plasmid pKLD1 to yield plasmid pKLD2. Finally, the transketolase gene including its

30
native ribosome binding site and native terminator sequences was inserted directly
downstream of the talB stop codon in plasmid pKLD2 to create the 3348 bp Peno-talB-
tktA synthetic operon and yielding plasmid pKLD3.
To construct a pentose-specific transporter plasmid, the xylE gene was amplified
from E. coli MG1655 genomic DNA by PCR synthesis using primers KD007F and
KD007R. The 500 bp strong and constitutive pyruvate decarboxylase (pdc) promoter was
also amplified from Z. mobilis ZM4 genomic DNA by PCR synthesis using primers
KD006F and KD006R. For expression in Z. mobilis, the xylE gene was then fused from
its start codon and including its native terminator sequence to the pdc promoter using
overlap extension PCR. The Ppdc-xylE construct was then inserted into plasmid pEZCm to
yield plasmid pKLD4. For overexpression of Glf in Z. mobilis, the pdc promoter and the
glf gene were amplified from Z. mobilis ZM4 genomic DNA using primers KD008F,
KD008R, KD009F, and KD009R, respectively. The glf gene was then fused from its start
codon and including its native terminator sequence to the pdc promoter using overlap
extension PCR. The Ppdc-glf construct was then inserted into plasmid pEZCm to yield
plasmid pKLD5.
To construct plasmids for analyzing promoter strengths, the Venus fluorescent
protein gene was amplified from its start codon and including its native terminator
sequence from plasmid pVenus by PCR synthesis using primers KD011F, KD011R,
KD013F, and KD013R. The 310 bp glf promoter and the 500 bp pdc promoter were also
amplified separately from Z. mobilis ZM4 genomic DNA by PCR synthesis using primers
KD010F, KD010R, KD012F, and KD012R, respectively. The Venus gene was then fused
separately to the glf and pdc promoters using overlap extension PCR. The resulting Pglf-

31
Venus and Ppdc-Venus constructs were then inserted into plasmid pEZCm to yield
plasmids pKLD6 and pKLD7, respectively.
To construct a plasmid for analyzing transporter localization, the 310 bp glf
promoter was amplified from Z. mobilis ZM4 genomic DNA by PCR synthesis using
primers KD014F and KD014R. The xylE gene was amplified without its stop codon and
including a polypeptide linker sequence from E. coli MG1655 genomic DNA by PCR
synthesis using primers KD015F and KD015R. The Venus fluorescent protein gene was
amplified from its start codon and including its native terminator sequence from plasmid
pVenus by PCR synthesis using primers KD016F and KD016R. The glf promoter, the
xylE gene, and the Venus gene were then fused together using overlap extension PCR,
yielding the Pglf-xylE-venus translational fusion construct. The fusion was then inserted
into plasmid pEZCm to yield plasmid pKLD8. Z. mobilis expressing plasmid pKLD6 was
used as a control in localization experiments.
2.2.3 Plasmid transformation into Z. mobilis using electroporation
Z. mobilis cells grown in RM containing 20 g/L glucose to an OD600nm of at least
0.8 were harvested and washed once with water and once with 10% (v/v) glycerol. Cells
were then resuspended in 10% glycerol to 1/100 of the original volume. For each
transformation, 2.5 µg of TypeOne Restriction Inhibitor (Epicentre) and at least 1 µg of
unmethylated plasmid DNA isolated from E. coli strain GM119 or GM2929 were added
to 40 µL of competent cells. Cuvettes with 0.1 cm gaps were used in a BioRad XCell
gene pulser set at 1.6 kV, 200 Ω, and 25 µF. Following transformation, cells were
resuspended in RM and allowed to recover at 30°C for 1 to 3 h. Transformants were
isolated after growth for 3 to 7 days on RM plates containing the appropriate antibiotic.

32
Plasmid presence was confirmed by plasmid isolation and subsequent transformation into
E. coli DH5α. For strains harboring two plasmids, transformations were performed
sequentially.
2.2.4 Fermentations
Pre-seed cultures were prepared by inoculating a single colony from an RM agar
plate into liquid RM in a 5 mL glass tube. Pre-seed cultures were grown with shaking
until stationary phase, when they were used to inoculate seed cultures. Seed cultures were
grown with shaking in 15 mL sealed conical tubes filled with RM to 60% volume until
late exponential phase. All pre-seed and seed cultures were grown in RM medium
containing 20 g/L glucose as carbon source. Fermentation cultures used for analyses were
inoculated with seed culture to an OD600nm of 0.02. They were then grown with shaking at
140 rpm in 50 mL sealed conical tubes filled with RM to 90% volume and containing the
appropriate antibiotics and the appropriate amounts of glucose and/or xylose as carbon
source(s).
2.2.5 Analytical methods
Cell growth was determined by the optical density (OD) at 600 nm. Fermentation
samples were passed through 0.22 µm polyethersulfone syringe filters and the resulting
supernatant was analyzed for glucose, xylose, xylitol, and ethanol concentrations using
high-performance liquid chromatography (HPLC). A Shimadzu HPLC system with a
refractive index detector (Shimadzu RID-10A) was used together with an Aminex HPX-
87H carbohydrate analysis column and a cation H micro-guard cartridge (Bio-Rad) kept
at a temperature of 65°C. The mobile phase was 5 mM H2SO4, pumped at a flow rate of
0.6 mL/min. Peaks were identified and quantified by retention time comparison to

33
authentic standards (glucose, xylose, xylitol, and ethanol). All data reported are the
average and standard deviation of three replicates.
For promoter strength analyses, pre-seed, seed, and fermentation cultures were
prepared as described above, except fermentation cultures were filled to 40% volume to
allow for the aeration necessary for proper expression of the Venus protein. At late
exponential phase, 150 µL was withdrawn from the fermentation culture and transferred
to a single well of a black-walled, clear-bottomed Costar 96-well microtiter plate. The
fluorescence (Ex/Em: 515/528 nm) and optical density (600 nm) were measured using a
Tecan Safire2 microplate reader. Fluorescence measurements are reported as the relative
fluorescence normalized to the optical density of the sample to correct for variation in
cell density and given in terms of relative fluorescence units (RFU).
For transporter localization analyses, cells were mounted on an agar pad on a #1.5
coverslip. Total internal reflection fluorescence (TIRF) microscopy images were captured
on a Leica AF6000 microscope fitted with a Hamamatsu EM-1K EM-CCD camera,
automated stage, adaptive focus control (AFC), and a 488 nm laser. A 3x magnification,
100x/1.47 oil-immersion objective was used, and the evanescent field penetration depth
was set to 250 nm.
2.3 Results
2.3.1 Construction of a Z. mobilis strain expressing a xylose-specific
transporter
We hypothesized that transport limits the rate of xylose utilization in Z. mobilis.
To test this hypothesis, we expressed XylE, the low-affinity xylose transporter from E.
coli (Davis and Henderson 1987), in a xylose-utilizing strain of Z. mobilis (Figure 2.1).

34
We tested both the native pdc and glf promoters for expressing XylE and found that the
pdc promoter yielded significantly better results. As a consequence, we only report the
results for this promoter. We separately tested the expression of these two promoters
using transcriptional fusions to the fluorescent protein Venus (Nagai et al. 2002) and
found that the pdc promoter is significantly stronger than the glf promoter (7582 ± 661
RFU versus 3032 ± 255 RFU). In addition, we confirmed that the XylE protein is able to
localize to the Z. mobilis membrane using a translational fusion to Venus (Figure 2.2).
We also tested AraE, the low-affinity arabinose transporter from E. coli, which is
known to also transport xylose (Desai and Rao 2010; Novotny and Englesberg 1966;
Shamanna and Sanderson 1979). However, we found that the expression of AraE
inhibited growth in Z. mobilis. Specifically, we observed a large lag in the growth (~72
hours) of strains expressing AraE. We also tested a xylose-utilizing strain of Z. mobilis
expressing the native Glf transporter using the pdc promoter from a plasmid. These
results are included in our analysis. Our control was a xylose-utilizing strain of Z. mobilis
containing the empty plasmid used to express the transporter.
The xylose-utilizing strain was made by expressing the xylose isomerase (xylA),
xylulokinase (xylB), transaldolase (talB) and transketolase (tktA) genes from E. coli in Z.
mobilis. This design is based on the one previously developed by Zhang and coworkers
(Zhang et al. 1995). In order for this strain to consume xylose as its sole carbon source,
we needed to first grow the strain in glucose and then slowly adapt it to xylose as
reported previously (Agrawal et al. 2011a).

35
2.3.2 Expression of XylE and Glf improves xylose fermentation in xylose-only
media
We first tested our different strains using xylose as the sole carbon source. We
examined three different xylose concentrations: 10 g/L, 50 g/L, and 100 g/L (Table 2.3).
At a xylose concentration of 10 g/L, we did not observe any improvement in xylose
utilization or ethanol production when XylE or Glf were expressed. At a xylose
concentration of 50 g/L, we observed a small increase in the rate of xylose utilization
(~30%) and production of ethanol (~20%). The increases were similar for both XylE and
Glf. At a concentration of 100 g/L (Figure 2.3), we observed significant increases in both
the rate of xylose utilization (~50-140%) and production of ethanol (~70-100%) when
XylE or Glf were expressed in the cells. The increases were greater for XylE than Glf.
Collectively, these results demonstrate that transport limits xylose metabolism at high
sugar concentrations. These results also demonstrate that the expression of xylose
transporters, including the promiscuous Glf transporter, can improve the rate of xylose
utilization and ethanol production.
We also measured xylitol as it has previously been shown to inhibit xylose
utilization in Z. mobilis. Consistent with increased rates of xylose utilization, we found
that cells expressing XylE or Glf produced more xylitol than those that did not. This
increase, however, was seen only at the higher xylose concentrations (Table 2.3).
2.3.3 Expression of XylE improves xylose fermentation in glucose-xylose
mixed media
We next tested whether the expression of XylE or Glf would improve xylose
utilization when glucose is also present. Once again, we tested the performance of these

36
strains at three sugar concentrations (Table 2.4). At a concentration of 10 g/L xylose and
10 g/L glucose, we did not observe any increase in the rate of xylose utilization or
ethanol production. In fact, the utilization and productivity rates were less than the wild-
type control though the final ethanol titers were the same. At a concentration of 25 g/L
and 25 g/L glucose, we again observed no increase in the rate of xylose utilization or
ethanol production: rates and final titers were roughly the same for the different strains.
Once again, only at the highest concentrations of sugars (50 g/L xylose and 50 g/L
glucose) did we see an effect (Figure 2.4). In this case, expression of XylE increased the
xylose utilization rate by approximately 50% and final ethanol titer by 15%. Interestingly,
expression of XylE reduced the glucose consumption rate. Even then, however, sugar
utilization was not simultaneous as xylose metabolism is still far slower than glucose
metabolism. We also note that less xylitol is produced in strains expressing XylE for
reasons unknown. Finally, we did not observe any difference in strains expressing Glf
relative to the control.
2.4 Discussion
We investigated whether the expression of xylose-specific transporters improves
the rate of xylose metabolism in Z. mobilis. We found that XylE expression improves
ethanol production in both xylose only and glucose-xylose mixed media, but only when
the starting sugar concentrations are high (>50 g/L). Despite these increases, no
significant improvements in glucose-xylose co-consumption were observed. In addition,
we found that overexpression of the native Glf transporter improves ethanol production in
the pentose-fermenting strain, but only in xylose-only media and again only when the

37
starting sugar concentrations are high. Collectively, these results suggest that transport is
a bottleneck in xylose metabolism but only at high sugar concentrations.
To potentially explain this result, we note that Glf is expected to saturate at a
xylose concentration of approximately 45 g/L (assuming KM=30 mM and the transport
rate is 90% maximal). This value is roughly the concentration where transporter
expression increases xylose utilization. This would suggest that the expression of XylE
simply increases the overall capacity for transport rather than increasing the specific
affinity for xylose. Consistent with this conclusion, we also found that over-expression of
Glf yielded similar results as those involving the xylose-specific transporter XylE.
Moreover, with regards to glucose-xylose co-utilization, our data would suggest that
glucose does not inhibit xylose uptake – rather, the Glf transporter simply saturates at
higher xylose concentrations. The apparent diauxie is likely due to inefficiencies in
xylose metabolism and not transport. In other words, the sugars are consumed
sequentially because xylose is consumed much more slowly than glucose.
Interestingly, the KM of XylE for xylose is approximately 100 μM (Sumiya et al.
1995), which suggests that the transporter would be saturated at concentrations less than
1 g/L. This would suggest that expression of XylE should increase xylose utilization at
low concentrations, something we did not observe. While we cannot fully explain this
result, it may be worth noting that XylE is a proton-linked sugar transporter. It utilizes the
proton-motive force as the energy source for sugar transport. Glf, on the other hand, is
not energy dependent and transport is due solely to concentration differences (Parker et
al. 1995). One possibility is that the membrane is not equivalently energized in Z.
mobilis, leading to a higher effective KM.

38
We conclude by first noting that similar strategies have also been employed in
xylose-fermenting strains of yeast (Du et al. 2010b; Leandro et al. 2006a; Runquist et al.
2009b; Saloheimo et al. 2007; Subtil and Boles 2011; Young et al. 2011). Of note is the
work of Young and coworkers, where they applied directed evolution to xylose-specific
transporters for expression in yeast (Young et al. 2012a; Young et al. 2014a). In
principle, analogous approaches can be employed to further improve xylose transport in
Z. mobilis. Second, we note that transporter expression complements other approaches
employed to improve xylose metabolism in Z. mobilis including serial adaptation and
targeted deletions (Agrawal and Chen 2011; Agrawal et al. 2011a; Agrawal et al. 2012a).
These strategies can potentially be combined to further improve xylose metabolism in Z.
mobilis.

39
2.5 Tables and figures
Table 2.1 Plasmids used in this study.
Plasmid Genotype or relevant characteristics Source or reference
pEZCm CmR, pACYC184-pZMO1 Jeff Skerker
pJS71 SpR, pBBR1MCS-lacZα (Skerker and Shapiro 2000a)
pVenus KanR, MCS yfp(venus) t0 attλ, oriR6K (Saini et al. 2009)
pKLD1 pJS71-PgapxylAB This study
pKLD2 pJS71-PgapxylAB-PenotalB This study
pKLD3 pJS71-PgapxylAB-PenotalB-tktA This study
pKLD4 pEZCm-PpdcxylE This study
pKLD5 pEZCm-Ppdcglf This study
pKLD6 pEZCm-Pglfvenus This study
pKLD7 pEZCm-Ppdcvenus This study
pKLD8 pEZCm-PglfxylE-venus This study

40
Table 2.2 Oligonucleotide primers used in this study.
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD001F Pgap, for ATGCCTCGAGACTTTGTTCGATCAACAACCCG
KD001R xylAB GTTTATTCTCCTAACTTATTAAGTAGCTACTATATTCC
KD002F xylAB, for GTTAGGAGAATAAACATGCAAGCCTATTTTGACCAGCTCGATCGCG
KD002R Pgap ATGCTCTAGAGCCATTAACAAATGATTTTCAGAATA
KD003F Peno, for ATGCTCTAGACCTTCATGTTTTGCTTCATG
KD003R talB ATCGAAACCTTTCTTAAAATCTTTTAGAC
KD004F talB, for AAGAAAGGTTTCGATATGACGGACAAATTGACCTC
KD004R Peno ATGCGGGCCCTTACAGCAGATCGCCGATCA
KD005F tktA ATGCGGGCCCTCATCCGATCTGGAGTCAAAATGTCCTCACG
KD005R ATGCGAGCTCCCGCAAACGGACATATCAAGG
KD006F PPDC, for ATGCTCTAGATGCGGTATAATATCTGTAACAGCTCATTGAT
KD006R xylE TGCTTACTCCATATATTCAAAAC
KD007F xylE, for TATATGGAGTAAGCAATGAATACCCAGTATAATTCCAG
KD007R PPDC ATGCTCTAGAGAAACGGCTGAAGCGGG
KD008F PPDC, for ATGCTCTAGATGCGGTATAATATCTGTAACAGCTCATTGAT
KD008R glf CAGAACTCATTGCTTACTCCATATATTCAAAACACTATGTCTGAA
KD009F glf, for GTGTTTTGAATATATGGAGTAAGCAATGAGTTCTGAAAGTAGTCAGGGTCT
AG
KD009R PPDC ATGCTCTAGAAGTATAATTGGTTAAGACTTATCTAAAAAAGACAAAAGGA
TTC
KD010F Pglf, for ATGCTCTAGATTTTTTAAAAAGAAAACTGTTTTTTTAAACACTTATGTTGC
KD010R Venus CTTCTCCTTTACTCATGGCGATTCCTCTCCCGCC
KD011F Venus, for AGGAATCGCCATGAGTAAAGGAGAAGAACTTTTCACTGGAG
KD011R Pglf ATGCCTCGAGACCGAGCGTTCTGAACAAATCCAG
KD012F Ppdc, for ATGCTCTAGATGCGGTATAATATCTGTAACAGCTCATTGAT
KD012R Venus TTCTTCTCCTTTACTCATTGCTTACTCCATATATTCAAAACACTATGTCTGA
KD013F Venus, for TGGAGTAAGCAATGAGTAAAGGAGAAGAACTTTTCACTGGAG
KD013R Ppdc ATGCCTCGAGACCGAGCGTTCTGAACAAATCCAG
KD014F Pglf, for ATGCTCTAGATTTTTTAAAAAGAAAACTGTTTTTTTAAACACTTATGTTGCC
KD014R xylE GGCGATTCCTCTCCCGCC
KD015F xylE, for Pglf GGCGGGAGAGGAATCGCCATGAATACCCAGTATAATTCCAGTTATATATT
TTC
KD015R and Venus CTCCTTTACTCATGCTACCGCTGCCGCTACCCAGCGTAGCAGTTTGTTGTGT
TTTCT
KD016F Venus, for GCAGCGGTAGCATGAGTAAAGGAGAAGAACTTTTCACTGGA
KD016R xylE ATGCCTCGAGACCGAGCGTTCTGAACAAATCCA
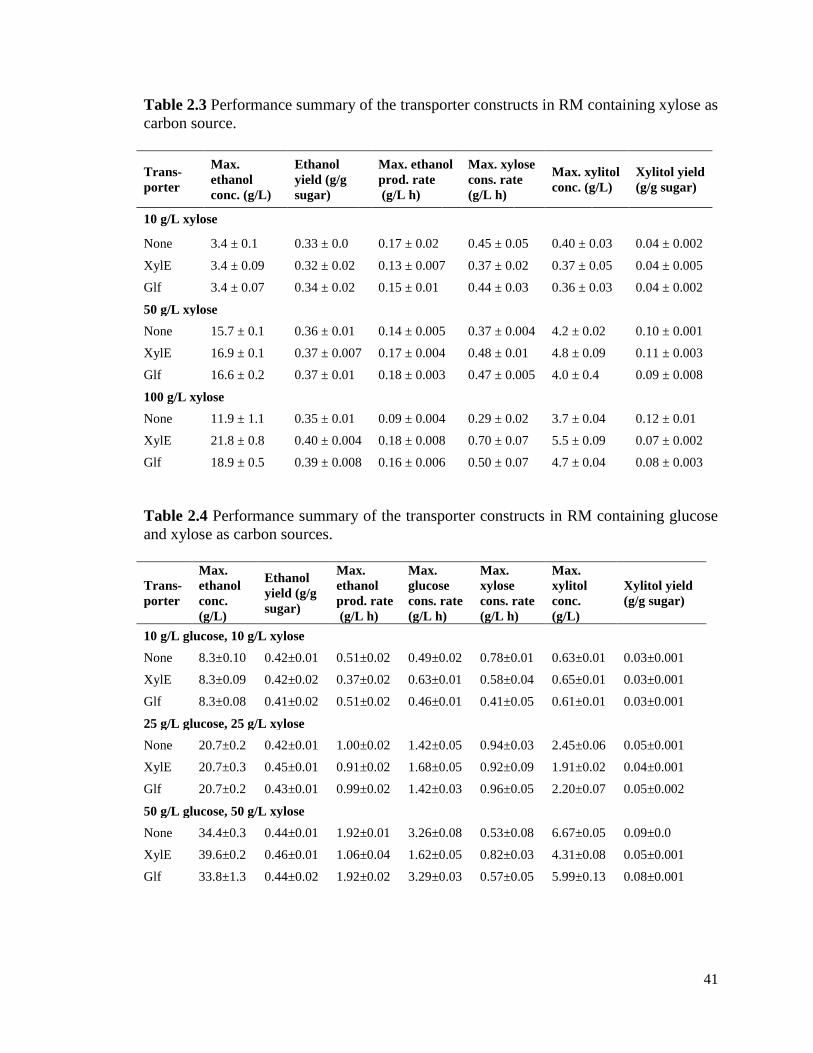
41
Table 2.3 Performance summary of the transporter constructs in RM containing xylose as
carbon source.
Trans-
porter
Max.
ethanol
conc. (g/L)
Ethanol
yield (g/g
sugar)
Max. ethanol
prod. rate
(g/L h)
Max. xylose
cons. rate
(g/L h)
Max. xylitol
conc. (g/L)
Xylitol yield
(g/g sugar)
10 g/L xylose
None 3.4 ± 0.1 0.33 ± 0.0 0.17 ± 0.02 0.45 ± 0.05 0.40 ± 0.03 0.04 ± 0.002
XylE 3.4 ± 0.09 0.32 ± 0.02 0.13 ± 0.007 0.37 ± 0.02 0.37 ± 0.05 0.04 ± 0.005
Glf 3.4 ± 0.07 0.34 ± 0.02 0.15 ± 0.01 0.44 ± 0.03 0.36 ± 0.03 0.04 ± 0.002
50 g/L xylose
None 15.7 ± 0.1 0.36 ± 0.01 0.14 ± 0.005 0.37 ± 0.004 4.2 ± 0.02 0.10 ± 0.001
XylE 16.9 ± 0.1 0.37 ± 0.007 0.17 ± 0.004 0.48 ± 0.01 4.8 ± 0.09 0.11 ± 0.003
Glf 16.6 ± 0.2 0.37 ± 0.01 0.18 ± 0.003 0.47 ± 0.005 4.0 ± 0.4 0.09 ± 0.008
100 g/L xylose
None 11.9 ± 1.1 0.35 ± 0.01 0.09 ± 0.004 0.29 ± 0.02 3.7 ± 0.04 0.12 ± 0.01
XylE 21.8 ± 0.8 0.40 ± 0.004 0.18 ± 0.008 0.70 ± 0.07 5.5 ± 0.09 0.07 ± 0.002
Glf 18.9 ± 0.5 0.39 ± 0.008 0.16 ± 0.006 0.50 ± 0.07 4.7 ± 0.04 0.08 ± 0.003
Table 2.4 Performance summary of the transporter constructs in RM containing glucose
and xylose as carbon sources.
Trans-
porter
Max.
ethanol
conc.
(g/L)
Ethanol
yield (g/g
sugar)
Max.
ethanol
prod. rate
(g/L h)
Max.
glucose
cons. rate
(g/L h)
Max.
xylose
cons. rate
(g/L h)
Max.
xylitol
conc.
(g/L)
Xylitol yield
(g/g sugar)
10 g/L glucose, 10 g/L xylose
None 8.3±0.10 0.42±0.01 0.51±0.02 0.49±0.02 0.78±0.01 0.63±0.01 0.03±0.001
XylE 8.3±0.09 0.42±0.02 0.37±0.02 0.63±0.01 0.58±0.04 0.65±0.01 0.03±0.001
Glf 8.3±0.08 0.41±0.02 0.51±0.02 0.46±0.01 0.41±0.05 0.61±0.01 0.03±0.001
25 g/L glucose, 25 g/L xylose
None 20.7±0.2 0.42±0.01 1.00±0.02 1.42±0.05 0.94±0.03 2.45±0.06 0.05±0.001
XylE 20.7±0.3 0.45±0.01 0.91±0.02 1.68±0.05 0.92±0.09 1.91±0.02 0.04±0.001
Glf 20.7±0.2 0.43±0.01 0.99±0.02 1.42±0.03 0.96±0.05 2.20±0.07 0.05±0.002
50 g/L glucose, 50 g/L xylose
None 34.4±0.3 0.44±0.01 1.92±0.01 3.26±0.08 0.53±0.08 6.67±0.05 0.09±0.0
XylE 39.6±0.2 0.46±0.01 1.06±0.04 1.62±0.05 0.82±0.03 4.31±0.08 0.05±0.001
Glf 33.8±1.3 0.44±0.02 1.92±0.02 3.29±0.03 0.57±0.05 5.99±0.13 0.08±0.001

42
Figure 2.1 Schematic representations of plasmids. a Plasmid used to express the xylose
metabolism genes. b Plasmid used to express the transporters.
Figure 2.2 Transporter localization analysis. a Z. mobilis expressing fluorescently-
labeled XylE. b Z. mobilis expressing free fluorescent protein. Note that fluorescence is
concentrated at the cell’s edges in a, showing that XylE is correctly expressed in the
membrane (bar = 5 µm).

43
Figure 2.3 Analysis of the Ppdc-xylE transporter construct in sugar-rich xylose-only
media. a,b Xylose and ethanol concentrations for Z. mobilis with pKLD3 and pEZCm
(Control) and Z. mobilis with pKLD3 and pKLD4 (+XylE). c Growth of the strains as
determined by OD600 nm.
Figure 2.4 Analysis of the Ppdc-xylE transporter construct in sugar-rich glucose-xylose
mixed media. a-c Glucose, xylose, and ethanol concentrations for Z. mobilis with pKLD3
and pEZCm (Control), and Z. mobilis with pKLD3 and pKLD4 (+XylE). d Growth of the
strains as determined by OD600 nm.

2Portions of this chapter were reprinted with permission from “High-throughput sequencing reveals
adaptation-induced mutations in pentose-fermenting strains of Zymomonas mobilis” in the journal
Biotechnology and Bioengineering, 2015, by Dunn and Rao. This article is protected by copyright. All
rights reserved.
44
Chapter 3: High-throughput sequencing reveals adaptation-
induced mutations in pentose-fermenting strains of
Zymomonas mobilis2
3.1 Introduction
In chapter 2, we found that pentose-specific transporter expression can improve
xylose fermentations in metabolically-engineered Z. mobilis (Dunn and Rao 2014).
However, this result was only true at high sugar concentrations, primarily in media
containing xylose as the sole carbon source. Therefore, we wanted to elucidate additional
bottlenecks in pentose metabolism. To do so, we used adaptive evolution and high-
throughput sequencing.
Adaptation is a powerful technique for strain development, because it does not
require knowledge of the specific metabolic bottlenecks in need of targeting (Lawford et
al. 1998; Rosenberg 2001). In a notable study, Agrawal and coworkers used adaptive
evolution to isolate a strain of Z. mobilis capable of consuming xylose up to three times
faster than its parent (Agrawal et al. 2011b). Previous studies had identified xylitol
production as an important bottleneck in xylose metabolism due to the loss of carbon and
energy (Feldmann et al. 1992; Kim et al. 2000a). Aware of this finding, Agrawal and
coworkers determined that reduced xylitol production was partially responsible for their
strain’s improved performance. Additionally, they found that the activity of the
heterologously-expressed xylose isomerase enzyme was increased by roughly fivefold in
their adapted strain. They did not, however, determine all of the genetic mutations
collectively responsible for the observed phenotype (Agrawal et al. 2011b; Agrawal et al.
2012b). Similarly, Deanda and coworkers noted that their arabinose-fermenting strain of

45
Z. mobilis performed better after adaptation in arabinose-only media, but the authors were
unable to attribute the improvement to any obvious differences in arabinose-related
enzyme activities (Deanda et al. 1996b).
Intrigued by these studies, we also adapted xylose and arabinose-fermenting
strains of Z. mobilis to growth on these pentose sugars as their sole carbon sources (Dunn
and Rao 2015). We isolated strains with rapid growth, sugar consumption, and ethanol
production rates. In order to identify additional bottlenecks in pentose metabolism, we
then used high-throughput sequencing to pinpoint the genetic changes responsible for the
improved phenotypes. In both the xylose and the arabinose-adapted strains, we found
mutations in the coding region of the primary sugar transporter, Glf, and in the DNA
repair protein AddB. Consistent with the results of Agrawal and coworkers, we found a
mutation in the xylose reductase gene in the xylose-fermenting strain. In the arabinose-
fermenting strain, we found mutations in the native glyceraldehyde-3-phosphate
dehydrogenase and transketolase coding sequences. Collectively, our results reveal
potential targets for the future engineering of pentose metabolism in Z. mobilis.
3.2 Materials and methods
3.2.1 Bacterial strains, media, and growth conditions
All bacterial strains used in this study are listed in Table 3.1. Liquid and solid
rich medium (RM) with 10 g/L yeast extract, 2 g/L potassium phosphate, dibasic
supplemented with glucose, xylose, or arabinose at the designated concentrations were
used for all Z. mobilis experiments. All Z. mobilis cultures were grown at 30°C and
antibiotics were used at the following concentrations: spectinomycin, 100 µg/mL; and
chloramphenicol, 100 µg/mL.

46
3.2.2 Plasmid construction
All plasmids used in this study are listed in Table 3.1. All primers used in this
study are listed in Table 3.2. The construction of a plasmid containing the xylose
fermentation genes (Figure 3.1a) was described previously (Dunn and Rao 2014). To
construct a plasmid containing the arabinose metabolism and pentose phosphate pathway
genes, the L-arabinose isomerase (araA), L-ribulokinase (araB), L-ribulose-5-phosphate
4-epimerase (araD), transaldolase (talB), and transketolase (tktA) genes were isolated
from E. coli MG1655 genomic DNA by PCR synthesis using primers KD024F, KD024R,
KD025F, KD025R, KD021F, KD021R, KD022F, and KD022R, respectively. To express
the transaldolase and transketolase genes in Z. mobilis, the transaldolase gene from its
start to stop codon was first fused to the strong and constitutive 308 bp Z. mobilis enolase
(eno) promoter, amplified from Z. mobilis ZM4 genomic DNA using primers KD020F
and KD020R, using overlap extension PCR (Horton et al. 1989). The constructed 1264
bp Peno-talB fragment was then inserted into plasmid pJS71 to yield plasmid pKLD9.
Next, the transketolase gene including its native ribosome binding site and native
terminator sequences was inserted directly downstream of the talB stop codon in the
plasmid pKLD9 to create the 3348 bp Peno-talB-tktA synthetic operon, yielding plasmid
pKLD10. To express the araBAD genes in Z. mobilis, repetitive palindromic sequences
were first removed from between araA and araD, as was done by Deanda and coworkers
(Deanda et al. 1996b). The araBA and araD gene fragments were then fused from the
araB start codon to the araD terminator sequence to the strong and constitutive 310 bp Z.
mobilis glyceraldehyde-3-phosphate dehydrogenase (gap) promoter, amplified from Z.
mobilis ZM4 genomic DNA using primers KD023F and KD023R, using overlap

47
extension PCR. The resulting 4397 bp Pgap-araBAD synthetic operon was then inserted
into plasmid pKLD10 to form plasmid pKLD11 (Figure 3.1b).
To construct plasmids for mutant transporter analyses, glf including its native
promoter and terminator sequences was amplified from Z. mobilis ZM4 genomic DNA,
glfA18T including its native promoter and terminator sequences was amplified from the
genomic DNA of strain KLD1, and glfV275F including its native promoter and
terminator sequences was amplified from the genomic DNA of strain KLD2, by PCR
synthesis using primers KD026F and KD026R. The glf genes were then inserted
separately into plasmid pEZCm to yield plasmids pKLD12, pKLD13, and pKLD14,
respectively. To construct a vector containing the glfA18TV275F allele, the first half of
the glf gene containing the A18T mutation was amplified from the genomic DNA of
strain KLD1 by PCR synthesis using primers KD027F and KD027R. The second half of
the glf gene containing the V275F mutation was amplified from the genomic DNA of
strain KLD2 by PCR synthesis using primers KD028F and KD028R. The two glf
fragments were then fused together using overlap extension PCR. The constructed
glfA18TV275F fragment was then inserted into plasmid pEZCm to yield plasmid
pKLD15.
To construct a plasmid for the purification of xylulokinase from E. coli, the XKS1
coding region was amplified from Saccharomyces cerevisiae BY4742 genomic DNA by
PCR synthesis using primers KD029F and KD029R. The XKS1 gene was then inserted
into plasmid pPROTet.E in-frame with the vector’s 6xHN tag to yield plasmid pKLD16.
To construct a vector for the inactivation of addB, the region 1 kb upstream of the
addB gene was amplified from Z. mobilis ZM4 genomic DNA by PCR synthesis using

48
primers KD030F and KD030R. The region 1 kb downstream of the addB gene was also
amplified from Z. mobilis ZM4 genomic DNA by PCR synthesis using primers KD031F
and KD031R. The spectinomycin resistance gene was amplified from plasmid pJS71 by
PCR synthesis using primers KD032F and KD032R to include flippase recognition target
(FRT) sequences for the future removal of the resistance from the chromosome, if
necessary (Datsenko and Wanner 2000; Martinez-Morales et al. 1999). The pPROTet.E
vector backbone was amplified from the pPROTet.E vector by PCR synthesis using
primers KD033F and KD033R. The four DNA fragments were then phosphorylated using
T4 polynucleotide kinase (New England Biolabs), and combined with Ampligase
thermostable DNA ligase and buffer (epicentre) and bridging oligonucleotides KD034F,
KD034R, KD035F, and KD035R. The fragments were then joined together such that the
addB upstream and downstream regions were flanking the spectinomycin gene in the
pPROTet.E vector using the optimized ligase cycling reaction parameters reported by de
Kok and coworkers (de Kok et al. 2014) to yield plasmid pKLD17.
To construct a pBBR1-based vector for plasmid stability analyses, the 500 bp
pyruvate decarboxylase (pdc) promoter was amplified from Z. mobilis ZM4 genomic
DNA by PCR synthesis using primers KD012F and KD012R (Dunn and Rao 2014). The
Venus fluorescent protein gene was amplified from its start codon and including its native
terminator sequence from plasmid pVenus by PCR synthesis using primers KD013F and
KD013R (Dunn and Rao 2014). The pdc promoter was then fused to the Venus gene
using overlap extension PCR. The resulting Ppdc-Venus construct was then inserted into
plasmid pJS14 to yield plasmid pKLD18.

49
3.2.3 Plasmid transformation into Z. mobilis using electroporation
Z. mobilis cells grown to an OD600nm of at least 0.8 in RM containing 20 g/L
glucose were harvested and washed once with water and once with 10% (v/v) glycerol.
Cells were then resuspended in 10% glycerol to 1% of the original culture volume. For
each transformation, at least 1 µg of unmethylated plasmid DNA isolated from E. coli
strain GM119 and 2.5 µg of TypeOne Restriction Inhibitor (epicentre) were added to 40
µL of competent cells. Cuvettes with 0.1 cm gaps were used in a BioRad XCell gene
pulser set at 1.6 kV, 200 Ω, and 25 µF. Following transformation, cells were allowed to
recover in RM at 30°C for 1 to 3 h. Transformants were isolated after growth for 3 to 7
days on RM plates containing the appropriate antibiotic. Plasmid presence was confirmed
by plasmid isolation and subsequent transformation into E. coli DH5α. When two
plasmids were used, transformations were performed sequentially.
3.2.4 Strain adaptation
Z. mobilis containing plasmid pKLD3 was grown with shaking in liquid RM
containing 50 g/L xylose as sole carbon source in a 5 mL glass tube until stationary phase
to select for cells able to grow in conditions of low xylose, high ethanol, and high xylitol.
Cells were then subcultured in fresh RM containing 50 g/L xylose for a total of 30
transfers. After this initial adaptation, cells were subcultured into fresh RM containing
100 g/L xylose for an additional 60 transfers (total: roughly 300 generations). The
resulting strain was denoted KLD1. Z. mobilis containing plasmid pKLD11 was grown
with shaking in liquid RM containing 25 g/L arabinose as sole carbon source in a 5 mL
glass tube until stationary phase to select for cells able to grow in conditions of low
arabinose and high ethanol. Cells were then subcultured into fresh RM containing 25 g/L

50
arabinose for a total of 60 transfers (roughly 200 generations). The resulting strain was
denoted KLD2.
3.2.5 Fermentations
Cultures used for analyses were grown as was described previously (Dunn and
Rao 2014). Briefly, to prepare pre-seed cultures, a single colony from an RM agar plate
was inoculated into liquid RM in a 5 mL glass tube and grown until stationary phase. Pre-
seed cultures were then used to inoculate seed cultures. Seed cultures were grown with
shaking until late exponential phase in 15 mL sealed conical tubes filled with RM to 60%
volume. All pre-seed and seed cultures were grown in RM containing 20 g/L glucose as
carbon source. Cultures used for analyses were inoculated with seed culture to an
OD600nm of 0.02. They were then grown with shaking at 140 rpm in 50 mL sealed conical
tubes filled with RM to 90% volume and containing the appropriate amounts of xylose or
arabinose as carbon source and the appropriate amount of antibiotic(s).
3.2.6 Analytical methods
Fermentation cultures were analyzed as was described previously (Dunn and Rao
2014). Briefly, the optical density (OD) at 600 nm was used to monitor cell growth.
Culture samples were purified using 0.22 µm polyethersulfone syringe filters and the
resulting supernatant was analyzed for xylose, arabinose, xylitol, and ethanol
concentrations using high-performance liquid chromatography (HPLC). A Shimadzu
HPLC system with a Shimadzu RID-10A refractive index detector was used together
with an Aminex HPX-87H carbohydrate analysis column and a cation H micro-guard
cartridge (Bio-Rad) kept at a temperature of 65°C. The 5 mM H2SO4 mobile phase was
pumped at a flow rate of 0.6 mL/min. Peaks were identified and quantified by retention

51
time comparison to authentic xylose, arabinose, xylitol, and ethanol standards. For cell
aggregation analyses, cells grown to late exponential phase were mounted on an agar pad.
Phase contrast images were captured on a Leica AF6000 microscope fitted with a
Hamamatsu EM-1K EM-CCD camera, automated stage, and adaptive focus control. A
x100/1.47 oil immersion objective was used.
3.2.7 Sequencing reactions
Chromosomal DNA was isolated from Z. mobilis strains using the Qiagen
QIAamp DNA mini kit according to the manufacturer’s instructions. Shotgun genomic
libraries were prepared at the Roy J. Carver Biotechnology Center (CBC) at the
University of Illinois at Urbana-Champaign using the TruSeq DNA sample preparation
kit (Illumina). Libraries were sequenced by the CBC on an Illumina HiSeq 2500
instrument. Paired-end reads were generated for all samples. Both the forward and
reverse reactions for each sample generated over 23 million reads. Fastq files were
generated using Casava 1.8.2 (Illumina). Fastq files were analyzed using the CLC
Genomics Workbench 7.0.3 (http://www.clcbio.com).
3.2.8 Quantitative PCR (qPCR) reactions
For qPCR reactions carried out using DNA, total DNA was isolated from Z.
mobilis strains using the Qiagen QIAamp DNA mini kit according to the manufacturer’s
instructions. PCR amplification of this DNA using primers KD037F- KD043R, designed
using the Primer3Plus software (Untergasser et al. 2007), was visualized using the
HotStart-IT SYBR Green qPCR Master Mix with UDG (Affymetrix) and a Bio-Rad
MiniOpticon Real-Time PCR System. For qPCR reactions carried out using RNA, total
RNA was isolated from Z. mobilis strains grown to mid-exponential phase using the

52
Qiagen RNeasy Mini Kit according to the manufacturer’s instructions. cDNA was
generated from RNA using the Qiagen QuantiTect Reverse Transcription Kit according
to the manufacturer’s instructions. PCR amplification of cDNA using primers KD037F-
KD049R, designed using the Primer3Plus software (Untergasser et al. 2007), was
visualized using the HotStart-IT SYBR Green qPCR Master Mix with UDG (Affymetrix)
and a Bio-Rad MiniOpticon Real-Time PCR System. All results were quantified by PCR
cycle-number comparison to standard curves prepared using E. coli MG1655 or Z.
mobilis ZM4 genomic DNA. The Z. mobilis rrsA gene was used as a control for
normalizing differences in total DNA or RNA quantities (He et al. 2012).
3.2.9 Enzymatic assays
Enzymatic assay methods were adapted from Agrawal and coworkers (Agrawal et
al. 2011b). Briefly, Z. mobilis cells grown in RM containing 50 g/L xylose and 50 g/L
glucose for 48 h or 25 g/L arabinose and 25 g/L glucose for 24 h were harvested by
centrifugation and washed once with freshly prepared extraction buffer (10 mM Tris-HCl
pH 7.5, 5 mM MgCl2, 1 mM dithiothreitol, and 1 mM phenylmethylsulfonyl fluoride).
Cell pellets were resuspended in extraction buffer to an OD600nm of 100 and sonicated for
8 10 s cycles. Sonicated cells were spun at 52,000 x g for 20 min at 4°C and the resulting
supernatant was used for enzymatic assays. Reactions were carried out in quartz cuvettes
in a final volume of 400 µL. The absorbance of NADH was measured at 340 nm using a
Shimadzu BioSpec-1601 DNA/ Protein/ Enzyme Analyzer. Glyceraldehyde-3-phosphate
dehydrogenase activity was assayed in 42 mM Tris-HCl buffer, pH 8.5, containing 10
mM trisodium phosphate, 1 mM NAD, 3mM β-mercaptoethanol, 1 mM DL-
glyceraldehyde-3-phosphate, and 20 µL cell-free extract. Transketolase activity was

53
measured using the coupling assay reported by Lee and coworkers (Lee et al. 2008).
Briefly, xylulokinase from S. cerevisiae was overexpressed in E. coli using plasmid
pKLD16. Cells were harvested and washed once with phosphate buffered saline (PBS).
The cell pellet was resuspended in 3 mL lysis buffer (50 mM NaH2PO4, 300 mM NaCl,
and 10 mM imidazole, pH 8.0) for every gram of cells and sonicated for 6 15 s cycles.
Sonicated cells were spun at 2,934 x g for 20 min at 4°C. The resulting supernatant was
then passed through a gravity chromatography column containing Ni-NTA resin to yield
purified 6xHN-Xks1. The purified 6xHN-Xks1 was used to create xylulose 5-phosphate
for the transketolase assay in 100 mM Tris-HCl buffer, pH 7.8, containing 2.5 mM
xylulose, 2.5 mM ATP, and 20 μg/mL purified 6xHN-Xks1. Transketolase activity was
assayed in 42 mM Tris-HCl buffer, pH 8.5, containing 5mM MgCl2, 1 mM thiamine
pyrophosphate, 0.5 mM ribose-5-phosphate, 0.2 mM NADH, 2.5 U/mL glycerophosphate
dehydrogenase, 25 U/mL triose phosphate isomerase, 80 μL of the 6xHN-Xks1 reaction
mixture, and 40 μL cell-free extract. All chemicals and commercial enzymes used for
assays were purchased from Sigma-Aldrich (St. Louis, MO). Results were normalized to
the total protein content of the lysates, determined using the PierceTM
BCA Protein Assay
Kit (Thermo Scientific) according to the manufacturer’s instructions. Relative activities
were then determined by setting the activity of the unadapted strain lysate to 1. Statistical
significance of the enzyme assay data was determined using the Microsoft Excel t-test,
two-sample with unequal variance. Assay results were significantly different (P < 0.05).

54
3.2.10 Construction of an addB knockout strain
Plasmid pKLD17 was linearized by digestion with EcoRI and KpnI to reduce the
likelihood of single crossover events. At least 1 µg of the digested vector was
transformed into Z. mobilis as described above. Cells were allowed to recover for 3 h
then plated on spectinomycin to select for transformants. Insertional inactivation of addB
was confirmed by PCR using primers KD036F and KD036R and genomic DNA
chemically extracted using standard protocols. Successful inactivation yielded a PCR
product 1.5 kb in length. Unsuccessful inactivation yielded a PCR product 3.0 kb in
length. Single crossover events yielded two PCR products, one 1.5 kb in length and
another 3.0 kb in length.
3.2.11 Plasmid stability experiments
Plasmid stability was determined using a reporter gene method similar to that of
Meima and coworkers (Meima et al. 1995). Briefly, unadapted Z. mobilis containing
plasmid pKLD3 and pKLD7 (Dunn and Rao 2014), unadapted Z. mobilis containing
plasmid pKLD11 and pKLD7, and strains KLD1, KLD2, and KLD3 containing plasmid
pKLD7 were grown in RM containing spectinomycin and chloramphenicol for a total of
20 media transfers. Unadapted strains of Z. mobilis containing plasmids pKLD3 and
pKLD11 and strains KLD1 and KLD2 were also transformed with plasmid pKLD18 and
grown in RM containing chloramphenicol only for a total of 20 media transfers (because
pKLD3, pKLD11, and pKLD18 have the same origin of replication and therefore cannot
be stably co-maintained). Strain KLD3 containing pKLD18 was grown in RM containing
spectinomycin and chloramphenicol for a total of 20 media transfers. After this brief
adaptation, cells were analyzed using flow cytometry. For a typical flow cytometry

55
experiment, cells were grown overnight in RM containing antibiotic(s), then subcultured
to an OD600nm of 0.05 and again grown overnight. At late-exponential phase cells were
harvested by centrifugation and washed once with PBS. Cells were then resuspended in
PBS and analyzed using a BD LSR II flow cytometer. Fluorescence values for
approximately 500,000 cells were recorded using the fluorescein isothiocyanate (FITC)
channel (excitation, 488 nm; emission, 530/30 nm). Separately, cells were stained with
4’,6-diamidino-2-phenylindole (DAPI) to distinguish them from other debris. For a
typical flow cytometry experiment with DAPI stain, cells were grown as reported above,
harvested and washed once with PBS, then resuspended in 14.3 µM DAPI buffer
containing 1 mg/mL chloramphenicol. Cells were incubated at room temperature for 30
minutes then analyzed as reported above, except the Pacific Blue channel (excitation, 405
nm; emission, 450/50 nm) was used. The value of fluorescence at which cells were
considered fluorescent versus non-fluorescent was determined by comparison to a Z.
mobilis wild-type control. FCS Express version 4 (De Novo Software) was used to
extract and analyze the data, which was then exported to Microsoft Excel for the final
determination of the percentage of fluorescent and non-fluorescent cells in each sample.
Statistical significance of the differences in plasmid stability in the unadapted versus the
adapted strains for xylose and arabinose was determined using the Microsoft Excel t-test,
two-sample with unequal variance. Plasmid stabilities were significantly different (P <
0.05).

56
3.3 Results
3.3.1 Construction, adaptation, and sequencing of pentose-fermenting strains
of Z. mobilis
Adaptation can be used to quickly isolate bacterial strains that have improved
performance under controlled environmental conditions. We used adaptation to isolate
strains of Z. mobilis that perform better when consuming either xylose or arabinose as
sole carbon source. The development of a xylose-fermenting strain of Z. mobilis was
described previously (Dunn and Rao 2014). An arabinose-fermenting strain was made by
expressing the L-arabinose isomerase (araA), L-ribulokinase (araB), L-ribulose-5-
phosphate 4-epimerase (araD), transaldolase (talB), and transketolase (tktA) genes from
E. coli in Z. mobilis. Our design is based on the one previously developed by Deanda and
coworkers (Deanda et al. 1996b). In order for this strain to consume arabinose as its sole
carbon source, we needed to first grow the strain on glucose and then slowly adapt it to
arabinose, as reported previously for xylose-fermenting strains (Agrawal et al. 2011b)
except cells from the final glucose-containing culture were plated on an RM agar plate
containing arabinose as sole carbon source. Attempts to isolate an arabinose-fermenting
strain from liquid RM containing arabinose as sole carbon source were unsuccessful.
Following the initial adaptation periods necessary to isolate the pentose-
fermenting strains, subculturing was continued for 30 transfers in media containing 5%
xylose followed by 60 transfers in media containing 10% xylose, or for 60 transfers in
media containing 2.5% arabinose. Sugar concentrations used during adaptation periods
were chosen to emulate the conditions the bacterium could experience in industrial
fermentations with concentrated lignocellulosic hydrolysates (Dominguez et al. 1996;

57
Huang et al. 2009). Following adaptation, the pentose-fermenting strains showed
improved fermentation characteristics (Figure 3.2). Specifically, we observed significant
increases in the maximum rates of pentose sugar utilization (~50-250%) and in the final
ethanol titers (~60-80%). To determine additional bottlenecks in pentose fermentations
by Z. mobilis, we then used high-throughput sequencing to determine the genetic
mutations responsible for the altered phenotypes. The mutations we found compared to
the sequence of our wild-type Z. mobilis are listed in Table 3.3. From these, several
mutations were selected from each strain for further analysis. The plasmids from the
adapted strains were also sequenced but no mutations were found.
3.3.2 Improved pentose sugar transport improves xylose and arabinose
fermentation in Z. mobilis
We found mutations in the coding sequence of the native sugar transporter, the
glucose facilitated diffusion protein (Glf), in both strain KLD1 and strain KLD2 (Table
3.3). Quantitative PCR results revealed that the mutations did not affect the expression
levels of this protein in the adapted strains (Figure 3.4b). Therefore, to predict the effect
the mutations had on the activity of the protein, we used the HMMTOP software to
perform a sequence-based analysis (Tusnady and Simon 1998; Tusnady and Simon
2001). Consistent with previous reports, we found that the Glf protein likely consists of
12 transmembrane helices and one major cytoplasmic loop (Ren et al. 2009). The A18T
mutation likely appears in the first transmembrane helix, while the V275F mutation likely
appears in the seventh transmembrane helix. Previous studies have shown that these
helices in particular are important to the sugar specificity of Glf and other transporters
that also contain 12 transmembrane helices (Arbuckle et al. 1996; Eriksen et al. 2013;

58
Hashiramoto et al. 1992; Ren et al. 2009; Seatter et al. 1998; Young et al. 2012b). To
then confirm that the mutant transporters improve pentose fermentations in Z. mobilis, we
expressed the glfA18T and glfV275F alleles from vectors in the xylose and arabinose
unadapted strains. We also expressed glfA18TV275F, an allele containing both
mutations, in the unadapted strains. We then compared these strains to a strain expressing
the wild-type glf from a vector. Strains expressing glfA18TV275F performed poorly;
therefore, this construct was removed from further analysis. However, cells expressing
glfA18T and glfV275F outperformed the strain expressing the wild-type transporter.
Specifically, glfA18T expression led to a 44% higher ethanol titer in the unadapted
xylose-fermenting strain than did expression of the wild-type glf (Figure 3.3). In the
unadapted arabinose-fermenting strain, expression of glf from a vector led to an
extremely long lag phase, while the expression of glfV275F did not (Figure 3.3). The
GlfA18T xylose-adapted transporter also improved ethanol production in the unadapted
arabinose-fermenting strain, and the GlfV275F arabinose-adapted transporter improved
ethanol production in the unadapted xylose-fermenting strain. Interestingly, the
improvements seen were greater when the expressed transporter was adapted on the
opposite sugar, for reasons unknown (Figure 3.3). Consistent with our previous findings
(Dunn and Rao 2014), these results demonstrate that sugar transport is a bottleneck in
pentose fermentations by Z. mobilis. They also suggest that the sugar transport limitation
is more pronounced during growth on arabinose than during growth on xylose, because
the mutant transporters lead to substantially better performance than does the wild-type
Glf during growth on this sugar.

59
3.3.3 Improved plasmid stability improves xylose and arabinose fermentation
in Z. mobilis
We found mutations in the coding sequence of the double-strand break repair
protein, AddB, in both strain KLD1 and strain KLD2 (Table 3.3). In other bacteria,
AddB has been shown to affect concatemer formation, typically from plasmids that
replicate via a rolling-circle mechanism. Specifically, long repeats of plasmid DNA have
been found in cells expressing a mutant form of addB (Viret and Alonso 1987).
Concurrently, the copy number of certain vectors was also affected (Viret and Alonso
1987). Because plasmids pKLD3 and pKLD11 do not replicate via rolling-circle
replication (Antoine and Locht 1992), it was unlikely that concatemer formation and
plasmid copy number were affected in adapted Z. mobilis due to mutations in addB.
Quantitative PCR reactions confirmed this hypothesis—no significant differences were
seen in the DNA copy numbers of plasmid-bound genes in the unadapted versus the
adapted strains (Figure 3.4a). Additionally, no significant differences in the transcript
levels of plasmid-bound genes were seen (Figure 3.4b).
AddB has also been shown to influence structural plasmid stability, specifically
affecting the frequency of illegitimate recombination of plasmid DNA (Meima et al.
1997; Meima et al. 1995). To determine if the addB mutations improve plasmid stability
in the adapted strains of Z. mobilis, mutation frequency was monitored by the expression
of the Venus fluorescent protein on two vectors: one that replicates via rolling-circle
replication (pKLD7), and one that replicates by the same mechanism as pKLD3 and
pKLD11 (pKLD18). Antibiotic-resistant progeny that no longer fluoresce are assumed to

60
have lost fluorescence due to deletion or inversion mutations on the vector (Meima et al.
1995).
For the vector that replicates via a rolling-circle mechanism, pKLD7, we found
that strains KLD1 and KLD2 had the lowest frequency of fluorescence loss after 20
transfers in media containing antibiotics (~2%), and therefore had the highest plasmid
stability. The unadapted strains had slightly higher frequencies of fluorescence loss (~4-
8%) while an addB knockout strain of Z. mobilis had a significantly higher frequency of
fluorescence loss (~83%) and therefore had the lowest plasmid stability of all of the
strains tested (Figure 3.5). These results indicate that the AddB protein plays an
important role in the stability of vectors that replicate using a rolling-circle mechanism in
Z. mobilis.
In the case of the vector that does not replicate via rolling-circle replication,
pKLD18, we found that strains KLD1 and KLD2 had lower frequencies of fluorescence
loss (~3-5%) after 20 transfers in media containing antibiotics compared to the unadapted
strains (~11%), suggesting again that structural plasmid stability was increased in these
strains (Figure 3.5). Interestingly, however, the addB knockout strain of Z. mobilis
performed differently with pKLD18—the fluorescence of nearly all of the cells increased
after 20 transfers in media containing antibiotics (Figure 3.5), a result not seen with
plasmid pKLD7. An increase in fluorescence is consistent with the formation of cell
aggregates. Flow cytometry results also suggested aggregation in the unadapted strains,
while suggesting no significant aggregation in the adapted strains, KLD1 and KLD2
(Figure 3.5). Phase-contrast microscopy confirmed these aggregation patterns (Figure
3.6). To potentially explain why aggregation is occurring, we note that in addition to not

61
replicating via rolling circle replication, the vector pKLD18 has another feature that
distinguishes it from pKLD7—it contains a functional origin of transfer and is therefore
mobilizable. We also note that the RecBCD protein complex commonly replaces the
AddAB complex in other bacteria (Chedin et al. 1998b). RecBCD deletion mutants have
been shown to acquire foreign DNA through horizontal gene transfer at a higher
frequency than wild-type cells (Harms and Wackernagel 2008). Further, a phylogenetic
tree analysis suggests that horizontal gene transfer is promoted when RecBCD or AddAB
are lost from the genome (Cromie 2009). Additionally, it has been shown that the Z.
mobilis strain used in the present study, ZM4, contains at least two genes necessary for
the conjugal transfer of mobilizable plasmids (Seo et al. 2005). Therefore, we
hypothesize that in AddB deletion mutants and to a lesser extent in wild-type Z. mobilis,
the cells aggregate in an attempt to carry out conjugation for the purpose of horizontal
gene transfer. This is possibly in an attempt to obtain a better-performing analogue of the
AddB protein. The mutated forms of AddB present in strains KLD1 and KLD2 may
therefore prevent the cells from attempting a futile conjugation process, thereby relieving
this metabolic burden.
3.3.4 Reduced xylitol production improves xylose fermentation in Z. mobilis
Agrawal and coworkers found that reduced xylitol production due to a mutation in
the Z. mobilis xylose reductase (xr) gene, ZMO0976, was partially responsible for their
adapted strain’s improved performance on xylose (Agrawal et al. 2012b). We, too, found
a mutation at this locus in strain KLD1 (Table 3.3). We measured the amount of xylitol
produced by the unadapted and the adapted xylose-fermenting strains. While strain KLD1
consumed more xylose and therefore produced more total xylitol, ~18% less carbon

62
became xylitol on a gram per gram basis compared to the unadapted strain (Table 3.4).
While the functionality of the enzyme was altered in the adapted strain, quantitative PCR
showed that the xr expression level was not affected (Figure 3.4b). These results confirm
the importance of xylitol production in xylose fermentation by Z. mobilis.
3.3.5 Reduced glyceraldehyde-3-phosphate dehydrogenase activity and
increased transketolase activity improve arabinose fermentation in Z. mobilis
In strain KLD2, we found a mutation in the coding region of the glyceraldehyde-
3-phosphate dehydrogenase (GAPDH) enzyme (Table 3.3). GAPDH is a glycolytic
enzyme that reversibly catalyzes the phosphorylation of glyceraldehyde-3-phosphate to
yield 1,3-diphosphoglycerate (Conway et al. 1987d). Therefore, this enzyme has the same
substrate as the heterologously-expressed transaldolase enzyme, which converts
glyceraldehyde-3-phosphate and sedoheptulose-7-phosphate into fructose-6-phosphate
and erythrose-4-phosphate (Sprenger et al. 1995). We found that the glyceraldehyde-3-
phosphate dehydrogenase activity in the unadapted arabinose-fermenting strain grown in
glucose-arabinose mixed media until the arabinose phase of growth was ~67% higher
than in the adapted strain, KLD2 (Table 3.5). Quantitative PCR showed that this was not
due to differences in expression levels (Figure 3.4b). The reduced GAPDH activity in
the adapted strain may increase the access of transaldolase to its substrate, thereby
increasing the amount of carbon source that is able to enter central metabolism to become
ethanol and ATP.
In strain KLD2, we also found a mutation in the coding region of the Z. mobilis
native transketolase (Tkt) enzyme (Table 3.3). Tkt is an enzyme of the pentose phosphate
pathway that, together with transaldolase, converts xylulose-5-phosphate into

63
intermediates of central metabolism (Zhang et al. 1995). The activity of this enzyme in
wild-type Z. mobilis is low (Feldmann et al. 1992). We found that the Tkt activity in
strain KLD2 grown in glucose-arabinose mixed media until the arabinose phase of
growth was ~70% higher than in the unadapted arabinose-fermenting strain (Table 3.5).
Quantitative PCR showed that this was not due to differences in expression levels
(Figure 3.4b). These results suggest that low Tkt activity limits arabinose fermentations
in Z. mobilis.
3.4 Discussion
We used adaptive evolution and high-throughput sequencing to determine
bottlenecks in xylose and arabinose fermentations by Z. mobilis. We found that mutations
in the native sugar transporter, Glf, improve both xylose and arabinose fermentations by
this organism, suggesting that sugar transport is a bottleneck in pentose metabolism. We
also found that mutations in the AddB protein improve structural plasmid stability and
reduce the aggregation of cells expressing mobilizable vectors, revealing that the wild-
type version of this protein limits pentose fermentations carried out using vector DNA.
Additionally, we found that a mutation in the xylose reductase gene led to reduced xylitol
production and improved xylose fermentations by Z. mobilis. Finally, we found mutations
in the glyceraldehyde-3-phosphate dehydrogenase and transketolase coding regions that
decreased and increased the activity of these enzymes, respectively, and led to improved
arabinose fermentations by Z. mobilis.
We previously investigated sugar transport as a bottleneck in xylose fermentations
by Z. mobilis. We found that sugar transport was a process bottleneck, but only at high
xylose concentrations (Dunn and Rao 2014). In this study, we adapted Z. mobilis to

64
growth at high xylose concentrations. Again, we found that improved xylose transport led
to improved xylose fermentation performance, confirming our previous results. Previous
reports have also predicted that sugar transport is a limiting factor in arabinose
fermentations by Z. mobilis (Deanda et al. 1996b). The Glf protein likely transports this
sugar into the cell, but the affinity of Glf for arabinose is much lower than its affinity for
other sugars, including glucose and xylose (Weisser et al. 1996b). Consistent with this
finding, our results suggest that sugar transport is a more important bottleneck in
arabinose fermentations than in xylose fermentations, as mutant forms of Glf lead to
more pronounced improvements in fermentation performance by arabinose-fermenting
strains. The overexpression of the wild-type Glf nearly abolishes growth by these strains,
likely due to the combined modest transport rate of arabinose through this protein and the
additional metabolic burden presented by its overexpression. In general, however, the
presence of mutations in glf in both the xylose and the arabinose-adapted strains of Z.
mobilis suggests that sugar transport engineering is an important strategy toward
expanding the substrate range of this organism. In support of this conclusion, sugar
transport engineering has been an effective approach toward the improvement of
substrate utilization in many microorganisms (Du et al. 2010a; Kuyper et al. 2005;
Leandro et al. 2006b; Runquist et al. 2009a; Young et al. 2012b; Young et al. 2014b).
It is well-known that plasmid stability is an issue in Z. mobilis (Afendra and
Drainas 1987a; Byun et al. 1986; Dong et al. 2011; Kerr et al. 2011). Prior studies have
shown that members of the DNA repair system and the DNA restriction-modification
systems play important roles in plasmid stability and maintenance in Z. mobilis (Kerr et
al. 2011; Vartholomatos et al. 1993). In this study, we found another member of the DNA

65
repair system that is important to plasmid stability: the double-strand break repair protein,
AddB. AddB is the nuclease portion of the AddAB helicase-nuclease protein complex
(Chedin et al. 1998a). Using a vector that replicates using a rolling-circle mechanism and
one that does not, we found that strains expressing mutant forms of the AddB protein
show increased structural plasmid stability. Consistent with this result, several studies
have shown that this protein affects plasmid stability in other bacteria (Kupsch et al.
1989; Meima et al. 1997; Meima et al. 1995; Meima et al. 1996; Peijnenburg et al. 1987).
The precise mechanism by which the AddB protein affects structural plasmid stability in
Z. mobilis is currently unknown. However, in general, the presence of mutations in addB
that improve plasmid stability in both the xylose and the arabinose-fermenting strains
suggests that the chromosomal integration of heterologous genes is an important step in
the development of industrial strains of Z. mobilis. In cases where vector DNA is desired,
however, strains expressing the mutant forms of AddB reported here may be viable
options. Further, using a mobilizable vector that does not replicate using a rolling-circle
mechanism, we found that the loss of the AddB protein leads to the aggregation of cells,
possibly to encourage horizontal gene transfer through conjugation. Cells expressing
mutant forms of AddB reported here, however, did not aggregate. This result suggests
that removing the origin of transfer or mobilization element from pBBR1-based vectors
before electroporation into Z. mobilis may improve the performance of strains utilizing
these plasmids. If conjugation must be used for plasmid insertion into the cells, however,
strains expressing the mutant forms of AddB reported here can serve as viable hosts.
Several previous reports have found that xylitol production and the concurrent
production of the toxic by-product xylitol-5-phosphate limit xylose fermentations in Z.

66
mobilis (Agrawal et al. 2011b; Feldmann et al. 1992; Jeon et al. 2005a; Kim et al. 2000a).
We found that reduced xylitol production by Z. mobilis due to a mutation in the xylose
reductase gene, ZMO0976, improves xylose fermentations in this organism. Agrawal and
Chen’s strain of Z. mobilis adapted to growth on xylose also contained a mutation at this
locus that led to a significant reduction in xylitol production (Agrawal and Chen 2011;
Agrawal et al. 2011b). Consequently, the authors deleted this gene from the chromosome,
and found that an even greater reduction in xylitol production could be achieved
compared to their adapted strain when xylose isomerase was overexpressed in the
knockout cells (Agrawal et al. 2012b). Potentially, a xylose reductase knockout can be
combined with the additional mutations reported here to further improve xylose
metabolism in Z. mobilis.
Recent studies have suggested that the heterologously-expressed metabolic
enzymes may be rate-limiting in xylose fermentations by Z. mobilis (De Graaf et al.
1999a; Kim et al. 2000a). In this study, we found that the same may be true for arabinose
fermentations—the native Z. mobilis transketolase gene contained a mutation that
increased its activity by ~70%, suggesting that the activity of the heterologous
transketolase is insufficient for proper arabinose assimilation. Also in support of this
hypothesis, the Z. mobilis glyceraldehyde-3-phosphate dehydrogenase (GAPDH) enzyme
contained a mutation that reduced its activity in the adapted cells by ~40%. Because the
substrate of GAPDH and the heterologous transaldolase are the same, it is likely that the
reduced GAPDH activity increases the access of transaldolase to its substrate. This
suggests that the activity of the heterologous transaldolase is also insufficient for proper

67
arabinose assimilation. Interestingly, the transketolase and transaldolase enzymes were
not found to be rate-limiting in the xylose-fermenting strain of Z. mobilis.
Collectively, our results reveal additional bottlenecks in pentose fermentations by
Z. mobilis. These bottlenecks are potential targets for the future engineering of pentose
metabolism in the strain. Additionally, several of the mutations reported here are not
specific to xylose and arabinose metabolism—they can be applied toward the metabolic
engineering of Z. mobilis to consume additional substrates and to produce additional
products.

68
3.5 Tables and figures
Table 3.1 Bacterial strains and plasmids used in this study.
Genotype or relevant characteristics Source or reference
Strains KLD1 ATCC 31821 [pKLD3], adapted to growth on xylose This study
KLD2 ATCC 31821 [pKLD11], adapted to growth on arabinose This study
KLD3 ATCC 31821 ΔaddB::SpR This study
Plasmids pEZCm Cm
R, pACYC184-pZMO1 (Skerker and Shapiro 2000b)
pJS71 SpR, pBBR1MCS-lacZα (Skerker and Shapiro 2000b)
pJS14 CmR, pBBR1MCS-lacZα (Skerker and Shapiro 2000b)
pPROTet.E CmR, PLTetO-1-RBS-6xHN-MCS-T1 Clontech
pVenus KanR, MCS yfp (Venus) t0 attλ, oriR6K (Saini et al. 2009)
pKLD3 pJS71-PgapxylAB-PenotalB-tktA (Dunn and Rao 2014)
pKLD7 pEZCm-PpdcVenus (Dunn and Rao 2014)
pKLD9 pJS71-PenotalB This study
pKLD10 pJS71-PenotalB-tktA This study
pKLD11 pJS71-PenotalB-tktA-PgaparaBAD This study
pKLD12 pEZCm-glf This study
pKLD13 pEZCm-glf A18T This study
pKLD14 pEZCm-glf V275F This study
pKLD15 pEZCm-glf A18TV275F This study
pKLD16 pPROTet.E-XKS1 This study
pKLD17 pPROTet.E-addBUP-SpR-addBDOWN This study
pKLD18 pJS14-PpdcVenus This study

69
Table 3.2 Oligonucleotide primers used in this study.
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD020F Peno, for ATGCTCTAGACCTTCATGTTTTGCTTCATG
KD020R talB ATCGAAACCTTTCTTAAAATCTTTTAGAC
KD021F talB, for AAGAAAGGTTTCGATATGACGGACAAATTGACCTC
KD021R Peno ATGCGGGCCCTTACAGCAGATCGCCGATCA
KD022F tktA ATGCGGGCCCTCATCCGATCTGGAGTCAAAATGTCCTCACG
KD022R ATGCGAGCTCCCGCAAACGGACATATCAAGG
KD023F Pgap, for ATGCCTCGAGTTTGTTCGATCAACAACCCGA
KD023R araBA GTTTATTCTCCTAACTTATTAAGT
KD024F araBA, for ACTTAATAAGTTAGGAGAATAAACATGGCGATTGCAATTGGCCT
KD024R Pgap and araD TTAGCGACGAAACCCGTAATA
KD025F araD, for TATTACGGGTTTCGTCGCTAAGGAAGGAGTCAACATGTTAGA
KD025R araBA ATGCTCTAGAGGCCCGTTGTCCGTCGC
KD026F glf ATGCCTCGAGTTGTTGTCGCGGGAGAGGC
KD026R ATGCGGTACCAAGCAAACCATCGGCATCAAGAC
KD027F glfA18T, for ATGCCTCGAGTTGTTGTCGCGGGAGAGGC
KD027R glfV275F CCTTCTGAAGCCGGAGACCAG
KD028F glfV275F, for CTGGTCTCCGGCTTCAGAAGG
KD028R glfA18T ATGCGGTACCAAGCAAACCATCGGCATCAAGAC
KD029F XKS1 ATGCGTCGACTTGTGTTCAGTAATTCAGAGACAGACAAGAG
KD029R ATGCTCTAGATTAGATGAGAGTCTTTTCCAGTTCGCTTA
KD030F addB, 1 kb TCGTAGAATGGCCAGAAAGATTGGG
KD030R upstream ATTCAAAATTAACCGCGTCTAAGAATGGTATC
KD031F addB, 1 kb GGGAATCCTATGGCTCATCCACAG
KD031R downstream TTAGGCTGATAATCTGGTGAATCGCC
KD032F SpR GAAGTTCCTATACTTTCTAGAGAATAGGAACTTCGGAATAGGAACTT
CCGAACCACTTCATCCGGGGT
KD032R GAAGTTCCTATTCCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC
CAATTATGTGCTTAGTGCATCTAACGCTT
KD033F pPROTet.E CCTAGGGCGTTCGGCTGC
KD033R TCTAGGGCGGCGGATTTGT
KD034F pPROTet.E-addB
up bridge
ACAAATCCGCCGCCCTAGATCGTAGAATGGCCAGAAAGATTGGG
KD034R addB up- SpR
bridge
ACCATTCTTAGACGCGGTTAATTTTGAATGAAGTTCCTATACTTTCTA
GAGAATAGGAAC
KD035F SpR- addB down
bridge
AGAGAATAGGAACTTCGGAATAGGAACTTCGGGAATCCTATGGCTCA
TCCACA
KD035R addB down
bridge-
pPROTet.E
GGCGATTCACCAGATTATCAGCCTAACCTAGGGCGTTCGGCTGC
KD036F ΔaddB::SpR GATGTCAGCACGCCCAAAGC
KD036R check GTTCTGCCTCCAAAGGCATTAATCG

70
Table 3.2 (cont.)
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD037F xylA qPCR TGCTACTGGCACACCTTCTG
KD037R TCCACATCGTGGAAGCAATA
KD038F xylB qPCR AGCATTCTCTGCAGGACGTT
KD038R CGCTGAACCCATAGCAATTT
KD039F talB qPCR CGTTTGTTGGCCGTATTCTT
KD039R AGAATTTCGCCGATGTTACG
KD040F tktA qPCR GAGCATGGACGCAGTACAGA
KD040R GCAGCTGACGGAAGTTTTTC
KD041F araB qPCR CCTCGATTTTGGCAGTGATT
KD041R GCTGTTCGACGCTAAGCTCT
KD042F araA qPCR GTCACCGATGGCGATAAAGT
KD042R TCATGGTGTAGCAGCTTTCG
KD043F araD qPCR AACCGGTGAAGTGGTTGAAG
KD043R GATTTCTGCGTCGGTCATTT
KD044F addB qPCR ACAGCCACCCAGTGAAAAAC
KD044R GAGCGAAGGCTGTTTAATGC
KD045F rrsA qPCR GTTCGGAATTACTGGGCGTA
KD045R CCACTGGTGTTCTTCCGAAT
KD046F gapdh, qPCR TCCGGATTCTGGACTTGAAC
KD046R GGTGAAGATACCCGTGCATT
KD047F tklB qPCR AAGGCTCAGTTCGGTGAAGA
KD047R ATGACCATCACAAGCCACAA
KD048F xr qPCR CGTTAGGCACATGGGCTATT
KD048R CCGACTTTGGTCGCAATAAT
KD049F glf qPCR AGTCACGCGACTAGCCCTAA
KD049R ACAACGACCATCCCAGAAAG
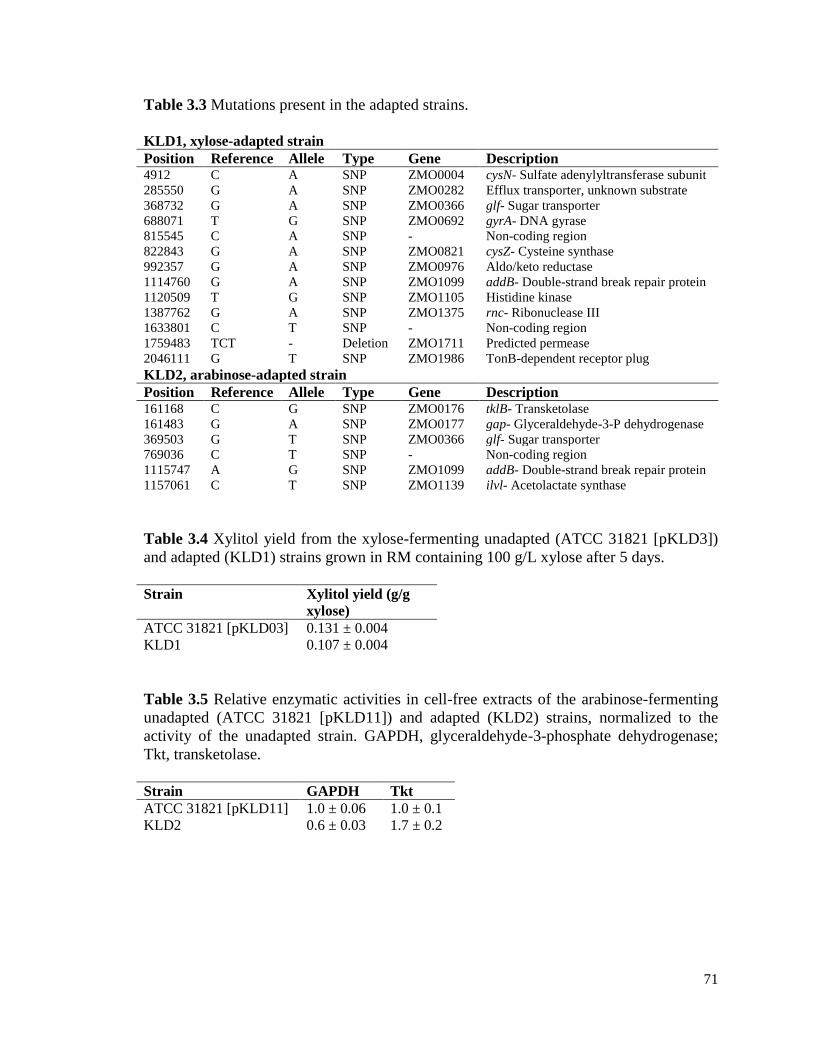
71
Table 3.3 Mutations present in the adapted strains.
KLD1, xylose-adapted strain
Position Reference Allele Type Gene Description 4912 C A SNP ZMO0004 cysN- Sulfate adenylyltransferase subunit
285550 G A SNP ZMO0282 Efflux transporter, unknown substrate
368732 G A SNP ZMO0366 glf- Sugar transporter
688071 T G SNP ZMO0692 gyrA- DNA gyrase
815545 C A SNP - Non-coding region
822843 G A SNP ZMO0821 cysZ- Cysteine synthase
992357 G A SNP ZMO0976 Aldo/keto reductase
1114760 G A SNP ZMO1099 addB- Double-strand break repair protein
1120509 T G SNP ZMO1105 Histidine kinase
1387762 G A SNP ZMO1375 rnc- Ribonuclease III
1633801 C T SNP - Non-coding region
1759483 TCT - Deletion ZMO1711 Predicted permease
2046111 G T SNP ZMO1986 TonB-dependent receptor plug
KLD2, arabinose-adapted strain
Position Reference Allele Type Gene Description 161168 C G SNP ZMO0176 tklB- Transketolase
161483 G A SNP ZMO0177 gap- Glyceraldehyde-3-P dehydrogenase
369503 G T SNP ZMO0366 glf- Sugar transporter
769036 C T SNP - Non-coding region
1115747 A G SNP ZMO1099 addB- Double-strand break repair protein
1157061 C T SNP ZMO1139 ilvl- Acetolactate synthase
Table 3.4 Xylitol yield from the xylose-fermenting unadapted (ATCC 31821 [pKLD3])
and adapted (KLD1) strains grown in RM containing 100 g/L xylose after 5 days.
Strain Xylitol yield (g/g
xylose)
ATCC 31821 [pKLD03] 0.131 ± 0.004
KLD1 0.107 ± 0.004
Table 3.5 Relative enzymatic activities in cell-free extracts of the arabinose-fermenting
unadapted (ATCC 31821 [pKLD11]) and adapted (KLD2) strains, normalized to the
activity of the unadapted strain. GAPDH, glyceraldehyde-3-phosphate dehydrogenase;
Tkt, transketolase.
Strain GAPDH Tkt
ATCC 31821 [pKLD11] 1.0 ± 0.06 1.0 ± 0.1
KLD2 0.6 ± 0.03 1.7 ± 0.2

72
Figure 3.1 Schematic representations of plasmids. a Plasmid used to express the xylose
metabolism genes. b Plasmid used to express the arabinose metabolism genes.
Figure 3.2 Adapted strain analyses. a-b Xylose and ethanol concentrations for strain
ATCC 31821 [pKLD3] (unadapted) and strain KLD1 (adapted). c Growth of the strains
as determined by OD600nm. d-e Arabinose and ethanol concentrations for strain ATCC
31821 [pKLD11] (unadapted) and strain KLD2 (adapted). f Growth of the strains as
determined by OD600nm.
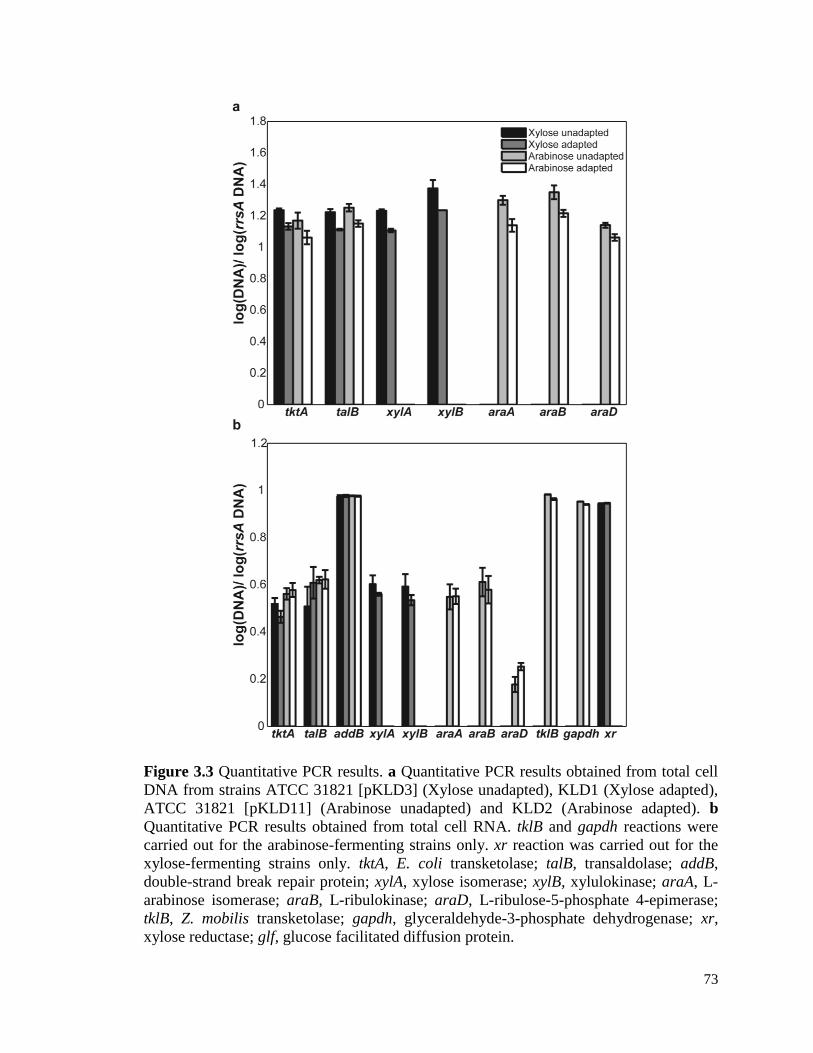
73
Figure 3.3 Quantitative PCR results. a Quantitative PCR results obtained from total cell
DNA from strains ATCC 31821 [pKLD3] (Xylose unadapted), KLD1 (Xylose adapted),
ATCC 31821 [pKLD11] (Arabinose unadapted) and KLD2 (Arabinose adapted). b
Quantitative PCR results obtained from total cell RNA. tklB and gapdh reactions were
carried out for the arabinose-fermenting strains only. xr reaction was carried out for the
xylose-fermenting strains only. tktA, E. coli transketolase; talB, transaldolase; addB,
double-strand break repair protein; xylA, xylose isomerase; xylB, xylulokinase; araA, L-
arabinose isomerase; araB, L-ribulokinase; araD, L-ribulose-5-phosphate 4-epimerase;
tklB, Z. mobilis transketolase; gapdh, glyceraldehyde-3-phosphate dehydrogenase; xr,
xylose reductase; glf, glucose facilitated diffusion protein.

74
Figure 3.4 Mutant transporter analyses. a-b Xylose and ethanol concentrations for strain
ATCC 31821 [pKLD3] [pKLD12] (+Glf), strain ATCC 31821 [pKLD3] [pKLD13]
(+Xyl adapted Glf), and strain ATCC 31821[pKLD3] [pKLD14] (+Ara adapted Glf). c
Growth of the strains as determined by OD600nm. d-e Arabinose and ethanol
concentrations for strain ATCC 31821 [pKLD11] [pKLD12] (+Glf), strain ATCC 31821
[pKLD11] [pKLD13] (+Xyl adapted Glf), and strain ATCC 31821 [pKLD11] [pKLD14]
(+Ara adapted Glf). f Growth of the strains as determined by OD600nm.
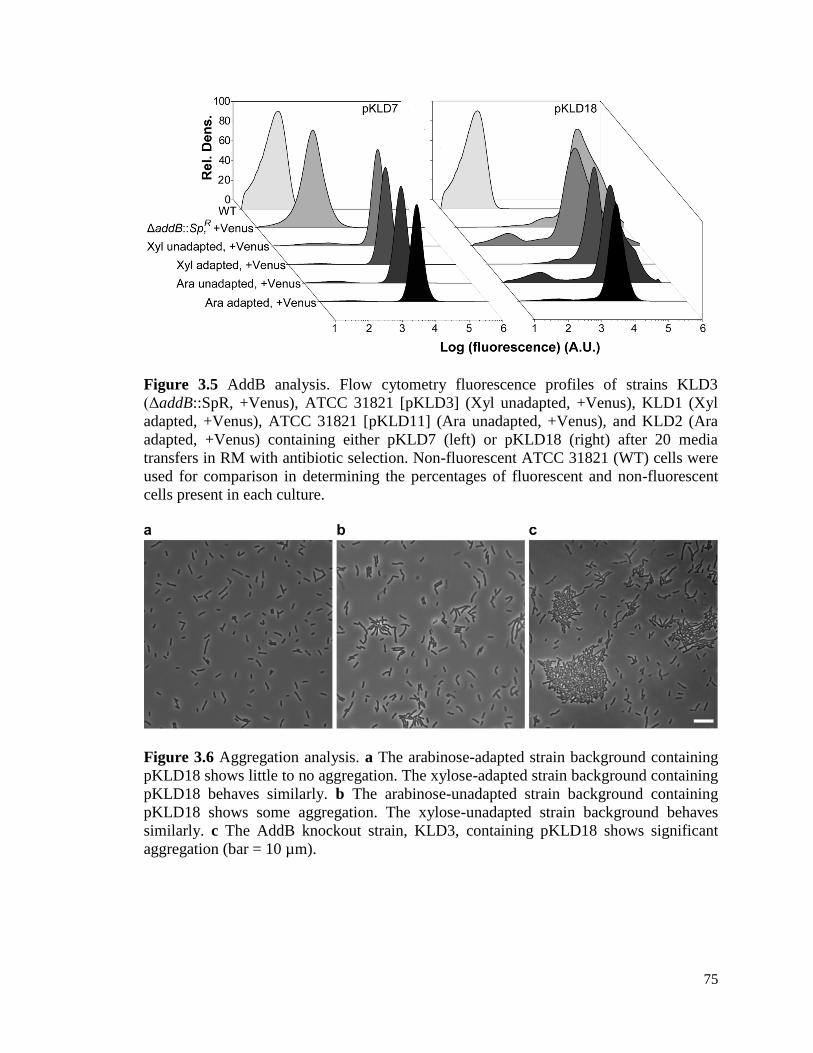
75
Figure 3.5 AddB analysis. Flow cytometry fluorescence profiles of strains KLD3
(ΔaddB::SpR, +Venus), ATCC 31821 [pKLD3] (Xyl unadapted, +Venus), KLD1 (Xyl
adapted, +Venus), ATCC 31821 [pKLD11] (Ara unadapted, +Venus), and KLD2 (Ara
adapted, +Venus) containing either pKLD7 (left) or pKLD18 (right) after 20 media
transfers in RM with antibiotic selection. Non-fluorescent ATCC 31821 (WT) cells were
used for comparison in determining the percentages of fluorescent and non-fluorescent
cells present in each culture.
Figure 3.6 Aggregation analysis. a The arabinose-adapted strain background containing
pKLD18 shows little to no aggregation. The xylose-adapted strain background containing
pKLD18 behaves similarly. b The arabinose-unadapted strain background containing
pKLD18 shows some aggregation. The xylose-unadapted strain background behaves
similarly. c The AddB knockout strain, KLD3, containing pKLD18 shows significant
aggregation (bar = 10 µm).

76
Chapter 4: Genetically modifying Z. mobilis using the λ Red
recombinase system and a CRISPR-associated nuclease
4.1 Introduction
Although Z. mobilis is primarily known for its ability to produce the biofuel
ethanol, this bacterium also holds promise for the production of other value-added
products (Belauich and Senez 1965; Johns et al. 1991; McGill and Dawes 1971). In order
for alternative product yields and titers to be economical, however, further engineering of
the strain to shift metabolism away from ethanol production must be done. Making the
necessary chromosomal modifications to Z. mobilis for alternative product synthesis
using the outdated and inefficient techniques available to researchers today would be
difficult. Specifically, techniques for the construction of gene knockouts and
chromosomal integrations, as well as techniques for genome regulation need to be
improved if Z. mobilis is to be accepted as an industrial host for products other than
biofuels.
Gene knockouts and chromosomal integrations have historically been constructed
in Z. mobilis using suicide vectors that require hundreds or thousands of base pairs of
homology to the target region but still yield only a few transformants (Agrawal et al.
2012b; Delgado et al. 2002; Kerr et al. 2011; Senthilkumar et al. 2004). Alternatively,
bacteriophage recombination systems have been shown to effectively induce homologous
recombination between linear DNA fragments and bacterial chromosomes (Datsenko and
Wanner 2000). These systems can therefore be used to introduce a variety of genetic
alterations to a bacterial host, including gene knockouts, insertions, and point mutations
(Baba et al. 2006; Datsenko and Wanner 2000; Lemuth et al. 2011; Posfai et al. 2006;
Wang and Pfeifer 2008).

77
The recombination system that has received the most attention for gene knockouts
and integrations is the bacteriophage λ Red system, which contains the proteins Gamma,
Beta, and Exo (Murphy 1998). The Gamma protein is able to inhibit RecBCD-type host
nucleases from degrading linear DNA, protecting the cassette (Karu et al. 1975). Exo acts
as an exonuclease on double-stranded cassettes to create single-stranded sticky ends that
are then bound by the Beta protein and directed to anneal and recombine with
complementary DNA on the chromosome (Sharan et al. 2009). When expressed in E.
coli, these three proteins can easily induce a linear selectable marker DNA cassette
flanked by only 35 bp of homology to crossover into the chromosome (Datsenko and
Wanner 2000). When the selectable marker is flanked by flippase recognition target
(FRT) sites, it can then be easily removed from the chromosome by yeast flippase,
leaving behind only a FRT scar (Cherepanov and Wackernagel 1995). This system of
using the λ Red proteins to introduce a selectable marker in place of a gene of interest
followed by removing the selectable marker gene with flippase has allowed for the rapid
construction of gene knockouts and integrations in several species of bacteria (Datsenko
and Wanner 2000; Katashkina et al. 2009; Lesic and Rahme 2008; Yamamoto et al.
2009). The system may therefore be effective in Z. mobilis.
The bacteriophage λ protein Beta is also capable of mediating the modification of
chromosomal genes using single stranded DNA (ssDNA) oligonucleotides. These
oligonucleotides are incorporated into the host genome at the replication fork during
DNA synthesis, leading to allelic replacement (Ellis et al. 2001; Wang et al. 2009; Zhang
et al. 1998). The process works most efficiently when the mutS gene, responsible for
mismatch repair, has been removed from the chromosome (Costantino and Court 2003).

78
The oligonucleotides can be designed to target any chromosomal locus or loci. When
several cycles of ssDNA incorporation are performed to accumulate mutations, the
process is called multiplex automated genome engineering, or MAGE (Wang et al. 2009).
MAGE enables rapid, large-scale evolution of cells, allowing for the isolation of
mutagenic strains with improved phenotypes rapidly, efficiently, and inexpensively
(Wang et al. 2009). Theoretically, if the λ Red proteins successfully mediate gene
knockouts and integrations in Z. mobilis, MAGE should also be possible in this strain.
While the λ Red recombinase system is a very powerful tool for constructing gene
knockouts and integrations with the use of selectable markers, researchers have also
recently discovered the power of another system for modifying genomes without
selection. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
adaptive immunity system utilizes small RNAs for sequence-specific silencing of foreign,
invading nucleic acids (Jinek et al. 2012; Wiedenheft et al. 2012). The type II
CRISPR/Cas9 system from Streptococcus pyogenes has been widely utilized for marker-
free genome engineering (Cong et al. 2013; Hwang et al. 2013; Jiang et al. 2013; Mali et
al. 2013). This system has three primary components: the CRISPR RNA (crRNA), the
trans-activating CRISPR RNA (tracrRNA), and the CRISPR associated (Cas) nuclease,
Cas9 (Jinek et al. 2012). The tracrRNA acts as a scaffold for the crRNA, which contains
about 20 to 30 base pairs of sequence homology to the invading or target DNA (Mali et
al. 2013). This homology is known as the protospacer sequence, and it is flanked by
direct repeats that are recognized by the tracrRNA (Ishino et al. 1987; Jinek et al. 2012;
Makarova et al. 2011). The tracrRNA and crRNA can also be fused into a single
transcript called guide RNA (gRNA) (Jinek et al. 2012). The tracrRNA:crRNA complex

79
or gRNA transcript recruit the Cas9 protein to the homologous base pairs in the target
DNA (Jinek et al. 2012). In order for Cas9 to successfully bind, however, the target
sequence must also contain a Protospacer Adjacent Motif (PAM) sequence at its 3’ end
(Deveau et al. 2008). For Cas9, the optimal PAM sequence is NGG (Mojica et al. 2009).
After successful binding to DNA containing an adjacent PAM, Cas9 is able to cleave,
leading to a double-stranded break (Jinek et al. 2012).
For the generation of chromosomal knockouts or integrations utilizing the
CRISPR-Cas system in bacteria, double-stranded DNA flanked by regions of homology
to the target gene are introduced into the cells (Jiang et al. 2013). The dsDNA cassette
does not need to be a marker gene, thereby allowing for the replacement of chromosomal
genes with any gene of interest. DNA coding for the Cas9 protein and gRNA or
tracrRNA:crRNA complementary to the unmutated target gene is simultaneously
transformed into the cells. After crossover into the chromosome, the cassette will prevent
recognition of the target by the Cas9-tracrRNA:crRNA complex, allowing mutated cells
to evade cleavage by Cas9. The DNA of unmutated cells, however, will be targeted by
the endonuclease and the resulting double-stranded break will likely lead to cell death
(Gudbergsdottir et al. 2011; Jiang et al. 2013; Marraffini and Sontheimer 2010). Due to
the broad applicability of the CRISPR system and its proven functionality in a wide
variety of cell types and historically recalcitrant organisms (Sander and Joung 2014), it is
likely that this system can also be applied for genome engineering in Z. mobilis.
In this chapter, we constructed a λ Red expression system for use in Z. mobilis.
We tested the effectiveness of this system for the development of gene knockouts
compared to the effectiveness of natural homologous recombination. We found that

80
although the number of colony forming units was relatively unchanged when the λ Red
proteins were expressed, a larger fraction of these colony forming units displayed the
desired mutant genotype. We also constructed a vector for the expression of yeast
flippase in Z. mobilis to enable marker removal from the chromosome. We found that this
enzyme was highly effective in the strain. Because our knockout results suggested
activity of the λ Beta protein in Z. mobilis, we constructed a host strain for MAGE.
Unfortunately, however, we were unable to provide evidence for ssDNA incorporation in
the strain under the conditions tested here. Finally, we also constructed an expression
system for the CRISPR small RNAs and the Cas9 protein in Z. mobilis. We tested the
system in Z. mobilis and found that the Cas9 protein is able to induce cell death when
directed to a chromosomal gene by the tracrRNA:crRNA complex. We also found that
the Cas9 system is likely able to induce dsDNA crossover into the chromosome.
Collectively, our work provides proof-of-concept for the use of modern genome
engineering techniques in Z. mobilis and sets the stage for the acceptance of this strain as
an alternative industrial host.
4.2 Materials and methods
4.2.1 Bacterial strains, media, and growth conditions
All experiments were performed in Zymomonas mobilis subsp. mobilis ZM4
(ATCC 31821). RM liquid and solid medium (10 g/L yeast extract and 2 g/L potassium
phosphate, dibasic) supplemented with 20 g/L glucose was used for all Z. mobilis
experiments. All Z. mobilis cultures were grown at 30°C. Antibiotics were used at the
following concentrations: spectinomycin, 100 µg/mL; chloramphenicol, 100 µg/mL; and
tetracycline, 25 µg/mL.

81
4.2.2 Plasmid construction
All plasmids used in this study are listed in Table 4.1. All primers used in this
study are listed in Table 4.2. To construct a plasmid to express the λ Red genes in Z.
mobilis, the gam-beta-exo operon was amplified to include its native ribosome binding
site and terminator sequences from plasmid pKD46 by PCR synthesis using primers
KD050F and KD050R. The fragment was then inserted into plasmid pSRKTc directly
downstream of the vector’s engineered lac promoter to form plasmid pKLD19.
To construct a plasmid for the expression of yeast flippase in Z. mobilis, the
flippase (flp) gene was amplified from its start codon and including its native terminator
sequence from S. cerevisiae BY4742 DNA by PCR synthesis using primers KD051F and
KD051R or KD053F and KD053R. The 500 bp pdc promoter was amplified from Z.
mobilis ZM4 genomic DNA by PCR synthesis using primers KD054F and KD054R. The
two fragments were then joined together using overlap extension PCR (Horton et al.
1989). The Ppdc-flp gene fragment was then inserted into plasmid pJS14 to form plasmid
pKLD20. Separately, the lacIq-Plac fragment was amplified from plasmid pSRKTc by
PCR synthesis using primers KD052F and KD052R. The lacIq-Plac and flp fragments
were then joined together using overlap extension PCR, and the resulting lacIq-Placflp
fragment was inserted into plasmid pJS14 to yield plasmid pKLD21.
To construct vectors for the generation of linear dsDNA cassettes, the
spectinomycin resistance gene including its native promoter and terminator sequences
was amplified by PCR synthesis from plasmid pJS71 using primers KD055F and
KD055R, such that FRT sites were added at both ends of the gene. When FRT sites were
not added, primers KD056F and KD056R were used. The pPROTet.E vector was also

82
amplified by PCR synthesis from plasmid pPROTet.E using primers KD057F and
KD057R. One kilobase fragments both upstream and downstream of five of the six target
genes (Table 4.3) were amplified using primers KD058F-KD062R and KD063F-
KD067R, respectively. The appropriate DNA fragments were then phosphorylated using
T4 polynucleotide kinase (New England Biolabs), and combined with Ampligase
thermostable DNA ligase and buffer (epicentre) and bridging oligonucleotides KD068F-
KD077R, as necessary. The fragments were then joined together such that the target gene
upstream and downstream regions were flanking the spectinomycin gene in the
pPROTet.E vector using the optimized ligase cycling reaction parameters reported by de
Kok and coworkers (de Kok et al. 2014) to yield plasmids pKLD22-pKLD26,
respectively. For the construction of vector pKLD29, the sacB upstream and sacC
downstream regions were amplified from Z. mobilis ZM4 genomic DNA by PCR
synthesis using primers KD078F, KD078R, KD079F, and KD079R, respectively, and
inserted sequentially into pPROTet.E to form plasmids pKLD27 and pKLD28. The
spectinomycin gene was amplified from plasmid pJS71 by PCR synthesis using primers
KD080F and KD080R and inserted between the sacB upstream and sacC downstream
regions in plasmid pKLD28 to form plasmid pKLD29.
To construct a vector for the development of an ssDNA recombineering host
strain, the regions 1 kb upstream and 1 kb downstream of the zmrr gene were amplified
using primers KD062F, KD062R, KD067F, and KD067R, respectively. The pUC19
plasmid was amplified from pUC19 using primers KD081F and KD081R. The
spectinomycin resistance gene was amplified by PCR synthesis to contain upstream KpnI
and BamHI restriction sites from plasmid pJS71 using primers KD082F and KD082R.

83
The four fragments were then joined together as was described above using primers
KD083F-KD084R and the optimized ligase cycling reaction parameters reported by de
Kok and coworkers to yield plasmid pKLD30. Separately, the plasmid pKD3 was
amplified using primers KD085F and KD085R such that a premature stop codon was
introduced into the chloramphenicol resistance gene. This PCR product was then
phosphorylated using T4 polynucleotide kinase (New England Biolabs) and self-ligated
to yield plasmid pKLD31. The chloramphenicol resistance gene containing a premature
stop codon was then amplified from plasmid pKLD31 by PCR synthesis using primers
KD086F and KD086R. This allele was then inserted at the KpnI and BamHI sites of
pKLD30 to yield pKLD32.
To construct a vector for constitutive expression of the λ Red genes in Z. mobilis,
the gam beta exo operon was amplified from its start codon and including its native
terminator sequence from plasmid pKD46 by PCR synthesis using primers KD087F and
KD087R. Separately, the strong and constitutive 500 bp pdc promoter was amplified
from Z. mobilis ZM4 genomic DNA by PCR synthesis using primers KD088F and
KD088R. The two fragments were then joined together using overlap extension PCR.
The resulting Ppdc-gam-beta-exo DNA was then inserted into plasmid pSRKTc to form
plasmid pKLD33.
To construct all-inclusive CRISPR/Cas expression vectors for use in Z. mobilis,
the 1.6 kb plasmid pZMO1 maintenance region from pEZCm was amplified by PCR
synthesis using primers KD089F and KD089R. The resulting fragment was then inserted
into plasmid pCas9 to form plasmid pKLD34. Plasmid pCas9 was a gift from Luciano
Marraffini (Addgene plasmid # 42876). Separately, the 5.3 kb region of pCas9

84
comprising the tracrRNA, cas9, and direct repeat coding regions was amplified by PCR
synthesis using primers KD090F and KD090R and inserted into plasmid pSRKTc to form
plasmid pKLD35.
To construct co-expression vectors for the CRISPR/Cas system in Z. mobilis, the
CRISPR direct repeats including the 100 bp upstream and downstream regions were
amplified by PCR synthesis using primers KD091F and KD091R from plasmid pCas9. A
635 bp fragment containing the tracrRNA coding sequence was also amplified by PCR
synthesis from plasmid pCas9 using primers KD092F and KD092R. The two fragments
were then joined together using overlap extension PCR and inserted into plasmid pEZCm
to yield plasmid pKLD36. Separately, the cas9 gene including its native promoter and
terminator sequences was amplified by PCR synthesis using primers KD093F and
KD093R from plasmid pCas9 and inserted into plasmid pSRKTc to form plasmid
pKLD37, into plasmid pEZCm to form plasmid pKLD38, and into plasmid pJS71 to form
plasmid pKLD39. To insert a zmrr targeting spacer into plasmid pKLD36, a method
similar to that of Jiang and coworkers was used (Jiang et al. 2013). Briefly, 2 μM of each
primer KD094F and KD094R were first phosphorylated using T4 polynucleotide kinase
(New England Biolabs) in a total reaction volume of 50 μL. The primers were then
annealed by adding 50 mM NaCl, incubating for 5 minutes at 95°C, and then slowly
cooling to room temperature. The annealed primers were then diluted and ligated into
BsaI-digested pKLD36 to form plasmid pKLD40.
4.2.3 Plasmid transformation into Z. mobilis using electroporation
For standard plasmid transformations, Z. mobilis cells grown to an OD600nm of at
least 0.8 in RM containing 20 g/L glucose were harvested and washed once with water

85
and once with 10% (v/v) glycerol. Cells were then resuspended in 10% glycerol to 1% of
the original culture volume. For each transformation, at least 1 µg of unmethylated
plasmid DNA isolated from E. coli strain GM119 and 2.5 µg of TypeOne Restriction
Inhibitor (epicentre) were added to 40 µL of competent cells. Cuvettes with 0.1 cm gaps
were used in a BioRad XCell gene pulser set at 1.6 kV, 200 Ω, and 25 µF. Following
transformation, cells were allowed to recover in RM at 30°C for 1 to 3 h. Transformants
were isolated after growth for 3 to 7 days on RM plates containing the appropriate
antibiotic. Plasmid presence was confirmed by plasmid isolation and subsequent
transformation into E. coli DH5α. When two plasmids were used, transformations were
performed sequentially unless otherwise indicated.
4.2.4 Z. mobilis λ Red-mediated knockout experiments
Z. mobilis containing plasmid pKLD19 and Z. mobilis without pKLD19 were
inoculated from single colonies on agar plates in liquid RM (containing antibiotic as
necessary) in 5 mL glass tubes and grown with shaking overnight. Cells were then
subcultured to an OD600nm of 0.05 in 50 mL RM containing 500 μM IPTG in 250 mL
flasks and grown for 16 hours. Cells were then induced a second time with 500 μM
IPTG. After 3 h, cells were prepared as described above and electroporated with at least 1
μg of cassette plasmid (pKLD22-pKLD26 or pKLD29) linearized by a double restriction
digest with enzymes that cut in the vector backbone. Cells were allowed to recover for 3
h, at which time they were plated on RM plates containing spectinomycin and allowed to
grow for 3 to 7 days.
Insertional inactivation of target genes was confirmed by PCR using primers
KD095F-KD100R and genomic DNA extracted using standard protocols. For all target

86
genes, successful inactivation through a double crossover event yielded a single PCR
product 1.5 kb in length. Unsuccessful inactivation through a single crossover event
yielded two PCR products, one 1.5 kb in length and the other the length of the target
gene. Transformants obtained through single crossover events were also resistant to
chloramphenicol, due to the insertion of the entire cassette vector backbone into the
chromosome. False positive spectinomycin-resistant cells yielded a single PCR product
the length of the target gene.
4.2.5 Motility plate assays
The FliA protein is a transcriptional regulator of motility (Seo et al. 2005). When
it is absent, cells are no longer motile (Chen and Helmann 1992). Motility loss can be
detected using a simple plate assay. Specifically, cells were inoculated from single
colonies on agar plates in liquid RM in 5 mL glass tubes and grown with shaking until
late exponential phase. Roughly 5 x 105 cells were then spotted in the center of an RM
agar plate containing 3 g/L agar to allow the cells to swim. Plated cells were allowed to
grow overnight, at which time the size of the halo was used as an indicator of motility.
4.2.6 Marker removal from Z. mobilis
Cells with confirmed insertional inactivation of a target gene due to replacement
with spectinomycin flanked by FRT sites were transformed with plasmid pKLD20 as was
described above. Successful transformants isolated on chloramphenicol agar plates were
streaked for individual colonies again on an RM agar plate containing chloramphenicol
and grown for 2 days at 30°C. Colonies were then tested for the loss of spectinomycin
resistance by streaking on an RM agar plate and an RM agar plate containing
spectinomycin. Plasmid pKLD21 was tested in a similar manner, except cells were

87
induced with 500 μM IPTG 3 h before competent cells were prepared. RM agar plates
containing chloramphenicol also contained 500 μM IPTG when pKLD21 was used.
4.2.7 ssDNA recombineering experiments
To construct an ssDNA recombineering host strain, vector pKLD32 was
linearized with the NdeI and AatII restriction enzymes then used to construct a zmrr
knockout in strain KLD10. This led to the chromosomal integration of a non-functional
chloramphenicol resistance gene containing a premature stop codon. This strain was
designated KLD11. Plasmid pKLD19 and plasmid pKLD33 were then separately
electroporated into this strain. These cells were then grown in RM containing
spectinomycin and tetracycline, prepared as was described above, and electroporated with
6 μM oligonucleotide KD101F. Cells were allowed to grow for 3 hours before plating on
RM agar plates containing chloramphenicol to select for transformants. When IPTG was
necessary, cells were induced as was described above for λ Red-mediated knockout
experiments. Experiments with oligonucleotides KD101R and KD102F were similar
except Z. mobilis containing plasmid pKLD33 was used as the host strain. All
oligonucleotides were designed to target the lagging strand during DNA replication.
4.2.8 Z. mobilis CRISPR/Cas experiments
Z. mobilis cells containing plasmid pKLD37 and Z. mobilis without pKLD37
were inoculated from single colonies on agar plates in liquid RM (containing antibiotic as
necessary) in 5 mL glass tubes and grown with shaking overnight. Cells were then
prepared and electroporated as was described above, except both plasmid pKLD40 and
linearized plasmid pKLD26 were transformed at once. After recovery for 3 h, cells were
plated on RM agar plates containing spectinomycin and chloramphenicol and allowed to

88
grow for 3 to 7 days to select for transformants. Insertional inactivation of the target gene
was confirmed by PCR using primers KD099F and KD099R as was described above. In a
separate experiment, plasmid pKLD40 was transformed individually into Z. mobilis with
and without pKLD39. The entire transformations were plated on RM agar plates
containing chloramphenicol and the resulting number of colony forming units was
quantified.
4.3 Results
4.3.1 Construction of Z. mobilis λ Red recombinase and CRISPR system host
strains
Genetic techniques available for use in Z. mobilis are inefficient and unreliable. In
order to ensure that the strain reaches its full industrial potential, we wanted to develop
protocols for modern genome engineering strategies in Z. mobilis. Specifically, to
improve the construction efficiency of gene knockouts and integrations, we developed a
strain of Z. mobilis that expresses the phage λ Red proteins Beta, Exo, and Gamma under
the control of an inducible lac promoter with a low basal level of expression from a
vector (Figure 4.1a). These proteins are able to mediate homologous recombination
between linear dsDNA and the chromosome (Datsenko and Wanner 2000). Similarly, we
also developed a strain that expresses the three proteins constitutively under the control
of the strong Z. mobilis pdc promoter. Exclusively, the Beta protein is also able to
mediate the incorporation of ssDNA oligonucleotides into bacterial chromosomes (Ellis
et al. 2001). To test if it could do so in Z. mobilis, we constructed a Beta-expressing strain
deficient in the MutS protein. MutS is responsible for DNA mismatch repair—when it is

89
not present, the efficiency of ssDNA recombineering is greatly improved (Costantino and
Court 2003).
Another system for genome engineering that has received considerable attention
in recent years is the CRISPR/Cas system for marker-free gene knockouts, insertions, and
point mutations. To test this system in Z. mobilis, we constructed a strain that expresses
the Cas9 protein under the control of its native promoter from a broad-host range vector
(Figure 4.1c). We also constructed a vector for the expression of the small crRNA and
tracrRNA transcripts (Figure 4.1b).
4.3.2 The λ Red recombinase system increases double crossover events in Z.
mobilis
To test the λ Red recombinase system in our strain of Z. mobilis expressing
Gamma, Beta, and Exo, we first needed to generate linear dsDNA cassettes with regions
of homology to the chromosome. Recombineering in alternative hosts frequently requires
homology regions of up to 1,000 bp for the best results (Lesic and Rahme 2008; Pontes
and Dale 2011; van Kessel and Hatfull 2007). Additionally, Z. mobilis requires at least 1
μg of DNA for a typical transformation to be successful. We attempted to use a 2-step
PCR process to construct cassettes with the required 1,000 bp of homology, but we found
that cassette yields were not high enough to ensure transformation success. Therefore, we
utilized the optimized ligase cycling reaction parameters reported by de Kok and
coworkers (de Kok et al. 2014) to quickly construct high-quality cassettes in a high-copy
vector, which we then linearized by restriction double digest. These linear cassettes were
then electroporated into Z. mobilis containing plasmid pKLD19 that had been induced
with IPTG, and Z. mobilis containing plasmid pKLD19 that had not been induced with

90
IPTG. After selection, we found roughly the same number of colony forming units on the
induced and uninduced plates. However, we had previously observed that single
crossover events that led to the chromosomal integration of the entire cassette vector
(including its resistance gene) in Z. mobilis were relatively common (Figure 4.2a).
Therefore, we wanted to determine if the expression of the λ Red proteins increased the
likelihood of desirable double crossover events (Figure 4.2b). Using PCR verification
(Figure 4.3) and antibiotic resistance checks, we found that a higher percentage of
colonies tested were in fact knockout mutants when the λ Red proteins were induced.
Specifically, in the best case, ~90% of colonies tested displayed the desired ΔsacBC::SpR
genotype when the λ Red proteins were induced, while only ~19% of colonies displayed
this genotype when they were not (Table 4.4). The constitutive λ Red expression vector
pKLD33 yielded similar results as pKLD19 compared to a wild-type Z. mobilis control.
When possible, we also utilized phenotypic characterization to confirm our knockouts
(Figure 4.4).
In an attempt to increase the overall efficiency of the process, λ Red-mediated
knockouts were also attempted using KLD5 as the host strain. Strain KLD5 is deficient in
AddA, the Z. mobilis analogue of RecB. However, the addA knockout strain carrying the
recombinase vector pKLD19 or pKLD33 grew slowly, did not revive well from freezer
stock, and led to only one or two colony forming units after cassette transformation. This
made it difficult to obtain meaningful results with this strain.
4.3.3 Yeast flippase removes marker genes from the Z. mobilis chromosome
When FRT sites are placed at either end of an integrated resistance gene, the yeast
flippase enzyme can be used to remove the gene from the chromosome. We wanted to

91
determine if flippase was functional in Z. mobilis. We constructed a Z. mobilis flippase
expression system by placing this enzyme under the control of an inducible lac promoter
with a low basal level of expression from a broad-host range vector. However, very few
colonies actually lost resistance when flp expression was induced. To attempt to improve
this system, we then constructed a constitutive expression vector with flp placed under
the control of the strong and constitutive pdc promoter. With this vector, we found
consistently that nearly all candidates tested lost the FRT-flanked spectinomycin
resistance cassette, proving that flippase is an effective tool for genome engineering in Z.
mobilis.
4.3.4 The λ Red Beta protein does not mediate ssDNA recombineering in a
mutS-deficient strain of Z. mobilis
While we did not see λ Red knockout efficiencies in Z. mobilis that were close to
those that have been reported in E. coli, our results did suggest that the proteins were
active in the strain. Therefore, we wanted to attempt ssDNA recombineering in Z.
mobilis. ssDNA recombineering only requires the action of the λ Red Beta protein, but it
is common practice to utilize strains expressing all three λ Red proteins during ssDNA
oligonucleotide incorporation (Copeland et al. 2001; Wang et al. 2009). The process is
also much more efficient in strains deficient in the MutS protein, which is responsible for
mismatch repair (Costantino and Court 2003). Therefore, we first constructed a mutS
knockout strain of Z. mobilis. To this strain we added a non-functional chloramphenicol
resistance cassette containing a premature stop codon at the zmrr locus. This strain,
designated KLD11, was confirmed to be sensitive to chloramphenicol. Vector pKLD19
or pKLD33 was then electroporated into strain KLD11 to yield Z. mobilis ssDNA

92
recombineering host strains. The 60 bp ssDNA oligonucleotide KD101F was then
electroporated into these strains to attempt to correct the premature stop codon by
oligonucleotide incorporation into the chromosome. However, after several attempts, no
transformants were recovered on chloramphenicol agar plates. Separately, 60 bp
oligonucleotides KD101R and KD102F were transformed into Z. mobilis containing
plasmid pKLD33. If incorporated, these oligonucleotides would lead to a loss of motility
or a loss of the ability to grow on fructose, respectively, but no fliA or frk null mutants
could be found using motility plate assays and fructose growth tests. While ssDNA
recombineering may still be possible in Z. mobilis, we found that it is likely not possible
under the precise conditions tested here.
4.3.5 The Cas9 protein induces cell death when directed to a Z. mobilis
chromosomal gene
As a simple initial test of the CRISPR/Cas system in Z. mobilis, we wanted to see
if the Cas9 protein could be directed to create a double-stranded break in the chromosome
by a tracrRNA:crRNA complex containing a spacer with genomic homology and the
required adjacent PAM sequence (Figure 4.5). We attempted to transform Z. mobilis
with two different all-inclusive Cas expression vectors, pKLD34 and pKLD35. pKLD34
contains the tracrRNA-cas9-dr coding region and the pZMO1 native vector replication
origin. pKLD35 contains the tracrRNA-cas9-dr coding region and the pBBR1 replication
origin. When we attempted to transform these vectors into Z. mobilis, we found no
colony forming units on chloramphenicol or tetracycline RM agar plates, respectively,
despite several attempts. Because it was possible that simply the expression level of the
Cas9 protein was lethal to the cells, we also constructed three vectors containing the cas9

93
coding region only—pKLD37, pKLD38, and pKLD39. Without the tracrRNA:crRNA
complex, the Cas9 protein would no longer be directed to the chromosome, so if these
vectors were also unable to be transformed into the cells then we knew Cas9 expression
levels and not chromosomal cleavage were responsible for cell death. The three vectors
also contained the pBBR1 or pZMO1 vector origins to account for copy number effects.
When we transformed these vectors into Z. mobilis, we found many colony forming units
on tetracycline, chloramphenicol, and spectinomycin plates, respectively.
A similar test was carried out using two separate vectors, one carrying the Cas9
protein and the other carrying the small RNAs and a spacer to target the Z. mobilis
chromosome at a site with the required adjacent PAM sequence. Cells containing
pKLD39 and cells without pKLD39 were transformed with plasmid pKLD40. With Cas9
present on pKLD39, we most frequently saw no colony forming units on an RM agar
plate containing chloramphenicol, because the tracrRNA:crRNA complex directed Cas9
to cleave the chromosome (Table 4.5). When Cas9 was not present, we saw many colony
forming units on an RM agar plate containing chloramphenicol. Consistent with previous
results, we did find a single colony expressing Cas9 that was able to evade cleavage by
the nuclease, likely due to a mutation near the PAM site (Jiang et al. 2013). Collectively,
these results provide preliminary proof that the tracrRNA:crRNA complex is able to
direct Cas9 cleavage of the Z. mobilis chromosome.
4.3.6 The Cas system mediates dsDNA crossover into the Z. mobilis
chromosome
To prove the utility of the CRISPR/Cas system in Z. mobilis, we wanted to use it
to mediate a gene knockout in the strain. Therefore, we simultaneously transformed cells

94
containing pKLD37 and cells without pKLD37 with pKLD40 and pKLD26 linearized by
double restriction digest. In the cells expressing Cas9, the Cas9 protein would be directed
to cut the chromosome at the wild-type zmrr allele if the cassette was incorporated
through a single crossover event or not incorporated at all, while cells with a successful
knockout genotype would avoid cleavage by Cas9 and represent the majority of
transformants. In cells not expressing Cas9, undesirable single crossover events would
likely represent the majority of the transformants. As expected, PCR verification showed
the presence of the wild-type zmrr gene on the chromosome of cells not expressing Cas9,
while cells that were expressing Cas9 no longer contained the wild-type zmrr allele
(Figure 4.6). However, when Cas9 was expressed, the chloramphenicol resistance gene
was incorporated at the zmrr allele instead of the spectinomycin resistance gene, as
expected, for reasons unknown. Collectively, because cells expressing Cas9 lost the wild-
type allele and cells without Cas9 did not, our results suggest that the CRISPR/Cas
system is likely useful for genome editing in Z. mobilis.
4.4 Discussion
We attempted to apply modern genetic engineering techniques for use in Z.
mobilis. We found that the proteins of the λ Red recombinase system are able to increase
the frequency of desirable double crossover events when used to mediate gene knockouts
in the strain. We also found that the yeast flippase enzyme is very efficient at removing
genes flanked by FRT sites from the Z. mobilis chromosome. Unfortunately, we were
unable to provide evidence for ssDNA oligonucleotide incorporation into the
chromosome of the strain. We did, however, provide evidence of tracrRNA:crRNA-
directed Cas9 cleavage of the chromosome.

95
The proteins of the λ Red system have been used to mediate allelic exchange in a
variety of organisms (Datsenko and Wanner 2000; Katashkina et al. 2009; Lesic and
Rahme 2008; Yamamoto et al. 2009). While our results suggest that the proteins are
active in Z. mobilis, they also suggest that the system is not highly efficient in the strain.
To potentially explain this result, we first note that Z. mobilis is known to contain highly
active type I and type IV DNA restriction-modification (R-M) systems (Kerr et al. 2011).
These systems efficiently degrade any foreign DNA that enters the cell, including our
dsDNA cassettes, and are the primary reason why the general transformation efficiency
of Z. mobilis is so low (Kerr et al. 2011). In this study, we constructed a strain of Z.
mobilis (KLD8) deficient in a putative methylated adenine recognition and restriction
(mrr) gene, zmrr. The zmrr gene is a member of the type IV R-M system. It is possible
that using KLD9 as a host strain for λ Red-mediated knockouts would improve the
efficiency of the system in Z. mobilis, by decreasing the likelihood that the dsDNA
cassette is degraded immediately upon entry into the cell. Further, strains deficient in the
type I R-M genes identified by Kerr and coworkers (Kerr et al. 2011) may also serve as
better Z. mobilis λ Red recombineering hosts.
More importantly, we also note that the λ Red Gamma protein is responsible for
inhibiting the host RecBCD helicase-nuclease complex from degrading linear DNA
(Karu et al. 1975). Z. mobilis does not contain RecBCD and instead contains the
functionally similar AddAB complex (Cromie 2009). The Gamma protein is specifically
able to inhibit the RecB portion of RecBCD (Marsic et al. 1993). The AddA protein of
the AddAB complex shares homology with RecB (Haijema et al. 1996). Additionally,
chemical inhibitors of the RecBCD complex are also able to inhibit the AddAB complex

96
(Amundsen et al. 2012), and recombineering in other alternative hosts was shown to
occur without a Gamma-equivalent function (Datta et al. 2008). Therefore, it was
reasonable to believe that the λ Red Gamma protein might also display activity toward
AddA, or that the system would still function efficiently if it did not. Our results,
however, suggest that this is likely not the case. Attempts to use the λ Red recombinase
system in Bacillus subtilis, another bacterium that contains AddAB instead of RecBCD,
were also met with minimal success (Wang et al. 2012). Therefore, an obvious step in
using the λ Red system in Z. mobilis was to attempt the process in our addA knockout
strain, KLD5. Unfortunately, however, attempts to do so were met with limited success
due to the low viability of cells deficient in AddA. Therefore, adding small-molecule
inhibitors of AddAB, such as those identified by Amundsen and coworkers (Amundsen et
al. 2012), to the λ Red recombinase transformation mixture might be a viable alternative
to removing addA from the Z. mobilis chromosome.
Although the λ Red expression system constructed here did not lead to knockout
efficiencies in Z. mobilis that rival those seen in E. coli, it is quite possible that the system
can be used to improve knockout and integration efficiencies in other bacterial hosts. In
our design, the three λ Red proteins are expressed on a broad-host range vector, whose
origin has been shown to be stably maintained in E. coli, Bordetella pertussis, Vibrio
cholera, Rhizobium meliloti, and Pseudomonas putida, among others (Antoine and Locht
1992). On pKLD19, the proteins are expressed from an engineered lac promoter that has
also been shown to function in a great diversity of bacteria (Khan et al. 2008).
Frequently, the λ Red proteins are lethal when expressed too highly (Katashkina et al.

97
2009; Sergueev et al. 2001). Because the lac promoter is inducible, the protein expression
level can also be optimized for the host of interest using our system.
Efficiently removing antibiotic resistance cassettes from the Z. mobilis
chromosome using yeast flippase is a very useful technique that came from this work. Z.
mobilis is naturally resistant to many commonly used antibiotics, including ampicillin,
gentamycin, and kanamycin, among others (Swings and De Ley 1977). Our experience
shows that only three antibiotics can be reliably used in the strain: chloramphenicol,
spectinomycin, and tetracycline. Another lab reports that even fewer antibiotics can be
used for their strain of Z. mobilis (Kerr et al. 2011). This is an issue when multiple
genetic modifications are to be made to a single chromosome; the number of
modifications is limited to the number of available antibiotics, in general. However, when
antibiotic resistance cassettes are removed from the chromosome with flippase, they can
be recycled and used again for the construction of additional chromosomal modifications.
Further, the removal of antibiotic resistances is also very important if a strain is going to
be used industrially (de Lorenzo 1992). Separately, Zou and coworkers also showed that
flippase is highly functional in Z. mobilis, thereby confirming our positive results (Zou et
al. 2012).
While the efficiency of λ Red-mediated chromosomal modifications using
dsDNA is low in certain species of bacteria, attempts to modify genomes using ssDNA
oligonucleotides have been met with greater rates of success. For example, the genome of
B. subtilis has been successfully modified with ssDNA and the λ Red Beta protein but not
with dsDNA and the three λ Red proteins (Wang et al. 2012). In general, the efficiency of
ssDNA incorporation into the chromosome is 10-100 times greater than the efficiency of

98
dsDNA incorporation (Ellis et al. 2001). Therefore, it was reasonable to believe that
ssDNA recombineering would work in Z. mobilis. However, we were unable to provide
evidence that this was the case. To attempt to explain this result, we note that the
efficiency of ssDNA incorporation in E. coli was improved after endogenous nucleases
were removed from the chromosome (Mosberg et al. 2012). This implies that expression
of the λ Red Beta protein alone is not enough for total ssDNA evasion of host nucleases,
even in E. coli. The low transformation efficiency as well as the to-date phage resistance
of Z. mobilis suggests that host nucleases in this strain are highly active, making it even
less likely that the λ Red Beta protein will be sufficient at protecting ssDNA until it is
incorporated into the chromosome. To overcome this issue and improve host nuclease
evasion, phosphorothioation of oligonucleotides has proven useful (Wang et al. 2011),
and alternatives to the Beta protein have been used that are more efficient at protecting
ssDNA in alternative hosts (Datta et al. 2008). Secondly, we also note that it is possible
that 60 bp ssDNA oligonucleotides are too short for incorporation into the Z. mobilis
chromosome. Previous research has shown that longer oligonucleotides are incorporated
with higher efficiencies (DiCarlo et al. 2013a; Wang et al. 2009), and incorporation into
the B. subtilis chromosome required single-stranded PCR products with two regions of
homology (Wang et al. 2012). Attempting ssDNA recombineering in Z. mobilis with
longer oligonucleotides may lead to positive results.
A system with even broader applicability compared to the λ Red recombinase
system is the CRISPR/Cas adaptive immunity system. This system does not require the
use of selective markers, and it has been shown to function in an even greater range of
species than λ Red—Cas systems have been used successfully in bacteria, fungi, plants,

99
mice, and even human cells (DiCarlo et al. 2013b; Jiang et al. 2013; Mali et al. 2013;
Shan et al. 2013; Wang et al. 2013). The broad functionality of the CRISPR-associated
system arises because the Cas proteins and small RNAs do not interact with specific host
proteins, and instead only interact with DNA (Hwang et al. 2013). Here, we showed
evidence that two small RNAs are able to direct Cas9 cleavage of the Z. mobilis
chromosome. We also showed that the Cas system is likely able to mediate dsDNA
crossover into the Z. mobilis chromosome. These results are promising for the future
genome editing of Z. mobilis—nearly all genetic techniques can be improved through the
action of Cas9. Specifically, gene knockouts, integrations, point mutations, inversions,
and internal deletions and insertions can all be mediated with CRISPR/Cas genes. An
important next step for the system in Z. mobilis is to show that the efficiency is high
enough to allow for marker-free genome engineering. Combining the action of the λ Red
proteins with the CRISPR/Cas system may prove useful for constructing these marker-
free mutants (Jiang et al. 2013).
Collectively, our results provide a strong groundwork for the future engineering
of Z. mobilis for the production of biofuels and other value-added products. We have
attempted dsDNA λ Red recombineering, ssDNA λ Red recombineering, and
CRISPR/Cas-mediated genome engineering in the strain and have provided positive
results for two of the three. It is imperative that Z. mobilis be made more amenable to
genetic manipulation if it is ever to be accepted as an industrial host. The systems and
results reported here can be used toward achieving this goal.
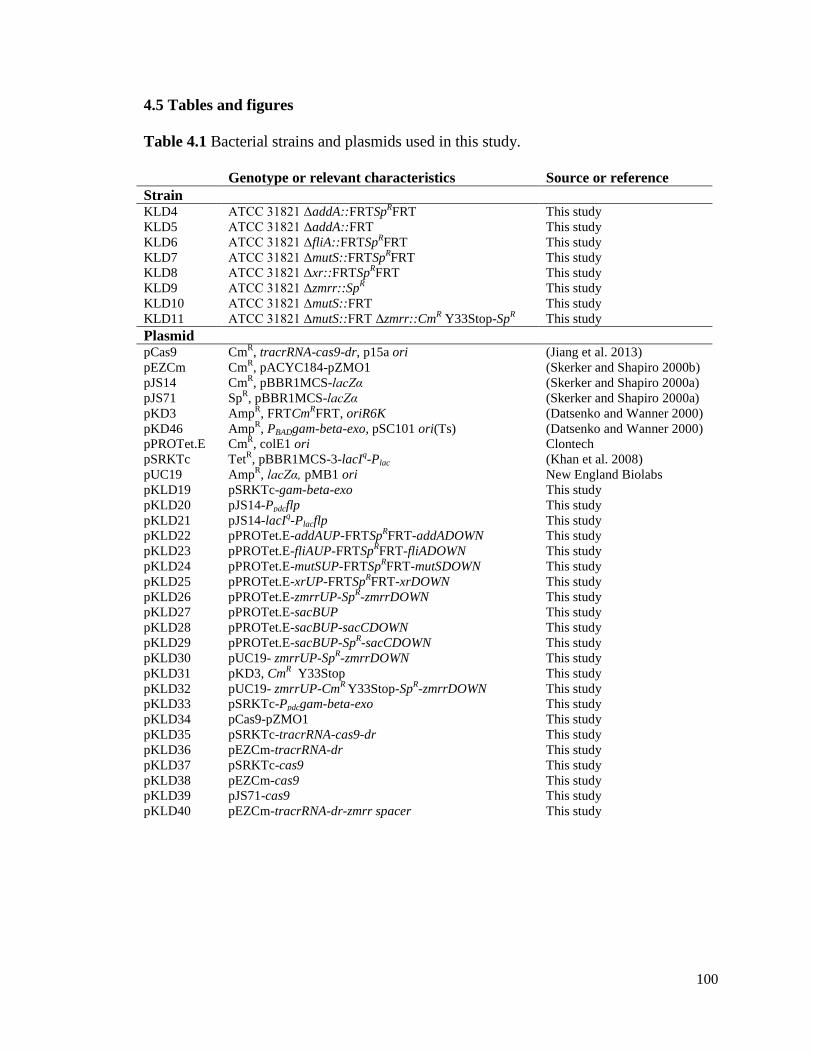
100
4.5 Tables and figures
Table 4.1 Bacterial strains and plasmids used in this study.
Genotype or relevant characteristics Source or reference
Strain KLD4 ATCC 31821 ΔaddA::FRTSp
RFRT This study
KLD5 ATCC 31821 ΔaddA::FRT This study
KLD6 ATCC 31821 ΔfliA::FRTSpRFRT This study
KLD7 ATCC 31821 ΔmutS::FRTSpRFRT This study
KLD8 ATCC 31821 Δxr::FRTSpRFRT This study
KLD9 ATCC 31821 Δzmrr::SpR This study
KLD10 ATCC 31821 ΔmutS::FRT This study
KLD11 ATCC 31821 ΔmutS::FRT Δzmrr::CmR Y33Stop-Sp
R This study
Plasmid pCas9 Cm
R, tracrRNA-cas9-dr, p15a ori (Jiang et al. 2013)
pEZCm CmR, pACYC184-pZMO1 (Skerker and Shapiro 2000b)
pJS14 CmR, pBBR1MCS-lacZα (Skerker and Shapiro 2000a)
pJS71 SpR, pBBR1MCS-lacZα (Skerker and Shapiro 2000a)
pKD3 AmpR, FRTCm
RFRT, oriR6K (Datsenko and Wanner 2000)
pKD46 AmpR, PBADgam-beta-exo, pSC101 ori(Ts) (Datsenko and Wanner 2000)
pPROTet.E CmR, colE1 ori Clontech
pSRKTc TetR, pBBR1MCS-3-lacI
q-Plac (Khan et al. 2008)
pUC19 AmpR, lacZα, pMB1 ori New England Biolabs
pKLD19 pSRKTc-gam-beta-exo This study
pKLD20 pJS14-Ppdcflp This study
pKLD21 pJS14-lacIq-Placflp This study
pKLD22 pPROTet.E-addAUP-FRTSpRFRT-addADOWN This study
pKLD23 pPROTet.E-fliAUP-FRTSpRFRT-fliADOWN This study
pKLD24 pPROTet.E-mutSUP-FRTSpRFRT-mutSDOWN This study
pKLD25 pPROTet.E-xrUP-FRTSpRFRT-xrDOWN This study
pKLD26 pPROTet.E-zmrrUP-SpR-zmrrDOWN This study
pKLD27 pPROTet.E-sacBUP This study
pKLD28 pPROTet.E-sacBUP-sacCDOWN This study
pKLD29 pPROTet.E-sacBUP-SpR-sacCDOWN This study
pKLD30 pUC19- zmrrUP-SpR-zmrrDOWN This study
pKLD31 pKD3, CmR Y33Stop This study
pKLD32 pUC19- zmrrUP-CmR
Y33Stop-SpR-zmrrDOWN This study
pKLD33 pSRKTc-Ppdcgam-beta-exo This study
pKLD34 pCas9-pZMO1 This study
pKLD35 pSRKTc-tracrRNA-cas9-dr This study
pKLD36 pEZCm-tracrRNA-dr This study
pKLD37 pSRKTc-cas9 This study
pKLD38 pEZCm-cas9 This study
pKLD39 pJS71-cas9 This study
pKLD40 pEZCm-tracrRNA-dr-zmrr spacer This study
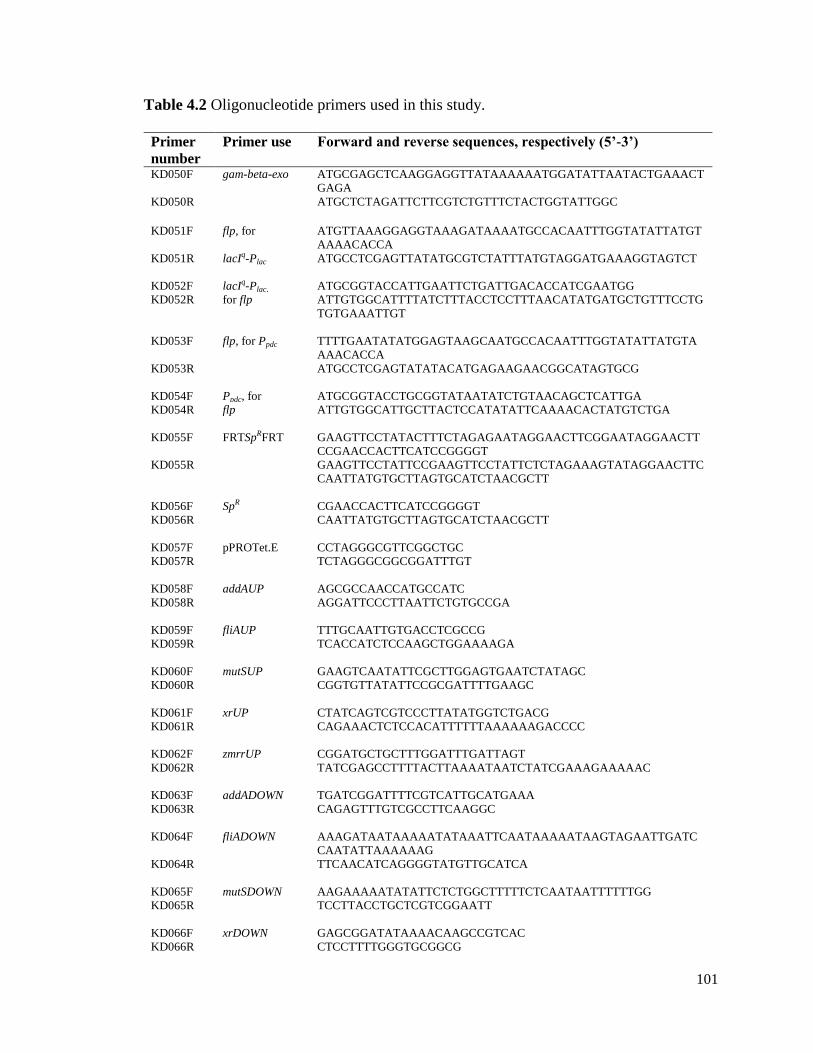
101
Table 4.2 Oligonucleotide primers used in this study.
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD050F gam-beta-exo ATGCGAGCTCAAGGAGGTTATAAAAAATGGATATTAATACTGAAACT
GAGA
KD050R ATGCTCTAGATTCTTCGTCTGTTTCTACTGGTATTGGC
KD051F flp, for ATGTTAAAGGAGGTAAAGATAAAATGCCACAATTTGGTATATTATGT
AAAACACCA
KD051R lacIq-Plac ATGCCTCGAGTTATATGCGTCTATTTATGTAGGATGAAAGGTAGTCT
KD052F lacIq-Plac, ATGCGGTACCATTGAATTCTGATTGACACCATCGAATGG
KD052R for flp ATTGTGGCATTTTATCTTTACCTCCTTTAACATATGATGCTGTTTCCTG
TGTGAAATTGT
KD053F flp, for Ppdc TTTTGAATATATGGAGTAAGCAATGCCACAATTTGGTATATTATGTA
AAACACCA
KD053R ATGCCTCGAGTATATACATGAGAAGAACGGCATAGTGCG
KD054F Ppdc, for ATGCGGTACCTGCGGTATAATATCTGTAACAGCTCATTGA
KD054R flp ATTGTGGCATTGCTTACTCCATATATTCAAAACACTATGTCTGA
KD055F FRTSpRFRT GAAGTTCCTATACTTTCTAGAGAATAGGAACTTCGGAATAGGAACTT
CCGAACCACTTCATCCGGGGT
KD055R GAAGTTCCTATTCCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC
CAATTATGTGCTTAGTGCATCTAACGCTT
KD056F SpR CGAACCACTTCATCCGGGGT
KD056R CAATTATGTGCTTAGTGCATCTAACGCTT
KD057F pPROTet.E CCTAGGGCGTTCGGCTGC
KD057R TCTAGGGCGGCGGATTTGT
KD058F addAUP AGCGCCAACCATGCCATC
KD058R AGGATTCCCTTAATTCTGTGCCGA
KD059F fliAUP TTTGCAATTGTGACCTCGCCG
KD059R TCACCATCTCCAAGCTGGAAAAGA
KD060F mutSUP GAAGTCAATATTCGCTTGGAGTGAATCTATAGC
KD060R CGGTGTTATATTCCGCGATTTTGAAGC
KD061F xrUP CTATCAGTCGTCCCTTATATGGTCTGACG
KD061R CAGAAACTCTCCACATTTTTTAAAAAAGACCCC
KD062F zmrrUP CGGATGCTGCTTTGGATTTGATTAGT
KD062R TATCGAGCCTTTTACTTAAAATAATCTATCGAAAGAAAAAC
KD063F addADOWN TGATCGGATTTTCGTCATTGCATGAAA
KD063R CAGAGTTTGTCGCCTTCAAGGC
KD064F fliADOWN AAAGATAATAAAAATATAAATTCAATAAAAATAAGTAGAATTGATC
CAATATTAAAAAAG
KD064R TTCAACATCAGGGGTATGTTGCATCA
KD065F mutSDOWN AAGAAAAATATATTCTCTGGCTTTTTCTCAATAATTTTTTGG
KD065R TCCTTACCTGCTCGTCGGAATT
KD066F xrDOWN GAGCGGATATAAAACAAGCCGTCAC
KD066R CTCCTTTTGGGTGCGGCG

102
Table 4.2 (cont.)
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD067F zmrrDOWN GCTTTTTGAAAAAGCCGTTTCCTGTATTG
KD067R TTACCAGCATGTTTTATCAGGAATCGGA
KD068F pPROTet.E-
addAUP bridge
ACAAATCCGCCGCCCTAGAAGCGCCAACCATGCCATC
KD068R addAUP-
FRTSpRFRT
bridge
ATCGGCACAGAATTAAGGGAATCCTGAAGTTCCTATACTTTCTAGAG
AATAGGAACTTCG
KD069F FRTSpRFRT-
addADOWN
bridge
AGAGAATAGGAACTTCGGAATAGGAACTTCTGATCGGATTTTCGTCA
TTGCATGAAATG
KD069R addADOWN-
pPROTet.E
bridge
GCCTTGAAGGCGACAAACTCTGCCTAGGGCGTTCGGCTGC
KD070F pPROTet.E-
fliAUP bridge
ACAAATCCGCCGCCCTAGATTTGCAATTGTGACCTCGCCG
KD070R fliAUP-
FRTSpRFRT
bridge
TCTTTTCCAGCTTGGAGATGGTGAGAAGTTCCTATACTTTCTAGAGAA
TAGGAACTTCG
KD071F FRTSpRFRT-
fliADOWN bridge
AGAGAATAGGAACTTCGGAATAGGAACTTCAAAGATAATAAAAATA
TAAATTCAATAAAA
KD071R fliADOWN-
pPROTet.E
bridge
TGATGCAACATACCCCTGATGTTGAACCTAGGGCGTTCGGCTGC
KD072F pPROTet.E-
mutSUP bridge
ACAAATCCGCCGCCCTAGAGAAGTCAATATTCGCTTGGAGTGAATCT
ATAGC
KD072R mutSUP-
FRTSpRFRT
bridge
CTTCAAAATCGCGGAATATAACACCGGAAGTTCCTATACTTTCTAGA
GAATAGGAACTT
KD073F FRTSpRFRT-
mutSDOWN
bridge
AGAATAGGAACTTCGGAATAGGAACTTCAAGAAAAATATATTCTCTG
GCTTTTTCTCAAT
KD073R mutSDOWN-
pPROTet.E
bridge
AATTCCGACGAGCAGGTAAGGACCTAGGGCGTTCGGCTGC
KD074F pPROTet.E-xrUP
bridge
ACAAATCCGCCGCCCTAGACTATCAGTCGTCCCTTATATGGTCTGAC
G
KD074R xrUP-SpR bridge GGGGTCTTTTTTAAAAAATGTGGAGAGTTTCTGCGAACCACTTCATCC
GGGGT
KD075F SpR-xrDOWN
bridge
AAGCGTTAGATGCACTAAGCACATAATTGGAGCGGATATAAAACAA
GCCGTCAC
KD075R xrDOWN-
pPROTet.E
bridge
CGCCGCACCCAAAAGGAGCCTAGGGCGTTCGGCTGC
KD076F pPROTet.E-
zmrrUP bridge
ACAAATCCGCCGCCCTAGACGGATGCTGCTTTGGATTTGATTAGT
KD076R zmrrUP-SpR
bridge
TTTTTCTTTCGATAGATTATTTTAAGTAAAAGGCTCGATACGAACCAC
TTCATCCGGGGT

103
Table 4.2 (cont.)
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD077F SpR-zmrrDOWN
bridge
AAGCGTTAGATGCACTAAGCACATAATTGGCTTTTTGAAAAAGCCGT
TTCCTGTATTG
KD077R zmrrDOWN-
pPROTet.E
bridge
TCCGATTCCTGATAAAACATGCTGGTAACCTAGGGCGTTCGGCTGC
KD078F sacBUP ATGCGAATTCCAAGATTTTTGACAGACATCACCGAATGAT
KD078R ATGCGGTACCAAGAACTATCCTAATAAATGATGAAATAAAGCCGAA
AA
KD079F sacCDOWN ATGCGTCGACTTTTTGACGACAAAAAATTGCGCTGAAA
KD079R ATGCGGATCCCTAAAAATCTGAGCGATTTTTCGGGTGT
KD080F SpR ATGCGGTACCCGAACCACTTCATCCGGGGT
KD080R ATGCGTCGACCAATTATGTGCTTAGTGCATCTAACGCTT
KD081F pUC19 GGCGTAATCATGGTCATAGCTGTTTC
KD081R ACTGGCCGTCGTTTTACAACGT
KD082F KpnI BamHI GGTACCGGATCCCGAACCACTTCATCCGGGGT
KD082R SpR CAATTATGTGCTTAGTGCATCTAACGCTT
KD083F pUC19-zmrrUP
bridge
ACGTTGTAAAACGACGGCCAGTCGGATGCTGCTTTGGATTTGATTAG
TG
KD083R zmrrUP-KpnI
BamHI SpR
bridge
TTTTTCTTTCGATAGATTATTTTAAGTAAAAGGCTCGATAGGTACCGG
ATCCCGAACCAC
KD084F KpnI BamHI SpR-
zmrrDOWN
bridge
AAGCGTTAGATGCACTAAGCACATAATTGGCTTTTTGAAAAAGCCGT
TTCCTGTATTG
KD084R zmrrDOWN-
pUC19 bridge
ATCCGATTCCTGATAAAACATGCTGGTAAGGCGTAATCATGGTCATA
GCTGTTTC
KD085F pKD3 CmR TGTACCTAAAACCAGACCGTTCAGC
KD085R Y33Stop TTGAGCAACTGACTGAAATGCCTCA
KD086F CmRY33Stop ATGCGGTACCTGAGACGTTGATCGGCACGTA
KD086R ATGCGGATCCATTTAAATGGCGCGCCTTACGC
KD087F gam-beta-exo, for
Ppdc
TGTTTTGAATATATGGAGTAAGCAATGGATATTAATACTGAAACTGA
GATCAAGCAAAAG
KD087R ATGCCTCGAGTTCTTCGTCTGTTTCTACTGGTATTGGC
KD088F Ppdc, for ATGCGGTACCTGCGGTATAATATCTGTAACAGCTCATTGA
KD088R gam-beta-exo TAATATCCATTGCTTACTCCATATATTCAAAACACTATGTCTGA
KD089F pZMO1 ATGCTCTAGACGCGAACGGACAAGCCC
KD089R ATGCATCGATGGCAGGAGTGCAGGAAATAGCT
KD090F tracrRNA- ATGCTCTAGATATCTCTTCAAATGTAGCACCTGAAGTCAG
KD090R cas9-dr ATGCCTCGAGGCGATGCTGTCGGAATGGAC
KD091F dr, for ATGCTCTAGACTCTTAATAAATGCAGTAATACAGGGGCTTTTC
KD091R tracrRNA TAACTGTGATTAACCCTCTTTCTCAAGTTATCATCGGC

104
Table 4.2 (cont.)
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD092F tracrRNA, for dr TGATAACTTGAGAAAGAGGGTTAATCACAGTTAAATTGCTAACGCA
GTCAG
KD092R ATGCTCTAGATGATGATTTCAATAATAGTTTTAATGACCTCCGAAAT
T
KD093F cas9 ATGCTCTAGAAAAAAGAAAAATTAAAGCATATTAAACTAATTTCGG
AGGTC
KD093R ATGCCTCGAGATACCATATTTTTAGTTATTAAGAAATAATCTTCATC
TAAAATATACTTC
KD094F zmrr spacer AAACTGGTGGCTATGGGCTATGGCGGCAATCATGG
KD094R AAAACCATGATTGCCGCCATAGCCCATAGCCACCA
KD095F ΔaddA::FRT
SpRFRT
TTTTGATGAATGGCGCAGTAATCCC
KD095R check AGTATGATCTAATAGAAATACCATTTCATGCAATGACG
KD096F ΔfliA::FRT
SpRFRT
GCTGTCTGGATTTATTCCCCAGATCA
KD096R check AAATAAATCTTTTGATAGATAATAGCTTTTTTAATATTGGATCAATT
CTACT
KD097F ΔmutS::FRT
SpRFRT
ACTGTCAAAAAGACTAAAGCTGCAAAGAA
KD097R check TCACTTATTCCAAAAAATTATTGAGAAAAAGCCAGA
KD098F Δxr::SpR TTAAAGGCGGATCAACAGGCCA
KD098R check CGACAGTGACGGCTTGTTTTATATCC
KD099F Δzmrr::SpR ATTCTGTCGCCGGATAGGCG
KD099R check GCCAGAATCCTAACAATACAGGAAACG
KD100F ΔsacBC::SpR CTTTTTTCGGCTTTATTTCATCATTTATTAGGATAGTTCTT
KD100R check TTCAGCGCAATTTTTTGTCGTCAAAAA
KD101F CmRY33Stop
repair
GGCATTTCAGTCAGTTGCTCAATGTACCTATAACCAGACCGTTCAGC
TGGATATTACGGC
KD101R fliAL33Stop AAGAATGTCGAAGCGTTAATCAAAACCCATTAACAACTGGTTCGGA
AAATTGCATGGCAT
KD102F frkE33Stop ATTGATTCTGACCGGAAGATGCTGGCTGTGTAACGTGTTCCGACCAC
AACCCCTGAAGAA

105
Table 4.3 Z. mobilis λ Red-mediated knockout target genes.
Gene Description Protein significance ZMO1098 addA- double-strand break repair helicase RecB analogue
ZMO0626 fliA- flagellar-specific sigma factor Regulates motility
ZMO1907 mutS- DNA mismatch repair protein Reduces ssDNA recombineering efficiency
ZMO0374-5 sacBC- levansucrases Required for sucrose utilization
ZMO0976 xr- aldo/keto reductase Contributes to xylitol production
ZMO0028 zmrr- restriction endonuclease Decreases transformation efficiency
Table 4.4 Frequency of ΔsacBC::SpR double crossover events in cells expressing
pKLD19 with IPTG added (Induced) or without IPTG (Uninduced).
Percent of colonies tested
displaying the correct genotype
Induced 90 ± 2
Uninduced 19 ± 5
Table 4.5 Colony forming units obtained in the presence or absence of Cas9. Cells with
pKLD39 (+Cas9) and without pKLD39 (-Cas9) were transformed with pKLD40. When
Cas9 is present, the tracrRNA:crRNA complex directs it to cleave the chromosome,
leading to cell death.
Colony forming units
+Cas9 0 or 1
-Cas9 48 ± 11

106
Figure 4.1 Schematic representations of plasmids. a Plasmid used to express the λ Red
recombinase system. b Plasmid used to express the tracrRNA:crRNA complex. c One of
three plasmids used to express the Cas9 protein.

107
Figure 4.2 Single versus double crossover events. a When DNA is incorporated into the
chromosome through a single crossover event, the entire circular vector is integrated into
the chromosome. b When DNA is incorporated into the chromosome through a double
crossover event, the gene of interest (zmrr) is removed from the chromosome, leading to
a gene knockout.

108
Figure 4.3 PCR verification of knockouts. Control DNA yielded a PCR product the
length of the wild-type allele. Desirable double knockout mutants yielded a PCR product
the length of the spectinomycin resistance gene, 1.5 kb. Single crossover mutants yielded
two PCR products, one the length of the wild-type allele and one the length of the
spectinomycin resistance gene, 1.5 kb. Results shown are for the zmrr gene, whose wild-
type PCR product is 1 kb in length.
Figure 4.4 Phenotypic verification of the Z. mobilis ΔfliA::FRTSpRFRT strain. The FliA
protein is a transcriptional regulator of motility. When it is absent, cells are no longer
motile. Motility loss can be detected using a simple plate assay. a Wild-type Z. mobilis
control cells swim toward the edge of the plate. b Z. mobilis ΔfliA::FRTSpRFRT cells
cannot swim.
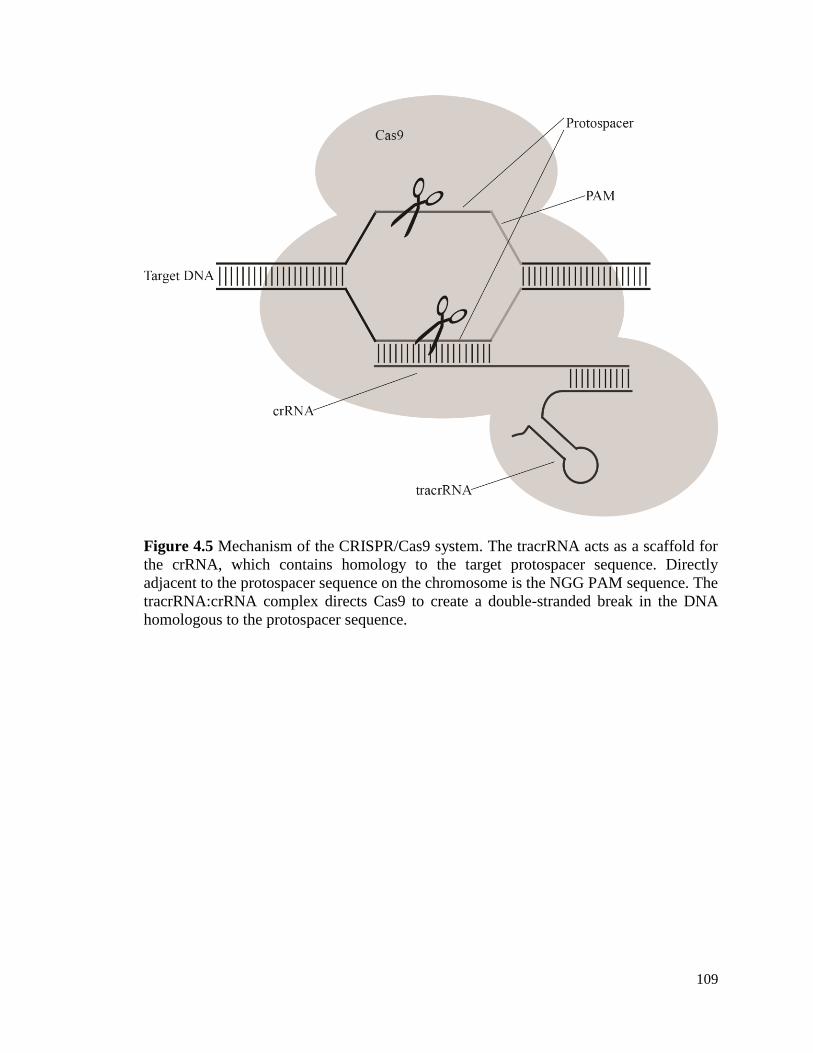
109
Figure 4.5 Mechanism of the CRISPR/Cas9 system. The tracrRNA acts as a scaffold for
the crRNA, which contains homology to the target protospacer sequence. Directly
adjacent to the protospacer sequence on the chromosome is the NGG PAM sequence. The
tracrRNA:crRNA complex directs Cas9 to create a double-stranded break in the DNA
homologous to the protospacer sequence.

110
Figure 4.6 PCR verification of CRISPR/Cas9-mediated knockouts. Cells expressing
pKLD37 (+Cas9) no longer contain the wild-type zmrr allele, confirmed by the lack of a
PCR product at 1 kb. Cells lacking pKLD37 (-Cas9) are unsuccessful knockouts and still
yield a 1 kb zmrr PCR product.

111
Chapter 5: Development of a promoter library for use in Z.
mobilis
5.1 Introduction
In chapter 4, we attempted to improve upon the genetic techniques used for
modifying the chromosome of Z. mobilis. In addition to chromosomal modifications, the
efficient production of heterologous proteins will also be important if Z. mobilis is ever to
be accepted as a host for alternative product synthesis. Efficient protein production
requires the precise control of heterologous gene expression at the transcriptional level.
Currently, no set of Z. mobilis gene promoters of varying strengths exists for this
purpose.
Typically, Z. mobilis native promoters, such as the gap, eno, and pdc promoters
are used to express heterologous enzymes in this host (Deanda et al. 1996b; Zhang et al.
1995). These promoters are strong and constitutive, so expression from them is
uncontrolled and not optimized. The inducible tac promoter, which is a hybrid of the trp
and lac UV5 promoters, has been shown to function in Z. mobilis for the controlled
expression of heterologous genes (de Boer et al. 1983). However, the system showed
only partial control and led to a high level of basal expression (Arfman et al. 1992). In
previous chapters, we successfully utilized an engineered lac promoter for protein
expression in Z. mobilis. We saw tightly regulatable expression from this promoter, but
the maximal expression level was considerably lower than what can be achieved with Z.
mobilis native promoters. Therefore, a set of gene promoters of varying strengths
designed for use in Z. mobilis would be a very useful tool set for the future engineering of
the strain.

112
In this chapter, we used error-prone PCR to construct a large promoter library
based on the strong and constitutive Z. mobilis pdc promoter. We then used flow
cytometry to select the candidates displaying the highest level of single-cell homogeneity.
These stable candidates were then characterized using transcriptional fusions to the
Venus and mCherry fluorescent proteins, and transcriptional-level control was confirmed
using qPCR. The final library candidates identified here are useful for pathway
optimization and heterologous protein expression in Z. mobilis.
5.2 Materials and methods
5.2.1 Bacterial strains, media, and growth conditions
All experiments were performed in Zymomonas mobilis subsp. mobilis ZM4
(ATCC 31821). Liquid and solid rich medium (RM) with 10 g/L yeast extract, 2 g/L
potassium phosphate, dibasic supplemented with 20 g/L glucose was used for all Z.
mobilis experiments. All Z. mobilis cultures were grown at 30°C and antibiotics were
used at the following concentrations: spectinomycin, 100 µg/mL; chloramphenicol, 100
µg/mL; and tetracycline, 25 µg/mL.
5.2.2 Plasmid construction
All strains and plasmids used in this study are listed in Table 5.1. All primers
used in this study are listed in Table 5.2. To construct a plasmid for library development,
the pEZCm vector was first amplified by PCR synthesis using primers KD103F and
KD103R such that it contained XbaI and BamHI restriction sites, the Z. mobilis gap
ribosome binding site sequence, and the KpnI and XhoI restriction sites, respectively.
This PCR product was digested with BamHI and self-ligated to form plasmid pKLD40.
The venus fluorescent protein gene was amplified from plasmid pVenus by PCR

113
synthesis using primers KD104F and KD104R and inserted at the KpnI and XhoI sites of
pKLD40 to form plasmid pKLD41. Separately, to construct a vector for additional library
analyses, the mCherry fluorescent protein gene was amplified from plasmid pSZ by PCR
synthesis using primers KD105F and KD105R and also inserted at the KpnI and XhoI
sites of pKLD40 to form plasmid pKLD42.
To construct a vector containing the pdc promoter for use as an error-prone PCR
template, the pdc promoter was amplified by PCR synthesis using primers KD106F and
KD106R from Z. mobilis ZM4 genomic DNA. This promoter was inserted into the
plasmid pJS71 to yield plasmid pKLD43.
To construct vectors for pdc promoter analysis upstream of mCherry, promoters
pdc 1-8 were amplified by PCR synthesis from vectors pKLD44-pKLD51, respectively,
using primers KD107F and KD107R. The promoters were then inserted separately into
plasmid pKLD42 to form plasmids pKLD52-pKLD59, respectively.
To construct vectors for testing inducible promoters in Z. mobilis, the venus
fluorescent protein gene was amplified by PCR synthesis to include its ribosome binding
site using primers KD108F and KD108R. The venus gene fragment was then inserted into
pSRKTc downstream of the engineered lac promoter to form plasmid pKLD60.
Separately, the araC-ParaBAD region was amplified from E. coli MG1655 genomic DNA
by PCR synthesis using primers KD109F and KD109R. The venus gene was amplified
from pVenus by PCR synthesis using primers KD110F and KD110R. The araC-ParaBAD
and venus gene fragements were then joined together by overlap extension PCR to yield
the araC-ParaBADvenus gene fragment. This fragment was then inserted into plasmid
pJS14 to yield plasmid pKLD61.

114
5.2.3 Library construction
A library of pdc promoter mutants was constructed using error-prone PCR.
Specifically, plasmid pKLD43 was used as template for nucleotide analogue mutagenesis
in the presence of 20 μM 8-oxo-2’deoxyguanosine (8-oxo-dGTP) and 6-(2-deoxy-β-D-
ribofuranosyl)-3,4-dihydro-8H-pyrimido-[4,5-c][1,2]oxazin-7-one (dPTP) (TriLink
Biotechnologies) (Zaccolo and Gherardi 1999) using primers KD111F and KD111R.
These primers bound to pKLD43 such that the entire 150 bp pdc promoter length could
be mutagenized. Reactions were carried out in a 50 μL volume using GoTaq® master
mix (Promega). An annealing temperature of 55ºC was used and 30 amplification cycles
were performed. The PCR products were then cleaned and digested for insertion between
the XbaI and BamHI sites of plasmid pKLD41. After ligation, the product was
transformed into electrocompetent DH5α. The entire library of colonies was transferred
to 50 mL liquid LB medium using a sterile loop and grown to mid-exponential phase, at
which time 10 mL was withdrawn from the culture for plasmid isolation. The isolated
library plasmid miniprep was then transformed into electrocompetent E. coli strain
GM119. The entire library was again transferred to 50 mL liquid LB medium using a
sterile loop and grown to mid-exponential phase, at which time 10 mL was withdrawn
from the culture for plasmid isolation and subsequent transformation into
electrocompetent Z. mobilis.
5.2.4 Fluorescence measurements
Colonies containing library candidates were transferred to 1 mL RM or LB media
in a single well of a polypropylene 2.2 mL 96-well deep microtiter plate (VWR) and
grown with shaking at 600 rpm overnight. At late exponential phase, cells were

115
subcultured 1:100 in 1 mL RM or LB media in a single well of a polypropylene 2.2 mL
96-well deep microtiter plate and grown with shaking at 600 rpm for 18 h (Z. mobilis) or
8 h (E. coli), at which time 150 μL of culture was transferred to a single well of a black-
walled, clear-bottomed Costar 96-well microtiter plate. For final analysis of the refined 8-
member library, cells were grown in 2 mL RM in 5 mL glass tubes with shaking for 18 h
instead of in a 96-well deep microtiter plate. The fluorescence (Venus Ex/Em: 515/528
nm, mCherry Ex/Em: 587/610 nm) and optical density (600 nm) were measured using a
Tecan Safire2 microplate reader. Fluorescence measurements are reported as the relative
fluorescence normalized to the optical density of the sample to correct for variation in
cell density. Fluorescences normalized to the optical density were then normalized to the
fluorescence value obtained from the wild-type pdc promoter.
5.2.5 Flow cytometry experiments
A subset of 25 Z. mobilis library members were selected with a wide range of
Venus fluorescence levels for flow cytometry analysis, to ensure that bulk culture
behavior accurately represented the fluorescence seen at the single-cell level. For a
typical flow cytometry experiment, cells were grown overnight in RM containing
chloramphenicol, then subcultured to an OD600nm of 0.05 and again grown overnight. At
late-exponential phase, cells were harvested by centrifugation and washed once with
PBS. Cells were then resuspended in PBS and analyzed using a BD LSR II flow
cytometer. Fluorescence values for approximately 500,000 cells were recorded using the
fluorescein isothiocyanate (FITC) channel (excitation, 488 nm; emission, 530/30 nm).
The eight candidates with clean monovariate distributions were selected from the original

116
25 for further analysis. These candidates were designated promoters pdc 1-8, in vectors
pKLD44-pKLD51, respectively.
5.2.6 Quantitative PCR (qPCR) reactions
Total RNA was isolated from Z. mobilis strains grown to mid-exponential phase
using the Qiagen RNeasy Mini Kit according to the manufacturer’s instructions. cDNA
was generated from RNA using the Qiagen QuantiTect Reverse Transcription Kit
according to the manufacturer’s instructions. PCR amplification of cDNA using primers
KD112F, KD112R, KD045F, and KD045R (Dunn and Rao 2015), designed using the
Primer3Plus software (Untergasser et al. 2007), was visualized using the HotStart-IT
SYBR Green qPCR Master Mix with UDG (Affymetrix) and a Bio-Rad MiniOpticon
Real-Time PCR System. All results were quantified by PCR cycle-number comparison to
standard curves prepared using E. coli strain AB273 or Z. mobilis ZM4 genomic DNA.
The Z. mobilis rrsA gene was used as a control for normalizing differences in total RNA
quantities (He et al. 2012).
5.3 Results
5.3.1 Error-prone PCR leads to a library of promoters with varying
strengths in Z. mobilis
It is important that gene expression levels be precisely controlled when producing
heterologous proteins in a bacterial host. Inducible promoters are often useful for this
purpose. We tested two inducible promoter systems in Z. mobilis—the lac promoter,
inducible with IPTG, and the araBAD promoter, inducible with arabinose. Using
transcriptional fusions to the venus fluorescent protein gene, we found that even at full
induction these promoters led to only ~30% and ~5% of the activity of the wild-type pdc

117
promoter, respectively. Therefore, to cover the whole range of promoter strengths, we
used nucleotide analogue mutagenesis to construct a library of promoters based on the
strong and constitutive Z. mobilis pdc promoter. This approach is similar to the one used
by Alper and coworkers (Alper et al. 2005). The promoters were cloned upstream of the
venus fluorescent protein gene in a vector designed to facilitate library construction and
analysis (Figure 5.1). The resulting promoter library led to a range of Venus fluorescence
levels in both E. coli and Z. mobilis (Figure 5.2). While the library initially contained
hundreds of members, we found that very few promoter variants led to consistent results
in Z. mobilis. Cells initially displaying a very high level of fluorescence in Z. mobilis
were particularly unstable. Through repeated fluorescence tests and by using flow
cytometry to identify those candidates with homogeneous single-cell fluorescence
distributions, the library was refined to contain 8 variants that led to reproducible results
in Z. mobilis. The DNA sequences of these candidates can be found in Table 5.3. The
promoter strengths of the 8 library members were found to span a 160-fold range when
Venus was used as the reporter (Figure 5.3).
5.3.2 Expression from the library of promoters is constitutive
To verify the constitutive nature of the 8 pdc promoter variants, each was cloned
upstream of the mCherry fluorescent protein gene in the same library vector. Again, we
found that the promoters led to reproducible fluorescence results and behavior consistent
with that seen when venus was used as the reporter gene. It is important to note, however,
that not all Z. mobilis cells will produce mCherry when the vector is present in the cells,
for reasons unknown. Therefore, results shown are for colonies with confirmed

118
fluorescence. With mCherry, the promoter strengths of the 8 library members were found
to span a 140-fold range (Figure 5.4).
5.3.3 Expression-level variation is due to transcriptional differences
To characterize the promoter library at the transcriptional level, we used
quantitative PCR to measure the relative venus transcript levels in Z. mobilis cells
expressing the 8 different library candidates. The mRNA levels (ng) spanned a 256-fold
range, and normalized results showed a high level of correlation with fluorescence results
(Figure 5.5). This confirms that expression-level variation between the 8 library
members is due to transcriptional differences.
5.4 Discussion
Expressing heterologous genes in Z. mobilis under the control of strong and
constitutive native promoters is not always the best option. Therefore, we used error-
prone PCR to construct a library of promoter variants based on the strong and constitutive
Z. mobilis pdc promoter. We found that the library led to a range of venus fluorescence
levels in both E. coli and Z. mobilis. Transcriptional fusions of library members to the
mCherry fluorescent protein led to similar results, confirming the constitutive nature of
the promoters. qPCR results confirmed that fluorescence differences were due to control
at the transcriptional level.
The promoter library constructed here is a very useful toolkit for heterologous
protein production in Z. mobilis. While inducible promoters can also be used to control
gene expression levels, the level of inducer is not homogeneous at the single-cell level,
and adding inducer to large cultures can be expensive (Siegele and Hu 1997). Further, we
have found that typical inducible promoters do not lead to high expression levels in Z.

119
mobilis, even at full induction. Although we only isolated one stable candidate with an
expression level that is higher than that of the wild-type pdc promoter, this result has also
been observed in other alternative hosts (Markley et al. 2015), and the wild-type pdc
promoter expression level is already considerably high. Regardless, the promoter library
is useful for determining the precise impact of gene dosage on a specific phenotype. For
example, by cloning the E. coli transketolase and transaldolase genes under the control of
the 8 different library members and then analyzing the effect each expression level has on
arabinose fermentation by Z. mobilis, we can discern exactly how limiting each enzyme
was in our unadapted arabinose-fermenting strain constructed in chapter 3.
As Z. mobilis gains popularity for alternative product synthesis, the ability to fine-
tune protein expression levels will become increasingly important. The promoter library
constructed here will be useful for this purpose. Should more promoter options be
necessary, the initial library containing hundreds of candidates can be scanned again to
isolate additional stable candidates. Further, error-prone PCR can be performed on other
Z. mobilis native promoters if needed. The entire library can also be scanned for variants
that respond to distinct environmental conditions, if needed for a specific application. As
a whole, our promoter library facilitates the process of constructing synthetic gene
networks in Z. mobilis.

120
5.5 Tables and figures
Table 5.1 Strains and plasmids used in this study.
Genotype or relevant characteristics Source or reference
Strain
AB273 E. coli MG1655 attλ::[kanR PptsGvenus] Ahmet Badur
Plasmid
pEZCm CmR, pACYC184-pZMO1 Jeff Skerker
pJS71 SpR, pBBR1MCS-lacZα (Skerker and Shapiro
2000a)
pSZ CmR, pPROTet.E-PLTet.0-mCherry Shuyan Zhang
pSRKTc TetR, pBBR1MCS-3-lacI
q-Plac (Khan et al. 2008)
pVenus KanR, MCS yfp(venus) t0 attλ, oriR6K (Saini et al. 2009)
pKLD40 pEZCm-gapRBS This study
pKLD41 pEZCm-gapRBSvenus This study
pKLD42 pEZCm-gapRBSmCherry This study
pKLD43 pJS71-Ppdc This study
pKLD44 pEZCm-Ppdc1gapRBSvenus This study
pKLD45 pEZCm-Ppdc2gapRBSvenus This study
pKLD46 pEZCm-Ppdc3gapRBSvenus This study
pKLD47 pEZCm-Ppdc4gapRBSvenus This study
pKLD48 pEZCm-Ppdc5gapRBSvenus This study
pKLD49 pEZCm-Ppdc6gapRBSvenus This study
pKLD50 pEZCm-Ppdc7gapRBSvenus This study
pKLD51 pEZCm-Ppdc8gapRBSvenus This study
pKLD52 pEZCm-Ppdc1gapRBSmCherry This study
pKLD53 pEZCm-Ppdc2gapRBSmCherry This study
pKLD54 pEZCm-Ppdc3gapRBSmCherry This study
pKLD55 pEZCm-Ppdc4gapRBSmCherry This study
pKLD56 pEZCm-Ppdc5gapRBSmCherry This study
pKLD57 pEZCm-Ppdc6gapRBSmCherry This study
pKLD58 pEZCm-Ppdc7gapRBSmCherry This study
pKLD59 pEZCm-Ppdc8gapRBSmCherry This study
pKLD60 pSRKTc-venus This study
pKLD61 pJS14-araC-ParaBADvenus This study
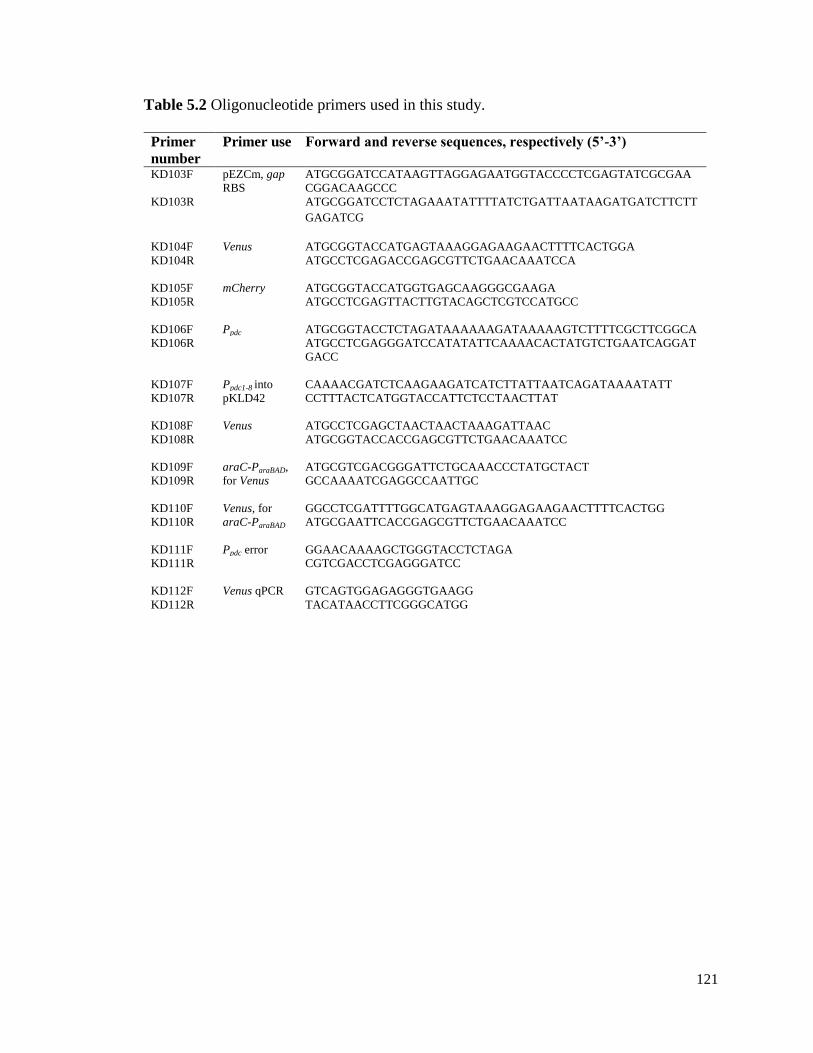
121
Table 5.2 Oligonucleotide primers used in this study.
Primer
number
Primer use Forward and reverse sequences, respectively (5’-3’)
KD103F pEZCm, gap
RBS
ATGCGGATCCATAAGTTAGGAGAATGGTACCCCTCGAGTATCGCGAA
CGGACAAGCCC
KD103R ATGCGGATCCTCTAGAAATATTTTATCTGATTAATAAGATGATCTTCTT
GAGATCG
KD104F Venus ATGCGGTACCATGAGTAAAGGAGAAGAACTTTTCACTGGA
KD104R ATGCCTCGAGACCGAGCGTTCTGAACAAATCCA
KD105F mCherry ATGCGGTACCATGGTGAGCAAGGGCGAAGA
KD105R ATGCCTCGAGTTACTTGTACAGCTCGTCCATGCC
KD106F Ppdc ATGCGGTACCTCTAGATAAAAAAGATAAAAAGTCTTTTCGCTTCGGCA
KD106R ATGCCTCGAGGGATCCATATATTCAAAACACTATGTCTGAATCAGGAT
GACC
KD107F Ppdc1-8 into CAAAACGATCTCAAGAAGATCATCTTATTAATCAGATAAAATATT
KD107R pKLD42 CCTTTACTCATGGTACCATTCTCCTAACTTAT
KD108F Venus ATGCCTCGAGCTAACTAACTAAAGATTAAC
KD108R ATGCGGTACCACCGAGCGTTCTGAACAAATCC
KD109F araC-ParaBAD, ATGCGTCGACGGGATTCTGCAAACCCTATGCTACT
KD109R for Venus GCCAAAATCGAGGCCAATTGC
KD110F Venus, for GGCCTCGATTTTGGCATGAGTAAAGGAGAAGAACTTTTCACTGG
KD110R araC-ParaBAD ATGCGAATTCACCGAGCGTTCTGAACAAATCC
KD111F Ppdc error GGAACAAAAGCTGGGTACCTCTAGA
KD111R CGTCGACCTCGAGGGATCC
KD112F Venus qPCR GTCAGTGGAGAGGGTGAAGG
KD112R TACATAACCTTCGGGCATGG

122
Table 5.3 DNA sequences of the wild-type pdc promoter and the 8 library candidates.
Mutated oligonucleotides are shown in red.
Promoter Sequence (5’-3’)
Ppdc TAAAAAAGATAAAAAGTCTTTTCGCTTCGGCAGAAGAGGTTCATCATGAACAAAAA
TTCGGCATTTTTAAAAATGCCTATAGCTAAATCCGGAACGACACTTTAGAGGTTTCT
GGGTCATCCTGATTCAGACATAGTGTTTTGAATATAT
Ppdc1 TAAAAAAGATAAAAAGTCTTTTCGCTTCGGCAGAAGAGGTTCCTCATGAACGAAAA
TTCGGCATCTTTGGAAGTGCCTACAGCTGAATTCGGAACGACACTTCAGAGGTTCCT
GGGTCATCCTGATTCAGACATAGTGTTTTGAGTATAT
Ppdc2 TAAAAAAGATAAAAAGTCTTTTCGCTTCGGCAGAAGAGGTCCATCATGAACGAAAA
TTCGGCATCTTTAAAAGTGCCTATAGCTAGATCCGGAACGACACTTTAGAGGTTTCT
GGGTCATCCTGATTCAGACATAGTGTTTTGAATATAT
Ppdc3 AAAAAAAGATAAAAAGTCTTTTCGCTCCGGCAGAAGAGACTCATCATGAACAAAAG
TTCGGCATCTTTAAAAATGCCTATAGCTAAATCCGGAACGACACTTTAGAGGTTTCT
GGGTCACCCTGATTCAAACATAGTGTTTTGAATATAT
Ppdc4 TAAGAAAGATAAAAAGTCTTTTCGCTTCGGCAGAAGAGGTTCATCACGAACAAAAA
TTCGGCATCTTTAAAAATGCCTATAGCTAAATCCGGAACGACACTTTAGAGGTTTCT
GGGCCGCCCTGATTCAGACATAGTGTTTTGAGTATAT
Ppdc5 TAAAAAAGATAAGAAGTCTTTTCGCTTCGGCAGAAGAGGTTCATCATGAGCAAAAA
TTCGGCATTTTTAAAAATGCCTATAGCTAAATCCGGGACGACACTTTAGAGGTTTCT
GGGTCATCCTGATTCAGACATAGTGTTTTGAATATAT
Ppdc6 TAAAAAAGATAAAAAGTCTTTTCGCTTCGGCAGAAGAGGTTCATCATGAACAAAAA
TTCGGCATTCTTAAAAATGCCTATAGCTAAATCCGGAACGACACTTTAGAGGTTTCT
GGGTCATCCTGATTCAGACATAGTGTTTTGAATATAT
Ppdc7 TAAAAAAGATAAAAAGTCTTTTCGCTTCGGCAGAAGAGGTTCGTCATGAACAAAAA
TTCGACATTTTTAAAAATGCCTATAGCTAAATCCGGAGCGACACTTTAGAGGTTTCT
GGGTCATCCTGATTCAGACATAGTGTCTTGAATATAT
Ppdc8 TAAAAAGGATAGAAAGTCTTTTCGCTTCGGCAGAAGAGGTTCATCATGAACAAAAA
TTCGGCATTTTTAAAAATGCCTATAGCTAAATCCGGAACGACACTTTAGAGGTCTCT
GGGTCATCCTGATTCAGACATAGTGTTTTGAATATGT

123
Figure 5.1 Schematic representation of plasmid constructed for library characterization.
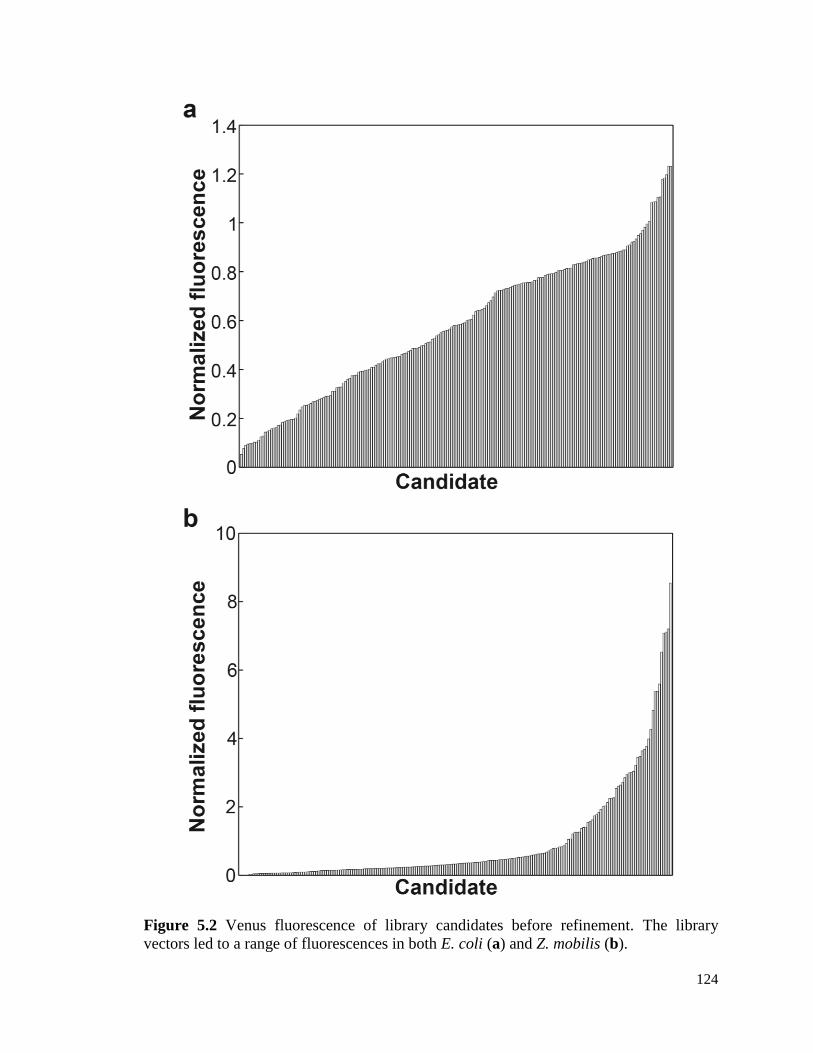
124
Figure 5.2 Venus fluorescence of library candidates before refinement. The library
vectors led to a range of fluorescences in both E. coli (a) and Z. mobilis (b).

125
Figure 5.3 Venus fluorescence of the wild-type pdc promoter and the 8 stable library
candidates in Z. mobilis.

126
Figure 5.4 mCherry fluorescence of the wild-type pdc promoter and the 8 stable library
candidates in Z. mobilis.

127
Figure 5.5 Quantitative PCR results showing relative venus transcript levels in Z.
mobilis.

128
Chapter 6: Conclusions and recommendations
6.1 Contributions
Z. mobilis has received considerable attention for its ability to produce the biofuel
ethanol at near theoretical yields using unique metabolic pathways. In this work, we
made improvements to Z. mobilis and Z. mobilis genetic techniques to render the strain a
feasible industrial host, not only for the production of ethanol but for the production of
other value-added products as well. In chapter 2, we used sugar transport engineering to
improve the rate at which Z. mobilis produces ethanol from the five-carbon sugar xylose.
In chapter 3, we found several chromosomal mutations in xylose and arabinose-
fermenting strains of Z. mobilis that improved the performance of these strains during
growth on these sugars as sole carbon sources; we then thoroughly characterized several
of these mutations to elucidate their individual contributions to the global phenotypes. In
chapter 4, we constructed expression systems for the λ Red recombinase proteins and the
bacterial CRISPR/Cas9 system for use in Z. mobilis. We proceeded to use these systems
to facilitate genome editing in the strain. In chapter 5, we constructed a promoter library
to be used for the precise control of gene expression levels in Z. mobilis, which is useful
for heterologous protein production in the strain. Overall, we have contributed
significantly to the advancement of Z. mobilis as an industrial workhorse. However, there
is more work to be done.
6.2 Future directions
6.2.1 Cellulosic ethanol production
One of the primary limitations associated with Z. mobilis for bioethanol
production is that the substrate range of this organism is limited to the six carbon sugars

129
glucose, fructose, and sometimes sucrose (Dawes et al. 1966). For ethanol production
from cellulosic feedstocks, pentose sugar digestion is also important. While previous
studies have developed engineered strains of Z. mobilis capable of fermenting the five
carbon sugars xylose and arabinose (Deanda et al. 1996b; Zhang et al. 1995), and
additional studies have improved upon these strains (Agrawal et al. 2011b; Agrawal et al.
2012b; Feldmann et al. 1992), pentose sugar digestion still lags far behind glucose
consumption in Z. mobilis. Our work contributed greatly to determining why this is the
case, but our engineered strains still do not consume xylose and arabinose as rapidly as
they do glucose. Therefore, more work is needed to construct a strain of Z. mobilis that
truly ferments pentoses and hexoses simultaneously. Toward this goal, further analysis of
the mutations we found in adapted strains of Z. mobilis in chapter 3 may uncover
additional metabolic pathways that inhibit pentose fermentations. Exploiting these
pathways further may yield even greater improvements in the performance of Z. mobilis
on these sugars. It is also possible that mutations found in efficient xylose-fermenting
strains could improve arabinose fermentations, and vice versa. Moreover, we have
determined in both chapters 2 and 3 that sugar transport is very important to both xylose
and arabinose fermentations in Z. mobilis. Therefore, additional experiments focused on
improving sugar transport should be done. Specifically, the mutant forms of the native Z.
mobilis sugar transporter, Glf, that we discovered in chapter 3 can be combined with the
wild-type Glf or the E. coli XylE in a single adapted strain to ensure that all types of
sugars are being transported efficiently. Because xylose transport by XylE has been
shown to be subject to a certain degree of “dead-end” glucose repression (Lam et al.
1980), mutant forms of XylE unable to bind glucose may be useful for expression in Z.

130
mobilis. Finally, because our xylose-fermenting strain still produced large amounts of the
inhibiting by-product xylitol, and because removal of the associated aldo/keto reductase
gene from the chromosome has been shown to improve xylose fermentation by Z. mobilis
(Agrawal et al. 2012b), a strain deficient in this gene but still containing the other
adaptation-induced mutations should be constructed.
In addition to modifications that stem directly from our results, there are other
modifications that can be made to Z. mobilis to improve it further for cellulosic ethanol
production. The sugars mannose, galactose, and rhamnose are also common in cellulosic
hydrolysates (Dwivedi et al. 2009). Therefore, to make Z. mobilis completely ideal for
industrial fuel production, it should be engineered to ferment these sugars in addition to
glucose, xylose, and arabinose. Adaptation can then be used to improve the growth of Z.
mobilis on these sugars, and high-throughput sequencing can be used to reveal the genes
implicated in the improved growth phenotypes. Additionally, Z. mobilis is known to be
highly sensitive to the acetic acid that is present in lignocellulosic hydrolysates.
Adaptation on acetic acid has been performed to increase the strain’s tolerance to this
inhibitor (Agrawal et al. 2012b), but a superior approach may be to engineer Z. mobilis to
convert acetic acid into ethanol. Acetic acid consumption has proven successful in yeast,
but it required coupling with an NADH-producing xylose utilization pathway to maintain
redox balance (Wei et al. 2013). Because Z. mobilis displays the uncoupled growth
phenomenon, it is possible that NADH and ATP are readily available for acetic acid
consumption in wild-type cells. We constructed a vector to express the E. coli acetic acid
consumption genes, acetyl-coenzyme A synthetase (acs) and acetaldehyde
dehydrogenase (mhpF), in Z. mobilis, but the plasmid would not transform into the cells.

131
This suggests that redox balance was in fact an issue. However, we did utilize strong and
constitutive Z. mobilis promoters for gene expression, so the expression level may have
simply been too high for the proteins to only consume extra ATP and reducing
equivalents that were not needed for growth. Further optimization of the protein levels
using the promoter library we constructed in chapter 5 may lead to the successful
consumption of acetic acid by Z. mobilis.
6.2.2 Genetic techniques
The highly active restriction-modification system in Z. mobilis has limited the
development of new, efficient genetic techniques for use in this organism for many years.
We have attempted to overcome this issue by applying the λ Red recombinase system and
the CRISPR/Cas bacterial adaptive immunity system for genome editing in Z. mobilis.
While our attempts were met with some success, there is still room for improvement. The
λ Red proteins increased the likelihood that DNA would be incorporated into the Z.
mobilis chromosome through double crossover events, but the number of colony forming
units we obtained upon completion of our knockout trials was still low. Optimizing
protocol parameters only increased the likelihood of double crossover events and did not
increase the number of colony forming units to the levels seen when these proteins are
used in E. coli. We also were not able to utilize these proteins for the incorporation of
ssDNA oligonucleotides into the Z. mobilis chromosome. On the other hand, our results
using the CRISPR/Cas9 system are promising and may lead to the development of a
high-efficiency knockout/integration protocol for Z. mobilis. Regardless, to ensure that all
protocols developed in the future for genome editing in the strain are functional and
efficient, the Z. mobilis restriction-modification system must be very thoroughly

132
investigated. Three genes of the type I and type IV R-M systems were identified and
characterized by Kerr and coworkers, but a third plasmid-bound type I system has not
been characterized at all (Kerr et al. 2011). The fact that no bacteriophage capable of
attacking Z. mobilis has surfaced shows just how robust the organism’s R-M system
really is. While this is an excellent reason to pursue Z. mobilis for use in future
biorefineries, phage pose a huge threat to industrial processes currently carried out using
other bacteria. Therefore, further investigation into the R-M proteins present in Z. mobilis
may be useful for engineering phage-resistance into other industrial hosts.
Because the results we obtained using the CRISPR/Cas9 system were the most
promising for Z. mobilis genome editing, future work should focus on improving this
system. We expressed the Cas9 protein under the control of its native S. pyogenes
promoter. We also constructed a vector to express Cas9 under the control of the strong
and constitutive Z. mobilis pdc promoter, but have not yet analyzed this construct. It is
well-known that the expression level of the Cas9 protein is very important to the
functionality of the process in other hosts (Duda et al. 2014), so optimization of the Cas9
expression level in Z. mobilis is an important next step in improving the process.
Additionally, using the system for a marker-free knockout or integration will shed a lot of
light on the true potential of this tool in Z. mobilis. Further, software should be developed
to assist with the design of the Z. mobilis spacer sequence to reduce off-target effects.
This is especially important in bacteria, because Cas9 cleavage of the chromosome
typically leads to cell death in these hosts. Another way to diminish the effects of off-
target binding by Cas9 is to use a catalytically-inactive form of Cas9, known as dCas9, to
carry out CRISPR interference (Qi et al. 2013). CRISPR interference occurs when dCas9

133
binds to a target gene without actually cleaving the chromosome. In this way, it blocks
transcription and leads to gene silencing. A benefit of dCas9 over Cas9 is that the effects
of dCas9 are reversible. We constructed an expression system for dCas9 in Z. mobilis,
including a vector expressing dCas9 under the control of an inducible promoter, a strain
of Z. mobilis with the venus fluorescent protein reporter integrated into the chromosome,
and a vector containing the DNA coding for the gRNA complex and a spacer to target the
promoter of the integrated venus gene. However, at the time of publication, we had not
yet thoroughly tested the system in Z. mobilis. Future experiments will therefore reveal if
dCas9 is a useful tool for genome regulation in the strain.
6.2.3 Alternative product synthesis
The interesting physiology displayed by Z. mobilis is worth manipulating for the
production of alternative products. We worked to improve genetic techniques in the strain
for this purpose, and our results on improving pentose sugar consumption by Z. mobilis
can also be applied to alternative production processes. Central to the development of Z.
mobilis strains capable of producing economical amounts of alternative products is
shifting metabolic flux away from ethanol production. Specifically, the pyruvate
decarboxylase gene must be silenced or removed from the chromosome. We attempted to
remove pdc from the chromosome of a strain of Z. mobilis that we engineered to
overproduce lactate for redox balance. However, our attempts were unsuccessful, even in
pH-controlled media. We also attempted to use pdc antisense RNA to silence the gene in
a lactate-overproducing strain, but no significant decrease in ethanol production was
seen. It is clear that the successful development of a strain of Z. mobilis deficient in pdc
would be extremely useful for alternative product synthesis—the high metabolic flux

134
could be directed toward a new, non-ethanol product. With current industry trends
suggesting that cellulosic ethanol may not be the fuel of the future, it is likely that
researchers will start focusing on the potential of Z. mobilis for the production of fatty
acids, sorbitol, gluconate, fructose, butanol, and other value-added products. We hope
that our work will be helpful in making an industrial production process utilizing Z.
mobilis a reality.

135
References
Afendra AS, Drainas C (1987a) Expression and Stability of a Recombinant Plasmid in
Zymomonas mobilis and Escherichia-Coli. Journal of General Microbiology
133:127-134
Afendra AS, Drainas C (1987b) Expression and stability of a recombinant plasmid in
Zymomonas mobilis and Escherichia coli. J Gen Microbiol 133(1):127-34
Afendra AS, Vartholomatos G, Arvanitis N, Drainas C (1999) Characterization of the
mobilization region of the Zymomonas mobilis ATCC10988 plasmid pZMO3.
Plasmid 41(1):73-7 doi:S0147-619X(98)91374-9 [pii] 10.1006/plas.1998.1374
Agrawal M, Chen RR (2011) Discovery and characterization of a xylose reductase from
Zymomonas mobilis ZM4. Biotechnology Letters 33(11):2127-33
doi:10.1007/s10529-011-0677-6
Agrawal M, Mao Z, Chen RR (2011a) Adaptation yields a highly efficient xylose-
fermenting Zymomonas mobilis strain. Biotechnol Bioeng 108(4):777-85
doi:10.1002/bit.23021
Agrawal M, Wang Y, Chen RR (2012a) Engineering efficient xylose metabolism into an
acetic acid-tolerant Zymomonas mobilis strain by introducing adaptation-induced
mutations. Biotechnol Lett 34(10):1825-32 doi:10.1007/s10529-012-0970-z
Akinterinwa O, Cirino PC (2009) Heterologous expression of D-xylulokinase from
Pichia stipitis enables high levels of xylitol production by engineered Escherichia
coli growing on xylose. Metab Eng 11(1):48-55 doi:10.1016/j.ymben.2008.07.006
S1096-7176(08)00046-3 [pii]
Algar EM, Scopes RK (1985) Studies on Cell-Free Metabolism - Ethanol-Production by
Extracts of Zymomonas mobilis. Journal of Biotechnology 2(5):275-287 doi:Doi
10.1016/0168-1656(85)90030-6
Alper H, Fischer C, Nevoigt E, Stephanopoulos G (2005) Tuning genetic control through
promoter engineering. Proc Natl Acad Sci U S A 102(36):12678-83
doi:0504604102 [pii] 10.1073/pnas.0504604102
Amundsen SK, Spicer T, Karabulut AC, Londono LM, Eberhart C, Fernandez Vega V,
Bannister TD, Hodder P, Smith GR (2012) Small-molecule inhibitors of bacterial
AddAB and RecBCD helicase-nuclease DNA repair enzymes. ACS Chem Biol
7(5):879-91 doi:10.1021/cb300018x
An H, Scopes RK, Rodriguez M, Keshav KF, Ingram LO (1991) Gel electrophoretic
analysis of Zymomonas mobilis glycolytic and fermentative enzymes:
identification of alcohol dehydrogenase II as a stress protein. J Bacteriol
173(19):5975-82
Antoine R, Locht C (1992) Isolation and molecular characterization of a novel broad-
host-range plasmid from Bordetella bronchiseptica with sequence similarities to
plasmids from gram-positive organisms. Mol Microbiol 6(13):1785-99
Arbuckle MI, Kane S, Porter LM, Seatter MJ, Gould GW (1996) Structure-function
analysis of liver-type (GLUT2) and brain-type (GLUT3) glucose transporters:
expression of chimeric transporters in Xenopus oocytes suggests an important role
for putative transmembrane helix 7 in determining substrate selectivity.
Biochemistry 35(51):16519-27 doi:10.1021/bi962210nbi962210n [pii]

136
Arfman N, Worrell V, Ingram LO (1992) Use of the tac promoter and lacIq for the
controlled expression of Zymomonas mobilis fermentative genes in Escherichia
coli and Zymomonas mobilis. J Bacteriol 174(22):7370-8
Armengaud J, Trapp J, Pible O, Geffard O, Chaumot A, Hartmann EM (2014) Non-
model organisms, a species endangered by proteogenomics. J Proteomics
doi:S1874-3919(14)00019-0 [pii]10.1016/j.jprot.2014.01.007
Arvanitis N, Pappas KM, Kolios G, Afendra AS, Typas MA, Drainas C (2000)
Characterization and replication properties of the Zymomonas mobilis ATCC
10988 plasmids pZMO1 and pZMO2. Plasmid 44(2):127-37
doi:10.1006/plas.2000.1480S0147-619X(00)91480-X [pii]
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M,
Wanner BL, Mori H (2006) Construction of Escherichia coli K-12 in-frame,
single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006 0008
doi:msb4100050 [pii]10.1038/msb4100050
Baratti JC, Bu'lock JD (1986) Zymomonas mobilis: a bacterium for ethanol production.
Biotechnol Adv 4(1):95-115 doi:0734975086900066 [pii]
Barrow KD, Collins JG, Rogers PL, Smith GM (1984) The structure of a novel
polysaccharide isolated from Zymomonas mobilis determined by nuclear magnetic
resonance spectroscopy. Eur J Biochem 145(1):173-9
Baumler DJ, Hung KF, Bose JL, Vykhodets BM, Cheng CM, Jeong KC, Kaspar CW
(2006) Enhancement of acid tolerance in Zymomonas mobilis by a proton-
buffering peptide. Appl Biochem Biotechnol 134(1):15-26 doi:ABAB:134:1:15
[pii]
Bekers M, Vigants A, Laukevics J, Toma M, Rapoports A, Zikmanis P (2000) The effect
of osmo-induced stress on product formation by Zymomonas mobilis on sucrose.
Int J Food Microbiol 55(1-3):147-50
Belauich JP, Senez JC (1965) Influence of Aeration and of Pantothenate on Growth
Yields of Zymomonas mobilis. J Bacteriol 89:1195-200
Benschoter AS, Ingram LO (1986) Thermal Tolerance of Zymomonas mobilis:
Temperature-Induced Changes in Membrane Composition. Appl Environ
Microbiol 51(6):1278-84
Bothast RJ, Nichols NN, Dien BS (1999) Fermentations with new recombinant
organisms. Biotechnol Prog 15(5):867-75 doi:bp990087w [pii]
10.1021/bp990087w
Bresticgoachet N, Gunasekaran P, Cami B, Baratti J (1990) Transfer and Expression of a
Bacillus-Licheniformis Alpha-Amylase Gene in Zymomonas mobilis. Archives of
Microbiology 153(3):219-225 doi:Doi 10.1007/Bf00249071
Bringer S, Sahm H, Swyzen W (1984) Ethanol-Production by Zymomonas mobilis and Its
Application on an Industrial-Scale. Biotechnology and Bioengineering:311-319
Bringermeyer S, Sahm H (1989) Junctions of Catabolic and Anabolic Pathways in
Zymomonas mobilis - Phosphoenolpyruvate Carboxylase and Malic Enzyme.
Appl Microbiol Biot 31(5-6):529-536
Browne GM, Skotnicki ML, Goodman AE, Rogers PL (1984) Transformation of
Zymomonas mobilis by a hybrid plasmid. Plasmid 12(3):211-4
Buchholz SE, Eveleigh DE (1986) Transfer of Plasmids to an Antibiotic-Sensitive
Mutant of Zymomonas mobilis. Appl Environ Microb 52(2):366-370

137
Byun MOK, Kaper JB, Ingram LO (1986) Construction of a New Vector for the
Expression of Foreign Genes in Zymomonas mobilis. Journal of Industrial
Microbiology 1(1):9-15 doi:Doi 10.1007/Bf01569411
Carey VC, Walia SK, Ingram LO (1983) Expression of a Lactose Transposon (Tn951) in
Zymomonas mobilis. Appl Environ Microbiol 46(5):1163-8
Carroll A, Somerville C (2009) Cellulosic biofuels. Annu Rev Plant Biol 60:165-82
doi:10.1146/annurev.arplant.043008.092125
Chedin F, Noirot P, Biaudet V, Ehrlich SD (1998a) A five-nucleotide sequence protects
DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of
Bacillus subtilis. Mol Microbiol 29(6):1369-77
Chedin F, Noirot P, Biaudet V, Ehrlich SD (1998b) A five-nucleotide sequence protects
DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of
Bacillus subtilis. Molecular Microbiology 29(6):1369-77
Chen RR, Wang Y, Shin HD, Agrawal M, Mao Z (2009) Improved strains of Zymomonas
mobilis for fermentation of biomass. Google Patents
Chen YF, Helmann JD (1992) Restoration of motility to an Escherichia coli fliA flagellar
mutant by a Bacillus subtilis sigma factor. Proc Natl Acad Sci U S A
89(11):5123-7
Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and
KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-
resistance determinant. Gene 158(1):9-14 doi:037811199500193A [pii]
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W,
Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas
systems. Science 339(6121):819-23 doi:10.1126/science.1231143
science.1231143 [pii]
Conway T (1992) The Entner-Doudoroff pathway: history, physiology and molecular
biology. FEMS Microbiol Rev 9(1):1-27
Conway T, Byun MO, Ingram LO (1987a) Expression Vector for Zymomonas mobilis.
Appl Environ Microbiol 53(2):235-41
Conway T, Osman YA, Ingram LO (1987b) Gene expression in Zymomonas mobilis:
promoter structure and identification of membrane anchor sequences forming
functional lacZ' fusion proteins. J Bacteriol 169(6):2327-35
Conway T, Osman YA, Konnan JI, Hoffmann EM, Ingram LO (1987c) Promoter and
nucleotide sequences of the Zymomonas mobilis pyruvate decarboxylase. J
Bacteriol 169(3):949-54
Conway T, Sewell GW, Ingram LO (1987d) Glyceraldehyde-3-Phosphate
Dehydrogenase Gene from Zymomonas mobilis - Cloning, Sequencing, and
Identification of Promoter Region. Journal of Bacteriology 169(12):5653-5662
Cook GM, Russell JB (1994) Energy-spilling reactions of Streptococcus bovis and
resistance of its membrane to proton conductance. Appl Environ Microbiol
60(6):1942-8
Copeland NG, Jenkins NA, Court DL (2001) Recombineering: a powerful new tool for
mouse functional genomics. Nat Rev Genet 2(10):769-79 doi:10.1038/35093556
35093556 [pii]

138
Costantino N, Court DL (2003) Enhanced levels of lambda Red-mediated recombinants
in mismatch repair mutants. Proc Natl Acad Sci U S A 100(26):15748-53
doi:10.1073/pnas.2434959100 2434959100 [pii]
Cromie GA (2009) Phylogenetic ubiquity and shuffling of the bacterial RecBCD and
AddAB recombination complexes. Journal of Bacteriology 191(16):5076-84
doi:10.1128/JB.00254-09 JB.00254-09 [pii]
da Silva TL, Gouveia L, Reis A (2013) Integrated microbial processes for biofuels and
high value-added products: the way to improve the cost effectiveness of biofuel
production. Appl Microbiol Biotechnol doi:10.1007/s00253-013-5389-5
Dally EL, Stokes HW, Eveleigh DE (1982) A Genetic Comparison of Strains of
Zymomonas mobilis by Analysis of Plasmid DNA. Biotechnology Letters 4(2):91-
96 doi:Doi 10.1007/Bf01091343
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in
Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A
97(12):6640-5 doi:10.1073/pnas.120163297 120163297 [pii]
Datta S, Costantino N, Zhou X, Court DL (2008) Identification and analysis of
recombineering functions from Gram-negative and Gram-positive bacteria and
their phages. Proceedings of the National Academy of Sciences 105(5):1626-1631
doi:10.1073/pnas.0709089105
Davis EO, Henderson PJ (1987) The cloning and DNA sequence of the gene xylE for
xylose-proton symport in Escherichia coli K12. J Biol Chem 262(29):13928-32
Dawes EA, Large PJ (1970) Effect of starvation on the viability and cellular constituents
of Zymomonas anaerobia and Zymomonas mobilis. J Gen Microbiol 60(1):31-42
Dawes EA, Ribbons DW, Large PJ (1966) The route of ethanol formation in Zymomonas
mobilis. Biochem J 98(3):795-803
de Boer HA, Comstock LJ, Vasser M (1983) The tac promoter: a functional hybrid
derived from the trp and lac promoters. Proc Natl Acad Sci U S A 80(1):21-5
De Graaf AA, Striegel K, Wittig RM, Laufer B, Schmitz G, Wiechert W, Sprenger GA,
Sahm H (1999a) Metabolic state of Zymomonas mobilis in glucose-, fructose-,
and xylose-fed continuous cultures as analysed by 13C- and 31P-NMR
spectroscopy. Arch Microbiol 171(6):371-85 doi:91710371.203 [pii]
de Kok S, Stanton LH, Slaby T, Durot M, Holmes VF, Patel KG, Platt D, Shapland EB,
Serber Z, Dean J, Newman JD, Chandran SS (2014) Rapid and Reliable DNA
Assembly via Ligase Cycling Reaction. Acs Synth Biol 3(2):97-106 doi:Doi
10.1021/Sb4001992
de Lorenzo V (1992) Genetic engineering strategies for environmental applications. Curr
Opin Biotechnol 3(3):227-31
Deanda K, Zhang M, Eddy C, Picataggio S (1996a) Development of an arabinose-
fermenting Zymomonas mobilis strain by metabolic pathway engineering. Appl
Environ Microbiol 62(12):4465-70
Deley J, Swings J (1976) Phenotypic Description, Numerical-Analysis, and Proposal for
an Improved Taxonomy and Nomenclature of Genus Zymomonas Kluyver and
Van Niel 1936. Int J Syst Bacteriol 26(2):146-157
Delgado OD, Martinez MA, ABate CM, Sineriz F (2002) Chromosomal integration and
expression of green fluorescent protein in Zymomonas mobilis. Biotechnology
Letters 24(15):1285-1290 doi:Doi 10.1023/A:1016266110391

139
Demirbas MF (2009) Biorefineries for biofuel upgrading: A critical review. Appl Energ
86:S151-S161 doi:DOI 10.1016/j.apenergy.2009.04.043
Desai TA, Rao CV (2010) Regulation of arabinose and xylose metabolism in Escherichia
coli. Appl Environ Microbiol 76(5):1524-32 doi:10.1128/AEM.01970-09
Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA,
Horvath P, Moineau S (2008) Phage response to CRISPR-encoded resistance in
Streptococcus thermophilus. J Bacteriol 190(4):1390-400 doi:JB.01412-07 [pii]
10.1128/JB.01412-07
DiCarlo JE, Conley AJ, Penttila M, Jantti J, Wang HH, Church GM (2013a) Yeast oligo-
mediated genome engineering (YOGE). Acs Synth Biol 2(12):741-9
doi:10.1021/sb400117c
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM (2013b) Genome
engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic
Acids Res 41(7):4336-43 doi:10.1093/nar/gkt135 gkt135 [pii]
Dien BS, Cotta MA, Jeffries TW (2003) Bacteria engineered for fuel ethanol production:
current status. Appl Microbiol Biotechnol 63(3):258-66 doi:10.1007/s00253-003-
1444-y
Dimarco AA, Romano AH (1985) d-Glucose Transport System of Zymomonas mobilis.
Appl Environ Microbiol 49(1):151-7
Doelle HW, Greenfield PF (1985) The Production of Ethanol from Sucrose Using
Zymomonas mobilis. Appl Microbiol Biot 22(6):405-410
Doelle MB, Doelle HW (1990) Sugarcane Molasses Fermentation by Zymomonas
mobilis. Appl Microbiol Biot 33(1):31-35
Doelle MB, Greenfield PF, Doelle HW (1990) The Relationship between Sucrose
Hydrolysis, Sorbitol Formation and Mineral Ion Concentration during Bioethanol
Formation Using Zymomonas mobilis 2716. Appl Microbiol Biot 34(2):160-167
Dominguez JM, Gong CS, Tsao GT (1996) Pretreatment of sugar cane bagasse
hemicellulose hydrolysate for xylitol production by yeast. Appl Biochem
Biotechnol 57-58:49-56
Dong HW, Bao J, Ryu DD, Zhong JJ (2011) Design and construction of improved new
vectors for Zymomonas mobilis recombinants. Biotechnol Bioeng 108(7):1616-27
doi:10.1002/bit.23106
Drainas C, Slater AA, Coggins L, Montague P, Costa RG, Ledingham WM, Kinghorn JR
(1983) Electron-Microscopic Analysis of Zymomonas mobilis, Strain Atcc-10988
Plasmid DNA. Biotechnology Letters 5(6):405-408 doi:Doi 10.1007/Bf00131281
Du J, Li S, Zhao H (2010a) Discovery and characterization of novel d-xylose-specific
transporters from Neurospora crassa and Pichia stipitis. Mol Biosyst 6(11):2150-
6 doi:10.1039/c0mb00007h
Duda K, Lonowski LA, Kofoed-Nielsen M, Ibarra A, Delay CM, Kang Q, Yang Z,
Pruett-Miller SM, Bennett EP, Wandall HH, Davis GD, Hansen SH, Frodin M
(2014) High-efficiency genome editing via 2A-coupled co-expression of
fluorescent proteins and zinc finger nucleases or CRISPR/Cas9 nickase pairs.
Nucleic Acids Res 42(10):e84 doi:10.1093/nar/gku251 gku251 [pii]
Dunn KL, Rao CV (2014) Expression of a xylose-specific transporter improves ethanol
production by metabolically engineered Zymomonas mobilis. Appl Microbiol
Biotechnol doi:10.1007/s00253-014-5812-6

140
Dunn KL, Rao CV (2015) High-throughput sequencing reveals adaptation-induced
mutations in pentose-fermenting strains of Zymomonas mobilis. Biotechnol
Bioeng doi:10.1002/bit.25631
Dwivedi P, Alavalapati JRR, Lal P (2009) Cellulosic ethanol production in the United
States: Conversion technologies, current production status, economics, and
emerging developments. Energy Sustain Dev 13(3):174-182 doi:DOI
10.1016/j.esd.2009.06.003
Ellis HM, Yu D, DiTizio T, Court DL (2001) High efficiency mutagenesis, repair, and
engineering of chromosomal DNA using single-stranded oligonucleotides. Proc
Natl Acad Sci U S A 98(12):6742-6 doi:10.1073/pnas.121164898 121164898
[pii]
Eriksen DT, Hsieh PC, Lynn P, Zhao H (2013) Directed evolution of a cellobiose
utilization pathway in Saccharomyces cerevisiae by simultaneously engineering
multiple proteins. Microb Cell Fact 12:61 doi:10.1186/1475-2859-12-61 1475-
2859-12-61 [pii]
Feldmann SD, Sahm H, Sprenger GA (1992) Pentose Metabolism in Zymomonas mobilis
Wild-Type and Recombinant Strains. Appl Microbiol Biot 38(3):354-361
Fieschko J, Humphrey RE (1983) Effects of temperature and ethanol concentration on the
maintenance and yield coefficient of Zymomonas mobilis. Biotechnol Bioeng
25(6):1655-60 doi:10.1002/bit.260250618
Forrest WW (1967) Energies of activation and uncoupled growth in Streptococcus
faecalis and Zymomonas mobilis. J Bacteriol 94(5):1459-63
Gao Q, Zhang M, McMillan JD, Kompala DS (2002) Characterization of heterologous
and native enzyme activity profiles in metabolically engineered Zymomonas
mobilis strains during batch fermentation of glucose and xylose mixtures. Appl
Biochem Biotechnol 98-100:341-55
Gibbs M, Demoss RD (1954) Anaerobic dissimilation of C14-labeled glucose and
fructose by Pseudomonas lindneri. J Biol Chem 207(2):689-94
Goetz G, Iwan P, Hauer B, Breuer M, Pohl M (2001) Continuous production of (R)-
phenylacetylcarbinol in an enzyme-membrane reactor using a potent mutant of
pyruvate decarboxylase from Zymomonas mobilis. Biotechnology and
Bioengineering 74(4):317-325 doi:Doi 10.1002/Bit.1122
Goldman D, Lavid N, Schwartz A, Shoham G, Danino D, Shoham Y (2008) Two active
forms of Zymomonas mobilis levansucrase. An ordered microfibril structure of the
enzyme promotes levan polymerization. J Biol Chem 283(47):32209-17
doi:10.1074/jbc.M805985200 M805985200 [pii]
Gudbergsdottir S, Deng L, Chen Z, Jensen JV, Jensen LR, She Q, Garrett RA (2011)
Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems
when challenged with vector-borne viral and plasmid genes and protospacers.
Mol Microbiol 79(1):35-49 doi:10.1111/j.1365-2958.2010.07452.x
Gunasekaran P, Karunakaran T, Cami B, Mukundan AG, Preziosi L, Baratti J (1990)
Cloning and sequencing of the sacA gene: characterization of a sucrase from
Zymomonas mobilis. J Bacteriol 172(12):6727-35
Haijema BJ, Venema G, Kooistra J (1996) The C terminus of the AddA subunit of the
Bacillus subtilis ATP-dependent DNase is required for the ATP-dependent
exonuclease activity but not for the helicase activity. J Bacteriol 178(17):5086-91

141
Harms K, Wackernagel W (2008) The RecBCD and SbcCD DNases suppress homology-
facilitated illegitimate recombination during natural transformation of
Acinetobacter baylyi. Microbiol-Sgm 154:2437-2445 doi:DOI
10.1099/mic.0.2008/018382-0
Hashiramoto M, Kadowaki T, Clark AE, Muraoka A, Momomura K, Sakura H, Tobe K,
Akanuma Y, Yazaki Y, Holman GD, et al. (1992) Site-directed mutagenesis of
GLUT1 in helix 7 residue 282 results in perturbation of exofacial ligand binding.
J Biol Chem 267(25):17502-7
Hayashi T, Kato T, Furukawa K (2012) Respiratory chain analysis of Zymomonas mobilis
mutants producing high levels of ethanol. Appl Environ Microbiol 78(16):5622-9
doi:10.1128/AEM.00733-12 AEM.00733-12 [pii]
He MX, Wu B, Qin H, Ruan ZY, Tan FR, Wang JL, Shui ZX, Dai LC, Zhu QL, Pan K,
Tang XY, Wang WG, Hu QC (2014) Zymomonas mobilis: a novel platform for
future biorefineries. Biotechnology for Biofuels 7 doi:Artn 101Doi 10.1186/1754-
6834-7-101
He MX, Wu B, Shui ZX, Hu QC, Wang WG, Tan FR, Tang XY, Zhu QL, Pan K, Li Q,
Su XH (2012) Transcriptome profiling of Zymomonas mobilis under ethanol
stress. Biotechnol Biofuels 5(1):75 doi:10.1186/1754-6834-5-75 1754-6834-5-75
[pii]
Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes
without the use of restriction enzymes: gene splicing by overlap extension. Gene
77(1):61-8 doi:0378-1119(89)90359-4 [pii]
Huang C, Zong MH, Wu H, Liu QP (2009) Microbial oil production from rice straw
hydrolysate by Trichosporon fermentans. Bioresour Technol 100(19):4535-8
doi:10.1016/j.biortech.2009.04.022 S0960-8524(09)00387-3 [pii]
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR,
Joung JK (2013) Efficient genome editing in zebrafish using a CRISPR-Cas
system. Nat Biotechnol 31(3):227-9 doi:10.1038/nbt.2501 nbt.2501 [pii]
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence
of the iap gene, responsible for alkaline phosphatase isozyme conversion in
Escherichia coli, and identification of the gene product. J Bacteriol 169(12):5429-
33
Iwan P, Goetz G, Schmitz S, Hauer B, Breuer M, Pohl M (2001) Studies on the
continuous production of (R)-(-)-phenylacetylcarbinol in an enzyme-membrane
reactor. J Mol Catal B-Enzym 11(4-6):387-396 doi:Doi 10.1016/S1381-
1177(00)00029-1
Jeon YJ, Svenson CJ, Joachimsthal EL, Rogers PL (2002) Kinetic analysis of ethanol
production by an acetate-resistant strain of recombinant Zymomonas mobilis.
Biotechnology Letters 24(10):819-824 doi:Doi 10.1023/A:1015546521000
Jeon YJ, Svenson CJ, Rogers PL (2005a) Over-expression of xylulokinase in a xylose-
metabolising recombinant strain of Zymomonas mobilis. Fems Microbiology
Letters 244(1):85-92 doi:DOI 10.1016/j.femsle.2005.01.025
Jeon YJ, Svenson CJ, Rogers PL (2005b) Over-expression of xylulokinase in a xylose-
metabolising recombinant strain of Zymomonas mobilis. FEMS Microbiol Lett
244(1):85-92 doi:S0378-1097(05)00039-X [pii] 10.1016/j.femsle.2005.01.025

142
Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA (2013) RNA-guided editing of
bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31(3):233-9
doi:10.1038/nbt.2508 nbt.2508 [pii]
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A
programmable dual-RNA-guided DNA endonuclease in adaptive bacterial
immunity. Science 337(6096):816-21 doi:10.1126/science.1225829
science.1225829 [pii]
Joachimsthal EL, Rogers PL (2000a) Characterization of a high-productivity recombinant
strain of Zymomonas mobilis for ethanol production from glucose/xylose
mixtures. Appl Biochem Biotechnol 84-86:343-56
Joachimsthal EL, Rogers PL (2000b) Characterization of a high-productivity recombinant
strain of Zymomonas mobilis for ethanol production from glucose/xylose
mixtures. Appl Biochem Biotechnol 84-86:343-56
Jobses IM, Egberts GT, van Baalen A, Roels JA (1985) Mathematical modelling of
growth and substrate conversion of Zymomonas mobilis at 30 and 35 degrees C.
Biotechnol Bioeng 27(7):984-95 doi:10.1002/bit.260270709
Johns MR, Greenfield PF, Doelle HW (1991) Byproducts from Zymomonas mobilis
Bioreactor Systems and Effects. Advances in Biochemical
Engineering/Biotechnology, vol 44. Springer Berlin Heidelberg, pp 97-121
Kalnenieks U (2006) Physiology of Zymomonas mobilis: some unanswered questions.
Adv Microb Physiol 51:73-117 doi:S0065-2911(06)51002-1 [pii] 10.1016/S0065-
2911(06)51002-1
Kalnenieks U, Degraaf AA, Bringermeyer S, Sahm H (1993) Oxidative-Phosphorylation
in Zymomonas mobilis. Archives of Microbiology 160(1):74-79
Kalnenieks U, Galinina N, Bringer-Meyer S, Poole RK (1998) Membrane D-lactate
oxidase in Zymomonas mobilis: evidence for a branched respiratory chain. FEMS
Microbiol Lett 168(1):91-7 doi:S0378-1097(98)00424-8 [pii]
Kalnenieks U, Galinina N, Strazdina I, Kravale Z, Pickford JL, Rutkis R, Poole RK
(2008) NADH dehydrogenase deficiency results in low respiration rate and
improved aerobic growth of Zymomonas mobilis. Microbiology 154(Pt 3):989-94
doi:10.1099/mic.0.2007/012682-0 154/3/989 [pii]
Kalnenieks U, Galinina N, Toma MM, Poole RK (2000) Cyanide inhibits respiration yet
stimulates aerobic growth of Zymomonas mobilis. Microbiology 146 ( Pt 6):1259-
66
Kalnenieks UZ, Pankova LM, Shvinka YE (1987) Proton Motive Force in the Bacterium
Zymomonas mobilis. Biochemistry-Moscow+ 52(5):617-620
Kannan R, Mukundan G, Ait-Abdelkader N, Augier-Magro V, Baratti J, Gunasekaran P
(1995a) Molecular cloning and characterization of the extracellular sucrase gene
(sacC) of Zymomonas mobilis. Arch Microbiol 163(3):195-204
Kannan R, Pitchaimani K, Gunasekaran P, Ait-Abdelkader N, Baratti J (1995b)
Overexpression of extracellular sucrase (SacC) of Zymomonas mobilis in
Escherichia coli. FEMS Microbiol Lett 133(1-2):29-33
Karu AE, Sakaki Y, Echols H, Linn S (1975) The gamma protein specified by
bacteriophage gamma. Structure and inhibitory activity for the recBC enzyme of
Escherichia coli. J Biol Chem 250(18):7377-87

143
Katashkina JI, Hara Y, Golubeva LI, Andreeva IG, Kuvaeva TM, Mashko SV (2009) Use
of the lambda Red-recombineering method for genetic engineering of Pantoea
ananatis. BMC Mol Biol 10:34 doi:10.1186/1471-2199-10-34 1471-2199-10-34
[pii]
Kerr AL, Jeon YJ, Svenson CJ, Rogers PL, Neilan BA (2011) DNA restriction-
modification systems in the ethanologen, Zymomonas mobilis ZM4. Appl
Microbiol Biotechnol 89(3):761-9 doi:10.1007/s00253-010-2936-1
Khan SR, Gaines J, Roop RM, 2nd, Farrand SK (2008) Broad-host-range expression
vectors with tightly regulated promoters and their use to examine the influence of
TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ
Microbiol 74(16):5053-62 doi:10.1128/AEM.01098-08 AEM.01098-08 [pii]
Kim IS, Barrow KD, Rogers PL (2000a) Kinetic and nuclear magnetic resonance studies
of xylose metabolism by recombinant Zymomonas mobilis ZM4(pZB5). Appl
Environ Microbiol 66(1):186-93
Kim IS, Barrow KD, Rogers PL (2000c) Nuclear magnetic resonance studies of acetic
acid inhibition of rec Zymomonas mobilis ZM4(pZB5). Appl Biochem Biotechnol
84-86:357-70
Kita K, Konishi K, Anraku Y (1984a) Terminal oxidases of Escherichia coli aerobic
respiratory chain. I. Purification and properties of cytochrome b562-o complex
from cells in the early exponential phase of aerobic growth. J Biol Chem
259(5):3368-74
Kita K, Konishi K, Anraku Y (1984b) Terminal oxidases of Escherichia coli aerobic
respiratory chain. II. Purification and properties of cytochrome b558-d complex
from cells grown with limited oxygen and evidence of branched electron-carrying
systems. J Biol Chem 259(5):3375-81
Kremer TA, LaSarre B, Posto AL, McKinlay JB (2015) N-2 gas is an effective fertilizer
for bioethanol production by Zymomonas mobilis. P Natl Acad Sci USA
112(7):2222-2226 doi:DOI 10.1073/pnas.1420663112
Kupsch J, Alonso JC, Trautner TA (1989) Analysis of Structural and Biological
Parameters Affecting Plasmid Deletion Formation in Bacillus subtilis. Molecular
& General Genetics 218(3):402-408 doi:Doi 10.1007/Bf00332402
Kuyper M, Toirkens MJ, Diderich JA, Winkler AA, van Dijken JP, Pronk JT (2005)
Evolutionary engineering of mixed-sugar utilization by a xylose-fermenting
Saccharomyces cerevisiae strain. Fems Yeast Res 5(10):925-934 doi:DOI
10.1016/j.femsyr.2005.04.004
Kyono K, Yanase H, Tonomura K, Kawasaki H, Sakai T (1995) Cloning and
characterization of Zymomonas mobilis genes encoding extracellular levansucrase
and invertase. Biosci Biotechnol Biochem 59(2):289-93
Lam VMS, Daruwalla KR, Henderson PJF, Jonesmortimer MC (1980) Proton-Linked D-
Xylose Transport in Escherichia coli. Journal of Bacteriology 143(1):396-402
Lawford HG, Rousseau JD (1999) The effect of glucose on high-level xylose
fermentations by recombinant Zymomonas in batch and fed-batch fermentations.
Appl Biochem Biotech 77-9:235-249 doi:Doi 10.1385/Abab:77:1-3:235
Lawford HG, Rousseau JD (2000a) Comparative energetics of glucose and xylose
metabolism in recombinant Zymomonas mobilis. Appl Biochem Biotechnol 84-
86:277-93

144
Lawford HG, Rousseau JD (2000b) Comparative energetics of glucose and xylose
metabolism in recombinant Zymomonas mobilis. Appl Biochem Biotech 84-
6:277-293 doi:Doi 10.1385/Abab:84-86:1-9:277
Lawford HG, Rousseau JD (2002) Performance testing of Zymomonas mobilis
metabolically engineered for cofermentation of glucose, xylose, and arabinose.
Appl Biochem Biotechnol 98-100:429-48
Lawford HG, Rousseau JD, Mohagheghi A, McMillan JD (1998) Continuous culture
studies of xylose-fermenting Zymomonas mobilis. Appl Biochem Biotechnol 70-
72:353-67 doi:10.1007/BF02920151
Lazdunsk.A, Belaich JP (1972) Uncoupling in Bacterial Growth - Atp Pool Variation in
Zymomonas mobilis Cells in Relation to Different Uncoupling Conditions of
Growth. Journal of General Microbiology 70(Apr):187-&
Leandro MJ, Goncalves P, Spencer-Martins I (2006a) Two glucose/xylose transporter
genes from the yeast Candida intermedia: first molecular characterization of a
yeast xylose-H+ symporter. Biochem J 395(3):543-9 doi:10.1042/BJ20051465
Lee JY, Cheong DE, Kim GJ (2008) A novel assay system for the measurement of
transketolase activity using xylulokinase from Saccharomyces cerevisiae.
Biotechnol Lett 30(5):899-904 doi:10.1007/s10529-007-9616-y
Lemuth K, Steuer K, Albermann C (2011) Engineering of a plasmid-free Escherichia coli
strain for improved in vivo biosynthesis of astaxanthin. Microb Cell Fact 10:29
doi:10.1186/1475-2859-10-29 1475-2859-10-29 [pii]
Leonelli S, Ankeny RA (2013) What makes a model organism? Endeavour 37(4):209-12
doi:10.1016/j.endeavour.2013.06.001 S0160-9327(13)00037-9 [pii]
Lesic B, Rahme LG (2008) Use of the lambda Red recombinase system to rapidly
generate mutants in Pseudomonas aeruginosa. BMC Mol Biol 9:20
doi:10.1186/1471-2199-9-20 1471-2199-9-20 [pii]
Liu C, Dong H, Zhong J, Ryu DD, Bao J (2010) Sorbitol production using recombinant
Zymomonas mobilis strain. J Biotechnol 148(2-3):105-12
doi:10.1016/j.jbiotec.2010.04.008 S0168-1656(10)00193-8 [pii]
Liu CQ, Goodman AE, Dunn NW (1988) Expression of Cloned Xanthomonas D-Xylose
Catabolic Genes in Zymomonas mobilis. Journal of Biotechnology 7(1):61-70
doi:Doi 10.1016/0168-1656(88)90035-1
Loos H, Kramer R, Sahm H, Sprenger GA (1994) Sorbitol promotes growth of
Zymomonas mobilis in environments with high concentrations of sugar: evidence
for a physiological function of glucose-fructose oxidoreductase in
osmoprotection. J Bacteriol 176(24):7688-93
Lynd LR, Cushman JH, Nichols RJ, Wyman CE (1991) Fuel ethanol from cellulosic
biomass. Science 251(4999):1318-23 doi:251/4999/1318 [pii]
10.1126/science.251.4999.1318
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S,
Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV (2011) Evolution
and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9(6):467-77
doi:10.1038/nrmicro2577 nrmicro2577 [pii]
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM
(2013) RNA-guided human genome engineering via Cas9. Science
339(6121):823-6 doi:10.1126/science.1232033 science.1232033 [pii]

145
Markley AL, Begemann MB, Clarke RE, Gordon GC, Pfleger BF (2015) Synthetic
Biology Toolbox for Controlling Gene Expression in the Cyanobacterium
Synechococcus sp. strain PCC 7002. Acs Synth Biol 4(5):595-603
doi:10.1021/sb500260k
Marraffini LA, Sontheimer EJ (2010) Self versus non-self discrimination during CRISPR
RNA-directed immunity. Nature 463(7280):568-71 doi:10.1038/nature08703
nature08703 [pii]
Marsic N, Roje S, Stojiljkovic I, Salaj-Smic E, Trgovcevic Z (1993) In vivo studies on
the interaction of RecBCD enzyme and lambda Gam protein. J Bacteriol
175(15):4738-43
Martinez-Morales F, Borges AC, Martinez A, Shanmugam KT, Ingram LO (1999)
Chromosomal integration of heterologous DNA in Escherichia coli with precise
removal of markers and replicons used during construction. Journal of
Bacteriology 181(22):7143-8
McGill DJ, Dawes EA (1971) Glucose and fructose metabolism in Zymomonas
anaerobia. Biochem J 125(4):1059-68
Meima R, Haijema BJ, Dijkstra H, Haan GJ, Venema G, Bron S (1997) Role of enzymes
of homologous recombination in illegitimate plasmid recombination in Bacillus
subtilis. J Bacteriol 179(4):1219-29
Meima R, Haijema BJ, Venema G, Bron S (1995) Overproduction of the ATP-dependent
nuclease AddAB improves the structural stability of a model plasmid system in
Bacillus subtilis. Mol Gen Genet 248(4):391-8
Meima R, Venema G, Bron S (1996) A positive selection vector for the analysis of
structural plasmid instability in Bacillus subtilis. Plasmid 35(1):14-30 doi:S0147-
619X(96)90002-5 [pii] 10.1006/plas.1996.0002
Misawa N, Nakamura K (1989) Nucleotide-Sequence of the 2.7 Kb Plasmid of
Zymomonas mobilis Atcc10988. Journal of Biotechnology 12(1):63-70 doi:Doi
10.1016/0168-1656(89)90129-6
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C (2009) Short motif
sequences determine the targets of the prokaryotic CRISPR defence system.
Microbiology 155(3):733-740 doi:10.1099/mic.0.023960-0
Mosberg JA, Gregg CJ, Lajoie MJ, Wang HH, Church GM (2012) Improving lambda red
genome engineering in Escherichia coli via rational removal of endogenous
nucleases. PLoS One 7(9):e44638 doi:10.1371/journal.pone.0044638 PONE-D-
12-12936 [pii]
Murphy KC (1998) Use of bacteriophage lambda recombination functions to promote
gene replacement in Escherichia coli. J Bacteriol 180(8):2063-71
Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of
yellow fluorescent protein with fast and efficient maturation for cell-biological
applications. Nat Biotechnol 20(1):87-90 doi:10.1038/nbt0102-87
Nevoigt E (2008) Progress in metabolic engineering of Saccharomyces cerevisiae.
Microbiol Mol Biol Rev 72(3):379-412 doi:10.1128/MMBR.00025-07 72/3/379
[pii]
Novotny CP, Englesberg E (1966) The L-arabinose permease system in Escherichia coli
B/r. Biochim Biophys Acta 117(1):217-30

146
Osman YA, Conway T, Bonetti SJ, Ingram LO (1987) Glycolytic flux in Zymomonas
mobilis: enzyme and metabolite levels during batch fermentation. J Bacteriol
169(8):3726-36
Osman YA, Ingram LO (1985) Mechanism of ethanol inhibition of fermentation in
Zymomonas mobilis CP4. J Bacteriol 164(1):173-80
Pankova LM, Shvinka JE, Beker MJ (1988) Regulation of Intracellular H+ Balance in
Zymomonas mobilis 113 during the Shift from Anaerobic to Aerobic Conditions.
Appl Microbiol Biot 28(6):583-588
Pappas KM, Galani I, Typas MA (1997) Transposon mutagenesis and strain construction
in Zymomonas mobilis. J Appl Microbiol 82(3):379-88
Parker C, Barnell WO, Snoep JL, Ingram LO, Conway T (1995) Characterization of the
Zymomonas mobilis glucose facilitator gene product (glf) in recombinant
Escherichia coli: examination of transport mechanism, kinetics and the role of
glucokinase in glucose transport. Mol Microbiol 15(5):795-802
Peijnenburg AA, Bron S, Venema G (1987) Structural plasmid instability in
recombination- and repair-deficient strains of Bacillus subtilis. Plasmid
17(2):167-70 doi:0147-619X(87)90023-0 [pii]
Pohl M (1997) Protein design on pyruvate decarboxylase (PDC) by site-directed
mutagenesis. Application to mechanistical investigations, and tailoring PDC for
the use in organic synthesis. Advances in biochemical engineering/biotechnology
58:15-43
Pontes MH, Dale C (2011) Lambda red-mediated genetic modification of the insect
endosymbiont Sodalis glossinidius. Appl Environ Microbiol 77(5):1918-20
doi:10.1128/AEM.02166-10 AEM.02166-10 [pii]
Poole RK (1994) Oxygen reactions with bacterial oxidases and globins: binding,
reduction and regulation. Antonie Van Leeuwenhoek 65(4):289-310
Posfai G, Plunkett G, 3rd, Feher T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V,
Stahl B, Sharma SS, de Arruda M, Burland V, Harcum SW, Blattner FR (2006)
Emergent properties of reduced-genome Escherichia coli. Science
312(5776):1044-6 doi:1126439 [pii] 10.1126/science.1126439
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA (2013)
Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control
of Gene Expression. Cell 152(5):1173-1183 doi:DOI 10.1016/j.cell.2013.02.022
Ranatunga TD, Jervis J, Helm RF, McMillan JD, Hatzis C (1997) Identification of
inhibitory components toxic toward Zymomonas mobilis CP4(pZB5) xylose
fermentation. Appl Biochem Biotech 67(3):185-198 doi:Doi 10.1007/Bf02788797
Raps S, Demoss RD (1962) Glycolytic enzymes in Zymomonas mobilis. J Bacteriol
84:115-8
Ren C, Chen T, Zhang J, Liang L, Lin Z (2009) An evolved xylose transporter from
Zymomonas mobilis enhances sugar transport in Escherichia coli. Microb Cell
Fact 8:66 doi:10.1186/1475-2859-8-66 1475-2859-8-66 [pii]
Reyes L, Scopes RK (1991) Membrane-associated ATPase from Zymomonas mobilis;
purification and characterization. Biochim Biophys Acta 1068(2):174-8 doi:0005-
2736(91)90207-O [pii]

147
Reynen M, Reipen I, Sahm H, Sprenger GA (1990) Construction of expression vectors
for the gram-negative bacterium Zymomonas mobilis. Mol Gen Genet 223(2):335-
41
Rogers PL, Jeon YJ, Lee KJ, Lawford HG (2007a) Zymomonas mobilis for Fuel Ethanol
and Higher Value Products. In: Olsson L (ed) Biofuels. Advances in Biochemical
Engineering/Biotechnology, vol 108. Springer Berlin Heidelberg, pp 263-288
Rogers PL, Jeon YJ, Lee KJ, Lawford HG (2007b) Zymomonas mobilis for fuel ethanol
and higher value products. Adv Biochem Eng Biotechnol 108:263-88
doi:10.1007/10_2007_060
Rogers PL, Lee KJ, Tribe DE (1979) Kinetics of Alcohol Production by Zymomonas
mobilis at High Sugar Concentrations. Biotechnology Letters 1(4):165-170
doi:Doi 10.1007/Bf01388142
Rogers PL, Shin HS, Wang B (1997) Biotransformation for L-ephedrine production. Adv
Biochem Eng Biotechnol 56:33-59
Rosenberg SM (2001) Evolving responsively: adaptive mutation. Nat Rev Genet
2(7):504-15 doi:10.1038/35080556 35080556 [pii]
Ruhrmann J, Kramer R (1992) Mechanism of glutamate uptake in Zymomonas mobilis. J
Bacteriol 174(23):7579-84
Runquist D, Fonseca C, Radstrom P, Spencer-Martins I, Hahn-Hagerdal B (2009a)
Expression of the Gxf1 transporter from Candida intermedia improves
fermentation performance in recombinant xylose-utilizing Saccharomyces
cerevisiae. Appl Microbiol Biotechnol 82(1):123-30 doi:10.1007/s00253-008-
1773-y
Saez LP, Garcia P, Martinez-Luque M, Klipp W, Blasco R, Castillo F (2001) Role for
draTG and rnf genes in reduction of 2,4-dinitrophenol by Rhodobacter
capsulatus. J Bacteriol 183(5):1780-3 doi:10.1128/JB.183.5.1780-1783.2001
Sahm H, Bringer-Meyer S, Sprenger GA (2006) The Genus Zymomonas. Prokaryotes: A
Handbook on the Biology of Bacteria, Vol 5, Third Edition:201-221 doi:Doi
10.1007/0-387-30745-1_10
Saini S, Pearl JA, Rao CV (2009) Role of FimW, FimY, and FimZ in regulating the
expression of type i fimbriae in Salmonella enterica serovar Typhimurium.
Journal of Bacteriology 191(9):3003-10 doi:10.1128/JB.01694-08 JB.01694-08
[pii]
Saloheimo A, Rauta J, Stasyk OV, Sibirny AA, Penttila M, Ruohonen L (2007) Xylose
transport studies with xylose-utilizing Saccharomyces cerevisiae strains
expressing heterologous and homologous permeases. Appl Microbiol Biot
74(5):1041-52 doi:10.1007/s00253-006-0747-1
Sander JD, Joung JK (2014) CRISPR-Cas systems for editing, regulating and targeting
genomes. Nat Biotechnol 32(4):347-55 doi:10.1038/nbt.2842 nbt.2842 [pii]
Schmehl M, Jahn A, Meyer zu Vilsendorf A, Hennecke S, Masepohl B, Schuppler M,
Marxer M, Oelze J, Klipp W (1993) Identification of a new class of nitrogen
fixation genes in Rhodobacter capsulatus: a putative membrane complex involved
in electron transport to nitrogenase. Mol Gen Genet 241(5-6):602-15
Schmidt W, Schugerl K (1987) Continuous Ethanol-Production by Zymomonas mobilis
on a Synthetic Medium. Chem Eng J Bioch Eng 36(3):B39-B48 doi:Doi
10.1016/0300-9467(87)80030-8

148
Schubert C (2006) Can biofuels finally take center stage? Nature Biotechnology
24(7):777-784 doi:Doi 10.1038/Nbt0706-777
Scordaki A, Drainas C (1987) Analysis of Natural Plasmids of Zymomonas mobilis Atcc-
10988. Journal of General Microbiology 133:2547-2556
Scordaki A, Drainas C (1990) Analysis and stability of Zymomonas mobilis ATCC 10988
plasmid pZMO3. Plasmid 23(1):59-66 doi:0147-619X(90)90044-D [pii]
Seatter MJ, De la Rue SA, Porter LM, Gould GW (1998) QLS motif in transmembrane
helix VII of the glucose transporter family interacts with the C-1 position of D-
glucose and is involved in substrate selection at the exofacial binding site.
Biochemistry 37(5):1322-6 doi:10.1021/bi972322u bi972322u [pii]
Senthilkumar V, Rajendhran J, Busby SJ, Gunasekaran P (2009) Characterization of
multiple promoters and transcript stability in the sacB-sacC gene cluster in
Zymomonas mobilis. Arch Microbiol 191(6):529-41 doi:10.1007/s00203-009-
0479-6
Senthilkumar V, Rameshkumar N, Busby SJ, Gunasekaran P (2004) Disruption of the
Zymomonas mobilis extracellular sucrase gene (sacC) improves levan production.
J Appl Microbiol 96(4):671-6 doi:2169 [pii]
Seo JS, Chong H, Park HS, Yoon KO, Jung C, Kim JJ, Hong JH, Kim H, Kim JH, Kil JI,
Park CJ, Oh HM, Lee JS, Jin SJ, Um HW, Lee HJ, Oh SJ, Kim JY, Kang HL, Lee
SY, Lee KJ, Kang HS (2005) The genome sequence of the ethanologenic
bacterium Zymomonas mobilis ZM4. Nat Biotechnol 23(1):63-8 doi:nbt1045 [pii]
10.1038/nbt1045
Sergueev K, Yu D, Austin S, Court D (2001) Cell toxicity caused by products of the p(L)
operon of bacteriophage lambda. Gene 272(1-2):227-35 doi:S0378111901005352
[pii]
Shamanna DK, Sanderson KE (1979) Uptake and catabolism of D-xylose in Salmonella
typhimurium LT2. J Bacteriol 139(1):64-70
Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C
(2013) Targeted genome modification of crop plants using a CRISPR-Cas system.
Nat Biotechnol 31(8):686-8 doi:10.1038/nbt.2650 nbt.2650 [pii]
Sharan SK, Thomason LC, Kuznetsov SG, Court DL (2009) Recombineering: a
homologous recombination-based method of genetic engineering. Nat Protoc
4(2):206-23 doi:10.1038/nprot.2008.227 nprot.2008.227 [pii]
Shuai L, Yang Q, Zhu JY, Lu FC, Weimer PJ, Ralph J, Pan XJ (2010) Comparative study
of SPORL and dilute-acid pretreatments of spruce for cellulosic ethanol
production. Bioresour Technol 101(9):3106-14
doi:10.1016/j.biortech.2009.12.044 S0960-8524(09)01709-X [pii]
Siegele DA, Hu JC (1997) Gene expression from plasmids containing the araBAD
promoter at subsaturating inducer concentrations represents mixed populations.
Proc Natl Acad Sci U S A 94(15):8168-72
Silbir S, Dagbagli S, Yegin S, Baysal T, Goksungur Y (2014) Levan production by
Zymomonas mobilis in batch and continuous fermentation systems. Carbohydr
Polym 99:454-61 doi:10.1016/j.carbpol.2013.08.031 S0144-8617(13)00815-1
[pii]
Skerker JM, Leon D, Price MN, Mar JS, Tarjan DR, Wetmore KM, Deutschbauer AM,
Baumohl JK, Bauer S, Ibanez AB, Mitchell VD, Wu CH, Hu P, Hazen T, Arkin

149
AP (2013) Dissecting a complex chemical stress: chemogenomic profiling of
plant hydrolysates. Mol Syst Biol 9:674 doi:10.1038/msb.2013.30 msb201330
[pii]
Skerker JM, Shapiro L (2000a) Identification and cell cycle control of a novel pilus
system in Caulobacter crescentus. Embo J 19(13):3223-34
doi:10.1093/emboj/19.13.3223
Skotnicki ML, Lee KJ, Tribe DE, Rogers PL (1981) Comparison of ethanol production
by different zymomonas strains. Appl Environ Microbiol 41(4):889-93
Skotnicki ML, Lee KJ, Tribe DE, Rogers PL (1982) Genetic alteration of Zymomonas
mobilis for ethanol production. Basic Life Sci 19:271-90
Skotnicki ML, Tribe DE, Rogers PL (1980) R-Plasmid Transfer in Zymomonas mobilis.
Appl Environ Microbiol 40(1):7-12
Song KB, Joo HK, Rhee SK (1993) Nucleotide sequence of levansucrase gene (levU) of
Zymomonas mobilis ZM1 (ATCC10988). Biochim Biophys Acta 1173(3):320-4
Song KB, Lee SK, Joo HK, Rhee SK (1994) Nucleotide and derived amino acid
sequences of an extracellular sucrase gene (invB) of Zymomonas mobilis ZM1
(ATCC10988). Biochim Biophys Acta 1219(1):163-6 doi:0167-4781(94)90262-3
[pii]
Sootsuwan K, Thanonkeo P, Keeratirakha N, Thanonkeo S, Jaisil P, Yamada M (2013)
Sorbitol required for cell growth and ethanol production by Zymomonas mobilis
under heat, ethanol, and osmotic stresses. Biotechnol Biofuels 6(1):180
doi:10.1186/1754-6834-6-180 1754-6834-6-180 [pii]
Sprenger GA (1993) Approaches to Broaden the Substrate and Product Range of the
Ethanologenic Bacterium Zymomonas mobilis by Genetic-Engineering. Journal of
Biotechnology 27(3):225-237 doi:Doi 10.1016/0168-1656(93)90087-4
Sprenger GA (1996) Carbohydrate metabolism in Zymomonas mobilis: A catabolic
highway with some scenic routes. Fems Microbiology Letters 145(3):301-307
Sprenger GA, Schorken U, Sprenger G, Sahm H (1995) Transaldolase B of Escherichia
coli K-12: cloning of its gene, talB, and characterization of the enzyme from
recombinant strains. Journal of Bacteriology 177(20):5930-6
Stanley GA, Hobley TJ, Pamment NB (1997) Effect of acetaldehyde on Saccharomyces
cerevisiae and Zymomonas mobilis subjected to environmental shocks. Biotechnol
Bioeng 53(1):71-8 doi:10.1002/(SICI)1097-0290(19970105)53:1<71::AID-
BIT10>3.0.CO;2-C
Strohdeicher M, Bringermeyer S, Neuss B, Vandermeer R, Duine JA, Sahm H (1989)
Glucose-Dehydrogenase from Zymomonas mobilis - Evidence for a Quinoprotein.
Pqq and Quinoproteins:103-105
Strohdeicher M, Neuss B, Bringermeyer S, Sahm H (1990) Electron-Transport Chain of
Zymomonas mobilis - Interaction with the Membrane-Bound Glucose-
Dehydrogenase and Identification of Ubiquinone-10. Archives of Microbiology
154(6):536-543
Strohdeicher M, Schmitz B, Bringermeyer S, Sahm H (1988) Formation and Degradation
of Gluconate by Zymomonas mobilis. Appl Microbiol Biot 27(4):378-382
Strzelecki AT, Goodman AE, Cail RG, Rogers PL (1990) Behavior of the hybrid plasmid
pNSW301 in Zymomonas mobilis grown in continuous culture. Plasmid
23(3):194-200

150
Strzelecki AT, Goodman AE, Rogers PL (1987) Behavior of the IncW plasmid Sa in
Zymomonas mobilis. Plasmid 18(1):46-53 doi:0147-619X(87)90077-1 [pii]
Subtil T, Boles E (2011) Improving L-arabinose utilization of pentose fermenting
Saccharomyces cerevisiae cells by heterologous expression of L-arabinose
transporting sugar transporters. Biotechnol Biofuels 4:38 doi:10.1186/1754-6834-
4-38
Sumiya M, Davis EO, Packman LC, McDonald TP, Henderson PJ (1995) Molecular
genetics of a receptor protein for D-xylose, encoded by the gene xylF, in
Escherichia coli. Receptors Channels 3(2):117-28
Swings J, De Ley J (1977) The biology of Zymomonas. Bacteriol Rev 41(1):1-46
Tanaka H, Ishikawa H, Osuga K, Takagi Y (1990) Fermentation Performance of
Zymomonas mobilis against Oxygen-Supply .1. Fermentative Ability of
Zymomonas mobilis under Various Oxygen-Supply Conditions in Batch Culture. J
Ferment Bioeng 69(4):234-239 doi:Doi 10.1016/0922-338x(90)90219-M
Toh H, Doelle H (1997a) Changes in the growth and enzyme level of Zymomonas mobilis
under oxygen-limited conditions at low glucose concentration. Arch Microbiol
168(1):46-52
Toh H, Doelle H (1997b) Changes in the growth and enzyme level of Zymomonas
mobilis under oxygen-limited conditions at low glucose concentration. Archives
of Microbiology 168(1):46-52 doi:DOI 10.1007/s002030050468
Tusnady GE, Simon I (1998) Principles governing amino acid composition of integral
membrane proteins: application to topology prediction. J Mol Biol 283(2):489-
506 doi:S0022-2836(98)92107-6 [pii] 10.1006/jmbi.1998.2107
Tusnady GE, Simon I (2001) The HMMTOP transmembrane topology prediction server.
Bioinformatics 17(9):849-50
Typas MA, Galani I (1992) Chemical and Uv Mutagenesis in Zymomonas mobilis.
Genetica 87(1):37-45 doi:Doi 10.1007/Bf00128771
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA (2007)
Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35(Web
Server issue):W71-4 doi:gkm306 [pii] 10.1093/nar/gkm306
van Kessel JC, Hatfull GF (2007) Recombineering in Mycobacterium tuberculosis. Nat
Methods 4(2):147-52 doi:nmeth996 [pii] 10.1038/nmeth996
Vartholomatos G, Typas MA, Drainas C (1993) An ultraviolet-sensitive mutant of
Zymomonas mobilis affecting the stability of its natural plasmid pZMO2. Plasmid
29(1):10-8 doi:S0147-619X(83)71002-4 [pii] 10.1006/plas.1993.1002
Viikari L, Gisler R (1986) By-Products in the Fermentation of Sucrose by Different
Zymomonas-Strains. Appl Microbiol Biot 23(3-4):240-244
Viret JF, Alonso JC (1987) Generation of linear multigenome-length plasmid molecules
in Bacillus subtilis. Nucleic Acids Res 15(16):6349-67
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013)
One-step generation of mice carrying mutations in multiple genes by
CRISPR/Cas-mediated genome engineering. Cell 153(4):910-8
doi:10.1016/j.cell.2013.04.025 S0092-8674(13)00467-4 [pii]
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM (2009)
Programming cells by multiplex genome engineering and accelerated evolution.
Nature 460(7257):894-8 doi:10.1038/nature08187 nature08187 [pii]

151
Wang L, Chen S, Vergin KL, Giovannoni SJ, Chan SW, DeMott MS, Taghizadeh K,
Cordero OX, Cutler M, Timberlake S, Alm EJ, Polz MF, Pinhassi J, Deng Z,
Dedon PC (2011) DNA phosphorothioation is widespread and quantized in
bacterial genomes. Proc Natl Acad Sci U S A 108(7):2963-8
doi:10.1073/pnas.1017261108 1017261108 [pii]
Wang Y, Pfeifer BA (2008) 6-deoxyerythronolide B production through chromosomal
localization of the deoxyerythronolide B synthase genes in E. coli. Metab Eng
10(1):33-8 doi:S1096-7176(07)00050-X [pii] 10.1016/j.ymben.2007.09.002
Wang Y, Weng J, Waseem R, Yin X, Zhang R, Shen Q (2012) Bacillus subtilis genome
editing using ssDNA with short homology regions. Nucleic Acids Res 40(12):e91
doi:10.1093/nar/gks248 gks248 [pii]
Wecker MS, Zall RR (1987) Production of Acetaldehyde by Zymomonas mobilis. Appl
Environ Microbiol 53(12):2815-20
Wei N, Quarterman J, Kim SR, Cate JHD, Jin YS (2013) Enhanced biofuel production
through coupled acetic acid and xylose consumption by engineered yeast. Nat
Commun 4 doi:Artn 2580 Doi 10.1038/Ncomms3580
Weisser P, Kramer R, Sprenger GA (1996a) Expression of the Escherichia coli pmi gene,
encoding phosphomannose-isomerase in Zymomonas mobilis, leads to utilization
of mannose as a novel growth substrate, which can be used as a selective marker.
Appl Environ Microbiol 62(11):4155-61
Wheals AE, Basso LC, Alves DMG, Amorim HV (1999) Fuel ethanol after 25 years.
Trends Biotechnol 17(12):482-487 doi:Doi 10.1016/S0167-7799(99)01384-0
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems
in bacteria and archaea. Nature 482(7385):331-8 doi:10.1038/nature10886
nature10886 [pii]
Wyman CE (2001) Twenty years of trials, tribulations, and research progress in
bioethanol technology: selected key events along the way. Appl Biochem
Biotechnol 91-93:5-21
Yamamoto S, Izumiya H, Morita M, Arakawa E, Watanabe H (2009) Application of
lambda Red recombination system to Vibrio cholerae genetics: simple methods
for inactivation and modification of chromosomal genes. Gene 438(1-2):57-64
doi:10.1016/j.gene.2009.02.015 S0378-1119(09)00107-3 [pii]
Yanase H, Kurii J, Tonomura K (1988) Fermentation of Lactose by Zymomonas mobilis
Carrying a Lac+ Recombinant Plasmid. Journal of Fermentation Technology
66(4):409-415 doi:Doi 10.1016/0385-6380(88)90007-6
Yang S, Land ML, Klingeman DM, Pelletier DA, Lu TY, Martin SL, Guo HB, Smith JC,
Brown SD (2010a) Paradigm for industrial strain improvement identifies sodium
acetate tolerance loci in Zymomonas mobilis and Saccharomyces cerevisiae. Proc
Natl Acad Sci U S A 107(23):10395-400 doi:10.1073/pnas.0914506107
0914506107 [pii]
Young E, Poucher A, Comer A, Bailey A, Alper H (2011) Functional survey for
heterologous sugar transport proteins, using Saccharomyces cerevisiae as a host.
Appl Environ Microbiol 77(10):3311-9 doi:10.1128/AEM.02651-10
Young EM, Comer AD, Huang H, Alper HS (2012a) A molecular transporter engineering
approach to improving xylose catabolism in Saccharomyces cerevisiae. Metab
Eng 14(4):401-11 doi:10.1016/j.ymben.2012.03.004

152
Young EM, Tong A, Bui H, Spofford C, Alper HS (2014a) Rewiring yeast sugar
transporter preference through modifying a conserved protein motif. Proc Natl
Acad Sci U S A 111(1):131-6 doi:10.1073/pnas.1311970111
Zaccolo M, Gherardi E (1999) The effect of high-frequency random mutagenesis on in
vitro protein evolution: a study on TEM-1 beta-lactamase. J Mol Biol 285(2):775-
83 doi:S0022-2836(98)92262-8 [pii] 10.1006/jmbi.1998.2262
Zhang M, Eddy C, Deanda K, Finkelstein M, Picataggio S (1995) Metabolic Engineering
of a Pentose Metabolism Pathway in Ethanologenic Zymomonas mobilis. Science
267(5195):240-3 doi:267/5195/240 [pii] 10.1126/science.267.5195.240
Zhang X, Chen G, Liu W (2009) Reduction of xylose to xylitol catalyzed by glucose-
fructose oxidoreductase from Zymomonas mobilis. FEMS Microbiol Lett
293(2):214-9 doi:10.1111/j.1574-6968.2009.01529.x FML1529 [pii]
Zhang Y, Buchholz F, Muyrers JP, Stewart AF (1998) A new logic for DNA engineering
using recombination in Escherichia coli. Nat Genet 20(2):123-8 doi:10.1038/2417
Zikmanis P, Kruce R, Auzina L (1997) An elevation of the molar growth yield of
Zymomonas mobilis during aerobic exponential growth. Arch Microbiol
167(2/3):167-71 doi:71670167.203 [pii]
Zikmanis P, Kruce R, Auzina L (1999) Molar growth yields of Zymomonas mobilis on
glucose after the transition from anaerobic to aerobic continuous growth. Acta
Biotechnol 19(1):69-75 doi:DOI 10.1002/abio.370190111
Zou SL, Hong LF, Wang C, Jing X, Zhang MH (2012) Construction of an Unmarked
Zymomonas mobilis Mutant Using a Site-Specific FLP Recombinase. Food
Technol Biotech 50(4):406-411


















